Wilate 900 Ui/Ml/ 800 Ui Fvw, Polvo Y Disolvente Para Solucion Inyectable
Información obsoleta, busque otroDE SANIDAD, POLÍTICA SOCIAL E IGUALDAD
|
I | |
|
Oí |
k agencia española de 1 medicamentos y I productos sanitarios |
NOMBRE DEL MEDICAMENTO
Wilate, 450 UI FVIII/400 UI FvW, polvo y disolvente para solución inyectable Wilate, 900 UI FVIII800 UI FvW, polvo y disolvente para solución inyectable
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Wilate se presenta como polvo y disolvente para solución inyectable, con un contenido nominal de 450 UI/900 UI de factor VIII de coagulación humano y 400 UI/800 UI de factor de von Willebrand humano (FvW) por vial.
El producto contiene aproximadamente 80 UI/ml de factor de von Willebrand humano cuando se reconstituye con 5 ml/10 ml de agua para preparaciones inyectables con un 0,1 % de polisorbato 80.
La actividad específica de Wilate es aproximadamente > 53 UI FvW:RCo/mg proteína.
La potencia del FVW (UI) se mide teniendo en cuenta la actividad del cofactor de ristocetina (FvW:RCo) comparado con el Estándar Internacional del Concentrado del Factor de von Willebrand (OMS).
El producto contiene aproximadamente 90 UI/ml de factor VIII de coagulación humano cuando se reconstituye con 5 ml/10 ml de agua para preparaciones inyectables con un 0,1% de polisorbato 80.
La potencia del FVIII (UI) se determina usando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de Wilate es aproximadamente > 60 UI FVIII:C/mg proteína.
Para consultar la lista completa de excipientes ver sección 6.1.
FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
DATOS CLÍNICOS
Indicaciones terapéuticas
Enfermedad de von Willebrand (EvW)
Tratamiento y profilaxis de la hemorragia o sangrados en cirugías en pacientes con EvW, cuando el tratamiento con desmopresina (DDAVP) es ineficaz o está contraindicado.
Correo electronicoI
C/ CAMPEZO, 1 - EDIFICIO 8 28022 MADRID
Se atenderán exclusivamente incidencias informáticas sobre la aplicación CIMA (http://www.aemps.gob.es/cima)

Hemofilia A
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de FVIII).
Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico especialista en el tratamiento de trastornos de la coagulación. El producto es de un solo uso y hay que administrar todo el contenido del vial. En caso de que quede algo de contenido, éste debe eliminarse de acuerdo con las normativas locales.
Enfermedad de von Willebrand (EvW)
La relación entre FVIII:C y FvW:RCo es aproximadamente 1:1. Generalmente, 1 UI/kg PC FVIII:C y FvW:RCo aumenta el nivel plasmático 1,5-2% de la actividad normal de la proteína respectiva. Normalmente, se necesita alrededor de 20 a 50 UI Wilate/kg PC para alcanzar una hemostasis adecuada. Esto aumentará el FVIII:C y FvW:RCo en los pacientes aproximadamente de un 30 a un 100%.
Puede requerirse una dosis inicial de 50 a 80 UI Wilate/kg PC, especialmente en pacientes con EvW tipo 3, en los que para mantener los niveles adecuados de plasma pueden ser necesarias dosis más altas que para otros tipos de EvW.
Prevención de la hemorragia en caso de cirugía o trauma grave:
Para la prevención de la hemorragia en caso de cirugía debe administrarse Wilate 1-2 horas antes de comenzar el procedimiento quirúrgico. Deben alcanzarse niveles de FvW:RCo > 60 UI/dl (> 60%) y los niveles de FVIII:C > 40 UI/dl (> 40%).
Hay que volver a administrar una dosis adecuada cada 12-24 horas de tratamiento. La dosis y la duración del tratamiento depende del estado clínico del paciente, el tipo y la gravedad de la hemorragia, así como de los niveles de FVIII:C y FvW:RCo.
En pacientes tratados con productos de FvW que contienen FVIII, hay que monitorizar los niveles plasmáticos de FVIII:C para revelar los niveles plasmáticos excesivos y sostenidos de FVIII:C, los cuales pueden incrementar el riesgo de episodios trombóticos, especialmente en pacientes con factores de riesgo conocidos clínicos o de laboratorio. En caso de observarse niveles plasmáticos excesivos de FVIII:C, hay que considerar la reducción de las dosis y/o prolongar el intervalo de administración o el uso de un producto de FvW que contenga un nivel bajo de FVIII.
Profilaxis:
Para una profilaxis antihemorrágica a largo plazo en pacientes EvW, deben administrarse dosis de 20-40 UI/kg de peso corporal 2 ó 3 veces por semana. En algunos casos, como en pacientes con sangrado gastrointestinal, pueden ser necesarias dosis más altas.
Hemofilia A
La dosis y la duración de la terapia de sustitución dependen de la gravedad de la deficiencia de FVIII, de la localización y de la intensidad de la hemorragia, así como del estado clínico del paciente.
El número de unidades de FVIII administradas se expresa en Unidades Internacionales (UI), que están relacionadas con el estándar actual de la OMS para productos de FVIII. La actividad del FVIII en plasma se expresa en porcentaje (respecto al plasma humano normal) ó en UI (respecto a un Estándar Internacional para el FVIII en plasma).
Una UI de actividad del FVIII es equivalente a esa cantidad de FVIII en un mililitro de plasma humano normal.
El cálculo de la dosis requerida de FVIII se basa en la observación empírica de que 1 UI FVIII:C/kg PC aumenta el nivel plasmático en un 1,5-2% de la actividad normal. La dosis requerida se determina utilizando la siguiente fórmula:
UI requeridas = PC (kg) x aumento deseado del FVIII (%) (UI/dl) x 0,5 Ul/kg
La cantidad a administrar y la frecuencia de administración debe estar siempre orientada a la eficacia clínica en cada caso individual. En caso de los siguientes episodios hemorrágicos, la actividad del factor VIII no debe caer por debajo del nivel de la actividad plasmática dada (en % del plasma normal o en UI/dl) en el periodo correspondiente.
Se puede utilizar la siguiente tabla como guía de dosificación para episodios hemorrágicos y cirugía.
Pautas de tratamiento en hemorragias y cirugía
|
Grado de hemorragia/ Tipo de procedimiento quirúrgico |
Nivel de FVIII requerido (%) (UI/dl) |
Frecuencia de administración/Duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis temprana, hemorragia muscular u oral., |
20 - 40 |
Repetir cada 12 a 24 horas. Al menos 1 día, hasta que el episodio hemorrágico, como indica el dolor se resuelva o se consiga la cicatrización |
|
Hemartrosis más extensa, hemorragia muscular o hematoma |
30 - 60 |
Repetir la perfusión cada 12 a 24 horas durante 3 ó 4 días ó más hasta que el dolor y la discapacidad se hayan resuelto. |
|
Hemorragia amenazante para la vida |
60 - 100 |
Repetir la perfusión cada 8 a 24 horas hasta que remita el peligro. |
|
Cirugía | ||
|
Menor incluyendo extracción dental |
30 - 60 |
Cada 24 horas, al menos 1 día, hasta que se consiga la cicatrización. |
|
Mayor |
80 - 100 |
Repetir la perfusión cada 8 a 24 horas hasta que la herida |
|
Grado de hemorragia/ Tipo de procedimiento quirúrgico |
Nivel de FVIII requerido (%) (UI/dl) |
Frecuencia de administración/Duración de la terapia (días) |
|
(pre- y postoperatorio) |
cicatrice adecuadamente, después continuar la terapia al menos durante otros 7 días para mantener la actividad del FVIII del 30% al 60% (UI/dl). |
Profilaxis:
Para la profilaxis a largo plazo frente a las hemorragias en pacientes con hemofilia A grave, deben administrarse dosis de 20 a 40 UI Wilate/kg PC a intervalos de 2 a 3 días. En algunos casos, especialmente en pacientes más jóvenes, pueden ser necesarios intervalos de administración más cortos o dosis más elevadas.
Perfusión continua:
Antes de una operación quirúrgica, hay que realizar un análisis farmacocinético para obtener una estimación del aclaramiento. La velocidad inicial de perfusión puede calcularse de la siguiente manera:
Velocidad de perfusión (Ul/kg/h) = aclaramiento (mL/kg/h) x nivel deseado en estado estacionario (UI/mL)
Después de las primeras 24 horas de perfusión continua, debe calcularse de nuevo el aclaramiento cada día usando la ecuación del estado estacionario con el nivel que se ha determinado y la velocidad de perfusión conocida.
Durante el curso del tratamiento, se recomienda una determinación adecuada de los niveles de FVIII:C para poder orientar la dosis a administrar y la frecuencia de las perfusiones repetidas. En el caso particular de intervenciones quirúrgicas mayores, es indispensable una monitorización precisa de la terapia de sustitución mediante pruebas de coagulación (FVIII:C). Puede variar la respuesta individual de los pacientes al tratamiento con FVIII, consiguiendo diferentes niveles de recuperación in vivo y demostrando diferente semivida.
Hay que monitorizar el desarrollo de anticuerpos neutralizantes del FVIII en los pacientes (inhibidores). Si no se consiguen los niveles esperados de actividad plasmática de FVIII, o si no se controla la hemorragia con una dosis adecuada, se realizará un análisis para determinar la posible presencia de un inhibidor del FVIII. En pacientes con niveles elevados de inhibidor, la terapia con FVIII puede no ser eficaz y habrá que considerar otras opciones terapéuticas. El tratamiento de esos pacientes debe estar dirigido por un médico especialista en atención a pacientes con alteraciones hemostáticas. Ver también 4.4.

Los datos para recomendar el uso de Wilate en niños menores de 6 años son insuficientes. Forma de administración
Para administración intravenosa tras la reconstitución con el disolvente que le acompaña. Ver 6.6.
La velocidad de inyección o de perfusión no debe superar los 2-3 ml por minuto.
Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes.
Advertencias y precauciones especiales de empleo
Al igual que con cualquier producto proteico derivado del plasma para administración intravenosa, es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico. Hay que monitorizar estrechamente a los pacientes y observar cuidadosamente cualquier síntoma durante todo el periodo de perfusión.
Se informará a los pacientes de los primeros signos de las reacciones de hipersensibilidad que incluyen erupciones cutáneas, urticaria generalizada, opresión en el pecho, dificultad para respirar, hipotensión y anafilaxis. Si aparecen estos síntomas, los pacientes deben interrumpir la administración inmediatamente y ponerse en contacto con su médico.
En caso de shock, se aplicarán los estándares médicos vigentes para el tratamiento del shock.
Para prevenir la transmisión de enfermedades infecciosas cuando se administran medicamentos derivados de la sangre o plasma humanos, se toman medidas estándar como la selección de los donantes, cribaje de las donaciones individuales y mezclas de plasma para marcadores específicos de infección, así como la inclusión de procedimientos efectivos para la inactivación/eliminación viral en el proceso de producción. A pesar de ello, cuando se administran medicamentos derivados de sangre o plasma humano, no se puede descartar por completo la posibilidad de transmisión de enfermedades infecciosas. Esto también es aplicable a virus desconocidos o emergentes y otros patógenos.
Estos procedimientos se consideran efectivos para virus envueltos como VIH, VHB y VHC, así como para el virus sin cubierta VHA. Estos procedimientos pueden tener un valor limitado frente a los virus sin cubierta tales como parvovirus B19.
La infección por parvovirus B19 puede resultar seria en mujeres embarazadas (infección fetal) y en pacientes con inmunodeficiencia o con eritropoyesis incrementada (p.e. en anemia hemolítica).
Es altamente recomendable que cada vez que se administre Wilate se registre el nombre y el número de lote del producto con objeto de mantener una trazabilidad entre el paciente y el lote del producto.
Se deberá considerar una vacunación adecuada (hepatitis A y B) para los pacientes que estén recibiendo de forma regular/repetida concentrados de factor VIII/FvW derivado de plasma.
Enfermedad de von Willebrand (EvW)
Cuando se usa un producto de FvW que contiene FVIII, hay que advertir al médico que el tratamiento continuado puede producir un aumento excesivo de FVIII:C. En pacientes tratados con productos FvW que contienen FVIII, hay que monitorizar los niveles plasmáticos de FVIII:C para evitar niveles excesivos y sostenidos de FVIII: C, que pueden incrementar el riesgo de episodios trombóticos.
Existe un riesgo de aparición de episodios trombóticos cuando se administran productos FvW que contienen FVIII, en especial en pacientes con factores de riesgo conocidos clínicos o de laboratorio. Por tanto, hay que monitorizar a los pacientes de riesgo para observar la presencia de cualquier signo precoz de trombosis. Debe instituirse una profilaxis contra la tromboembolia venosa, de acuerdo con las recomendaciones actuales.
Los pacientes con EvW, especialmente los pacientes tipo 3, pueden desarrollar anticuerpos neutralizantes (inhibidores) del FvW. Si no se alcanzan los niveles previstos de actividad plasmática de FvW:Rco, o si no se controla la hemorragia con una dosis adecuada, habrá que llevar a cabo un análisis adecuado para determinar la posible presencia de un inhibidor FvW. En pacientes con niveles elevados de inhibidor, la terapia FvW puede no ser eficaz y hay que considerar otras opciones terapéuticas. El tratamiento de estos pacientes debe estar dirigido por un médico con experiencia en el cuidado de pacientes con alteraciones hemostáticas.
Hemofilia A
La formación de anticuerpos neutralizantes (inhibidores) del FVIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores suelen ser inmunoglobulinas IgG dirigidas contra la actividad procoagulante del FVIII, que se cuantifica en Unidades Bethesda Modificadas (UB) por mL de plasma utilizando el análisis modificado. El riesgo de desarrollar inhibidores está relacionado con la exposición a FVIII antihemofílico, siendo el mayor riesgo dentro de los 20 primeros días de exposición. En raras ocasiones los inhibidores pueden desarrollarse después de los 100 primeros días de exposición. Los pacientes tratados con FVIII deben ser cuidadosamente monitorizados en cuanto al desarrollo de inhibidores mediante las observaciones clínicas y las pruebas de laboratorio adecuadas. (Ver también sección 4.8.)
Se han observado casos de inhibidores recurrentes (de bajo título) tras el cambio deun producto con factor VIII a otro, en pacientes previamente tratados con más de 100 días de exposición con una historia clínica previa de desarrollo de inhibidores. Por tanto, se recomienda monitorizar a los pacientes cuidadosamente para la aparición de inhibidores tras cualquier cambio de producto.
Este medicamento contiene hasta 2,55 mmol de sodio (58,7 mg) por dosis para 450 UI FVIII y 400 UI FvW/vial y hasta 5,1 mmol sodio (117,3 mg) por dosis para 900 UI FVIII y 800 UI FvW/vial. Lo que deberá tenerse en cuenta en el tratamiento de pacientes con dietas pobres en sodio.
Interacción con otros medicamentos y otras formas de interacción
No se conocen interacciones con otros medicamentos.
Embarazo y lactancia

No se han llevado a cabo estudios de reproducción en animales con FVIII/FvW.
Enfermedad de von Willebrand (EvW)
No se dispone de experiencia en el tratamiento de mujeres embarazadas o en periodo de lactancia.
Wilate solo debe administrarse a mujeres embarazadas o en periodo de lactancia con deficiencia de FvW si está claramente indicado, teniendo en cuenta que el parto aumenta el riesgo de episodios hemorrágicos en estas pacientes.
Hemofilia A
Debido a la escasa frecuencia de hemofilia A en mujeres, no se dispone de experiencia sobre el tratamiento durante el embarazo y la lactancia. Por tanto, Wilate solo debe utilizarse durante el embarazo y la lactancia si está claramente indicado.
Efectos sobre la capacidad para conducir y utilizar máquinas
No se han observado efectos sobre la capacidad para conducir o utilizar máquinas.
Reacciones adversas
De forma poco frecuente se han observado reacciones de hipersensibilidad o alérgicas (que pueden incluir angioedema, sensación de irritación y escozor en el punto de la perfusión, escalofríos, rubor, urticaria generalizada, cefaleas, eczemas, hipotensión, letargia, náuseas, cansancio, taquicardia, opresión en el pecho, hormigueo, vómitos y disnea), y en algunos casos pueden progresar a anafilaxis severa (incluyendo shock). En raras ocasiones, se ha observado fiebre.
|
Clasificación de oréanos del sistema |
Poco frecuentes |
Raras |
Muy raras |
|
Trastornos del sistema inmunológico |
reacción de hipersensibilidad |
shock anafiláctico | |
|
Trastornos generales y alteraciones en el lugar de administración |
fiebre | ||
|
Investigaciones |
inhibidores del FVIII |
inhibidores del FvW |
Poco frecuentes (> 1/1.000, < 1/100) raras (>1/10.000, <1/1.000)

muy raras (<1/10.000), incluyendo informes aislados
Enfermedad de von Willebrand (EvW)
Los pacientes con EvW, especialmente los pacientes tipo 3, en muy raras ocasiones pueden desarrollar anticuerpos neutralizantes del FvW. Si aparecen estos inhibidores, la enfermedad se manifestará en si misma como una respuesta clínica inadecuada. Tales anticuerpos pueden precipitar y puede ocurrir de forma concomitante a las reacciones anafilácticas. Por tanto, debe evaluarse la presencia de un inhibidor en aquellos pacientes que sufran reacciones anafilácticas.
En todos esos casos, se recomienda contactar con un centro especializado en hemofilia.
No se han notificado casos de inhibodores para el factor von Willebrand en los ensayos clínicos ni tras su comercialización.
Existe riesgo de aparición de episodios trombóticos, en particular en pacientes con factores de riesgo conocidos clínicos o de laboratorio. Por tanto, los pacientes de riesgo deben ser monitorizados para observar los primeros signos de trombosis. Debe instituirse una profilaxis frente a la tromboembolia venosa, de acuerdo con las recomendaciones actuales.
En pacientes tratados con productos FvW que contienen FVIII los niveles plasmáticos excesivos y sostenidos de FVIII:C pueden incrementar el riesgo de episodios trombóticos..
Hemofilia A
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) del FVIII. La presencia de tales inhibidores se manifestará como una respuesta clínica insuficiente. En ese caso, se recomienda contactar con un centro especializado en hemofilia.
La experiencia con Wilate en pacientes no tratados previamente (PnTP) es limitada. En un ensayo clínico con24 PnTP con un mínimo de 50 días de exposición tratados con Wilate, solo tres pacientes con un inhibidor mayor a 5 UB/ml persistente y clínicamente manifiesto fueron detectados. Tres pacientes desarrollaron bajos títulos sin ninguna manifestación clínica y dos pacientes tuvieron un bajo título de inhibidor en una sola ocasión sin resultados en el seguimiento.
Ver también la sección 4.2. No se observaron desarrollo de inhibidores en los pacientes tratados previamente.
Para información sobre seguridad frente a agentes transmisibles, ver 4.4
Sobredosis
No se han notificado casos de sobredosis con FVIII ó FvW humanos. En caso de sobredosis mayor pueden producirse episodios tromboembólicos.
PROPIEDADES FARMACOLÓGICAS
Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antihemorrágicos: factores de coagulación: factor de Von Willebrand y factor VIII en combinación
Código ATC: B02BD06
Enfermedad de von Willebrand (EvW)
El FvW (del concentrado) es un componente normal del plasma humano y se comporta de la misma manera que el FvW endógeno.
La administración de FvW permite la corrección de los trastornos hemostáticos presentes en pacientes que sufren deficiencia de FvW (EvW) a dos niveles:
- El FvW restablece la adhesión plaquetaria al subendotelio vascular en el lugar del daño vascular (ya que se une tanto al subendotelio como a la membrana plaquetaria), proporcionando hemostasis primaria como muestra el acortamiento del tiempo de sangrado. Este efecto se produce inmediatamente y se sabe que depende en gran medida del nivel de polimerización de la proteína;
- FvW produce una corrección retardada de la deficiencia asociada al FVIII. El FvW administrado por vía intravenosa se une al FVIII endógeno (que normalmente es producido por el paciente) y estabilizando este factor, evita su rápida degradación. Debido a esto, la administración de FvW puro (producto FvW con un bajo nivel de FVIIIl) restablece el nivel de FVIII:C a la normalidad como un efecto secundario tras la primera perfusión.
La administración de una preparación de FvW que contiene FVIII restablece el nivel FVIII:C a la normalidad inmediatamente después de la primera perfusión.
Además de este papel como una proteína protectora del FVIII, el FvW media la adhesión plaquetaria en las zonas del daño vascular y juega un papel en la agregación plaquetaria.
Hemofilia A
El complejo FVIII/FvW consiste en dos moléculas (FVIII y FvW) con diferentes funciones fisiológicas. Cuando se administra a un paciente hemofílico, el FVIII se une al FvW en la circulación del paciente. El FVIII activado (FVIIIa) actúa como un cofactor del factor IX activado (FIXa), acelerando la conversión del factor X a factor X activado (FXa). El FXa convierte la protrombina a trombina. Luego la trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo.
La hemofilia A es un trastorno de la coagulación hereditario ligado al sexo debido a bajos niveles de FVIII:C y da como resultado un sangrado excesivo en las articulaciones, los músculos ó los órganos internos, producido de forma expontánea o como resultado de un trauma accidental o quirúrgico. Los niveles plasmáticos de FVIII son restituidos con la terapia de sustitución, consiguiéndose así corregir temporalmente la deficiencia del factor y las tendencias hemorrágicas.
Propiedades farmacocinéticas

Enfermedad de von Willebrand (EvW)
FvW (del concentrado) es un componente normal del plasma humano y actúa como el FvW endógeno.
Se observaron los siguientes resultados basados en el metanálisis de tres estudios farmacocinéticos que incluían a 24 pacientes evaluables con EvW de todos los tipos.
|
Todos los tipos EvW |
EvW tipo 1 |
EvW tipo 2 |
EvW tipo 3 | |||||||||||||||||
|
Parametro |
N |
Media |
SD |
Min. |
Max. |
N |
Media |
SD |
Min. |
Max. |
N |
Media |
SD |
Min. |
Max. |
N |
Media |
SD |
Min. |
Max. |
|
Recuperación(%/IU/kg) |
24 |
1.56 |
0.48 |
0.90 |
2.93 |
2 |
1.19 |
0.07 |
1.14 |
1.24 |
5 |
1.83 |
0.86 |
0.98 |
2.93 |
17 |
1.52 |
0.32 |
0.90 |
2.24 |
|
AUC (0-inf) (h*%) |
23 |
1981 |
960 |
593 |
4831 |
2 |
2062 |
510 |
1701 |
2423 |
5 |
2971 |
1383 |
1511 |
4831 |
16 |
1662 |
622 |
593 |
2606 |
|
Semivida (h) |
24 |
23.3 |
12.6 |
7.4 |
58.4 |
2 |
39.7 |
18.3 |
26.7 |
52.7 |
5 |
34.9 |
16 |
17.5 |
58.4 |
17 |
18 |
6.2 |
7.4 |
30.5 |
|
MRT (h) |
24 |
33.1 |
19 |
10.1 |
89.7 |
2 |
53.6 |
25.9 |
35.3 |
71.9 |
5 |
53.5 |
24.6 |
27.8 |
89.7 |
17 |
24.7 |
8.5 |
10.1 |
37.7 |
|
Aclaramiento (mL/h/kg) |
24 |
3.29 |
1.67 |
0.91 |
7.41 |
2 |
2.66 |
0.85 |
2.06 |
3.27 |
5 |
1.95 |
1.02 |
0.91 |
3.31 |
17 |
3.76 |
1.69 |
1.83 |
7.41 |
Clave: AUC = área bajo la curva; MRT = tiempo medio de residencia Hemofilia A
El FVIII (del concentrado) es un componente normal del plasma humano y actúa como el FVIII endógeno. Tras la inyección del producto, aproximadamente entre dos terceras partes y tres cuartas partes del FVIII permanecen en la circulación. El nivel de FVIII:C alcanzado en el plasma debe estar entre un 80-120% del FVIII:C previsto.
FVIIFC disminuye de modo exponencial en dos fases. En las fase inicial, la distribución entre el compartimiento intravascular y otros compartimentos (fluidos corporales) se produce con una semivida de eliminación del plasma de 3 a 6 horas. En la siguiente fase más lenta, la semivida varía entre 8 a 18 horas, con una media de 15 horas, Esto se corresponde con la semivida biológica.
Se observaron los siguientes resultados en un estudio clínico en 12 pacientes (ensayo cromogénico, doble medición):
|
Parametro |
Visita basal |
Visita a los 6 meses | ||
|
Media |
SD |
Media |
SD | |
|
Recuperación %/IU/kg |
FVIII:C 2.27 |
1.20 |
FVIII:C 2.26 |
1.19 |
|
AUC norm % * h/IU/kg |
FVIII:C 31.3 |
7.31 |
FVIII:C 33.8 |
10.9 |
|
Semivida (h) |
FVIII:C 11.2 |
2.85 |
FVIII:C 11.8 |
3.37 |
|
MRT (h) |
FVIII:C 15.3 |
3.5 |
FVIII:C 16.3 |
4.6 |
|
Aclarameinto mL/h/kg |
FVIII:C 3.37 |
0.86 |
FVIII:C 3.24 |
1.04 |

Clave: AUC = área bajo la curva; MRT = tiempo medio de residencia; SD = desviación estándar
Datos preclínicos sobre seguridad
El FVIII y el FvW en Wilate son componentes normales del plasma humano y actúan como el FVIII/FvW endógeno.
Los análisis convencionales de seguridad de este tipo de compuesos en animales de laboratorio no aportan información útil a la experiencia clínica, y por tanto, no son requeridos.
DATOS FARMACÉUTICOS
Lista de excipientes
Polvo: cloruro de sodio, glicina, sacarosa, citrato de sodio y cloruro de calcio Disolvente: agua para preparaciones inyectables con un 0,1 % de polisorbato 80
Incompatibilidades
Wilate no debe mezclarse con otros medicamentos ni administrarse simultáneamente con otros preparados intravenosos en el mismo equipo de perfusión.
Solo debe utilizarse el equipo de inyección/perfusión que se proporciona ya que puede fracasar el tratamiento como consecuencia de la adsorción de FVIII/FvW en la superficie interna de algunos equipos de perfusión.
Periodo de validez
3 años
Se ha demostrado que la solución reconstituida tiene una estabilidad de 12 horas a temperatura ambiente (max. +25°C). No obstante, para evitar la contaminación microbiológica, la solución reconstituida debe utilizarse inmediatamente.
Precauciones especiales de conservación

Conservar el polvo y el vial del disolvente en nevera (+2-8°C). Conservar los viales en el embalaje exterior para protegerlos de la luz. No congelar.
El producto puede conservarse a temperatura ambiente (max. +25°C) durante 2 meses. En este caso el periodo de validez finaliza 2 meses después de que el producto ha sido sacado de la nevera por primera vez. El paciente debe anotar el nuevo periodo de validez en el embalaje exterior. La solución reconstituida debe utilizarse solo en una ocasión. Se eliminará la solución que sobra.
Naturaleza y contenido del envase
Tamaños de envase:
Wilate, 450 UI FVIII y 400 UI FVW 1 caja contiene:
1 vial de polvo, de vidrio tipo I, cerrado con un tapón (bromobutilo) y sellado con una cápsula
flip off.
1 vial con disolvente (5 ml agua para preparaciones inyectables con un 0,1% polisorbato 80), vidrio tipo I, cerrado con un tapón (clorobutilo) y sellado con una cápsula flip off
1 caja con los dispositivos de administración (1 jeringa desechable, 1 equipo de transferencia [1 aguja de doble punta y 1 aguja con filtro], 1 equipo de perfusión)
2 apósitos con alcohol
Wilate, 900 UI FVIII y 800 UI FvW 1 caja contiene:
1 vial de polvo, de vidrio tipo I, cerrado con un tapón (bromobutilo) y sellado con una cápsula flip off.
1 vial con disolvente (10ml de agua para preparaciones inyectables con un 0,1% polisorbato 80), vidrio tipo I, cerrado con un tapón (clorobutilo) y sellado con una cápsula flip off
1 caja con los dispositivos de administración (1 jeringa desechable, 1 equipo de transferencia [1 aguja de doble punta y 1 aguja con filtro], 1 equipo de perfusión)
2 apósitos con alcohol
6.6 Precauciones especiales de eliminación y otras manipulaciones
1. Atemperar el disolvente y el polvo en los viales cerrados hasta alcanzar la temperatura ambiente. Si se utiliza un baño de agua para atemperar, debe evitarse que el agua entre en contacto con los tapones de goma (no contienen látex) o las cápsulas de los viales. La temperatura del baño de agua no debe ser superior a +37°C.
2. Quitar las cápsulas del vial de polvo y del vial de disolvente (Fig. A) y desinfectar los tapones de goma con un apósito con alcohol.
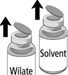
- . A
3. Colocar el vial de disolvente sobre una superficie plana. Introducir la aguja de doble punta con el extremo ondulado en el vial de disolvente (“Wave to Water”) y presionar lo máximo posible (Fig. B).

4. Colocar el vial de concentrado sobre una superficie plana. Retirar la cubierta protectora de la aguja de doble punta, asegurándose de que no toque la punta expuesta de la aguja. Mantener el vial de disolvente con la aguja de doble punta en posición vertical y perforar deprisa el centro del tapón de goma del vial de concentrado con la aguja y presionar lo máximo posible (Fig. C). El vacío en el interior del vial de concentrado extrae el disolvente.

5. Retirar el vial del disolvente y la aguja de doble punta del vial de polvo (Fig. D). Wilate se disuelve muy deprisa por tanto solo hay que girar muy despacio el vial.

La solución es límpida o ligeramente opalescente. No deben utilizarse soluciones turbias o con precipitados.
Inyección:
1. Quitar la cubierta protectora del filtro y perforar el tapón de goma del vial de concentrado (Fig. E).
2. Tirar del pistón de la jeringa para extraer el aire.
3. Extraer el cierre del filtro y acoplar la jeringa al filtro (Fig. Fa).
4. Inyectar el aire dentro del vial (Fig. Fb).

5. Poner el vial con la jeringa acoplada en posición vertical y extraer la solución al interior de la jeringa (Fig. G).

6. Retirar la jeringa del filtro.
7. Limpiar la zona donde se va a poner la inyección con un apósito con alcohol.
8. Acoplar la mariposa a la jeringa (Fig. H) e inyectar inmediatamente la preparación por vía intravenosa. Velocidad de inyección: 2-3 ml por minuto.
*JL '
Jt _ü_l
9. Si el paciente recibe más de un vial de concentrado, puede utilizarse la misma mariposa. También puede utilizarse la jeringa para varios viales de concentrado. Cada vez que se extrae la solución hay que utilizar un filtro nuevo.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Octapharma S.A.
Parq. Em. S. Fernando, Avda. Castilla 2 28830 San Fernando de Henares (Madrid), España
NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Agosto 2009
FECHA DE LA REVISIÓN DEL TEXTO
Agencia española de
medicamentos y
productos sanitarios