Vancomicina Sandoz 1.000 Mg Polvo Para Solucion Para Perfusion Efg
Información obsoleta, busque otroFICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Vancomicina Sandoz 500 mg polvo para solución para perfusión EFG Vancomicina Sandoz 1.000 mg polvo para solución para perfusión EFG
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Vancomicina Sandoz 500 mg polvo para solución para perfusión:
Cada vial contiene 500 mg de vancomicina (hidrocloruro) equivalente a 500.000 UI.
Vancomicina Sandoz 1.000 mg polvo para solución para perfusión:
Cada vial contiene 1.000 mg vancomicina (hidrocloruro) equivalente a 1.000.000 UI.
3. FORMA FARMACÉUTICA
Polvo para solución para perfusión.
Polvo blanco o casi blanco.
Tras la reconstitución se obtiene una solución con un pH de aproximadamente 3.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
La solución de vancomicina administrada por vía intravenosa está indicada en el tratamiento de infecciones graves, con potencial peligro para la vida, debidas a microorganismos sensibles Gram-positivos que no pueden ser tratados, o que no han respondido a otros medicamentos antimicrobianos menos tóxicos, tales como penicilinas y cefalosporinas.
Vancomicina debe reservarse para aquellos casos en los que hay una indicación específica, con el fin de minimizar la aparición de resistencias.
Vancomicina es útil en el tratamiento de las siguientes infecciones graves causadas por microorganismos sensibles (ver sección 5.1):
• Endocarditis,
• Infecciones óseas (osteomielitis),
• Neumonía,
• Infecciones de los tejidos blandos.
La endocarditis causada por los enterococos Streptococcus viridans o S. bovis debe tratarse con una combinación de vancomicina y un aminoglucósido.
Vancomicina se puede utilizar en la profilaxis perioperativo contra la endocarditis bacteriana, en aquellos pacientes que presenten un riesgo elevado de desarrollar endocarditis bacteriana al someterse a procedimientos quirúrgicos mayores (p. ej.; cirugía cardiaca y vascular, etc) y que no pueden tratarse con los agentes antibacterianos beta-lactámicos adecuados.
Deben tenerse en cuenta las directrices oficiales relativas al uso y prescripción adecuados de los antibióticos.
4.2 Posología y forma de administración
Vancomicina polvo para solución para perfusión debe administrarse por vía intravenosa. Cada dosis debe administrarse a una velocidad que no supere los 10 mg/min o durante un periodo de tiempo de al menos 60 minutos (lo que sea más largo).
La dosis deberá adaptarse individualmente según el peso, la edad y la función renal.
Se recomiendan los siguientes regímenes de dosificación:
Pacientes con la función renal normal
Adultos y adolescentes mayores de 12 años
La dosis diaria intravenosa recomendada es de 2.000 mg dividida en dosis de 500 mg cada 6 horas, o 1.000 mg cada 12 horas.
Para la endocarditis bacteriana, el régimen generalmente aceptado es de 1.000 mg de vancomicina por vía intravenosa cada 12 horas durante 4 semanas, ya sea sola o en combinación con otros antibióticos (gentamicina más rifampina, gentamicina, estreptomicina). La endocarditis enterocócica se trata durante 6 semanas con vancomicina en combinación con un aminoglucósido - siguiendo las recomendaciones nacionales.
Profilaxis perioperativa contra la endocarditis bacteriana: antes de la cirugía, a los adultos se les administran 1.000 mg de vancomicina por vía intravenosa (antes de la inducción de la anestesia) y dependiendo del tiempo y tipo de cirugía, se puede administrar una dosis de 1.000 mg de vancomicina i.v. 12 horas después de la misma.
Niños de un mes a 12 años de edad
12 horas.
La dosis intravenosa recomendada es de 10 mg/kg, cada 6 horas o 20 mg/kg cada
Recién nacidos y lactantes
horas durante la primera Se recomienda un control
La dosis inicial recomendada es de 15 mg/kg, seguida de 10 mg/kg cada 12 semana de vida, y cada ocho horas después de esta edad y hasta un mes de vida. cuidadoso de la concentración en suero de vancomicina (ver posteriormente).
Pacientes de edad avanzada
ser necesarias dosis de
Debido a la reducción en la función renal por causa de la edad, pueden mantenimiento más bajas.
Pacientes obesos
Puede ser necesario modificar las dosis diarias normales.
Pacientes con insuficiencia hepática No hay evidencias de que deba reducirse la dosis en los pacientes con insuficiencia hepática.
Pacientes con insuficiencia renal
En los pacientes con insuficiencia renal, la dosis debe ajustarse, pudiendo servir de guía el siguiente nomograma. Se recomienda un control cuidadoso de la concentración de vancomicina en suero (ver posteriormente).
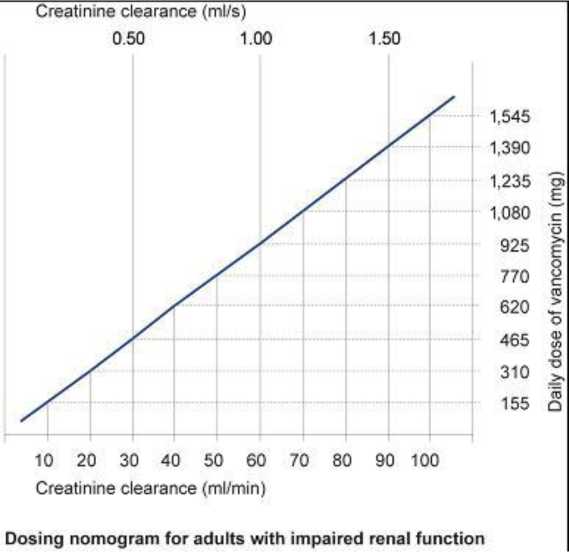
En los pacientes con una insuficiencia renal leve o moderada, la dosis de inicio no debe ser inferior a 15 mg/kg. En los pacientes con una insuficiencia renal grave, es preferible administrar una dosis de mantenimiento entre 250 mg y 1.000 mg con un intervalo de varios días, más que administrar dosis diarias más bajas.
Los pacientes con anuria (con una función renal prácticamente inexistente) deben recibir una dosis de 15 mg/kg de peso corporal hasta alcanzar la concentración terapéutica en suero. Las dosis de mantenimiento son de 1,9 mg/kg de peso corporal por cada 24 horas. Con el fin de facilitar el procedimiento, en los pacientes adultos con una función renal muy alterada se puede seguir una dosis de mantenimiento de 250 -1.000 mg a intervalos de varios días, en lugar de una dosis diaria.
Dosificación en caso de hemodiálisis
Es también posible la siguiente dosificación para los pacientes con una función renal nula, incluso bajo régimen de hemodiálisis regular:
Dosis de saturación de 1.000 mg, dosis de mantenimiento de 1.000 mg cada 7 - 10 días.
En caso de utilizar membranas de polisulfonas (diálisis de alto flujo) en la hemodiálisis la semivida de vancomicina se reduce. En los pacientes que siguen una hemodiálisis regular, puede que sean necesarias dosis adicionales de mantenimiento.
Monitorización de las concentraciones séricas de vancomicina
Debe monitorizarse la concentración sérica de vancomicina en el segundo día de tratamiento inmediatamente antes de la próxima dosis, y una hora después de la perfusión. Los niveles sanguíneos terapéuticos de vancomicina deben situarse entre 30 y 40 mg/l (máximo 50 mg/l) una hora después del final de la perfusión, siendo los niveles mínimos (justo antes de la próxima administración) entre 5 y 10 mg/l.
Normalmente, las concentraciones deben monitorizarse dos o tres veces por semana.
Forma de administración
Vancomicina por vía parenteral solamente debe administrarse como una perfusión intravenosa lenta (no más de 10 mg/min - durante por lo menos 60 min) que esté suficientemente diluida (por lo menos 100 ml por 500 mg o por lo menos 200 ml por 1.000 mg).
A los pacientes que requieren una restricción de fluidos, se les puede administrar una solución de 500 mg/50 ml ó 1.000 mg/100 ml. El riesgo de aparición de efectos adversos relacionados con la perfusión puede estar aumentado al utilizar estas altas concentraciones.
Para información respecto a la preparación de la solución, ver sección 6.6.
Duración del tratamiento
La duración del tratamiento depende de la gravedad de la infección, así como del progreso clínico y bacteriológico.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo Advertencias
Vancomicina solamente debe utilizarse en presencia de anuria aguda o lesión de la cóclea cuando sea absolutamente necesario y no se disponga de otras alternativas más seguras.
En caso de reacciones de hipersensibilidad graves agudas (p.ej., anafilaxis), se debe parar inmediatamente el tratamiento con vancomicina e iniciar las medidas de urgencia adecuadas normales (p.ej., antihistamínicos, corticosteroides, y - si es necesario - respiración artificial).
La administración en bolo rápido (es decir, durante algunos minutos) puede estar asociada con hipotensión (incluyendo shock y paro cardiaco), respuestas tipo histamina y erupción maculopapular o eritematosa (“síndrome de hombre rojo” o “síndrome del cuello rojo”). Vancomicina debe ser perfundida lentamente en una solución diluida (2,5 a 5,0 g/l) a una velocidad no superior a 10 mg/min y durante un período de tiempo no inferior a 60 minutos, a fin de evitar reacciones relacionadas con la perfusión rápida. Estas reacciones normalmente cesan rápidamente al parar la perfusión.
Debido al riesgo de necrosis, vancomicina se debe administrar solamente por vía intravenosa. Se minimiza el riesgo de una irritación venosa administrando vancomicina como perfusión diluida y cambiando el lugar de aplicación de la inyección.
La administración de vancomicina mediante inyección intraperitoneal en diálisis peritoneal ambulatoria continua se ha asociado con un síndrome de peritonitis química.
Nefrotoxicidad: vancomicina debe utilizarse con precaución en los pacientes con fallo renal dado que la posibilidad de que se desarrollen efectos tóxicos es mucho más alta cuando se está en presencia de concentraciones sanguíneas altas prolongadas. A fin de reducir al mínimo el riesgo de nefrotoxicidad, se deben realizar tests seriados de la función renal y adecuar los regímenes de dosis de forma apropiada en el tratamiento de estos pacientes y de los que están recibiendo tratamiento concomitante con otros principios activos nefrotóxicos (p.ej., aminoglucósidos) (ver sección 4.2).
Ototoxicidad: se ha observado ototoxicidad, que puede ser transitoria o permanente (ver seccion 4.8), en pacientes que presentaban sordera previa y que habían recibido dosis intravenosas excesivas o un tratamiento concomitante con otros principios activos ototóxicos, como un aminoglucósido. La sordera puede estar precedida por tinnitus. La experiencia con otros antibióticos sugiere que la sordera puede ser progresiva aunque cese el tratamiento. A fin de reducir la ototoxicidad, se recomienda determinar de forma periódica los niveles en sangre y llevar a cabo periódicamente ensayos sobre la función auditiva.
Precauciones
Vancomicina es muy irritante para los tejidos y causa necrosis en el lugar de la inyección si se inyecta intramuscularmente. En la mayoría de pacientes tratados con vancomicina puede aparecer dolor y tromboflebitis que pueden ser ocasionalmente graves. Pueden minimizarse la frecuencia y gravedad de la
tromboflebitis mediante la administración del medicamento de forma lenta y como solución diluida (ver sección 6.6) y cambiando de forma regular los lugares de aplicación de la perfusión. La frecuencia de las reacciones relacionadas con la perfusión (hipotensión, rubor, eritema, urticaria y prurito) aumenta con la administración concomitante de agentes anestésicos. Este hecho puede reducirse mediante la administración de vancomicina mediante perfusión como mínimo 60 minutos antes de la inducción anestésica.
Vancomicina debe utilizarse con precaución en pacientes con reacciones alérgicas a teicoplanina, dado que se han observado reacciones cruzadas de hipersensibilidad entre vancomicina y teicoplanina.
La anestesia induce una depresión del miocardio que puede estar aumentada por vancomicina. Durante la anestesia, las dosis deben estar bien diluidas y se deben administrar de forma lenta manteniendo una estrecha monitorización cardiaca. A fin de permitir un ajuste postural, se deben retrasar los cambios de posición hasta que la perfusión haya finalizado.
Se debe controlar a intervalos regulares el recuento de leucocitos en los pacientes tratados con vancomicina durante un período de tiempo prolongado o que son tratados concomitantemente con otros medicamentos que pueden causar neutropenia o agranulocitosis.
Todos los pacientes tratados con vancomicina deben hacerse de forma periódica estudios hematológicos, análisis de orina y tests sobre la función hepática y renal.
El uso prolongado de vancomicina puede dar lugar a superinfecciones con microorganismos resistentes, por lo que dichos pacientes deben ser monitorizados de forma regular. En caso de ocurrir una superinfección durante la terapia, deberán adoptarse las medidas adecuadas.
Se ha informado de la aparición de colitis pseudomembranosa con casi todos los agentes antibacterianos incluyendo vancomicina, con gravedad desde leve hasta capaz de poner en peligro la vida. Como consecuencia, es importante considerar este diagnóstico en los pacientes que presentan diarrea tras la administración de vancomicina. Está contraindicado el uso de antiperistálticos.
La monitorización regular de los niveles en sangre de vancomicina está indicada en la utilización a largo plazo, en particular en los pacientes con trastorno renal o con la facultad de audición alterada, así como cuando se administran de forma concurrente sustancias nefrotóxicas u ototóxicas, respectivamente.
Las dosis deben ajustarse en función de los niveles en suero. Regularmente, se deben monitorizar los niveles en sangre y realizar ensayos sobre la función renal.
Como recomendación general, se deben monitorizar las concentraciones 2-3 veces por semana.
Los pacientes de edad avanzada son particularmente susceptibles a las lesiones auditivas y deben seguir tests seriados si tienen más de 60 años. Debe evitarse el uso secuencial o concomitante de sustancias neurotóxicas.
Vancomicina debe utilizarse con especial cuidado en los niños y prematuros debido a su inmadurez renal y al posible aumento en las concentraciones séricas del medicamento. Como consecuencia, se deben monitorizar cuidadosamente las concentraciones de vancomicina. El uso concomitante en niños de vancomicina y agentes anestésicos se ha asociado con eritema y con reacciones anafilácticas. En caso de que sea necesaria la administración de vancomicina para la profilaxis quirúrgica, es aconsejable administrar los agentes anestésicos tras finalizar la administración de la perfusión de vancomicina.
4.5 Interacción con otros medicamentos y otras formas de interacción
Otros medicamentos potencialmente nefrotóxicos u ototóxicos
La administración concomitante o secuencial de vancomicina con otros principios activos potencialmente neurotóxicos y/o nefrotóxicos, en especial gentamicina, amfotericina B, estreptomicina, neomicina,
kanamicina, amikacina, tobramicina, viomicina, bacitracina, polimixina B, colistina y cisplatino puede potenciar la nefrotoxicidad y/u ototoxicidad de vancomicina, por lo que se requiere una monitorización cuidadosa del paciente.
Debido a la acción sinérgica, (p.ej., con gentamicina) en estos casos debe restringirse la dosis máxima de vancomicina a 500 mg cada 8 horas.
Anestésicos
La administración concomitante de vancomicina y agentes anestésicos se ha asociado con eritema, rubefacción histamínica y reacciones anafilactoides. Éstas pueden reducirse si se administra vancomicina como mínimo 60 minutos antes de la inducción anestésica.
Relajantes musculares
Si se administra vancomicina durante o inmediatamente después de la cirugía, puede aumentar y prolongarse el efecto (bloqueo neuromuscular) de los relajantes musculares (tales como succinilcolina) que se usen concomitantemente.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No se dispone de suficiente experiencia sobre la seguridad respecto a la utilización de vancomicina durante el embarazo en humanos. Los estudios toxicológicos sobre reproducción llevados a cabo con animales no sugieren ningún efecto sobre el desarrollo del embrión, feto o del periodo de gestación (ver sección 5.3).
Sin embargo, como vancomicina penetra en la placenta y no se puede excluir un potencial riesgo de nefrotoxicidad y ototoxicidad embrional. Por tanto, vancomicina solamente debe administrarse durante el embarazo en caso de que sea claramente necesario y tras una cuidadosa evaluación sobre el riesgo/beneficio.
Lactancia
Vancomicina se excreta en la leche materna y, por tanto, solamente debería utilizarse durante la lactancia en el caso de que otros antibióticos hayan fracasado. Vancomicina debe administrarse cuidadosamente a las mujeres en periodo de lactancia debido a la posibilidad de aparición de efectos adversos en el niño (alteraciones en la flora intestinal con diarrea, colonización con hongos tipo levaduras y posible sensibilización).
Debe considerarse la decisión de interrupir la lactancia teniendo en cuenta la importancia de este medicamento para la madre.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Vancomicina Sandoz sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Las reacciones adversas que se indican a continuación se definen utilizando la frecuenciay clasificación por órganos y sistemas de MedDRA:
Muy frecuentes (> 1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Las reacciones adversas más frecuentes son flebitis y reacciones pseudoalérgicas relacionadas con una administración intravenosa demasiado rápida de vancomicina.
Trastornos de la sangre y del sistema linfático
Raras (>1/10.000 a <1/1.000): trombocitopenia, neutropenia, agranulocitosis, eosinofilia.
Trastornos del sistema inmunológico
Raras (>1/10.000 a <1/1.000): reacciones anafilácticas, reacciones de hipersensibilidad.
Trastornos del oído y del laberinto
Poco frecuentes (>1/1.000 a <1/100): pérdida de audición transitoria o permanente.
Raras (>1/10.000 a <1/1.000): tinnitus, mareos.
Trastornos cardiacos
Muy raras (<1/10.000): parada cardiaca.
Trastornos vasculares
Frecuentes (> 1/100 a <1/10): disminución de la presión arterial, tromboflebitis.
Raras (>1/10.000 a <1/1.000): vasculitis.
Trastornos respiratorios, torácicos y mediastínicos Frecuentes (> 1/100 a <1/10): disnea, estridor.
Trastornos gastrointestinales
Raras (>1/10.000 a <1/1.000): nauseas.
Muy raras (<1/10.000): enterocolitis pseudomembranosa.
Trastornos de la piel y del tejido subcutáneo
Frecuentes (>1/100 a <1/10): exantema e inflamación de las mucosas, inflamación, prurito, urticaria. Muy raras (<1/10.000): dermatitis exfoliativa, síndrome de Stevens-Johnson, síndrome de Lyell, dermatitis vesicular IgA inducida.
Trastornos renales y urinarios
Frecuentes (> 1/100 a <1/10): insuficiencia renal manifestada principalmente por un aumento de las concentraciones de creatinina o de urea en suero.
Raras (>1/10.000 a <1/1.000): nefritis intersticial, fallo renal agudo.
Trastornos generales y alteraciones en el lugar de administración
Frecuentes (> 1/100 a <1/10): enrojecimiento de la parte superior del cuerpo y de la cara, dolor y espasmo de los músculos del pecho y espalda.
Raras (>1/10.000 a <1/1.000): fiebre medicamentosa, estremecimientos.
Pueden ocurrir reacciones anafilácticas durante o poco después de una perfusión rápida. Las reacciones disminuyen al parar la administración, generalmente entre 20 minutos y 2 horas después de su finalización.
La ototoxicidad se ha observado principalmente en los pacientes a los que se les administran altas dosis o en los que siguen un tratamiento concomitante con otros medicamentos ototóxicos, o en aquellos con una reducción preexistente de la función renal o de la audición.
Notificación de sospechas de reaciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaram.es
4.9 Sobredosis
Se ha informado de toxicidad debida a sobredosis. 500 mg IV a un niño de dos años dio lugar a una intoxicación letal. La administración de un total de 56 g durante 10 días a un adulto, ocasionó una insuficiencia renal. En ciertas condiciones de alto riesgo (p.ej., en caso de insuficiencia renal grave) pueden darse altos niveles en suero y efectos oto y nefrotóxicos.
Medidas en caso de sobredosis
• Se desconoce un antídoto específico.
• Es necesario un tratamiento sintomático mientras se mantiene la función renal.
• Vancomicina se elimina lentamente de la sangre mediante hemodiálisis o diálisis peritoneal. Se ha utilizado la hemofiltración o la hemoperfusión con resinas de polisulfona a fin de reducir las concentraciones en suero de vancomicina.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas Propiedades generales
Clasificación ATC
Grupo farmacoterapéutico: antibacterianos glucopéptidos, código ATC: J01XA01.
Mecanismo de acción
Vancomicina es un antibiótico glucopéptido. Vancomicina tiene un efecto bactericida sobre la proliferación de las bacterias mediante la inhibición de la biosíntesis de la pared celular. Además, altera la permeabilidad de la membrana celular bacteriana y de la síntesis de ARN.
Mecanismos de resistencia
La resistencia adquirida a los glucopéptidos está basada en la adquisición de varios complejos genéticos van. Se han encontrado raramente los genes van en Staphylococcus aureus, donde los cambios en la estructura de la pared celular dan lugar a una sensibilidad “intermedia”, que es más frecuentemente heterogénea.
No hay una resistencia cruzada entre vancomicina y otros antibióticos, pero puede ocurrir una resistencia cruzada con otros antibióticos glucopéptidos, como teicoplanina. Es raro el desarrollo de una resistencia secundaria durante el tratamiento.
En algunos países se han observado aumentos de casos de resistencias en particular en enterococos; son especialmente alarmantes cepas multiresistentes de Enterococcus faecium.
Sinergismo
La combinación de vancomicina con un antibiótico aminoglucósido tiene un efecto sinérgico frente a la mayoría de cepas de Staphylococcus aureus, D-streptococos no enterococos, enterocococos y estreptococos del grupo Viridans. La combinación de vancomicina con una cefalosporina tiene un efecto sinérgico frente algunas cepas de Staphylococcus epidermidis resistentes a oxacilina, y la combinación de vancomicina con rifampicina tiene un efecto sinérgico frente a Staphylococcus epidermidis y un parcial efecto sinérgico frente a algunas cepas de Staphylococcus aureus. Es de utilidad efectuar ensayos previos de sinergismo dado que vancomicina en combinación con una cefalosporina puede también tener un efecto antagónico frente a algunas cepas de Staphylococcus epidermidis y, en combinación con rifampicina, frente a algunas cepas de Staphylococcus aureus.
Deben obtenerse muestras para los cultivos bacterianos a fin de aislar e identificar los organismos causantes y determinar su sensibilidad a vancomicina.
Puntos de corte
Los puntos críticos de concentración mínima inhibitoria (CMI) establecidos por el European Committee on Antimicrobial Susceptibility Testing (EUCAST) para Staphylococcus spp. y Streptococcus spp. son Sensibles = 2 mg/l y Resistentes > 2 mg/l; para Enterococcus spp. son Sensibles = 4 mg/l y Resistentes > 4 mg/l; y para las especies no relacionadas son Sensibles = 2 mg/l y Resistentes > 4 mg/l.
Sensibilidad
La prevalencia de la resistencia adquirida puede variar geográficamente y con el tiempo para especies concretas, siendo deseable la información local sobre resistencias, en particular cuando se están tratando infecciones graves. En el caso de dudas sobre la utilidad del agente en algunos tipos de infección, es necesario el consejo de un experto sobre la prevalencia local de resistencias.
Vancomicina tiene un estrecho espectro de acción.
Especies frecuentemente sensibles
Staphylococcusspp Streptococcus pneumoniae Streptococcus spp Corynebacterium spp Enterococcusspp
5.2 Propiedades farmacocinéticas
Especies para las cuales la resistencia adquirida puede ser un problema
Enterococcus ^ faecium_
Organismos inherentemente resistentes_
Bacterias Gram-negativas, micobacterias, hongos_
Distribución : tras la administración intravenosa, vancomicina se distribuye a casi todos los tejidos y difunde en los fluidos pleural, pericardial, ascítico y sinovial, así como en el músculo cardiaco y las válvulas del corazón. Se alcanzan altas concentraciones similares a las del plasma sanguíneo. Los datos respecto a las concentraciones de vancomicina en el tejido óseo (esponjoso, compacto) varían ampliamente. Se ha determinado el volumen de distribución aparente en el estado de equilibrio en 0,43 (hasta 0,9) l/kg. En las meninges no inflamadas, vancomicina atraviesa solamente en pequeña cantidad la barrera hematoencefálica. Vancomicina se une a las proteínas plasmáticas en 30 a 55 % e incluso en cantidad superior.
Eliminación: vancomicina solamente se metaboliza en pequeña proporción. Tras la administración parenteral, se elimina casi completamente como la sustancia microbiológicamente activa (aprox. 75-90% en 24 horas) a través de la filtración glomerular por los riñones. La eliminación biliar es insignificante (menos del 5% de la dosis).
En los pacientes con una función renal normal, la semivida en suero es de aproximadamente 4-6 (5-11) horas, en niños 2,2-3 horas. En la insuficiencia renal, la semivida de vancomicina puede estar prolongada considerablemente (hasta 7,5 días). En estos casos y debido a la ototoxicidad de vancomicina en tratamiento adyuvante, se deben monitorizar las concentraciones plasmáticas.
Las concentraciones plasmáticas medias después de la perfusión i.v. de 1.000 mg de vancomicina durante 60 minutos fueron de aproximadamente 63 mg/l al final de la perfusión, de aproximadamente 23 mg/l transcurridas 2 horas y aproximadamente 8 mg/l después de 11 horas.
El aclaramiento de vancomicina del plasma se correlaciona estrechamente con la tasa de filtración glomerular.
El aclaramiento renal y sistémico total de vancomicina puede estar reducido en los pacientes de edad avanzada.
En los estudios con pacientes anéfricos se ha observado que el aclaramiento metabólico parece ser muy bajo.
Hasta el momento no se han identificado metabolitos de vancomicina en los humanos.
En el caso de que vancomicina se administre durante la diálisis peritoneal por vía intraperitoneal, aprox. el 60% alcanza la circulación sistémica durante 6 horas. Después de la administración i.p. de 30 mg/kg de peso corporal, se consiguen niveles en suero de aprox. 10 mg/l.
En caso de administración por vía oral, virtualmente no se absorbe vancomicina altamente polar. Después de la administración oral, aparece en las heces en la forma activa, siendo por tanto adecuada para la quimioterapia de la colitis pseudomembranosa y la colitis estafilocócica.
Vancomicina difunde fácilmente a través de la placenta y se distribuye en la sangre del cordón.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad y toxicidad a dosis repetidas.
Los datos limitados sobre los efectos mutagénicos muestran resultados negativos, no disponiéndose de estudios a largo plazo realizados en animales sobre el potencial carcinogénico. No se observaron efectos teratógenos directos o indirectos en los estudios realizados con ratas y conejos en los cuales los animales recibieron dosis que correspondían aproximadamente a las dosis humanas en base a la superficie corporal (mg/m2).
No se dispone de estudios en animales en cuanto a los efectos sobre la fertilidad o en relación al uso durante el período perinatal/postnatal.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Ninguno.
6.2 Incompatibilidades
Las soluciones de vancomicina tienen un valor de pH bajo. Esto puede ocasionar una inestabilidad física o química en el caso de que se mezcle con otras sustancias. En consecuencia, cada solución parenteral, antes de su utilización, debe comprobarse visualmente en cuanto a la presencia de precipitaciones y decoloraciones.
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
Terapia de combinación
En caso de una terapia de combinación de vancomicina con otros antibióticos/quimioterápicos, los preparados deberán administrarse por separado.
Se ha observado que son físicamente incompatibles las mezclas de soluciones de vancomicina y antibióticos beta-lactámicos. La probabilidad de precipitación aumenta con las concentraciones más altas de vancomicina. Se recomienda enjuagar adecuadamente las líneas intravenosas entre la administración de estos antibióticos. También se recomienda diluir las soluciones de vancomicina a 5 mg/ml o menos.
6.3 Periodo de validez
Polvo:
3 años.
Solución reconstituida:
Se ha demostrado la estabilidad en uso química y física durante 24 horas a 25°C y 96 horas a 2 - 8°C. Solución diluida:
Las soluciones para perfusión que están diluidas a 5 mg/ml con inyección de glucosa al 5% o inyección de cloruro de sódio 9 mg/ml (0,9%) son química y físicamente estables si se conservan en nevera (2°C - 8°C) durante 48 horas, o a 25°C durante 24 horas.
Las soluciones para perfusión que están diluidas a 5 mg/ml con inyección de glucosa al 5% + inyección de cloruro de sódio 9 mg/ml (0,9%) son química y físicamente estables si se conservan en nevera (2°C - 8°C) durante 48 horas, o a 25°C durante 24 horas.
Desde un punto de vista microbiológico, el medicamento debe utilizarse inmediatamente, a menos que la reconstitución y dilución se hayan llevado a cabo en condiciones asépticas controladas y validadas.
En caso de no utilizarse inmediatamente, los tiempos y condiciones de almacenamiento en uso son responsabilidad del usuario.
6.4 Precauciones especiales de conservación
Polvo:
No conservar a temperatura superior a 25°C.
Para las condiciones de conservación tras la reconstitución y dilución, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Vancomicina Sandoz 500 mg polvo para solución para perfusión EFG: 1 vial de vidrio incoloro de tipo I de 15 ml, con un tapón de goma de bromobutilo y una cápsula de aluminio/plástico tipo “flip-off’.
Vancomicina Sandoz 1.000 mg polvo para solución para perfusión EFG: 1 vial de vidrio incoloro de tipo I de 25 ml, , con un tapón de goma de bromobutilo y una cápsula de aluminio/plástico tipo “flip-off’.
Tamaños de envase: 1, 5, 10 y 100 viales.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El producto debe reconstituirse y el concentrado resultante debe diluirse posteriormente antes de su uso. Preparación de la solución reconstituida
Disolver Vancomicina Sandoz 500 mg polvo para solución para perfusión en 10 ml de agua para preparaciones inyectables.
Disolver Vancomicina Sandoz 1.000 mg polvo para solución para perfusión en 20 ml de agua para preparaciones inyectables.
1 ml de la solución reconstituida contiene 50 mg de vancomicina.
Aspecto de la solución reconstituida
Después de la reconstitución, la solución es transparente e incolora a ligeramente marrón amarillenta sin partículas visibles.
Para las condiciones de conservación del medicamento reconstituido, ver sección 6.3.
Preparación final de la solución para perfusión
Las soluciones reconstituidas que contienen 50 mg/ml de vancomicina deben diluirse posteriormente.
Los diluyentes adecuados son:
Inyección de glucosa al 5% o,
Inyección de cloruro de sodio 9 mg/ml (0,9%) o,
Inyección de glucosa al 5% con inyección de cloruro de sodio 9 mg/ml (0,9%).
Perfusión intermitente
diluirse posteriormente diluirse posteriormente
La solución reconstituida conteniendo 500 mg de vancomicina (50 mg/ml) debe con por lo menos 100 ml de diluyente (a 5mg/ml).
La solución reconstituida conteniendo 1.000 mg de vancomicina (50 mg/ml) debe con por lo menos 200 ml de diluyente (a 5mg/ml).
La concentración de la solución para perfusión de vancomicina no debe superar los 5 mg/ml.
La dosis deseada debe administrarse de forma lenta por vía intravenosa a una velocidad no superior a los 10 mg/minuto, durante por lo menos 60 minutos o incluso más.
Perfusión continua
Este tipo de perfusión solamente debe utilizarse si no es posible el tratamiento con una perfusión intermitente. Diluir 1.000 mg a 2.000 mg de vancomicina disuelta en una cantidad suficiente de los diluyentes adecuados indicados anteriormente y administrar en forma de perfusión gota a gota, de forma que el paciente reciba la dosis diaria prescrita en 24 horas.
Aspecto de la solución diluida
Después de la dilución, la solución es transparente e incolora sin partículas visibles.
Para las condiciones de conservación del medicamento diluido, ver sección 6.3.
Antes de la administración, las soluciones reconstituidas y diluidas deben ser inspeccionadas visualmente por si hay signos de decoloración y partículas. Solamente debe utilizarse una solución transparente y sin partículas.
Eliminación
Los viales son para un único uso. El medicamento sin utilizar debe ser eliminado.
La eliminación del medicamento no utilizado y de los materiales de desecho, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz Farmacéutica, S.A.
Centro Empresarial Osa Mayor Avda. Osa Mayor, n° 4 28023 (Aravaca) Madrid España
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Septiembre 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
Octubre 2009
La información detallada y actualizada de este medicamento está disponible en la página web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.gob.es/
13 de 13