Vancomicina Kabi 500 Mg Polvo Para Concentrado Para Solucion Para Perfusion Efg
Información obsoleta, busque otroam
FICHA TECNICA
1. NOMBRE DEL MEDICAMENTO
Vancomicina Kabi 500 mg polvo para concentrado para solución para perfusión EFG Vancomicina Kabi 1.000 mg polvo para concentrado para solución para perfusión EFG
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Vancomicina Kabi 500mg
Cada vial contiene 500 mg de vancomicina (en forma de hidrocloruro de vancomicina equivalente a 500.000 UI).
Vancomicina Kabi 1.000 mg
Cada vial contiene 1.000 mg de vancomicina (en forma de hidrocloruro de vancomicina equivalente a 1.000.000 UI).
3. FORMA FARMACÉUTICA
Polvo para concentrado para solución para perfusión Pastilla de polvo compacto, poroso, de color blanco a crema.
Tras la reconstitución se obtiene una solución con un pH de aproximadamente 3.
4. DATOS CLÍNICOS
4.1. Indicaciones terapéuticas
La vancomicina intravenosa está indicada en las siguientes infecciones graves causadas por microorganismos grampositivos sensibles a vancomicina que no puedan ser tratadas o hayan fracasado o sean resistentes a otros antibióticos como penicilinas y cefalosporinas (ver sección 5.1).
-endocarditis
-infección de huesos (osteomielitis)
-neumonía
-infecciones de tejidos blandos.
En los casos en que se considere adecuado, vancomicina se debe administrar con otros agentes antibacterianos, especialmente en el tratamiento de la endocarditis.
La vancomicina puede ser usada para la profilaxis perioperatoria contra la endocarditis bacteriana, en pacientes con un elevado riesgo de desarrollar endocarditis bacteriana cuando se someten a procedimientos quirúrgicos mayores (por ejemplo procedimientos cardiacos y vasculare) y no se les puede administrar un agente antibacteriano de tipo beta- lactámico apropiado.
Deben tenerse en cuenta las recomendaciones oficiales sobre el uso adecuado de agentes antibacterianos.
4.2. Posología y forma de administración
1 de 11
Posología
Perfusión en pacientes con función renal normal:
Adultos y adolescentes mayores de 12 años.
Se recomienda una dosis intravenosa diaria de 2.000 mg. Dividida en dosis de 500mg cada 6 horas o de 1000mg cada 12 horas. O bien 30-40 mg/kg/día en 2-4 administraciones diarias.
Para endocarditis bacteriana, se acepta 1.000 mg de vancomicina intravenosa cada 12 horas durante 4 semanas, sola o en combinación con otros antibióticos (gentamicina y rifampicina, gentamicina, estreptomicina).
La endocarditis enterocócica se trata con vancomicina en combinación con un aminoglucósido durante 6 semanas. Se recomienda consultar las guías oficiales.
Población pediátrica Niños de 1 mes-12 años:
La dosis habitual intravenosa es de 10mg/kg cada 6 horas (40mg diarios por kilo de peso corporal). Cada dosis debe ser administrada durante al menos un período de 60 minutos.
Recién nacidos (a término):
0-7 días: Una dosis de inicio de 15mg/kg seguido de 10mg/kg cada 12 horas.
7-30 días: Una dosis de inicio de 15mg/kg, seguido de 10mg/kg cada 8 horas.
Cada dosis debe ser administrada durante 60 minutos. Se aconseja una vigilancia cuidadosa de las concentraciones séricas de vancomicina.
Embarazo:
Se ha notificado que pueden ser necesarios incrementos significativos de dosis para obtener concentraciones séricas terapéuticas en pacientes embarazadas (ver sección 4.6).
Pacientes de edad avanzada:
Puede ser necesaria una reducción de la dosis mayor de la esperada debido a la disminución de la función renal.
Pacientes obesos:
Es posible que sea necesario modificar la dosis diaria habitual.
Pacientes con insuficiencia hepática:
No hay evidencia de que haya que reducir la dosis en pacientes con insuficiencia hepática.
Pacientes con la función renal reducida:
Se deben realizar ajustes de dosis para evitar niveles séricos tóxicos. En lactantes prematuros y en pacientes de edad avanzada, es posible que sea necesario reducir la dosis en mayor medida de lo esperado debido a la disminución de la función renal. Se aconseja la monitorización regular de los niveles séricos de estos pacientes, ya que se han notificado casos de acumulación, especialmente después de terapias prolongadas.
Las concentraciones séricas de vancomicina pueden determinarse mediante ensayos microbiológico, radioinmunoensayo, inmunoensayo de polarización por fluorescencia, inmunoensayos de fluorescencia o cromatografía líquida de alta resolución. El nomograma siguiente, basado en los valores de aclaramiento de creatinina, se ofrece como una guía para ajustar la dosis.
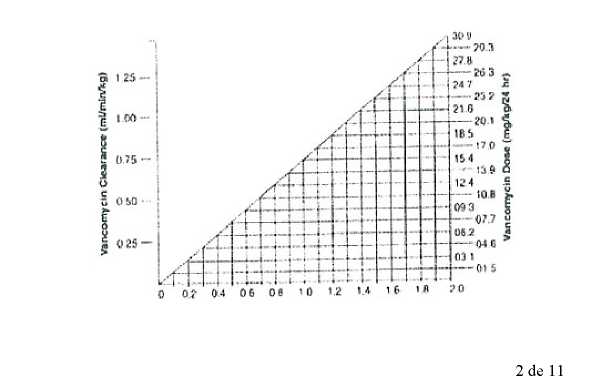
ÍTT1
CltltmliW Cl«r»nc« (mumln/ko)
Dosafle nomogram lor vancomycin in patients with ¡mpaired renal lunction
El nomograma no resulta válido en pacientes con diálisis, funcionalmente anéfricos.
Para estos pacientes se recomienda una dosis de carga de 15mg/kg de peso corporal para alcanzar rápidamente niveles séricos terapéuticos, y una dosis de mantenimiento de 1.9mg/kg/24horas. En pacientes con insuficiencia renal severa es preferible administrar una dosis de mantenimiento entre 250 mg y 1 g cada varios días, que administrar diariamente dosis más bajas.
En casos de anuria, se recomienda una dosis de 1g cada siete o diez días.
Si se dispone únicamente del nivel de creatinina sérica, puede emplearse la siguiente fórmula para calcular el aclaramiento de creatinina aproximado:
Hombre: Peso(kg)x (140 - edad (años))
72x creatinina sérica (mg/100ml)
Mujer: 0,85 multiplicado por el valor calculado en la fórmula anterior.
Monitorización de las concentraciones séricas de Vancomicina:
Debe supervisarse la concentración sérica de vancomicina el segundo día de tratamiento, antes de la siguiente dosis, y una hora después de la perfusión. Los niveles terapéuticos de vancomicina en sangre deben estar entre 30-40 mg/l (máximo 50mg/l) una hora después del final de la infusión, el nivel mínimo (antes de la siguiente administración) entre 5-10mg/l.
Se recomienda monitorizar la concentración dos o tres veces a la semana.
Forma de administración
La vancomicina parenteral debe ser administrada por perfusión intravenosa lenta (no más de 10mg/min - durante al menos 60 min) y suficientemente diluida (al menos 100 ml por cada 500 mg o 200 ml cada 1000 mg)
En pacientes que requieran restricción de líquidos pueden recibir una solución de 500 mg/50 ml o 1000 mg/100 ml. Con esta mayor concentración el riesgo de efectos secundarios relacionados con la infusión puede verse aumentado. Los efectos secundarios relacionados con la perfusión pueden aparecer con cualquier grado de concentración.
La dosis debe ser individualizada de acuerdo con el peso, edad y función renal del paciente. Los niveles de vancomicina se pueden medir para tener la dosis ajustada correctamente.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
Duración del tratamiento:
La duración del tratamiento depende de la gravedad de la infección, así como de la evolución clínica y bacteriológica.
4.3. Contraindicaciones
Hipersensibilidad al principio activo vancomicina.
4.4. Advertencias y precauciones especiales de empleo
La administración intravenosa rápida (por ejemplo en varios minutos) está asociada con hipotensión exacerbada, shock y, raramente, parada cardíaca, reacciones de tipo histamínico y exantema máculopapular o rash eritematoso (“síndrome del hombre rojo” o “síndrome de cuello rojo”). La vancomicina debe ser administrada en una solución diluida durante un período de inferior a 60 minutos, con el fin de evitar las reacciones derivadas de una perfusión rápida. Tras la interrupción de la infusión normalmente desaparecen estas reacciones (ver secciones 4.2 y 4.8).
Debido a su potencial ototoxicidad y nefrotoxicidad, vancomicina debe ser utilizada con precaución en pacientes con
3 de 11
MINISTB?K)DE SANIDAD, POLIT1C A SOCIAL E IGUALDAD Agencia esparcía óe medKarnentos y productos san oros
an
insuficiencia renal y la dosis debe ser reducida de acuerdo con el grado de insuficiencia renal. El riesgo de toxicidad se ve aumentado por altas concentraciones en sangre o tratamientos prolongados. Regularmente se deben monitorizar los niveles en sangre y deben realizarse controles periódicos de la función renal.
Se debe evitar vancomicina en pacientes con pérdida audición previa. Si se administra en dichos pacientes, se debe ajustar la dosis, si es posible en función de determinaciones periódicas de las concentraciones en sangre. La sordera puede ser precedida por tinnitus.
Los pacientes de edad avanzada son más susceptibles al daño auditivo. La experiencia con otros antibióticos sugiere que la sordera puede ser progresiva a pesar del cese del tratamiento.
Población pediátrica: En los recién nacidos prematuros y lactantes, puede ser conveniente confirmar las concentraciones séricas de vancomicina deseadas.
La administración concomitante de vancomicina y agentes anestésicos se ha asociado con eritema y rubefacción seudohistaminíca.
Uso en pacientes de edad avanzada La disminución natural de la filtración glomerular propia de la edad puede provocar concentraciones séricas de vancomicina elevadas si no se ajusta la dosis (ver sección 4.2).
Precauciones:
La monitorización regular de los niveles en sangre de vancomicina está indicada en el uso a largo plazo, particularmente en pacientes con disfunción renal o deterioro auditivo, así como en la administración simultánea de sustancias neurotóxicas u ototóxicas, respectivamente.
La dosis debe ser ajustada en función de los niveles séricos. Deben monitorizarse los niveles en sangre y realizar controles regulares de la función renal.
Los pacientes con una función renal que está en el límite de la normalidad y las personas con más de 60 años, deben someterse a pruebas seriadas de la función auditiva y de niveles en sangre de vancomicina. Todos los pacientes en tratamiento con el medicamento deben realizarse análisis hematológicos periódicos, análisis de orina y pruebas de la función renal.
Vancomicina resulta muy irritante para los tejidos, y causa necrosis en lugar de la administración cuando se administra por vía intramuscular; se debe perfundir por vía intravenosa. Dolor en el lugar de inyección y tromboflebitis se dan en muchos de los pacientes que reciben vancomicina y pueden ser ocasionalmente graves.
La frecuencia y la severidad de la tromboflebitis puede ser minimizada por la administración lenta del medicamento como una solución diluida (2.5 a 5.0 g/l) y rotando los lugares de administración.
El uso prolongado de vancomicina puede provocar el crecimiento excesivo de los microorganismos no susceptibles. La cuidadosa observación de los pacientes es esencial. Si se produce una sobreinfección durante el tratamiento, se deben tomar las medidas oportunas. En raras ocasiones se ha informado de colitis pseudomembranosa, por C. difficile, desarrollada en pacientes que recibieron tratamiento con vancomicina.
También se han detectado casos de hipersensibilidad cruzada.
Se han notificado casos de hipersensibilidad cruzada, por tanto vancomicina debe administrarse con cuidado en pacientes con hipersensibilidad conocida a teicoplanina.
4.5. Interacción con otros medicamentos y otras formas de interacción
La administración concomitante de vancomicina con agentes anestésicos, se ha asociado con eritema, r ubefacción de tipo histamínico y reacciones anafilactoides.
Se ha observado que la frecuencia de las reacciones asociadas a la perfusión aumenta al administrar concomitantemente con agentes anestésicos. Las reacciones asociadas a la perfusión se pueden minimizar administrando vancomicina en perfusión 60 minutos antes de la inducción anestésica.
4 de 11
MINISTB?K)DE SANIDAD, POLIT1C A SOCIAL E IGUALDAD Ageticaesparicídóe medcamenios y productos untarlos
El uso sistémico o tópico concomitante o secuencial de otros medicamentos potencialmente ototóxicos, neurotóxicos o neurotóxicos, tales como amfotericina B, aminoglucósidos, bacitracina, polimixina B, colistina, viomicina o cisplatino, requiere la monitorización del paciente, cuando esté indicado.
Existe un elevado potencial del bloqueo neuromuscular con la administración conjunta de vancomicina y agentes bloqueantes neuromusculares.
4.6. Fertilidad, embarazo y lactancia
Embarazo
No hay experiencia suficiente sobre la seguridad de vancomicina en relación al embarazo. Los estudios de toxicidad para la reproducción en animales no sugieren ningún efecto sobre el desarrollo del embrión, feto o período de gestación (ver sección 5.3).
Sin embargo, la vancomicina penetra en la placenta y no puede excluirse un riesgo potencial de ototoxicidad y nefrotoxicidad embrionario y neonatal. Por lo tanto vancomicina debe administrarse en el embarazo sólo si es claramente necesario y tras una cuidadosa evaluación del beneficio-riesgo.
Lactancia
Dado que la vancomicina se excreta en la leche humana, únicamente se deberá administrar a las mujeres en periodo de lactancia si otros antibióticos no han sido efectivos. Vancomicina debe administrarse con precaución a las madres en periodo de lactancia por el riesgo potencial de reacciones adversas en el bebé (alteraciones en la flora intestinal con diarrea, colonización de hongos tipo levaduras y posible sensibilización).
Se debe decidir si es necesario interrumpir la lactancia tras considerar el beneficio de este medicamento para la madre
4.7. Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de las vancomicina sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8. Reacciones adversas
En cada uno de los grupos de frecuencia, las reacciones adversas se presentan por orden descendente de gravedad.
Las reacciones adversas enumeradas a continuación se definen según la siguiente clasificación MedDRA: muy frecuentes (>1/10), frecuentes (>1/100, <1/10), poco frecuentes (>1/1.000, <1/100), raras (>1/10.000, <1/1.000); muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Perfusión intravenosa:
Las reacciones adversas más comunes son la flebitis y reacciones seudoalérgicas en relación con la perfusión intravenosa demasiado rápida de vancomicina.
Trastornos de la sangre y del sistema linfático:
Raras : Trombocitopenia, neutropenia, agranulocitosis, eosinofília.
Trastornos del sistema inmunológico:
Raras : Reacciones anafilácticas ,reacciones alérgicas.
Trastornos del oído y del laberinto:
Poco frecuentes : pérdida de audición temporal o permanente. .
Raras: Tinnitus, mareos.
Trastornos cardíacos:
Muy raras: paro cardíaco.
5 de 11
Trastornos Vasculares:
Frecuentes: Descenso de la presión sanguínea.
Raras: Vasculitis.
Trastornos respiratorios, torácicos y mediastínicos:
Frecuentes: Disnea y estridor
Trastornos gastrointestinales:
Raras : Náuseas.
Muy raras: Enterocolitis seudomembranosa.
Trastornos de la piel y del tejido subcutáneo :
Frecuentes: Exantema e inflamación de las mucosas, prurito, urticaria.
Muy raras: Dermatitis exfoliativa, Síndrome de Stevens-Johnson, Síndrome de Lyell dermatosis ampollosa por IgA lineal. Trastornos renales y urinarios :
Frecuentes: Insuficiencia renal que se manifiesta fundamentalmente por el aumento de la creatinina sérica.
Raros : nefritis intersticial, insuficiencia renal aguda.
Trastornos generales y alteraciones en el lugar de administración:
Frecuentes: Flebitis y rubefacción de la parte superior del cuerpo y la cara.
Raras : fiebre medicamentosa, temblor. Dolor en el pecho y músculos de la espalda.
Reacciones relacionadas con la perfusión:
Durante o poco después de una perfusión rápida, puede ocurrir una reacción anafilactoide, incluyendo hipotensión, disnea, urticaria o prurito. Se puede producir enrojecimiento de la piel en la parte superior el cuerpo (“síndrome del hombre rojo”), dolor y calambres en el pecho o los músculos de la espalda.
Las reacciones disminuyen cuando la administración se detiene, generalmente entre 20 minutos y 2 horas después. La vancomicina debe perfundirse lentamente, durante más de 60 minutos (ver sección 4.4).
La ototoxicidad puede ser reversible o permanente, y se ha detectado principalmente en pacientes que han sido tratados con una sobredosis, en pacientes con un historial de reducción de la audición y en pacientes con tratamiento concomitante con otros fármacos ototóxicos, como los aminoglucósidos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de medicamentos de Uso Humano: https://www.notificaram.es
4.9. Sobredosis
Se recomienda tratamiento de soporte, con mantenimiento de la filtración glomerular. La vancomicina no se elimina de forma adecuada mediante hemodiálisis o diálisis peritoneal. Se ha indicado que la hemoperfusión con resina Amberlite XAD-4 produce un beneficio limitado.
5. PROPIEDADES FARMACOLÓGICAS
5.1. Propiedades farmacodinámicas
Grupo farmacéutico: “antibacterianos para uso sistémico, antibacterianos glucopeptídicos” .
Código ATC: JO1XA01
6 de 11
MINISTB?K)DE SANIDAD, POLIT1C A SOCIAL E IGUALDAD Ageticaesparicídóe m«icamentosy productos untarlos
Mecanismo de acción:
La vancomicina es un antibiótico glucopéptido tricíclico que inhibe la síntesis de la pared celular de las bacterias sensibles al unirse con alta afinidad al D-alanil-D-alanina terminal de las unidades precursoras de la pared celular. El medicamento es bactericida para los microorganismos en división.
Relación PK/PD
La actividad de vancomicina se considera que es dependiente del tiempo.
Mecanismo de resistencia
La resistencia adquirida a glucopéptidos es más común en los enterococcos y se basa en la adquisición de varios complejos de genes van que modifican el objetivo D-alanil-D-alanina convirtiéndolo en D-alanil-D-lactato o D-alanil-D-serina, el cual se une muy poco a vancomicina. La resistencia cruzada con teicoplanina se ha detectado para algunos genes van. Los genes van, se han encontado raramente en Staphylococcus aureus, dónde los cambios en la estructura de la pared celular producen una sensibilidad intermedia que suele ser heterogénea.
Sensibilidad:
Vancomicina es particularmente activa contra bacterias grampositivas, como estafilococos, estreptococos, enterococos, neumococos, clostridios y difteroides . Las bacterias gramnegativas son resistentes.
La prevalencia de la resistencia adquirida puede variar geográficamente y con el tiempo para determinadas especies y toda la información local sobre la resistencia es deseable, sobre todo cuando se tratan infecciones graves . Debería solicitarse el consejo de un experto cuando la prevalencia local de resistencias es tal que la utilidad de la sustancia en al menos algunos tipos de infección es cuestionable.
Puntos de corte:
Recomendaciones EUCAST (Comité europeo sobre pruebas de susceptibilidad antimicrobiana) (del inglés, “European Committee on Antimicrobial Susceptibility Testing”)
|
Sensible |
Resistente | |
|
Staphylococcus spp. |
< 2 mg/L |
> 2 mg/L |
|
Enterococcus spp. |
< 4 mg/L |
> 4 mg/L |
|
Streptococcus spp. |
< 2 mg/L |
> 2 mg/L |
|
Streptococcus pneumoniae |
< 2 mg/L |
> 2 mg/L |
|
Anaerobios gram positivos |
< 2 mg/L |
< 2 mg/L |
|
No relacionado con la especie* |
< 2 mg/L |
> 4 mg/L |
Los puntos de corte no relacionados con la especie se han determinado principalmente sobre la base de los datos del PK/PD y son independientes de la distribución del CMI de especies específicas. Sólo se utilizan para especies para las que no se dispone de un punto crítico específico de especie y no para aquellas para las que no se recomiendan pruebas de sensibilidad.
Clases
EspeciesE Especies frecuEEspecies GramGGram posi
EnterococcusEEntsrooc StaphylococcuSSStaphy StaphylococcuSSStaphylocSStaphylo Streptococcus spp.
StreptococcuSSStreptococS
7 de 11
Clostridium s p p .
EspeciesF. EspeEEy EEspEts E Especies paE Especies EEspecEEsp EE sEEspeciesE
EnterococcusEEnteroc
Intrínsecamente resistentes
BacteriasBBa c tBBacterias
Chlamydia spp.
Mycobacteria
Mycoplasma spp.
Rickettsia spp._
5.2. Propiedades farmacocinéticas
Absorción
La vancomicina se administra por vía intravenosa para el tratamiento de infecciones sistémicas. En el caso de pacientes con función renal normal, la perfusión intravenosa de dosis múltiples de 1g de vancomicina (15 mg/kg) durante 60 minutos produce unas concentraciones plasmáticas medias aproximadas de 50-60 microgramos/ml, 20-25 microgramos/ml y 5-10 microgramos/ml, inmediatamente, 2 horas y 11 horas tras finalizar la perfusión, respectivamente. La perfusión intravenosa de dosis múltiples de 500 mg durante 30 minutos produce concentraciones plasmáticas medias de unos 40-50 mg/ml, 19-20 mg/ml y 10-11mg/ml, inmediatamente, 2 horas y 6 horas tras la finalización de la perfusión, respectivamente. Los niveles plasmáticos obtenidos tras dosis múltiples son similares a los obtenidos tras dosis múltiples son similares a los obtenidos tras una dosis única.
En caso de uso oral, la vancomicina de alta polaridad prácticamente no se absorbe. Tras la administración oral aparece la forma activa en las heces, por lo que es una quimioterapia adecuada para la colitis pseudomembranosa y colitis estafilocócica .
Distribución
A concentraciones séricas de vancomicina de 10 mg/l hasta 100 mg/l, la unión del fármaco a las proteínas plasmáticas es de aproximadamente 30-55%, medida por ultrafiltración.
Después de la administración intravenosa de hidrocloruro de vancomicina, se observan concentraciones inhibitorias en los líquidos pleural, pericárdico, ascítico y sinovial, en la orina y el líquido de diálisis peritoneal y en el tejido de la orejuela auricular.
En las meninges no están inflamadas, la vancomicina atraviesa muy poco la barrera hematoencefálica.
Eliminación
La vida media de eliminación de vancomicina es de 4 a 6 horas en pacientes con función renal normal. En las primeras 24 horas, aproximadamente el 80% de la dosis administrada de vancomicina se excreta en la orina por filtración glomerular. La alteración renal retrasa la excreción de vancomicina. En pacientes anéfricos, la vida media es de 7,5 días. Hay muy poco metabolismo del medicamento. Aproximadamente el 35-65% de una dosis intraperitoneal de vancomicina administrada durante la diálisis peritoneal se absorbe de forma sistémica en seis horas. Las concentraciones séricas de aproximadamente 8 mg/l se logran a través de la inyección intraperitoneal de 30 mg/kg de vancomicina. Aunque la vancomicina no se elimina eficientemente por hemodiálisis o diálisis peritoneal, se han detectado un aumento en el aclaramiento de vancomicina con hemoperfusión y hemofiltración. El aclaramiento total sistémico y renal de vancomicina puede estar disminuido en las personas de edad avanzada.
8 de 11
5.3. Datos preclínicos sobre seguridad
Los datos preclínicos no revelan ningún riesgo especial para los humanos, basándose en estudios convencionales de seguridad farmacológica y toxicidad a dosis repetida.
Los datos limitados de que se dispone sobre los efectos mutagénicos muestran resultados negativos. No se dispone de estudios a largo plazo en relación al potencial carcinogénico. En los estudios teratogénicos, donde ratas y conejos recibieron dosis que corresponden aproximadamente a las dosis humanas basándose en la superficie corporal (mg/m2), no se observaron efectos teratogénicos directos ni indirectos.
No se dispone de estudios con animales durante el periodo perinatal/postnatal, ni de los efectos en la fertilidad.
6 . DATOS FARMACÉUTICOS
6.1. Lista de excipientes
No contiene excipientes.
6.2. Incompatibilidades
La solución de vancomicina tiene un pH bajo que puede causar inestabilidad química o física cuando se mezcla con otros compuestos. Se debe evitar la mezcla con soluciones alcalinas. Comprobar visualmente antes de usar si hay precipitación o decoloración.
Este medicamento no debe mezclarse con otros medicamentos excepto los mencionados en la sección 6.6.
6.3. Periodo de validez
Polvo envasado para su comercialización:
2 años
Concentrado reconstituido:
El concentrado reconstituido debe diluirse inmediatamente después de su reconstitución.
Medicamento diluido:
Desde el punto de vista microbiológico y físicoquímico, el producto debe ser usado inmediatamente.
6.4. Precauciones especiales de conservación
Polvo envasado para su comercialización:
Conservar por debajo de 25°C
Mantener el vial en el embalaje exterior para protegerlo de la luz.
Concentrado reconstituido y producto diluido:
Para las condiciones de almacenamiento del concentrado reconstituido y el producto diluido, ver sección 6.3.
6.5. Naturaleza y contenido del envase
Vial de vidrio de 20 ml con tapón de clorobutilo recubierto de silicona tipo 1 y cápsula flip-off de aluminio/polipropileno de color gris la dosis de 500mg y de color verde la dosis de 1.000 mg.
9 de 11
MINISTB?K)DE SANIDAD, POLIT1C A SOCIAL E IGUALDAD Ageticaesparicídóe medKarnentos y productos untarlos
Tamaños de envase: 1 vial, 10 viales
Puede que no todos los tamaños de envase estén comercializados.
6.6. Precauciones especiales de eliminación y otras manipulaciones
El producto debe ser reconstituido y el concentrado resultante debe ser diluido antes de su uso.
Preparación del concentrado reconstituido:
Vancomicina Kabi 500mg : Disolver el contenido de cada vial de 500mg en 10ml de agua para preparaciones inyectables.
Vancomicina Kabi 1.000 mg : Disolver el contenido de cada vial de 1g en 20ml de agua para preparaciones inyectables.
Aspecto del concentrado reconstituido:
Solución transparente e incolora sin partículas.
Un ml de concentrado reconstituido contiene 50mg de vancomicina.
Para las condiciones de almacenamiento del concentrado reconstituido, ver sección 6.3.
Preparación de la solución diluida final para perfusión :
El concentrado reconstituido que contiene 50mg/ml de vancomicina, debe diluirse inmediatamente después de su reconstitución.
Los diluyentes adecuados son:
Cloruro de sodio 9mg/ml (0,9%) para inyección, glucosa 50mg/ml (5%) para inyección, cloruro de sodio 9mg/ml (0,9%) y glucosa 50mg/ml (5%) para inyección o Acetato de Ringer para inyección.
Antes de la administración, las soluciones reconstituidas y diluidas deben ser inspeccionadas visualmente para detectar partículas y decoloración. Sólo se deben utilizar soluciones claras incoloras y libres de partículas.
Perfusión intermitente:
Vancomicina Kabi 500mg : Concentrado reconstituido que contiene 500mg de vancomicina (50 mg/ml) debe ser diluido en al menos 100 ml de diluyente inmediatamente después de la reconstitución.
Vancomicina Kabi 1.000 mg : Concentrado reconstituido que contiene 1g de vancomicina (50 mg/ml) debe ser diluido en al menos 200 ml de diluyente inmediatamente después de la reconstitución.
La concentración de vancomicina en solución para perfusión no debe exceder los 5 mg/ml.
La dosis deseada debe ser administrada lentamente por perfusión intravenosa a una velocidad de no más de 10 mg/minuto, por lo menos durante 60 minutos o incluso más.
Para las condiciones de almacenamiento del producto diluido, ver sección 6.3.
Eliminación
Los viales son para un solo uso. Deben descartarse los productos no utilizados. .
Cualquier producto no utilizado o material de desecho debe eliminarse de acuerdo con las normativas locales.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN Fresenius Kabi España, S.A.U.
10 de 11
on
Marina, 16-18 08005 Barcelona España
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Febrero 2015
10. FECHA DE LA REVISIÓN DEL TEXTO
Diciembre 2012
11 de 11