Suprecur Depot 3,6 Mg
Información obsoleta, busque otroDE SANIDAD, POLÍTICA SOCIAL E IGUALDAD
|
1 | |
|
Oí |
k agencia española de 1 medicamentos y I productos sanitarios |
FICHA TÉCNICA
1. Denominación de la especialidad
Suprecur® depot 3,6 mg
2. Composición cualitativa y cuantitativa
Una jeringa precargada de doble cámara contiene 3,6 mg de acetato de buserelina, equivalentes a 3,4 mg de buserelina (cámara 1).
3. Forma farmacéutica
Polvo y disolvente para suspensión para inyección.
4. Datos clínicos
4.1 Indicaciones terapéuticas
Tratamiento preoperatorio sintomático de los fibromas uterinos.
4.2 Posología y método de administración
El tratamiento con Suprecur Depot se debe iniciar en el primer o segundo día de la menstruación (ver "4.4 Advertencias y precauciones especiales de uso").
Cada mes se administra una dosis de 3,6 mg de acetato de buserelina mediante inyección subcutánea. Suprecur Depot se inyectará de preferencia en la pared lateral del abdomen. Para cada inyección se elegirá un punto diferente.
Se recomienda insertar la aguja 1 ó 2 cm en el tejido subcutáneo. Antes de la inyección es preciso cerciorarse de que la aguja no se ha introducido en un vaso sanguíneo. La suspensión de micropartículas se debe inyectar inmediatamente después de su reconstitución.
Suprecur Depot se debe administrar a intervalos de 1 mes durante los 3 meses anteriores a la intervención quirúrgica programada. El intervalo entre dos inyecciones consecutivas deberá ser de 28 hasta un máximo de 35 días. La intervención quirúrgica se deberá practicar antes de transcurridas 5 semanas desde la última inyección.
4.3 Contraindicaciones
Suprecur Depot no se debe administrar a pacientes con hipersensibilidad al acetato de buserelina, a los análogos de la LHRH o a cualquiera de los excipientes contenidos en Suprecur Depot.
Para su utilización en mujeres embarazadas o en período de lactancia, consultar "4.4 Advertencias y precauciones especiales de uso" y "4.6 Embarazo y lactancia ".
Correo electrúnicoI
C / CAMPEZO, 1 - EDIFICIO 8 28022 MADRID
4.4
Advertencias y precauciones especiales de uso
El uso durante el embarazo no está indicado. Con objeto de excluir un posible embarazo al comenzar el tratamiento, éste se deberá iniciar en el primer o el segundo día de la menstruación. En caso de duda, se recomienda realizar un test de embarazo.
Antes de comenzar el tratamiento con Suprecur Depot se deberá interrumpir la administración de contraceptivos orales.
No es probable que se produzca un embarazo durante el tratamiento con Suprecur Depot si éste se administra de forma periódica, cada mes. Por razones de seguridad se deberían utilizar métodos alternativos de contracepción, no hormonales (p.ej. preservativos) durante el tratamiento. Si a pesar de ello se produjera un embarazo durante el tratamiento, deberá suspenderse la administración de Suprecur Depot.
El tratamiento con agonistas de la LHRH puede ocasionar una pérdida de masa y matriz óseas. Este efecto aumenta al aumentar la duración del tratamiento. Por consiguiente, el beneficio del tratamiento con Suprecur Depot se deberá valorar cuidadosamente frente al riesgo de un aumento de la pérdida de masa y matriz óseas:
- cuando se consideren ciclos de tratamiento repetidos con Suprecur Depot.
- en pacientes con osteoporosis manifiesta y aquéllos particularmente sensibles a la pérdida ósea (p.ej. pacientes bajo tratamiento de larga duración con corticosteroides).
En pacientes con hipertensión se deberá controlar periódicamente la presión arterial (riesgo de deterioro de los valores de presión arterial). En pacientes diabéticos, se deberá controlar periódicamente la glucemia (riesgo de deterioro del control metabólico debido a una reducción de la tolerancia a la glucosa).
Los pacientes con antecedentes de depresión deberán ser controlados cuidadosamente y, en caso necesario, deberán ser tratados (riesgo de recurrencia o agravamiento de la depresión).
4.5 Interacción con otros medicamentos y otras formas de interacción
Durante el tratamiento con buserelina, el efecto de los antidiabéticos puede verse atenuado (ver "4.8 Reacciones adversas"). En el tratamiento concomitante con hormonas sexuales ("efecto añadido") se recomienda limitar la dosis de estrógenos de forma que no se vea afectado el éxito global del tratamiento.
4.6 Embarazo y lactancia
La buserelina no está indicada en el embarazo. En estudios en animales no evidenció efectos de embriotoxicidad, fetotoxicidad o teratogenicidad de relevancia para el ser humano. Ver "4.4 Advertencias y precauciones especiales de uso ".

La buserelina pasa a la leche materna en pequeñas cantidades. Aun cuando no se han observado efectos hormonales en los niños, no se deberá administrar Suprecur depot durante la lactancia.
4.7 Efectos sobre la capacidad para conducir vehículos y utilizar maquinaria
Determinados efectos indeseables (p.ej. vértigo) pueden alterar la capacidad de reacción o de concentración del paciente y por consiguiente constituyen un riesgo en situaciones en las que éstas son de especial importancia como es el caso de la conducción de vehículos y el manejo de maquinaria.
4.8. Reacciones adversas
La mayoría de los efectos que se detallan a continuación se encuentran directa o indirectamente relacionados con la acción farmacológica de buserelina sobre la producción estrogénica ovárica.
Frecuentes: Hemorragias uterinas (“período”) por lo general en las primeras semanas de tratamiento, quistes ováricos (en la fase inicial de tratamiento), disminución de la masa y matriz óseas (tras repetidos ciclos de tratamiento), flujo vaginal, sequedad vaginal, dispareunia, aumento o disminución del tamaño de los senos con mayor sensibilidad a la presión, disminución de la líbido, sofocos; aumento de la sudoración, sequedad cutánea, acné, aumento o pérdida de cabello; cefalea (en raras ocasiones de tipo migraña), palpitaciones, nerviosismo, alteraciones del sueño, cansancio, somnolencia, vértigo, inestabilidad emocional; dolor abdominal, náuseas, vómitos, diarrea, estreñimiento; aumento o disminución del peso corporal, dolor de espalda y dolor en las extremidades, dolor y rigidez muscular, molestias articulares; dolor o reacciones locales en el punto de inyección.
Ocasionales: Sequedad ocular (que puede dar lugar a irritación ocular en pacientes que usan lentes de contacto), alteración visual (p.ej. visión borrosa), sensación de presión retroocular; fragilidad de las uñas, aumento o disminución del vello corporal; galactorrea; edema (en cara y extremidades); alteraciones de la memoria y la concentración, sensación de ansiedad, depresión o agravamiento de una depresión preexistente; aumento de la sed, cambios en el apetito, parestesia; aumento de los niveles séricos de enzimas hepáticos (transaminasas) o la bilirrubina; reacciones de hipersensibilidad tales como enrojecimiento de la piel, picor, rash cutáneo (incluyendo urticaria).
Raros: Alteraciones de los lípidos hemáticos; tinnitus, alteraciones del oído; cistitis.
Muy raros: Aumento de la presión arterial y reducción de la tolerancia a la glucosa (posible deterioro del control metabólico en los pacientes diabéticos), trombopenia, leucopenia, reacciones de hipersensibilidad graves con broncospasmo y disnea.
En casos aislados: Reacciones de hipersensibilidad graves con shock; necrosis de los fibromas uterinos (que pueden requerir intervención quirúrgica).
El efecto de estimulación inicial sobre la secreción hormonal de la buserelina puede llevar a un agravamiento transitorio de los síntomas y signos clínicos.
El tratamiento de larga duración con agonistas de la LHRH puede desencadenar, en casos aislados, el desarrollo de adenomas hipofisarios. No obstante, hasta el presente no se ha observado este fenómeno en los pacientes tratados con buserelina.
4.9.
Sobredosificación
Hasta el presente no se ha observado ningún caso de intoxicación con buserelina. Los efectos descritos con la sobredosis de buserelina no difieren de los efectos adversos observados durante su utilización normal. En caso necesario, el tratamiento de la sobredosis será sintomático.
5. Propiedades farmacológicas
5.1. Propiedades farmacodinámicas
Clasificación terapéutica/Código ATC Código ATC L02AE01
La buserelina es un análogo de la hormona liberadora de gonadotropinas natural (gonadorrelina; GnRH, LHRH), con una marcada actividad biológica. El efecto farmacológico inicial de la buserelina consiste en la estimulación de la liberación de gonadotropinas y de la secreción gonadal de hormonas, seguido de un descenso progresivo de la liberación pulsátil de LH-FSH. Durante esta fase, en las mujeres, se produce una disminución de la secreción ovárica de esteroides hasta niveles postmenopáusicos. Por consiguientes, la administración continuada de buserelina da lugar a la supresión de la secreción estrogénica y, como consecuencia, a la supresión de la función ovárica. Estos efectos, por su parte, conllevan en muchos pacientes una disminución del tamaño del útero, atrofia del endometrio con disminución del sangrado y reducción de fibromas (leiomioma).
Mientras que durante el tratamiento continuado con buserelina resulta inhibida la liberación de gonadotropina, la secreción de otras hormonas hipofisarias (hormona del crecimiento, prolactina, ACTH, TSH) no se ve influenciada directamente. Sin embargo, el déficit estrogénico puede producir una disminución de la secreción de hormona del crecimiento y prolactina. La secreción de esteroides corticoadrenales permanece inalterada.
Tras administración de una dosis única de 3,6 mg de acetato de buserelina en micropartículas, la supresión de la secreción de estradiol tuvo una duración aproximada de 42 días (intervalo: 21 a 56 días). Tras la inyección, se observó una elevación del nivel de progesterona (indicativa de ovulación) transcurridos 83 días, aproximadamente (intervalo: 62 a 99 días) y la menstruación se reinstauró transcurridos aproximadamente 98 días de la inyección.
5.2 Propiedades farmacocinéticas
Suprecur Depot contiene acetato de buserelina en forma de micropartículas. Estas están compuestas por un polímero totalmente biodegradable [poli-(D,L-láctido-co-glicólido) 1:1]. Tras la inyección subcutánea, el polímero libera el principio activo.
La liberación de buserelina a partir de las micropartículas es controlada por la degradación de la matriz polimérica. El perfil de liberación es bifásico: la fase de
liberación inicial es seguida por una fase de liberación lenta continuada a lo largo del intervalo de dosificación. A continuación se observa una disminución progresiva de la liberación hasta haberse completado la biodegradación de la matriz del polímero. La vida media terminal de liberación en humanos es de 10 a 12 días; del 8 al 27% del contenido original de buserelina de las micropartículas permanece sin liberar transcurrido el intervalo de 4 semanas (reserva de liberación).
La biodisponibilidad de buserelina a partir de la formulación de micropartículas es del 72 %.
La buserelina es soluble en agua y se absorbe rápidamente tras su inyección subcutánea. Se acumula de preferencia en hígado, riñones y lóbulo anterior de la hipófisis, que constituye el órgano biológico diana. La buserelina circula en el suero predominantemente en forma activa inalterada. La unión a proteínas es de aprox. el 15%. La buserelina y sus metabolitos inactivos se excretan por vía renal y biliar.
La buserelina es inactivada mediante peptidasas (piroglutamil peptidasa y endopeptidasas de tipo quimotripsina) en el hígado y los riñones. En la hipófisis la buserelina unida al receptor se inactiva mediante enzimas localizados en la membrana.
5.3 Datos preclínicos sobre seguridad
No se detectaron signos de toxicidad ni alteraciones histopatológicas en los estudios de farmacología y toxicología realizados con buserelina en ratas, perros y monos; los efectos endocrinos observados se restringieron a las gónadas. Se produjeron adenomas hipofisarios durante el tratamiento de larga duración en ratas; este fenómeno no se produjo en perros y monos.
No se observaron indicaciones de potencial mutagénico o carcinogénico en ninguno de los estudios realizados.
La tolerancia local de buserelina micropartículas es buena, siendo de escasa importancia las reacciones observadas en el punto de inyección.
6. Datos farmacéuticos
6.1 Relación de excipientes
Cámara 1
Poli-(D,L-láctido-co-glicólido) 1:1, Dextrano 40, Polisorbato 40, Cloruro sódico Cámara 2
Agua para inyección
6.2 Incompatibilidades
Suprecur Depot se presenta en jeringa precargada para inyección subcutánea y no se debe mezclar con otros fármacos.
Período de validez
3 años. La suspensión se administrará inmediatamente tras la reconstitución.

6.4 Precauciones especiales de conservación
No almacenar a temperatura superior a + 25 °C. Proteger de la luz.
6.5 Naturaleza y contenido del recipiente
Envase con 1 ó 3 jeringas precargadas de doble cámara, conteniendo cada una de ellas 3,6 mg de acetato de buserelina en forma de polvo en una de las cámaras y 1,5 ml de agua para inyección como disolvente (para la resuspensión) en la otra cámara. Aguja de inyección (tamaño G23).
6.6 Instrucciones de uso y manipulación
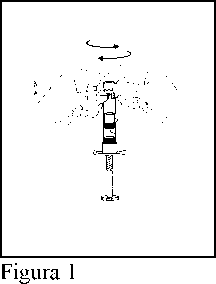
Preparar buserelina micropartículas para inyección en la forma siguiente (7 ilustraciones):
1. Sujetar la jeringa por la banda (A), seguidamente girar el precinto (B).
(Figura 1)
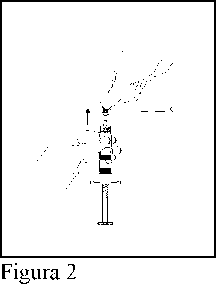
2. Retirar el tapón gris (C). (Figura 2)
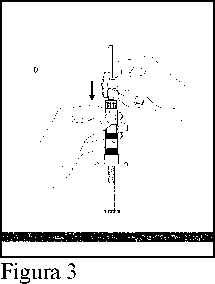
3. Sin retirar el capuchón protector, insertar la aguja en la jeringa (D) (Figura 3)
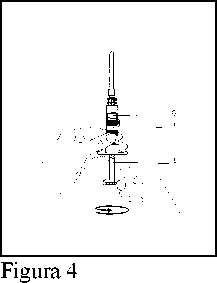
4. Girar el émbolo (E) hasta que la totalidad del agua para inyección haya pasado desde la cámara posterior (F) a la cámara anterior (G) de la jeringa. El émbolo debe girar libremente de modo que más tarde pueda ser presionado hacia delante para realizar la inyección.
(Figura 4)
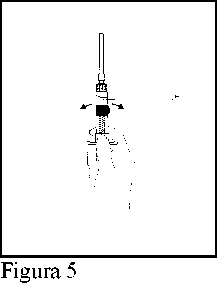
5. Suspender las micropartículas presentes en la mezcla, formadas a partir de la sustancia seca y el agua (H) dando ligeros golpecitos en la jeringa de uno y otro lado hasta que se forma una suspensión uniforme.
(Figura 5)
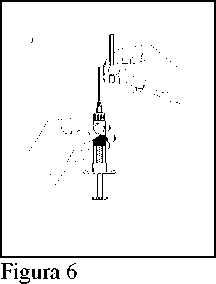
6. Retirar el capuchón protector de la aguja (J). (Figura 6)
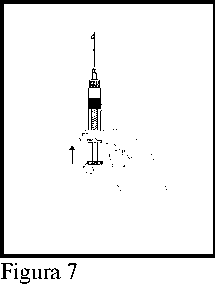
7. Expeler el aire restante de la cámara anterior y la aguja. La jeringa está preparada para efectuar la inyección de forma inmediata.
(Figura 7).
7. Titular de la autorización de comercialización
Hoechst Marion Roussel, S.A.
Ronda General Mitre, 72-74 08017 - Barcelona
8. Número de autorización de comercialización
9. Fecha de la primera autorización/revalidación de la autorización
21 de Enero de 1998
10. Fecha de revisión del texto
Febrero 2000
Especialidad de diagnóstico hospitalario
H00000FT02
Agencia española de
medicamentos y
productos sanitarios