Iluvien 190 Microgramos Implante Intravitreo En Aplicador
Información obsoleta, busque otroFICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
ILUVIEN 190 microgramos implante intravítreo en aplicador.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada implante contiene 190 microgramos de acetónido de fluocinolona.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Implante intravítreo en aplicador.
Cilindro de color castaño claro, de aproximadamente 3,5 mm x 0,37 mm de tamaño. Aplicador del implante con aguja de calibre 25.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
ILUVIEN está indicado para el tratamiento del deterioro visual asociado al edema macular diabético crónico, cuando la respuesta a las terapias disponibles se considera insuficiente.
4.2 Posología y forma de administración
Posología
La dosis recomendada es un implante de ILUVIEN en el ojo afectado. No se recomienda la administración concurrente en ambos ojos (ver sección 4.4).
Puede colocarse un implante adicional a los 12 meses si el paciente experimenta una diminución de la visión o un aumento del espesor de la retina secundario a la recurrencia o al empeoramiento del edema macular diabético (ver sección 5.1).
No deberá repetirse el tratamiento a menos que los beneficios potenciales superen a los riesgos.
Únicamente debe tratarse con ILUVIEN a los pacientes que han tenido una respuesta insuficiente al tratamiento previo con fotocoagulación con láser u otras terapias disponibles para el edema macular diabético.
Población pediátrica
No existe una indicación específica para ILUVIEN en la población pediátrica para edema macular diabético (EMD).
Poblaciones especiales
No es necesario ajustar las dosis en pacientes de edad avanzada, ni en aquellos afectados por insuficiencia hepática o renal.
Forma de administración
EXCLUSIVAMENTE PARA USO INTRAVÍTREO.
El tratamiento con ILUVIEN es exclusivamente para uso intravítreo, y deberá ser administrado por un oftalmólogo experimentado en la aplicación de inyecciones intravítreas.
La inserción del implante intravítreo deberá realizarse en condiciones asépticas controladas, las que incluyen el uso de guantes estériles, una venda estéril y un espéculo palpebral (o equivalente) estéril. Antes de la inserción deberá administrarse una anestesia adecuada y un microbicida de amplio espectro.
1. Puede administrarse un antibiótico en gotas antes de la operación, a criterio del oftalmólogo tratante.
2. Inmediatamente antes de la inserción, administrar anestesia tópica sobre el lugar de la aplicación (se recomienda el cuadrante inferotemporal) en una gota, seguido por un aplicador con punta de algodón embebido en anestésico, o bien la administración subconjuntiva de un anestésico adecuado.
3. Administrar 2-3 gotas de un antiséptico tópico apropiado en el fórnix inferior. Pueden frotarse los párpados con un aplicador con punta de algodón embebido en el antiséptico apropiado. Colocar un espéculo palpebral estéril. Hacer que el paciente mire hacia arriba, y apoyar un aplicador con punta de algodón embebido en el antiséptico apropiado en el lugar de la inserción. Dejar secar la aplicación de antiséptico tópico durante 30-60 segundos antes de la inserción de ILUVIEN.
4. El exterior de la bandeja no debe considerarse estéril. Un asistente (no estéril) deberá extraer la bandeja de la caja y retirará la tapa sin tocar la superficie interior. Inspeccionar visualmente por la ventana del sistema de aplicador para verificar que el implante farmacológico esté en el interior.
5. Extraer el aplicador de la bandeja con los guantes estériles colocados, tocando únicamente el aplicador y la superficie estériles.
6. Para reducir la cantidad de aire administrado con el implante, la administración requiere un proceso de dos pasos. Antes de insertar la aguja en el ojo, empujar el botón hacia abajo y deslizarlo hasta el primer tope (las marcas negras curvas). En el primer tope, soltar el botón y éste se desplazará hasta la posición UP (arriba).
7. La ubicación ideal del implante es inferior con respecto al disco óptico, y posterior al ecuador. Esto puede lograrse dirigiendo la aguja hacia la cara inferior del disco óptico. Medir 4 milímetros en sentido inferotemporal desde el limbo, con la ayuda de un calibre.
8. Quitar la tapa protectora de la aguja.
9. Desplazar suavemente la conjuntiva de modo tal que, al retirar la aguja, los lugares de entrada de la aguja en la conjuntiva y la esclerótica no queden alineados. Deberán tomarse recaudos para evitar el contacto entre la aguja y el borde del párpado o las pestañas. Insertar la aguja en el ojo. Para liberar el implante, con el botón en la posición superior, presionar el botón hacia adelante hasta el final y retirar la aguja.
10. Retirar el espéculo palpebral y realizar una oftalmoscopia indirecta para verificar la ubicación del implante, la perfusión adecuada de la arteria central de la retina y la inexistencia de cualquier otra complicación.
Tras la inserción intravítrea, se deberá realizar un examen de oftalmoscopia indirecta en el cuadrante de inserción, a fin de verificar la ubicación correcta. La depresión esclerótica puede mejorar la visualización del implante. El examen deberá incluir un control de la perfusión de la cabeza del nervio óptico inmediatamente después de la inserción. A criterio del oftalmólogo, puede hacerse una medición inmediata de la PIO.
Tras el procedimiento, se deberá controlar a los pacientes para detectar posibles complicaciones tales como endoftalmitis, aumento de la presión intraocular, desprendimientos de retina y hemorragias o desprendimientos del vítreo. Deberá realizarse una biomicroscopía con tonometría dentro de los dos a siete días posteriores a la inserción del implante.
Posteriormente se recomienda monitorizar a los pacientes como mínimo en forma trimestral para detectar posibles complicaciones, dada la prolongada duración de la liberación de acetónido de fluocinolona, de aproximadamente 36 meses (ver sección 4.4).
4.3 Contraindicaciones
El implante intravítreo con ILUVIEN está contraindicado en presencia de glaucoma preexistente o infección ocular o periocular activa o presunta, incluidas la mayoría de las enfermedades virales de la córnea y la conjuntiva, entre ellas queratitis epitelial activa por herpes simplex (queratitis dendrítica), vaccinia, varicela, infecciones micobacterianas y micosis.
ILUVIEN está contraindicado en pacientes con hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Las inyecciones intravítreas han sido asociadas con endoftalmitis, elevación de la presión intraocular, desprendimiento de retina y hemorragias o desprendimientos del vítreo. Se debe indicar a los pacientes que comuniquen sin demoras cualquier síntoma que sugiera endoftalmitis. Un control de los pacientes dentro de los dos a siete días posteriores a la inyección puede permitir la identificación temprana y el tratamiento de una infección ocular, del aumento de la presión intraocular o de cualquier otra complicación. Posteriormente, se recomienda controlar la presión intraocular como mínimo en forma trimestral
El uso de corticosteroides por vía intravítrea puede provocar cataratas, aumento de la presión intraocular y glaucoma, y puede incrementar el riesgo de infecciones secundarias.
No se ha estudiado la seguridad y la eficacia de la administración concurrente de ILUVIEN en ambos ojos. No se recomienda el tratamiento concurrente en ambos ojos hasta no conocer la respuesta ocular y sistémica del paciente al primer implante.
En los estudios FAME, la incidencia de cirugía de cataratas en la totalidad de los pacientes fáquicos fue aproximadamente 3 veces mayor en el grupo tratado con ILUVIEN (80,0%) que en el grupo sometido a tratamiento simulado (27,3%). La mediana del tiempo hasta la catarata comunicada como acontecimiento adverso fue de aproximadamente 14 meses, y los sujetos fáquicos presentaron deterioro de la visión desde el desarrollo de la catarata, desde aproximadamente el mes 9 hasta el mes 18 después de la implantación, antes de la extirpación de la catarata (ver sección 4.8).
En la población general de los estudios clínicos, que excluían a sujetos con una PIO inicial >21 mmHg, el porcentaje de pacientes tratados con ILUVIEN que requirieron tratamiento con medicamentos reductores de la PIO fue de 38%, frente a 14% en el grupo de tratamiento simulado. Esta proporción se elevaba a 47% en aquellos sujetos con una PIO superior a la media al inicio (>15 mmHg). Se requirieron intervenciones quirúrgicas para el tratamiento de la hipertensión ocular en el 4,8% de los sujetos tratados con ILUVIEN, frente al 0,5% de los sujetos con tratamiento simulado (ver sección 4.8). En consecuencia, en pacientes con una PIO elevada al inicio ILUVIEN debe ser utilizado con precaución, y la PIO deberá vigilarse cuidadosamente.
En el caso de un aumento de la PIO que no responda a los medicamentos o a los procedimientos para reducirla, es posible retirar el implante de ILUVIEN por vitrectomía.
En los estudios FAME se investigó a una cantidad limitada de pacientes con diabetes tipo 1, si bien la respuesta a ILUVIEN en estos sujetos no difirió significativamente de los sujetos con diabetes tipo 2.
La experiencia del efecto de ILUVIEN en los ojos tras una vitrectomía es limitada. Es probable que la eliminación del fármaco se acelere tras la vitrectomía, aunque no se espera que se vean afectadas las concentraciones en estado de equilibrio. Esto puede abreviar la duración de la acción del implante.
En los estudios FAME hubo un 24% de los sujetos en el grupo de tratamiento simulado a los que se trató en algún momento con anticoagulantes o bien antiplaquetarios, frente a un 72% en los sujetos tratados con ILUVIEN. Los sujetos tratados con ILUVIEN concomitantemente o dentro de los 30 días posteriores al cese del tratamiento con medicamentos anticoagulantes o antiplaquetarios tuvieron una incidencia ligeramente mayor de hemorragia conjuntival frente a los sujetos con tratamiento simulado (0,5% en el grupo con tratamiento simulado y 2,7% en los tratados con ILUVIEN. El otro único acontecimiento comunicado con una tasa de incidencia más alta en los sujetos tratados con ILUVIEN fueron las complicaciones oculares por la operación (0% con el tratamiento simulado y 0,3% en los tratados con ILUVIEN).
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones con otros medicamentos.
4.6 Fertilidad, embarazo y lactancia Embarazo
No existen datos suficientes del uso de acetónido de fluocinolona administrado en forma intravítrea en mujeres embarazadas. El tratamiento sistémico a largo plazo con glucocorticoides durante el embarazo aumenta el riesgo de retrasos en el crecimiento intrauterino e insuficiencia suprarrenal en el neonato. Los estudios realizados en animales han mostrado efectos teratógenos tras la administración sistémica (ver sección 5.3). En consecuencia, si bien se esperaría que la exposición sistémica del acetónido de fluocinolona sea muy baja tras el tratamiento intraocular local, no se recomienda el uso de ILUVIEN durante el embarazo a menos que el potencial beneficio justifique el riesgo potencial para el feto.
Lactancia
El acetónido de fluocinolona se excreta en la leche materna. No se esperan efectos sobre el niño debido a la vía de administración y los niveles sistémicos resultantes. Sin embargo, no se recomienda el uso de ILUVIEN durante la lactancia a menos que sea claramente necesario.
Fertilidad
No hay datos disponibles sobre fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Los pacientes pueden sufrir una disminución temporal de la visión tras la administración de ILUVIEN, y deberán abstenerse de conducir o utilizar máquinas hasta que esto se haya resuelto.
4.8 Reacciones adversas
Resumen del perfil de seguridad
ILUVIEN fue evaluado en 768 sujetos con edema macular diabético en los estudios clínicos FAME. Las reacciones adversas al fármaco notificadas más frecuentemente fueron operación de cataratas, cataratas y aumento de la presión intraocular.
En los estudios de fase 3, el 38,4% de los sujetos tratados con ILUVIEN requirió medicamentos reductores de la PIO, y el 4.8% requirió cirugía para reducir la PIO. El uso de medicamentos para reducir la PIO fue similar en los sujetos que recibieron dos o más tratamientos con ILUVIEN.
Se notificaron dos casos de endoftalmitis en sujetos tratados con ILUVIEN durante los estudios de fase 3. Esto representa una tasa de incidencia del 0,2% (2 casos dividido por 1022 inyecciones).
Mientras que la mayoría de los sujetos participantes en los estudios clínicos FAME recibieron sólo un implante (ver sección 5.1), se desconocen las implicaciones de seguridad a largo plazo de la retención del implante no bioerosionable dentro del ojo. En los estudios clínicos FAME, los datos de 3 años muestran que los acontecimientos como cataratas, aumento de la presión intraocular y formas flotantes se produjeron con una frecuencia sólo ligeramente superior en los sujetos que habían recibido 2 o más implantes. Esto se considera una función del aumento de la exposición al fármaco, más que un efecto del implante en sí. En los estudios no clínicos no hubo ninguna indicación de un aumento en los problemas de seguridad, a excepción de cambios de lentes en los ojos de los conejos con 2-4 implantes en un periodo de 24 meses. El implante está hecho de poliimida, y es esencialmente similar a un háptico de lente intraocular; en consecuencia, se espera que se mantenga inerte dentro del ojo.
Listado tabulado de reacciones adversas
Las reacciones adversas que aparecen a continuación se consideraron relacionadas con el tratamiento, y se clasifican según la siguiente convención: muy frecuentes (> 1/10); frecuentes (>1/100 a < 1/10); poco frecuentes (>1/1.000 a < 1/100); raras (>1/10.000 a < 1/1.000); y muy raras (< 1/10.000). Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Infecciones e infestaciones |
Poco frecuentes: endoftalmitis |
|
Trastornos del sistema nervioso |
Poco frecuentes: cefalea |
|
Trastornos oculares |
Muy frecuentes: cirugía de cataratas, catarata1, presión intraocular aumentada2, formas flotantes (miodesopsia) Frecuentes: glaucoma3, trabeculectomía, dolor ocular4, hemorragia del vítreo, hemorragia conjuntival, visión borrosa5, cirugía del glaucoma, agudeza visual disminuida, vitrectomía, trabeculoplastia Poco frecuentes: oclusión vascular retiniana6, trastorno del nervio óptico, maculopatía, atrofia óptica, úlcera conjuntival, neurovascularización del iris, exudados retinianos, degeneración vítrea, desprendimiento del cuerpo vítreo, opacificación de la cápsula posterior, adherencias del iris, hiperemia ocular, adelgazamiento de la esclerótica, extracción del implante extruido de la esclerótica, secreción ocular, prurito en el ojo |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Poco frecuentes: extrusión del implante, implante en la línea de visión, complicación de una intervención, dolor asociado a procedimiento terapéutico |
1 Incluye los términos de MedDRA para catarata (NEOM), catarata subcapsular, catarata cortical, catarata nuclear y catarata diabética.
2 Incluye los términos de MedDRA para presión intraocular aumentada e hipertensión ocular.
3 Incluye los términos de MedDRA para glaucoma, glaucoma de ángulo abierto, glaucoma dudoso, excavación del nervio óptico y relación fovea/papila del nervio óptico aumentada.
4 Incluye los términos de MedDRA para dolor ocular, irritación ocular y molestia ocular.
5 Incluye los términos de MedDRA para visión borrosa y deterioro visual.
6 Incluye los términos de MedDRA para oclusión venosa retiniana, oclusión arterial retiniana y oclusión vascular retiniana.
Descripción de reacciones adversas seleccionadas
El uso a largo plazo de corticosteroides puede provocar cataratas y aumento de la presión intraocular. Las frecuencias que aparecen a continuación reflejan los hallazgos en todos los pacientes participantes en los estudios FAME. Las frecuencias observadas en los pacientes con EMD no difirieron de forma significativa de las observadas en la población general.
La incidencia de cataratas en sujetos fáquicos fue de aproximadamente 82% en los sujetos tratados con ILUVIEN y del 50% en los tratados en forma simulada en los estudios clínicos de fase 3. El 80% de los sujetos fáquicos tratados con ILUVIEN requirió cirugía de cataratas al año 3, frente al 27% de los que recibieron tratamiento simulado, con una mayoría de sujetos que requirió cirugía a los 21 meses. La catarata subcapsular posterior es el tipo más frecuente de catarata relacionada con corticosteroides. La cirugía para este tipo de cataratas es más dificultosa, y puede estar asociada a un mayor riesgo de complicaciones quirúrgicas.
En los estudios FAME se excluyó a los sujetos con una PIO inicial >21 mmHg. La incidencia del aumento de la presión intraocular fue del 37%, y el 38% de los sujetos requirió medicamentos para reducir la PIO, de los cuales la mitad requirió como mínimo dos medicamentos para controlarla. El uso de medicamentos reductores de la PIO fue similar en los sujetos que recibieron tratamiento con un implante adicional durante el estudio. Además, el 5,6% (21/375) de los sujetos que recibieron un implante requirió un procedimiento quirúrgico o con láser para controlar la PIO (trabeculoplastia 5 (1,3%), trabeculectomía 10 (2,7%), endocicloablación 2 (0,5%) y otros procedimientos quirúrgicos 6 (1,6%)).
En el subgrupo de sujetos con PIO superior a la mediana al inicio (>15 mmHg), el 47% requirió medicamentos para reducir la PIO, y el porcentaje de procedimientos quirúrgicos o con láser aumentó a 7,1%. En este subgrupo, se trató a 5 (2,2%) sujetos con trabeculoplastia, a 7 (3,1%) con trabeculectomía, a 2 (0,9%) con endocicloablación y a 4 (1,8%) con otros procedimientos quirúrgicos para glaucoma.
4.9 Sobredosis
No se ha notificado ningún caso de sobredosis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: AGENTES ANTINFLAMATORIOS, corticosteroides, monofármacos Código ATC: S01BA15
Los corticosteroides inhiben la respuesta inflamatoria a diversos agentes desencadenantes. Inhiben el edema, la deposición de fibrina, la dilatación capilar, la migración de leucocitos, la proliferación capilar, la proliferación de fibroblastos, la deposición de colágeno y la formación de cicatrices asociadas con la inflamación.
Se piensa que los corticosteroides actúan por la inducción de las proteínas inhibidoras de la fosfolipasa A, denominadas colectivamente lipocortinas. Se postula que estas proteínas controlan la biosíntesis de mediadores potentes de la inflamación, tales como las prostaglandinas y los leucotrienos, al inhibir la liberación del precursor común ácido araquidónico. El ácido araquidónico es liberado de los fosfolípidos de la membrana por la fosfolipasa A2. Los corticosteroides también han demostrado reducir los niveles de factor de crecimiento endotelial vascular, una proteína que aumenta la permeabilidad vascular y provoca edema.
Se evaluó la eficacia de ILUVIEN en dos estudios multicéntricos aleatorizados, doble ciego, en grupos paralelos, que incorporaron a sujetos con edema macular diabético que previamente habían sido tratados con fotocoagulación con láser como mínimo una vez, y cada uno involucró tres años de seguimiento. Hubo un 74,4% de sujetos tratados con 1 implante, 21,6% con 2 implantes, 3,5% con 3 implantes, 0,5% con 4 implantes y 0% con >4 implantes. En ambos estudios, el criterio primario de valoración de eficacia fue la proporción de sujetos cuya visión había mejorado en 15 letras o más tras 24 meses. En cada uno de estos estudios, ILUVIEN satisfizo el criterio de valoración primario (en la Figura 1 pueden verse los resultados integrados del criterio de valoración primario).
Figura 1: Porcentaje de sujetos con una mejoría > 15 letras con respecto al inicio, estudios FAME integrados
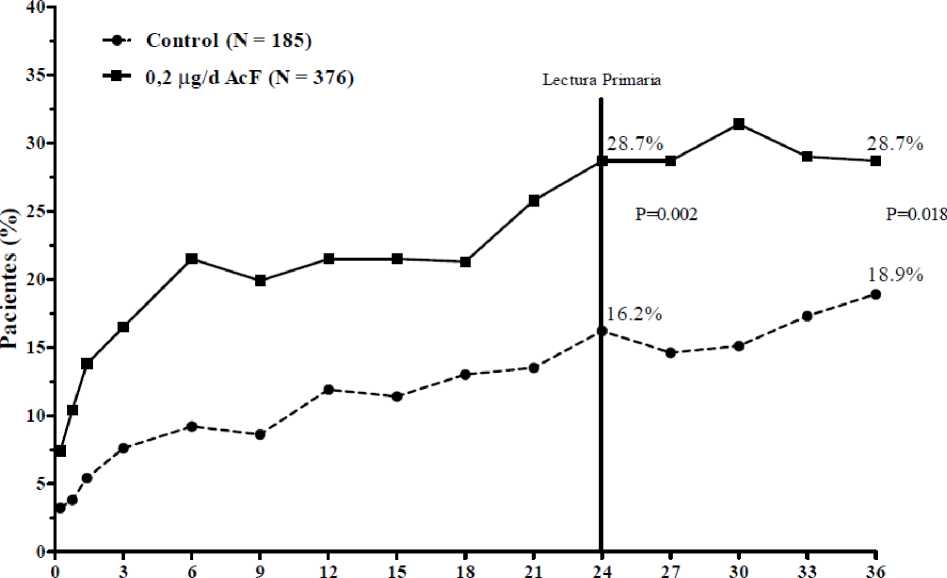
Meses
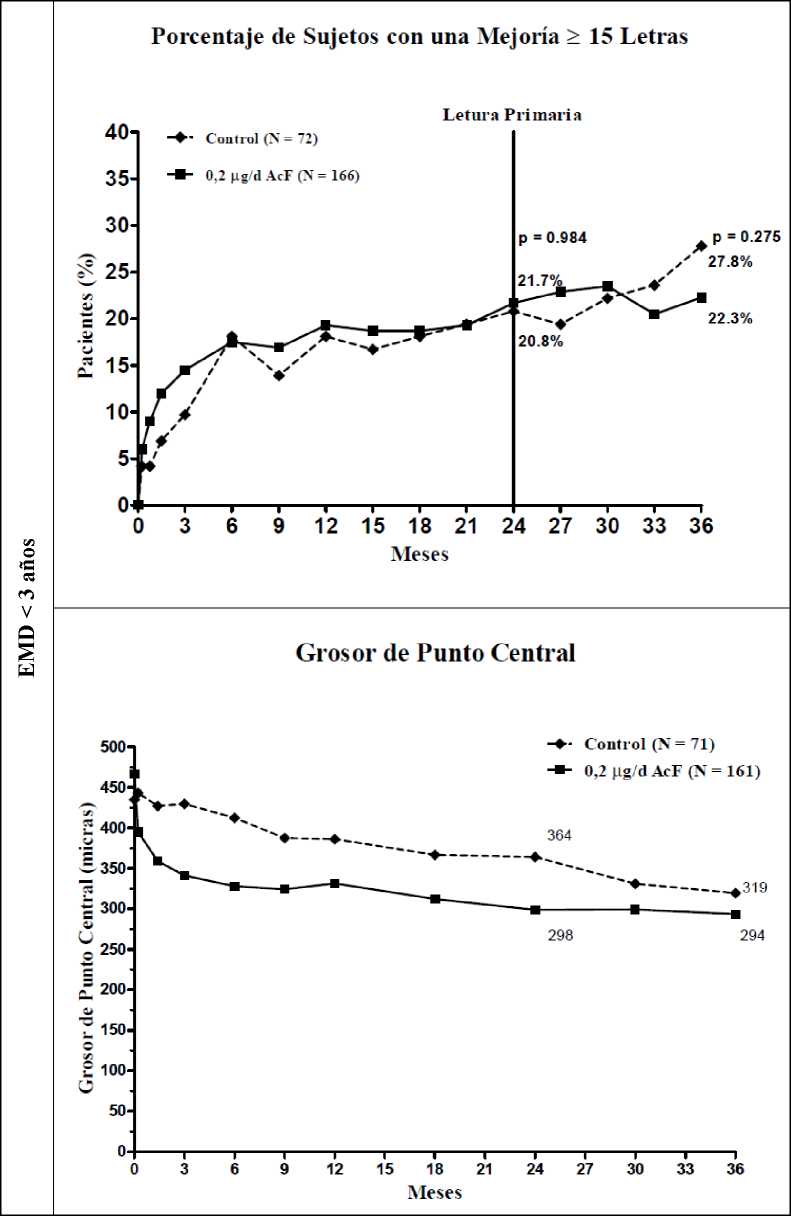
Al evaluar la eficacia como función de la duración de la enfermedad, los sujetos con una duración del EMD superior a la mediana (>3 años) tuvieron una significativa respuesta beneficiosa a ILUVIEN, mientras que aquellos con un EMD de menor duración no mostraron un beneficio adicional durante el tratamiento de control en lo que concierne a la mejoría visual (Figuras 2 y 3). Los datos de este subgrupo respaldan la indicación en la sección 4.1, sobre el uso en pacientes con EMD crónico (es decir, una duración de 3 años como mínimo).
Figura 2: Comparación del porcentaje de sujetos con una mejoría >15 letras con respecto a la MAVC inicial y cambio medio con respecto al grosor de punto central excesivo al inicio por subgrupo de duración del EMD > 3 años
4Ch
35-
30-
> 25-
20-
Porcentaje de Sujetos con una Mejoría >15 Letras
Lectura Primaria
Control (N - 112)
0,’ ng/tl AcF (A = 209)
34.4%
♦—
p < 0.001
34.0%
13.4%
13.4%
Meses
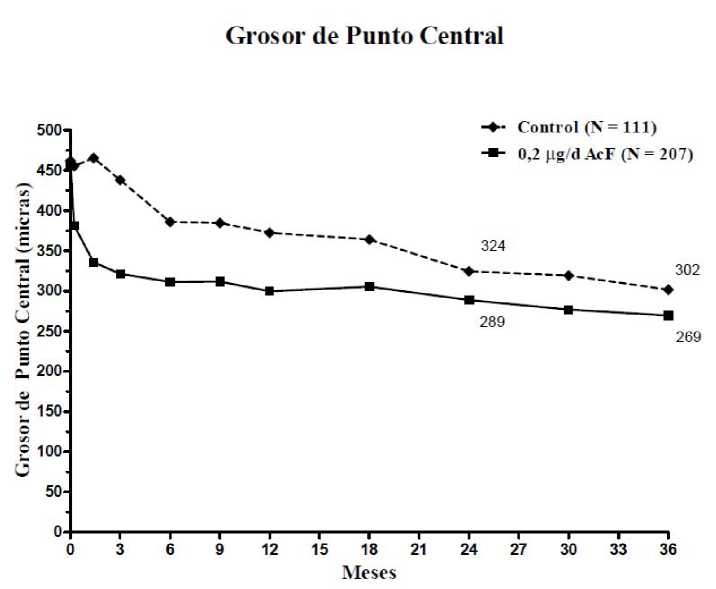
Figura 3: Comparación del cambio medio con respecto al grosor de punto central al inicio y porcentaje de sujetos con una mejoría >15 letras con respecto a la MAVC inicial por subgrupo de duración
del EMD < 3 años
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los estudios realizados con ILUVIEN en los diferentes grupos de la población pediátrica para el tratamiento del edema macular diabético (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
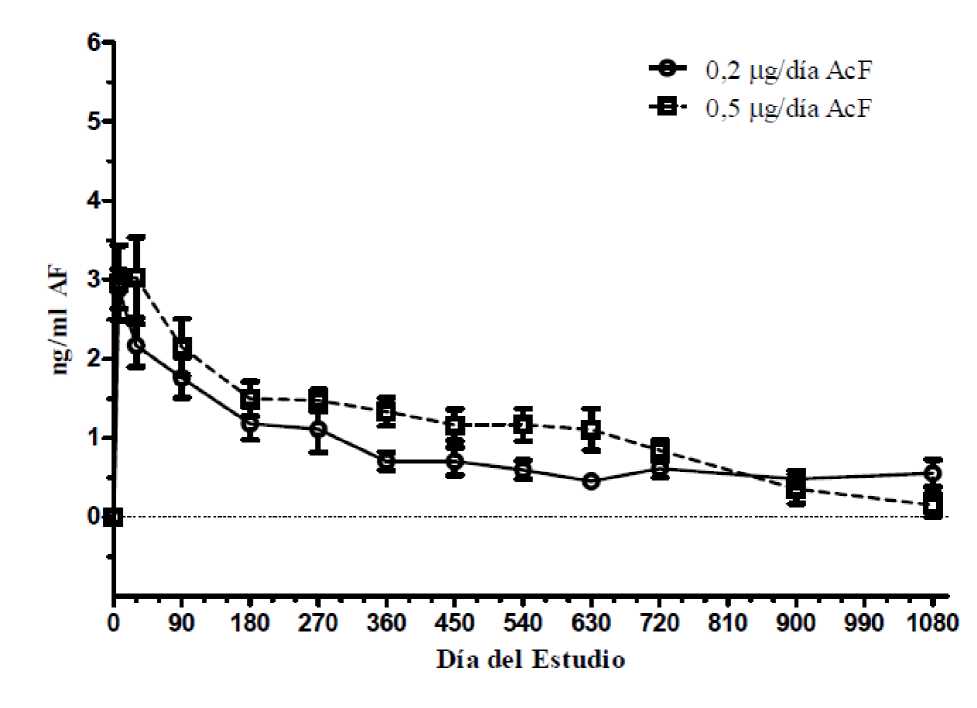
En un estudio de farmacocinética en humanos (C-01-06-002, el estudio FAMOUS), las concentraciones plasmáticas de acetónido de fluocinolona estuvieron por debajo del límite inferior de cuantificación del ensayo (100 pg/ml) en todos los puntos temporales, del día 1 al mes 36, lo que indica una exposición sistémica insignificante. Las concentraciones máximas de acetónido de fluocinolona en el humor acuoso se observaron el día 7 para la mayoría de los sujetos. Las concentraciones de acetónido de fluocinolona en el humor acuoso disminuyeron en los primeros 3-6 meses, y permanecieron básicamente en los mismos valores hasta el mes 36 en los sujetos a los que no se les repitió el tratamiento. Los sujetos a los que se les repitió el tratamiento experimentaron una segunda concentración máxima de acetónido de fluocinolona, similar a la que había seguido a la dosis inicial. Tras la repetición del tratamiento, las concentraciones de acetónido de fluocinolona en el humor acuoso volvieron a niveles aproximadamente similares a los observados en oportunidad del primer tratamiento.
Figura 4: Niveles de AF en el humor acuoso humano en sujetos que recibieron 1 implante
(Estudio FAMOUS)
5.3 Datos preclínicos sobre seguridad
Unicamente se observaron reacciones en los estudios no clínicos con exposiciones consideradas suficientemente superiores a la máxima humana, lo que indica poca relevancia para su uso clínico.
No se dispone de datos sobre mutagenia, carcinogenia ni toxicidad para la reproducción o el desarrollo para ILUVIEN. El acetónido de fluocinolona ha demostrado ser teratógeno en ratones y conejos tras la administración sistémica.
Evaluación del Riesgo Medioambiental (ERA)
El riesgo medioambiental como resultado del uso prescrito del implante de ILUVIEN es insignificante, en vista de la dosis extremadamente baja de acetónido de fluocinolona que se implanta.
El aplicador especialmente diseñado para la inyección de ILUVIEN no presenta ningún riesgo ambiental, ya que tras su único uso específico se lo descarta de inmediato en un recipiente para elementos medicinales/quirúrgicos cortantes desechables.
Ver sección 6.6 para consultar las instrucciones de manipulación y eliminación.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Alcohol polivinílico Tubo de poliimida Adhesivo de silicona
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
2 años
Tras la primera apertura de las tapas, usar de inmediato.
6.4 Precauciones especiales de conservación
Conservar por debajo de 30°C. No refrigerar o congelar.
No abrir la bandeja sellada hasta inmediatamente antes de la aplicación.
6.5 Naturaleza y contenido del envase
El implante se suministra en un aplicador para un único uso, con una aguja de calibre 25. Cada aplicador estéril contiene un implante cilíndrico de color castaño claro, de 3,5 mm de largo. El aplicador está envasado en una bandeja plástica sellada con tapa.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Desechar el aplicador en forma segura en un contenedor para elementos cortantes biopeligrosos.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Alimera Sciences Limited Royal Pavilion Wellesley Road Aldershot Hampshire GU11 1PZ Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Enero de 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
02/2012
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos, http://www.ema.europa.eu.