Canur 35 Microgramos/Hora Parches Transdermicos
Información obsoleta, busque otro"I
an
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Canur 35 microgramos/hora parches transdérmicos Canur 52,5 microgramos/hora parches transdérmicos Canur 70 microgramos/hora parches transdérmicos
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Canur 35 microgramos/hora parches transdérmicos Cada parche transdérmico contiene 20 mg de buprenorfina.
Área que contiene el principio activo: 25 cm2.
Velocidad nominal de liberación: 35 microgramos de buprenorfina por hora. Excipiente: aceite de soja 16 mg.
Canur 52,5 microgramos/hora parches transdérmicos Cada parche transdérmico contiene 30 mg buprenorfina.
Área que contiene el principio activo: 37.5 cm2.
Velocidad nominal de liberación: 52,5 microgramos de buprenorfina por hora. Excipiente: aceite de soja 24 mg.
Canur 70 microgramos/hora parches transdérmicos Cada parche transdérmico contiene 40 mg buprenofina.
Área que contiene el principio activo: 50 cm2.
Velocidad nominal de liberación: 70 microgramos de buprenorfina por hora. Excipiente: aceite de soja 32 mg.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Parche transdérmico.
Parche color marrón, rectangular, con esquinas redondeadas, identificado como:
Buprenorphin 35 pg/h Buprenorphin 52.5 pg/h Buprenorphin 70 pg/h
Cada parche está acondicionado en sobres sellados individuales.
.Uí1.
"I
an
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Dolor moderado a intenso oncológico y dolor intenso que no responda a analgésicos no opioides.
Canur no es idóneo para el tratamiento del dolor agudo.
4.2 Posología y forma de administración
Posología
• Pacientes mayores de 18 años
La dosis de Canur debe ser adaptada a las condiciones individuales de cada paciente (intensidad del dolor, sufrimiento, reacción individual).Debe administrarse la dosis más baja posible que proporcione alivio adecuado del dolor. Existen parches transdérmicos de tres concentraciones para facilitar la adaptación de este tratamiento: Canur 35 microgramos/hora, Canur 52,5 microgramos/hora y Canur 70 microgramos/hora.
- Selección de la dosis inicial:
Los pacientes que no han recibido previamente ningún analgésico deben empezar con la dosis más baja del parche transdérmico (Canur 35 microgramos/hora ).Los pacientes a los que se les haya administrado un analgésico (no opiode) incluido en el primer escalón de la OMS o un analgésico (opioide débil) incluido en el segundo escalón de la OMS deben empezar también con Canur 35 microgramos/hora. Según las recomendaciones de OMS, dependiendo de la situación médica global del paciente, se puede continuar con la administración de un analgésico no opioide.
Cuando se cambie de un analgésico de tercer escalón (opioide fuerte) a Canur y se haya elegido la concentración inicial del parche transdérmico, debe tenerse en cuenta la naturaleza de la medicación previa, la vía de administración y la dosis diaria media para evitar la recurrencia del dolor. En general, es aconsejable ajustar la dosis de forma individual, comenzando por la menor dosis disponible del parche transdérmico (Canur 35 microgramos/hora). La experiencia clínica ha mostrado que pacientes que fueron tratados previamente con dosis diarias más altas de un opioide fuerte (en el rango de dosis correspondiente a 120 mg de morfina oral aproximadamente) pueden comenzar el tratamiento con la dosis superior siguiente del parche transdérmico (ver sección 5.1).
Con el fin de permitir la adaptación a la dosis individual en un periodo de tiempo adecuado, deberá disponerse durante el ajuste de dosis de suficientes analgésicos de liberación inmediata suplementarios.
La concentración necesaria de Canur debe adaptarse a las necesidades individuales de cada paciente, y debe revisarse a intervalos regulares.
Después de la aplicación del primer parche transdérmico de Canur las concentraciones séricas de buprenorfina aumentan lentamente tanto en pacientes que han sido tratados previamente con analgésicos como en los que no. Por lo tanto, es improbable que inicialmente se produzca una aparición rápida del efecto. En consecuencia, solo debe realizarse una primera evaluación del efecto analgésico después de 24 horas.
Durante las primeras 12 horas después de cambiar a Canur debe administrarse la misma dosis de la medicación analgésica previa (a excepción de los opioides por vía transdérmica) Durante las siguientes 12 horas debe proporcionarse una adecuada medicación de rescate a demanda.
- Ajuste de la dosis y tratamiento de mantenimiento:
Canur puede ser aplicada por un máximo de 72 horas.
ÍTTI
Canur debe reemplazarse como máximo a las 72 horas (3 días). Debe ajustarse la dosis de forma individual hasta que se obtenga una eficacia analgésica. Si al final del periodo inicial de aplicación la analgesia es insuficiente, debe aumentarse la dosis aplicando más de un parche transdérmico de Canur de la misma concentración o cambiando a la siguiente concentración de parche transdérmico. No deben aplicarse al mismo tiempo más de dos parches transdérmicos sea cual sea su concentración.
Antes de la aplicación de la siguiente concentración de Canur debe tenerse en consideración la cantidad total de opioides administrados además del parche transdérmico previo, es decir, la cantidad total de opioides necesaria, y ajustar la dosis en consecuencia.
Los pacientes que necesiten analgésicos adicionales (por ejemplo para el dolor irruptivo) durante el tratamiento de mantenimiento pueden tomar, por ejemplo, uno o dos comprimidos sublinguales de buprenorfina 0,4 mg cada 24 horas además del parche transdérmico. Si habitualmente es necesario añadir entre 0,4-0,6 mg de buprenorfina sublingual debe utilizarse la siguiente concentración.
• Pacientes menores de 18 años
No se han realizado estudios en pacientes menores de 18 años, por tanto, no se recomienda el uso de Canur en este grupo de pacientes.
• Pacientes ancianos
No es necesario un ajuste de la dosis de Canur en pacientes ancianos.
• Pacientes con insuficiencia renal
Puede usarse en pacientes con insuficiencia renal, incluyendo pacientes dializados, debido a que la farmacocinética de buprenorfina no se ve alterada durante la evolución de la misma.
• Pacientes con insuficiencia hepática
La buprenorfina se metaboliza en el hígado. La intensidad y la duración de su acción pueden verse afectadas en pacientes con alteración de la función hepática. Por lo tanto, los pacientes con insuficiencia hepática deben ser controlados cuidadosamente durante el tratamiento con Canur.
Método de aplicación
Canur debe aplicarse sobre la piel limpia no irritada, sobre una superficie lisa exenta de vello. No debe aplicarse en ninguna parte de la piel donde haya grandes cicatrices. Los lugares preferibles de la parte superior del cuerpo son: la parte superior de la espalda o sobre el pecho, bajo la clavícula. Debe cortarse el vello que quede con unas tijeras (no afeitarlo). Si el lugar de aplicación necesita lavarse debe hacerse con agua. No utilizar jabón ni ningún otro agente de limpieza. Deben evitarse los preparados para la piel que puedan afectar la adhesión del parche transdérmico en el área seleccionada para la aplicación de Canur.
La piel debe estar completamente seca antes de la aplicación. Debe aplicarse Canur inmediatamente después de sacarlo del sobre.
Canur debe llevarse puesto de forma continuada hasta un máximo de 3 días. Al reemplazar el parche transdérmico previo, el nuevo parche transdérmico de Canur debe aplicarse en un lugar diferente de la piel. Deben transcurrir al menos 1 semana antes de poder aplicar un parche transdérmico nuevo en el mismo área de la piel.
• Como poner el parche
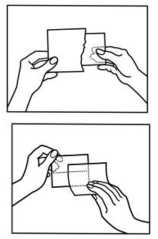
parte de aluminio.
1. No abra el sobre hasta el momento del uso del parche
2. Primero se retira la lámina protectora que cubre la
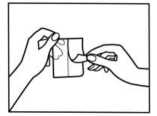
3. Despegue cuidadosamente la mitad de la lámina. Intente no tocar la la parte adhesiva del parche.

4. Pegue el parche en el área de la piel que haya elegido y retire el resto de la lámina.
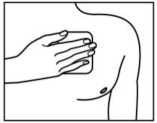
5. Presione el parche contra su piel con la palma de su mano durante 30-60 segundos. Asegúrese que todo el parche está en contacto con su piel, especialmente los bordes.
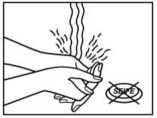
6. Debe aclararse las manos con abundante agua después de usar el parche. No usar ningún producto de limpieza.
Siempre que el parche se aplique correctamente, es muy difícil que se despegue. El baño, la ducha o la natación no afecta al parche pero no debe exponerse a excesivo calor (por ejemplo sauna o radiación infrarroja).
Sin embargo si el parche comienza a retirarse antes del siguiente cambio no puede usarse este mismo parche de nuevo. Pegue uno nuevo inmediatamente (ver “cambio de parche”).
• Cambio de parche
- Retirar el parche antiguo.
- Dóblelo por la mitad con el lado adhesivo hacía dentro.
- El parche se debe eliminar cuidadosamente.
- Se debe aplicar un nuevo parche en otra área adecuada (detallado anteriormente). Deben transcurrir al menos 2 aplicaciones antes de poder aplicar un parche nuevo en la misma área de la piel.
• Duración del tratamiento
Canur no debe aplicarse bajo ninguna circunstancia durante más tiempo que el estrictamente necesario. Si fuera preciso un tratamiento del dolor a largo plazo con Canur debido a la naturaleza y gravedad de la enfermedad, debe llevarse a cabo un control regular y cuidadoso (si fuera preciso con interrupciones del tratamiento) para establecer si es necesario alargar el tratamiento y en qué medida.
• Supresión del tratamiento con el parche Canur
Después de retirar Canur las concentraciones séricas de buprenorfina disminuyen gradualmente por lo que el efecto analgésico se mantiene durante algún tiempo. Esto se debe considerar cuando el tratamiento con Canur vaya a ir seguido de otros opioides.
Como regla general, no debe administrase otro opioide en las 24 horas posteriores a la retirada de Canur. En estos momentos solo tenemos cierta información disponible sobre la dosis de inicio de otros opioides administrados después de la interrupción de Canur.
4.3 Contraindicaciones
Canur está contraindicado en:
• Hipersensibilidad al principio activo, soja o cacahuete o a alguno de los excipientes
incluidos en la sección 6.1.
• En pacientes opioide-dependientes y en el tratamiento de abstinencia de narcóticos.
• Situaciones en las que la función y el centro repiratorio están gravemente dañadas o puedan estarlo.
• Pacientes que están recibiendo inhibidores de la MAO o que los hayan tomado en las
dos últimas semanas (ver sección 4.5).
• Pacientes que padezcan miastenia grave.
• Pacientes que padezcan delirium tremens.
4.4 Advertencias y precauciones especiales de empleo
Canur sólo debe utilizarse con precaución especial en caso de intoxicación etílica aguda, trastornos convulsivos, en pacientes con traumatismmo craneoencefálico, shock, disminución del grado de conciencia de origen desconocido, aumento de la presión intracraneal sin posibilidad de ventilación.
La buprenorfina produce ocasionalmente depresión respiratoria. Por lo tanto, debe tenerse precaución en el tratamiento de pacientes con alteración de la función respiratoria o en pacientes que estén recibiendo medicamentos que puedan originar depresión respiratoria.
La buprenorfina presenta una dependencia sustancialmente menor que los agonistas opiodes puros. En estudios realizados con Canur en pacientes y en voluntarios sanos no se han observado reacciones de abstinencia. Sin embargo, después del uso prolongado de Canur no pueden descartarse síntomas de abstinencia similares a los producidos por la retirada de opioides (ver sección 4.8). Estos síntomas son: agitación, ansiedad, nerviosismo, insomnio, hipercinesia, temblores y alteraciones gastrointestinales.
En los pacientes que presentan abuso de opioides la sustitución con buprenorfina puede prevenir los síntomas de abstinencia. A raíz de esto, se ha producido algún caso de abuso de buprenorfina, por lo que debe tenerse precaución a la hora de prescribírselo a pacientes sospechosos de tener problemas de drogodependencia.
La buprenorfina se metaboliza en el hígado. La intensidad y la duración del efecto pueden verse modificadas en pacientes con alteraciones de la función hepática. Por lo
ÍTTI
tanto dichos pacientes deben ser controlados cuidadosamente durante el tratamiento con Canur.
Se debe advertir a los deportistas que este medicamento puede producir un resultado analítico positivo en las pruebas de control del dopaje.
Pacientes con fiebre/calor externo
La fiebre y la presencia de calor pueden incrementar la permeabilidad de la piel.
Teóricamente en dichas situaciones las concentraciones séricas de buprenorfina pueden aumentar durante el tratamiento con Canur. Por lo tanto, durante el tratamiento con Canur debe prestarse atención al aumento de la posibilidad de reacciones opioides en pacientes febriles o en aquellos con incremento de temperatura de la piel debido a otras causas.
4.5 Interacción con otros medicamentos y otras formas de interacción
Cuando se administraron inhibidores de la MAO dentro de los últimos 14 días previos a la administración del opioide petidina se observaron interacciones potencialmente mortales que afectaban al sistema nervioso central y a las funciones respiratoria y cardiovascular. No se pueden descartar las mismas interacciones entre los inhibidores de la MAO y Canur (ver sección 4.3).
Cuando se aplica Canur conjuntamente con otros opioides, anestésicos, hipnóticos, sedantes, antidepresivos, neurolépticos y, en general, medicamentos que depriman el sistema respiratorio y el sistema nervioso central, los efectos sobre éste último pueden verse intensificados. Esto es aplicable también al alcohol.
Si se administra conjuntamente con inhibidores o inductores del CYP 3A4 la eficacia de Canur puede verse intensificada (en el caso de los inhibidores como ketoconazol) o debilitada (en el caso de los inductores, como fenobarbital, carbamazepina, fenitoina y rifampicina).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No existen datos suficientes sobre el uso de Canur en mujeres gestantes. Los estudios realizados en animales han mostrado toxicidad sobre la reproducción (ver sección 5.3). No se conoce el riesgo potencial en humanos.
Hacia el final del embarazo dosis altas de buprenorfina pueden inducir depresión respiratoria en el neonato incluso después de un periodo corto de administración. La administración prolongada de buprenorfina durante los tres últimos meses de embarazo puede producir síndrome de abstinencia en el neonato.
Por lo tanto, Canur no debe usarse durante el embarazo y en mujeres en edad fértil que no estén empleando métodos anticonceptivos apropiados.
Lactancia
Buprenorfina se excreta en la leche materna. Se ha observado que en ratas, buprenorfina inhibe la lactancia.
Canur no debe utilizarse durante la lactancia.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Incluso si se utiliza siguiendo las instrucciones, Canur puede afectar las reacciones del paciente hasta el punto de que la seguridad vial y la capacidad para utilizar maquinaria puedan verse disminuidas. Esto es aplicable especialmente cuando se utiliza junto con otras sustancias que actúan a nivel central incluyendo el alcohol, tranquilizantes, sedantes e hipnóticos.
Los pacientes que lleven un parche transdérmico de Canur no deben conducir ni utilizar maquinaria, por lo menos durante las 24 horas posteriores a la retirada del parche.
4.8 Reacciones adversas
Se comunicaron las siguientes reacciones adversas tras la administración de Canur en ensayos clínicos y tras la comercialización.
Las frecuencias son las siguientes:
Muy frecuentes (>1/10)
Frecuentes (>1/100 a <1/10)
Poco frecuentes (>1/1.000 a <1/100)
Raras (>1/10.000 a <1/1.000)
Muy raras (<1/10.000)
Desconocidas (no se pueden estimar a partir de los datos disponibles)
Las reacciones adversas sistémicas comunicadas más frecuentes fueron náuseas y vómitos.
Las reacciones adversas locales comunicadas más frecuentemente fueron eritema y prurito.
• Trastornos del sistema inmune
Muy raras: reacciones alérgicas graves.
• Trastornos metabólicos y nutricionales
Raras: pérdida de apetito.
• Trastornos psiquiátricos
Poco frecuentes: confusión, trastornos del sueño, inquietud.
Raras: efectos psicoticomiméticos (por ejemplo.
alucinaciones, ansiedad, pesadillas), disminución de la libido.
Muy raras: dependencia, cambios de humor.
• Trastornos del sistema nervioso
Frecuentes: dolor de cabeza, mareos.
Poco frecuentes: sedación, somnolencia.
Raras: dificultad en la concentración, trastornos del habla,
adormecimiento, desequilibrio, parestesia (sensación de calor u hormigueo en la piel).
Muy raras: contracción muscular, alteraciones del gusto.
• Trastornos oculares
Raras: alteraciones visuales, visión borrosa, edema palpebral.
*
A** A
Muy raras: miosis.
• Trastorno del oído y laberinto
Muy raras: otalgia (dolor de oídos).
• Trastornos cardiacos y vasculares
Poco frecuentes:
trastornos circulatorios (tales como hipotensión o incluso,
raramente, colapso circulatorio).
Raras: sofocos.
• Trastornos respiratorios, del tórax y mediastino
Frecuentes: disnea.
Raras: depresión respiratoria.
Muy raras: hiperventilación, hipo.
• Trastornos gastrointestinales
Muy frecuentes: Frecuentes: Poco frecuentes:
Muy raras:
náuseas.
vómitos, estreñimiento. sequedad de boca. Raras: pirosis. nauseas.
Trastornos de la piel y tejidos subcutáneos
Muy frecuentes: Frecuentes: Poco frecuentes: Raras:
Muy raras:
eritema, prurito. exantema, diaforesis. rash
urticarias. pústulas, vesículas.
• Trastornos renales y urinarios
Poco frecuentes: retención urinaria, alteraciones de la micción.
• Trastorno del aparato reproductor y de la mama
Raras: disminución de la erección.
• Trastornos generales y alteraciones en el lugar de administración
edema, cansancio. debilidad.
síntomas de abstinencia, reacciones en el lugar de
Frecuentes: Poco frecuentes:
Raras:
administración.
Muy raras:
dolor torácico.
En algunos casos tienen lugar reacciones alérgicas tardías con marcados signos de inflamación. En estos casos se debe retirar el tratamiento con Canur.
Buprenorfina tiene un bajo riesgo de dependencia. Después del tratamiento con Canur, es improbable que aparezcan síntomas de abstinencia. Este hecho es debido a la muy lenta disociación de buprenorfina de los receptores opioides y a la disminución gradual de las concentraciones séricas de buprenorfina (normalmente durante un periodo de 30 horas después de la eliminación del último
MI!
SAI
50
Tfl*
ílMt
ÍTTI
parche transdérmico). Sin embargo, después del tratamiento a largo plazo con Canur no se puede excluir la aparición de síntomas de abstinencia, similares a los que aparecen durante la retirada de un opioide. Estos síntomas incluyen: agitación, ansiedad, nerviosismo, insomnio, hipercinesia, temblor y trastornos gastrointestinales.
Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: https://www.notificaram.es
4.9 Sobredosis
La buprenorfina tiene un amplio margen de seguridad. Debido a la tasa de liberación controlada de pequeñas cantidades de buprenorfina en la circulación sanguínea es improbable que se produzcan concentraciones altas o tóxicas de buprenorfina en la sangre.
La concentración sérica máxima de buprenorfina tras la aplicación del parche transdérmico de 70 microgramos/hora de Canur es seis veces menor que la concentración alcanzada tras la administración intravenosa de la dosis terapéutica de 0,3 mg de buprenorfina.
• Síntomas
Principalmente, en la sobredosis de buprenorfina se esperan síntomas similares a los de otros analgésicos que actúan a nivel central (opioides). Estos son: depresión respiratoria, sedación, somnolencia, náuseas, vómitos, colapso cardiovascular y miosis acusada.
• Tratamiento
Aplicar las medidas de emergencia generales. Mantener las vías aéreas libres (aspiración), mantener la respiración y circulación dependiendo de los síntomas. La naloxona tiene un efecto limitado sobre los efectos depresores respiratorios causados por la buprenorfina. Se necesita la administración de altas dosis, bien en bolos repetidos, o bien en infusión intravenosa (por ejemplo empezando con una administración en bolos de 1-2 mg intravenosa). Una vez se ha conseguido un efecto antagonista adecuado, se recomienda la administración por infusión intravenosa para mantener constantes los niveles plasmáticos de naloxona. Por lo tanto se debe establecer una ventilación adecuada.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: opioides, derivados de oripavina.
Código ATC: N02AE0I.
La buprenorfina es un opioide potente con actividad agonista sobre receptores mu-opioides y actividad antagonista sobre los receptores kappa opioides. La buprenorfina se asemeja a la morfina, pero tiene su propia farmacología específica y sus propias características clínicas.
Adicionalmente, numerosos factores, por ejemplo: indicación y situación clínica, vía de administración y variabilidad interindividual, influyen sobre la analgesia y por lo tanto, deben ser tenidos en cuenta cuando se comparen analgésicos.
En la práctica clínica diaria, los diferentes opioides de clasifican en función de su potencia relativa, aunque generalmente se considera una simplificación.
La potencia relativa de buprenorfina en las diferentes formas de aplicación y en diferentes
ÍTTI
ámbitos clínicos ha sido descrita en la literatura de la siguiente manera:
• Morfina v.o : BUP i.m como 1: 67-150 (dosis única: modelo de dolor agudo)
• Morfina v.o : BUP s.l como 1: 60-100 (dosis única, modelo de dolor
agudo; dosis múltiple, dolor crónico, dolor oncológico)
• Morfina v.o : BUP TDS como 1: 75-115 (dosis múltiple, dolor crónico)
Los efectos adversos son similares a los de otros analgésicos opioides potentes. La buprenorfina parece tener una tendencia menor a la dependencia que la morfina.
5.2 Propiedades farmacocinéticas
• Características generales del principio activo
La buprenorfina se une a proteínas plasmáticas en un 96%.
La buprenorfina se metaboliza en el hígado en N-dealquilbuprenorfina (norbuprenorfina) y en metabolitos glucuronido-conjugados. Dos tercios del principio activo se eliminan inalterados por las heces y un tercio se elimina a través la orina como buprenorfina conjugada o de-alquilada. Existen indicios de recirculación enterohepática.
Los estudios en ratas gestantes y no gestantes han mostrado que la buprenorfina atraviesa la barrera hematoencefálica y placentaria. Las concentraciones en el cerebro (que contenían solamente buprenorfina inalterada) después de la administración parenteral fueron de 2 a 3 veces mayores que tras la administración oral.
Después de la administración intramuscular u oral la buprenorfina se acumula aparentemente en la luz gastrointestinal fetal, presumiblemente debido a la excreción biliar, ya que la circulación enterohepática no se ha desarrollado totalmente.
• Características de Canur en voluntarios sanos
Tras la aplicación de Canur, la buprenorfina se absorbe a través de la piel. La liberación continua de buprenorfina a la circulación sistémica se realiza a través de la liberación controlada del sistema matricial basado en un polímero adhesivo.
Tras la aplicación inicial de Canur las concentraciones plasmáticas de buprenorfina se incrementan gradualmente, y después de 4 a 12 horas las concentraciones plasmáticas alcanzan la concentración mínima eficaz de 100 pg/ml. A partir de los estudios realizados en voluntarios sanos con Canur 35 microgramos/hora se ha determinado una Cmáx media de 273 pg/ml y una tmáx media de 34 h y con Canur 70 microgramos/h una Cmáx media de 425 pg/ml y una tmáx media de 29 h.
Después de haber retirado Canur, las concentraciones plasmáticas de buprenorfina disminuyen de forma regular y se eliminan con una vida media de aproximadamente 25 horas (en el intervalo 24-27). Debido a la continua absorción de buprenorfina procedente del depósito en la piel la eliminación es más lenta que después de la administración intravenosa.
5.3 Datos preclínicos sobre seguridad
Los estudios toxicológicos normalizados no han mostrado indicios de ningún riesgo potencial particular en humanos. En ensayos en los que se utilizaron dosis repetidas de buprenorfina en ratas se redujo el aumento del peso corporal.
Los estudios sobre la fertilidad y la capacidad reproductora general en ratas no mostraron
ÍTTI
efectos perjudiciales. Los estudios en ratas y conejos revelaron signos de fetotoxicidad y un incremento de la pérdida postimplantación.
Los estudios en ratas mostraron una disminución del crecimiento intrauterino, retrasos en el desarrollo de algunas funciones neurológicas y una alta mortalidad peri-postnatal en neonatos después del tratamiento de las madres durante la gestación o la lactancia.
Existen indicios de que el parto complicado y una lactancia reducida contribuyen a estos efectos. No hubo evidencia de embriotoxicidad incluida la teratogenicidad en ratas o conejos.
Los ensayos in-vitro e in-vivo sobre el potencial mutagénico de buprenorfina no indicaron ningún efecto clínico relevante.
En estudios a largo plazo en ratas y ratones no hubo indicios de potencial carcinogénico relevante en humanos.
Los datos toxicológicos disponibles no indican un potencial sensibilizador de los aditivos del parche transdérmico.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Matriz adhesiva (con buprenorfina): estireno-butadieno-estireno (SBS) y grupo de copolímeros de estireno-butadieno, resinas colophonium, antioxidantes (2,4-bis (1,1-dimetiletil)fenil fosfito (3:1), Tris (2,4-di-terc-butilfenil) fosfato), aceite de extracto de hoja de aloe vera (también contiene aceite refinado de soja y acetato de alfa-tocoferol).
Lámina separadora de polietileno-pigmentado, resina termoplástica y poliéster recubierto de vapor de aluminio.
Tinta de impresión azul.
Capa protectora de liberación: capa de poliéster, por un lado siliconado (para retirar antes de aplicar).
6.2 Incompatibilidades No procede
6.3 Periodo de validez
18 meses
6.4 Precauciones especiales de conservación
No conservar a temperatura superior de 25°C.
No congelar.
6.5 Naturaleza y contenido del envase
Cada parche transdérmico se cubre con una amplia lámina siliconada protectora de aluminio PETP y se envasa individualmente en un sobre precintado. Cada sobre está compuesto por PETP, lámina de aluminio y polietileno de baja densidad.
Los envases contienen 4, 5, 8, 10, 16, 20 y 24 parches (6 cajas de 4 parches) precintados individualmente.
Puede que solamente estén comercializadas algunas presentaciones.
ÍTTI
6.6 Precauciones especiales de eliminación
Grandes cantidades de buprenorfina permanecen en los parches transdérmicos incluso después de su uso.
Los parches transdérmicos usados deberían doblarse con la parte adhesiva hacía el interior y eliminarlos o si es posible devolverse a la farmacia. Cualquier medicamento que no haya sido usado debería devolverse a la farmacia.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Laboratorios Gebro Pharma, S.A.
Avda. Tibidabo, 29 - 08022 Barcelona (España)
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
10. FECHA DE LA REVISIÓN DEL TEXTO
Febrero 2014
12 de 12