Zeffix 100 Mg Comprimidos Con Cubierta Pelicular
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Zeffix 100 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 100 mg de lamivudina. Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido)
Comprimidos de color caramelo, recubiertos con película, con forma de cápsulas, biconvexos, de unas dimensiones aproximadamente de 11 mm x 5 mm y marcados “GX CG5” en una cara.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Zeffix está indicado para el tratamiento de la hepatitis B crónica en adultos con:
• enfermedad hepática compensada con evidencia de replicación viral activa, niveles de alanina aminotransferasa (ALT) sérica elevados de forma persistente y evidencia histológica de inflamación hepática activa y/o fibrosis. Sólo se debe considerar el comienzo del tratamiento con lamivudina cuando no está disponible o no sea apropiado el uso de un agente antiviral alternativo con una barrera genética más alta a resistencia (ver sección 5.1).
• enfermedad hepática descompensada en combinación con un segundo agente sin resistencia cruzada a lamivudina (ver sección 4.2).
4.2 Posología y forma de administración
El tratamiento con Zeffix debe ser iniciado por un médico con experiencia en el tratamiento de la
hepatitis B crónica.
Posología
Adultos
La dosis recomendada de Zeffix es de 100 mg una vez al día.
En pacientes con enfermedad hepática descompensada, lamivudina se debe usar siempre en combinación con un segundo agente, sin resistencia cruzada a lamivudina, para reducir el riesgo de resistencia y conseguir una supresión viral rápida.
Duración del tratamiento
Se desconoce la duración óptima del tratamiento. 1
haya pérdida de eficacia (ver sección 4.4). El ALT sérico y los niveles del ADN del VHB deben ser controlados regularmente después de la discontinuación de tratamiento para percatarse de cualquier recaída virológica tardía.
• En pacientes con HBC HBeAg negativos (mutante pre-core), sin cirrosis, el tratamiento debe ser administrado por lo menos hasta la seroconversión de HBs o haya evidencia de pérdida de eficacia. Con tratamiento prolongado, se recomienda un control regular para confirmar que la continuación de la terapia seleccionada sigue siendo apropiada para el paciente.
• No se recomienda la interrupción del tratamiento en pacientes con enfermedad hepática descompensada o cirrosis y en receptores de trasplante hepático (ver sección 5.1).
Si se interrumpe el tratamiento con lamivudina los pacientes deben ser controlados periódicamente para comprobar la existencia de una hepatitis recurrente (ver sección 4.4).
Resistencia clínica
En pacientes con HBC tanto HBeAg positivos como HBeAg negativos, el desarrollo del mutante YMDD (tirosina-metionina-aspartato-aspartato) del VHB puede dar lugar a una menor respuesta terapéutica a lamivudina, indicada por un aumento del ADN del VHB y de ALT con respecto a los niveles anteriores durante el tratamiento. Con el fin de reducir el riesgo de resistencia en pacientes que reciben lamivudina en monoterapia, si el ADN del VHB continúa siendo detectable en suero a las 24 semanas de tratamiento o más se debe considerar el cambio o la adición de un agente alternativo sin resistencia cruzada a lamivudina en base a las guías terapéuticas (ver sección 5.1).
Para el tratamiento de pacientes coinfectados con el VIH y que están recibiendo o que planean recibir tratamiento con lamivudina o con la combinación lamivudina-zidovudina, se debe mantener la dosis prescrita de lamivudina para la infección por el VIH (normalmente 150 mg/dos veces al día en combinación con otros antirretrovirales).
Poblaciones especiales
Insuficiencia renal
Las concentraciones séricas de lamivudina (AUC) aumentan en pacientes con insuficiencia renal moderada a grave, debido a una disminución del aclaramiento renal. Por lo tanto, debe reducirse la dosis en pacientes con un aclaramiento de creatinina inferior a 50 ml/minuto. Cuando se requieran dosis inferiores a 100 mg, debe utilizarse Zeffix solución oral (ver Tabla 1 a continuación).
Tabla 1: Posología de Zeffix en pacientes con aclaramiento renal disminuido.
|
Aclaramiento de creatinina |
Primera dosis de Zeffix |
Dosis de mantenimiento |
|
ml/min |
solución oral 1 |
una vez al día |
|
30 a < 50 |
20 ml (100 mg) |
10 ml (50 mg) |
|
15 a < 30 |
20 ml (100 mg) |
5 ml (25 mg) |
|
5 a < 15 |
7 ml (35 mg) |
3 ml (15 mg) |
|
< 5 |
7 ml (35 mg) |
2 ml (10 mg) |
*Zeffix solución oral conteniendo 5 mg/ml de lamivudina.
Los datos disponibles de pacientes sometidos a hemodiálisis intermitente (diálisis menor o igual a 4 h 2-3 veces a la semana), indican que tras la reducción de la dosis inicial de lamivudina para ajustarse al aclaramiento de creatinina del paciente, no se precisan ajustes de dosis adicionales mientras se practique la diálisis.
Insuficiencia hepática
Los datos obtenidos en pacientes con insuficiencia hepática, incluyendo aquellos con enfermedad hepática en fase terminal a la espera de trasplante, demuestran que la farmacocinética de lamivudina no se ve afectada de forma significativa por la disfunción hepática. Según estos datos no es necesario ajustar la dosis en pacientes con insuficiencia hepática, a menos que esté acompañada de insuficiencia renal.
Pacientes de edad avanzada
En los pacientes de edad avanzada, el envejecimiento con la correspondiente disminución de la función renal no tiene efecto clínicamente significativo en la exposición a lamivudina, excepto en pacientes con un aclaramiento de creatinina de < 50 ml/min.
Población pediátrica
No se ha establecido la seguridad y eficacia de Zeffix en niños y adolescentes menores de 18 años. Los datos disponibles en la actualidad, que se describen en las secciones 4.4 y 5.1, no permiten hacer una recomendación posológica.
Forma de administración Vía oral.
Zeffix puede tomarse con o sin alimentos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Acidosis láctica y hepatomegalia grave con esteatosis
Con el uso de análogos de nucleósidos se ha comunicado la aparición de acidosis láctica (en ausencia de hipoxemia), a veces mortal, habitualmente asociada a hepatomegalia grave y esteatosis hepática. Este riesgo no puede excluirse dado que lamivudina es un análogo de nucleósido. El tratamiento con análogos de nucleósido se debe interrumpir si aparece una elevación rápida de los niveles de aminotransferasas, hepatomegalia progresiva o acidosis metabólica/láctica de etiología desconocida. Síntomas digestivos benignos, como náuseas, vómitos y dolor abdominal, pueden ser indicativos del desarrollo de una acidosis láctica. Los casos graves, a veces con resultado de muerte, fueron asociados con pancreatitis, insuficiencia hepática/esteatosis hepática, insuficiencia renal y altos niveles de lactato en suero. Se debe tener precaución cuando se administren análogos de nucleósidos a pacientes (en especial mujeres obesas) con hepatomegalia, hepatitis u otros factores de riesgo conocidos de enfermedad hepática y esteatosis hepática (incluyendo ciertos medicamentos y el alcohol). Los pacientes co-infectados con hepatitis C y tratados con interferón alfa y ribavirina puede constituir un riesgo especial. Estos pacientes deben estar sujetos a una estrecha vigilancia.
Exacerbaciones de la hepatitis
Exacerbaciones en el tratamiento
Las exacerbaciones espontáneas en hepatitis B crónica son relativamente comunes y se caracterizan por aumentos transitorios de ALT sérica. Después de iniciar el tratamiento antiviral, en algunos pacientes puede aumentar la ALT sérica, mientras los valores séricos del ADN del VHB disminuyen. En pacientes con enfermedad hepática compensada, estos aumentos de ALT sérica normalmente no iban acompañados de un aumento en las concentraciones de bilirrubina sérica ni de signos de descompensación hepática.
Con el tratamiento prolongado, se han identificado subpoblaciones virales del VHB con una sensibilidad reducida a lamivudina (mutante YMDD del VHB). En algunos pacientes el desarrollo del mutante YMDD del VHB puede dar lugar a exacerbación de la hepatitis, detectada principalmente por elevaciones de ALT sérica y reaparición del ADN del VHB (ver sección 4.2). En pacientes con el mutante YMDD del VHB, se debe considerar el cambio o la adición de un agente alternativo que no tenga resistencia cruzada a lamivudina en base a las guías terapéuticas (ver sección 5.1).
Exacerbaciones tras la interrupción del tratamiento
Se ha observado exacerbación aguda de la hepatitis en pacientes que han interrumpido el tratamiento para la hepatitis B y normalmente se detecta por elevaciones de ALT sérica y reaparición del ADN del VHB. En los ensayos fase III controlados con seguimiento sin tratamiento activo, la incidencia de elevaciones de ALT tras el tratamiento (más de 3 veces la línea basal), fue mayor en los pacientes tratados con lamivudina (21 %) comparada con los que recibieron placebo (8 %). Sin embargo, la proporción de pacientes que tuvieron elevaciones post-tratamiento asociadas a un aumento en la bilirrubina fue baja y similar en ambos brazos (ver Tabla 3 en la sección 5.1). Para los pacientes tratados con lamivudina, la mayoría de elevaciones de ALT post-tratamiento tuvieron lugar entre las semanas 8 y 12 tras el tratamiento. La mayoría de los acontecimientos han sido autolimitados, aunque se produjeron algunas muertes. Si se interrumpe el tratamiento con Zeffix, los pacientes deben controlarse periódicamente tanto desde el punto de vista clínico, como a través de la evaluación de las pruebas de la función hepática en suero (niveles de ALT y bilirrubina) durante al menos cuatro meses y posteriormente según esté clínicamente indicado.
Exacerbaciones en pacientes con cirrosis descompensada
Los receptores de trasplante y los pacientes con cirrosis descompensada presentan un mayor riesgo de replicación viral activa. Debido a la reducción de la función hepática en estos pacientes, la reactivación de la hepatitis al interrumpir el tratamiento con lamivudina o la pérdida de eficacia durante el tratamiento puede inducir una descompensación grave e incluso mortal. En estos pacientes se debería controlar los parámetros clínicos, virológicos y serológicos asociados a la hepatitis B, funciones renal y hepática y respuesta antiviral durante el tratamiento (al menos cada mes) y, si se interrumpe el tratamiento por alguna razón, durante al menos 6 meses después del tratamiento. Los parámetros de laboratorio a controlar deberían incluir (como mínimo) ALT en suero, bilirrubina, albúmina, nitrógeno ureico en sangre, creatinina y carga viral: antígeno/anticuerpo VHB y, si es posible, concentraciones séricas de ADN del VHB. Los pacientes que experimenten signos de insuficiencia hepática durante o después del tratamiento deben ser controlados con más frecuencia cuando sea conveniente.
En pacientes que desarrollen evidencia de hepatitis recurrente tras el tratamiento, no existen datos suficientes sobre los beneficios de reiniciar el tratamiento con lamivudina.
Disfunción mitocondrial
Se ha demostrado in vitro e in vivo que los análogos de nucleósido y de nucleótido causan un grado variable de daño mitocondrial. Se han notificado casos de disfunción mitocondrial en niños expuestos en el útero y/o posparto a los análogos de nucleósidos. Los principales acontecimientos adversos notificados son trastornos hematológicos (anemia, neutropenia), trastornos metabólicos (hiperlactemia, hiperlipasemia). Se han notificado algunos trastornos neurológicos de aparición tardía (hipertonía, convulsión, comportamiento anormal). Los trastornos neurológicos pueden ser transitorios o permanentes. Cualquier niño expuesto in utero a análogos de nucleósido o de nucleótido, debe ser sometido a un seguimiento clínico y de laboratorio y en caso de signos o síntomas relevantes, debe ser minuciosamente investigada una posible disfunción mitocondrial.
Pacientes pediátricos
Se ha administrado lamivudina a niños (de 2 años de edad y mayores) y adolescentes con hepatitis B crónica compensada. Sin embargo, debido a las limitaciones de los datos, la administración de lamivudina en esta población de pacientes no está recomendada actualmente (ver sección 5.1).
Hepatitis Delta o hepatitis C
No se ha establecido la eficacia de lamivudina en pacientes coinfectados con hepatitis Delta o hepatitis C, por lo que se recomienda tener precaución.
Tratamientos inmunodepresores
Los datos en relación al empleo de lamivudina en pacientes HBeAg negativos (mutante pre-core) y en aquellos que reciben tratamientos inmunodepresores concurrentes, incluyendo quimioterapia en cáncer son limitados. Lamivudina debe ser usada con precaución en estos pacientes.
Monitorización
Durante el tratamiento con Zeffix, los pacientes deben ser controlados de forma regular. Cada 3 meses se deben monitorizar los niveles de ALT sérica y el ADN del VHB y cada 6 meses se debe determinar los HBeAg en pacientes HBeAg positivos.
Coinfección con el VIH
Para el tratamiento de pacientes coinfectados con el VIH y que estén recibiendo o esté previsto que reciban tratamiento con lamivudina o con la combinación lamivudina/zidovudina, debe mantenerse la dosis de lamivudina prescrita para la infección por el VIH (normalmente 150 mg/dos veces al día en combinación con otros antirretrovirales). En los pacientes coinfectados con el VIH que no precisen tratamiento antirretroviral, existe un riesgo de mutación del VIH al utilizar sólo lamivudina para el tratamiento de la hepatitis B crónica.
Transmisión de hepatitis B
No se dispone de información sobre la transmisión materno fetal del virus de la hepatitis B en mujeres embarazadas que estén en tratamiento con lamivudina. Se deben seguir los procedimientos estándar recomendados de inmunización frente al virus de la hepatitis B en niños.
Debe informarse a los pacientes de que el tratamiento con lamivudina no ha demostrado reducir el riesgo de transmisión del virus de la hepatitis B a otras personas, por lo que deben tomarse las precauciones adecuadas.
Interacción con otros medicamentos
Zeffix no debe ser tomado con cualquier otro medicamento que contenga lamivudina o medicamentos que contengan emtricitabina (ver sección 4.5).
No se recomienda la combinación de lamivudina con cladribina (ver sección 4.5).
4.5 Interacción con otros medicamentos y otras formas de interacción
Los estudios de interacción se han realizado sólo en adultos.
La probabilidad de interacciones metabólicas es baja debido a un metabolismo y unión a proteínas plasmáticas limitado y a la casi completa eliminación renal de la sustancia inalterada.
La lamivudina se elimina predominantemente por secreción catiónica orgánica activa. Debe considerarse la posibilidad de interacciones con otros medicamentos administrados al mismo tiempo, especialmente cuando su principal vía de eliminación sea la secreción renal activa por el sistema de transporte catiónico, por ejemplo trimetoprim. Otros medicamentos (por ej. ranitidina, cimetidina) se eliminan sólo en parte por este mecanismo y demostraron no interaccionar con lamivudina.
No es probable que sustancias que hayan demostrado excretarse principalmente bien por la vía aniónica orgánica activa o por filtración glomerular, den lugar a interacciones clínicamente significativas con lamivudina. La administración de trimetoprim/sulfametoxazol 160 mg/800 mg incrementó la exposición a lamivudina en alrededor de un 40 %. Lamivudina no ejerció ningún efecto sobre la farmacocinética de trimetoprim o sulfametoxazol. No obstante, a menos que el paciente presente insuficiencia renal, no es necesario ajustar la dosis de lamivudina.
Se observó un modesto incremento en el valor de Cmax (28 %) para zidovudina al administrarse con lamivudina, aunque la exposición general (AUC) no se alteró de forma significativa. Zidovudina carece de efecto sobre la farmacocinética de lamivudina (ver sección 5.2.).
Lamivudina no ejerce interacciones farmacocinéticas con interferón-alfa cuando ambos medicamentos se administran al mismo tiempo. No se observaron interacciones adversas clínicamente significativas en pacientes que toman al mismo tiempo lamivudina con medicamentos inmunodepresores comúnmente utilizados (p. ej. ciclosporina A). No obstante, no se han realizado estudios formales de interacción.
Emtricitabina
Debido a sus similitudes, Zeffix no debe ser administrado de forma concomitante con otros análogos de citidina, como emtricitabina. Además, Zeffix no se debe tomar con ningún otro medicamento que contenga lamivudina (ver sección 4.4).
Cladribina
Lamivudina in vitro inhibe la fosforilación intracelular de cladribina dando lugar a un potencial riesgo de pérdida de eficacia de cladribina si se toma en combinación durante el manejo clínico. Algunos hallazgos clínicos también sugieren una posible interacción entre lamivudina y cladribina. Por tanto, no se recomienda el uso concomitante de lamivudina con cladribina (ver sección 4.4).
4.6 Fertilidad, embarazo y lactancia
Embarazo
Una gran cantidad de datos obtenidos en mujeres embarazadas (resultados de más 1.000 mujeres expuestas) indica que no hay toxicidad malformativa. Si es clínicamente necesario Zeffix puede usarse durante el embarazo.
En pacientes que están siendo tratadas con lamivudina y posteriormente se quedan embarazadas se debe considerar la posibilidad de una reaparición de la hepatitis al discontinuar el tratamiento con lamivudina.
Lactancia
Basándose en más de 200 parejas madre/hijo tratadas para el VIH, las concentraciones séricas de lamivudina en niños lactantes de madres tratadas para el VIH es muy baja (menos de un 4% de las concentraciones de suero materno) y disminuye progresivamente a niveles indetectables cuando los lactantes alcanzan las 24 semanas de edad. La cantidad total de lamivudina ingerida por un lactante es muy baja y por tanto es probable que dé lugar a exposiciones con un efecto antiviral sub-óptimo. La hepatitis B materna no es una contraindicación para la lactancia si se controla la prevención de la infección por hepatitis B por parte del recién nacido en el nacimiento y no hay evidencia de que las bajas concentraciones de lamivudina en leche materna puedan producir reacciones adversas en lactantes. Por tanto, la lactancia se puede considerar en madres tratadas con lamivudina para VHB teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento para la mujer. Cuando hay transmisión materna de VHB, a pesar de una adecuada profilaxis, se debe considerar interrumpir la lactancia para reducir el riesgo de que aparezcan mutantes resistentes a lamivudina en el niño.
Fertilidad
Los estudios de reproducción en animales no han mostrado efecto en la fertilidad masculina y femenina (ver sección 5.3).
Disfunción mitocondrial
Se ha demostrado in vitro e in vivo que los análogos de nucleósidos y nucleótidos causan daño mitocondrial de grado variable. Se han notificado casos de disfunción mitocondrial en niños expuestos en el útero y/o post-parto a análogos de nucleósidos (ver sección 4.4).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Se debe informar a los pacientes que se han notificado malestar y fatiga durante el tratamiento con lamivudina. El estado clínico del paciente y el perfil de reacciones adversas de lamivudina se deben tener en cuenta al considerar la capacidad de que el paciente conduzca o maneje máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
La incidencia de reacciones adversas y anormalidades de laboratorio (a excepción de los incrementos de ALT y CPK, ver a continuación) fue similar entre los pacientes tratados con placebo y los tratados con lamivudina. Las reacciones adversas comunicadas con más frecuencia fueron malestar y fatiga, infecciones del tracto respiratorio, molestias en la garganta y amígdalas, cefalea, dolor y molestias abdominales, náuseas, vómitos y diarrea.
Tabla de reacciones adversas
A continuación se presentan las reacciones adversas clasificadas por órganos, sistemas y frecuencias. Las categorías de frecuencia se asignan únicamente a aquellas reacciones adversas consideradas al menos posiblemente relacionadas con lamivudina. Las frecuencias se definen como: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raras (> 1/10.000 a < 1/1000), muy raras (< 1/10.000) y de frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Las categorías de frecuencias asignadas a las reacciones adversas se basan principalmente en la experiencia de los ensayos clínicos que incluyeron un total de 1.171 pacientes con hepatitis B crónica que recibieron 100 mg de lamivudina.
|
Trastornos de la sangre y del sistema linfático | |
|
Desconocida |
T rombocitopenia |
|
Trastornos del sistema inmunológico | |
|
Raras |
Angioedema |
|
Trastornos hepatobiliares | |
|
Muy frecuentes |
Incrementos de ALT (ver sección 4.4) |
|
Durante el tratamiento y tras la retirada de lamivudina se han notificado exacerbaciones de la hepatitis, detectadas principalmente por aumento en las concentraciones de ALT en suero. La mayoría han remitido, aunque muy raramente se han observado muertes (ver sección 4.4). | |
|
Trastornos de la piel y del tejido subcutáneo | |
|
Frecuentes |
Erupción, prurito |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |
|
Frecuentes |
Incrementos de CPK |
|
Frecuentes |
Trastornos musculares, incluyendo mialgia y calambres* |
|
Frecuencia no conocida |
Rabdomiolisis |
|
*En los ensayos fase I |
II la frecuencia observada en el grupo tratado con lamivudina no fue mayor que |
la observada en el grupo placebo Población pediátrica
Basándose en los datos limitados en niños de 2 a 17 años de edad, no se identificaron nuevas cuestiones de seguridad en comparación a las observadas en adultos.
Otras poblaciones especiales
En pacientes con infección por VIH, se han comunicado casos de pancreatitis y neuropatía periférica (o parestesia) En pacientes con hepatitis B crónica no se observaron diferencias en la incidencia de estos eventos entre los pacientes tratados con placebo y los tratados con lamivudina.
Se han notificado casos de acidosis láctica, a veces mortales, normalmente asociados con hepatomegalia grave y esteatosis hepática, con el uso de terapia de combinación de análogos de nucleósidos en pacientes con VIH. Raramente se han comunicado casos de acidosis láctica en pacientes que recibían lamivudina para el tratamiento de hepatitis B.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los
profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V
4.9 Sobredosis
En estudios de toxicidad aguda en animales, la administración de dosis muy elevadas de lamivudina no produjo toxicidad en ningún órgano. Se dispone de datos limitados sobre las consecuencias de la ingestión de sobredosis agudas en humanos. No hubo fallecimientos y todos los pacientes se recuperaron. No se han identificado signos o síntomas específicos después de tal sobredosis.
En caso de sobredosis, se vigilará al paciente y se aplicará el tratamiento de apoyo estándar que sea necesario. Dado que lamivudina es dializable, puede emplearse una hemodiálisis continua para el tratamiento de la sobredosificación, aunque ello no se ha estudiado.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antivirales para uso sistémico, nucleósidos y nucleótidos inhibidores de la transcriptasa inversa, código ATC: J05AF05.
Lamivudina es un agente antiviral muy activo frente al virus de la hepatitis B en todas las líneas celulares estudiadas y en animales infectados experimentalmente.
Lamivudina es metabolizada tanto por células infectadas como por no infectadas al derivado trifosfato (TP) que es la forma activa del compuesto original. La semivida intracelular del trifosfato en los hepatocitos es de 17-19 horas in vitro. Lamivudina -TP actúa como sustrato para la polimerasa viral del VHB.
La formación de más ADN viral se bloquea por incorporación de lamivudina-TP a la cadena y posterior terminación de la misma.
Lamivudina-TP no interfiere con el metabolismo celular normal de los deoxinucleótidos. También es un débil inhibidor de las alfa y beta ADN polimerasas de mamíferos. Además, lamivudina-TP tiene un escaso efecto sobre el contenido de ADN en las células de mamíferos.
En los estudios relativos a los potenciales efectos de la sustancia sobre la estructura mitocondrial y sobre el contenido y función del ADN, se vió que lamivudina carecía de efectos tóxicos apreciables. Presenta un potencial muy bajo para disminuir el contenido de ADN mitocondrial, no se incorpora de forma permanente al ADN mitocondrial y no actúa como inhibidor de la ADN gamma polimerasa mitocondrial.
Experiencia, clínica.
Experiencia en pacientes con HBC HBeAg positivos y enfermedad hepática compensada En estudios controlados, el tratamiento durante 1 año con lamivudina suprimió de forma significativa la replicación del ADN del VHB [ 34-57 % de los pacientes estaban por debajo de los límites de detección (ensayo de hibridación en solución de Abbott Genostics, LLOD < 1,6pg/ml)], normalizó el nivel de ALT (40-72 % de los pacientes), indujo la seroconversión del HBeAg (pérdida de HBeAg y detección de HBeAc con pérdida de ADN del VHB [por ensayo convencional], 16-18 % de pacientes), mejoró la histología (38-52 % de los pacientes tuvieron un descenso > a 2 puntos en el Índice de Actividad Histológica de Knodell [IAH]), y redujo la progresión de la fibrosis (en 3-17 % de los pacientes) y la progresión a cirrosis.
El tratamiento continuo con lamivudina durante 2 años más en pacientes que no lograron una seroconversión de HBeAg en el primer año de los estudios controlados, dio lugar a una mejoría añadida de la fibrosis en puente. En pacientes con el mutante YMDD del VHB, 41/82 (50 %) pacientes mejoraron en la inflamación hepática y 40/56 (71 %) pacientes sin el mutante YMDD del VHB mejoraron. La mejoría en la fibrosis en puente se produjo en 19/30 (63 %) pacientes sin el mutante YMDD y en 22/44 (50 %) pacientes con el mutante. El 5% de los pacientes (3/56) sin el mutante YMDD y el 13 % (11/82) pacientes con el mutante YMDD mostraron empeoramiento en la inflamación hepática con respecto a los niveles pre-tratamiento. La progresión a cirrosis se produjo en 4/68 (6%) de los pacientes con el mutante YMDD, mientras que ningún paciente sin el mutante progresó a cirrosis.
En un estudio de tratamiento prolongado en pacientes asiáticos (NUCB3018) la tasa de seroconversión de HBeAg y la velocidad de normalización de ALT al final del periodo de tratamiento de 5 años, fueron de 48 % (28/58) y 47 % (15/32), respectivamente. La seroconversión de HBeAg aumentó en pacientes con niveles elevados de ALT; seroconvirtieron el 77 % (20/26) de los pacientes con niveles de ALT previos al tratamiento > 2x LSN. Al final de los 5 años, todos los pacientes tuvieron niveles de ADN del VHB indetectables o menores que los niveles previos al tratamiento.
En la Tabla 2 se resumen los últimos resultados del ensayo según el estado del mutante YMDD.
Tabla 2: Resultados de eficacia de 5 años según el estado de YMDD (Estudio asiático)
NUCB3018
|
Pacientes, % (n°.) | ||||
|
YMDD1 |
No-YMDD1 | |||
|
Estado VHB mutante YMDD | ||||
|
Seroconversión de HBeAg | ||||
|
- Todos los pacientes |
38 |
(15/40) |
72 |
(13/18) |
|
- ALT basal < 1 x LSN2 |
9 |
(1/11) |
33 |
(2/6) |
|
- ALT basal > 2 x LSN |
60 |
(9/15) |
100 |
(11/11) |
|
ADN de VHB no detectable | ||||
|
- Basal 3 |
5 |
(2/40) |
6 |
(1/18) |
|
- Semana 260 4 | ||||
|
negativo |
8 |
(2/25) |
0 | |
|
positivo < nivel basal |
92 |
(23/25) |
100 |
(4/4) |
|
positivo > nivel basal |
0 |
0 | ||
|
Normalización ALT | ||||
|
- Basal | ||||
|
normal |
28 |
(11/40) |
33 |
(6/18) |
|
por encima de lo normal |
73 |
(29/40) |
67 |
(12/18) |
|
- Semana 260 | ||||
|
normal |
46 |
(13/28) |
50 |
(2/4) |
|
por encima de lo normal < nivel basal |
21 |
(6/28) |
0 | |
|
por encima de lo normal > nivel basal |
32 |
(9/28) |
50 |
(2/4) |
1 Los pacientes categorizados como mutante YMDD fueron aquellos con >5% de mutante YMDD del VHB en cualquier instante del año durante un período de 5 años. Los pacientes categorizados como no mutante -YMDD fueron aquellos con > 95 % de tipo salvaje de VHB para cualquier momento del año durante el período de 5 años del estudio
2 Límite normal superior
3 Ensayo de hibridación en solución Abbott Genostics (LLOD < 1,6 pg/ml)
4 Ensayo Chiron Quantiplex (LLOD 0,7 Meq/ml)
También se dispone de datos comparativos según el status YMDD para su evaluación histológica, pero sólo hasta 3 años. En pacientes con el mutante YMDD del VHB, 18/39 (46 %) hubo mejoría en la actividad necroinflamatoria y en 9/39 (23 %) un empeoramiento. En los pacientes sin el mutante,
20/27 (74 %) hubo mejoría en la actividad necroinflamatoria, y en 2/27 (7 %) un empeoramiento.
Después de la seroconversión del HBeAg, la respuesta serológica y la remisión clínica son generalmente duraderas tras interrumpir el tratamiento con lamivudina. Sin embargo, pueden ocurrir recaídas tras la seroconversión. En un estudio a largo plazo de seguimiento de pacientes que tuvieron seroconversión e interrumpieron el tratamiento con lamivudina, la recaída virológica posterior ocurrió en un 39 % de los sujetos. Por tanto, tras la seroconversión de HBeAg, los pacientes deben ser controlados periódicamente para determinar si las respuestas serológica y clínica se mantienen. En pacientes que no mantengan una respuesta serológica constante, debería considerarse un retratamiento, o bien con lamivudina, o bien con un agente antiviral alternativo para la reanudación del control clínico del VHB.
En pacientes bajo seguimiento hasta 16 semanas tras la interrupción del tratamiento un año después de iniciarlo, se han observado con mayor frecuencia elevaciones de ALT post-tratamiento en pacientes que habían recibido lamivudina que en pacientes que habían recibido placebo. En la Tabla 3 se muestra una comparación de las elevaciones de ALT post-tratamiento entre las semanas 52 y 68 en pacientes que interrumpieron el tratamiento con lamivudina en la semana 52 y pacientes en los mismos estudios que recibieron placebo a lo largo del período de tratamiento. La proporción de pacientes que tuvieron elevaciones de ALT post-tratamiento junto con un aumento de los niveles de bilirrubina fue baja y similar en pacientes que recibieron tanto lamivudina como placebo.
Tabla 3: Elevaciones de ALT post-tratamiento en 2 estudios controlados con placebo en adultos
|
Valor Anormal |
Pacientes con Elevación de ALT/ Pacientes con Observaciones* | |
|
Lamivudina |
Placebo | |
|
ALT >2 x valor basal |
37/137 (27 %) |
22/116 (19 %) |
|
ALT >3 x valor basal ^ |
29/137 (21 %) |
9/116 (8 %) |
|
ALT >2 x valor basal y ALT absoluta>500 Ul/l |
21/137 (15 %) |
8/116 (7 %) |
|
ALT >2 x valor basal; y bilirrubina >2 x LSN y >2 x valor basal |
1/137 (0,7 %) |
1/116 (0,9 %) |
* Cada paciente puede estar representado en una o más categorías.
^ Comparable a Toxicidad de Grado 3 de acuerdo al criterio modificado de la OMS. LSN = Límite superior de normalidad.
Experiencia en pacientes con HBC HBeAg negativos
Los datos iniciales indican que la eficacia de lamivudina en pacientes con HBC HBeAg negativos es similar a la de pacientes con HBC HBeAg positivos, obteniendo el 71 % de los pacientes una supresión de ADN del VHB por debajo del límite de detección del ensayo; el 67 % normalización de ALT; y el 38 %, una mejoría del IAH tras un año de tratamiento. Cuando se interrumpió el tratamiento con lamivudina, la mayoría de los pacientes (70 %) volvieron a presentar replicación viral. Se dispone de datos procedentes de un estudio de tratamiento prolongado en pacientes HBeAg negativos, tratados con lamivudina (NUCAB3017). Tras dos años de tratamiento en este estudio, se observó la normalización de los niveles de ALT y no se detectó ADN del VHB en 30/69 (43 %) y 32/68 (47 %) pacientes, respectivamente, así como una mejoría en el grado de necroinflamación en 18/49 (37 %) pacientes. En pacientes sin el mutante YMDD del VHB, 14/22 (64 %) mostraron mejoría en el grado de necroinflamación y 1/22 (5 %) empeoraron con respecto a los niveles pre-tratamiento. En pacientes con el mutante, 4/26 (15 %) mostraron mejoría en el grado de necroinflamación, y 8/26 (31 %) empeoraron con respecto a los niveles pre-tratamiento. Ningún paciente en ninguno de los grupos progresó a cirrosis.
Frecuencia de aparición del muíante YMDD del VHB e impacto en la respuesta al tratamiento La monoterapia con lamivudina da lugar a la aparición del mutante YMDD del VHB en aproximadamente el 24% de los pacientes tras un año de terapia, aumentando hasta el 69% tras 5 años de terapia. El desarrollo del mutante YMDD del VHB está asociado con una reducción en la respuesta al tratamiento en algunos pacientes, lo cual se evidencia por un aumento de los niveles de ADN del VHB y por elevación de los niveles de ALT respecto a los niveles previos durante la terapia, progresión de signos y síntomas de enfermedad hepática y/o empeoramiento de hallazgos necroinflamatorios hepáticos. Dado el riesgo del mutante YMDD del VHB, no es adecuado el mantenimiento de la monoterapia con lamivudina en pacientes con ADN del VHB detectable en suero al cabo de 24 semanas o más de tratamiento (ver sección 4.4).
En un estudio doble ciego en pacientes con HBC con el mutante YMDD del VHB y enfermedad hepática compensada (NUC20904), con una respuesta virológica y bioquímica reducida a lamivudina (n=95), la adición de 10 mg de adefovir dipivoxilo una vez al día al tratamiento con 100 mg de lamivudina durante 52 semanas, dio lugar a una disminución media del ADN del VHB de 4,6 logio copias/ml en comparación con un aumento medio de 0,3 log10 copias/ml en aquellos pacientes que recibieron lamivudina en monoterapia. La normalización de los niveles de ALT se produjo en un 31% (14/45) de los pacientes que recibieron terapia combinada, frente a un 6 % (3/47) que recibieron lamivudina sola. Se mantuvo la supresión viral (estudio de seguimiento NUC20917) con terapia combinada durante el segundo año de tratamiento hasta la semana 104 con pacientes que continuaron mejorando en las respuestas virológica y bioquímica.
En un estudio retrospectivo para determinar los factores asociados con la evolución del ADN del VHB se trataron con lamivudina 159 HBeAg pacientes positivos asiáticos y se siguieron durante un periodo cuya mediana fue de casi 30 meses. Aquellos con niveles ADN del VHB mayores de 200 copias/ml a los 6 meses (24 semanas) de tratamiento con lamivudina tuvieron un 60 % de posibilidad de desarrollar el mutante YMDD en comparación con un 8 % de aquellos con un ADN del VHB menores de 200 copias/ml a las 24 semanas de tratamiento con lamivudina. El riesgo de desarrollar el mutante YMDD fue de 63 % versus el 13 % con un corte de 1000 copias/ml (NUCB3009 y NUCB3018).
Experiencia en pacientes con enfermedad hepática descompensada
En pacientes con descompensación hepática no se han realizado estudios controlados con placebo por considerarse inapropiados. En estudios no controlados, en los que se administró lamivudina antes y durante el trasplante, se demostró la supresión eficaz del ADN del VHB y la normalización de los valores de ALT. Cuando se continuaba administrando lamivudina después del trasplante, la reinfección del injerto por el VHB era menor, tenía lugar una mayor pérdida del HBsAg y la tasa de supervivencia al cabo de 1 año era del 76-100 %.
Como era de esperar, debido a la inmunodepresión concomitante, la tasa de aparición del mutante YMDD del VHB, al cabo de 52 semanas de tratamiento, fue superior (36 % - 64 %) en la población con trasplante de hígado que en los pacientes inmunocompetentes con HBC (14 % - 32 %).
Se incluyeron en un brazo abierto del estudio NUC20904 cuarenta pacientes (HBeAg negativos o HBeAg positivos), bien con enfermedad hepática descompensada o con VHB recurrente tras un trasplante hepático, y con el mutante YMDD. La adición de 10 mg de adefovir dipivoxilo una vez al día al tratamiento en curso con 100 mg de lamivudina durante 52 semanas dio lugar a un descenso medio en el ADN del VHB de 4,6 log10 copias/ml. También se observó una mejoría en la función hepática tras un año de tratamiento. Este grado de supresión viral se mantuvo (estudio de seguimiento NUC20917) con terapia combinada durante el segundo año de tratamiento hasta la semana 104 y la mayoría de pacientes mejoraron en cuanto a los marcadores de función hepática y continuaron hacia un beneficio clínico.
Experiencia en pacientes con HBC con fibrosis avanzada o cirrosis
En un estudio controlado con placebo en 651 pacientes con hepatitis B crónica clínicamente compensada y fibrosis o cirrosis confirmadas histológicamente, el tratamiento con lamivudina (32 meses de duración media) redujo significativamente la tasa de progresión global de la enfermedad (34/436, 7,8 % para lamivudina, frente a 38/215, 17,7 % para placebo, p=0,001), demostrado por una
reducción significativa en el porcentaje de pacientes que experimentaron un aumento en la puntuación de Child-Pugh (15/436, 3,4 % frente a 19/215, 8,8 %, p=0,023), o bien un desarrollo de carcinoma hepatocelular (17/436, 3,9 % frente a 16/215, 7,4 %, p=0,047). La tasa de progresión global de la enfermedad en el grupo tratado con lamivudina fue mayor en aquellos pacientes en los que se detectó el mutante YMDD del ADN del VHB (23/209, 11 %), en comparación con aquellos en los que no se detectó el mutante (11/221, 5 %). Sin embargo, la progresión de la enfermedad en pacientes con YMDD en el grupo tratado con lamivudina fue menor que la progresión de la enfermedad en el grupo tratado con placebo (23/209, 11 %, frente a 38/214, 18 % respectivamente). La seroconversión de HBeAg tuvo lugar en el 47 % (118/252) de los pacientes tratados con lamivudina y el 93 % (320/345) de los pacientes que recibieron lamivudina se convirtieron en ADN del VHB negativos durante el estudio (VERSANT [versión 1], ensayo bDNA, LLOD < 0,7 mEq/ml).
Experiencia en niños y adolescentes
Se ha administrado lamivudina a niños y adolescentes con HBC compensada en un estudio controlado con placebo en 286 pacientes de edades comprendidas entre los 2 y los 17 años. La población estaba compuesta principalmente por niños con hepatitis B de grado mínimo. Se empleó una dosis de 3mg/kg una vez al día (hasta un máximo de 100 mg diarios) en niños de edades comprendidas entre 2 y 11 años y una dosis de 100 mg una vez al día en adolescentes de 12 años y mayores. Se necesita conocer más datos acerca de esta dosis. La diferencia en los índices de seroconversión de HBeAg (pérdida de HBeAg y ADN del VHB con detección de HBeAc) entre placebo y lamivudina no fue estadísticamente significativa en esta población (los índices después de un año fueron del 13 %
(12/95) para placebo frente al 22 % (42/191) para la^nivudina; p 0,057). La incidencia del anulante YMDD del VHB fue similar a la observada en adultos, variando del 19 % en la semana 52 hasta el 45 % en pacientes tratados de forma continuada durante 24 meses.
5.2 Propiedades farmacocinéticas
Absorción
Lamivudina se absorbe bien en el tracto gastrointestinal y la biodisponibilidad de lamivudina por vía oral en adultos está normalmente entre el 80-85 %. Tras la administración por vía oral, el tiempo medio (tmax) hasta las concentraciones séricas máximas (Cmax) es de aproximadamente una hora. A dosis terapéuticas, es decir, 100 mg una vez al día, la Cmax es del orden de 1,1-1,5 pg/ml siendo los niveles mínimos de 0,015-0,020 pg/ml.
La administración de lamivudina con alimentos dio lugar a un retraso de tmax y a una Cmax inferior (reducida en un 47 %). No obstante, la cantidad de lamivudina absorbida (en función del AUC) no se vio afectada, por lo que lamivudina se puede administrar con o sin alimentos.
Distribución
Según los estudios realizados con lamivudina por vía intravenosa, el volumen medio de distribución es de 1,3 l/kg. Lamivudina presenta una farmacocinética lineal a lo largo del intervalo de dosis terapéuticas y su unión a la proteína plasmática albúmina es baja.
Los datos limitados de que se dispone muestran que lamivudina penetra en el sistema nervioso central y que alcanza el líquido cefalorraquídeo (LCR). El valor medio del ratio LCR/concentración sérica de lamivudina a las 2-4 horas de la administración por vía oral fue, aproximadamente, de 0,12.
Biotransformación
Lamivudina se elimina principalmente por excreción renal de la sustancia inalterada. La probabilidad de interacciones metabólicas de lamivudina con otras sustancias es baja, debido al pequeño grado de metabolismo hepático (5-10 %) y a la escasa unión a proteínas plasmáticas.
Eliminación
El aclaramiento sistémico medio de lamivudina es de, aproximadamente, 0,3 l/h/kg. La semivida de eliminación observada es de 5 a 7 horas. Lamivudina se excreta en su mayor parte inalterada en orina por filtración glomerular y secreción activa (sistema de transporte catiónico orgánico). El aclaramiento renal representa aproximadamente el 70 % de la eliminación de lamivudina.
Poblaciones especiales
Los estudios en pacientes con insuficiencia renal, demuestran que la eliminación de lamivudina se ve afectada por la disfunción renal. Es necesario reducir la dosis en pacientes con un aclaramiento de creatinina inferior a 50 ml/min (ver sección 4.2).
La farmacocinética de lamivudina no se ve afectada por la insuficiencia hepática. Los datos limitados de que se dispone en pacientes sometidos a trasplante hepático demuestran que la insuficiencia en la función hepática no tiene un impacto significativo sobre la farmacocinética de lamivudina a menos que esté acompañada de disfunción renal.
En pacientes ancianos, el perfil farmacocinético de lamivudina indica que un envejecimiento normal con la disminución de la función renal que lo acompaña, carece de efecto clínicamente significativo sobre la exposición a lamivudina, excepto en pacientes con un aclaramiento de creatinina inferior a 50 ml/min (ver sección 4.2).
5.3 Datos preclínicos sobre seguridad
La administración de lamivudina en los estudios de toxicidad con animales a dosis elevadas no se asoció con toxicidad en ningún órgano principal. A las dosis más altas se observaron efectos menores sobre los indicadores de las funciones hepática y renal, junto a reducciones ocasionales en el peso del hígado. Los efectos clínicamente importantes apreciados fueron una reducción en el recuento de eritrocitos y neutropenia. Estos efectos se observaron con poca frecuencia en los estudios clínicos.
Lamivudina no fue mutagénica en los ensayos con bacterias pero, como muchos análogos de nucleósidos, mostró actividad en un ensayo citogenético in vitro y en el ensayo de linfoma de ratón. Lamivudina no fue genotóxica in vivo a dosis que dieron lugar a concentraciones plasmáticas aproximadamente 60-70 veces más elevadas que los niveles plasmáticos clínicos previstos. Como la actividad mutagénica in vitro de lamivudina no pudo confirmarse en las pruebas in vivo, se concluye que lamivudina no representa un riesgo de aparición de genotoxicidad en pacientes sometidos a tratamiento.
Los estudios sobre la reproducción en animales no han mostrado evidencia de teratogenicidad y tampoco han mostrado efectos sobre la fertilidad en machos o hembras. Lamivudina induce embrioletalidad temprana cuando se administra a conejos preñados a niveles de exposición comparables a los alcanzados en el hombre, pero no en la rata incluso a exposiciones sistémicas muy altas.
Los resultados de los estudios de carcinogenicidad a largo plazo con lamivudina realizados con ratas y ratones mostraron la ausencia de potencial carcinogénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido:
Celulosa microcristalina Carboximetilalmidón de sodio Estearato de magnesio.
Recubrimiento del comprimido: Hipromelosa Dióxido de titanio Macrogol 400 Polisorbato 80
Óxidos de hierro amarillo y rojo sintéticos
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30°C.
6.5 Naturaleza y contenido del recipiente
Estuches conteniendo 28 u 84 comprimidos recubiertos con película en embalaje alveolar (blíster) de doble lámina, laminados con policloruro de vinilo.
Pueden no estar comercializados todos los tamaños de envases.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Ltd 980 Great West Road Brentford Middlesex
TW8 9GS Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/99/114/001 EU/1/99/114/002
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización 29/julio/1999 Fecha de la última renovación 23/junio/2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
NOMBRE DEL MEDICAMENTO
1.
Zeffix 5 mg/ml solución oral
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada ml de la solución oral contiene 5 mg de lamivudina.
Excipientes con efecto conocido:
Sacarosa 20 % p/p (4 g/20 ml)
Parahidroxibenzoato de metilo (E218) 1,5 mg/ml Parahidroxibenzoato de propilo (E216) 0,18 mg/ml
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución oral.
Límpida, incolora o de color amarillo pálido.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Zeffix está indicado para el tratamiento de la hepatitis B crónica en adultos con:
• enfermedad hepática compensada con evidencia de replicación viral activa, niveles de alanina aminotransferasa (ALT) sérica elevados de forma persistente y evidencia histológica de inflamación hepática activa y/o fibrosis. Sólo se debe considerar el comienzo del tratamiento con lamivudina cuando no está disponible o no sea apropiado el uso de un agente antiviral alternativo con una barrera genética más alta a resistencia (ver sección 5.1).
• enfermedad hepática descompensada en combinación con un segundo agente sin resistencia cruzada a lamivudina (ver sección 4.2).
4.2 Posología y forma de administración
El tratamiento con Zeffix debe ser iniciado por un médico con experiencia en el tratamiento de la
hepatitis B crónica.
Posología
Adultos
La dosis recomendada de Zeffix es de 100 mg una vez al día.
En pacientes con enfermedad hepática descompensada, lamivudina se debe usar siempre en combinación con un segundo agente, sin resistencia cruzada a lamivudina, para reducir el riesgo de resistencia y conseguir una supresión viral rápida.
Duración del tratamiento
Se desconoce la duración óptima del tratamiento. 2
HBeAg (pérdida de HBeAg y ADN del VHB con detección de HBeAc) con el fin de reducir el riesgo de recaída virológica, o continuar hasta que tenga lugar la seroconversión de HbsAg o haya pérdida de eficacia (ver sección 4.4). El ALT sérico y los niveles del ADN del VHB deben ser controlados regularmente después de la discontinuación de tratamiento para percatarse de cualquier recaída virológica tardía.
• En pacientes con HBC HBeAg negativos (mutante pre-core), sin cirrosis, el tratamiento debe ser administrado por lo menos hasta la seroconversion de HBs o haya evidencia de pérdida de eficacia. Con tratamiento prolongado, se recomienda un control regular para confirmar que la continuación de la terapia seleccionada sigue siendo apropiada para el paciente.
• No se recomienda la interrupción del tratamiento en pacientes con enfermedad hepática descompensada o cirrosis y en receptores de trasplante hepático (ver sección 5.1).
Si se interrumpe el tratamiento con lamivudina los pacientes deben ser controlados periódicamente para comprobar la existencia de una hepatitis recurrente (ver sección 4.4).
Resistencia clínica
En pacientes con HBC tanto HBeAg positivos como HBeAg negativos, el desarrollo del mutante YMDD (tirosina-metionina-aspartato-aspartato) del VHB puede dar lugar a una menor respuesta terapéutica a lamivudina, indicada por un aumento del ADN del VHB y de ALT con respecto a los niveles anteriores durante el tratamiento. Con el fin de reducir el riesgo de resistencia en pacientes que reciben lamivudina en monoterapia, si el ADN del VHB continúa siendo detectable en suero a las 24 semanas de tratamiento o más debería considerarse el cambio o la adición de un agente alternativo sin resistencia cruzada a lamivudina en base a las guías terapéuticas (ver sección 5.1).
Para el tratamiento de pacientes coinfectados con el VIH y que están recibiendo o que planean recibir tratamiento con lamivudina o con la combinación lamivudina-zidovudina, se debe mantener la dosis prescrita de lamivudina para la infección por el VIH (normalmente 150 mg/dos veces al día en combinación con otros antirretrovirales).
Poblaciones especiales
Insuficiencia renal
Las concentraciones séricas de lamivudina (AUC) aumentan en pacientes con insuficiencia renal moderada a grave, debido a una disminución del aclaramiento renal. Por lo tanto, debe reducirse la dosis en pacientes con un aclaramiento de creatinina inferior a 50 ml/minuto. Cuando se requieran dosis inferiores a 100 mg debe utilizarse Zeffix solución oral (ver Tabla 1 a continuación).
Tabla 1: Posología de Zeffix en pacientes con aclaramiento renal disminuido.
|
Aclaramiento de creatinina ml/min |
Primera dosis de Zeffix solución oral |
Dosis de mantenimiento una vez al día |
|
30 a < 50 |
20 ml (100 mg) |
10 ml (50 mg) |
|
15 a < 30 |
20 ml (100 mg) |
5 ml (25 mg) |
|
5 a < 15 |
7 ml (35 mg) |
3 ml (15 mg) |
|
<5 |
7 ml (35 mg) |
2 ml (10 mg) |
Los datos disponibles de pacientes sometidos a hemodiálisis intermitente (diálisis menor o igual a 4 h 2-3 veces a la semana), indican que tras la reducción de la dosis inicial de lamivudina para ajustarse al aclaramiento de creatinina del paciente, no se precisan ajustes de dosis adicionales mientras se practique la diálisis.
Insuficiencia hepática
Los datos obtenidos en pacientes con insuficiencia hepática, incluyendo aquellos con enfermedad hepática en fase terminal a la espera de trasplante, demuestran que la farmacocinética de lamivudina no se ve afectada de forma significativa por la disfunción hepática. Según estos datos no es necesario ajustar la dosis en pacientes con insuficiencia hepática, a menos que esté acompañada de insuficiencia renal.
Pacientes de edad avanzada
En los pacientes de edad avanzada, el envejecimiento con la correspondiente disminución de la función renal no tiene efecto clínicamente significativo en la exposición a lamivudina, excepto en pacientes con un aclaramiento de creatinina de <50 ml/min.
Población pediátrica
No se ha establecido la seguridad y eficacia de Zeffix en niños y adolescentes menores de 18 años.
Los datos disponibles en la actualidad, que se describen en las secciones 4.4 y 5.1, no permiten hacer una recomendación posológica.
Forma de administración Vía oral.
Zeffix puede tomarse con o sin alimentos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Acidosis láctica y hepatomegalia grave con esteatosis
Con el uso de análogos de nucleósidos se ha comunicado la aparición de acidosis láctica (en ausencia de hipoxemia), a veces mortal, habitualmente asociada a hepatomegalia grave y esteatosis hepática. Este riesgo no puede excluirse dado que lamivudina es un análogo de nucleósido. El tratamiento con análogos de nucleósido se debe interrumpir si aparece una elevación rápida de los niveles de aminotransferasas, hepatomegalia progresiva o acidosis metabólica/láctica de etiología desconocida. Síntomas digestivos benignos, como náuseas, vómitos y dolor abdominal, pueden ser indicativos del desarrollo de una acidosis láctica. Los casos graves, a veces con resultado de muerte, fueron asociados con pancreatitis, insuficiencia hepática/esteatosis hepática, insuficiencia renal y altos niveles de lactato en suero. Se debe tener precaución cuando se administren análogos de nucleósidos a pacientes (en especial mujeres obesas) con hepatomegalia, hepatitis u otros factores de riesgo conocidos de enfermedad hepática y esteatosis hepática (incluyendo ciertos medicamentos y el alcohol). Los pacientes co-infectados con hepatitis C y tratados con interferón alfa y ribavirina puede constituir un riesgo especial. Estos pacientes deben estar sujetos a una estrecha vigilancia.
Exacerbaciones de la hepatitis
Exacerbaciones en el tratamiento
Las exacerbaciones espontáneas en hepatitis B crónica son relativamente comunes y se caracterizan por aumentos transitorios de ALT sérica. Después de iniciar el tratamiento antiviral, en algunos pacientes puede aumentar la ALT sérica, mientras los valores séricos del ADN del VHB disminuyen. En pacientes con enfermedad hepática compensada, estos aumentos de ALT sérica normalmente no iban acompañados de un aumento en las concentraciones de bilirrubina sérica ni de signos de descompensación hepática.
Con el tratamiento prolongado, se han identificado subpoblaciones virales del VHB con una sensibilidad reducida a lamivudina (mutante YMDD del VHB). En algunos pacientes el desarrollo del mutante YMDD del VHB puede dar lugar a exacerbación de la hepatitis, detectada principalmente por elevaciones de ALT sérica y reaparición del ADN del VHB (ver sección 4.2). En pacientes con el mutante YMDD del VHB, se debe considerar el cambio o la adición de un agente alternativo que no tenga resistencia cruzada a lamivudina en base a las guías terapéuticas (ver sección 5.1).
Exacerbaciones tras la interrupción del tratamiento
Se ha observado exacerbación aguda de la hepatitis en pacientes que han interrumpido el tratamiento para la hepatitis B y normalmente se detecta por elevaciones de ALT sérica y reaparición del ADN del VHB. En los ensayos fase III controlados con seguimiento sin tratamiento activo, la incidencia de elevaciones de ALT tras el tratamiento (más de 3 veces la línea basal), fue mayor en los pacientes tratados con lamivudina (21 %) comparada con los que recibieron placebo (8%). Sin embargo, la proporción de pacientes que tuvieron elevaciones post-tratamiento asociadas a un aumento en la bilirrubina fue baja y similar en ambos brazos (ver Tabla 3 en la sección 5.1). Para los pacientes tratados con lamivudina, la mayoría de elevaciones de ALT post-tratamiento tuvieron lugar entre las semanas 8 y 12 tras el tratamiento. La mayoría de los acontecimientos han sido autolimitados, aunque se produjeron algunas muertes. Si se interrumpe el tratamiento con Zeffix, los pacientes deben controlarse periódicamente tanto desde el punto de vista clínico, como a través de la evaluación de las pruebas de la función hepática en suero (niveles de ALT y bilirrubina) durante al menos cuatro meses y posteriormente según esté clínicamente indicado.
Exacerbaciones en pacientes con cirrosis descompensada
Los receptores de trasplante y los pacientes con cirrosis descompensada presentan un mayor riesgo de replicación viral activa. Debido a la reducción de la función hepática en estos pacientes, la reactivación de la hepatitis al interrumpir el tratamiento con lamivudina o la pérdida de eficacia durante el tratamiento puede inducir una descompensación grave e incluso mortal. En estos pacientes se debería controlar los parámetros clínicos, virológicos y serológicos asociados a la hepatitis B, funciones renal y hepática y respuesta antiviral durante el tratamiento (al menos cada mes) y, si se interrumpe el tratamiento por alguna razón, durante al menos 6 meses después del tratamiento. Los parámetros de laboratorio a controlar deberían incluir (como mínimo) ALT en suero, bilirrubina, albúmina, nitrógeno ureico en sangre, creatinina y carga viral: antígeno/anticuerpo VHB y, si es posible, concentraciones séricas de ADN del VHB. Los pacientes que experimenten signos de insuficiencia hepática durante o después del tratamiento deben ser controlados con más frecuencia cuando sea conveniente.
En pacientes que desarrollen evidencia de hepatitis recurrente tras el tratamiento, no existen datos suficientes sobre los beneficios de reiniciar el tratamiento con lamivudina.
Disfunción mitocondrial
Se ha demostrado in vitro e in vivo que los análogos de nucleósido y de nucleótido causan un grado variable de daño mitocondrial. Se han notificado casos de disfunción mitocondrial en niños expuestos en el útero y/o posparto a los análogos de nucleósidos. Los principales acontecimientos adversos notificados son trastornos hematológicos (anemia, neutropenia), trastornos metabólicos (hiperlactemia, hiperlipasemia). Se han notificado algunos trastornos neurológicos de aparición tardía (hipertonía, convulsión, comportamiento anormal). Los trastornos neurológicos pueden ser transitorios o permanentes. Cualquier niño expuesto in utero a análogos de nucleósido o de nucleótido, debe ser sometido a un seguimiento clínico y de laboratorio y en caso de signos o síntomas relevantes, debe ser minuciosamente investigada una posible disfunción mitocondrial.
Pacientes pediátricos
Se ha administrado lamivudina a niños (de 2 años de edad y mayores) y adolescentes con hepatitis B crónica compensada. Sin embargo, debido a las limitaciones de los datos, la administración de lamivudina en esta población de pacientes no está recomendada actualmente (ver sección 5.1.).
Hepatitis Delta o hepatitis C
No se ha establecido la eficacia de lamivudina en pacientes coinfectados con hepatitis Delta o hepatitis C, por lo que se recomienda tener precaución.
Tratamientos inmunodepresores
Los datos en relación al empleo de lamivudina en pacientes HBeAg negativos (con mutante pre-core) y en aquellos que reciben tratamientos inmunodepresores concurrentes, incluyendo quimioterapia en cáncer son limitados. Lamivudina debe ser usada con precaución en estos pacientes.
Monitorización
Durante el tratamiento con Zeffix, los pacientes deben ser controlados de forma regular. Cada 3 meses se debe monitorizar los niveles de ALT sérica el ADN del VHB y cada 6 meses se debe determinar los HBeAg en pacientes HBeAg positivos.
Coinfección con el VIH
Para el tratamiento de pacientes coinfectados con el VIH y que estén recibiendo o esté previsto que reciban tratamiento con lamivudina o con la combinación lamivudina/zidovudina, debe mantenerse la dosis de lamivudina prescrita para la infección por el VIH (normalmente 150 mg/dos veces al día en combinación con otros antirretrovirales). En los pacientes coinfectados con el VIH que no precisen tratamiento antirretroviral, existe un riesgo de mutación del VIH al utilizar sólo lamivudina para el tratamiento de la hepatitis B crónica.
Transmisión de la hepatitis B
No se dispone de información sobre la transmisión materno fetal del virus de la hepatitis B en mujeres embarazadas que estén en tratamiento con lamivudina. Se deben seguir los procedimientos estándar recomendados de inmunización frente al virus de la hepatitis B en niños.
Debe informarse a los pacientes de que el tratamiento con lamivudina no ha demostrado reducir el riesgo de transmisión del virus de la hepatitis B a otras personas, por lo que deben tomarse las precauciones adecuadas.
Interacciones con otros medicamentos
Zeffix no debe ser tomado con cualquier otro medicamento que contenga lamivudina o medicamentos que contengan emtricitabina (ver sección 4.5).
No se recomienda la combinación de lamivudina con cladribina (ver sección 4.5).
Intolerancia a excipientes
Los pacientes con problemas raros de intolerancia hereditaria a la fructosa, problemas de absorción a la glucosa o galactosa, o insuficiencia de sacarasa-isomaltasa, no deben tomar este medicamento.
Los pacientes diabéticos deben ser informados de que cada dosis de solución oral (100 mg = 20 ml) contiene 4 g de sacarosa.
La solución oral contiene parahidroxibenzoato de metilo y de propilo. Estos productos pueden provocar una reacción alérgica en algunos individuos. Esta reacción puede ser retardada.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los estudios de interacción se han realizado sólo en adultos.
La probabilidad de interacciones metabólicas es baja debido a un metabolismo y unión a proteínas plasmaticas limitado y a la casi completa eliminación renal de la sustancia inalterada.
La lamivudina se elimina predominantemente por secreción catiónica orgánica activa. Debe considerarse la posibilidad de interacciones con otros medicamentos administrados al mismo tiempo, especialmente cuando su principal vía de eliminación sea la secreción renal activa por el sistema de transporte catiónico, por ejemplo trimetoprim. Otros medicamentos (por ej. ranitidina, cimetidina) se eliminan sólo en parte por este mecanismo y demostraron no interaccionar con lamivudina.
No es probable que sustancias que hayan demostrado excretarse principalmente bien por la vía aniónica orgánica activa o por filtración glomerular, den lugar a interacciones clínicamente significativas con lamivudina. La administración de trimetoprim/sulfametoxazol 160 mg/800 mg incrementó la exposición a lamivudina en alrededor de un 40 %. Lamivudina no ejerció ningún efecto sobre la farmacocinética de trimetoprim o sulfametoxazol. No obstante, a menos que el paciente presente insuficiencia renal, no es necesario ajustar la dosis de lamivudina.
Se observó un modesto incremento en el valor de Cmax (28 %) para zidovudina al administrarse con lamivudina, aunque la exposición general (AUC) no se alteró de forma significativa. Zidovudina carece de efecto sobre la farmacocinética de lamivudina (ver sección 5.2.).
Lamivudina no ejerce interacciones farmacocinéticas con interferón-alfa cuando ambos medicamentos se administran al mismo tiempo. No se observaron interacciones adversas clínicamente significativas en pacientes que toman al mismo tiempo lamivudina con medicamentos inmunodepresores comúnmente utilizados (p. ej. ciclosporina A). No obstante, no se han realizado estudios formales de interacción.
Emtricitabina
Debido a sus similitudes, Zeffix no debe ser administrado de forma concomitante con otros análogos de citidina, como emtricitabina. Además, Zeffix no se debe tomar con ningún otro medicamento que contenga lamivudina (ver sección 4.4).
Cladribina
Lamivudina in vitro inhibe la fosforilación intracelular de cladribina dando lugar a un potencial riesgo de pérdida de eficacia de cladribina si se toma en combinación durante el manejo clínico. Algunos hallazgos clínicos también sugieren una posible interacción entre lamivudina y cladribina. Por tanto, no se recomienda el uso concomitante de lamivudina con cladribina (ver sección 4.4).
4.6 Fertilidad, embarazo y lactancia
Embarazo
Una gran cantidad de datos obtenidos en mujeres embarazadas (resultados de más 1.000 mujeres expuestas) indica que no hay toxicidad malformativa. Si es clínicamente necesario Zeffix puede usarse durante el embarazo.
En pacientes que están siendo tratadas con lamivudina y posteriormente se quedan embarazadas se debe considerar la posibilidad de una reaparición de la hepatitis al discontinuar el tratamiento con lamivudina.
Lactancia
Basándose en más de 200 parejas madre/hijo tratadas para el VIH, las concentraciones séricas de lamivudina en niños lactantes de madres tratadas para el VIH es muy baja (menos de un 4% de las concentraciones de suero materno) y disminuye progresivamente a niveles indetectables cuando los lactantes alcanzan las 24 semanas de edad. La cantidad total de lamivudina ingerida por un lactante es muy baja y por tanto es probable que dé lugar a exposiciones con un efecto antiviral sub-óptimo. La hepatitis B materna no es una contraindicación para la lactancia si se controla la prevención de la infección por hepatitis B por parte del recién nacido en el nacimiento y no hay evidencia de que las bajas concentraciones de lamivudina en leche materna puedan producir reacciones adversas en lactantes. Por tanto, la lactancia se puede considerar en madres tratadas con lamivudina para VHB teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento para la mujer. Cuando hay transmisión materna de VHB, a pesar de una adecuada profilaxis, se debe considerar interrumpir la lactancia para reducir el riesgo de que aparezcan mutantes resistentes a lamivudina en el niño.
Fertilidad
Los estudios de reproducción en animales no han mostrado efecto en la fertilidad masculina y femenina (ver sección 5.3).
Disfunción mitocondrial
Se ha demostrado in vitro e in vivo que los análogos de nucleósidos y nucleótidos causan daño mitocondrial de grado variable. Se han notificado casos de disfunción mitocondrial en niños expuestos en el útero y/o post-parto a análogos de nucleósidos (ver sección 4.4).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Se debe informar a los pacientes que se han notificado malestar y fatiga durante el tratamiento con lamivudina. El estado clínico del paciente y el perfil de reacciones adversas de lamivudina se deben tener en cuenta al considerar la capacidad de que el paciente conduzca o maneje máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
La incidencia de reacciones adversas y anormalidades de laboratorio (a excepción de los incrementos de ALT y CPK, ver a continuación) fue similar entre los pacientes tratados con placebo y los tratados con lamivudina. Las reacciones adversas comunicadas con más frecuencia fueron malestar y fatiga, infecciones del tracto respiratorio, molestias en la garganta y amígdalas, cefalea, dolor y molestias abdominales, náuseas, vómitos y diarrea.
Tabla de reacciones adversas
A continuación se presentan las reacciones adversas clasificadas por órganos, sistemas y frecuencias. Las categorías de frecuencia se asignan únicamente a aquellas reacciones adversas consideradas al menos posiblemente relacionadas con lamivudina. Las frecuencias se definen como: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raras (> 1/10.000 a < 1/1000), muy raras (< 1/10.000) y de frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Las categorías de frecuencias asignadas a las siguientes reacciones adversas se basan principalmente en la experiencia de los ensayos clínicos que incluyeron un total de 1.171 pacientes con hepatitis B crónica que recibieron 100 mg de lamivudina.
|
Trastornos de la sangre y del sistema linfático | |
|
Desconocida |
T rombocitopenia |
|
Trastornos del sistema inmunológico | |
|
Raras |
Angioedema |
|
Trastornos hepatobiliares | |
|
Muy frecuentes |
Incrementos de ALT (ver sección 4.4) |
|
Durante el tratamiento y |
tras la retirada de lamivudina se han notificado exacerbaciones de la |
|
hepatitis, detectadas principalmente por aumento en las concentraciones de ALT en suero. La | |
|
mayoría han remitido, aunque muy raramente se han observado muertes (ver sección 4.4). | |
|
Trastornos de la piel y del tejido subcutáneo | |
|
Frecuentes |
Erupción, prurito |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |
|
Frecuentes |
Incrementos de CPK |
|
Frecuentes |
Trastornos musculares, incluyendo mialgia y calambres |
|
Frecuencia no conocida |
Rabdomiolisis |
*En los ensayos fase III la frecuencia observada en el grupo tratado con lamivudina no fue mayor que la observada en el grupo placebo
Población pediátrica
Basándose en los datos limitados en niños de 2 a 17 años de edad, no se identificaron nuevas cuestiones de seguridad en comparación a las observadas en adultos.
Otras poblaciones especiales
En pacientes con infección por el VIH, se han comunicado casos de pancreatitis y neuropatía periférica (o parestesia). En pacientes con hepatitis B crónica no se observaron diferencias en la incidencia de estos eventos entre los pacientes tratados con placebo y los tratados con lamivudina.
Se han notificado casos de acidosis láctica, a veces mortales, normalmente asociados con hepatomegalia grave y esteatosis hepática, con el uso de terapia de combinación de análogos de nucleósidos en pacientes con VIH. Raramente se han comunicado casos de acidosis láctica en pacientes que recibían lamivudina para el tratamiento de hepatitis B.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V
4.9 Sobredosis
En estudios de toxicidad aguda en animales, la administración de dosis muy elevadas de lamivudina no produjo toxicidad en ningún órgano. Se dispone de datos limitados sobre las consecuencias de la ingestión de sobredosis agudas en humanos. No hubo fallecimientos y todos los pacientes se recuperaron. No se han identificado signos o síntomas específicos después de tal sobredosis.
En caso de sobredosis, se vigilará al paciente y se aplicará el tratamiento de apoyo estándar que sea necesario. Dado que lamivudina es dializable, puede emplearse una hemodiálisis continua para el tratamiento de la sobredosificación, aunque ello no se ha estudiado.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antivirales para uso sistémico, nucleósidos y nucleótidos inhibidores de la transcriptasa inversa, código ATC: J05AF05.
Lamivudina es un agente antiviral muy activo frente al virus de la hepatitis B en todas las líneas celulares estudiadas y en animales infectados experimentalmente.
Lamivudina es metabolizada tanto por células infectadas como por no infectadas al derivado trifosfato (TP) que es la forma activa del compuesto original. La semivida intracelular del trifosfato en los hepatocitos es de 17-19 horas in vitro. Lamivudina -TP actúa como sustrato para la polimerasa viral del VHB.
La formación de más ADN viral se bloquea por incorporación de lamivudina-TP a la cadena y posterior terminación de la misma.
Lamivudina-TP no interfiere con el metabolismo celular normal de los deoxinucleótidos. También es un débil inhibidor de las alfa y beta ADN polimerasas de mamíferos. Además, lamivudina-TP tiene un escaso efecto sobre el contenido de ADN en las células de mamíferos.
En los estudios relativos a los potenciales efectos de la sustancia sobre la estructura mitocondrial y sobre el contenido y función del ADN, se vió que lamivudina carecía de efectos tóxicos apreciables. Presenta un potencial muy bajo para disminuir el contenido de ADN mitocondrial, no se incorpora de forma permanente al ADN mitocondrial y no actúa como inhibidor de la ADN gamma polimerasa mitocondrial.
Experiencia clínica
Experiencia en pacientes con HBC HBeAg positivos y enfermedad hepática compensada En estudios controlados, el tratamiento durante 1 año con lamivudina suprimió de forma significativa la replicación del ADN del VHB [34-57 % de los pacientes estaban por debajo de los límites de detección (ensayo de hibridación en solución de Abbott Genostics, LLOD < 1,6 pg/ml)], normalizó el nivel de ALT (40-72 % de los pacientes), indujo la seroconversión del HBeAg (pérdida de HBeAg y detección de HBeAc con pérdida de ADN del VHB [por ensayo convencional], 16-18 % de pacientes), mejoró la histología (38-52 % de los pacientes tuvieron un descenso > a 2 puntos en el Índice de Actividad Histológica de Knodell [IAH]), y redujo la progresión de la fibrosis (en 3-17 % de los pacientes) y la progresión a cirrosis.
El tratamiento continuo con lamivudina durante 2 años más en pacientes que no lograron una seroconversión de HBeAg en el primer año de los estudios controlados, dio lugar a una mejoría añadida de la fibrosis en puente. En pacientes con el mutante YMDD del VHB, 41/82 (50 %) pacientes mejoraron en la inflamación hepática, y 40/56 (71 %) pacientes sin el mutante YMDD del VHB mejoraron. La mejoría en la fibrosis en puente se produjo en 19/30 (63 %) pacientes sin el mutante YMDD y en 22/44 (50 %) pacientes con el mutante. El 5 % de los pacientes (3/56) sin el mutante YMDD y el 13 % (11/82) pacientes con el mutante YMDD mostraron un empeoramiento en la inflamación hepática con respecto a los niveles pre-tratamiento. La progresión a cirrosis se produjo en 4/68 (6 %) de los pacientes con el mutante YMDD, mientras que ningún paciente sin el mutante progresó a cirrosis.
En un estudio de tratamiento prolongado en pacientes asiáticos (NUCB3018), la tasa de seroconversión de HBeAg y la velocidad de normalización de ALT al final del periodo de tratamiento de 5 años, fueron de 48 % (28/58) y 47 % (15/32), respectivamente. La seroconversión de HBeAg aumentó en pacientes con niveles elevados de ALT; seroconvirtieron el 77 % (20/26) de los pacientes con niveles de ALT previos al tratamiento >2x LSN. Al final de los 5 años, todos los pacientes tuvieron niveles de ADN del VHB indetectables o menores que los niveles previos al tratamiento.
En la Tabla 2 se resumen los últimos resultados del ensayo según el estado del mutante YMDD.
Tabla 2: Resultados de eficacia de 5 años según el estado de YMDD (Estudio asiático) NUCB3018
|
Pacientes, % (n°.) | ||||
|
Estado VHB mutante YMDD |
YMDD1 |
No-YMDD1 | ||
|
Seroconversión de HBeAg - Todos los pacientes |
38 |
(15/40) |
72 |
(13/18) |
|
- ALT basal < 1 x LSN2 |
9 |
(1/11) |
33 |
(2/6) |
|
- ALT basal > 2 x LSN |
60 |
(9/15) |
100 |
(11/11) |
|
ADN de VHB no detectable | ||||
|
- Basal 3 |
5 |
(2/40) |
6 |
(1/18) |
|
- Semana 260 4 | ||||
|
negativo |
8 |
(2/25) |
0 | |
|
positivo < nivel basal |
92 |
(23/25) |
100 |
(4/4) |
|
positivo > nivel basal |
0 |
0 | ||
|
Normalización ALT | ||||
|
- Basal | ||||
|
normal |
28 |
(11/40) |
33 |
(6/18) |
|
por encima de lo normal |
73 |
(29/40) |
67 |
(12/18) |
|
- Semana 260 | ||||
|
normal |
46 |
(13/28) |
50 |
(2/4) |
|
por encima de lo normal < nivel basal |
21 |
(6/28) |
0 | |
|
por encima de lo normal > nivel basal |
32 |
(9/28) |
50 |
(2/4) |
1. Los pacientes categorizados como mutante YMDD fueron aquellos con >5% de variante YMDD del VHB en cualquier instante del año durante un período de 5 años. Los pacientes categorizados como no mutante -YMDD fueron aquellos con > 95% de tipo salvaje de VHB para cualquier momento del año durante el período de 5 años del estudio
2. Límite normal superior
3. Ensayo de hibridación en solución Abbott Genostics (LLOD < 1,6 pg/ml)
4. Ensayo Chiron Quantiplex (LLOD 0,7 Meq/ml)
También se dispone de datos comparativos según el status YMDD para su evaluación histológica, pero sólo hasta 3 años. En pacientes con el mutante YMDD del VHB, 18/39 (46 %) hubo mejoría en la actividad necroinflamatoria, y en 9/39 (23 %) un empeoramiento. En los pacientes sin el mutante, 20/27 (74 %) hubo mejoría en la actividad necroinflamatoria, y en 2/27 (7 %) un empeoramiento.
Después de la seroconversión del HBeAg, la respuesta serológica y la remisión clínica son generalmente duraderas tras interrumpir el tratamiento con lamivudina. Sin embargo, pueden ocurrir recaídas tras la seroconversión. En un estudio a largo plazo de seguimiento de pacientes que tuvieron seroconversión e interrumpieron el tratamiento con lamivudina, la recaída virológica posterior ocurrió en un 39 % de los sujetos. Por tanto, tras la seroconversión de HBeAg, los pacientes deben ser controlados periódicamente para determinar si las respuestas serológica y clínica se mantienen. En pacientes que no mantengan una respuesta serológica constante, debería considerarse un retratamiento, o bien con lamivudina, o bien con un agente antiviral alternativo para la reanudación del control clínico del VHB.
En pacientes bajo seguimiento hasta 16 semanas tras la interrupción del tratamiento un año después de iniciarlo, se han observado con mayor frecuencia elevaciones de ALT post-tratamiento en pacientes que habían recibido lamivudina que en pacientes que habían recibido placebo. En la Tabla 3 se muestra una comparación de las elevaciones de ALT post-tratamiento entre las semanas 52 y 68 en pacientes que interrumpieron el tratamiento con lamivudina en la semana 52 y pacientes en los mismos estudios que recibieron placebo a lo largo del período de tratamiento. La proporción de pacientes que tuvieron elevaciones de ALT post-tratamiento junto con un aumento de los niveles de bilirrubina fue baja y similar en pacientes que recibieron tanto lamivudina como placebo.
Tabla 3: Elevaciones de ALT post-tratamiento en 2 estudios controlados con placebo en adultos
|
Valor Anormal |
Pacientes con Elevación de ALT/ Pacientes con Observaciones* | |
|
Lamivudina |
Placebo | |
|
ALT >2 x valor basal |
37/137 (27 %) |
22/116 (19 %) |
|
ALT >3 x valor basal ^ |
29/137 (21 %) |
9/116 (8 %) |
|
ALT >2 x valor basal y ALT absoluta>500 Ul/l |
21/137 (15 %) |
8/116 (7 %) |
|
ALT >2 x valor basal; y bilirrubina >2 x LSN y >2 x valor basal |
1/137 (0,7%) |
1/116 (0,9%) |
* Cada paciente puede estar representado en una o más categorías.
^ Comparable a Toxicidad de Grado 3 de acuerdo al criterio modificado de la OMS. LSN = Límite superior de normalidad.
Experiencia en pacientes con HBC HBeAg negativos
Los datos iniciales indican que la eficacia de lamivudina en pacientes con HBC HBeAg negativos es similar a la de pacientes con HBC HBeAg positivos, obteniendo el 71 % de los pacientes una supresión de ADN del VHB por debajo del límite de detección del ensayo; el 67 % normalización de ALT; y el 38 %, una mejoría del IAH tras un año de tratamiento. Cuando se interrumpió el tratamiento con lamivudina, la mayoría de los pacientes (70 %) volvieron a presentar replicación viral. Se dispone de datos procedentes de un estudio de tratamiento prolongado en pacientes HBeAg negativos, tratados con lamivudina (NUCAB3017). Tras dos años de tratamiento en este estudio, se observó la normalización de los niveles de ALT, y no se detectó ADN del VHB en 30/69 (43 %) y 32/68 (47 %) pacientes, respectivamente, así como una mejoría en el grado de necroinflamación en 18/49 (37 %) pacientes. En pacientes sin el mutante YMDD del VHB, 14/22 (64 %) mostraron mejoría en el grado de necroinflamación y 1/22 (5 %) empeoraron con respecto a los niveles pre-tratamiento. En pacientes con el mutante, 4/26 (15 %) mostraron mejoría en el grado de necroinflamación, y 8/26 (31 %) empeoraron con respecto a los niveles pre-tratamiento. Ningún paciente en ninguno de los grupos progresó a cirrosis.
Frecuencia de aparición del mutante YMDD del VHB e impacto en la respuesta al tratamiento La monoterapia con lamivudina da lugar a la aparición del mutante YMDD del VHB en aproximadamente el 24 % de los pacientes tras un año de terapia, aumentando hasta el 69 % tras 5 años de terapia. El desarrollo del mutante YMDD del VHB está asociado con una reducción en la respuesta al tratamiento en algunos pacientes, lo cual se evidencia por un aumento de los niveles de ADN del VHB y por elevación de los niveles de ALT, con respecto a los niveles previos durante la terapia, progresión de signos y síntomas de enfermedad hepática y/o empeoramiento de hallazgos necroinflamatorios hepáticos. Dado el riesgo del mutante YMDD del VHB, no es adecuado el mantenimiento de la monoterapia con lamivudina en pacientes con ADN del VHB detectable en suero al cabo de 24 semanas o más de tratamiento (ver sección 4.4).
En un estudio doble ciego en pacientes con HBC con el mutante YMDD del VHB y enfermedad hepática compensada (NUC20904), con una respuesta virológica y bioquímica reducida a lamivudina (n=95), la adición de 10 mg de adefovir dipivoxilo una vez al día al tratamiento con 100 mg de lamivudina durante 52 semanas, dio lugar a una disminución media del ADN del VHB de 4,6 logi0 copias/ml, en comparación con un aumento medio de 0,3 logJ0 copias/ml en aquellos pacientes que recibieron lamivudina en monoterapia. La normalización de los niveles de ALT se produjo en un 31 % (14/45) de los pacientes que recibieron terapia combinada, frente a un 6 % (3/47) que recibieron lamivudina sola. Se mantuvo la supresión viral (estudio de seguimiento NUC20917) con terapia combinada durante el segundo año de tratamiento hasta la semana 104 con pacientes que continuaron mejorando en las respuestas virológica y bioquímica.
En un estudio retrospectivo para determinar los factores asociados con la evolución del ADN del VHB se trataron con lamivudina 159 HBeAg pacientes positivos asiáticos y se siguieron durante un periodo cuya mediana fue de casi 30 meses. Aquellos con niveles ADN del VHB mayores de 200 copias/ml a los 6 meses (24 semanas) de tratamiento con lamivudina tuvieron un 60 % de posibilidad de desarrollar el mutante YMDD en comparación con un 8% de aquellos con un ADN del VHB menores de 200 copias/ml a las 24 semanas de tratamiento con lamivudina. El riesgo de desarrollar el mutante YMDD fue de 63 % versus el 13 % con un corte de 1.000 copias/ml (NUCB3009 y NUCB3018).
Experiencia en pacientes con enfermedad hepática descompensada
En pacientes con descompensación hepática no se han realizado estudios controlados con placebo por considerarse inapropiados. En estudios no controlados, en los que se administró lamivudina antes y durante el trasplante, se demostró la supresión eficaz del ADN del VHB y la normalización de los valores de ALT. Cuando se continuaba administrando lamivudina después del trasplante, la reinfección del injerto por el VHB era menor, tenía lugar una mayor pérdida del HBsAg y la tasa de supervivencia al cabo de 1 año era del 76-100 %.
Como era de esperar, debido a la inmunodepresión concomitante, la tasa de aparición del mutante YMDD del VHB, al cabo de 52 semanas de tratamiento, fue superior (36 % - 64 %) en la población con trasplante de hígado que en los pacientes inmunocompetentes con HBC (14 % - 32 %).
Se incluyeron en un brazo abierto del estudio NUC20904 cuarenta pacientes (HBeAg negativos o HBeAg positivos), bien con enfermedad hepática descompensada o con VHB recurrente tras un trasplante hepático, y con el mutante YMDD. La adición de 10 mg de adefovir dipivoxilo una vez al día al tratamiento en curso con 100 mg de lamivudina durante 52 semanas dio lugar a un descenso medio en el ADN del VHB de 4,6 logi0 copias/ml. También se observó una mejoría en la función hepática tras un año de tratamiento. Este grado de supresión viral se mantuvo (estudio de seguimiento NUC20917) con terapia combinada durante el segundo año de tratamiento hasta la semana 104 y la mayoría de pacientes mejoraron en cuanto a los marcadores de función hepática y continuaron hacia un beneficio clínico.
Experiencia en pacientes con HBC con fibrosis avanzada o cirrosis
En un estudio controlado con placebo en 651 pacientes con hepatitis B crónica clínicamente compensada y fibrosis o cirrosis confirmadas histológicamente, el tratamiento con lamivudina (32 meses de duración media) redujo significativamente la tasa de progresión global de la enfermedad (34/436, 7,8 % para lamivudina, frente a 38/215, 17,7 % para placebo, p=0,001), demostrado por una reducción significativa en el porcentaje de pacientes que experimentaron un aumento en la puntuación de Child-Pugh (15/436, 3,4 % frente a 19/215, 8,8 %, p=0,023), o bien un desarrollo de carcinoma hepatocelular (17/436, 3,9 % frente a 16/215, 7,4 %, p=0,047). La tasa de progresión global de la enfermedad en el grupo tratado con lamivudina fue mayor en aquellos pacientes en los que se detectó el mutante YMDD del ADN del VHB (23/209, 11 %), en comparación con aquéllos en los que no se detectó el mutante (11/221, 5 %). Sin embargo, la progresión de la enfermedad en pacientes con YMDD en el grupo tratado con lamivudina fue menor que la progresión de la enfermedad en el grupo tratado con placebo (23/209, 11 %, frente a 38/214, 18 %, respectivamente). La seroconversión del HBeAg tuvo lugar en el 47 % (118/252) de los pacientes tratados con lamivudina, y el 93 % (320/345) de los pacientes que recibieron lamivudina se convirtieron en ADN del VHB negativos durante el estudio (VERSANT [versión 1], ensayo bDNA, LLOD < 0,7 mEq/ml).
Experiencia en niños y adolescentes
Se ha administrado lamivudina a niños y adolescentes con HBC compensada en un estudio controlado con placebo en 286 pacientes de edades comprendidas entre los 2 y los 17 años. La población estaba compuesta principalmente por niños con hepatitis B de grado mínimo. Se empleó una dosis de 3 mg/kg una vez al día (hasta un máximo de 100 mg diarios) en niños de edades comprendidas entre 2 y 11 años y una dosis de 100 mg una vez al día en adolescentes de 12 años y mayores. Se necesita conocer más datos acerca de esta dosis. La diferencia en los índices de seroconversión de HBeAg (pérdida de HBeAg y ADN del VHB con detección de HBeAc) entre placebo y lamivudina no fue estadísticamente significativa en esta población (los índices después de un año fueron del 13 % (12/95) para placebo, frente al 22 % (42/191) para la^mvudina, p 0,057). La incidencia del pautante YMDD del VHB fue similar a la observada en adultos, variando del 19 % en la semana 52, hasta el 45 % en pacientes tratados de forma continuada durante 24 meses.
5.2 Propiedades farmacocinéticas
Absorción
Lamivudina se absorbe bien en el tracto gastrointestinal y la biodisponibilidad de lamivudina por vía oral en adultos está normalmente entre el 80-85 %. Tras la administración por vía oral, el tiempo medio (tmax) hasta las concentraciones séricas máximas (Cmax) es de aproximadamente una hora. A dosis terapéuticas, es decir, 100 mg una vez al día, la Cmax es del orden de 1,1-1,5 pg/ml siendo los niveles mínimos de 0,015-0,020 pg/ml.
La administración de lamivudina con alimentos dio lugar a un retraso de tmax y a una Cmax inferior (reducida en un 47 %). No obstante, la cantidad de lamivudina absorbida (en función del AUC) no se vio afectada, por lo que lamivudina se puede administrar con o sin alimentos.
Distribución
Según los estudios realizados con lamivudina por vía intravenosa, el volumen medio de distribución es de 1,3 l/kg. Lamivudina presenta una farmacocinética lineal a lo largo del intervalo de dosis terapéuticas y su unión a la proteína plasmática albúmina es baja.
Los datos limitados de que se dispone muestran que lamivudina penetra en el sistema nervioso central y que alcanza el líquido cefalorraquídeo (LCR). El valor medio del ratio LCR/concentración sérica de lamivudina a las 2-4 horas de la administración por vía oral fue, aproximadamente, de 0,12.
Biotransformación
Lamivudina se elimina principalmente por excreción renal de la sustancia inalterada. La probabilidad de interacciones metabólicas de lamivudina con otras sustancias es baja, debido al pequeño grado de metabolismo hepático (5-10 %) y a la escasa unión a proteínas plasmáticas.
Eliminación
El aclaramiento sistémico medio de lamivudina es de, aproximadamente, 0,3 l/h/kg. La semivida de eliminación observada es de 5 a 7 horas. Lamivudina se excreta en su mayor parte inalterada en orina por filtración glomerular y secreción activa (sistema de transporte catiónico orgánico). El aclaramiento renal representa aproximadamente el 70 % de la eliminación de lamivudina.
Poblaciones especiales
Los estudios en pacientes con insuficiencia renal, demuestran que la eliminación de lamivudina se ve afectada por la disfunción renal. Es necesario reducir la dosis en pacientes con un aclaramiento de creatinina inferior a 50 ml/min (ver sección 4.2).
La farmacocinética de lamivudina no se ve afectada por la insuficiencia hepática. Los datos limitados de que se dispone en pacientes sometidos a trasplante hepático, demuestran que la insuficiencia en la función hepática no tiene un impacto significativo sobre la farmacocinética de lamivudina a menos que esté acompañada de disfunción renal.
En pacientes ancianos, el perfil farmacocinético de lamivudina indica que un envejecimiento normal con la disminución de la función renal que lo acompaña, carece de efecto clínicamente significativo sobre la exposición a lamivudina, excepto en pacientes con un aclaramiento de creatinina inferior a 50 ml/min (ver sección 4.2).
5.3 Datos preclínicos sobre seguridad
La administración de lamivudina en los estudios de toxicidad con animales a dosis elevadas no se asoció con toxicidad en ningún órgano principal. A las dosis más altas se observaron efectos menores sobre los indicadores de las funciones hepática y renal, junto a reducciones ocasionales en el peso del
hígado. Los efectos clínicamente importantes apreciados fueron una reducción en el recuento de eritrocitos y neutropenia. Estos efectos se observaron con poca frecuencia en los estudios clínicos.
Lamivudina no fue mutagénica en los ensayos con bacterias pero, como muchos análogos de nucleósidos, mostró actividad en un ensayo citogenético in vitro y en el ensayo de linfoma de ratón. Lamivudina no fue genotóxica in vivo a dosis que dieron lugar a concentraciones plasmáticas aproximadamente 60-70 veces más elevadas que los niveles plasmáticos clínicos previstos. Como la actividad mutagénica in vitro de lamivudina no pudo confirmarse en las pruebas in vivo, se concluye que lamivudina no representa un riesgo de aparición de genotoxicidad en pacientes sometidos a tratamiento.
Los estudios sobre la reproducción en animales no han mostrado evidencia de teratogenicidad y tampoco han mostrado efectos sobre la fertilidad en machos o hembras. Lamivudina induce embrioletalidad temprana cuando se administra a conejos preñados a niveles de exposición comparables a los alcanzados en el hombre, pero no en la rata incluso a exposiciones sistémicas muy altas.
Los resultados de los estudios de carcinogenicidad a largo plazo con lamivudina realizados con ratas y ratones mostraron la ausencia de potencial carcinogénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Sacarosa (20 % p/v)
Parahidroxibenzoato de metilo (E218)
Parahidroxibenzoato de propilo (E216)
Ácido cítrico (anhidro)
Propilenglicol Citrato de sodio Saborizante artificial de fresa Saborizante artificial de plátano Agua purificada
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
2 años.
Desde que se abre el envase por primera vez: 1 mes
6.4 Precauciones especiales de conservación No conservar a temperatura superior a 25°C.
6.5 Naturaleza y contenido del recipiente
Estuches conteniendo un frasco opaco de polietileno de alta densidad (HDPE) con cierre de polipropileno resistente a los niños con 240 ml de solución oral de lamivudina. El envase también incluye un aplicador-adaptador de polietileno y un aplicador de 10 ml para la dosificación oral, compuesta por un cilindro de polipropileno (con graduaciones en ml) y un émbolo de polietileno.
Se incluye un aplicador para la medición exacta de la dosis prescrita de solución oral.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Ltd
980 Great West Road
Brentford
Middlesex
TW8 9GS
Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/99/114/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización 29/julio/1999 Fecha de la última renovación 23/junio/2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
A. FABRICANTE(S) RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE(S) RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de los fabricantes responsables de la liberación de los lotes
Comprimido recubierto con película:
Glaxo Wellcome Operations
Priory Street, Ware
Hertfordshire
SG12 0DJ
Reino Unido
o
GlaxoSmithKline Pharmaceuticals S.A. ul. Grunwaldzka 189 60-322 Poznan Polonia
Solución oral:
Aspen Bad Oldesloe GmbH Industriestrasse 32-36 23843 Bad Oldesloe Alemania
o
Glaxo Operations UK Limited
(operando como Glaxo Wellcome Operations)
Harmire Road Barnard Castle County Durham DL12 8DT Reino Unido
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quarter, apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
No procede.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
EMBALAJE EXTERIOR PARA COMPRIMIDOS: Estuche de 28 comprimidos, estuche de 84 comprimidos
1. NOMBRE DEL MEDICAMENTO
Zeffix 100 mg comprimidos recubiertos con película Lamivudina
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 100 mg de lamivudina
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
28 comprimidos recubiertos con película 84 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30°C
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Ltd
980 Great West Road
Brentford
Middlesex
TW8 9GS
Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/99/114/001 28 comprimidos EU/1/99/114/002 84 comprimidos
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
zeffix 100 mg
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS: Estuche de 28 comprimidos, estuche de 84 comprimidos_
1. NOMBRE DEL MEDICAMENTO
Zeffix 100 mg comprimidos Lamivudina
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Ltd
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DEL LOTE
Lot
5. OTROS
1. NOMBRE DEL MEDICAMENTO
Zeffix 5 mg/ml solución oral Lamivudina
2. PRINCIPIO(S) ACTIVO(S)
Cada ml de solución oral contiene 5 mg de lamivudina
3. LISTA DE EXCIPIENTES
Contiene entre otros:
azúcar (sacarosa), conservantes: parahidroxibenzoato de metilo (E218) y parahidroxibenzoato de propilo (E216)
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cada frasco contiene 240 ml de solución oral El envase contiene un aplicador oral.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Desechar el frasco transcurrido un mes tras la primera apertura
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 25°C
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Ltd 980 Great West Road Brentford Middlesex
TW8 9GS Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/99/114/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
zeffix 5 mg/ml
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
1. NOMBRE DEL MEDICAMENTO
Zeffix 5 mg/ml solución oral Lamivudina
2. PRINCIPIO(S) ACTIVO(S)
Cada ml de solución oral contiene 5 mg de lamivudina
3. LISTA DE EXCIPIENTES
Contiene entre otros:
azúcar (sacarosa), conservantes: parahidroxibenzoato de metilo (E218) y parahidroxibenzoato de propilo (E216)
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cada frasco contiene 240 ml de solución oral
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral
|
6. |
ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS |
|
Mantener fuera de la vista y del alcance de los niños | |
|
7. |
OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO |
|
8. |
FECHA DE CADUCIDAD |
|
CAD | |
|
Desechar el frasco transcurrido un mes tras la primera apertura | |
|
9. |
CONDICIONES ESPECIALES DE CONSERVACIÓN |
No conservar a temperatura superior a 25°C
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL PRODUCTO NO
UTILIZADO O DE LOS MATERIALES QUE ESTÉN EN CONTACTO DIRECTO CON EL PRODUCTO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Ltd 980 Great West Road Brentford Middlesex
TW8 9GS Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/99/114/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
B. PROSPECTO
Prospecto: información para el paciente
Zeffix 100 mg comprimidos recubiertos con película
lamivudina
Lea todo el prospecto detenidamente antes de empezar a tomar el medicamento.
- Conserve este prospecto ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Zeffix y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Zeffix
3. Cómo tomar Zeffix
4. Posibles efectos adversos
5. Conservación de Zeffix
6. Contenido del envase e información adicional
1. Qué es Zeffix y para qué se utiliza
El principio activo de Zeffix es lamivudina.
Zeffix se utiliza para tratar la infección de larga duración (crónica) por hepatitis B en adultos.
Zeffix es un medicamento antiviral que inhibe el virus de la hepatitis B y pertenece a un grupo de medicamentos denominados inhibidores de la transcriptasa inversa análogos de nucleósidos (INTIs).
El virus de la hepatitis B infecta al hígado, causa una infección de larga duración (crónica), y puede ocasionar un daño hepático. Zeffix puede ser utilizado en pacientes cuyo hígado está dañado pero todavía funciona con normalidad (enfermedad hepática compensada) y en combinación con otros medicamentos en pacientes cuyo hígado está dañado y no funciona de forma normal (enfermedad hepática descompensada).
El tratamiento con Zeffix puede reducir la cantidad de virus de hepatitis B en su organismo. Esto daría lugar a una reducción del daño hepático y a una mejoría en el funcionamiento de su hígado. No todo el mundo responde al tratamiento con Zeffix de la misma manera. Su médico controlará la efectividad del tratamiento con análisis de sangre periódicos.
2. Qué necesita saber antes de empezar a tomar Zeffix No tome Zeffix:
• si es alérgico a lamivudina o a cualquiera de los demás componentes de este medicamento
(incluidos en la sección 6).
Consulte con su médico si cree que esto le afecta.
Advertencias y precauciones
Algunas personas que toman Zeffix u otros medicamentos similares tienen un mayor riesgo de sufrir efectos adversos graves. Usted necesita saber que hay un mayor riesgo:
• si alguna vez ha tenido otros tipos de enfermedad hepática, como la hepatitis C
• si tiene un sobrepeso importante (especialmente si es mujer)
+ Consulte a su médico si padece alguna de estas circustancias. Puede necesitar pruebas adicionales, como análisis de sangre, mientras toma este medicamento. Para más información sobre los riesgos ver Sección 4.
No deje de tomar Zeffix sin el consejo de su médico, ya que existe un riesgo de que su hepatitis empeore. Cuando deje de tomar Zeffix su médico lo controlará durante al menos cuatro meses para comprobar si existe algún problema. Esto implicará tomar muestras de sangre para comprobar si hay elevaciones en los niveles de las enzimas hepáticas que puedan indicar lesión hepática. Ver sección 3 para más información sobre cómo tomar Zeffix.
Esté atento a los síntomas importantes
Algunas personas que toman medicamentos para la infección por hepatitis B desarrollan otros trastornos, que pueden ser graves. Usted necesita conocer a qué signos y síntomas importantes debe prestar atención mientras está tomando Zeffix.
^ Lea la información sobre “Otros posibles efectos adversos de la terapia para la hepatitis B” en la Sección 4 de este prospecto.
Proteja a otras personas
La hepatitis B se transmite por mantener contacto sexual con alguien que padezca la enfermedad o por transferencia de sangre infectada (por ejemplo, por compartir agujas). Zeffix no evita el riesgo de contagio de la infección por hepatitis B a los demás. Para prevenir que otras personas se infecten por hepatitis B:
• Utilice preservativo cuando mantenga sexo oral o con penetración.
• Evite el riesgo de transferencias de sangre — por ejemplo, no comparta agujas.
Toma de Zeffix con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento, incluyendo aquellos con plantas medicinales o medicamentos sin receta.
Recuerde decirle a su médico o farmacéutico si empieza a tomar algún otro medicamento mientras está tomando Zeffix.
Estos medicamentos no deben tomarse con Zeffix:
• otros medicamentos que contengan lamivudina, usado para tratar la infección por el VIH (a veces también llamado virus del SIDA)
• emtricitabina, usado para tratar la infección por el VIH o por hepatitis B
• cladribina, usado para el tratamiento de la leucemia de células pilosas ^Informe a su médico si está siendo tratado con alguno de estos medicamentos.
Embarazo
Si está embarazada, cree que podría estar embarazada o tiene intención de quedarse embarazada:
+ Hable con su médico sobre los riesgos y beneficios de tomar Zeffix durante el embarazo.
No interrumpa el tratamiento con Zeffix sin el consejo de su médico.
Lactancia
Zeffix puede pasar a la leche materna. Si está en periodo de lactancia, o pensando en dar el pecho:
^ Hable con su médico antes de tomar Zeffix.
Conducción y uso de máquinas
Zeffix puede hacer que se sienta cansado, lo que podría afectar a su capacidad para conducir o utilizar máquinas.
+ No conduzca o utilice máquinas a no ser que esté seguro de que esto no le afecta.
3. Cómo tomar Zeffix
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. Consulte a su médico o farmacéutico si tiene dudas.
Mantenga un contacto regular con su médico
Zeffix ayuda a controlar su infección por hepatitis B. Necesita continuar tomándolo cada día para controlar la infección y evitar que su infección empeore.
^ Mantenga el contacto con su médico y no deje de tomar Zeffix sin el consejo de su médico. Cuánto tomar
La dosis habitual de Zeffix es de un comprimido (100 mg de lamivudina) una vez al día.
Su médico puede recetarle una dosis más baja si tiene problemas de riñón. Está disponible Zeffix solución oral para personas que necesitan una dosis más baja de lo habitual, o que no puedan tomar comprimidos.
+ Hable con su médico si se encuentra en esta situación.
Si ya está tomando otro medicamento que contenga lamivudina para el tratamiento de la infección por el VIH, su médico le continuará tratando con la dosis más alta (normalmente 150 mg dos veces al día), porque la dosis de lamivudina de Zeffix (100 mg) no es suficiente para tratar la infección por el VIH. Si está planeando cambiar su tratamiento para el VIH, discuta este cambio con su médico antes.
Trague el comprimido entero con agua. Zeffix puede tomarse con o sin alimentos.
Si toma más Zeffix del que debe
Es improbable que la ingestión accidental de una cantidad excesiva de Zeffix, pueda causar problemas serios. Si accidentalmente toma demasiado, comuníquelo a su médico o farmacéutico, o acuda al servicio de urgencias del hospital más próximo para que le aconsejen.
Si olvidó tomar Zeffix
Si olvida tomar una dosis, tómela tan pronto como se acuerde. Luego continúe tomándolo como antes. No tome una dosis doble para compensar las dosis olvidadas.
No interrumpa el tratamiento con Zeffix
No deje de tomar Zeffix sin consultar a su médico. Hay riesgo de que su hepatitis empeore (ver sección 2). Cuando deje de tomar Zeffix su médico le monitorizará durante al menos cuatro meses para comprobar que no hay ningún problema. Esto significa que le realizarán análisis de sangre para comprobar si hay aumento en los niveles de enzimas del hígado, lo que puede indicar daño hepático.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Además de los efectos adversos listados a continuación para Zeffix, se pueden desarrollar otras condiciones durante el tratamiento de combinación para la hepatitis B.
+ Es importante leer la información bajo “Otros posibles efectos de la terapia de combinación para la hepatitis B”.
Los efectos adversos recogidos más comúnmente en los ensayos clínicos con Zeffix fueron cansancio, infecciones del tracto respiratorio, molestias en la garganta, cefalea, molestias y dolor de estómago, náuseas, vómitos y diarrea, aumento en las enzimas hepáticas y en las enzimas producidas en los músculos (ver abajo).
Reacción alérgica
Es rara (puede afectar hasta 1 de cada 1.000 personas). Los signos incluyen:
* hinchazón de los párpados, cara o labios
* dificultad para tragar o respirar.
+ Contacte con un médico inmediatamente si tiene estos síntomas. Deje de tomar Zeffix.
Efectos adversos que se cree están causados por Zeffix:
Un efecto adverso muy frecuente (puede afectar a más de 1 de cada 10 pacientes)
• que puede aparecer en los análisis de sangre es un aumento en los niveles de algunas enzimas producidas por el hígado (transaminasas), que puede ser un signo de inflamación o daño en el hígado.
Un efecto adverso frecuente (puede afectar hasta 1 de cada 10 pacientes) es:
• calambres y dolores musculares
• erupción cutánea o “habones” en cualquier parte del cuerpo.
Un efecto adverso frecuente que puede aparecer en los análisis de sangre es:
• Un aumento en el nivel de una enzima producida en los músculos (creatinfosfoquinasa) que puede ser un síntoma de que el hígado está dañado.
Otros efectos adversos
Se han notificado otros efectos adversos en un número muy pequeño de personas, pero su frecuencia exacta es desconocida:
• rotura muscular.
• empeoramiento de la enfermedad hepática si el virus hepatitis B se hace resistente a Zeffix o se interrumpe el tratamiento. Esto puede ser mortal en algunas personas.
• acidosis láctica (ver la siguiente sección “Otros posibles efectos adversos del tratamiento para la hepatitis B”)
Un efecto adverso que puede aparecer en los análisis de sangre es:
• una reducción en el número de células implicadas en la coagulación sanguínea (trombocitopenia).
Si tiene efectos adversos
^ Hable con su médico o farmacéutico. Esto incluye cualquiera de los posibles efectos adversos no incluidos en este prospecto.
Otros posibles efectos adversos del tratamiento para la hepatitis B
Zeffix y los medicamentos relacionados (INTIs) pueden causar otras condiciones que se desarrollan durante el tratamiento para la hepatitis B.
Acidosis láctica es un efecto adverso raro pero grave
Algunas personas que toman Zeffix u otros medicamentos similares (INTIs) desarrollan un trastorno denominado acidosis láctica, junto con un aumento del tamaño del hígado.
La acidosis láctica se debe a un aumento de ácido láctico en el organismo. Es raro, y si aparece, normalmente se desarrolla después de unos pocos meses de tratamiento. Puede suponer un riesgo para la vida, al causar fallos en órganos internos.
Es más probable que la acidosis láctica se desarrolle en personas que tienen alguna afección hepática o en personas obesas (sobrepeso importante), especialmente mujeres.
Los signos de la acidosis láctica incluyen:
• respiración dificultosa, rápida y profunda
• somnolencia
• entumecimiento o debilidad de las extremidades
• malestar (náuseas), vómitos
• dolor de estómago.
Durante su tratamiento, su médico controlará cualquier signo que indique que puede estar desarrollando acidosis láctica. Si tiene cualquiera de los síntomas mencionados anteriormente o le preocupa algún otro síntoma:
^ Acuda a su médico tan pronto como le sea posible.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Zeffix
Mantener este medicamento fuera de la vista y del alcance de los niños.
No tome este medicamento después de la fecha de caducidad que aparece en el embalaje y blíster.
No conservar a temperatura superior a 30°C.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico como deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Zeffix
El principio activo es lamivudina. Cada comprimido recubierto con película contiene 100 mg de lamivudina.
Los demás componentes son: celulosa microcristalina, carboximetilalmidón de sodio, estearato de magnesio, hipromelosa, dióxido de titanio, macrogol 400, polisorbato 80, óxido de hierro rojo y amarillo sintético.
Aspecto del producto y contenido del envase
Zeffix comprimidos recubiertos con película se presenta en blísters inviolables conteniendo 28 u 84 comprimidos. Los comprimidos son de color caramelo, con forma de cápsula, biconvexos y marcados “GX CG5” en una cara.
Puede que solamente estén comercializados algunos tamaños de envases.
Responsable de la fabricación
Titular de la Autorización de Comercialización
Glaxo Group Ltd
980 Great West Road
Brentford
Middlesex
TW8 9GS
Reino Unido
GlaxoWellcome Operations
Priory Street
Ware
Herts SG12 0DJ Reino Unido
o
GlaxoSmithKline Pharmaceuticals S.A. ul. Grunwaldzka 189 60-322 Poznan Polonia
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
GlaxoSmithKline Pharmaceuticals s.a./n.v. Tél/Tel: + 32 (0)10 85 52 00
Etarapna
raaKcoCMHTKaanH EOOfl Tea.: + 359 2 953 10 34
Ceská republika
GlaxoSmithKline s.r.o.
Tel: + 420 222 001 111 cz.info@gsk.com
Danmark
GlaxoSmithKline Pharma A/S Tlf: + 45 36 35 91 00 dk-info@gsk.com
Deutschland
GlaxoSmithKline GmbH & Co. KG Tel.: + 49 (0)89 36044 8701 produkt.info@gsk.com
Eesti
GlaxoSmithKline Eesti OÜ Tel: + 372 6676 900 estonia@gsk.com
EXXáóa
GlaxoSmithKline A.E.B.E.
Tnh + 30 210 68 82 100
España
GlaxoSmithKline, S.A.
Tel: + 34 902 202 700 es-ci@gsk.com
Luxembourg/Luxemburg
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Belgique/Belgien
Tél/Tel: + 32 (0) 10 85 52 00
Magyarország
GlaxoSmithKline Kft.
Tel.: + 36 1 225 5300
Malta
GlaxoSmithKline (Malta) Limited Tel: + 356 21 238131
Nederland
GlaxoSmithKline BV Tel: + 31 (0)30 6938100 nlinfo@gsk.com
Norge
GlaxoSmithKline AS Tlf: + 47 22 70 20 00 firmapost@gsk.no
Osterreich
GlaxoSmithKline Pharma GmbH Tel: + 43 (0)1 97075 0 at.info@gsk.com
Polska
GSK Services Sp.zo.o.
Tel.: + 48 (0)22 576 9000
Portugal
Glaxo Wellcome Farmacéutica, Lda. Tel: + 351 21 412 95 00 FI.PT@gsk.com
France
Laboratoire GlaxoSmithKline Tél.: + 33 (0)1 39 17 84 44 diam@gsk.com
Hrvatska
GlaxoSmithKline d.o.o.
Tel: + 385 1 6051 999
Slovenija
GlaxoSmithKline d.o.o.
Tel: + 386 (0)1 280 25 00 medical.x.si@gsk.com
Slovenská republika
GlaxoSmithKline Slovakia s. r. o. Tel: + 421 (0)2 48 26 11 11 recepcia.sk@gsk.com
Kúnpoq
GlaxoSmithKline (Cyprus) Ltd T@: + 357 22 39 70 00 gskcyprus@gsk.com
Latvija
GlaxoSmithKline Latvia SIA Tel: + 371 67312687 lv-epasts@gsk.com
Lietuva
GlaxoSmithKline Lietuva UAB Tel: + 370 5 264 90 00 info.lt@gsk.com
Suomi/Finland
GlaxoSmithKline Oy Puh/Tel: + 358 (0)10 30 30 30 finland.tuoteinfo@gsk.com
Sverige
GlaxoSmithKline AB Tel: + 46 (0)8 638 93 00 info.produkt@gsk.com
United Kingdom
GlaxoSmithKline UK Ltd Tel: + 44 (0)800 221441 customercontactuk@gsk.com
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema. europa.eu
Prospecto: información para el paciente
Zeffix 5 mg/ml solución oral
lamivudina
Lea todo el prospecto detenidamente antes de empezar a tomar el medicamento.
- Conserve este prospecto ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted personalmente y no debe dárselo a otras personas, aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Zeffix y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Zeffix
3. Cómo tomar Zeffix
4. Posibles efectos adversos
5. Conservación de Zeffix
6. Contenido del envase e información adicional
1. Qué es Zeffix y para qué se utiliza
El principio activo de Zeffix es lamivudina.
Zeffix se utiliza para tratar la infección de larga duración (crónica) por hepatitis B en adultos.
Zeffix es un medicamento antiviral que inhibe el virus de la hepatitis B y pertenece a un grupo de medicamentos denominados inhibidores de la transcriptasa inversa análogos de nucleósidos (INTIs).
El virus de la hepatitis B infecta al hígado, causa una infección de larga duración (crónica) y puede ocasionar un daño hepático. Zeffix puede ser utilizado en pacientes cuyo hígado está dañado pero todavía funciona con normalidad (enfermedad hepática compensada) y en combinación con otros medicamentos en pacientes cuyo hígado está dañado y no funciona de forma normal (enfermedad
hepática descompensada).
El tratamiento con Zeffix puede reducir la cantidad de virus de hepatitis B en su organismo. Esto daría lugar a una reducción del daño hepático y a una mejoría en el funcionamiento de su hígado. No todo el mundo responde al tratamiento con Zeffix de la misma manera. Su médico controlará la efectividad del tratamiento con análisis de sangre periódicos.
2. Qué necesita saber antes de empezar a tomar Zeffix No tome Zeffix:
• si es alérgico a lamivudina o a cualquiera de los demás componentes de este medicamento
(incluidos en la sección 6).
^ Consulte con su médico si cree que esto le afecta.
Advertencias y precauciones
Algunas personas que toman Zeffix u otros medicamentos similares tienen mayor riesgo de sufrir efectos adversos graves. Usted necesita saber que hay un mayor riesgo:
• si alguna vez ha tenido otros tipos de enfermedad hepática, como la hepatitis C
• si tiene un sobrepeso importante (especialmente si es mujer)
^ Consulte a su médico si padece alguna de estas circunstancias. Puede necesitar pruebas adicionales, como análisis de sangre, mientras toma este medicamento. Para más información sobre los riesgos ver Sección 4.
No deje de tomar Zeffix sin el consejo de su médico, ya que existe un riesgo de que su hepatitis empeore. Cuando deje de tomar Zeffix su médico lo controlará durante al menos cuatro meses para comprobar si existe algún problema. Esto implicará tomar muestras de sangre para comprobar si hay elevaciones en los niveles de las enzimas hepáticas que puedan indicar lesión hepática. Ver sección 3 para más información sobre cómo tomar Zeffix.
Esté atento a los síntomas importantes
Algunas personas que toman medicamentos para la infección por hepatitis B desarrollan otros trastornos, que pueden ser graves. Usted necesita conocer a qué signos y síntomas importantes debe prestar atención mientras está tomando Zeffix.
^ Lea la información sobre “Otros posibles efectos adversos de la terapia para la hepatitis B” en la Sección 4 de este prospecto.
Proteja a otras personas
La hepatitis B se transmite por mantener contacto sexual con alguien que padezca la enfermedad o por transferencia de sangre infectada (por ejemplo, por compartir agujas). Zeffix no evita el riesgo de contagio de la infección por hepatitis B a los demás. Para prevenir que otras personas se infecten por hepatitis B:
• Utilice preservativo cuando mantenga sexo oral o con penetración.
• Evite el riesgo de transferencias de sangre — por ejemplo, no comparta agujas.
Toma de Zeffix con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento, incluyendo aquellos con plantas medicinales o medicamentos sin receta.
Recuerde decirle a su médico o farmacéutico si empieza a tomar algún otro medicamento mientras está tomando Zeffix.
Estos medicamentos no deben tomarse con Zeffix
• otros medicamentos que contengan lamivudina, usado para tratar la infección por el VIH (a veces también llamado virus del SIDA)
• emtricitabina, usado para tratar la infección por el VIH o por hepatitis B
• cladribina, usado para el tratamiento de la leucemia de células pilosas
+ Informe a su médico si está siendo tratado con alguno de estos medicamentos.
Embarazo
Si está embarazada, cree que podría estar embarazada o tiene intención de quedarse embarazada: ■♦Hable con su médico sobre los riesgos y beneficios de tomar Zeffix durante el embarazo.
No interrumpa el tratamiento con Zeffix sin el consejo de su médico.
Lactancia
Zeffix puede pasar a la leche materna. Si está en periodo de lactancia, o pensando en dar el pecho:
♦ Hable con su médico antes de tomar Zeffix.
Conducción y uso de máquinas
Zeffix puede hacer que se sienta cansado, lo que podría afectar a su capacidad para conducir o utilizar máquinas.
+ No conduzca o utilice máquinas a no ser que esté seguro de que esto no le afecta.
Zeffix contiene azúcar y conservantes.
Si usted es diabético, tenga en cuenta que cada dosis de Zeffix (100 mg - 20 ml) contiene 4 g de azúcar.
Zeffix contiene sacarosa. Si su médico le ha indicado que padece una intolerancia a ciertos azúcares, consulte con él antes de tomar Zeffix. La sacarosa puede ser perjudicial para sus dientes.
Zeffix también contiene conservantes (parahidroxibenzoatos) que pueden causar reacciones alérgicas (posiblemente retardadas).
3. Cómo tomar Zeffix
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. Consulte a su médico o farmacéutico si tiene dudas.
Mantenga un contacto regular con su médico
Zeffix ayuda a controlar su infección por hepatitis B. Necesita continuar tomándolo cada día para controlar la infección y evitar que su infección empeore.
^ Mantenga el contacto con su médico y no deje de tomar Zeffix sin el consejo de su médico. Cuánto tomar
La dosis habitual de Zeffix es de 20 ml (100 mg de lamivudina) una vez al día.
Su médico puede recetarle una dosis más baja si tiene problemas de riñón.
^ Hable con su médico si se encuentra en esta situación.
Si ya está tomando otro medicamento que contenga lamivudina para el tratamiento de la infección por el VIH, su médico le continuará tratando con la dosis más alta (normalmente 150 mg dos veces al día), porque la dosis de lamivudina de Zeffix (100 mg) no es suficiente para tratar la infección por el VIH. Si está planeando cambiar su tratamiento para el VIH, discuta este cambio con su médico antes.
Zeffix puede tomarse con o sin alimentos.
Vea el diagrama e instrucciones para medir y tomar una dosis de medicamento tras la sección 6 de este prospecto.
Si toma más Zeffix del que debe
Es improbable que la ingestión accidental de una cantidad excesiva de Zeffix, pueda causar problemas serios. Si accidentalmente toma demasiado, comuníquelo a su médico o farmacéutico, o acuda al servicio de urgencias del hospital más próximo para que le aconsejen.
Si olvidó tomar Zeffix
Si olvida tomar una dosis, tómela tan pronto como se acuerde. Luego continúe tomándolo como antes. No tome una dosis doble para compensar las dosis olvidadas.
No interrumpa el tratamiento con Zeffix
No deje de tomar Zeffix sin consultar a su médico. Hay riesgo de que su hepatitis empeore (ver sección 2). Cuando deje de tomar Zeffix su médico le monitorizará durante al menos cuatro meses para comprobar que no hay ningún problema. Esto significa que le realizarán análisis de sangre para comprobar si hay aumento en los niveles de enzimas del hígado, lo que puede indicar daño hepático.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Además de los efectos adversos listados a continuación para Zeffix, se pueden desarrollar otras condiciones durante el tratamiento de combinación para la hepatitis B.
+ Es importante leer la información bajo “Otros posibles efectos de la terapia de combinación para la hepatitis B”.
Los efectos adversos recogidos más comúnmente en los ensayos clínicos con Zeffix fueron cansancio, infecciones del tracto respiratorio, molestias en la garganta, cefalea, molestias y dolor de estómago, náuseas, vómitos y diarrea, aumento en las enzimas hepáticas y en las enzimas producidas en los músculos (ver abajo).
Reacción alérgica
Es rara (puede afectar hasta 1 de cada 1.000 personas). Los signos incluyen:
* hinchazón de los párpados, cara o labios
* dificultad para tragar o respirar.
^ Contacte con un médico inmediatamente si tiene estos síntomas. Deje de tomar Zeffix.
Efectos adversos que se cree están causados por Zeffix:
Un efecto adverso muy frecuente (puede afectar a más de 1 de cada 10 pacientes) es
• un aumento en los niveles de algunas enzimas producidas por el hígado (transaminasas), que puede ser un signo de inflamación o daño en el hígado.
Un efecto adverso frecuente (puede afectar hasta 1 de cada 10 pacientes) es:
• calambres y dolores musculares
• erupción cutánea o “habones” en cualquier parte del cuerpo.
Un efecto adverso frecuente que puede aparecer en los análisis de sangre es:
• Un aumento en el nivel de una enzima producida en los músculos (creatininfosfoquinasa) que puede ser un síntoma de que su hígado está dañado.
Otros efectos adversos
Se han notificado otros efectos adversos en un número muy pequeño de personas, pero su frecuencia exacta es desconocida:
• rotura muscular.
• empeoramiento de la enfermedad hepática si el virus hepatitis B se hace resistente a Zeffix o se interrumpe el tratamiento. Esto puede ser mortal en algunos pacientes.
• acidosis láctica (ver la siguiente sección “Otros posibles efectos adversos del tratamiento para la hepatitis B”)
Un efecto adverso que puede aparecer en sus análisis de sangre es:
• una reducción en el número de células implicadas en la coagulación sanguínea (trombocitopenia).
Si tiene efectos adversos
^ Hable con su médico o farmacéutico. Esto incluye cualquiera de los posibles efectos adversos no incluidos en este prospecto.
Otros posibles efectos adversos del tratamiento para la hepatitis B
Zeffix y los medicamentos relacionados (INTIs) pueden causar otras condiciones que se desarrollan durante el tratamiento para la hepatitis B.
Acidosis láctica es un efecto adverso raro pero grave
Algunas personas que toman Zeffix u otros medicamentos similares (INTIs) desarrollan un trastorno denominado acidosis láctica, junto con un aumento del tamaño del hígado.
La acidosis láctica se debe a un aumento de ácido láctico en el organismo. Es raro, y si aparece, normalmente se desarrolla después de unos pocos meses de tratamiento. Puede suponer un riesgo para la vida, al causar fallos en órganos internos.
Es más probable que la acidosis láctica se desarrolle en personas que tienen alguna afección hepática o en personas obesas (sobrepeso importante), especialmente mujeres.
Los signos de la acidosis láctica incluyen:
• respiración dificultosa, rápida y profunda
• somnolencia
• entumecimiento o debilidad de las extremidades
• malestar (náuseas), vómitos
• dolor de estómago.
Durante su tratamiento, su médico controlará cualquier signo que indique que puede estar desarrollando acidosis láctica. Si tiene cualquiera de los síntomas mencionados anteriormente o le preocupa algún otro síntoma:
^ Acuda a su médico tan pronto como le sea posible.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V*. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Zeffix
Mantener este medicamento fuera de la vista y del alcance de los niños.
No tome este medicamento después de la fecha de caducidad que aparece en el embalaje y frasco.
No conservar a temperatura superior a 25°C.
Desechar transcurrido un mes desde que se abrió por primera vez.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Zeffix
El principio activo es lamivudina. Cada ml de solución oral contiene 5 mg de lamivudina.
Los demás componentes son: sacarosa, parahidroxibenzoato de metilo (E218), parahidroxibenzoato de propilo (E216), ácido cítrico, propilenglicol, citrato de sodio, saborizante artificial de fresa, saborizante artificial de plátano, agua purificada.
Aspecto del producto y contenido del envase
Zeffix solución oral se presenta en embalajes que contienen un frasco de polietileno blanco provisto de cierre resistente a los niños. La solución es límpida, incolora o de color amarillo pálido con sabor a fresa/plátano. El frasco contiene 240 ml de solución de lamivudina (5 mg/ml). El envase incluye un aplicador oral graduado en ml y un adaptador del aplicador que debe encajarse en el frasco antes de uso.
Responsable de la fabricación
Aspen Bad Oldesloe GmbH Industriestrasse 32-36 23843 Bad Oldesloe Alemania
Titular de la Autorización de Comercialización
Glaxo Group Ltd
980 Great West Road
Brentford
Middlesex
TW8 9GS
Reino Unido o
Glaxo Operations UK Limited (operando como Glaxo Wellcome Operations) Harmire Road Barnard Castle County Durham DL12 8DT Reino Unido
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
GlaxoSmithKline Pharmaceuticals s.a./n.v. Tél/Tel: + 32 (0)10 85 52 00
Etarapna
raaKcoCMHTKaaHH EOOfl Tea.: + 359 2 953 10 34
Ceská republika
GlaxoSmithKline s.r.o.
Tel: + 420 222 001 111 cz.info@gsk.com
Danmark
GlaxoSmithKline Pharma A/S Tlf: + 45 36 35 91 00 dk-info@gsk.com
Deutschland
GlaxoSmithKline GmbH & Co. KG Tel.: + 49 (0)89 36044 8701 produkt.info@gsk.com
Luxembourg/Luxemburg
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Belgique/Belgien
Tél/Tel: + 32 (0) 10 85 52 00
Magyarország
GlaxoSmithKline Kft.
Tel.: + 36 1 225 5300
Malta
GlaxoSmithKline (Malta) Limited Tel: + 356 21 238131
Nederland
GlaxoSmithKline BV Tel: + 31 (0)30 6938100 nlinfo@gsk.com
Norge
GlaxoSmithKline AS Tlf: + 47 22 70 20 00 firmapost@gsk.no
|
Eesti GlaxoSmithKline Eesti OÜ Tel: + 372 6676 900 estonia@gsk.com |
Osterreich GlaxoSmithKline Pharma GmbH Tel: + 43 (0)1 97075 0 at.info@gsk.com |
|
EkkúSa GlaxoSmithKline A.E.B.E. T@: + 30 210 68 82 100 |
Polska GSK Services Sp.zo.o. Tel.: + 48 (0)22 576 9000 |
|
España GlaxoSmithKline, S.A. Tel: + 34 902 202 700 es-ci@gsk.com |
Portugal Glaxo Wellcome Farmacéutica, Lda. Tel: + 351 21 412 95 00 FI.PT@gsk.com |
|
France Laboratoire GlaxoSmithKline Tél.: + 33 (0)1 39 17 84 44 diam@gsk.com |
Romania GlaxoSmithKline (GSK) S.R.L. Tel: + 4021 3028 208 |
|
Hrvatska GlaxoSmithKline d.o.o. Tel: + 385 1 6051 999 |
Slovenija GlaxoSmithKline d.o.o. Tel: + 386 (0)1 280 25 00 medical.x.si@gsk.com |
|
Ireland GlaxoSmithKline (Ireland) Limited Tel: + 353 (0)1 4955000 |
Slovenská republika GlaxoSmithKline Slovakia s. r. o. Tel: + 421 (0)2 48 26 11 11 recepcia.sk@gsk.com |
|
Ísland Vistor hf. Sími: + 354 535 7000 |
Suomi/Finland GlaxoSmithKline Oy Puh/Tel: + 358 (0)10 30 30 30 finland.tuoteinfo@gsk.com |
|
Italia GlaxoSmithKline S.p.A. Tel: + 39 (0)45 9218 111 |
Sverige GlaxoSmithKline AB Tel: + 46 (0)8 638 93 00 info.produkt@gsk.com |
|
Kúrcpog GlaxoSmithKline (Cyprus) Ltd T@: + 357 22 39 70 00 gskcyprus@gsk.com Latvija GlaxoSmithKline Latvia SIA Tel: + 371 67312687 lv-epasts@gsk.com Lietuva GlaxoSmithKline Lietuva UAB Tel: + 370 5 264 90 00 info.lt@gsk.com |
United Kingdom GlaxoSmithKline UK Ltd Tel: + 44 (0)800 221441 customercontactuk@gsk.com |
|
Fecha de la última revisión de este prospecto: |
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema. europa.eu
Cómo medir la dosis y tomar el medicamento
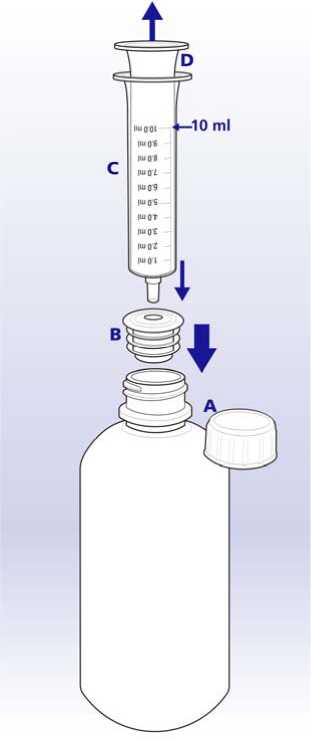
Para medir su dosis con exactitud, emplee el aplicador oral que se incluye en el envase (ver también
Sección 3).
Cuando está llena, el aplicador contiene 10 ml de solución.
1. Quite el tapón a prueba de niños (A) al frasco. Consérvelo en un lugar seguro.
2. Sujete el frasco. Introduzca el adaptador de plástico (B) en el cuello de la botella, tanto como pueda.
3. Inserte el aplicador (C) firmemente en el adaptador.
4. Coloque el frasco boca abajo.
5. Tire del émbolo del aplicador (D) hasta que se haya retirado la primera porción de su dosis completa.
6. Ponga el frasco en posición correcta. Retire el aplicador del adaptador.
7. Ponga el aplicador en su boca, colocando la punta del aplicador contra el interior de la mejilla. Apriete lentamente el émbolo, dejando tiempo para tragar. No apriete muy fuerte, y vierta el líquido hacia la parte de atrás de la garganta o de lo contrario puede que se atragante.
8. Repita los pasos 3 a 7 de la misma forma hasta que se haya tomado la dosis completa. Por ejemplo, si su dosis es de 20 ml, necesita tomar 2 aplicadores completos de medicamento.
9. Retire el aplicador del frasco y lávelo bien en agua limpia. Dejar secar completamente antes de utilizar de nuevo. Deje el adaptador en el frasco.
10. Cierre firmemente el frasco con el tapón.
ANEXO IV
CONCLUSIONES CIENTÍFICAS Y MOTIVOS PARA LA MODIFICACIÓN DE LAS CONDICIONES DE LAS AUTORIZACIONES DE COMERCIALIZACIÓN
Conclusiones científicas
Teniendo en cuenta lo dispuesto en el Informe de Evaluación del Comité para la Evaluación de Riesgos en Farmacovigilancia (PRAC) sobre los informes periódicos de seguridad (IPSs) para lamivudina (hepatitis B crónica) las conclusiones científicas del Comité de Medicamentos de Uso Humano (CHMP) son las siguientes:
La información de producto de ZEFFIX se debe modificar para reflejar mejor las guías actuales de resistencia a lamivudina. Por esta razón, el PRAC recomendó cambios en la sección 4.2 de la ficha técnica para el cambio o adición de un agente alternativo sin resistencia cruzada a lamivudina en base a las guías terapéuticas. Se consideró necesario realizar cambios consecuentes en las secciones 4.4 y 5.1 de la ficha técnica para apoyar esta información. No se introdujeron cambios en el prospecto en relación con la resistencia a lamivudina.
El titular también ha aprovechado la oportunidad para actualizar la información de producto de acuerdo con la última versión de las plantillas QRD (v10).
Por tanto, en base a los datos de los informes periódicos de seguridad presentados (IPSs), el PRAC consideró que los cambios en la información del producto de los productos que contenían lamivudina estaban justificados.
El CHMP está de acuerdo con las conclusiones científicas del PRAC.
Motivos para la modificación de las condiciones de la(s) Autorización(es) de Comercialización
De acuerdo con las conclusiones científicas para lamivudina (hepatitis B crónica), el CHMP considera que el balance beneficio-riesgo del medicamento o medicamentos que contiene(n) dicho(s) principio(s) activo(s) no se modifica sujeto a los cambios propuestos en la información del producto.
El CHMP recomienda que se modifiquen las condiciones de la(s) Autorización(es) de Comercialización.
60
En pacientes con hepatitis B crónica (HBC) HBeAg positivos, sin cirrosis, debe administrarse el tratamiento durante al menos 6-12 meses después de que se confirme la seroconversión de HBeAg (pérdida de HBeAg y ADN del VHB con detección de HBeAc) con el fin de reducir el riesgo de recaída virológica, o continuar hasta que tenga lugar la seroconversión de HBsAg o
En pacientes con hepatitis B crónica (HBC) HBeAg positivos, sin cirrosis, debe administrarse el tratamiento durante al menos 6-12 meses después de que se confirme la seroconversión de