Xyrem 500 Mg/Ml Solucion Oral
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Xyrem 500 mg/ml solución oral.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada ml de solución contiene 500 mg de oxibato de sodio.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución oral.
La solución oral es clara a ligeramente opalescente.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento de la narcolepsia con cataplejía en pacientes adultos.
4.2 Posología y forma de administración
El tratamiento debe iniciarse y mantenerse bajo la dirección de un médico con experiencia en el tratamiento de trastornos del sueño.
Posología
La dosis inicial recomendada es de 4,5 g/día de oxibato de sodio repartida en dos dosis iguales de 2,25 g/dosis. La dosis debe ajustarse de acuerdo a la eficacia y tolerabilidad obtenida con el fármaco (ver sección 4.4) hasta un máximo de 9 g/día repartida en dos dosis iguales de 4,5 g/dosis aumentando o disminuyendo la dosis en 1,5 g/día (por ejemplo 0,75 g/dosis). Se recomienda un mínimo de una a dos semanas entre incrementos de dosis. No debe excederse la dosis de 9 g/día debido a la posible aparición de síntomas graves a dosis de 18 g/día o superiores (ver sección 4.4).
No se deben administrar inicialmente dosis individuales de 4,5 g a menos que el paciente se haya ajustado previamente a este nivel.
Cuando oxibato de sodio se administra de forma concomitante con valproato (ver sección 4.5), se recomienda disminuir la dosis de oxibato de sodio un 20%. La dosis de inicio recomendada de oxibato de sodio en este caso es de 3,6 g por noche, administrada por vía oral y dividida a su vez en dos dosis iguales de aproximadamente 1,8 g. Si la administración concomitante está justificada, se debe vigilar la respuesta y tolerabilidad del paciente y se debe adaptar la dosis de acuerdo a ello (ver sección 4.4).
Interrupción del tratamiento con Xyrem
Los efectos tras la interrupción del tratamiento con oxibato de sodio no han sido específicamente evaluados en los ensayos clínicos controlados realizados con el producto (ver sección 4.4).
Si el paciente interrumpe la toma del medicamento durante más de 14 días, el ajuste deberá comenzarse desde la dosis más baja.
Poblaciones especiales Pacientes de edad avanzada
En pacientes de edad avanzada es preciso vigilar estrechamente posibles alteraciones en las funciones motora y/o cognitiva que pudieran aparecer al recibir oxibato de sodio (ver sección 4.4).
Insuficiencia hepática
La dosis inicial debe reducirse a la mitad en todos los pacientes que presentan insuficiencia hepática, monitorizando estrechamente la respuesta a incrementos de dosis (ver sección 4.4).
Insuficiencia renal
En todos los pacientes que presentan insuficiencia renal se deben tomar medidas dietéticas para reducir la ingesta de sodio (ver sección 4.4).
Población pediátrica
No se ha establecido la seguridad y eficacia de oxibato de sodio en niños de 0 a 18 años. No hay datos disponibles.
Forma de administración
Xyrem debe tomarse por vía oral antes de acostarse y de nuevo entre 2,5 y 4 horas después. Se recomienda que ambas dosis de Xyrem se preparen al mismo tiempo antes de acostarse.
Xyrem viene provisto de una jeringa graduada y dos vasos dosificadores de 90 ml con tapón de seguridad resistente a niños. Cada dosis medida de Xyrem debe dispensarse en el vaso dosificador y diluirse en 60 ml de agua antes de la toma.
Dado que los alimentos reducen significativamente la biodisponibilidad del oxibato de sodio, los pacientes deben tratar de comer varias horas (2-3) antes de acostarse, momento en el que deben tomar la primera dosis de Xyrem. Los pacientes deben tratar de mantener siempre constante el momento de administrar las dosis en relación con las comidas. Las dosis se deben tomar dentro de las 24 horas posteriores a su preparación, o sino desecharlas.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
En pacientes con depresión grave.
En pacientes que presentan déficit de succinato-semialdehído-deshidrogenasa.
En pacientes que están siendo tratados con fármacos opioides o barbitúricos.
4.4 Advertencias y precauciones especiales de empleo
Xyrem tiene potencial para inducir depresión respiratoria.
Depresión respiratoria y del sistema nervioso central (SNC)
Además, el oxibato de sodio tiene potencial para inducir depresión respiratoria. La apnea y la depresión respiratoria se han observado en pacientes sanos en ayunas tras una dosis única de 4,5 g (dos veces la dosis inicial recomendada). Se deben valorar en los pacientes posibles signos de depresión respiratoria o del SNC. Deben tomarse precauciones especiales en pacientes con trastornos respiratorios subyacentes. Debido al mayor riesdo de apnea del sueño, los pacientes con un IMC >
40 kg/m2, deben ser vigilados cuando tomen oxibato de sodio.
Aproximadamente el 80 % de los pacientes a los que se administró oxibato de sodio durante los ensayos clínicos recibieron tratamiento concomitante con estimulantes del SNC. No se sabe si hubo un efecto de estos fármacos sobre la respiración nocturna. Antes de incrementar la dosis de oxibato de sodio (ver sección 4.2), los especialistas deben tener en cuenta que más del 50 % de los pacientes que presentan narcolepsia presentan episodios de apnea del sueño.
• Benzodiazepinas
Se deberá evitar el uso concomitante de benzodiazepinas y oxibato de sodio, dada la posibilidad de incremento del riesgo de depresión respiratoria.
• Alcohol y otros depresores del SNC
El uso combinado de alcohol o cualquier medicamento depresor del SNC con oxibato de sodio puede potenciar los efectos depresores del SNC de oxibato de sodio, así como aumentar el riesgo de depresión respiratoria. Por lo tanto, los pacientes deberán ser advertidos de los efectos producidos por el uso combinado de alcohol con oxibato de sodio.
• Inhibidores de la gamma-hidroxibutirato (GHB) deshidrogenasa
Debido a que se han observado interacciones farmacocinéticas y farmacodinámicas cuando se han administrado de forma conjunta oxibato de sodio y valproato, se requiere precaución en pacientes tratados de forma concomitante con valproato y otros inhibidores de la GHB deshidrogenasa (ver sección 4.5). Si la administración concomitante está justificada, se debe considerar un ajuste de dosis (ver sección 4.2). Además, se debe vigilar cuidadosamente la respuesta y tolerabilidad del paciente y se debe adaptar la dosis de acuerdo a ello.
• Topiramato
Tras la administración conjunta de oxibato de sodio y topiramato se notificaron observaciones clínicas de coma y aumento de la concentración plasmática de GHB. Por tanto, se debe advertir a los pacientes acerca de no utilizar de forma conjunta topiramato y oxibato de sodio (sección 4.5).
Potencial de abuso y dependencia
El oxibato de sodio, sal sódica de GHB, es un principio activo depresor del SNC con un conocido potencial de abuso. Antes del inicio del tratamiento, el especialista deberá analizar en la historia clínica del paciente los posibles antecedentes o la predisposición al consumo de drogas para realizar un estrecho seguimiento de los casos positivos. Debe vigilarse rutinariamente a los pacientes y, en caso de sospecha de abuso, el tratamiento con oxibato sódico debe interrumpirse. Se han notificado casos de dependencia tras un uso ilícito de GHB con una frecuencia de dosis repetida (de 18 a 250 g/día) superior al rango de dosis terapéutico. Aunque no existe una evidencia clara de aparición de dependencia en pacientes que reciben oxibato de sodio a dosis terapéuticas no se puede excluir esta posibilidad.
Pacientes con porfiria
El oxibato de sodio se considera inseguro en pacientes que presentan porfiria, ya que ha demostrado actividad porfirogénica en animales o modelos in vitro.
Efectos neuropsiquiátricos
Los pacientes pueden percibir cierto grado de confusión mientras permanecen en tratamiento con oxibato de sodio. Si esto ocurre, deberán ser observados, y se considerará una intervención individual apropiada. Otros efectos neuropsiquiátricos incluyen ansiedad, psicosis, paranoia, alucinaciones y agitación. La aparición de trastornos de pensamiento y/o anomalías en la conducta en pacientes tratados con oxibato de sodio requieren una observación inmediata y minuciosa.
La aparición de depresión en pacientes tratados con oxibato de sodio requiere una cuidadosa e inmediata observación. Los pacientes con historial previo de enfermedad depresiva y/o intento de suicidio deberán ser supervisados vigilando especialmente la aparición de estos síntomas mientras reciben oxibato de sodio. El uso de Xyrem está contraindicado en casos de depresión grave (sección 4.3).
Si un paciente experimenta incontinencia urinaria o fecal durante la terapia con oxibato de sodio, se deberán realizar las pruebas pertinentes para excluir etiologías subyacentes.
Se ha notificado sonambulismo en pacientes tratados con oxibato de sodio en los ensayos clínicos. No está claro si algunos de ellos o todos estos episodios corresponden a verdadero sonambulismo (parasomnia ocurrida durante el sueño no-REM) o a cualquier otro trastorno clínico específico. El riesgo de herida o autolesión deberá tenerse en cuenta en cualquier paciente con sonambulismo, por lo tanto, los episodios de sonambulismo deberán ser analizados adecuadamente, tomando las medidas que correspondan.
Ingesta de sodio
Los pacientes en tratamiento con oxibato de sodio recibirán una ingesta diaria adicional de sodio de entre 0,82 g (para una dosis de Xyrem de 4,5 g/día) a 1,6 g (dosis de Xyrem de 9 g/día). Se deberán considerar modificaciones dietéticas para reducir la ingesta de sodio en pacientes que presentan insuficiencia cardíaca, hipertensión arterial o función renal comprometida (ver sección 4.2).
Pacientes de edad avanzada
Existe una experiencia muy limitada con oxibato de sodio en pacientes de edad avanzada. Por lo tanto, en pacientes de edad avanzada que estén recibiendo oxibato de sodio, deberá vigilarse estrechamente la aparición de trastornos de la función motora y/o cognitiva.
Pacientes epilépticos
Se han observado crisis epilépticas en pacientes tratados con oxibato de sodio. No se ha establecido la seguridad y la eficacia de oxibato de sodio en pacientes con epilepsia, por lo que no se recomienda su uso.
Efecto rebote y síndrome de retirada
Los efectos tras la interrupción del tratamiento con oxibato de sodio no han sido específicamente valorados en los ensayos clínicos controlados. En algunos pacientes, la cataplejía puede reaparecer con una frecuencia superior al abandonar la terapia con oxibato de sodio, sin embargo esto puede deberse a la propia historia natural de la enfermedad. Aunque la experiencia en ensayos clínicos con oxibato de sodio a dosis terapéuticas en pacientes que presentan narcolepsia/cataplejía no muestra una evidencia clara de síndrome de abstinencia, en raras ocasiones se observaron efectos como insomnio, dolor de cabeza, ansiedad, vértigo, trastornos del sueño, somnolencia, alucinación y trastornos psicóticos, tras la interrupción del GHB.
4.5 Interacción con otros medicamentos y otras formas de interacción
El uso combinado de alcohol con oxibato de sodio puede potenciar los efectos depresores del oxibato de sodio sobre el sistema nervioso central. Los pacientes deberán ser advertidos en contra del uso de cualquier bebida alcohólica con oxibato de sodio.
El oxibato de sodio no debe usarse en combinación con hipnóticos sedantes u otros depresores del SNC.
Hipnóticos sedantes
Los estudios de interacción farmacológica, en adultos sanos con oxibato sódico (una dosis única de 2,25 g) y lorazepam (una dosis única de 2 mg) y tartrato de zolpidem (una dosis única de 5 mg) no demostraron interacción farmacocinética. Se observó un aumento de la somnolencia después de la administración concomitante de oxibato de sodio (2,25 g) y lorazepam (2 mg). No se ha evaluado la interacción farmacodinámica con zolpidem. Cuando se combinan dosis más altas de hasta 9 g/día de oxibato de sodio con dosis más altas de hipnóticos (dentro de la dosis recomendada) no se pueden excluir interacciones farmacodinámicas asociadas con síntomas de depresión del SNC y/o depresión del sistema respiratorio (ver sección 4.3).
Tramadol
Un estudio de interacción farmacológica, en adultos sanos con oxibato sódico (una dosis única de 2,25 g) y tramadol (una dosis única de 100 mg), no demostró interacción farmacocinética /farmacodinámica. Cuando se combinan dosis más altas de hasta 9 g/día de oxibato de sodio con dosis más altas de opioides (dentro de la dosis recomendada) no se pueden excluir interacciones farmacodinámicas asociadas con síntomas de depresión del SNC y/o depresión del sistema respiratorio (ver sección 4.3).
Antidepresivos
Los estudios de interacción farmacológica en adultos sanos no demostraron interacciones farmacocinéticas entre el oxibato de sodio (una dosis única de 2,25 g) y el antidepresivo clorhidrato de protriptilina (una dosis única de 10 mg) y duloxetina (60 mg en el estado de equilibrio). No se observó efecto adicional sobre la somnolencia cuando se compara con dosis únicas de oxibato de sodio solo (2,25 g) y oxibato de sodio (2,25 g) en combinación con duloxetina (60 mg en el estado de equilibrio). Se han usado antidepresivos en el tratamiento de la cataplejía. No se puede excluir un posible efecto aditivo entre los antidepresivos y el oxibato de sodio. La frecuencia de las reacciones adversas se ha incrementado cuando se administra oxibato de sodio conjuntamente con antidepresivos tricíclicos.
Modafinilo
Un estudio de interacción farmacológica en adultos sanos no demostró interacción farmacocinética entre el oxibato sódico (una única dosis de 4,5 g) y modafinilo (una dosis única de 200 mg). El oxibato de sodio se ha administrado de manera asociada con medicamentos estimulantes del SNC en aproximadamente el 80 % de pacientes en los estudios clínicos en narcolepsia. Se desconoce si esto afecta la respiración durante la noche.
Omeprazol
La administración combinada con omeprazol no tiene un efecto clínicamente significativo sobre la farmacocinética de oxibato de sodio. La dosis de oxibato de sodio, por lo tanto, no requiere ajuste cuando se administra de manera concomitante con inhibidores de la bomba de protones.
Ibuprofeno
Estudios de interacción farmacológica en adultos sanos no demostraron interacciones farmacocinéticas entre el oxibato de sodio y el ibuprofeno.
Diclofenaco
Estudios de interacción farmacológica en adultos sanos no demostraron interacciones farmacocinéticas entre el oxibato de sodio y el diclofenaco. La administración conjunta de oxibato de sodio y diclofenaco en voluntarios sanos redujo el déficit de atención provocado por la administración de Xyrem únicamente, tal como se midió mediante tests psicométricos.
Inhibidores de la GHB deshidrogenasa
Dado que el oxibato de sodio es metabolizado por la GHB deshidrogenasa, hay un riesgo potencial de interacción con medicamentos que estimulan o inhiben esta enzima (por ejemplo valproato, fenitoina o etosuximida) (ver sección 4.4).
La administración conjunta de oxibato de sodio (6 g al día) y valproato (1250 mg al día) produjo un aumento en la exposición a oxibato de sodio de aproximadamente un 25% sin cambio significativo en la Cmax. No se observó efecto en la farmacocinética de valproato. Los efectos farmacodinámicos resultantes, incluyendo el aumento de los trastornos de la función cognitiva y de la somnolencia fueron mayores con la administración conjunta que los observados con cualquiera de los medicamentos por separado. Si la administración concomitante está justificada, se debe vigilar la respuesta y tolerabilidad del paciente y se deben realizar ajustes de dosis si se requiere (ver sección 4.2).
Topiramato
Cuando se utiliza oxibato de sodio de forma concomitante con topiramato no se pueden excluir posibles interacciones farmacodinámicas y farmacocinéticas, ya que se han notificado observaciones clínicas de coma y aumento de la concentración plasmática de GHB en paciente(s) en tratamiento concomitante con oxibato de sodio y topiramato (sección 4.4).
Estudios in-vitro con agregados de microsomas hepáticos humanos indican que el oxibato de sodio no inhibe de forma significativa las actividades de las isoenzimas hepáticas (ver sección 5.2).
4.6 Fertilidad, embarazo y lactancia
Embarazo
Estudios en animales no han mostrado evidencias de teratogenicidad, sin embargo se ha observado embrioletalidad en estudios en ratas y conejos (ver sección 5.3).
Los datos obtenidos de un número limitado de mujeres embarazadas expuestas durante el primer trimestre indican un posible incremento de abortos espontáneos. Hasta la fecha no hay otros datos epidemiológicos relevantes disponibles. Datos limitados en mujeres embarazadas durante el segundo y tercer trimestre de embarazo no indican toxicidad malformativa ni fetal/neonatal del oxibato de sodio.
No se recomienda el uso de oxibato de sodio durante el embarazo.
Lactancia
No se conoce si el oxibato de sodio y/o sus metabolitos se excretan en la leche materna. No se recomienda la lactancia durante el tratamiento con oxibato de sodio.
Fertilidad
No hay datos clínicos disponibles sobre el efecto del oxibato de sodio sobre la fertilidad. Estudios en ratas macho y hembra a dosis de hasta 1.000 mg/kg/día de GHB no mostraron evidencia de efecto adverso sobre la fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de oxibato de sodio sobre la capacidad para conducir y utilizar máquinas es importante.
Los pacientes no deben emprender actividades que requieran una alerta mental completa o coordinación motora, como utilizar maquinaria o conducir, durante al menos 6 horas después de la toma de oxibato de sodio.
Desde que los pacientes comienzan a recibir oxibato de sodio, hasta que descubren si el medicamento les induce algún otro efecto al día siguiente, deberán mantener un cuidado extremo en la conducción de vehículos, manejo de maquinaria pesada, o al realizar cualquier otra tarea que pudiera resultar peligrosa o requerir una alerta mental completa.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Las reacciones adversas notificadas con más frecuencia son mareo, náuseas y dolor de cabeza, presentes en un 10% a 20% de los pacientes. Las reacciones adversas más graves son intento de suicidio, psicosis, depresión respiratoria y convulsión.
La seguridad y eficacia del oxibato de sodio en el tratamiento de los síntomas de la narcolepsia se establecieron en cuatro ensayos multicéntricos, aleatorizados, doble ciego, controlados con placebo y de grupos paralelos, en pacientes con narcolepsia con cataplejía, excepto para un ensayo en el que no se requirió la cataplejía para participar. Se llevaron a cabo dos estudios fase 3 y uno fase 2, doble ciego, de grupos paralelos, controlados con placebo para evaluar la indicación de oxibato de sodio en
fíbromialgia. Además, se llevaron a cabo estudios aleatorizados, doble ciego, controlados con placebo de interacción cruzada fármaco-fármaco con ibuprofeno, diclofenaco y valproato en sujetos sanos, que se resumen en la sección 4.5.
Además de las reacciones adversas notificadas durante los estudios clínicos, se han notificado reacciones adversas en la experiencia post-comercialización. No siempre es posible estimar de forma fiable la frecuencia de su incidencia en la población a tratar.
Tabla de reacciones adversas
Las reacciones adversas se incluyen de acuerdo a la Clasificación de órganos del sistema MedDRA.
Frecuencia estimada: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1000 a < 1/100); raras (> 1/10000 a < 1/1000); muy raras (< 1/10000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Infecciones e infestaciones Frecuentes: nasofaringitis, sinusitis
Trastornos del sistema inmunológico Poco frecuentes: hipersensibilidad
Trastornos del metabolismo y de la nutrición Frecuentes: anorexia, disminución del apetito Frecuencia no conocida: deshidratación
Trastornos psiquiátricos
Frecuentes: depresión, cataplejía, ansiedad, sueños anormales, estado de confusión, desorientación, pesadillas, sonambulismo, trastornos del sueño, insomnio, insomnio medio o de mantenimiento, nerviosismo
Poco frecuentes: intento de suicidio, psicosis, paranoia, alucinaciones, pensamiento anormal, agitación, insomnio inicial
Frecuencia no conocida: ideación suicida, estado de ánimo eufórico, trastornos alimenticios relacionados con el sueño, ataque de pánico, manía/trastorno bipolar, delirio, bruxismo
Trastornos del sistema nervioso Muy frecuentes: mareo, cefalea
Frecuentes: parálisis del sueño, somnolencia, temblor, trastornos del equilibrio, alteración de atención, hipoestesia, parestesia, sedación, disgeusia
Poco frecuentes: mioclonía, amnesia, síndrome de piernas inquietas Frecuencia no conocida: convulsión, pérdida de conciencia
Trastornos oculares Frecuentes: visión borrosa
Trastornos del oído y del laberinto
Frecuentes: vértigo
Frecuencia no conocidas: tinnitus
Trastornos cardiacos Frecuentes: palpitaciones
Trastornos vasculares Frecuentes: hipertensión
Trastornos respiratorios, torácicos y mediastínicos
Frecuentes: disnea, ronquidos, congestión nasal
Frecuencia no conocida: depresión respiratoria, apnea del sueño
Trastornos gastrointestinales
Muy frecuentes: náuseas (la frecuencia de náuseas es mayor en mujeres que en hombres) Frecuentes: vómitos, diarrea, dolor abdominal superior Poco frecuentes: incontinencia fecal Frecuencia no conocida: boca seca
Trastornos de la piel y del tejido subcutáneo Frecuentes: hiperhidrosis, erupción Frecuencia no conocida: urticaria, angioedema
Trastornos musculoesqueléticos y del tejido conjuntivo Frecuentes: artralgia, espasmos musculares, dolor de espalda
Trastornos renales y urinarios
Frecuentes: enuresis nocturna, incontinencia urinaria Frecuencia no conocida: polaquiuria, urgencia urinaria
Trastornos generales y alteraciones en el lugar de administración Frecuentes: astenia, fatiga, sensación de embriaguez, edema periférico
Exploraciones complementarias
Frecuentes: aumento de la presión sanguínea, pérdida de peso
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos
Frecuentes: caídas
Descripción de reacciones adversas seleccionadas
En algunos pacientes, la cataplejía puede reaparecer con una frecuencia más elevada al abandonar la terapia con oxibato de sodio, sin embargo esto puede deberse a la propia variabilidad normal de la enfermedad. Aunque la experiencia en ensayos clínicos con oxibato de sodio a dosis terapéuticas en pacientes que presentan narcolepsia/cataplejía no muestra una clara evidencia de síndrome de abstinencia, en raras ocasiones se observaron reacciones adversas como insomnio, dolor de cabeza, ansiedad, vértigo, trastornos del sueño, somnolencia, alucinaciones, y trastornos psicóticos, tras la interrupción de GHB.
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación incluido en el Anexo V.
4.9 Sobredosis
La información sobre signos y síntomas asociados a la sobredosis con oxibato de sodio es limitada. La mayor parte de datos derivan del uso ilícito de GHB. El oxibato de sodio es la sal sódica de GHB. Los
efectos adversos asociados al síndrome de abstinencia se han observado con dosis superiores al rango terapéutico.
Síntomas
Los pacientes han mostrado diferentes grados de alteraciones de la conciencia que pueden fluctuar rápidamente entre un estado confusional combativo, agitado con ataxia y coma. Se puede observar emesis (incluso con estado de conciencia alterado), sudoración, cefalea, y habilidades psicomotrices disminuidas. También se ha notificado visión borrosa. A dosis más elevadas se ha observado un incremento en la profundidad del coma. Se han notificado mioclonías y ataques tónico-clónicos. Hay casos donde la frecuencia y profundidad de la respiración se han visto comprometidas y situaciones de depresión respiratoria donde la vida podría verse amenazada, haciendo necesaria la intubación y ventilación artificial. Se ha notificado respiración de Cheyne-Stokes y apnea. La bradicardia e hipotermia pueden acompañarse de inconsciencia, así como de hipotonía muscular, pero con los reflejos tendinosos intactos. La bradicardia responde a la administración de atropina intravenosa.
Tratamiento
Si se sospecha de ingesta asociada de otras sustancias, debe tenerse en cuenta el lavado gástrico. Dado que la emesis puede producirse durante la pérdida de conciencia, se deberá garantizar la posición adecuada (decúbito lateral izquierdo) y la protección de la vía aérea mediante intubación. Aunque el reflejo nauseoso puede no aparecer en pacientes en coma profundo, hasta los pacientes inconscientes pueden oponerse de manera activa a la intubación, por lo que debe considerarse una inducción rápida de la misma (sin administración previa de sedantes).
Tras la administración de flumazenilo no cabe esperar reversión alguna de los efectos depresores centrales del oxibato de sodio. No hay pruebas suficientes para recomendar el empleo de naloxona en el tratamiento de la sobredosis con GHB. No se ha estudiado el empleo de hemodiálisis u otras formas de eliminación extracorpórea del medicamento en sobredosis de oxibato de sodio. Sin embargo, no se pueden recomendar estas medidas debido al rápido metabolismo del oxibato de sodio.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Otros fármacos que actúan sobre el sistema nervioso, código ATC: N07XX04
El oxibato de sodio es un depresor del SNC que reduce la excesiva somnolencia diurna y la cataplejía en pacientes que presentan narcolepsia y modifica la arquitectura del sueño reduciendo el sueño nocturno fragmentado. No se conoce el mecanismo exacto por el cual el oxibato de sodio produce un efecto, sin embargo parece que el oxibato de sodio actúa promoviendo el sueño de onda lenta (delta) y consolidando el sueño nocturno. El oxibato de sodio administrado antes del sueño nocturno, aumenta las fases 3 y 4 del sueño y aumenta la latencia de sueño, mientras reduce la frecuencia de inicio de períodos de sueño REM (SOREMPs). También pueden estar implicados otros mecanismos que permanecen aún sin aclarar.
Más del 80% de los pacientes incluidos en los ensayos clínicos, recibían tratamiento concomitante con fármacos estimulantes.
La efectividad del oxibato de sodio en el tratamiento de los síntomas de la narcolepsia se ha establecido en cuatro ensayos multicéntricos, aleatorizados, a doble ciego de grupos paralelos y controlados con placebo (Ensayos 1, 2, 3 y 4) en pacientes con narcolepsia con cataplejía excepto el Ensayo 2, donde la cataplejía no fue un requisito para incluirse en el ensayo. Se permitió el uso concomitante de estimulantes en todos los ensayos (excepto en la fase de tratamiento activo del Ensayo 2); se retiraron los antidepresivos antes del tratamiento activo en todos los ensayos con la
excepción del Ensayo 2. En cada ensayo, la dosis diaria se dividía en dos dosis iguales. La primera dosis se administraba cada noche antes de acostarse y la segunda dosis entre 2,5 y 4 horas más tarde.
Tabla 1 Resumen de resultados de ensayos clínicos realizados utilizando el oxibato de sodio para el tratamiento de la narcolepsia
|
Ensayo |
Eficacia Primaria |
N |
Eficacia Secundaria |
Duración |
Tratamiento activo y Dosis (g/día) |
|
Ensayo 1 |
EDS (ESS); CGI-c |
246 |
MWT/Arquitectura del sueño/ Cataplejía /Siestas/FOSQ |
8 semanas |
Xyrem 4,5 - 9 |
|
Ensayo 2 |
EDS (MWT) |
231 |
Arquitectura del sueño / ESS/CGI-c/ Siestas |
8 semanas |
Xyrem 6 - 9 Modafinilo200 -600 mg |
|
Ensayo 3 |
Cataplejía |
136 |
EDS (ESS)/CGI-c/ Siestas |
4 semanas |
Xyrem 3 - 9 |
|
Ensayo 4 |
Cataplejía |
55 |
Ninguno |
4 semanas |
Xyrem 3 - 9 |
EDS - Somnolencia diurna excesiva; ESS - Escala de Somnolencia Epworth; MWT - Test de mantenimiento de la vigilia; Siestas - Número de siestas diurnas involuntarias; CGI-c - Impresión Global Clínica de Cambio; FOSQ - Resultados Funcionales del Cuestionario del sueño
El Ensayo 1 incluyó 246 pacientes con narcolepsia e incorporó un periodo de una semana de escalada de dosis. Las medidas primarias de la eficacia fueron cambios en la somnolencia diurna excesiva medida mediante la Escala de Somnolencia Epworth (EES), y el cambio en la severidad general de los síntomas de narcolepsia del paciente evaluados por el investigador usando la medida de la Impresión Global Clínica de Cambio (CGI-c).
Tabla 2 Resumen de la EES en el Ensayo 1
|
Escala de Somnolencia Epworth (ESS; rango 0-24) | ||||
|
Grupo de dosis [g/día (n)] |
Punto de referencia |
Punto final |
Cambio medio desde el punto de referencia |
Cambio desde el punto de referencia comparado con placebo (valor p) |
|
Placebo (60) |
17,3 |
16,7 |
-0,5 |
- |
|
4,5 (68) |
17,5 |
15,7 |
-1,0 |
0,119 |
|
6 (63) |
17,9 |
15,3 |
-2,0 |
0,001 |
|
9 (55) |
17,9 |
13,1 |
-2,0 |
<0,001 |
Tabla 3 Resumen de CGI-c en el Ensayo 1
|
Impresión Clínica Global de cambio (CGI-c) | ||
|
Grupo de dosis [g/día (n)] |
Respondedores1 N (%) |
Cambio desde el punto de referencia comparado con placebo (valor p) |
|
Placebo (60) |
13 (21,7) |
- |
|
4,5 (68) |
32 (47,1) |
0,002 |
|
6 (63) |
30 (47,6) |
<0,001 |
|
9 (55) |
30 (54,4) |
<0,001 |
En el Ensayo 2 se compararon los efectos del oxibato de sodio administrado por vía oral, modafinilo y oxibato de sodio + modafinilo, con placebo en el tratamiento de la somnolencia diurna en la narcolepsia. Durante el periodo doble ciego de 8 semanas, los pacientes tomaron modafinilo a la dosis establecida o el placebo equivalente. La dosis de oxibato de sodio o placebo equivalente fue 6 g/día durante las 4 primeras semanas y se aumentó hasta 9 g/día durante las 4 semanas restantes. La medida primaria de la eficacia fue la somnolencia diurna excesiva medida por respuesta objetiva en MWT.
Tabla 4 Resumen de MWT en el Ensayo 2
|
ENSAYO 2 | ||||
|
Grupo de dosis |
Punto de referencia |
Punto final |
Cambio medio desde el punto de referencia |
Punto final comparado con placebo |
|
Placebo (56) |
9,9 |
6,9 |
-2,7 |
- |
|
Oxibato de sodio (55) |
11,5 |
11,3 |
0,16 |
<0,001 |
|
Modafinilo (63) |
10,5 |
9,8 |
-0,6 |
0,004 |
|
Oxibato de sodio + Modafinilo (57) |
10,4 |
12,7 |
2,3 |
<0,001 |
En el Ensayo 3 se incluyeron 136 pacientes narcolépticos con cataplejía de moderada a severa (media de 21 ataques de cataplejía por semana) en el punto de referencia. La medida primaria de la eficacia en este ensayo fue la frecuencia de los ataques de cataplejía.
Tabla 5 Resumen de Resultados en el Ensayo 3
|
Dosis |
Número de sujetos |
Ataques de Cataplejía | ||
|
Ensayo 3 |
Punto de referencia |
Cambio medio desde el punto de referencia |
Cambio desde el punto de referencia comparado con Placebo (valor p) | |
|
Ataques medios/semana | ||||
|
Placebo |
33 |
20,5 |
-4 |
- |
|
3,0 g/día |
33 |
20,0 |
-7 |
0,5235 |
|
6,0 g/día |
31 |
23,0 |
-10 |
0,0529 |
|
9,0 g/día |
33 |
23,5 |
-16 |
0,0008 |
En el Ensayo 4 se incluyeron 55 pacientes narcolépticos que habían estado tomando oxibato de sodio durante 7 - 44 meses. Los pacientes fueron aleatorizados para continuar el tratamiento con oxibato de sodio a su dosis estable o placebo. El Ensayo 4 se designó específicamente para evaluar la eficacia continuada del oxibato de sodio tras el uso prolongado. La medida primaria de la eficacia en este ensayo fue la frecuencia de los ataques de cataplejía.
Tabla 6 Resumen de Resultados en el Ensayo 4
|
Grupo |
Número de |
Ataques de cataplejía |
|
Tratamiento |
sujetos |
|
Ensayo 4 |
Punto de referencia |
Cambio medio desde el punto de referencia |
Cambio desde el punto de referencia comparado con Placebo (valor p) | ||
|
Ataques medios/2 semanas | |||||
|
Placebo |
29 |
4,0 |
21,0 |
- | |
|
Oxibato de sodio |
26 |
1,9 |
0 |
p<0,001 | |
En el Ensayo 4, la respuesta fue numéricamente similar para los pacientes tratados con dosis de 6 a 9 g/día, pero no se observó ningún efecto en pacientes tratados con dosis inferiores a 6 g/día.
5.2 Propiedades farmacocinéticas
El oxibato de sodio se absorbe rápidamente y casi completamente tras su administración oral; las comidas ricas en grasas retrasan y disminuyen su absorción. Se elimina principalmente en forma de metabolitos con una semivida de 0,5 a 1 hora. La farmacocinética no es lineal con un área bajo la curva frente a tiempo que se incrementa en 3,8 veces cuando se duplica la dosis (de 4,5 g hasta 9 g) La farmacocinética no se altera con dosis repetidas.
Absorción
El oxibato de sodio se absorbe rápidamente tras su administración oral con una biodisponibilidad absoluta aproximada del 88 %. Las concentraciones plasmáticas máximas medias (1er y 2° pico) tras la administración de una dosis diaria de 9 g, repartida en dos dosis equivalentes, administradas cada cuatro horas, fueron 78 y 142 pg ( microgramos)/ml, respectivamente. El tiempo medio para alcanzar la concentración plasmática máxima (Tmax) osciló de 0,5 a 2 horas en ocho estudios farmacocinéticos. Tras la administración oral, los niveles plasmáticos de oxibato de sodio aumentan más con el incremento de la dosis de lo que cabría esperar proporcionalmente. No se han estudiado dosis únicas superiores a 4,5 g. La administración de oxibato de sodio inmediatamente después de una comida rica en grasa tuvo como resultado una absorción retrasada (la Tmax media aumentó de 0,75 h a 2,0 h) y una reducción de la concentración plasmática máxima (Cmax) - un 58% de media - y de la exposición sistémica (AUC) - un 37% de media.
Distribución
El oxibato de sodio es un compuesto hidrófilo con un volumen aparente de distribución promedio de 190-384 ml/kg. Con concentraciones de oxibato de sodio entre 3 y 300 pg (microgramos)/ml, menos del 1% se presenta unido a proteínas plasmáticas.
Biotransformación
Estudios en animales indican que el metabolismo es la vía principal de eliminación del oxibato de sodio, produciendo dióxido de carbono y agua mediante el ciclo del ácido tricarboxílico (Krebs) y secundariamente por p-oxidación. La vía primaria implica una enzima citosólica ligada a NADP+, la GHB deshidrogenasa, que cataliza la conversión de oxibato de sodio a succinato semialdehído, que es biotransformado a ácido succínico por la succinato semialdehído deshidrogenasa. El ácido succínico entra en el ciclo Krebs donde es metabolizado a dióxido de carbono y agua. Una segunda enzima oxidoreductasa mitocondrial, una transhidrogenasa, también cataliza la conversión a semialdehido succínico en presencia de a-cetoglutarato. Una vía alternativa de biotransformación supone la p-oxidación vía 3,4-dihidroxibutirato a Acetil CoA, que también se incorpora al ciclo del ácido cítrico para dar lugar a la formación de dióxido de carbono y agua. No se han identificado metabolitos activos.
Los estudios in vitro con agregados de microsomas hepáticos humanos indican que el oxibato de sodio no inhibe significativamente las actividades de las isoenzimas hepáticas: CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, o CYP3A, hasta concentraciones de 3 mM [378 pg (microgramos)/ml]. Estos niveles son bastante más elevados que los alcanzados con dosis terapéuticas.
Eliminación
El aclaramiento de oxibato de sodio se realiza casi completamente por biotransformación a dióxido de carbono, que es eliminado a través de la respiración. Por regla general, menos del 5% de medicamento aparece inalterado en la orina humana, entre 6 y 8 horas después de recibir la medicación. La excreción fecal no es significativa.
Pacientes de edad avanzada
La farmacocinética del oxibato de sodio en un número limitado de pacientes mayores de 65 años no fue diferente comparada con pacientes más jóvenes de 65 años.
Población pediátrica
No se ha estudiado la farmacocinética del oxibato de sodio en pacientes menores de 18 años. Insuficiencia renal
Dado que el riñón no tiene un papel significativo en la excreción de oxibato de sodio, no se ha realizado ningún estudio farmacocinético en pacientes con alteraciones de la función renal; en principio no es de esperar ningún efecto de la función renal sobre la farmacocinética del oxibato de sodio.
Insuficiencia hepática
El oxibato de sodio sufre un metabolismo presistémico significativo (metabolismo hepático de primer paso). Tras una dosis única oral de 25 mg/kg, los valores de AUC fueron el doble en pacientes cirróticos, con un aclaramiento oral aparente reducido, de 9,1 ml/min/kg en adultos sanos a 4,5 y 4,1 ml/min/kg en pacientes de la Clase A (sin ascitis) y pacientes de la Clase C (con ascitis), respectivamente. La semivida de eliminación fue considerablemente más larga en pacientes de la Clase C y Clase A que en los sujetos control (t1/2 media de 59 y 32 frente 22 minutos). La dosis inicial se debe reducir a la mitad en todos los pacientes con insuficiencia hepática y se deben vigilar estrechamente la respuesta a los incrementos de dosis (ver sección 4.2).
Raza
No se ha evaluado el efecto de la raza sobre el metabolismo del oxibato de sodio.
5.3 Datos preclínicos sobre seguridad
La administración repetida del oxibato de sodio a ratas (90 días y 26 semanas) y perros (52 semanas) no produjo ningún hallazgo significativo en la bioquímica clínica o en la micro- y macro anatomía patológica. Los signos clínicos relacionados con el tratamiento estuvieron principalmente relacionados con sedación, consumo reducido de alimentos y cambios secundarios en el peso corporal, aumento de peso corporal y peso de los órganos. Las exposiciones en ratas y perros al NOEL fueron menores (~50%) que en humanos. El oxibato de sodio no fue mutagénico ni clastogénico en ensayos in vivo e in vitro.
La Gamma Butironolactona (GBL), profármaco del GHB, ensayado a las mismas dosis que las esperadas en humanos (1,21 - 1,64 veces), ha sido clasificado por la NTP como no carcinogénico en ratas y carcinógeno potencial en ratones, debido a un ligero aumento de los feocromocitomas que dificultan la interpretación debido a la elevada mortalidad en los grupos de altas dosis. En un estudio de carcinogenicidad en ratas con oxibato de sodio no se identificaron tumores relacionados con el compuesto.
GHB no tiene efecto sobre el apareamiento, fertilidad general o parámetros del esperma y no produce toxicidad embrio-fetal en ratas expuestas a más de 1000 mg/kg/día de GHB (1,64 veces la exposición humana calculada en animales no gestantes). La mortalidad perinatal se incrementó y el peso medio de la descendencia disminuyó durante el periodo de lactancia en animales Fi a dosis altas. No se puede establecer la asociación entre estos efectos sobre el desarrollo y la toxicidad materna. En conejos se observó una ligera fetotoxicidad.
Estudios de discriminación del fármaco muestran que GHB produce un estímulo discriminativo único que en muchos aspectos es similar al del alcohol, morfina y determinados medicamentos GABA-miméticos. Estudios de auto-administración en ratas, ratones y monos han producido resultados conflictivos, mientras que la tolerancia a GHB así como la tolerancia cruzada al alcohol y baclofeno se ha mostrado evidente en roedores.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Agua purificada
Ácido málico para ajustar el pH
Hidróxido de sodio para ajustar el pH
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
5 años
Tras la primera apertura: 40 días
Tras la dilución en los vasos dosificadores, la preparación debe ser utilizada en 24 horas.
6.4 Precauciones especiales de conservación
Este medicamento no requiere condiciones especiales de conservación.
Para las condiciones de conservación tras la primera apertura del medicamento, ver sección 6.3.
Para las condiciones de conservación tras la dilución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase y de los equipos especiales para su utilización
Frasco ámbar, de 240 ml, ovalado, de PET, que contiene 180 ml de solución, y se presenta con una lámina de sellado de plástico y con un cierre de seguridad a prueba de niños, compuesto por HDPE/polipropileno con un revestimiento interno de cartón absorbente.
Cada envase contiene un frasco, un adaptador a presión del frasco que consta de un alojamiento de LDPE, una válvula silástica biomédica elastómera ETR, una válvula de retención de termopolímero de estireno, butadieno acrilonitrilo, y tubo de LDPE, un aparato de medida graduado (jeringa de polipropileno), dos vasos dosificadores de polipropileno y dos cierres de rosca resistentes a niños de HDPE.
6.6 Precauciones especiales de eliminación y otras manipulaciones
No precisa requerimientos especiales.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
UCB Pharma Ltd 208 Bath Road
Slough Berkshire SL1 3WE Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/312/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 13 Octubre 2005 Fecha de la última revalidación: 08 Septiembre 2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes
UCB Pharma Ltd 208 Bath Road Slough
Berkshire SL1 3WE Reino Unido
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica especial y restringida (Ver anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
• Medidas adicionales de minimización de riesgos
El Titular de la Autorización de Comercialización (TAC) desarrollará un programa educacional para Xyrem con el fin de asegurar que los médicos que quieran prescribir Xyrem estén informados sobre la posología de Xyrem y los riesgos importantes. Los cuatro componentes de este exhaustivo programa son:
• Lista de verificación para el Profesional Sanitario (por ej. formularios de iniciación del
tratamiento): para recordar a los médicos que verifiquen las contraindicaciones, advertencias y precauciones en la Ficha Técnica, señalando específicamente que Xyrem puede causar depresión respiratoria y del SNC, que el alcohol puede potenciar la depresión del SNC y que Xyrem tiene potencial de abuso.
• Hoja de Información al Paciente con las preguntas frecuentes (para entregar al paciente): para dar a los pacientes respuestas a algunas preguntas que puedan tener sobre la toma de Xyrem.
• Manual de Cómo tomar Xyrem (para entregar al paciente): para dar a los pacientes información sobre el uso de Xyrem.
• Tarjeta de alerta para el paciente (para entregar al paciente): para recordar a los pacientes, a los médicos y/o farmacéuticos la importancia de la información de seguridad relacionada con el uso de Xyrem.
El TAC ha establecido un programa de distribución controlado que mejora los controles existentes para Xyrem con el fin de permitir su alcance a la población prevista de pacientes con narcolepsia, mientras que minimiza el riesgo de que Xyrem sea desviado a aquellos que buscan su mal uso.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR Y EN EL ACONDICIONAMIENTO PRIMARIO
Caja y frasco
1. NOMBRE DEL MEDICAMENTO
Xyrem 500 mg/ml solución oral Oxibato de sodio
2. PRINCIPIO(S) ACTIVO(S)
Cada ml de solución contiene 500 mg de oxibato de sodio
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Frasco de 180 ml de solución oral
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD:
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
El medicamento debe utilizarse en los 40 días siguientes tras la primera apertura.
Tras la dilución en los vasos dosificadores, la preparación debe ser utilizada en 24 horas.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
UCB Pharma Ltd 208 Bath Road Slough Berkshire SL1 3WE.
Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/312/001
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
MEDICAMENTO SUJETO A PRESCRIPCIÓN MÉDICA
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Xyrem 500 mg/ml (afecta solo a la caja)
B. PROSPECTO
Prospecto: información para el usuario
Xyrem 500 mg/ml solución oral
Oxibato de sodio
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Xyrem y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Xyrem
3. Cómo tomar Xyrem
4. Posibles efectos adversos
5. Conservación de Xyrem
6. Contenido del envase e información adicional
1. Qué es Xyrem y para qué se utiliza
Xyrem contiene el principio activo oxibato de sodio. Xyrem actúa consolidando el sueño nocturno, aunque se desconoce su mecanismo de acción exacto.
Xyrem se usa para tratar la narcolepsia con cataplejía en adultos.
La narcolepsia es un trastorno del sueño que puede incluir ataques de sueño durante las horas en que normalmente se está despierto, así como cataplejía, parálisis del sueño, alucinaciones e insomnio. La cataplejía es aparición repentina de debilidad o parálisis muscular sin pérdida de consciencia, en respuesta a una reacción emocional repentina como rabia, miedo, alegría, risa o sorpresa.
2. Qué necesita saber antes de empezar a tomar Xyrem No tome Xyrem
- si es alérgico al oxibato de sodio o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6);
- si presenta un trastorno metabólico raro caracterizado por el déficit de succinato-semialdehído-deshidrogenasa;
- si sufre una depresión grave;
- si está recibiendo tratamiento con medicamentos opioides o barbitúricos.
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar Xyrem.
- si tiene problemas respiratorios o pulmonares (y especialmente si es obeso), ya que Xyrem tiene el potencial para causar dificultad para respirar;
- si tiene o ha tenido depresión;
- si padece insuficiencia cardíaca, hipertensión (presión arterial elevada), problemas de hígado o riñón, es posible que le deban ajustar su dosis;
- si previamente ha consumido drogas o ha abusado de medicamentos;
- si sufre epilepsia, ya que no se recomienda el uso de Xyrem en esta enfermedad;
- si padece porfiria (trastorno metabólico raro).
Si padece alguno de estos problemas, informe a su médico antes de tomar Xyrem.
Si mientras toma Xyrem, sufre pérdidas de orina nocturnas e incontinencia (tanto urinaria como fecal), confusión, alucinaciones, episodios de sonambulismo o pensamiento anormal, deberá comunicárselo inmediatamente a su médico. Aunque estos efectos son poco frecuentes, si aparecen, son por lo general de naturaleza leve a moderada.
En las personas mayores, el médico seguirá cuidadosamente su evolución para comprobar si Xyrem produce los efectos deseados.
Xyrem tiene un potencial de abuso bien conocido. Se han dado casos de dependencia tras un uso ilícito del oxibato de sodio.
Su médico le preguntara si ha consumido cualquier droga antes de comenzar a tomar Xyrem y mientras esté tomando este medicamento.
Niños y adolescentes
No dé este medicamento a niños y adolescentes.
Uso de Xyrem con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
En particular, Xyrem no se debe usar junto con medicamentos que inducen el sueño y medicamentos que reducen la actividad del Sistema Nervioso Central (el Sistema Nervioso Central es la parte del cuerpo compuesto por el cerebro y la médula espinal):
También debe informar a su médico o farmacéutico si está usando alguno de los siguientes tipos de medicamentos:
• medicamentos que incrementan la actividad del sistema nervioso central y antidepresivos
• medicamentos que pueden metabolizarse de forma similar por el organismo (ej., valporato, fenitoína o etosuximida, que se utilizan para el tratamiento de las crisis epilépticas)
• topiramato (utilizado para el tratamiento de la epilepsia)
• si está tomando valproato, su dosis diaria de Xyrem tendrá que ser ajustada (ver sección 3) ya que puede dar lugar a interacciones.
Toma de Xyrem con los alimentos, bebidas y alcohol
No debe beber alcohol mientras esté tomando Xyrem, ya que sus efectos pueden verse incrementados.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Ha habido muy pocas mujeres que hayan tomado Xyrem durante su embarazo y algunas de ellas sufrieron abortos espontáneos. No se conoce el riesgo de tomar Xyrem durante el embarazo, por lo que no se recomienda su uso en mujeres embarazadas o mujeres que estén tratando de quedarse embarazadas.
No se sabe si Xyrem pasa a la leche materna. Las pacientes que tomen Xyrem deberán interrumpir la lactancia.
Conducción y uso de máquinas
Xyrem puede afectarle si usted conduce o utiliza máquinas. No conduzca, no utilice maquinaria pesada, ni realice cualquier actividad que pueda ser peligrosa o que requiera alerta mental, durante al menos 6 horas después de la ingesta de Xyrem. Cuando empiece a tomar Xyrem por primera vez y hasta que sepa si le produce somnolencia al día siguiente, tenga especial cuidado cuando conduzca, opere con maquinaria pesada o haga cualquier otra actividad que pudiera resultar peligrosa o necesite un estado de alerta mental completo.
Xyrem contiene sodio
Dado que Xyrem contiene sodio (que se encuentra en la sal de mesa), necesita vigilar la cantidad de sal que ingiere, ya que puede afectarle si anteriormente tuvo problemas de hipertensión, trastornos cardíacos o renales. Si toma cada noche 2 dosis de oxibato de sodio de 2,25 g cada una, estará ingiriendo 0,82 g de sodio, o si toma 2 dosis de 4,5 g de oxibato de sodio cada noche, estará ingiriendo
1,6 g de sodio. Debe moderar la ingesta de sal durante el tratamiento con Xyrem.
3. Cómo tomar Xyrem
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
La dosis inicial recomendada es 4,5 g/día, repartida en dos dosis iguales de 2,25 g/dosis cada una. Su médico podrá aumentar gradualmente su dosis hasta un máximo de 9 g/día repartidos en dos dosis de 4,5 g/dosis.
Tome Xyrem vía oral dos veces cada noche. Tome la primera dosis al ir acostarse y la segunda dosis de 2,5 a 4 horas más tarde. Puede necesitar un despertador para asegurarse que se despertará para tomar la segunda dosis. Los alimentos disminuyen la cantidad de Xyrem que absorbe su organismo. Por lo tanto, es mejor tomar Xyrem siempre a una hora determinada dos - tres horas después de las comidas. Prepare ambas dosis antes de acostarse. Tome las dosis dentro de las 24 horas posteriores a su preparación.
Si está tomando valproato junto con Xyrem, su médico le ajustará la dosis de Xyrem. La dosis inicial recomendada de Xyrem cuando se utiliza junto con valproato es de 3,6 g/día, dividida en dos dosis iguales de 1,8 g. Tome la primera dosis cuando se vaya a acostar y la segunda dosis de 2,5 a 4 horas después.
Si tiene problemas de riñón, debe tener en cuenta las recomendaciones dietéticas para reducir la ingesta de sodio.
Si tiene problemas de hígado, la dosis inicial se debe reducir a la mitad. Su médico puede aumentar su dosis gradualmente.
Instrucciones para diluir Xyrem
Las siguientes instrucciones explican cómo preparar Xyrem. Lea detenidamente las instrucciones y sígalas paso a paso.
Para ayudarle, el envase de Xyrem contiene 1 frasco de medicamento, una jeringa graduada y dos vasos dosificadores con tapones de seguridad a prueba de niños.
1. Quite el tapón del frasco presionando hacia abajo, y desenrosque en sentido contrario a las agujas del reloj (hacia la izquierda). Después de quitar el tapón, ponga el frasco vertical sobre una mesa. Se debe retirar la lámina de sellado de plástico de la boca del frasco, antes de utilizarlo por primera vez. Manteniendo el frasco en posición vertical, inserte en el cuello del frasco el adaptador a presión. Esto sólo debe hacerse la primera vez que se abra el frasco. El adaptador se puede dejar puesto en el frasco para los usos siguientes.
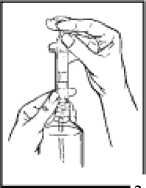
2. A continuación, inserte la punta de la jeringa graduada en el centro de la apertura del frasco y haga presión firmemente (Ver Figura 1).

3. Manteniendo el frasco y la jeringa en una mano, prepare la dosis prescrita con la otra mano
tirando del émbolo. NOTA: El medicamento no fluirá en la jeringa a no ser que usted mantenga la botella en posición vertical (Ver Figura 2).
■ . ■ 2
4. Quite la jeringa del centro de la apertura del frasco. Vacíe el medicamento de la jeringa en uno de los vasos dosificadores proporcionados empujando el émbolo (Ver Figura 3). Repita este paso para el segundo vaso dosificador. Agregue entonces aproximadamente 60 ml de agua a cada vaso dosificador (60 ml son aproximadamente 4 cucharadas).
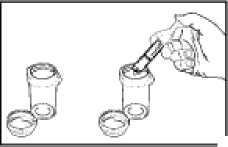
5. Ponga los tapones de los vasos dosificadores y gire cada tapón en el sentido de las agujas del reloj (a la derecha) hasta sentir el click y ciérrelo en la posición a prueba de niños. (Ver Figura 4). Aclare la jeringa con agua.
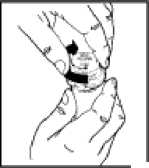
6. Coloque su segunda dosis cerca de su cama justo antes ir a dormir. Puede necesitar un despertador para asegurarse que se levantará para tomar su segunda dosis, no antes de 2,5 horas y no más tarde de 4 horas después de su primera dosis. Quite el tapón del primer vaso dosificador haciendo presión sobre el tapón de seguridad a prueba de niños y gírelo en sentido contrario a las agujas del reloj (hacia la izquierda). Beba la primera dosis sentado en la cama, tape el vaso, y luego acuéstese enseguida.
7. Cuando se despierte entre 2,5 y 4 horas más tarde, quite el tapón del segundo vaso dosificador. Sentado en la cama, beba la segunda dosis justo antes de volver a acostarse para seguir durmiendo. Tape el segundo vaso.
Si considera que el efecto de Xyrem es demasiado intenso o demasiado débil, comuníqueselo a su médico o farmacéutico.
Si toma más Xyrem del que debiera
Los síntomas de sobredosis por Xyrem pueden incluir agitación, confusión, movilidad alterada, dificultad respiratoria, visión borrosa, sudoración excesiva, dolor de cabeza, vómitos, conciencia disminuida que puede conducir a coma y crisis epiléptica. Si usted toma más Xyrem del que debiera, o lo toma por accidente, solicite inmediatamente ayuda médica de urgencia. Debe llevar con usted la caja del medicamento, incluso si está vacío.
Si olvidó tomar Xyrem
Si olvidó tomar la primera dosis, tómela en cuanto lo recuerde y continúe con el procedimiento descrito previamente. Si omite la segunda dosis, salte esa dosis y no tome Xyrem de nuevo hasta la próxima noche. No tome una dosis doble para compensar las dosis olvidadas.
Si interrumpe el tratamiento con Xyrem
Deberá seguir tomando Xyrem mientras su médico se lo continúe prescribiendo. Si se interrumpe la medicación, los ataques de cataplejía pueden volver y puede experimentar insomnio, dolor de cabeza, ansiedad, vértigo, trastornos del sueño, somnolencia, alucinaciones y pensamiento anormal.
Si interrumpe el tratamiento con Xyrem durante más de 14 días, debe consultar con su médico ya que debe volver a empezar el tratamiento con Xyrem a partir de una dosis menor.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Éstos con frecuencia son de intensidad leve a moderada. Informe a su médico si tiene alguno de los efectos adversos siguientes.
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
Náuseas, mareo, dolor de cabeza.
Frecuentes (pueden afectar hasta 1 de cada 10personas):
Trastornos del sueño incluyendo insomnio, visión borrosa, palpitaciones, vómitos, dolor de estómago, diarrea, anorexia, disminución del apetito, pérdida de peso, debilidad, sueños anormales, cansancio, sensación de embriaguez, parálisis del sueño, somnolencia, temblores, confusión/desorientación, pesadillas, sonambulismo, pérdidas nocturnas de orina, sudoración, depresión, calambres musculares, hinchazón, caídas, dolor de articulaciones, dolor de espalda, excesiva somnolencia durante el día, trastornos de equilibrio, trastornos de atención, alteraciones de la sensibilidad particularmente del tacto, sensación táctil anormal, sedación, alteración en el gusto, ansiedad, dificultad para quedarse dormido a medianoche, nerviosismo, sensación de mareo (vértigo), incontinencia urinaria, dificultad de la respiración, ronquidos, congestión de la nariz, erupción cutánea, inflamación de los senos nasales, inflamación de la nariz y de la garganta, aumento de la presión sanguínea.
Poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
Psicosis (un trastorno mental que puede producir alucinaciones, discurso incoherente, o comportamiento desorganizado o agitado), paranoia, pensamiento anormal, alucinaciones, agitación, intento de suicidio, dificultad para conciliar el sueño, piernas inquietas, amnesia (pérdidas de memoria), mioclonías (contracciones musculares involuntarias), pérdidas involuntarias de heces, alergia.
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles):
Convulsión, frecuencia o profundidad respiratoria disminuida, urticaria, pensamientos suicidas, interrupciones breves de la respiración durante la noche, estado de ánimo eufórico, boca seca, hinchazón de la cara (angioedema), deshidratación, ataque de pánico, manía/trastorno bipolar, delirio, bruxismo (rechinar los dientes y apretar la mandíbula), polaquiuria/urgencia urinaria (aumento de la necesidad de orinar), tinnitus (ruido en los oídos como pitidos o zumbidos), trastornos alimenticios relacionados con el sueño y pérdida de conciencia.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Xyrem
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el frasco después de (CAD). La fecha de caducidad es el último día del mes que se indica.
Tras la dilución en los vasos dosificadores, la preparación se debe utilizar dentro de las 24 horas posteriores.
Una vez abierto el frasco de Xyrem, cualquier contenido que no se haya usado en 40 días tras su apertura deberá ser desechado.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Xyrem
- El principio activo es oxibato de sodio. Cada ml contiene 500 mg de oxibato sódico.
- Los demás componentes son agua purificada, ácido málico e hidróxido de sodio.
Aspecto del producto y contenido del envase
Xyrem se presenta en un frasco de plástico de color ámbar de 240 ml que contiene 180 ml de solución oral, cerrado con un tapón a prueba de niños. El frasco tiene una lámina de sellado de plástico en la boca del mismo, debajo del tapón. Cada envase contiene un frasco, un adaptador al frasco a presión (PIBA), una jeringa graduada de plástico y dos vasos dosificadores con tapones a prueba de niños.
Xyrem es una solución clara a ligeramente opalescente.
Titular de la autorización de comercialización y responsable de la fabricación
UCB Pharma Ltd, 208 Batch Road, Slough, Berkshire, SL1 3WE, Reino Unido.
Su médico le tiene que haber entregado un paquete de información de Xyrem, que incluye un folleto sobre cómo tomar el medicamento, una hoja de información al paciente con las Preguntas Frecuentes y una tarjeta de alerta para el paciente.
Puede solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien UCB Pharma SA/NV Tel/Tél: +32 / (0)2 559 92 00 |
Lietuva UCB Pharma Oy Finland Tel: + 358 10 234 6800 (Suomija) |
|
Etnrapun ro CH EH Etnrapna EOOfl Ten.: + 359 (0) 2 962 99 20 |
Luxembourg/Luxemburg UCB Pharma SA/NV Tél/Tel: +32 / (0)2 559 92 00 |
|
Ceská republika UCB s.r.o. Tel: + 420 221 773 411 |
Magyarország UCB Magyarország Kft. Tel.: + 36-(1) 391 0060 |
|
Danmark UCB Nordic A/S Tlf: + 45 / 32 46 24 00 |
Malta Pharmasud Ltd. Tel: +356 / 21 37 64 36 |
|
Deutschland UCB Pharma GmbH Tel: + 49 /(0) 2173 48 4848 |
Nederland UCB Pharma B.V. Tel.: +31 / (0)76-573 11 40 |
|
Eesti UCB Pharma Oy Finland Tel: + 358 10 234 6800 (Soome) |
Norge UCB Nordic A/S Tel: +45 / 32 46 24 00 |
|
EXláóa UCB A.E. TpU +30 / 2109974000 |
Osterreich UCB Pharma GmbH Tel: + 43 (1) 291 80 00 |
|
España UCB Pharma, S.A. Tel: + 34 / 91 570 34 44 |
Polska UCB Pharma Sp. z o.o. Tel.: + 48 22 696 99 20 |
|
France UCB Pharma S.A. Tél: + 33 / (0)1 47 29 44 66 |
Portugal UCB Pharma (Produtos Farmacéuticos), Lda Tel: + 351 / 21 302 5300 |
|
Hrvatska Medis Adria d.o.o. Tel: +385 (0) 1 230 34 46 |
Romania UCB Pharma Romania S.R.L. Tel: +40 21 300 29 04 |
Ireland
UCB (Pharma) Ireland Ltd.
Tel: + 353 / (0)1-46 37 395
Ísland
Vistor hf.
Tel: +354 535 7000 Italia
UCB Pharma S.p.A.
Tel: + 39 / 02 300 791
Kúrcpog
Lifepharma (Z.A.M.) Ltd Tnk: + 357 22 34 74 40
Latvija
UCB Pharma Oy Finland Tel: + 358 10 234 6800 (Somija)
Slovenija
Medis, d.o.o.
Tel: + 386 1 589 69 00
Slovenská republika
UCB s.r.o., organizacná zlozka Tel: + 421 (0) 2 5920 2020
Suomi/Finland
UCB Pharma Oy Finland Puh/ Tel: + 358 10 234 6800
Sverige
UCB Nordic A/S
Tel: + 46 / (0) 40 29 49 00
United Kingdom UCB Pharma Ltd.
Tel : +44 / (0)1753 534 655
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu/
32
Los datos CGI-c se analizaron considerando a los respondedores como pacientes que mejoraron muchísimo o mucho.