Xalkori 250 Mg Capsulas Duras
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
XALKORI200 mg cápsulas duras XALKORI250 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
XALKORI 200 mg cápsulas duras
Cada cápsula dura contiene 200 mg de crizotinib.
XALKORI 250 mg cápsulas duras
Cada cápsula dura contiene 250 mg de crizotinib.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsulas duras.
XALKORI 200 mg cápsulas duras
Cápsulas duras, de color blanco opaco y rosa opaco, con “Pfizer” impreso en la tapa y “CRZ 200” en el cuerpo.
XALKORI 250 mg cápsulas duras
Cápsulas duras, de color rosa opaco, con “Pfizer” impreso en la tapa y “CRZ 250” en el cuerpo.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
XALKORI está indicado como tratamiento de primera línea para adultos con cáncer de pulmón no microcítico (CPNM) avanzado, positivo para la quinasa del linfoma anaplásico (ALK).
XALKORI está indicado para el tratamiento de adultos con cáncer de pulmón no microcítico (CPNM) avanzado, positivo para la quinasa del linfoma anaplásico (ALK), previamente tratado.
XALKORI está indicado para el tratamiento de adultos con cáncer de pulmón no microcítico (CPNM) avanzado, positivo para ROS 1.
4.2 Posología y forma de administración
El tratamiento con XALKORI debe instaurarse y administrarse bajo la supervisión de un médico con experiencia en el uso de medicamentos antineoplásicos.
Determinación de ALK y ROS1
Para seleccionar a los pacientes que van a recibir tratamiento con XALKORI se precisa un método exacto y validado de determinación de ALK o de ROS1 (para la información sobre los métodos utilizados en los ensayos, ver sección 5.1).
Antes de instaurarse el tratamiento con crizotinib, debe confirmarse la presencia de CPNM ALK-positivo o ROS1-positivo. La evaluación correrá a cargo de laboratorios con competencia demostrada en la técnica específica que se está utilizando (ver sección 4.4).
Posología
La pauta posológica recomendada de XALKORI es de 250 mg dos veces al día (500 mg diarios), administrado de manera continua.
Si se olvida una dosis, el paciente deberá tomarla en cuanto se acuerde, salvo que queden menos de 6 horas hasta la dosis siguiente, en cuyo caso el paciente no tomará la dosis olvidada. Los pacientes no deben tomar dos dosis al mismo tiempo para compensar una dosis olvidada.
Ajustes de dosis
Podría ser necesario interrumpir el tratamiento y/o reducir la dosis en función de la seguridad y la tolerabilidad individuales. En 1.722 pacientes con CPNM ALK-positivo o ROS1-positivo tratados con crizotinib en estudios clínicos, las reacciones adversas más frecuentes (> 3%) asociadas a interrupciones del tratamiento fueron neutropenia, transaminasas elevadas, vómitos y náuseas. Las reacciones adversas más frecuentes (> 3%) asociadas a reducciones de dosis fueron transaminasas elevadas y neutropenia. Si se precisa una reducción de dosis, la dosis de XALKORI se reducirá a 200 mg tomados dos veces al día. Si se necesita una reducción adicional, entonces la dosis debe modificarse a 250 mg una vez al día en función de la seguridad y la tolerabilidad individuales. En las tablas 1 y 2 se presentan las guías de reducción de la dosis por efectos tóxicos hematológicos y no hematológicos.
Tabla 1. Modificación de la dosis de XALKORI por toxicidad hematológicaa b
|
Grado CTCAEc |
Tratamiento con XALKORI |
|
Grado 3 |
Interrumpir hasta recuperación a grado < 2, luego reanudar a la misma dosis |
|
Grado 4 |
Interrumpir hasta recuperación a grado < 2, luego reiniciar con 200 mg dos veces al díad |
a Excepto linfopenia (salvo que esté asociada con acontecimientos clínicos, por ejemplo, infecciones oportunistas).
b En el caso de los pacientes que presenten neutropenia y leucopenia, ver también secciones 4.4 y 4.8. c Criterios terminológicos comunes para acontecimientos adversos (Common Terminology Criteria for Adverse Events) del Instituto Nacional del Cáncer (NCI, National Cáncer Institute). d En caso de que reaparezca, el tratamiento se debe interrumpir hasta recuperación a grado < 2 y después se debe reiniciar con 250 mg una vez al día. XALKORI debe suspenderse de manera permanente en caso de recurrencia con grado 4.
|
Tabla 2. Modificación de la dosis de XA |
^KORI por toxicidad no hematológica |
|
Grado CTCAEa |
Tratamiento con XALKORI |
|
Elevación de alanina-aminotransferasa (ALT) o de aspartato-aminotransferasa (AST) de grado 3 ó 4, con elevación de bilirrubina total grado < 1 |
Interrumpir hasta recuperación a grado < 1 o a la situación basal, luego reiniciar con 250 mg una vez al día y aumentar a 200 mg dos veces al día si existe tolerancia clínicab |
|
Elevación de ALT o de AST de grado 2, 3 ó 4, con elevación concomitante de bilirrubina total (en ausencia de colestasis o hemólisis) de grado 2, 3 ó 4 |
Suspender permanentemente |
|
Grado CTCAEa |
Tratamiento con XALKORI |
|
Enfermedad pulmonar intersticial (EPI)/neumonitis de cualquier grado |
Interrumpir si se sospecha EPI/neumonitis y suspender permanentemente si se diagnostica EPI/neumonitis relacionada con el tratamiento0 |
|
Prolongación del QTc de grado 3 |
Interrumpir hasta recuperación a grado < 1, comprobar los electrolitos y corregir si es necesario, luego reiniciar con 200 mg dos veces al díab |
|
Prolongación del QTc de grado 4 |
Suspender permanentemente |
|
Bradicardia de grado 2, 3c d Sintomática, puede ser grave y con relevancia médica, está indicada una intervención médica |
Interrumpir hasta recuperación a grado < 1 o a una frecuencia cardiaca igual o superior a 60 Evaluar los medicamentos concomitantes con efecto conocido de bradicardia, así como los medicamentos antihipertensivos Si se identifica e interrumpe algún medicamento concomitante que contribuya a la bradicardia, o se ajusta su dosis, reiniciar con la dosis anterior cuando se produzca una recuperación a grado < 1 o a una frecuencia cardiaca igual o superior a 60 Si no se identifica ningún medicamento concomitante que contribuya a la bradicardia o si no se interrumpen los medicamentos concomitantes que contribuyen a la bradicardia ni se modifica su dosis, reiniciar con una dosis menor cuando se produzca una recuperación a grado < 1 o a una frecuencia cardiaca igual o superior a 60 |
|
Bradicardia de grado 4c,d,e Consecuencias potencialmente mortales, está indicada una intervención de urgencia |
Suspender permanentemente si no se identifica ningún medicamento concomitante que contribuya a la bradicardia Si se identifica e interrumpe algún medicamento concomitante que contribuya a la bradicardia, o se ajusta su dosis, reiniciar con 250 mg una vez al día cuando se produzca una recuperación a grado < 1 o a una frecuencia cardiaca igual o superior a 60, y someter al paciente a una monitorización frecuente |
|
Trastorno ocular de grado 4 (pérdida de visión) |
Interrumpir durante la evaluación de la pérdida de visión grave |
a Criterios terminológicos comunes para acontecimientos adversos del NCI.
b XALKORI debe suspenderse de manera permanente en caso de recurrencia con grado > 3. Ver secciones 4.4 y
4.8.
c Ver secciones 4.4 y 4.8.
d Frecuencia cardiaca inferior a 60 latidos por minuto (l.p.m.) e Suspensión permanente en caso de recurrencia
Insuficiencia hepática
No se ha estudiado crizotinib en pacientes con insuficiencia hepática. En los estudios clínicos realizados se excluyó a los pacientes con unos valores de AST o ALT > 2,5 veces el límite superior normal (LSN), o > 5,0 veces el LSN si se debía a neoplasia maligna subyacente, o con bilirrubina total >1,5 veces el LSN. El tratamiento con crizotinib debe utilizarse con precaución en los pacientes con insuficiencia hepática leve o moderada. No debe administrarse crizotinib en pacientes con alteración hepática grave (ver secciones 4.3, 4.4 y 4.8).
Insuficiencia renal
No se recomienda ajustar la dosis inicial en pacientes con insuficiencia renal leve (aclaramiento de creatinina [CLcr] igual o superior a 60 e inferior a 90 ml/min) o moderada (CLcr igual o superior a 30 e inferior a 60 ml/min), ya que el análisis farmacocinético de la población indicó ausencia de cambios clínicamente significativos en la exposición a concentraciones de estado estacionario de crizotinib en estos pacientes. En pacientes con insuficiencia renal grave (CLcr < 30 ml/min), las concentraciones plasmáticas de crizotinib pueden aumentar. En pacientes con insuficiencia renal grave que no requieren diálisis peritoneal ni hemodiálisis, la dosis inicial de crizotinib debe ajustarse a 250 mg administrados de forma oral una vez al día. Después de al menos 4 semanas de tratamiento, la dosis se puede aumentar a 200 mg dos veces al día en función de la seguridad y tolerabilidad individual (ver secciones 4.4 y 5.2).
Pacientes de edad avanzada
No es necesario ajustar la dosis inicial (ver las secciones 5.1 y 5.2).
Población pediátrica
No se ha establecido la seguridad ni la eficacia de crizotinib en pacientes pediátricos. No se dispone de datos.
Forma de administración
Las cápsulas deben tragarse enteras, preferiblemente con agua, sin aplastarlas, disolverlas ni abrirlas. Pueden tomarse con o sin comida. Debe evitarse el pomelo y el zumo de pomelo porque pueden aumentar la concentración plasmática de crizotinib; también debe evitarse la hierba de San Juan porque puede disminuir la concentración plasmática de crizotinib (ver sección 4.5).
4.3 Contraindicaciones
Hipersensibilidad a crizotinib o a alguno de los excipientes incluidos en la sección 6.1.
Alteración hepática grave (ver las secciones 4.2, 4.4 y 4.8).
4.4 Advertencias y precauciones especiales de empleo
Evaluación del estado de ALK y de ROS1
Cuando se evalúe el estado de ALK o de ROS1 de un paciente, es importante elegir una metodología sólida y adecuadamente validada con el fin de evitar falsos negativos o falsos positivos.
Hepatotoxicidad
Se han notificado casos de hepatotoxicidad inducida por el medicamento (incluidos casos con desenlace mortal) en pacientes tratados con crizotinib en estudios clínicos (ver sección 4.8). No debe utilizarse crizotinib en los pacientes con insuficiencia hepática grave (incluidos los pacientes con bilirrubina total por encima de 3 veces el LSN con independencia de la ALT/AST) (ver secciones 4.2, 4.3 y 4.8). Se deben realizar pruebas de la función hepática, incluyendo ALT, AST y bilirrubina total, una vez a la semana durante los dos primeros meses de tratamiento, y posteriormente una vez al mes y cuando esté indicado clínicamente, con una repetición más frecuente de las determinaciones en caso de aumentos de grado 2, 3 ó 4. Para los pacientes que presenten aumento de transaminasas, ver sección 4.2.
Enfermedad pulmonar intersticial/neumonitis
Pueden producirse casos de enfermedad pulmonar intersticial (EPI)/neumonitis de carácter grave, potencialmente mortal o mortal en pacientes tratados con crizotinib. Se debe monitorizar a los pacientes con síntomas pulmonares indicativos de EPI/neumonitis. Debe interrumpirse el tratamiento con crizotinib si se sospecha EPI/neumonitis. Debe tenerse en cuenta la EPI/neumonitis inducida por el medicamento en el diagnóstico diferencial de los pacientes con afecciones similares a la EPI, tales como neumonitis, neumonitis por radiación, neumonitis por hipersensibilidad, neumonitis intersticial, fibrosis pulmonar, síndrome de insuficiencia respiratoria aguda (SDRA), alveolitis, infiltración pulmonar, neumonía, edema pulmonar, enfermedad pulmonar obstructiva crónica, derrame pleural, neumonía aspirativa, bronquitis, bronquiolitis obliterante y bronquiectasia. Se deben excluir otras causas posibles de EPI/neumonitis, y se ha de suspender permanentemente el tratamiento con crizotinib en aquellos pacientes diagnosticados de EPI/neumonitis relacionada con el tratamiento (ver secciones 4.2 y 4.8).
Prolongación del intervalo QT
Se ha observado prolongación del intervalo QTc en estudios clínicos con pacientes tratados con crizotinib (ver secciones 4.8 y 5.2), que puede dar lugar a un incremento en el riesgo de taquiarritmias ventriculares (por ejemplo, Torsade de Pointes) o muerte súbita. Deben sopesarse los beneficios y los posibles riesgos de crizotinib antes de comenzar el tratamiento en pacientes con bradicardia preexistente, con antecedentes o predisposición a la prolongación del intervalo QTc, que estén recibiendo antiarrítmicos u otros medicamentos con un efecto conocido de prolongación del intervalo QT y en pacientes con enfermedad cardiaca relevante pre-existente y/o alteraciones electrolíticas. Crizotinib debe administrarse con precaución en dichos pacientes y es necesario un control periódico de electrocardiogramas (ECG), electrolitos y función renal. Cuando se utilice crizotinib, deben realizarse ECG y determinaciones de electrolitos (por ejemplo, calcio, magnesio, potasio) lo más cerca posible del momento de la primera administración, y se recomienda realizar un control periódico mediante ECG y determinación de electrolitos, especialmente al inicio del tratamiento en caso de vómitos, diarrea, deshidratación o función renal alterada. Deben corregirse los electrolitos según sea necesario. Si el QTc aumenta en 60 ms o más en relación con la situación basal, pero es inferior a 500 ms, debe interrumpirse crizotinib y consultarse a un cardiólogo. Si el QTc aumenta a 500 ms o más, debe consultarse de inmediato a un cardiólogo. Para los pacientes que desarrollen una prolongación del QTc, ver secciones 4.2, 4.8 y 5.2.
Bradicardia
En los estudios clínicos se notificaron casos de bradicardia atribuibles a cualquier causa en el 13% de los pacientes tratados con crizotinib. Los pacientes que reciben crizotinib pueden presentar bradicardia sintomática (por ejemplo, síncope, mareo, hipotensión). El efecto completo de crizotinib sobre el descenso de la frecuencia cardiaca puede no aparecer hasta varias semanas después del inicio del tratamiento. Debe evitarse, en la medida de lo posible, el empleo de crizotinib en combinación con otros fármacos con efecto bradicárdico (por ejemplo, betabloqueantes, bloqueantes de los canales del calcio no dihidropiridínicos, como verapamilo y diltiazem, clonidina, digoxina), ya que dicha combinación aumenta el riesgo de bradicardia sintomática. Deben controlarse la frecuencia cardiaca y la tensión arterial con regularidad. En los casos de bradicardia asintomática, no es necesario modificar la dosis. Para el tratamiento de los pacientes que presentan bradicardia sintomática, ver secciones Modificación de la dosis y Reacciones adversas (ver secciones 4.2 y 4.8).
Insuficiencia cardiaca
En los estudios clínicos con crizotinib y durante la vigilancia posterior a su comercialización se han notificado reacciones adversas de insuficiencia cardiaca de carácter grave, potencialmente mortales o mortales (ver sección 4.8).
Se debe vigilar a los pacientes con o sin trastornos cardiacos preexistentes en tratamiento con crizotinib por si presentan signos y síntomas de fallo cardiaco (disnea, edema, aumento rápido de peso por retención de líquidos). Si se observan dichos síntomas, se debe considerar la interrupción de la administración, la reducción de la dosis o la suspensión definitiva del tratamiento según sea pertinente.
Neutropenia y leucopenia
En los estudios clínicos con crizotinib en pacientes con CPNM ALK-positivo o ROSl-positivo se ha notificado con mucha frecuencia (12%) neutropenia de grado 3 o 4. Se ha notificado con frecuencia (3%) leucopenia de grado 3 o 4 (ver sección 4.8). Menos del 0,5% de los pacientes sufrió neutropenia febril en los estudios clínicos con crizotinib. Deberán realizarse hemogramas completos con fórmula leucocítica según esté clínicamente indicado y repetirse las pruebas con mayor frecuencia si se observan anomalías de grado 3 o 4 o si se produce fiebre o infección (ver sección 4.2).
Perforación gastrointestinal
En estudios clínicos con crizotinib se han notificado casos de perforación gastrointestinal. Ha habido notificaciones de casos de perforación gastrointestinal con desenlace mortal durante el uso de crizotinib tras su comercialización (ver sección 4.8).
Crizotinib debe utilizarse con precaución en pacientes con riesgo de perforación gastrointestinal (por ejemplo, antecedentes de diverticulitis, metástasis del tracto gastrointestinal, empleo concomitante de otros medicamentos con riesgo reconocido de perforación gastrointestinal).
La administración de crizotinib debe suspenderse en pacientes que desarrollen perforación gastrointestinal. Debe informarse a los pacientes de los primeros signos de perforación intestinal y aconsejarles que consulten rápidamente en caso de que ocurran.
Efectos renales
En los estudios clínicos con crizotinib se observó un aumento de la creatinina en sangre y una disminución del aclaramiento de creatinina en los pacientes. Se notificó insuficiencia renal e insuficiencia renal aguda en pacientes tratados con crizotinib en los ensayos clínicos y durante el periodo posterior a la comercialización. También se observaron casos con desenlace mortal, casos que requirieron hemodiálisis y casos de hiperpotasemia de grado 4. Se recomienda el seguimiento de los pacientes para evaluar su función renal al inicio y durante el tratamiento con crizotinib, prestando especial atención a aquellos que tienen factores de riesgo o antecedentes de insuficiencia renal (ver sección 4.8).
Insuficiencia renal
En el caso de pacientes con insuficiencia renal grave que no requieren diálisis peritoneal ni hemodiálisis, debe ajustarse la dosis de crizotinib (ver secciones 4.2 y 5.2).
Efectos sobre la visión
En los estudios clínicos con crizotinib en pacientes con CPNM ALK-positivo o ROS1-positivo (N=1.722), se ha notificado defecto del campo visual de grado 4 con pérdida de visión en 4 (0,2%) pacientes. La atrofia óptica y el trastorno del nervio óptico se han identificado como causas potenciales de la pérdida de visión.
En pacientes con una nueva aparición de pérdida de visión grave (mejor agudeza visual corregida inferior a 6/60 en uno o ambos ojos), se debe interrumpir el tratamiento con crizotinib (ver sección 4.2). Se debe llevar a cabo una evaluación oftalmológica que incluya la evaluación de la mejor agudeza visual corregida, fotografías retinales, evaluación del campo visual, tomografía de coherencia óptica (TCO) y otras evaluaciones, según proceda, para la nueva aparición de la pérdida de visión grave. No hay información suficiente para caracterizar los riesgos que conlleva la reanudación del tratamiento con crizotinib en pacientes con una pérdida de visión grave. A la hora de decidir la reanudación del tratamiento con crizotinib se debe tener en cuenta el beneficio potencial para el paciente.
Se recomienda una evaluación oftalmológica si los trastornos en la visión persisten o empeoran en gravedad (ver sección 4.8).
Interacciones medicamentosas
Debe evitarse el uso concomitante de crizotinib con inhibidores potentes o inductores potentes y moderados de CYP3A4 (ver sección 4.5).
Se debe evitar el uso concomitante de crizotinib con sustratos de CYP3A4 de estrecho margen terapéutico (ver sección 4.5). Evítese el uso de crizotinib en combinación con otros fármacos con efecto bradicárdico, medicamentos con un efecto conocido de prolongación del intervalo QTc y/o antiarrítmicos (ver sección 4.4 Prolongación del intervalo QT, Bradicardia, y sección 4.5).
Pacientes sin histología de adenocarcinoma
La información disponible en pacientes con CPNM ALK-positivo y ROS1-positivo sin histología de adenocarcinoma, incluido el carcinoma de células escamosas (CCE), es limitada (ver sección 5.1).
4.5 Interacción con otros medicamentos y otras formas de interacción
Interacciones farmacocinéticas
Asentes que_pueden aumentar las concentraciones_plasmáticas de crizotinib
La administración concomitante de crizotinib con inhibidores potentes de CYP3A puede aumentar las concentraciones plasmáticas de crizotinib. La administración concomitante de una dosis oral única de 150 mg de crizotinib en presencia de ketoconazol (200 mg dos veces al día), un potente inhibidor de CYP3A, aumentó la exposición sistémica a crizotinib, con unos valores de área bajo la curva de la concentración plasmática frente al tiempo desde el tiempo cero hasta el infinito (AUCinf) y de concentración plasmática máxima observada (Cmax) de crizotinib aproximadamente 3,2 veces y 1,4 veces, respectivamente, mayores que los observados tras la administración de crizotinib solo.
En consecuencia, debe evitarse el uso concomitante de inhibidores potentes de CYP3A (ciertos inhibidores de la proteasa como atazanavir, indinavir, nelfinavir, ritonavir, saquinavir, y ciertos antimicóticos azólicos como itraconazol, ketoconazol y voriconazol, y ciertos macrólidos como claritromicina, telitromicina y troleandomicina). El pomelo y el zumo de pomelo también pueden aumentar las concentraciones plasmáticas de crizotinib, por lo que deben evitarse (ver secciones 4.2 y 4.4). Además, no se ha establecido el efecto de los inhibidores de CYP3A sobre la exposición a crizotinib en estado estacionario.
Asentes que_pueden disminuir las concentraciones_plasmáticas de crizotinib
La administración concomitante de dosis repetidas de crizotinib (250 mg dos veces al día) con dosis repetidas de rifampicina (600 mg una vez al día), un potente inductor de CYP3A4, produjo una disminución del 84% y el 79%, respectivamente, en los valores de AUCtau y Cmax de crizotinib en el estado estacionario, respecto a los obtenidos tras la administración de crizotinib solo. Se debe evitar el uso concomitante de inductores potentes de CYP3A, entre los que se pueden citar carbamazepina, fenobarbital, fenitoína, rifampicina y hierba de San Juan (ver sección 4.4).
El efecto de un inductor moderado, como podrían ser efavirenz o rifabutina, entre otros, no se ha establecido de forma clara, por tanto, su combinación con crizotinib se debe evitar (ver sección 4.4).
Administración concomitante con medicamentos que aumentan el _pH gástrico
La solubilidad acuosa de crizotinib es pH dependiente, un pH bajo (ácido) produce una mayor solubilidad. La administración de una sola dosis de 250 mg de crizotinib después del tratamiento con esomeprazol, 40 mg una vez al día durante 5 días, produjo una disminución aproximada del 10% en la exposición total a crizotinib (AUCinf) y ningún cambio en la concentración máxima (Cmáx); la extensión del cambio en la exposición total no fue clínicamente significativa. Por tanto, no se requiere ajuste de la dosis inicial cuando se administra crizotinib junto con agentes que aumentan el pH gástrico (como los inhibidores de la bomba de protones, los H2 bloqueantes o los antiácidos).
Agentes cuyas concentraciones_plasmáticas_pueden verse alteradas_por crizotinib
En pacientes oncológicos, después de 28 días de tratamiento con crizotinib a dosis de 250 mg dos veces al día, el AUC de midazolam oral fue 3,7 veces mayor que el observado tras la administración de midazolam solo, lo que sugiere que crizotinib es un inhibidor moderado de CYP3A. En consecuencia, la administración concomitante de crizotinib con sustratos del CYP3A con estrecho margen terapéutico, entre los que se pueden citar alfentanilo, cisaprida, ciclosporina, derivados ergóticos, fentanilo, pimozida, quinidina, sirolimus y tacrolimus, debe evitarse (ver sección 4.4). Si la combinación fuera necesaria, se deberá realizar una monitorización estrecha.
Estudios in vitro han indicado que crizotinib es un inhibidor de CYP2B6. Por tanto, la administración de crizotinib junto con medicamentos que sean metabolizados por CYP2B6 (por ejemplo, bupropión, efavirenz) podría aumentar las concentraciones plasmáticas de éstos últimos.
Estudios in vitro en hepatocitos humanos indicaron que crizotinib puede inducir enzimas reguladas por el receptor X de pregnano (PXR) y por el receptor constitutivo de androstano (CAR) (por ejemplo, CYP3A4, CYP2B6, CYP2C8, CYP2C9, UGT1A1). Sin embargo, no se observó inducción in vivo cuando se administró crizotinib conjuntamente con midazolam, un sustrato de CYP3A. Debe actuarse con precaución cuando se administre crizotinib en combinación con medicamentos que sean metabolizados mayoritariamente por estas enzimas. Conviene destacar que puede verse reducida la eficacia de los anticonceptivos orales administrados de forma concomitante.
Los estudios in vitro indicaron que crizotinib es un inhibidor débil de uridina difosfato glucuronosiltransferasa (UGT)1A1 y UGT2B7. Por tanto, crizotinib puede tener el potencial de aumentar las concentraciones plasmáticas de los medicamentos administrados simultáneamente que sean metabolizados mayoritariamente por UGT1A1 (p.ej. raltegravir, irinotecán) o UGT2B7 (morfina, naloxona).
Según un estudio in vitro, crizotinib podría ser un inhibidor de la glicoproteína P intestinal (gp-P). En consecuencia, la administración de crizotinib con medicamentos que sean sustratos de la gp-P (por ejemplo, digoxina, dabigatrán, colchicina, pravastatina) podría aumentar su efecto terapéutico y sus reacciones adversas. Se recomienda una estrecha vigilancia en caso de que crizotinib se administre con estos medicamentos.
Crizotinib es un inhibidor de OCT1 y OCT2 in vitro. Por consiguiente, crizotinib puede tener el potencial de aumentar las concentraciones plasmáticas de medicamentos coadministrados que sean sustratos de OCT1 u OCT2 (por ejemplo, metformina, procainamida).
Interacciones farmacodinámicas
En los estudios clínicos, se observó una prolongación del intervalo QT con crizotinib. Por tanto, el uso concomitante de crizotinib con medicamentos con efecto conocido de prolongación del intervalo QT o con medicamentos que puedan inducir Torsades de pointes (por ejemplo, agentes de clase IA [quinidina, disopiramida] o clase III [por ejemplo, amiodarona, sotalol, dofetilida, ibutilida], metadona, cisaprida, moxifloxacino, antipsicóticos, etc.) deberá realizarse de forma cuidadosa. En el caso de combinaciones de este tipo de medicamentos se debe realizar una monitorización del intervalo QT (ver secciones 4.2 y 4.4).
Se ha notificado bradicardia durante los estudios clínicos; por tanto, debido al riesgo de una bradicardia excesiva, crizotinib debe utilizarse con precaución si se administra en combinación con otros productos bradicárdicos (por ejemplo, bloqueantes de los canales del calcio no dihidropiridínicos, como verapamilo y diltiazem, betabloqueantes, clonidina, guanfacina, digoxina, mefloquina, anticolinesterasas, pilocarpina) (ver secciones 4.2 y 4.4).
4.6 Fertilidad, embarazo y lactancia
Anticoncepción en hombres y mujeres
Se debe advertir a las mujeres en edad fértil que eviten quedarse embarazadas mientras estén en tratamiento con XALKORI.
Se deben utilizar métodos anticonceptivos adecuados durante todo el tratamiento y hasta 90 días tras finalizarlo (ver sección 4.5).
Embarazo
XALKORI puede provocar efectos perjudiciales en el feto cuando se administra durante el embarazo. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3).
No hay datos relativos al uso de crizotinib en mujeres embarazadas. No debe utilizarse este medicamento durante el embarazo a no ser que la situación clínica de la mujer requiera tratamiento. Las mujeres embarazadas o pacientes que se queden embarazadas durante el tratamiento con crizotinib, o los pacientes varones en tratamiento cuyas parejas se hayan quedado embarazadas, deberán ser informados del posible riesgo para el feto.
Lactancia
Se desconoce si crizotinib y sus metabolitos se excretan en la leche materna. Ante el posible riesgo para el lactante, se indicará a las madres que no deben amamantar durante el tratamiento con XALKORI (ver sección 5.3).
Fertilidad
De acuerdo a los datos de seguridad de estudios no clínicos, la fertilidad masculina y femenina podría verse comprometida por el tratamiento con XALKORI (ver sección 5.3). Tanto hombres como mujeres deben recibir asesoramiento sobre la conservación de la fertilidad antes del tratamiento.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Debe tenerse precaución al conducir o utilizar máquinas porque los pacientes pueden presentar bradicardia sintomática (por ejemplo, síncope, mareo, hipotensión), trastornos de la visión o cansancio durante el tratamiento con XALKORI (ver secciones 4.2, 4.4 y 4.8).
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los datos que se describen a continuación reflejan la exposición a XALKORI de 1.669 pacientes con CPNM avanzado ALK-positivo que participaron en 2 estudios fase III aleatorizados (estudios 1007 y 1014) y en dos estudios de un único brazo de tratamiento (estudios 1001 y 1005), y de 53 pacientes con CPNM avanzado ROS1-positivo que participaron en el estudio 1001 de un único brazo de tratamiento, con un total de 1.722 pacientes (ver sección 5.1). Estos pacientes recibieron una dosis oral inicial de 250 mg tomados dos veces al día de manera continua. En el estudio 1014, la mediana de duración del tratamiento del estudio fue de 47 semanas para los pacientes del brazo de crizotinib (N=171); la mediana de duración del tratamiento fue de 23 semanas para los pacientes que pasaron de estar en el brazo de quimioterapia a recibir tratamiento con crizotinib (N=109). En el estudio 1007, la mediana de duración del tratamiento del estudio fue de 48 semanas para los pacientes del brazo de crizotinib (N=172). Para los pacientes con CPNM ALK-positivo en los estudios 1001 (N=154) y 1005 (N=1.063), la mediana de duración del tratamiento fue de 57 y 45 semanas, respectivamente. Para los pacientes con CPNM ROS1-positivo en el estudio 1001 (N=53), la mediana de duración del tratamiento fue de 101 semanas.
Las reacciones adversas de mayor gravedad en 1.722 pacientes con CPNM avanzado ALK-positivo o ROS1-positivo fueron hepatotoxicidad, EPI/neumonitis, neutropenia y prolongación del intervalo QT (ver sección 4.4). Las reacciones adversas más frecuentes (> 25%) en pacientes con CPNM ALK-positivo o ROS1-positivo fueron trastornos de la visión, náuseas, diarrea, vómitos, edema, estreñimiento, elevación de las transaminasas, cansancio, disminución del apetito, mareo y neuropatía.
Tabla de reacciones adversas
En la tabla 3 se presentan las reacciones adversas notificadas en 1.722 pacientes con CPNM avanzado ALK-positivo o ROS1-positivo que recibieron crizotinib en 2 estudios en fase III aleatorizados (estudios 1007 y 1014) y en 2 estudios clínicos de un único brazo de tratamiento (estudios 1001 y 1005) (ver sección 5.1).
Las reacciones adversas más frecuentes (> 3%, frecuencia atribuible a cualquier causa) asociadas a la interrupción de la administración fueron neutropenia (11%), elevación de las transaminasas (7%), vómitos (5%) y náuseas (4%). Las reacciones adversas más frecuentes (> 3%, frecuencia atribuible a cualquier causa) asociadas a reducciones de la dosis fueron la elevación de las transaminasas (4%) y la neutropenia (3%). En 302 (18%) pacientes se produjeron acontecimientos adversos atribuibles a cualquier causa y asociados a la suspensión permanente del tratamiento, siendo los más frecuentes (> 1%) la enfermedad pulmonar intersticial (1%) y la elevación de las transaminasas (1%).
Las reacciones adversas enumeradas en la tabla 3 se presentan por categorías de sistema de clasificación de órganos y de frecuencia, definidas mediante la siguiente convención: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000), muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada grupo de frecuencia, los efectos no deseados se presentan en orden decreciente de gravedad.
Tabla 3._Reacciones adversas notificadas en estudios clínicos con crizotinib (N=1.722)
|
Sistema de clasificación de órganos |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Trastornos de la sangre y del sistema linfático |
Neutropenia3 (22%) Anemiab (15%) Leucopeniac (15%) | ||
|
Trastornos del metabolismo y de la nutrición |
Disminución del apetito (30%) |
Hipofosfatemia (6%) | |
|
Trastornos del sistema nervioso |
Neuropatíad (25%) Disgeusia (21%) | ||
|
Trastornos oculares |
Trastornos de la visióne (63%) | ||
|
Trastornos cardiacos |
Mareo1 (26%) Bradicardiag (13%) |
Insuficiencia cardiaca11 (1%) Prolongación del intervalo electrocardiográfico QT (4%) Síncope (3%) |
|
Trastornos respiratorios, torácicos y mediastínicos |
Enfermedad pulmonar intersticial1 (3%) | ||
|
Trastornos gastrointestinales |
Vómitos (51%) Diarrea (54%) Náuseas (57%) Estreñimiento (43%) Dolor abdominal (21%) |
Dispepsia (8%) Esofagitisk (2%) |
Perforación gastrointestinal1 (< 1%) |
|
Trastornos hepatobiliares |
Elevación de las transaminasasm (32%) |
Aumento de la fosfatasa alcalina sanguínea (7%) |
Insuficiencia hepática (< 1%) |
|
Trastornos de la piel y del tejido subcutáneo |
Erupción (13%) | ||
|
Trastornos renales y urinarios |
Quiste renaln (3%) Aumento de la creatinina en sangreo (8%) |
Insuficiencia renal aguda (< 1%) Insuficiencia renal (< 1%) | |
|
Trastornos generales y alteraciones en el lugar de administración |
Edemap (47%) Cansancio (30%) | ||
|
Exploraciones complementarias |
Disminución de la testosterona en sangreq (2%) |
Los términos que representan el mismo concepto o afección médica se agrupan y presentan como una única
reacción adversa en la tabla 3. Los términos presentados en el estudio hasta la fecha de corte de datos y que
contribuyen a la reacción adversa pertinente se indican entre paréntesis, como se indica más abajo.
a. Neutropenia (neutropenia febril, neutropenia, recuento disminuido de neutrófilos).
b. Anemia (anemia, hemoglobina disminuida, anemia hipocrómica).
c. Leucopenia (leucopenia, recuento disminuido de leucocitos).
d. Neuropatía, (sensación de ardor, disestesia, hormigueo, alteración de la marcha, hiperestesia, hipoestesia, hipotonía, disfunción motora, atrofia muscular, pérdida de fuerza muscular, neuralgia, neuritis, neuropatía periférica, neurotoxicidad, parestesia, neuropatía periférica motora, neuropatía sensorimotora periférica, neuropatía sensitivo periférica, parálisis del nervio peroneo, polineuropatía, alteración sensitiva, sensación de ardor en piel).
e. Trastornos de la visión (diplopia, visión con efecto de halo, fotofobia, fotopsia, visión borrosa, agudeza visual disminuida, claridad visual, alteración de la visión, perseveración visual, moscas volantes).
f. Mareo (alteración del equilibrio, mareo, mareo postural, presíncope).
g. Bradicardia (bradicardia, frecuencia cardiaca disminuida, bradicardia sinusal).
h. Insuficiencia cardiaca (insuficiencia cardiaca, insuficiencia cardiaca congestiva, disminución de la fracción de eyección, insuficiencia ventricular izquierda, edema pulmonar). En los estudios clínicos (n=1.722), 19 (1,1%) pacientes en tratamiento con crizotinib presentaron algún grado de insuficiencia cardiaca, 8 (0,5%) pacientes presentaron grado 3 o 4, y en 3 (0,2%) pacientes el desenlace fue mortal.
i. Enfermedad pulmonar intersticial (síndrome de insuficiencia respiratoria aguda, alveolitis, enfermedad pulmonar intersticial, neumonitis).
j. Dolor abdominal (molestia abdominal, dolor abdominal, dolor en la zona inferior del abdomen, dolor en la zona superior del abdomen, dolor a la palpación abdominal).
k. Esofagitis (esofagitis, úlcera esofágica).
l. Perforación gastrointestinal (perforación gastrointestinal, perforación intestinal, perforación del intestino grueso).
m. Elevación de las transaminasas (aumento de la alanino aminotransferasa, aumento de la aspartato aminotransferasa, gamma glutamiltransferasa elevada, aumento de las enzimas hepáticas, función hepática anormal, prueba anormal de función hepática, transaminasas elevadas).
n. Quiste renal (absceso renal, quiste renal, hemorragia de quiste renal, infección de un quiste renal).
o. Aumento de la creatinina en sangre (aumento de la creatinina en sangre, disminución del aclaramiento renal de la creatinina).
p. Edema (edema facial, edema generalizado, hinchazón local, edema localizado, edema, edema periférico, edema periorbital).
q. Disminución de la testosterona en sangre (disminución de la testosterona en sangre, hipogonadismo, hipogonadismo secundario).
Descripción de determinadas reacciones adversas
Hepatotoxicidad
Se produjeron casos de hepatotoxicidad inducida por el medicamento con desenlace mortal en el 0,1% de los 1.722 pacientes tratados con crizotinib en ensayos clínicos. En menos del 1% de los pacientes tratados con crizotinib se han observado elevaciones concomitantes de la ALT y/o la AST > 3 veces el LSN y de la bilirrubina total > 2 veces el LSN sin elevaciones significativas de la fosfatasa alcalina (> 2 veces el LSN).
Se observaron elevaciones de la ALT o la AST a grado 3 ó 4 en 187 (11%) y 95 (6%) pacientes, respectivamente. Diecisiete (1%) pacientes precisaron una suspensión permanente del tratamiento asociada a la elevación de las transaminasas, lo que sugiere que dichos acontecimientos pudieron tratarse, en general, mediante las modificaciones de la dosis definidas en la tabla 2 (ver sección 4.2). En el estudio 1014 de fase III aleatorizado se observaron elevaciones de la ALT o la AST a grado 3 o 4 en el 15% y el 8% de los pacientes tratados con crizotinib frente al 2% y el 1% de los pacientes que recibieron quimioterapia. En el estudio 1007 de fase III aleatorizado se observaron elevaciones de la ALT o la AST a grado 3 o 4 en el 18% y el 9% de los pacientes que recibieron crizotinib y en el 5% y < 1% de los pacientes que recibieron quimioterapia.
En general, las elevaciones de las transaminasas se produjeron en los 2 primeros meses de tratamiento. En los estudios con crizotinib en pacientes con CPNM ALK-positivo o ROS1-positivo, la mediana de tiempo hasta la aparición de las elevaciones de las transaminasas de grado 1o 2 fue de 23 días. La mediana de tiempo hasta la aparición de las elevaciones de las transaminasas de grado 3 o 4 fue de 43 días.
Las elevaciones de las transaminasas de grado 3 y 4 generalmente fueron reversibles con la interrupción del tratamiento. En los estudios con crizotinib en pacientes con CPNM ALK-positivo o ROS1-positivo (N=1.722), se produjeron reducciones de la dosis asociadas con las elevaciones de las transaminasas en 76 (4%) pacientes. Diecisiete (1%) pacientes precisaron la suspensión permanente del tratamiento.
No debe utilizarse crizotinib en los pacientes con insuficiencia hepática grave (ver secciones 4.2, 4.3 y 4.4). Debe monitorizarse a los pacientes por si se produce hepatotoxicidad y someterlos al tratamiento recomendado en las secciones 4.2 y 4.4.
Efectos gastrointestinales
Los acontecimientos gastrointestinales atribuibles a cualquier causa comunicados con mayor frecuencia fueron náuseas (57%), diarrea (54%), vómitos (51%) y estreñimiento (43%). La mayoría de los acontecimientos fueron de gravedad leve a moderada. La mediana de tiempo hasta la aparición de náuseas y vómitos fue de 3 días y estos acontecimientos disminuyeron en frecuencia 3 semanas más tarde. El tratamiento de soporte debe incluir el uso de antieméticos. La mediana de tiempo hasta la aparición de diarrea y estreñimiento fue de 13 y 17 días, respectivamente. El tratamiento de soporte para la diarrea y el estreñimiento debe incluir el uso de antidiarreicos y laxantes convencionales, respectivamente.
En estudios clínicos con crizotinib se han notificado casos de perforación gastrointestinal. Se han notificado casos de perforación gastrointestinal con desenlace mortal durante el uso de crizotinib tras su comercialización (ver sección 4.4).
Prolongación del intervalo QT
En los estudios en pacientes con CPNM avanzado ALK-positivo o ROSl-positivo se registró un QTcF (QT corregido mediante el método de Fridericia) > 500 ms en 34 (2,1%) de 1.619 pacientes con al menos una evaluación electrocardiografía posbasal, y se observó un aumento máximo en el QTcF > 60 ms en relación con el momento basal en 79 (5,0%) de 1.585 pacientes con una evaluación electrocardiografía basal y al menos una evaluación posbasal. Se notificó una prolongación del intervalo electrocardiográfico QT de grado 3 o 4 atribuible a cualquier causa en 27 (1,6%) de 1.722 pacientes (ver secciones 4.2, 4.4, 4.5 y 5.2).
En un subestudio electrocardiográfico con un solo brazo (ver sección 5.2) en el que se emplearon mediciones electrocardiografías manuales con enmascaramiento, once (21%) pacientes presentaron un aumento del valor del QTcF > 30 y < 60 ms en relación con el momento basal y un (2%) paciente presentó un aumento del valor del QTcF > 60 ms en relación con el momento basal. Ningún paciente presentó un QTcF máximo > 480 ms. El análisis de tendencia central indicó que el mayor cambio promedio desde el momento basal en el QTcF fue de 12,3 ms (95% IC 5,1-19,5 ms media de mínimos cuadrados [MC] a partir del análisis de la varianza [ANOVA]) y se produjo 6 horas después de la administración de la dosis el día 1 del ciclo 2. Todos los límites superiores del IC del 90% para el cambio promedio de MC en el QTcF en relación con el momento basal en todos los puntos temporales del día 1 del ciclo 2 fueron <20 ms.
La prolongación del intervalo QT puede dar lugar a arritmias y es un factor de riesgo de muerte súbita. La prolongación del intervalo QT puede manifestarse clínicamente como bradicardia, mareo y síncope. Alteraciones electrolíticas, deshidratación y bradicardia pueden además aumentar el riesgo de prolongación del intervalo QTc y, por lo tanto, se recomienda una monitorización periódica del electrocardiograma y de los niveles de electrolitos en pacientes con toxicidad gastrointestinal (ver sección 4.4).
Bradicardia
En los estudios con crizotinib en pacientes con CPNM avanzado ALK-positivo o ROS1-positivo, 219 (13%) de 1.722 pacientes tratados con crizotinib sufrieron bradicardia atribuible a cualquier causa. La mayoría de los acontecimientos fueron de gravedad leve. Un total de 259 (16%) de 1.666 pacientes con al menos una evaluación posbasal de las constantes vitales presentaron una frecuencia cardiaca < 50 l.p.m.
Debe evaluarse cuidadosamente el empleo de medicamentos concomitantes asociados con la bradicardia. Los pacientes que padezcan bradicardia sintomática deben recibir el tratamiento recomendado en las secciones Modificación de la dosis y Advertencias y precauciones (ver secciones 4.2, 4.4 y 4.5).
Enfermedad pulmonar intersticial/neumonitis
Los pacientes en tratamiento con crizotinib pueden presentar enfermedad pulmonar intersticial (EPI)/neumonitis de carácter grave, potencialmente mortal o mortal. En los estudios en pacientes con CPNM ALK-positivo o ROS1-positivo (N=1.722), 50 (3%) pacientes tratados con crizotinib presentaron EPI atribuible a cualquier causa y de cualquier grado, incluyendo 18 (1%) pacientes con grado 3 o 4, y 8 (< 1%) pacientes con desenlace mortal. De acuerdo con la evaluación del Comité de Revisión Independiente de pacientes con CPNM ALK-positivo (N=1.669), 20 (1,2%) pacientes presentaron EPI/neumonitis, incluyendo 10 (<1%) pacientes con desenlace mortal. Por lo general, dichos casos se produjeron en los 3 meses siguientes al inicio del tratamiento. Deberá monitorizarse a los pacientes con síntomas pulmonares indicativos de EPI/neumonitis. Deben descartarse otras causas posibles de EPI/neumonitis (ver secciones 4.2 y 4.4).
Efectos sobre la visión
En los estudios clínicos con crizotinib en pacientes con CPNM avanzado ALK-positivo o ROS1-positivo (N=1.722), se ha notificado defecto del campo visual de grado 4 con pérdida de visión en 4 (0,2%) pacientes. La atrofia óptica y el trastorno del nervio óptico se han identificado como causas potenciales de la pérdida de visión (ver sección 4.4).
De 1.722 pacientes tratados con crizotinib, 1.084 (63%) presentaron trastornos de la visión, de todos los grados y atribuibles a cualquier causa, de los cuales los más frecuentes fueron alteración de la visión, fotopsia, visión borrosa y moscas volantes. De 1.084 pacientes que presentaron trastornos de la visión, el 95% presentaron acontecimientos de carácter leve. A 7 (0,4%) de los pacientes se les suspendió temporalmente el tratamiento y a 2 (0,1%) se les redujo la dosis debido a la alteración de la visión. No se produjeron suspensiones permanentes asociadas a la alteración de la visión en ninguno de los 1.722 pacientes tratados con crizotinib.
De acuerdo con el Cuestionario de Evaluación de los Síntomas Visuales (VSAQ-ALK), los pacientes tratados con crizotinib en el estudio 1007 y el estudio 1014 notificaron una incidencia de alteraciones visuales mayor que la notificada por los pacientes tratados con quimioterapia. Por lo general, la aparición de trastornos de la visión comenzó en la primera semana de la administración del fármaco. La mayoría de los pacientes del brazo de crizotinib de los estudios 1007 y 1014 en fase III aleatorizados (>50%) notificó alteraciones visuales, que se produjeron con una frecuencia de entre 4 y 7 días a la semana, tuvieron una duración de hasta 1 minuto y tuvieron un impacto leve o nulo (puntuaciones de entre 0 y 3 de una puntuación máxima de 10) en las actividades cotidianas, según lo recogido en el cuestionario VSAQ-ALK.
Se realizó un subestudio oftalmológico, en el que se emplearon evaluaciones oftalmológicas específicas en puntos temporales especificados, en 54 pacientes con CPNM que recibieron crizotinib 250 mg dos veces al día. Treinta y ocho (70,4%) de los 54 pacientes presentaron un acontecimiento adverso surgido durante el tratamiento y atribuible a cualquier causa perteneciente a la clase de órganos y sistemas de trastornos oculares, treinta de los cuales tenían una exploración oftalmológica. De estos treinta pacientes, en 14 (36,8%) de ellos se notificó una anormalidad oftalmológica de cualquier tipo y en 16 (42,1%) no se observó ningún hallazgo oftalmológico. Los hallazgos más frecuentes estaban relacionados con la biomicroscopia con lámpara de hendidura (21,1%), la fundoscopia (15,8%) y la agudeza visual (13,2%). En muchos pacientes se observaron anormalidades oftalmológicas preexistentes y trastornos médicos concomitantes que podrían coadyuvar a los hallazgos oculares, por lo que no se ha podido establecer una relación causal concluyente con crizotinib. No se observaron hallazgos relacionados con el recuento de células en el humor acuoso ni con el examen de la turbidez del humor acuoso de la cámara anterior. Ninguna de las alteraciones visuales asociadas a crizotinib pareció guardar relación con cambios en la mejor agudeza visual corregida, el vítreo, la retina ni el nervio óptico.
En pacientes con una nueva aparición de pérdida de visión de grado 4, se debe interrumpir el tratamiento con crizotinib y se debe llevar a cabo una evaluación oftalmológica. Se recomienda una evaluación oftalmológica si el trastorno de la visión persiste o empeora en cuanto a gravedad (ver secciones 4.2 y 4.4).
Efectos en el sistema nervioso
Cuatrocientos treinta y cinco (25%) de 1.722 pacientes tratados con crizotinib presentaron neuropatía atribuible a cualquier causa, tal y como se define en la tabla 3. En estos estudios también se notificó como muy frecuente disgeusia, principalmente con gravedad de grado 1.
Quiste renal
Cincuenta y dos (3%) de 1.722 pacientes tratados con crizotinib presentaron quistes renales complejos atribuibles a cualquier causa. En algunos pacientes se observó invasión quística local fuera del riñón. En el caso de los pacientes que presenten quistes renales, deberá considerarse una monitorización periódica con técnicas de diagnóstico por la imagen y análisis de orina.
Neutropenia y leucopenia
En los estudios en pacientes con CPNM avanzado ALK-positivo o ROS1-positivo (N=1.722) se observó neutropenia de grado 3 o 4 en 212 (12%) pacientes tratados con crizotinib. La mediana de tiempo hasta la aparición de neutropenia de cualquier grado fue de 89 días. La neutropenia se asoció a una reducción de la dosis o a una suspensión permanente del tratamiento en el 3% y < 1% de los pacientes, respectivamente. Menos del 0,5% de los pacientes presentó neutropenia febril en los estudios clínicos con crizotinib.
En los estudios en pacientes con CPNM avanzado ALK-positivo o ROS1-positivo (N=1.722) se observó leucopenia de grado 3 o 4 en 48 (3%) pacientes tratados con crizotinib. La mediana de tiempo hasta la aparición de leucopenia de cualquier grado fue de 85 días.
La leucopenia se asoció a una reducción de la dosis en < 0,5% de los pacientes y ningún paciente sufrió una suspensión permanente del tratamiento debido a la leucopenia.
En los estudios clínicos con crizotinib en pacientes con CPNM avanzado ALK-positivo o ROS1-positivo se observaron disminuciones en los leucocitos y los neutrófilos de grado 3 o 4 con frecuencias del 4% y del 13%, respectivamente.
Se realizarán hemogramas completos, con fórmula y recuento leucocítico, cuando esté clínicamente indicado y con una repetición más frecuente si se observan alteraciones de grado 3 ó 4, o en caso de fiebre o infección. Para los pacientes que presenten alteraciones del hemograma, ver sección 4.2.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
El tratamiento de una sobredosis de este medicamento consistirá en cuidados generales de apoyo. No hay un antídoto para XALKORI.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes antineoplásicos - inhibidor de la proteína tirosina-quinasa, código ATC: L01XE16.
Mecanismo de acción
Crizotinib es una molécula pequeña, inhibidor selectivo del receptor tirosina-quinasa (RTK) ALK y sus variantes oncogénicas (es decir, eventos de fusión de ALK y mutaciones seleccionadas de ALK). Crizotinib inhibe también la actividad tirosina-quinasa del receptor del factor de crecimiento de los hepatocitos (HGFR, c-Met), ROS1 (c-ros) y del receptor de Nantes (RON). Crizotinib demostró en ensayos bioquímicos una inhibición de la actividad quinasa de ALK, ROS1 y c-Met dependiente de la concentración, y en ensayos celulares inhibió la fosforilación y moduló fenotipos dependientes de quinasas. Crizotinib demostró una actividad inhibitoria del crecimiento, potente y selectiva, e indujo la apoptosis de líneas celulares tumorales que mostraban acontecimientos de fusión de ALK (tales como proteína 4 asociada al microtúbulo del equinodermo [EML4]-ALK y nucleofosmina [NPM]-ALK), acontecimientos de fusión de ROS1, o que mostraban amplificación del locus génico MET o ALK. Crizotinib demostró eficacia antitumoral, incluida una marcada actividad citorreductora, en ratones portadores de heteroinjertos tumorales que expresaban proteínas de fusión ALK. La eficacia antitumoral de crizotinib fue dependiente de la dosis y mostró una correlación con la inhibición farmacodinámica de la fosforilación de proteínas de fusión ALK (tales como EML4-ALK y NPM-ALK) en tumores in vivo. Crizotinib también demostró una notable actividad antitumoral en estudios de xenoinjerto en ratones, en los que los tumores se generaron usando un panel de líneas celulares
NIH-3T3 diseñadas para expresar fusiones de ROS1 clave identificadas en tumores humanos. La eficacia antitumoral de crizotinib fue dependiente de la dosis y mostró una correlación con la inhibición de la fosforilación de ROS1 in vivo.
Estudios clínicos
CPNM avanzado ALK-positivo sin tratamiento previo: estudio 1014 en fase III aleatorizado En el estudio 1014, aleatorizado, abierto y a escala mundial, se demostró la eficacia y la seguridad de crizotinib en el tratamiento de pacientes con CPNM metastásico ALK-positivo que no habían recibido un tratamiento sistémico previo para el cáncer avanzado.
La población total analizada estuvo compuesta por 343 pacientes con CPNM avanzado ALK-positivo identificado mediante hibridación fluorescente in situ (FISH) antes de la aleatorización: se aleatorizó a 172 pacientes a recibir crizotinib y a 171 pacientes a recibir quimioterapia (pemetrexed + carboplatino o cisplatino; hasta 6 ciclos de tratamiento). Las características demográficas y de la enfermedad de la población global del estudio fueron las siguientes: el 62% eran mujeres, la mediana de edad era de 53 años, el estado funcional del Grupo Oncológico Cooperativo de la Costa Este (ECOG) en el momento basal era de 0 o 1 (95%), el 51% era de raza blanca y el 46% asiática, el 4% fumaba, el 32% eran exfumadores y el 64% nunca había fumado. En cuanto a las características de la enfermedad de dicha población, el 98% de los pacientes presentaba metástasis, en el 92% de los pacientes los tumores presentaban histología de adenocarcinoma y el 27% de los pacientes presentaba metástasis cerebral.
Los pacientes pudieron continuar el tratamiento con crizotinib después de producirse progresión de la enfermedad, definida según los Criterios de evaluación de respuesta en tumores sólidos (RECIST), a criterio del investigador, si seguían experimentando una mejoría clínica. Sesenta y cinco de 89 (73%) pacientes tratados con crizotinib y 11 de 132 (8,3%) pacientes tratados con quimioterapia continuaron el tratamiento durante al menos 3 semanas después de la progresión objetiva de la enfermedad. Los pacientes aleatorizados a recibir quimioterapia tuvieron la posibilidad de cambiar al tratamiento con crizotinib cuando se produjo progresión de la enfermedad, definida según los criterios RECIST y confirmada mediante una revisión radiológica independiente (RRI). Ciento veinte (70%) pacientes del brazo de quimioterapia recibieron posteriormente tratamiento con crizotinib.
Crizotinib aumentó significativamente la supervivencia libre de progresión (PFS), el objetivo principal del estudio, en comparación con la quimioterapia, según la evaluación realizada mediante una RRI. La mejoría de la PFS obtenida con crizotinib fue homogénea entre los subgrupos de pacientes atendiendo a sus características basales, tales como la edad, el sexo, la raza, el hábito tabáquico, el tiempo transcurrido desde el diagnóstico, el estado funcional ECOG y la presencia de metástasis cerebral. En la tabla 4 se resumen los datos de la eficacia correspondientes al estudio 1014 en fase III aleatorizado y en las figuras 1 y 2 se muestran las curvas de Kaplan-Meier para la PFS y la supervivencia global (OS) (por sus siglas en inglés), respectivamente. Los datos de supervivencia global (OS) no eran maduros en el momento del análisis de la PFS.
analizada) en pacientes con CPNM avanzado ALK-positivo sin tratamiento previo
|
Parámetro de respuesta |
Crizotinib N=172 |
Quimioterapia N=171 |
|
Supervivencia libre de progresión (basada en una RRI) | ||
|
Número de pacientes con evento, n (%) |
100 (58%) |
137 (80%) |
|
Mediana de PFS en meses (IC del 95%) |
10,9 (8,3; 13,9) |
7,0a (6,8; 8,2) |
|
HR(IC del 95%)b |
0,45 (0,35; 0,60) | |
|
Valor de pc |
< 0,0001 | |
|
Supervivencia globald | ||
|
Número de muertes, n (%) |
44 (26%) |
46 (27%) |
|
Mediana de OS en meses (IC del 95%) |
NSA |
NSA |
|
HR (IC del 95%)b |
0,82 (0,54; 1,26) | |
|
Valor de pc |
0,1804 | |
|
Probabilidad de supervivencia a los 12 meses,d % (IC del 95%) |
83,5 (76,7; 88,5) |
78,6 (71,3; 84,2) |
|
Probabilidad de supervivencia a los 18 meses,d % (IC del 95%) |
68,6 (59,5; 76,1) |
67,3 (58,1; 74,9) |
|
Tasa de respuesta objetiva (ORR) (basada en una RRI) | ||
|
Tasa de respuesta objetiva % (IC del 95%) |
74% (67; 81) |
45%e (37; 53) |
|
Valor de pf |
< 0,0001 | |
|
Duración de la respuesta | ||
|
Meses8 (IC del 95%) |
11,3 (8,1; 13,8) |
5,3 (4,1; 5,8) |
Abreviaturas: IC = intervalo de confianza; HR = Hazard Ratio; RRI = revisión radiológica independiente; N/n = número de pacientes; NSA = no se alcanzó; PFS = supervivencia libre de progresión; OS = supervivencia global. a. Las medianas de PFS fueron de 6,9 meses (IC del 95%: 6,6; 8,3) para pemetrexed/cisplatino (HR = 0,49;
valor de p < 0,0001 para crizotinib en comparación con pemetrexed/cisplatino) y de 7,0 meses (IC del 95%: 5,9; 8,3) para pemetrexed/carboplatino (HR = 0,45; valor de p < 0,0001 para crizotinib en comparación con pemetrexed/carboplatino).
b. Basado en el análisis estratificado de riesgos proporcionales de Cox.
c. Basado en la prueba del orden logarítmico estratificada (unilateral).
d. El análisis de la OS no se ajustó para contemplar los posibles efectos de confusión derivados del cambio de grupo.
e. Las ORR fueron del 47% (IC del 95%: 37; 58) para pemetrexed/cisplatino (valor de p < 0,0001 en comparación con crizotinib) y del 44% (IC del 95%: 32; 55) para pemetrexed/carboplatino (valor de p < 0,0001 en comparación con crizotinib).
f. Basado en la prueba de Cochran-Mantel-Haenszel estratificada (bilateral).
g. Calculados utilizando el método de Kaplan-Meier.
Figura 1. Curvas de Kaplan-Meier para la supervivencia libre de progresión (basada en una RRI) por brazo de tratamiento en el estudio 1014 en fase III aleatorizado (población analizada) en pacientes con CPNM avanzado ALK-positivo sin tratamiento previo
• oo
80 -
Hazard Ratio = 0.45
p < 0,0001
_
XALKORI (N=172)
Mediana 10,9 meses
Quimioterapia (N=171)
Mediana 7.0 meses
Numero de pacientes con riesgo
XALKORI 172
Quimioterapia 171
Tiempo (meses)
Figura 2. Curvas de Kaplan-Meier para la OS por brazo de tratamiento en el estudio 1014 en fase III aleatorizado (población total analizada) en pacientes con CPNM avanzado ALK-positivo sin tratamiento previo
100
Numero de pacientes con riesgo XALKORI 172 Quimioterapia 171
XALKORI (N=172)
Mediana no alcanzada
Quimioterapia (N=171)
Mediana no alcanzada
Hazard Ratio
IC del 95% (0,54; 1,26)
0.1804
Tiempo (meses)
La mediana del tiempo hasta la progresión intracraneal (TTP-IC) en los pacientes que presentaban en la visita inicial metástasis cerebral previamente tratada fue de 15,7 meses en el brazo de crizotinib (N = 39) y de 12,5 meses en el brazo de quimioterapia (N = 40) (HR = 0,45 [IC del 95%: 0,19; 1,07]; valor de p unilateral = 0,0315). En el caso de los pacientes sin metástasis cerebral en la visita inicial, no se alcanzó la mediana de TTP-IC ni en el brazo de crizotinib (N = 132) ni en el de quimioterapia (N= 131) (HR = 0,69 [IC del 95%: 0,33; 1,45]; valor de p unilateral = 0,1617).
Los síntomas y la calidad de vida global notificados por los pacientes se recopilaron mediante el cuestionario EORTC QLQ-C30 y su módulo para el cáncer de pulmón (EORTC QLQ-LC13). Un total
de 166 pacientes del brazo de crizotinib y de 163 pacientes del brazo de quimioterapia habían contestado los cuestionarios EORTC QLQ-C30 y LC-13 en la visita inicial y al menos en una visita posterior a la inicial. La mejoría observada en la calidad de vida global fue significativamente mayor en el brazo de crizotinib que en el brazo de quimioterapia (diferencia global en el cambio en relación con las puntuaciones de la visita inicial: 13,8; valor de p < 0,0001).
El tiempo hasta el empeoramiento (THE) se definió previamente como la primera aparición de un aumento >10 puntos en las puntuaciones de los síntomas de dolor torácico, tos o disnea evaluados mediante el cuestionario EORTC QLQ-LC13.
En comparación con la quimioterapia, crizotinib produjo una mejoría de los síntomas al aumentar significativamente el THE (mediana de 2,1 meses frente a 0,5 meses; HR = 0,59; IC del 95%: 0,45; 0,77; valor de p bilateral del orden logarítmico ajustada por el método de Hochberg = 0,0005).
CPNM avanzado ALK-positivo previamente tratado: estudio 1007 en fase III aleatorizado En el estudio 1007, aleatorizado, abierto y a escala mundial, se demostró la eficacia y la seguridad de crizotinib en el tratamiento de pacientes con CPNM metastásico ALK-positivo que habían recibido un tratamiento sistémico previo para el cáncer avanzado.
La población total analizada estuvo compuesta por 347 pacientes con CPNM avanzado ALK-positivo identificado mediante hibridación fluorescente in situ (FISH) antes de la aleatorización. Se aleatorizó a 173 pacientes a recibir crizotinib y a 174 pacientes a recibir quimioterapia (pemetrexed o docetaxel). Las características demográficas y de la enfermedad de la población global del estudio fueron las siguientes: el 56% eran mujeres, la mediana de edad era de 50 años, el estado funcional ECOG en el momento basal era de 0 (39%) o 1 (52%), el 52% era de raza blanca y el 45% asiática, el 4% fumaba, el 33% eran exfumadores y el 63% nunca había fumado, el 93% presentaba metástasis y en el 93% los tumores presentaban histología de adenocarcinoma.
Los pacientes pudieron continuar con el tratamiento asignado después de producirse progresión de la enfermedad, definida según los criterios RECIST, a criterio del investigador, si se observaba que seguían experimentando una mejoría clínica. Cincuenta y ocho de los 84 (69%) pacientes tratados con crizotinib y 17 de los 119 (14%) pacientes tratados con quimioterapia continuaron el tratamiento durante al menos 3 semanas después de la progresión objetiva de la enfermedad. Los pacientes aleatorizados a recibir quimioterapia tuvieron la posibilidad de cambiar al tratamiento con crizotinib cuando se produjo progresión de la enfermedad, definida según los criterios RECIST y confirmada mediante una RRI. Ciento doce (64%) pacientes recibieron posteriormente tratamiento con crizotinib.
Crizotinib aumentó significativamente la PFS, el objetivo principal del estudio, en comparación con la quimioterapia, según la evaluación realizada mediante una RRI. El aumento de la PFS obtenida con crizotinib fue homogénea entre los subgrupos de pacientes atendiendo a sus características basales, tales como la edad, el sexo, la raza, el hábito tabáquico, el tiempo transcurrido desde el diagnóstico, el estado funcional ECOG, la presencia de metástasis cerebral y el tratamiento anterior con un TKI del EGFR.
En la tabla 5 se resumen los datos de eficacia correspondientes al estudio 1007 y en las figuras 3 y 4 se muestran las curvas de Kaplan-Meier para la PFS y la OS, respectivamente. Los datos sobre la supervivencia global (OS) no eran maduros en el momento del análisis de la PFS.
analizada) en pacientes con CPNM avanzado ALK-positivo previamente tratado
|
Parámetro de respuesta |
Crizotinib N=173 |
Quimioterapia N=174 |
|
Supervivencia libre de progresión (basada en una RRI) | ||
|
Número de pacientes con evento, n (%) |
100 (58%) |
127 (73%) |
|
Tipo de evento, n (%) | ||
|
Progresión tumoral |
84 (49%) |
119 (68%) |
|
Muerte sin progresión objetiva |
16 (9%) |
8 (5%) |
|
Mediana de PFS en meses (IC del 95%) |
7,7 (6,0; 8,8) |
3,0a (2,6; 4,3) |
|
HR(IC del 95%)b |
0,49(0,37; 0,64) | |
|
Valor de pc |
<0,0001 | |
|
Supervivencia globald | ||
|
Número de muertes, n (%) |
49 (28%) |
47 (27%) |
|
Mediana de OS en meses (IC del 95%) |
20,3 (18,1; NSA) |
22,8 (18,6; NSA) |
|
HR (IC del 95%)b |
1,02 (0,68; 1,54) | |
|
Valor de pc |
0,5394 | |
|
Probabilidad de supervivencia a los 6 meses,e % (IC del 95%) |
86,8 (80,4; 91,2) |
83,8 (77,0; 88,7) |
|
Probabilidad de supervivencia a 1 año,e % (IC del 95%) |
69,5 (60,6; 76,8) |
71,8 (63,3; 78,7) |
|
Tasa de respuesta objetiva (ORR) (basada en una RRI) | ||
|
Tasa de respuesta objetiva % (IC del 95%) |
65% (58; 72) |
20%f (14; 26) |
|
Valor de pg |
<0,0001 | |
|
Duración de la respuesta | ||
|
Medianae, meses (IC del 95%) |
7,4 (6,1; 9,7) |
5,6 (3,4; 8,3) |
Abreviaturas: IC = intervalo de confianza; HR = Hazard Ratio; RRI = revisión radiológica independiente; N/n = número de pacientes; NSA= no se alcanzó; PFS = supervivencia libre de progresión; OS = supervivencia global. a. Las medianas de PFS fueron de 4,2 meses (IC del 95%: 2,8; 5,7) para pemetrexed (HR = 0,59; valor de
p = 0,0004 para crizotinib en comparación con pemetrexed) y de 2,6 meses (IC del 95%: 1,6; 4,0) para docetaxel (HR = 0,30; valor de p < 0,0001 para crizotinib en comparación con docetaxel).
b. Basado en el análisis estratificado de riesgos proporcionales de Cox.
c. Basado en la prueba del orden logarítmico estratificada (unilateral).
d. Análisis intermedio de la OS realizado con el 40% de los eventos totales necesarios para el análisis final. El análisis de la OS no se ajustó para contemplar los posibles efectos de confusión derivados del cambio de grupo.
e. Calculada utilizando el método de Kaplan-Meier.
f. Las ORR fueron del 29% (IC del 95%: 21; 39) para pemetrexed (valor de p < 0,0001 en comparación con crizotinib) y del 7% (IC del 95%: 2; 16) para docetaxel (valor de p< 0,0001 en comparación con crizotinib).
g. Basado en la prueba de Cochran-Mantel-Haenszel estratificada (bilateral).
Figura 3. Curvas de Kaplan-Meier para la supervivencia libre de progresión (basada en una RRI) por brazo de tratamiento en el estudio 1007 en fase III aleatorizado (población total analizada) en pacientes con CPNM avanzado ALK-positivo previamente tratado
100
80-
60-
40-
XALKORI (N=173)
Mediana 7,7 meses
Quimioterapia (N=174)
Mediana 3.0 meses
'«A
20-
Hazard Ratio = 0,49
IC del 95% (0,37; 0,64)
Q.
o
p < 0,0001
Tiempo (meses)
173
174
Numero de pacientes con riesgo XALKORI Quimioterapia
Figura 4. Curvas de Kaplan-Meier para la supervivencia global por brazo de tratamiento en el estudio 1007 en fase III aleatorizado (población total analizada) en pacientes con CPNM avanzado ALK-positivo previamente tratado
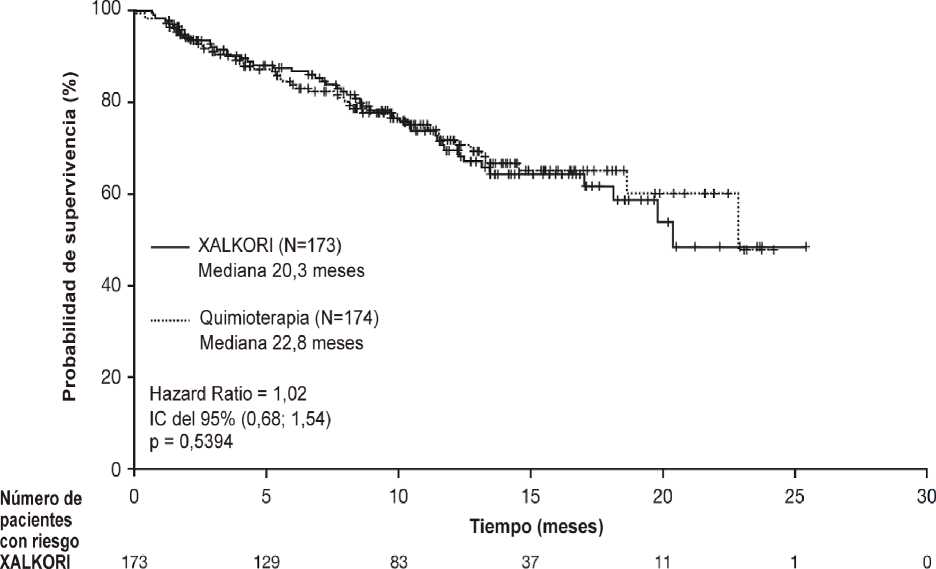
Quimioterapia 174 129 84 34 10 0
pacientes del brazo de crizotinib y de 151 pacientes del brazo de quimioterapia había contestado los cuestionarios EORTC QLQ-C30 y LC13 en la visita inicial y al menos en 1 visita posterior a la inicial.
Crizotinib produjo una mejoría de los síntomas al aumentar significativamente el tiempo hasta el empeoramiento (mediana de 4,5 meses frente a 1,4 meses) del dolor torácico, la disnea o la tos reportados por los pacientes, en comparación con la quimioterapia (HR 0,50; IC del 95%: 0,37; 0,66; valor de p bilateral del orden logarítmico ajustada por el método de Hochberg < 0,0001).
Crizotinib demostró una mejoría desde la visita inicial significativamente mayor que la quimioterapia en la alopecia (ciclos 2 a 15; valor de p < 0,05), la tos (ciclos 2 a 20; valor de p < 0,0001), la disnea (ciclos 2 a 20; valor de p < 0,0001), la hemoptisis (ciclos 2 a 20; valor de p < 0,05), el dolor en brazo u hombro (ciclos 2 a 20; valor de p < 0,0001), el dolor torácico (ciclos 2 a 20; valor de p < 0,0001) y el dolor en otras zonas (ciclos 2 a 20; valor de p < 0,05). Crizotinib produjo un empeoramiento desde la visita inicial significativamente menor que con la quimioterapia en la neuropatía periférica (ciclos 6 a 20; valor de p < 0,05), la disfagia (ciclos 5 a 11; valor de p < 0,05) y el dolor bucal (ciclos 2 a 20; valor de p < 0,05).
Crizotinib mejoró la calidad de vida global total, y se observó una mejoría desde la visita inicial significativamente mayor en el brazo de crizotinib que en el brazo de quimioterapia (ciclos 2 a 20; valor de p < 0,05).
Estudios de un solo brazo en pacientes con CPNM avanzado ALK-positivo
El uso de crizotinib en monoterapia en el tratamiento del CPNM avanzado ALK-positivo se investigó en dos estudios multinacionales, de un solo brazo de tratamiento (estudios 1001 y 1005). De los pacientes incluidos en estos estudios, los que se describen a continuación habían recibido tratamiento sistémico previo para enfermedad localmente avanzada o metastásica. La variable principal de eficacia en ambos estudios fue la tasa de respuesta objetiva (ORR, por sus siglas en inglés) con arreglo a los criterios RECIST.
Un total de 149 pacientes con CPNM avanzado ALK-positivo, entre los que se encontraban 125 pacientes con CPNM avanzado ALK-positivo previamente tratado, fueron incluidos en el estudio 1001 en el momento de la fecha del punto de corte para el análisis de la PFS y la ORR. Las características demográficas y de la enfermedad eran las siguientes: el 50% eran mujeres, la mediana de edad era de 51 años, el estado funcional ECOG en el momento basal era de 0 (32%) o 1 (55%), el 61% era de raza blanca y el 30% asiática, menos del 1% fumaba, el 27% eran exfumadores, el 72% nunca había fumado, el 94% tenía metástasis, y el 98% de los cánceres presentaba una histología de adenocarcinoma. La mediana de la duración del tratamiento fue de 42 semanas.
Un total de 934 pacientes con CPNM avanzado ALK-positivo recibieron tratamiento con crizotinib en el estudio 1005 en la fecha del corte de datos para el análisis de la PFS y la ORR. Las características demográficas y de la enfermedad eran las siguientes: el 57% eran mujeres, la mediana de edad era de 53 años, el estado funcional ECOG en el momento basal era de 0/1 (82%) o 2/3 (18%), el 52% era de raza blanca y el 44% asiática, el 4% fumaba, el 30% eran exfumadores, el 66% nunca había fumado, el 92% tenía metástasis, y el 94% de los cánceres presentaba una histología de adenocarcinoma. La mediana de duración del tratamiento de estos pacientes fue de 23 semanas. Los pacientes pudieron continuar con el tratamiento después de producirse progresión de la enfermedad, definida según los criterios RECIST, a criterio del investigador. Setenta y siete de 106 pacientes (73%) continuaron el tratamiento con crizotinib durante al menos 3 semanas después de la progresión objetiva de la enfermedad.
Tabla 6. Resultados de eficacia en CPNM avanzado ALK-positivo de los estudios 1001 y 1005
|
Parámetro de eficacia |
Estudio 1001 |
Estudio 1005 |
|
N=125a |
N=765a | |
|
Tasa de respuesta objetivab [% (intervalo de confianza, IC, del 95%)] |
60 (51; 69) |
48 (44; 51) |
|
Tiempo hasta la respuesta tumoral [mediana (intervalo)] en semanas |
7,9 (2,1; 39,6) |
6,1 (3; 49) |
|
Duración de la respuesta0 [mediana (IC del 95%)] en semanas |
48,1 (35,7; 64,1) |
47,3 (36; 54) |
|
Supervivencia libre de progresiónc [mediana (IC del 95%)] en meses |
9,2 (7,3; 12,7) |
7,8 (6,9; 9,5)d |
|
N=154e |
N=905e | |
|
Número de muertes, n (%) |
83 (54%) |
504 (56%) |
|
Supervivencia globalc (mediana [IC del 95%]) en meses |
28,9 (21,1; 40,1) |
21,5 (19,3; 23,6) |
Abreviaturas: IC = intervalo de confianza; N/n = número de pacientes.
a En las fechas de corte de datos de 1 de junio de 2011 (estudio 1001) y 15 de febrero de 2012 (estudio 1005). b No fueron evaluables para la respuesta 3 pacientes del estudio 1001 y 42 pacientes del estudio 1005. c Estimado con el método de Kaplan-Meier.
d Los datos de PFS del estudio 1005 se obtuvieron de 807 pacientes de la población de análisis de seguridad identificados mediante la prueba de FISH (fecha de corte de datos 15 de febrero de 2012). eEn la fecha de corte de datos del 30 de noviembre de 2013.
CPNM avanzado ROS1 -positivo
El uso de crizotinib en monoterapia en el tratamiento del CPNM avanzado ROS1-positivo se investigó en el estudio 1001, multicéntrico, multinacional, de un solo brazo. En el estudio se incluyeron un total de 53 pacientes con CPNM avanzado ROS1-positivo en el momento del corte de datos, incluyendo a 46 pacientes con CPNM avanzado ROS1-positivo previamente tratados y un número limitado de pacientes (N=7) que no habían recibido tratamiento sistémico previo. La variable principal de eficacia fue la ORR con arreglo a los criterios RECIST. Las variables secundarias incluyeron el tiempo hasta la respuesta tumoral, la duración de la respuesta, la PFS y la OS. Los pacientes recibieron 250 mg de crizotinib por vía oral dos veces al día.
Las características demográficas fueron las siguientes: el 57% eran mujeres, la mediana de edad era de 55 años, el estado funcional ECOG en el momento basal era de 0 o 1 (98%) o 2 (2%), el 57% era de raza blanca y el 40% asiática, el 25% eran exfumadores y el 75% nunca había fumado. En cuanto a las características de la enfermedad, el 91% presentaba metástasis, el 96% de los cánceres presentaba una histología de adenocarcinoma y el 13% no había recibido tratamiento sistémico previo para la enfermedad metastásica.
En el estudio 1001, se requería que los pacientes tuvieran CPNM avanzado ROS1-positivo para poder participar en el ensayo clínico. En la mayoría de los pacientes, el CPNM ROS1-positivo se identificó mediante la prueba de FISH. La mediana de duración del tratamiento fue de 101 semanas. Se observaron 5 respuestas completas y 32 respuestas parciales para una ORR del 70% (IC del 95%:
56%, 82%). No se alcanzó la mediana de la duración de la respuesta (IC del 95%: 15,2 meses, NSA). El 51% de las respuestas objetivas tumorales se alcanzaron durante las primeras 8 semanas de tratamiento. La mediana de PFS en el momento del corte de datos fue de 19,3 meses (IC del 95%:
14,8, NSA). Los datos de supervivencia global no eran maduros en el momento del corte de datos.
En la tabla 7 se presentan los datos de eficacia en los pacientes con CPNM avanzado ROSl-positivo del estudio 1001.
|
Tabla 7. Resultados de eficacia en CPNM avanzar |
o ROSl-positivo del estudio 1001 |
|
Parámetro de eficacia |
Estudio 1001 N=53a |
|
Tasa de respuesta objetiva [% (IC del 95%)] |
70 (56; 82) |
|
Tiempo hasta la respuesta tumoral [mediana (intervalo)] en semanas |
8 (4; 32) |
|
Duración de la respuestab [mediana (IC del 95%)] en semanas |
NSA (15,2; NSA) |
|
Supervivencia libre de progresiónb [mediana (IC del 95%)] en meses |
19,3 (14,8; NSA) |
Abreviaturas: IC = intervalo de confianza; N = número de pacientes, NSA = no se alcanzó.
a. En la fecha de corte de datos del 30 de noviembre de 2014.
b. Estimado con el método de Kaplan-Meier.
Pacientes sin histología de adenocarcinoma
En los estudios 1014 y 1007 en fase III aleatorizados se incluyeron, respectivamente, 21 pacientes con CPNM avanzado ALK-positivo sin histología de adenocarcinoma sin tratamiento previo y 12 previamente tratados. Los subgrupos en estos estudios eran demasiado pequeños para poder extraer conclusiones fiables. Cabe destacar que no se aleatorizaron pacientes con histología de CCE al brazo de crizotinib en el estudio 1007 ni se incluyeron pacientes con CCE en el estudio 1014 debido al empleo de un tratamiento con pemetrexed como comparador.
La información disponible procede de 45 pacientes evaluables para respuesta, con CPNM del tipo no adenocarcinoma (incluidos 22 pacientes con CCE) previamente tratado en el estudio 1005. Se observaron respuestas parciales en 20 de 45 pacientes con CPNM del tipo no adenocarcinoma y una ORR del 44% y en 9 de 22 pacientes con CPNM del tipo CCE y una ORR del 41%, ambas inferiores a las ORR observadas en el estudio 1005 (54%) para todos los pacientes.
Re-tratamiento con crizotinib
No se dispone de datos de seguridad y eficacia sobre el re-tratamiento con crizotinib en pacientes que lo habían recibido en líneas previas de tratamiento.
Pacientes de edad avanzada
De los 171 pacientes con CPNM ALK-positivo tratados con crizotinib en el estudio 1014 en fase III aleatorizado, 22 (13%) tenían 65 años de edad o más, y de los 109 pacientes ALK-positivo tratados con crizotinib que cambiaron desde el brazo de quimioterapia, 26 (24%) tenían 65 años de edad o más. De los 172 pacientes ALK-positivo tratados con crizotinib en el estudio 1007 en fase III aleatorizado, 27 (16%) tenían 65 años de edad o más. De los 154 y 1.063 pacientes con CPNM ALK-positivo en los estudios de un único brazo 1001 y 1005, 22 (14%) y 173 (16%) tenían 65 años de edad o más, respectivamente. En pacientes con CPNM ALK-positivo, la frecuencia de las reacciones adversas fue, por lo general, similar en los pacientes < 65 años de edad y en los pacientes > 65 años de edad, salvo en el caso del edema y el estreñimiento, que se notificaron con mayor frecuencia (diferencia > 15%) en el estudio 1014 en los pacientes tratados con crizotinib > 65 años de edad. No hubo pacientes > 85 años de edad en el brazo de crizotinib de los estudios 1007 y 1014 de fase III aleatorizados ni en el estudio 1005 con un único brazo de tratamiento. De los 154 pacientes del estudio 1001 con un único brazo de tratamiento, uno era ALK-positivo > 85 años de edad (ver también las secciones 4.2 y 5.2). De los 53 pacientes con CPNM ROS1-positivo en el estudio 1001 de un único brazo, 15 (28%) tenían 65 años de edad o mayores. No hubo pacientes ROS1-positivo > 85 años de edad en el estudio 1001.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con XALKORI en todos los grupos de la población pediátrica en el CPNM (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
Aprobación condicional
Este medicamento se ha autorizado con una «aprobación condicional».
Esta modalidad de aprobación significa que se espera obtener más información sobre este medicamento. La Agencia Europea de Medicamentos revisará la información nueva de este medicamento al menos una vez al año y esta Ficha Técnica o Resumen de las Características del Producto (RCP) se actualizará cuando sea necesario.
5.2 Propiedades farmacocinéticas
Absorción
Tras la administración de una dosis oral única en ayunas, crizotinib se absorbe con una mediana de tiempo hasta alcanzar las concentraciones máximas de 4 a 6 horas. Con la administración dos veces al día, se alcanzó el estado estacionario en un plazo de 15 días. Se determinó una biodisponibilidad absoluta de crizotinib del 43% tras la administración de una dosis oral única de 250 mg.
Una comida rica en grasas redujo los valores de AUCinf y Cmax de crizotinib en aproximadamente el 14% tras la administración de una dosis única de 250 mg en voluntarios sanos. Crizotinib puede administrarse con o sin comida (ver sección 4.2).
Distribución
La media geométrica del volumen de distribución (Vss) de crizotinib fue de 1772 litros tras la administración intravenosa de una dosis de 50 mg, lo que indica una amplia distribución desde plasma al interior de los tejidos.
La unión de crizotinib a las proteínas plasmáticas humanas in vitro es del 91% y es independiente de la concentración del medicamento. Los estudios in vitro sugieren que crizotinib es un sustrato de la glicoproteína P (gp-P).
Biotransformación
Los estudios in vitro demostraron que CYP3A4/5 fueron las principales enzimas involucradas en la eliminación metabólica de crizotinib. Las principales vías metabólicas en el ser humano fueron la oxidación del anillo de piperidina para formar crizotinib lactámico y la O-dealquilación, con una posterior conjugación de fase 2 de los metabolitos O-dealquilados.
Los estudios in vitro en microsomas hepáticos humanos demostraron que crizotinib es un inhibidor de CYP2B6 y CYP3A dependiente del tiempo (ver sección 4.5). Los estudios in vitro indicaron que es improbable que se produzcan interacciones medicamentosas clínicas como consecuencia de una inhibición mediada por crizotinib del metabolismo de medicamentos que son sustratos de CYP1A2, CYP2C8, CYP2C9, CYP2C19 o CYP2D6.
Estudios in vitro mostraron que crizotinib es un inhibidor débil de UGT1A1 y UGT2B7 (ver sección 4.5). No obstante, estudios in vitro indicaron que es improbable que se produzcan interacciones medicamentosas clínicas como consecuencia de la inhibición mediada por crizotinib del metabolismo de los medicamentos que son sustratos de UGT1A4, UGT1A6 o UGT1A9.
Estudios in vitro en hepatocitos humanos indicaron que es improbable que se produzcan interacciones medicamentosas clínicas como consecuencia de una inducción mediada por crizotinib del metabolismo de medicamentos que son sustratos de CYP1A2.
Eliminación
Tras la administración a pacientes de dosis únicas, la semivida terminal plasmática aparente de crizotinib fue de 42 horas.
Tras la administración a voluntarios sanos de una dosis única de 250 mg de crizotinib marcado radioactivamente, el 63% y el 22% de la dosis administrada se recuperó en heces y orina, respectivamente. El crizotinib sin modificar representó en heces y orina alrededor del 53% y el 2,3% de la dosis administrada, respectivamente.
Administración concomitante con medicamentos que son sustratos de transportadores
Crizotinib es un inhibidor de la glicoproteína P (gp-P) in vitro. En consecuencia, crizotinib podría incrementar las concentraciones plasmáticas de medicamentos que son sustratos de la gp-P si se administran de manera concomitante (ver sección 4.5).
Crizotinib es un inhibidor de OCT1 y OCT2 in vitro. Por consiguiente, crizotinib puede tener el potencial de aumentar las concentraciones plasmáticas de medicamentos administrados simultáneamente que sean sustratos de OCT1 u OCT2 (ver sección 4.5).
In vitro, crizotinib no inhibió las proteínas humanas transportadoras con actividad de captación hepática polipéptido de transporte de anión orgánico (OATP)1B1 u OATP1B3 ni las proteínas transportadoras de captación renal transportador de anión orgánico (OAT) 1 u OAT3 a concentraciones clínicamente relevantes. Por tanto, es improbable que se produzcan interacciones medicamentosas clínicas como consecuencia de una inhibición mediada por crizotinib de la captación hepática o renal de medicamentos que son sustratos de estos transportadores.
Efecto sobre otras proteínas transportadoras
In vitro, crizotinib no es un inhibidor de BSEP a concentraciones clínicamente relevantes. Farmacocinética en grupos especiales de pacientes
Insuficiencia hepática
Dado que crizotinib es ampliamente metabolizado en el hígado, es probable que el deterioro hepático aumente las concentraciones plasmáticas de crizotinib. Sin embargo, no se ha estudiado crizotinib en pacientes con alteración hepática. En los estudios clínicos realizados se excluyó a los pacientes con unos valores de ALT o AST > 2,5 veces el LSN, o > 5,0 veces el LSN si se debía a una neoplasia maligna subyacente, o con una bilirrubina total >1,5 veces el LSN (ver sección 4.2). El análisis farmacocinético de la población utilizando los datos procedentes de esos estudios indicaba que las concentraciones basales de bilirrubina total o de AST no tenían un efecto clínicamente significativo sobre la farmacocinética de crizotinib.
Insuficiencia renal
En los estudios 1001 y 1005 de un solo brazo se incluyeron pacientes con deterioro renal leve (CLcr igual o superior a 60 e inferior a 90 ml/min) y moderado (CLcr igual o superior a 30 e inferior a 60 ml/min). Se evaluó el efecto de la función renal, medida por el CLcr basal sobre las concentraciones mínimas de estado estacionario de crizotinib (Cmínima, ss) observadas. En el estudio 1001, la media geométrica ajustada de la Cmínima, ss plasmática en los pacientes con deterioro renal leve (N=35) y en los pacientes con deterioro moderado (N=8) fue respectivamente un 5,1% y un 11% más elevada que en los pacientes con función renal normal. En el estudio 1005, la media geométrica ajustada de la Cmínima, ss de crizotinib en el grupo con deterioro renal leve (N=191) y en el grupo de deterioro moderado (N=65) fue respectivamente un 9,1% y un 15% superior a la observada en pacientes con función renal normal. Además, los análisis farmacocinéticos de la población realizados utilizando datos de los estudios 1001, 1005 y 1007 indicaron que el CLcr no tenía un efecto clínicamente significativo sobre la farmacocinética de crizotinib. Debido al pequeño tamaño de los aumentos en la exposición a crizotinib (5%-15%), no se recomienda un ajuste de la dosis inicial para los pacientes con deterioro renal leve o moderado.
Tras una única dosis de 250 mg en sujetos con alteración renal grave (CLcr <30 ml/min) que no requerían diálisis peritoneal ni hemodiálisis, el AUC y la Cmax de crizotinib aumentaron en un 79% y un 34% respectivamente, en comparación con aquellos cuya función renal era normal. Se recomienda un ajuste de la dosis de crizotinib cuando se administre a pacientes con alteración renal grave que no requieran diálisis peritoneal ni hemodiálisis (ver secciones 4.2 y 4.4).
Edad
Según el análisis farmacocinético poblacional de los datos procedentes de los estudios 1001, 1005 y 1007, la edad no tiene efecto sobre la farmacocinética de crizotinib (ver secciones 4.2 y 5.1).
Peso corporal y sexo
Según el análisis farmacocinético poblacional de los datos procedentes de los estudios 1001, 1005 y 1007, no hubo un efecto clínicamente significativo del peso corporal o del sexo sobre la farmacocinética de crizotinib.
Etnia
Según el análisis farmacocinético poblacional de los datos procedentes de los estudios 1001, 1005 y 1007, el AUC prevista en estado estacionario (IC del 95%) fue entre un 23% y un 37% mayor en los pacientes asiáticos (N=523) que en los no asiáticos (N=691).
En los estudios en pacientes con CPNM avanzado ALK-positivo (N=1.669) se notificaron las siguientes reacciones adversas con una diferencia absoluta > 10% entre los pacientes asiáticos (N=753) y los no asiáticos (N=916): elevación de las transaminasas, disminución del apetito, neutropenia y leucopenia. No se notificaron reacciones adversas al fármaco con una diferencia absoluta > 15%.
Geriatría
Los datos disponibles en este subgrupo de pacientes son limitados (ver secciones 4.2 y 5.1). Según el análisis farmacocinético poblacional de los datos procedentes de los estudios 1001, 1005 y 1007, la edad no tiene efecto sobre la farmacocinética de crizotinib.
Electrofisiología cardiaca
El potencial de crizotinib para prolongar el intervalo QT se evaluó en los pacientes con CPNM ALK-positivo o ROS1-positivo que recibieron 250 mg de crizotinib dos veces al día. Se obtuvieron electrocardiogramas consecutivos por triplicado después de una dosis única y en estado estacionario, con el fin de evaluar el efecto de crizotinib sobre los intervalos QT. Se observó que 34 de 1.619 pacientes (2,1%) con al menos una evaluación electrocardiografía posbasal presentaban un QTcF > 500 ms, y 79 de 1.585 pacientes (5,0%) con una evaluación electrocardiografía basal y al menos una posbasal presentaban un aumento respecto al valor basal de QTcF > 60 ms, según lectura automática del aparato electrocardiográfico (ver sección 4.4).
Se realizó un subestudio electrocardiográfico, en el que se emplearon mediciones electrocardiográficas manuales con enmascaramiento, en 52 pacientes con CPNM ALK-positivo que recibieron 250 mg de crizotinib dos veces al día. Once (21%) pacientes presentaron un aumento del valor del QTcF >30 y <60 ms en relación con el momento basal y un paciente (2%) presentó un aumento del valor del QTcF >60 ms en relación con el momento basal. Ningún paciente presentó un QTcF máximo >480 ms. El análisis de tendencia central indicó que todos los límites superiores del IC del 90% para el cambio promedio de MC en el QTcF en relación con el momento basal en todos los puntos temporales del día 1 del ciclo 2 fueron <20 ms. Un análisis de farmacocinética/farmacodinamia sugirió una relación entre la concentración plasmática de crizotinib y el QTc. Además, se concluyó que la disminución de la frecuencia cardiaca estaba asociada con el aumento de la concentración plasmática de crizotinib (ver sección 4.4), con una reducción promedio máxima de 17,8 latidos por minuto (l.p.m.) a las 8 horas el día 1 del ciclo 2.
5.3 Datos preclínicos sobre seguridad
En los estudios de toxicidad a dosis repetidas en ratas y perros de hasta 3 meses de duración, los efectos principales en órganos afectados estuvieron relacionados con el sistema gastrointestinal (emesis, alteraciones de las heces, congestión), sistema hematopoyético (hipocelularidad de la médula ósea), sistema cardiovascular (bloqueo mixto de canales iónicos, disminución de la frecuencia cardiaca y la presión arterial, aumento de la presión telediastólica de ventrículo izquierdo y de los intervalos QRS y PR, y disminución de la contractilidad miocárdica) o sistema reproductor (degeneración de espermatocitos paquitenos, necrosis unicelular de folículos ováricos). El nivel sin efectos adversos observados (NOAEL, por sus siglas en inglés) de estos hallazgos fue o bien subterapéutico o bien de hasta 2,6 veces la exposición clínica en humanos según el AUC. También se observaron efectos sobre la función hepática (elevación de las transaminasas hepáticas) y la función retiniana, y la posibilidad de fosfolipidosis en múltiples órganos sin efectos tóxicos correspondientes.
Crizotinib no fue mutagénico in vitro en el ensayo de mutación inversa bacteriana (Ames). Fue aneugénico en un ensayo in vitro de micronúcleos de células de ovario de hámster chino y en un ensayo in vitro de aberraciones cromosómicas en linfocitos humanos. A concentraciones citotóxicas, se observó un ligero aumento de aberraciones cromosómicas estructurales en linfocitos humanos. El NOAEL de aneugenicidad fue de aproximadamente 1,8 veces la exposición clínica en humanos según el AUC.
No se han realizado estudios de carcinogenicidad con crizotinib.
No se han efectuado estudios específicos con crizotinib en animales para evaluar el efecto sobre la fertilidad; sin embargo, se considera que crizotinib puede afectar la función reproductora y la fertilidad de los seres humanos, a juzgar por las observaciones de los estudios de toxicidad a dosis repetidas en ratas. Los hallazgos observados en el aparato reproductor masculino consistieron en degeneración de espermatocitos paquitenos testiculares en ratas que recibieron >50 mg/kg/día durante 28 días (aproximadamente 1,1 veces la exposición clínica en humanos según el AUC). Los hallazgos en el aparato reproductor femenino consistieron en necrosis unicelular de folículos ováricos en ratas que recibieron 500 mg/kg/día durante 3 días.
Crizotinib no demostró ser teratogénico en ratas y conejas gestantes. La pérdida postimplantación aumentó a dosis >50 mg/kg/día (aproximadamente 0,4 veces el AUC de la dosis recomendada en humanos) en ratas, y la disminución del peso corporal fetal se consideró un efecto adverso en ratas y conejas tratadas con 200 y 60 mg/kg/día, respectivamente (aproximadamente 1,2 veces la exposición clínica en humanos según el AUC).
Se observó una disminución de la formación ósea en los huesos largos en crecimiento de ratas inmaduras tratadas con 150 mg/kg/día después de un tratamiento diario durante 28 días (aproximadamente 3,3 veces la exposición clínica humana según el AUC). No se han evaluado en animales jóvenes otros efectos tóxicos de posible significación para los pacientes pediátricos.
Los resultados de un estudio de fototoxicidad in vitro demostraron que crizotinib podría tener potencial fototóxico.
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Contenido de la cápsula Sílice coloidal anhidra Celulosa microcristalina Hidrogenofosfato de calcio anhidro Carboximetilalmidón sódico (tipo A)
Estearato de magnesio
Cápsula
Gelatina
Dióxido de titanio (E171)
Óxido de hierro rojo (E172)
Tinta de impresión
Shellac
Propilenglicol
Hidróxido de potasio
Óxido de hierro negro (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase XALKORI200 mg cápsulas duras
Frascos de polietileno de alta densidad (HDPE) con un cierre de polipropileno conteniendo 60 cápsulas duras.
Blísteres de PVC-aluminio conteniendo 10 cápsulas duras.
Cada envase contiene 60 cápsulas duras.
XALKORI 250 mg cápsulas duras
Frascos de polietileno de alta densidad (HDPE) con un cierre de polipropileno conteniendo 60 cápsulas duras.
Blísteres de PVC-aluminio conteniendo 10 cápsulas duras.
Cada envase contiene 60 cápsulas duras.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
XALKORI200 mg cápsulas duras
EU/1/12/793/001
EU/1/12/793/002
XALKORI 250 mg cápsulas duras
EU/1/12/793/003
EU/1/12/793/004
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 23/octubre/2012 Fecha de la última renovación: 29/julio/2016
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
ANEXO II
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POSTAUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN CONDICIONAL
Nombre y dirección del (de los) fabricante(s) responsable(s) de la liberación de los lotes
Pfizer Manufacturing Deutschland GmbH Mooswaldallee 1 79090 Freiburg Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Medidas adicionales de minimización de riesgos
Antes del lanzamiento del producto en cada Estado Miembro, el TAC deberá acordar el contenido y formato de los materiales educacionales con las Autoridades Nacionales Competentes. La redacción final que se utilice en los materiales educacionales debe estar alineada con la información aprobada para el producto.
El TAC debe asegurar que, en el lanzamiento y posteriormente, todos los profesionales sanitarios que se espere que vayan a utiliar y/o prescribir XALKORI dispongan de un material informativo.
El material informativo debe contener lo siguiente:
1. Resumen de las Características del Producto y Prospecto.
2. Material informativo para profesionales sanitarios.
3. Carpeta para el paciente incluyendo una Tarjeta de Información para el Paciente (texto acordado por el CHMP).
El material informativo para los profesionales sanitarios debe contener los siguientes elementos clave:
• XALKORI prolonga el intervalo QTc lo que puede conducir a un incremento en el riesgo de taquiarritmias ventriculares (por ejemplo, Torsade de Pointes) o muerte súbita.
• El riesgo de prolongación del intervalo QTc puede aumentar en pacientes en tratamiento concomitante con antiarrítmicos y en pacientes con enfermedad cardiaca relevante pre-existente, bradicardia o alteraciones electrolíticas (por ejemplo, secundaria a diarrea o vómitos).
• XALKORI debe administrarse con precaución en pacientes:
a. Que tengan antecedentes o predisposición a la prolongación del intervalo QTc.
b. Que estén recibiendo medicamentos con un efecto conocido de prolongación del intervalo QT.
• Cuando se utilice XALKORI en estos pacientes, deberá realizarse un control periódico mediante electrocardiograma y determinación de electrolitos.
• Los pacientes que desarrollen una prolongación del intervalo QTc de Grado 3, deben interrumpir el tratamiento con XALKORI hasta recuperación a grado < 1, luego reiniciar con 200 mg dos veces al día.
• Los pacientes que desarrollen una prolongación del intervalo QTc de Grado 4, deben interrumpir el tratamiento con XALKORI de forma permanente.
• Que XALKORI puede causar trastornos de la visión, incluyendo defecto del campo visual de Grado 4 con pérdida de visión. Sin embargo, los trastornos de la visión observados con más frecuencia fueron diplopia, fotopsia, visión borrosa, alteración visual y moscas volantes y en la mayor parte de los casos fueron leves en cuanto a su severidad.
• En los pacientes con trastornos visuales se debe considerar una evaluación oftalmológica si dichos trastornos persisten o empeoran en cuanto a su gravedad. En el caso de pacientes que experimenten pérdida visual grave (mejor agudeza visual corregida inferior a 6/60 en uno o en ambos ojos), se debe interrumpir el tratamiento con XALKORI y se ha de realizar una adecuada evaluación. XALKORI puede causar hepatotoxicidad, bradicardia sintomática (por ejemplo, síncope, mareo, hipotensión), enfermedad pulmonar intersticial/neumonitis, neutropenia y leucopenia, quiste renal, edema, neuropatía y toxicidad reproductiva. Se proporcionarán recomendaciones sobre la forma de mitigar dichos riesgos mediante una monitorización y un tratamiento adecuados.
• Debe evitarse el uso concomitante de XALKORI con fuertes inhibidores/inductores del CYP3A4 y sustratos del CYP3A4 con estrecho margen terapéutico.
• Que XALKORI podría causar perforación gastrointestinal. Por tanto, el medicamento se debe utilizar con precaución en pacientes con riesgo de desarrollar perforación gastrointestinal; y suspenderlo en pacientes que experimenten esta reacción adversa. Los pacientes deben estar informados acerca de los primeros signos de perforación gastrointestinal y se les debe indicar que han de consultar rápidamente con su médico en caso de que les ocurra.
• Que XALKORI podría causar fallo cardiaco grave, mortal o potencialmente mortal. Por tanto, los pacientes con o sin trastornos cardiacos pre-existentes, en tratamiento con crizotinib, deben ser monitorizados en cuanto a los signos y síntomas de fallo cardiaco. Si tales síntomas se observan, se debe considerar la interrupción del tratamiento, la reducción de la dosis o la suspensión, en función de lo que sea más adecuado.
• La necesidad de asesorar a los pacientes sobre los riesgos relativos a la administración de XALKORI e informarles de los signos y síntomas que tienen que conocer y las acciones a tomar.
• El papel y el uso de la Tarjeta de Información para el Paciente.
Al ser esta una autorización de comercialización condicional y según lo que establece el Artículo 14(7) del Reglamento (CE) 726/2004, el TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
Descripción |
Fecha límite |
|
Se solicita que el TAC actualice el estado de OS del estudio A8081007 y facilite los datos definitivos en un plazo de 9 meses desde que se hayan alcanzado los 238 acontecimientos de OS necesarios. El CSR debe incluir también un análisis detallado de la seguridad. |
30 de junio de 2016 |
ANEXO III
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
XALKORI200 mg cápsulas duras Crizotinib
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene 200 mg de crizotinib
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
60 cápsulas duras
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/793/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
XALKORI 200 mg
1. NOMBRE DEL MEDICAMENTO
XALKORI200 mg cápsulas duras Crizotinib
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene 200 mg de crizotinib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
60 cápsulas duras
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/793/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
XALKORI 200 mg
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLÍSTER
1. NOMBRE DEL MEDICAMENTO
XALKORI200 mg cápsulas duras Crizotinib
|
2. |
NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN | ||
|
Pfizer Ltd |
(en forma de logo del titular de la autorización de comercialización) | ||
|
3. |
FECHA DE CADUCIDAD | ||
|
EXP | |||
|
4. |
NÚMERO DE LOTE | ||
|
Lot | |||
|
5. |
OTROS | ||
1. NOMBRE DEL MEDICAMENTO
XALKORI250 mg cápsulas duras Crizotinib
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene 250 mg de crizotinib
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
60 cápsulas duras
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/793/004
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
XALKORI 250 mg
1. NOMBRE DEL MEDICAMENTO
XALKORI250 mg cápsulas duras Crizotinib
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene 250 mg de crizotinib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
60 cápsulas duras
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/793/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
XALKORI 250 mg
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLÍSTER
1. NOMBRE DEL MEDICAMENTO
XALKORI250 mg cápsulas duras Crizotinib
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Ltd (en forma de logo del titular de la autorización de comercialización)
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
B. PROSPECTO
Prospecto: información para el usuario
XALKORI 200 mg cápsulas duras XALKORI 250 mg cápsulas duras
Crizotinib
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es XALKORI y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar XALKORI
3. Cómo tomar XALKORI
4. Posibles efectos adversos
5. Conservación de XALKORI
6. Contenido del envase e información adicional
1. Qué es XALKORI y para qué se utiliza
XALKORI es un medicamento contra el cáncer, que contiene crizotinib como principio activo, usado para tratar adultos con un tipo de cáncer de pulmón denominado cáncer de pulmón no microcítico, que tiene una determinada alteración o defecto en un gen denominado quinasa del linfoma anaplásico (ALK) o en un gen llamado ROS1.
XALKORI le puede ser prescrito para el tratamiento inicial si su cáncer de pulmón está en una fase avanzada.
XALKORI le puede ser prescrito si su enfermedad está en una fase avanzada y el tratamiento previo no ha ayudado a detener su enfermedad.
XALKORI puede retardar o detener el crecimiento del cáncer de pulmón. Esto puede ayudar a que el tumor se reduzca.
Si tiene alguna duda sobre cómo actúa XALKORI o por qué le ha sido recetado, consulte a su médico.
2. Qué necesita saber antes de empezar a tomar XALKORI No tome XALKORI
• Si es alérgico al crizotinib o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6, "Composición de XALKORI").
• Si tiene una enfermedad grave en el hígado.
Advertencias y precauciones
Consulte a su médico antes de empezar a tomar XALKORI:
• Si alguna vez ha tenido una enfermedad de tipo leve o moderada en el hígado.
• Si alguna vez ha tenido cualquier otro problema en el pulmón. Algunos problemas en el pulmón pueden empeorar durante el tratamiento con XALKORI, ya que XALKORI puede causar inflamación de los pulmones durante el tratamiento. Estos síntomas pueden ser similares a los del cáncer de pulmón. Consulte a su médico de inmediato si tiene un síntoma nuevo o si empeora alguno de los síntomas incluyendo dificultad al respirar, falta de aliento, o tos con o sin mucosidad, o fiebre.
• Si después de realizarle un electrocardiograma (ECG), le han informado de que tiene una alteración en el corazón conocida como prolongación del intervalo QT.
• Si tiene disminución de la frecuencia cardiaca.
• Si ha tenido alguna vez problemas estomacales o intestinales, como orificios (perforación), o ha padecido enfermedades que provoquen inflamación en el interior del abdomen (diverticulitis) o si el cáncer se ha extendido al abdomen (metástasis).
• Si tiene alteraciones en la visión (ve flashes de luz, visión borrosa o visión doble).
• Si tiene una enfermedad grave en el riñón.
• Si está siendo tratado actualmente con cualquier otro medicamento incluido en la sección Toma de XALKORI con otros medicamentos.
Hable con su médico de inmediato tras haber tomado XALKORI:
• Si experimenta intenso dolor de estómago o abdominal, fiebre, escalofríos, falta de aliento, pulso acelerado, pérdida de visión parcial o completa (en uno o ambos ojos) o cambios del hábito intestinal.
La mayor parte de la información disponible es para pacientes con alguno de los tipos específicos de la histología del carcinoma de pulmón no microcítico (adenocarcinoma) ALK-positivo, mientras que la información disponible para otras histologías es limitada.
Niños y adolescentes
No se recomienda el tratamiento de niños y adolescentes con este medicamento. La indicación no incluye a niños y adolescentes.
Toma de XALKORI con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento, incluyendo plantas medicinales y medicamentos adquiridos sin receta.
En particular, los siguientes medicamentos pueden aumentar el riesgo de efectos adversos con XALKORI:
• Claritromicina, telitromicina, troleandomicina, antibióticos utilizados para tratar infecciones por bacterias.
• Ketoconazol, itraconazol, voriconazol, utilizados para tratar infecciones por hongos.
• Atazanavir, indinavir, nelfinavir, ritonavir, saquinavir, utilizados para tratar infecciones por VIH/SIDA.
Los siguientes medicamentos pueden disminuir la eficacia de XALKORI:
• Fenitoína, carbamacepina o fenobarbital, antiepilépticos utilizados para tratar convulsiones o ataques epilépticos.
• Rifabutina, rifampicina, utilizados para el tratamiento de la tuberculosis.
• Hierba de San Juan (Hypericum perforatum), una planta medicinal utilizada para tratar la depresión.
XALKORI puede aumentar los efectos adversos asociados a los siguientes medicamentos:
• Alfentanilo y otros opiáceos de corta duración como el fentanilo (analgésicos utilizados para procedimientos quirúrgicos).
• Quinidina, digoxina, disopiramida, amiodarona, sotalol, dofetilida, ibutilida, verapamilo, diltiazem, utilizados para tratar enfermedades del corazón.
• Medicamentos para la hipertensión denominados betabloqueantes, como atenolol, propranolol, labetolol.
• Pimozida, utilizada para tratar enfermedades mentales.
• Metformina, utilizada para tratar la diabetes.
• Procainamida, utilizada para tratar las arritmias cardiacas.
• Cisaprida, utilizada para tratar enfermedades gástricas.
• Ciclosporina, sirolimus y tacrolimus, utilizados en pacientes trasplantados.
• Alcaloides ergóticos (por ejemplo, ergotamina, dihidroergotamina) utilizados para tratar migrañas.
• Dabigatrán, anticoagulante utilizado para reducir la coagulación de la sangre.
• Colchicina, utilizada para tratar la gota.
• Pravastatina, utilizada para reducir los niveles de colesterol.
• Clonidina, guanfacina, utilizadas para tratar la hipertensión.
• Mefloquina, utilizada para la prevención de la malaria.
• Pilocarpina, utilizada para tratar el glaucoma (enfermedad grave del ojo).
• Anticolinesterasas, utilizadas para restaurar la función muscular.
• Antipsicóticos, utilizados para tratar enfermedades mentales.
• Moxifloxacino, utilizado para tratar infecciones por bacterias.
• Metadona, utilizada para tratar el dolor y para el tratamiento de la dependencia de opiáceos.
• Bupropión, utilizado para tratar la depresión y para dejar de fumar.
• Efavirenz, raltegravir, utilizados para tratar la infección por VIH.
• Irinotecán, un medicamento quimioterápico utilizado para el tratamiento del cáncer de colon y recto.
• Morfina, utilizada para tratar el dolor agudo y por cáncer.
• Naloxona, utilizado para el tratamiento de la adicción a opiáceos y su retirada.
Estos medicamentos se deben evitar durante el tratamiento con XALKORI.
Anticonceptivos orales
Si está tomando anticonceptivos orales mientras toma XALKORI, los anticonceptivos orales podrían ser ineficaces.
Toma de XALKORI con alimentos y bebidas
XALKORI puede tomarse con o sin alimentos; sin embargo, debe evitar beber zumo de pomelo o comer pomelo mientras esté en tratamiento con XALKORI, ya que pueden alterar las cantidades de XALKORI en su cuerpo.
Embarazo y lactancia
Si está embarazada, puede quedarse embarazada o está en periodo de lactancia, consulte con su médico o farmacéutico antes de tomar este medicamento.
Se recomienda que las mujeres eviten quedarse embarazadas y que los hombres no sean padres durante el tratamiento con XALKORI, ya que este medicamento puede dañar al feto. Se deberá usar un método anticonceptivo adecuado durante el tratamiento y durante al menos 90 días después de haber completado el tratamiento, si hubiera alguna posibilidad de que la persona que está utilizando este medicamento se quede embarazada o conciba un hijo, ya que los anticonceptivos orales podrían ser ineficaces mientras toma XALKORI.
No dé el pecho durante el tratamiento con XALKORI. XALKORI podría dañar al bebé lactante.
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Conducción y uso de máquinas
Tenga especial cuidado cuando conduzca o utilice máquinas, ya que los pacientes en tratamiento con XALKORI pueden experimentar trastornos visuales, mareos y cansancio.
3. Cómo tomar XALKORI
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
• La dosis recomendada es una cápsula de 250 mg, vía oral, dos veces al día (cantidad total 500 mg).
• Tome una cápsula por la mañana y otra por la tarde.
• Tome las cápsulas aproximadamente a las mismas horas cada día.
• Puede tomar las cápsulas con o sin comida evitando siempre el pomelo.
• Las cápsulas deben tragarse enteras, sin aplastarlas, disolverlas ni abrirlas.
Si es preciso, su médico puede reducir la dosis a 200 mg tomados por vía oral dos veces al día (cantidad total 400 mg) y si se necesitase una nueva reducción, se administrarían 250 mg por vía oral una vez al día.
Si toma más XALKORI del que debe
Si de forma accidental toma más cápsulas, consulte de inmediato con su médico o farmacéutico. Puede requerir atención médica.
Si olvidó tomar XALKORI
El modo de proceder si olvidó tomar una cápsula, depende de cuánto tiempo falte hasta la siguiente dosis:
• Si la siguiente dosis es dentro de 6 horas o más, tome la cápsula olvidada lo antes posible. Después tome la siguiente cápsula a la hora habitual.
• Si la siguiente dosis es en menos de 6 horas, no tome la cápsula olvidada. Después tome la siguiente cápsula a la hora habitual.
Informe a su médico de la dosis olvidada en su siguiente visita.
No tome una dosis doble (dos cápsulas al mismo tiempo) para compensar la cápsula olvidada.
Si vomita tras tomar una dosis de XALKORI, no tome una dosis adicional; tome la dosis siguiente a la hora habitual.
Si interrumpe el tratamiento con XALKORI
Es importante que tome XALKORI todos los días, durante el tiempo que su médico se lo haya recetado. Si no es capaz de tomar este medicamento tal y como su médico se lo ha recetado, o cree que ya no lo necesita más, contacte inmediatamente con su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Si experimenta efectos adversos, consulte con su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto.
Algunos efectos adversos pueden ser graves. Usted deberá contactar inmediatamente con su médico si experimenta alguno de los siguientes efectos adversos graves (ver también sección 2 “Qué necesita saber antes de empezar a tomar XALKORI”):
• Insuficiencia hepática
Consulte con su médico inmediatamente si se siente más cansado de lo habitual, si su piel y las zonas blancas de sus ojos se vuelven amarillas, si su orina se vuelve oscura o marrón (color té), si tiene náuseas, vómitos, o menos apetito, si le duele la parte derecha del estómago, o si tiene picores o si le salen moratones con más facilidad de lo normal. Su médico podrá hacerle análisis de sangre para comprobar su función hepática, y si los resultados de estos análisis fueran anormales, podría reducir la dosis de XALKORI o suspender el tratamiento.
• Inflamación del pulmón
Consulte con su médico inmediatamente si experimenta dificultad para respirar, especialmente si está asociada con tos o fiebre.
• Reducción en el número de células blancas de la sangre (incluyendo neutrófilos)
Consulte con su médico inmediatamente si experimenta fiebre o infección. Su médico puede hacerle análisis de sangre y si los resultados fueran anómalos, su médico puede tomar la decisión de reducir la dosis de XALKORI.
• Sensación de mareo, desmayo o dolor en el pecho
Consulte con su médico inmediatamente si padece alguno de estos síntomas ya que podrían ser signos de cambios en la actividad eléctrica (se observan en un electrocardiograma) o de un ritmo anormal del corazón. Su médico podría hacerle electrocardiogramas para comprobar que no hay problemas en su corazón durante el tratamiento con XALKORI.
• Pérdida de visión parcial o completa en uno o ambos ojos
Consulte a su médico inmediatamente si experimenta pérdida de visión o algún cambio en la visión, como dificultad para ver con uno o ambos ojos. Su médico puede interrumpir el tratamiento con XALKORI y derivarlo a un oftalmólogo.
Otros efectos adversos con XALKORI pueden incluir:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas)
• Efectos visuales (ver flashes de luz, visión borrosa o visión doble, normalmente aparecen pronto después de comenzar el tratamiento con XALKORI).
• Problemas gástricos, incluyendo vómitos, diarrea, náuseas.
• Edema (exceso de líquido en el tejido corporal que causa inflamación de las manos y de los pies).
• Estreñimiento.
• Anomalías en las pruebas del hígado en los análisis de sangre.
• Disminución del apetito.
• Cansancio.
• Mareo.
• Neuropatías (sensación de entumecimiento u hormigueo en las articulaciones o extremidades).
• Alteración del sentido del gusto.
• Dolor en el abdomen.
• Reducción en el número de glóbulos rojos de la sangre (anemia).
• Erupción cutánea.
• Reducción del ritmo cardiaco.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas)
• Indigestión.
• Aumento de los niveles de creatinina en sangre (puede indicar que los riñones no funcionan
adecuadamente).
• Aumento de los niveles de la enzima fosfatasa alcalina en sangre (indicador de una disfunción o lesión de un órgano, especialmente del hígado, páncreas, huesos, glándula tiroides o vesícula biliar).
• Hipofosfatemia (niveles bajos de fosfato en sangre que pueden provocar confusión o debilidad muscular).
• Líquido encapsulado dentro del riñón (quistes renales).
• Desmayo.
• Inflamación del esófago (conducto de la deglución).
• Disminución de los niveles de testosterona, una hormona sexual masculina.
• Fallo cardiaco.
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas)
• Orificio (perforación) en el estómago o el intestino.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de XALKORI
• Mantener este medicamento fuera de la vista y del alcance de los niños.
• No utilice este medicamento después de la fecha de caducidad que aparece en el frasco, en el
blíster o en el estuche después de “CAD” o “EXP”. La fecha de caducidad es el último día del mes que se indica.
• No requiere condiciones especiales de conservación.
• No utilice este medicamento si está dañado o tiene signos de deterioro.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de XALKORI
- El principio activo de XALKORI es crizotinib.
XALKORI 200 mg: cada cápsula contiene 200 mg de crizotinib XALKORI 250 mg: cada cápsula contiene 250 mg de crizotinib
- Los demás componentes son:
Contenido de la cápsula: sílice coloidal anhidra, celulosa microcristalina, hidrogenofosfato de calcio anhidro, carboximetilalmidón sódico (tipo A), estearato de magnesio.
Cápsula: gelatina, dióxido de titanio (E171) y óxido de hierro rojo (E172).
Tinta de impresión: shellac, propilenglicol, hidróxido de potasio y óxido de hierro negro (E172).
Aspecto del producto y contenido del envase
XALKORI200 mg se presenta en forma de cápsulas duras de gelatina con la tapa de color rosa y el cuerpo de color blanco, con “Pfizer” impreso en tinta negra en la tapa y “CRZ 200” en el cuerpo.
XALKORI 250 mg se presenta en forma de cápsulas duras de gelatina con la tapa y el cuerpo de color rosa, con “Pfizer” impreso en tinta negra en la tapa y “CRZ 250” en el cuerpo.
Están disponibles en envases de 60 cápsulas duras y en frascos de plástico de 60 cápsulas duras.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Pfizer Limited Ramsgate Road Sandwich, Kent CT13 9NJ Reino Unido
Responsable de la fabricación
Pfizer Manufacturing Deutschland GmbH Betriebsstatte Freiburg Mooswaldallee 1 79090 Freiburg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Lietuva
Pfizer Luxembourg SARL filialas Lietuvoje Tel. + 370 52 51 4000
Luxembourg/Luxemburg
Pfizer S.A.
Tél/Tel: +32 (0)2 554 62 11
Magyarország
Pfizer Kft.
Tel.: +36-1-488-37-00 Malta
V.J. Salomone Pharma Ltd.
Tel. +356 21220174
Nederland
Pfizer BV
Tel: +31 (0)10 406 43 01
Belgique/ Belgie /Belgien
Pfizer S.A. / N.V.
Tél/Tel: +32 (0)2 554 62 11
Etarapna
n^afeep HroKceMÓypr CAPH, KaoH Etarapua Tea.: +359 2 970 4333
Ceská republika
Pfizer PFE, spol. s r.o.
Tel.: +420 283 004 111
Danmark
Pfizer ApS Tlf: +45 44 20 11 00
Deutschland
Pfizer Pharma GmbH Tel: +49 (0)30 550055 51000
|
Eesti Pfizer Luxembourg SARL Eesti filiaal Tel.: +372 666 7500 |
Norge Pfizer Norge AS Tlf: +47 67 52 61 00 |
|
EXláSa Pfizer EAAág A.E. TqA.: +30 210 6785 800 |
Osterreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0 |
|
España Pfizer S.L. Tél: +34 91 490 99 00 |
Polska Pfizer Polska Sp.z.o.o Tel.:+48 22 335 61 00 |
|
France Pfizer Tél: +33 (0)1 58 07 34 40 |
Portugal Pfizer Biofarmaceutica, Sociedade Unipessoal Lda Tel: +351 21 423 5500 |
|
Hrvatska Pfizer Croatia d.o.o. Tel: +385 1 3908 777 |
Romania Pfizer Romania S.R.L. Tel: +40 (0) 21 207 28 00 |
|
Ireland Pfizer Healthcare Ireland Tel: 1800 633 363 (toll free) +44 (0)1304 616161 |
Slovenija Pfizer Luxembourg SARL Pfizer, podruznica za svetovanje s podrocja farmacevtske dejavnosti, Ljubljana Tel.: + 386 (0)1 52 11 400 |
|
Ísland Icepharma hf. Sími: +354 540 8000 |
Slovenská republika Pfizer Luxembourg SARL, organizacná zlozka Tel.: + 421 2 3355 5500 |
|
Italia Pfizer S.r.l. Tel: +39 06 33 18 21 |
Suomi/Finland Pfizer Oy Puh./Tel: +358 (0)9 43 00 40 |
|
Kúnpoq Pfizer EAAág A.E. (Cyprus Branch) TnA: +357 22 817690 |
Sverige Pfizer Innovations AB Tel: +46 (0)8 550 520 00 |
|
Latvija Pfizer Luxembourg SARL filiale Latvija Tel.: + 371 670 35 775 |
United Kingdom Pfizer Limited Tel: +44 (0) 1304 616161 |
Fecha de la última revisión de este prospecto: MM/AAAA
Este medicamento se ha autorizado con una «aprobación condicional».
Esta modalidad de aprobación significa que se espera obtener más información de este medicamento. La Agencia Europea de Medicamentos revisará la información nueva de este medicamento al menos una vez al año y este prospecto se actualizará cuando sea necesario.
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu/.
56