Vedrop 50 Mg/Ml En Solucion Oral
ANEXO I
RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Vedrop 50 mg/ml en solución oral
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada ml contiene 50 mg de d-alfa-tocoferol, en forma de tocofersolán, que equivalen a 74,5 UI de tocoferol. Excipientes:
Cada ml contiene 6 mg de metil-parahidroxibenzoato sódico (E219), 4 mg de etil-parahidroxibenzoato sódico (E215) y 0,18 mmoles (4,1 mg) de sodio.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución oral.
Solución amarillo pálida, ligeramente viscosa.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Vedrop está indicado en el tratamiento de déficit de vitamina E producido por la malabsorción intestinal en pacientes pediátricos con colestasis congénita crónica o colestasis hereditaria crónica, desde el nacimiento (en los recién nacidos a término) hasta los 18 años de edad.
4.2 Posología y forma de administración
El tratamiento con Vedrop deberá ser iniciado y supervisado por un médico experimentado en el control de pacientes con colestasis congénita crónica o colestasis hereditaria crónica.
La biodisponibilidad de la vitamina E proporcionada por el medicamento Vedrop difiere de la de otros medicamentos. La dosis deberá prescribirse en mg de d-alfa tocoferol en forma de tocofersolán. La vitamina E en plasma deberá vigilarse mensualmente durante al menos los primeros meses de terapia y, posteriormente, a intervalos regulares, ajustándose la dosis si fuera necesario.
Posología
La dosis diaria total recomendada en pacientes pediátricos con colestasis congénita crónica o colestasis hereditaria crónica es de 0,34 ml/kg/día (17 mg/kg de d-alfa tocoferol en forma de tocofersolán). La dosis se debe prescribir en ml.
La dosis deberá ajustarse en función de los niveles plasmáticos de vitamina E.
Para calcular la dosis de Vedrop a administrar, divida la dosis prescrita de d-alfa-tocoferol (en mg) por 50. El resultado corresponde al volumen de Vedrop en ml:
Dosis de Vedrop (en ml) = dosis de d-alfa tocoferol (en mg)
50
La tabla siguiente muestra el volumen de solución oral en función del peso de los pacientes.
|
Peso (kg) |
Volumen de solución oral (ml) |
|
3 |
1,0 |
|
4 |
1,4 |
|
5 |
1,7 |
|
6 |
2,0 |
|
7 |
2,4 |
|
8 |
2,7 |
|
9 |
3,1 |
|
10 |
3,4 |
|
15 |
5,1 |
Poblaciones especiales Insuficiencia hepática o renal
La experiencia con las terapias con tocofersolán en pacientes con insuficiencia renal o insuficiencia hepática subyacente ha demostrado que no es necesario adaptar la pauta posológica de Vedrop (ver sección 4.4).
Forma de administración
Vedrop se administra por vía oral con o sin agua. Las jeringas de 1 ml ó 2 ml incluidas en el envase están diseñadas para ayudar a medir la dosis exacta de acuerdo con la posología prescrita.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Vedrop no debe administrarse a recién nacidos prematuros.
4.4 Advertencias y precauciones especiales de empleo
Se han comunicado que las dosis elevadas de vitamina E aumentan la tendencia hemorrágica en pacientes con déficit de vitamina K o en aquellos sometidos a tratamiento oral con antivitamina K, en consecuencia se recomienda vigilar el tiempo de protrombina y el índice internacional normalizado (INR). Puede ser necesario realizar un ajuste de la dosis de anticoagulante oral durante y después del tratamiento con Vedrop.
Puesto que los datos disponibles sobre pacientes con insuficiencia renal son limitados, Vedrop deberá administrarse con precaución y monitorizando cuidadosamente la función renal en los pacientes con insuficiencia renal, por ejemplo, pacientes deshidratados (ver sección 4.2).
Vedrop deberá administrarse con precaución en pacientes con insuficiencia hepática subyacente y monitorizando cuidadosamente la función hepática en dichos pacientes (ver sección 4.2).
Vedrop contiene metil-parahidroxibenzoato sódico (E219) y etil-parahidroxibenzoato sódico (E215) que pueden causar reacciones alérgicas (posiblemente tardías).
Este medicamento contiene sodio, lo que debe tenerse en cuenta en el caso de pacientes con dieta pobre en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones.
Se recomienda monitorizar el funcionamiento de la coagulación cuando se administre junto con un tratamiento con antagonistas de la vitamina K (ver sección 4.4).
Debido a la inhibición de la proteína transportadora de la P-glicoproteína, tocofersolán puede también aumentar la absorción intestinal de otras vitaminas liposolubles administradas concomitantemente (A, D, E, K) o de medicamentos altamente lipofílicos (como esteroides, antibióticos, antihistamínicos, ciclosporina, tacrolimus). Por lo tanto, debe realizarse una monitorización y, en caso necesario, hacer un ajuste de las dosis.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No se dispone de datos clínicos de embarazos expuestos a tocofersolán. Los estudios en animales no muestran efectos perjudiciales directos ni indirectos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo postnatal (ver sección 5.3). Hay que tener precaución en la prescripción a mujeres embarazadas.
Lactancia
Se desconoce si tocofersolán se excreta en la leche materna. La excreción de tocofersolán en la leche no ha sido estudiada en animales. Para decidir la continuación/interrupción de la lactancia materna o la continuación/interrupción de la terapia con Vedrop deberán tenerse en cuenta el beneficio de la lactancia materna para el niño y el beneficio de la terapia con tocofersolán para la mujer.
Fertilidad
No se dispone de datos.
4.7 Efectos sobre la capacidad para conducir y utilizar maquinaria
La influencia de Vedrop sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Resumen del perfil de seguridad
La reacción adversa que se ha notificado con mayor frecuencia durante el tratamiento es diarrea.
Tabla de reacciones adversas
A continuación se enumeran las reacciones adversas notificadas, según la clasificación por órganos y sistemas, y por frecuencia. Las frecuencias se definieron como: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1 000 a < 1/100), raras (> 1/10 000 a < 1/1 000), muy raras (< 1/10 000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Sistema de Clasificación de Órganos |
Reacciones adversas |
|
Trastornos gastrointestinales |
Frecuente: diarrea Frecuencia no conocida: dolor abdominal |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes: alopecia, prurito, erupciones cutáneas |
|
Trastornos generales y alteraciones en el lugar de administración |
Poco frecuentes: astenia, cefaleas |
|
Exploraciones complementarias |
Poco frecuentes: niveles de sodio sérico anormales, niveles de potasio sérico anormales, aumento de las transaminasas |
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Dosis muy elevadas de vitamina E pueden causar diarrea, dolor abdominal, y otros trastornos gastrointestinales.
En caso de sobredosis, debe proponerse un tratamiento sintomático.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Vitaminas. Otros preparados de vitaminas; código ATC: A11HA08
La vitamina E es el principal antioxidante liposoluble en el organismo. Actúa fraccionando cadenas de radicales libres, deteniendo la peroxidación de los ácidos grasos poliinsaturados e interviene en el mantenimiento de la estabilidad y la integridad de las membranas celulares.
Este medicamento se ha autorizado en “circunstancias excepcionales”. Esta modalidad de aprobación significa que, debido a la rareza de la enfermedad, no ha sido posible obtener información completa sobre este medicamento.
La Agencia Europea de Medicamentos revisará anualmente la información nueva del medicamento que pueda estar disponible y esta Ficha Técnica o Resumen de las Características del Producto (RCP) se actualizará cuando sea necesario.
5.2 Propiedades farmacocinéticas
Absorción
El principio activo d-alfa-tocoferol-polietilenglicol 1000 succinato (tocofersolán) es un profármaco, cuyo metabolito activo es el d-alfa tocoferol. A bajas concentraciones, tocofersolán forma micelas que aumentan la absorción de lípidos no polares como las vitaminas liposolubles. Su concentración micelar crítica es baja (de 0,04 a 0,06 mmol/l).
La hidrólisis de tocofersolán tiene lugar en la luz intestinal. Una vez captado por las células, la fracción alfa-tocoferol aparece en quilomicrones en la linfa de forma idéntica a la vitamina E absorbida a partir de la
dieta. La captación celular no requiere la presencia de receptores, proteínas fijadoras ni procesos metabólicos, y no ocurre por pinocitosis. La absorción de tocofersolán deuterado mostró un patrón normal de lipoproteínas: en primer lugar, el alfa-tocoferol presentó un pico en los quilomicrones, a continuación, en las lipoproteínas de muy baja densidad (VLDL) y, finalmente, en las lipoproteínas de baja densidad (LDL) y las lipoproteínas de alta densidad (HDL), y las porciones de desaparición de las curvas mostraron correlación con las de los sujetos control.
Un estudio realizado en 12 voluntarios sanos comparó tocofersolán con una vitamina E de referencia miscible en agua tras una dosis de carga oral única de 1.200 UI. La biodisponibilidad relativa de tocofersolán tendió a ser superior (Frel de 1,01 ± 1,74) con un ABC0-t de 0,383 ± 0,203pM.h/mg, una Cmáx de 0,013 ± 0,006, un tmáx de 6,0 h (6,0 - 24,0) y un tJ/2 de 29,7 h (16,0 - 59,5).
En un estudio similar, tocofersolán mostró una mayor biodisponibilidad que una vitamina E de referencia miscible en agua en pacientes pediátricos con colestasis crónica (n = 6); la absorción fue significativamente superior tanto durante el aumento máximo de la concentración plasmática (p = 0,008) como en el ABC
(p = 0,0026).
Distribución
Localizada principalmente en las membranas celulares, en el interior de las mitocondrias y los microsomas, la vitamina E presenta una distribución ubicuitaria (glóbulos rojos, cerebro, músculo, hígado, plaquetas) siendo el tejido graso su reservorio principal.
Eliminación
La vitamina E se elimina mayoritariamente por la bilis (75%) y en las heces, ya sea como tocoferol libre, ya en sus formas oxidadas. La orina constituye una vía menor de eliminación de la vitamina E (conjugada con ácido glucorónico).
5.3 Datos preclínicos sobre seguridad
Los datos no clínicos publicados en la literatura no revelan ningún riesgo especial para el hombre según los estudios convencionales de toxicidad a dosis repetidas, genotoxicidad y toxicidad para la reproducción.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Sorbato potásico
Metil-parahidroxibenzoato sódico (E219)
Etil-parahidroxibenzoato sódico (E215)
Glicerol
Fosfato disódico dodecahidrato Ácido clorhídrico concentrado Agua ultrapurificada
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
Una vez abierto: 1 mes.
6.4 Precauciones especiales de conservación
Este medicamento no requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Frasco de cristal marrón de tipo III con tapón a rosca de HDPE y precinto de LDPE. Jeringas de uso oral con cuerpo de LDPE y émbolo de polistirol. Cada frasco contiene 10 ml, 20 ml ó 60 ml de solución oral.
Las cajas contienen:
■ un frasco de 10 ml y una jeringa de uso oral de 1 ml
■ un frasco de 20 ml y una jeringa de uso oral de 1 ml
■ un frasco de 60 ml y una jeringa de uso oral de 2 ml
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Las dosis a administrar deberán ser extraídas del frasco usando las jeringas para uso oral incluidas en el envase.
La jeringa de uso oral de 1 ml contiene una escala graduada de 0,05 a 1 ml en incrementos de 0,05 ml. Una marca de graduación de la jeringa de uso oral de 1 ml corresponde a 2,5 mg de d-alfa tocoferol en forma de tocofersolán.
La jeringa de uso oral de 2 ml contiene una escala graduada de 0,1 a 2 ml en incrementos de 0,1 ml. Una marca de graduación de la jeringa de uso oral de 2 ml corresponde a 5 mg de d-alfa tocoferol en forma de tocofersolán.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Orphan Europe SARL Immeuble “le Wilson”
70, avenue du Général de Gaulle
F-92800 Puteaux
Francia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/09/533/001 frasco de 10 ml EU/1/09/533/002 frasco de 20 ml EU/1/09/533/003 frasco de 60 ml
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 24 de julio de 2009 Fecha de la última renovación: 23 de abril de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada sobre este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POSAUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN EN CIRCUNSTANCIAS EXCEPCIONALES
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F-92800 Puteaux
Francia
o
Orphan Europe SARL
Pare d’Activités des Peupliers
39, rue des Peupliers, Batiment K
F-92000 Nanterre
Francia
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POSAUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN EN CIRCUNSTANCIAS EXCEPCIONALES
Al ser esta una autorización de comercialización en circustancias excepcionales y según lo que establece el artículo 14(8) del Reglamento (CE) 726/2004, el TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
Descripción |
Fecha límite |
|
El TAC deberá presentar, en el marco de cada reevaluación anual, informes preliminares anuales a partir del registro establecido en pacientes con colestasis congénita crónica o colestasis hereditaria. |
Informes preliminares anuales en el marco de cada reevaluación anual |
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR Caja de cartón - frasco de 10 ml, 20 ml y 60 ml_
1. NOMBRE DEL MEDICAMENTO
Vedrop 50 mg/ml en solución oral Tocofersolán
2. PRINCIPIO(S) ACTIVO(S)
Cada ml contiene 50 mg de d-alfa tocoferol en forma de tocofersolán, que equivale a 74,5 UI de tocoferol.
3. LISTA DE EXCIPIENTES
Excipientes: metil-parahidroxibenzoato sódico (E219), etil-parahidroxibenzoato sódico (E215). Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución oral.
Frasco de 10 ml y jeringa de uso oral de 1 ml. Frasco de 20 ml y jeringa de uso oral de 1 ml. Frasco de 60 ml y jeringa de uso oral de 2 ml.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Uso oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA
DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
EXP
Desechar un mes después de la apertura.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F-92800 Puteaux
Francia
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/09/533/001 frasco de 10 ml EU/1/09/533/002 frasco de 20 ml EU/1/09/533/003 frasco de 60 ml
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Vedrop 50 mg/ml
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR Y EL ACONDICIONAMIENTO PRIMARIO
Etiqueta del frasco - frascos de 10 ml, 20 ml y 60 ml
1. NOMBRE DEL MEDICAMENTO
Vedrop 50 mg/ml solución oral. Tocofersolán
2. PRINCIPIO(S) ACTIVO(S)
Cada ml contiene 50 mg de d-alfa tocoferol en forma de tocofersolán, que equivalen a 74,5 UI de tocoferol.
3. LISTA DE EXCIPIENTES
Excipientes: metil-parahidroxibenzoato sódico (E219), etil-parahidroxibenzoato sódico (E215). Para obtener más información consulte el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución oral
10 ml 20 ml 60 ml
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Uso oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
EXP
Desechar un mes después de la apertura..
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Orphan Europe SARL Immeuble “Le Wilson”
70 avenue du Général de Gaulle
F-92800 Puteaux
Francia
13. NÚMERO DE LOTE
EU/1/09/533/001 frasco de 10 ml EU/1/09/533/002 frasco de 20 ml EU/1/09/533/003 frasco de 60 ml
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Vedrop 50 mg/ml
B. PROSPECTO
Prospecto: información para el usuario
Vedrop 50 mg/ml en solución oral
Tocofersolán
'VEste medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto.Ver sección 4.
Contenido del prospecto:
1. Qué es Vedrop y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Vedrop
3. Cómo tomar Vedrop
4. Posibles efectos adversos
5. Conservación de Vedrop
6. Contenido del envase e información adicional
1. Qué es Vedrop y para qué se utiliza
Vedrop contiene vitamina E (en forma de tocofersolán). Este medicamento se utiliza para tratar la carencia de vitamina E producida por la malabsorción digestiva (por la que los nutrientes de los alimentos no se absorben fácilmente durante la digestión) en pacientes con edades comprendidas entre el nacimiento (recién nacidos a término) hasta los 18 años con colestasis crónica (enfermedad hereditaria o congénita en la que la bilis no puede pasar del hígado al intestino).
2. Qué necesita saber antes de empezar a tomar Vedrop
No tome Vedrop
- Si es alérgico a la vitamina E (d-alfa tocoferol o a cualquiera de los demás componentes de este
medicamento (incluidos en la sección 6).
- Vedrop no debe administrarse a bebés prematuros.
Advertencias y precauciones
Consulte a su médico antes de empezar a tomar Vedrop si tiene:
■ Problemas de riñón o deshidratación. Vedrop deberá usarse con precaución y deberá vigilarse cuidadosamente la función renal, ya que el polietilenglicol, que forma parte del principio activo tocofersolán, puede dañar los riñones.
■ Problemas de hígado. Vedrop deberá usarse con precaución y las funciones del hígado deberán vigilarse estrictamente.
Toma de Vedrop con otros medicamentos
Informe a su médico o farmacéutico si está tomando o ha tomado recientemente o podría tener que tomar
cualquier otro medicamento.
Informe a su médico o farmacéutico si está tomando:
■ Ciertos medicamentos que disminuyen la viscosidad de la sangre (anticoagulantes orales tales como la warfarina). Su médico le pedirá que se someta a análisis de sangre regularmente y podrá ajustar la dosis para evitar un mayor riesgo de hemorragia.
■ Vitaminas liposolubles (como las vitaminas A, D, E o K) o medicamentos altamente liposolubles (como los corticoides, ciclosporina, tacrolimus, antihistamínicos). Vedrop puede aumentar su absorción durante la digestión, por lo que el médico supervisará el efecto del tratamiento y ajustará la dosis cuando sea necesario.
Embarazo y lactancia
No existen datos clínicos disponibles de la utilización de este medicamento durante el embarazo. Informe a su médico si está embarazada para que decida si es conveniente utilizar el medicamento.
No existen datos acerca de si este medicamento se excreta en la leche materna. Informe a su médico si usted desea amamantar a su hijo para que decida si es conveniente utilizar el medicamento. Su médico le ayudará a tomar la mejor decisión para usted y su hijo.
Consulte a su médico o farmacéutico antes de tomar cualquier medicamento.
Conducción y uso de máquinas
Es poco probable que Vedrop afecte a su capacidad para conducir y utilizar máquinas.
Vedrop contiene metil-parahidroxibenzoato sódico (E219) y etil-parahidroxibenzoato sódico (E215),
que pueden producir reacciones alérgicas (posiblemente retardadas).
Vedrop contiene 0,18 mmoles (4,1 mg) de sodio por ml. Consulte a su médico si sigue una dieta pobre en sodio.
3. Cómo tomar Vedrop
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
La dosis habitual es de 0,34 ml/kg/día.
Su médico le recetará la dosis en ml.
El médico le modificará la dosis de este medicamento de acuerdo con sus niveles de vitamina E en sangre. Forma de administración
Trague la solución con o sin agua. Úsela únicamente con la jeringa para uso oral que se incluye en el envase. Puede tomar Vedrop antes o durante las comidas, con o sin agua.
Para medir la dosis:
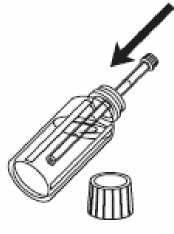
1- Abra el frasco.
2- Introduzca la jeringa de uso oral en el frasco.
3-Llene la jeringa de uso oral con el líquido tirando del émbolo hasta la marca de referencia correspondiente a la cantidad en mililitros (ml) que le ha prescrito el médico.
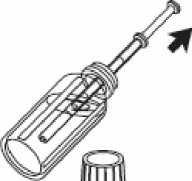
*
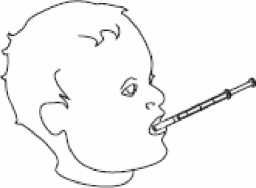
4- Retire la jeringa de uso oral del frasco.
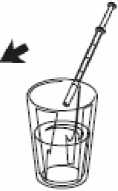
5- Vacíe el contenido de la jeringa presionando sobre el émbolo hasta el fondo ya sea:
- directamente en la boca, o
- viértalo en un vaso de agua y, posteriormente, beba el contenido del mismo.

Si toma más Vedrop del que debe
Si toma dosis altas de vitamina E, puede padecer diarrea y dolor de estómago transitorios. Consulte a su médico o farmacéutico si los síntomas persisten más de dos días.
Si olvidó tomar Vedrop
No tome la dosis que ha olvidado y vuelva a la pauta de administración regular programada. No tome una dosis doble para compensar las dosis olvidadas.
Si interrumpe el tratamiento con Vedrop
No interrumpa el tratamiento sin consultar a su médico porque puede volver a producirse una carencia de vitamina E que afecte a su salud. Póngase en contacto con su médico o farmacéutico antes de interrumpirlo.
Si tiene cualquier otra duda sobre el uso de este producto, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Se comunicaron los siguientes efectos adversos:
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas)
• Diarrea
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas)
• Astenia (sensación de debilidad)
• Cefalea
• Pérdida del cabello
• Picores
• Erupción cutánea
• Niveles anormales de sodio en sangre
• Niveles anormales de potasio en sangre
• Aumento de las transaminasas (enzimas hepáticas)
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
• Dolor de estómago
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5 Conservación de Vedrop
• Mantener este medicamento fuera de la vista y del alcance de los niños.
• No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en el frasco después de EXP. La fecha de caducidad es el último día del mes que se indica.
• Este medicamento no requiere condiciones especiales de conservación.
• Desechar el medicamento un mes después que se haya abierto, aunque quede algo de solución.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Vedrop
- El principio activo es tocofersolán. Cada ml de solución contiene 50 mg de d-alfa-tocoferol en forma de tocofersolán, que equivalen a 74,5 UI de tocoferol.
- Los demás componentes son: sorbato potásico, metil-parahidroxibenzoato sódico (E219) y etil-parahidroxibenzoato sódico (E215) (ver el final de la sección 2 para obtener más información sobre estos 2 excipientes), glicerol, fosfato disódico dodecahidrato, ácido clorhídrico concentrado, agua ultrapurificada.
Aspecto de Vedrop y contenido del envase
Vedrop es una solución oral de color amarillo pálido ligeramente viscosa contenida en un frasco de cristal de color marrón. Los frascos contienen 10 ml, 20 ml ó 60 ml de solución oral. Cada envase contiene un frasco y una jeringa de uso oral (una jeringa de 1 ml con un frasco de 10 ml ó 20 ml, una jeringa de 2 ml con un frasco de 60 ml).
Titular de la autorización de comercialización
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du General de Gaulle
F-92800 Puteaux
Francia
Responsable de la fabricación
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
92800 Puteaux
Francia
o
Orphan Europe SARL
Pare d’Activités des Peupliers
39, rue des Peupliers, Batiment K
F-92000 Nanterre
Francia
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización.
Belgique/Belgie/Belgien Lietuva
Orphan Europe Benelux Orphan Europe AB
Koning Albert I laan 48 bus 3 Isafjordsgatan 30C, plan 3
BE-1780 Wemmel (Brussels) S-164 40 Kista
Tél/Tel: +32 2 46101 36 Svedija
Tel: + 46 8 545 80 230
Bt^rapnn
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm repMaHHH
Tel: +49 731 140 554 0
Ceská republika
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm Nemecko
Tel: +49 731 140 554 0
Danmark
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Sverige
Tlf : +46 8 545 80 230
Deutschland
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm Tel: +49 731 140 554 0
Luxembourg/Luxemburg
Orphan Europe Benelux Koning Albert I laan 48 bus 3 BE-1780 Wemmel(Brussels) Belgique/Belgien Tél/Tel: +32 2 46101 36
Magyarország
Orphan Europe (Germany) GmbH
Eberhard-Finckh-StraBe 55
D-89075 Ulm
Németország
Tel: +49 731 140 554 0
Malta
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F-92800 Puteaux
Franza
Tel: +33 1 47 73 64 58 Nederland
Orphan Europe Benelux Koning Albert I Iaan 48 bus 3 BE-1780 Wemmel (Brussels) Belgie
Tel: +32 2 46101 36
Eesti
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Rootsi
Tel: + 46 8 545 80 230 EXXúda
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F-92800 Puteaux
rakkía
Tpk: +33 1 47 73 64 58 España
Orphan Europe, S.L.
C/ Isla de la Palma, 37, 2a planta E-28700 San Sebastián de los Reyes, Madrid Tel: + 34 91 659 28 90
France
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F-92800 Puteaux
Tél: +33 (0)1 47 73 64 58
Hrvatska
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F-92800 Puteaux
Francuska
Tél: +33 (0)1 47 73 64 58 Ireland
Orphan Europe (UK) Ltd.
Isis House, 43 Station road Henley-on-Thames Oxfordshire RG9 1AT - UK United Kingdom Tel: +44 1491 414333
Ísland
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Sví^jóó
Simi:+46 8 545 80 230
Norge
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Sverige
Tlf : +46 8 545 80 230 Osterreich
Orphan Europe (Germany) GmbH
Eberhard-Finckh-StraBe 55
D-89075 Ulm
Deutschland
Tel: +49 731 140 554 0
Polska
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm Niemcy
Tel: +49 731 140 554 0 Portugal
Orphan Europe, S.L.
C/ Isla de la Palma, 37, 2a planta
E-28700 San Sebastián de los Reyes, Madrid
Espanha
Tel: +34 91 659 28 90 Romania
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm Germania
Tel: +49 731 140 554 0
Slovenija
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm Nemcija
Tel: +49 731 140 554 0
Slovenská republika
Orphan Europe (Germany) GmbH Eberhard-Finckh-StraBe 55 D-89075 Ulm Nemecko
Tel: +49 731 140 554 0
Italia
Orphan Europe (Italy) Srl Via Marostica, 1 I-20146 Milano Tel: +39 02 487 87 173
Kúrcpog
Orphan Europe SARL Immeuble “Le Wilson”
70, avenue du Général de Gaulle
F - 92800 Puteaux
Ta^Ma
Tr(k : +33 1 47 73 64 58 Latvija
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Zviedrija
Tel: + 46 8 545 80 230
Suomi/Finland
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Sverige
Puh/Tel : +46 8 545 80 230 Sverige
Orphan Europe AB Isafjordsgatan 30C, plan 3 S-164 40 Kista Tel : +46 8 545 80 230
United Kingdom
Orphan Europe (UK) Ltd. Isis House, 43 Station road Henley-on-Thames Oxfordshire RG9 1AT - UK Tel: +44 (0)1491 414333
Fecha de la última revisión de este prospecto:
Este medicamento se ha autorizado en “circunstancias excepcionales”. Esta modalidad de aprobación significa que, debido a la rareza de su enfermedad, no ha sido posible obtener información completa de este medicamento.
La Agencia Europea de Medicamentos revisará anualmente la información nueva de este medicamento que pueda estar disponible y este prospecto se actualizará cuando sea necesario.
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu/.
24