Vancomicina Pfizer 1000 Mg Polvo Para Concentrado Para Solucion Para Perfusion Efg
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Vancomicina Pfizer 1.000 mg polvo para concentrado para solución para perfusión EFG.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene 1.000 mg de vancomicina (en forma de hidrocloruro de vancomicina) equivalentes a 1.000.000 UI.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para concentrado para solución para perfusión ‘Pasta porosa de color blanco crema’
Después de la reconstitución se obtiene una solución con un pH de aproximadamente 3.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
La vancomicina intravenosa está indicada en las siguientes infecciones graves causadas por bacterias grampositivas sensibles a la vancomicina que no pueden tratarse, no responden o son resistentes a otros antibióticos, como penicilinas o cefalosporinas (ver sección 5.1).
- endocarditis
- infecciones óseas (osteomielitis)
- neumonía
- infecciones de tejidos blandos
En los casos en que se considere adecuado, la vancomicina se debe administrar junto con otros agentes antibacterianos. Esto se aplica especialmente en el tratamiento de la endocarditis.
La vancomicina puede utilizarse para la profilaxis perioperatoria contra la endocarditis bacteriana en pacientes con alto riesgo de desarrollar endocarditis bacteriana cuando son sometidos a intervenciones quirúrgicas mayores (por ejemplo, intervenciones cardiacas y vasculares) y no se les puede administrar un agente antibacteriano betalactámico apropiado.
Deben tenerse en cuenta las consideraciones oficiales sobre el uso adecuado de agentes antibacterianos.
4.2 Posología y forma de administración
Forma de administración:
La vancomicina únicamente deberá administrarse por vía parenteral como perfusión intravenosa (no más de 10 mg/min durante al menos 60 min) que esté suficientemente diluida (al menos 100 ml por 500 mg o al menos 200 ml por 1.000 mg).
En el caso de pacientes que precisen restringir el volumen de líquido perfundido, se puede administrar una solución de 500 mg/50 ml o 1.000 mg/100 ml500 mg/50 ml o 1.000 mg/100 ml. La administración de estas concentraciones elevadas puede aumentar el riesgo de efectos adversos asociados a la perfusión. No obstante, pueden producirse efectos adversos asociados a la perfusión independientemente de la concentración y la velocidad.
La dosis debe adaptarse a cada persona en función del peso, la edad y la función renal. Se pueden medir los niveles de vancomicina para ayudar a ajustar la dosis.
Para consultar las instrucciones de preparación de la solución, ver sección 6.6.
Perfusión intravenosa en pacientes con función renal normal:
Adultos y adolescentes mayores de 12 años:
La dosis intravenosa diaria recomendada es 2000 mg, divididos en dosis de 500 mg cada 6 horas o 1000 mg cada 12 horas. O bien, de 30 a 40 mg/kg/día en 2 a 4 administraciones diarias.
En el caso de la endocarditis bacteriana, la pauta generalmente aceptada es 1.000 mg de vancomicina por vía intravenosa cada 12 horas durante 4 semanas, sola o combinada con otros antibióticos (gentamicina más rifampicina, gentamicina, estreptomicina). La endocarditis enterocócica se trata durante 6 semanas con vancomicina combinada con un aminoglucósido. Deben consultarse las guías oficiales.
Niños de un mes a 12 años de edad:
La dosis intravenosa habitual es 10 mg/kg por dosis, administrada cada seis horas (dosis diaria total de 40 mg/kg de peso corporal). Cada dosis debe administrarse durante un periodo de tiempo mínimo de 60 minutos.
Recién nacidos (a término):
0 a 7 días de edad: una dosis inicial de 15 mg/kg, seguida de dosis de 10 mg/kg cada 12 horas.
7 a 30 días de edad: una dosis inicial de 15 mg/kg, seguida de dosis de 10 mg/kg cada 8 horas.
Cada dosis debe administrarse durante un periodo de tiempo de 60 minutos. La supervisión minuciosa de las concentraciones séricas de vancomicina está justificada en este tipo de pacientes.
Embarazo:
Se ha observado que es posible que se precisen dosis considerablemente superiores para obtener concentraciones séricas terapéuticas en pacientes embarazadas; ver sección 4.6.
Pacientes de edad avanzada:
Es posible que sea necesario reducir la dosis en mayor medida de lo esperado debido a la disminución de la función renal (ver sección siguiente).
Pacientes obesos:
Es posible que sea necesario modificar la dosis diaria habitual.
Pacientes con insuficiencia hepática
No hay evidencias de que sea necesario reducir la dosis en pacientes con insuficiencia hepática.
Pacientes con función renal reducida:
La dosis debe ajustarse para evitar niveles séricos tóxicos. En lactantes prematuros y pacientes de edad avanzada, es posible que sea necesario reducir la dosis en mayor medida de lo esperado debido a la disminución de la función renal. En el caso de estos pacientes, es conveniente supervisar de forma periódica los niveles séricos ya que se conocen casos de acumulación, especialmente después de un tratamiento prolongado.
Las concentraciones séricas de vancomicina pueden determinarse mediante ensayo microbiológico, radioinmunoensayo, inmunoensayo de polarización de fluorescencia, inmunoensayo de fluorescencia o cromatografía de líquidos de alta presión. Como guía para ajustar la dosis, se proporciona el nomograma siguiente basado en los valores de aclaramiento de creatinina:
Creslinlne CI«ar»neo (ml/min/kg)
Dosage nomogrnm for vancomycin in patients with impaired renal function
|
Inglés |
Español |
|
Vancomycin Clearance (ml/min/kg) |
Aclaramiento de vancomicina (ml/min/kg) |
|
Creatinine Clearance (ml/min/kg) |
Aclaramiento de creatinina (ml/min/kg) |
|
Vancomycin Dose (mg/kg/24 hr) |
Dosis de vancomicina (mg/kg/24 horas) |
|
Dosage nomogram for vancomycin in patients with impaired renal function |
Nomograma de dosificación para vancomicina en pacientes con función renal disminuida |
Este nomograma no es válido para pacientes funcionalmente anéfricos en tratamiento de diálisis. En el caso de estos pacientes, es preciso administrar una dosis inicial de 15 mg/kg de peso corporal con el fin de alcanzar rápidamente los niveles séricos terapéuticos y la dosis necesaria para mantener niveles estables es 1,9 mg/kg/24 horas. Dado que es conveniente administrar dosis de mantenimiento individuales entre 250 mg y 1 g, en el caso de los pacientes con insuficiencia renal pronunciada se puede administrar una dosis cada varios días en lugar de diariamente. En casos de anuria, se recomienda administrar una dosis de 1 g cada 7-10 días.
Si se dispone únicamente del nivel de creatinina sérica, se puede aplicar la fórmula siguiente para calcular el aclaramiento de creatinina:
Hombres: Peso (kg) x (140 - edad (años))
72 x creatinina sérica (mg/100 ml)
Mujeres: 0,85 multiplicado por el valor obtenido del cálculo anterior
Para obtener las instrucciones para la preparación de las soluciones, ver sección 6.6.
Supervisión de las concentraciones séricas de vancomicina:
Debe supervisarse la concentración sérica de vancomicina en el segundo día de tratamiento inmediatamente antes de la siguiente dosis, y una hora después de la perfusión. Los niveles terapéuticos de vancomicina en sangre deben ser entre 30 y 40 mg/l (máximo 50 mg/l) una hora después de finalizar la perfusión, y el nivel mínimo (poco antes de la siguiente administración) entre 5 y 10 mg/l.
Normalmente, las concentraciones se supervisarán dos o tres veces por semana.
Duración del tratamiento
no y
-I 21 8
2Ó 3
i oo -
0 75 —
0 25 —
10 H
093
US
-01
La duración del tratamiento depende de la gravedad de la infección así como de la evolución clínica y bacteriológica.
4.2.1 Posología
Perfusión intravenosa en pacientes con función renal normal:
Adultos y adolescentes mayores de 12 años:
La dosis intravenosa diaria recomendada es 2000 mg, divididos en dosis de 500 mg cada 6 horas o 1.000 mg cada 12 horas. O bien, de 30 a 40 mg/kg/día en 2 a 4 administraciones diarias.
En el caso de la endocarditis bacteriana, la pauta generalmente aceptada es 1000 mg de vancomicina por vía intravenosa cada 12 horas durante 4 semanas, sola o combinada con otros antibióticos (gentamicina más rifampicina, gentamicina, estreptomicina). La endocarditis enterocócica se trata durante 6 semanas con vancomicina combinada con un aminoglucósido. Deben consultarse las guías oficiales.
Población pediátrica
Niños de un mes a 12 años de edad:
La dosis intravenosa habitual es 10 mg/kg por dosis, administrada cada seis horas (dosis diaria total de 40 mg/kg de peso corporal). Cada dosis debe administrarse durante un periodo de tiempo mínimo de 60 minutos.
Recién nacidos (a término):
0 a 7 días de edad: una dosis inicial de 15 mg/kg, seguida de dosis de 10 mg/kg cada 12 horas.
7 a 30 días de edad: una dosis inicial de 15 mg/kg, seguida de dosis de 10 mg/kg cada 8 horas.
Cada dosis debe administrarse durante un periodo de tiempo de 60 minutos. La supervisión minuciosa de las concentraciones séricas de vancomicina está justificada en este tipo de pacientes.
Poblaciones especiales
Embarazo:
Se ha observado que es posible que se precisen dosis considerablemente superiores para obtener concentraciones séricas terapéuticas en pacientes embarazadas; ver sección 4.6.
Pacientes de edad avanzada:
Es posible que sea necesario reducir la dosis en mayor medida de lo esperado debido a la disminución de la función renal (ver sección siguiente).
Pacientes obesos:
Es posible que sea necesario modificar la dosis diaria habitual.
Pacientes con insuficiencia hepática:
No hay evidencias de que sea necesario reducir la dosis en pacientes con insuficiencia hepática.
Pacientes con función renal reducida:
La dosis debe ajustarse para evitar niveles séricos tóxicos. En lactantes prematuros y pacientes de edad avanzada, es posible que sea necesario reducir la dosis en mayor medida de lo esperado debido a la disminución de la función renal. En el caso de estos pacientes, es conveniente supervisar de forma periódica los niveles séricos ya que se conocen casos de acumulación, especialmente después de un tratamiento prolongado.
Las concentraciones séricas de vancomicina pueden determinarse mediante ensayo microbiológico, radioinmunoensayo, inmunoensayo de polarización de fluorescencia, inmunoensayo de fluorescencia o cromatografía de líquidos de alta presión. Como guía para ajustar la dosis, se proporciona el nomograma siguiente basado en los valores de aclaramiento de creatinina:
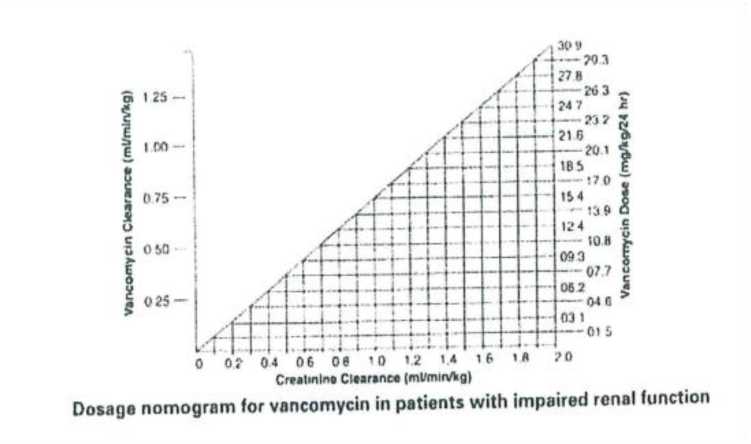
|
Inglés |
Español |
|
Vancomycin Clearance (ml/min/kg) |
Aclaramiento de vancomicina (ml/min/kg) |
|
Creatinine Clearance (ml/min/kg) |
Aclaramiento de creatinina (ml/min/kg) |
|
Vancomycin Dose (mg/kg/24 hr) |
Dosis de vancomicina (mg/kg/24 horas) |
|
Dosage nomogram for vancomycin in patients with impaired renal function |
Nomograma de dosificación para vancomicina en pacientes con función renal disminuida |
Este nomograma no es válido para pacientes funcionalmente anéfricos en tratamiento de diálisis. En el caso de estos pacientes, es preciso administrar una dosis inicial de 15 mg/kg de peso corporal con el fin de alcanzar rápidamente los niveles séricos terapéuticos y la dosis necesaria para mantener niveles estables es 1,9 mg/kg/24 horas. Dado que es conveniente administrar dosis de mantenimiento individuales entre 250 mg y 1 g, en el caso de los pacientes con insuficiencia renal pronunciada se puede administrar una dosis cada varios días en lugar de diariamente. En casos de anuria, se recomienda administrar una dosis de 1 g cada 7-10 días.
Si se dispone únicamente del nivel de creatinina sérica, se puede aplicar la fórmula siguiente para calcular el aclaramiento de creatinina:
Hombres: Peso (kg) x (140 - edad (años))
72 x creatinina sérica (mg/100 ml)
Mujeres: 0,85 multiplicado por el valor obtenido del cálculo anterior
Para obtener las instrucciones para la preparación de las soluciones, ver sección 6.6.
Supervisión de las concentraciones séricas de vancomicina:
Debe supervisarse la concentración sérica de vancomicina en el segundo día de tratamiento inmediatamente antes de la siguiente dosis, y una hora después de la perfusión. Los niveles terapéuticos de vancomicina en sangre deben ser entre 30 y 40 mg/l (máximo 50 mg/l) una hora después de finalizar la perfusión, y el nivel mínimo (poco antes de la siguiente administración) entre 5 y 10 mg/l.
Normalmente, las concentraciones se supervisarán dos o tres veces por semana.
Duración del tratamiento
La duración del tratamiento depende de la gravedad de la infección así como de la evolución clínica y bacteriológica.
4.2.2 Forma de administración
La vancomicina únicamente deberá administrarse por vía parenteral como perfusión intravenosa lenta (no más de 10 mg/min durante al menos 60 min) que esté suficientemente diluida (al menos 100 ml para 500 mg o al menos 200 ml para 1.000 mg).
En el caso de pacientes que precisen restringir el volumen de líquido perfundido, se puede administrar una solución de 500 mg/50 ml o 1.000 mg/100 ml. La administración de estas concentraciones elevadas puede aumentar el riesgo de efectos adversos asociados a la perfusión. No obstante, pueden producirse efectos adversos asociados a la perfusión independientemente de la concentración y la velocidad.
La dosis debe adaptarse a cada persona en función del peso, la edad y la función renal. Se pueden medir los niveles de vancomicina para ayudar a ajustar la dosis.
Para consultar las instrucciones de preparación de la solución, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
La administración intravenosa rápida (por ejemplo, en pocos minutos) puede provocar: hipotensión exacerbada , incluyendo shock y, raramente, parada cardiaca, reacciones del tipo histamínico y exantema máculopapular o rash eritematoso (“síndrome del hombre rojo” o “síndrome del cuello rojo”). La vancomicina debe perfundirse en una solución diluida durante un período de tiempo no inferior a 60 minutos, con el fin de evitar reacciones derivadas de una perfusión rápida. En un caso así, estas reacciones normalmente remiten en cuanto se interrumpe la perfusión (ver secciones 4.2 y 4.8).
Debido a que la vancomicina puede causar ototoxicidad y nefrotoxicidad, en el caso de los pacientes con insuficiencia renal es preciso administrarla con precaución y reducir la dosis en función del grado de insuficiencia renal. El riesgo de toxicidad aumenta notablemente si las concentraciones en sangre son elevadas o el tratamiento es prolongado. Es necesario supervisar los niveles sanguíneos y realizar pruebas para evaluar la función renal con regularidad.
Asimismo, se debe evitar el uso de la vancomicina en pacientes con pérdida de audición previa. Si se administra a estos pacientes, la dosis debe regularse, en la medida de lo posible, mediante la determinación periódica del nivel de medicamento en sangre. La sordera puede estar precedida de acúfenos.
Los pacientes de edad avanzada son más susceptibles de experimentar daño auditivo. La experiencia con otros antibióticos sugiere que el desarrollo de la sordera puede continuar incluso después de la interrupción del tratamiento.
Uso en pacientes pediátricos: en pacientes neonatos y lactantes, puede ser conveniente confirmar las concentraciones séricas de vancomicina deseadas. La administración simultánea de vancomicina y agentes anestésicos en niños se ha asociado a eritema y rubefacción seudohistamínica.
í>.
n
Uso en pacientes de edad avanzada: la disminución natural de la filtración glomerular propia de la edad puede derivar en concentraciones séricas de vancomicina elevadas si la dosis no se ajusta (ver sección 4.2).
Precauciones
Se recomienda la supervisión regular de los niveles en sangre de vancomicina cuando se administre durante un período de tiempo prolongado, especialmente en pacientes con disfunción renal o con función auditiva reducida, así como en la administración simultánea de sustancias nefrotóxicas u ototóxicas, respectivamente.
La dosis debe ajustarse en función de los niveles séricos. Es necesario supervisar los niveles sanguíneos y realizar pruebas para evaluar la función renal con regularidad.
Los pacientes cuya función renal está en el límite de la normalidad y las personas mayores de 60 años deben someterse a pruebas seriadas de la función auditiva y de los niveles de vancomicina en sangre. Además, es preciso realizar periódicamente análisis hematológicos y de orina y pruebas de la función renal a todos los pacientes a los que se administre el medicamento.
La vancomicina es muy irritante para los tejidos y, si se inyecta por vía intramuscular, provoca la necrosis del sitio de inyección; por tanto, debe infundirse por vía intravenosa. Muchos pacientes en tratamiento con vancomicina desarrollan tromboflebitis y experimentan dolor en el sitio de perfusión, aunque esto no suele revestir gravedad.
La frecuencia y la gravedad de la tromboflebitis puede reducirse mediante la administración lenta del medicamento en una solución diluida (2,5-5,0 g/l) y la alternancia del sitio de perfusión.
El uso prolongado de la vancomicina puede causar la proliferación de microorganismos no sensibles. Es crucial observar minuciosamente la evolución del paciente. En caso de que se produzca una sobreinfección durante el tratamiento, se deberán tomar las medidas oportunas. Se han comunicado casos poco frecuentes de desarrollo de colitis seudomembranosa causada por Clostridium difficile en pacientes en tratamiento de vancomicina intravenosa.
Debido a que se han comunicado casos de hipersensibilidad cruzada, la vancomicina debe administrarse con precaución a pacientes con hipersensibilidad conocida a la teicoplanina.
4.5 Interacción con otros medicamentos y otras formas de interacción
La administración simultánea de vancomicina y agentes anestésicos se ha asociado a eritema, rubefacción tipo histamínica y reacciones anafilactoides.
Se ha observado que la frecuencia de efectos adversos asociados a la perfusión aumenta con la administración simultánea de agentes anestésicos. Los efectos adversos asociados a la perfusión pueden reducirse mediante la administración de la vancomicina como perfusión durante 60 minutos antes de la inducción anestésica.
Es preciso monitorizar minuciosamente el uso tópico o sistémico simultáneo o secuencial de otros medicamentos potencialmente ototóxicos, neurotóxicos o nefrotóxicos indicados, como amfotericina B, aminoglucósidos, bacitracina, polimixina B, colistina, viomicina o cisplatino.
La administración simultánea de vancomicina y bloqueantes neuromusculares aumenta el riesgo de bloqueo neuromuscular.
4.6 Fertilidad, embarazo y lactancia
4.6.1 Embarazo
No hay datos, o estos son limitados, relativos al uso de vancomicina en mujeres embarazadas. Los estudios realizados en animales no sugieren efectos perjudiciales en el desarrollo del embrión, feto o período de gestación en términos de toxicidad para la reproducción (ver sección 5.3).
No obstante, la vancomicina penetra en la placenta y no se puede excluir el riesgo de ototoxicidad y nefrotoxicidad embrionaria o neonatal. Por consiguiente, la vancomicina debe administrarse durante el embarazo únicamente en caso de necesidad evidente y después de evaluar minuciosamente los riesgos y beneficios.
4.6.2 Lactancia
La vancomicina se excreta en la leche materna y, por tanto, se debe administrar durante la lactancia únicamente si otros antibióticos no han sido efectivos. La vancomicina debe administrarse con precaución a las madres en período de lactancia debido a posibles reacciones adversas en el bebé (alteración de la flora intestinal con diarrea, colonización de levaduras y posible sensibilización).
Se debe decidir si es necesario interrumpir la lactancia tras considerar el beneficio de este medicamento para la madre.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de la vancomicina sobre la capacidad para conducir y utilizar máquinas es insignificante.
4.8 Reacciones adversas
En cada uno de los grupos de frecuencia, las reacciones adversas se presentan por orden descendente de gravedad.
Las reacciones adversas enumeradas a continuación se definen según la siguiente clasificación MedDRA:
Muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000), muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Perfusión intravenosa:
Las reacciones adversas más comunes son la flebitis y las reacciones seudoalérgicas relacionadas con una perfusión demasiado rápida de la vancomicina.
Trastornos de la sangre y del sistema linfático:
Raras: trombocitopenia, neutrocitopenia, agranulocitosis, eosinofilia.
Trastornos del sistema inmunológico
Raras: reacciones anafilácticas, reacciones alérgicas.
Trastornos del oído y del laberinto:
Poco frecuentes: pérdida de audición temporal o permanente.
Raras: acúfenos y mareos.
LITTCA ALDAD ota efe
Trastornos cardiacos:
Muy raras: paro cardiaco.
Trastornos vasculares:
Frecuentes: descenso de la presión sanguínea.
Raras: vasculitis.
Trastornos respiratorios, torácicos y mediastínicos:
Frecuentes: disnea y estridor.
Trastornos gastrointestinales:
Raras: náuseas.
Muy raras: enterocolitis seudomembranosa.
Trastornos de la piel y del tejido subcutáneo:
Frecuentes: exantema e inflamación de las mucosas, prurito y urticaria.
Muy raras: dermatitis exfoliativa, síndrome de Stevens-Johnson,
Síndrome de Lyell, dermatosis ampollosa IgA lineal,
Trastornos renales y urinarios:
Frecuentes: insuficiencia renal manifestada principalmente por un aumento de la creatinina sérica.
Raras: nefritis intersticial y disfunción renal aguda.
Trastornos generales y alteraciones en el lugar de administración:
Frecuentes: flebitis y rubefacción de la parte superior del cuerpo y la cara.
Raras: fiebre medicamentosa y escalofríos. Dolor muscular en el pecho y la espalda.
Efectos asociados a la perfusión:
Durante o poco después de una perfusión rápida pueden producirse reacciones anafilactoides, como hipotensión, disnea, urticaria o prurito. Puede producirse rubefacción de la parte superior del cuerpo (síndrome del hombre rojo), dolor y espasmos musculares en el pecho o la espalda.
Estas reacciones remiten una vez que se interrumpe la administración, normalmente entre 20 minutos y 2 horas después. La vancomicina debe perfundirse lentamente (durante más de 60 minutos; ver sección 4.4).
La ototoxicidad puede ser reversible o permanente, y se ha informado de casos debidos principalmente a sobredosis en pacientes con historial de audición reducida y con tratamientos simultáneos con otros medicamentos ototóxicos, como aminoglucósidos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de medicamentos de Uso Humano: https://www.notificaram.es.
4.9 Sobredosis
Se recomienda tratamiento de soporte, con mantenimiento de la filtración glomerular. La hemodiálisis y la diálisis peritoneal no son métodos eficaces para la eliminación adecuada de la vancomicina en sangre. Sí se han observado beneficios limitados de la hemoperfusión con resina Amberlite XAD-4
5. PROPIEDADES FARMACOLÓGICAS
Grupo farmacoterapéutico, “antibacterianos para uso sistémico, antibacterianos glucopeptídicos”, código ATC: JO1X A01”.
5.1 Propiedades farmacodinámicas
5.1.1 Mecanismo de acción
La vancomicina es un antibiótico glucopéptido tricíclico que inhibe la síntesis de la pared celular en bacterias sensibles mediante una unión de gran afinidad con el extremo D-alanil-D-alanina de las unidades precursoras de la pared celular. El medicamento es bactericida para la división de microorganismos.
5.1.2 Efectos farmacodinámicos
La actividad de la vancomicina se considera una función dependiente del tiempo.
Mecanismo de resistencia:
La resistencia adquirida a glucopéptidos es muy común en los enterococos y se basa en la adquisición de distintos complejos de genes van que modifica la D-alanil-D-alanina objetivo convirtiéndola en D-alanil-D-lactato o D-alanil-D-serina que difícilmente se unen a la vancomicina. Se han comunicado casos de resistencia cruzada con la teicoplanina en el caso de algunos genes van. Los genes van son poco frecuentes en Staphylococcus aureus, en el que los cambios en la estructura de la pared celular dan como resultado una sensibilidad “intermedia”, que la mayoría de las veces es heterogénea.
Sensibilidad:
La vancomicina es particularmente activa contra bacterias grampositivas, como estafilococos, estreptococos, enterococos, neumococos, clostridios y difteroides. Las bacterias gramnegativas son resistentes.
La prevalencia de la resistencia adquirida de las especies seleccionadas puede variar geográficamente y con el tiempo, por lo que sería deseable disponer de información local acerca de la resistencia, especialmente en el tratamiento de infecciones graves. Si fuera necesario, se solicitará la opinión de expertos cuando la prevalencia local de la resistencia sea tal que la utilidad del agente, al menos en algunos tipos de infecciones, sea cuestionable.
Puntos de corte
Recomendaciones del EUCAST (Comité europeo sobre pruebas de susceptibilidad antimicrobiana) (del inglés, “European Committee on Antimicrobial Susceptibility Testing”)
|
Sensible |
Resistente | |
|
Staphylococcus spp. |
< 2 mg/L |
> 2 mg/L |
|
Enterococcus spp. |
< 4 mg/L |
> 4 mg/L |
|
Streptococcus spp |
< 2 mg/L |
> 2 mg/L |
|
Streptococcus pneumoniae |
< 2 mg/L |
> 2 mg/L |
|
Anaerobios Gram positivos |
< 2 mg/L |
< 2 mg/L |
|
Especies no relacionadas * |
< 2 mg/L |
> 4 mg/L |
*Se han determinado los puntos de corte no relacionados con la especie principalmente sobre la base de datos de PK/PD y son independientes de las distribuciones de valores CMI de especies específicas. Son para usar solamente en el caso de especies a las que no se les ha dado valores críticos específicos de especie y para no usar en el caso de las especies en las que no se recomienda utilizar pruebas de sensibilidad.
Clases_
Especies frecuentemente sensibles Gram positivas
Enterococcus faecalis Staphylococcus aureus Staphylococcus coagulasa-negativos Streptococcus spp.
Streptococcus pneumoniae
Clostridium spp._
Especies para las que la resistencia adquirida puede ser un problema
Enterococcus ^ faecium_
Intrínsecamente resistentes_
Bacterias Gram negativas Chlamydia spp.
Mycobacteria Mycoplasma spp.
Rickettsia spp._
5.2 Propiedades farmacocinéticas
5.2.1 Absorción
Vancomicina se administra por vía intravenosa para el tratamiento de infecciones sistémicas. En el caso de pacientes con función renal normal, la perfusión intravenosa de dosis múltiples de 1 g de vancomicina (15 mg/kg) durante 60 minutos produce aproximadamente concentraciones plasmáticas medias de 5060 mcg/mL, 20-25 mcg/mL y 5-10 mcg/mL, inmediatamente, transcurridas 2 horas y transcurridas 11 horas tras la finalización de la perfusión, respectivamente. La perfusión intravenosa de múltiples dosis de 500 mg de vancomicina durante 30 minutos produce aproximadamente concentraciones plasmáticas medias de 4050 mg/l, 19-20 mg/l y 10-11 mg/l, inmediatamente, transcurridas 2 horas y transcurridas 6 horas tras la finalización de la perfusión, respectivamente. Los niveles plasmáticos obtenidos tras múltiples dosis son similares a los obtenidos tras una sola dosis.
En el caso de la administración oral, la vancomicina de gran polaridad prácticamente no se absorbe.
Después de la administración oral, aparece en su forma activa en las heces y, por consiguiente, es una quimioterapia apropiada para la colitis seudomembranosa y la colitis estafilocócica
5.2.2 Distribución
A concentraciones séricas de vancomicina entre 10 mg/l y 100 mg/l, la tasa de unión del medicamento a proteínas plasmáticas, determinada mediante ultrafiltración, es del orden del 30-55 %.
Tras la administración de clorhidrato de vancomicina por vía intravenosa, se observan concentraciones inhibidoras en los líquidos pleural, pericárdico, ascítico y sinovial, en la orina y en el líquido de diálisis peritoneal, y en el tejido de la orejuela auricular.
En las meninges no inflamadas, la vancomicina no traspasa fácilmente la barrera hematoencefálica.
5.2.4 Eliminación
En el caso de los pacientes con función renal normal, el organismo tarda entre cuatro y seis horas en eliminar la mitad de una dosis de vancomicina. Durante las primeras 24 horas, aproximadamente el 80 % de la dosis de vancomicina administrada se excreta en la orina por filtración glomerular. La disfunción renal retrasa la excreción de la vancomicina. En pacientes anéfricos, el tiempo medio de eliminación de la mitad de una dosis es de 7,5 días. El medicamento apenas se metaboliza. Aproximadamente el 30-60 % de una dosis intraperitoneal de vancomicina administrada durante la diálisis peritoneal se absorbe sistémicamente en seis horas. Mediante la inyección intraperitoneal de 30 mg/kg de vancomicina se logran concentraciones séricas del orden de 8 mg/litro. Aunque la vancomicina no se elimina eficazmente mediante hemodiálisis ni diálisis peritoneal, se han comunicado casos de aumento del aclaramiento de la vancomicina mediante hemoperfusión y hemofiltración. El aclaramiento sistémico y renal total de la vancomicina puede ser menor en personas de edad avanzada.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad y toxicidad a dosis repetidas.
Aunque no se han realizado estudios de larga duración en animales sobre el potencial carcinogénico, los pocos datos existentes sobre los efectos mutagénicos muestran resultados negativos. En estudios de teratogenicidad en los que se administró a ratas y conejos dosis similares a las que corresponderían a humanos en función de la superficie corporal (mg/m2), no se observaron efectos teratogénicos directos ni indirectos.
No se han realizado estudios en animales sobre los efectos de su uso durante el período perinatal y postnatal, y sobre la fertilidad.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Ninguno.
6.2 Incompatibilidades
La solución de vancomicina tiene un pH bajo, por lo que puede causar inestabilidad química o física cuando se mezcla con otras sustancias. Debe evitarse mezclarla con soluciones alcalinas. Debe realizarse una inspección visual de cada solución parenteral antes de su uso para comprobar si se ha producido precipitación y decoloración.
]£
Este medicamento no puede mezclarse con otros excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez
Polvo envasado para su comercialización:
2 años
Concentrado reconstituido:
El concentrado reconstituido debe volver a diluirse inmediatamente después de la reconstitución. Medicamento diluido:
Desde el punto de vista microbiológico y fisicoquímico, el producto se debe utilizar de forma inmediata.
6.4 Precauciones especiales de conservación
Polvo envasado para su comercialización:
Conservar por debajo de 25 C.
Guardar el vial en el envase exterior para protegerlo de la luz.
Concentrado reconstituido y producto diluido:
Para obtener información sobre las condiciones de conservación del concentrado reconstituido y del producto diluido, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Vial de vidrio incoloro tipo 1, de 20 ml, con tapón de clorobutilo tipo 1 recubierto de silicona y precinto gris de aluminio/polipropileno.
Tamaños de envases: 1y 10 viales.
Envases múltiples con 100 viales (10 estuches con 10 viales).
Puede que solamente estén comercializados algunos tamaños de envases
6.6 Precauciones especiales de eliminación <y otras manipulaciones>
El producto debe reconstituirse y, a continuación, el concentrado resultante debe diluirse antes de utilizarlo. Preparación del concentrado reconstituido:
Disolver el contenido de cada vial de 1000 mg en 20 ml de agua estéril para inyecciones.
Aspecto del concentrado reconstituido:
Solución clara e incolora sin partículas.
Un ml de concentrado reconstituido contiene 50 mg de vancomicina.
Para las condiciones de conservación del concentrado reconstituido, ver sección 6.3.
Preparación de la solución diluida final para perfusión:
El concentrado reconstituido que contiene 50 mg/ml de vancomicina debe volver a diluirse inmediatamente después de la reconstitución.
.<ítp.
Los diluyentes apropiados son:
Cloruro de sodio 9 mg/ml (al 0,9%) para inyección, glucosa 50 mg/ml (al 5%) para inyección, mezcla de cloruro de sodio 9 mg/ml (al 0,9%) y glucosa 50 mg/ml (al 0,5%) para inyección, o Ringer-acetato para inyección.
Antes de su administración, se deben inspeccionar visualmente las soluciones reconstituidas y diluidas para descartar la presencia de partículas o decoloración. Únicamente deben utilizarse soluciones claras e incoloras sin partículas.
Perfusión intermitente:
El concentrado reconstituido que contiene 1000 mg de vancomicina (50 mg/ml) debe volver a diluirse con al menos 200 ml de diluyente inmediatamente después de la reconstitución.
La concentración de vancomicina en solución para perfusión no debe superar los 5 mg/ml.
La dosis deseada debe administrarse lentamente mediante perfusión intravenosa a una velocidad máxima de 10 mg/minuto durante al menos 60 minutos o más.
Para las condiciones de conservación del medicamento diluido, ver sección 6.3.
Eliminación
Los viales son de un solo uso. Deben descartarse los productos no utilizados.
Todo producto no utilizado y otros residuos deben desecharse de acuerdo con la normativa local aplicable.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer, S.L.
Avda. de Europa 20B
Parque Empresarial La Moraleja, 28108
Alcobendas, Madrid
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
73785
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 30 de agosto de 2011 Fecha de la última renovación: 18 de marzo de 2015
10. FECHA DE LA REVISIÓN DEL TEXTO
11/2015
¡y
taños
14 de 14