Vancomicina Kern Pharma 500 Mg Polvo Para Concentrado Para Solucion Para Perfusión Efg
Información obsoleta, busque otroFICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Vancomicina Kem Pharma 500 mg polvo para concentrado para solución para perfusión EFG
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene 500 mg de vancomicina (equivalentes a 525.000 UI) (como hidrocloruro de vancomicina). Una vez reconstituido con 10 ml de agua para preparaciones inyectables, el concentrado resultante para la solución para perfusión contiene 50 mg/ml de vancomicina.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para concentrado para solución para perfusión Polvo blanco cristalino.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
La vancomicina intravenosa está indicada en las siguientes infecciones graves causadas por bacterias gram-positivas sensibles a vancomicina que no puedan tratarse con o que no respondan o sean resistentes a otros antibióticos, como las penicilinas y las cefalosporinas.
■ endocarditis
■ infecciones óseas (osteítis, osteomielitis)
■ neumonía
■ infecciones de tejidos blandos.
La endocarditis causada por enterococos, Streptococcus viridans o S. bovis se tratará con una combinación de vancomicina y un aminoglucósido.
La vancomicina puede usarse como profilaxis perioperatoria frente a la endocarditis bacteriana, en pacientes con un alto riesgo de sufrir endocarditis bacteriana cuando se sometan a una intervención quirúrgica mayor (p.ej., intervenciones cardíacas y vasculares, etc.) y no puedan recibir un antibacteriano beta-lactámico apropiado.
Deberán tenerse en cuenta las guías oficiales sobre el uso adecuado de antibacterianos.
4.2 Posología y forma de administración
Vancomicina Kern Pharma 500 mg polvo para concentrado para solución para perfusión debe administrarse por vía intravenosa (mediante perfusión).
Cada dosis intravenosa se administrará a una velocidad que no supere los 10 mg/min y
durante un período de tiempo de al menos 60 minutos a concentraciones que no superen los 5 mg/ml (500 mg diluidos en al menos 100 ml y 1.000 mg en al menos 200 ml del disolvente apropiado). En algunos pacientes que requieran una ingesta de fluidos limitada, podrán usarse soluciones de hasta 10 mg/ml (500 mg/50 ml o 1.000 mg/100 ml). Sin embargo, el uso de concentraciones más elevadas puede incrementar el riesgo de reacciones adversas relacionadas con la perfusión (ver sección 4.8).
La dosis se adaptará individualmente según el peso, edad y función renal.
Se recomiendan las siguientes pautas posológicas:
Administración intravenosa
Pacientes con función renal normal
Adultos y adolescentes ( a partir de 12 años de edad):
La dosis intravenosa diaria recomendada es de 2 g, dividida en dosis de 500 mg cada 6 horas o 1.000 mg cada 12 horas.
Para la endocarditis bacteriana, el régimen aceptado generalmente es de 1.000 mg de vancomicina por vía intravenosa cada 12 horas durante 4 semanas solo o en combinación con otros antibióticos (gentamicina más rifampicina, gentamicina, estreptomicina). La endocarditis enterocócica se trata durante 6 semanas con vancomicina en combinación con un aminoglucósido - según las recomendaciones nacionales.
Profilaxis perioperatoria frente a la endocarditis bacteriana: Los adultos recibirán 1.000 mg de vancomicina por vía intravenosa antes de la cirugía (antes de la inducción de anestesia) y dependiendo del tiempo y tipo de cirugía, puede administrarse postoperatoriamente una dosis de 1.000 mg de vancomicina i.v. tras 12 horas.
Población pediátrica
Niños de un mes a 12 años de edad
La dosis intravenosa recomendada es de 10 mg/kg cada 6 horas.
Recién nacidos (hasta 1 mes de edad)
La dosis inicial recomendada es de 15 mg/kg, seguida de 10 mg/kg cada 12 horas durante la primera semana de vida y cada 8 horas después de esa edad y hasta 1 mes de edad. Se recomienda una estrecha vigilancia de la concentración sérica de vancomicina (ver a continuación).
Pacientes de edad avanzada
Pueden necesitarse dosis de mantenimiento inferiores debido a la reducción en la función renal relacionada con la edad.
Pacientes obesos
Puede que sea necesario modificar las dosis diarias habituales.
Pacientes con alteración de la función renal
La dosis debe ajustarse en pacientes con insuficiencia renal y el nomograma siguiente puede servir como guía. Se recomienda una estrecha vigilancia de la concentración sérica
de vancomicina (ver a continuación)._
POSOLOGIA DE VANCOMICINA EN PACIENTES CON INSUFICIENCIA RENAL
|
Aclaramiento de creatinina (ml/min) |
Dosis de vancomicina (mg/24 horas) |
|
100 |
1.545 |
|
90 |
1.390 |
|
80 |
1.235 |
|
70 |
1.080 |
|
60 |
925 |
|
50 |
770 |
|
40 |
620 |
|
30 |
465 |
|
20 |
310 |
|
10 |
155 |
Aclaramiento de creatinina (ml/s)
0 50 1 00 1 50
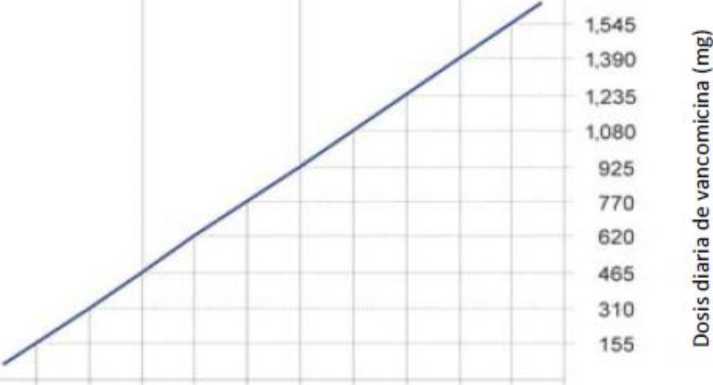
10 20 30 40 50 60 70 80 90 100
Aclaramiento de creatinina (ml/m¡n)
Nomograma de dosis para adultos con insuficiencia renal
En pacientes con insuficiencia renal leve o moderada, la dosis inicial no debe ser inferior a 15 mg/kg. En pacientes con insuficiencia renal grave, es preferible administrar una dosis de mantenimiento entre 250 mg y 1.000 mg espaciada varios días en vez de administrar dosis diarias inferiores.
Los pacientes con anuria (prácticamente sin función renal) recibirán una dosis de 15 mg/kg
|
de peso corporal hasta llegar a la concentración sérica terapéutica. Las dosis de mantenimiento son de 1,9 mg/kg de peso corporal cada 24 horas. Para facilitar la intervención, los pacientes adultos con una grave insuficiencia renal pueden obtener una dosis de mantenimiento de 250 - 1.000 mg a intervalos de varios días en vez de una dosis diaria. Posología en caso de hemodiálisis En pacientes sin función renal, incluso bajo hemodiálisis habitual, también es posible la posología siguiente: Dosis de inicio 1.000 mg, dosis de mantenimiento 1.000 mg cada 7 - 10 días. Si se usan membranas de polisulfona para hemodiálisis (“diálisis de alto flujo“), se acorta la semivida de vancomicina. En pacientes sometidos a hemodiálisis periódicamente, puede ser necesaria una dosis de mantenimiento adicional. Pacientes con insuficiencia hepática No hay pruebas de que la dosis tenga que reducirse en pacientes con insuficiencia hepática. Medida de las concentraciones séricas de vancomicina La concentración sérica de vancomicina se vigilará el segundo día de tratamiento inmediatamente antes de la dosis siguiente, y una hora después de la perfusión. Los niveles terapéuticos de vancomicina en sangre oscilarán entre 30 y 40 mg/l (máximo 50 mg/l) una hora después de finalizar la perfusión, el nivel mínimo (poco antes de la siguiente administración) entre 5 y 10 mg/l. Las concentraciones normalmente se medirán dos o tres veces a la semana. Forma de administración La vancomicina parenteral sólo se administrará por vía intravenosa mediante perfusión lenta (no más de 10 mg/min - durante al menos 60 min) que esté suficientemente diluida (al menos 100 ml por 500 mg o al menos 200 ml por 1.000 mg). Los pacientes que requieran una restricción de líquidos pueden recibir una solución de 500 mg / 50 ml o 1.000 mg / 100 ml. Con estas concentraciones más elevadas, puede aumentar el riesgo de reacciones adversas relacionadas con la perfusión. Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6. Duración del tratamiento La duración del tratamiento depende de la gravedad de la infección y de la evolución clínica y bacteriológica. 4.3 Contraindicaciones Hipersensibilidad a vancomicina. 4.4 Advertencias y precauciones especiales de empleo Advertencias En presencia de anuria aguda o daño coclear, la vancomicina sólo debe usarse en indicaciones vitales. |
*lí¡¡S* |
|
Página 4 |
En caso de reacciones de hipersensibilidad aguda graves (p.ej., anafilaxia), debe suspenderse inmediatamente el tratamiento con vancomicina e instituirse las medidas de emergencia apropiadas (p.ej., antihistamínicos, corticoesteroides, y - si es necesario -respiración artificial).
La administración rápida de bolos está relacionada con hipotensión, choque y en raras ocasiones parada cardíaca (ver sección 4.8) y, por tanto, debe administrarse diluida en un período no inferior a 60 minutos y a una velocidad no mayor de 10 mg/min. Estas reacciones cesan tras la interrupción de la perfusión.
Nefrotoxicidad: la vancomicina debe usarse con precaución en pacientes con insuficiencia renal ya que la posibilidad de aparición de efectos tóxicos es mucho mayor en presencia de concentraciones elevadas en sangre prolongadas. En el tratamiento de estos pacientes y en los que estén recibiendo tratamiento concomitante con otros medicamentos nefrotóxicos (p.ej., aminoglucósidos), deben realizarse pruebas en serie de la función renal y seguirse las pautas posológicas adecuadas para reducir el riesgo de nefrotoxicidad al mínimo (ver sección 4.2).
Ototoxicidad: se ha comunicado ototoxicidad, que puede ser pasajera o permanente (ver sección 4.8), en pacientes con sordera previa, que hayan recibido dosis intravenosas excesivas o que reciban tratamiento concomitante con otros medicamentos ototóxicos como un aminoglucósido. El inicio de sordera puede ir precedido de acúfenos. El riesgo de daños auditivos es mayor en pacientes de edad avanzada. Para reducir el riesgo de ototoxicidad, deben medirse periódicamente los niveles en sangre y se recomienda realizar pruebas periódicas de la función auditiva. La sordera puede ser progresiva pese al cese del tratamiento.
La vancomicina debe administrarse sólo por vía intravenosa (mediante perfusión), debido al riesgo de necrosis. El riesgo de irritación venosa se reduce al mínimo administrando vancomicina en forma de una perfusión diluida (2,5 a 5 g/l) y cambiando el punto de inyección.
La administración de vancomicina por inyección intraperitoneal durante la diálisis peritoneal ambulatoria continua se ha relacionado con un síndrome de peritonitis química.
Precauciones
Puede aparecer tromboflebitis, cuya frecuencia y gravedad pueden reducirse al mínimo administrando el medicamento lentamente y en forma diluida (2,5 a 5 g/l), así como cambiando los puntos de inyección.
La frecuencia de reacciones relacionadas con la perfusión (hipotensión, enrojecimiento, eritema, urticaria y prurito) aumenta con la administración concomitante de anestésicos. Ésta puede reducirse administrando vancomicina por perfusión durante 60 minutos, antes de la inducción anestésica.
La vancomicina debe administrarse con precaución en pacientes alérgicos a la teicoplanina, ya que se han detectado reacciones alérgicas cruzadas entre la vancomicina y la teicoplanina.
Se ha comunicado colitis pseudomembranosa con casi todos los antibacterianos, incluida la vancomicina, y su gravedad puede oscilar de leve a potencialmente mortal. Por tanto, es
importante considerar este diagnóstico en los pacientes que presenten diarrea posterior a la administración de vancomicina. Están contraindicados los antiperistálticos.
Igual que con otros antibióticos, el uso prolongado de vancomicina puede producir un crecimiento excesivo de bacterias y hongos no sensibles. Es esencial una estrecha vigilancia del paciente. Deben adoptarse las medidas oportunas si se produce sobreinfección durante el tratamiento.
Se ha comunicado la aparición de neutropenia reversible (ver sección 4.8.). Debe realizarse un hemograma periódico en los pacientes que reciban tratamientos prolongados con vancomicina o tratamiento concomitante con medicamentos que puedan provocar neutropenia.
La vancomicina puede potenciar la depresión del miocardio inducida por anestésicos. Durante la anestesia, las dosis deben diluirse bien y administrarse lentamente, con una estrecha monitorización cardíaca. Los cambios de posición se retrasarán hasta que se termine la perfusión para poder ajustar la postura.
Uso en pacientes de edad avanzada
Debido a su ototoxicidad y nefrotoxicidad, la vancomicina se usará con precaución en los pacientes con insuficiencia renal o pérdida auditiva previa. Los pacientes de edad avanzada tienen un riesgo especial. Las dosis se ajustarán sobre la base de los niveles séricos. Se vigilarán los niveles en sangre y se realizarán pruebas de función renal periódicamente. Los pacientes de edad avanzada son especialmente propensos a las lesiones auditivas y se someterán a pruebas en serie de la función auditiva si tienen más de 60 años. Debe evitarse el uso concomitante o secuencial de otras sustancias neurotóxicas.
Uso en lactantes/niños
La vancomicina se usará con especial precaución en lactantes y niños prematuros, por su inmadurez renal y el posible aumento de la concentración sérica de vancomicina. Por tanto, se vigilarán atentamente las concentraciones de vancomicina en la sangre. El uso concomitante de vancomicina y anestésicos en niños se ha relacionado con eritema y reacciones anafilactoides. Si fuese necesario administrar vancomicina como profilaxis quirúrgica, es aconsejable administrar los anestésicos después de terminar la perfusión de vancomicina (ver sección 4.8).
La vigilancia periódica de los niveles de vancomicina en sangre está indicada en uso a largo plazo, especialmente en pacientes con alteración de la función renal o deterioro de la función auditiva, así como en la administración concomitante de sustancias nefrotóxicas u ototóxicas, respectivamente.
4.5 Interacción con otros medicamentos y otras formas de interacción
Otros medicamentos potencialmente nefrotóxicos u ototóxicos
La administración concomitante o secuencial de vancomicina con otras sustancias activas potencialmente neurotóxicas o nefrotóxicas, especialmente gentamicina, anfotericina B, estreptomicina, neomicina, kanamicina, amikacina, tobramicina, viomicina, bacitracina, polimixina B, colistina y cisplatino, puede potenciar la nefrotoxicidad y la ototoxicidad de la vancomicina y en consecuencia requiere una atenta vigilancia del paciente.
Debido a la acción sinérgica (p.ej., con gentamicina) en estos casos la dosis máxima de
vancomicina debe limitarse a 500 mg cada 8 horas.
Anestésicos
La administración concomitante de vancomicina y anestésicos se ha relacionado con eritema, sofocos de tipo histamina y reacciones anafilactoides. Esto puede reducirse si la vancomicina se administra por perfusión en 60 minutos, antes de la inducción anestésica.
Relajantes musculares
Si la vancomicina se administra durante o directamente después de la cirugía, el efecto (bloqueo neuromuscular) de los relajantes musculares (como succinilcolina) usados de forma concomitante puede potenciarse y prolongarse.
4.6 Embarazo y lactancia
Embarazo
No hay experiencia suficiente con vancomicina durante el embarazo en humanos. Los estudios de toxicología en la reproducción animal no indican efectos en el desarrollo del embrión, feto o el período de gestación (ver sección 5.3).
Sin embargo, la vancomicina penetra en la placenta y no puede excluirse un posible riesgo de ototoxicidad y nefrotoxicidad embrionaria y neonatal. Por tanto, la vancomicina se administrará en el embarazo sólo si está claramente indicado y tras una minuciosa evaluación del riesgo/beneficio.
Lactancia
Vancomicina se excreta en la leche humana y por tanto sólo debe usarse en el período de lactancia si otros antibióticos han fallado. La vancomicina debe administrarse con precaución a las madres lactantes debido a las posibles reacciones adversas en lactantes (alteraciones de la flora intestinal con diarrea, colonización con un hongo de tipo levadura y posiblemente sensibilización).
Teniendo en cuenta la importancia de este medicamento para la madre en período de lactancia, se considerará la decisión de interrumpir la lactancia.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de vancomicina sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Las reacciones adversas más frecuentes son flebitis y reacciones pseudoalérgicas relacionadas con un uso intravenoso demasiado rápido (por perfusión) de vancomicina.
En cada grupo de frecuencia, las reacciones adversas se presentan por orden de gravedad decreciente.
Las reacciones adversas que se mencionan a continuación se definen mediante la siguiente convención de MedDRA y base de datos de clasificación de órganos del sistema: muy frecuentes (1/10); frecuentes (1/100 a < 1/10); poco frecuentes (1/1.000 a < 1/100); raras (1/10.000 a < 1/1.000); muy raras (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Trastornos de la sangre y del sistema linfático:
Raros (> 10.000 a <1/1.000): Trombocitopenia,
eosinofilia.
neutropenia, agranulocitosis,
Trastornos del sistema inmunológico:
Raros (> 10.000 a <1/1.000): Reacciones anafilácticas,
hipersensibilidad.
reacciones
de
Trastornos del oído y del laberinto:
Poco frecuentes (> 1.000 a <1/1 00): Pérdida auditiva pasajera o permanente.
Raros (> 10.000 a <1/1.000):
Trastornos cardíacos Muy raros (< 1/10.000)):
Trastornos vasculares: Frecuentes (> 1/100 a < 1/10): Raros (> 10.000 a <1/1.000):
Acúfenos, vértigo.
Parada cardíaca
Reducción de la presión arterial, tromboflebitis. Vasculitis.
Trastornos respiratorios, torácicos y mediastínicos: Frecuentes (> 1/100 a < 1/10): Disnea, estridor.
Trastornos gastrointestinales: Raros (> 10.000 a <1/1.000): Muy raros (< 1/10.000))
Náuseas
Enterocolitis pseudomembranosa.
Trastornos de la piel y del tejido subcutáneo:
Exantema e inflamación de la mucosa, prurito, urticaria.
Dermatitis exfoliativa, síndrome de Stevens-Johnson, síndrome de Lyell, vasculitis, dermatitis bullosa inducida por IgA
Frecuentes (> 1/100 a < 1/10): Muy raros (< 1/10.000):
Trastornos renales y urinarios: Frecuentes (> 1/100 a < 1/10):
Raros (> 10.000 a <1/1.000):
Insuficiencia renal que se manifiesta principalmente por un aumento de la creatinina sérica o concentraciones séricas de urea.
Nefritis intersticial, insuficiencia renal aguda.
Trastornos generales y alteraciones en el lugar de administración:
Frecuentes (> 1/100 a < 1/10): Enrojecimiento en la parte superior del cuerpo y de la
cara. Dolor y espasmo del tórax y músculos de la espalda.
Raros (> 10.000 a <1/1.000): Fiebre medicamentosa, temblores.
Durante o poco después de la perfusión rápida, pueden producirse reacciones anafilácticas. Las reacciones remiten cuando se suspende la administración, generalmente entre 20 minutos y 2 horas después de terminar la administración.
La ototoxicidad se ha comunicado principalmente en pacientes que reciben dosis elevadas o tratamiento concomitante con otros medicamentos ototóxicos o con una reducción preexistente de la función renal o la audición.
4.9 Sobredosis
Se ha comunicado toxicidad por sobredosis. 500 mg IV a un niño de 2 años de edad provocaron una intoxicación mortal. La administración de un total de 56 g durante 10 días a un adulto produjo una intoxicación mortal. En ciertos grupos de riesgo (p.ej., en caso de insuficiencia renal grave), pueden aparecer niveles séricos elevados y efectos oto- y nefrotóxicos.
Medidas en caso de sobredosis
• No se conoce un antídoto específico.
• Las concentraciones elevadas pueden reducirse de forma eficaz por hemodiálisis utilizando membranas de polisulfona o por hemofiltrado o hemoperfusión utilizando resina de polisulfona.
• Además, en caso de sobredosis, es necesario un tratamiento sintomático bajo mantenimiento de la función renal.
5. PROPIEDADES FARMACOLÓGICAS 5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antibacterianos glucopéptidos Código ATC: J01XA01 Mecanismo de acción
La vancomicina es un antibiótico glucopéptido. La vancomicina tiene un efecto bactericida en los patógenos proliferantes inhibiendo la biosíntesis de la pared celular. Además, altera la permeabilidad de la membrana de la célula bacteriana y la síntesis de ARN.
Relación FC/FD
La actividad de la vancomicina se considera dependiente del tiempo - es decir, la actividad antimicrobiana depende de la duración en que el nivel del fármaco supera la concentración mínima inhibitoria (CMI) del organismo diana.
Mecanismo de resistencia
La resistencia adquirida a los glucopéptidos se basa en la adquisición de distintos complejos de genes van y la alteración del objetivo D-alanil-D-alanina a D-alanil-D-lactato o D-alanil-D-serina que se une mal a la vancomicina, porque falta un punto crítico para las uniones de hidrógeno. Esta forma de resistencia se observa especialmente en Enterococcus faecium.
No se conoce bien la reducción de la sensibilidad o resistencia a la vancomicina en Staphylococcus. Se necesitan varios elementos genéticos y numerosas mutaciones.
Se ha comunicado resistencia cruzada con teicoplanina.
Sensibilidad
La vancomicina es activa frente a las bacterias gram-positivas. Las bacterias gram-negativas son resistentes.
Los puntos límite de la CMI que separan los microorganismos sensibles de los resistentes son:
sobre la base de los datos de FC/FD y son independientes de las distribuciones de la CMI de especies específicas. Son para uso sólo para especies a las que no se ha adjudicado un punto límite específico de especie y no para especies en las que no se recomienden pruebas de sensibilidad.
|
Recomendaciones del EUCAST (Comité |
europeo de |
pruebas de sensibilidad | |
|
antimicrobiana) |
Sensible |
Resistente | |
|
Staphylococcus spp. |
< 2 mg/l |
>2mg/l | |
|
Enterococcus spp. |
< 4 mg/l |
>4mg/l | |
|
Streptococcus spp |
< 2 mg/l |
>2mg/l | |
|
Streptococcus pneumoniae |
< 2 mg/l |
>2mg/l | |
|
Anaerobios gram-positivos |
< 2 mg/l |
>2mg/l | |
|
Puntos de corte no |
< 2 mg/l |
>4mg/l | |
|
relacionados con especies | |||
|
específicas* | |||
|
* Se han determinado los puntos de corte no relacionados |
con especies principalmente | ||
La prevalencia de resistencia adquirida podrá variar geográficamente y con el tiempo en especies seleccionadas y es deseable una información local sobre la resistencia, especialmente al tratar infecciones graves. Cuando sea necesario, debe buscarse el asesoramiento de un experto si la prevalencia local de la resistencia es tal que la utilidad del fármaco sea cuestionable, al menos en algunos tipos de infecciones.
Clases
Especies frecuentemente sensibles Bacterias Gram-positivas Enterococcus faecalis.
Staphylococcus aureus Staphylococcus coagulasa negativo Staphylococcus epidermidis Streptococcus spp.
Streptococcus pneumoniae
Especies para las que la resistencia adquirida puede suponer un problema
Enterococcus faecium Streptococcus bovis Streptococcus viridans Especies con resistencia intrínseca Bacterias Gram-negativas Chlamydia spp.
Mycobacteria Mycoplasma spp.
Rickettsia spp.
5.2 Propiedades farmacocinéticas
La vancomicina no se absorbe de manera significativa desde el aparato gastrointestinal normal.
Distribución
Tras su administración intravenosa, la vancomicina se distribuye a casi todos los tejidos y se difunde en el líquido pleural, pericárdico, ascítico y sinovial así como en el músculo cardíaco y las válvulas del corazón. Se logran concentraciones elevadas comparables a las
|
del plasma. Los datos sobre las concentraciones de vancomicina en hueso (esponjoso, compacto) varían ampliamente. El volumen de distribución aparente en estado estacionario es de 0,43 (hasta 0,9) l/kg. En las meninges no inflamadas, la vancomicina atraviesa la barrera hematoencefálica sólo en poca medida. La vancomicina se une a las proteínas plasmáticas en un 30 al 55% e incluso más. Eliminación La vancomicina se metaboliza sólo en bajo grado. Tras la administración parenteral se excreta casi totalmente como principio microbiológicamente activo (aprox. 75-90% antes de 24 horas) por filtración glomerular a través de los riñones. La excreción biliar es insignificante (menos del 5% de una dosis). En pacientes con función renal normal, la semi-vida en suero es de alrededor de 4-6 (5-11) horas y en niños de 2,2-3 horas. En la insuficiencia renal, la semivida de la vancomicina puede prolongarse considerablemente (hasta 7,5 días). Debido a la ototoxicidad del tratamiento con vancomicina, en estos casos está indicada la vigilancia complementaria de las concentraciones plasmáticas. Las concentraciones plasmáticas medias tras la perfusión i.v. de 1.000 mg de vancomicina en 60 minutos fueron de alrededor de 63 mg/l al final de la perfusión, alrededor de 23 mg/l a las 2 horas y alrededor de 8 mg/l a las 11 horas. El aclaramiento de vancomicina del plasma es casi el mismo que la velocidad de filtración glomerular. El aclaramiento sistémico y renal total de vancomicina puede reducirse en los pacientes de edad avanzada. Como mostraron los estudios en pacientes anéfricos, el aclaramiento metabólico parece ser muy bajo. No se han detectado hasta ahora metabolitos de la vancomicina en el hombre. Si la vancomicina se administra durante una diálisis peritoneal por vía intraperitoneal, aproximadamente el 60% llega a la circulación sistémica durante 6 horas. Tras la administración i.p. de 30 mg/kg de peso, se logran niveles séricos de alrededor de 10 mg/l. La vancomicina difunde enseguida a través de la placenta y se distribuye a la sangre del cordón. 5.3 Datos preclínicos sobre seguridad Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad y toxicidad a dosis repetidas. Los datos limitados sobre efectos mutágenos muestran resultados negativos, no se dispone de estudios a largo plazo en animales sobre carcinogenicidad. En estudios de teratogenicidad, en los que ratas y conejos recibieron dosis aproximadamente correspondientes a la dosis humana sobre la base de la superficie corporal (mg/m2), no se observaron efectos teratógenos directos o indirectos. No se dispone de estudios en animales sobre su uso durante el período perinatal/postnatal ni respecto a los efectos sobre la fertilidad. |
*lí¡¡S* |
|
Página 11 |
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Ácido clorhídrico (para ajuste del pH).
6.2 Incompatibilidades
La solución de vancomicina presenta un pH bajo que puede provocar inestabilidad química o física cuando se mezcla con otros compuestos. Debe evitarse la mezcla con soluciones alcalinas. Cada solución parenteral debe inspeccionarse visualmente en cuanto a precipitado y decoloración antes de su uso.
Se ha demostrado que las mezclas de soluciones de vancomicina y antibióticos beta-lactámicos son físicamente incompatibles. La posibilidad de precipitado aumenta con las mayores concentraciones de vancomicina. Se recomienda aclarar debidamente las vías intravenosas entre las administraciones de estos antibióticos. Se recomienda también diluir las soluciones de vancomicina a 5 mg/ml o menos.
Aunque la inyección intravítrea no es una vía de administración aprobada para la vancomicina, se ha comunicado precipitado tras la inyección intravítrea de vancomicina y ceftazidima por endoftalmitis utilizando distintas jeringas y agujas. Los precipitados se disuelven progresivamente, aclarándose totalmente la cavidad vítrea en dos meses y con una mejora de la agudeza visual.
Este medicamento no debe mezclarse con otros medicamentos, excepto los que se mencionan en la sección 6.6.
6.3 Período de validez
Polvo en su envase original sin reconstituir Frasco vial de 500 mg: 3 años.
Tras la reconstitución/dilución
Desde un punto de vista microbiológico, el producto debe usarse inmediatamente. Si no se usa inmediatamente, los tiempos de conservación durante el uso y las condiciones previas al uso son responsabilidad del usuario y normalmente no deberán superar las 24 horas a 2-8°C.
6.4 Precauciones especiales de conservación
Polvo
Conservar por debajo de 25°C. Conservar el vial en el embalaje exterior para protegerlo de la luz.
Tras la reconstitución / dilución
Para las condiciones de conservación del medicamento reconstituido y diluido, ver sección 6.3.
Antes de su administración, los medicamentos parenterales se inspeccionarán visualmente
en cuanto a partículas y decoloración siempre que la solución o el recipiente lo permitan.
6.5 Naturaleza y contenido del envase
tapados con cierres de caucho (tipo I) y con cápsula de aluminio y un tapón de
Viales transparentes de vidrio (tipo I) de 19 ml, cerrados con tapón de caucho (tipo I) y sellado plástico azul sobrepuesto (flip-off).
Tamaños de envase: 1, 5, 10 y 20 unidades.
Puede que solamente están comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El polvo debe reconstituirse y el concentrado resultante debe diluirse antes de su uso.
Preparación del concentrado reconstituido
En el momento de su uso, el contenido de cada vial de 500 mg se disuelve en 10 ml de agua para inyección. Un ml de solución reconstituida contiene 50 mg de vancomicina.
Para las condiciones de conservación del medicamento reconstituido, véase la sección 6.3.
Preparación de la solución diluida final para perfusión
Las soluciones reconstituidas que contienen 50 mg/ml de vancomicina se diluirán más, dependiendo del método de administración.
Los diluyentes adecuados son:
Solución de glucosa al 5% (50 mg/ml) o Solución de cloruro sódico al 0,9% (9 mg/ml).
Perfusión intermitente
La concentración de vancomicina en solución para perfusión no debe superar los 5 mg/ml.
La dosis deseada se administrará por vía intravenosa, mediante perfusión lenta a una velocidad de no más de 10 mg/minuto, durante al menos 60 minutos o incluso más.
Si se administra durante un período de tiempo más breve o a mayores concentraciones, existe la posibilidad de inducir una notable hipotensión además de tromboflebitis. La administración rápida también produce enrojecimiento y un eczema pasajero en el cuello y los hombros.
Perfusión continua
Se usará sólo si no es posible el tratamiento con perfusión intermitente. Se diluyen 1.000 mg a 2 g de vancomicina disuelta en una cantidad suficiente del diluyente anterior adecuado y se administra en forma de perfusión por goteo, de forma que el paciente recibirá la dosis diaria prescrita en 24 horas.
Para las condiciones de conservación del medicamento diluido, ver sección 6.3.
Antes de la administración, se inspeccionarán visualmente las soluciones reconstituidas y
diluidas en cuanto a partículas y decoloración. Sólo se usará una solución transparente e incolora libre de partículas.
Eliminación
Los viales son para un solo uso. Debe desecharse el producto no utilizado.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
KERN PHARMA, S.L.
Pol. Ind. Colón II, C/ Venus 72 08228 Terrassa (Barcelona)
España
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Febrero de 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
16 de Diciembre de 2010
Página 14