Translarna 1000 Mg Granulado Para Suspension Oral
Información obsoleta, busque otroANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Translarna 125 mg granulado para suspensión oral
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada sobre contiene 125 mg de atalureno.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Granulado para suspensión oral.
Granulado de color entre blanco y blanquecino.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Translarna está indicado para el tratamiento de la distrofia muscular de Duchenne (DMD) debida a una mutación sin sentido en el gen de la distrofina, en pacientes ambulantes a partir de 5 o más años de edad (ver sección 5.1). No se ha demostrado eficacia en pacientes no ambulantes.
La presencia de una mutación sin sentido en el gen de la distrofina se debe determinar mediante pruebas genéticas (ver sección 4.4).
4.2 Posología y forma de administración
El tratamiento con Translarna se debe iniciar solo por médicos especialistas con experiencia en el control de la distrofia muscular de Duchenne/Becker.
Posología
Atalureno se debe administrar por vía oral, 3 veces al día.
La primera toma se debe administrar por la mañana, la segunda al mediodía y la tercera por la noche. Los intervalos posológicos recomendados son de 6 horas entre la toma de la mañana y la del mediodía; de 6 horas entre la del mediodía y la de la noche, y de 12 horas entre la de la noche y la primera toma del día siguiente.
La dosis recomendada es de 10 mg/kg de peso corporal por la mañana, 10 mg/kg de peso corporal a mediodía y 20 mg/kg de peso corporal por la noche (para una dosis diaria total de 40 mg/kg de peso corporal).
Translarna se presenta en sobres de 125 mg, 250 mg o 1.000 mg. La siguiente tabla ofrece información sobre la o las dosis del sobre que se deben emplear para preparar la posología recomendada según el intervalo de peso corporal.
|
Intervalo de peso (kg) |
Número de sobres | |||||||||
|
Mañana |
Mediodía |
Noche | ||||||||
|
Sobres de 125 mg |
Sobres de 250 mg |
Sobres de 1.000 mg |
Sobres de 125 mg |
Sobres de 250 mg |
Sobres de 1.000 mg |
Sobres de 125 mg |
Sobres de 250 mg |
Sobres de 1.000 mg | ||
|
12 |
14 |
1 |
0 |
0 |
1 |
0 |
0 |
0 |
1 |
0 |
|
15 |
16 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
1 |
0 |
|
17 |
20 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
|
21 |
23 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
1 |
0 |
|
24 |
26 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
0 |
|
27 |
31 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
0 |
|
32 |
35 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
0 |
|
36 |
39 |
1 |
1 |
0 |
1 |
1 |
0 |
0 |
3 |
0 |
|
40 |
44 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
3 |
0 |
|
45 |
46 |
0 |
2 |
0 |
0 |
2 |
0 |
1 |
3 |
0 |
|
47 |
55 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
0 |
1 |
|
56 |
62 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
1 |
1 |
|
63 |
69 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
1 |
1 |
|
70 |
78 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
2 |
1 |
|
79 |
86 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
3 |
1 |
|
87 |
93 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
3 |
1 |
|
94 |
105 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
|
106 |
111 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
|
112 |
118 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
|
119 |
125 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
2 |
2 |
Retraso u olvido de la toma de la dosis
Si hay un retraso en la administración de atalureno de menos de 3 horas después de la toma habitual de la mañana o del mediodía, o de menos de 6 horas después de la de la noche, se deberá tomar la dosis sin realizar cambios en la pauta posológica. Si hay un retraso de más de 3 horas después de la toma habitual de la mañana o del mediodía, o de más de 6 horas después de la de la noche, no se debe tomar la dosis, y el paciente debe reanudar su pauta posológica habitual. Los pacientes no deben tomar una dosis doble o adicional para compensar una dosis olvidada. Es importante administrar la dosis correcta. El aumento de la dosis por encima de la dosis recomendada puede tener como resultado una reducción de la eficacia.
Poblaciones especiales
Pacientes de edad avanzada
No se ha establecido todavía la seguridad y eficacia de atalureno en pacientes de 65 años o más. (Ver sección 5.2).
Insuficiencia renal y hepática
No se ha establecido ni la seguridad ni la eficacia de atalureno en pacientes con insuficiencia renal o hepática. (Ver sección 4.4).
Población pediátrica
No se ha establecido todavía ni la seguridad ni la eficacia de Translama en niños con edades comprendidas entre los 6 meses y los 5 años de edad. No se dispone de datos.
Forma de administración
Translarna se debe administrar por vía oral después de mezclarlo para formar una suspensión en líquido o con un alimento semisólido.
No se deben abrir los sobres hasta el momento de la preparación de la dosis. Se debe mezclar todo el contenido de cada sobre con un mínimo de 30 ml de líquido (agua, leche o zumo de fruta) o 3 cucharadas soperas de un alimento semisólido (yogur o compota de manzana). La dosis preparada se debe mezclar bien antes de su administración. Se puede aumentar la cantidad de líquido o alimento semisólido según las preferencias del paciente. Los pacientes deben tomar la dosis completa.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Uso concomitante de aminoglucósidos por vía intravenosa (ver sección 4.4 y 4.5).
4.4 Advertencias y precauciones especiales de empleo Pacientes que no tienen una mutación sin sentido
Los pacientes deben tener una mutación sin sentido en el gen de la distrofina, determinada mediante análisis genéticos, como parte de su estado patológico subyacente. No se debe administrar atalureno a pacientes que no tengan una mutación sin sentido.
Insuficiencia renal y hepática
Los pacientes con insuficiencia renal y hepática deben ser sometidos a un estrecho control.
Cambios en el perfil lipídico
Puesto que se han notificados cambios en el perfil lipídico (aumento de triglicéridos y colesterol) de algunos pacientes en ensayos clínicos, se recomienda realizar, en los pacientes con distrofia muscular de Duchene debida a una mutación sin sentido (DMDmss) que reciban atalureno, un control del colesterol total, LDL, HDL y triglicéridos cada año, o con mayor frecuencia si es necesario por el estado clínico del paciente.
Hipertensión con el uso concomitante de corticosteroides sistémicos
Puesto que se han notificado casos de hipertensión con el uso concomitante de corticosteroides sistémicos en algunos pacientes en ensayos clínicos, se recomienda realizar, en los pacientes con DMDmss que reciban atalureno de forma concomitante con corticosteroides, un control de la tensión arterial sistólica y diastólica en reposo cada 6 meses, o con mayor frecuencia si es necesario por el estado clínico del paciente.
Monitorización de la función renal
Puesto que se han notificados ligeros aumentos de las concentraciones medias de creatinina sérica, nitrógeno ureico en sangre ( BUN, por sus siglas en inglés) y de cistatina C en el estudio comparativo de DMDmss, se recomienda realizar, en los pacientes con DMDmss que reciban atalureno, un control de la creatinina sérica, el BUN y la cistatina C cada 6-12 meses, o con mayor frecuencia si es necesario por el estado clínico del paciente.
Interacciones potenciales con otros medicamentos
Se debe actuar con precaución al administrar atalureno de forma conjunta con medicamentos que sean sustratos o inductores de UGT1A9, inhibidores de BCRP o sustratos de OAT1, OAT3 u OATP1B3 (ver sección 4.5).
Aminoglucósidos
Se ha demostrado que los aminoglucósidos reducen la actividad de lectura ribosómica del atalureno in vitro. Asimismo, se encontró que el atalureno aumenta la nefrotoxicidad de los aminoglucósidos intravenosos. Se debe evitar la administración conjunta de esos medicamentos con atalureno (ver sección 4.3). Dado que se desconoce el mecanismo mediante el cual el atalureno aumenta la nefrotoxicidad de los aminoglucósidos intravenosos, no se recomienda el uso concomitante de otros medicamentos nefrotóxicos con atalureno. En caso de que esto sea inevitable, (por ejemplo: vancomicina para el tratamiento de SARM) se recomienda una monitorización minuciosa de la función renal (ver sección 4.5).
4.5 Interacción con otros medicamentos y otras formas de interacción
Aminoglucósidos
No se debe administrar Atalureno de forma conjunta con aminoglucósidos intravenosos, pues se han observado casos de disminución de la función renal en un ensayo clínico en pacientes con nmCF (ver sección 4.3).
En varios pacientes tratados con atalureno y aminoglucósidos intravenosos junto con otros antibióticos para los brotes de la fibrosis quística se produjeron elevaciones de la creatinina sérica. Las elevaciones de la creatinina sérica remitieron en todos los casos cuando se interrumpió la administración del aminoglucósido intravenoso, tanto si se continuó como si se interrumpió la administración de Translarna. Estos datos sugieren que la administración conjunta de Translama y aminoglucósidos intravenosos podría potenciar el efecto nefrotóxico de los aminoglucósidos. En consecuencia, si es necesario el tratamiento con aminoglucósidos intravenosos se debe interrumpir el tratamiento con Translarna, que se podrá reanudar 2 días después de finalizar la administración del aminoglucósido. Se desconoce el efecto de la administración conjunta de ataluren con otros medicamentos nefrotóxicos.
La deshidratación podría ser un factor que contribuyera en alguno de estos casos. Los pacientes se deben mantener adecuadamente hidratados mientras tomen atalureno. Ver sección 4.4.
Efectos de otros medicamentos en la farmacocinética de atalureno
Los datos obtenidos en estudios in vitro indican que atalureno es un sustrato de UGT1A9 y de la proteína resistente al cáncer de mama (BCRP). Se debe actuar con precaución cuando se administra de forma conjunta atalureno con medicamentos inductores de UGT1A9 (como el mofetil micofenolato) o inhibidores de BCRP (por ejemplo, ciclosporina).
Efectos de atalureno sobre la farmacocinética de otros medicamentos
Los datos obtenidos en estudios in vitro indican que atalureno es un inhibidor de UGT1A9, del transportador de aniones orgánico 1 (OAT1), del transportador de aniones orgánico 3 (OAT3) y del polipéptido de transporte del anión orgánico 1B3 (OATP1B3). Se debe actuar con precaución cuando se administra de forma conjunta atalureno con medicamentos sustratos de UGT1A9, OAT1, OAT3 u OATP1B3 (por ejemplo, oseltamivir, aciclovir, ciprofloxacino, captoprilo, furosemida, bumetanida, valsartán, pravastatina, rosuvastatina, atorvastatina o pitavastatina), debido al riesgo del aumento de la concentración de estos medicamentos .
Basándose en estudios in vitro no se espera que atalureno sea un inhibidor del transporte mediado por P-gp ni del metabolismo mediado por el citocromo P450. Del mismo modo, resulta poco probable que atalureno sea in vivo un inductor de las isoenzimas del citocromo P450.
La administración conjunta de corticosteroides (deflazacort, prednisona o prednisolona) con atalureno no afectó a las concentraciones plasmáticas de atalureno. No se observó ningún cambio clínicamente relevante en las concentraciones plasmáticas de los corticoides con la administración conjunta de atalureno. Estos datos indican que no existe interacción farmacológica aparente entre los corticosteroides y atalureno, no siendo necesario ningún ajuste de la dosis.
Medicamentos que afectan el transportador de glucoproteína P
In vitro, atalureno no es un sustrato del transportador de glucoproteína P. Es poco probable que la farmacocinética de atalureno resulte afectada por medicamentos que inhiben el transportador de glucoproteína P.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos suficientes relativos al uso de atalureno en mujeres embarazadas. Los estudios realizados en animales han revelado una toxicidad para la reproducción solamente con dosis tóxicas para la madre (ver sección 5.3).
Como medida de precaución no se recomienda utilizar atalureno durante el embarazo.
Lactancia
Se desconoce si el atalureno/sus metabolitos se excretan en la leche materna. Los datos farmacodinámicos/toxicológicos disponibles en animales muestran que el atalureno/ sus metabolitos se excretan en la leche (ver sección 5.3). No se puede excluir el riesgo en recién nacidos/niños lactantes.
Debe interrumpirse la lactancia durante el tratamiento con atalureno.
Fertilidad
Los datos preclínicos no muestran riesgos para los seres humanos en base a los estudios de fertilidad estándar con machos y hembras de rata (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se ha estudiado el efecto de atalureno sobre la conducción y la utilización de máquinas. Los pacientes que experimenten mareos deben tener cuidado si conducen o utilizan máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
En ensayos clínicos en pacientes con distrofia muscular de Duchenne debida a una mutación sin sentido (DMDmss), las reacciones adversas más frecuentes con la dosis recomendada fueron náuseas, vómitos y cefaleas. Esas reacciones adversas no requirieron por lo general una intervención médica, y ningún paciente interrumpió el tratamiento con atalureno a causa de una reacción adversa.
Tabla de reacciones adversas
Las reacciones adversas notificadas en el estudio clínico de pacientes predominantemente pediátricos con DMDmss tratados con la dosis recomendada de 10, 10 y 20 mg/kg se clasifican de acuerdo con la Clasificación por órganos y sistemas del MedDRA y la frecuencia. Los grupos de frecuencia se definen según la siguiente convención: muy frecuentes (> 1/10) frecuentes (> 1/100 a < 1/10) y frecuencia no conocida (no puede estimarse a partir de datos disponibles). Las reacciones adversas se presentan por orden decreciente de gravedad dentro de cada grupo de frecuencias.
Tabla 1. Reacciones adversas al Translarna en el estudio controlado de DMDmss
|
Sistema de clasificación de órganos |
Muy frecuentes |
Frecuentes |
Frecuencia no conocida |
|
Trastornos del metabolismo y de la nutrición |
Disminución del apetito |
Cambio en el perfil lipídico (aumento de triglicéridos y colesterol) | |
|
Trastornos del sistema nervioso |
Cefalea |
Mareo | |
|
Trastornos vasculares |
Hipertensión | ||
|
Trastornos respiratorios, torácicos y mediastínicos |
Tos, epistaxis | ||
|
Trastornos gastrointestinales |
Náuseas, vómitos |
Dolor abdominal alto, flatulencia, diarrea, molestias en el estómago, dolor abdominal, estreñimiento, regurgitación | |
|
Trastornos de la piel y del tejido subcutáneo |
Eritema | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor en las extremidades | ||
|
Trastornos renales y urinarios |
Enuresis, quiste renal, polaquiuria, coloración de la orina anormal |
Cambio en las pruebas de función renal (aumento de la creatinina, nitrógeno ureico en sangre, cistatina C) | |
|
Trastornos generales y alteraciones en el lugar de administración |
Pirexia, fatiga, pérdida de peso |
Descripción de reacciones adversas seleccionadas
Lípidos séricos
Durante el estudio controlado de DMDmss, la concentración media de colesterol total y de triglicéridos fueron normales en el momento inicial y aumentó, alcanzando el umbral de los valores límite alto o valores elevados. Los valores tendían a estabilizarse en la parte inicial del estudio y no aumentaron más con la continuación del tratamiento.
Pruebas de función renal
Durante el estudio controlado de DMDmss se observaron ligeros aumentos en el valor medio de la creatinina sérica, nitrógeno ureico en sangre (BUN) y cistatina C. Los valores tendían a estabilizarse en la parte inicial del estudio y no aumentaron más con la continuación del tratamiento.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continua de la relación del beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Algunos voluntarios sanos que recibieron una única dosis por vía oral de 200 mg/kg de atalureno experimentaron síntomas transitorios leves de cefalea, náuseas, vómitos y diarrea. No se observaron reacciones adversas graves en esos sujetos. En el caso de sospecha de sobredosis se debe proporcionar tratamiento de soporte, incluyendo la consulta con un profesional sanitario y una estrecha observación del estado clínico del paciente.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: {grupo}, código ATC: no se ha asignado aún Mecanismo de acción
Una mutación sin sentido en el ADN tiene como resultado un codón de parada prematuro en un ARNm. Este codón de parada prematuro en el ARNm provoca la enfermedad al finalizar la traducción antes de que se genere una proteína funcional. Atalureno facilita la lectura ribosómica del ARNm que contiene un codón de parada prematuro de ese tipo, lo que tiene como resultado la producción de una proteína funcional.
Efectos farmacodinámicos
Los experimentos no clínicos in vitro realizados en cultivos de células con mutaciones sin sentido y en larvas de peces cultivadas en una solución de atalureno han demostrado que atalureno facilita la lectura ribosómica con una relación concentración/respuesta en forma de campana (forma de U invertida). Se propone la hipótesis de que la relación dosis/respuesta in vivo también podría tener forma de campana, pero los datos de estudios in vivo realizados en un modelo de ratón para DMDmss y en humanos son demasiado limitados para confirmar esa hipótesis.
Los estudios no clínicos in vitro sugieren que puede ser importante la exposición continua a atalureno para maximizar su actividad, y que los efectos del principio activo sobre la lectura ribosómica de los codones de parada prematuros revierten poco después de la retirada de atalureno.
Eficacia clínica y seguridad
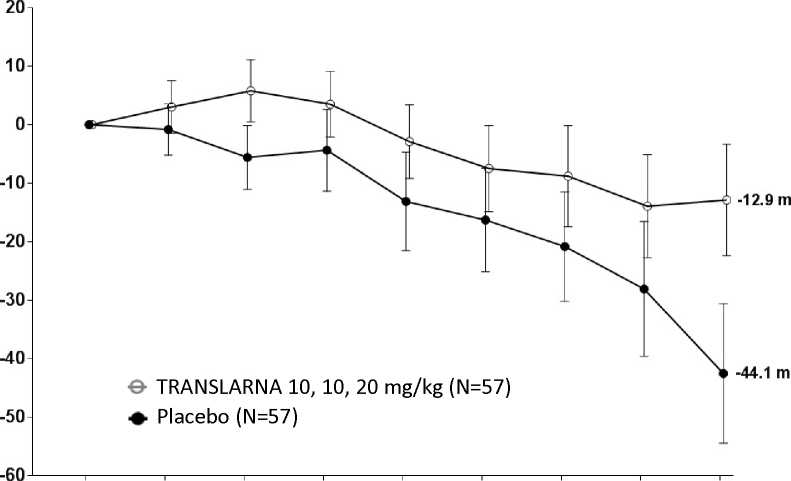
La eficacia y la seguridad de Translarna se evaluaron en un estudio aleatorizado, doble ciego, controlado con placebo, multicéntrico, sobre la distrofia muscular de Duchenne causada por una mutación sin sentido (nonsense mutation Duchenne muscular dystrophy, DMDmss) en 174 pacientes varones de entre 5 y 20 años de edad. Se exigió que todos los pacientes fueran pacientes ambulantes, definida como la capacidad de caminar > 75 metros sin necesidad de dispositivos de asistencia durante una prueba de marcha de 6 minutos (6-minute Walk Test, 6MWT) realizada durante el cribado. También fue necesario que los pacientes tuvieran una confirmación documentada de la presencia de una mutación sin sentido en el gen de la distrofina, determinada por secuenciación genética. La mayoría de los pacientes de todos los grupos de tratamiento eran de origen caucásico (90 %). Se asignó aleatoriamente a los pacientes en una proporción de 1:1:1 para recibir atalureno o placebo 3 veces al día (mañana, mediodía y noche) durante 48 semanas: 57 recibieron placebo, 57 recibieron atalureno (10, 10 y 20 mg/kg) y 60 recibieron atalureno (20, 20 y 40 mg/kg). Finalizaron el estudio 173 pacientes. La variable principal de eficacia era el Atalureno sobre la deambulación, evaluada por medio del cambio en la distancia (6MWD) recorrida durante una prueba 6MWT. El análisis post hoc reveló que desde el periodo basal hasta la semana 48, en los pacientes tratados con atalureno 10, 10 y 20 mg/kg se observó una disminución media de la 6MWD de 12,9 metros y los pacientes que recibían placebo se observó una disminución media de la 6MWD de 44,1 metros (figura 1). Por lo tanto, el cambio medio en la 6MWD observada desde el momento inicial hasta la semana 48 fue de 31,3 metros mejor en el grupo de atalureno 10, 10 y 20 mg/kg que en el grupo de placebo (p=0,056). En una estimación de modelo estadístico, la diferencia media fue de 31,7 metros (p ajustada = 0,0367). No hubo ninguna diferencia entre atalureno 20, 20 y 40 mg/kg y placebo. Estos resultados indican que atalureno 10, 10 y 20 mg/kg disminuye el ritmo de la pérdida de la capacidad ambulante en los pacientes con DMDmss.
Figura 1. Cambio medio en la distancia recorrida en 6 minutos
Periodo basal E> V IB M S Üfi 7? 7i\
Semanas
Un análisis post hoc del tiempo transcurrido hasta el empeoramiento persistente del 10 % en la 6MWD mostró que en el 26 % de los pacientes del grupo de atalureno 10, 10 y 20 mg/kg había habido progresión en la semana 48, frente al 44 % de los del grupo de placebo (p=0,0652) (figura 2). No hubo ninguna diferencia entre atalureno 20, 20 y 40 mg/kg y placebo. Estos resultados indican que un menor número de pacientes que reciben atalureno 10, 10 y 20 mg/kg empeoraron en 6MWD a lo largo de 48 semanas.
Figura 2. Curva de Kaplan-Meier del tiempo transcurrido hasta el empeoramiento persistente del 10 % en la 6MWD
I
100
0
4-»
c
O) g 80
5
S 70 Q.
1 60
(D
^ 50
oN
® 40
90
26 % con empeoramiento - 44 % con empeoramiento
c
55
ai
'i?
c
0)
o
o
Q.
30
20
10
TRANSLARNA 10, 10, 20 mg/kg (N=57) Placebo (N=57)
0 6 12 1 8 24 30 36 42 48 54 60
Semanas
En las pruebas de tiempo funcionales (timed function test, TFT), las pruebas de tiempo para correr/caminar 10 metros, el tiempo para ascender 4 escalones y el tiempo para descender 4 escalones, los pacientes tratados con atalureno demostraron incrementos menores en el tiempo necesario para correr/caminar 10 metros, ascender 4 escalones y descender 4 escalones, lo que indica una disminución de la progresión DMDmss en relación con el placebo.
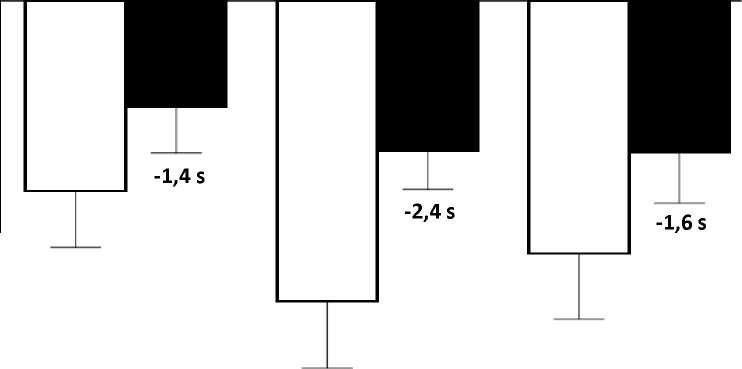
El cambio de la media en pruebas de tiempo funcionales desde el periodo basal hasta la semana 48 fue mejor en el grupo de atalureno 10, 10 y 20 mg/kg que en el grupo de placebo en el tiempo necesario para correr/caminar 10 metros (mejor en 1,4 segundos), el tiempo para ascender cuatro escalones (mejor en 2,4 segundos), y el tiempo para descender 4 escalones (mejor en 1,6 segundos), Figura 3.
Figura 3, cambio de la media en pruebas de tiempo funcionales
10m Correr/Caminar 4-Ascender escaleras 4-Descender escaleras
4
RJ
-Q .
1/1
C
O)
E
2
o
<D
Q.
E
a>
O o -O -a o =
“ 5>
ai cuO ai Qj'
ai </> ■a +i
■o T3
•2 £ .2 E,
£
RJ
u
■ TRANSLARNA 10, 10, 20 mg/kg (N=57) Placebo (N=57)
Resultados de 6MWD en pacientes con periodo basal de 6MWD < 350 metros.
En el caso de pacientes con un periodo basal de 6MWD < 350 metros, el cambio de media observado en 6MWD desde el periodo basal a la semana 48 fue de un mejoramiento en 68 metros del grupo de atalureno 10, 10 y 20 mg/kg respecto al el grupo de placebo (p=0.0053).
En el caso de estos pacientes, el cambio de la media en pruebas de tiempo funcionales desde el periodo basal hasta la semana 48 fue mejor en el grupo de atalureno 10, 10 y 20 mg/kg que en el grupo de placebo en el tiempo necesario para correr/caminar 10 metros (mejor en 3,5 segundos), el tiempo para ascender cuatro escalones (mejor en 6,4 segundos), y el tiempo para descender 4 escalones (mejor en 5,0 segundos).
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con atalureno en DMDmss en dos diferentes grupos de la población pediátrica, desde el nacimiento hasta menos de 28 días y lactantes de entre 28 días y menos de 6 meses, conforme a la decisión sobre el Plan de Investigación Pediátrica (PIP) en la indicación autorizada (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con atalureno en DMDmss en un diferente grupo de la población pediátrica, de entre 6 meses y menos de 5 años de edad, conforme a la decisión sobre el Plan de Investigación Pediátrica (PIP) en la indicación autorizada (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
Este medicamento se ha autorizado con una «aprobación condicional». Esta modalidad de aprobación significa que se espera obtener más información sobre este medicamento. La Agencia Europea de Medicamentos revisará la información nueva de este medicamento al menos una vez al año y esta Ficha Técnica o Resumen de las Características del Producto (RCP) se actualizará cuando sea necesario.
5.2 Propiedades farmacocinéticas
La administración de atalureno con una dosis ajustada al peso corporal (mg/kg) tuvo como resultado exposiciones similares en estado estacionario o (AUC) entre niños y adolescentes con DMDmss en un amplio intervalo de pesos corporales. Aunque atalureno es prácticamente insoluble en agua, se absorbe con facilidad tras su administración por vía oral en suspensión.
Características generales de atalureno después de la administración
Absorción
Los niveles plasmáticos máximos de atalureno se alcanzan aproximadamente 1,5 horas después de la administración en personas que han recibido el medicamento en los 30 minutos siguientes a una comida. Basándose en la recuperación de radiactividad en la orina observada en un estudio de dosis única de atalureno radiomarcado, se calcula que la biodisponibilidad oral de atalureno es de > 55 %. Las concentraciones plasmáticas de atalureno en estado estacionario aumentan proporcionalmente a la dosis. Las concentraciones plasmáticas en estado estacionario son proporcionales para dosis de atalureno de entre 10 y 50 mg/kg, no observándose ninguna acumulación tras su administración repetida.
Distribución
In vitro, atalureno está unido en un 99,6 % a las proteínas plasmáticas humanas, y esa unión es independiente de la concentración plasmática. Atalureno no se distribuye en los glóbulos rojos.
Biotransformación
Atalureno se metaboliza mediante conjugación por la acción de las enzimas uridina difosfato glucuronosiltransferasa (UGT), principalmente UGT1A9, en el hígado y en el intestino.
In vivo, el único metabolito detectado en el plasma tras la administración por vía oral de atalureno radiomarcado fue el O-1p-acil glucurónido de atalureno; la exposición a este metabolito en seres humanos fue de aproximadamente el 8 % de la AUC plasmática de atalureno.
Eliminación
La semivida plasmática de atalureno oscila entre 2 y 6 horas, y no se ve afectada ni por la dosis ni por la administración repetida. La eliminación de atalureno depende probablemente de la glucuronidación hepática e intestinal de atalureno seguida de la excreción renal del metabolito glucurónido resultante.
Después de una dosis oral única de atalureno radiomarcado, aproximadamente la mitad de la dosis radiactiva administrada se recupera en las heces, y el resto se recupera en la orina. En la orina, el atalureno inalterado y el metabolito acil-glucurónido suponen, respectivamente, < 1% y el 49 % de la dosis administrada.
Linealidad/no linealidad
Las concentraciones plasmáticas en estado estacionario son proporcionales a la dosis para dosis de atalureno de entre 10 y 50 mg/kg, no observándose ninguna acumulación tras una administración repetida. Los datos obtenidos con voluntarios sanos indican que la biodisponibilidad relativa de atalureno es aproximadamente un 40 % más baja en estado estacionario que después de la dosis inicial. Se calcula que el inicio de la reducción de la biodisponibilidad relativa tiene lugar aproximadamente 60 horas después de la primera dosis. El estado estacionario se establece después de aproximadamente dos semanas de tres administraciones al día.
Características en grupos específicos de sujetos o pacientes
Edad
Los datos obtenidos en personas de entre 5 y 57 años de edad indican que la edad no tiene un efecto claro sobre la exposición plasmática de atalureno. No es necesario ajustar la dosis por la edad.
Sexo
En los ensayos clínicos de DMDmss no se estudiaron mujeres. Sin embargo, en otras poblaciones no existe un efecto claro del sexo sobre la exposición plasmática de atalureno.
Raza
Es poco probable que la farmacocinética de atalureno se vea afectada de forma significativa por los polimorfismos de UTG1A9 en la población de origen caucásico. A causa del bajo número de pacientes de otras razas incluidos en los estudios clínicos, no se pueden establecer conclusiones sobre el efecto de UTG1A9 en otros grupos étnicos.
Insuficiencia renal o hepática
No se han realizado estudios con Translama en pacientes con insuficiencia renal o hepática. Los pacientes con insuficiencia renal o hepática deben ser sometidos a un estrecho control.
Pacientes no ambulantes
No existen diferencias claras debidas a la pérdida de la capacidad ambulante, ni en la biodisponibilidad relativa en estado estacionario ni en el aclaramiento aparente. No es necesario ajustar las dosis para los pacientes que han perdido la capacidad ambulante.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de seguridad farmacológica y genotoxicidad.
Se ha dispuesto de un conjunto estándar de estudios sobre toxicidad reproductiva. No se han observado efectos sobre la fertilidad masculina ni femenina, pero no se han investigado los efectos sobre la fertilidad durante la edad adulta del tratamiento en la primera juventud. Se ha observado toxicidad embrio-fetal (p. ej. aumento de reabsorciones prematuras, pérdidas después de la implantación, disminución del número de fetos viables) y signos de retraso en el desarrollo (aumento de variaciones esqueléticas) en ratas y conejos en presencia de toxicidad materna. La exposición, en el nivel de exposición sin efectos adversos observados (no observed adverse effect level, NOAEL), fue similar (en el conejo) o 4 veces superior (en la rata) a la exposición sistémica en seres humanos (10,
10, 20 mg/kg/día). Se demostró la existencia de una transferencia placentaria de atalureno radiomarcado en las ratas. Con una dosis materna única ensayada, relativamente baja, de 30 mg/kg, la concentración de radiactividad fetal fue < 27 % de la concentración materna. En el estudio de toxicidad para el desarrollo pre/posnatal, con una exposición de aproximadamente 5 veces la exposición en seres humanos se observó una toxicidad materna significativa y efectos sobre el peso corporal y el desarrollo de la actividad ambulante en las crías. La exposición sistémica materna en el nivel de exposición sin efectos adversos observados (no observed effect level, NOEL) para la toxicidad neonatal fue 3 veces superior a la exposición en seres humanos. Con una dosis materna única, relativamente baja, de 30 mg/kg de atalureno radiomarcado, la concentración más alta medida de radiactividad en la leche de rata fue del 37 % de la concentración plasmática materna. La presencia de radiactividad en el plasma de las crías confirmó la absorción por las crías a partir de la leche.
Se produjo toxicidad renal (nefrosis de la nefrona distal) en estudios de dosis orales repetidas en ratones con una exposición sistémica equivalente a 0,3 veces la AUC en equilibrio en pacientes a quienes se administró Translarna con las dosis respectivas, por la mañana, al mediodía y por la noche, de 10, 10 y 20 mg/kg y superiores.
En un modelo de ratón transgénico de 26 semanas para la carcinogenicidad no se encontraron indicios de carcinogenicidad. En un estudio de carcinogenicidad en ratas de 2 años se observó un caso de hibemoma. Además de eso, con una exposición mucho más alta que en los pacientes se observó un aumento de los tumores de la vejiga urinaria (raros). Se considera poco probable la importancia de los tumores de la vejiga urinaria en seres humanos.
En uno de dos estudios de administración repetida en ratas de 26 semanas de duración, realizado con ratas de 4-5 semanas de edad, se observó un aumento relacionado con la dosis de la incidencia del hibernoma maligno, un tumor raro en las ratas. Además de eso, se encontró un caso de hibernoma maligno con la dosis más alta en un estudio de carcinogenicidad con ratas de 2 años de duración. La tasa histórica de incidencia de este tipo de tumor en ratas y en seres humanos es muy baja, y se desconoce el mecanismo que causa ese tumor en los estudios con ratas (incluida su relación con el tratamiento con atalureno). Se desconoce la importancia que puedan tener estos datos para los seres humanos.
En un estudio de 1 año de duración realizado con perros de 10-12 semanas de edad se observaron hallazgos en las glándulas suprarrenales (inflamación localizada y degeneración en las regiones de la corteza que producen glucocorticoides) y una cierta dificultad para la producción de cortisol tras la estimulación exógena con hormona adrenocorticotropa. Estos hallazgos se observaron en perros con una exposición sistémica equivalente a 0,8 veces la AUC en equilibrio en pacientes a quienes se administró Translama con las dosis respectivas, por la mañana, al mediodía y por la noche, de 10, 10 y 20 mg/kg y superiores. En un estudio de distribución con ratas se observó una elevada concentración suprarrenal de atalureno.
Además de los efectos antes mencionados, en los estudios de administración repetida se observaron algunos otros efectos menos adversos; en concreto, la disminución del aumento de peso y de la ingesta de alimento y el aumento del peso del hígado sin una correlación histológica y con una relevancia clínica poco clara. También en estudios con ratas y perros se encontraron cambios en los lípidos plasmáticos (colesterol y triglicéridos) que sugieren cambios en el metabolismo de los lípidos.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polidextrosa (E1200)
Macrogol Poloxámero Manitol (E421)
Crospovidona
Hidroxietilcelulosa
Aroma artificial de vainilla: maltodextrina, aromas artificiales y propilenglicol.
Dióxido de sílice coloidal (E551)
Estearato de magnesio
6.2 Incompatibilidades
No procede
6.3 Período de validez
4 años
Se recomienda administrar cada dosis preparada inmediatamente después de su preparación. Si la dosis preparada se mantiene refrigerada (2-8 °C) se debe desechar si no se utiliza en las 24 horas siguientes a su preparación o en las 3 horas siguientes si se mantiene a temperatura ambiente (15 -30 °C).
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
Para las condiciones de conservación tras la dilución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Sobre de una hoja de aluminio laminada y termosellada : tereftalato de polietileno (seguridad infantil), polietileno (color y adhesión por poliester/lámina), lámina de aluminio, (barrera contra humedad), adhesivo (de la clase de los poliuretanos), copolímero de etileno y ácido metacrílico (resina selladora para la integridad del envoltorio).
Envase con 30 sobres.
6.6 Precauciones especiales de eliminación y otras manipulaciones
No se deben abrir los sobres hasta el momento de la preparación de la dosis. Se debe mezclar todo el contenido de cada sobre con un mínimo de 30 ml de líquido (agua, leche o zumo de fruta) o 3 cucharadas soperas de un alimento semisólido (yogur o compota de manzana). La dosis preparada se debe mezclar bien antes de su administración. Puede aumentarse la cantidad de líquido o alimento semisólido según las preferencias del paciente.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
PTC Therapeutics International Limited 77 Sir John Rogerson’s Quay Dublin 2 Irlanda
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/902/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 31 de julio 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Translarna 250 mg granulado para suspensión oral
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada sobre contiene 250 mg de atalureno.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Granulado para suspensión oral.
Granulado de color entre blanco y blanquecino.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Translarna está indicado para el tratamiento de la distrofia muscular de Duchenne (DMD) debida a una mutación sin sentido en el gen de la distrofina, en pacientes ambulantes a partir de 5 o más años de edad (ver sección 5.1). No se ha demostrado eficacia en pacientes no ambulantes.
La presencia de una mutación sin sentido en el gen de la distrofina se debe determinar mediante pruebas genéticas (ver sección 4.4).
4.2 Posología y forma de administración
El tratamiento con Translarna se debe iniciar solo por médicos especialistas con experiencia en el control de la distrofia muscular de Duchenne/Becker.
Posología
Atalureno se debe administrar por vía oral, 3 veces al día.
La primera toma se debe administrar por la mañana, la segunda al mediodía y la tercera por la noche. Los intervalos posológicos recomendados son de 6 horas entre la toma de la mañana y la del mediodía; de 6 horas entre la del mediodía y la de la noche, y de 12 horas entre la de la noche y la primera toma del día siguiente.
La dosis recomendada es de 10 mg/kg de peso corporal por la mañana, 10 mg/kg de peso corporal a mediodía y 20 mg/kg de peso corporal por la noche (para una dosis diaria total de 40 mg/kg de peso corporal).
Translarna se presenta en sobres de 125 mg, 250 mg o 1.000 mg. La siguiente tabla ofrece información sobre la o las dosis del sobre que se deben emplear para preparar la posología recomendada según el intervalo de peso corporal.
|
Intervalo de peso (kg) |
Número de sobres | |||||||||
|
Mañana |
Mediodía |
Noche | ||||||||
|
Sobres de 125 mg |
Sobres de 250 mg |
Sobres de 1.000 mg |
Sobres de 125 mg |
Sobres de 250 mg |
Sobres de 1.000 mg |
Sobres de 125 mg |
Sobres de 250 mg |
Sobres de 1.000 mg | ||
|
12 |
14 |
1 |
0 |
0 |
1 |
0 |
0 |
0 |
1 |
0 |
|
15 |
16 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
1 |
0 |
|
17 |
20 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
|
21 |
23 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
1 |
0 |
|
24 |
26 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
0 |
|
27 |
31 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
0 |
|
32 |
35 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
0 |
|
36 |
39 |
1 |
1 |
0 |
1 |
1 |
0 |
0 |
3 |
0 |
|
40 |
44 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
3 |
0 |
|
45 |
46 |
0 |
2 |
0 |
0 |
2 |
0 |
1 |
3 |
0 |
|
47 |
55 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
0 |
1 |
|
56 |
62 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
1 |
1 |
|
63 |
69 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
1 |
1 |
|
70 |
78 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
2 |
1 |
|
79 |
86 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
3 |
1 |
|
87 |
93 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
3 |
1 |
|
94 |
105 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
|
106 |
111 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
|
112 |
118 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
|
119 |
125 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
2 |
2 |
Retraso u olvido de la toma de la dosis
Si hay un retraso en la administración de atalureno de menos de 3 horas después de la toma habitual de la mañana o del mediodía, o de menos de 6 horas después de la de la noche, se deberá tomar la dosis sin realizar cambios en la pauta posológica. Si hay un retraso de más de 3 horas después de la toma habitual de la mañana o del mediodía, o de más de 6 horas después de la de la noche, no se debe tomar la dosis, y el paciente debe reanudar su pauta posológica habitual. Los pacientes no deben tomar una dosis doble o adicional para compensar una dosis olvidada. Es importante administrar la dosis correcta. El aumento de la dosis por encima de la dosis recomendada puede tener como resultado una reducción de la eficacia.
Poblaciones especiales
Pacientes de edad avanzada
No se ha establecido todavía la seguridad y eficacia de atalureno en pacientes de 65 años o más. (Ver sección 5.2).
Insuficiencia renal y hepática
No se ha establecido ni la seguridad ni la eficacia de atalureno en pacientes con insuficiencia renal o hepática. (Ver sección 4.4).
Población pediátrica
No se ha establecido todavía ni la seguridad ni la eficacia de Translama en niños con edades comprendidas entre los 6 meses y los 5 años de edad. No se dispone de datos.
Forma de administración
Translarna se debe administrar por vía oral después de mezclarlo para formar una suspensión en líquido o con un alimento semisólido.
No se deben abrir los sobres hasta el momento de la preparación de la dosis. Se debe mezclar todo el contenido de cada sobre con un mínimo de 30 ml de líquido (agua, leche o zumo de fruta) o 3 cucharadas soperas de un alimento semisólido (yogur o compota de manzana). La dosis preparada se debe mezclar bien antes de su administración. Se puede aumentar la cantidad de líquido o alimento semisólido según las preferencias del paciente. Los pacientes deben tomar la dosis completa.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Uso concomitante de aminoglucósidos por vía intravenosa (ver sección 4.4 y 4.5).
4.4 Advertencias y precauciones especiales de empleo Pacientes que no tienen una mutación sin sentido
Los pacientes deben tener una mutación sin sentido en el gen de la distrofina, determinada mediante análisis genéticos, como parte de su estado patológico subyacente. No se debe administrar atalureno a pacientes que no tengan una mutación sin sentido.
Insuficiencia renal y hepática
Los pacientes con insuficiencia renal y hepática deben ser sometidos a un estrecho control.
Cambios en el perfil lipídico
Puesto que se han notificados cambios en el perfil lipídico (aumento de triglicéridos y colesterol) de algunos pacientes en ensayos clínicos, se recomienda realizar, en los pacientes con distrofia muscular de Duchene debida a una mutación sin sentido (DMDmss) que reciban atalureno, un control del colesterol total, LDL, HDL y triglicéridos cada año, o con mayor frecuencia si es necesario por el estado clínico del paciente.
Hipertensión con el uso concomitante de corticosteroides sistémicos
Puesto que se han notificado casos de hipertensión con el uso concomitante de corticosteroides sistémicos en algunos pacientes en ensayos clínicos, se recomienda realizar, en los pacientes con DMDmss que reciban atalureno de forma concomitante con corticosteroides, un control de la tensión arterial sistólica y diastólica en reposo cada 6 meses, o con mayor frecuencia si es necesario por el estado clínico del paciente.
Monitorización de la función renal
Puesto que se han notificados ligeros aumentos de las concentraciones medias de creatinina sérica, nitrógeno ureico en sangre (BUN, por sus siglas en inglés) y de cistatina C en el estudio comparativo de DMDmss, se recomienda realizar, en los pacientes con DMDmss que reciban atalureno, un control de la creatinina sérica, el BUN y la cistatina C cada 6-12 meses, o con mayor frecuencia si es necesario por el estado clínico del paciente.
Interacciones potenciales con otros medicamentos
Se debe actuar con precaución al administrar atalureno de forma conjunta con medicamentos que sean sustratos o inductores de UGT1A9, inhibidores de BCRP o sustratos de OAT1, OAT3 u OATP1B3 (ver sección 4.5).
Aminoglucósidos
Se ha demostrado que los aminoglucósidos reducen la actividad de lectura ribosómica del atalureno in vitro. Asimismo, se encontró que el atalureno aumenta la nefrotoxicidad de los aminoglucósidos intravenosos. Se debe evitar la administración conjunta de esos medicamentos con atalureno (ver sección 4.3). Dado que se desconoce el mecanismo mediante el cual el atalureno aumenta la nefrotoxicidad de los aminoglucósidos intravenosos, no se recomienda el uso concomitante de otros medicamentos nefrotóxicos con atalureno. En caso de que esto sea inevitable, (por ejemplo: vancomicina para el tratamiento de SARM) se recomienda una monitorización minuciosa de la función renal (ver sección 4.5).
4.5 Interacción con otros medicamentos y otras formas de interacción
Aminoglucósidos
No se debe administrar Atalureno de forma conjunta con aminoglucósidos intravenosos, pues se han observado casos de disminución de la función renal en un ensayo clínico en pacientes con nmCF (ver sección 4.3).
En varios pacientes tratados con atalureno y aminoglucósidos intravenosos junto con otros antibióticos para los brotes de la fibrosis quística se produjeron elevaciones de la creatinina sérica. Las elevaciones de la creatinina sérica remitieron en todos los casos cuando se interrumpió la administración del aminoglucósido intravenoso, tanto si se continuó como si se interrumpió la administración de Translarna. Estos datos sugieren que la administración conjunta de Translama y aminoglucósidos intravenosos podría potenciar el efecto nefrotóxico de los aminoglucósidos. En consecuencia, si es necesario el tratamiento con aminoglucósidos intravenosos se debe interrumpir el tratamiento con Translarna, que se podrá reanudar 2 días después de finalizar la administración del aminoglucósido. Se desconoce el efecto de la administración conjunta de ataluren con otros medicamentos nefrotóxicos.
La deshidratación podría ser un factor que contribuyera en alguno de estos casos. Los pacientes se deben mantener adecuadamente hidratados mientras tomen atalureno. Ver sección 4.4.
Efectos de otros medicamentos en la farmacocinética de atalureno
Los datos obtenidos en estudios in vitro indican que atalureno es un sustrato de UGT1A9 y de la proteína resistente al cáncer de mama (BCRP). Se debe actuar con precaución cuando se administra de forma conjunta atalureno con medicamentos inductores de UGT1A9 (como el mofetil micofenolato) o inhibidores de BCRP (por ejemplo, ciclosporina).
Efectos de atalureno sobre la farmacocinética de otros medicamentos
Los datos obtenidos en estudios in vitro indican que atalureno es un inhibidor de UGT1A9, del transportador de aniones orgánico 1 (OAT1), del transportador de aniones orgánico 3 (OAT3) y del polipéptido de transporte del anión orgánico 1B3 (OATP1B3). Se debe actuar con precaución cuando se administra de forma conjunta atalureno con medicamentos sustratos de UGT1A9, OAT1, OAT3 u OATP1B3 (por ejemplo, oseltamivir, aciclovir, ciprofloxacino, captoprilo, furosemida, bumetanida, valsartán, pravastatina, rosuvastatina, atorvastatina o pitavastatina), debido al riesgo del aumento de la concentración de estos medicamentos .
Basándose en estudios in vitro no se espera que atalureno sea un inhibidor del transporte mediado por P-gp ni del metabolismo mediado por el citocromo P450. Del mismo modo, resulta poco probable que atalureno sea in vivo un inductor de las isoenzimas del citocromo P450.
La administración conjunta de corticosteroides (deflazacort, prednisona o prednisolona) con atalureno no afectó a las concentraciones plasmáticas de atalureno. No se observó ningún cambio clínicamente relevante en las concentraciones plasmáticas de los corticoides con la administración conjunta de atalureno. Estos datos indican que no existe interacción farmacológica aparente entre los corticosteroides y atalureno, no siendo necesario ningún ajuste de la dosis.
Medicamentos que afectan el transportador de glucoproteína P
In vitro, atalureno no es un sustrato del transportador de glucoproteína P. Es poco probable que la farmacocinética de atalureno resulte afectada por medicamentos que inhiben el transportador de glucoproteína P.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos suficientes relativos al uso de atalureno en mujeres embarazadas. Los estudios realizados en animales han revelado una toxicidad para la reproducción solamente con dosis tóxicas para la madre (ver sección 5.3).
Como medida de precaución no se recomienda utilizar atalureno durante el embarazo.
Lactancia
Se desconoce si el atalureno/sus metabolitos se excretan en la leche materna. Los datos farmacodinámicos/toxicológicos disponibles en animales muestran que el atalureno/ sus metabolitos se excretan en la leche (ver sección 5.3). No se puede excluir el riesgo en recién nacidos/niños lactantes.
Debe interrumpirse la lactancia durante el tratamiento con atalureno.
Fertilidad
Los datos preclínicos no muestran riesgos para los seres humanos en base a los estudios de fertilidad estándar con machos y hembras de rata (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se ha estudiado el efecto de atalureno sobre la conducción y la utilización de máquinas. Los pacientes que experimenten mareos deben tener cuidado si conducen o utilizan máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
En ensayos clínicos en pacientes con distrofia muscular de Duchenne debida a una mutación sin sentido (DMDmss), las reacciones adversas más frecuentes con la dosis recomendada fueron náuseas, vómitos y cefaleas. Esas reacciones adversas no requirieron por lo general una intervención médica, y ningún paciente interrumpió el tratamiento con atalureno a causa de una reacción adversa.
Tabla de reacciones adversas
Las reacciones adversas notificadas en el estudio clínico de pacientes predominantemente pediátricos con DMDmss tratados con la dosis recomendada de 10, 10 y 20 mg/kg se clasifican de acuerdo con la Clasificación por órganos y sistemas del MedDRA y la frecuencia. Los grupos de frecuencia se definen según la siguiente convención: muy frecuentes (> 1/10) , frecuentes (> 1/100 a < 1/10) y frecuencia no conocida ( no puede estimarse a partir de datos disponibles). Las reacciones adversas se presentan por orden decreciente de gravedad dentro de cada grupo de frecuencias.
Tabla 2. Reacciones adversas al Translama en el estudio controlado de DMDmss
|
Sistema de clasificación de órganos |
Muy frecuentes |
Frecuentes |
Frecuencia no conocida |
|
Trastornos del metabolismo y de la nutrición |
Disminución del apetito |
Cambio en el perfil lipídico (aumento de triglicéridos y colesterol) | |
|
Trastornos del sistema nervioso |
Cefalea |
Mareo | |
|
Trastornos vasculares |
Hipertensión | ||
|
Trastornos respiratorios, torácicos y mediastínicos |
Tos, epistaxis | ||
|
Trastornos gastrointestinales |
Náuseas, vómitos |
Dolor abdominal alto, flatulencia, diarrea, molestias en el estómago, dolor abdominal, estreñimiento, regurgitación | |
|
Trastornos de la piel y del tejido subcutáneo |
Eritema | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor en las extremidades | ||
|
Trastornos renales y urinarios |
Enuresis, quiste renal, polaquiuria, coloración de la orina anormal |
Cambio en las pruebas de función renal (aumento de la creatinina, nitrógeno ureico en sangre, cistatina C) | |
|
Trastornos generales y alteraciones en el lugar de administración |
Pirexia, fatiga, pérdida de peso |
Descripción de reacciones adversas seleccionadas
Lípidos séricos
Durante el estudio controlado de DMDmss, la concentración media de colesterol total y de triglicéridos fueron normales en el momento inicial y aumentó, alcanzando el umbral de los valores límite alto o valores elevados. Los valores tendían a estabilizarse en la parte inicial del estudio y no aumentaron más con la continuación del tratamiento.
Pruebas de función renal
Durante el estudio controlado de DMDmss se observaron ligeros aumentos en el valor medio de la creatinina sérica, nitrógeno ureico en sangre (BUN) y cistatina C. Los valores tendían a estabilizarse en la parte inicial del estudio y no aumentaron más con la continuación del tratamiento.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continua de la relación del beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Algunos voluntarios sanos que recibieron una única dosis por vía oral de 200 mg/kg de atalureno experimentaron síntomas transitorios leves de cefalea, náuseas, vómitos y diarrea. No se observaron reacciones adversas graves en esos sujetos. En el caso de sospecha de sobredosis se debe proporcionar tratamiento de soporte, incluyendo la consulta con un profesional sanitario y una estrecha observación del estado clínico del paciente.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: {grupo}, código ATC: no se ha asignado aún Mecanismo de acción
Una mutación sin sentido en el ADN tiene como resultado un codón de parada prematuro en un ARNm. Este codón de parada prematuro en el ARNm provoca la enfermedad al finalizar la traducción antes de que se genere una proteína funcional. Atalureno facilita la lectura ribosómica del ARNm que contiene un codón de parada prematuro de ese tipo, lo que tiene como resultado la producción de una proteína funcional.
Efectos farmacodinámicos
Los experimentos no clínicos in vitro realizados en cultivos de células con mutaciones sin sentido y en larvas de peces cultivadas en una solución de atalureno han demostrado que atalureno facilita la lectura ribosómica con una relación concentración/respuesta en forma de campana (forma de U invertida). Se propone la hipótesis de que la relación dosis/respuesta in vivo también podría tener forma de campana, pero los datos de estudios in vivo realizados en un modelo de ratón para DMDmss y en humanos son demasiado limitados para confirmar esa hipótesis.
Los estudios no clínicos in vitro sugieren que puede ser importante la exposición continua a atalureno para maximizar su actividad, y que los efectos del principio activo sobre la lectura ribosómica de los codones de parada prematuros revierten poco después de la retirada de atalureno.
Eficacia clínica y seguridad
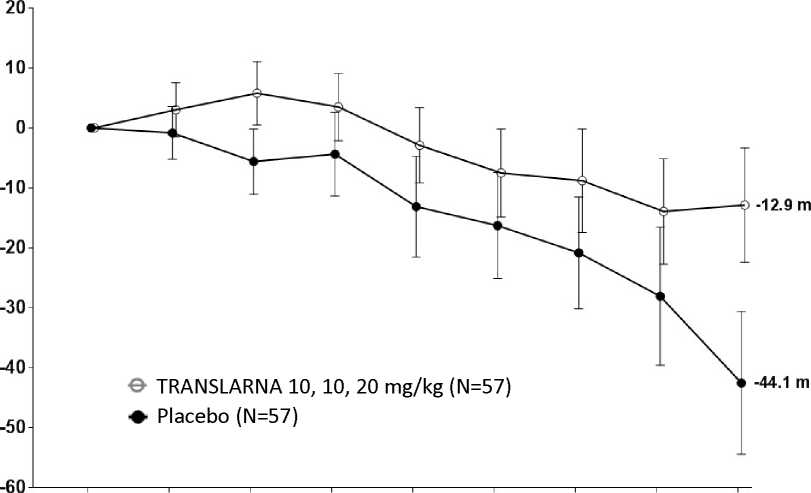
La eficacia y la seguridad de Translarna se evaluaron en un estudio aleatorizado, doble ciego, controlado con placebo, multicéntrico, sobre la distrofia muscular de Duchenne causada por una mutación sin sentido (nonsense mutation Duchenne muscular dystrophy, DMDmss) en 174 pacientes varones de entre 5 y 20 años de edad. Se exigió que todos los pacientes fueran pacientes ambulantes, definida como la capacidad de caminar > 75 metros sin necesidad de dispositivos de asistencia durante una prueba de marcha de 6 minutos (6-minute Walk Test, 6MWT) realizada durante el cribado. También fue necesario que los pacientes tuvieran una confirmación documentada de la presencia de una mutación sin sentido en el gen de la distrofina, determinada por secuenciación genética. La mayoría de los pacientes de todos los grupos de tratamiento eran de origen caucásico (90 %). Se asignó aleatoriamente a los pacientes en una proporción de 1:1:1 para recibir atalureno o placebo 3 veces al día (mañana, mediodía y noche) durante 48 semanas: 57 recibieron placebo, 57 recibieron atalureno (10, 10 y 20 mg/kg) y 60 recibieron atalureno (20, 20 y 40 mg/kg). Finalizaron el estudio 173 pacientes. La variable principal de eficacia era el Atalureno sobre la deambulación, evaluada por medio del cambio en la distancia (6MWD) recorrida durante una prueba 6MWT. El análisis post hoc reveló que desde el periodo basal hasta la semana 48, en los pacientes tratados con atalureno 10, 10 y 20 mg/kg se observó una disminución media de la 6MWD de 12,9 metros y los pacientes que recibían placebo se observó una disminución media de la 6MWD de 44,1 metros (figura 1). Por lo tanto, el cambio medio en la 6MWD observada desde el momento inicial hasta la semana 48 fue de 31,3 metros mejor en el grupo de atalureno 10, 10 y 20 mg/kg que en el grupo de placebo (p=0,056). En una estimación de modelo estadístico, la diferencia media fue de 31,7 metros (p ajustada = 0,0367). No hubo ninguna diferencia entre atalureno 20, 20 y 40 mg/kg y placebo. Estos resultados indican que atalureno 10, 10 y 20 mg/kg disminuye el ritmo de la pérdida de la capacidad ambulante en los pacientes con DMDmss.
Figura 1. Cambio medio en la distancia recorrida en 6 minutos
Periodo basal Ei 12 1S¡ 7-1 3E) 3Eí 12 4S¡
Semanas
Un análisis post hoc del tiempo transcurrido hasta el empeoramiento persistente del 10 % en la 6MWD mostró que en el 26 % de los pacientes del grupo de atalureno 10, 10 y 20 mg/kg había habido progresión en la semana 48, frente al 44 % de los del grupo de placebo (p=0,0652) (figura 2). No hubo ninguna diferencia entre atalureno 20, 20 y 40 mg/kg y placebo. Estos resultados indican que un menor número de pacientes que reciben atalureno 10, 10 y 20 mg/kg empeoraron en 6MWD a lo largo de 48 semanas.
Figura 2. Curva de Kaplan-Meier del tiempo transcurrido hasta el empeoramiento persistente del 10 % en la 6MWD
100n
o
4-»
c
<D
E
2
o
0)
Q.
E
0)
0)
■c
v?
o
c
55
ai
i?
c
0)
o
o
Q.
90
80
70
60
50
40
30
20
10
26 % con empeoramiento ■ 44 % con empeoramiento
— TRANSLARNA 10, 10, 20 mg/kg (N=57)
- - Placebo (N=57)
0 6 12 18 24 30 36 42 48 54 60
Semanas
En las pruebas de tiempo funcionales (timed function test, TFT), las pruebas de tiempo para correr/caminar 10 metros, el tiempo para ascender 4 escalones y el tiempo para descender 4 escalones, los pacientes tratados con atalureno demostraron incrementos menores en el tiempo necesario para correr/caminar 10 metros, ascender 4 escalones y descender 4 escalones, lo que indica una disminución de la progresión DMDmss en relación con el placebo.
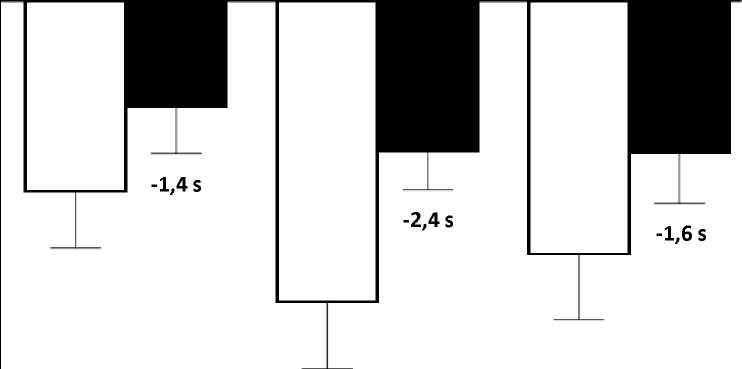
El cambio de la media en pruebas de tiempo funcionales desde el periodo basal hasta la semana 48 fue mejor en el grupo de atalureno 10, 10 y 20 mg/kg que en el grupo de placebo en el tiempo necesario para correr/caminar 10 metros (mejor en 1,4 segundos), el tiempo para ascender cuatro escalones (mejor en 2,4 segundos), y el tiempo para descender 4 escalones (mejor en 1,6 segundos), Figura 3.
Figura 3, cambio de la media en pruebas de tiempo funcionales
10m Correr/Caminar 4-Ascender escaleras 4-Descender escaleras
in
O
T3
m CuO Q. ®
QJ uj1
<U to
+i
.2
'S
<u
E
íO
-Q
O
T3
O
T3
V)
Q)
■o
O
5
E
ÍO
U
C
O)
E
2
o
<D
Q.
E
a>
■ TRANSLARNA 10, 10, 20 mg/kg (N=57) Placebo (N=57)
Resultados de 6MWD en pacientes con periodo basal de 6MWD < 350 metros.
En el caso de pacientes con un periodo basal de 6MWD < 350 metros, el cambio de media observado en 6MWD desde el periodo basal a la semana 48 fue de un mejoramiento en 68 metros del grupo de atalureno 10, 10 y 20 mg/kg respecto al el grupo de placebo (p=0.0053).
En el caso de estos pacientes, el cambio de la media en pruebas de tiempo funcionales desde el periodo basal hasta la semana 48 fue mejor en el grupo de atalureno 10, 10 y 20 mg/kg que en el grupo de placebo en el tiempo necesario para correr/caminar 10 metros (mejor en 3,5 segundos), el tiempo para ascender cuatro escalones (mejor en 6,4 segundos), y el tiempo para descender 4 escalones (mejor en 5,0 segundos).
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con atalureno en DMDmss en dos diferentes grupos de la población pediátrica, desde el nacimiento hasta menos de 28 días y lactantes de entre 28 días y menos de 6 meses, conforme a la decisión sobre el Plan de Investigación Pediátrica (PIP) en la indicación autorizada (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con atalureno en DMDmss en un diferente grupo de la población pediátrica, de entre 6 meses y menos de 5 años de edad, conforme a la decisión sobre el Plan de Investigación Pediátrica (PIP) en la indicación autorizada (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
Este medicamento se ha autorizado con una «aprobación condicional». Esta modalidad de aprobación significa que se espera obtener más información sobre este medicamento. La Agencia Europea de Medicamentos revisará la información nueva de este medicamento al menos una vez al año y esta Ficha Técnica o Resumen de las Características del Producto (RCP) se actualizará cuando sea necesario.
5.2 Propiedades farmacocinéticas
La administración de atalureno con una dosis ajustada al peso corporal (mg/kg) tuvo como resultado exposiciones similares en estado estacionario o (AUC) entre niños y adolescentes con DMDmss en un amplio intervalo de pesos corporales. Aunque atalureno es prácticamente insoluble en agua, se absorbe con facilidad tras su administración por vía oral en suspensión.
Características generales de atalureno después de la administración
Absorción
Los niveles plasmáticos máximos de atalureno se alcanzan aproximadamente 1,5 horas después de la administración en personas que han recibido el medicamento en los 30 minutos siguientes a una comida. Basándose en la recuperación de radiactividad en la orina observada en un estudio de dosis única de atalureno radiomarcado, se calcula que la biodisponibilidad oral de atalureno es de > 55 %. Las concentraciones plasmáticas de atalureno en estado estacionario aumentan proporcionalmente a la dosis. Las concentraciones plasmáticas en estado estacionario son proporcionales para dosis de atalureno de entre 10 y 50 mg/kg, no observándose ninguna acumulación tras su administración repetida.
Distribución
In vitro, atalureno está unido en un 99,6 % a las proteínas plasmáticas humanas, y esa unión es independiente de la concentración plasmática. Atalureno no se distribuye en los glóbulos rojos.
Biotransformación
Atalureno se metaboliza mediante conjugación por la acción de las enzimas uridina difosfato glucuronosiltransferasa (UGT), principalmente UGT1A9, en el hígado y en el intestino.
In vivo, el único metabolito detectado en el plasma tras la administración por vía oral de atalureno radiomarcado fue el O-1p-acil glucurónido de atalureno; la exposición a este metabolito en seres humanos fue de aproximadamente el 8 % de la AUC plasmática de atalureno.
Eliminación
La semivida plasmática de atalureno oscila entre 2 y 6 horas, y no se ve afectada ni por la dosis ni por la administración repetida. La eliminación de atalureno depende probablemente de la glucuronidación hepática e intestinal de atalureno seguida de la excreción renal del metabolito glucurónido resultante.
Después de una dosis oral única de atalureno radiomarcado, aproximadamente la mitad de la dosis radiactiva administrada se recupera en las heces, y el resto se recupera en la orina. En la orina, el atalureno inalterado y el metabolito acil-glucurónido suponen, respectivamente, < 1% y el 49 % de la dosis administrada.
Linealidad/no linealidad
Las concentraciones plasmáticas en estado estacionario son proporcionales a la dosis para dosis de atalureno de entre 10 y 50 mg/kg, no observándose ninguna acumulación tras una administración repetida. Los datos obtenidos con voluntarios sanos indican que la biodisponibilidad relativa de atalureno es aproximadamente un 40 % más baja en estado estacionario que después de la dosis inicial. Se calcula que el inicio de la reducción de la biodisponibilidad relativa tiene lugar aproximadamente 60 horas después de la primera dosis. El estado estacionario se establece después de aproximadamente dos semanas de tres administraciones al día.
Características en grupos específicos de sujetos o pacientes
Edad
Los datos obtenidos en personas de entre 5 y 57 años de edad indican que la edad no tiene un efecto claro sobre la exposición plasmática de atalureno. No es necesario ajustar la dosis por la edad.
Sexo
En los ensayos clínicos de DMDmss no se estudiaron mujeres. Sin embargo, en otras poblaciones no existe un efecto claro del sexo sobre la exposición plasmática de atalureno.
Raza
Es poco probable que la farmacocinética de atalureno se vea afectada de forma significativa por los polimorfismos de UTG1A9 en la población de origen caucásico. A causa del bajo número de pacientes de otras razas incluidos en los estudios clínicos, no se pueden establecer conclusiones sobre el efecto de UTG1A9 en otros grupos étnicos.
Insuficiencia renal o hepática
No se han realizado estudios con Translarna en pacientes con insuficiencia renal o hepática. Los pacientes con insuficiencia renal o hepática deben ser sometidos a un estrecho control.
Pacientes no ambulantes
No existen diferencias claras debidas a la pérdida de la capacidad ambulante, ni en la biodisponibilidad relativa en estado estacionario ni en el aclaramiento aparente. No es necesario ajustar las dosis para los pacientes que han perdido la capacidad ambulante.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de seguridad farmacológica y genotoxicidad.
Se ha dispuesto de un conjunto estándar de estudios sobre toxicidad reproductiva. No se han observado efectos sobre la fertilidad masculina ni femenina, pero no se han investigado los efectos sobre la fertilidad durante la edad adulta del tratamiento en la primera juventud. Se ha observado toxicidad embrio-fetal (p. ej. aumento de reabsorciones prematuras, pérdidas después de la implantación, disminución del número de fetos viables) y signos de retraso en el desarrollo (aumento de variaciones esqueléticas) en ratas y conejos en presencia de toxicidad materna. La exposición, en el nivel de exposición sin efectos adversos observados (no observed adverse effect level, NOAEL), fue similar (en el conejo) o 4 veces superior (en la rata) a la exposición sistémica en seres humanos (10,
10, 20 mg/kg/día). Se demostró la existencia de una transferencia placentaria de atalureno radiomarcado en las ratas. Con una dosis materna única ensayada, relativamente baja, de 30 mg/kg, la concentración de radiactividad fetal fue < 27 % de la concentración materna. En el estudio de toxicidad para el desarrollo pre/posnatal, con una exposición de aproximadamente 5 veces la exposición en seres humanos se observó una toxicidad materna significativa y efectos sobre el peso corporal y el desarrollo de la actividad ambulante en las crías. La exposición sistémica materna en el nivel de exposición sin efectos adversos observados (no observed effect level, NOEL) para la toxicidad neonatal fue 3 veces superior a la exposición en seres humanos. Con una dosis materna única, relativamente baja, de 30 mg/kg de atalureno radiomarcado, la concentración más alta medida de radiactividad en la leche de rata fue del 37 % de la concentración plasmática materna. La presencia de radiactividad en el plasma de las crías confirmó la absorción por las crías a partir de la leche.
Se produjo toxicidad renal (nefrosis de la nefrona distal) en estudios de dosis orales repetidas en ratones con una exposición sistémica equivalente a 0,3 veces la AUC en equilibrio en pacientes a quienes se administró Translarna con las dosis respectivas, por la mañana, al mediodía y por la noche, de 10, 10 y 20 mg/kg y superiores.
En un modelo de ratón transgénico de 26 semanas para la carcinogenicidad no se encontraron indicios de carcinogenicidad. En un estudio de carcinogenicidad en ratas de 2 años se observó un caso de hibemoma. Además de eso, con una exposición mucho más alta que en los pacientes se observó un aumento de los tumores de la vejiga urinaria (raros). Se considera poco probable la importancia de los tumores de la vejiga urinaria en seres humanos.
En uno de dos estudios de administración repetida en ratas de 26 semanas de duración, realizado con ratas de 4-5 semanas de edad, se observó un aumento relacionado con la dosis de la incidencia del hibernoma maligno, un tumor raro en las ratas. Además de eso, se encontró un caso de hibernoma maligno con la dosis más alta en un estudio de carcinogenicidad con ratas de 2 años de duración. La tasa histórica de incidencia de este tipo de tumor en ratas y en seres humanos es muy baja, y se desconoce el mecanismo que causa ese tumor en los estudios con ratas (incluida su relación con el tratamiento con atalureno). Se desconoce la importancia que puedan tener estos datos para los seres humanos.
En un estudio de 1 año de duración realizado con perros de 10-12 semanas de edad se observaron hallazgos en las glándulas suprarrenales (inflamación localizada y degeneración en las regiones de la corteza que producen glucocorticoides) y una cierta dificultad para la producción de cortisol tras la estimulación exógena con hormona adrenocorticotropa. Estos hallazgos se observaron en perros con una exposición sistémica equivalente a 0,8 veces la AUC en equilibrio en pacientes a quienes se administró Translama con las dosis respectivas, por la mañana, al mediodía y por la noche, de 10, 10 y 20 mg/kg y superiores. En un estudio de distribución con ratas se observó una elevada concentración suprarrenal de atalureno.
Además de los efectos antes mencionados, en los estudios de administración repetida se observaron algunos otros efectos menos adversos; en concreto, la disminución del aumento de peso y de la ingesta de alimento y el aumento del peso del hígado sin una correlación histológica y con una relevancia clínica poco clara. También en estudios con ratas y perros se encontraron cambios en los lípidos plasmáticos (colesterol y triglicéridos) que sugieren cambios en el metabolismo de los lípidos.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polidextrosa (E1200)
Macrogol Poloxámero Manitol (E421)
Crospovidona
Hidroxietilcelulosa
Aroma artificial de vainilla: maltodextrina, aromas artificiales y propilenglicol.
Dióxido de sílice coloidal (E551)
Estearato de magnesio
6.2 Incompatibilidades
No procede
6.3 Período de validez
4 años
Se recomienda administrar cada dosis preparada inmediatamente después de su preparación. Si la dosis preparada se mantiene refrigerada (2-8 °C) se debe desechar si no se utiliza en las 24 horas siguientes a su preparación,o en las 3 horas siguientes si se mantiene a temperatura ambiente (15 -30 °C).
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
Para las condiciones de conservación tras la dilución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Sobre de una hoja de aluminio laminada y termosellada : tereftalato de polietileno (seguridad infantil), polietileno (color y adhesión por poliester/lámina), lámina de aluminio, (barrera contra humedad), adhesivo (de la clase de los poliuretanos), copolímero de etileno y ácido metacrílico (resina selladora para la integridad del envoltorio).
Envase con 30 sobres.
6.6 Precauciones especiales de eliminación y otras manipulaciones
No se deben abrir los sobres hasta el momento de la preparación de la dosis. Se debe mezclar todo el contenido de cada sobre con un mínimo de 30 ml de líquido (agua, leche o zumo de fruta) o 3 cucharadas soperas de un alimento semisólido (yogur o compota de manzana). La dosis preparada se debe mezclar bien antes de su administración. Puede aumentarse la cantidad de líquido o alimento semisólido según las preferencias del paciente.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
PTC Therapeutics International Limited 77 Sir John Rogerson’s Quay Dublin 2 Irlanda
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/902/002
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 31 de julio 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Translama 1.000 mg granulado para suspensión oral
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada sobre contiene 1.000 mg de atalureno.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Granulado para suspensión oral.
Granulado de color entre blanco y blanquecino.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Translarna está indicado para el tratamiento de la distrofia muscular de Duchenne (DMD) debida a una mutación sin sentido en el gen de la distrofina, en pacientes ambulantes a partir de 5 o más años de edad (ver sección 5.1). No se ha demostrado eficacia en pacientes no ambulantes.
La presencia de una mutación sin sentido en el gen de la distrofina se debe determinar mediante pruebas genéticas (ver sección 4.4).
4.2 Posología y forma de administración
El tratamiento con Translarna se debe iniciar solo por médicos especialistas con experiencia en el control de la distrofia muscular de Duchenne/Becker.
Posología
Atalureno se debe administrar por vía oral, 3 veces al día.
La primera toma se debe administrar por la mañana, la segunda al mediodía y la tercera por la noche. Los intervalos posológicos recomendados son de 6 horas entre la toma de la mañana y la del mediodía; de 6 horas entre la del mediodía y la de la noche, y de 12 horas entre la de la noche y la primera toma del día siguiente.
La dosis recomendada es de 10 mg/kg de peso corporal por la mañana, 10 mg/kg de peso corporal a mediodía y 20 mg/kg de peso corporal por la noche (para una dosis diaria total de 40 mg/kg de peso corporal).
Translarna se presenta en sobres de 125 mg, 250 mg o 1.000 mg. La siguiente tabla ofrece información sobre la o las dosis del sobre que se deben emplear para preparar la posología recomendada según el intervalo de peso corporal.
|
Intervalo de peso (kg) |
Número de sobres | |||||||||
|
Mañana |
Mediodía |
Noche | ||||||||
|
Sobres de 125 mg |
Sobres de 250 mg |
Sobres de 1.000 mg |
Sobres de 125 mg |
Sobres de 250 mg |
Sobres de 1.000 mg |
Sobres de 125 mg |
Sobres de 250 mg |
Sobres de 1.000 mg | ||
|
12 |
14 |
1 |
0 |
0 |
1 |
0 |
0 |
0 |
1 |
0 |
|
15 |
16 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
1 |
0 |
|
17 |
20 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
|
21 |
23 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
1 |
0 |
|
24 |
26 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
0 |
|
27 |
31 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
0 |
|
32 |
35 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
0 |
|
36 |
39 |
1 |
1 |
0 |
1 |
1 |
0 |
0 |
3 |
0 |
|
40 |
44 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
3 |
0 |
|
45 |
46 |
0 |
2 |
0 |
0 |
2 |
0 |
1 |
3 |
0 |
|
47 |
55 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
0 |
1 |
|
56 |
62 |
0 |
2 |
0 |
0 |
2 |
0 |
0 |
1 |
1 |
|
63 |
69 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
1 |
1 |
|
70 |
78 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
2 |
1 |
|
79 |
86 |
0 |
3 |
0 |
0 |
3 |
0 |
0 |
3 |
1 |
|
87 |
93 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
3 |
1 |
|
94 |
105 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
0 |
2 |
|
106 |
111 |
0 |
0 |
1 |
0 |
0 |
1 |
0 |
1 |
2 |
|
112 |
118 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
1 |
2 |
|
119 |
125 |
0 |
1 |
1 |
0 |
1 |
1 |
0 |
2 |
2 |
Retraso u olvido de la toma de la dosis
Si hay un retraso en la administración de atalureno de menos de 3 horas después de la toma habitual de la mañana o del mediodía, o de menos de 6 horas después de la de la noche, se deberá tomar la dosis sin realizar cambios en la pauta posológica. Si hay un retraso de más de 3 horas después de la toma habitual de la mañana o del mediodía, o de más de 6 horas después de la de la noche, no se debe tomar la dosis, y el paciente debe reanudar su pauta posológica habitual. Los pacientes no deben tomar una dosis doble o adicional para compensar una dosis olvidada. Es importante administrar la dosis correcta. El aumento de la dosis por encima de la dosis recomendada puede tener como resultado una reducción de la eficacia.
Poblaciones especiales
Pacientes de edad avanzada
No se ha establecido todavía la seguridad y eficacia de atalureno en pacientes de 65 años o más. (Ver sección 5.2).
Insuficiencia renal y hepática
No se ha establecido ni la seguridad ni la eficacia de atalureno en pacientes con insuficiencia renal o hepática. (Ver sección 4.4).
Población pediátrica
No se ha establecido todavía ni la seguridad ni la eficacia de Translama en niños con edades comprendidas entre los 6 meses y los 5 años de edad. No se dispone de datos.
Forma de administración
Translarna se debe administrar por vía oral después de mezclarlo para formar una suspensión en líquido o con un alimento semisólido.
No se deben abrir los sobres hasta el momento de la preparación de la dosis. Se debe mezclar todo el contenido de cada sobre con un mínimo de 30 ml de líquido (agua, leche o zumo de fruta) o 3 cucharadas soperas de un alimento semisólido (yogur o compota de manzana). La dosis preparada se debe mezclar bien antes de su administración. Se puede aumentar la cantidad de líquido o alimento semisólido según las preferencias del paciente. Los pacientes deben tomar la dosis completa.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Uso concomitante de aminoglucósidos por vía intravenosa (ver sección 4.4 y 4.5).
4.4 Advertencias y precauciones especiales de empleo Pacientes que no tienen una mutación sin sentido
Los pacientes deben tener una mutación sin sentido en el gen de la distrofina, determinada mediante análisis genéticos, como parte de su estado patológico subyacente. No se debe administrar atalureno a pacientes que no tengan una mutación sin sentido.
Insuficiencia renal y hepática
Los pacientes con insuficiencia renal y hepática deben ser sometidos a un estrecho control.
Cambios en el perfil lipídico
Puesto que se han notificados cambios en el perfil lipídico (aumento de triglicéridos y colesterol) de algunos pacientes en ensayos clínicos, se recomienda realizar, en los pacientes con distrofia muscular de Duchene debida a una mutación sin sentido (DMDmss) que reciban atalureno, un control del colesterol total, LDL, HDL y triglicéridos cada año, o con mayor frecuencia si es necesario por el estado clínico del paciente.
Hipertensión con el uso concomitante de corticosteroides sistémicos
Puesto que se han notificado casos de hipertensión con el uso concomitante de corticosteroides sistémicos en algunos pacientes en ensayos clínicos, se recomienda realizar, en los pacientes con DMDmss que reciban atalureno de forma concomitante con corticosteroides, un control de la tensión arterial sistólica y diastólica en reposo cada 6 meses, o con mayor frecuencia si es necesario por el estado clínico del paciente.
Monitorización de la función renal
Puesto que se han notificados ligeros aumentos de las concentraciones medias de creatinina sérica, nitrógeno ureico en sangre (BUN, por sus siglas en inglés) y de cistatina C en el estudio comparativo de DMDmss, se recomienda realizar, en los pacientes con DMDmss que reciban atalureno, un control de la creatinina sérica, el BUN y la cistatina C cada 6-12 meses, o con mayor frecuencia si es necesario por el estado clínico del paciente.
Interacciones potenciales con otros medicamentos
Se debe actuar con precaución al administrar atalureno de forma conjunta con medicamentos que sean sustratos o inductores de UGT1A9, inhibidores de BCRP o sustratos de OAT1, OAT3 u OATP1B3 (ver sección 4.5).
Aminoglucósidos
Se ha demostrado que los aminoglucósidos reducen la actividad de lectura ribosómica del atalureno in vitro. Asimismo, se encontró que el atalureno aumenta la nefrotoxicidad de los aminoglucósidos intravenosos. Se debe evitar la administración conjunta de esos medicamentos con atalureno (ver sección 4.3). Dado que se desconoce el mecanismo mediante el cual el atalureno aumenta la nefrotoxicidad de los aminoglucósidos intravenosos, no se recomienda el uso concomitante de otros medicamentos nefrotóxicos con atalureno. En caso de que esto sea inevitable, (por ejemplo: vancomicina para el tratamiento de SARM) se recomienda una monitorización minuciosa de la función renal (ver sección 4.5).
4.5 Interacción con otros medicamentos y otras formas de interacción
Aminoglucósidos
No se debe administrar Atalureno de forma conjunta con aminoglucósidos intravenosos, pues se han observado casos de disminución de la función renal en un ensayo clínico en pacientes con nmCF (ver sección 4.3).
En varios pacientes tratados con atalureno y aminoglucósidos intravenosos junto con otros antibióticos para los brotes de la fibrosis quística se produjeron elevaciones de la creatinina sérica. Las elevaciones de la creatinina sérica remitieron en todos los casos cuando se interrumpió la administración del aminoglucósido intravenoso, tanto si se continuó como si se interrumpió la administración de Translarna. Estos datos sugieren que la administración conjunta de Translama y aminoglucósidos intravenosos podría potenciar el efecto nefrotóxico de los aminoglucósidos. En consecuencia, si es necesario el tratamiento con aminoglucósidos intravenosos se debe interrumpir el tratamiento con Translarna, que se podrá reanudar 2 días después de finalizar la administración del aminoglucósido. Se desconoce el efecto de la administración conjunta de ataluren con otros medicamentos nefrotóxicos.
La deshidratación podría ser un factor que contribuyera en alguno de estos casos. Los pacientes se deben mantener adecuadamente hidratados mientras tomen atalureno. Ver sección 4.4.
Efectos de otros medicamentos en la farmacocinética de atalureno
Los datos obtenidos en estudios in vitro indican que atalureno es un sustrato de UGT1A9 y de la proteína resistente al cáncer de mama (BCRP). Se debe actuar con precaución cuando se administra de forma conjunta atalureno con medicamentos inductores de UGT1A9 (como el mofetil micofenolato) o inhibidores de BCRP (por ejemplo, ciclosporina).
Efectos de atalureno sobre la farmacocinética de otros medicamentos
Los datos obtenidos en estudios in vitro indican que atalureno es un inhibidor de UGT1A9, del transportador de aniones orgánico 1 (OAT1), del transportador de aniones orgánico 3 (OAT3) y del polipéptido de transporte del anión orgánico 1B3 (OATP1B3). Se debe actuar con precaución cuando se administra de forma conjunta atalureno con medicamentos sustratos de UGT1A9, OAT1, OAT3 u OATP1B3 (por ejemplo, oseltamivir, aciclovir, ciprofloxacino, captoprilo, furosemida, bumetanida, valsartán, pravastatina, rosuvastatina, atorvastatina o pitavastatina), debido al riesgo del aumento de la concentración de estos medicamentos .
Basándose en estudios in vitro no se espera que atalureno sea un inhibidor del transporte mediado por P-gp ni del metabolismo mediado por el citocromo P450. Del mismo modo, resulta poco probable que atalureno sea in vivo un inductor de las isoenzimas del citocromo P450.
La administración conjunta de corticosteroides (deflazacort, prednisona o prednisolona) con atalureno no afectó a las concentraciones plasmáticas de atalureno. No se observó ningún cambio clínicamente relevante en las concentraciones plasmáticas de los corticoides con la administración conjunta de atalureno. Estos datos indican que no existe interacción farmacológica aparente entre los corticosteroides y atalureno, no siendo necesario ningún ajuste de la dosis.
Medicamentos que afectan el transportador de glucoproteína P
In vitro, atalureno no es un sustrato del transportador de glucoproteína P. Es poco probable que la farmacocinética de atalureno resulte afectada por medicamentos que inhiben el transportador de glucoproteína P.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos suficientes relativos al uso de atalureno en mujeres embarazadas. Los estudios realizados en animales han revelado una toxicidad para la reproducción solamente con dosis tóxicas para la madre (ver sección 5.3).
Como medida de precaución no se recomienda utilizar atalureno durante el embarazo.
Lactancia
Se desconoce si el atalureno/sus metabolitos se excretan en la leche materna. Los datos farmacodinámicos/toxicológicos disponibles en animales muestran que el atalureno/ sus metabolitos se excretan en la leche (ver sección 5.3). No se puede excluir el riesgo en recién nacidos/niños lactantes.
Debe interrumpirse la lactancia durante el tratamiento con atalureno.
Fertilidad
Los datos preclínicos no muestran riesgos para los seres humanos en base a los estudios de fertilidad estándar con machos y hembras de rata (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se ha estudiado el efecto de atalureno sobre la conducción y la utilización de máquinas. Los pacientes que experimenten mareos deben tener cuidado si conducen o utilizan máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
En ensayos clínicos en pacientes con distrofia muscular de Duchenne debida a una mutación sin sentido (DMDmss), las reacciones adversas más frecuentes con la dosis recomendada fueron náuseas, vómitos y cefaleas. Esas reacciones adversas no requirieron por lo general una intervención médica, y ningún paciente interrumpió el tratamiento con atalureno a causa de una reacción adversa.
Tabla de reacciones adversas
Las reacciones adversas notificadas en el estudio clínico de pacientes predominantemente pediátricos con DMDmss tratados con la dosis recomendada de 10, 10 y 20 mg/kg se clasifican de acuerdo con la Clasificación por órganos y sistemas del MedDRA y la frecuencia. Los grupos de frecuencia se definen según la siguiente convención: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10) y frecuencia no conocida (no puede estimarse a partir de datos disponibles). Las reacciones adversas se presentan por orden decreciente de gravedad dentro de cada grupo de frecuencias.
Tabla 3. Reacciones adversas al Translarna en el estudio controlado de DMDmss
|
Sistema de clasificación de órganos |
Muy frecuentes |
Frecuentes |
Frecuencia no conocida |
|
Trastornos del metabolismo y de la nutrición |
Disminución del apetito |
Cambio en el perfil lipídico (aumento de triglicéridos y colesterol) | |
|
Trastornos del sistema nervioso |
Cefalea |
Mareo | |
|
Trastornos vasculares |
Hipertensión | ||
|
Trastornos respiratorios, torácicos y mediastínicos |
Tos, epistaxis | ||
|
Trastornos gastrointestinales |
Náuseas, vómitos |
Dolor abdominal alto, flatulencia, diarrea, molestias en el estómago, dolor abdominal, estreñimiento, regurgitación | |
|
Trastornos de la piel y del tejido subcutáneo |
Eritema | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor en las extremidades | ||
|
Trastornos renales y urinarios |
Enuresis, quiste renal, polaquiuria, coloración de la orina anormal |
Cambio en las pruebas de función renal (aumento de la creatinina, nitrógeno ureico en sangre, cistatina C) | |
|
Trastornos generales y alteraciones en el lugar de administración |
Pirexia, fatiga, pérdida de peso |
Descripción de reacciones adversas seleccionadas
Lípidos séricos
Durante el estudio controlado de DMDmss, la concentración media de colesterol total y de triglicéridos fueron normales en el momento inicial y aumentó, alcanzando el umbral de los valores límite alto o valores elevados. Los valores tendían a estabilizarse en la parte inicial del estudio y no aumentaron más con la continuación del tratamiento.
Pruebas de función renal
Durante el estudio controlado de DMDmss se observaron ligeros aumentos en el valor medio de la creatinina sérica, nitrógeno ureico en sangre (BUN) y cistatina C. Los valores tendían a estabilizarse en la parte inicial del estudio y no aumentaron más con la continuación del tratamiento.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continua de la relación del beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Algunos voluntarios sanos que recibieron una única dosis por vía oral de 200 mg/kg de atalureno experimentaron síntomas transitorios leves de cefalea, náuseas, vómitos y diarrea. No se observaron reacciones adversas graves en esos sujetos. En el caso de sospecha de sobredosis se debe proporcionar tratamiento de soporte, incluyendo la consulta con un profesional sanitario y una estrecha observación del estado clínico del paciente.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: {grupo}, código ATC: no se ha asignado aún Mecanismo de acción
Una mutación sin sentido en el ADN tiene como resultado un codón de parada prematuro en un ARNm. Este codón de parada prematuro en el ARNm provoca la enfermedad al finalizar la traducción antes de que se genere una proteína funcional. Atalureno facilita la lectura ribosómica del ARNm que contiene un codón de parada prematuro de ese tipo, lo que tiene como resultado la producción de una proteína funcional.
Efectos farmacodinámicos
Los experimentos no clínicos in vitro realizados en cultivos de células con mutaciones sin sentido y en larvas de peces cultivadas en una solución de atalureno han demostrado que atalureno facilita la lectura ribosómica con una relación concentración/respuesta en forma de campana (forma de U invertida). Se propone la hipótesis de que la relación dosis/respuesta in vivo también podría tener forma de campana, pero los datos de estudios in vivo realizados en un modelo de ratón para DMDmss y en humanos son demasiado limitados para confirmar esa hipótesis.
Los estudios no clínicos in vitro sugieren que puede ser importante la exposición continua a atalureno para maximizar su actividad, y que los efectos del principio activo sobre la lectura ribosómica de los codones de parada prematuros revierten poco después de la retirada de atalureno.
Eficacia clínica y seguridad
La eficacia y la seguridad de Translarna se evaluaron en un estudio aleatorizado, doble ciego, controlado con placebo, multicéntrico, sobre la distrofia muscular de Duchenne causada por una mutación sin sentido (nonsense mutation Duchenne muscular dystrophy, DMDmss) en 174 pacientes varones de entre 5 y 20 años de edad. Se exigió que todos los pacientes fueran pacientes ambulantes, definida como la capacidad de caminar > 75 metros sin necesidad de dispositivos de asistencia durante una prueba de marcha de 6 minutos (6-minute Walk Test, 6MWT) realizada durante el cribado. También fue necesario que los pacientes tuvieran una confirmación documentada de la presencia de una mutación sin sentido en el gen de la distrofina, determinada por secuenciación genética. La mayoría de los pacientes de todos los grupos de tratamiento eran de origen caucásico (90 %). Se asignó aleatoriamente a los pacientes en una proporción de 1:1:1 para recibir atalureno o placebo 3 veces al día (mañana, mediodía y noche) durante 48 semanas: 57 recibieron placebo, 57 recibieron atalureno (10, 10 y 20 mg/kg) y 60 recibieron atalureno (20, 20 y 40 mg/kg). Finalizaron el estudio 173 pacientes. La variable principal de eficacia era el Atalureno sobre la deambulación, evaluada por medio del cambio en la distancia (6MWD) recorrida durante una prueba 6MWT. El análisis post hoc reveló que desde el periodo basal hasta la semana 48, en los pacientes tratados con atalureno 10, 10 y 20 mg/kg se observó una disminución media de la 6MWD de 12,9 metros y los pacientes que recibían placebo se observó una disminución media de la 6MWD de 44,1 metros (figura 1). Por lo tanto, el cambio medio en la 6MWD observada desde el momento inicial hasta la semana 48 fue de 31,3 metros mejor en el grupo de atalureno 10, 10 y 20 mg/kg que en el grupo de placebo (p=0,056). En una estimación de modelo estadístico, la diferencia media fue de 31,7 metros (p ajustada = 0,0367). No hubo ninguna diferencia entre atalureno 20, 20 y 40 mg/kg y placebo. Estos resultados indican que atalureno 10, 10 y 20 mg/kg disminuye el ritmo de la pérdida de la capacidad ambulante en los pacientes con DMDmss.
Figura 1. Cambio medio en la distancia recorrida en 6 minutos
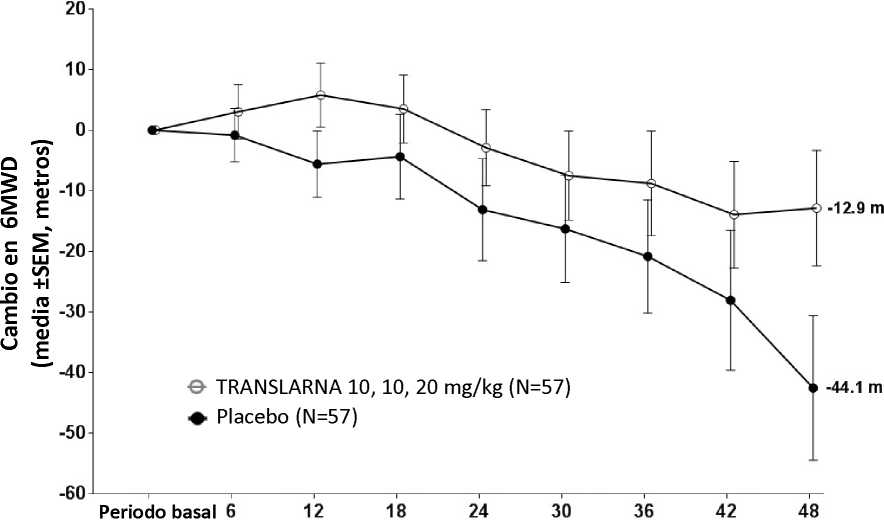
Semanas
Un análisis post hoc del tiempo transcurrido hasta el empeoramiento persistente del 10 % en la 6MWD mostró que en el 26 % de los pacientes del grupo de atalureno 10, 10 y 20 mg/kg había habido progresión en la semana 48, frente al 44 % de los del grupo de placebo (p=0,0652) (figura 2). No hubo ninguna diferencia entre atalureno 20, 20 y 40 mg/kg y placebo. Estos resultados indican que un menor número de pacientes que reciben atalureno 10, 10 y 20 mg/kg empeoraron en 6MWD a lo largo de 48 semanas.
Figura 2. Curva de Kaplan-Meier del tiempo transcurrido hasta el empeoramiento persistente del 10 % en la 6MWD
100i
90
80
0)
■c
oN