Stivarga 40Mg Comprimidos Recubiertos Con Pelicula
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Stivarga 40 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 40 mg de regorafenib.
Excipientes con efecto conocido:
Cada dosis diaria de 160 mg contiene 2,427 mmol (o 55,8 mg) de sodio (ver sección 4.4).
Cada dosis diaria de 160 mg contiene 1,68 mg de lecitina (derivada de la soja) (ver sección 4.4).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película.
Comprimidos recubiertos con película de color rosa claro y forma oval, de 16 mm de largo y 7 mm de ancho, marcados con "BAYER" en una cara y "40" en la otra.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Stivarga está indicado para el tratamiento de pacientes adultos con:
- cáncer colorrectal (CCR) metastásico que han sido previamente tratados con las terapias disponibles o no se les considera candidatos adecuados a dichas terapias. Esto incluye quimioterapia basada en fluoropirimidinas, terapia anti-VEGF y terapia anti-EGFR (ver sección 5.1).
- tumores del estroma gastrointestinal (gastrointestinal stromal tumors, GIST) irresecables o metastásicos que progresaron durante el tratamiento previo con imatinib y sunitinib o son intolerantes al mismo.
4.2 Posología y forma de administración
Stivarga debe ser prescrito por médicos con experiencia en la administración de terapias
anticancerosas.
Posología
La dosis recomendada de regorafenib es 160 mg (4 comprimidos de 40 mg), administrados una vez al
día durante 3 semanas seguidas de 1 semana sin tratamiento. Este período de 4 semanas se considera
un ciclo de tratamiento.
En caso de omisión de una dosis, el paciente debe tomarla ese mismo día en cuanto se acuerde. El paciente no debe tomar una dosis doble el mismo día para compensar una dosis olvidada. En caso de vómitos tras la administración de regorafenib, el paciente no debe tomar comprimidos adicionales.
El tratamiento debe continuar mientras se observen beneficios o hasta la aparición de toxicidad inaceptable (ver sección 4.4).
Los pacientes con estado funcional (PS) 2 o mayor se excluyeron de los estudios clínicos. Se dispone de datos limitados en pacientes con PS > 2.
Ajustes de la posología
Puede ser necesario realizar interrupciones y/o reducciones de la dosis en función de la seguridad y la tolerabilidad de cada paciente. Las modificaciones de dosis deben efectuarse en escalones de 40 mg (un comprimido). La dosis diaria mínima recomendada es 80 mg. La dosis diaria máxima es 160 mg.
En la Tabla 1 se indican las modificaciones de la dosis y las medidas recomendadas que es preciso adoptar en caso de reacción cutánea mano-pie (Hand-foot skin reaction, HFSR) / síndrome de eritrodisestesia palmoplantar.
Tabla 1: Modificaciones de la dosis y medidas recomendadas para la HFSR
|
Grado de toxicidad cutánea |
Aparición |
Modificaciones de la dosis y medidas recomendadas |
|
Grado 1 |
Cualquiera |
Mantener la dosis e instaurar inmediatamente medidas de soporte para el alivio de los síntomas. |
|
1a aparición |
Reducir la dosis en 40 mg (un comprimido) e instaurar inmediatamente medidas de soporte. Si no se produce mejoría a pesar de la reducción de la dosis, interrumpir el tratamiento durante un mínimo de 7 días hasta que la toxicidad disminuya a grado 0-1. Se permite un escalado de la dosis según criterio médico. | |
|
Grado 2 |
Ausencia de mejoría en un plazo de 7 días o 2a aparición |
Interrumpir el tratamiento hasta que la toxicidad disminuya a grado 0-1. Al reiniciar el tratamiento, reducir la dosis en 40 mg (un comprimido). Se permite un escalado de la dosis según criterio médico. |
|
3a aparición |
Interrumpir el tratamiento hasta que la toxicidad disminuya a grado 0-1. Al reiniciar el tratamiento, reducir la dosis en 40 mg (un comprimido). Se permite un escalado de la dosis según criterio médico. | |
|
4a aparición |
Suspender permanentemente el tratamiento con Stivarga. |
|
Grado de toxicidad cutánea |
Aparición |
Modificaciones de la dosis y medidas recomendadas |
|
Grado 3 |
1a aparición |
Instaurar inmediatamente medidas de soporte. Interrumpir el tratamiento durante un mínimo de 7 días hasta que la toxicidad disminuya a grado 0-1. Al reiniciar el tratamiento, reducir la dosis en 40 mg (un comprimido). Se permite un escalado de la dosis según criterio médico. |
|
2a aparición |
Instaurar inmediatamente medidas de soporte. Interrumpir el tratamiento durante un mínimo de 7 días hasta que la toxicidad disminuya a grado 0-1. Al reiniciar el tratamiento, reducir la dosis en 40 mg (un comprimido). | |
|
3a aparición |
Suspender permanentemente el tratamiento con Stivarga. |
En la Tabla 2 se indican las medidas y las modificaciones de las dosis recomendadas en caso de empeoramiento de las pruebas de la función hepática que se consideran relacionadas con el tratamiento con Stivarga (ver también la sección 4.4).
Tabla 2: Medidas y modificaciones de las dosis recomendadas en caso de anomalías en las pruebas de la función hepática relacionadas con el medicamento
|
Elevaciones observadas de ALT y/o AST |
Aparición |
Medidas y modificaciones de la dosis recomendadas |
|
<5 veces el límite superior de la normalidad (ULN) (máximo grado 2) |
Cualquier aparición |
Continuar el tratamiento con Stivarga. Monitorizar la función hepática semanalmente hasta que las transaminasas retornen a < 3 veces el ULN (grado 1) o al nivel basal. |
|
>5 veces el ULN <20 veces el ULN (grado 3) |
1a aparición |
Interrumpir el tratamiento con Stivarga. Monitorizar las transaminasas semanalmente hasta que retornen a < 3 veces el ULN o al nivel basal. Reinicio: Si el beneficio potencial supera al riesgo de hepatotoxicidad, reiniciar el tratamiento con Stivarga, reducir la dosis en 40 mg (un comprimido) y monitorizar la función hepática semanalmente durante al menos 4 semanas. |
|
Reaparición |
Suspender permanentemente el tratamiento con Stivarga. | |
|
>20 veces el ULN (grado 4) |
Cualquier aparición |
Suspender permanentemente el tratamiento con Stivarga. |
|
Elevaciones observadas de ALT y/o AST |
Aparición |
Medidas y modificaciones de la dosis recomendadas |
|
>3 veces el ULN (grado 2 o mayor) con bilirrubina concurrente >2 veces el ULN |
Cualquier aparición |
Suspender permanentemente el tratamiento con Stivarga. Monitorizar la función hepática semanalmente hasta la resolución o el retorno al nivel basal. Excepción: los pacientes con síndrome de Gilbert que presenten una elevación de las transaminasas deben tratarse según las recomendaciones previamente indicadas para la respectiva elevación observada de ALT y/o AST. |
Insuficiencia hepática
Regorafenib se elimina principalmente por vía hepática.
En los estudios clínicos, no se observaron diferencias importantes en cuanto a exposición, seguridad o eficacia entre los pacientes con insuficiencia hepática leve (Child-Pugh A) y los pacientes con función hepática normal. No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. Dado que se dispone de escasos datos para los pacientes con insuficiencia hepática moderada (Child-Pugh B), no es posible proporcionar una recomendación posológica. Se recomienda efectuar una estrecha monitorización de la seguridad global en estos pacientes (ver las secciones 4.4 y 5.2).
No se recomienda el uso de Stivarga en los pacientes con insuficiencia hepática grave (Child-Pugh C), dado que Stivarga no se ha estudiado en esta población.
Insuficiencia renal
En los estudios clínicos, no se observaron diferencias importantes en cuanto a exposición, seguridad o eficacia entre los pacientes con insuficiencia renal leve (tasa de filtración glomerular estimada [eGFR] 60-89 ml/min/1,73 m2) y los pacientes con función renal normal. Datos farmacocinéticos limitados indican que no existen diferencias en cuanto a exposición en los pacientes con insuficiencia renal moderada (eGFR 30-59 ml/min/1,73 m2). No es necesario ajustar la dosis en los pacientes con insuficiencia renal leve o moderada (ver también la sección 5.2). No se dispone de datos clínicos en pacientes con insuficiencia renal grave (eGFR < 30 ml/min/1,73 m2).
Pacientes de edad avanzada
En los estudios clínicos, no se observaron diferencias importantes en cuanto a exposición, seguridad o eficacia entre los pacientes de edad avanzada (de edad igual o superior a 65 años) y los de edades más jóvenes (ver también la sección 5.2).
Sexo
En los estudios clínicos, no se observaron diferencias importantes en cuanto a exposición, seguridad o eficacia entre los hombres y las mujeres. No es necesario ajustar la dosis en función del sexo (ver también la sección 5.2).
Diferencias étnicas
En los estudios clínicos, no se observaron diferencias importantes en cuanto a exposición o eficacia entre los pacientes de distintos grupos étnicos. Se observó una incidencia más alta de reacción cutánea mano-pie (Hand-foot skin reaction, HFSR, por sus siglas en inglés) / síndrome de eritrodisestesia palmoplantar, anomalías graves en las pruebas de función hepática y disfunción hepática en los pacientes asiáticos (en especial los japoneses) tratados con Stivarga en comparación con los caucásicos. Los pacientes asiáticos tratados con Stivarga en los estudios clínicos eran principalmente de asia oriental (aproximadamente el 90%). Se dispone de datos limitados sobre regorafenib en la población de pacientes de raza negra.
No es necesario ajustar la dosis en función de la etnia (ver sección 5.2).
Población pediátrica
No hay un uso relevante de Stivarga en la población pediátrica para la indicación de cáncer colorrectal mestastásico.
No se ha establecido la seguridad y eficacia de regorafenib en pacientes menores de 18 años para la indicación de tumores del estroma gastrointestinal (GIST). No hay datos disponibles.
Forma de administración Stivarga es para uso por vía oral.
Stivarga debe tomarse todos los días a la misma hora. Los comprimidos deben tomarse enteros con agua después de una comida ligera con un contenido menor del 30% en grasa. Un ejemplo de comida ligera (baja en grasa) incluiría 1 porción de cereales (alrededor de 30 g), 1 vaso de leche descremada,
1 tostada con mermelada, 1 vaso de zumo de manzana y 1 taza de café o té (520 calorías, 2 g de grasa).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Efectos hepáticos
Se han observado con frecuencia anomalías en las pruebas de la función hepática (alanina aminotransferasa [ALT], aspartato aminotransferasa [AST] y bilirrubina) en los pacientes tratados con Stivarga. Se han notificado anomalías graves en las pruebas de la función hepática (de grado 3 a 4) y disfunción hepática con manifestaciones clínicas (incluidos casos con desenlace fatal) en un pequeño porcentaje de pacientes (ver sección 4.8). En los ensayos clínicos, se observó una incidencia más alta de anomalías graves en las pruebas de función hepática y disfunción hepática en los pacientes asiáticos (en especial los japoneses) tratados con Stivarga en comparación con los caucásicos (ver sección 4.2).
Se recomienda realizar pruebas de función hepática (ALT, AST y bilirrubina) antes del inicio del tratamiento con Stivarga y mantener bajo una estrecha monitorización (al menos cada dos semanas) durante los 2 primeros meses de tratamiento. A partir de entonces, la monitorización debe proseguir con una periodicidad al menos mensual y cuando esté clínicamente indicado.
Regorafenib es un inhibidor de la uridindifosfato glucuronosil transferasa (UGT) 1A1 (ver sección 4.5). En los pacientes con síndrome de Gilbert puede producirse una hiperbilirrubinemia (no conjugada) indirecta de grado leve.
En los pacientes en los que se observe un empeoramiento de las pruebas de función hepática que se considere relacionado con el tratamiento con Stivarga (es decir, en los que no existe una causa alternativa evidente, como colestasis poshepática o progresión de la enfermedad), deben seguirse las recomendaciones en cuanto a modificación de la dosis y monitorización indicadas en la Tabla 2 (ver sección 4.2).
Regorafenib se elimina principalmente por vía hepática.
Se recomienda efectuar una estrecha monitorización de la seguridad global en los pacientes con insuficiencia hepática leve o moderada (ver también las secciones 4.2 y 5.2). No se recomienda el uso de Stivarga en los pacientes con insuficiencia hepática grave (Child-Pugh C), dado que Stivarga no se ha estudiado en esta población y la exposición podría estar aumentada en estos pacientes.
Hemorragia
Stivarga se ha asociado a un incremento de la incidencia de acontecimientos hemorrágicos, algunos de ellos fatales (ver sección 4.8). Deben monitorizarse los recuentos sanguíneos y los parámetros de la coagulación en los pacientes con enfermedades que les predispongan a sufrir hemorragias y en los tratados con anticoagulantes (por ejemplo, warfarina y fenprocumón) u otros medicamentos concomitantes que aumenten el riesgo de hemorragias. En caso de hemorragia grave que precise intervención médica urgente, debe plantearse la suspensión permanente del tratamiento con Stivarga.
Isquemia cardiaca e infarto
Stivarga se ha asociado a un incremento de la incidencia de isquemia e infarto de miocardio (ver sección 4.8). Los pacientes con angina inestable o angina de nueva aparición (en los 3 meses previos al inicio del tratamiento con Stivarga), infarto de miocardio reciente (en los 6 meses previos al inicio del tratamiento con Stivarga) y con insuficiencia cardiaca grado 2 o mayor de la New York Heart Association (NYHA) se excluyeron de los estudios clínicos.
Los pacientes con antecedentes de cardiopatía isquémica deben monitorizarse en busca de signos y síntomas clínicos de isquemia miocárdica. En los pacientes que presenten isquemia cardiaca y/o infarto, se recomienda la interrupción del tratamiento con Stivarga hasta la resolución. La decisión de reiniciar el tratamiento con Stivarga debe basarse en una cuidadosa evaluación de los beneficios y riesgos potenciales para cada paciente en concreto. Stivarga debe suspenderse permanentemente si no se produce una resolución.
Síndrome de leucoencefalopatía posterior reversible (PRES)
Se han notificado casos de PRES en relación con el tratamiento con Stivarga (ver sección 4.8). Entre los signos y síntomas se encuentran convulsiones, cefalea, alteración del estado mental, alteraciones visuales o ceguera cortical, con o sin hipertensión asociada. El diagnóstico del PRES requiere confirmación mediante pruebas de imagen cerebrales. En los pacientes que presenten un PRES, se recomienda la suspensión del tratamiento con Stivarga, además del control de la hipertensión y el tratamiento médico de soporte para el resto de los síntomas.
Perforación y fístulas gastrointestinales
Se han notificado perforación (incluidos casos con desenlace mortal) y fístulas gastrointestinales en pacientes tratados con Stivarga (ver sección 4.8). Se sabe también que estos acontecimientos son complicaciones frecuentes, relacionadas con la enfermedad, en los pacientes con enfermedades malignas intraabdominales. Se recomienda la suspensión del tratamiento con Stivarga en los pacientes que presenten perforación o fístulas gastrointestinales.
Hipertensión arterial
Stivarga se ha asociado a un incremento de la incidencia de hipertensión arterial (ver sección 4.8). Debe realizarse un control de la presión arterial antes del inicio del tratamiento con Stivarga. Se recomienda monitorizar la presión arterial y tratar la hipertensión de acuerdo a la práctica médica estándar. En los casos de hipertensión grave o persistente a pesar del tratamiento médico adecuado, se debe interrumpir temporalmente el tratamiento y/o reducir su dosis según criterio médico (ver sección 4.2). Stivarga debe suspenderse en caso de crisis hipertensiva.
Complicaciones en la cicatrización de heridas
Dado que los medicamentos con propiedades antiangiogénicas pueden suprimir o alterar el proceso de curación de heridas, se recomienda interrumpir temporalmente el tratamiento con Stivarga como medida de precaución en los pacientes que vayan a someterse a procedimientos quirúrgicos mayores. La decisión de reanudar el tratamiento con Stivarga tras estas intervenciones debe basarse en el criterio clínico sobre la adecuada cicatrización de las heridas.
Toxicidad dermatológica
La reacción cutánea mano-pie (HFSR) o síndrome de eritrodisestesia palmoplantar y el exantema son las reacciones adversas dermatológicas más frecuentemente observadas con Stivarga (ver sección 4.8). En los ensayos clínicos, se observó una incidencia más alta de HFSR en los pacientes asiáticos (en especial los japoneses) tratados con Stivarga en comparación con los caucásicos (ver sección 4.2). Entre las medidas para la prevención del HFSR se encuentran el control de las callosidades y el uso de guantes y almohadillas en los zapatos para evitar el estrés sobre palmas y plantas. El tratamiento del HFSR puede incluir el uso de cremas queratolíticas (p. ej., cremas con urea, ácido salicílico o alfahidroxiácidos aplicadas con moderación, solamente en las zonas afectadas) y cremas hidratantes (aplicadas generosamente) para el alivio de los síntomas. Debe plantearse una reducción de la dosis y/o una interrupción temporal del tratamiento con Stivarga o, en los casos graves o persistentes, la suspensión permanente del mismo (ver sección 4.2).
Anomalías de las pruebas metabólicas y bioquímicas de laboratorio
Stivarga se ha asociado a un incremento de la incidencia de anomalías electrolíticas (incluidas hipofosfatemia, hipocalcemia, hiponatremia e hipopotasemia) y metabólicas (incluidos niveles aumentados de la hormona estimulante del tiroides, la lipasa y la amilasa). Estas anomalías son generalmente de intensidad leve a moderada, no se asocian con manifestaciones clínicas y no suelen precisar interrupciones ni reducciones de la dosis. Se recomienda monitorizar los parámetros bioquímicos y metabólicos durante el tratamiento con Stivarga e instaurar la terapia sustitutiva adecuada de acuerdo con las prácticas clínicas estándar, si es necesario. Debe plantearse una interrupción o reducción de la dosis o la suspensión permanente del tratamiento con Stivarga en caso de anomalías significativas persistentes o recurrentes (ver sección 4.2).
Información importante sobre algunos de los componentes
Cada dosis diaria de 160 mg contiene 2,427 mmol (o 55,8 mg) de sodio, lo que debe ser tenido en cuenta en pacientes con dietas pobres en sodio. Cada dosis diaria de 160 mg contiene 1,68 mg de lecitina (derivada de la soja).
4.5 Interacción con otros medicamentos y otras formas de interacción
Inhibidores de CYP3A4 y UGT1A9 / inductores de CYP3A4
Los datos in vitro indican que regorafenib es metabolizado a través del citocromo CYP3A4 y la uridindifosfato glucuronosil transferasa UGT1A9.
La administración de ketoconazol (400 mg durante 18 días), un potente inhibidor de CYP3A4, con una dosis única de regorafenib (160 mg el día 5) dio lugar a un aumento de la exposición media (AUC) a regorafenib de aproximadamente el 33% y a una disminución de la exposición media a los metabolitos activos, M-2 (N-óxido) y M-5 (N-óxido y N-desmetilo), de alrededor del 90%. Se recomienda evitar el uso concomitante de inhibidores potentes de la actividad de CYP3A4 (por ejemplo, claritromicina, zumo de pomelo, itraconazol, ketoconazol, posaconazol, telitromicina y voriconazol), ya que no se ha estudiado su influencia sobre la exposición a regorafenib y sus metabolitos en el estado estacionario.
Debe evitarse la administración concomitante de un inhibidor potente de UGT1A9 (por ejemplo, ácido mefenámico, diflunisal y ácido niflúmico) durante el tratamiento con regorafenib, ya que no se ha estudiado su influencia sobre la exposición a regorafenib y sus metabolitos en el estado estacionario.
La administración de rifampicina (600 mg durante 9 días), un potente inductor de CYP3A4, con una dosis única de regorafenib (160 mg el día 7) dio lugar a una reducción de la AUC de regorafenib de aproximadamente el 50% y a un aumento de tres a cuatro veces en la exposición media al metabolito activo M-5, pero no provocó cambios en la exposición al metabolito activo M-2. Es posible que otros inductores potentes de CYP3A4 (por ejemplo, fenitoína, carbamazepina, fenobarbital y hierba de San Juan) también puedan aumentar el metabolismo de regorafenib. Debe evitarse el uso de inductores potentes de CYP3A4 o plantearse la selección de un medicamento concomitante alternativo sin capacidad o con una capacidad mínima de inducción de CYP3A4.
Sustratos de UGT1A1 y UGT1A9
Los datos in vitro indican que regorafenib y su metabolito activo M-2 inhiben la glucuronidación mediada por UGT1A1 y UGT1A9, mientras que M-5 sólo inhibe UGT1A1, en concentraciones que se alcanzan in vivo en el estado estacionario. La administración de regorafenib con un intervalo de 5 días de descanso antes de la administración de irinotecán generó un aumento de aproximadamente el 44% en la AUC de SN-38, un sustrato de UGT1A1 y un metabolito activo del irinotecán. También se observó un incremento de la AUC de irinotecán de alrededor del 28%. Esto indica que la administración concomitante de regorafenib puede aumentar la exposición sistémica a los sustratos de UGT1A1 y UGT1A9.
Sustratos de la proteína de resistencia al cáncer de mama (BCRP) y la glicoproteína P La administración de regorafenib (160 mg durante 14 días) previa a la administración de una dosis única de rosuvastatina (5 mg), en sustrato BCRP, resultó en un aumento de 3,8 veces la exposición media (AUC) de rosuvastatina y en un aumento de 4,6 veces la Cmax.
Esto indica que la administración concomitante de regorafenib puede aumentar las concentraciones plasmáticas de otros sustratos concomitantes de la BCRP (por ejemplo, metotrexato, fluvastatina, atorvastatina). Por lo tanto, se recomienda monitorizar de cerca a los pacientes en busca de signos y síntomas de un aumento de la exposición a los sustratos de la BCRP.
Los datos clínicos indican que regorafenib no tiene efecto en la farmacocinética de digoxina por lo tanto, puede ser administrado concomitantemente con sustratos de la glicoproteína P, como digoxina, sin una interacción clínicamente significativa.
Inhibidores de la glicoproteína P y la BCRP / inductores de la glicoproteína P y la BCRP Los estudios in vitro indican que los metabolitos activos M-2 y M-5 son sustratos de la glicoproteína P y la BCRP. Los inhibidores e inductores de la BCRP y la glicoproteína P pueden interferir con la exposición a M-2 y M-5. Se desconoce la significación clínica de estos hallazgos.
Sustratos selectivos de isoformas del CYP
Los datos in vitro indican que regorafenib es un inhibidor competitivo de los citocromos CYP2C8 (valor Ki de 0,6 micromolar), CYP2C9 (valor Ki de 4,7 micromolar) y CYP2B6 (valor Ki de 5,2 micromolar) en concentraciones que se alcanzan in vivo en el estado estacionario (concentración plasmática máxima de 8,1 micromolar). La potencia inhibitoria in vitro frente a CYP3A4 (valor Ki de 11,1 micromolar) y CYP2C19 (valor Ki de 16,4 micromolar) fue menos pronunciada.
Se realizó un estudio clínico de sustratos sonda para evaluar el efecto de 14 días de administración de 160 mg de regorafenib sobre la farmacocinética de los sustratos sonda de CYP2C8 (rosiglitazona), CYP2C9 (S-warfarina), CYP2C19 (omeprazol) y CYP3A4 (midazolam).
Los datos farmacocinéticos indican que regorafenib puede administrarse concomitantemente con sustratos de CYP2C8, CYP2C9, CYP3A4 y CYP2C19 sin interacciones medicamentosas clínicamente significativas (ver también la sección 4.4).
Antibióticos
La curva concentración-tiempo indica que regorafenib y sus metabolitos pueden pasar a la circulación enterohepática (ver sección 5.2). La administración concomitante con neomicina, un agente antimicrobiano que se absorbe poco, usado para erradicar la microflora gastrointestinal (que puede interferir con la circulación enterohepática de regorafenib) no tuvo ningún efecto en la exposición al mismo, pero se dió una disminución de aproximadamente el 80% en la exposición de los metabolitos activos M-2 y M-5 que mostró una actividad farmacológica in vivo e in vitro comparable a regorafenib. Se desconoce la relevancia clínica de esta interacción con neomicina, pero es posible que de lugar a una disminución de la eficacia de regorafenib. No se han estudiado las interacciones farmacocinéticas de otros antibióticos.
Agentes secuestradores de sales biliares
Regorafenib, M-2 y M-5 son propensos a someterse a la circulación enterohepática (ver sección 5.2). Los agentes secuestradores de sales biliares como colestiramina y colestagel pueden interactuar con regorafenib mediante la formación de complejos insolubles que podrían afectar la absorción (o la reabsorción), lo que resultaría en la exposición potencialmente disminuida. La importancia clínica de estas interacciones potenciales es desconocida, pero podría resultar en una disminución de la eficacia de regorafenib.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil / Anticoncepción en hombres y mujeres
Debe informarse a las mujeres en edad fértil que regorafenib puede causar daño fetal.
Las mujeres en edad fértil y los hombres deben asegurarse de utilizar métodos anticonceptivos efectivos durante el tratamiento y hasta 8 semanas tras finalizar el tratamiento.
Embarazo
No hay datos relativos al uso de regorafenib en mujeres embarazadas.
Debido a su mecanismo de acción, se sospecha que puede causar daño fetal si se administra durante el embarazo. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3).
Stivarga no debe usarse durante el embarazo a menos que sea claramente necesario y tras una cuidadosa evaluación de los beneficios para la madre y los riesgos para el feto.
Lactancia
Se desconoce si regorafenib o sus metabolitos se excretan en la leche materna.
En ratas, regorafenib o sus metabolitos se excretan en la leche. No se puede excluir el riesgo en el lactante. Regorafenib podría alterar el crecimiento y desarrollo del lactante (ver sección 5.3).
Debe interrumpirse la lactancia durante el tratamiento con Stivarga.
Fertilidad
No hay datos relativos al efecto de Stivarga sobre la fertilidad humana. Los resultados de los estudios realizados en animales indican que regorafenib puede afectar a la fertilidad masculina y femenina (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios de los efectos de Stivarga sobre la capacidad para conducir y utilizar máquinas. Si los pacientes presentan síntomas que afecten a su capacidad para concentrarse y reaccionar durante el tratamiento con Stivarga, se recomienda que no conduzcan ni utilicen máquinas hasta que remita el efecto.
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad global de Stivarga se basa en los datos obtenidos de más de 1.200 pacientes tratados en ensayos clínicos, incluidos los datos de fase III controlados con placebo de 500 pacientes con cáncer colorrectal (CCR) metastásico y 132 pacientes con tumores del estroma gastrointestinal (GIST).
El perfil de seguridad de regorafenib en estos estudios fue consistente con los resultados de seguridad del estudio fase III B realizado en 2872 pacientes con cáncer colorrectal metastásico cuya enfermedad había progresado tras el tratamiento con las terapias estándar.
Las reacciones adversas más graves en los pacientes tratados con Stivarga son lesión hepática grave, hemorragia y perforación gastrointestinal.
Las reacciones adversas más frecuentemente observadas (> 30%) en los pacientes tratados con Stivarga son astenia/fatiga, reacción cutánea mano-pie, diarrea, disminución del apetito y de la ingesta de alimentos, hipertensión, disfonía e infección.
Tabla de reacciones adversas
Las reacciones adversas notificadas en los ensayos clínicos en los pacientes tratados con Stivarga se indican en la Tabla 3, según la clasificación por órganos y sistemas, y se utiliza la terminología MedDRA más apropiada para describir una reacción determinada y sus sinónimos y problemas relacionados.
Las reacciones adversas se agrupan conforme a sus frecuencias. Los grupos de frecuencia se definen de acuerdo con la siguiente convención: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); y raras (> 1/10.000 a < 1/1.000).
Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Tabla 3: Reacciones Adversas al Medicamento (RAM) notificadas en los ensayos clínicos en los pacientes tratados con Stivarga
|
Clasificación de órganos del sistema (MedDRA) |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
Infecciones e infestaciones |
Infección | |||
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Queratoacantoma / Carcinoma cutáneo de células escamosas | |||
|
Trastornos de la sangre y del sistema linfático |
Trombocitopenia Anemia |
Leucopenia | ||
|
Trastornos del sistema inmunológico |
Reacción de hipersensibili- dad | |||
|
Trastornos endocrinos |
Hipotiroidismo | |||
|
Trastornos del metabolismo y de la nutrición |
Disminución del apetito y de la ingesta de alimentos |
Hipopotasemia Hipofosfatemia Hipocalcemia Hiponatremia Hipomagnesemia Hiperuricemia | ||
|
Trastornos del sistema nervioso |
Cefalea |
Temblor |
Síndrome de leucoencefalopatí a posterior |
|
Clasificación de órganos del sistema (MedDRA) |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
reversible (PRES) | ||||
|
Trastornos cardiacos |
Infarto de miocardio Isquemia miocárdica | |||
|
Trastornos vasculares |
Hemorragia* Hipertensión |
Crisis hipertensiva | ||
|
Trastornos respiratorios, torácicos y mediastínicos |
Disfonía | |||
|
Trastornos gastrointestinales |
Diarrea Estomatitis Vómitos Náuseas |
Trastornos del gusto Sequedad de boca Reflujo gastroesofágico Gastroenteritis |
Perforación gastrointestinal* Fístula gastrointestinal | |
|
Trastornos hepatobiliares |
Hiperbilirrubi- nemia |
Aumento de las transaminasas |
Lesión hepática grave*# | |
|
Trastornos de la piel y del tejido subcutáneo |
Reacción cutánea mano-pie** Exantema Alopecia |
Sequedad cutánea Exantema exfoliativo |
Trastorno ungueal Eritema multiforme |
Síndrome de Stevens-Johnson Necrolisis epidérmica tóxica |
|
Trastornos musculoesqueléti-cos y del tejido conjuntivo |
Rigidez musculoesqueléti- ca | |||
|
Trastornos renales y urinarios |
Proteinuria | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Astenia / fatiga Dolor Fiebre Mucositis | |||
|
Exploraciones complementarias |
Pérdida de peso |
Aumento de la amilasa Aumento de la lipasa Relación internacional normalizada (INR) anómala |
* Se han notificado casos fatales
** Síndrome de eritrodisestesia palmoplantar según la terminología MedDRA
# De acuerdo con los criterios de lesión hepática inducida por fármacos (LHIF) del grupo de trabajo
de expertos sobre LHIF
Descripción de reacciones adversas seleccionadas
En la mayoría de los casos de lesiones hepáticas graves, la disfunción hepática se inició dentro de los primeros 2 meses de tratamiento, y se caracterizó por un patrón de lesión hepatocelular con elevación de las transaminasas > 20xULN, seguido de un aumento de la bilirrubina. En los ensayos clínicos, se observó una mayor incidencia de lesiones hepáticas graves con desenlace mortal en pacientes japoneses (~ 1,5%) tratados con Stivarga en comparación con los pacientes no japoneses (<0,1%).
En los dos ensayos de fase III controlados con placebo, la incidencia global de hemorragia fue del 19,3% en los pacientes tratados con Stivarga. La mayoría de los casos de acontecimientos hemorrágicos en los pacientes tratados con Stivarga fueron de intensidad leve a moderada (grados 1 y 2: 16,9%), con la epistaxis como acontecimiento más notorio (7,6%). Los acontecimientos fatales en los pacientes tratados con Stivarga fueron poco frecuentes (0,6%) y correspondieron a problemas en los tractos respiratorio, gastrointestinal y genitourinario.
En los dos ensayos de fase III controlados con placebo, se observaron infecciones con mayor frecuencia en los pacientes tratados con Stivarga que en los que recibieron placebo (todos los grados: 31,0% frente a 14,4 %). La mayoría de las infecciones de los pacientes tratados con Stivarga fueron de intensidad leve a moderada (grados 1 y 2: 22,9%) e incluyeron infecciones del tracto urinario (6,8%), nasofaringitis (4,2%) e infecciones micóticas mucocutáneas y sistémicas (2,4%). No se observaron diferencias en cuanto a desenlaces fatales en relación con la infección entre los grupos de tratamiento (0,6% en el grupo tratado con Stivarga frente a 0,6% en el que recibió placebo).
En el ensayo de fase III en CCR metastásico controlado con placebo, la incidencia global de reacción cutánea mano-pie fue del 45,2% en los pacientes tratados con Stivarga, frente al 7,1% en los que recibieron placebo. En el ensayo de fase III en GIST controlado con placebo, la incidencia global de reacción cutánea mano-pie fue del 66,7% en los pacientes tratados con Stivarga, frente al 15,2% en los que recibieron placebo. En ambos ensayos, la mayoría de los casos de reacción cutánea mano-pie en los pacientes tratados con Stivarga aparecieron durante el primer ciclo de tratamiento y fueron de intensidad leve a moderada (grados 1 y 2: 28,6% en los pacientes con CCR y 44,7% en los pacientes con GIST). La incidencia de reacción cutánea mano-pie de grado 3 fue del 16,6% (CCR) y el 22,0% (GIST). En ambos ensayos, la incidencia global de reacción cutánea mano-pie (78,4% CCR y 88,2% GIST) fue superior en los pacientes asiáticos tratados con Stivarga comparados con otras etnias. La incidencia de reacción cutánea mano-pie de grado 3 en asiáticos fue del 28,4% (CCR) y el 23,5% (GIST) (ver secciones 4.2 y 4.4).
En el ensayo de fase III en CCR metastásico controlado con placebo, la incidencia global de hipertensión fue del 30,4% en los pacientes tratados con Stivarga, frente al 7,9% en los que recibieron placebo. En el ensayo de fase III en GIST controlado con placebo, la incidencia global de hipertensión fue del 59,1% en los pacientes tratados con Stivarga, frente al 27,3% en los que recibieron placebo. En ambos ensayos, la mayoría de los casos de hipertensión en los pacientes tratados con Stivarga aparecieron durante el primer ciclo de tratamiento y fueron de intensidad leve a moderada (grados 1 y 2: 22,8% en los pacientes con CCR y 31,1% en los pacientes con GIST). La incidencia de hipertensión de grado 3 fue del 7,6% (CCR) y el 27,3% (GIST). En el ensayo en GIST se notificó un caso de hipertensión de grado 4.
En el ensayo de fase III controlado con placebo en pacientes con CCR metastásico, la incidencia global de proteinuria surgida durante el tratamiento fue del 7,4% en los pacientes tratados con Stivarga, frente al 2,4% en los que recibieron el placebo. De estos acontecimientos, el 40,5% de los del grupo tratado con Stivarga y el 66,7% de los del grupo tratado con placebo se han notificado como no recuperados / no resueltos. En el ensayo de fase III en GIST controlado con placebo, la incidencia global de proteinuria fue del 6,8% en los pacientes tratados con Stivarga, frente al 1,5% en los que recibieron el placebo.
En todos los ensayos clínicos, los acontecimientos correspondientes a trastornos cardiacos (de todos los grados) se notificaron con mayor frecuencia (20,5% frente a 10,4%) en los pacientes tratados con Stivarga de edad igual o superior a 75 años (N=78) que en los pacientes tratados con Stivarga de edad inferior a 75 años (N=995).
Anomalías de las pruebas analíticas
En las Tablas 4, 4a y 5 se indican las anomalías analíticas observadas durante el tratamiento en los ensayos de fase III controlados con placebo (ver también la sección 4.4).
Tabla 4: Anomalías de las pruebas analíticas durante el tratamiento comunicadas en el ensayo de fase III controlado con placebo en pacientes con CCR metastásico (CORRECT)
|
Stivarga más BSC§ |
Placebo más BSC§ | |||||
|
Parámetro analítico (en % |
(N=500) |
(N=253) | ||||
|
Todos los grados* |
Grado 3 * |
Grado 4 * |
Todos los grados* |
Grado 3 * |
Grado 4 * | |
|
de muestras investigadas) | ||||||
|
Trastornos de la sangre y del sistema linfático Disminución de la |
78,5 |
4,7 |
0,6 |
66,3 |
2,8 |
0 |
|
hemoglobina Disminución del recuento de |
40,5 |
2,4 |
0,4 |
16,8 |
0,4 |
0 |
|
plaquetas Disminución del recuento de |
2,8 |
0,6 |
0 |
0 |
0 |
0 |
|
neutrófilos Disminución del recuento de linfocitos |
54,1 |
9,3 |
0 |
34,4 |
3,2 |
0 |
|
Trastornos del metabolismo y de la nutrición Disminución del calcio |
59,3 |
1,0 |
0,2 |
18,3 |
1,2 |
0 |
|
Disminución del potasio |
25,7 |
4,3 |
0 |
8,3 |
0,4 |
0 |
|
Disminución del fosfato |
57,4 |
30,5 |
0,6 |
11,1 |
3,6 |
0 |
|
Trastornos hepatobiliares Aumento de la bilirrubina |
44,6 |
9,6 |
2,6 |
17,1 |
5,2 |
3,2 |
|
Aumento de la AST |
65,0 |
5,3 |
0,6 |
45,6 |
4,4 |
0,8 |
|
Aumento de la ALT |
45,2 |
4,9 |
0,6 |
29,8 |
2,8 |
0,4 |
|
Trastornos renales y | ||||||
|
urinarios | ||||||
|
Proteinuria |
59,7 |
0,4 |
0 |
34,1 |
0,4 |
0 |
|
Exploraciones complementarias Aumento del INR** |
23,7 |
4,2 |
-# |
16,6 |
1,6 |
-# |
|
Aumento de la lipasa |
46,0 |
9,4 |
2,0 |
18,7 |
2,8 |
1,6 |
|
Aumento de la amilasa |
25,5 |
2,2 |
0,4 |
16,7 |
2,0 |
0,4 |
§ Mejor tratamiento de soporte (best supportive care, BSC)
* Common Terminology Criteria for Adverse Events (CTCAE), versión 3.0 ** Relación internacional normalizada
# El grado 4 no aparece establecido en los CTCAE, versión 3.0
En comparación con el ensayo global de fase III en CCR (CORRECT) con inclusión de pacientes predominantemente caucásicos (aproximadamente el 80%), se observó una incidencia más alta de aumento de las enzimas hepáticas en los pacientes tratados con Stivarga del ensayo asiático de fase III en CCR (CONCUR) con inclusión de pacientes predominantemente de asia oriental (> 90%).
Tabla 4a: Anomalías de las pruebas enzimáticas hepáticas durante el tratamiento comunicadas en el ensayo de fase III controlado con placebo en pacientes asiáticos con CCR metastásico (CONCUR)
|
Parámetro analítico (en % de muestras investigadas) |
Stivarga más BSC§ (N=136) |
Placebo más BSC§ (N=68) | ||||
|
Todos los grados* |
Grado 3 * |
Grado 4 * |
Todos los grados* |
Grado 3 * |
Grado 4 * | |
|
Aumento de la bilirrubina |
66,7 |
7,4 |
4,4 |
32,8 |
4,5 |
0,0 |
|
Aumento de la AST |
69,6 |
10,4 |
0,7 |
47,8 |
3,0 |
0,0 |
|
Aumento de la ALT |
54,1 |
8,9 |
0,0 |
29,9 |
1,5 |
0,0 |
§ Mejor tratamiento de soporte (best supportive care, BSC)
* Common Terminology Criteria for Adverse Events (CTCAE), versión 4.0
Tabla 5: Anomalías de las pruebas analíticas durante el tratamiento comunicadas en el ensayo de fase III controlado con placebo (fase a doble ciego) en pacientes con GIST (GRID)
|
Stivarga más BSC§ |
Placebo más BSC§ | |||||
|
(N=132) |
(N=66) | |||||
|
Parámetro analítico (en % de muestras investigadas) |
Todos los |
Grado 3 * |
Grado 4 * |
Todos los |
Grado 3 * |
Grado 4 * |
|
grados* |
grados* | |||||
|
Trastornos de la sangre y del sistema linfático Disminución de la |
75,0 |
3,0 |
0 |
72,7 |
1,5 |
0 |
|
hemoglobina Disminución del recuento de |
12,9 |
0,8 |
0 |
1,5 |
0 |
1,5 |
|
plaquetas Disminución del recuento de |
15,9 |
2,3 |
0 |
12,1 |
3,0 |
0 |
|
neutrófilos Disminución del recuento de linfocitos |
29,5 |
7,6 |
0 |
24,2 |
3,0 |
0 |
|
Trastornos del metabolismo y de la nutrición Disminución del calcio |
16,7 |
1,5 |
0 |
4,5 |
0 |
0 |
|
Disminución del potasio |
20,5 |
3,0 |
0 |
3,0 |
0 |
0 |
|
Disminución del fosfato |
54,5 |
19,7 |
1,5 |
3,1 |
1,5 |
0 |
|
Trastornos hepatobiliares Aumento de la bilirrubina |
33,3 |
3,0 |
0,8 |
12,1 |
1,5 |
0 |
|
Aumento de la AST |
58,3 |
3,0 |
0,8 |
47,0 |
3,0 |
0 |
|
Aumento de la ALT |
39,4 |
3,8 |
0,8 |
39,4 |
1,5 |
0 |
|
Stivarga más BSC§ (N=132) |
Placebo más BSC§ (N=66) | |||||
|
Parámetro analítico (en % de muestras investigadas) |
Todos los grados* |
Grado 3 * |
Grado 4 * |
Todos los grados* |
Grado 3 * |
Grado 4 * |
|
Trastornos renales y urinarios Proteinuria |
38,5 |
1,5 |
39,0 |
1,7 | ||
|
Exploraciones complementarias Aumento del INR** |
9,3 |
1,6 |
12,5 |
4,7 | ||
|
Aumento de la lipasa |
14,4 |
0 |
0,8 |
4,6 |
0 |
0 |
§ Mejor tratamiento de soporte (best supportive care, BSC)
* Common Terminology Criteria for Adverse Events (CTCAE), versión 4.0 ** Relación internacional normalizada
- El grado 4 no aparece establecido en los CTCAE, versión 4.0
En los dos ensayos de fase III controlados con placebo, los análisis de la hormona estimulante del tiroides (TSH) mostraron resultados > ULN con posterioridad a la situación basal en el 26,1% de los pacientes tratados con Stivarga y en el 15,1% de los que recibieron el placebo. Se notificaron valores de TSH > 4 veces por encima del ULN con posterioridad a la situación basal en el 6,9% de los pacientes tratados con Stivarga y en el 0,7% de los que recibieron el placebo. Se notificó una concentración de triyodotironina libre (FT3) por debajo del límite inferior de la normalidad (< LLN) con posterioridad a la situación basal en el 25,6% de los pacientes tratados con Stivarga y en el 20,9% de los que recibieron el placebo. Se notificó una concentración de tiroxina libre (FT4) < LLN con posterioridad a la situación basal en el 8,0% de los pacientes tratados con Stivarga y en el 6,6% de los que recibieron el placebo. En general, aproximadamente el 7% de los pacientes tratados con Stivarga desarrollaron hipotiroidismo requiriendo tratamiento de sustitución hormonal.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
La dosis más alta de Stivarga estudiada clínicamente fue 220 mg por día. Las reacciones adversas más frecuentemente observadas con esta dosis fueron alteraciones dermatológicas, disfonía, diarrea, mucositis, sequedad de boca, disminución del apetito, hipertensión y fatiga.
No existe un antídoto específico para la sobredosis de Stivarga. En caso de sospecha de sobredosis, se debe interrumpir inmediatamente la administración de Stivarga, con inicio del mejor tratamiento de soporte bajo las órdenes de un profesional médico, y vigilar al paciente hasta su estabilidad clínica.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: fármacos antineoplásicos, inhibidor de proteinquinasas;
Código ATC: L01XE21
Mecanismo de acción y efectos farmacodinámicos
Regorafenib es un fármaco antitumoral oral que bloquea de forma potente varias proteinquinasas, incluidas las quinasas implicadas en la angiogénesis tumoral (VEGFR1, -2, -3, TIE2), la oncogénesis (KIT, RET, rAf-1, BRAF, BRAFV600E) y el microambiente tumoral (PDGFR, FGFR). En concreto, regorafenib inhibe el KIT mutado, un importante controlador oncogénico en los tumores del estroma gastrointestinal, y, por lo tanto, bloquea la proliferación celular tumoral. En los estudios preclínicos, regorafenib ha demostrado una potente actividad antitumoral en una amplia gama de modelos tumorales, incluidos modelos tumorales colorrectales y del estroma gastrointestinal, mediada por sus efectos antiangiogénicos y antiproliferativos. Además, regorafenib ha mostrado efectos antimetastásicos in vivo. Los principales metabolitos humanos (M-2 y M-5) presentaron una eficacia similar a la de regorafenib en los modelos in vitro e in vivo.
Eficacia clínica y seguridad
Cáncer colorrectal (CCR) mestastásico
La eficacia clínica y la seguridad de Stivarga se han evaluado en un estudio de fase III internacional, multicéntrico, aleatorizado, doble ciego y controlado con placebo (CORRECT), en pacientes con cáncer colorrectal metastásico que han progresado tras el fracaso a terapias estándar.
El objetivo primario de eficacia fue la supervivencia global (SG). Los objetivos secundarios fueron la supervivencia libre de progresión (SLP), la tasa de respuesta tumoral objetiva y la tasa de control de la enfermedad.
En total, 760 pacientes fueron aleatorizados en una proporción 2:1 a recibir 160 mg de regorafenib (4 comprimidos de Stivarga con 40 mg de regorafenib cada uno) por vía oral una vez al día (N=505) más el mejor tratamiento de soporte (BSC) o placebo (N=255) más el mejor tratamiento de soporte (BSC) durante 3 semanas de tratamiento seguido de 1 semana sin tratamiento. La dosis diaria media de regorafenib administrada fue de 147 mg.
Los pacientes continuaron el tratamiento hasta la progresión de la enfermedad o la aparición de toxicidad inaceptable. Se realizó un análisis intermedio de la eficacia, planificado con antelación, una vez se produjeron 432 muertes. El estudio se desenmascaró después de que en este análisis intermedio planificado de la SG se hubiese pasado el límite preestablecido para la eficacia.
Entre los 760 pacientes aleatorizados, la mediana de edad era de 61 años, el 61% eran varones, el 78% eran caucásicos y todos ellos se encontraban en un estado funcional (PS) ECOG basal de 0 ó 1. Se notificó un PS > 2 durante el tratamiento con Stivarga en el 11,4% de los pacientes. La mediana de la duración del tratamiento y de la dosis diaria, así como la tasa de modificación de la dosis y de reducción de la dosis fueron similares a las observadas en los pacientes tratados con placebo con un PS notificado > 2 (8,3%). La mayoría de los pacientes con PS > 2 interrumpieron el tratamiento por progresión de enfermedad. La localización primaria de la enfermedad era el colon (65%), el recto (29%), o ambos (6%). Se notificó la mutación del gen KRAS en el 57% de los pacientes en el momento de su entrada en el estudio.
La mayoría de los pacientes (52%) habían recibido un máximo de 3 líneas de tratamiento previas para la enfermedad metastásica. Entre los tratamientos administrados se encontraban quimioterapia basadas
en fluoropirimidinas, terapia anti-VEGF y, si el paciente presentaba KRAS-wild type, terapia anti-EGFR.
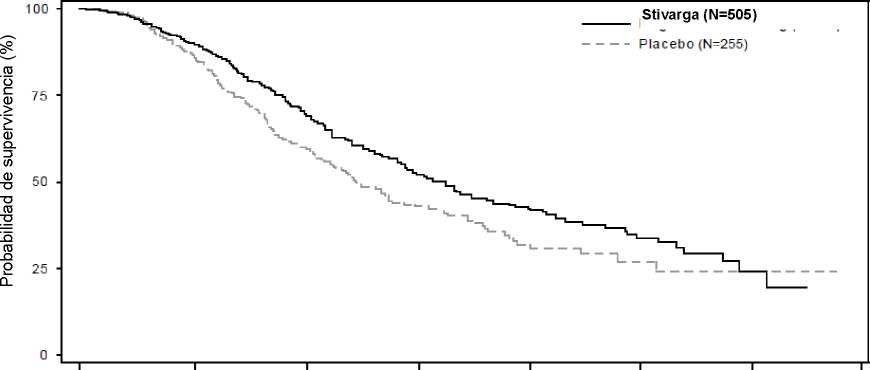
La adición de Stivarga al BSC generó una supervivencia significativamente más prolongada que la observada con el placebo más el BSC, con un hazard ratio (HR) de 0,774 (p=0,005178 stratified long Rank test) y una mediana de la SG de 6,4 meses frente a 5,0 meses [IC del 95%: 0,636; 0,942] (ver la Tabla 6 y la Figura 1). La SLP fue significativamente más prolongada en los pacientes tratados con Stivarga más el BSC (HR: 0,494, p<0,000001, ver la Tabla 6). La tasa de respuesta (respuesta completa o respuesta parcial) fue del 1% y el 0,4% para los pacientes tratados con Stivarga y placebo, respectivamente (p=0,188432, unilateral). La tasa de control de la enfermedad (respuesta completa o respuesta parcial o enfermedad estable) fue significativamente más alta en los pacientes tratados con Stivarga (41,0% frente a 14,9%, p<0,000001, unilateral).
Tabla 6: Resultados de eficacia del estudio CORRECT
|
Parámetro de eficacia |
Hazard ratio* (IC del 95%) |
Valor de p (unilateral) |
Mediana (IC ( |
el 95%) |
|
Stivarga más BSC§ (N=505) |
Placebo más BSC§ (N=255) | |||
|
Supervivencia global |
0,774 (0,636; 0,942) |
0,005178 |
6,4 meses (5,9; 7,3) |
5,0 meses (4,4; 5,8) |
|
Supervivencia libre de progresión** |
0,494 (0,419; 0,582) |
<0,000001 |
1,9 meses (1,9; 2,1) |
1,7 meses (1,7; 1,7) |
§ Mejor tratamiento de soporte (best supportive care, BSC)
* HR < 1 favorece a Stivarga
** basada en la evaluación de la respuesta tumoral por parte del investigador
Figura 1: Curva de Kaplan-Meier de la supervivencia global
|
0 |
2 |
4 |
6 8 |
10 |
12 |
14 |
|
Meses desde la aleatorización | ||||||
|
Pacientes en riesgo Stivarga Placebo |
452 221 |
352 150 |
187 93 75 32 |
33 9 |
7 3 |
El análisis de subgrupos para supervivencia global y supervivencia libre de progresión en función de la edad (<65, > 65 años), el sexo, el estado funcional ECOG, el sitio primario de la enfermedad, el tiempo desde el primer diagnóstico de la enfermedad metastásica, el tratamiento previo contra el cáncer, las líneas previas de tratamiento para la enfermedad metastásica y la mutación KRAS, mostró un efecto del tratamiento favoreciendo el régimen de regorafenib frente al régimen de placebo.
Los resultados del análisis por subgrupos según el estado histórico mutacional de KRAS demostraron un efecto del tratamiento para la supervivencia global a favor de regorafenib frente a placebo para los pacientes con tumores KRAS wild type mientras que se notificó un efecto numéricamente menor en los pacientes con tumores KRAS mutado; el efecto del tratamiento observado para la supervivencia libre de progresión (SLP) fue a favor de regorafenib independientemente del estado mutacional de KRAS. El hazard ratio (IC del 95%) para supervivencia global fue 0,653 (0,476-0,895) para pacientes con tumores KRAS wild type y 0,867 (0,670-1,123) para pacientes con tumores KRAS mutado, sin evidencia de heterogeneidad en el efecto del tratamiento (prueba de interacción no significativa). El hazard ratio (IC del 95%) para supervivencia libre de progresión fue de 0,475 (0,362-0,623) para pacientes con tumores KRAS wild type y 0,525 (0,425-0,649) para pacientes con tumores KRAS mutado.
En un segundo estudio de fase III internacional, multicéntrico, aleatorizado, doble ciego y controlado con placebo (CONCUR) se evaluó la eficacia y seguridad de Stivarga en 204 pacientes asiáticos (> 90% de asia oriental) previamente tratados, con cáncer colorrectal metastático que habían presentado una progresión de la enfermedad tras el fracaso de la quimioterapia basada en fluoropirimidinas. Solo el 59,5% de los pacientes incluidos en el estudio CONCUR habían sido tratados previamente también con fármacos dirigidos contra VEGF o EGFR. La variable principal de eficacia fue la SG. La adición de Stivarga al BSC generó una supervivencia significativamente más prolongada que la observada con el placebo más el BSC, con un HR de 0,550 (p = 0,000159 stratified log rank test) y una mediana de la SG de 8,8 meses frente a 6,3 meses [IC del 95%: 0,395; 0,765]. La SLP también fue significativamente más prolongada en los pacientes tratados con Stivarga más el BSC (HR: 0,311, p<0,000001), con una mediana de la SLP de 3,2 meses con Stivarga frente a 1,7 meses con placebo. El perfil de seguridad de Stivarga más BSC en el estudio CONCUR fue consistente con el observado en el estudio CORRECT.
Tumores del estroma gastrointestinal (GIST)
La eficacia clínica y la seguridad de Stivarga se han evaluado en un estudio de fase III internacional, multicéntrico, aleatorizado, doble ciego y controlado con placebo (GRID) en pacientes con tumores del estroma gastrointestinal (GIST) previamente tratados con 2 inhibidores de la tirosina quinasa (imatinib y sunitinib).
El análisis del objetivo primario de eficacia, supervivencia libre de progresión (SLP) se realizó tras 144 acontecimientos de SLP (evaluación centralizada ciega). También se evaluaron (análisis intermedio) los objetivos secundarios, entre los que se encontraban el tiempo transcurrido hasta la progresión (TTP) y la supervivencia global (SG).
En total, 199 pacientes con GIST fueron aleatorizados en una proporción 2:1 a recibir 160 mg de regorafenib más el mejor tratamiento de soporte (BSC; N=133) por vía oral una vez al día o placebo más BSC (N=66) durante 3 semanas de tratamiento seguido de 1 semana sin tratamiento. La dosis diaria media de regorafenib administrada fue de 140 mg.
Los pacientes continuaron el tratamiento hasta la progresión de la enfermedad o la aparición de toxicidad inaceptable. A los pacientes tratados con placebo que presentaron progresión de la enfermedad se les ofreció regorafenib en régimen abierto (opción de cruce). A los pacientes tratados con regorafenib que presentaron progresión de la enfermedad y para los que, en opinión del investigador, el tratamiento con regorafenib estaba proporcionando un beneficio clínico, se les ofreció la oportunidad de continuar con regorafenib en régimen abierto.
Entre los 199 pacientes aleatorizados, la edad media era de 58 años, el 64% eran varones, el 68% eran caucásicos y todos ellos se encontraban en un estado funcional ECOG basal de 0 ó 1. La mediana global del tiempo transcurrido desde la progresión o recidiva más reciente hasta la aleatorización era de 6 semanas.
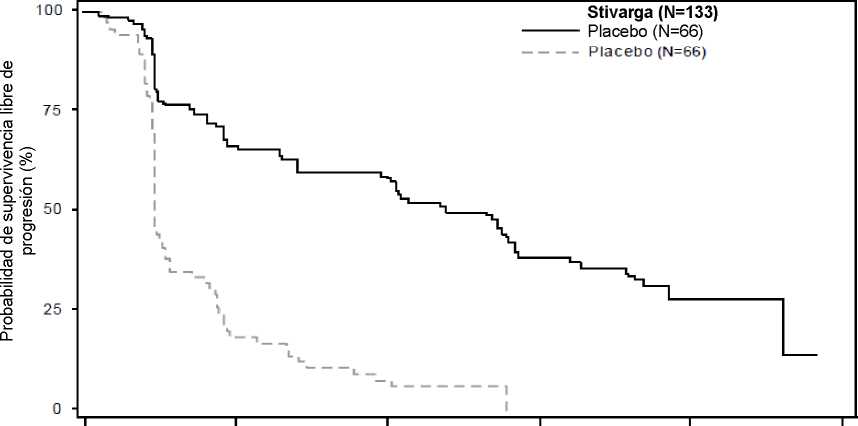
Regorafenib más el BSC alcanzó una SLP significativamente más larga que la observada con el placebo más el BSC, con un hazard ratio (HR) de 0,268 [IC del 95%: 0,185; 0,388] y una mediana de la SLP de 4,8 meses frente a 0,9 meses (p < 0,000001). El riesgo relativo de progresión de la enfermedad o muerte se redujo en aproximadamente un 73,2% en los pacientes tratados con regorafenib, frente a los tratados con placebo (ver la Tabla 7 y la Figura 2). El aumento de la SLP fue robusto, independientemente de la edad, el sexo, la región geográfica, las líneas previas de tratamiento y el estado funcional ECOG.
El TTP fue significativamente más largo en los pacientes tratados con regorafenib más BSC que en los tratados con placebo más BSC, con un hazard ratio de 0,248 [IC del 95%: 0,170; 0,364] y una mediana del TTP de 5,4 meses frente a 0,9 meses (p < 0,000001) (ver la Tabla 7).
El HR del análisis de la SG fue 0,772 (IC del 95%: 0,423, 1,408; p = 0,199; la mediana de la SG no se alcanzó en ninguno de los grupos); 85% de los pacientes inicialmente aleatorizados al grupo tratado con placebo, recibieron tratamiento con regorafenib tras progresión (ver la Tabla 7 y la Figura 3).
Tabla 7: Resultados de eficacia del estudio GRID
|
Parámetro de eficacia |
Hazard ratio* (IC del 95%) |
Valor de p (unilateral) |
Mediana (IC del 95%) | |
|
Stivarga más BSC§ (N=133) |
Placebo más BSC§ (N=66) | |||
|
Supervivencia libre de progresión |
0,268 (0,185; 0,388) |
< 0,000001 |
4,8 meses (4,0; 5,7) |
0,9 meses (0,9; 1,1) |
|
Tiempo hasta la progresión |
0,248 (0,170; 0,364) |
< 0,000001 |
5,4 meses (4,1; 5,7) |
0,9 meses (0,9; 1,1) |
|
Supervivencia global |
0,772 (0,423; 1,408) |
0,199 |
NA** |
NA** |
|
§ Mejor tratamiento d |
e soporte (best supportive care, BSC) | |||
* HR < 1 favorece a Stivarga ** NA: no alcanzada
Figura 2: Curvas de Kaplan-Meier de la supervivencia libre de progresión
: - Meses desde la aleatorización i :
72 27 9
5 0 0
Pacientes en riesgo Stivarga
Placebo : 7
■ IU V. uu 12
ro
-Q
o
c
<1)
>
£
(D
CL
3
ti)
ro
-Q
g
CL
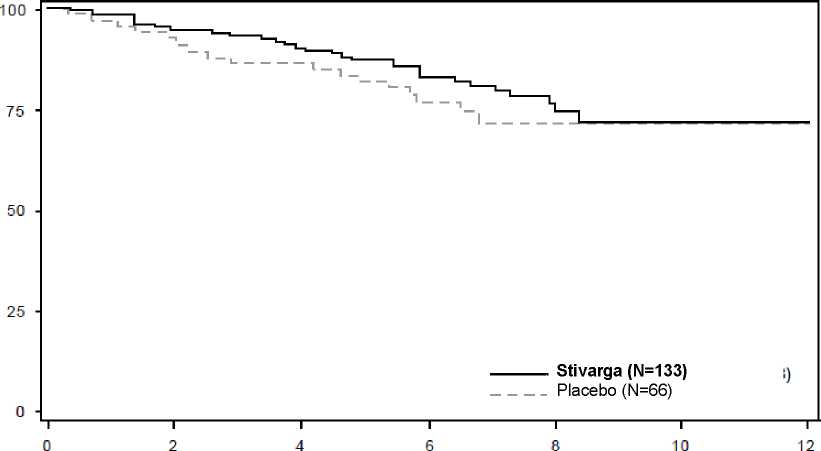
Meses desde la aleatorización
Pacientes en riesgo
Stivarga
Placebo
126
61
119
57
94
41
39
16
10
3
Además, 56 pacientes tratados con placebo más BSC recibieron Stivarga en régimen abierto al realizarse el cruce tras la progresión de la enfermedad y un total de 41 pacientes tratados con Stivarga más BSC continuaron el tratamiento con Stivarga tras la progresión de la enfermedad. La mediana de la SLP secundaria (medida conforme a la evaluación del investigador) fue de 5,0 y 4,5 meses, respectivamente.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Stivarga en los diferentes grupos de la población pediátrica en el tratamiento del adenocarcinoma de colon y recto (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Stivarga en uno o más grupos de la población pediátrica en el tratamiento de los tumores sólidos malignos (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Regorafenib alcanza las concentraciones plasmáticas máximas medias de aproximadamente 2,5 mg/l al cabo de unas 3 a 4 horas tras una dosis única por vía oral de 160 mg administrada en forma de 4 comprimidos de 40 mg cada uno. Tras dosis únicas de 60 mg o 100 mg, el promedio de la biodisponibilidad relativa de los comprimidos, en comparación con una solución oral, fue del 69% y el 83%, respectivamente.
Las concentraciones de regorafenib y sus principales metabolitos farmacológicamente activos (M-2 y M-5) alcanzaron sus máximos niveles cuando el fármaco se administró después de un desayuno (ligero) bajo en grasa, en comparación con un desayuno alto en grasa o un estado de ayuno. La exposición a regorafenib aumentó un 48% cuando se administró con un desayuno alto en grasa y un 36% cuando se administró con un desayuno bajo en grasa, en comparación con el estado de ayuno. La exposición a los metabolitos M-2 (N-óxido) y M-5 (N-óxido y N-desmetilo) es mayor cuando regorafenib se administra con un desayuno bajo en grasa en comparación con el estado de ayuno y menor cuando se administra con una comida alta en grasa en comparación con el estado de ayuno.
Distribución
Los perfiles de concentración plasmática - tiempo de regorafenib y sus principales metabolitos circulantes mostraron múltiples picos a lo largo del intervalo de administración de 24 horas, que se atribuyen a circulación enterohepática. La unión a proteínas in vitro de regorafenib a las proteínas plasmáticas humanas es alta (99,5%). La unión a proteínas in vitro de M-2 y M-5 es mayor (99,8% y 99,95%, respectivamente) que la de regorafenib. Los metabolitos M-2 y M-5 son sustratos débiles de la glicoproteína P. El metabolito M-5 es un sustrato débil de la BCRP.
Biotransformación
Regorafenib se metaboliza fundamentalmente en el hígado a través de un metabolismo oxidativo mediado por CYP3A4, así como por una glucuronidación mediada por UGT1A9. Se han identificado en plasma dos metabolitos principales y seis metabolitos menores de regorafenib. Los principales metabolitos circulantes de regorafenib en el plasma humano son M-2 (N-óxido) y M-5 (N-óxido y N-desmetilo), que son farmacológicamente activos y presentan concentraciones similares a las de regorafenib en el estado estacionario. Además, M-2 sufre metabolismo oxidativo mediado por CYP3A4, así como glucuronidación mediada por UGT1A9.
La flora microbiana del tracto gastrointestinal puede reducir o hidrolizar los metabolitos, lo que permite la reabsorción de la sustancia activa no conjugada y los metabolitos (circulación enterohepática).
Tras la administración oral, la semivida de eliminación media de regorafenib y su metabolito M-2 en plasma oscila entre 20 y 30 horas en diferentes estudios. La semivida de eliminación media del metabolito M-5 es de aproximadamente 60 horas (intervalo de 40 a 100 horas).
Alrededor del 90% de la dosis radiactiva se recuperó en un plazo de 12 días después de la administración, con aproximadamente el 71% de la dosis excretada en heces (47% en forma de compuesto original, 24% en forma de metabolitos) y alrededor del 19% de la dosis excretada en orina en forma de glucurónidos. La excreción urinaria de glucurónidos disminuyó por debajo del 10% en las condiciones del estado estacionario. El compuesto original encontrado en las heces podría derivarse de la degradación intestinal de los glucurónidos o la reducción del metabolito M-2 (N-óxido), así como de regorafenib no absorbido.
La flora microbiana puede reducir M-5 a M-4 en el tracto gastrointestinal, lo que permite la reabsorción de M-4 (circulación enterohepática). M-5 se excreta finalmente vía M-4 como M-6 (ácido carboxílico) en las heces.
Linealidad/No linealidad
La exposición sistémica a regorafenib en el estado estacionario aumenta proporcionalmente a la dosis hasta 60 mg y menos que proporcionalmente a dosis superiores a 60 mg. La acumulación de regorafenib en el estado estacionario genera un aumento aproximadamente del doble de las concentraciones plasmáticas, lo que concuerda con la semivida de eliminación y la frecuencia de dosificación. En el estado estacionario, regorafenib alcanza unas concentraciones plasmáticas máximas medias de 3,9 mg/l (8,1 micromolar) tras la administración oral de 160 mg de regorafenib y el cociente de la concentración plasmática media máxima/mínima es menor a 2.
Ambos metabolitos, M-2 y M-5, presentan acumulación no lineal, que puede deberse a reciclaje enterohepático o saturación de la ruta de UGT1A9. Mientras que las concentraciones plasmáticas de M-2 y M-5 tras una dosis única de regorafenib son mucho más bajas que las del compuesto original, las concentraciones plasmáticas en el estado estacionario de M-2 y M-5 son comparables a las de regorafenib.
Insuficiencia hepática
La exposición a regorafenib y sus metabolitos M-2 y M-5 es comparable entre los pacientes con insuficiencia hepática leve (Child-Pugh A) y los pacientes con función hepática normal.
Los escasos datos disponibles para los pacientes con insuficiencia hepática moderada (Child-Pugh B) indican una exposición similar a la observada en los pacientes con función hepática normal tras una dosis única de 100 mg de regorafenib. No se dispone de datos para los pacientes con insuficiencia hepática Child-Pugh C (grave). Regorafenib se elimina principalmente por vía hepática y la exposición puede estar aumentada en esta población de pacientes.
Insuficiencia renal
Los datos clínicos disponibles y los modelos farmacocinéticos fisiológicos indican una exposición a regorafenib y sus metabolitos M-2 y M-5 en el estado estacionario similar entre los pacientes con insuficiencia renal leve y moderada y los pacientes con función renal normal.
No se ha estudiado la farmacocinética de regorafenib en pacientes con insuficiencia renal grave ni nefropatía terminal. No obstante, los modelos farmacocinéticos fisiológicos no predicen ningún cambio relevante en cuanto a exposición en estos pacientes.
Pacientes de edad avanzada
La edad no afectó a la farmacocinética de regorafenib a lo largo del intervalo de edades estudiado (29-85 años).
Sexo
El sexo no influye sobre la farmacocinética de regorafenib.
Diferencias étnicas
La exposición a regorafenib en diversas poblaciones asiáticas (chinos, japoneses, coreanos) se encuentra dentro del mismo intervalo que el observado en los caucásicos.
Electrofisiología cardíaca/prolongación del intervalo QT
En un estudio específico sobre el QT en pacientes con cáncer de ambos sexos, no se observaron efectos de prolongación del intervalo QTc tras la administración de 160 mg de regorafenib en el estado estacionario.
5.3 Datos preclínicos sobre seguridad
Toxicidad sistémica
Tras la administración de dosis repetidas a ratones, ratas y perros, se observaron efectos adversos en varios órganos, fundamentalmente los riñones, el hígado, el tracto digestivo, la glándula tiroides, el sistema linfo/hematopoyético, el sistema endocrino, el aparato reproductor y la piel. Se observó una incidencia ligeramente aumentada de engrosamiento de las válvulas auriculoventriculares cardíacas en el estudio de toxicidad a dosis repetidas de 26 semanas de duración en ratas. Esto puede deberse a una aceleración de un proceso fisiológico relacionado con la edad. Estos efectos aparecieron con exposiciones sistémicas que se encontraban dentro del intervalo de exposición humana prevista o por debajo del mismo (de acuerdo con la comparación de AUC).
Las alteraciones dentales y óseas y los efectos adversos sobre el sistema reproductor fueron más pronunciados en animales de corta edad y en etapa de crecimiento y en ratas jóvenes e indican un posible riesgo para niños y adolescentes.
Toxicidad para la reproducción y el desarrollo
No se han realizado estudios específicos de fertilidad. No obstante, debe considerarse la posibilidad de que regorafenib afecte adversamente a la reproducción tanto masculina como femenina de acuerdo con los cambios morfológicos en los testículos, los ovarios y el útero observados tras la administración de dosis repetidas a ratas y perros con exposiciones más bajas que la exposición humana prevista (de acuerdo con la comparación de AUC). Los cambios observados solo fueron parcialmente reversibles.
Se constató un efecto de regorafenib sobre el desarrollo intrauterino en los conejos con exposiciones más bajas que la exposición humana prevista (de acuerdo con la comparación de AUC). Los principales hallazgos fueron malformaciones del aparato urinario, el corazón y los grandes vasos, así como del esqueleto.
Genotoxicidad y carcinogenicidad
No hubo datos indicativos de potencial genotóxico para regorafenib cuando se analizó mediante ensayos estándar in vitro e in vivo en ratones.
No se han realizado estudios sobre el potencial carcinogénico de regorafenib.
Evaluación del Riesgo Medioambiental (ERA)
Estudios sobre la evaluación del riesgo medioambiental han mostrado que regorafenib tiene el potencial de ser persistente, bioacumulativo y tóxico para el medio ambiente y puede suponer un riesgo para aguas superficiales y sedimentos (ver sección 6.6).
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Núcleo del comprimido Celulosa microcristalina Croscarmelosa sódica Estearato de magnesio Povidona (K-25)
Sílice coloidal anhidra
Cubierta películar Óxido de hierro rojo (E172)
Óxido de hierro amarillo (E172)
Lecitina (derivada de la soja)
Macrogol 3350
Alcohol polivinílico parcialmente hidrolizado Talco
Dióxido de titanio (E171)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años
Una vez abierto el frasco, se ha constatado que el medicamento es estable durante 7 semanas. A partir de entonces, el medicamento debe desecharse.
6.4 Precauciones especiales de conservación
Conservar en el embalaje original para protegerlo de la humedad.
Conservar el frasco perfectamente cerrado y mantener el desecante en el frasco.
6.5 Naturaleza y contenido del envase
Frasco de HDPE blanco opaco con un cierre de rosca de PP/PP (polipropileno) con junta de estanqueidad y un desecante de tamiz molecular.
Cada frasco contiene 28 comprimidos recubiertos con película.
Tamaños de envases:
Envase con 28 comprimidos recubiertos con película.
Envase con 84 (3 frascos de 28) comprimidos recubiertos con película.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Este medicamento puede suponer un riesgo para el medio ambiente (ver sección 5.3).
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Bayer Pharma AG 13342 Berlin Alemania
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/858/001
EU/1/13/858/002
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 26 de agosto de 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
Nombre y dirección del fabricante responsable de la liberación de los lotes
Bayer Pharma AG Kaiser-Wilhelm-Allee 51368 Leverkusen Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107c, párrafo 7, de la Directiva 2001/83/CE y posteriores actualizaciones, publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Obligación de llevar a cabo medidas posautorización
El TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
Descripción |
Fechas límite |
|
Presentar el análisis exploratorio de biomarcadores genético pre especificado (incluyendo NRAS, KRAS, BRAF y PIK3CA) y no genético (ANG-2, IL-6, IL-8, P1GF, VEGFR-1, TIE1, VEGF-A, VEGF-C, VEGF-D, VEGF-A-121, BMP-7, VWF, M-CSF, SDF-1) apropiado del estudio 15983 (ensayo fase III randomizado, doble ciego controlado con placebo de regorafenib adyuvante versus placebo para pacientes con cáncer colorrectal estadio IV tras tratamiento curativo de metástasis hepáticas). El análisis de biomarcadores genético y no genético debe ser implementado como obligatorio para todos los pacientes incluidos. |
31/12/2020 |
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR EMBALAJE EXTERIOR
1. NOMBRE DEL MEDICAMENTO
Stivarga 40 mg comprimidos recubiertos con película regorafenib
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 40 mg de regorafenib.
3. LISTA DE EXCIPIENTES
Contiene sodio y lecitina (derivada de la soja). Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
28 comprimidos recubiertos con película
84 (3 x 28) comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en el embalaje original para protegerlo de la humedad. Conservar el frasco perfectamente cerrado. Mantener el desecante en el frasco.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlin Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/858/001
EU/1/13/858/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
stivarga 40 mg
INFORMACIÓN QUE DEBE FIGURAR EN EL ACONDICIONAMIENTO PRIMARIO ETIQUETA
1. NOMBRE DEL MEDICAMENTO
Stivarga 40 mg comprimidos recubiertos con película regorafenib
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 40 mg de regorafenib
3. LISTA DE EXCIPIENTES
Contiene sodio y lecitina (derivada de la soja).
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
28 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
EXP
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en el embalaje original para protegerlo de la humedad. Conservar el frasco perfectamente cerrado. Mantener el desecante en el frasco.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlin Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/858/001
EU/1/13/858/002
13. NÚMERO DE LOTE
Lot
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
B. PROSPECTO
Prospecto: Información para el usuario
Stivarga 40 mg comprimidos recubiertos con película
regorafenib
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Stivarga y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Stivarga
3. Cómo tomar Stivarga
4. Posibles efectos adversos
5. Conservación de Stivarga
6. Contenido del envase e información adicional
1. Qué es Stivarga y para qué se utiliza
Stivarga contiene el principio activo regorafenib. Es un medicamento que se utiliza para tratar el cáncer y actúa retardando el crecimiento y la diseminación de las células cancerosas y eliminando el aporte de sangre que les permite seguir creciendo.
Stivarga se utiliza para tratar:
- el cáncer de colon o de recto que se ha diseminado a otras partes del organismo en pacientes adultos que han recibido otros tratamientos o que no pueden recibir tratamiento con otros medicamentos (quimioterapia basada en fluoropirimidinas, terapia anti-VEGF y terapia anti-EGFR).
- los tumores del estroma gastrointestinal (GIST), un tipo de cáncer de estómago e intestino, que se ha extendido a otras partes del organismo o no es tratable con cirugía, en pacientes adultos que han recibido tratamiento previo con otros medicamentos contra el cáncer (imatinib y sunitinib).
Si tiene alguna duda sobre cómo actúa Stivarga o el motivo por el que se le ha recetado este medicamento, consulte a su médico.
2. Qué necesita saber antes de empezar a tomar Stivarga No tome Stivarga
- si es alérgico al regorafenib o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar Stivarga.
Tenga especial cuidado con Stivarga
- si tiene cualquier problema hepático, incluido el síndrome de Gilbert, con signos tales como: decoloración amarillenta de la piel y del blanco de los ojos, orina oscura y confusión y/o desorientación. El tratamiento con Stivarga puede generar un mayor riesgo de problemas hepáticos. Su médico le realizará análisis de sangre para vigilar su función hepática antes y durante el transcurso del tratamiento con Stivarga. Si tiene un deterioro grave de la función hepática, no debe recibir tratamiento con Stivarga, ya que no se dispone de datos sobre el uso de Stivarga en pacientes con deterioro grave de la función hepática
- si tiene o ha tenido cualquier problema de sangrado y si está tomando warfarina, fenprocumón u otro medicamento que haga menos espesa la sangre para prevenir los coágulos sanguíneos. El tratamiento con Stivarga puede generar un mayor riesgo de sangrado. Su médico puede decidir que es necesario realizarle un análisis de sangre antes de comenzar a tomar Stivarga. Stivarga puede causar sangrados graves en el aparato digestivo, como, por ejemplo, en el estómago, la garganta, el recto o el intestino, o en los pulmones, los riñones, la boca, la vagina y/o el cerebro. Busque ayuda médica inmediatamente si presenta los siguientes síntomas: presencia de sangre en las heces o heces de color negro, presencia de sangre en la orina, dolor de estómago, tos / vómitos con sangre
- si tiene dolor en el pecho o cualquier problema cardíaco. Su médico comprobará el funcionamiento de su corazón antes de comenzar a tomar Stivarga y durante el transcurso del tratamiento. Busque ayuda médica inmediatamente si presenta los siguientes síntomas, ya que pueden ser signos de un ataque al corazón o de una reducción del flujo sanguíneo cardíaco: molestias o dolor en el pecho que pueden extenderse más allá del mismo a los hombros, los brazos, la espalda, el cuello, los dientes, la mandíbula o el estómago y que pueden ir y venir; sensación de falta de aire; aparición súbita de sudoración con piel fría y húmeda, sensación de mareo o desmayo
- si presenta un dolor de cabeza grave y persistente, alteraciones visuales, convulsiones o alteración del estado mental (como, por ejemplo, confusión, pérdida de la memoria o desorientación), póngase en contacto con su médico inmediatamente
- si presenta problemas gástricos e intestinales graves (perforación o fístulas gastrointestinales), su médico deberá tomar la decisión de interrumpir el tratamiento con Stivarga. Busque ayuda médica inmediatamente si presenta los siguientes síntomas: dolor de estómago grave o dolor de estómago que no desaparece, vómitos con sangre, heces rojas o negras
- si tiene hipertensión arterial. Stivarga puede elevar la tensión arterial. Su médico la vigilará antes y durante el transcurso del tratamiento y podría administrarle un medicamento para tratar la hipertensión arterial
- si se ha sometido recientemente o se va a someter a un procedimiento quirúrgico. Stivarga puede afectar al proceso de curación de sus heridas y es posible que haya que detener el tratamiento hasta que se cure la herida
- si sufre problemas en la piel. Stivarga puede causar enrojecimiento, dolor, hinchazón o ampollas en las palmas de las manos o las plantas de los pies. Si nota cualquier cambio, póngase en contacto con su médico. Para tratar los síntomas, puede que su médico le recomiende utilizar cremas y/o almohadillas en los zapatos y guantes. Si sufre este efecto adverso, es posible que su médico le cambie la dosis o interrumpa el tratamiento hasta que su situación mejore
Si se encuentra en alguna de estas circunstancias, informe a su médico antes de tomar Stivarga. Es posible que sea necesario tratarlas, así como realizar pruebas adicionales (consulte también la sección 4 "Posibles efectos adversos").
Niños y adolescentes
No hay un uso relevante de Stivarga en niños y adolescentes para la indicación de cáncer de colon o de recto que se ha diseminado a otras partes del organismo.
No se ha establecido la seguridad y eficacia de Stivarga en niños y adolescentes para la indicación de tumores del estroma gastrointestinal (GIST). No hay datos disponibles.
Otros medicamentos y Stivarga
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o pudiera tener que tomar cualquier otro medicamento. Esto incluye los adquiridos sin receta o los de venta libre, como vitaminas, suplementos dietéticos o medicamentos a base de plantas. Algunos medicamentos pueden afectar al modo en que Stivarga actúa o Stivarga puede afectar al modo de actuación de dichos medicamentos y causar efectos adversos graves. Informe a su médico en particular si está tomando alguno de los de esta lista o cualquier otro medicamento:
- ciertos medicamentos para tratar las infecciones por hongos (por ejemplo, ketoconazol, itraconazol, posaconazol y voriconazol)
- ciertos medicamentos para tratar el dolor (por ejemplo, ácido mefenámico, diflunisal y ácido niflúmico)
- ciertos medicamentos para tratar las infecciones bacterianas (por ejemplo, rifampicina, claritromicina, telitromicina)
- medicamentos habitualmente utilizados para tratar la epilepsia (convulsiones) (por ejemplo, fenitoína, carbamazepina o fenobarbital)
- metotrexato, un medicamento habitualmente utilizado para tratar el cáncer
- rosuvastatina, fluvastatina, atorvastatina, medicamentos habitualmente utilizados para tratar los niveles altos de colesterol
- warfarina o fenprocumón, medicamentos habitualmente utilizados para hacer menos espesa la sangre
- hierba de San Juan (medicamento adquirido sin receta), un tratamiento a base de plantas para la depresión
Consulte a su médico o farmacéutico antes de utilizar cualquier medicamento.
Toma de Stivarga con alimentos y bebidas
Evite beber zumo de pomelo mientras esté tomando Stivarga, ya que puede afectar al modo en que Stivarga actúa.
Embarazo, lactancia y fertilidad
Informe a su médico si cree que está embarazada, podría estarlo o tiene intención de quedarse embarazada, ya que Stivarga no debe usarse durante el embarazo a menos que sea claramente necesario. Su médico comentará con usted los posibles riesgos de tomar Stivarga durante el embarazo.
Evite quedarse embarazada durante el tratamiento con Stivarga, ya que este medicamento puede dañar al bebe nonato.
Tanto las mujeres como los hombres en edad fértil deben usar anticonceptivos efectivos durante el tratamiento y durante al menos ocho semanas después de la finalización del mismo.
No debe dar el pecho a su bebé durante el tratamiento con Stivarga, ya que este medicamento puede interferir con el crecimiento y el desarrollo del bebé. Informe a su médico si está dando el pecho o tiene intención de hacerlo.
Stivarga puede reducir la fertilidad tanto de hombres como de mujeres. Consulte a su médico antes de tomar Stivarga.
Conducción y uso de máquinas
Se desconoce si Stivarga altera la capacidad para conducir y utilizar máquinas. No conduzca ni use herramientas o máquinas si presenta síntomas relacionados con el tratamiento que afecten a su capacidad para concentrarse y reaccionar.
Información importante sobre algunos de los componentes de Stivarga
Este medicamento contiene 2,427 mmol (o 55,8 mg) de sodio por dosis diaria (4 comprimidos), lo que debe ser tenido en cuenta en pacientes con dietas pobres en sodio.
Este medicamento contiene 1,68 mg de lecitina (derivada de la soja) por dosis diaria (4 comprimidos).
3. Cómo tomar Stivarga
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
La dosis diaria recomendada en adultos es 4 comprimidos de Stivarga 40 mg (160 mg de regorafenib). Su médico puede cambiarle la dosis. Tome la dosis de Stivarga que le haya recetado su médico. Generalmente, su médico le pedirá que tome Stivarga durante 3 semanas y a continuación deje de tomarlo durante 1 semana. Esto es 1 ciclo de tratamiento.
Tome Stivarga a la misma hora todos los días, después de una comida ligera (baja en grasa). Tome el comprimido entero con agua después de una comida ligera con un contenido menor del 30% en grasa. Un ejemplo de comida ligera (baja en grasa) incluiría 1 porción de cereales (alrededor de 30 g), 1 vaso de leche descremada, 1 tostada con mermelada, 1 vaso de zumo de manzana y 1 taza de café o té (520 calorías, 2 g de grasa). No debe tomar Stivarga con zumo de pomelo (ver también la sección "Toma de Stivarga con alimentos y bebidas").
En caso de vómitos tras la administración de regorafenib, no debe tomar comprimidos adicionales y debe informar a su médico.
Es posible que su médico necesite reducirle la dosis o decida interrumpir o suspender el tratamiento permanentemente, si es preciso. En general, tomará Stivarga mientras obtenga beneficios del tratamiento y no sufra efectos adversos inaceptables.
No es necesario ajustar la dosis si tiene un deterioro leve de la función hepática. En caso de deterioro leve o moderado de la función hepática mientras se encuentra en tratamiento con Stivarga, su médico deberá mantenerle bajo estrecha vigilancia. Si tiene un deterioro grave de la función hepática, no debe recibir tratamiento con Stivarga, ya que no se dispone de datos sobre el uso de Stivarga en pacientes con deterioro grave de la función hepática.
No es necesario ajustar la dosis si tiene un deterioro leve o moderado de la función renal. No se dispone de datos sobre el uso de Stivarga en pacientes con deterioro grave de la función renal.
Si toma más Stivarga del que debe
Informe a su médico inmediatamente si ha tomado una dosis mayor que la que se le ha recetado. Es posible que necesite atención médica y que su médico le diga que deje de tomar Stivarga.
Tomar demasiado Stivarga puede aumentar la probabilidad de que aparezcan algunos efectos adversos o de que estos sean más graves, especialmente:
- reacciones cutáneas (exantema, ampollas, enrojecimiento, dolor, hinchazón, picor o descamación de la piel)
- cambios en la voz o ronquera (disfonía)
- movimientos intestinales frecuentes o de consistencia suelta (diarrea)
- llagas bucales (inflamación de la mucosa)
- sequedad de boca
- disminución del apetito
- tensión arterial elevada (hipertensión)
- cansancio excesivo (fatiga)
Si olvidó tomar Stivarga
Si olvida una dosis, tómela en cuanto se acuerde ese mismo día. No tome dos dosis el mismo día para compensar una dosis omitida el día anterior. Informe a su médico de toda dosis que haya omitido.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Este medicamento también puede alterar los resultados de algunos análisis de sangre.
Los efectos adversos más graves, para los que se ha observado un desenlace mortal, son:
- Problemas hepáticos graves, sangrado y perforación gastrointestinal.
Informe a su médico inmediatamente si presenta alguno de los siguientes síntomas:
Problemas hepáticos:
El tratamiento con Stivarga puede generar un mayor riesgo de problemas hepáticos graves. Busque ayuda médica inmediatamente si presenta los siguientes síntomas:
- decoloración amarillenta de la piel y del blanco de los ojos
- orina oscura
- confusión y/o desorientación
Estos pueden ser signos de una lesión hepática grave.
Sangrado:
Stivarga puede causar sangrados graves en el aparato digestivo, como, por ejemplo, en el estómago, la garganta, el recto o el intestino, o en los pulmones, los riñones, la boca, la vagina y/o el cerebro. Busque ayuda médica inmediatamente si presenta los siguientes síntomas:
- presencia de sangre en las heces o heces de color negro
- presencia de sangre en la orina
- dolor de estómago
- tos / vómitos con sangre Estos pueden ser signos de sangrado.
Problemas gástricos e intestinales graves (perforación o fístula gastrointestinal):
Busque ayuda médica inmediatamente si presenta los siguientes síntomas:
- dolor de estómago (abdominal) grave o dolor de estómago que no desaparece
- vómitos con sangre
- heces rojas o negras
Estos pueden ser signos de problemas gástricos o intestinales graves.
Otros efectos adversos de Stivarga indicados en función de su frecuencia:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 usuarios):
- infección
- reducción del número de plaquetas en sangre que se caracteriza por una fácil aparición de hematomas o sangrados (trombocitopenia)
- reducción del número de glóbulos rojos en sangre (anemia)
- disminución del apetito y de la ingesta de alimentos
- dolor de cabeza
- tensión arterial elevada (hipertensión)
- cambios en la voz o ronquera (disfonía)
- movimientos intestinales frecuentes o de consistencia suelta (diarrea)
- sequedad o dolor en la boca, dolor en la lengua, llagas bucales (estomatitis y/o inflamación de
la mucosa)
- sensación de mareo (náuseas)
- vómitos
- concentraciones sanguíneas elevadas de bilirrubina, una sustancia producida por el hígado
(hiperbilirrubinemia)
- enrojecimiento, dolor, ampollas e hinchazón de las palmas de las manos o las plantas de los pies (reacción cutánea mano-pie)
- exantema
- caída del cabello (alopecia)
- debilidad, falta de fuerza y energía, cansancio excesivo y somnolencia inusual (astenia/fatiga)
- dolor
- fiebre
- pérdida de peso
Efectos adversos frecuentes (pueden afectar a hasta 1 de cada 10 usuarios):
- disminución del número de glóbulos blancos (leucopenia)
- disminución de la actividad de la glándula tiroides (hipotiroidismo)
- concentraciones sanguíneas bajas de potasio, fosfato, calcio, sodio o magnesio (hipopotasemia, hipofosfatemia, hipocalcemia, hiponatremia e hipomagnesemia)
- concentraciones sanguíneas elevadas de ácido úrico (hiperuricemia)
- temblor
trastornos del gusto
sequedad de boca
ardores (reflujo gastroesofágico)
infección o irritación del estómago e intestinos (gastroenteritis)
cambios en enzimas producidas por el hígado, que pueden indicar una alteración hepática
(aumento de las transaminasas) sequedad cutánea
erupción cutánea con formación de escamas o descamación de la piel (exantema exfoliativo) rigidez de los músculos o las articulaciones proteínas en la orina (proteinuria)
concentraciones elevadas de ciertas enzimas implicadas en la digestión (aumento de la amilasa y la lipasa)
situación anómala en la coagulación de la sangre (INR anómalo)
Efectos adversos poco frecuentes (pueden afectar a hasta 1 de cada 100 usuarios):
- signos/síntomas de una reacción alérgica que podría incluir una erupción grave generalizada, sensación de mareo, fiebre, falta de aliento, ictericia, cambios en las sustancias químicas producidas por el hígado (reacción de hipersensibilidad).
- ataque al corazón, dolor en el pecho (infarto de miocardio e isquemia miocárdica)
- cifras de tensión arterial gravemente elevadas que provocan dolor de cabeza, confusión, visión borrosa, náuseas, vómitos y desmayos (crisis hipertensiva)
- trastornos ungueales (cambios en las uñas tales como surcos y/o uñas quebradizas)
- exantemas cutáneos múltiples (eritema multiforme)
Efectos adversos raros (pueden afectar a hasta 1 de cada 1.000 usuarios):
- ciertos cánceres de piel (queratoacantoma / carcinoma cutáneo de células escamosas)
- dolor de cabeza, confusión, convulsiones y pérdida visual asociada con o sin hipertensión arterial (síndrome de leucoencefalopatía posterior reversible / PRESS)
- reacciones graves en la piel y/o las membranas mucosas entre las que se pueden encontrar ampollas dolorosas y fiebre, incluido un desprendimiento extenso de la piel (síndrome de Stevens-Johnson y necrolisis epidérmica tóxica)
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Stivarga
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la caja y la etiqueta del frasco después de “EXP”. La fecha de caducidad es el último día del mes que se indica.
Conservar en el embalaje original para protegerlo de la humedad.
Conservar el frasco perfectamente cerrado y mantener el desecante en el frasco. El desecante es un material absorbente de la humedad introducido en un pequeño recipiente para proteger a los comprimidos de la misma.
Una vez abierto el frasco, el medicamento debe desecharse al cabo de 7 semanas.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Stivarga
- El principio activo es regorafenib. Cada comprimido recubierto con película contiene 40 mg de regorafenib.
- Los demás componentes son: celulosa microcristalina, croscarmelosa sódica, estearato de magnesio, povidona (K-25), sílice coloidal anhidra, óxido de hierro rojo (E172), óxido de hierro amarillo (E172), lecitina (derivada de la soja), macrogol 3350, alcohol polivinílico (parcialmente hidrolizado), talco y dióxido de titanio (E171).
Aspecto del producto y contenido del envase
Los comprimidos Stivarga 40 mg son de color rosa claro y forma oval, marcados con "BAYER" en una cara y "40" en la otra.
Cada frasco contiene 28 comprimidos recubiertos con película.
Los comprimidos de Stivarga 40 mg se presentan en envases de un frasco de 28 comprimidos o en un envase con 3 frascos, cada uno de los cuales contiene 28 comprimidos.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Bayer Pharma AG 13342 Berlin Alemania
Responsable de la fabricación
Bayer Pharma AG 51368 Leverkusen Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie / Belgique / Belgien
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Etarapun
Eaüep Etarapna EOOfl Tea. +359 02 81 401 01 Ceská republika Bayer s.r.o.
Tel: +420 266 101 111
Danmark
Bayer A/S
Tlf: +45-45 23 50 00
Deutschland
Bayer Vital GmbH
Tel: +49-(0)214-30 513 48
Eesti
Bayer OÜ
Tel: +372 655 85 65
EXláda
Bayer EA7ág ABEE Tqk+30 210 618 75 00 España
Bayer Hispania S.L.
Tel: +34-93-495 65 00 France
Bayer HealthCare Tél: +33-(0)800 87 54 54 Hrvatska Bayer d.o.o.
Tel: + 385-(0)1-6599 900
Ireland
Bayer Limited
Tel: +353 1 299 93 13
Ísland
Icepharma hf.
Sími: +354 540 80 00 Italia
Bayer S.p.A.
Tel: +39-02-397 81 Kúnpoq
NOVAGEM Limited Tp^: +357 22 48 38 58 Latvija SIA Bayer
Tel: +371 67 84 55 63
Lietuva
UAB Bayer
Tel. +370 5 23 36 868
Luxembourg / Luxemburg
Bayer SA-NV
Tél/Tel: +32-(0)2-535 63 11
Magyarország
Bayer Hungária Kft.
Tel.:+36-14 87-41 00
Malta
Alfred Gera and Sons Ltd. Tel: +356-21 44 62 05 Nederland Bayer B.V.
Tel: +31-(0)297-28 06 66 Norge
Bayer AS
Tlf. +47 23 13 05 00 Osterreich
Bayer Austria Ges. m. b. H. Tel: +43-(0)1-711 46-0 Polska
Bayer Sp. z o.o.
Tel.: +48-22-572 35 00 Portugal
Bayer Portugal, Lda.
Tel: +351-21-416 42 00
Romania
SC Bayer SRL
Tel: +40 21 528 59 00
Slovenija
Bayer d. o. o.
Tel.: +386-(0)1-58 14 400 Slovenská republika
Bayer, spol. s r.o.
Tel: +421 2 59 21 31 11 Suomi/Finland
Bayer Oy
Puh/Tel: +358-20 785 21
Sverige
Bayer AB
Tel: +46-(0)8-580 223 00 United Kingdom
Bayer plc
Tel: +44 (0)1 635-56 30 00
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos (EMA): http://www.ema.europa.eu.
45