Sprycel 80 Mg Comprimidos Recubiertos Con Pelicula
Información obsoleta, busque otroANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
SPRY CEL 20 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 20 mg de dasatinib (como monohidrato). Excipientes con efecto conocido:
Cada comprimido recubierto con película contiene 27 mg de lactosa monohidrato.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
Comprimido recubierto con película redondo, de color blanco a blanquecino, biconvexo, con “BMS” grabado en una cara y “527” en la otra.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
SPRYCEL está indicado para el tratamiento de pacientes adultos con:
■ leucemia mieloide crónica (LMC) en fase crónica de nuevo diagnóstico cromosoma Filadelfia positivo (Ph+).
■ leucemia mieloide crónica (LMC) en fase crónica, acelerada o blástica, con resistencia o intolerancia al tratamiento previo, incluido el mesilato de imatinib.
■ leucemia linfoblástica aguda (LLA) cromosoma Filadelfia positivo (Ph+) y crisis blástica linfoide procedente de LMC con resistencia o intolerancia al tratamiento previo.
4.2 Posología y forma de administración
El tratamiento debe iniciarlo un médico con experiencia en el diagnóstico y el tratamiento de pacientes con leucemia.
Posología
La dosis de inicio recomendada para LMC en fase crónica es de 100 mg de dasatinib una vez al día, administrada por vía oral.
La dosis de inicio recomendada para LMC en fase acelerada, crisis blástica mieloide o linfoide (fases avanzadas) o en LLA cromosoma Filadelfia positivo (Ph+) es de 140 mg una vez al día administrada por vía oral (ver sección 4.4).
Duración del tratamiento
En ensayos clínicos, el tratamiento con SPRY CEL se continuó hasta la progresión de la enfermedad o hasta que ya no se tolerara por el paciente. No se ha investigado el efecto de la suspensión del tratamiento sobre el resultado de la enfermedad a largo plazo después de conseguirse una respuesta citogenética o molecular [incluyendo respuesta citogenética completa (RCyC), respuesta molecular mayor (RMM) y RM4.5].
Para alcanzar la dosis recomendada, SPRYCEL está disponible como comprimidos recubiertos con película de 20 mg, 50 mg, 70 mg, 80 mg, 100 mg y 140 mg. Se recomienda el incremento o la reducción de la dosis en base a la respuesta del paciente y a la tolerabilidad.
Escalado de dosis
En ensayos clínicos en pacientes adultos con LMC y LLA Ph+, se permitió un aumento gradual de la dosis a 140 mg una vez al día (LMC en fase crónica) o 180 mg una vez al día (LMC en fases avanzadas o LLA Ph+) en pacientes que no alcanzaron una respuesta hematológica o citogenética a las dosis recomendadas de inicio.
Ajustes de la dosis por reacciones adversas
Mielosupresión
En los ensayos clínicos, la mielosupresión requirió una reducción de la dosis o la suspensión temporal o permanente del tratamiento en estudio. Se realizaron transfusiones de plaquetas y glóbulos rojos en los pacientes que lo requirieron. Se administraron factores de crecimiento hematopoyéticos en los pacientes con mielosupresión resistente. Las directrices para los ajustes de la dosis se resumen en la Tabla 1.
Tabla 1: Ajustes de la dosis por neutropenia y trombocitopenia
|
LMC en fase crónica (dosis inicial de 100 mg, una vez al día) |
RAN < 0,5 x 109/l y/o plaquetas < 50 x 109/l |
1 Suspender el tratamiento hasta recuperación de RAN > 1,0 x 109/l y plaquetas > 50 x 109/l. 2 Reanudar el tratamiento a la dosis inicial original. 3 Si las plaquetas < 25 x 109/l y/o un nuevo descenso del RAN < 0,5 x 109/l durante > 7 días, debe repetirse el paso 1 y reanudarse el tratamiento a una dosis reducida de 80 mg una vez al día para el segundo episodio. Para el tercer episodio, reducir dosis de modo adicional hasta 50 mg una vez al día (para pacientes recientemente diagnosticados) o interrumpir el tratamiento (para pacientes resistentes o intolerantes al tratamiento previo incluyendo imatinib). |
|
LMC en fase acelerada y crisis blástica y LLA Ph+ (dosis inicial de 140 mg, una vez al día) |
RAN < 0,5 x 109/l y/o plaquetas < 10 x 109/l |
1 Descartar que la citopenia esté relacionada con la leucemia (aspirado y/o biopsia medular). 2 Si la citopenia no está relacionada con la leucemia, suspender el tratamiento hasta recuperación del RAN > 1,0 x 109/l y plaquetas > 20 x 109/l y reanudar a la dosis inicial original. 3 Ante un nuevo episodio de citopenia, repetir el paso 1 y reanudar el tratamiento a una dosis reducida de 100 mg una vez al día (segundo episodio) u 80 mg una vez al día (tercer episodio). 4 Si la citopenia está relacionada con la leucemia, valorar aumentar la dosis a 180 mg una vez al día. |
RAN: recuento absoluto de neutrófilos
Reacciones adversas no hematológicas
Si se desarrolla una reacción adversa no hematológica, moderada, grado 2, con dasatinib, se interrumpirá el tratamiento hasta que el acontecimiento se haya resuelto o hasta que haya retornado al nivel basal. Continuar con la misma dosis si es la primera vez que ocurre y reducir la dosis si es un acontecimiento recurrente. Si se desarrolla una reacción adversa no hematológica grave, grado 3 ó 4, con dasatinib, el tratamiento debe interrumpirse hasta que el acontecimiento se haya resuelto. Posteriormente, si es conveniente, el tratamiento puede reanudarse, a una dosis reducida, dependiendo de la gravedad inicial del acontecimiento. Para pacientes con LMC en fase crónica que hayan recibido 100 mg una vez al día, se recomienda una reducción de dosis hasta 80 mg una vez al día, con una reducción de dosis adicional desde 80 mg una vez al día hasta 50 mg una vez al día, si fuese necesario. Para pacientes con fase avanzada de LMC o LLA cromosoma Filadelfia positivo (Ph+) que hayan recibido 140 mg una vez al día, se recomienda una reducción de dosis hasta 100 mg una vez al día, con una reducción de dosis adicional hasta 50 mg una vez al día, si fuese necesario.
Derrame pleural: si se diagnóstica un derrame pleural, interrumpir el tratamiento con dasatinib hasta que el paciente sea asintomático o haya retornado a su estado basal. Si el episodio no mejora dentro de aproximadamente una semana, considerar un tratamiento con diuréticos o corticosteroides o ambos al mismo tiempo (ver secciones 4.4 y 4.8). Una vez resuelto el primer episodio, considerar la reintroducción de dasatinib al mismo nivel de dosis. Tras la resolución de un episodio posterior, reintroducir dasatinib con un nivel de dosis reducido. Una vez resuelto un episodio grave (grado 3 ó 4), el tratamiento puede continuarse como proceda a un nivel de dosis reducido dependiendo de la gravedad inicial del acontecimiento.
Población pediátrica
No se ha establecido todavía, la seguridad y eficacia de SPRYCEL en niños y adolescentes menores de 18 años de edad. No hay datos disponibles (ver sección 5.1).
Pacientes de edad avanzada
No se han observado diferencias farmacocinéticas clínicamente relevantes relacionadas con la edad en estos pacientes. No es necesaria ninguna recomendación de dosis específica en los pacientes de edad avanzada.
Insuficiencia hepática
Los pacientes con insuficiencia hepática leve, moderada o grave pueden recibir la dosis de inicio recomendada. Sin embargo, SPRYCEL debe utilizarse con precaución en pacientes con insuficiencia hepática (ver secciones 4.4 y 5.2).
Insuficiencia renal
No se han realizado ensayos clínicos con SPRYCEL en pacientes con función renal reducida (el estudio en pacientes con LMC en fase crónica de nuevo diagnóstico, excluyó a los pacientes con una concentración de creatinina en suero > 3 veces el límite superior del rango normal, y ensayos en pacientes con LMC en fase crónica con resistencia o intolerancia al tratamiento previo con imatinib excluyó a pacientes con una concentración de creatinina sérica > 1,5 veces el límite superior del rango normal). Como el aclaramiento renal de dasatinib y sus metabolitos representa < 4%, en pacientes con insuficiencia renal no se espera una disminución del aclaramiento corporal total.
Forma de administración
SPRY CEL debe ser administrado por vía oral.
Los comprimidos recubiertos con película no deben aplastarse ni fraccionarse para minimizar el riesgo de exposición dérmica, deben tragarse enteros. Los comprimidos pueden tomarse con o sin comida y deben tomarse sistemáticamente o por la mañana o por la noche.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Interacciones clínicamente relevantes
Dasatinib es un sustrato y un inhibidor del citocromo P450 (CYP) 3A4. Por tanto, existe la posibilidad de interacción con otros medicamentos administrados simultáneamente, que se metabolizan fundamentalmente por CYP3A4 o que modulan su actividad (ver sección 4.5).
El uso concomitante de dasatinib y medicamentos o sustancias que inhiban de forma potente el CYP3A4 (p. ej., ketoconazol, itraconazol, eritromicina, claritromicina, ritonavir, telitromicina, zumo de pomelo) puede aumentar la exposición a dasatinib. Por tanto, no se recomienda la coadministración de inhibidores potentes de CYP3A4 en pacientes que reciben dasatinib (ver sección 4.5).
El uso concomitante de dasatinib con medicamentos inductores de CYP3A4 (p. ej., dexametasona, fenitoína, carbamazepina, rifampicina, fenobarbital o medicamentos a base de plantas que contienen Hypericum perforatum, también conocido como hierba de San Juan) puede reducir significativamente la exposición a dasatinib, incrementando potencialmente el riesgo de fracaso terapéutico. Por lo tanto, en pacientes que reciben dasatinib, deberá optarse por la coadministración con medicamentos alternativos con menor capacidad de inducción de CYP3A4 (ver sección 4.5).
El uso concomitante de dasatinib y un sustrato de CYP3A4 puede aumentar la concentración plasmática del sustrato de CYP3A4. Por tanto, se debe garantizar precaución cuando se coadministre dasatinib con sustratos de CYP3A4 de margen terapéutico estrecho como astemizol, terfenadina, cisaprida, pimozida, quinidina, bepridilo o alcaloides del cornezuelo del centeno (ergotamina, dihidroergotamina) (ver sección 4.5).
El uso concomitante de dasatinib y antagonistas de la histamina tipo 2 (H2) (p. ej., famotidina), o los inhibidores de la bomba de protones (p. ej., omeprazol) o hidróxido de aluminio/hidróxido de magnesio puede reducir la exposición a dasatinib. Por tanto, no se recomienda la utilización de los antagonistas-H2 o inhibidores de la bomba de protones. Sin embargo, pueden administrarse productos con hidróxido de aluminio/hidróxido de magnesio hasta 2 horas antes o 2 horas después de la administración de dasatinib (ver sección 4.5).
Poblaciones especiales
Basado en los hallazgos de un estudio farmacocinético de dosis única, los pacientes con insuficiencia hepática leve, moderada o grave pueden recibir la dosis de inicio recomendada (ver secciones 4.2 y 5.2). Debido a las limitaciones de este ensayo clínico, se recomienda precaución al administrar dasatinib a pacientes con insuficiencia hepática (ver sección 4.2).
Reacciones adversas importantes
Mielosupresión
El tratamiento con dasatinib se asocia a anemia, neutropenia y trombocitopenia. Su incidencia es más temprana y más frecuente en pacientes con LMC en fase avanzada o LLA Ph+ que en pacientes con LMC en fase crónica. En los pacientes con LMC en fase avanzada o LLA Ph+, deben realizarse hemogramas completos cada semana durante los 2 primeros meses, y posteriormente cada mes o con la frecuencia que le sea indicada clínicamente. En pacientes con leucemia mieloide crónica (LMC) en fase crónica deben realizarse hemogramas completos cada 2 semanas durante las primeras 12 semanas, después cada 3 meses y posteriormente cuando esté clínicamente indicado. La mielosupresión es generalmente reversible y normalmente se controla interrumpiendo temporalmente la administración de dasatinib o reduciendo la dosis (ver secciones 4.2 y 4.8).
Sangrado
En pacientes con LMC en fase crónica (n = 548), 5 pacientes (1%) que recibieron dasatinib tuvieron hemorragia grado 3 ó 4. En ensayos clínicos en pacientes con LMC en fase avanzada que recibieron la dosis recomendada de SPRYCEL (n = 304) ocurrieron hemorragias graves en el sistema nervioso central (SNC) en el 1% de los pacientes. Un caso fue mortal y se asoció según los Criterios de Toxicidad Común (CTC), a una trombocitopenia grado 4. Se produjo hemorragia gastrointestinal grado 3 ó 4 en el 6% de los pacientes con LMC en fase avanzada y generalmente requirieron suspensión del tratamiento y transfusiones. Se produjeron otras hemorragias grado 3 ó 4 en el 2% de los pacientes con LMC en fase avanzada . La mayoría de los sangrados relacionados en estos pacientes fueron típicamente asociados con trombocitopenia grado 3 ó 4 (ver sección 4.8). Adicionalmente, los estudios de función plaquetaria in vivo e in vitro sugieren que el tratamiento con SPRYCEL afecta de modo reversible a la activación de plaquetas.
Hay que tener precaución si los pacientes utilizan antiagregantes plaquetarios o anticoagulantes.
Retención de líquidos
Dasatinib se asocia con retención de líquidos. En el ensayo Fase III en pacientes con LMC en fase crónica de nuevo diagnóstico , se notificó retención de líquidos grado 3 ó 4 en 13 pacientes (5%) en el grupo de tratamiento con dasatinib y 2 pacientes (1%) en el grupo de tratamiento con imatinib tras un seguimiento mínimo de 60 meses (ver sección 4.8). En todos los pacientes tratados con SPRYCEL con LMC en fase crónica se produjo retención de líquidos grave en 32 pacientes (6%) de los que recibieron SPRYCEL a la dosis recomendada (n = 548). En ensayos clínicos en pacientes con LMC en fase avanzada que recibieron SPRYCEL a la dosis recomendada (n = 304) se notificó retención de líquidos grado 3 ó 4 en el 8% de los pacientes, incluyendo derrame pleural y pericárdico grado 3 ó 4 en el 7% y el 1% de los pacientes respectivamente. En estos pacientes se notificó edema pulmonar grado 3 ó 4 e hipertensión pulmonar en el 1% de los pacientes.
Los pacientes que desarrollen síntomas tales como disnea o tos seca que sugieran derrame pleural, deberán ser evaluados por radiografía de tórax. Los derrames pleurales grado 3 ó 4, pueden requerir toracocentesis y oxigenoterapia. La retención de líquidos se trató normalmente con medidas de apoyo que incluyeron la administración de diuréticos y tratamientos cortos con esteroides (ver secciones 4.2 y 4.8). Pacientes con 65 años o más tienen mayor probabilidad que los pacientes jóvenes de experimentar episodios de derrame pleural, disnea, tos, derrame pericárdico e insuficiencia cardiaca congestiva, y deben ser monitorizados cuidadosamente.
Hipertensión arterial pulmonar (HTPA)
La HTPA (hipertensión pulmonar arterial precapilar confirmada por cateterismo derecho) ha sido notificada asociada al tratamiento con dasatinib (ver sección 4.8). En estos casos, la HTPA se notificó después del inicio del tratamiento con dasatinib, incluyendo casos tras más de un año de tratamiento.
Antes de iniciar el tratamiento con dasatinib, debe evaluarse si el paciente presenta signos o síntomas de enfermedad cardiopulmonar subyacente. En caso positivo, deberá realizarse una ecocardiografía al inicio del tratamiento. Esta prueba deberá valorarse si el paciente presentara factores de riesgo de enfermedad cardíaca o pulmonar. En los pacientes que desarrollen disnea y fatiga tras el inicio del tratamiento se deberán evaluar las etiologías más comunes incluyendo derrame pleural, edema pulmonar, anemia o infiltrados pulmonares. De acuerdo con las recomendaciones para el manejo de las reacciones adversas no hematológicas (ver sección 4.2) deberá reducirse la dosis de dasatinib o interrumpir el tratamiento durante esta evaluación. Si no se encontrase explicación, o si no se produce una mejoría con la reducción de la dosis o la suspensión del tratamiento, debe considerarse el diagnóstico de HTPA. La aproximación diagnóstica debe seguir las directrices de la práctica clínica habitual. Si se confirma la HTPA, interrumpir permanentemente el tratamiento con dasatinib. El seguimiento de la HTPA deberá realizarse de acuerdo a las directrices de la práctica clínica habitual. Tras la suspensión del tratamiento en pacientes con HTPA, se han observado mejoras en los parámetros clínicos y hemodinámicos.
Prolongación de QT
Los datos in vitro sugieren que dasatinib tiene capacidad de prolongar la repolarización cardíaca ventricular (intervalo QT) (ver sección 5.3). En 258 pacientes tratados con dasatinib y 258 pacientes tratados con imatinib tras un seguimiento mínimo de 60 meses, en el ensayo Fase III en pacientes con LMC en fase crónica de nuevo diagnóstico, se notificó en 1 paciente (< 1%) en cada grupo una prolongación QTc como una reacción adversa. La mediana de cambio en el QTcF desde el nivel basal fue de 3,0 mseg en los pacientes tratados con dasatinib comparados con los 8,2 mseg en los pacientes tratados con imatinib. Un paciente (< 1%) en cada grupo experimentó un QTcF > 500 mseg. En 865 pacientes con leucemia, tratados con dasatinib, el cambio medio del intervalo QTc respecto a los valores basales, aplicando el método de Fridericia (QTcF) fue de 4-6 mseg, con un límite superior en el intervalo de confianza del 95% < 7 mseg (ver sección 4.8).
De los 2.182 pacientes con resistencia o intolerancia al tratamiento previo con imatinib, que recibieron dasatinib en los ensayos clínicos, 15 (1%) pacientes presentaron prolongación QTc como una reacción adversa. Veintiún de estos pacientes (1%) tuvieron un QTcF > 500 mseg.
Dasatinib debe administrarse con precaución en pacientes que tengan o puedan desarrollar prolongación del QTc. Esto incluye pacientes con hipopotasemia o hipomagnesemia, pacientes con síndrome congénito de QT largo, pacientes que toman medicamentos antiarrítmicos u otros medicamentos que induzcan prolongación de QT y pacientes en tratamiento con dosis altas acumulativas de antraciclinas. La hipopotasemia o hipomagnesemia debe corregirse antes de la administración de dasatinib.
Reacciones adversas cardíacas
Dasatinib fue estudiado en un ensayo aleatorizado de 519 pacientes con LMC en fase crónica de nuevo diagnóstico, que incluía pacientes con enfermedad cardíaca previa. Se notificaron reacciones adversas cardíacas, tipo insuficiencia cardiaca congestiva /insuficiencia cardiaca, derrame pericárdico, arritmias, palpitaciones, prolongación del intervalo QT e infarto de miocardio (incluyendo mortal) en pacientes que estaban tomando dasatinib. Los acontecimientos adversos cardíacos fueron más frecuentes en pacientes con factores de riesgo o con historial de enfermedad cardíaca. Los pacientes con factores de riesgo (p. ej. hipertensión, hiperlipidemia, diabetes) o con historial de enfermedad cardíaca (p.ej. intervención coronaria percutánea previa, enfermedad arterial coronaria documentada) deben ser monitorizados cuidadosamente para los signos y síntomas compatibles con insuficiencia cardíaca como dolor torácico, dificultad para respirar y diaforesis.
Sí aparecen estos signos o síntomas clínicos, se aconseja al médico interrumpir la administración de dasatinib. Después de su resolución, debe realizarse una evaluación funcional antes de continuar el tratamiento con dasatinib. Dasatinib puede reintroducirse a la dosis original si los acontecimientos adversos fueron leves/moderados (< grado 2) y reintroducirse a una dosis reducida si los acontecimientos fueron graves (> grado 3) (ver sección 4.2). Los pacientes que continúan el tratamiento deben ser monitorizados periódicamente.
En los ensayos clínicos no se incluyeron pacientes con enfermedades cardiovasculares importantes o no controladas.
Lactosa
Este medicamento contiene 135 mg de lactosa monohidrato en la dosis diaria de 100 mg y 189 mg de lactosa monohidrato en una dosis diaria de 140 mg. Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp o mala absorción de glucosa o galactosa no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Principios activos que pueden aumentar las concentraciones plasmáticas de dasatinib Los estudios in vitro indican que dasatinib es un sustrato de CYP3A4. El uso simultáneo de dasatinib con medicamentos o sustancias que pueden inhibir el CYP3A4 (p.ej.: ketoconazol, itraconazol, eritromicina, claritromicina, ritonavir, telitromicina, zumo de pomelo) puede aumentar la exposición a dasatinib. Por tanto, en pacientes que reciban dasatinib, no se recomienda la administración sistémica de inhibidores potentes de CYP3A4.
En base a los estudios in vitro, a concentraciones clínicamente relevantes, la unión de dasatinib a las proteínas plasmáticas es del 96% aproximadamente. No se han realizado estudios para evaluar la interacción de dasatinib con otros medicamentos que se unan a proteínas. Se desconoce el potencial de desplazamiento y su importancia clínica.
Principios activos que pueden reducir las concentraciones plasmáticas de dasatinib Cuando se administró dasatinib durante 8 días por la tarde en combinación con 600 mg de rifampicina, un potente inductor de CYP3A4, el AUC de dasatinib disminuyó en un 82%. Otros medicamentos que inducen la actividad de CYP3A4 (p.ej.: dexametasona, fenitoína, carbamazepina, fenobarbital o medicamentos a base de plantas que contengan Hypericum perforatum, también conocida como Hierba de San Juan) pueden también aumentar el metabolismo y disminuir las concentraciones plasmáticas de dasatinib. Por lo tanto, no se recomienda el uso simultáneo de inductores potentes de CYP3A4 con dasatinib. En pacientes en los que estén indicados la rifampicina u otros inductores de CYP3A4, deben usarse medicamentos alternativos con menor potencial de inducción enzimática.
Antagonistas de receptores histamina-2 e inhibidores de la bomba de protones Es probable que la supresión a largo plazo de la secreción de ácido gástrico por antagonistas-H2 o inhibidores de la bomba de protones (p. ej., famotidina y omeprazol) reduzca la exposición a dasatinib. En un ensayo de dosis única en sujetos sanos, la administración de famotidina 10 horas antes de una dosis única de SPRYCEL redujo la exposición a dasatinib en un 61%. En un ensayo de 14 sujetos sanos, la administración de una dosis única de 100 mg de SPRYCEL 22 horas después de una dosis de 40 mg de omeprazol durante 4 días, en el estado de equilibrio, redujo el AUC de dasatinib en un 43% y la Cmax en un 42%. Debe valorarse el uso de antiácidos en lugar de los antagonistas-H2 o los inhibidores de la bomba de protones en pacientes que reciban tratamiento con SPRYCEL (ver sección 4.4).
Antiácidos
Los datos preclínicos demuestran que la solubilidad de dasatinib es pH-dependiente. En sujetos sanos, el uso simultáneo de antiácidos con hidróxido de aluminio/magnesio y SPRYCEL redujo el AUC de una dosis única de SPRYCEL un 55% y la Cmáx un 58%. Sin embargo, cuando los antiácidos se administraron 2 horas antes de una dosis única de SPRYCEL, no se observaron cambios relevantes en la concentración o la exposición a dasatinib. Así pues, los antiácidos deben administrarse hasta 2 horas antes o 2 horas después de SPRYCEL (ver sección 4.4).
Principios activos cuya concentración plasmática puede verse alterada por dasatinib El uso concomitante de dasatinib y un sustrato de CYP3A4 puede aumentar la exposición al sustrato de CYP3A4. En un ensayo en sujetos sanos, una dosis única de 100 mg de dasatinib aumentó el AUC y la Cmáx de la simvastatina, un sustrato conocido de CYP3A4 un 20% y 37% respectivamente. No puede excluirse que el efecto sea superior después de dosis múltiples de dasatinib. Por tanto, los sustratos de CYP3A4 con margen terapéutico estrecho (p. ej., astemizol, terfenadina, cisaprida, pimozida, quinidina, bepridil o alcaloides ergóticos [ergotamina, dihidroergotamina]) deben administrarse con precaución en pacientes que están recibiendo dasatinib (ver sección 4.4).
La información in vitro indica un riesgo potencial de interacción con sustratos CYP2C8, tales como glitazonas.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Se debe advertir a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento.
Embarazo
En base a la experiencia en humanos, se sospecha que dasatinib pueda causar malformaciones congénitas incluyendo defectos del tubo neural y efectos farmacológicos perjudiciales en el feto cuando se administra durante el embarazo. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3).
SPRYCEL no debe utilizarse durante el embarazo excepto si la situación clínica de la mujer requiere tratamiento con dasatinib. Si SPRYCEL se utiliza durante el embarazo, la paciente debe estar informada del posible riesgo para el feto.
Lactancia
La información sobre la excreción de dasatinib en leche humana o animal es insuficiente/limitada. Los datos fisicoquímicos y los datos farmacodinámicos/toxicológicos disponibles sobre dasatinib apuntan a su excreción en la leche materna y no puede excluirse el riesgo para el niño lactante. Debe interrumpirse la lactancia durante el tratamiento con SPRYCEL.
Fertilidad
Se desconoce el efecto de dasatinib sobre los espermatozoides, por lo que tanto los hombres como las mujeres sexualmente activos deben utilizar métodos anticonceptivos eficaces durante el tratamiento.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas. Los pacientes deben ser informados de que pueden sufrir alguna reacción adversa como mareos o visión borrosa durante el tratamiento con dasatinib. Por lo tanto, se les debe recomendar que cuando conduzcan un coche o manejen máquinas lo hagan con precaución.
Resumen del perfil de seguridad
Los datos que se detallan a continuación corresponden a la exposición a SPRYCEL en 2.712 pacientes en ensayos clínicos incluyendo 324 pacientes con LMC en fase crónica de nuevo diagnóstico y 2.388 pacientes con LMC o LLA Ph+ con resistencia o intolerancia a imatinib. La mediana de duración del tratamiento en 2.712 pacientes tratados con SPRYCEL fue de 19,2 meses (rango 0- 93,2 meses).
En el ensayo Fase III en curso en pacientes con LMC en fase crónica de nuevo diagnóstico, con un mínimo de 5 años de seguimiento, la mediana de duración del tratamiento fue aproximadamente 60 meses para ambos, SPRYCEL (rango de 0,03-72,7 meses) e imatinib (rango de 0,3-74,6 meses. La mediana de la duración del tratamiento en 1.618 pacientes con LMC en fase crónica fue de 29 meses (rango de 0 -92,9 meses). En 1.094 pacientes con LMC en fase avanzada o LLA Ph+ , la mediana de la duración del tratamiento para pacientes fue de 6,2 meses (rango de 0-9,32 meses).
De los 2.712 pacientes tratados, el 18% tenían > 65 años, mientras que el 5% tenían >75 años.
La mayoría de los pacientes tratados con SPRYCEL experimentaron reacciones adversas en algún momento. En la población total de 2.712 pacientes tratados con SPRYCEL, 520 (19%) experimentaron reacciones adversas que condujeron a la suspensión del tratamiento. La mayoría de las reacciones fueron de grado leve a moderado.
En el ensayo Fase III, en pacientes con LMC en fase crónica de nuevo diagnóstico, el tratamiento se interrumpió por reacciones adversas en el 5% de los pacientes tratados con SPRYCEL y en el 4% de los pacientes tratados con imatinib tras un seguimiento mínimo de 12 meses. Después de un mínimo de 60 meses de seguimiento, las tasas acumuladas de suspensión fueron del 14 % y del 7 % respectivamente. Entre los 1.618 pacientes tratados con dasatinib con LMC en fase crónica, se notificaron reacciones adversas que condujeron a la suspensión del tratamiento en 329 (20,3%) pacientes y entre los 1.094 pacientes tratados con dasatinib con fase avanzada de la enfermedad, se notificaron reacciones adversas que condujeron a la suspensión del tratamiento en 191(17,5%) pacientes.
La mayoría de los pacientes intolerantes a imatinib con LMC en fase crónica fueron capaces de tolerar el tratamiento con SPRYCEL. En ensayos clínicos con 24 meses de seguimiento en LMC fase crónica, 10 de los 215 pacientes con intolerancia a imatinib presentaron la misma toxicidad no hematológica grado 3-4 con SPRYCEL que tuvieron con imatinib; 8 de los 10 pacientes se manejaron con reducción de dosis y fueron capaces de continuar el tratamiento con SPRYCEL.
Basadas en un seguimiento mínimo de 12 meses, las reacciones adversas notificadas con más frecuencia en pacientes tratados con SPRYCEL con LMC en fase crónica de nuevo diagnóstico fueron retención de líquidos (incluyendo derrame pleural) (19%), diarrea (17%), cefalea (12%), erupción cutánea (11%), dolor musculoesquelético (11%), náuseas (8%), fatiga (8%), mialgia (6%), vómitos (5%) e inflamación muscular (4%). Tras un seguimiento mínimo de 60 meses, las tasas acumuladas de erupción cutánea (14%), dolor músculo esquelético (14%), cefalea (13%), fatiga (11%) náuseas (10%), mialgias (7%), vómitos (5%), e inflamación muscular o espasmos (5%) se incrementaron < 3%. Las tasas acumuladas de retención de líquidos y diarrea fueron del 39% y del 22% respectivamente. Las reacciones adversas más frecuentemente notificadas en pacientes en tratamiento con SPRYCEL con resistencia o intolerancia al tratamiento previo con imatinib fueron retención de líquidos (incluyendo derrame pleural), diarrea, cefalea, náuseas, erupción cutánea, disnea, hemorragias, fatiga, dolor musculo esquelético, infección, vómitos, tos, dolor abdominal y fiebre. Neutropenia febril relacionada con el tratamiento se notificó en un 5% de los pacientes tratados con SPRYCEL con resistencia o intolerancia al tratamiento previo con imatinib.
En ensayos clínicos en pacientes con resistencia o intolerancia a tratamiento previo con imatinib, se recomendó que el tratamiento con imatinib fuese discontinuado al menos 7 días antes de empezar el tratamiento con SPRYCEL.
Las siguientes reacciones adversas, excluyendo anomalías de laboratorio, se notificaron en pacientes tratados con SPRYCEL, en ensayos clínicos y la experiencia post-comercialización (Tabla 2). Estas reacciones se presentan clasificadas por órganos y frecuencias. Se definen las frecuencias como: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); raras (> 1/10.000 a < 1/1.000); no conocida (no puede estimarse de los datos post-comercialización disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Infecciones e infestaciones | |
|
Muy frecuentes |
infección (incluyendo bacteriana, vírica, fúngica, no-específica) |
|
Frecuentes |
neumonía (incluyendo bacteriana, vírica, y fúngica), infecciones/inflamación del tracto respiratorio superior,infección por virus de herpes, enterocolitis, sépsis (incluyendo casos poco frecuentes con desenlaces mortales) |
|
Trastornos de la sangre y del sistema linfático | |
|
Muy frecuentes |
mielosupresión (incluyendo anemia, neutropenia, trombocitopenia) |
|
Frecuentes |
neutropenia febril |
|
Poco frecuentes |
linfadenoptía, linfopenia |
|
Raras |
aplasia pura de serie roja |
|
Trastornos del sistema inmunológico | |
|
Poco frecuentes |
hipersensibilidad (incluyendo eritema nodoso) |
|
Trastornos endocrinos | |
|
Poco frecuentes |
hipotiroidismo |
|
Raras |
hipertiroidismo, tiroiditis |
|
Trastornos del metabolismo y de la nutrición | |
|
Frecuentes |
alteraciones del apetito3, hiperuricemia |
|
Poco frecuentes |
síndrome de lisis tumoral, deshidratación, hipoalbuminemia, hipercolesterolemia |
|
Raras |
diabetes mellitus |
|
Trastornos psiquiátricos | |
|
Frecuentes |
depresión, insomnio |
|
Poco frecuentes |
ansiedad, estado confusional, alteración de la carga emocional, disminución de la líbido |
|
Trastornos del sistema nervioso | |
|
Muy frecuentes |
cefalea |
|
Frecuentes |
neuropatía (incluyendo neuropatía periférica), mareos, disgeusia, somnolencia |
|
Poco frecuentes |
sangrado del SNC*b, síncope, temblor, amnesia, alteraciones del equilibrio |
|
Raras |
accidente cerebrovascular, accidente isquémico transitorio, convulsiones, neuritis óptica, parálisis del VII par, demencia, ataxia |
|
Trastornos oculares | |
|
Frecuentes |
alteraciones visuales (incluyendo visión distorsionada, visión borrosa, y agudeza visual reducida), ojo seco |
|
Poco frecuentes |
insuficiencia visual, conjuntivitis, fotofobia, incremento del lagrimeo |
|
Trastornos del oído y el laberinto | |
|
Frecuentes |
tinnitus |
|
Poco frecuentes |
pérdida de audición, vértigo |
|
Trastornos Can |
íacos |
|
Frecuentes |
insuficiencia cardiaca congestiva/insuficiencia cardíaca*3, derrame pericárdico*, arritmia (incluyendo taquicardia), palpitaciones |
|
Poco frecuentes |
infarto de miocardio (incluyendo desenlace mortal)*, electrocardiograma con intervalo QT prolongado*, pericarditis, arritmia ventricular (incluyendo taquicardia ventricular), angina de pecho, cardiomegalia, electrocardiograma con onda T anormal, incremento de la troponina |
|
Raras |
cor pulmonale, miocarditis, síndrome coronario agudo, parada cardiaca electrocardiograma con prolongación del intervalo PR , enfermedad arterial coronaria, pleuropericarditis |
|
No conocida |
fibrilación auricular/flutter auricular |
|
Trastornos Vasculares | |
|
Muy frecuentes |
hemorragia*d |
|
Frecuentes |
hipertensión, rubor |
|
Poco frecuentes |
hipotensión, tromboflebitis |
|
Raras |
trombosis venosa profunda, embolismo, livedo reticularis |
|
Trastornos respiratorios , torácicos y mediastínicos | |
|
Muy frecuentes |
derrame pleural*, disnea |
|
Frecuentes |
edema pulmonar *, hipertensión pulmonar*, infiltración pulmonar, neumonitis, tos |
|
Poco frecuentes |
hipertensión arterial pulmonar, broncoespasmo, asma |
|
Raras |
embolismo pulmonar, síndrome de distrés respiratorio agudo |
|
No conocida |
enfermedad intersticial pulmonar |
|
Trastornos gastrointestinales | |
|
Muy frecuentes |
diarrea, vómitos, nausea, dolor abdominal |
|
Frecuentes |
hemorragia gastrointestinal*, colitis (incluyendo colitis neutropénica), gastritis, inflamación de la mucosa (incluyendo mucositis/estomatitis), dispepsia, distensión abdominal, estreñimiento, alteraciones de la mucosa oral |
|
Poco frecuentes |
pancreatitis (incluyendo pancreatitis aguda),úlcera del tracto gastrointestinal superior, esofagitis, ascitis*, fisura anal, disfagia, enfermedad por reflujo gastroesofágico |
|
Raras |
gastroenteropatía con pérdida de proteínas, íleo, fístula anal |
|
No conocida |
hemorragia gastrointestinal mortal* |
|
Trastornos hepatobiliares | |
|
Poco frecuentes |
hepatitis, colecistitis, colestasis |
|
Trastornos de la piel y del tejido subcutáneo | |
|
Muy frecuentes |
erupción cutáneae |
|
Frecuentes |
alopecia, dermatitis (incluyendo eczema), prurito, acné, sequedad de la piel, urticaria, hiperhidrosis |
|
Poco frecuentes |
dermatosis neutrofílica, fotosensibilidad, alteraciones de la pigmentación, paniculitis, úlcera cutánea, ampollas cutáneas, alteraciones en las uñas, síndrome de eritrodisestesia palmo-plantar, alteraciones del cabello |
|
Raras |
vasculitis leucocitoclástica, fibrosis cutánea |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |
|
Muy frecuentes |
dolor musculo esquelético |
|
Frecuentes |
artralgia, mialgia, debilidad muscular, rigidez musculo esquelética, espasmo muscular |
|
Poco frecuentes |
rabdomiolisis, osteonecrosis, inflamación muscular, tendinitis, artritis |
|
Trastornos rena |
es y urinarios |
|
Poco frecuentes |
insuficiencia renal (incluyendo fallo renal), frecuencia urinaria, proteinuria |
|
Embarazo, puerperio y alteraciones perinatales | |
|
Raras |
aborto |
|
Trastornos del aparto reproductor y de la mama | |
|
Poco frecuentes |
ginecomastia, alteraciones de la menstruación |
|
Trastornos generales y alteraciones en el lugar de administración | |
|
Muy frecuentes |
edema periférico f, fatiga, pirexia, edema facial8 |
|
Frecuentes |
astenia, dolor, dolor torácico, edema generalizado*h, escalofríos |
|
Poco frecuentes |
malestar, otros edemas superficiales1 |
|
Raras |
alteraciones de la marcha |
|
Exploraciones complementarias | |
|
Frecuentes |
pérdida de peso, aumento de peso |
|
Poco frecuentes |
aumento de la creatinfosfoquinasa en sangre aumento de la gamma-glutamil transferasa |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos | |
|
Frecuentes |
contusion |
d Excluye sangrado gastrointestinal, y sangrado del sistema nervioso central (SNC); estas reacciones adversas se han informado según la clasificación por órganos y sistemas dentro de trastornos gastrointestinales y dentro de trastornos del sistema nervioso respectivamente.
e Incluye erupción medicamentosa, eritema, eritema multiforme, eritrosis, erupción cutánea exfoliativa, eritema generalizado, erupción genital, miliaria, milio, psoriasis pustular miliaria , erupción, erupción eritematosa, erupción folicular, erupción generalizada, erupción macular, erupción maculopapulosa, erupción papulosa, erupción pruriginosa, erupción pustular, erupción vesicular, exfoliación cutánea, irritación de la piel erupción cutánea tóxica , urticaria vesiculosa y erupción vasculítica.
f edema gestacional, edema localizado, edema periférico.
g edema orbitario, edema peneano, edema periorbitario, edema con fóvea, edema escrotal, tumefacción cutánea, tumefacción facial y edema lingual.
h edema conjuntival, edema ocular, tumefacción ocular, edema palpebral, edema facial, edema labial, edema macular, edema bucal, edema orbital, edema periorbital, tumefacción facial.
i sobrecarga líquida, retención de líquidos, edema gastrointestinal, edema generalizado, edema, edema debido a enfermedad cardíaca, derrame perinéfrico, edema posterior a un procedimiento, edema visceral.
j tumefacción genital, edema en el lugar de la incisión, edema genital, edema peneano, tumefacción peneana, edema escrotal, tumefacción cutánea, tumefacción testicular, tumefacción vulvovginal.
*
para detalles adicionales, ver sección "Descripción de reacciones adversas seleccionadas"
Descripción de reacciones adversas seleccionadas Mielosupresión
El tratamiento con SPRYCEL se asocia con anemia, neutropenia y trombocitopenia. Su incidencia es más temprana y más frecuente en pacientes con LMC en fases avanzadas o LLA Ph+ que en pacientes con LMC en fase crónica (ver sección 4.4).
Sangrado
Se han notificado hemorragias relacionadas con el tratamiento en pacientes que tomaban SPRY CEL, desde petequias y epistaxis a hemorragia digestiva y del SNC grado 3 ó 4 (ver sección 4.4).
Retención de líquidos
Reacciones adversas variadas como derrame pleural, ascitis, edema pulmonar y derrame pericárdico con o sin edema superficial pueden describirse colectivamente como “retención de líquidos”. En un ensayo con pacientes con LMC en fase crónica de nuevo diagnóstico tras un seguimiento mínimo de 60 meses, la retención de líquidos relacionada con dasatinib incluyó derrame pleural (28%), edema superficial (14%), hipertensión pulmonar (5%), edema generalizado (4%) y derrame pericárdico (4%). Fallo cardíaco congestivo/insuficiencia cardíaca y edema pulmonar se notificaron en < 2% de pacientes.
La tasa acumulativa de derrame pleural relacionado con dasatinib (todos los grados) a lo largo del tiempo fue del 10% a 12 meses, 14% a 24 meses, 19% a 36 meses, 24% a 48 meses y 28% a 60 meses. Un total de 46 pacientes tratados con dasatinib tuvieron derrame pleural recurrente. Diecisiete pacientes tuvieron 2 eventos separados, 6 tuvieron 3 eventos, 18 tuvieron 4 a 8 y 5 tuvieron >8 episodios de derrame pleural. La mediana del tiempo del primer derrame pleural relacionado con dasatinib, grado 1 o 2 fue de 114 semanas (rango: 4 a 299 semanas). Menos del 10% de los pacientes tuvieron derrame pleural grave (grado 3 o 4) relacionado con dasatinib. La mediana del tiempo hasta la primera incidencia de derrame pleural de grado > 3 relacionado con dasatinib fue de 117 semanas (rango: 114 a 274 semanas). La mediana de duración del derrame pleural relacionado con dasatinib (todos los grados) fue 283 días (~ 40 semanas).
El derrame pleural fue generalmente reversible y se manejó suspendiendo el tratamiento con SPRYCEL y utilizando diuréticos u otras medidas de soporte adecuadas (ver secciones 4.2 y 4.4).
Entre los pacientes tratados con dasatinib con derrame pleural relacionado con él, (n=73), 45 (62%) tuvieron interrupciónes de la dosis y 30 (41%) tuvieron reducciones de dosis. Adicionalmente,
34(47%) recibieron diuréticos , 23 (32%) recibieron corticosteroides y 20(27%) recibió ambos
corticosteroides y diuréticos. A nueve (12%) de pacientes se les realizó una toracocentesis terapéutica.
Seis por ciento de pacientes tratados con dasatinib interrumpieron el tratamiento debido a derrame pleural relacionado. El derrame pleural no impidió a los pacientes obtener una respuesta. Entre los pacientes tratados con dasatinib con derrame pleural, un 96% alcanzó una RCyC , un 82% alcanzó RMM y un 50% alcanzó una RM4,5 a pesar de las interrupciones y ajustes de dosis.
Ver la sección 4.4 para más información sobre pacientes con LMC en fase crónica y LMC en fase avanzada o LLA Ph+.
Hipertensión Pulmonar Arterial (HTPA)
Se han notificado casos de hipertensión pulmonar arterial precapilar (HTPA) confirmada por cateterismo derecho asociados al tratamiento con dasatinib. En estos casos, la HTPA se notificó después del inicio del tratamiento con dasatinib, incluyendo en pacientes en tratamientos de más de un año de duración. En estas notificaciones, los pacientes con HTPA estaban tomando con frecuencia medicamentos concomitantes o tenían otras co-morbilidades además de la patología maligna de base. En algunos pacientes con HTPA se ha observado mejoría en los parámetros clínicos y hemodinámicos tras suspender el tratamiento con dasatinib.
Prolongación QT
En el ensayo Fase III en pacientes con LMC en fase crónica de nuevo diagnóstico, un paciente (< 1%) de los pacientes tratados con SPRYCEL y un paciente (< 1%) de los pacientes tratados con imatinib tuvieron un QTcF > 500 mseg tras un seguimiento mínimo de 12 meses. (ver sección 4.4). No se notificó un QTcF > 500 mseg en ningún paciente adicional tras un seguimiento mínimo de 60 meses.
En 5 estudios Fase II, en pacientes con resistencia o intolerancia a tratamiento previo con imatinib, se obtuvieron repetidamente ECGs en condiciones basales y a tiempos pre-especificados durante el tratamiento de 865 pacientes que recibieron 70 mg de SPRYCEL dos veces al día. Estos ECGs fueron valorados de forma centralizada. El intervalo QT se corrigió por la frecuencia cardíaca mediante el método Fridericia. Para todos los valores post-administración recogidos durante el día 8, el cambio medio respecto a los valores basales del intervalo QTcF estuvo entre 4-6 mseg, con un límite superior en el intervalo de confianza del 95% < 7 mseg. De los 2.182 pacientes que recibieron SPRYCEL en los ensayos clínicos, en 15 (1%) pacientes se notificó como reacción adversa la prolongación QTc informado como una reacción adversa. Veintiún pacientes (1%) presentaron un QTcF > 500 mseg (ver sección 4.4).
Reacciones adversas cardíacas
Los pacientes con factores de riesgo o con historial de enfermedad cardíaca deben ser monitorizados cuidadosamente para los signos y síntomas consistentes con insuficiencia cardíaca y deberán ser evaluados y tratados adecuadamente (ver sección 4.4).
En el ensayo Fase III de optimización de la dosis en pacientes con LMC en fase crónica con resistencia o intolerancia a tratamiento previo con imatinib (mediana de la duración del tratamiento de 30 meses), la incidencia de derrame pleural e insuficiencia cardiaca congestiva/insuficiencia cardíaca, fue menor en pacientes tratados con 100 mg de SPRYCEL una vez al día que en aquellos tratados con 70 mg de SPRYCEL dos veces al día. También se notificó mielosupresión con menor frecuencia en el grupo de tratamiento que recibió la apauta posológica de 100 mg una vez al día (ver anormalidades en Pruebas de laboratorio a continuación). La mediana de duración del tratamiento en el grupo de 100 mg una vez al día fue de 37 meses (rango: 1-91 meses). Las tasas acumuladas de reacciones adversas seleccionadas que se notificaron en el grupo de tratamiento con la pauta posológica de 100 mg una vez al día como dosis de inicio recomendada se muestran en la Tabla 3a.
Tabla 3a: Reacciones adversas seleccionadas notificadas en el ensayo clínico fase 3 de optimización
|
de dosis (LMC en Fase Crónica resistente o intolerante a Imatinib |
va | |||||
|
Mínimo 2 años de |
Mínimo 5 años de |
Mínimo 7 años de | ||||
|
Seguimiento |
Seguimiento |
Seguimiento | ||||
|
Todos los |
Grado 3/4 |
Todos los _ Grado 3/4 |
Tpodos los |
Grado | ||
|
grados |
grados |
grados |
3/4 | |||
|
Reacción adversa |
Porcentaje |
(%) de Pacientes | ||||
|
Diarrea |
27 |
2 |
28 |
2 |
28 |
2 |
|
Retención de líquidos |
34 |
4 |
42 |
6 |
48 |
7 |
|
Edema superficial |
18 |
0 |
21 |
0 |
22 |
0 |
|
Derrame pleural |
18 |
2 |
24 |
4 |
28 |
5 |
|
Edema generalizado |
3 |
0 |
4 |
0 |
4 |
0 |
|
Derrame pericárdico |
2 |
1 |
2 |
1 |
3 |
1 |
|
Hipertensión |
0 |
0 |
0 |
0 |
2 |
1 |
|
pulmonar | ||||||
|
Hemorragia |
11 |
1 |
11 |
1 |
12 |
1 |
|
Sangrado |
2 |
1 |
2 |
1 |
2 |
1 |
|
gastrointestinal | ||||||
a Resultados notificados en el ensayo Fase 3 de optimización de dosis, en la población (n=165) con la dosis inicial recomendada de 100 mg una vez al día.
En el ensayo Fase III de optimización de dosis en pacientes con LMC en fases avanzadas y LLA Ph+, la mediana de la duración del tratamiento fue de 14 meses para LMC en fase acelerada, 3 meses para LMC en crisis mieloide blástica, 4 meses para LMC en crisis linfoide blástica y 3 meses para LLA Ph+. Las reacciones adversas seleccionadas que se notificaron con la dosis inicial recomendada de 140 mg una vez al día se muestran en la Tabla 3b. La pauta posológica de 70 mg dos veces al día también se estudió. La pauta posológica de 140 mg una vez al día mostró un perfil de eficacia comparable a la pauta posológica de 70 mg dos veces al día pero un perfil de seguridad más favorable.
Tabla 3b: Reacciones adversas seleccionadas notificadas en el ensayo clínico fase III de
optimización de dosis : Fase avanzada de LMC y LLA Ph+a
|
140 mg una vez al día n = 304 | ||
|
Todos los grados |
Grado 3/4 | |
|
Reacción adversa |
Porcentaje (%) de pacientes | |
|
Diarrea |
28 |
3 |
|
Retención de líquidos |
33 |
7 |
|
Edema superficial |
15 |
< 1 |
|
Derrame pleural |
20 |
6 |
|
Edema generalizado |
2 |
0 |
|
Insuficiencia cardíaca |
1 |
0 |
|
congestiva/ insuficiencia | ||
|
cardíacab | ||
|
Derrame pericárdico |
2 |
1 |
|
Edema pulmonar |
1 |
1 |
|
Hemorragia |
23 |
8 |
|
Sangrado gastrointestinal |
8 |
6 |
a Resultados notificados del ensayo Fase 3 de optimización de dosis con la dosis inicial recomendada de 140 mg una vez al día en la población (n=304) al final de los 2 años de seguimiento del ensayo.
b Incluye insuficiencia ventricular, insuficiencia cardíaca, insuficiencia cardíaca congestiva, cardiomiopatía, cardiomiopatía congestiva, disfunción diastólica, descenso de la fracción de eyección y fracaso ventricular
Anomalías en las Pruebas de laboratorio
Hematología
En el ensayo Fase III en pacientes con LCM en fase crónica de nuevo diagnóstico, se notificaron las siguientes anomalías en las pruebas de laboratorio grado 3 ó 4 tras un seguimiento mínimo de 12 meses en pacientes que estaban tomando_SPRYCEL: neutropenia (21%), trombocitopenia (19%), y anemia (10%). Después de un mínimo de 60 meses de seguimiento, las tasas acumuladas de neutropenia, trombocitopenia y anemia fueron del 29%, 22% y 13%, respectivamente.
En pacientes tratados con SPRYCEL con LMC en fase crónica de nuevo diagnóstico que experimentaron mielosupresión grado 3 ó 4, generalmente se produjo recuperación después de una breve suspensión de la dosis y/o reducción, y la suspensión permanente del tratamiento se produjo en el 1,6% de los pacientes tras un seguimiento mínimo de 12 meses. Después de un mínimo de 60 meses de seguimiento, la tasa acumulada de suspensión permanente del tratamiento debido a mielosupresión grado 3 ó 4 fue del 2,3%.
En pacientes con LMC, con resistencia o intolerancia al tratamiento previo con imatinib, las citopenias (trombocitopenia, neutropenia y anemia) fueron un hallazgo consistente. Sin embargo, su aparición fue claramente dependiente del estadio de la enfermedad. La frecuencia de anormalidades hematológicas grados 3 y 4 se presentan en la Tabla 4.
Tabla 4: Alteraciones hematológicas de laboratorio grados 3 y 4 en ensayos
clínicos según los criterios comunes de toxicidad (CTC) en pacientes _con resistencia o intolerancia a tratamiento previo con imatiniba_
|
Crisis Blástica | ||
|
Crisis Blástica |
Linfoide y | |
|
Fase Crónica |
Fase Acelerada Mieloide |
Ph+ ALL |
|
(n= 165)b |
(n= 157)c (n= 74)c |
(n= 168)c |
|
Percent (%) of Patients | ||
|
Parámetros hematológicos | ||||
|
Neutropenia |
36 |
58 |
77 |
76 |
|
Trombocitopenia |
23 |
63 |
78 |
74 |
|
Anemia |
13 |
47 |
74 |
44 |
a Resultados del ensayo Fase 3 de optimización de dosis notificados al final de los 2 años de seguimiento del ensayo. . b CA180-034 resultados del ensayo con la dosis inicial recomendada de 100 mg una vez al día. c CA180-035 resultados del ensayo con la dosis inicial recomendada de 140 mg una vez al día.
CTC grados: neutropenia (Grado 3 > 0.5- < 1.0 x 109/l, Grado 4 < 0.5 x 109/l); trombocitopenia (Grade 3 > 25 - < 50 x 109/l, Grade 4 < 25 x 109/l); anemia (hemoglobina Grado 3 > 65 - < 80 g/l, Grade 4 < 65 g/l).
Las citopenias grado 3 ó 4 acumuladas en pacientes tratados con 100 mg una vez al día fueron similares a los 2 y 5 años, incluyendo: neutropenia (35% vs 36%), trombocitopenia (23% vs 24%) y anemia (13% vs 13%).
En los pacientes que desarrollaron una mielosupresión grado 3 ó 4, la recuperación se lograba habitualmente después de interrupciones del tratamiento breves y/o reducciones en la dosis. El tratamiento se interrumpió de forma permanente en el 5% de los pacientes. La mayoría de los pacientes continuó con el tratamiento sin nuevas evidencias de mielosupresión.
Bioquímica
En un ensayo en LMC en fase crónica de nuevo diagnóstico, se notificó hipofosfatemia grado 3 ó 4 en un 4% de los pacientes tratados con SPRYCEL, y una elevación de las transaminasas grado 3 ó 4, elevación de creatinina y de bilirrubina se notificaron en < 1% de los pacientes tras un seguimiento mínimo de 12 meses. Después de un mínimo de 60 meses de seguimiento, la tasa acumulada de hipofosfatemia grado 3 ó 4 fue del 7%, elevaciones de grado 3 ó 4 de creatinina y bilirrubina fue del 1% y elevaciones de las transaminasas grado 3 ó 4 permanecieron en el 1%. No hubo suspensión en el tratamiento con SPRYCEL debido a estos parámetros bioquímicos de laboratorio. 2
Se comunicaron elevaciones de transaminasas o bilirrubina grado 3 ó 4 en < 1% de los pacientes con LMC en fase crónica, (resistente o intolerante a imatinib), pero se notificaron elevaciones con frecuencias más elevadas del 1 al 7% de los pacientes con fases avanzadas de LMC y LLA Ph+. Habitualmente se controlaron mediante reducción de la dosis o suspensión del tratamiento. En el ensayo Fase III de optimización de la dosis, estudio en fase crónica de LMC, elevaciones de transaminasas o bilirrubina grado 3 ó 4 fueron notificadas en < 1% de los pacientes con incidencia baja similar en los cuatro grupos tratados. En el ensayo Fase III de optimización de dosis en fase avanzada de LMC y LLA Ph+ elevaciones de transaminasas o bilirrubina grado 3 ó 4 se notificaron en el 1% al 5% de los pacientes de los grupos tratados.
Aproximadamente un 5% de los pacientes tratados con SPRYCEL, que tenían niveles basales normales de calcio, experimentaron hipocalcemia transitoria grado 3 ó 4 en algún momento del ensayo. En general, no se asoció la disminución de calcio con síntomas clínicos. Los pacientes que desarrollaron hipocalcemia grado 3 ó 4 con frecuencia se recuperaban con la administración de suplementos orales. En pacientes con todas las fases de LMC se comunicaron casos de hipofosfatemia, hipocalcemia e hipopotasemia grado 3 ó 4 pero se detectó un incremento de la frecuencia en los pacientes con LMC en crisis blástica mieloide o crisis blástica linfoide y LLA Ph+. Aumentos de la creatinina grado 3 ó 4 se notificaron en < 1% de los pacientes con LMC en fase crónica con un aumento de la frecuencia del 1 al 4% en los pacientes con LMC en fase avanzada.
Otras poblaciones especiales
Mientras el perfil de seguridad de SPRYCEL en la población de edad avanzada fue similar al de la población joven, los pacientes con 65 años de edad y mayores tienen mayor probabilidad de experimentar las reacciones adversas comúnmente notificadas como fatiga, derrame pleural, disnea, tos, hemorragia en el tracto gastrointestinal inferior y alteraciones del apetito y mayor probabilidad de experimentar reacciones adversas menos frecuentemente notificada como distensión abdominal, mareos, derrame pericárdico, fallo cardíaco congestivo y disminución de peso, por lo que deben monitorizarse cuidadosamente (ver sección 4.4).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
La experiencia referente a la sobredosis de SPRYCEL en los ensayos clínicos está limitada a casos aislados. La sobredosis más alta de 280 mg por día durante una semana se notificó en dos pacientes y ambos desarrollaron un descenso significativo en el recuento de plaquetas. Considerando que dasatinib se asocia con mielosupresión grado 3 ó 4 (ver sección 4.4), los pacientes que ingieran una dosis mayor de la recomendada deben ser monitorizados cuidadosamente por la aparición de mielosupresión y ser tratados con la terapia de soporte adecuada.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: agente antineoplásico, inhibidores directos de la proteín quinasa, código ATC: L01XE06.
Dasatinib inhibe la actividad de la quinasa BCR-ABL y de las quinasas de la familia SRC junto con otras quinasas oncogénicas específicas incluyendo c-KIT, los receptores quinasa de las efrinas (EPH) y el receptor del PDGF-p. Dasatinib es un inhibidor potente, a concentraciones subnanomolares (0,6-0,8 nM), de la quinasa BCR-ABL. Se une no sólo a la conformación inactiva de la enzima BCR-ABL, sino también a la activa.
In vitro, dasatinib es activo en líneas celulares representativas de variantes de leucemia sensibles y resistentes a imatinib. Los estudios preclínicos demuestran que dasatinib puede superar la resistencia a imatinib resultante de la sobreexpresión de BCR-ABL, mutaciones del dominio de BCR-ABL quinasa, activación de las vías de señalización alternativas que afectan a las quinasas de la familia de SRC (LYN, HCK) y la sobreexpresión del gen (mdr) de resistencia múltiple. Además, dasatinib inhibe las quinasas de la familia SRC a concentraciones subnanomolares.
In vivo, en experimentos independientes usando modelos murinos de LMC, dasatinib previno la progresión de la LMC crónica a fase blástica y prolongó la supervivencia de los ratones implantados con líneas celulares de LMC obtenidas de pacientes, en diversas localizaciones, incluido el sistema nervioso central.
Eficacia clínica y seguridad En el ensayo clínico Fase I, se observaron respuestas hematológicas y citogenéticas en todas las fases de LMC y en LLA Ph+ en los primeros 84 pacientes tratados y seguidos hasta 27 meses. Las respuestas fueron duraderas en todas las fases de la LMC y en la LLA Ph+.
Se han realizado cuatro ensayos clínicos Fase II con un único brazo de estudio, no controlados y abiertos para determinar la seguridad y la eficacia de dasatinib en pacientes con LMC en fase crónica, acelerada o blástica mieloide, que eran resistentes o intolerantes a imatinib. Un ensayo aleatorizado no comparativo se realizó con pacientes que se encontraban en la fase crónica y habían fracasado a un tratamiento inicial con 400 ó 600 mg de imatinib. La dosis inicial de dasatinib fue de 70 mg dos veces al día. Se permitieron modificaciones de la dosis para mejorar la actividad o para el manejo de la toxicidad (ver sección 4.2).
Dos ensayos aleatorizados, abiertos Fase III, se realizaron para evaluar la eficacia de dasatinib administrado una vez al día comparado con dasatinib administrado dos veces al día. Además se realizó un ensayo de Fase III, comparativo abierto, aleatorizado en pacientes adultos con LMC en fase crónica de nuevo diagnóstico. La eficacia de dasatinib se basa en las tasas de respuesta hematológica y citogenética.
La duración de la respuesta y las tasas estimadas de supervivencia aportan una evidencia adicional del beneficio clínico de dasatinib.
Un total de 2.712 pacientes fueron evaluados en los ensayos clínicos: de estos, un 23% fueron > 65 años de edad y un 5% fueron > 75 años de edad.
LMC en Fase Crónica de nuevo diagnóstico
Se realizó un ensayo clínico internacional, abierto, multicéntrico, aleatorizado y comparativo de Fase III, en pacientes adultos con LMC en fase crónica de nuevo diagnóstico. Los pacientes fueron aleatorizados para recibir tratamiento con 100 mg de SPRYCEL, una vez al día o 400 mg de imatinib una vez al día. El objetivo primario fue la tasa de Respuesta Citogenética Completa confirmada (RCyCc) a 12 meses. Objetivos secundarios incluyeron tiempo en RCyCc (medida de la durabilidad de la respuesta), tiempo hasta RCyCc, tasa de Respuesta Molecular Mayor (RMM), tiempo hasta la RMM, supervivencia libre de progresión (PFS) y supervivencia global (OS). Otros resultados relevantes de eficacia incluyeron RCyC y tasas de Respuesta Molecular Completa (RMC). Este estudio continúa.
Un total de 519 pacientes fueron aleatorizados para un grupo de tratamiento: 259 al grupo de SPRYCEL y 260 al grupo de imatinib. Las características basales estuvieron bien equilibradas entre los dos grupos de tratamiento con respecto a la edad (mediana de edad de 46 años para el grupo de SPRYCEL y de 49 años para el grupo de imatinib con un 10% y un 11% de pacientes de 65 años de edad o mayores, respectivamente), género (un 44% de mujeres y un 37%, respectivamente) y raza (caucasiana 51% y 55%; asiática 42% y 37%, respectivamente). En el estado basal, la distribución del Índice de Hasford fue similar en los grupos de tratamiento con SPRY CEL y en el de imatinib (riesgo bajo: 33% y 34%; riesgo intermedio 48% y 47%; riesgo alto 19% y 19%, respectivamente).
Con un mínimo de 12 meses de seguimiento, un 85% de pacientes aleatorizados al grupo de SPRYCEL y un 81% de pacientes aleatorizados al grupo de imatinib estaban todavía recibiendo tratamiento de primera línea. La suspensión dentro de los 12 meses, debido a progresión de la enfermedad se produjo en un 3% de los pacientes tratados con SPRYCEL y un 5% de los pacientes tratados con imatinib.
Con un mínimo de 60 meses de seguimiento, un 60% de pacientes aleatorizados del grupo de SPRYCEL y un 63% de pacientes aleatorizados del grupo de Imatinib todavía estaban recibiendo tratamiento de primera línea. La suspensión dentro de los 60 meses debido a progresión de la enfermedad se produjo en un 11% de los pacientes tratados con SPRYCEL y un 14% de los pacientes tratados con imatinib.
Los resultados de eficacia se presentan en la Tabla 5. Una estadísticamente significativa mayor proporción de pacientes en el grupo de SPRYCEL alcanzó una RCyCc comparada con los pacientes del grupo de imatinib dentro de los primeros 12 meses de tratamiento. La eficacia de SPRYCEL se consideró demostrada a través de los diferentes subgrupos, incluyendo, edad, género, e Índice de Hasford basal.
Tabla 5: Resultados de eficacia de un ensayo fase 3 en pacientes con LMC
en fase crónica de nuevo diagnostico .
|
SPRYCEL n= 259 |
imatinib n= 260 |
p-value | |
|
Response rate (95% CI) | |||
|
Respuesta citogenética | |||
|
dentro de 12 meses | |||
|
RCyCca |
76,8% (71,2-81,8) |
66,2% (60,1-71,9) |
p< 0.007* |
|
RCyCb |
85,3% (80,4-89,4) |
73,5% (67,7-78,7) |
— |
|
dentro de 24 meses | |||
|
RCyCca |
80,3% |
74,2% |
— |
|
RCyCb |
87,3% |
82,3% |
— |
|
dentro de 36 meses | |||
|
RCyCca |
82,6% |
77,3% |
— |
|
RCyCb |
88,0% |
83,5% |
— |
|
dentro de 48 meses | |||
|
RCyCca |
82,6% |
78,5% |
— |
|
RCyCb |
87,6% |
83,8% |
— |
|
dentro de 60 meses | |||
|
RCyCca |
83,0% |
78,5% |
— |
|
RCyCb |
88,0% |
83,8% |
— |
|
Respuesta c Molecular Mayor | |||
|
12 meses |
52,1% (45,9-58,3) |
33,8% (28,1-39,9) |
p< 0.00003* |
|
24 meses |
64,5% (58,3-70,3) |
50% (43,8-56,2) |
— |
|
36 meses |
69,1% (63,1-74,7) |
56,2% (49,9-62,3) |
— |
|
48 meses |
75,7% (70,0-80,8) |
62,7% (56,5-68,6) |
— |
|
60 meses |
76,4% (70,8-81,5) |
64,2% (58,1-70,1) |
p=0,0021 |
|
Hazard Ratio (HR) | |||
|
dentro de 12 meses (99.99% CI) | |||
|
Tiempo hasta RCyCc |
1,55 (1,0-2,3) |
p< 0,0001* | |
|
Tiempo hasta MMR |
2,01 (1,2-3,4) |
p< 0,0001* | |
|
Durabilidad de RCyCc |
0,7 (0,4-1,4) |
p< 0,035 | |
|
dentro 24 meses (95% CI) | |||
|
Tiempo hasta RCyCc |
1,49 (1,22-1,82) |
— | |
|
Tiempo hasta MMR |
1,69 (1,34-2,12) |
— | |
|
Durabilidad de RCyCc |
0,77 (0,55-1,10) |
— | |
|
Tiempo hasta RCyCc |
1,48 (1,22-1,80) |
— |
|
Tiempo hasta MMR |
1,59 (1,28-1,99) |
— |
|
Durabilidad de RCyCc |
0,77 (0,53-1,11) |
— |
|
dentro de 48 meses (95% CI) | ||
|
Tiempo hasta RCyCc |
1,45 (1,20-1,77) |
— |
|
Tiempo hasta MMR |
1,55 (1,26-1,91) |
— |
|
Durabilidad de RCyCc |
0,81 (0,56-1,17) |
— |
|
dentro de 60 meses (95% CI) | ||
|
Tiempo hasta RCyCc |
1,46 (1,20-1,77) |
p=0,0001 |
|
Tiempo hasta MMR |
1,54 (1,25-1,89) |
p<0,0001 |
|
Durabilidad de RCyCc |
0,79 (0,55-1,13) |
p=0.1983 |
a Respuesta citogenética completa confirmada (RCyCc) se define como respuesta obtenida en dos ocasiones consecutivas (como mínimo, separadas por 28 días).
b Respuesta citogenética completa (RCyC) se basa en una única evaluación citogenética de la médula ósea. c Respuesta Molecular Mayor (en cualquier momento) se definió como tasas de BCR-ABL < 0.1% por RQ-PCR en muestras de sangre periférica estandarizadas en una escala internacional. Estas tasas son acumulativas representando el seguimiento mínimo para el espacio de tiempo especificado.
*Ajustado por Índice de Hasford y significación estadística indicada a un nivel nominal pre-definido de significación.
IC = intervalo de confianza
Después de 60 meses de seguimiento, la mediana de tiempo hasta RCyC fue de 3,1 meses en el grupo de SPRYCEL, y 5,8 meses en el grupo de imatinib en pacientes con una RCyC confirmada. La mediana de tiempo hasta RMM después de 60 meses de seguimiento fue de 9,3 meses en el grupo de SPRYCEL, y 15,0 meses en el grupo de imatinib en pacientes con RMM. Estos resultados son consistentes con los observados a 12, 24 y 36 meses.
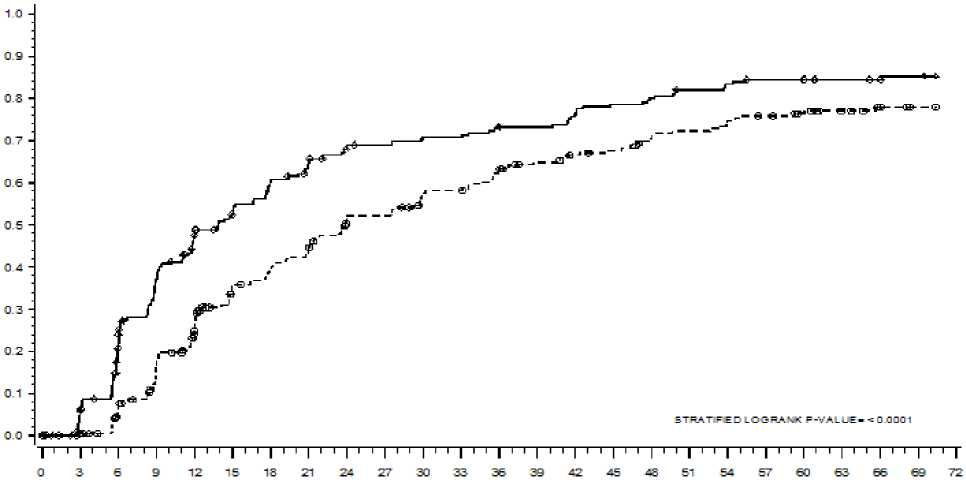
El tiempo hasta RMM se muestra gráficamente en la Figura 1. El tiempo hasta RMM fue consistentemente más corto en los pacientes tratados con dasatinib comparado con los pacientes tratados con imatinib.
Figura 1: Estimados de Kaplan-Meier del tiempo hasta respuesta molecular mayor (RMM)
PROPORCIÓN DE RESPONDEDORES
------Imatinib
: : Censurados
MESES
_Dasatinib
' ■' Censurados
GRUPO_# RESPONDEDORES / # ALEATORIZADOS HAZARD RATIO (95% IC)
DASATINIB 198/259
IMATINIB 167/260
DASATINIB SOBRE IMATINIB 1,54 (1,25 - 1,89)
Las tasas de RCyCc en los grupos de tratamiento con SPRY CEL e imatinib, respectivamente, en 3 meses de tratamiento (54% y 30%), 6 meses (70% y 56%), 9 meses (75% y 63%),
24 meses (80% y 74%) , 36 meses (83% y 77%) y 48 meses (83% y 79%) fueron consistentes con el objetivo primario. Las tasas de RMM en los grupos de tratamiento con SPRYCEL e imatinib, respectivamente en 3 meses de tratamiento (8% y 0,4%), 6 meses (27% y 8%), 9 meses (39% y 18%), 12 meses (46% y 28%), 24 meses (64% y 46%) ,
36 meses (67% y 55%), 48 meses (73% y 60%) y 60 meses (76% y 64%) también fueron consistentes con el objetivo primario.
Las tasas de RMM en punto específico de tiempo se representan gráficamente en la Figura 2. Las tasas de RMM fueron consistentemente más altas en los pacientes tratados con dasatinib comparados con los pacientes tratados con imatinib.
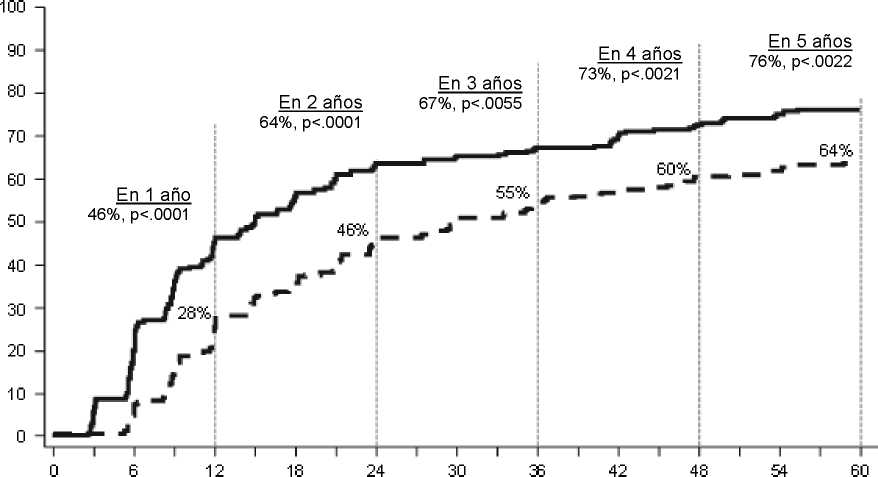
Meses desde Aleatorización
N
Dasatinib 100 mg una vez al día 259 Imatinib 400 mg una vez al día 260
La proporción de pacientes que alcanzaron un ratio BCR-ABL, < 0,01% (reducción de log 4) en cualquier momento fue más alta en el grupo de SPRYCEL, comparado con el grupo de imatinib (76% frente a 64 %). La proporción de pacientes que alcanzaron un ratio BCR-ABL < 0,0032% (reducción de log 4,5) en cualquier momento fue más alta en el grupo de SPRYCEL, comparado con el grupo de imatinib (44 % frente a 34%).
La tasa RM4,5 a lo largo del tiempo se representa gráficamente en la Figura 3. Las tasas RM4,5 a lo largo del tiempo fueron consistentemente más altas en el grupo de pacientes tratados con dasatinib comparadas con el grupo de pacientes tratados con imatinib.
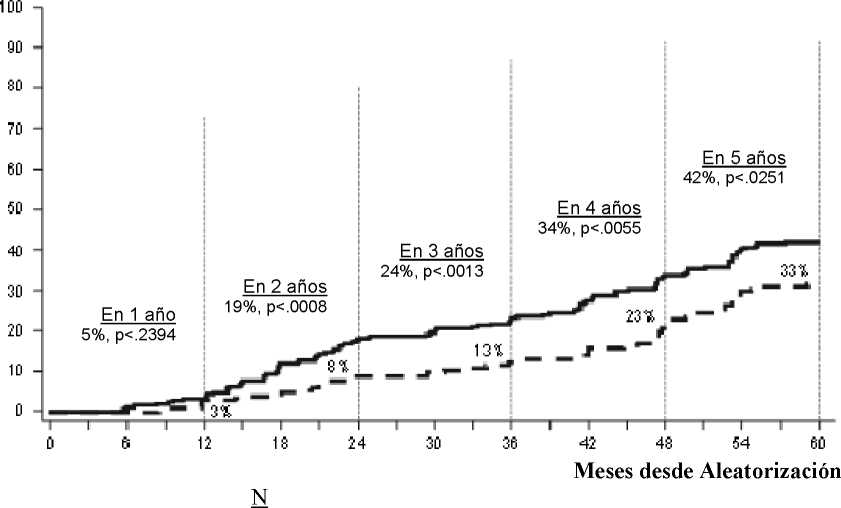
Dasatinib 100 mg una vez al día 259 Imatinib 400 mg una vez al día 260
La tasa de RMM en cualquier momento en cada grupo de riesgo determinada por el índice de Hasford fue más alta en el grupo de SPRYCEL, comparado con el grupo de imatinib (riesgo bajo: 90% y 69%; riesgo intermedio: 71% y 65%; riesgo alto: 67% y 54%, respectivamente).
En un análisis adicional, más pacientes tratados con dasatinib (84%) alcanzaron respuesta molecular temprana (definida como nivel BCR-ABL < 10% a 3 meses) comparado con los pacientes tratados con imatinib (64%). Los pacientes que alcanzaron respuesta molecular temprana tuvieron un riesgo de transformación bajo, tasa de supervivencia libre de progresión (PFS) alta y tasa de supervivencia global (OS) alta como se muestra en la Tabla 6.
Tabla 6: Pacientes en el brazo de dasatinib con BCR-ABL < 10% y > 10% a 3 meses
|
Dasatinib N = 235 |
Pacientes con BCR-ABL < 10% a 3 meses |
Pacientes con BCR-ABL > 10% a 3 meses |
|
Número de pacientes (%) |
198 (84,3) |
37 (15,7) |
|
Transformación a 60 meses, n/N (%) |
6/198 (3,0) |
5/37 (13,5) |
|
Tasa de PFS a 60 meses (95% CI) |
92,9% (89,6; 95,2) |
73,8% (52,0; 90,7) |
|
Tasa de OS a 60 meses (95% CI) |
93,8% (89,3; 96,4) |
80,6% (63,5; 90,2) |
La tasa de OS en momentos específicos se representa gráficamente en la Figura 4. La tasa de OS fue consistentemente más alta en los pacientes tratados con dasatinib que alcanzaron un nivel de BCR-ABL< 10% a 3 meses que en aquellos que no la alcanzaron.
Figura 4: Curva de Supervivencia Global para Dasatinib por nivel de BCR-ABL (< 10% ó > 10%) a 3 Meses en un ensayo Fase 3 en pacientes con LMC en Fase Crónica de nuevo diagnóstico
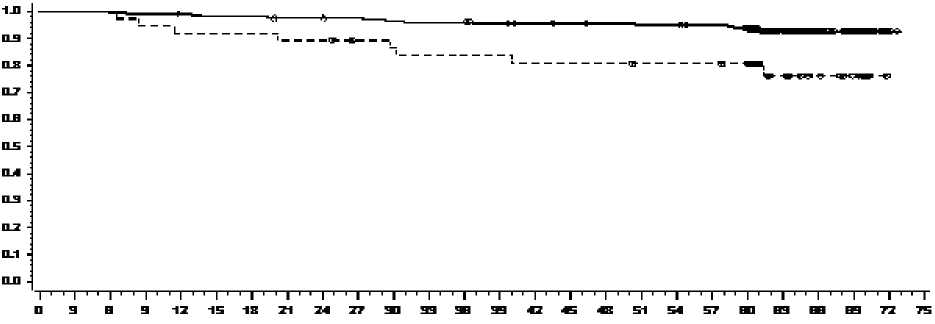
MESES
Pacientes en riesgo
<=10% 193 193 197 193 195 190 190 191 191 190 188 187 187 184 182 181 183 179 179 177 171 93 54 29 3 0
>10% 373737353434343333 31 3029292923232327272723 15 10 600
O
£
>
Z
■O
HH
V
g
a.
O
g
Q-
------>10%
0QO Censurados
_<10%
'■ ■' Censurados
GRUPO # MUERTES / # Pacientes <10% 14/198
>10% 8/37
MEDIANA (95% CI) HAZARD RATIO (95% CI)
.(. - .)
.(. - .)
0,29 (0,12 - 0,69)
La progresión de la enfermedad se definió como un incremento de glóbulos blancos sanguíneos a pesar de un manejo terapéutico adecuado, pérdida de RHC, RCy parcial, o RCyC, progresión a fase acelerada, fase blástica, o muerte. La tasa estimada de PFS a 60 meses fue del 88,9% (IC: 84 % -92,4%) y para ambos grupos de tratamiento con dasatinib e imatinib. A los 60 meses la transformación a la fase acelerada o blástica ocurrió en menos pacientes tratados con dasatinib (n= 8; 3%) comparados con los pacientes tratados con imatinib (n= 15; 5,8%). Las tasas de supervivencia estimadas a 60 meses para los sujetos tratados con dasatinib e imatinib fueron 90,9% (IC: 86,6% -93,8%) y 89,6% (IC:85,2%- 92,8%), respectivamente. No hubo diferencias en OS (HR 1,01; 95% IC: 0,58-1,73 p=0,9800) y PFS ( HR 1,00; 95%IC:0,58-1,72, p=0,9998) entre dasatinib e imatinib.
En los pacientes en los que se notificó progresión de la enfermedad o suspensión del tratamiento con dasatinib o imatinib, se realizó secuenciación de BCR-ABL en las muestras sanguíneas de los pacientes dónde éstas estaban disponibles. Se observaron tasas de mutación similares en ambos brazos de tratamiento. Las mutaciones detectadas en los pacientes tratados con dasatinib fueron T315I, F317I/L y V299L. Una mutación de espectro diferente se detectó en el brazo de tratamiento con imatinib. Dasatinib no parece ser activo frente a la mutación T315I, en base a los datos in vitro.
LMC en Fase Crónica- resistencia o intolerancia a tratamiento previo con imatinib
Se han realizado dos ensayos clínicos en pacientes resistentes o intolerantes a imatinib; el objetivo
primario de eficacia en estos ensayos fue la Respuesta Citogenética Mayor (RCyM)
1- Se realizó un ensayo multicéntrico abierto, aleatorizado, no-comparativo en pacientes que habían fallado a un tratamiento inicial con 400 ó 600 mg de imatinib. Los pacientes fueron aleatorizados (2:1) para recibir tratamiento con dasatinib (70 mg dos veces al día) o imatinib (400 mg dos veces al día).
Se permitieron cambios al grupo de tratamiento alternativo si los pacientes mostraban pruebas de progresión de la enfermedad o intolerancia que no pudiese controlarse mediante modificación de la dosis. La variable primaria fue RCyM a 12 semanas. Se dispone de resultados de 150 pacientes: 101 asignados aleatoriamente a tratamiento con dasatinib y 49 a imatinib (todos resistentes a imatinib).
La mediana de tiempo desde el diagnóstico a la randomización fue de 64 meses en el grupo de dasatinib y 52 meses en el grupo de imatinib. Todos los pacientes habían sido pretratados con diversas líneas terapéuticas. El 93% de la población global de pacientes había alcanzado una respuesta hematológica completa (RHC) previa con imatinib. El 28% y el 29% de los pacientes aleatorizados a dasatinib e imatinib, respectivamente, se había alcanzado una RCyM previa con imatinib.
La mediana de la duración del tratamiento fue de 23 meses para dasatinib (con un 44% de los pacientes tratados durante > 24 meses hasta la fecha) y 3 meses para imatinib (con 10% de pacientes tratados durante > 24 meses hasta la fecha). El 93% de los pacientes del grupo de dasatinib y el 82% de los pacientes del grupo de imatinib, alcanzaron una RHC antes de cambiar de grupo de tratamiento.
A los 3 meses, se obtuvo una RCyM con mayor frecuencia en el grupo tratado con dasatinib (36%) que en el grupo tratado con imatinib (29%). Es destacable que en el 22% de los pacientes se comunicó respuesta citogenética completa (RCyC) en el grupo tratado con dasatinib, mientras que sólo se alcanzó RCyC en el 8% del grupo tratado con imatinib. Con un tratamiento más largo y seguimiento (mediana de 24 meses) RCyM se alcanzó en un 53% de los pacientes tratados con dasatinib (RCyC en un 44%) y en un 33% de pacientes tratados con imatinib (RCyC en un 18%) antes de cambiar de grupo de tratamiento. Entre los pacientes que habían recibido 400 mg antes de entrar en el ensayo, RCyM se alcanzó en un 61% de pacientes en el grupo de dasatinib y un 50% en el grupo de imatinib.
Basado en los estimados de Kaplan-Meier, la proporción de pacientes que mantuvieron RCyM durante
1 año fue del 92% (IC del 95% [85%-100%]) para dasatinib (RCyC 97%, IC del 95% [92%-100%]) y un 74% (IC del 95% [49%-100%]) para imatinib (RCyC100%). La proporción de pacientes que mantuvieron RCyM durante 18 meses fue del 90%(IC del 95% [82%-98%]) para dasatinib RCyC del 94%, (IC del 95% [87%-100%]) y 74% (IC del 95% [49%-100%]) para imatinib (RCyC100%).
Basado en los estimados de Kaplan-Meier, la proporción de pacientes que tuvieron supervivencia libre de progresión (SLP) durante 1 año fue del 91% (IC del 95%: [85%-97%]) para dasatinib y un 73% (IC del 95%: [54%-91%]) para imatinib. La proporción de pacientes que tuvieron SLP a los 2 años fue del 86% (ICdel 95% [78%-93%]) para dasatinib y 65% (IC del 95%: [43%-87%]) para imatinib.
Un total de un 43% de los pacientes tratados con dasatinib y un 82% de los tratados con imatinib presentaron fracaso del tratamiento, definido como progresión de la enfermedad o cambio al otro tratamiento (falta de respuesta, intolerancia al medicamento en estudio, etc.).
La tasa de respuesta molecular mayor (definida como ratio de transcritos BCR- ABL/ control < 0,1% por RQ-PCR en muestras de sangre periférica) antes del cambio fue del 29% para dasatinib y un 12% para imatinib.
2- Se realizó un ensayo abierto, no controlado, multicéntrico, en pacientes intolerantes o resistentes a imatinib (p.ej. pacientes que experimentaron una toxicidad significativa durante el tratamiento con imatinib que impedía la continuación del mismo).
Un total de 387 pacientes recibieron dasatinib 70 mg dos veces al día (288 resistentes y 99 intolerantes). La mediana del tiempo desde el diagnóstico hasta el inicio del tratamiento fue de 61 meses. La mayoría de los pacientes (53%) habían recibido tratamiento previo con imatinib durante más de 3 años. La mayoría de los pacientes resistentes (72%) habían recibido > 600 mg de imatinib. Además del imatinib, el 35% de los pacientes habían recibido previamente quimioterapia citostática, 65% habían recibido tratamiento con interferón y 10% un trasplante de células madre. Un 38% de los pacientes tenían mutaciones basales conocidas relacionadas con la resistencia a imatinib. La mediana de duración del tratamiento con dasatinib fue de 24 meses con un 51% de los pacientes tratados > 24 meses hasta la fecha. En la Tabla 7 se presentan los resultados de eficacia. RCyM se alcanzó en un 55% de los pacientes resistentes a imatinib y en un 82% de los pacientes intolerantes a imatinib. Con un mínimo de 24 meses de seguimiento, 21 de 240 pacientes que alcanzaron una RCyM tuvieron progresión y la mediana de duración de la RCyM no fue alcanzada.
Basado en los estimados de Kaplan-Meier, un 95% (IC del 95%: [92%-98%]) de los pacientes mantuvieron RCyM durante 1 año y el 88% (IC del 95%: [83%-93%]) mantuvieron RCyM durante
2 años. La proporción de pacientes que mantuvieron RCyC durante 1 año fue del 97% (IC del 95%:
[94%-99%]) y durante 2 años fue del 90% (IC del 95%: [86%-95%]). El 42% de los pacientes resistentes a imatinib con RCyM no anterior a imatinib (n = 188) alcanzó una RCyM con dasatinib. Hubo 45 mutaciones BCR-ABL diferentes en un 38% de los pacientes incluidos en este ensayo. La respuesta hematológica completa o RCyM se alcanzó en pacientes que portan una gran variedad de mutaciones BCR-ABL asociadas con la resistencia a imatinib excepto T315I. Las tasas de RCyM a los dos años fueron similares a aquellos pacientes que tenían una mutación basal BCR-ABL, mutación P-loop, o que no tenían mutación (63%, 61% y 62% respectivamente).
Entre los pacientes resistentes a imatinib, la tasa estimada de SLP fue del 88% (IC del 95%: [84%-92%]) a 1 año y un 75% (IC del 95% [69%-81%]) a los 2 años. Entre los pacientes intolerantes a imatinib, la tasa estimada de SLP fue del 98% (IC del 95% [95%-100%]) a 1 año y del 94% (IC del 95% [88%-99%]) a los 2 años.
La tasa de respuesta molecular mayor a 24 meses fue del 45% (35% para pacientes resistentes a imatinib y un 74% para pacientes intolerantes a imatinib).
LMC en Fase Acelerada
Se realizó un ensayo abierto, no controlado, multicéntrico, en pacientes intolerantes o resistentes a imatinib. Un total de 174 pacientes recibieron dasatinib 70 mg dos veces al día (161 resistentes y 13 intolerantes a imatinib). La mediana de tiempo desde el diagnóstico hasta el inicio del tratamiento fue de 82 meses. La mediana de duración del tratamiento con dasatinib fue de 14 meses con un 31% de los pacientes tratados durante > 24 meses hasta la fecha. La tasa de respuesta molecular mayor (evaluada en 41 pacientes con una RCyC) fue del 46% a los 24 meses. En la Tabla 7 se presentan los resultados de eficacia adicional.
LMC en Crisis Blástica Mieloide
Se realizó un ensayo abierto, no controlado, multicéntrico, en pacientes intolerantes o resistentes a imatinib. Un total de 109 pacientes recibieron dasatinib 70 mg dos veces al día (99 resistentes y 10 intolerantes a imatinib). La mediana de tiempo desde el diagnóstico hasta el inicio del tratamiento fue de 48 meses. La mediana de duración del tratamiento con dasatinib fue de 3,5 meses con un 12% de los pacientes tratados > 24 meses hasta la fecha. La tasa de respuesta molecular mayor (evaluada en 19 pacientes con una RCyC) fue del 68% a los 24 meses. En la Tabla 7 se presentan los resultados de eficacia adicional.
LMC en Crisis Blástica Linfoide y LLA Ph+
Se realizó un ensayo abierto, no controlado, multicéntrico, en pacientes con LMC en crisis blástica linfoide o LLA Ph+ resistentes o intolerantes al tratamiento previo con imatinib. Un total de 48 pacientes con LMC blástica linfoide recibieron dasatinib 70 mg dos veces al día (42 resistentes y 6 intolerantes a imatinib). La mediana de tiempo desde el diagnóstico hasta el inicio del tratamiento fue de 28 meses. La mediana de duración del tratamiento con dasatinib fue de 3 meses con un 2% de pacientes tratados durante > 24 meses hasta la fecha. La tasa de respuesta molecular mayor (en todos los 22 pacientes tratados con una RCyC) fue del 50% a los 24 meses. Además, 46 pacientes con LLA Ph+ recibieron dasatinib 70 mg dos veces al día (44 resistentes y 2 intolerantes a imatinib). La mediana del tiempo desde el diagnóstico hasta el inicio del tratamiento fue de 18 meses. La mediana de la duración del tratamiento con dasatinib fue de 3 meses con un 7% de los pacientes tratados > 24 meses hasta la fecha. La tasa de respuesta molecular mayor (en todos los 25 pacientes tratados con una RCyC) fue del 52% a los 24 meses. En la Tabla 7 se presentan los resultados de eficacia. Además se puede destacar que las respuestas hematológicas mayores (RHM) fueron alcanzadas rápidamente (la mayoría dentro de los 35 días desde la primera administración de dasatinib en los pacientes con LMC en crisis blástica linfoide y dentro de los 55 días en los pacientes con LLA Ph+).
|
Blástica |
Blástica | ||||
|
Crónica |
Acelerada |
Mieloide |
Linfoide |
Ph+ ALL | |
|
(n = 387) |
(n = 174) |
(n = 109) |
(n = 48) |
(n = 46) | |
|
Tasa de respuesta hematológicab (%) | |||||
|
RHMa (95% CI) |
n/a |
64% (57-72) |
33% (24-43) |
35% (22-51) |
41% (27-57) |
|
RHC (95% CI) |
91% (88-94) |
50% (42-58) |
26% (18-35) |
29% (17-44) |
35% (21-50) |
|
NEL (95% CI) |
n/a |
14% (10-21) |
7% (3-14) |
6% (1-17) |
7% (1-18) |
|
Duración de MaHR (%; estimados Kaplan-Meier) | |||||
|
1 año |
n/a |
79% (71-87) |
71% (55-87) |
29% (3-56) |
32% (8-56) |
|
2 años |
n/a |
60% (50-70) |
41% (21-60) |
10% (0-28) |
24% (2-47) |
|
Respuesta citogenéticac (%) | |||||
|
RCyM (IC 95%) |
62% (57-67) |
40% (33-48) |
34% (25-44) |
52% (37-67) |
57%(41-71) |
|
RCyC (IC 95%) |
54% (48-59) |
33% (26-41) |
27% (19-36) |
46% (31-61) |
54% (39-69) |
|
Supervivencia (%; estimados Kaplan-Meier) | |||||
|
Libre de Progression | |||||
|
1 año |
91% (88-94) |
64% (57-72) |
35% (25-45) |
14% (3-25) |
21% (9-34) |
|
2 años |
80% (75-84) |
46% (38-54) |
20% (11-29) |
5% (0-13) |
12% (2-23) |
|
Global | |||||
|
1 año |
97% (95-99) |
83% (77-89) |
48% (38-59) |
30% (14-47) |
35% (20-51) |
|
2 años |
94% (91-97) |
72% (64-79) |
38% (27-50) |
26% (10-42) |
31% (16-47) |
Los datos descritos en esta tabla son de ensayos en los que se ha utilizado una dosis inicial de 70 mg dos veces al día. Ver sección 4.2 para la dosis inicial recomendada. a Números en negrita son los resultados de las variables primarias.
b Criterios de respuesta hematológica (todas las respuestas confirmadas después de 4 semanas): Respuesta hematológica mayor (RHMa)= respuesta hematológica completa (RHC) + no evidencia de leucemia (NEL).
RHC (LCM crónica: glóbulos blancos < institucional LSN, plaquetas < 450.000/mm3, no hay blastos ni promielocitos en sangre periférica, < 5% mielocitos más metamielocitos en sangre periférica, basófilos en sangre periférica < 20%, y no implicación extramedularmente.
RHC (LCM/Ph+ ALL avanzada): glóbulos blancos < institucional LSN, ANC > 1.000/mm3, plaquetas > 100.000/mm3, ni blastos ni promielocitos en sangre periférica, blastos en médula ósea < 5%, < 5% mielocitos más metamielocitos en sangre periférica, basófilos en sangre periférica < 20%, y no implicación extramedularmente.
NEL: mismo criterio que para RHC pero ANC > 500/mm3 y < 1.000/mm3, o plaquetas > 20.000/mm3 y < 100.000/mm3.
c Criterios de respuesta citogenética completa (0% Ph+ metafases) o parcial (> 0%-35%). RCyM (0%-35%) combinan ambas respuesta completa y parcial.
n/a = no aplicable, IC = intervalo de confianza, LSN = límite superior del rango normal.
El resultado de los pacientes sometidos a trasplantes de médula ósea, después del tratamiento con dasatinib no ha sido evaluado completamente.
Ensayos clínicos Fase III en pacientes con LMC en fase crónica o acelerada o crisis blástica mieloide y LLA Ph+ que fueron resistentes o intolerantes a imatinib
Dos ensayos abiertos, aleatorizados, se han llevado a cabo para evaluar la eficacia de dasatinib administrado una vez al día comparado con dasatinib administrado dos veces al día. Los resultados descritos abajo se basan en un mínimo de 2 años y 7 años de seguimiento después del comienzo del tratamiento con dasatinib.
1.- En el ensayo de LMC en fase crónica, la variable principal de eficacia fue la RCyM de los pacientes resistentes a imatinib. La variable de eficacia secundaria fue la tasa de RCyM alcanzada según la dosis diaria total en los pacientes resistentes a imatinib. Otras variables secundarias de eficacia incluyeron la duración de la RCyM, la SLP (supervivencia libre de progresión) y la supervivencia global. Un total de 670 pacientes, de los que 497 eran resistentes a imatinib, fueron aleatorizados a dasatinib 100 mg una vez al día, 140 mg una vez al día, 50 mg dos veces al día y
70 mg dos veces al día. La mediana de duración del tratamiento para todos los pacientes todavía en tratamiento con un mínimo de 5 años de seguimiento (n = 205) fue de 59 meses (rango de 2866 meses). La mediana de la duración del tratamiento para todos los pacientes a 7 años de seguimiento fue de 29,8 meses (rango < 1-92,9 meses).
La eficacia se alcanzó en todas las ramas del tratamiento con dasatinib siendo la eficacia comparable (no-inferior) con la toma una vez al día frente a la toma dos veces al día en términos de la variable principal de eficacia (diferencia en tasa de RCyM de 1,9%; IC 95% [-6,8%-10,6%]); sin embargo, la pauta posológica de 100 mg una vez al día demostró una mejora en la seguridad y tolerabilidad. Los resultados de eficacia se muestran en la Tablas 8 y 9.
Tabla 8: Eficacia de SPRYCEL en el ensayo Fase III de optimización de dosis en LMC en Fase Crónica resistente o intolerante a imatinib (resultados a 2 años)a
|
Todos los pacientes |
n=167 |
|
Pacientes resistentes a Imatinib |
n=124 |
|
Tasa de Respuesta Hematológicab (%) (95% CI) | |
|
RHC |
92% (86-95) |
|
Respuesta Citogenética c (%) (95% CI) | |
|
RCyM | |
|
Todos los pacientes |
63% (56-71) |
|
Pacientes Resistentes a Imatinib |
59% (50-68) |
|
RCyC Todos los pacientes |
50% (42-58) |
|
Pacientes Resistentes a Imatinib |
44% (35-53) |
|
Respuesta Molecular Mayor en pacientes que alcanzan RCyC |
d (%) (95% CI) |
|
Todos los pacientes |
69% (58-79) |
|
Pacientes Resistentes a Imatinib |
72% (58-83) |
Resultados notificados con la dosis inicial recomendada de 100 mg una vez al día.
b Criterios de respuesta hematológica (todas las respuestas confirmadas después de 4 semanas): Respuesta Hematológica Completa RHC (LMC crónica): WBC < LSN institucional, plaquetas < 450.000/mm3, no blastos o promielocitos en sangre periférica, < 5% mielocitos más metamielocitos en sangre periférica, basófilos en sangre periférica < 20%, y no infiltración extramedular. c Criterios de Respuesta Citogenética: completa (0% Ph+ metafases) o parcial (> 0%-35%). RCyM (0%-35%) que combina tanto respuesta completa como parcial.
d Criterios de Respuesta Molecular Mayor: definida como BCR-ABL/transcritos control < 0.1% por RQ-PCR en muestras de sangre periférica.
pacientes con LMC en Fase Crónica resistente o intolerante a Imatinib
|
Período Mínimo de Seguimiento | ||||
|
1 año |
2 años |
5 años |
7 años | |
|
Respuesta Molecular Mayor | ||||
|
Todos los pacientes |
NA |
37% (57/154) |
44% (71/160) |
46% (73/160) |
|
Pacientes resistentes a Imatinib |
NA |
35% (41/117) |
42% (50/120) |
43% (51/120) |
|
Pacientes intolerantes a Imatinib |
NA |
43% (16/37) |
53% (21/40) |
55% (22/40) |
|
Supervivencia Libre de Progresión |
b | |||
|
Todos los pacientes |
90% (86, 95) |
80% (73, 87) |
51% (41, 60) |
42% (33, 51) |
|
Pacientes resistentes a Imatinib |
88% (82, 94) |
77% (68, 85) |
49% (39, 59) |
39% (29, 49) |
|
Pacientes intolerantes a Imatinib |
97% (92, 100) |
87% (76, 99) |
56% (37, 76) |
51% (32, 67) |
|
Supervivencia Global | ||||
|
Todos los pacientes |
96% (93, 99) |
91% (86, 96) |
78% (72, 85) |
65% (56, 72) |
|
Pacientes resistentes a Imatinib |
94% (90, 98) |
89% (84, 95) |
77% (69, 85) |
63% (53, 71) |
|
Pacientes intolerantes a Imatinib |
100% (100, 100) |
95% (88, 100) |
82% (70, 94) |
70% (52, 82) |
a Resultados notificados con la dosis inicial recomendada de 100 mg una vez al día.
b Progresión se definió como un incremento en el recuento de WBC, pérdida de CHR o MCyR, >30% incremento en metafases Ph+, AP/BP confirmada enfermedad o muerte. PFS se analizó en base al principio de intención de tratamiento y los pacientes fueron seguidos con respecto a eventos incluyendo tratamiento posterior.
Basados en los estimados de Kaplan-Meier, la proporción de pacientes tratados con dasatinib 100 mg una vez al día que mantuvieron una RCyM durante 18 meses fue del 93% (IC del 95%: [88%-98%]) .
La eficacia también se evaluó en pacientes que eran intolerantes a imatinib. En esta población de pacientes que recibieron 100 mg una vez al día, la RCyM se alcanzó en un 77%, y la RCyC en un 67%.
2.- En un ensayo en fases avanzadas de la LMC y LLA Ph+, la variable principal fue RHM. Un total de 611 se aleatorizaron a dasatinib 140 mg/día o 70 mg dos veces al día. La mediana de duración del tratamiento fue aproximadamente de 6 meses (rango 0,03-31 meses).
La pauta posológica con “una vez al día” demostró una eficacia comparable (no-inferioridad) a la obtenida con “dos veces al día” respecto a la variable principal (diferencia en RHM 0,8%; IC 95% [-7,1% - 8,7%]), sin embargo la pauta posológica de 140 mg una vez al día demostró una mejora en la seguridad y tolerabilidad. Las tasas de respuesta se presentan en la Tabla 10.
Tabla 10: Eficacia de SPRYCEL en el ensayo fase III de optimización de dosis: Fase Avanzada de LMC y se CML y Ph+ LLA (resultados a 2 años)a
|
Acelerada (n= 158) |
Crisis Blástica Mieloide (n= 75) |
Crisis Blástica Linfoide (n= 33) |
Ph+ALL (n= 40) | |
|
RHMab |
66% |
28% |
42% |
38% |
|
(95% CI) |
(59-74) |
(18-40) |
(26-61) |
(23-54) |
|
RHCb |
47% |
17% |
21% |
33% |
|
(95% CI) |
(40-56) |
(10-28) |
(9-39) |
(19-49) |
|
NELb |
19% |
11% |
21% |
5% |
|
(95% CI) |
(13-26) |
(5-20) |
(9-39) |
(1-17) |
|
RCyMc |
39% |
28% |
52% |
70% |
|
(95% CI) |
(31-47) |
(18-40) |
(34-69) |
(54-83) |
|
RCyC |
32% |
17% |
39% |
50% |
|
(95% CI) |
(25-40) |
(10-28) |
(23-58) |
(34-66) |
a Resultados notificados con la dosis inicial recomendada de 140 mg una vez al día (ver sección 4.2).
b Criterios de Respuesta Hematológica (todas las respuestas confirmadas después de 4 semanas): Respuesta Hematológica Mayor (RHMa) = respuesta hematológica completa (RHC) + no evidencia de leucemia (NEL).
RHC: WBC< LSN institucional, ANC > 1.000/mm3, plaquetas > 100.000/mm3, sin blastos o promielocitos en sangre periférica, blastos de médula ósea < 5%, < 5% mielocitos más metamielocitos en sangre periférica, basófilos en sangre periférica < 20%, y no infiltración extramedular.NEL: mismo criterio como para RHC pero ANC > 500/mm3, y < 1.000/mm3, o plaquetas > 20.000/mm3 y < 100.000/mm3. c RCyM combina ambas respuestas completa (0% Ph+ metafase) y parcial (> 0%-35%).
IC = intervalo de confianza, LSN = Límite superior del rango normal.
incremento de la dosis en el rango de dosis de 25 mg a 120 mg dos veces al día. La media global de la semivida de eliminación terminal de dasatinib es, aproximadamente, de 5-6 horas en los pacientes.
Los datos de sujetos sanos que recibieron una dosis única, de 100 mg de dasatinib 30 minutos después de una comida rica en grasas indicaron un aumento del 14% en el ABC medio de dasatinib. Una dieta pobre en grasas 30 minutos antes de la administración de dasatinib produjo un aumento del 21% en la media de AUC para dasatinib. Los efectos debidos a los alimentos no representan cambios clínicamente relevantes en la exposición al fármaco.
Distribución
En pacientes, dasatinib tiene un volumen de distribución aparente muy grande (2.505 l), lo que sugiere que el fármaco se distribuye ampliamente por el espacio extravascular. Basándose en los resultados de ensayos in vitro la unión a proteínas plasmáticas de dasatinib a las concentraciones clínicas relevantes es del 96%.
Biotransformación
Dasatinib se ampliamente metabolizado en los seres humanos por múltiples enzimas implicadas en la transformación de los metabolitos. En sujetos sanos que recibieron 100 mg de dasatinib marcado con [14C], la fracción de dasatinib inalterada representó el 29% de la radiactividad circulante en el plasma. La concentración plasmática y la actividad medida in vitro indican que es poco probable que los metabolitos de dasatinib desempeñen un papel importante en la farmacología observada del producto. El CYP3A4 es una enzima importante responsable del metabolismo de dasatinib.
Eliminación
La eliminación se produce predominantemente por las heces, principalmente como metabolitos.
Después de una dosis oral única de dasatinib marcado con [14C], aproximadamente el 89% de la dosis se eliminó en 10 días recuperándose un 4% y 85% de la radioactividad en orina y heces, respectivamente. La fracción inalterada de dasatinib representó el 0,1% y el 19% de la dosis en orina y heces, respectivamente, mientras que el resto de la dosis se eliminó como metabolitos.
Insuficiencia hepática y renal
El efecto de la insuficiencia hepática en la farmacocinética de dosis única de dasatinib se evaluó en 8 pacientes con insuficiencia hepática moderada que recibieron una dosis de 50 mg y en 5 pacientes con insuficiencia hepática grave que recibieron una dosis de 20 mg y se compararon con voluntarios sanos que recibieron una dosis de 70 mg de dasatinib. La media de la C max y del ABC de dasatinib ajustadas a una dosis de 70 mg, disminuyeron un 47% y un 8% respectivamente en aquellos pacientes con insuficiencia hepática moderada comparados con aquellos que tienen una función hepática normal. En aquellos pacientes con insuficiencia hepática grave, la media de la C max y del ABC de dasatinib ajustadas a una dosis de 70 mg, disminuyeron un 43% y un 28% respectivamente comparados con aquellos que tienen una función hepática normal (ver secciones 4.2 y 4.4).
Dasatinib y sus metabolitos se excretan mínimamente por vía renal.
5.3 Datos preclínicos sobre seguridad
El perfil de seguridad preclínico de dasatinib fue valorado en una batería de estudios in vitro e in vivo en ratones, ratas, monos y conejos.
Las principales formas de toxicidad se presentaron en los sistemas gastrointestinal, hematopoyético y linfoide.
La toxicidad gastrointestinal fue dosis limitante en ratas y monos, siendo el intestino el órgano diana de forma consistente. En las ratas, descensos mínimos o leves en los parámetros eritrocitarios, se acompañaron de cambios en la médula ósea; en los monos se detectaron cambios similares pero con una incidencia menor. La toxicidad linfoide observada en ratas consistió en depleción linfoide de los ganglios linfáticos, el bazo y el timo, y disminución del peso de los órganos linfoides. Los cambios en los sistemas gastrointestinal, hematopoyético y linfoide fueron reversibles después de la suspensión del tratamiento.
Se observaron cambios renales en monos tratados hasta 9 meses y se limitaron a un aumento de la mineralización renal de fondo. Se observó hemorragia cutánea en un estudio de toxicidad aguda, de dosis única oral en monos, pero no se observó en estudios de dosis repetidas en monos o ratas. En ratas, dasatinib inhibió la agregación plaquetaria in vitro y prolongó el tiempo de hemorragia in vivo, pero no provocó hemorragias espontáneas.
La actividad in vitro de dasatinib en los ensayos hERG y fibras de Purkinje sugería un potencial de prolongación de la repolarización ventricular cardíaca (intervalo QT). Sin embargo, en un estudio in vivo de dosis únicas en monos conscientes monitorizados a distancia, no hubo cambios en el intervalo QT ni en la forma de la onda del ECG.
Dasatinib no fue mutagénico en ensayos de células bacterianas in vitro (test de Ames) y no fue genotóxico en un estudio de micronúcleos de la rata in vivo. Fue clastogénico in vitro en la división de las células de ovario de hámster (COH) chino.
Dasatinib no afectó a la fertilidad tanto de machos como de hembras en un estudio convencional de fertilidad y desarrollo embrionario temprano en ratas, pero provocó letalidad embrionaria a niveles de dosis que se aproximan a la exposición clínica en humanos. Asimismo en estudios de desarrollo embriofetal, dasatinib provocó letalidad embrionaria asociada con disminución en el tamaño de las ratas recién nacidas y también alteraciones esqueléticas en el feto tanto en las ratas como en las conejas. Estos efectos aparecieron a dosis que no producían toxicidad materna e indica que dasatinib es un tóxico reproductivo selectivo desde la implantación hasta que se completa la organogénesis.
En ratones, dasatinib produjo inmunodepresión relacionada con la dosis y controlada eficazmente mediante reducción de la dosis y/o cambios en la pauta posológica. Dasatinib tuvo potencial fototóxico en un estudio de fototoxicidad de captación de rojo neutro in vitro en fibroblastos de ratón. Se consideró que dasatinib no era fototóxico in vivo después de una única administración por vía oral a ratones hembra sin pelo con un nivel de exposición de hasta 3 veces la exposición en humanos después de la administración de las dosis terapéuticas recomendadas (basadas en el área bajo la curva, AUC).
En un estudio de carcinogenicidad a dos años, las ratas recibieron dasatinib a dosis orales de 0,3, 1 y 3 mg/kg/dia. La dosis más alta dió como resultado un nivel plasmático (AUC) generalmente equivalente a la exposición humana correspondiente al rango de dosis iniciales recomendadas desde 100 mg a 140 mg diarios. Se advirtió un incremenmto estadisticamente significativo en la incidencia combinada de carcinomas celulares escamosos y papilomas en útero y cervix en hembras a dosis altas y de adenomas de próstata en machos a dosis bajas. No se conoce la relevancia de los hallazgos de estudios de carcinogenicidad de ratas en los humanos.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido Lactosa monohidrato Celulosa microcristalina Croscarmelosa sódica Hidroxipropilcelulosa Estearato de magnesio
Cubierta pelicular Hipromelosa Dióxido de titanio Macrogol 400
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Blisteres Alu/Alu (blíster calendario o blíster precortado unidosis).
Frasco de polietileno de alta densidad (HDPE) con cierre de polipropileno a prueba de niños.Envase conteniendo 56 comprimidos recubiertos con película; en 4 blísteres de 14 comprimidos recubiertos con película cada uno.
Envase conteniendo 60 comprimidos recubiertos con película, en blísteres precortados unidosis. Envase conteniendo un frasco con 60 comprimidos recubiertos con película.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Los comprimidos recubiertos con película constan de un núcleo del comprimido, rodeado de una cubierta pelicular para prevenir la exposición de los profesionales sanitarios al principio activo. Sin embargo, si los comprimidos recubiertos con película se aplastan o se rompen accidentalmente, los profesionales sanitarios deben llevar guantes desechables de quimioterapia para minimizar el riesgo de la exposición dérmica durante la correcta eliminación.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
BRISTOL-MYERS SQUIBB PHARMA EEIG
Uxbridge Business Park
Sanderson Road
Uxbridge UB8 1DH
Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/363/004
EU/1/06/363/007
EU/1/06/363/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA
AUTORIZACIÓN
Fecha de la primera autorización: 20/noviembre/2006
Fecha de la última renovación: 20/noviembre/2011
10. FECHA DE LA REVISIÓN DEL TEXTO
Información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http: //www.ema. europa. eu/.
1. NOMBRE DEL MEDICAMENTO
SPRYCEL 50 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 50 mg de dasatinib (como monohidrato). Excipientes con efecto conocido:
Cada comprimido recubierto con película contiene 67,5 mg de lactosa monohidrato.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
Comprimido recubierto con película oval, de color blanco a blanquecino, biconvexo, con “BMS” grabado en una cara y “528” en la otra.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
SPRYCEL está indicado para el tratamiento de pacientes adultos con:
■ leucemia mieloide crónica (LMC) en fase crónica de nuevo diagnóstico cromosoma Filadelfia positivo (Ph+).
■ leucemia mieloide crónica (LMC) en fase crónica, acelerada o blástica, con resistencia o intolerancia al tratamiento previo, incluido el mesilato de imatinib.
■ leucemia linfoblástica aguda (LLA) cromosoma Filadelfia positivo (Ph+) y crisis blástica linfoide procedente de LMC con resistencia o intolerancia al tratamiento previo.
4.2 Posología y forma de administración
El tratamiento debe iniciarlo un médico con experiencia en el diagnóstico y el tratamiento de pacientes con leucemia.
Posología
La dosis de inicio recomendada para LMC en fase crónica es de 100 mg de dasatinib una vez al día, administrada por vía oral.
La dosis de inicio recomendada para LMC en fase acelerada, crisis blástica mieloide o linfoide (fases avanzadas) o en LLA cromosoma Filadelfia positivo (Ph+) es de 140 mg una vez al día administrada por vía oral (ver sección 4.4).
Duración del tratamiento
En ensayos clínicos, el tratamiento con SPRY CEL se continuó hasta la progresión de la enfermedad o hasta que ya no se tolerara por el paciente. No se ha investigado el efecto de la suspensión del tratamiento sobre el resultado de la enfermedad a largo plazo después de conseguirse una respuesta citogenética o molecular [incluyendo respuesta citogenética completa (RCyC), respuesta molecular mayor (RMM) y RM4.5].
Para alcanzar la dosis recomendada, SPRYCEL está disponible como comprimidos recubiertos con película de 20 mg, 50 mg, 70 mg, 80 mg, 100 mg y 140 mg. Se recomienda el incremento o la reducción de la dosis en base a la respuesta del paciente y a la tolerabilidad.
Escalado de dosis
En ensayos clínicos en pacientes adultos con LMC y LLA Ph+, se permitió un aumento gradual de la dosis a 140 mg una vez al día (LMC en fase crónica) o 180 mg una vez al día (LMC en fases avanzadas o LLA Ph+) en pacientes que no alcanzaron una respuesta hematológica o citogenética a las dosis recomendadas de inicio.
Ajustes de la dosis por reacciones adversas
Mielosupresión
En los ensayos clínicos, la mielosupresión requirió una reducción de la dosis o la suspensión temporal o permanente del tratamiento en estudio. Se realizaron transfusiones de plaquetas y glóbulos rojos en los pacientes que lo requirieron. Se administraron factores de crecimiento hematopoyéticos en los pacientes con mielosupresión resistente. Las directrices para los ajustes de la dosis se resumen en la Tabla 1.
Tabla 1: Ajustes de la dosis por neutropenia y trombocitopenia
|
LMC en fase crónica (dosis inicial de 100 mg, una vez al día) |
RAN < 0,5 x 109/l y/o plaquetas < 50 x 109/l |
1 Suspender el tratamiento hasta recuperación de RAN > 1,0 x 109/l y plaquetas > 50 x 109/l. 2 Reanudar el tratamiento a la dosis inicial original. 3 Si las plaquetas < 25 x 109/l y/o un nuevo descenso del RAN < 0,5 x 109/l durante > 7 días, debe repetirse el paso 1 y reanudarse el tratamiento a una dosis reducida de 80 mg una vez al día para el segundo episodio. Para el tercer episodio, reducir dosis de modo adicional hasta 50 mg una vez al día (para pacientes recientemente diagnosticados) o interrumpir el tratamiento (para pacientes resistentes o intolerantes al tratamiento previo incluyendo imatinib). |
|
LMC en fase acelerada y crisis blástica y LLA Ph+ (dosis inicial de 140 mg, una vez al día) |
RAN < 0,5 x 109/l y/o plaquetas < 10 x 109/l |
1 Descartar que la citopenia esté relacionada con la leucemia (aspirado y/o biopsia medular). 2 Si la citopenia no está relacionada con la leucemia, suspender el tratamiento hasta recuperación del RAN > 1,0 x 109/l y plaquetas > 20 x 109/l y reanudar a la dosis inicial original. 3 Ante un nuevo episodio de citopenia, repetir el paso 1 y reanudar el tratamiento a una dosis reducida de 100 mg una vez al día (segundo episodio) u 80 mg una vez al día (tercer episodio). 4 Si la citopenia está relacionada con la leucemia, valorar aumentar la dosis a 180 mg una vez al día. |
RAN: recuento absoluto de neutrófilos
Reacciones adversas no hematológicas
Si se desarrolla una reacción adversa no hematológica, moderada, grado 2, con dasatinib, se interrumpirá el tratamiento hasta que el acontecimiento se haya resuelto o hasta que haya retornado al nivel basal. Continuar con la misma dosis si es la primera vez que ocurre y reducir la dosis si es un acontecimiento recurrente. Si se desarrolla una reacción adversa no hematológica grave, grado 3 ó 4, con dasatinib, el tratamiento debe interrumpirse hasta que el acontecimiento se haya resuelto. Posteriormente, si es conveniente, el tratamiento puede reanudarse, a una dosis reducida, dependiendo de la gravedad inicial del acontecimiento. Para pacientes con LMC en fase crónica que hayan recibido 100 mg una vez al día, se recomienda una reducción de dosis hasta 80 mg una vez al día, con una reducción de dosis adicional desde 80 mg una vez al día hasta 50 mg una vez al día, si fuese necesario. Para pacientes con fase avanzada de LMC o LLA cromosoma Filadelfia positivo (Ph+) que hayan recibido 140 mg una vez al día, se recomienda una reducción de dosis hasta 100 mg una vez al día, con una reducción de dosis adicional hasta 50 mg una vez al día, si fuese necesario.
Derrame pleural: si se diagnóstica un derrame pleural, interrumpir el tratamiento con dasatinib hasta que el paciente sea asintomático o haya retornado a su estado basal. Si el episodio no mejora dentro de aproximadamente una semana, considerar un tratamiento con diuréticos o corticosteroides o ambos al mismo tiempo (ver secciones 4.4 y 4.8). Una vez resuelto el primer episodio, considerar la reintroducción de dasatinib al mismo nivel de dosis. Tras la resolución de un episodio posterior, reintroducir dasatinib con un nivel de dosis reducido. Una vez resuelto un episodio grave (grado 3 ó 4), el tratamiento puede continuarse como proceda a un nivel de dosis reducido dependiendo de la gravedad inicial del acontecimiento.
Población pediátrica
No se ha establecido todavía, la seguridad y eficacia de SPRYCEL en niños y adolescentes menores de 18 años de edad. No hay datos disponibles (ver sección 5.1).
Pacientes de edad avanzada
No se han observado diferencias farmacocinéticas clínicamente relevantes relacionadas con la edad en estos pacientes. No es necesaria ninguna recomendación de dosis específica en los pacientes de edad avanzada.
Insuficiencia hepática
Los pacientes con insuficiencia hepática leve, moderada o grave pueden recibir la dosis de inicio recomendada. Sin embargo, SPRYCEL debe utilizarse con precaución en pacientes con insuficiencia hepática (ver secciones 4.4 y 5.2).
Insuficiencia renal
No se han realizado ensayos clínicos con SPRYCEL en pacientes con función renal reducida (el estudio en pacientes con LMC en fase crónica de nuevo diagnóstico, excluyó a los pacientes con una concentración de creatinina en suero > 3 veces el límite superior del rango normal, y ensayos en pacientes con LMC en fase crónica con resistencia o intolerancia al tratamiento previo con imatinib excluyó a pacientes con una concentración de creatinina sérica > 1,5 veces el límite superior del rango normal). Como el aclaramiento renal de dasatinib y sus metabolitos representa < 4%, en pacientes con insuficiencia renal no se espera una disminución del aclaramiento corporal total.
Forma de administración
SPRY CEL debe ser administrado por vía oral.
Los comprimidos recubiertos con película no deben aplastarse ni fraccionarse para minimizar el riesgo de exposición dérmica, deben tragarse enteros. Los comprimidos pueden tomarse con o sin comida y deben tomarse sistemáticamente o por la mañana o por la noche.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Interacciones clínicamente relevantes
Dasatinib es un sustrato y un inhibidor del citocromo P450 (CYP) 3A4. Por tanto, existe la posibilidad de interacción con otros medicamentos administrados simultáneamente, que se metabolizan fundamentalmente por CYP3A4 o que modulan su actividad (ver sección 4.5).
El uso concomitante de dasatinib y medicamentos o sustancias que inhiban de forma potente el CYP3A4 (p. ej., ketoconazol, itraconazol, eritromicina, claritromicina, ritonavir, telitromicina, zumo de pomelo) puede aumentar la exposición a dasatinib. Por tanto, no se recomienda la coadministración de inhibidores potentes de CYP3A4 en pacientes que reciben dasatinib (ver sección 4.5).
El uso concomitante de dasatinib con medicamentos inductores de CYP3A4 (p. ej., dexametasona, fenitoína, carbamazepina, rifampicina, fenobarbital o medicamentos a base de plantas que contienen Hypericum perforatum, también conocido como hierba de San Juan) puede reducir significativamente la exposición a dasatinib, incrementando potencialmente el riesgo de fracaso terapéutico. Por lo tanto, en pacientes que reciben dasatinib, deberá optarse por la coadministración con medicamentos alternativos con menor capacidad de inducción de CYP3A4 (ver sección 4.5).
El uso concomitante de dasatinib y un sustrato de CYP3A4 puede aumentar la concentración plasmática del sustrato de CYP3A4. Por tanto, se debe garantizar precaución cuando se coadministre dasatinib con sustratos de CYP3A4 de margen terapéutico estrecho como astemizol, terfenadina, cisaprida, pimozida, quinidina, bepridilo o alcaloides del cornezuelo del centeno (ergotamina, dihidroergotamina) (ver sección 4.5).
El uso concomitante de dasatinib y antagonistas de la histamina tipo 2 (H2) (p. ej., famotidina), o los inhibidores de la bomba de protones (p. ej., omeprazol) o hidróxido de aluminio/hidróxido de magnesio puede reducir la exposición a dasatinib. Por tanto, no se recomienda la utilización de los antagonistas-H2 o inhibidores de la bomba de protones. Sin embargo, pueden administrarse productos con hidróxido de aluminio/hidróxido de magnesio hasta 2 horas antes o 2 horas después de la administración de dasatinib (ver sección 4.5).
Poblaciones especiales
Basado en los hallazgos de un estudio farmacocinético de dosis única, los pacientes con insuficiencia hepática leve, moderada o grave pueden recibir la dosis de inicio recomendada (ver secciones 4.2 y 5.2). Debido a las limitaciones de este ensayo clínico, se recomienda precaución al administrar dasatinib a pacientes con insuficiencia hepática (ver sección 4.2).
Reacciones adversas importantes
Mielosupresión
El tratamiento con dasatinib se asocia a anemia, neutropenia y trombocitopenia. Su incidencia es más temprana y más frecuente en pacientes con LMC en fase avanzada o LLA Ph+ que en pacientes con LMC en fase crónica. En los pacientes con LMC en fase avanzada o LLA Ph+, deben realizarse hemogramas completos cada semana durante los 2 primeros meses, y posteriormente cada mes o con la frecuencia que le sea indicada clínicamente. En pacientes con leucemia mieloide crónica (LMC) en fase crónica deben realizarse hemogramas completos cada 2 semanas durante las primeras 12 semanas, después cada 3 meses y posteriormente cuando esté clínicamente indicado. La mielosupresión es generalmente reversible y normalmente se controla interrumpiendo temporalmente la administración de dasatinib o reduciendo la dosis (ver secciones 4.2 y 4.8).
Sangrado
En pacientes con LMC en fase crónica (n = 548), 5 pacientes (1%) que recibieron dasatinib tuvieron hemorragia grado 3 ó 4. En ensayos clínicos en pacientes con LMC en fase avanzada que recibieron la dosis recomendada de SPRYCEL (n = 304) ocurrieron hemorragias graves en el sistema nervioso central (SNC) en el 1% de los pacientes. Un caso fue mortal y se asoció según los Criterios de Toxicidad Común (CTC), a una trombocitopenia grado 4. Se produjo hemorragia gastrointestinal grado 3 ó 4 en el 6% de los pacientes con LMC en fase avanzada y generalmente requirieron suspensión del tratamiento y transfusiones. Se produjeron otras hemorragias grado 3 ó 4 en el 2% de los pacientes con LMC en fase avanzada . La mayoría de los sangrados relacionados en estos pacientes fueron típicamente asociados con trombocitopenia grado 3 ó 4 (ver sección 4.8). Adicionalmente, los estudios de función plaquetaria in vivo e in vitro sugieren que el tratamiento con SPRYCEL afecta de&nb