Sebivo 20 Mg/Ml Solucion Oral
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Sebivo 600 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 600 mg de telbivudina. Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película
Comprimido oval recubierto con película, de color blanco a ligeramente amarillento con la inscripción «LDT» en una cara.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Sebivo está indicado para el tratamiento de la hepatitis B crónica en pacientes adultos con enfermedad hepática compensada y signos de replicación viral, niveles séricos de alanina aminotransferasa (ALT) persistentemente elevados y signos histológicos de inflamación activa y/o fibrosis.
El inicio del tratamiento con Sebivo sólo se debe considerar cuando el uso de un agente antiviral alternativo con una barrera genética a la resistencia más alta no se encuentre disponible o no sea apropiado.
Ver sección 5.1 para los detalles del ensayo y las características específicas de los pacientes en los cuales está basada esta indicación.
4.2 Posología y forma de administración
El tratamiento debe iniciarlo un médico con experiencia en el manejo de la infección por hepatitis B crónica.
Posología
Adultos
La dosis recomendada de Sebivo es de 600 mg (un comprimido) una vez al día.
Se puede valorar la utilización de Sebivo solución oral en pacientes con dificultad para tragar los comprimidos.
Monitorización durante el tratamiento
Se ha demostrado que la respuesta al tratamiento en la semana 24 es predictiva de la respuesta a largo plazo (ver Tabla 7 en la sección 5.1). Los niveles de ADN del VHB se deben monitorizar tras 24 semanas de tratamiento para asegurar la supresión viral completa (ADN del VHB inferior a 300 copias/ml). En el caso de pacientes con ADN del VHB detectable tras 24 semanas de terapia, se debe considerar una modificación del tratamiento.
Para asegurar una respuesta continuada se debe monitorizar el ADN del VHB cada 6 meses. Si en algún momento después de la respuesta inicial los pacientes obtienen un resultado positivo en la prueba del ADN del VHB, se debe considerar una modificación del tratamiento. La terapia óptima se debe guiar por la evaluación de las resistencias.
Duración del tratamiento
Se desconoce la duración óptima del tratamiento. La interrupción del tratamiento deberá considerarse de la siguiente forma:
• En pacientes HBeAg positivos sin cirrosis, el tratamiento deberá administrarse durante un mínimo de 6-12 meses después de que se confirme seroconversión de HBeAg (pérdida de HBeAg y de ADN del VHB con detección de anti-HBe) o hasta seroconversión de HBsAg o exista evidencia de pérdida de eficacia. Deberán controlarse regularmente los niveles séricos de ALT y ADN del VHB después de la interrupción del tratamiento para detectar cualquier recidiva virológica tardía.
• En pacientes HBeAg negativos sin cirrosis, el tratamiento deberá administrarse como mínimo hasta seroconversión de HBsAg o hasta que exista evidencia de falta de eficacia. En tratamientos prolongados de más de dos años, se recomienda reevaluar de forma regular para confirmar que la continuación del tratamiento seleccionado continúa siendo apropiado para el paciente.
Pacientes de edad avanzada (mayores de 65 años)
No se dispone de datos que apoyen una recomendación de dosis específica para pacientes mayores de 65 años (ver sección 4.4).
Insuficiencia renal
No es necesario ajustar la dosis recomendada de telbivudina en pacientes con un aclaramiento de creatinina >50 ml/min. Se requiere un ajuste de la dosis en pacientes que presenten un aclaramiento de creatinina <50 ml/min, incluyendo los pacientes con enfermedad renal terminal (ERT) en hemodiálisis. Tal y como se detalla a continuación en la Tabla 1, se recomienda una reducción de la dosis diaria cuando se utilice Sebivo solución oral. Si no es posible el uso de la solución oral se puede usar como alternativa Sebivo comprimidos recubiertos con película y en ese caso se deberá ajustar la dosis aumentando el intervalo de tiempo entre dosis, tal y como se detalla en la Tabla 1.
Tabla 1 Ajuste de la pauta de dosificación de Sebivo en pacientes con insuficiencia renal
|
Aclaramiento de creatinina (ml/min) |
Telbivudina 20 mg/ml solución oral Ajuste de dosis diario |
Telbivudina 600 mg comprimidos recubiertos con película Alternativa** ajuste de dosis mediante aumento del intervalo de tiempo entre dosis |
|
>50 |
600 mg (30 ml) una vez al día |
600 mg una vez al día |
|
30-49 |
400 mg (20 ml) una vez al día |
600 mg una vez cada 48 horas |
|
<30 (no requiere diálisis) |
200 mg (10 ml) una vez al día |
600 mg una vez cada 72 horas |
|
ERT* |
120 mg (6 ml) una vez al día |
600 mg una vez cada 96 horas |
* Enfermedad renal terminal
** En caso de que no sea posible el uso de la solución oral
Las modificaciones de dosis propuestas están basadas en la extrapolación y puede que no sean óptimas. No se ha evaluado clínicamente la seguridad y eficacia de estas recomendaciones de ajuste de dosis. Por lo tanto, se recomienda una cuidadosa monitorización clínica de estos pacientes.
Pacientes con enfermedad renal terminal
En los pacientes con ERT, Sebivo debe administrarse después de la hemodiálisis (ver sección 5.2).
Insuficiencia hepática
No es necesario ajustar la dosis recomendada de Sebivo en pacientes con insuficiencia hepática (ver sección 5.2).
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de Sebivo en la población pediátrica.
Forma de administración
Sebivo se administra por vía oral, con o sin alimentos. El comprimido no se debe masticar, dividir ni triturar.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
La combinación de telbivudina con interferón alfa pegilado o estándar (ver secciones 4.4 y 4.5).
4.4 Advertencias y precauciones especiales de empleo
Las exacerbaciones agudas graves de hepatitis B crónica son relativamente frecuentes y se caracterizan por una elevación transitoria de la ALT sérica. Tras el inicio del tratamiento antiviral, la ALT sérica puede aumentar en algunos pacientes mientras que los niveles séricos de ADN del VHB descienden (ver sección 4.8). Como promedio, transcurren entre 4-5 semanas antes de que aparezca una exacerbación en pacientes tratados con telbivudina. En general, los aumentos transitorios de ALT se producen más frecuentemente en pacientes HBeAg positivos que en pacientes HBeAg negativos.
En pacientes con enfermedad hepática compensada, estas elevaciones de ALT sérico no van generalmente acompañadas de niveles elevados de bilirrubina sérica o de otros signos de descompensación hepática. El riesgo de descompensación hepática y de una exacerbación de hepatitis posterior, puede ser elevado en pacientes con cirrosis. Por lo tanto, estos pacientes deberán ser controlados estrechamente.
También se han notificado exacerbaciones de la hepatitis en pacientes que han finalizado el tratamiento de la hepatitis B. Normalmente, los aumentos transitorios de ALT postratamiento se asocian con incrementos en los niveles séricos de ADN del VHB, y la mayoría de estos casos han demostrado ser autolimitados. No obstante, también se han notificado casos de exacerbaciones graves de la enfermedad (en ocasiones mortales) postratamiento. Por lo tanto, la función hepática deberá controlarse regularmente durante un periodo mínimo de 6 meses después de la interrupción del tratamiento de la hepatitis B mediante seguimiento clínico y de laboratorio.
Acidosis láctica
Se ha notificado la aparición de acidosis láctica (en ausencia de hipoxemia) en algunos casos mortal, y normalmente asociada con hepatomegalia grave con esteatosis, con el uso de análogos nucleósidos o nucleótidos. Como la telbivudina es un análogo nucleósido, no puede excluirse este riesgo. El tratamiento con análogos nucleósidos deberá interrumpirse cuando los niveles de aminotransferasa aumenten rápidamente o cuando se produzca hepatomegalia progresiva o acidosis metabólica o láctica de etiología desconocida. Síntomas digestivos benignos, tales como náuseas, vómitos y dolor abdominal, pueden ser indicativos del desarrollo de acidosis láctica. Casos graves, algunos mortales, se asociaron con pancreatitis, insuficiencia o esteatosis hepática, insuficiencia renal y niveles más elevados de lactato sérico. Deberá tenerse precaución cuando se prescriban análogos nucleósidos a cualquier paciente (particularmente mujeres obesas) con hepatomegalia, hepatitis u otros factores de riesgo conocidos para una enfermedad hepática. Estos pacientes deberán ser vigilados estrechamente.
Efectos musculares
Se han notificado casos de miopatía y mialgia con el uso de telbivudina desde varias semanas hasta meses después de iniciar el tratamiento (ver sección 4.8). Se han notificado casos de rabdomiolisis durante el uso postcomercialización de telbivudina (ver sección 4.8).
En cualquier paciente que presente mialgias difusas inexplicables, sensibilidad muscular, debilidad muscular o miositis (definida como miopatía con evidencia histológica de daño muscular), deberá considerarse la miopatía, definida como dolor muscular y/o debilidad muscular inexplicable persistente, independientemente del grado de incrementos en los niveles de creatinquinasa (CK). Deberá advertirse a los pacientes que comuniquen rápidamente cualquier achaque, dolor, sensibilidad o debilidad muscular inexplicable persistente. Si se notifica cualquiera de estos síntomas, deberá realizarse un examen muscular detallado con el fin de evaluar la función muscular. Deberá interrumpirse el tratamiento con telbivudina si se diagnostica miopatía.
Se desconoce si el riesgo de miopatía durante el tratamiento con telbivudina, incrementa con la administración concomitante de otros medicamentos asociados con miopatía (p.ej. estatinas, fibratos o ciclosporina). Los médicos que consideren el tratamiento concomitante con otros medicamentos asociados con miopatía deberán valorar cuidadosamente los beneficios y riesgos potenciales y deberán controlar a los pacientes para detectar cualquier signo o síntoma sugestivo de miopatía.
Neuropatía periférica
Se han notificado casos poco frecuentes de neuropatía periférica en pacientes tratados con telbivudina. Si existe sospecha de neuropatía periférica, debe reconsiderarse el tratamiento con telbivudina (ver sección 4.8).
Se ha observado en un ensayo un mayor riesgo de desarrollar neuropatía periférica cuando la telbivudina y el interferón alfa-2a pegilado se administraron conjuntamente (ver sección 4.5). No se puede excluir este aumento del riesgo con otro interferón alfa (pegilado o estándar). Asimismo, no se ha establecido hasta el momento el beneficio de telbivudina en combinación con interferón alfa (pegilado o estándar). Por lo tanto, la combinación de telbivudina con interferón alfa pegilado o estándar está contraindicada (ver sección 4.3).
Función renal
La telbivudina se elimina principalmente por vía renal, por lo tanto, se recomienda un ajuste del intervalo entre dosis en aquellos pacientes que presenten un aclaramiento de creatinina <50 ml/min, incluidos los pacientes en hemodiálisis. No se ha evaluado clínicamente la efectividad del ajuste del intervalo de tiempo entre dosis. Por lo tanto, deberá controlarse estrechamente la respuesta virológica en pacientes en los que se aumente el intervalo entre dosis (ver secciones 4.2 y 5.2).
Pacientes con cirrosis sin descompensación
Debido a que los datos disponibles son limitados (aproximadamente el 3% de los pacientes incluidos en los ensayos presentaron cirrosis), la telbivudina deberá utilizarse con especial precaución en pacientes con cirrosis. Estos pacientes deberán controlarse estrechamente en cuanto a los parámetros clínicos, bioquímicos y virológicos asociados con hepatitis B, durante el tratamiento y tras la interrupción del mismo.
Pacientes con cirrosis con descompensación
No hay datos de eficacia y seguridad adecuados en pacientes con cirrosis descompensada.
Pacientes con exposición previa a análogos nucleósidos/nucleótidos In vitro, la telbivudina no fue activa contra las cepas del VHB que contienen mutaciones en rtM204V/rtL180M o en rtM204I (ver sección 5.1). Telbivudina en monoterapia no es una opción para pacientes con infección confirmada por el virus de la hepatitis B resistente a lamivudina. Los pacientes que no lograron alcanzar respuesta virológica tras el tratamiento con lamivudina durante más de 24 semanas es improbable que obtengan beneficio de telbivudina en monoterapia.
Actualmente no existen datos clínicos para valorar adecuadamente el riesgo y beneficio de cambiar a telbivudina a los pacientes tratados con lamivudina que han alcanzado una supresión viral completa con este fármaco.
No se dispone de datos sobre el tratamiento de telbivudina en pacientes con el virus de la hepatitis B resistente a adefovir confirmado con mutaciones simples en rtN236T o A181V. Los resultados de valoraciones celulares mostraron que la resistencia a adefovir asociada a las sustituciones en A181V tenía una susceptibilidad a telbivudina reducida en 1,5 a 4 veces aproximadamente.
Receptores de trasplante hepático
Se desconoce la eficacia y seguridad de telbivudina en receptores de trasplante hepático.
Pacientes de edad avanzada
Los ensayos clínicos de telbivudina no incluyeron un número suficiente de pacientes >65 años de edad para determinar si responden de forma diferente a pacientes más jóvenes. En general, deberá tenerse precaución cuando se prescriba Sebivo a pacientes de edad avanzada debido a una mayor frecuencia de casos de función renal disminuida como consecuencia de una enfermedad concomitante o del uso simultáneo de otros medicamentos.
Otras poblaciones especiales
Sebivo no se ha investigado en pacientes con hepatitis B coinfectados (p.ej. pacientes coinfectados con el virus de la inmunodeficiencia humana [VIH], virus de la hepatitis C [VHC] o virus de la hepatitis D [VHD]).
General
Deberá advertirse a los pacientes que el tratamiento con Sebivo no ha demostrado que reduzca el riesgo de transmisión del VHB a otras personas a través de contacto sexual o por contaminación de la sangre.
No se recomienda utilizar conjuntamente telbivudina y lamivudina ya que en un ensayo de fase II se observó que la respuesta al tratamiento con la terapia combinada de telbivudina y lamivudina era inferior a la observada con telbivudina en monoterapia.
Actualmente no se dispone de datos de eficacia y seguridad sobre la combinación de otros antivirales con telbivudina.
4.5 Interacción con otros medicamentos y otras formas de interacción
Como la telbivudina se elimina principalmente por vía renal, la coadministración de Sebivo con medicamentos que alteren la función renal (como aminoglicósidos, diuréticos con acción sobre el asa de Henle, compuestos de platino, vancomicina, anfotericina B) puede alterar las concentraciones plasmáticas de telbivudina y/o del medicamento coadministrado. La combinación de telbivudina con estos medicamentos deberá utilizarse con precaución. La farmacocinética de telbivudina en estado estacionario no se alteró tras la administración de dosis múltiples en combinación con lamivudina, adefovir dipivoxil, tenofovir disoproxil fumarato, ciclosporina o interferón alfa-2a pegilado. Asimismo, telbivudina no modifica la farmacocinética de lamivudina, adefovir dipivoxil, tenofovir disoproxil fumarato o ciclosporina. No se puede extraer una conclusión definitiva acerca de los efectos de telbivudina sobre la farmacocinética del interferón pegilado debido a la elevada variabilidad interindividual de las concentraciones del interferón alfa-2a pegilado. En un ensayo clínico en el cual se investiga la combinación de telbivudina, 600 mg al día, con interferón alfa-2a pegilado, 180 microgramos una vez a la semana por vía subcutánea, se ha observado que esta combinación está asociada con un mayor riesgo de desarrollar neuropatía periférica. Se desconoce el mecanismo por el cual se producen estos acontecimientos (ver sección 4.4). La combinación de telbivudina con cualquier producto que contenga interferón alfa está contraindicada (ver sección 4.3).
La telbivudina no es un sustrato, ni es inhibidor o inductor del sistema enzimático del citocromo P450 (CYP450) (ver sección 5.2). Por lo tanto, el potencial de interacción de los fármacos mediados por el CYP450 con Sebivo es bajo.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Los estudios en animales no muestran efectos dañinos directos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo posnatal (ver sección 5.3). Los estudios en ratas y conejos gestantes demostraron que telbivudina atraviesa la placenta. Los estudios en conejos gestantes mostraron parto prematuro y/o aborto secundario a toxicidad materna.
Los datos clínicos limitados (menos de 300 resultados de embarazo) tras la exposición a telbivudina durante el primer trimestre del embarazo no indican toxicidad malformativa y un elevado número de datos (más de 1.000 resultados de embarazo) tras la exposición durante el segundo y tercer trimestre no indican toxicidad fetal/neonatal.
Sebivo únicamente deberá utilizarse durante el embarazo si el beneficio para la madre supera el riesgo potencial para el feto.
Las publicaciones muestran que la exposición a la telbivudina en el segundo y/o tercer trimestre del embarazo reduce el riesgo de transmisión del VHB de la madre al hijo si la telbivudina se administra junto a la inmunoglobulina de la Hepatitis B y la vacuna de la Hepatitis B.
Lactancia
La telbivudina se excreta en la leche de las ratas. Se desconoce si la telbivudina se excreta en la leche humana. Las mujeres no deberán amamantar durante el tratamiento con Sebivo.
Fertilidad
No existen datos clínicos acerca de los efectos de telbivudina sobre la fertilidad masculina o femenina. En los estudios de toxicidad reproductiva en animales adultos, la fertilidad se vio ligeramente reducida cuando ambas ratas, macho y hembra, recibieron telbivudina. Los efectos adversos sobre la fertilidad fueron superiores en un estudio separado en animales jóvenes cuando ambos sexos recibieron telbivudina (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios sobre la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas Resumen del perfil de seguridad
La valoración de las reacciones adversas está basada principalmente en dos ensayos, NV-02B-007 (GLOBE) y NV-02B-015, en los cuales 1.699 pacientes con hepatitis B crónica recibieron tratamiento doble ciego con 600 mg/día de telbivudina (n = 847) o lamivudina (n = 852) durante 104 semanas.
En los ensayos clínicos de 104 semanas, las reacciones adversas notificadas se clasificaron normalmente según la gravedad de leves a moderadas. Las reacciones adversas más frecuentes fueron elevaciones de creatinquinasa en sangre de grado 3 ó 4 (6,8%), fatiga (4,4%), cefalea (3,0%) y náuseas (2,6%).
Lista tabulada de reacciones adversas
En la Tabla 2 se enumeran las reacciones adversas de acuerdo con el sistema de clasificación de órganos y frecuencias MedDRA, utilizando las siguientes definiciones: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo frecuencia.
|
Trastornos del metabolismo y de la nutrición | |
|
Raras* |
Acidosis láctica como una reacción secundaria, a menudo asociada con estados graves (p.ej. fallo multiorgánico o sepsis) |
|
Trastornos del sistema nervioso | |
|
Frecuentes |
Mareos, cefalea |
|
Poco frecuentes |
Neuropatía periférica, disgeusia, hipoestesia, parestesia, ciática |
|
Trastornos respiratorios, torácicos y mediastínicos | |
|
Frecuentes |
Tos |
|
Trastornos gastrointestinales | |
|
Frecuentes |
Diarrea, aumento de lipasa sanguínea, náuseas, dolor abdominal |
|
Trastornos de la piel y del tejido subcutáneo | |
|
Frecuentes |
Erupción |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |
|
Poco frecuentes |
Miopatía/miositis, artralgia, mialgia, dolor en las extremidades, dolor de espalda, espasmo muscular, dolor en el cuello, dolor en el costado |
|
Raras* |
Rabdomiolisis |
|
Trastornos generales y alteraciones en el lugar de administración | |
|
Frecuentes |
Fatiga |
|
Poco frecuentes |
Malestar |
|
Exploraciones complementarias | |
|
Frecuentes |
Aumento de la creatinfosfoquinasa sanguínea, aumento de la alanina aminotransferasa sanguínea, aumento de la amilasa sanguínea |
|
Poco frecuentes |
Aumento de la aspartato aminotransferasa |
* Esta reacción adversa se identificó durante la vigilancia post-comercialización pero no se
observó en los ensayos clínicos controlados. La categoría de frecuencia se estimó a partir de un cálculo estadístico basado en el número total de pacientes expuestos a telbivudina en los ensayos clínicos (n = 8.914).
Descripción de reacciones adversas seleccionadas
Elevación de creatinquinasa
En el análisis en conjunto de NV-02B-007 (GLOBE) y NV-02B-015, en las 104 semanas de tratamiento se observaron elevaciones de CK de grado 3 ó 4 (>7x LSN) en el 12,6% de los pacientes tratados con telbivudina (n = 847) y en el 4,0% de los pacientes tratados con lamivudina (n = 846). La mayoría de las elevaciones de CK fueron asintomáticas y por lo general, los valores de CK disminuyeron en la siguiente visita con el tratamiento continuado.
Elevaciones transitorias de ALT
La incidencia durante el tratamiento de elevaciones transitorias de alanina aminotransferasa (ALT) en los dos grupos de tratamiento de acuerdo con la definición (elevación de ALT >2x valor basal y >10x LSN) de la AASLD (American Association for the Study of Liver Diseases) se describe además en la Tabla 3.
Tabla 3 Resumen de las elevaciones transitorias de ALT durante el tratamiento - Ensayos NV-02B-007 (GLOBE) y NV-02B-015 en conjunto
|
Elevación transitoria de ALT: Elevación de ALT >2x valor basal y >10x LSN |
Lamivudina n/N (%) |
Telbivudina n/N (%) |
|
Total |
67/852 (7,9) |
41/847 (4,8) |
|
Del valor basal a la semana 24 |
25/852 (2,9) |
25/847 (3,0) |
|
De la semana 24 al final del ensayo |
44/837 (5,3) |
17/834 (2,0) |
Se recomienda la monitorización periódica de la función hepática durante el tratamiento (ver sección 4.4).
Exacerbaciones de la hepatitis B tras la interrupción del tratamiento
Se han notificado exacerbaciones agudas graves de hepatitis B en pacientes que han interrumpido el tratamiento contra la hepatitis B, incluyendo telbivudina (ver sección 4.4).
La incidencia postratamiento de elevaciones transitorias de alanina aminotransferasa (ALT) en los dos grupos de tratamiento se describe además en la Tabla 4.
Tabla 4 Resumen de las elevaciones transitorias postratamiento de ALT - Ensayos NV-02B-007 (GLOBE) y NV-02B-015 en conjunto
|
Lamivudina |
Telbivudina | |
|
Elevación transitoria de ALT |
n/N (%) |
n/N (%) |
|
Elevación de ALT >2x valor basal y >10x LSN |
10/180 (5,6) |
9/154 (5,8) |
Resultados a las 208 semanas
Tras 104 semanas de tratamiento con telbivudina, el 78% de los pacientes (530/680) del ensayo NV-02B-007 (GLOBE) y el 82% (137/167) de los pacientes del ensayo NV-02B-015 se incluyeron en el ensayo de extensión CLDT600A2303 (ver sección 5.1) para continuar con el tratamiento durante un máximo de 208 semanas. La población de seguridad a largo plazo constaba de 655 pacientes incluyendo 518 del ensayo NV-02B-007 (GLOBE) y 137 del ensayo NV-02B-015. El perfil de seguridad global del análisis en conjunto de un máximo de 104 y de 208 semanas fue similar. El 15,9% de los pacientes tratados con telbivudina durante 208 semanas presentaron nuevas elevaciones de CK de grado 3 ó 4 ocurrieron. La mayoría de las elevaciones de CK de grado 3 ó 4 fueron asintomáticas y transitorias.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se dispone de información de sobredosis intencionada de telbivudina, pero un paciente recibió una sobredosis no intencionada la cual fue asintomática. Dosis ensayadas de hasta 1.800 mg/día, tres veces superior a la dosis diaria recomendada, han sido bien toleradas. No se ha determinado la dosis máxima tolerada de telbivudina. En caso de sobredosis, deberá interrumpirse el tratamiento con Sebivo y proporcionar un tratamiento de apoyo general adecuado, según proceda.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antivirales de uso sistémico, nucleósidos y nucleótidos inhibidores de la transcriptasa inversa, código ATC: J05AF11
Mecanismo de acción
La telbivudina es un análogo nucleósido sintético de la timidina, con actividad contra la ADN polimerasa del virus de la hepatitis B (VHB). Tras ser fosforilada eficientemente por cinasas celulares se convierte en el trifosfato activo, cuya semivida intracelular es de 14 horas. La telbivudina-5'-trifosfato inhibe la ADN polimerasa (transcriptasa inversa) del VHB al competir con el sustrato natural, timidina 5'-trifosfato. La incorporación de la telbivudina-5'-trifosfato en el ADN vírico causa la finalización de la cadena de ADN, dando como resultado la inhibición de la replicación del VHB.
Efectos farmacodinámicos
La telbivudina es un inhibidor de la síntesis de ambas cadenas del VHB, la primera (CI50 = 0,4-1,3 pM) y la segunda (CI50 = 0,12-0,24 pM) y muestra una preferencia distintiva por la inhibición de la producción de la segunda cadena. Por el contrario, la telbivudina-5'-trifosfato, a concentraciones de hasta 100 pM, no inhibe las ADN polimerasas celulares a, p o y. En los ensayos relacionados con la estructura miticondrial, con el contenido y la función del ADN, la telbivudina no ejerció efectos tóxicos apreciables a concentraciones de hasta 10 pM y no incrementó la producción de ácido láctico in vitro.
Se evaluó la actividad antivírica in vitro de la telbivudina en la línea celular 2.2.15 de hematoma humano expresada por el VHB. La concentración de telbivudina que inhibió eficazmente el 50% de la síntesis vírica (CI50) fue de 0,2 pM aproximadamente. La actividad antivírica de la telbivudina es específica frente al virus de la hepatitis B y los hepadnavirus relacionados. La telbivudina no fue activa frente al VIH in vitro. No se ha evaluado en ensayos clínicos la ausencia de actividad de telbivudina frente al VIH.
Experiencia clínica
La seguridad y eficacia del tratamiento a largo plazo (104 semanas) de Sebivo se evaluó en dos ensayos clínicos controlados con tratamiento activo que incluyeron 1.699 pacientes con hepatitis B crónica (NV-02B-007 (GLOBE) y NV-02B-015).
Ensayo NV-02B-007 (GLOBE)
El ensayo NV-02B-007 (GLOBE) es un ensayo de fase III multinacional, doble ciego, randomizado, de telbivudina comparado con lamivudina durante un periodo de tiempo de 104 semanas en 1.367 pacientes HBeAg positivos y HBeAg negativos con hepatitis B crónica que nunca antes habían recibido terapia nucleosídica. La mayoría de la población incluida en el ensayo era asiática. Los genotipos de VHB más frecuentes fueron el B (26%) y el C (51%). Se trataron con telbivudina un pequeño número de pacientes caucásicos (98 en total). El análisis principal de datos se realizó después de que todos los pacientes cumplieron la semana 52 de tratamiento.
Pacientes HBeAgpositivos: La edad media de los pacientes fue 32 años, el 74% eran de sexo masculino, el 82% asiáticos, el 12% caucásicos y el 6% habían recibido tratamiento previo con interferón alfa.
Pacientes HBeAg negativos: La edad media de los pacientes fue 43 años, el 79% eran de sexo masculino, el 65% asiáticos, el 23% caucásicos y el 11% habían recibido tratamiento previo con interferón alfa.
Resultados clínicos a la semana 52
Las variables de eficacia clínicas y virológicas se evaluaron por separado en las poblaciones de pacientes HBeAg positivos y HBeAg negativos. La variable principal de la respuesta terapéutica fue una variable serológica compuesta que requiere la supresión del ADN del VHB hasta <5 log10 copias/ml junto con la pérdida de HBeAg sérico o normalización de ALT. Las variables secundarias incluyeron respuesta histológica, normalización de ALT y varias mediciones de la eficacia antivírica.
Independientemente de las características basales, la mayoría de los pacientes que tomaron Sebivo mostraron respuesta histológica, virológica, bioquímica y serológica al tratamiento. Los niveles basales de ALT >2x LSN y ADN del VHB basales <9 log10 copias/ml se asociaron con índices más elevados de seroconversión del HBeAg en pacientes HBeAg positivos. Los pacientes que alcanzaron niveles de ADN del VHB <3 log10 copias/ml en la semana 24 presentaron respuesta óptima al tratamiento; por el contrario, pacientes con niveles de ADN del VHB >4 log10 copias/ml a las 24 semanas tuvieron resultados menos favorables a la semana 52.
En pacientes HBeAg positivos, la telbivudina fue superior a lamivudina en cuanto a la respuesta terapéutica (75,3% vs 67,0% de respondedores; p = 0,0047). En pacientes HBeAg negativos, la telbivudina resultó no inferior a lamivudina (75,2% y 77,2% de respondedores; p = 0,6187). La etnicidad caucásica se asoció con una respuesta inferior al tratamiento con los dos agentes antivirales utilizados en el ensayo NV-02B-007 (GLOBE); sin embargo, la población de pacientes caucásicos fue muy limitada (n = 98).
En la semana 24, 203 pacientes HBeAg positivos y 177 HBeAg negativos alcanzaron niveles de ADN del VHB no detectables. De los pacientes HBeAg positivos, el 95% alcanzó niveles de ADN del VHB no detectables, el 39% alcanzó seroconversión de HBeAg, el 90% alcanzó normalización de ALT en la semana 52 y el 0,5% presentó resistencia en la semana 48. De forma similar, de los pacientes HBeAg negativos, el 96% alcanzó niveles de ADN del VHB no detectables, el 79% alcanzó normalización de ALT en la semana 52 y el 0% presentó resistencia en la semana 48.
En la Tabla 5 se muestran los resultados de las mediciones virológicas, bioquímicas y serológicas seleccionadas y en la Tabla 6 se muestra la respuesta histológica.
|
Parámetro de respuesta |
HBeAg positivos (n = 921) |
HBeAg negativos (n = 446) | ||
|
Telbivudina 600 mg (n = 458) |
Lamivudina 100 mg (n = 463) |
Telbivudina 600 mg (n = 222) |
Lamivudina 100 mg (n = 224) | |
|
Reducción media del ADN del VHB respecto al valor basal (log10 copias/ml) Media ± EEM1,2,3 |
-6,45 (0,11) * |
-5,54 (0,11) |
-5,23 (0,13) * |
-4,40 (0,13) |
|
Pacientes con ADN del VHB indetectable por PCR (en %) |
60%* |
40% |
88%* |
71% |
|
Normalización de ALT4 |
77% |
75% |
74% |
79% |
|
Seroconversión de HBeAg4 |
23% |
22% |
- |
- |
|
Pérdida de HBeAg5 |
26% |
23% |
- |
- |
1 EEM: Error estándar de la media
2 Valoración de PCR COBAS Amplicor® de Roche (límite inferior de cuantificación <300 copias/ml).
3 HBeAg positivos n = 443 y 444, HBeAg negativos n = 219 y 219, para los grupos de telbivudina y lamivudina, respectivamente. Las diferencias de población se deben a interrupciones en el tratamiento de pacientes del ensayo y a la falta de valoración del ADN del VHB en la semana 52.
4 HBeAg positivos n = 440 y 446, HBeAg negativos n = 203 y 207, para los grupos de telbivudina y lamivudina, respectivamente. Se evaluó la normalización de ALT únicamente en pacientes con ALT > LSN en el periodo basal.
5 n = 432 y 442, para los grupos de telbivudina y lamivudina, respectivamente. Se evaluó la seroconversión y la pérdida de HBeAg únicamente en los pacientes con cantidades detectables de HBeAg en el periodo basal.
*p < 0,0001
Mejoría histológica y cambio de puntuación en la escala de Fibrosis de Ishak en la semana 52 en el ensayo NV-02B-007 (GLOBE)
|
HBeAg positivo (n = 921) |
HBeAg negativo (n = 446) | |||
|
Telbivudina |
Lamivudina |
Telbivudina |
Lamivudina | |
|
600 mg (n = 384)1 |
100 mg (n = 386)1 |
600 mg (n = 199)1 |
100 mg (n = 207)1 | |
|
Respuesta histológica2 | ||||
|
Mejoría |
71%* |
61% |
71% |
70% |
|
Ausencia de mejoría |
17% |
24% |
21% |
24% |
|
Puntuación en la escala de Fibrosis de Ishak3 | ||||
|
Mejoría |
42% |
47% |
49% |
45% |
|
Sin cambios |
39% |
32% |
34% |
43% |
|
Deterioro |
8% |
7% |
9% |
5% |
|
Ausencia de biopsia en la semana 52 |
12% |
15% |
9% |
7% |
|
1 Pacientes que recibieron > una dosis del medicamento de estudio, con biopsias hepáticas basales evaluables y puntuación basal del Índice de Actividad Histológica (IAH) de Knodell >3. 2 Respuesta histológica definida como disminución >2 puntos en la puntuación de actividad Necroinflamatoria de Knodell sin deterioro de la puntuación de Fibrosis de Knodell. 3 En la puntuación de Fibrosis de Ishak, la mejoría de definió como reducción >1 punto en la | ||||
|
puntuación de Fibrosis de Ishak desde el valor basal hasta la semana 52. | ||||
|
*p = 0,0024 | ||||
Resultados clínicos a la semana 104
En general, los resultados clínicos a la semana 104 en los pacientes tratados con telbivudina fueron consistentes con los obtenidos a la semana 52, demostrando durabilidad de las respuestas de eficacia para los pacientes tratados con telbivudina con tratamiento continuado.
Entre los pacientes HBeAg positivos, la respuesta terapéutica (63% vs 48%; p < 0,0001) y las variables secundarias principales (reducción media del ADN del VHB log10: -5,74 vs -4,42; p < 0,0001, indetectabilidad del ADN del VHB: 56% vs 39%; p < 0,0001 y normalización de ALT del 70% vs 62%) demostraron una mayor diferencia a la semana 104 entre telbivudina y lamivudina, respectivamente. También se observó para telbivudina una tendencia a presentar valores más elevados de pérdida de HBeAg (35% vs 29%) y seroconversión (30% vs 25%). Además, en el subgrupo de pacientes con niveles basales de ALT >2x LSN (320), una proporción significativamente superior de pacientes tratados con telbivudina en relación a los pacientes tratados con lamivudina alcanzaron seroconversión de HBeAg a la semana 104 (36% vs 28%, respectivamente).
Entre los pacientes HBeAg negativos, las diferencias en la respuesta terapéutica (78% vs 66%) y las variables secundarias principales (reducción media del ADN del VHB logJ0: -5,00 vs -4,17, e indetectabilidad del ADN del VHB: 82% vs 57%; p < 0,0001) fueron más elevadas para telbivudina hasta la semana 104. Las tasas de normalización de ALT (78% vs 70%) continuaron siendo más elevadas en la semana 104.
Predictibilidad a la semana 24
En la semana 24, 203 pacientes HBeAg positivos (44%) y 177 pacientes HBeAg negativos (80%) tratados con telbivudina, alcanzaron niveles indetectables de ADN del VHB.
Para ambos tipos de pacientes HBeAg positivos y HBeAg negativos, los resultados del ADN del VHB a la semana 24 fueron predecibles de los resultados favorables a largo plazo. Los pacientes tratados con telbivudina que obtuvieron ADN del VHB indetectable por PCR a la semana 24, presentaron las tasas más altas de indetectabilidad del ADN del VHB y seroconversión de HBeAg (en pacientes HBeAg positivos), y las tasas globales más bajas de reactivación virológica a la semana 104.
En la Tabla 7 se presentan los resultados finales a la semana 104, en base al nivel de ADN del VHB en la semana 24 para los pacientes tanto HBeAg positivos como HBeAg negativos.
Tabla 7 Variables de eficacia principales a la semana 104 en base a los niveles séricos de ADN del VHB a la semana 24, en pacientes tratados con telbivudina en el ensayo NV-02B-007 (GLOBE)
|
ADN del VHB a la semana 24 |
Resultados para las variables de eficacia principales a la semana 104 en base a los resultados a la semana 24 | ||||
|
Respuesta terapéutica n/N (%) |
Indetectabilida d del ADN del VHB n/N (%) |
Seroconversió n de HBeAg n/N (%) |
Normalización de ALT n/N (%) |
Reactivación virológica* n/N (%) | |
|
HBeAg positivo | |||||
|
<300 copias/ml |
172/203 (85) |
166/203 (82) |
84/183 (46) |
160/194 (82) |
22/203 (11) |
|
300 copias/ml a <3 logi0 copias/ml |
36/57 (63) |
35/57 (61) |
21/54 (39) |
40/54 (74) |
18/57 (32) |
|
>3 log10 copias/ml |
82/190 (43) |
54/190 (28) |
23/188 (12 ) |
106/184 (58) |
90/190 (47) |
|
HBeAg negativo | |||||
|
<300 copias/ml |
146/177 (82) |
156/177 (88) |
N/A |
131/159 (82) |
11/177 (6) |
|
300 copias/ml a <3 log10 copias/ml |
13/18 (72) |
14/18 (78) |
N/A |
13/17 (76) |
4/18 (22) |
|
>3 log10 copias/ml |
13/26 ( 50) |
12/26 (46) |
N/A |
14/26 (54) |
12/26 ( 46) |
N/A = no aplica
* Reactivación virológica: “1 log por encima de nadir” definición valorada a la semana 104
Ensayo NV-02B-015
En el ensayo NV-02B-015 se confirmaron los resultados de eficacia y seguridad del ensayo NV-02B-007 (GLOBE). Es un ensayo de fase III, doble ciego, randomizado, de telbivudina 600 mg una vez al día comparado con lamivudina 100 mg una vez al día durante un periodo de tratamiento de 104 semanas en 332 pacientes chinos HBeAg positivos y HBeAg negativos con hepatitis B crónica que nunca antes habían recibido terapia nucleosídica.
Ensayo CLDT600A2303 - Resultados clínicos de 208 semanas
El ensayo CLDT600A2303 es un ensayo abierto de extensión de 104 semanas de duración en pacientes con hepatitis B crónica compensada que habían sido tratados previamente con telbivudina durante 2 años, incluyendo pacientes de los ensayos NV-02B-007 (GLOBE) y NV-02B-015, y que aporta datos de eficacia y seguridad tras 156 y 208 semanas de tratamiento continuado con telbivudina. Los pacientes con ADN del VHB indetectable en la semana 24 obtuvieron mejores resultados a las semanas 156 y 208 (Tabla 8).
Tabla 8 Análisis de la eficacia de los datos conjuntos de los ensayos NV-02B-007 (GLOBE), NV-02B-015 y CLDT600A2303
|
Semana 52 |
Semana 104 |
Semana 156 |
Semana 208 | |
|
Pacientes HBeAg positivos (n = 293*) | ||||
|
ADN del VHB indetectable sostenido (<300 copias/ml) |
70,3% (206/293) |
77,3% (218/282) |
75,0% (198/264) |
76,2% (163/214) |
|
ADN del VHB indetectable sostenido (<300 copias/ml) con ADN del VHB indetectable a la semana 24 |
99,4% (161/162) |
94,9% (150/158) |
86,7% (130/150) |
87,9% (109/124) |
|
Tasas de seroconversión de HBeAg acumuladas (%) |
27,6% (81/293) |
41,6% (122/293) |
48,5% (142/293) |
53,2% (156/293) |
|
Tasas de seroconversión de HBeAg acumuladas en pacientes con ADN del VHB indetectable a la semana 24 (%) |
40,1% (65/162) |
52,5% (85/162) |
59,3% (96/162) |
65,4% (106/162) |
|
Normalización de ALT sostenida |
81,4% (228/280) |
87,5% (237/271) |
82,9% (209/252) |
86,4% (178/106) |
|
Pacientes HBeAg negativos (n = 209*) | ||||
|
ADN del VHB indetectable sostenido (<300 copias/ml) |
95,2% (199/209) |
96,5% (195/202) |
84,7% (160/189) |
86,0% (141/164) |
|
ADN del VHB indetectable sostenido (<300 copias/ml) con ADN del VHB indetectable a la semana 24 |
97,8% (175/179) |
96,5% (166/172) |
86,7% (143/165) |
87,5% (126/144) |
|
Normalización de ALT sostenida |
80,3% (151/188) |
89,0% (161/181) |
83,5% (142/170) |
89,6% (129/144) |
* La población sin resistencia viral a la entrada del ensayo CLDT600A2303 constaba de 502 pacientes (293 HBeAg positivos y 209 HBeAg negativos).
Ensayo CLDT600ACN04E1 - Impacto del tratamiento en la histología del hígado En el ensayo CLDT600ACN04E1, se evaluaron los cambios en la histología del hígado de 57 pacientes con biopsias hepáticas pareadas disponibles al inicio y tras un tratamiento de 260,8 semanas de media (38 pacientes HBeAg positivos y 19 HBeAg negativos).
• La puntuación media de actividad necroinflamatoria de Knodell de 7,6 (DE 2,9) al inicio mejoró (p < 0,0001) hasta 1,4 (DE 0,9) con un cambio medio de -6,3 (DE 2,8). En el 98,2% (56/57) de los pacientes se observó una puntuación de actividad necroinflamatoria de Knodell <3 (necroinflamación mínima o nula).
• La puntuación de Ishak media de 2,2 (DE 1,1) al inicio mejoró (p < 0,0001) hasta 0,9 (DE 1,0) con un cambio medio de -1,3 (DE 1,3). En el 84,2% (48/57) de los pacientes se observó una puntuación de fibrosis de Ishak <1 (fibrosis mínima o nula).
Los cambios en las puntuaciones de Ishak y de actividad necroinflamatoria de Knodell fueron similares para los pacientes HBeAg positivos y los HBeAg negativos.
CLDT600A2303 - Duración de las respuestas HBeAg una vez interrumpido el tratamiento El ensayo CLDT600A2303 incluyó pacientes HBeAg positivos de los ensayos NV-02B-007 (GLOBE) o NV-02B-015 para el seguimiento una vez interrumpido el tratamiento. Estos pacientes completaron >52 semanas de tratamiento con telbivudina y en la última visita durante el tratamiento mostraron pérdida de HBeAg durante >24 semanas con ADN del VHB <5 logi0 copias/ml. La duración media del tratamiento fue de 104 semanas. Tras un periodo medio de seguimiento sin tratamiento de 120 semanas, la mayoría de pacientes tratados con telbivudina HBeAg positivos mostraron una pérdida de HBeAg sostenida (83,3%; 25/30) y una seroconversión de HBeAg sostenida (79,2%; 19/24). Los pacientes con seroconversión de HBeAg sostenida presentaron un valor medio de ADN del VHB de 3,3 log10 copias/ml; y el 73,7% presentaron un ADN del VHB <4 log10 copias/ml.
Resistencia clínica
En el ensayo NV-02B-007 (GLOBE; n = 680) se realizó la prueba de resistencia genotípica en pacientes con rechazo virológico (incremento confirmado de >1 log10 copias/ml de ADN del VHB desde nadir).
Entre los pacientes HBeAg positivos y HBeAg negativos, el 5% (23/458) y 2% (5/222) respectivamente presentaron rechazo virológico con mutaciones de resistencia al VHB detectables en la semana 48.
Ensayos NV-02B-007 (GLOBE) y CLDT600A2303 - tasas de resistencia genotípica acumuladas El análisis original de la resistencia genotípica acumulada a las semanas 104 y 208 se basó en la población por ITT e incluyó a todos los pacientes que continuaron con el tratamiento durante 4 años, independientemente de los niveles de ADN del VHB. De los 680 pacientes tratados con telbivudina e incluidos inicialmente en el ensayo pivotal NV-02B-007 (GLOBE), 517 (76%) se incluyeron en el ensayo CLDT600A2303 para continuar el tratamiento con telbivudina durante un máximo de 208 semanas. De estos 517 pacientes, 159 pacientes (HBeAg positivo=135, HBeAg negativo=24) presentaron ADN del VHB detectable.
Las proporciones genotípicas acumuladas en la semana 104, fueron del 25,1% (115/458) para los pacientes HBeAg positivo y del 10,8% (24/222) para los pacientes HBeAg negativo.
En la población por ITT global, las tasas de resistencia acumuladas en el año 4 para los pacientes HBeAg positivos y HBeAg negativos fueron del 40,8% (131/321) y del 18,9% (37/196) respectivamente.
Las tasas de resistencia genotípica acumuladas también se evaluaron mediante la aplicación de un modelo matemático en el que sólo se consideran pacientes con ADN del VHB indetectable al inicio del año respectivo. En este análisis las tasas de resistencia acumuladas en el año 4 fueron del 22,3% para los pacientes HBeAg positivos y del 16,0% para los pacientes HBeAg negativos.
Cuando consideramos pacientes con reactivación viral en la semana 104 en el ensayo NV-02B-007 (GLOBE), la proporción de resistencias fue inferior en pacientes con ADN del VHB <300 copias/ml en la semana 24 que en pacientes con ADN del VHB >300 copias/ml en la semana 24. En pacientes HBeAg positivo con ADN del VHB <300 copias/ml en la semana 24, la resistencia fue del 1%
(3/203) a las 48 semanas y del 9% (18/203) en la semana 104, mientras que en pacientes con ADN del VHB >300 copias/ml, la resistencia fue del 8% (20/247) a las 48 semanas y del 39% (97/247) en la semana 104. En pacientes HBeAg negativo con ADN del VHB <300 copias/ml en la semana 24, la resistencia fue del 0% (0/177) a las 48 semanas y del 5% (9/177) en la semana 104, mientras que en pacientes con ADN del VHB >300 copias/ml, la resistencia fue del 11% (5/44) a las 48 semanas y del 34% (15/44) en la semana 104.
Patrón de mutación genotípica y resistencia cruzada
Los análisis genotípicos de 203 pares de muestras evaluables con ADN del VHB >1.000 copias/ml a la semana 104 (NV-02B-007 (GLOBE)) demostraron que la mutación primaria asociada con la resistencia a telbivudina fue rtM204I, a menudo asociada con mutaciones en rtL180M y rtL80I/V e infrecuentemente con rtV27A, rtL82M, rtV173L, rtT184I y rtA200V. Los factores basales asociados con el desarrollo de resistencia genotípica al fármaco, incluyeron: tratamiento con lamivudina, ADN del VHB basal más elevado, ALT sérico basal más bajo, y aumento del peso corporal/BMI. Los parámetros de respuesta en tratamiento a la semana 24, que predijeron la aparición del virus resistente al fármaco en la semana 104 fueron ADN del VHB >300 copias/ml y elevación del ALT sérico.
Los análisis genotípicos de 50 aislados del VHB de pacientes tratados con telbivudina en la semana 208 (CLDT600A2303) revelaron un perfil de resistencia similar al notificado en la semana 104. Las conversiones en la posición 80, 180 y posiciones polimórficas 91, 229 se detectaron siempre en secuencias que albergaban la mutación en M204I la cual confiere resistencia genotípica.
Lo más probable es que estas mutaciones sean compensatorias. Se notificó una mutación aislada en rtM204V y dos mutaciones en rtM204I/V/M en pacientes tratados con telbivudina que experimentaron reactivación viral hasta la semana 208. No se notificó ninguna mutación nueva.
Se ha observado resistencia cruzada entre los análogos nucleósidos del VHB (ver sección 4.4). En ensayos celulares, las cepas de VHB resistentes a lamivudina que contenían la mutación en rtM204I o la mutación doble rtL180M/rtM204V, presentaron una susceptibilidad a telbivudina reducida >1.000 veces. El VHB codificado con la resistencia a adefovir asociada a las sustituciones en rtN236T o rtA181V mostró cambios en la susceptibilidad a telbivudina alrededor de 0,3- y 4-veces en cultivo celular, respectivamente (ver sección 4.4).
5.2 Propiedades farmacocinéticas
Se ha evaluado la farmacocinética de telbivudina tras dosis únicas y múltiples en voluntarios sanos y en pacientes con hepatitis B crónica. No se ha evaluado la farmacocinética de telbivudina a la dosis recomendada de 600 mg en pacientes con hepatitis B crónica. Sin embargo, la farmacocinética de telbivudina es similar entre ambas poblaciones.
Absorción
Tras la administración oral de una dosis única de 600 mg de telbivudina a voluntarios sanos (n = 42), la concentración plasmática máxima (Cmax) de telbivudina fue de 3,2 ± 1,1 pg/ml (media ± DE) y se alcanzó a una media de 3,0 horas después de la dosis. El área bajo la curva de las concentraciones plasmáticas de telbivudina en función del tiempo (AUC0.^) fue de 28,0 ± 8,5 pg*h/ml (media ± DE). En las determinaciones de exposición sistémica (Cmax, AUC) la variabilidad interindividual por lo general fue del 30% aproximadamente.
Efecto de los alimentos sobre la absorción oral
La administración de una dosis única de 600 mg con alimentos no modificó ni a la absorción ni a la exposición a telbivudina.
Distribución
In vitro, la unión de telbivudina a las proteínas plasmáticas es baja (3,3%).
Biotransformación
No se detectaron metabolitos de telbivudina tras la administración de 14C-telbivudina a los seres humanos. La telbivudina no es un sustrato, inhibidor, ni inductor del sistema enzimático del citocromo P450 (CYP450).
Eliminación
Tras alcanzar la concentración máxima, la disposición plasmática de telbivudina disminuye de forma biexponencial con una semivida de eliminación terminal (ti/2) de 41,8 ± 11,8 horas. La telbivudina se elimina principalmente por excreción urinaria como fármaco inalterado. El aclaramiento renal de telbivudina se aproxima a la tasa de filtración glomerular normal, lo que sugiere que el principal mecanismo de excreción es la filtración. Tras la administración de una dosis oral única de 600 mg de telbivudina, en un plazo de 7 días se recupera en la orina aproximadamente el 42% de la dosis administrada. Dado que la vía de eliminación predominante es la vía renal, tanto los pacientes con insuficiencia renal de moderada a grave como los pacientes en hemodiálisis requerirán un ajuste del intervalo de tiempo entre dosis (ver sección 4.2).
Linealidad/No linealidad
La farmacocinética de telbivudina es proporcional a la dosis administrada en el rango de 25 a 1.800 mg. Después de 5 a 7 días con una única administración diaria se alcanzó el estado estacionario con una acumulación aproximada de 1,5 veces en la exposición sistémica, lo que sugiere una semivida de acumulación efectiva de aproximadamente 15 horas. Tras una única administración diaria de 600 mg de telbivudina, las concentraciones plasmáticas mínimas en estado estacionario fueron de 0,2-0,3 pg/ml aproximadamente.
Poblaciones especiales
Sexo
No existen diferencias significativas relacionadas con el sexo en la farmacocinética de telbivudina.
Raza
No existen diferencias significativas relacionadas con la raza en la farmacocinética de telbivudina.
Pediatría y pacientes de edad avanzada (65 años de edad o mayores)
No se han realizado estudios farmacocinéticos en sujetos pediátricos o de edad avanzada.
Insuficiencia renal
Se ha evaluado la farmacocinética de telbivudina tras dosis únicas (200, 400 y 600 mg) en pacientes (sin hepatitis B crónica) con diversos grados de insuficiencia renal (valorado con el aclaramiento de creatinina). Según los resultados que se muestran en la Tabla 9, se recomienda ajustar el intervalo de tiempo entre las dosis de telbivudina en los pacientes que presenten un aclaramiento de creatinina <50 ml/min (ver secciones 4.2 y 4.4).
Tabla 9 Parámetros farmacocinéticos (media ± DE) de telbivudina en sujetos con diversos grados de función renal
|
Función renal (aclaramiento de creatinina en ml/min' | |||||
|
Normal (>80) (n = 8) 600 mg |
Leve (50-80) (n = 8) 600 mg |
Moderada (30-49) (n = 8) 400 mg |
Grave(<30) (n = 6) 200 mg |
ERT/ Hemodiálisis (n = 6) 200 mg | |
|
Cmax (Pg/ml) |
3,4 ± 0,9 |
3,2 ± 0,9 |
2,8 ± 1,3 |
1,6 ± 0,8 |
2,1 ± 0,9 |
|
AUC0-» (pg^h/ml) |
28,5 ± 9,6 |
32,5 ± 10,1 |
36,0 ± 13,2 |
32,5 ± 13,2 |
67,4 ± 36,9 |
|
CLRENAL (ml/min) |
126,7 ± 48,3 |
83,3 ± 20,0 |
43,3 ± 20,0 |
11,7 ± 6,7 |
- |
Pacientes con insuficiencia renal en hemodiálisis
La hemodiálisis (de hasta 4 horas) reduce la exposición sistémica a telbivudina un 23% aproximadamente. Aparte del ajuste del intervalo de tiempo entre dosis en función del aclaramiento de creatinina, no es necesario realizar ninguna modificación adicional de la dosis durante la hemodiálisis de rutina (ver sección 4.2). La telbivudina debe administrarse después de la hemodiálisis.
Insuficiencia hepática
Se ha estudiado la farmacocinética de telbivudina en pacientes (sin hepatitis B crónica) con diversos grados de insuficiencia hepática y en algunos pacientes con insuficiencia hepática descompensada. No se observaron cambios significativos en la farmacocinética de telbivudina entre los sujetos con y sin insuficiencia hepática. Los resultados de estos estudios indican que no es necesario realizar un ajuste de la dosis en pacientes con insuficiencia hepática (ver sección 4.2).
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas y genotoxicidad.
La telbivudina no mostró potencial carcinogénico. No se observaron signos de efecto tóxico directo de telbivudina en estudios estándar de toxicidad reproductiva. En conejos, dosis de telbivudina equivalentes a niveles de exposición 37 veces superiores al observado en humanos con la dosis terapéutica (600 mg) se relacionaron con una mayor incidencia de abortos y partos prematuros. Este efecto se consideró secundario a la toxicidad materna.
La fertilidad se evaluó en estudios convencionales realizados en ratas adultas, y como parte de un estudio de toxicología juvenil.
En ratas adultas, la fertilidad se vio reducida cuando ambas ratas, macho y hembra, se trataron con telvibudina a dosis de 500 ó 1000 mg/kg/día (índice de fertilidad inferior comparado con los controles concurrentes). No se observaron anormalidades en la morfología o función del esperma, ni en la histología de testículos y ovarios.
No hubo evidencia de deterioro en la fertilidad en otros estudios cuando tanto las ratas macho como hembra se trataron con dosis de hasta 2000 mg/kg/día y se aparearon con ratas no tratadas (niveles de exposición sistémica 6-14 veces aproximadamente más elevado que los alcanzados en humanos).
En el estudio de toxicología juvenil, las ratas se trataron desde el día 14 hasta el día 70 postparto y se aparearon con ratas que recibieron el mismo tratamiento (no hubo apareamiento entre hermanos). La fertilidad se redujo en las parejas que recibieron >1000 mg/kg/día como se mostró por los descensos en la fertilidad y en los índices de apareamiento, y en la tasa de concepción reducida. Sin embargo, los parámetros ováricos y uterinos de las hembras que se aparearon con éxito no se vieron afectados.
El nivel de efecto adverso no observable (NOAEL) para efectos sobre la fertilidad o parámetros de apareamiento fue de 250 mg/kg/día, lo que proporcionó niveles de exposición 2,5 a 2,8 veces más elevados que los alcanzados en humanos con función renal normal a la dosis terapéutica.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido Celulosa microcristalina Povidona
Glicolato sódico de almidón Sílice coloidal anhidra Estearato de magnesio
Cubierta pelicular del comprimido Dióxido de titanio (E171)
Macrogol
Talco
Hipromelosa
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez 3 años
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase Blisters de PVC/aluminio
Tamaños del envase: 28 ó 98 comprimidos recubiertos con película Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación Ninguna especial para su eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/388/001
EU/1/07/388/002
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 24 Abril 2007 Fecha de la última renovación: 26 Abril 2012
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
1. NOMBRE DEL MEDICAMENTO
Sebivo 20 mg/ml solución oral
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un ml contiene 20 mg de telbivudina.
Excipiente con efecto conocido: Una dosis de 600 mg (30 ml) de solución oral contiene aproximadamente 47 mg de sodio.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución oral
Solución transparente, de incolora a amarillo pálido.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Sebivo está indicado para el tratamiento de la hepatitis B crónica en pacientes adultos con enfermedad hepática compensada y signos de replicación viral, niveles séricos de alanina aminotransferasa (ALT) persistentemente elevados y signos histológicos de inflamación activa y/o fibrosis.
El inicio del tratamiento con Sebivo sólo se debe considerar cuando el uso de un agente antiviral alternativo con una barrera genética a la resistencia más alta no se encuentre disponible o no sea apropiado.
Ver sección 5.1 para los detalles del ensayo y las características específicas de los pacientes en los cuales está basada esta indicación.
4.2 Posología y forma de administración
El tratamiento debe iniciarlo un médico con experiencia en el manejo de la infección por hepatitis B crónica.
Posología
Adultos
La dosis recomendada de Sebivo es de 30 ml, que proporciona una dosis equivalente a 600 mg, una vez al día.
Monitorización durante el tratamiento
Se ha demostrado que la respuesta al tratamiento en la semana 24 es predictiva de la respuesta a largo plazo (ver Tabla 7 en la sección 5.1). Los niveles de ADN del VHB se deben monitorizar tras 24 semanas de tratamiento para asegurar la supresión viral completa (ADN del VHB inferior a 300 copias/ml). En el caso de pacientes con ADN del VHB detectable tras 24 semanas de terapia, se debe considerar una modificación del tratamiento.
Para asegurar una respuesta continuada se debe monitorizar el ADN del VHB cada 6 meses. Si en algún momento después de la respuesta inicial los pacientes obtienen un resultado positivo en la prueba del ADN del VHB, se debe considerar una modificación del tratamiento. La terapia óptima se debe guiar por la evaluación de las resistencias.
Duración del tratamiento
Se desconoce la duración óptima del tratamiento. La interrupción del tratamiento deberá considerarse de la siguiente forma:
• En pacientes HBeAg positivos sin cirrosis, el tratamiento deberá administrarse durante un mínimo de 6-12 meses después de que se confirme seroconversión de HBeAg (pérdida de HBeAg y de ADN del VHB con detección de anti-HBe) o hasta seroconversión de HBsAg o exista evidencia de pérdida de eficacia. Deberán controlarse regularmente los niveles séricos de ALT y ADN del VHB después de la interrupción del tratamiento para detectar cualquier recidiva virológica tardía.
• En pacientes HBeAg negativos sin cirrosis, el tratamiento deberá administrarse como mínimo hasta seroconversión de HBsAg o hasta que exista evidencia de falta de eficacia. En tratamientos prolongados de más de dos años, se recomienda reevaluar de forma regular para confirmar que la continuación del tratamiento seleccionado continúa siendo apropiado para el paciente.
Pacientes de edad avanzada (mayores de 65 años)
No se dispone de datos que apoyen una recomendación de dosis específica para pacientes mayores de 65 años (ver sección 4.4).
Insuficiencia renal
No es necesario ajustar la dosis recomendada de telbivudina en pacientes con un aclaramiento de creatinina >50 ml/min. Se requiere un ajuste de la dosis en pacientes que presenten un aclaramiento de creatinina <50 ml/min, incluyendo los pacientes con enfermedad renal terminal (ERT) en hemodiálisis. Tal y como se detalla a continuación en la Tabla 1, se recomienda una reducción de la dosis diaria cuando se utilice Sebivo solución oral. Si no es posible el uso de la solución oral se puede usar como alternativa Sebivo comprimidos recubiertos con película y en ese caso se deberá ajustar la dosis aumentando el intervalo de tiempo entre dosis, tal y como se detalla en la Tabla 1.
Tabla 1 Ajuste de la pauta de dosificación de Sebivo en pacientes con insuficiencia renal
|
Aclaramiento de creatinina (ml/min) |
Telbivudina 20 mg/ml solución oral Ajuste de dosis diario |
Telbivudina 600 mg comprimidos recubiertos con película Alternativa** ajuste de dosis mediante aumento del intervalo de tiempo entre dosis |
|
>50 |
600 mg (30 ml) una vez al día |
600 mg una vez al día |
|
30-49 |
400 mg (20 ml) una vez al día |
600 mg una vez cada 48 horas |
|
<30 (no requiere diálisis) |
200 mg (10 ml) una vez al día |
600 mg una vez cada 72 horas |
|
ERT* |
120 mg (6 ml) una vez al día |
600 mg una vez cada 96 horas |
* Enfermedad renal terminal
** En caso de que no sea posible el uso de la solución oral
Las modificaciones de dosis propuestas están basadas en la extrapolación y puede que no sean óptimas. No se ha evaluado clínicamente la seguridad y eficacia de estas recomendaciones de ajuste de dosis. Por lo tanto, se recomienda una cuidadosa monitorización clínica de estos pacientes.
Pacientes con enfermedad renal terminal
En los pacientes con ERT, Sebivo debe administrarse después de la hemodiálisis (ver sección 5.2).
Insuficiencia hepática
No es necesario ajustar la dosis recomendada de Sebivo en pacientes con insuficiencia hepática (ver sección 5.2).
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de Sebivo en la población pediátrica.
Forma de administración
Sebivo se administra por vía oral, con o sin alimentos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
La combinación de telbivudina con interferón alfa pegilado o estándar (ver secciones 4.4 y 4.5).
4.4 Advertencias y precauciones especiales de empleo
Las exacerbaciones agudas graves de hepatitis B crónica son relativamente frecuentes y se caracterizan por una elevación transitoria de la ALT sérica. Tras el inicio del tratamiento antiviral, la ALT sérica puede aumentar en algunos pacientes mientras que los niveles séricos de ADN del VHB descienden (ver sección 4.8). Como promedio, transcurren entre 4-5 semanas antes de que aparezca una exacerbación en pacientes tratados con telbivudina. En general, los aumentos transitorios de ALT se producen más frecuentemente en pacientes HBeAg positivos que en pacientes HBeAg negativos.
En pacientes con enfermedad hepática compensada, estas elevaciones de ALT sérico no van generalmente acompañadas de niveles elevados de bilirrubina sérica o de otros signos de descompensación hepática. El riesgo de descompensación hepática y de una exacerbación de hepatitis posterior, puede ser elevado en pacientes con cirrosis. Por lo tanto, estos pacientes deberán ser controlados estrechamente.
También se han notificado exacerbaciones de la hepatitis en pacientes que han finalizado el tratamiento de la hepatitis B. Normalmente, los aumentos transitorios de ALT postratamiento se asocian con incrementos en los niveles séricos de ADN del VHB, y la mayoría de estos casos han demostrado ser autolimitados. No obstante, también se han notificado casos de exacerbaciones graves de la enfermedad (en ocasiones mortales) postratamiento. Por lo tanto, la función hepática deberá controlarse regularmente durante un periodo mínimo de 6 meses después de la interrupción del tratamiento de la hepatitis B mediante seguimiento clínico y de laboratorio.
Acidosis láctica
Se ha notificado la aparición de acidosis láctica (en ausencia de hipoxemia) en algunos casos mortal, y normalmente asociada con hepatomegalia grave con esteatosis, con el uso de análogos nucleósidos o nucleótidos. Como la telbivudina es un análogo nucleósido, no puede excluirse este riesgo. El tratamiento con análogos nucleósidos deberá interrumpirse cuando los niveles de aminotransferasa aumenten rápidamente o cuando se produzca hepatomegalia progresiva o acidosis metabólica o láctica de etiología desconocida. Síntomas digestivos benignos, tales como náuseas, vómitos y dolor abdominal, pueden ser indicativos del desarrollo de acidosis láctica. Casos graves, algunos mortales, se asociaron con pancreatitis, insuficiencia o esteatosis hepática, insuficiencia renal y niveles más elevados de lactato sérico. Deberá tenerse precaución cuando se prescriban análogos nucleósidos a cualquier paciente (particularmente mujeres obesas) con hepatomegalia, hepatitis u otros factores de riesgo conocidos para una enfermedad hepática. Estos pacientes deberán ser vigilados estrechamente.
Efectos musculares
Se han notificado casos de miopatía y mialgia con el uso de telbivudina desde varias semanas hasta meses después de iniciar el tratamiento (ver sección 4.8). Se han notificado casos de rabdomiolisis durante el uso postcomercialización de telbivudina (ver sección 4.8).
En cualquier paciente que presente mialgias difusas inexplicables, sensibilidad muscular, debilidad muscular o miositis (definida como miopatía con evidencia histológica de daño muscular), deberá considerarse la miopatía, definida como dolor muscular y/o debilidad muscular inexplicable persistente, independientemente del grado de incrementos en los niveles de creatinquinasa (CK). Deberá advertirse a los pacientes que comuniquen rápidamente cualquier achaque, dolor, sensibilidad o debilidad muscular inexplicable persistente. Si se notifica cualquiera de estos síntomas, deberá realizarse un examen muscular detallado con el fin de evaluar la función muscular. Deberá interrumpirse el tratamiento con telbivudina si se diagnostica miopatía.
Se desconoce si el riesgo de miopatía durante el tratamiento con telbivudina, incrementa con la administración concomitante de otros medicamentos asociados con miopatía (p.ej. estatinas, fibratos o ciclosporina). Los médicos que consideren el tratamiento concomitante con otros medicamentos asociados con miopatía deberán valorar cuidadosamente los beneficios y riesgos potenciales y deberán controlar a los pacientes para detectar cualquier signo o síntoma sugestivo de miopatía.
Neuropatía periférica
Se han notificado casos poco frecuentes de neuropatía periférica en pacientes tratados con telbivudina. Si existe sospecha de neuropatía periférica, debe reconsiderarse el tratamiento con telbivudina (ver sección 4.8).
Se ha observado en un ensayo un mayor riesgo de desarrollar neuropatía periférica cuando la telbivudina y el interferón alfa-2a pegilado se administraron conjuntamente (ver sección 4.5). No se puede excluir este aumento del riesgo con otro interferón alfa (pegilado o estándar). Asimismo, no se ha establecido hasta el momento el beneficio de telbivudina en combinación con interferón alfa (pegilado o estándar). Por lo tanto, la combinación de telbivudina con interferón alfa pegilado o estándar está contraindicada (ver sección 4.3).
Función renal
La telbivudina se elimina principalmente por vía renal, por lo tanto, se recomienda un ajuste del intervalo entre dosis en aquellos pacientes que presenten un aclaramiento de creatinina <50 ml/min, incluidos los pacientes en hemodiálisis. No se ha evaluado clínicamente la efectividad del ajuste del intervalo de tiempo entre dosis. Por lo tanto, deberá controlarse estrechamente la respuesta virológica en pacientes en los que se aumente el intervalo entre dosis (ver secciones 4.2 y 5.2).
Pacientes con cirrosis sin descompensación
Debido a que los datos disponibles son limitados (aproximadamente el 3% de los pacientes incluidos en los ensayos presentaron cirrosis), la telbivudina deberá utilizarse con especial precaución en pacientes con cirrosis. Estos pacientes deberán controlarse estrechamente en cuanto a los parámetros clínicos, bioquímicos y virológicos asociados con hepatitis B, durante el tratamiento y tras la interrupción del mismo.
Pacientes con cirrosis con descompensación
No hay datos de eficacia y seguridad adecuados en pacientes con cirrosis descompensada.
Pacientes con exposición previa a análogos nucleósidos/nucleótidos In vitro, la telbivudina no fue activa contra las cepas del VHB que contienen mutaciones en rtM204V/rtL180M o en rtM204I (ver sección 5.1). Telbivudina en monoterapia no es una opción para pacientes con infección confirmada por el virus de la hepatitis B resistente a lamivudina. Los pacientes que no lograron alcanzar respuesta virológica tras el tratamiento con lamivudina durante más de 24 semanas es improbable que obtengan beneficio de telbivudina en monoterapia.
Actualmente no existen datos clínicos para valorar adecuadamente el riesgo y beneficio de cambiar a telbivudina a los pacientes tratados con lamivudina que han alcanzado una supresión viral completa con este fármaco.
No se dispone de datos sobre el tratamiento de telbivudina en pacientes con el virus de la hepatitis B resistente a adefovir confirmado con mutaciones simples en rtN236T o A181V. Los resultados de valoraciones celulares mostraron que la resistencia a adefovir asociada a las sustituciones en A181V tenía una susceptibilidad a telbivudina reducida en 1,5 a 4 veces aproximadamente.
Receptores de trasplante hepático
Se desconoce la eficacia y seguridad de telbivudina en receptores de trasplante hepático.
Pacientes de edad avanzada
Los ensayos clínicos de telbivudina no incluyeron un número suficiente de pacientes >65 años de edad para determinar si responden de forma diferente a pacientes más jóvenes. En general, deberá tenerse precaución cuando se prescriba Sebivo a pacientes de edad avanzada debido a una mayor frecuencia de casos de función renal disminuida como consecuencia de una enfermedad concomitante o del uso simultáneo de otros medicamentos.
Otras poblaciones especiales
Sebivo no se ha investigado en pacientes con hepatitis B coinfectados (p.ej. pacientes coinfectados con el virus de la inmunodeficiencia humana [VIH], virus de la hepatitis C [VHC] o virus de la hepatitis D [VHD]).
General
Deberá advertirse a los pacientes que el tratamiento con Sebivo no ha demostrado que reduzca el riesgo de transmisión del VHB a otras personas a través de contacto sexual o por contaminación de la sangre.
No se recomienda utilizar conjuntamente telbivudina y lamivudina ya que en un ensayo de fase II se observó que la respuesta al tratamiento con la terapia combinada de telbivudina y lamivudina era inferior a la observada con telbivudina en monoterapia.
Actualmente no se dispone de datos de eficacia y seguridad sobre la combinación de otros antivirales con telbivudina.
Sebivo solución oral contiene aproximadamente 47 mg de sodio por cada dosis de 600 mg (30 ml), lo que deberá tenerse en cuenta en el tratamiento de pacientes con dietas pobres en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción
Como la telbivudina se elimina principalmente por vía renal, la coadministración de Sebivo con medicamentos que alteren la función renal (como aminoglicósidos, diuréticos con acción sobre el asa de Henle, compuestos de platino, vancomicina, anfotericina B) puede alterar las concentraciones plasmáticas de telbivudina y/o del medicamento coadministrado. La combinación de telbivudina con estos medicamentos deberá utilizarse con precaución. La farmacocinética de telbivudina en estado estacionario no se alteró tras la administración de dosis múltiples en combinación con lamivudina, adefovir dipivoxil, tenofovir disoproxil fumarato, ciclosporina o interferón alfa-2a pegilado. Asimismo, telbivudina no modifica la farmacocinética de lamivudina, adefovir dipivoxil, tenofovir disoproxil fumarato o ciclosporina. No se puede extraer una conclusión definitiva acerca de los efectos de telbivudina sobre la farmacocinética del interferón pegilado debido a la elevada variabilidad interindividual de las concentraciones del interferón alfa-2a pegilado. En un ensayo clínico en el cual se investiga la combinación de telbivudina, 600 mg al día, con interferón alfa-2a pegilado, 180 microgramos una vez a la semana por vía subcutánea, se ha observado que esta combinación está asociada con un mayor riesgo de desarrollar neuropatía periférica. Se desconoce el mecanismo por el cual se producen estos acontecimientos (ver sección 4.4). La combinación de telbivudina con cualquier producto que contenga interferón alfa está contraindicada (ver sección 4.3).
La telbivudina no es un sustrato, ni es inhibidor o inductor del sistema enzimático del citocromo P450 (CYP450) (ver sección 5.2). Por lo tanto, el potencial de interacción de los fármacos mediados por el CYP450 con Sebivo es bajo.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Los estudios en animales no muestran efectos dañinos directos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo posnatal (ver sección 5.3). Los estudios en ratas y conejos gestantes demostraron que telbivudina atraviesa la placenta. Los estudios en conejos gestantes mostraron parto prematuro y/o aborto secundario a toxicidad materna.
Los datos clínicos limitados (menos de 300 resultados de embarazo) tras la exposición a telbivudina durante el primer trimestre del embarazo no indican toxicidad malformativa y un elevado número de datos (más de 1.000 resultados de embarazo) tras la exposición durante el segundo y tercer trimestre no indican toxicidad fetal/neonatal.
Sebivo únicamente deberá utilizarse durante el embarazo si el beneficio para la madre supera el riesgo potencial para el feto.
Las publicaciones muestran que la exposición a la telbivudina en el segundo y/o tercer trimestre del embarazo reduce el riesgo de transmisión del VHB de la madre al hijo si la telbivudina se administra junto a la inmunoglobulina de la Hepatitis B y la vacuna de la Hepatitis B.
Lactancia
La telbivudina se excreta en la leche de las ratas. Se desconoce si la telbivudina se excreta en la leche humana. Las mujeres no deberán amamantar durante el tratamiento con Sebivo.
Fertilidad
No existen datos clínicos acerca de los efectos de telbivudina sobre la fertilidad masculina o femenina. En los estudios de toxicidad reproductiva en animales adultos, la fertilidad se vio ligeramente reducida cuando ambas ratas, macho y hembra, recibieron telbivudina. Los efectos adversos sobre la fertilidad fueron superiores en un estudio separado en animales jóvenes cuando ambos sexos recibieron telbivudina (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios sobre la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas Resumen del perfil de seguridad
La valoración de las reacciones adversas está basada principalmente en dos ensayos, NV-02B-007 (GLOBE) y NV-02B-015, en los cuales 1.699 pacientes con hepatitis B crónica recibieron tratamiento doble ciego con 600 mg/día de telbivudina (n = 847) o lamivudina (n = 852) durante 104 semanas.
En los ensayos clínicos de 104 semanas, las reacciones adversas notificadas se clasificaron normalmente según la gravedad de leves a moderadas. Las reacciones adversas más frecuentes fueron elevaciones de creatinquinasa en sangre de grado 3 ó 4 (6,8%), fatiga (4,4%), cefalea (3,0%) y náuseas (2,6%).
Lista tabulada de reacciones adversas
En la Tabla 2 se enumeran las reacciones adversas de acuerdo con el sistema de clasificación de órganos y frecuencias MedDRA, utilizando las siguientes definiciones: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo frecuencia.
Tabla 2 Reacciones adversas
|
Trastornos del metabolismo y de la nutrición | |
|
Raras* |
Acidosis láctica como una reacción secundaria, a menudo asociada con estados graves (p.ej. fallo multiorgánico o sepsis) |
|
Trastornos del sistema nervioso | |
|
Frecuentes |
Mareos, cefalea |
|
Poco frecuentes |
Neuropatía periférica, disgeusia, hipoestesia, parestesia, ciática |
|
Trastornos respiratorios, torácicos y mediastínicos | |
|
Frecuentes |
Tos |
|
Trastornos gastrointestinales | |
|
Frecuentes |
Diarrea, aumento de lipasa sanguínea, náuseas, dolor abdominal |
|
Trastornos de la piel y del tejido subcutáneo | |
|
Frecuentes |
Erupción |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |
|
Poco frecuentes |
Miopatía/miositis, artralgia, mialgia, dolor en las extremidades, dolor de espalda, espasmo muscular, dolor en el cuello, dolor en el costado |
|
Raras* |
Rabdomiolisis |
|
Trastornos generales y alteraciones en el lugar de administración | |
|
Frecuentes |
Fatiga |
|
Poco frecuentes |
Malestar |
|
Exploraciones complementarias | |
|
Frecuentes |
Aumento de la creatinfosfoquinasa sanguínea, aumento de la alanina aminotransferasa sanguínea, aumento de la amilasa sanguínea |
|
Poco frecuentes |
Aumento de la aspartato aminotransferasa |
* Esta reacción adversa se identificó durante la vigilancia post-comercialización pero no se
observó en los ensayos clínicos controlados. La categoría de frecuencia se estimó a partir de un cálculo estadístico basado en el número total de pacientes expuestos a telbivudina en los ensayos clínicos (n = 8.914).
Descripción de reacciones adversas seleccionadas
Elevación de creatinquinasa
En el análisis en conjunto de NV-02B-007 (GLOBE) y NV-02B-015, en las 104 semanas de tratamiento se observaron elevaciones de CK de grado 3 ó 4 (>7x LSN) en el 12,6% de los pacientes tratados con telbivudina (n = 847) y en el 4,0% de los pacientes tratados con lamivudina (n = 846). La mayoría de las elevaciones de CK fueron asintomáticas y por lo general, los valores de CK disminuyeron en la siguiente visita con el tratamiento continuado.
Elevaciones transitorias de ALT
La incidencia durante el tratamiento de elevaciones transitorias de alanina aminotransferasa (ALT) en los dos grupos de tratamiento de acuerdo con la definición (elevación de ALT >2x valor basal y >10x LSN) de la AASLD (American Association for the Study of Liver Diseases) se describe además en la Tabla 3.
Tabla 3 Resumen de las elevaciones transitorias de ALT durante el tratamiento - Ensayos NV-02B-007 (GLOBE) y NV-02B-015 en conjunto
|
Elevación transitoria de ALT: Elevación de ALT >2x valor basal y >10x LSN |
Lamivudina n/N (%) |
Telbivudina n/N (%) |
|
Total |
67/852 (7,9) |
41/847 (4,8) |
|
Del valor basal a la semana 24 |
25/852 (2,9) |
25/847 (3,0) |
|
De la semana 24 al final del ensayo |
44/837 (5,3) |
17/834 (2,0) |
Se recomienda la monitorización periódica de la función hepática durante el tratamiento (ver sección 4.4).
Exacerbaciones de la hepatitis B tras la interrupción del tratamiento
Se han notificado exacerbaciones agudas graves de hepatitis B en pacientes que han interrumpido el tratamiento contra la hepatitis B, incluyendo telbivudina (ver sección 4.4).
La incidencia postratamiento de elevaciones transitorias de alanina aminotransferasa (ALT) en los dos grupos de tratamiento se describe además en la Tabla 4.
Tabla 4 Resumen de las elevaciones transitorias postratamiento de ALT - Ensayos NV-02B-007 (GLOBE) y NV-02B-015 en conjunto
|
Lamivudina |
Telbivudina | |
|
Elevación transitoria de ALT |
n/N (%) |
n/N (%) |
|
Elevación de ALT >2x valor basal y >10x LSN |
10/180 (5,6) |
9/154 (5,8) |
Resultados a las 208 semanas
Tras 104 semanas de tratamiento con telbivudina, el 78% de los pacientes (530/680) del ensayo NV-02B-007 (GLOBE) y el 82% (137/167) de los pacientes del ensayo NV-02B-015 se incluyeron en el ensayo de extensión CLDT600A2303 (ver sección 5.1) para continuar con el tratamiento durante un máximo de 208 semanas. La población de seguridad a largo plazo constaba de 655 pacientes incluyendo 518 del ensayo NV-02B-007 (GLOBE) y 137 del ensayo NV-02B-015. El perfil de seguridad global del análisis en conjunto de un máximo de 104 y de 208 semanas fue similar. El 15,9% de los pacientes tratados con telbivudina durante 208 semanas presentaron nuevas elevaciones de CK de grado 3 ó 4 ocurrieron. La mayoría de las elevaciones de CK de grado 3 ó 4 fueron asintomáticas y transitorias.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se dispone de información de sobredosis intencionada de telbivudina, pero un paciente recibió una sobredosis no intencionada la cual fue asintomática. Dosis ensayadas de hasta 1.800 mg/día, tres veces superior a la dosis diaria recomendada, han sido bien toleradas. No se ha determinado la dosis máxima tolerada de telbivudina. En caso de sobredosis, deberá interrumpirse el tratamiento con Sebivo y proporcionar un tratamiento de apoyo general adecuado, según proceda.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antivirales de uso sistémico, nucleósidos y nucleótidos inhibidores de la transcriptasa inversa, código ATC: J05AF11
Mecanismo de acción
La telbivudina es un análogo nucleósido sintético de la timidina, con actividad contra la ADN polimerasa del virus de la hepatitis B (VHB). Tras ser fosforilada eficientemente por cinasas celulares se convierte en el trifosfato activo, cuya semivida intracelular es de 14 horas. La telbivudina-5'-trifosfato inhibe la ADN polimerasa (transcriptasa inversa) del VHB al competir con el sustrato natural, timidina 5'-trifosfato. La incorporación de la telbivudina-5'-trifosfato en el ADN vírico causa la finalización de la cadena de ADN, dando como resultado la inhibición de la replicación del VHB.
Efectos farmacodinámicos
La telbivudina es un inhibidor de la síntesis de ambas cadenas del VHB, la primera (CI50 = 0,4-1,3 pM) y la segunda (CI50 = 0,12-0,24 pM) y muestra una preferencia distintiva por la inhibición de la producción de la segunda cadena. Por el contrario, la telbivudina-5'-trifosfato, a concentraciones de hasta 100 pM, no inhibe las ADN polimerasas celulares a, p o y. En los ensayos relacionados con la estructura miticondrial, con el contenido y la función del ADN, la telbivudina no ejerció efectos tóxicos apreciables a concentraciones de hasta 10 pM y no incrementó la producción de ácido láctico in vitro.
Se evaluó la actividad antivírica in vitro de la telbivudina en la línea celular 2.2.15 de hematoma humano expresada por el VHB. La concentración de telbivudina que inhibió eficazmente el 50% de la síntesis vírica (CI50) fue de 0,2 pM aproximadamente. La actividad antivírica de la telbivudina es específica frente al virus de la hepatitis B y los hepadnavirus relacionados. La telbivudina no fue activa frente al VIH in vitro. No se ha evaluado en ensayos clínicos la ausencia de actividad de telbivudina frente al VIH.
Experiencia clínica
La seguridad y eficacia del tratamiento a largo plazo (104 semanas) de Sebivo se evaluó en dos ensayos clínicos controlados con tratamiento activo que incluyeron 1.699 pacientes con hepatitis B crónica (NV-02B-007 (GLOBE) y NV-02B-015).
Ensayo NV-02B-007 (GLOBE)
El ensayo NV-02B-007 (GLOBE) es un ensayo de fase III multinacional, doble ciego, randomizado, de telbivudina comparado con lamivudina durante un periodo de tiempo de 104 semanas en 1.367 pacientes HBeAg positivos y HBeAg negativos con hepatitis B crónica que nunca antes habían recibido terapia nucleosídica. La mayoría de la población incluida en el ensayo era asiática. Los genotipos de VHB más frecuentes fueron el B (26%) y el C (51%). Se trataron con telbivudina un pequeño número de pacientes caucásicos (98 en total). El análisis principal de datos se realizó después de que todos los pacientes cumplieron la semana 52 de tratamiento.
Pacientes HBeAgpositivos: La edad media de los pacientes fue 32 años, el 74% eran de sexo masculino, el 82% asiáticos, el 12% caucásicos y el 6% habían recibido tratamiento previo con interferón alfa.
Pacientes HBeAg negativos: La edad media de los pacientes fue 43 años, el 79% eran de sexo masculino, el 65% asiáticos, el 23% caucásicos y el 11% habían recibido tratamiento previo con interferón alfa.
Resultados clínicos a la semana 52
Las variables de eficacia clínicas y virológicas se evaluaron por separado en las poblaciones de pacientes HBeAg positivos y HBeAg negativos. La variable principal de la respuesta terapéutica fue una variable serológica compuesta que requiere la supresión del ADN del VHB hasta <5 logi0 copias/ml junto con la pérdida de HBeAg sérico o normalización de ALT. Las variables secundarias incluyeron respuesta histológica, normalización de ALT y varias mediciones de la eficacia antivírica.
Independientemente de las características basales, la mayoría de los pacientes que tomaron Sebivo mostraron respuesta histológica, virológica, bioquímica y serológica al tratamiento. Los niveles basales de ALT >2x LSN y ADN del VHB basales <9 logi0 copias/ml se asociaron con índices más elevados de seroconversión del HBeAg en pacientes HBeAg positivos. Los pacientes que alcanzaron niveles de ADN del VHB <3 logi0 copias/ml en la semana 24 presentaron respuesta óptima al tratamiento; por el contrario, pacientes con niveles de ADN del VHB >4 log10 copias/ml a las 24 semanas tuvieron resultados menos favorables a la semana 52.
En pacientes HBeAg positivos, la telbivudina fue superior a lamivudina en cuanto a la respuesta terapéutica (75,3% vs 67,0% de respondedores; p = 0,0047). En pacientes HBeAg negativos, la telbivudina resultó no inferior a lamivudina (75,2% y 77,2% de respondedores; p = 0,6187). La etnicidad caucásica se asoció con una respuesta inferior al tratamiento con los dos agentes antivirales utilizados en el ensayo NV-02B-007 (GLOBE); sin embargo, la población de pacientes caucásicos fue muy limitada (n = 98).
En la semana 24, 203 pacientes HBeAg positivos y 177 HBeAg negativos alcanzaron niveles de ADN del VHB no detectables. De los pacientes HBeAg positivos, el 95% alcanzó niveles de ADN del VHB no detectables, el 39% alcanzó seroconversión de HBeAg, el 90% alcanzó normalización de ALT en la semana 52 y el 0,5% presentó resistencia en la semana 48. De forma similar, de los pacientes HBeAg negativos, el 96% alcanzó niveles de ADN del VHB no detectables, el 79% alcanzó normalización de ALT en la semana 52 y el 0% presentó resistencia en la semana 48.
En la Tabla 5 se muestran los resultados de las mediciones virológicas, bioquímicas y serológicas seleccionadas y en la Tabla 6 se muestra la respuesta histológica.
|
Parámetro de respuesta |
HBeAg positivos (n = 921) |
HBeAg negativos (n = 446) | ||
|
Telbivudina 600 mg (n = 458) |
Lamivudina 100 mg (n = 463) |
Telbivudina 600 mg (n = 222) |
Lamivudina 100 mg (n = 224) | |
|
Reducción media del ADN del VHB respecto al valor basal (log10 copias/ml) Media ± EEM1,2,3 |
-6,45 (0,11) * |
-5,54 (0,11) |
-5,23 (0,13) * |
-4,40 (0,13) |
|
Pacientes con ADN del VHB indetectable por PCR (en %) |
60%* |
40% |
88%* |
71% |
|
Normalización de ALT4 |
77% |
75% |
74% |
79% |
|
Seroconversión de HBeAg4 |
23% |
22% |
- |
- |
|
Pérdida de HBeAg5 |
26% |
23% |
- |
- |
1 EEM: Error estándar de la media
2 Valoración de PCR COBAS Amplicor® de Roche (límite inferior de cuantificación <300 copias/ml).
3 HBeAg positivos n = 443 y 444, HBeAg negativos n = 219 y 219, para los grupos de telbivudina y lamivudina, respectivamente. Las diferencias de población se deben a interrupciones en el tratamiento de pacientes del ensayo y a la falta de valoración del ADN del VHB en la semana 52.
4 HBeAg positivos n = 440 y 446, HBeAg negativos n = 203 y 207, para los grupos de telbivudina y lamivudina, respectivamente. Se evaluó la normalización de ALT únicamente en pacientes con ALT > LSN en el periodo basal.
5 n = 432 y 442, para los grupos de telbivudina y lamivudina, respectivamente. Se evaluó la seroconversión y la pérdida de HBeAg únicamente en los pacientes con cantidades detectables de HBeAg en el periodo basal.
*p < 0,0001
Mejoría histológica y cambio de puntuación en la escala de Fibrosis de Ishak en la semana 52 en el ensayo NV-02B-007 (GLOBE)
|
HBeAg positivo (n = 921) |
HBeAg negativo (n = 446) | |||
|
Telbivudina |
Lamivudina |
Telbivudina |
Lamivudina | |
|
600 mg (n = 384)1 |
100 mg (n = 386)1 |
600 mg (n = 199)1 |
100 mg (n = 207)1 | |
|
Respuesta histológica2 | ||||
|
Mejoría |
71%* |
61% |
71% |
70% |
|
Ausencia de mejoría |
17% |
24% |
21% |
24% |
|
Puntuación en la escala de Fibrosis de Ishak3 | ||||
|
Mejoría |
42% |
47% |
49% |
45% |
|
Sin cambios |
39% |
32% |
34% |
43% |
|
Deterioro |
8% |
7% |
9% |
5% |
|
Ausencia de biopsia en la semana 52 |
12% |
15% |
9% |
7% |
|
1 Pacientes que recibieron > una dosis del medicamento de estudio, con biopsias hepáticas basales evaluables y puntuación basal del Índice de Actividad Histológica (IAH) de Knodell >3. 2 Respuesta histológica definida como disminución >2 puntos en la puntuación de actividad Necroinflamatoria de Knodell sin deterioro de la puntuación de Fibrosis de Knodell. 3 En la puntuación de Fibrosis de Ishak, la mejoría de definió como reducción >1 punto en la puntuación de Fibrosis de Ishak desde el valor basal hasta la semana 52. | ||||
|
*p = 0,0024 | ||||
Resultados clínicos a la semana 104
En general, los resultados clínicos a la semana 104 en los pacientes tratados con telbivudina fueron consistentes con los obtenidos a la semana 52, demostrando durabilidad de las respuestas de eficacia para los pacientes tratados con telbivudina con tratamiento continuado.
Entre los pacientes HBeAg positivos, la respuesta terapéutica (63% vs 48%; p < 0,0001) y las variables secundarias principales (reducción media del ADN del VHB log10: -5,74 vs -4,42; p < 0,0001, indetectabilidad del ADN del VHB: 56% vs 39%; p < 0,0001 y normalización de ALT del 70% vs 62%) demostraron una mayor diferencia a la semana 104 entre telbivudina y lamivudina, respectivamente. También se observó para telbivudina una tendencia a presentar valores más elevados de pérdida de HBeAg (35% vs 29%) y seroconversión (30% vs 25%). Además, en el subgrupo de pacientes con niveles basales de ALT >2x LSN (320), una proporción significativamente superior de pacientes tratados con telbivudina en relación a los pacientes tratados con lamivudina alcanzaron seroconversión de HBeAg a la semana 104 (36% vs 28%, respectivamente).
Entre los pacientes HBeAg negativos, las diferencias en la respuesta terapéutica (78% vs 66%) y las variables secundarias principales (reducción media del ADN del VHB logJ0: -5,00 vs -4,17, e indetectabilidad del ADN del VHB: 82% vs 57%; p < 0,0001) fueron más elevadas para telbivudina hasta la semana 104. Las tasas de normalización de ALT (78% vs 70%) continuaron siendo más elevadas en la semana 104.
Predictibilidad a la semana 24
En la semana 24, 203 pacientes HBeAg positivos (44%) y 177 pacientes HBeAg negativos (80%) tratados con telbivudina, alcanzaron niveles indetectables de ADN del VHB.
Para ambos tipos de pacientes HBeAg positivos y HBeAg negativos, los resultados del ADN del VHB a la semana 24 fueron predecibles de los resultados favorables a largo plazo. Los pacientes tratados con telbivudina que obtuvieron ADN del VHB indetectable por PCR a la semana 24, presentaron las tasas más altas de indetectabilidad del ADN del VHB y seroconversión de HBeAg (en pacientes HBeAg positivos), y las tasas globales más bajas de reactivación virológica a la semana 104.
En la Tabla 7 se presentan los resultados finales a la semana 104, en base al nivel de ADN del VHB en la semana 24 para los pacientes tanto HBeAg positivos como HBeAg negativos.
Tabla 7 Variables de eficacia principales a la semana 104 en base a los niveles séricos de ADN del VHB a la semana 24, en pacientes tratados con telbivudina en el ensayo NV-02B-007 (GLOBE)
|
ADN del VHB a la semana 24 |
Resultados para las variables de eficacia principales a la semana 104 en base a los resultados a la semana 24 | ||||
|
Respuesta terapéutica n/N (%) |
Indetectabilida d del ADN del VHB n/N (%) |
Seroconversió n de HBeAg n/N (%) |
Normalización de ALT n/N (%) |
Reactivación virológica* n/N (%) | |
|
HBeAg positivo | |||||
|
<300 copias/ml |
172/203 (85) |
166/203 (82) |
84/183 (46) |
160/194 (82) |
22/203 (11) |
|
300 copias/ml a <3 logi0 copias/ml |
36/57 (63) |
35/57 (61) |
21/54 (39) |
40/54 (74) |
18/57 (32) |
|
>3 log10 copias/ml |
82/190 (43) |
54/190 (28) |
23/188 (12 ) |
106/184 (58) |
90/190 (47) |
|
HBeAg negativo | |||||
|
<300 copias/ml |
146/177 (82) |
156/177 (88) |
N/A |
131/159 (82) |
11/177 (6) |
|
300 copias/ml a <3 log10 copias/ml |
13/18 (72) |
14/18 (78) |
N/A |
13/17 (76) |
4/18 (22) |
|
>3 log10 copias/ml |
13/26 ( 50) |
12/26 (46) |
N/A |
14/26 (54) |
12/26 ( 46) |
N/A = no aplica
* Reactivación virológica: “1 log por encima de nadir” definición valorada a la semana 104
Ensayo NV-02B-015
En el ensayo NV-02B-015 se confirmaron los resultados de eficacia y seguridad del ensayo NV-02B-007 (GLOBE). Es un ensayo de fase III, doble ciego, randomizado, de telbivudina 600 mg una vez al día comparado con lamivudina 100 mg una vez al día durante un periodo de tratamiento de 104 semanas en 332 pacientes chinos HBeAg positivos y HBeAg negativos con hepatitis B crónica que nunca antes habían recibido terapia nucleosídica.
Ensayo CLDT600A2303 - Resultados clínicos de 208 semanas
El ensayo CLDT600A2303 es un ensayo abierto de extensión de 104 semanas de duración en pacientes con hepatitis B crónica compensada que habían sido tratados previamente con telbivudina durante 2 años, incluyendo pacientes de los ensayos NV-02B-007 (GLOBE) y NV-02B-015, y que aporta datos de eficacia y seguridad tras 156 y 208 semanas de tratamiento continuado con telbivudina. Los pacientes con ADN del VHB indetectable en la semana 24 obtuvieron mejores resultados a las semanas 156 y 208 (Tabla 8).
Tabla 8 Análisis de la eficacia de los datos conjuntos de los ensayos NV-02B-007 (GLOBE), NV-02B-015 y CLDT600A2303
|
Semana 52 |
Semana 104 |
Semana 156 |
Semana 208 | |
|
Pacientes HBeAg positivos (n = 293*) | ||||
|
ADN del VHB indetectable sostenido (<300 copias/ml) |
70,3% (206/293) |
77,3% (218/282) |
75,0% (198/264) |
76,2% (163/214) |
|
ADN del VHB indetectable sostenido (<300 copias/ml) con ADN del VHB indetectable a la semana 24 |
99,4% (161/162) |
94,9% (150/158) |
86,7% (130/150) |
87,9% (109/124) |
|
Tasas de seroconversión de HBeAg acumuladas (%) |
27,6% (81/293) |
41,6% (122/293) |
48,5% (142/293) |
53,2% (156/293) |
|
Tasas de seroconversión de HBeAg acumuladas en pacientes con ADN del VHB indetectable a la semana 24 (%) |
40,1% (65/162) |
52,5% (85/162) |
59,3% (96/162) |
65,4% (106/162) |
|
Normalización de ALT sostenida |
81,4% (228/280) |
87,5% (237/271) |
82,9% (209/252) |
86,4% (178/106) |
|
Pacientes HBeAg negativos (n = 209*) | ||||
|
ADN del VHB indetectable sostenido (<300 copias/ml) |
95,2% (199/209) |
96,5% (195/202) |
84,7% (160/189) |
86,0% (141/164) |
|
ADN del VHB indetectable sostenido (<300 copias/ml) con ADN del VHB indetectable a la semana 24 |
97,8% (175/179) |
96,5% (166/172) |
86,7% (143/165) |
87,5% (126/144) |
|
Normalización de ALT sostenida |
80,3% (151/188) |
89,0% (161/181) |
83,5% (142/170) |
89,6% (129/144) |
* La población sin resistencia viral a la entrada del ensayo CLDT600A2303 constaba de 502 pacientes (293 HBeAg positivos y 209 HBeAg negativos).
Ensayo CLDT600ACN04E1 - Impacto del tratamiento en la histología del hígado En el ensayo CLDT600ACN04E1, se evaluaron los cambios en la histología del hígado de 57 pacientes con biopsias hepáticas pareadas disponibles al inicio y tras un tratamiento de
260,8 semanas de media (38 pacientes HBeAg positivos y 19 HBeAg negativos).
• La puntuación media de actividad necroinflamatoria de Knodell de 7,6 (DE 2,9) al inicio mejoró (p < 0,0001) hasta 1,4 (DE 0,9) con un cambio medio de -6,3 (DE 2,8). En el 98,2% (56/57) de los pacientes se observó una puntuación de actividad necroinflamatoria de Knodell <3 (necroinflamación mínima o nula).
• La puntuación de Ishak media de 2,2 (DE 1,1) al inicio mejoró (p < 0,0001) hasta 0,9 (DE 1,0) con un cambio medio de -1,3 (DE 1,3). En el 84,2% (48/57) de los pacientes se observó una puntuación de fibrosis de Ishak <1 (fibrosis mínima o nula).
Los cambios en las puntuaciones de Ishak y de actividad necroinflamatoria de Knodell fueron similares para los pacientes HBeAg positivos y los HBeAg negativos.
CLDT600A2303 - Duración de las respuestas HBeAg una vez interrumpido el tratamiento El ensayo CLDT600A2303 incluyó pacientes HBeAg positivos de los ensayos NV-02B-007 (GLOBE) o NV-02B-015 para el seguimiento una vez interrumpido el tratamiento. Estos pacientes completaron >52 semanas de tratamiento con telbivudina y en la última visita durante el tratamiento mostraron pérdida de HBeAg durante >24 semanas con ADN del VHB <5 logi0 copias/ml. La duración media del tratamiento fue de 104 semanas. Tras un periodo medio de seguimiento sin tratamiento de 120 semanas, la mayoría de pacientes tratados con telbivudina HBeAg positivos mostraron una pérdida de HBeAg sostenida (83,3%; 25/30) y una seroconversión de HBeAg sostenida (79,2%; 19/24). Los pacientes con seroconversión de HBeAg sostenida presentaron un valor medio de ADN del VHB de 3,3 log10 copias/ml; y el 73,7% presentaron un ADN del VHB <4 log10 copias/ml.
Resistencia clínica
En el ensayo NV-02B-007 (GLOBE; n = 680) se realizó la prueba de resistencia genotípica en pacientes con rechazo virológico (incremento confirmado de >1 log10 copias/ml de ADN del VHB desde nadir).
Entre los pacientes HBeAg positivos y HBeAg negativos, el 5% (23/458) y 2% (5/222) respectivamente presentaron rechazo virológico con mutaciones de resistencia al VHB detectables en la semana 48.
Ensayos NV-02B-007 (GLOBE) y CLDT600A2303 - tasas de resistencia genotípica acumuladas El análisis original de la resistencia genotípica acumulada a las semanas 104 y 208 se basó en la población por ITT e incluyó a todos los pacientes que continuaron con el tratamiento durante 4 años, independientemente de los niveles de ADN del VHB. De los 680 pacientes tratados con telbivudina e incluidos inicialmente en el ensayo pivotal NV-02B-007 (GLOBE), 517 (76%) se incluyeron en el ensayo CLDT600A2303 para continuar el tratamiento con telbivudina durante un máximo de 208 semanas. De estos 517 pacientes, 159 pacientes (HBeAg positivo=135, HBeAg negativo=24) presentaron ADN del VHB detectable.
Las proporciones genotípicas acumuladas en la semana 104, fueron del 25,1% (115/458) para los pacientes HBeAg positivo y del 10,8% (24/222) para los pacientes HBeAg negativo.
En la población por ITT global, las tasas de resistencia acumuladas en el año 4 para los pacientes HBeAg positivos y HBeAg negativos fueron del 40,8% (131/321) y del 18,9% (37/196) respectivamente.
Las tasas de resistencia genotípica acumuladas también se evaluaron mediante la aplicación de un modelo matemático en el que sólo se consideran pacientes con ADN del VHB indetectable al inicio del año respectivo. En este análisis las tasas de resistencia acumuladas en el año 4 fueron del 22,3% para los pacientes HBeAg positivos y del 16,0% para los pacientes HBeAg negativos.
Cuando consideramos pacientes con reactivación viral en la semana 104 en el ensayo NV-02B-007 (GLOBE), la proporción de resistencias fue inferior en pacientes con ADN del VHB <300 copias/ml en la semana 24 que en pacientes con ADN del VHB >300 copias/ml en la semana 24. En pacientes HBeAg positivo con ADN del VHB <300 copias/ml en la semana 24, la resistencia fue del 1%
(3/203) a las 48 semanas y del 9% (18/203) en la semana 104, mientras que en pacientes con ADN del VHB >300 copias/ml, la resistencia fue del 8% (20/247) a las 48 semanas y del 39% (97/247) en la semana 104. En pacientes HBeAg negativo con ADN del VHB <300 copias/ml en la semana 24, la resistencia fue del 0% (0/177) a las 48 semanas y del 5% (9/177) en la semana 104, mientras que en pacientes con ADN del VHB >300 copias/ml, la resistencia fue del 11% (5/44) a las 48 semanas y del 34% (15/44) en la semana 104.
Patrón de mutación genotípica y resistencia cruzada
Los análisis genotípicos de 203 pares de muestras evaluables con ADN del VHB >1.000 copias/ml a la semana 104 (NV-02B-007 (GLOBE)) demostraron que la mutación primaria asociada con la resistencia a telbivudina fue rtM204I, a menudo asociada con mutaciones en rtL180M y rtL80I/V e infrecuentemente con rtV27A, rtL82M, rtV173L, rtT184I y rtA200V. Los factores basales asociados con el desarrollo de resistencia genotípica al fármaco, incluyeron: tratamiento con lamivudina, ADN del VHB basal más elevado, ALT sérico basal más bajo, y aumento del peso corporal/BMI. Los parámetros de respuesta en tratamiento a la semana 24, que predijeron la aparición del virus resistente al fármaco en la semana 104 fueron ADN del VHB >300 copias/ml y elevación del ALT sérico.
Los análisis genotípicos de 50 aislados del VHB de pacientes tratados con telbivudina en la semana 208 (CLDT600A2303) revelaron un perfil de resistencia similar al notificado en la semana 104. Las conversiones en la posición 80, 180 y posiciones polimórficas 91, 229 se detectaron siempre en secuencias que albergaban la mutación en M204I la cual confiere resistencia genotípica.
Lo más probable es que estas mutaciones sean compensatorias. Se notificó una mutación aislada en rtM204V y dos mutaciones en rtM204I/V/M en pacientes tratados con telbivudina que experimentaron reactivación viral hasta la semana 208. No se notificó ninguna mutación nueva.
Se ha observado resistencia cruzada entre los análogos nucleósidos del VHB (ver sección 4.4). En ensayos celulares, las cepas de VHB resistentes a lamivudina que contenían la mutación en rtM204I o la mutación doble rtL180M/rtM204V, presentaron una susceptibilidad a telbivudina reducida >1.000 veces. El VHB codificado con la resistencia a adefovir asociada a las sustituciones en rtN236T o rtA181V mostró cambios en la susceptibilidad a telbivudina alrededor de 0,3- y 4-veces en cultivo celular, respectivamente (ver sección 4.4).
5.2 Propiedades farmacocinéticas
Se ha evaluado la farmacocinética de telbivudina tras dosis únicas y múltiples en voluntarios sanos y en pacientes con hepatitis B crónica. No se ha evaluado la farmacocinética de telbivudina a la dosis recomendada de 600 mg en pacientes con hepatitis B crónica. Sin embargo, la farmacocinética de telbivudina es similar entre ambas poblaciones.
Absorción
Tras la administración oral de una dosis única de 600 mg de telbivudina a voluntarios sanos (n = 42), la concentración plasmática máxima (Cmax) de telbivudina fue de 3,2 ± 1,1 pg/ml (media ± DE) y se alcanzó a una media de 3,0 horas después de la dosis. El área bajo la curva de las concentraciones plasmáticas de telbivudina en función del tiempo (AUC0.^) fue de 28,0 ± 8,5 pg*h/ml (media ± DE). En las determinaciones de exposición sistémica (Cmax, AUC) la variabilidad interindividual por lo general fue del 30% aproximadamente. Los comprimidos recubiertos con película que contienen 600 mg de telbivudina son bioequivalentes con 30 ml de telbivudina solución oral (20 mg/ml).
Efecto de los alimentos sobre la absorción oral
La administración de una dosis única de 600 mg con alimentos no modificó ni a la absorción ni a la exposición a telbivudina.
Distribución
In vitro, la unión de telbivudina a las proteínas plasmáticas es baja (3,3%).
Biotransformación
No se detectaron metabolitos de telbivudina tras la administración de 14C-telbivudina a los seres humanos. La telbivudina no es un sustrato, inhibidor, ni inductor del sistema enzimático del citocromo P450 (CYP450).
Eliminación
Tras alcanzar la concentración máxima, la disposición plasmática de telbivudina disminuye de forma biexponencial con una semivida de eliminación terminal (ti/2) de 41,8 ± 11,8 horas. La telbivudina se elimina principalmente por excreción urinaria como fármaco inalterado. El aclaramiento renal de telbivudina se aproxima a la tasa de filtración glomerular normal, lo que sugiere que el principal mecanismo de excreción es la filtración. Tras la administración de una dosis oral única de 600 mg de telbivudina, en un plazo de 7 días se recupera en la orina aproximadamente el 42% de la dosis administrada. Dado que la vía de eliminación predominante es la vía renal, tanto los pacientes con insuficiencia renal de moderada a grave como los pacientes en hemodiálisis requerirán un ajuste del intervalo de tiempo entre dosis (ver sección 4.2).
Linealidad/No linealidad
La farmacocinética de telbivudina es proporcional a la dosis administrada en el rango de 25 a 1.800 mg. Después de 5 a 7 días con una única administración diaria se alcanzó el estado estacionario con una acumulación aproximada de 1,5 veces en la exposición sistémica, lo que sugiere una semivida de acumulación efectiva de aproximadamente 15 horas. Tras una única administración diaria de 600 mg de telbivudina, las concentraciones plasmáticas mínimas en estado estacionario fueron de 0,2-0,3 pg/ml aproximadamente.
Poblaciones especiales
Sexo
No existen diferencias significativas relacionadas con el sexo en la farmacocinética de telbivudina.
Raza
No existen diferencias significativas relacionadas con la raza en la farmacocinética de telbivudina.
Pediatría y pacientes de edad avanzada (65 años de edad o mayores)
No se han realizado estudios farmacocinéticos en sujetos pediátricos o de edad avanzada.
Insuficiencia renal
Se ha evaluado la farmacocinética de telbivudina tras dosis únicas (200, 400 y 600 mg) en pacientes (sin hepatitis B crónica) con diversos grados de insuficiencia renal (valorado con el aclaramiento de creatinina). Según los resultados que se muestran en la Tabla 9, se recomienda ajustar el intervalo de tiempo entre las dosis de telbivudina en los pacientes que presenten un aclaramiento de creatinina <50 ml/min (ver secciones 4.2 y 4.4).
Tabla 9 Parámetros farmacocinéticos (media ± DE) de telbivudina en sujetos con diversos grados de función renal
|
Función renal (aclaramiento de creatinina en ml/min) | |||||
|
Normal (>80) (n = 8) 600 mg |
Leve (50-80) (n = 8) 600 mg |
Moderada (30-49) (n = 8) 400 mg |
Grave(<30) (n = 6) 200 mg |
ERT/ Hemodiálisis (n = 6) 200 mg | |
|
Cmax (Pg/ml) |
3,4 ± 0,9 |
3,2 ± 0,9 |
2,8 ± 1,3 |
1,6 ± 0,8 |
2,1 ± 0,9 |
|
AUC0-» (pg^h/ml) |
28,5 ± 9,6 |
32,5 ± 10,1 |
36,0 ± 13,2 |
32,5 ± 13,2 |
67,4 ± 36,9 |
|
CLRENAL (ml/min) |
126,7 ± 48,3 |
83,3 ± 20,0 |
43,3 ± 20,0 |
11,7 ± 6,7 |
- |
Pacientes con insuficiencia renal en hemodiálisis
La hemodiálisis (de hasta 4 horas) reduce la exposición sistémica a telbivudina un 23% aproximadamente. Aparte del ajuste del intervalo de tiempo entre dosis en función del aclaramiento de creatinina, no es necesario realizar ninguna modificación adicional de la dosis durante la hemodiálisis de rutina (ver sección 4.2). La telbivudina debe administrarse después de la hemodiálisis.
Insuficiencia hepática
Se ha estudiado la farmacocinética de telbivudina en pacientes (sin hepatitis B crónica) con diversos grados de insuficiencia hepática y en algunos pacientes con insuficiencia hepática descompensada. No se observaron cambios significativos en la farmacocinética de telbivudina entre los sujetos con y sin insuficiencia hepática. Los resultados de estos estudios indican que no es necesario realizar un ajuste de la dosis en pacientes con insuficiencia hepática (ver sección 4.2).
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas y genotoxicidad.
La telbivudina no mostró potencial carcinogénico. No se observaron signos de efecto tóxico directo de telbivudina en estudios estándar de toxicidad reproductiva. En conejos, dosis de telbivudina equivalentes a niveles de exposición 37 veces superiores al observado en humanos con la dosis terapéutica (600 mg) se relacionaron con una mayor incidencia de abortos y partos prematuros. Este efecto se consideró secundario a la toxicidad materna.
La fertilidad se evaluó en estudios convencionales realizados en ratas adultas, y como parte de un estudio de toxicología juvenil.
En ratas adultas, la fertilidad se vio reducida cuando ambas ratas, macho y hembra, se trataron con telvibudina a dosis de 500 ó 1000 mg/kg/día (índice de fertilidad inferior comparado con los controles concurrentes). No se observaron anormalidades en la morfología o función del esperma, ni en la histología de testículos y ovarios.
No hubo evidencia de deterioro en la fertilidad en otros estudios cuando tanto las ratas macho como hembra se trataron con dosis de hasta 2000 mg/kg/día y se aparearon con ratas no tratadas (niveles de exposición sistémica 6-14 veces aproximadamente más elevado que los alcanzados en humanos).
En el estudio de toxicología juvenil, las ratas se trataron desde el día 14 hasta el día 70 postparto y se aparearon con ratas que recibieron el mismo tratamiento (no hubo apareamiento entre hermanos). La fertilidad se redujo en las parejas que recibieron >1000 mg/kg/día como se mostró por los descensos en la fertilidad y en los índices de apareamiento, y en la tasa de concepción reducida. Sin embargo, los parámetros ováricos y uterinos de las hembras que se aparearon con éxito no se vieron afectados.
El nivel de efecto adverso no observable (NOAEL) para efectos sobre la fertilidad o parámetros de apareamiento fue de 250 mg/kg/día, lo que proporcionó niveles de exposición 2,5 a 2,8 veces más elevados que los alcanzados en humanos con función renal normal a la dosis terapéutica.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Ácido benzoico (E210)
Sacarina sódica Saborizante fruta de la pasión Hidróxido de sodio Ácido cítrico anhidro Agua purificada
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años
Utilizar en los 2 meses siguientes a la apertura del frasco.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30°C. No congelar.
6.5 Naturaleza y contenido del envase
Frasco de vidrio marrón de 300 ml con cierre a prueba de niños, que incluye un disco de sellado de polietileno y un anillo de seguridad, un vaso dosificador de polipropileno con graduaciones grabadas de 5 a 30 ml en aumentos de 5 ml y una jeringa para uso oral de polipropileno con graduaciones de 1 ml a 10 ml en incrementos de 0,5 ml.
6.6 Precauciones especiales de eliminación
Ninguna especial para su eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/388/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 24 Abril 2007 Fecha de la última renovación: 26 Abril 2012
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes
Novartis Pharma GmbH Roonstrasse 25 90429 Nuremberg Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (Ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107 ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos;
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CARTON
1. NOMBRE DEL MEDICAMENTO
Sebivo 600 mg comprimidos recubiertos con película telbivudina
2. PRINCIPIO(S) ACTIVO(S)
Un comprimido contiene 600 mg de telbivudina.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
28 comprimidos recubiertos con película 98 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento.
Vía oral. No debe masticar, dividir ni triturar el comprimido.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/388/001 28 comprimidos recubiertos con película
EU/1/07/388/002 98 comprimidos recubiertos con película
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Sebivo 600 mg
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLÍSTERS
1. NOMBRE DEL MEDICAMENTO
Sebivo 600 mg comprimidos recubiertos con película telbivudina
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
Lunes
Martes
Miércoles
Jueves
Viernes
Sábado
Domingo
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR Y EL ACONDICIONAMIENTO PRIMARIO
CARTONAJE Y ETIQUETA DEL FRASCO_
1. NOMBRE DEL MEDICAMENTO
Sebivo 20 mg/ml solución oral Telbivudina
2. PRINCIPIO(S) ACTIVO(S)
Cada ml contiene 20 mg de telbivudina.
3. LISTA DE EXCIPIENTES
También contiene sodio. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 frasco con 300 ml de solución oral [únicamente en cartonaje]
1 vaso dosificador + 1 jeringa para uso oral [únicamente en cartonaje]
300 ml [únicamente en la etiqueta del frasco]
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Utilizar en los 2 meses siguientes a la apertura del frasco.
No conservar a temperatura superior a 30°C. No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/388/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Sebivo 20 mg/ml [únicamente en cartonaje]
B. PROSPECTO
Prospecto: información para el usuario
Sebivo 600 mg comprimidos recubiertos con película
Telbivudina
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Sebivo y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Sebivo
3. Cómo tomar Sebivo
4. Posibles efectos adversos
5. Conservación de Sebivo
6. Contenido del envase e información adicional
1. Qué es Sebivo y para qué se utiliza
Sebivo contiene el principio activo telbivudina. Sebivo pertenece a un grupo de medicamentos conocidos como antivirales, que se utilizan para tratar infecciones causadas por virus.
Sebivo se utiliza para tratar la hepatitis B crónica en adultos.
La causa de la hepatitis B es la infección por el virus de la hepatitis B, que se multiplica en el hígado y causa lesiones en el hígado. El tratamiento con Sebivo reduce la cantidad de virus de la hepatitis B que hay en el cuerpo al impedir su desarrollo, lo que produce menos lesión en el hígado y una mejora de la función del hígado.
2. Qué necesita saber antes de empezar a tomar Sebivo No tome Sebivo
- si es alérgico a telbivudina o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si está siendo tratado con interferón alfa pegilado o estándar (ver “Uso de otros medicamentos”).
Si éste es su caso, no tome Sebivo. Consulte con su médico.
Advertencias y precauciones
Consulte a su médico antes de empezar a tomar Sebivo:
- si tiene o ha tenido problemas de riñón. Su médico puede solicitar pruebas de laboratorio para comprobar que sus riñones funcionan adecuadamente, antes y durante el tratamiento. Dependiendo de los resultados de estos análisis su médico puede aconsejarle que cambie la frecuencia con la que toma Sebivo.
- si tiene cirrosis hepática (una enfermedad grave que causa la aparición de cicatrices en el hígado). En este caso su médico le controlará más de cerca.
- si ha recibido un trasplante de hígado.
- si está utilizando otros medicamentos que puedan causarle problemas musculares (consulte a su médico o farmacéutico si tiene dudas).
- si está infectado con el virus VIH, el de la hepatitis C o D, o está tratado con cualquier antiviral. Si este es su caso, informe a su médico antes de tomar Sebivo.
Durante el tratamiento con Sebivo:
- Sebivo puede causar dolor muscular o debilidad muscular persistente e inexplicable (miopatía). Los síntomas musculares pueden progresar y convertirse en graves, conduciendo a veces a necrosis muscular (rabdomiolisis), la cual puede producir daño renal.
- Sebivo puede producir de forma poco frecuente adormecimiento, hormigueo, dolor y/o sensación de quemazón en brazos y/o piernas (neuropatía periférica).
Si usted nota cualquiera de estos síntomas durante su tratamiento con Sebivo, informe a su médico inmediatamente.
Otros efectos adversos de esta clase de medicamentos
Sebivo pertenece a una clase de medicamentos (un análogo nucleósido) que pueden causar un exceso de ácido láctico en la sangre (acidosis láctica) y aumento del tamaño del hígado (hepatomegalia) con hígado graso (esteatosis). La acidosis láctica es un efecto adverso raro pero grave que ocasionalmente puede resultar mortal. La acidosis láctica se produce más habitualmente en mujeres, en particular si tienen mucho sobrepeso. Su médico le controlará regularmente mientras esté recibiendo Sebivo. Si usted sufre dolor muscular, dolor estomacal grave y persistente con náuseas y vómitos, tiene dificultad para respirar de forma grave y persistente, cansancio e hinchazón y/o molestia abdominal mientras esté tomando Sebivo, informe a su médico inmediatamente.
Algunas personas pueden presentar síntomas de hepatitis muy graves cuando interrumpen el tratamiento con medicamentos como Sebivo. Después de que usted interrumpa el tratamiento con Sebivo, su médico controlará su estado de salud y efectuará análisis de sangre regulares para controlar el estado de su hígado. Informe a su médico inmediatamente si usted advierte algún síntoma nuevo o inusual después de interrumpir el tratamiento (ver “Si interrumpe el tratamiento con Sebivo” en la sección 3 de este prospecto).
Tenga cuidado de no infectar a otras personas
Sebivo no reduce el riesgo de infectar a otras personas con el virus de la hepatitis B (VHB) a través de contacto sexual o por exposición a sangre contaminada o a otros fluidos corporales. Si mantiene relaciones sexuales con una pareja que no es inmune a la hepatitis B, utilice siempre preservativos y evite cualquier otro intercambio de fluídos corporales. Nunca comparta agujas. No comparta artículos personales que puedan contener sangre o fluidos corporales, como el cepillo de dientes o las cuchillas de afeitar. Existe una vacuna disponible para prevenir la infección por el VHB.
Niños y adolescentes
No se recomienda el uso de Sebivo en niños y adolescentes.
Uso de Sebivo con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Su médico o farmacéutico deben saber si usted está tomando otros medicamentos porque algunos pueden afectar a sus riñones y porque además, Sebivo se elimina del organismo fundamentalmente a través de los riñones en la orina.
No tome Sebivo si está usando interferón alfa pegilado o estándar (ver “No tome Sebivo”), ya que la combinación de estos medicamentos puede incrementar su riesgo de desarrollar neuropatía periférica (adormecimiento, hormigueo, y/o sensación de quemazón en brazos y/o piernas). Informe a su médico o farmacéutico si está siendo tratado con interferón.
Embarazo y lactancia
- No use Sebivo durante el embarazo a no ser que su médico se lo recomiende. Si está embarazada o cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento. Su médico le informará de los riesgos potenciales de tomar Sebivo durante el embarazo.
- Si usted sufre de hepatitis B y se queda embarazada, consulte con su médico acerca de como puede proteger mejor a su bebé. Sebivo puede reducir el riesgo de transmitir su virus de la hepatitis B a su futuro bebé si se toma en combinación con la inmunoglobulina de la Hepatitis B y la vacuna de la Hepatitis B.
- No debe amamantar durante el tratamiento con Sebivo. Informe a su médico si usted está en período de lactancia.
3. Cómo tomar Sebivo
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Cantidad de Sebivo que debe tomar
La dosis recomendada de Sebivo es de un comprimido de 600 mg una vez al día. Tome el comprimido aproximadamente a la misma hora cada día.
El comprimido puede tomarlo con o sin alimentos. Tráguelo entero con un poco de agua. No lo mastique, divida, ni triture.
Si tiene problemas de riñón puede que necesite tomar Sebivo con menos frecuencia. Informe a su médico si tiene o ha tenido alguna vez, cualquier problema de riñón.
Durante cuanto tiempo debe tomar Sebivo
Continúe tomando Sebivo cada día durante el tiempo que le indique su médico. No cambie su dosis ni interrumpa el tratamiento con Sebivo sin consultarlo con su médico. Este medicamento está destinado para un tratamiento de larga duración, que posiblemente dure meses o años. Su médico hará un seguimiento de su enfermedad de forma regular para controlar que el tratamiento está teniendo el efecto deseado.
Si toma más Sebivo del que debe
Si usted ha tomado más Sebivo del que le haya indicado su médico, o si alguien toma accidentalmente sus comprimidos, acuda inmediatamente al hospital o consulte con su médico. Lleve el envase del medicamento y muéstrelo a su médico.
Si olvidó tomar Sebivo
- Si olvidó tomar Sebivo, tómelo tan pronto como se acuerde y luego tome la siguiente dosis según la pauta establecida.
- No obstante, si quedan menos de 4 horas para la siguiente dosis, no tome la dosis olvidada y tómela cuando corresponda.
No tome una dosis doble para compensar las dosis olvidadas. Esto puede aumentar el riesgo de que usted padezca efectos adversos no deseados. Consulte a su médico o farmacéutico si no está seguro de cómo proceder.
Si interrumpe el tratamiento con Sebivo
La interrupción del tratamiento con Sebivo puede producir un empeoramiento de su infección por el virus de la hepatitis B, es decir, progresión de la enfermedad y resultados anómalos de las pruebas (aumento de la carga viral, incremento de ALT). No interrumpa el tratamiento con Sebivo salvo que se lo indique su médico. Mientras esté tomando Sebivo, asegúrese que no se queda sin él.
Después de que usted interrumpa el tratamiento con Sebivo, su médico controlará su estado de salud y efectuará análisis de sangre regulares para controlar el estado de su hígado, ya que su infección por el virus de la hepatitis B puede empeorar o llegar a ser muy grave después de interrumpir el tratamiento. Informe a su médico inmediatamente si usted advierte algún síntoma nuevo o inusual después de interrumpir el tratamiento.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Algunos efectos adversos pueden ser graves:
- Dolor muscular o debilidad muscular persistente
- Adormecimiento, hormigueo, dolor y/o sensación de quemazón en brazos y/o piernas Si usted nota cualquiera de ellos, informe a su médico inmediatamente.
Sebivo también puede causar otros efectos adversos:
Frecuentes (pueden afectar hasta 1 de cada 10 pacientes)
- Mareo, dolor de cabeza
- Tos
- Diarrea, sensación de mareo (náuseas), dolor de estómago (abdominal)
- Erupción cutánea
- Cansancio (fatiga)
- Resultados de análisis de sangre que muestran niveles elevados de algunos enzimas hepáticos (p. ej. ALT, AST), amilasa, lipasa o creatinquinasa
Poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes)
- Dolor en articulaciones
- Dolor o debilidad muscular persistente (miopatía/miositis), calambre muscular
- Dolor de espalda, cuello y costado
- Adormecimiento, hormigueo, dolor y/o sensación de quemazón en brazos y/o piernas o alrededor de la boca
- Dolor en la parte inferior de la espalda o cadera que puede irradiar hacia la pierna (ciática)
- Alteración en el gusto
- Sensación de encontrarse mal (malestar)
Raros (pueden afectar hasta 1 de cada 1.000 pacientes)
- Exceso de ácido láctico en la sangre (acidosis láctica)
- Lesión muscular (rabdomiolisis)
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Sebivo
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de CAD. La fecha de caducidad es el último día del mes que se indica.
No requiere condiciones especiales de conservación.
No utilice este medicamento si el envase está dañado o muestra signos de deterioro.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Sebivo
- El principio activo es telbivudina. Cada comprimido contiene 600 mg de telbivudina.
- Los demás componentes son: celulosa microcristalina; povidona; glicolato sódico de almidón; sílice coloidal anhidra; estearato de magnesio; hipromelosa; dióxido de titanio (E171); talco y macrogol.
Aspecto de Sebivo y contenido del envase
Sebivo son comprimidos recubiertos con película ovales, de color blanco o ligeramente amarillento, con la inscripción «LDT» en una cara.
Sebivo comprimidos recubiertos con película se presenta en envases de 28 ó 98 comprimidos. Puede que en su país solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Etarapun Novartis Pharma Services Inc. Tea.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma GmbH Tel: +49 911 273 0 |
|
Ceská republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXXáda Novartis (Hellas) A.E.B.E. Tr(k: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
Prospecto: información para el usuario
Sebivo 20 mg/ml solución oral
Telbivudina
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Sebivo y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Sebivo
3. Cómo tomar Sebivo
4. Posibles efectos adversos
5. Conservación de Sebivo
6. Contenido del envase e información adicional
1. Qué es Sebivo y para qué se utiliza
Sebivo contiene el principio activo telbivudina. Sebivo pertenece a un grupo de medicamentos conocidos como antivirales, que se utilizan para tratar infecciones causadas por virus.
Sebivo se utiliza para tratar la hepatitis B crónica en adultos.
La causa de la hepatitis B es la infección por el virus de la hepatitis B, que se multiplica en el hígado y causa lesiones en el hígado. El tratamiento con Sebivo reduce la cantidad de virus de la hepatitis B que hay en el cuerpo al impedir su desarrollo, lo que produce menos lesión en el hígado y una mejora de la función del hígado.
2. Qué necesita saber antes de empezar a tomar Sebivo No tome Sebivo
- si es alérgico a telbivudina o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si está siendo tratado con interferón alfa pegilado o estándar (ver “Uso de otros medicamentos”).
Si éste es su caso, no tome Sebivo. Consulte con su médico.
Advertencias y precauciones
Consulte a su médico antes de empezar a tomar Sebivo:
- si tiene o ha tenido problemas de riñón. Su médico puede solicitar pruebas de laboratorio para comprobar que sus riñones funcionan adecuadamente, antes y durante el tratamiento. Dependiendo de los resultados de estos análisis su médico puede aconsejarle que cambie la frecuencia con la que toma Sebivo.
- si tiene cirrosis hepática (una enfermedad grave que causa la aparición de cicatrices en el hígado). En este caso su médico le controlará más de cerca.
- si ha recibido un trasplante de hígado.
- si está utilizando otros medicamentos que puedan causarle problemas musculares (consulte a su médico o farmacéutico si tiene dudas).
- si está infectado con el virus VIH, el de la hepatitis C o D, o está tratado con cualquier antiviral. Si este es su caso, informe a su médico antes de tomar Sebivo.
Durante el tratamiento con Sebivo:
- Sebivo puede causar dolor muscular o debilidad muscular persistente e inexplicable (miopatía). Los síntomas musculares pueden progresar y convertirse en graves, conduciendo a veces a necrosis muscular (rabdomiolisis), la cual puede producir daño renal.
- Sebivo puede producir de forma poco frecuente adormecimiento, hormigueo, dolor y/o sensación de quemazón en brazos y/o piernas (neuropatía periférica).
Si usted nota cualquiera de estos síntomas durante su tratamiento con Sebivo, informe a su médico inmediatamente.
Otros efectos adversos de esta clase de medicamentos
Sebivo pertenece a una clase de medicamentos (un análogo nucleósido) que pueden causar un exceso de ácido láctico en la sangre (acidosis láctica) y aumento del tamaño del hígado (hepatomegalia) con hígado graso (esteatosis). La acidosis láctica es un efecto adverso raro pero grave que ocasionalmente puede resultar mortal. La acidosis láctica se produce más habitualmente en mujeres, en particular si tienen mucho sobrepeso. Su médico le controlará regularmente mientras esté recibiendo Sebivo. Si usted sufre dolor muscular, dolor estomacal grave y persistente con náuseas y vómitos, tiene dificultad para respirar de forma grave y persistente, cansancio e hinchazón y/o molestia abdominal mientras esté tomando Sebivo, informe a su médico inmediatamente.
Algunas personas pueden presentar síntomas de hepatitis muy graves cuando interrumpen el tratamiento con medicamentos como Sebivo. Después de que usted interrumpa el tratamiento con Sebivo, su médico controlará su estado de salud y efectuará análisis de sangre regulares para controlar el estado de su hígado. Informe a su médico inmediatamente si usted advierte algún síntoma nuevo o inusual después de interrumpir el tratamiento (ver “Si interrumpe el tratamiento con Sebivo” en la sección 3 de este prospecto).
Tenga cuidado de no infectar a otras personas
Sebivo no reduce el riesgo de infectar a otras personas con el virus de la hepatitis B (VHB) a través de contacto sexual o por exposición a sangre contaminada o a otros fluidos corporales. Si mantiene relaciones sexuales con una pareja que no es inmune a la hepatitis B, utilice siempre preservativos y evite cualquier otro intercambio de fluídos corporales. Nunca comparta agujas. No comparta artículos personales que puedan contener sangre o fluidos corporales, como el cepillo de dientes o las cuchillas de afeitar. Existe una vacuna disponible para prevenir la infección por el VHB.
Niños y adolescentes
No se recomienda el uso de Sebivo en niños y adolescentes.
Uso de Sebivo con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Su médico o farmacéutico deben saber si usted está tomando otros medicamentos porque algunos pueden afectar a sus riñones y porque además, Sebivo se elimina del organismo fundamentalmente a través de los riñones en la orina.
No tome Sebivo si está usando interferón alfa pegilado o estándar (ver “No tome Sebivo”), ya que la combinación de estos medicamentos puede incrementar su riesgo de desarrollar neuropatía periférica (adormecimiento, hormigueo, y/o sensación de quemazón en brazos y/o piernas). Informe a su médico o farmacéutico si está siendo tratado con interferón.
Toma de Sebivo con alimentos y bebidas
Puede tomar Sebivo con o sin alimentos.
Embarazo y lactancia
- No use Sebivo durante el embarazo a no ser que su médico se lo recomiende. Si está embarazada o cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento. Su médico le informará de los riesgos potenciales de tomar Sebivo durante el embarazo.
- Si usted sufre de hepatitis B y se queda embarazada, consulte con su médico acerca de como puede proteger mejor a su bebé. Sebivo puede reducir el riesgo de transmitir su virus de la hepatitis B a su futuro bebé si se toma en combinación con la inmunoglobulina de la Hepatitis B y la vacuna de la Hepatitis B.
- No debe amamantar durante el tratamiento con Sebivo. Informe a su médico si usted está en período de lactancia.
Sebivo contiene sodio
Los pacientes con dietas pobres en sodio deben tener en cuenta que este medicamento contiene aproximadamente 47 mg de sodio por cada dosis de 600 mg (30 ml).
3. Cómo tomar Sebivo
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Cantidad de Sebivo que debe tomar
La dosis recomendada de Sebivo es de 30 ml de solución oral (600 mg de telbivudina) una vez al día. Tome Sebivo aproximadamente a la misma hora cada día. Puede tomarlo con o sin alimentos.
Para las instrucciones completas sobre cómo tomar Sebivo, ver sección “Instrucciones de uso” al final de este prospecto.
Retire el vaso dosificador y abra el frasco. Vierta la solución lentamente y con cuidado desde el frasco al vaso dosificador hasta que alcance la cantidad prescrita. Trague inmediatamente todo el contenido del vaso dosificador.
Si no puede medir de forma precisa la cantidad prescrita utilizando únicamente el vaso dosificador, deberá utilizar la jeringa para uso oral. Se proporcionan instrucciones detalladas sobre como usarla en la sección “Instrucciones de uso”.
Si tiene problemas de riñón puede que le reduzcan la dosis. Informe a su médico si tiene o ha tenido alguna vez, cualquier problema de riñón.
Durante cuanto tiempo debe tomar Sebivo
Continúe tomando Sebivo cada día durante el tiempo que le indique su médico. No cambie su dosis ni interrumpa el tratamiento con Sebivo sin consultarlo con su médico. Este medicamento está destinado para un tratamiento de larga duración, que posiblemente dure meses o años. Su médico hará un seguimiento de su enfermedad de forma regular para controlar que el tratamiento está teniendo el efecto deseado.
Si toma más Sebivo del que debe
Si usted ha tomado más Sebivo del que le haya indicado su médico, o si alguien toma accidentalmente su solución oral, acuda inmediatamente al hospital o consulte con su médico. Lleve el envase con usted y muéstrelo al médico.
Si olvidó tomar Sebivo
- Si olvidó tomar Sebivo, tómelo tan pronto como se acuerde y luego tome la siguiente dosis según la pauta establecida.
- No obstante, si quedan menos de 4 horas para la siguiente dosis, no tome la dosis olvidada y tómela cuando corresponda.
No tome una dosis doble para compensar las dosis olvidadas. Esto puede aumentar el riesgo de que usted padezca efectos adversos no deseados. Consulte a su médico o farmacéutico si no está seguro de cómo proceder.
Si interrumpe el tratamiento con Sebivo
La interrupción del tratamiento con Sebivo puede producir un empeoramiento de su infección por el virus de la hepatitis B, es decir, progresión de la enfermedad y resultados anómalos de las pruebas (aumento de la carga viral, incremento de ALT). No interrumpa el tratamiento con Sebivo salvo que se lo indique su médico. Mientras esté tomando Sebivo, asegúrese que no se queda sin él.
Después de que usted interrumpa el tratamiento con Sebivo, su médico controlará su estado de salud y efectuará análisis de sangre regulares para controlar el estado de su hígado, ya que su infección por el virus de la hepatitis B puede empeorar o llegar a ser muy grave después de interrumpir el tratamiento. Informe a su médico inmediatamente si usted advierte algún síntoma nuevo o inusual después de interrumpir el tratamiento.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Algunos efectos adversos pueden ser graves:
- Dolor muscular o debilidad muscular persistente
- Adormecimiento, hormigueo, dolor y/o sensación de quemazón en brazos y/o piernas
Si usted nota cualquiera de ellos, informe a su médico inmediatamente.
Sebivo también puede causar otros efectos adversos:
Frecuentes (pueden afectar hasta 1 de cada 10 pacientes)
- Mareo, dolor de cabeza
- Tos
- Diarrea, sensación de mareo (náuseas), dolor de estómago (abdominal)
- Erupción cutánea
- Cansancio (fatiga)
- Resultados de análisis de sangre que muestran niveles elevados de algunos enzimas hepáticos (p. ej. ALT, AST), amilasa, lipasa o creatinquinasa
Poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes)
- Dolor en articulaciones
- Dolor o debilidad muscular persistente (miopatía/miositis), calambre muscular
- Dolor de espalda, cuello y costado
- Adormecimiento, hormigueo, dolor y/o sensación de quemazón en brazos y/o piernas o alrededor de la boca
- Dolor en la parte inferior de la espalda o cadera que puede irradiar hacia la pierna (ciática)
- Alteración en el gusto
- Sensación de encontrarse mal (malestar)
Raros (pueden afectar hasta 1 de cada 1.000 pacientes)
- Exceso de ácido láctico en la sangre (acidosis láctica)
- Lesión muscular (rabdomiolisis)
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Sebivo
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en la etiqueta del frasco. La fecha de caducidad es el último día del mes que se indica.
No conservar a temperatura superior a 30°C. No congelar.
Utilizar en los 2 meses siguientes a la apertura del frasco.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Sebivo
- El principio activo es telbivudina. 30 ml de solución oral contienen 600 mg de telbivudina.
- Los demás componentes son: ácido benzoico, sacarina sódica, saborizante fruta de la pasión, hidróxido de sodio, ácido cítrico anhidro y agua purificada.
Aspecto de Sebivo y contenido del envase
Sebivo 20 mg/ml solución oral se presenta como 300 ml de solución transparente, de incolora a amarillo pálido en un frasco de vidrio marrón con un cierre de polipropileno blanco a prueba de niños, que incluye un disco de sellado de polietileno y un anillo de seguridad. El envase contiene un vaso dosificador para uso oral de polipropileno con graduaciones grabadas de 5 a 30 ml en aumentos de 5 ml y una jeringa para uso oral de polipropileno con graduaciones de 1 ml a 10 ml en incrementos de 0,5 ml.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Btnrapun Novartis Pharma Services Inc. Tea.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma GmbH Tel: +49 911 273 0 |
|
Ceská republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EkXúóa Novartis (Hellas) A.E.B.E. Tr(k: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A. Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
Ireland
Novartis Ireland Limited Tel: +353 1 260 12 55
Ísland
Vistor hf.
Sími: +354 535 7000 Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Slovenija
Novartis Pharma Services Inc.
Tel: +386 1 300 75 50
Slovenská republika
Novartis Slovakia s.r.o.
Tel: +421 2 5542 5439
Suomi/Finland
Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd.
Tel: +44 1276 698370
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
INSTRUCCIONES DE USO
Lea estas instrucciones detenidamente para que sepa como usar la solución correctamente.
1.
2.
3.
4.
Frasco con la solución oral.
Tapón de rosca a prueba de niños con un anillo de seguridad. Después de usar cierre siempre el frasco con el tapón.
Vaso dosificador para uso oral para medir la dosis. Después de usarlo y limpiarlo vuelva a colocar siempre el vaso dosificador sobre el tapón.
Jeringa para uso oral para medir las dosis que no se puedan medir de forma precisa usando el vaso dosificador.
Preparación de una dosis de medicamento usando el vaso dosificador
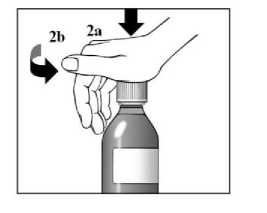
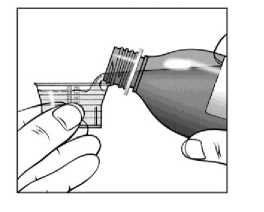
1. Retire el vaso dosificador.
2. Simultáneamente presione hacia abajo (2a) y gire el tapón a prueba de niños (2b) hacia la izquierda para abrir el frasco.
3. Antes de verter la solución en el vaso, compruebe la posición de la graduación asignada para evitar cualquier posible pérdida o derrame.
Sosteniendo el vaso a nivel de los ojos, vierta lentamente y con cuidado la cantidad prescrita de solución desde el frasco al vaso dosificador hasta que la solución alcance la parte superior de la graduación indicada.
Nota: Si la cantidad vertida en el vaso excede la dosis requerida, desechar el exceso en el fregadero. No lo vuelva a verter en el frasco.
4. Beba la solución o adminístresela al paciente inmediatamente.
5. Vuelva a cerrar el frasco herméticamente con el tapón de rosca.
Lave inmediatamente el vaso dosificador con agua. Elimine el agua del vaso dosificador, séquelo con un trapo limpio y vuelva a colocarlo sobre el tapón.
6.
7.
Preparación de una dosis de 6
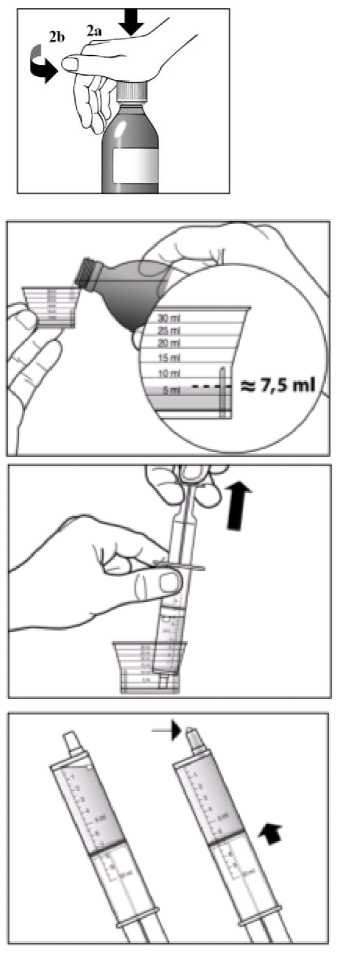
ml de medicamento usando la jeringa para uso oral
1. Retire el vaso dosificador.
2. Simultáneamente presione hacia abajo (2a) y gire el tapón a prueba de niños (2b) hacia la izquierda para abrir el frasco.
3. Antes de verter la solución en el vaso, compruebe la posición de las marcas de 5 y 10 ml para evitar cualquier posible pérdida o derrame.
Sosteniendo el vaso a nivel de los ojos, vierta lentamente y con cuidado la solución desde el frasco al vaso dosificador hasta que alcance aproximadamente la mitad entre las marcas de 5 ml y 10 ml.
4. Extraiga toda la solución desde el vaso dosificador a la jeringa.
5. Gire la jeringa en posición vertical e inclínela ligeramente con el fin de que las burbujas de aire asciendan a la parte superior.
6. Presione el émbolo lentamente y con cuidado para eliminar el aire hasta que aparezca una gota de la solución.
7. Mantenga la j eringa encima del vaso dosificador.
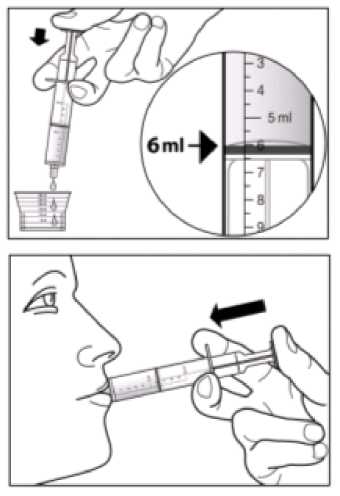
8. Presione el émbolo lentamente y con cuidado hasta que la solución alcance la marca de 6 ml.
9. Trague inmediatamente la solución directamente de la jeringa.
10. Deseche la solución sobrante del vaso dosificador en el fregadero. No lo vuelva a colocar en el frasco pues podría causar contaminación.
11. Cierre el frasco fuertemente.
12. Lave el vaso dosificador y la jeringa con agua clara.
13. Seque el vaso dosificador con un trapo limpio y vuelva a colocarlo sobre el tapón del frasco.
14. Deje secar al aire la jeringa y guárdela con el frasco.
66