Relvar Ellipta 184Mcg/22Mcg Polvo Para Inhalacion (Unidosis)
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Relvar Ellipta 92 microgramos/22 microgramos polvo para inhalación (unidosis).
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada inhalación proporciona una dosis liberada (la dosis que sale por la boquilla) de 92 microgramos de furoato de fluticasona y 22 microgramos de vilanterol (como trifenatato). Esto se corresponde con una dosis de 100 microgramos de furoato de fluticasona y 25 microgramos de vilanterol (como trifenatato).
Excipientes con efecto conocido:
Cada dosis liberada contiene aproximadamente 25 mg de lactosa (como monohidrato).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para inhalación (unidosis)
(Polvo para inhalación).
Polvo blanco en un inhalador de color gris claro con un protector de la boquilla de color amarillo y un contador de dosis.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Asma
Relvar Ellipta está indicado para el tratamiento regular del asma en adultos y adolescentes de 12 años de edad y mayores cuando la administración de una combinación (un agonista p2 de acción prolongada y un corticosteroide por vía inhalatoria) sea apropiada:
• pacientes no controlados adecuadamente con corticosteroides inhalados y agonistas p2 inhalados de acción corta administrados "a demanda".
EPOC (Enfermedad Pulmonar Obstructiva Crónica)
Relvar Ellipta está indicado para el tratamiento sintomático de adultos con EPOC, con un FEVi < 70% del normal (post-broncodilatador) y una historia clínica de exacerbaciones a pesar del uso regular de una terapia broncodilatadora.
4.2 Posología y forma de administración
Posología
Asma
Adultos y adolescentes de 12 años de edad y mayores
Una inhalación de Relvar Ellipta 92/22 microgramos una vez al día.
Los pacientes normalmente experimentan una mejora de la función pulmonar en los 15 minutos tras la inhalación de Relvar Ellipta. Sin embargo, se debe informar al paciente de que es necesario el uso regular diario para mantener el control de los síntomas del asma y que debe continuar usándolo aun cuando no tenga síntomas.
Si aparecen síntomas en los periodos entre dosis, se debe usar un agonista p2 inhalado de acción corta para un alivio inmediato.
En adultos y adolescentes de 12 años de edad y mayores que requieran una dosis de baja a intermedia de corticosteroide inhalado en combinación con un agonista p2 de acción prolongada se debe considerar una dosis de inicio de Relvar Ellipta 92/22 microgramos. Si los pacientes no están adecuadamente controlados con Relvar Ellipta 92/22 microgramos, se puede aumentar la dosis a 184/22 microgramos, lo que puede proporcionar una mejora adicional en el control del asma.
Los pacientes deben ser reevaluados regularmente por un médico, de manera que la concentración de furoato de fluticasona/vilanterol que reciban siga siendo la óptima y solo se modifique a criterio médico. Se debe ajustar la dosis de forma que se administre la dosis más baja que mantenga un control eficaz de los síntomas.
En adultos y adolescentes de 12 años de edad y mayores que requieran una dosis más alta de corticosteroide inhalado en combinación con un agonista p2 de acción prolongada se debe considerar la dosis de Relvar Ellipta 184/22 microgramos.
Los pacientes con asma deben recibir la concentración de Relvar Ellipta que contenga la dosis apropiada de furoato de fluticasona (FF) en base a la gravedad de su enfermedad. Los prescriptores deben saber que en los pacientes con asma, una dosis diaria de 100 microgramos de furoato de fluticasona (FF) es aproximadamente equivalente a 250 microgramos de propionato de fluticasona (PF) dos veces al día, mientras que 200 microgramos de FF una vez al día es aproximadamente equivalente a 500 microgramos de PF dos veces al día.
Niños menores de 12 años de edad
No se ha establecido la seguridad y eficacia de Relvar Ellipta en niños menores de 12 años de edad para la indicación en asma.
No hay datos disponibles.
EPOC
Adultos de 18 años de edad y mayores
Una inhalación de Relvar Ellipta 92/22 microgramos una vez al día.
Relvar Ellipta 184/22 microgramos no está indicado para pacientes con EPOC. No hay un beneficio adicional con la dosis de 184/22 microgramos en comparación con la dosis de 92/22 microgramos y hay un posible aumento en el riesgo de desarrollar neumonía y de reacciones adversas sistémicas debidas a los corticosteroides (ver secciones 4.4 y 4.8).
Los pacientes normalmente experimentan una mejora de la función pulmonar en los 16-17 minutos tras la inhalación de Relvar Ellipta.
Población pediátrica
No hay un uso relevante de Relvar Ellipta en la población pediátrica para la indicación de EPOC. Poblaciones especiales:
Pacientes de edad avanzada (>65 años)
(ver sección 5.2).
No se requiere ajuste de dosis en esta población Insuficiencia renal
(ver sección 5.2).
No se requiere ajuste de dosis en esta población Insuficiencia hepática
En estudios con sujetos con insuficiencia hepática leve, moderada y grave se observó un aumento en la exposición sistémica a furoato de fluticasona (ambos Cmax y AUC) (ver sección 5.2).
Se debe tener precaución cuando se prescriben dosis a pacientes con insuficiencia hepática ya que pueden tener un mayor riesgo de reacciones adversas sistémicas asociadas con los corticosteroides.
La dosis máxima para pacientes con insuficiencia hepática moderada o grave es de 92/22 microgramos (ver sección 4.4).
Forma de administración
Relvar Ellipta se administra solo por vía inhalatoria.
Debe administrarse a la misma hora del día, cada día.
La decisión final sobre si la administración debe ser por la mañana o por la noche se deja a elección del médico.
Si se olvida una dosis, la siguiente dosis debe administrarse al día siguiente a la hora habitual.
Si se conserva en nevera, se debe dejar que el inhalador vuelva a temperatura ambiente durante por lo menos una hora antes de utilizarlo.
Tras la inhalación, los pacientes deben aclararse la boca con agua sin tragarla.
Cuando el inhalador se utiliza por primera vez, no es necesario comprobar que funciona correctamente, ni prepararlo de ninguna forma especial para su uso. Se deben seguir las instrucciones de uso paso a paso.
El inhalador Ellipta está envasado en una bandeja que contiene una bolsa desecante para reducir la humedad. Una vez abierto se debe desechar la bolsa desecante.
Se debe advertir al paciente de que no abra la bandeja hasta que esté preparado para inhalar la dosis.
Cuando se saca el inhalador de la bandeja, estará en la posición “cerrado”. La fecha de “desechar el”, debe escribirse en el espacio designado para ello en la etiqueta del inhalador. La fecha de “desechar el” es de 6 semanas desde la fecha de apertura de la bandeja. Después de esta fecha, el inhalador debe desecharse. La bandeja se puede desechar después de la primera apertura.
Las instrucciones de uso paso a paso que se muestran a continuación para el inhalador Ellipta de 30 dosis también aplican para el inhalador Ellipta de 14 dosis.
Instrucciones de uso
1. Leer las siguientes instrucciones antes de utilizar el inhalador
Si la tapa del inhalador se abre y cierra sin que se inhale el medicamento, se perderá la dosis. La dosis perdida quedará retenida de forma segura dentro del inhalador, pero no estará disponible para ser inhalada.
No es posible administrarse accidentalmente una dosis adicional o una dosis doble mediante una inhalación.
Contador de dosis
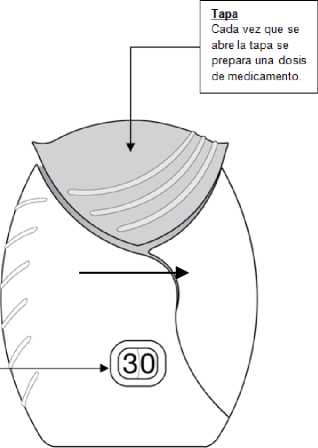
El contador de dosis indica cuántas dosis de medicamento quedan en el dispositivo. Antes de usar el inhalador, debe indicar exactamente 30 dosis.
Cada vez que se abre la tapa, el contador disminuye en 1 unidad.
Cuando quedan menos de 10 dosis, la mitad del contador de dosis se pone de color rojo.
Una vez se utiliza la última dosis, la mitad del contador de dosis se pone de color rojoe indica el número 0. El inhalador ahora está vacío.
Si se abre la tapa cuando el inhalador está vacío, el contador de dosis pasa de estar la mitad de color rojo a estarlo completamente.
2. Cómo preparar una dosis
Abrir la tapa cuando esté preparado para administrarse una dosis. No agitar el inhalador.
Deslizar la tapa hacia abajo hasta oír un ‘clic’.
El medicamento está ahora preparado para ser inhalado. Como confirmación, el contador de dosis disminuye en 1 unidad.
Si el contador de dosis no disminuye al oír el ‘clic’, el inhalador no liberará el medicamento. Llévelo
al farmacéutico y solicite ayuda.
Cómo inhalar el medicamento
3.
Mantener el inhalador alejado de la boca y espirar lo que razonablemente pueda.
No espirar dentro del inhalador.
Colocar la boquilla entre los labios y cerrarlos firmemente alreadedor de la boquilla.
No bloquear las ranuras de ventilación con los dedos.
Realizar una inspiración prolongada, continua y profunda. Mantener la respiración tanto tiempo como sea posible (al menos 3-4 segundos).
• Retirar el inhalador de la boca.
• Espirar suave y lentamente.
Puede que no sea capaz de distinguir el sabor o notar el medicamento, incluso cuando utiliza el inhalador de forma correcta.
4. Cerrar el inhalador y enjuagarse la boca
Si quiere limpiar la boquilla utilice un pañuelo seco antes de cerrar la tapa.
Deslizar la tapa hacia arriba hasta el tope para proteger la boquilla.
Enjuagarse la boca con agua, una vez utilizado el inhalador.
Esto hará que sea menos probable que se produzcan efectos adversos como ulceraciones en la boca o garganta.
4.3 Contraindicaciones
Hipersensibilidad a los principios activos o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Empeoramiento de la enfermedad
El furoato de fluticasona/vilanterol no se debe utilizar para tratar los síntomas agudos del asma o una exacerbación aguda en la EPOC, para lo que se requiere un broncodilatador de acción corta. El aumento de empleo de broncodilatadores de acción corta para aliviar los síntomas indica un empeoramiento en el control y los pacientes deben ser examinados por un médico.
Los pacientes no deben interrumpir el tratamiento con furoato de fluticasona/vilanterol en asma o EPOC, sin la supervisión de un médico ya que los síntomas pueden reaparecer tras interrumpir el tratamiento.
Durante el tratamiento con furoato de fluticasona/vilanterol pueden producirse efectos adversos relacionados con el asma y exacerbaciones de la enfermedad. Se debe pedir a los pacientes que continúen el tratamiento, pero que acudan a su médico si los síntomas del asma siguen sin estar controlados o empeoran tras comenzar el tratamiento con Relvar Ellipta.
Broncoespasmo paradójico
Tras la administración de la dosis puede aparecer broncoespasmo paradójico con un aumento inmediato en las sibilancias. Se debe de tratar inmediatamente con un broncodilatador inhalado de acción corta. Se debe interrumpir el tratamiento con Relvar Ellipta inmediatamente, evaluar al paciente e instaurar un tratamiento alternativo si es necesario.
Efectos cardiovasculares
Con el uso de medicamentos simpaticomiméticos, incluido Relvar Ellipta, se pueden observar efectos cardiovasculares como arritmias cardiacas, por ejemplo taquicardia supraventricular y extrasístoles.
Por lo tanto, furoato de fluticasona/vilanterol se debe usar con precaución en pacientes con enfermedad cardiovascular grave o anomalías en el ritmo cardiaco, tirotoxicosis, hipopotasemia no corregida o pacientes con predisposición a niveles séricos de potasio bajos.
Pacientes con insuficiencia hepática
En pacientes con insuficiencia hepática de moderada a grave se debe usar la dosis de
92/22 microgramos y se debe controlar a los pacientes por las reacciones adversas sistémicas debidas a
los corticosteroides (ver sección 5.2).
Efectos sistémicos de los corticosteroides
Pueden aparecer efectos sistémicos con cualquier corticosteroide administrado por vía inhalatoria, especialmente a dosis elevadas prescritas durante largos periodos. La probabilidad de que estos efectos aparezcan es mucho menor que con el uso de corticosteroides administrados por vía oral. Los posibles efectos sistémicos incluyen Síndrome de Cushing, aspecto Cushingoideo, supresión suprarrenal, disminución de la densidad mineral ósea, retraso en el crecimiento de niños y adolescentes, cataratas y glaucoma y más raramente, una serie de efectos psicológicos o del comportamiento que incluyen hiperactividad psicomotora, trastornos del sueño, ansiedad, depresión o agresividad (especialmente en niños).
Furoato de fluticasona/vilanterol se debe administrar con precaución en pacientes con tuberculosis pulmonar o en pacientes con infecciones crónicas o no tratadas.
Se han notificado casos dc aumento dc los niveles dc glucosa cn sangre cn pacientes diabéticos, lo cual dcbc tenerse cn cuenta cuando se prescriba a pacientes con antecedentes dc diabetes mellitus.
Neumonía en pacientes con EPOC
Se ha observado un aumento dc la incidencia dc neumonía, incluyendo neumonía que requiere hospitalización, en pacientes con EPOC que reciben corticosteroides inhalados. Existe alguna evidencia dc un mayor riesgo dc neumonía con el aumento dc la dosis dc cstcroidcs, pero esto no ha sido demostrado dc manera concluyente cn todos los estudios.
No hay evidencia clínica concluyente de diferencias intra-clase en la magnitud del riesgo de neumonía entre los corticoides inhalados.
Los médicos deben permanecer vigilantes ante el posible desarrollo dc neumonía cn pacientes con EPOC, ya que las características clínicas de estas infecciones se superponen con los síntomas de exacerbación de la EPOC.
Los factores de riesgo de neumonía en pacientes con EPOC incluyen el tabaquismo habitual, pacientes dc edad avanzada, bajo índice dc masa corporal (IMC) y EPOC grave.
Rclvar Ellipta 184/22 microgramos no está indicado para pacientes con EPOC. No hay un beneficio adicional con la dosis dc 184/22 microgramos cn comparación con la dosis dc 92/22 microgramos y hay un posible aumento cn el riesgo dc que se produzcan reacciones adversas sistémicas debidas a los corticosteroides (ver sección 4.8).
Neumonía en pacientes con Asma
La incidencia dc neumonía cn pacientes con asma fue frecuente con la dosis más alta. La incidencia dc neumonía cn pacientes con asma que utilizaban la dosis dc 184/22 microgramos dc furoato dc fluticasona/vilanterol fue numéricamente mayor cn comparación con los que recibían la dosis dc 92/22 microgramos dc furoato dc fluticasona/vilanterol o placebo (ver sección 4.8). No se identificaron factores dc riesgo.
Excipientes
Los pacientes con intolerancia hereditaria a galactosa, insuficiencia dc lactasa dc Lapp o malabsorción dc glucosa o galactosa no deben usar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Las interacciones clínicamente significativas mediadas por furoato dc fluticasona/vilanterol a las dosis clínicas se consideran poco probables debido a las bajas concentraciones plasmáticas que se alcanzan tras la inhalación de la dosis.
Interacción con betabloqueantes
Los bloqueantes p2-adrcnérgicos pueden disminuir o antagonizar el efecto dc los agonistas p2-adrcnérgicos. Se dcbc evitar el uso concomitante dc bloqueantes p2-adrenérgicos no selectivos y selectivos a menos que existan razones de peso para su uso.
Interacción con inhibidores del CYP3A4
Furoato dc fluticasona y vilantcrol sufren ambos un rápido aclaramiento debido a un intenso metabolismo dc primer paso mediado por la enzima hepática CYP3A4.
Se recomienda tener precaución cuando se administra este medicamento junto con inhibidores potentes del CYP3A4 (por ejemplo ketoconazol, ritonavir), ya que es posible un aumento en la exposición sistémica tanto de furoato de fluticasona como de vilanterol, por lo que se recomienda evitar el uso concomitante. Se realizó un estudio de interacción con CYP3A4 a dosis repetidas en sujetos sanos con la combinación furoato de fluticasona/vilanterol (184/22 microgramos) y ketoconazol (400 mg), potente inhibidor del CYP3A4. La administración concomitante aumentó la media del AUC(0-24) y Cmax de furoato de fluticasona en un 36% y 33%, respectivamente. El aumento de la exposición de furoato de fluticasona se asoció con una reducción del 27% en la media ponderada entre 0-24 horas del cortisol sérico. La administración concomitante aumentó la media del AUC(0-t) y Cmax de vilanterol en un 65% y 22%, respectivamente. El aumento de la exposición de vilanterol no se asoció con un aumento de los efectos sistémicos relacionados con los agonistas p2 como el ritmo cardiaco, los niveles de potasio en sangre o el intervalo QTcF.
Interacción con inhibidores de la glicoproteína-P
Furoato de fluticasona y vilanterol son ambos sustratos de la glicoproteína-P (P-gp). En un estudio clínico farmacológico realizado en sujetos sanos a los que se administró conjuntamente vilanterol y verapamilo, potente inhibidor de la P-gp e inhibidor moderado del CYP3A4, no se observó ningún efecto significativo en la farmacocinética de vilanterol. No se han realizado estudios clínicos farmacológicos con un inhibidor específico P-gp y furoato de fluticasona.
Medicamentos simpaticomiméticos
La administración concomitante con otros medicamentos simpaticomiméticos (en monoterapia o como parte de una combinación) pueden potenciar las reacciones adversas de furoato de fluticasona/vilanterol. Relvar Ellipta no debe utilizarse en combinación con otros agonistas p2-adrenérgicos de acción prolongada o medicamentos que contengan agonistas p2-adrenérgicos de acción prolongada.
Población pediátrica
Los estudios de interacción se han realizado solo en adultos.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Los estudios realizados en animales han mostrado toxicidad para la reproducción a exposiciones que no son relevantes clínicamente (ver sección 5.3). No hay datos o éstos son limitados relativos al uso de furoato de fluticasona y vilanterol trifenatato en mujeres embarazadas.
Solo se debe considerar la administración de furoato de fluticasona/vilanterol en mujeres embarazadas si el beneficio esperado para la madre es mayor que cualquier posible riesgo para el feto.
Lactancia
No se dispone de información suficiente relativa a la excreción de furoato de fluticasona o vilanterol trifenatato y/o sus metabolitos en la leche materna. Sin embargo, otros corticosteroides y p2 agonistas fueron detectados en la leche materna (ver sección 5.3). No se puede excluir el riesgo en recién nacidos/niños lactantes.
Se debe decidir si es necesario interrumpir la lactancia o interrumpir el tratamiento con furoato de fluticasona/vilanterol tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Fertilidad
No hay datos de fertilidad en humanos. Los estudios realizados en animales no han mostrado efectos de furoato de fluticasona/vilanterol trifenatato sobre la fertilidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de furoato de fluticasona o vilanterol sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los resultados de ensayos clínicos amplios en asma y en EPOC fueron utilizados para determinar la frecuencia de las reacciones adversas asociadas a furoato de fluticasona/vilanterol. En el programa de desarrollo clínico en asma fueron incluidos un total de 7.034 pacientes en una evaluación integrada de reacciones adversas. En el programa de desarrollo clínico en EPOC se incluyeron un total de 6.237 sujetos en una evaluación integrada de reacciones adversas.
Las reacciones adversas de furoato de fluticasona y vilanterol notificadas con más frecuencia fueron cefalea y nasofaringitis. A excepción de la neumonía y las fracturas, el perfil de seguridad fue similar en pacientes con asma y EPOC. Durante los ensayos clínicos, la neumonía y las fracturas se observaron generalmente con mayor frecuencia en pacientes con EPOC.
Tabla de reacciones adversas
Las reacciones adversas se enumeran clasificadas por órganos y frecuencia. Para la clasificación de frecuencias se utiliza el siguiente convenio: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000).
Dentro de cada grupo de frecuencias, las reacciones adversas se presentan por orden decreciente de gravedad.
|
Sistema de clasificación de órganos |
Reacciones adversas |
Frecuencia |
|
Infecciones e infestaciones |
Neumonía* Infecciones del tracto respiratorio superior Bronquitis Gripe Candidiasis en la boca y garganta |
Frecuentes |
|
Trastornos del sistema |
Reacciones de hipersensibilidad, |
Raras |
|
inmunológico |
incluyendo anafilaxia, angioedema, erupción, y urticaria | |
|
Trastornos del sistema |
Cefalea |
Muy frecuentes |
|
nervioso |
Temblor |
Raras |
|
Trastornos psiquiátricos |
Ansiedad |
Raras |
|
Trastornos cardiacos |
Extrasístoles |
Poco frecuentes |
|
Palpitaciones |
Raras | |
|
Taquicardia |
Raras | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Nasofaringitis Dolor orofaríngeo Sinusitis Faringitis Rinitis Tos Disfonía |
Muy frecuentes Frecuentes |
|
Trastornos gastrointestinales |
Dolor abdominal |
Frecuentes |
|
Trastornos |
Artralgia |
Frecuentes |
|
musculoesqueléticos y del tejido conjuntivo |
Dolor de espalda Fracturas** Espasmos musculares | |
|
Trastornos generales y alteraciones en el lugar de administración |
Pirexia |
Frecuentes |
*, ** Ver a continuación “Descripción de reacciones adversas seleccionadas”
Descripción de reacciones adversas seleccionadas
Neumonía
En un análisis integrado de dos estudios replicados de un año de duración en pacientes con EPOC con una exacerbación en el año anterior (n = 3.255), el número de casos de neumonía por 1.000 pacientes-año fue 97,9 con FF/VI 184/22, 85,7 en el grupo FF/VI 92/22 y 42,3 en el grupo VI 22. En el caso de neumonía severa, el número de casos correspondiente por 1.000 pacientes-año fueron 33,6, 35,5 y 7,6 respectivamente, mientras que los casos de neumonía grave por 1.000 pacientes-año fueron 35,1 para
FF/VI 184/22, 42,9 con FF/VI 92/22, 12,1 con VI 22. Por último, los casos de neumonía con desenlace mortal (ajustados por exposición) fueron 8,8 para FF/VI 184/22 frente a 1,5 para FF/VI 92/22 y 0 para VI 22.
En un análisis integrado de 11 estudios en asma (7.034 pacientes), la incidencia de neumonía por 1.000 pacientes-año fue 18,4 para FF/VI 184/22 frente a 9,6 para FF/VI 92/22 y 8,0 en el grupo placebo.
Fracturas
En dos estudios replicados de 12 meses de duración en el que participaron un total de 3.255 pacientes con EPOC, la incidencia de fracturas óseas fue baja de forma global en todos los grupos de tratamiento, con una incidencia mayor en todos los grupos con Relvar Ellipta (2%) en comparación con el grupo vilanterol 22 microgramos (<1%). Aunque hubo más fracturas en los grupos de tratamiento con Relvar Ellipta en comparación con el grupo vilanterol 22 microgramos, las fracturas típicamente asociadas al uso de corticosteroides (por ejemplo compresión espinal/fracturas vertebrales toracolumbares, fracturas de cadera y acetabulares) se produjeron en <1% en los brazos de tratamiento con Relvar Ellipta y vilanterol.
En un análisis integrado de 11 estudios en asma (7.034 pacientes), la incidencia de fracturas fue <1%, y normalmente se asociaban con traumatismos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Síntomas y signos
Una sobredosis de furoato de fluticasona/vilanterol puede producir signos y síntomas debido a la acción de los componentes por separado, incluyendo aquellos que se observan con una sobredosis de otros p2-agonistas y en consistencia con los efectos de clase conocidos de los corticosteroides inhalados (ver sección 4.4).
Tratamiento
No hay un tratamiento específico para la sobredosis con furoato de fluticasona/vilanterol. En caso de sobredosis, el paciente debe recibir el tratamiento de soporte necesario y un seguimiento apropiado.
Solo se debe considerar la administración de betabloqueantes cardioselectivos cuando se produzcan efectos clínicamente relevantes debidos a una sobredosis grave de vilanterol y que no respondan a las medidas de soporte. Los betabloqueantes cardioselectivos se deben usar con precaución en pacientes con antecedentes de broncoespasmo.
Para un manejo adicional se deben seguir las recomendaciones clínicas indicadas o las recomendaciones del Centro Nacional de Toxicología, si estuvieran disponibles.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes para las enfermedades obstructivas de las vías respiratorias. Adrenérgicos y otros agentes para las enfermedades obstructivas de las vías respiratorias, código ATC: R03AK10.
Mecanismo de acción
Furoato de fluticasona y vilanterol son dos tipos de medicamentos (un corticosteroide sintético y un agonista selectivo del receptor p2 de acción prolongada).
Efectos farmacodinámicos
Furoato de _fluticasona
Furoato de fluticasona es un corticosteroide sintético trifluorinado con una potente actividad antiinflamatoria. Se desconoce el mecanismo exacto por el cual furoato de fluticasona actúa sobre los síntomas del asma y EPOC. Los corticosteroides han demostrado tener una amplia variedad de acciones sobre diversos tipos celulares (por ejemplo, eosinófilos, macrófagos, linfocitos) y mediadores (por ejemplo, citoquinas y quimioquinas involucradas en la inflamación).
Vilanterol trifenatato
Vilanterol trifenatato es un agonista selectivo p2-adrenérgico de acción prolongada (LABA).
El efecto farmacológico de los fármacos agonistas p2-adrenérgicos, incluido vilanterol trifenatato, son al menos en parte atribuibles a la estimulación de la adenilato ciclasa intracelular, la enzima que cataliza la transformación de la adenosín trifosfato (ATP) a la adenosín monofosfato - 3’,5’ cíclico (AMP cíclico). El aumento en los niveles del AMP cíclico produce la relajación del músculo liso bronquial y la inhibición de la liberación de mediadores de la hipersensibilidad inmediata de las células, especialmente de los mastocitos.
Se producen interacciones moleculares entre los corticosteroides y LABAs, por las que los esteroides activan el gen del receptor p2 aumentando el número de receptores y la sensibilidad, y los LABAs preparan al receptor glucocorticoide para la activación dependiente de esteroides y aumentan la translocación nuclear celular. Estas interacciones sinérgicas se reflejan en un aumento de la actividad anti-inflamatoria, que se ha demostrado in vitro e in vivo en una variedad de células inflamatorias relevantes para la fisiopatología del asma y EPOC. En estudios de biopsias de las vías respiratorias con furoato de fluticasona y vilanterol se ha demostrado también la sinergia entre corticosteroides y LABAs a las dosis clínicas de los medicamentos en pacientes con EPOC.
Eficacia clínica y seguridad
Asma
Tres estudios fase III aleatorizados, doble ciego (HZA106827, HZA106829 y HZA106837) de diferente duración evaluaron la seguridad y eficacia de furoato de fluticasona/vilanterol en pacientes adultos y adolescentes con asma persistente. Todos los sujetos estaban utilizando CSI (corticosteroides inhalados) con o sin LABA durante al menos 12 semanas antes de la visita 1. En el estudio HZA106837 todos los pacientes tuvieron al menos una exacerbación que requirió tratamiento con corticosteroides orales en el año anterior a la visita 1. HZA106827 tuvo una duración de 12 semanas y evaluó la eficacia de furoato de fluticasona/vilanterol 92/22 microgramos [n=201] y FF 92 microgramos [n=205] en comparación con placebo [n=203], todos ellos administrados una vez al día. HZA106829 tuvo una duración de 24 semanas y evaluó la eficacia de furoato de fluticasona/vilanterol 184/22 microgramos [n=197] y FF 184 microgramos [n=194] ambos administrados una vez al día en comparación con 500 microgramos de PF dos veces al día [n=195].
En HZA106827/HZA106829 las variables co-primarias de eficacia fueron el cambio respecto a los valores basales en la visita clínica del FEVi valle (pre-broncodilatador y pre-dosis) al final del periodo de tratamiento en todos los sujetos y la media ponderada de los valores seriados del FEV1 durante 0 a 24 horas después de la administración de la dosis calculado en un subconjunto de los sujetos al final del periodo de tratamiento. El cambio respecto a los valores basales en el porcentaje de días sin medicación de rescate durante el tratamiento fue una variable secundaria robusta. Los resultados de las variables primarias y de las principales variables secundarias de estos estudios se describen en la Tabla 1.
Tabla 1 - Resultados de las variables primarias y principales variables secundarias en HZA106827 y HZA106829
|
N° de estudio |
HZA106829 |
HZA] |
106827 | |
|
Dosis de tratamiento de |
FF/VI 184/22 |
FF/VI 184/22 |
FF/VI 92/22 |
FF/VI/92/22 |
|
FF/VI*(microgramos) |
una vez al día |
una vez al día |
una vez al día |
una vez al día |
|
vs FF 184 una |
vs PF 500 dos |
vs FF 92 una |
vs placebo una | |
|
vez al día |
veces al día |
vez al día |
vez al día | |
|
Cambios respecto a los valores basales en el FEV1 valle por el método de Última Observación Realizada (LOCF) | ||||
|
Diferencia de tratamiento |
193 ml |
210 ml |
36 ml |
172 ml |
|
Valor p |
p<0,001 |
p<0,001 |
p=0,405 |
p<0,001 |
|
(IC 95%) |
(108; 277) |
(127; 294) |
(-48; 120) |
(87; 258) |
|
Media ponderada de los |
valores seriados del FEV1 en las 0-24 horas después de la dosis | |||
|
Diferencia de tratamiento |
136 ml |
206 ml |
116 ml |
302 ml |
|
Valor p |
p=0,048 |
p=0,003 |
p=0,06 |
p<0,001 |
|
(IC 95%) |
(1; 270) |
(73; 339) |
(-5; 236) |
(178; 426) |
|
Cambios respecto a los valores basales en el porcentaje t |
e días sin medicación de rescate | |||
|
Diferencia entre tratamientos |
11,7% |
6,3% |
10,6% |
19,3% |
|
Valor p |
p<0,001 |
p=0,067 |
p<0,001 |
p<0,001 |
|
(IC 95%) |
(4,9; 18,4) |
(-0,4; 13,1) |
(4,3; 16,8) |
(13,0; 25,6) |
|
Cambios respecto a los valores basales en el porcentaje d |
e días sin síntomas | |||
|
Diferencia entre tratamientos |
8,4% |
4,9% |
12,1% |
18,0% |
|
Valor p |
p=0,010 |
p=0,137 |
p<0,001 |
p<0,001 |
|
(IC 95%) |
(2,0; 14,8) |
(-1,6; 11,3) |
(6,2; 18,1) |
(12,0; 23,9) |
|
Cambios respecto a los valores basales en el flujo espiratorio máximo por la mañana | ||||
|
Diferencia entre tratamientos |
33,5 l/min |
32,9 l/min |
14,6 l/min |
33,3 l/min |
|
Valor p |
p<0,001 |
p<0,001 |
p<0,001 |
p<0,001 |
|
(IC 95%) |
(22,3; 41,7) |
(24,8; 41,1) |
(7,9; 21,3) |
(26,5; 40,0) |
|
Cambios respecto a los valores basales en el flujo espiratorio máximo por la noche | ||||
|
Diferencia entre tratamientos |
30,7 l/min |
26,2 l/min |
12,3 l/min |
28,2 l/min |
|
Valor p |
p<0,001 |
p<0,001 |
p<0,001 |
p<0,001 |
|
(IC 95%) |
(22,5; 38,9) |
(18,0; 34,3) |
(5,8; 18,8) |
(21,7; 34,8) |
*FF/VI = furoato de fluticasona/vilanterol
HZA106837 tuvo una duración de tratamiento variable (desde un mínimo de 24 semanas a un máximo de 76 semanas, en el que la mayoría de los pacientes fueron tratados durante al menos 52 semanas). En HZA106837, los pacientes fueron aleatorizados para recibir bien furoato de fluticasona/vilanterol
92/22 microgramos [n=1.009] o FF 92 microgramos [n=1.010] ambos administrados una vez al día.
En HZA106837 la variable primaria fue el tiempo hasta la primera exacerbación asmática grave. Una exacerbación asmática grave se definió como un empeoramiento del asma que requirió el uso de corticosteroides sistémicos durante al menos 3 días, o una hospitalización o visita a urgencias como consecuencia de que el asma requirió el uso de corticosteroides sistémicos. Se evaluó también como variable secundaria el cambio medio ajustado respecto los valores basales en el FEV1 valle.
En HZA106837 el riesgo de tener una exacerbación asmática grave en pacientes que recibieron furoato de fluticasona/vilanterol 92/22 microgramos se redujo en un 20% en comparación con FF 92 microgramos en monoterapia (hazard ratio 0,795; p=0,036 IC 95% 0,642; 0,985). La tasa de exacerbaciones asmáticas graves por paciente y año fue 0,19 en el grupo FF 92 microgramos (aproximadamente 1 cada 5 años) y 0,14 en el grupo furoato de fluticasona/vilanterol 92/22 microgramos (aproximadamente 1 cada 7 años). La proporción de la tasa de exacerbaciones para furoato de fluticasona/vilanterol 92/22 microgramos frente a FF 92 microgramos fue 0,755 (IC 95% 0,603; 0,945). Esto representa una reducción del 25% en la tasa de exacerbaciones asmáticas graves para los sujetos tratados con furoato de fluticasona/vilanterol 92/22 microgramos en comparación con FF 92 microgramos (p=0,014). El efecto broncodilatador de 24-horas de furoato de fluticasona/vilanterol se mantuvo durante un año de tratamiento en el que no hubo evidencia de pérdida de eficacia (sin taquifilaxia). Furoato de fluticasona/vilanterol 92 /22 microgramos demostró de forma consistente la mejora en el FEVi valle de 83 ml a 95 ml en las semanas 12, 36 y 52 y al final del estudio en comparación con FF 92 microgramos (p<0,001 IC 95% 52; 126 ml al final del estudio). El cuarenta y cuatro por ciento de los pacientes en el grupo furoato de fluticasona/vilanterol 92/22 microgramos estuvieron bien controlados (ACQ7 <0,75) al final del tratamiento en comparación con el 36% de los sujetos en el grupo FF 92 microgramos (p<0,001 IC 95% 1,23; 1,82).
Estudios frente a las combinaciones con salmeterol/propionato de fluticasona
En un estudio de 24 semanas de duración (HZA113091) en pacientes adultos y adolescentes con asma persistente se demostró mejoras en la función pulmonar respecto a los valores basales tanto con furoato de fluticasona/vilanterol 92/22 microgramos administrado una vez al día por la noche como con salmeterol/PF 50/250 microgramos administrado dos veces al día. El aumento medio ajustado entre tratamientos respecto a los valores basales en la media ponderada durante 0 a 24 horas del FEV1 de 341 ml (furoato de fluticasona/vilanterol) y 377 ml (salmeterol/PF) demostró una mejora global en la función pulmonar durante 24 horas para ambos tratamientos. La diferencia media ajustada entre tratamientos de 37 ml entre los grupos no fue estadísticamente significativa (p=0,162). En el FEV1 valle los sujetos en el grupo furoato de fluticasona/vilanterol alcanzaron un cambio medio en LS de 281 ml respecto a los valores basales y los sujetos en el grupo salmeterol/PF un cambio de 300 ml; (la diferencia media ajustada entre tratamientos de 19 ml (IC 95%:-0,073; 0,034) no fue estadísticamente significativa (p=0,485).
No se han realizado estudios comparativos frente a salmeterol/PF o frente a otras combinaciones CSI/LABA que comparen de forma adecuada los efectos de las exacerbaciones en asma.
Furoato de fluticasona en monoterapia
Un estudio de 24 semanas de duración aleatorizado, doble ciego, controlado con placebo (FFA112059) evaluó la seguridad y eficacia de FF 92 microgramos una vez al día [n=114] y PF 250 microgramos dos veces al día [n=114] frente a placebo [n=115] en pacientes adultos y adolescentes con asma persistente. Todos los sujetos tuvieron que estar tratados con una dosis estable de un CSI durante al menos 4 semanas antes de la visita 1 (visita de selección) y no se permitía el uso de LABAs en las 4 semanas anteriores a la visita 1. La variable primaria de eficacia fue el cambio respecto a los valores basales en la visita clínica del FEV1 valle (pre-broncodilatador y pre-dosis) al final del periodo de tratamiento. Una variable secundaria robusta fue el cambio respecto a los valores basales en el porcentaje de días sin medicación de rescate durante el periodo de tratamiento de 24 semanas. En la semana 24, aumentó el FEV1 valle en 146 ml (IC 95% 36; 257 ml; p=0,009) y 145 ml (IC 95% 33; 257 ml; p=0,011) con FF y PF respectivamente en comparación con placebo. Se aumentó el porcentaje de días sin medicación de rescate en 14,8% (IC 95% 6,9; 22,7; p<0,001) y 17,9% (IC 95% 10,0; 25,7; p<0,001) con FF y PF respectivamente frente a placebo.
Estudio de exposición a alérgenos
En un estudio (HZA113126) cruzado de cuatro brazos a dosis repetidas, controlado con placebo en pacientes con asma leve se evaluó el efecto broncoprotector de furoato de fluticasona/vilanterol 92/22 microgramos en la respuesta asmática temprana y tardía a alérgenos inhalados. Los pacientes fueron aleatorizados para recibir furoato de fluticasona/vilanterol 92/22 microgramos, FF 92 microgramos, vilanterol 22 microgramos o placebo una vez al día durante 21 días seguidos de una exposición a alérgenos 1 hora después de la dosis final. Los alérgenos fueron ácaros de polvo, caspa de gato, polen de abedul; la selección se basó en las pruebas de detección individual. Las mediciones de los valores seriados del FEV1 se compararon con los valores previos a la exposición a alérgenos medidos tras la inhalación de solución salina (valores iniciales). En general, se observaron efectos mayores sobre la respuesta asmática temprana con furoato de fluticasona/vilanterol 92/22 microgramos en comparación con FF 92 microgramos o vilanterol 22 microgramos en monoterapia. Tanto furoato de fluticasona/vilanterol 92/22 microgramos como FF 92 microgramos suprimieron prácticamente la respuesta asmática tardía en comparación con vilanterol en monoterapia. Furoato de fluticasona/vilanterol 92/22 microgramos proporcionó una mayor protección frente a la hiperreactividad bronquial inducida por alérgenos en comparación con FF y vilanterol en monoterapia evaluada el Día 22 mediante la prueba de provocación con metacolina.
Enfermedad Pulmonar Obstructiva Crónica
El programa de desarrollo clínico en EPOC incluyó un estudio de 12 semanas (HZC113107), dos estudios de 6 meses (HZC112206, HZC112207) y dos estudios de un año de duración (HZC102970, HZC102871) aleatorizados y controlados en pacientes con un diagnóstico clínico de EPOC. Estos estudios incluían medidas de función pulmonar, disnea y exacerbaciones moderadas y graves.
Estudios de seis meses
Los estudios HZC112206 y HZC112207 fueron de 24 semanas de duración, aleatorizados, doble ciego, controlados con placebo, de grupos paralelos que comparaban el efecto de la combinación frente a vilanterol y FF en monoterapia y frente a placebo. HZC112206 evaluó la eficacia de furoato de fluticasona/vilanterol 46 /22 microgramos [n=206] y furoato de fluticasona/vilanterol 92/22 microgramos [n=206] en comparación con FF 92 microgramos [n=206], vilanterol 22 microgramos [n=205] y placebo [n = 207], todos administrados una vez al día. HZC112207 evaluó la eficacia de furoato de fluticasona/vilanterol 92/22 microgramos [n=204] y furoato de fluticasona/vilanterol 184/22 microgramos [n=205] en comparación con FF 92 microgramos [n=204], FF 184 microgramos [n=203] y vilanterol 22 microgramos [n=203] y placebo [n = 205], todos administrados una vez al día.
Todos los pacientes debían tener antecedentes de haber fumado por lo menos 10 paquetes-año; que el cociente FEVi/FVC post-salbutamol fuera menor o igual a 0,70; que el FEVi post-salbutamol fuera menor o igual a 70% del normal y tuvieran una puntuación de disnea por la Escala Modificada de Evaluación de la Disnea (mMRC) >2 (escala de 0 a 4) en la fase de selección. En la fase de selección, el FEV1 medio pre-broncodilatador fue 42,6% y 43,6% del normal, y la reversibilidad media fue 15,9% y 12,0% en HZC112206 y HZC112207, respectivamente. Las variables co-primarias en ambos estudios fueron la media ponderada del FEV1 desde 0 a 4 horas tras la dosis el Día 168 y el cambio respecto a los valores basales del FEV1 valle pre-dosis el Día 169.
En un análisis integrado de ambos estudios, furoato de fluticasona/vilanterol 92/22 microgramos demostró mejoras clínicamente relevantes en la función pulmonar. El Día 169 furoato de fluticasona/vilanterol 92/22 microgramos y vilanterol aumentaron la media ajustada del FEV1 valle en 129 ml (IC 95%: 91; 167 ml; p<0,001) y 83 ml (IC 95%: 46; 121 ml; p<0,001) respectivamente en comparación con placebo. Furoato de fluticasona/vilanterol 92/22 microgramos aumentó el FEV1 valle en 46 ml en comparación con vilanterol (IC 95%: 8; 83 ml, p= 0,017). El Día 168 furoato de fluticasona/vilanterol 92 /22 microgramos y vilanterol aumentaron la media ajustada de la media ponderada del FEV1 durante 0 a 4 horas en 193 ml (IC 95%: 156; 230 ml; p<0,001) y 145 ml (IC 95%: 108; 181 ml, p<0,001) respectivamente en comparación con placebo. Furoato de fluticasona/vilanterol 92/22 microgramos aumentó la media ajustada de la media ponderada del FEVi durante 0 a 4 horas en 148 ml en comparación con FF en monoterapia (IC 95%: 112; 184 ml; p< 0,001).
Estudios de 12 meses
Los estudios HZC102970 y HZC102871 fueron estudios de 52 semanas de duración aleatorizados, doble ciego, de grupos paralelos que comparaban el efecto de furoato de fluticasona/vilanterol 184/22 microgramos, furoato de fluticasona/vilanterol 92/22 microgramos, furoato de fluticasona/vilanterol 46/22 microgramos con vilanterol 22 microgramos, todos administrados una vez al día, sobre la tasa anual de exacerbaciones moderadas/graves en sujetos con EPOC que tenían antecedentes de haber fumado por lo menos 10 paquetes-año y un cociente FEV1/FVC post-salbutamol menor o igual que 0,70 y un FEV1 post-salbutamol menor o igual al 70% del normal y un historial documentado de > 1 exacerbación de EPOC que requirió antibióticos y/o corticosteroides orales u hospitalización en los 12 meses anteriores a la visita 1. La variable primaria fue la tasa anual de exacerbaciones moderadas y graves. Las exacerbaciones moderadas/ graves se definieron como empeoramiento de los síntomas que requirieron tratamiento con corticosteroides orales y/o antibióticos u hospitalización de los pacientes. Ambos estudios tuvieron un periodo de pre-inclusión de 4 semanas durante el cual todos los sujetos recibieron de forma abierta salmeterol/PF 50/250 dos veces al día para estandarizar el tratamiento farmacológico de la EPOC y estabilizar la enfermedad antes de asignar al azar la medicación del estudio ciego durante 52 semanas. Antes del periodo de pre-inclusión, los sujetos interrumpieron el uso de medicación previa para la EPOC a excepción de los broncodilatadores de acción corta. Durante el periodo de tratamiento no estaba permitido el uso concomitante de broncodilatadores inhalados de acción prolongada (agonistas p2 y anticolinérgicos), medicamentos con la combinación ipratropio/salbutamol, agonistas orales p2, y las preparaciones de teofilina. Los corticosteroides orales y antibióticos estaban permitidos bajo directrices específicas de uso para el tratamiento de exacerbaciones agudas de la EPOC. Los sujetos utilizaron salbutamol en función de las necesidades a lo largo de los estudios.
Los resultados de ambos estudios demostraron que el tratamiento con furoato de fluticasona/vilanterol 92/22 microgramos una vez al día conllevaba una tasa anual de exacerbaciones moderadas/graves de la EPOC menor que con vilanterol (Tabla 2).
Tabla 2: Análisis de tasas de exacerbaciones tras 12 meses de tratamiento
|
HZC102970 |
HZC102871 |
HZC102970 y HZC102871 integrado | ||||
|
Vilanterol (n=409) |
furoato de fluticasona/ |
Vilanterol (n=409) |
furoato de fluticasona/ |
Vilanterol (n=818) |
furoato de fluticasona/ | |
|
Variable |
vilanterol 92/22 (n=403) |
vilanterol 92/22 (n=403) |
vilanterol 92/22 (n=806) | |||
|
Exacerbaciones moderadas y graves | ||||||
|
Tasa media anual ajustada |
1,14 |
0,90 |
1,05 |
0,70 |
1,11 |
0,81 |
|
Ratio vs VI (IC 95%) Valor p % reducción (IC 95%) |
0,79 (0,64; 0,97) 0,024 21 (3; 36) |
0,66 (0,54; 0,81) <0,001 34 (19; 46) |
0,73 (0,63; 0,84) <0,001 27 (16; 37) | |||
|
Diferencia absoluta en número por año vs VI (IC 95%) |
0,24 (0,03; 0,41) |
0,36 (0,20; 0,48) |
0,30 (0,18; 0,41) | |||
|
Tiempo hasta la primera exacerbación: Hazard ratio (IC 95%) % reducción |
0,80 (0,66; 0,99) 20 |
0,72 (0,59; 0,89) 28 |
0,76 (0,66; 0,88) 24 | |||
|
riesgo | ||||||
|
Valor p |
0,036 |
0,002 |
p<0,001 | |||
En un análisis integrado de HZC102970 y HZC102871 a la semana 52, se observó una mejora en la media ajustada del FEV1 valle (42 ml IC 95%: 19; 64 ml; p<0,001) al comparar furoato de fluticasona/vilanterol 92/22 microgramos frente a vilanterol 22 microgramos. El efecto broncodilatador de 24 horas de furoato de fluticasona/vilanterol se mantuvo desde la primera dosis y a lo largo de un año de tratamiento sin evidencia de pérdida de eficacia (sin taquifilaxia).
En general, a través de los dos estudios combinados 2.009 pacientes (62%) tenían antecedentes/factores de riesgo cardiovasculares en la fase de selección. La incidencia de antecedentes/factores de riesgo cardiovasculares fue similar en todos los grupos de tratamiento en los que los pacientes tenían más frecuentemente hipertensión (46%), seguidos de hipercolesterolemia (29%) y diabetes mellitus (12%). Se observaron efectos similares en la reducción de exacerbaciones moderadas y graves en este subgrupo en comparación con la población total. En pacientes con antecedentes/factores de riesgo cardiovasculares, furoato de fluticasona/vilanterol 92/22 microgramos produjo una disminución significante en la tasa anual de exacerbaciones moderadas/graves de la EPOC en comparación con vilanterol (tasa anual media ajustada de 0,83 y 1,18 respectivamente, 30% de reducción (IC 95%: 16; 42%; p<0,001)). También se observaron mejoras en la media ajustada del FEVi valle (44 ml IC 95%: 15; 73ml, (p=0,003) en este subgrupo en la semana 52 cuando se comparaba el furoato de fluticasona/vilanterol 92/22 microgramos frente a vilanterol 22 microgramos.
Estudios frente a las combinaciones con salmeterol/propionato de fluticasona
En un estudio de 12 semanas de duración (HZC113107) en pacientes con EPOC ambos tratamientos, furoato de fluticasona/vilanterol 92/22 microgramos administrado una vez al día por la mañana y salmeterol/PF 50/500 microgramos administrado dos veces al día, demostraron mejoras respecto a los valores basales en la función pulmonar. El aumento de la media ajustada entre tratamientos respecto a los valores basales en la media ponderada del FEV1 durante 0 a 24 horas de 130 ml (furoato de fluticasona/vilanterol) y 108 ml (salmeterol/PF) demostraron una mejora global en la función pulmonar durante las 24 horas en ambos tratamientos. La diferencia media ajustada entre tratamientos de 22 ml (IC 95%: -18; 63 ml) entre los grupos no fue estadísticamente significativa (p=0,282). El cambio medio ajustado respecto a los valores basales del FEVi valle en el Día 85 fue 111 ml en el grupo furoato de fluticasona/vilanterol y 88 ml en el grupo salmeterol/PF; la diferencia de 23 ml (IC 95%: -20; 66) entre los grupos de tratamiento no fue clínicamente relevante o estadísticamente significativa (p=0,294). No se han realizado estudios comparativos frente a salmeterol/PF o frente a otros broncodilatadores establecidos que permitan comparar de forma adecuada los efectos sobre las exacerbaciones en EPOC.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Relvar Ellipta en los diferentes grupos de la población pediátrica en EPOC ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Relvar Ellipta en uno o más grupos de la población pediátrica en asma (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
La biodisponibilidad absoluta de furoato de fluticasona y vilanterol administrados por vía inhalatoria como furoato de fluticasona/vilanterol fue de promedio de 15,2% y 27,3%, respectivamente. La biodisponibilidad oral tanto de furoato de fluticasona como de vilanterol fue baja, un promedio de 1,26% y <2%, respectivamente. Teniendo en cuenta esta baja biodisponibilidad oral, la exposición sistémica de furoato de fluticasona y vilanterol tras la inhalación es debida principalmente a la absorción de la porción inhalada de la dosis liberada al pulmón.
Distribución
Tras la administración intravenosa, tanto furoato de fluticasona como vilanterol se distribuyen ampliamente con unos volúmenes de distribución promedios en estado estacionario de 661 L y 165 L, respectivamente.
Tanto furoato de fluticasona como vilanterol tienen una baja asociación con los glóbulos rojos. La unión a proteínas plasmáticas in vitro de furoato de fluticasona y vilanterol en plasma humano fue alta, un promedio >99,6% y 93,9%, respectivamente. No hubo una disminución en el alcance de la unión a proteínas plasmáticas in vitro en sujetos con insuficiencia renal o hepática.
Furoato de fluticasona y vilanterol son sustratos de la glicoproteína-P (P-gp), sin embargo, se considera poco probable que la administración concomitante de furoato de fluticasona/vilanterol con inhibidores P-gp altere la exposición sistémica de furoato de fluticasona o vilanterol ya que ambas son moléculas que se absorben bien.
Biotransformación
En base a los resultados in vitro, las principales rutas del metabolismo de furoato de fluticasona y vilanterol en humanos están mediadas principalmente por el CYP3A4.
Furoato de fluticasona se metaboliza principalmente mediante hidrólisis del grupo carbotiato S-fluorometil a metabolitos con una actividad corticosteroidea significativamente reducida. Vilanterol se metaboliza principalmente por la O-desaquilación a una serie de metabolitos con actividad p1- y p2-agonista significativamente reducida.
Eliminación
Tras la administración oral, furoato de fluticasona se eliminó en humanos principalmente mediante metabolismo a través de metabolitos que se excretan casi exclusivamente en las heces, con <1% de la dosis radioactiva recuperada eliminada en la orina.
Tras la administración oral, vilanterol se eliminó principalmente por metabolismo seguido de la excreción de los metabolitos en orina y heces, aproximadamente 70% y 30% de la dosis radioactiva respectivamente en un estudio en humanos radiomarcado realizado por vía oral. La semivida plasmática de eliminación aparente de vilanterol tras una administración única inhalada de furoato de fluticasona/vilanterol fue, de promedio, 2,5 horas. La semivida de acumulación efectiva de vilanterol, determinada por la administración inhalada de dosis repetidas de vilanterol 25 microgramos, es de 16,0 horas en sujetos con asma y de 21,3 horas en sujetos con EPOC.
Población pediátrica
No se recomiendan modificaciones de las dosis en adolescentes (12 años o mayores).
No se ha estudiado la farmacocinética de furoato de fluticasona/vilanterol en pacientes menores de 12 años de edad. No se ha establecido la seguridad y eficacia de furoato de fluticasona/vilanterol en niños menores de 12 años.
Poblaciones especiales
Pacientes de edad avanzada (> 65 años)
Los efectos de la edad sobre la farmacocinética de furoato de fluticasona y vilanterol se determinaron en los estudios fase III en asma y EPOC. No hubo evidencia de que la edad (12-84) afecte a la farmacocinética de furoato de fluticasona y vilanterol en sujetos con asma.
No hubo evidencia de que la edad afecte a la farmacocinética de furoato de fluticasona en sujetos con EPOC, mientras que hubo un aumento (37%) en el AUC(0_24) de vilanterol en todos los rangos de edad observados de 41 a 84 años. Para sujetos de edad avanzada (84 años de edad) con bajo peso corporal (35 kg) se prevé que sea un 35% superior que en la población estimada (sujetos con EPOC de 60 años y peso corporal de 70 kg), mientras que la Cmax se mantuvo sin cambios. No es probable que estas diferencias sean clínicamente relevantes.
No se recomiendan modificaciones de las dosis en sujetos con asma y EPOC.
Insuficiencia renal
Un estudio clínico farmacológico de furoato de fluticasona/vilanterol mostró que la insuficiencia renal grave (aclaramiento de creatinina <30 ml/min) no supuso una exposición significativamente mayor a furoato de fluticasona o vilanterol o unos efectos sistémicos más marcados de los agonistas p2 o corticosteroides en comparación con los sujetos sanos.
No se requiere ajuste de dosis en pacientes con insuficiencia renal.
No se han estudiado los efectos de la hemodiálisis.
Insuficiencia hepática
Tras la administración de dosis repetidas de furoato de fluticasona/vilanterol durante 7 días, hubo un aumento de la exposición sistémica a furoato de fluticasona (hasta tres veces lo medido mediante AUC(0-24)) en sujetos con insuficiencia hepática (Child-Pugh A, B o C) en comparación con sujetos sanos. El aumento de la exposición sistémica de furoato de fluticasona en sujetos con insuficiencia hepática moderada (Child-Pugh B; furoato de fluticasona/vilanterol 184/22 microgramos) se asoció con una reducción promedio de 34% en el cortisol sérico en comparación con sujetos sanos. La exposición sistémica de la dosis normalizada de furoato de fluticasona fue similar en sujetos con insuficiencia hepática moderada y grave (Child-Pugh B o C).
Tras la administración de dosis repetidas de furoato de fluticasona/vilanterol durante 7 días, no hubo un aumento significativo en la exposición sistémica a vilanterol (Cmax y AUC) en sujetos con insuficiencia hepática leve, moderada o grave (Child-Pugh A, B o C).
No hubo efectos clínicamente relevantes de la combinación furoato de fluticasona/vilanterol sobre los efectos sistémicos de los p-adrenérgicos (ritmo cardiaco o potasio sérico) en sujetos con insuficiencia hepática leve o moderada (vilanterol, 22 microgramos) o con insuficiencia hepática grave (vilanterol, 12,5 microgramos) en comparación con sujetos sanos.
Otras poblaciones especiales
En sujetos con asma, el AUC(0-24) estimado de furoato de fluticasona para sujetos del Este asiático, japoneses y Sudeste asiático (12-13% de los sujetos) fue un promedio de 33% a 53% mayor en comparación con otros grupos raciales. Sin embargo, no hubo evidencia de que una mayor exposición sistémica en esta población esté asociada con un efecto mayor en la excreción de cortisol en orina de 24 horas. En promedio, se prevé que la Cmax de vilanterol sea de 220 a 287% mayor y el AUC(0-24) comparable para aquellos sujetos con antecedentes asiáticos en comparación con otros grupos raciales. Sin embargo, no hubo evidencia de que esta Cmax de vilanterol conllevara efectos clínicamente relevantes en el ritmo cardiaco.
En sujetos con EPOC el AUC(0-24) estimado de furoato de fluticasona para sujetos del Este asiático, japoneses y Sudeste asiático (13-14% de los sujetos) fue un promedio de 23% a 30% mayor en comparación con sujetos caucásicos. Sin embargo, no hubo evidencia de que este aumento de exposición sistémica en esta población esté asociada con un mayor efecto en la excreción de cortisol en orina de 24 horas. No hubo efectos de raza en los parámetros farmacocinéticos estimados de vilanterol en sujetos con EPOC.
Género, peso e IMC
No hubo evidencia de que el género, el peso o el IMC (índice de masa corporal) influyeran en la farmacocinética de furoato de fluticasona en base al análisis farmacocinético poblacional de los resultados de fase III en 1.213 sujetos con asma (712 mujeres) y 1.225 sujetos con EPOC (392 mujeres).
No hubo evidencia de que el género, el peso o el IMC influyeran en la farmacocinética de vilanterol en base a un análisis farmacocinético poblacional en 856 sujetos con asma (500 mujeres) y 1.091 sujetos con EPOC (340 mujeres).
No es necesario el ajuste de dosis por género, peso o IMC.
5.3 Datos preclínicos sobre seguridad
Los efectos farmacológicos y toxicológicos observados con furoato de fluticasona o vilanterol en los estudios no clínicos fueron aquellos típicamente asociados con glucocorticoides o p2-agonistas. La administración de furoato de fluticasona en combinación con vilanterol no conllevaba ninguna nueva toxicidad significativa.
Genotoxicidad y carcinogenicidad
Furoato de fluticasona
Furoato de fluticasona no resultó genotóxico en una batería estándar de estudios ni carcinogénico en estudios de inhalación a tiempo real realizados en ratas o ratones a exposiciones similares a las dosis máximas recomendadas en humanos basadas en el AUC.
Trifenatato de vilanterol
En estudios de toxicidad genética, vilanterol (como a-fenilcinamato) y el ácido trifenilacético no resultaron genotóxicos, lo cual indica que vilanterol (como trifenatato) no representa un peligro genotóxico para humanos.
De acuerdo con los resultados identificados en otros agonistas p2, en los estudios de inhalación a tiempo real trifenatato de vilanterol produjo efectos proliferativos en el aparato reproductor de ratas y ratones hembras y en la glándula pituitaria de las ratas. No hubo un aumento en la incidencia de tumores en ratas o ratones expuestos a dosis 2 o 30 veces la dosis máxima recomendada en humanos, respectivamente, en base al AUC.
Toxicidad reproductiva
Furoato de fluticasona
Los efectos observados tras la administración inhalada de furoato de fluticasona en combinación con vilanterol en ratas fueron similares a los que se observaron con furoato de fluticasona en monoterapia. El furoato de fluticasona no resultó teratogénico en ratas ni conejos, pero produjo retraso en el desarrollo en ratas y produjo abortos en conejos a dosis tóxicas para la madre. No se observaron efectos sobre el desarrollo en ratas expuestas a dosis aproximadamente 3 veces superiores a la dosis máxima recomendada, basada en el AUC.
Trifenatato de vilanterol
El trifenatato de vilanterol no fue teratogénico en ratas. En estudios de inhalación en conejos, el trifenatato de vilanterol produjo efectos similares a los que se observaban en otros p2-agonistas (paladar hendido, párpados abiertos, fusión esternebral y malrotación/flexión de extremidades).
Cuando se administró por vía subcutánea no hubo efectos a exposiciones 84 veces superiores a la dosis máxima recomendada, basada en el AUC.
Ni el furoato de fluticasona ni el trifenatato de vilanterol tuvieron efectos adversos sobre la fertilidad o sobre el desarrollo pre o post-natal en ratas.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Lactosa monohidrato Estearato de magnesio
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez 2 años.
Periodo de validez una vez abierta la bandeja: 6 semanas.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 25°C. Si se conserva el dispositivo en nevera se debe permitir que el inhalador alcance la temperatura ambiente durante al menos una hora antes de su uso.
Conservar en el embalaje original para protegerlo de la humedad.
Debe utilizarse en un plazo de 6 semanas tras la apertura de la bandeja.
Escribir la fecha en la que el inhalador se debe desechar en el espacio designado para ello, que aparece en la etiqueta del inhalador. La fecha se debe anotar tan pronto como el inhalador se saque de la bandeja.
6.5 Naturaleza y contenido del envase
El dispositivo inhalador está formado por un cuerpo gris claro, un protector de la boquilla amarillo, y un contador de dosis, envasado en una bandeja de aluminio laminada que contiene una bolsa desecante. La bandeja está sellada con una tapa de aluminio desplegable.
El dispositivo inhalador contiene dos tiras de aluminio laminado de 14 o 30 dosis.
El dispositivo inhalador es un dispositivo multi-componente compuesto de polipropileno, polietileno de alta densidad, polioximetileno, polibutileno tereftalato, acrilonitrilo butadieno estireno, policarbonato y acero inoxidable.
Tamaños de envases de 14 o 30 dosis por inhalador. Envase clínico de 3 x 30 dosis por inhalador. Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Para las instrucciones de uso, ver sección 4.2.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Limited.
980 Great West Road, Brentford, Middlesex TW8 9GS, Reino Unido.
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/886/001
EU/1/13/886/002
EU/1/13/886/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 13 noviembre 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Relvar Ellipta 184 microgramos/22 microgramos polvo para inhalación (unidosis).
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada inhalación proporciona una dosis liberada (la dosis que sale por la boquilla) de 184 microgramos de furoato de fluticasona y 22 microgramos de vilanterol (como trifenatato). Esto se corresponde con una dosis de 200 microgramos de furoato de fluticasona y 25 microgramos de vilanterol (como trifenatato).
Excipientes con efecto conocido:
Cada dosis liberada contiene aproximadamente 25 mg de lactosa (como monohidrato).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para inhalación (unidosis)
(Polvo para inhalación).
Polvo blanco en un inhalador de color gris claro con un protector de la boquilla de color amarillo y un contador de dosis.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Asma
Relvar Ellipta está indicado para el tratamiento regular del asma en adultos y adolescentes de 12 años de edad y mayores cuando la administración de una combinación (un agonista p2 de acción prolongada y un corticosteroide por vía inhalatoria) sea apropiada:
• pacientes no controlados adecuadamente con corticosteroides inhalados y agonistas p2 inhalados de acción corta administrados "a demanda".
4.2 Posología y forma de administración
Posología
Asma
Adultos y adolescentes de 12 años de edad y mayores
Una inhalación de Relvar Ellipta 184/22 microgramos una vez al día.
Los pacientes normalmente experimentan una mejora de la función pulmonar en los 15 minutos tras la inhalación de Relvar Ellipta. Sin embargo, se debe informar al paciente de que es necesario el uso
regular diario para mantener el control de los síntomas del asma y que debe continuar usándolo aun cuando no tenga síntomas.
Si aparecen síntomas en los periodos entre dosis, se debe usar un agonista p2 inhalado de acción corta para un alivio inmediato.
En adultos y adolescentes de 12 años de edad y mayores que requieran una dosis de baja a intermedia de corticosteroide inhalado en combinación con un agonista p2 de acción prolongada se debe considerar una dosis de inicio de Relvar Ellipta 92/22 microgramos. Si los pacientes no están adecuadamente controlados con Relvar Ellipta 92/22 microgramos, se puede aumentar la dosis a 184/22 microgramos, lo que puede proporcionar una mejora adicional en el control del asma.
Los pacientes deben ser reevaluados regularmente por un médico, de manera que la concentración de furoato de fluticasona/vilanterol que reciban siga siendo la óptima y solo se modifique a criterio médico. Se debe ajustar la dosis de forma que se administre la dosis más baja que mantenga un control eficaz de los síntomas.
En adultos y adolescentes de 12 años de edad y mayores que requieran una dosis más alta de corticosteroide inhalado en combinación con un agonista p2 de acción prolongada se debe considerar la dosis de Relvar Ellipta 184/22 microgramos.
La dosis máxima recomendada de Relvar Ellipta es de 184/22 microgramos una vez al día.
Los pacientes con asma deben recibir la concentración de Relvar Ellipta que contenga la dosis apropiada de furoato de fluticasona (FF) en base a la gravedad de su enfermedad. Los prescriptores deben saber que en los pacientes con asma, una dosis diaria de 100 microgramos de furoato de fluticasona (FF) es aproximadamente equivalente a 250 microgramos de propionato de fluticasona (PF) dos veces al día, mientras que 200 microgramos de FF una vez al día es aproximadamente equivalente a 500 microgramos de PF dos veces al día.
Niños menores de 12 años de edad
No se ha establecido la seguridad y eficacia de Relvar Ellipta en niños menores de 12 años de edad para la indicación en asma.
No hay datos disponibles.
Poblaciones especiales:
Pacientes de edad avanzada (>65 años)
No se requiere ajuste de dosis en esta población (ver sección 5.2).
Insuficiencia renal
No se requiere ajuste de dosis en esta población (ver sección 5.2).
Insuficiencia hepática
En estudios con sujetos con insuficiencia hepática leve, moderada y grave se observó un aumento en la exposición sistémica a furoato de fluticasona (ambos Cmax y AUC) (ver sección 5.2).
Se debe tener precaución cuando se prescriben dosis a pacientes con insuficiencia hepática ya que pueden tener un mayor riesgo de reacciones adversas sistémicas asociadas con los corticosteroides.
La dosis máxima para pacientes con insuficiencia hepática moderada o grave es de 92/22 microgramos (ver sección 4.4).
Forma de administración
Relvar Ellipta se administra solo por vía inhalatoria.
Debe administrarse a la misma hora del día, cada día.
La decisión final sobre si la administración debe ser por la mañana o por la noche se deja a elección del médico.
Si se olvida una dosis, la siguiente dosis debe administrarse al día siguiente a la hora habitual.
Si se conserva en nevera, se debe dejar que el inhalador vuelva a temperatura ambiente durante por lo menos una hora antes de utilizarlo.
Tras la inhalación, los pacientes deben aclararse la boca con agua sin tragarla.
Cuando el inhalador se utiliza por primera vez, no es necesario comprobar que funciona correctamente, ni prepararlo de ninguna forma especial para su uso. Se deben seguir las instrucciones de uso paso a paso.
El inhalador Ellipta está envasado en una bandeja que contiene una bolsa desecante para reducir la humedad. Una vez abierto se debe desechar la bolsa desecante.
Se debe advertir al paciente de que no abra la bandeja hasta que esté preparado para inhalar la dosis.
Cuando se saca el inhalador de la bandeja, estará en la posición “cerrado”. La fecha de “desechar el”, debe escribirse en el espacio designado para ello en la etiqueta del inhalador. La fecha de “desechar el” es de 6 semanas desde la fecha de apertura de la bandeja. Después de esta fecha, el inhalador debe desecharse. La bandeja se puede desechar después de la primera apertura.
Las instrucciones de uso paso a paso que se muestran a continuación para el inhalador Ellipta de 30 dosis también aplican para el inhalador Ellipta de 14 dosis.
Instrucciones de uso
1. Leer las siguientes instrucciones antes de utilizar el inhalador
Si la tapa del inhalador se abre y cierra sin que se inhale el medicamento, se perderá la dosis. La dosis perdida quedará retenida de forma segura dentro del inhalador, pero no estará disponible para ser inhalada.
No es posible administrarse accidentalmente una dosis adicional o una dosis doble mediante una inhalación.
Contador d& dosis
El contadorde dosis indica cuántas dosis de medicamento quedan en el dispositivo. Antes de usar el inhalador, debe indicar exactamente 30 dosis.
Cada vez que se abre la tapa, el contador disminuye en 1 unidad.
Cuando quedan menos de 10 dosis, la mitad del contador de dosis se pone de color rojo.
Una vez se utiliza la última dosis: la mitad del contador de dosis se pone de color rojoe indica el número 0. El inhalador
ahora está vacío.
Si se abre la tapa cuando el inhalador está vacío, el contadorde dosis pasa de estar la mitad de color rojo a estarlo completamente.
Tapa
Cada vez que se abre la tapa se prepara una dosis de medicamento.
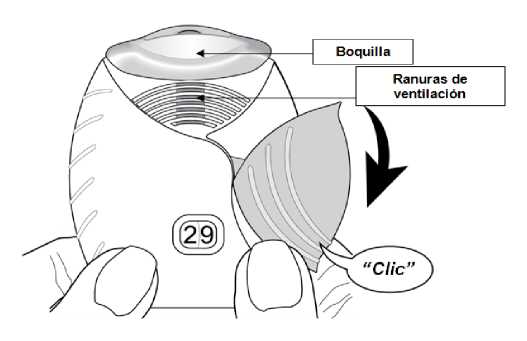
2. Cómo preparar una dosis
Abrir la tapa cuando esté preparado para administrarse una dosis. No agitar el inhalador.
Deslizar la tapa hacia abajo hasta oír un ‘clic’.
El medicamento está ahora preparado para ser inhalado. Como confirmación, el contador de dosis disminuye en 1 unidad.
Si el contador de dosis no disminuye al oír el ‘clic’, el inhalador no liberará el medicamento. Llévelo al farmacéutico y solicite ayuda.
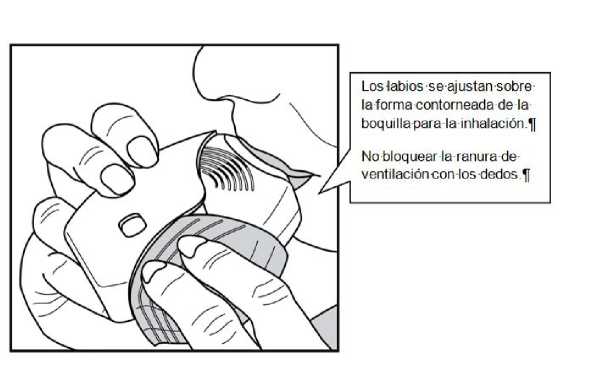
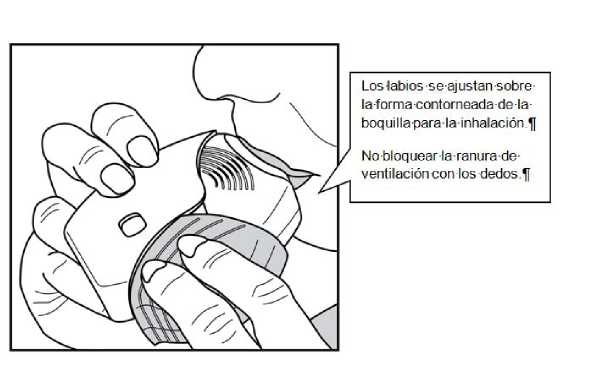
3. Cómo inhalar el medicamento
Mantener el inhalador alejado de la boca y espirar lo que razonablemente pueda.
No espirar dentro del inhalador.
Colocar la boquilla entre los labios y cerrarlos firmemente alreadedor de la boquilla.
No bloquear las ranuras de ventilación con los dedos.
Realizar una inspiración prolongada, continua y profunda. Mantener la respiración tanto tiempo como sea posible (al menos 3-4 segundos).
• Retirar el inhalador de la boca.
• Espirar suave y lentamente.
Puede que no sea capaz de distinguir el sabor o notar el medicamento, incluso cuando utiliza el inhalador de forma correcta.
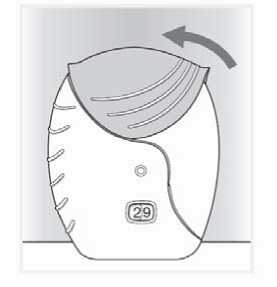
4. Cerrar el inhalador y enjuagarse la boca
Si quiere limpiar la boquilla utilice un pañuelo seco antes de cerrar la tapa.
Deslizar la tapa hacia arriba hasta el tope para proteger la boquilla.
Enjuagarse la boca con agua, una vez utilizado el inhalador.
Esto hará que sea menos probable que se produzcan efectos adversos como ulceraciones en la boca o garganta.
4.3 Contraindicaciones
Hipersensibilidad a los principios activos o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo Empeoramiento de la enfermedad
El furoato de fluticasona/vilanterol no se debe utilizar para tratar los síntomas agudos del asma, para lo que se requiere un broncodilatador de acción corta. El aumento de empleo de broncodilatadores de acción corta para aliviar los síntomas indica un empeoramiento en el control y los pacientes deben ser examinados por un médico.
Los pacientes no deben interrumpir el tratamiento con furoato de fluticasona/vilanterol en asma, sin la supervisión de un médico ya que los síntomas pueden reaparecer tras interrumpir el tratamiento.
Durante el tratamiento con furoato de fluticasona/vilanterol pueden producirse efectos adversos relacionados con el asma y exacerbaciones de la enfermedad. Se debe pedir a los pacientes que continúen el tratamiento, pero que acudan a su médico si los síntomas del asma siguen sin estar controlados o empeoran tras comenzar el tratamiento con Relvar Ellipta.
Broncoespasmo paradójico
Tras la administración de la dosis puede aparecer broncoespasmo paradójico con un aumento inmediato en las sibilancias. Se debe de tratar inmediatamente con un broncodilatador inhalado de acción corta. Se debe interrumpir el tratamiento con Relvar Ellipta inmediatamente, evaluar al paciente e instaurar un tratamiento alternativo si es necesario.
Efectos cardiovasculares
Con el uso de medicamentos simpaticomiméticos, incluido Relvar Ellipta, se pueden observar efectos cardiovasculares como arritmias cardiacas, por ejemplo taquicardia supraventricular y extrasístoles.
Por lo tanto, furoato de fluticasona/vilanterol se debe usar con precaución en pacientes con enfermedad cardiovascular grave o anomalías en el ritmo cardiaco, tirotoxicosis, hipopotasemia no corregida o pacientes con predisposición a niveles séricos de potasio bajos.
Pacientes con insuficiencia hepática
En pacientes con insuficiencia hepática de moderada a grave se debe usar la dosis de
92/22 microgramos y se debe controlar a los pacientes por las reacciones adversas sistémicas debidas a
los corticosteroides (ver sección 5.2).
Efectos sistémicos de los corticosteroides
Pueden aparecer efectos sistémicos con cualquier corticosteroide administrado por vía inhalatoria, especialmente a dosis elevadas prescritas durante largos periodos. La probabilidad de que estos efectos aparezcan es mucho menor que con el uso de corticosteroides administrados por vía oral. Los posibles efectos sistémicos incluyen Síndrome de Cushing, aspecto Cushingoideo, supresión suprarrenal, disminución de la densidad mineral ósea, retraso en el crecimiento de niños y adolescentes, cataratas y glaucoma y más raramente, una serie de efectos psicológicos o del comportamiento que incluyen hiperactividad psicomotora, trastornos del sueño, ansiedad, depresión o agresividad (especialmente en niños).
Furoato de fluticasona/vilanterol se debe administrar con precaución en pacientes con tuberculosis pulmonar o en pacientes con infecciones crónicas o no tratadas.
Hiperglucemia
Se han notificado casos de aumento de los niveles de glucosa en sangre en pacientes diabéticos, lo cual debe tenerse en cuenta cuando se prescriba a pacientes con antecedentes de diabetes mellitus.
Neumonía en pacientes con EPOC
Se ha observado un aumento en la aparición de neumonías en pacientes con EPOC que reciben furoato de fluticasona/vilanterol. También hubo un aumento en la incidencia de neumonías que requirieron hospitalización. En algunos casos, estos acontecimientos de neumonía fueron mortales (ver sección 4.8). Los médicos deben permanecer atentos ante un posible desarrollo de neumonía en pacientes con EPOC, ya que las características clínicas de estas infecciones se solapan con los síntomas de las exacerbaciones por EPOC. Entre los factores de riesgo de neumonía en pacientes con EPOC que reciben furoato de fluticasona/vilanterol se incluyen fumadores actuales, pacientes con antecedentes de neumonía previa, pacientes con un índice de masa corporal <25 kg/m2 y pacientes con un FEV1 (volumen espiratorio forzado) <50% del normal. Estos factores se deben considerar cuando se prescribe furoato de fluticasona/vilanterol y se debe volver a evaluar el tratamiento si se produce neumonía.
Relvar Ellipta 184/22 microgramos no está indicado para pacientes con EPOC. No hay un beneficio adicional con la dosis de 184/22 microgramos en comparación con la dosis de 92/22 microgramos y hay un posible aumento en el riesgo de que se produzcan reacciones adversas sistémicas debidas a los corticosteroides (ver sección 4.8).
La incidencia de neumonía en pacientes con asma fue frecuente con la dosis más alta. La incidencia de neumonía en pacientes con asma que utilizaban la dosis de 184/22 microgramos de furoato de fluticasona/vilanterol fue numéricamente mayor en comparación con los que recibían la dosis de 92/22 microgramos de furoato de fluticasona/vilanterol o placebo (ver sección 4.8). No se identificaron factores de riesgo.
Excipientes
Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp o malabsorción de glucosa o galactosa no deben usar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Las interacciones clínicamente significativas mediadas por furoato de fluticasona/vilanterol a las dosis clínicas se consideran poco probables debido a las bajas concentraciones plasmáticas que se alcanzan tras la inhalación de la dosis.
Interacción con betabloqueantes
Los bloqueantes p2-adrenérgicos pueden disminuir o antagonizar el efecto de los agonistas p2-adrenérgicos. Se debe evitar el uso concomitante de bloqueantes p2-adrenérgicos no selectivos y selectivos a menos que existan razones de peso para su uso.
Interacción con inhibidores del CYP3A4
Furoato de fluticasona y vilanterol sufren ambos un rápido aclaramiento debido a un intenso metabolismo de primer paso mediado por la enzima hepática CYP3A4.
Se recomienda tener precaución cuando se administra este medicamento junto con inhibidores potentes del CYP3A4 (por ejemplo ketoconazol, ritonavir), ya que es posible un aumento en la exposición sistémica tanto de furoato de fluticasona como de vilanterol, por lo que se recomienda evitar el uso concomitante. Se realizó un estudio de interacción con CYP3A4 a dosis repetidas en sujetos sanos con la combinación furoato de fluticasona/vilanterol (184/22 microgramos) y ketoconazol (400 mg), potente inhibidor del CYP3A4. La administración concomitante aumentó la media del AUC(0-24) y Cmax de furoato de fluticasona en un 36% y 33%, respectivamente. El aumento de la exposición de furoato de fluticasona se asoció con una reducción del 27% en la media ponderada entre 0-24 horas del cortisol sérico. La administración concomitante aumentó la media del AUC(0-t) y Cmax de vilanterol en un 65% y 22%, respectivamente. El aumento de la exposición de vilanterol no se asoció con un aumento de los efectos sistémicos relacionados con los agonistas p2 como el ritmo cardiaco, los niveles de potasio en sangre o el intervalo QTcF.
Interacción con inhibidores de la glicoproteína-P
Furoato de fluticasona y vilanterol son ambos sustratos de la glicoproteína-P (P-gp). En un estudio clínico farmacológico realizado en sujetos sanos a los que se administró conjuntamente vilanterol y verapamilo, potente inhibidor de la P-gp e inhibidor moderado del CYP3A4, no se observó ningún efecto significativo en la farmacocinética de vilanterol. No se han realizado estudios clínicos farmacológicos con un inhibidor específico P-gp y furoato de fluticasona.
Medicamentos simpaticomiméticos
La administración concomitante con otros medicamentos simpaticomiméticos (en monoterapia o como parte de una combinación) pueden potenciar las reacciones adversas de furoato de fluticasona/vilanterol. Relvar Ellipta no debe utilizarse en combinación con otros agonistas p2-adrenérgicos de acción prolongada o medicamentos que contengan agonistas p2-adrenérgicos de acción prolongada.
Población pediátrica
Los estudios de interacción se han realizado solo en adultos.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Los estudios realizados en animales han mostrado toxicidad para la reproducción a exposiciones que no son relevantes clínicamente (ver sección 5.3). No hay datos o éstos son limitados relativos al uso de furoato de fluticasona y vilanterol trifenatato en mujeres embarazadas.
Solo se debe considerar la administración de furoato de fluticasona/vilanterol en mujeres embarazadas si el beneficio esperado para la madre es mayor que cualquier posible riesgo para el feto.
Lactancia
No se dispone de información suficiente relativa a la excreción de furoato de fluticasona o vilanterol trifenatato y/o sus metabolitos en la leche materna. Sin embargo, otros corticosteroides y p2 agonistas fueron detectados en la leche materna (ver sección 5.3). No se puede excluir el riesgo en recién nacidos/niños lactantes.
Se debe decidir si es necesario interrumpir la lactancia o interrumpir el tratamiento con furoato de fluticasona/vilanterol tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Fertilidad
No hay datos de fertilidad en humanos. Los estudios realizados en animales no han mostrado efectos de furoato de fluticasona/vilanterol trifenatato sobre la fertilidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de furoato de fluticasona o vilanterol sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los resultados de ensayos clínicos amplios en asma y en EPOC fueron utilizados para determinar la frecuencia de las reacciones adversas asociadas a furoato de fluticasona/vilanterol. En el programa de desarrollo clínico en asma fueron incluidos un total de 7.034 pacientes en una evaluación integrada de reacciones adversas. En el programa de desarrollo clínico en EPOC se incluyeron un total de 6.237 sujetos en una evaluación integrada de reacciones adversas.
Las reacciones adversas de furoato de fluticasona y vilanterol notificadas con más frecuencia fueron cefalea y nasofaringitis. A excepción de la neumonía y las fracturas, el perfil de seguridad fue similar
en pacientes con asma y EPOC. Durante los ensayos clínicos, la neumonía y las fracturas se observaron generalmente con mayor frecuencia en pacientes con EPOC.
Tabla de reacciones adversas
Las reacciones adversas se enumeran clasificadas por órganos y frecuencia. Para la clasificación de frecuencias se utiliza el siguiente convenio: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000).
Dentro de cada grupo de frecuencias, las reacciones adversas se presentan por orden decreciente de gravedad.
|
Sistema de clasificación de órganos |
Reacciones adversas |
Frecuencia |
|
Infecciones e infestaciones |
Neumonía* Infecciones del tracto respiratorio superior Bronquitis Gripe Candidiasis en la boca y garganta |
Frecuentes |
|
Trastornos del sistema inmunológico |
Reacciones de hipersensibilidad, incluyendo anafilaxia, angioedema, erupción, y urticaria |
Raras |
|
Trastornos del sistema nervioso |
Cefalea Temblor |
Muy frecuentes Raras |
|
Trastornos psiquiátricos |
Ansiedad |
Raras |
|
Trastornos cardiacos |
Extrasístoles |
Poco frecuentes |
|
Palpitaciones |
Raras | |
|
Taquicardia |
Raras | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Nasofaringitis Dolor orofaríngeo Sinusitis Faringitis Rinitis Tos Disfonía |
Muy frecuentes Frecuentes |
|
Trastornos gastrointestinales |
Dolor abdominal |
Frecuentes |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia Dolor de espalda Fracturas** Espasmos musculares |
Frecuentes |
|
Trastornos generales y alteraciones en el lugar de administración |
Pirexia |
Frecuentes |
*, ** Ver a continuación “Descripción de reacciones adversas seleccionadas”
Descripción de reacciones adversas seleccionadas
Neumonía
En un análisis integrado de dos estudios replicados de un año de duración en pacientes con EPOC con una exacerbación en el año anterior (n = 3.255), el número de casos de neumonía por 1.000 pacientes-año fue 97,9 con FF/VI 184/22, 85,7 en el grupo FF/VI 92/22 y 42,3 en el grupo VI 22. En el caso de neumonía severa, el número de casos correspondiente por 1.000 pacientes-año fueron 33,6, 35,5 y 7,6 respectivamente, mientras que los casos de neumonía grave por 1.000 pacientes-año fueron 35,1 para FF/VI 184/22, 42,9 con FF/VI 92/22, 12,1 con VI 22. Por último, los casos de neumonía con desenlace mortal (ajustados por exposición) fueron 8,8 para FF/VI 184/22 frente a 1,5 para FF/VI 92/22 y 0 para VI 22.
En un análisis integrado de 11 estudios en asma (7.034 pacientes), la incidencia de neumonía por 1.000 pacientes-año fue 18,4 para FF/VI 184/22 frente a 9,6 para FF/VI 92/22 y 8,0 en el grupo placebo.
Fracturas
En dos estudios replicados de 12 meses de duración en el que participaron un total de 3.255 pacientes con EPOC, la incidencia de fracturas óseas fue baja de forma global en todos los grupos de tratamiento, con una incidencia mayor en todos los grupos con Relvar Ellipta (2%) en comparación con el grupo vilanterol 22 microgramos (<1%). Aunque hubo más fracturas en los grupos de tratamiento con Relvar Ellipta en comparación con el grupo vilanterol 22 microgramos, las fracturas típicamente asociadas al uso de corticosteroides (por ejemplo compresión espinal/fracturas vertebrales toracolumbares, fracturas de cadera y acetabulares) se produjeron en <1% en los brazos de tratamiento con Relvar Ellipta y vilanterol.
En un análisis integrado de 11 estudios en asma (7.034 pacientes), la incidencia de fracturas fue <1%, y normalmente se asociaban con traumatismos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Síntomas y signos
Una sobredosis de furoato de fluticasona/vilanterol puede producir signos y síntomas debido a la acción de los componentes por separado, incluyendo aquellos que se observan con una sobredosis de otros p2-agonistas y en consistencia con los efectos de clase conocidos de los corticosteroides inhalados (ver sección 4.4).
Tratamiento
No hay un tratamiento específico para la sobredosis con furoato de fluticasona/vilanterol. En caso de sobredosis, el paciente debe recibir el tratamiento de soporte necesario y un seguimiento apropiado.
Solo se debe considerar la administración de betabloqueantes cardioselectivos cuando se produzcan efectos clínicamente relevantes debidos a una sobredosis grave de vilanterol y que no respondan a las medidas de soporte. Los betabloqueantes cardioselectivos se deben usar con precaución en pacientes con antecedentes de broncoespasmo.
Para un manejo adicional se deben seguir las recomendaciones clínicas indicadas o las recomendaciones del Centro Nacional de Toxicología, si estuvieran disponibles.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes para las enfermedades obstructivas de las vías respiratorias. Adrenérgicos y otros agentes para las enfermedades obstructivas de las vías respiratorias, código ATC: R03AK10.
Mecanismo de acción
Furoato de fluticasona y vilanterol son dos tipos de medicamentos (un corticosteroide sintético y un agonista selectivo del receptor p2 de acción prolongada).
Efectos farmacodinámicos
Furoato de _fluticasona
Furoato de fluticasona es un corticosteroide sintético trifluorinado con una potente actividad antiinflamatoria. Se desconoce el mecanismo exacto por el cual furoato de fluticasona actúa sobre los síntomas del asma y EPOC. Los corticosteroides han demostrado tener una amplia variedad de acciones sobre diversos tipos celulares (por ejemplo, eosinófilos, macrófagos, linfocitos) y mediadores (por ejemplo, citoquinas y quimioquinas involucradas en la inflamación).
Vilanterol trifenatato
Vilanterol trifenatato es un agonista selectivo p2-adrenérgico de acción prolongada (LABA).
El efecto farmacológico de los fármacos agonistas p2-adrenérgicos, incluido vilanterol trifenatato, son al menos en parte atribuibles a la estimulación de la adenilato ciclasa intracelular, la enzima que cataliza la transformación de la adenosín trifosfato (ATP) a la adenosín monofosfato - 3’,5’ cíclico (AMP cíclico). El aumento en los niveles del AMP cíclico produce la relajación del músculo liso bronquial y la inhibición de la liberación de mediadores de la hipersensibilidad inmediata de las células, especialmente de los mastocitos.
Se producen interacciones moleculares entre los corticosteroides y LABAs, por las que los esteroides activan el gen del receptor p2 aumentando el número de receptores y la sensibilidad, y los LABAs preparan al receptor glucocorticoide para la activación dependiente de esteroides y aumentan la translocación nuclear celular. Estas interacciones sinérgicas se reflejan en un aumento de la actividad anti-inflamatoria, que se ha demostrado in vitro e in vivo en una variedad de células inflamatorias relevantes para la fisiopatología del asma y EPOC. En estudios de biopsias de las vías respiratorias con furoato de fluticasona y vilanterol se ha demostrado también la sinergia entre corticosteroides y LABAs a las dosis clínicas de los medicamentos en pacientes con EPOC.
Eficacia clínica y seguridad
Asma
Tres estudios fase III aleatorizados, doble ciego (HZA106827, HZA106829 y HZA106837) de diferente duración evaluaron la seguridad y eficacia de furoato de fluticasona/vilanterol en pacientes adultos y adolescentes con asma persistente. Todos los sujetos estaban utilizando CSI (corticosteroides inhalados) con o sin LABA durante al menos 12 semanas antes de la visita 1. En el estudio HZA106837 todos los pacientes tuvieron al menos una exacerbación que requirió tratamiento con corticosteroides orales en el año anterior a la visita 1. HZA106827 tuvo una duración de 12 semanas y evaluó la eficacia de furoato de fluticasona/vilanterol 92/22 microgramos [n=201] y FF 92 microgramos [n=205] en comparación con placebo [n=203], todos ellos administrados una vez al día. HZA106829 tuvo una duración de 24 semanas y evaluó la eficacia de furoato de fluticasona/vilanterol 184/22 microgramos [n=197] y FF 184 microgramos [n=194] ambos administrados una vez al día en comparación con 500 microgramos de PF dos veces al día [n=195].
En HZA106827/HZA106829 las variables co-primarias de eficacia fueron el cambio respecto a los valores basales en la visita clínica del FEVi valle (pre-broncodilatador y pre-dosis) al final del periodo de tratamiento en todos los sujetos y la media ponderada de los valores seriados del FEV1 durante 0 a 24 horas después de la administración de la dosis calculado en un subconjunto de los sujetos al final del periodo de tratamiento. El cambio respecto a los valores basales en el porcentaje de días sin medicación de rescate durante el tratamiento fue una variable secundaria robusta. Los resultados de las variables primarias y de las principales variables secundarias de estos estudios se describen en la Tabla 1.
Tabla 1 - Resultados de las variables primarias y principales variables secundarias en HZA106827 y HZA106829__
|
N° de estudio |
HZA106829 |
HZA106827 | ||
|
Dosis de tratamiento de |
FF/VI 184/22 |
FF/VI 184/22 |
FF/VI 92/22 |
FF/VI/92/22 |
|
FF/VI*(microgramos) |
una vez al día |
una vez al día |
una vez al día |
una vez al día |
|
vs FF 184 una |
vs PF 500 dos |
vs FF 92 una |
vs placebo una | |
|
vez al día |
veces al día |
vez al día |
vez al día | |
|
Cambios respecto a los valores basales en el FEV1 valle por el método de Última Observación Realizada (LOCF) | ||||
|
Diferencia entre tratamientos |
193 ml |
210 ml |
36 ml |
172 ml |
|
Valor p |
p<0,001 |
p<0,001 |
p=0,405 |
p<0,001 |
|
(IC 95%) |
(108; 277) |
(127; 294) |
(-48; 120) |
(87; 258) |
|
Media ponderada de los |
valores seriados del FEV1 en las 0-24 horas después de la dosis | |||
|
Diferencia entre tratamientos |
136 ml |
206 ml |
116 ml |
302 ml |
|
Valor p |
p=0,048 |
p=0,003 |
p=0,06 |
p<0,001 |
|
(IC 95%) |
(1; 270) |
(73; 339) |
(-5; 236) |
(178; 426) |
|
Cambios respecto a los valores basales en el porcentaje t |
e días sin medicación de rescate | |||
|
Diferencia entre tratamientos |
11,7% |
6,3% |
10,6% |
19,3% |
|
Valor p |
p<0,001 |
p=0,067 |
p<0,001 |
p<0,001 |
|
(IC 95%) |
(4,9; 18,4) |
(-0,4; 13,1) |
(4,3; 16,8) |
(13,0; 25,6) |
|
Cambios respecto a los valores basales en el porcentaje d |
e días sin síntomas | |||
|
Diferencia entre tratamientos |
8,4% |
4,9% |
12,1% |
18,0% |
|
Valor p |
p=0,010 |
p=0,137 |
p<0,001 |
p<0,001 |
|
(IC 95%) |
(2,0; 14,8) |
(-1,6; 11,3) |
(6,2; 18,1) |
(12,0; 23,9) |
|
Cambios respecto a los valores basales en el flujo espiratorio máximo por la mañana | ||||
|
Diferencia entre tratamientos |
33,5 l/min |
32,9 l/min |
14,6 l/min |
33,3 l/min |
|
Valor p |
p<0,001 |
p<0,001 |
p<0,001 |
p<0,001 |
|
(IC 95%) |
(22,3; 41,7) |
(24,8; 41,1) |
(7,9; 21,3) |
(26,5; 40,0) |
|
Cambios respecto a los valores basales en el flujo espiratorio máximo por la noche | ||||
|
Diferencia entre tratamientos |
30,7 l/min |
26,2 l/min |
12,3 l/min |
28,2 l/min |
|
Valor p |
p<0,001 |
p<0,001 |
p<0,001 |
p<0,001 |
|
(IC 95%) |
(22,5; 38,9) |
(18,0; 34,3) |
(5,8; 18,8) |
(21,7; 34,8) |
*FF/VI = furoato de fluticasona/vilanterol
HZA106837 tuvo una duración de tratamiento variable (desde un mínimo de 24 semanas a un máximo de 76 semanas, en el que la mayoría de los pacientes fueron tratados durante al menos 52 semanas). En HZA106837, los pacientes fueron aleatorizados para recibir bien furoato de fluticasona/vilanterol 92/22 microgramos [n=1.009] o FF 92 microgramos [n=1.010] ambos administrados una vez al día.
En HZA106837 la variable primaria fue el tiempo hasta la primera exacerbación asmática grave. Una exacerbación asmática grave se definió como un empeoramiento del asma que requirió el uso de corticosteroides sistémicos durante al menos 3 días, o una hospitalización o visita a urgencias como consecuencia de que el asma requirió el uso de corticosteroides sistémicos. Se evaluó también como variable secundaria el cambio medio ajustado respecto los valores basales en el FEV1 valle.
En HZA106837 el riesgo de tener una exacerbación asmática grave en pacientes que recibieron furoato de fluticasona/vilanterol 92/22 microgramos se redujo en un 20% en comparación con FF 92 microgramos en monoterapia (hazard ratio 0,795; p=0,036 IC 95% 0,642; 0,985). La tasa de exacerbaciones asmáticas graves por paciente y año fue 0,19 en el grupo FF 92 microgramos (aproximadamente 1 cada 5 años) y 0,14 en el grupo furoato de fluticasona/vilanterol 92/22 microgramos (aproximadamente 1 cada 7 años). La proporción de la tasa de exacerbaciones para furoato de fluticasona/vilanterol 92/22 microgramos frente a FF 92 microgramos fue 0,755 (IC 95% 0,603; 0,945). Esto representa una reducción del 25% en la tasa de exacerbaciones asmáticas graves para los sujetos tratados con furoato de fluticasona/vilanterol 92/22 microgramos en comparación con FF 92 microgramos (p=0,014). El efecto broncodilatador de 24-horas de furoato de fluticasona/vilanterol se mantuvo durante un año de tratamiento en el que no hubo evidencia de pérdida de eficacia (sin taquifilaxia). Furoato de fluticasona/vilanterol 92 /22 microgramos demostró de forma consistente la mejora en el FEVi valle de 83 ml a 95 ml en las semanas 12, 36 y 52 y al final del estudio en comparación con FF 92 microgramos (p<0,001 IC 95% 52; 126 ml al final del estudio). El cuarenta y cuatro por ciento de los pacientes en el grupo furoato de fluticasona/vilanterol 92/22 microgramos estuvieron bien controlados (ACQ7 <0,75) al final del tratamiento en comparación con el 36% de los sujetos en el grupo FF 92 microgramos (p<0,001 IC 95% 1,23; 1,82).
Estudios frente a las combinaciones con salmeterol/propionato de fluticasona
En un estudio de 24 semanas de duración (HZA113091) en pacientes adultos y adolescentes con asma persistente se demostró mejoras en la función pulmonar respecto a los valores basales tanto con furoato de fluticasona/vilanterol 92/22 microgramos administrado una vez al día por la noche como con salmeterol/PF 50/250 microgramos administrado dos veces al día. El aumento medio ajustado entre tratamientos respecto a los valores basales en la media ponderada durante 0 a 24 horas del FEV1 de 341 ml (furoato de fluticasona/vilanterol) y 377 ml (salmeterol/PF) demostró una mejora global en la función pulmonar durante 24 horas para ambos tratamientos. La diferencia media ajustada entre tratamientos de 37 ml entre los grupos no fue estadísticamente significativa (p=0,162). En el FEVi valle los sujetos en el grupo furoato de fluticasona/vilanterol alcanzaron un cambio medio en LS de 281 ml respecto a los valores basales y los sujetos en el grupo salmeterol/PF un cambio de 300 ml; (la diferencia media ajustada entre tratamientos de 19 ml (IC 95%:-0,073; 0,034) no fue estadísticamente significativa (p=0,485).
No se han realizado estudios comparativos frente a salmeterol/PF o frente a otras combinaciones CSI/LABA que comparen de forma adecuada los efectos de las exacerbaciones en asma.
Furoato de fluticasona en monoterapia
Un estudio de 24 semanas de duración aleatorizado, doble ciego, controlado con placebo (FFA112059) evaluó la seguridad y eficacia de FF 92 microgramos una vez al día [n=114] y PF 250 microgramos dos veces al día [n=114] frente a placebo [n=115] en pacientes adultos y adolescentes con asma persistente. Todos los sujetos tuvieron que estar tratados con una dosis estable de un CSI durante al menos 4 semanas antes de la visita 1 (visita de selección) y no se permitía el uso de LABAs en las 4 semanas anteriores a la visita 1. La variable primaria de eficacia fue el cambio respecto a los valores basales en la visita clínica del FEV1 valle (pre-broncodilatador y pre-dosis) al final del periodo de tratamiento. Una variable secundaria robusta fue el cambio respecto a los valores basales en el porcentaje de días sin medicación de rescate durante el periodo de tratamiento de 24 semanas. En la semana 24, aumentó el FEV1 valle en 146 ml (IC 95% 36; 257 ml; p=0,009) y 145 ml (IC 95% 33;
257 ml; p=0,011) con FF y PF respectivamente en comparación con placebo. Se aumentó el porcentaje de días sin medicación de rescate en 14,8% (IC 95% 6,9; 22,7; p<0,001) y 17,9% (IC 95% 10,0; 25,7; p<0,001) con FF y PF respectivamente frente a placebo.
Estudio de exposición a alérgenos
En un estudio (HZA113126) cruzado de cuatro brazos a dosis repetidas, controlado con placebo en pacientes con asma leve se evaluó el efecto broncoprotector de furoato de fluticasona/vilanterol 92/22 microgramos en la respuesta asmática temprana y tardía a alérgenos inhalados. Los pacientes fueron aleatorizados para recibir furoato de fluticasona/vilanterol 92/22 microgramos, FF 92 microgramos, vilanterol 22 microgramos o placebo una vez al día durante 21 días seguidos de una exposición a alérgenos 1 hora después de la dosis final. Los alérgenos fueron ácaros de polvo, caspa de gato, polen de abedul; la selección se basó en las pruebas de detección individual. Las mediciones de los valores seriados del FEV1 se compararon con los valores previos a la exposición a alérgenos medidos tras la inhalación de solución salina (valores iniciales). En general, se observaron efectos mayores sobre la respuesta asmática temprana con furoato de fluticasona/vilanterol 92/22 microgramos en comparación con FF 92 microgramos o vilanterol 22 microgramos en monoterapia. Tanto furoato de fluticasona/vilanterol 92/22 microgramos como FF 92 microgramos suprimieron prácticamente la respuesta asmática tardía en comparación con vilanterol en monoterapia. Furoato de fluticasona/vilanterol 92/22 microgramos proporcionó una mayor protección frente a la hiperreactividad bronquial inducida por alérgenos en comparación con FF y vilanterol en monoterapia evaluada el Día 22 mediante la prueba de provocación con metacolina.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Relvar Ellipta en uno o más grupos de la población pediátrica en asma (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
La biodisponibilidad absoluta de furoato de fluticasona y vilanterol administrados por vía inhalatoria como furoato de fluticasona/vilanterol fue de promedio de 15,2% y 27,3%, respectivamente. La biodisponibilidad oral tanto de furoato de fluticasona como de vilanterol fue baja, un promedio de 1,26% y <2%, respectivamente. Teniendo en cuenta esta baja biodisponibilidad oral, la exposición sistémica de furoato de fluticasona y vilanterol tras la inhalación es debida principalmente a la absorción de la porción inhalada de la dosis liberada al pulmón.
Distribución
Tras la administración intravenosa, tanto furoato de fluticasona como vilanterol se distribuyen ampliamente con unos volúmenes de distribución promedios en estado estacionario de 661 L y 165 L, respectivamente.
Tanto furoato de fluticasona como vilanterol tienen una baja asociación con los glóbulos rojos. La unión a proteínas plasmáticas in vitro de furoato de fluticasona y vilanterol en plasma humano fue alta, un promedio >99,6% y 93,9%, respectivamente. No hubo una disminución en el alcance de la unión a proteínas plasmáticas in vitro en sujetos con insuficiencia renal o hepática.
Furoato de fluticasona y vilanterol son sustratos de la glicoproteína-P (P-gp), sin embargo, se considera poco probable que la administración concomitante de furoato de fluticasona/vilanterol con inhibidores P-gp altere la exposición sistémica de furoato de fluticasona o vilanterol ya que ambas son moléculas que se absorben bien.
Biotransformación
En base a los resultados in vitro, las principales rutas del metabolismo de furoato de fluticasona y vilanterol en humanos están mediadas principalmente por el CYP3A4.
Furoato de fluticasona se metaboliza principalmente mediante hidrólisis del grupo carbotiato S-fluorometil a metabolitos con una actividad corticosteroidea significativamente reducida. Vilanterol se metaboliza principalmente por la O-desaquilación a una serie de metabolitos con actividad Pi- y p2-agonista significativamente reducida.
Eliminación
Tras la administración oral, furoato de fluticasona se eliminó en humanos principalmente mediante metabolismo a través de metabolitos que se excretan casi exclusivamente en las heces, con <1% de la dosis radioactiva recuperada eliminada en la orina.
Tras la administración oral, vilanterol se eliminó principalmente por metabolismo seguido de la excreción de los metabolitos en orina y heces, aproximadamente 70% y 30% de la dosis radioactiva respectivamente en un estudio en humanos radiomarcado realizado por vía oral. La semivida plasmática de eliminación aparente de vilanterol tras una administración única inhalada de furoato de fluticasona/vilanterol fue, de promedio, 2,5 horas. La semivida de acumulación efectiva de vilanterol, determinada por la administración inhalada de dosis repetidas de vilanterol 25 microgramos, es de 16,0 horas en sujetos con asma y de 21,3 horas en sujetos con EPOC.
Población pediátrica
No se recomiendan modificaciones de las dosis en adolescentes (12 años o mayores).
No se ha estudiado la farmacocinética de furoato de fluticasona/vilanterol en pacientes menores de 12 años de edad. No se ha establecido la seguridad y eficacia de furoato de fluticasona/vilanterol en niños menores de 12 años.
Poblaciones especiales
Pacientes de edad avanzada (> 65 años)
Los efectos de la edad sobre la farmacocinética de furoato de fluticasona y vilanterol se determinaron en los estudios fase III en asma y EPOC. No hubo evidencia de que la edad (12-84) afecte a la farmacocinética de furoato de fluticasona y vilanterol en sujetos con asma.
No se recomiendan modificaciones de las dosis en sujetos con asma y EPOC.
Insuficiencia renal
Un estudio clínico farmacológico de furoato de fluticasona/vilanterol mostró que la insuficiencia renal grave (aclaramiento de creatinina <30 ml/min) no supuso una exposición significativamente mayor a furoato de fluticasona o vilanterol o unos efectos sistémicos más marcados de los agonistas p2 o corticosteroides en comparación con los sujetos sanos.
No se requiere ajuste de dosis en pacientes con insuficiencia renal.
No se han estudiado los efectos de la hemodiálisis.
Insuficiencia hepática
Tras la administración de dosis repetidas de furoato de fluticasona/vilanterol durante 7 días, hubo un aumento de la exposición sistémica a furoato de fluticasona (hasta tres veces lo medido mediante AUC(0-24)) en sujetos con insuficiencia hepática (Child-Pugh A, B o C) en comparación con sujetos sanos. El aumento de la exposición sistémica de furoato de fluticasona en sujetos con insuficiencia hepática moderada (Child-Pugh B; furoato de fluticasona/vilanterol 184/22 microgramos) se asoció con una reducción promedio de 34% en el cortisol sérico en comparación con sujetos sanos. La exposición sistémica de la dosis normalizada de furoato de fluticasona fue similar en sujetos con insuficiencia hepática moderada y grave (Child-Pugh B o C).
Tras la administración de dosis repetidas de furoato de fluticasona/vilanterol durante 7 días, no hubo un aumento significativo en la exposición sistémica a vilanterol (Cmax y AUC) en sujetos con insuficiencia hepática leve, moderada o grave (Child-Pugh A, B o C).
No hubo efectos clínicamente relevantes de la combinación furoato de fluticasona/vilanterol sobre los efectos sistémicos de los p-adrenérgicos (ritmo cardiaco o potasio sérico) en sujetos con insuficiencia hepática leve o moderada (vilanterol, 22 microgramos) o con insuficiencia hepática grave (vilanterol,
12,5 microgramos) en comparación con sujetos sanos.
Otras poblaciones especiales
En sujetos con asma, el AUC(0-24) estimado de furoato de fluticasona para sujetos del Este asiático, japoneses y Sudeste asiático (12-13% de los sujetos) fue un promedio de 33% a 53% mayor en comparación con otros grupos raciales. Sin embargo, no hubo evidencia de que una mayor exposición sistémica en esta población esté asociada con un efecto mayor en la excreción de cortisol en orina de 24 horas. En promedio, se prevé que la Cmax de vilanterol sea de 220 a 287% mayor y el AUC(0-24) comparable para aquellos sujetos con antecedentes asiáticos en comparación con otros grupos raciales. Sin embargo, no hubo evidencia de que esta Cmax de vilanterol conllevara efectos clínicamente relevantes en el ritmo cardiaco.
Género, peso e IMC
No hubo evidencia de que el género, el peso o el IMC (índice de masa corporal) influyeran en la farmacocinética de furoato de fluticasona en base al análisis farmacocinético poblacional de los resultados de fase III en 1.213 sujetos con asma (712 mujeres).
No hubo evidencia de que el género, el peso o el IMC influyeran en la farmacocinética de vilanterol en base a un análisis farmacocinético poblacional en 856 sujetos con asma (500 mujeres).
No es necesario el ajuste de dosis por género, peso o IMC.
5.3 Datos preclínicos sobre seguridad
Los efectos farmacológicos y toxicológicos observados con furoato de fluticasona o vilanterol en los estudios no clínicos fueron aquellos típicamente asociados con glucocorticoides o p2-agonistas. La administración de furoato de fluticasona en combinación con vilanterol no conllevaba ninguna nueva toxicidad significativa.
Genotoxicidad y carcinogenicidad
Furoato de fluticasona
Furoato de fluticasona no resultó genotóxico en una batería estándar de estudios ni carcinogénico en estudios de inhalación a tiempo real realizados en ratas o ratones a exposiciones similares a las dosis máximas recomendadas en humanos basadas en el AUC.
Trifenatato de vilanterol
En estudios de toxicidad genética, vilanterol (como a-fenilcinamato) y el ácido trifenilacético no resultaron genotóxicos, lo cual indica que vilanterol (como trifenatato) no representa un peligro genotóxico para humanos.
De acuerdo con los resultados identificados en otros agonistas p2, en los estudios de inhalación a tiempo real trifenatato de vilanterol produjo efectos proliferativos en el aparato reproductor de ratas y ratones hembras y en la glándula pituitaria de las ratas. No hubo un aumento en la incidencia de tumores en ratas o ratones expuestos a dosis 2 o 30 veces la dosis máxima recomendada en humanos, respectivamente, en base al AUC.
Toxicidad reproductiva
Furoato de fluticasona
Los efectos observados tras la administración inhalada de furoato de fluticasona en combinación con vilanterol en ratas fueron similares a los que se observaron con furoato de fluticasona en monoterapia. El furoato de fluticasona no resultó teratogénico en ratas ni conejos, pero produjo retraso en el desarrollo en ratas y produjo abortos en conejos a dosis tóxicas para la madre. No se observaron efectos sobre el desarrollo en ratas expuestas a dosis aproximadamente 3 veces superiores a la dosis máxima recomendada, basada en el AUC.
Trifenatato de vilanterol
El trifenatato de vilanterol no fue teratogénico en ratas. En estudios de inhalación en conejos, el trifenatato de vilanterol produjo efectos similares a los que se observaban en otros p2-agonistas (paladar hendido, párpados abiertos, fusión esternebral y malrotación/flexión de extremidades).
Cuando se administró por vía subcutánea no hubo efectos a exposiciones 84 veces superiores a la dosis máxima recomendada, basada en el AUC.
Ni el furoato de fluticasona ni el trifenatato de vilanterol tuvieron efectos adversos sobre la fertilidad o sobre el desarrollo pre o post-natal en ratas.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Lactosa monohidrato Estearato de magnesio
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
2 años
Periodo de validez una vez abierta la bandeja: 6 semanas.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 25°C. Si se conserva el dispositivo en nevera se debe permitir que el inhalador alcance la temperatura ambiente durante al menos una hora antes de su uso.
Conservar en el embalaje original para protegerlo de la humedad.
Debe utilizarse en un plazo de 6 semanas tras la apertura de la bandeja.
Escribir la fecha en la que el inhalador se debe desechar en el espacio designado para ello, que aparece en la etiqueta del inhalador. La fecha se debe anotar tan pronto como el inhalador se saque de la bandeja.
6.5 Naturaleza y contenido del envase
El dispositivo inhalador está formado por un cuerpo gris claro, un protector de la boquilla amarillo y un contador de dosis, envasado en una bandeja de aluminio laminada que contiene una bolsa desecante. La bandeja está sellada con una tapa de aluminio desplegable.
El dispositivo inhalador contiene dos tiras de aluminio laminado de 14 o 30 dosis.
El dispositivo inhalador es un dispositivo multi-componente compuesto de polipropileno, polietileno de alta densidad, polioximetileno, polibutileno tereftalato, acrilonitrilo butadieno estireno, policarbonato y acero inoxidable.
Tamaños de envases de 14 o 30 dosis por inhalador. Envase clínico de 3 x 30 dosis por inhalador. Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Para las instrucciones de uso, ver sección 4.2.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Limited.
980 Great West Road, Brentford, Middlesex TW8 9GS, Reino Unido.
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/886/004
EU/1/13/886/005
EU/1/13/886/006
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA
AUTORIZACIÓN
Fecha de la primera autorización: 13 noviembre 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes
Glaxo Operations UK Ltd. (operando como Glaxo Wellcome Operations)
Priory Street
Ware, Hertfordshire SG12 0DJ Reino Unido
Glaxo Operations UK Ltd (operando como GlaxoWellcome Operations), Harmire Road, Barnard Castle, County Durham DL12 8DT, Reino Unido.
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica.
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 quater, apartado 7, de la Directiva 2001/83/CE y cualquier actualización subsecuente publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo
acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de
Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Obligación de llevar a cabo medidas post-autorización
El TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
Descripción |
Fecha límite |
|
Presentación del informe final del estudio experimental de seguridad postautorización realizado para investigar más el riesgo de neumonía con Relvar Ellipta en comparación con otras combinaciones de dosis fijas de ICS/LABA en el tratamiento de la EPOC, de acuerdo a un protocolo acordado por el Comité. |
31 diciembre 2016 |
|
Presentación del informe final del estudio experimental de seguridad postautorización realizado para investigar más el riesgo de neumonía con Relvar Ellipta en comparación con otras combinaciones de dosis fijas de ICS/LABA en el tratamiento del asma, de acuerdo a un protocolo acordado por el Comité. |
30 Junio 2018 |
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
92/22 microgramos
1. NOMBRE DEL MEDICAMENTO
Relvar Ellipta 92 microgramos/22 microgramos polvo para inhalación (unidosis). furoato de fluticasona/vilanterol
2. PRINCIPIO(S) ACTIVO(S)
Cada dosis liberada contiene 92 microgramos de furoato de fluticasona y 22 microgramos de vilanterol (como trifenatato).
3. LISTA DE EXCIPIENTES
También contiene lactosa y estearato de magnesio.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para inhalación (unidosis).
14 dosis 30 dosis 3 x 30 dosis
1 inhalador de 14 dosis 1 inhalador de 30 dosis
Envase clínico: 90 dosis (3 estuches de 30 dosis)
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía inhalatoria UNA VEZ AL DÍA
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
CAD
Periodo de validez tras la apertura: 6 semanas.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 25°C.
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Limited, 980 Great West Road, Brentford, Middlesex TW8 9GS, Reino Unido.
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/886/001
EU/1/13/886/002
EU/1/13/886/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
relvar ellipta 92/22
184/22 microgramos
1. NOMBRE DEL MEDICAMENTO
Relvar Ellipta 184 microgramos/22 microgramos polvo para inhalación (unidosis). furoato de fluticasona/vilanterol
2. PRINCIPIO(S) ACTIVO(S)
Cada dosis liberada contiene 184 microgramos de furoato de fluticasona y 22 microgramos de vilanterol (como trifenatato).
3. LISTA DE EXCIPIENTES
También contiene lactosa y estearato de magnesio.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para inhalación (unidosis).
14 dosis 30 dosis 3 x 30 dosis
1 inhalador de 14 dosis 1 inhalador de 30 dosis
Envase clínico: 90 dosis (3 estuches de 30 dosis)
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía inhalatoria UNA VEZ AL DÍA
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
CAD
Periodo de validez tras la apertura: 6 semanas.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 25°C.
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Limited, 980 Great West Road, Brentford, Middlesex TW8 9GS, Reino Unido.
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/886/004
EU/1/13/886/005
EU/1/13/886/006
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
relvar ellipta 184/22
92/22 microgramos
1. NOMBRE DEL MEDICAMENTO
Relvar Ellipta 92 microgramos/22 microgramos polvo para inhalación (unidosis). furoato de fluticasona/vilanterol
2. PRINCIPIO(S) ACTIVO(S)
Cada dosis liberada contiene 92 microgramos de furoato de fluticasona y 22 microgramos de vilanterol (como trifenatato).
3. LISTA DE EXCIPIENTES
También contiene lactosa y estearato de magnesio.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
30 dosis
1 inhalador de 30 dosis.
ENVASE CLÍNICO. Prohibida su venta al detalle.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN_
Vía inhalatoria UNA VEZ AL DÍA
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS_
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO_
8. FECHA DE CADUCIDAD_
CAD
Periodo de validez tras la apertura: 6 semanas.
No conservar a temperatura superior a 25°C.
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Limited, 980 Great West Road, Brentford, Middlesex TW8 9GS, Reino Unido.
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/886/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
relvar ellipta 92/22
184/22 microgramos
1. NOMBRE DEL MEDICAMENTO
Relvar Ellipta 184 microgramos/22 microgramos polvo para inhalación (unidosis). furoato de fluticasona/vilanterol
2. PRINCIPIO(S) ACTIVO(S)
Cada dosis liberada contiene 184 microgramos de furoato de fluticasona y 22 microgramos de vilanterol (como trifenatato).
3. LISTA DE EXCIPIENTES
También contiene lactosa y estearato de magnesio.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
30 dosis
1 inhalador de 30 dosis.
ENVASE CLÍNICO. Prohibida su venta al detalle.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN_
Vía inhalatoria UNA VEZ AL DÍA
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS_
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO_
8. FECHA DE CADUCIDAD_
CAD
Periodo de validez tras la apertura: 6 semanas.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 25°C.
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Limited, 980 Great West Road, Brentford, Middlesex TW8 9GS, Reino Unido.
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/886/006
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
relvar ellipta 184/22
ETIQUETA DE LA BANDEJA 92/22 microgramos
1. NOMBRE DEL MEDICAMENTO
Relvar Ellipta 92 microgramos/22 microgramos polvo para inhalación (unidosis). furoato de fluticasona/vilanterol
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Logo GSK
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
No abrir hasta que esté preparado para inhalar. Periodo de validez tras la apertura: 6 semanas.
14 dosis 30 dosis
ETIQUETA DE LA BANDEJA 184/22 microgramos
1. NOMBRE DEL MEDICAMENTO
Relvar Ellipta 184 microgramos/22 microgramos polvo para inhalación (unidosis). furoato de fluticasona/vilanterol
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Logo GSK
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
No abrir hasta que esté preparado para inhalar. Periodo de validez tras la apertura: 6 semanas.
14 dosis 30 dosis
ETIQUETA DEL DISPOSITIVO
92/22 microgramos
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Relvar Ellipta 92 microgramos/22 microgramos polvo para inhalación furoato de fluticasona/vilanterol Vía inhalatoria
2. FECHA DE CADUCIDAD
CAD
Periodo de validez tras la apertura: 6 semanas Desechar el:
3. NÚMERO DE LOTE
Lote
4. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
14 dosis 30 dosis
ETIQUETA DEL DISPOSITIVO
184/22 microgramos
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Relvar Ellipta 184 microgramos/22 microgramos polvo para inhalación furoato de fluticasona/vilanterol Vía inhalatoria
2. FECHA DE CADUCIDAD
CAD
Periodo de validez tras la apertura: 6 semanas Desechar el:
3. NÚMERO DE LOTE
Lote
4. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
14 dosis 30 dosis
B. PROSPECTO
Prospecto: información para el usuario
Relvar Ellipta 92 microgramos/22 microgramos polvo para inhalación (unidosis)
Relvar Ellipta 184 microgramos/22 microgramos polvo para inhalación (unidosis)
furoato de fluticasona/vilanterol
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Relvar Ellipta y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Relvar Ellipta
3. Cómo usar Relvar Ellipta
4. Posibles efectos adversos
5. Conservación de Relvar Ellipta
6. Contenido del envase e información adicional
Instrucciones de uso paso a paso
1. Qué es Relvar Ellipta y para qué se utiliza
Relvar Ellipta contiene dos principios activos: furoato de fluticasona y vilanterol. Existen dos concentraciones diferentes de Relvar Ellipta: furoato de fluticasona 92 microgramos/vilanterol 22 microgramos y furoato de fluticasona 184 microgramos/vilanterol 22 microgramos.
La concentración de 92/22 microgramos se utiliza para el tratamiento regular de la enfermedad pulmonar obstructiva crónica (EPOC) en adultos, así como para el tratamiento del asma en adultos y adolescentes de 12 años de edad y mayores.
La concentración de 184/22 microgramos se utiliza para el tratamiento del asma en adultos y adolescentes de 12 años de edad y mayores.
Relvar Ellipta se debe utilizar todos los días y no únicamente cuando tiene dificultad para respirar u otros síntomas del asma y la EPOC. No se debe utilizar para aliviar un ataque repentino de ahogo o sibilancias. Si tiene este tipo de ataques debe utilizar un inhalador de “rescate” de acción rápida (como salbutamol).
Furoato de fluticasona pertenece a un grupo de medicamentos llamados corticosteroides, a menudo llamados simplemente esteroides. Los corticosteroides reducen la inflamación. Además reducen la hinchazón (inflamación) e irritación de las pequeñas vías aéreas en los pulmones y alivian de forma gradual los problemas respiratorios. Los corticosteroides también ayudan a prevenir los ataques de asma y el empeoramiento de la EPOC.
Vilanterol pertenece a un grupo de medicamentos llamados broncodilatadores de acción prolongada. Actúa relajando los músculos de las pequeñas vías aéreas en los pulmones. Esto ayuda a abrir las vías respiratorias y facilita la entrada y salida de aire de los pulmones. Cuando se usa de forma regular, ayuda a que las pequeñas vías aéreas de los pulmones permanezcan abiertas.
El uso regular de estos dos principios activos juntos, le ayudará a controlar sus dificultades respiratorias, más que cualquiera de los medicamentos por separado.
El asma, es una enfermedad pulmonar crónica grave en la que los músculos que rodean las vías respiratorias más pequeñas se estrechan (broncoconstricción) y se hinchan e irritan (inflamación). Los síntomas van y vienen e incluyen dificultad para respirar, sibilancias, opresión en el pecho y tos.
La enfermedad pulmonar obstructiva crónica (EPOC), es una enfermedad pulmonar crónica grave en donde las vías respiratorias se inflaman y se engrosan. Los síntomas incluyen dificultad para respirar, tos, molestias en el pecho y tos acompañada de mucosidad. Relvar Ellipta ha demostrado reducir los brotes de los síntomas que acompañan a la EPOC.
2. Qué necesita saber antes de empezar a usar Relvar Ellipta No use Relvar Ellipta
- si es alérgico a furoato de fluticasona, vilanterol o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6)
- si piensa que lo anterior le aplica, no use Relvar Ellipta hasta haber consultado con su médico.
Tenga especial cuidado con Relvar Ellipta
Consulte a su médico antes de empezar a usar Relvar Ellipta:
- si tiene problemas hepáticos, ya que puede ser más propenso a tener efectos adversos. Si tiene problemas hepáticos moderados o graves, su médico limitará su dosis a la concentración más baja de Relvar Ellipta (92/22 microgramos una vez al día)
- si tiene problemas cardiacos o tensión arterial alta
- si tiene tuberculosis (TB) pulmonar o cualquier otra infección desde hace tiempo o que no haya sido tratada
- si tiene antecedentes de diabetes
- si tiene problemas de la glándula tiroides
- si tiene el potasio de la sangre bajo.
Consulte con su médico antes de utilizar este medicamento si piensa que cualquiera de las condiciones anteriores le aplican.
Dificultades respiratorias inmediatas
Si su respiración o las sibilancias empeoran inmediatamente después de utilizar Relvar Ellipta, deje de usarlo y busque ayuda médica inmediatamente.
Infección pulmonar
Si está utilizando este medicamento para el tratamiento de la EPOC, puede presentar un mayor riesgo de desarrollar una infección de los pulmones conocida como neumonía. Consulte la sección 4 "Posibles efectos adversos" para obtener información sobre los síntomas a los que debe estar atento mientras esté usando este medicamento. Consulte con su médico tan pronto como sea posible si desarrolla cualquiera de esos síntomas.
Niños y adolescentes
Este medicamento no está recomendado para el tratamiento del asma en niños menores de 12 años de edad, o en niños y adolescentes de cualquier edad para el tratamiento de la EPOC.
Uso de Relvar Ellipta con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Algunos medicamentos pueden afectar a la forma de actuar de este medicamento, o hacer que sea más probable que presente efectos adversos. Estos incluyen:
- betabloqueantes, como metoprolol, utilizado en el tratamiento de la tensión arterial alta o enfermedades del corazón
- ketoconazol, para tratar infecciones por hongos
- ritonavir, para tratar el VIH
- agonistas p2-adrenérgicos de acción prolongada, como salmeterol.
Consulte con su médico o farmacéutico si está tomando alguno de estos medicamentos. Embarazo
El uso de Relvar Ellipta en mujeres embarazadas solo se debe considerar si los beneficios esperados superan los riesgos.
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
Lactancia
Se desconoce si este medicamento se excreta en la leche materna. Por lo tanto, no se puede descartar que exista riesgo para los lactantes alimentados con leche materna.
Si está en periodo de lactancia, consulte con su médico antes de utilizar Relvar Ellipta.
Conducción y uso de máquinas
Es poco problable que este medicamento afecte a su capacidad para conducir o utilizar máquinas. Relvar Ellipta contiene lactosa
Si le han diagnosticado una intolerancia a ciertos azúcares, o a la proteína de la leche, consulte con su médico antes de utilizar este medicamento.
3. Cómo usar Relvar Ellipta
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte con su médico, farmacéutico o enfermero.
Cuánto usar
Asma
La dosis recomendada en el tratamiento del asma es de una inhalación (92 microgramos de furoato de fluticasona y 22 microgramos de vilanterol) una vez al día, administrada a la misma hora cada día.
Si tiene asma grave, su médico puede decidir que se administre una inhalación del inhalador que contiene la concentración más alta (184 microgramos de furoato de fluticasona y 22 microgramos de vilanterol). Esta dosis también se utiliza una vez al día, a la misma hora cada día.
EPOC
La dosis recomendada en el tratamiento de la EPOC es de una inhalación (92 microgramos de furoato de fluticasona y 22 microgramos de vilanterol) una vez al día, administrada a la misma hora cada día.
La concentración más alta de Relvar Ellipta no es adecuada para el tratamiento de la EPOC.
Use Relvar Ellipta a la misma hora cada día, ya que es eficaz durante 24 horas
Es muy importante que utilice este medicamento todos los días, como le haya recomendado su médico. Esto le ayudará a no tener síntomas ni durante el día ni durante la noche.
Relvar Ellipta no se debe utilizar para aliviar un ataque repentino de ahogo o sibilancias. Si tiene este tipo de ataques debe utilizar un inhalador de “rescate” de acción rápida (como salbutamol).
Si siente que se queda sin respiración o tiene sibilancias más frecuentemente de lo normal, o si está utilizando su inhalador de “rescate” de acción rápida más a menudo de lo habitual, acuda a su médico.
Cómo usar Relvar Ellipta
Para obtener la información completa lea las “Instrucciones de uso paso a paso” incluidas tras la sección 6 de este prospecto.
No es necesario preparar Relvar Ellipta de ninguna forma especial, ni siquiera la primera vez que se va a utilizar.
Si los síntomas no mejoran
Si los síntomas (ahogo, sibilancias, tos) no mejoran o empeoran, o si está utilizando su inhalador de “rescate” de acción rápida más a menudo de lo habitual: contacte con su médico lo antes posible.
Si usa más Relvar Ellipta del que debe
Si accidentalmente toma más Relvar Ellipta de lo recomendado por su médico, consulte con su médico o farmacéutico. Podría notar que su corazón late más rápido de lo normal, sentirse tembloroso o tener dolor de cabeza.
Si ha utilizado más medicamento de lo indicado durante un periodo de tiempo prolongado, es especialmente importante que reciba asesoramiento de su médico o farmacéutico. Esto se debe a que dosis mayores de Relvar Ellipta pueden reducir la cantidad de hormonas esteroideas producidas de forma natural por su cuerpo.
Si olvidó usar Relvar Ellipta
No tome una dosis doble para compensar las dosis olvidadas. Tome la siguiente dosis a su hora habitual.
Si tiene sibilancias o ahogo, o desarrolla cualquier otro síntoma de un ataque de asma, utilice su inhalador de “rescate” de acción rápida (por ejemplo salbutamol), y busque asesoramiento médico.
No deje de utilizar Relvar Ellipta sin consultar
Utilice este medicamento durante el tiempo que le haya recomendado su médico. Sólo será eficaz durante el tiempo que siga utilizándolo. No deje de utilizarlo hasta que su médico se lo indique, incluso si se encuentra mejor.
Si tiene alguna duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Reacciones alérgicas
Es raro que Relvar Ellipta provoque reacciones alérgicas (afectan a menos de 1 de cada 1.000 personas)
Si tras la administración de Relvar Ellipta, tiene alguno de los síntomas que figuran a continuación, deje de utilizar este medicamento y póngase en contacto con su médico inmediatamente:
• erupción en la piel (habones) o enrojecimiento
• hinchazón, algunas veces de la cara o la boca (angioedema)
• aumento de las sibilancias (sonido agudo al respirar), tos o dificultad para respirar
• sensación de debilidad repentina o mareo (que puede provocar colapso o pérdida de consciencia).
Dificultades respiratorias inmediatas
Si su respiración o las sibilancias empeoran inmediatamente tras el uso de Relvar Ellipta, deje de usarlo y busque ayuda médica inmediatamente.
Neumonía (infección de los pulmones) en pacientes con EPOC (efecto adverso frecuente).
Si tiene alguno de los síntomas que figuran a continuación mientras está utilizando Relvar Ellipta, consulte con su médico. Podrían ser síntomas de una infección pulmonar:
• fiebre o escalofríos
• aumento de la producción de la mucosidad, cambio en el color del moco
• aumento de la tos o aumento de la dificultad para respirar.
Otros efectos adversos incluyen:
Efectos adversos muy frecuentes
Pueden afectar a más de 1 de cada 10 personas:
• dolor de cabeza
• resfriado común.
Efectos adversos frecuentes
Pueden afectar hasta 1 de cada 10 personas:
• aftas, protuberancias en la boca o en la garganta causadas por una infección por hongos (candidiasis). Enjuagar la boca con agua inmediatamente después de usar Relvar Ellipta puede ayudar a que este efecto adverso no se produzca
• inflamación de los pulmones (bronquitis)
• infección de los senos nasales o garganta
• gripe
• dolor e irritación en la parte posterior de la boca y garganta
• inflamación de los senos
• picor, moqueo o nariz taponada
• tos
• alteraciones en la voz
• debilitamiento de los huesos que puede producir fracturas
• dolor de estómago
• dolor de espalda
• temperatura elevada (fiebre)
• dolor en las articulaciones
• espasmos musculares.
Efectos adversos poco frecuentes Pueden afectar hasta 1 de cada 100 personas:
• latido del corazón irregular.
Efectos adversos raros
Pueden afectar hasta 1 de cada 1.000 personas
• reacciones alérgicas
• latido rápido del corazón (taquicardia)
• nota el latido de su corazón (palpitaciones)
• temblor
ansiedad.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Relvar Ellipta
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de CAD. La fecha de caducidad es el último día del mes que se indica.
No conservar a temperatura superior a 25°C.
Conservar en el embalaje original para protegerlo de la humedad, y no abrir la tapa de aluminio hasta que esté preparado para inhalar. Una vez abierta la bandeja, el inhalador puede utilizarse durante un plazo de 6 semanas, contando desde la fecha de apertura de la bandeja. Escribir la fecha en la que se debe desechar el inhalador en el espacio designado para ello en la etiqueta del inhalador. La fecha se debe anotar tan pronto como el inhalador se saque de la bandeja.
Si lo conserva en la nevera, deje que el inhalador vuelva a la temperatura ambiente durante por lo menos una hora antes de utilizarlo.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Relvar Ellipta
- Los principios activos son furoato de fluticasona y vilanterol. Cada inhalación proporciona una dosis liberada (dosis que sale por la boquilla) de 92 o 184 microgramos de furoato de fluticasona y 22 microgramos de vilanterol (como trifenatato).
- Los demás componentes son lactosa monohidrato y estearato de magnesio.
Aspecto del producto y contenido del envase
Relvar Ellipta es un polvo blanco. El dispositivo Ellipta es un inhalador de color gris claro con un protector de la boquilla de color amarillo y un contador de dosis. Está envasado en una bandeja de aluminio laminado con una tapa de aluminio desplegable. La bandeja contiene una bolsa desecante para reducir la humedad en el envase. Una vez abierta la tapa de la bandeja, tire el desecante, no lo ingiera o lo inhale. El dispositivo no necesita ser conservado en la bandeja de aluminio laminado una vez que se ha abierto.
El inhalador contiene dos tiras de aluminio laminado de 14 o 30 dosis. El envase clínico contiene 3 x 30 dosis.
Instrucciones de uso paso a paso ¿Qué es el inhalador Ellipta?
La primera vez que utilice el inhalador Ellipta, no necesita comprobar que funciona correctamente, ya viene preparado para ser utilizado directamente. Sólo siga estas instrucciones de uso paso a paso.
El inhalador está envasado en una bandeja que contiene una bolsa desecante, para reducir la humedad. Tire el desecante, no lo ingiera o lo inhale.
Cuando saque el inhalador de la bandeja, estará en la posición de "cerrado". No lo abra hasta que esté preparado para inhalar una dosis del medicamento. Cuando se abre la bandeja, se debe anotar la fecha de “desechar el” en el espacio designado para ello que aparece en la etiqueta del inhalador. La fecha de “desechar el” es de 6 semanas desde la fecha de apertura de la bandeja. Después de esta fecha el inhalador no debe utilizarse más. La bandeja se puede desechar después de la primera apertura.
1. Leer las siguientes instrucciones antes de utilizar el inhalador Si abre y cierra la tapa sin inhalar el medicamento, perderá la dosis.
La dosis perdida quedará retenida de forma segura dentro del inhalador, pero no estará disponible para ser inhalada.
No es posible administrarse accidentalmente una dosis adicional o una dosis doble mediante una inhalación.
Contador de dosis
El contador de dosis indica cuántas dosis de medicamento quedan en el dispositivo. Antes de usar el inhalador, debe indicar exactamente 30 dosis.
Cada vez que se abre la tapa, el contador disminuye en 1 unidad.
Cuando quedan menos de 10 dosis, la mitad del contador de dosis se pone de color rojo.
Una vez se utiliza la última dosis, la mitad del contador de dosis se pone de color rojoe indica el número 0. El inhalador ahora está vacío.
Si se abre la tapa cuando el inhalador está vacío, el contador de dosis pasa de estar la mitad de color rojo a estarlo completamente.

2. Preparar una dosis
Antes de abrir la tapa, espere a estar preparado para administrarse una dosis. No agite el inhalador.
• Deslice la tapa hacia abajo hasta que oiga un “clic”.

El medicamento está ahora preparado para ser inhalado.
Como confirmación, el contador de dosis disminuye en 1 unidad.
• Si el contador de dosis no disminuye al oír el “clic”, el inhalador no liberará el medicamento. Llévelo a su farmacéutico y solicite ayuda.
3. Inhalar el medicamento
• Mantenga el inhalador alejado de la boca, espire lo que razonablemente le sea posible.
No espire dentro del inhalador.
• Coloque la boquilla entre sus labios, y ciérrelos firmemente alrededor de la boquilla. No
bloquee las ranuras de ventilación con los dedos.
Lostabios se ajustan sobre la forma contorneada de la boquilla-para-la-mhalacion.TÍ
No-bloquear-la-ranura-de-ventilacioncon-los-dedos ^
• Realice una inspiración prolongada, continua y profunda. Mantenga la respiración tanto tiempo como le sea posible (al menos 3-4 segundos).
• Retire el inhalador de la boca.
• Espire suave y lentamente.
Puede que no sea capaz de distinguir el sabor o de notar el medicamento, incluso cuando utilice el inhalador de forma correcta.
4. Cerrar el inhalador y si es posible enjuagarse la boca
Si quiere limpiar la boquilla utilice un pañuelo seco antes de cerrar la tapa.
• Deslice la tapa hacia arriba hasta el tope para proteger la boquilla.
• Enjuáguese la boca con agua, una vez utilizado el inhalador.
Esto hará que sea menos probable que se produzcan efectos adversos como ulceraciones en la boca o garganta.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización:
Glaxo Group Limited,
980 Great West Road,
Brentford,
Middlesex TW8 9GS,
Reino Unido.
Fabricante:
Glaxo Operations UK Limited (operando como Glaxo Wellcome Operations),
Priory Street,
Ware,
Hertfordshire, SG12 0DJ Reino Unido
Glaxo Operations UK Limited (operando como GlaxoWellcome Operations),
Harmire Road,
Barnard Castle,
County Durham, DL12 8DT Reino Unido.
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Lietuva
GlaxoSmithKline Lietuva UAB Tel: + 370 5 264 90 00 info.lt@gsk.com

Belgie/Belgique/Belgien
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Tél/Tel: + 32 (0) 10 85 52 00
|
Etnrapna rnaKcoCMmKnaHH EOOfl Ten.: + 359 2 953 10 34 |
Luxembourg/Luxemburg GlaxoSmithKline Pharmaceuticals s.a./n.v. Belgique/Belgien Tél/Tel: + 32 (0) 10 85 52 00 |
|
Ceská republika GlaxoSmithKline s.r.o. Tel: + 420 222 001 111 cz.info@gsk.com |
Magyarország GlaxoSmithKline Kft. Tel.: + 36 1 225 5300 |
|
Danmark GlaxoSmithKline Pharma A/S Tlf: + 45 36 35 91 00 dk-info@gsk.com |
Malta GlaxoSmithKline Malta Tel: + 356 21 238131 |
|
Deutschland GlaxoSmithKline GmbH & Co. KG Tel.: + 49 (0)89 36044 8701 produkt.info@gsk.com |
Nederland GlaxoSmithKline BV Tel: + 31 (0)30 6938100 nlinfo@gsk.com |
|
Eesti GlaxoSmithKline Eesti OÜ Tel: + 372 6676 900 estonia@gsk.com |
Norge GlaxoSmithKline AS Tlf: + 47 22 70 20 00 firmapost@gsk.no |
|
EXXáSa GlaxoSmithKline A.E.B.E. T^: + 30 210 68 82 100 |
Osterreich GlaxoSmithKline Pharma GmbH Tel: + 43 (0)1 97075 0 at.info@gsk.com |
|
España GlaxoSmithKline, S.A. Tel: + 34 902 202 700 es-ci@gsk.com |
Polska GSK Services Sp. z o.o. Tel.: + 48 (0)22 576 9000 |
|
France Laboratoire GlaxoSmithKline Tél.: + 33 (0)1 39 17 84 44 diam@gsk.com |
Portugal GlaxoSmithKline - Produtos Farmacéuticos, Lda. Tel: + 351 21 412 95 00 |
|
Hrvatska GlaxoSmithKline d.o.o. Tel: + 385 1 6051 999 |
Romania GlaxoSmithKline (GSK) S.R.L. Tel: + 4021 3028 208 |
|
Ireland GlaxoSmithKline (Ireland) Limited Tel: + 353 (0)1 4955000 |
Slovenija GlaxoSmithKline d.o.o. Tel: + 386 (0)1 280 25 00 medical.x.si@gsk.com |
|
Ísland Vistor hf. Sími: + 354 535 7000 |
Slovenská republika GlaxoSmithKline Slovakia s. r. o. Tel: + 421 (0)2 48 26 11 11 recepcia.sk@gsk.com |
Kúrcpog
GlaxoSmithKline (Cyprus) Ltd Tpk: + 357 22 39 70 00 gskcyprus@gsk.com
Latvija
GlaxoSmithKline Latvia SIA Tel: + 371 67312687 lv-epasts@gsk.com
Suomi/Finland
GlaxoSmithKline Oy Puh/Tel: + 358 (0)10 30 30 30 Finland.tuoteinfo@gsk.com
Sverige
GlaxoSmithKline AB Tel: + 46 (0)8 638 93 00 info.produkt@gsk.com
United Kingdom
GlaxoSmithKline UK Tel: + 44 (0)800 221441 customercontactuk@gsk.com
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
71