Pixuvri 29Mg Polvo Para Concentrado Para Solucion Para Perfusion
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Pixuvri 29 mg polvo para concentrado para solución para perfusión.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un vial contiene dimaleato de pixantrona equivalente a 29 mg de pixantrona.
Después de la reconstitución, cada ml de concentrado contiene dimaleato de pixantrona equivalente a 5,8 mg de pixantrona.
Excipientes con efecto conocido:
Un vial contiene 39 mg de sodio.
Para consultar la lista completa de excipientes, ver sección 6.1
3. FORMA FARMACÉUTICA
Polvo para concentrado para solución para perfusión. Polvo liofilizado de color azul oscuro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Pixuvri está indicado como monoterapia para el tratamiento de pacientes adultos con linfomas no hodgkinianos (LNH) de linfocitos B agresivos, multirrecidivantes o resistentes al tratamiento. No se ha establecido el beneficio del tratamiento con pixantrona en pacientes cuando se utiliza como quimioterapia de quinta línea o más en pacientes refractarios al tratamiento anterior.
4.2 Posología y forma de administración
Pixuvri debe ser administrado por médicos que estén familiarizados con el uso de agentes antineoplásicos y cuenten con instalaciones para el seguimiento periódico de los parámetros clínicos, hematológicos y bioquímicos durante y después del tratamiento (ver sección 6.6).
Posología
La dosis recomendada es de 50 mg/m2 de pixantrona los días 1, 8 y 15 de cada ciclo de 28 días hasta un máximo de 6 ciclos.
Atención:
En la UE la dosis recomendada se refiere al principio activo (pixantrona). El cálculo de la dosis que se debe administrar a un paciente concreto se debe basar en la concentración de la solución reconstituida que contiene 5,8 mg/ml de pixantrona y en la dosis recomendada de 50 mg/m2. En algunos ensayos y publicaciones, la dosis recomendada se basó en la sal (dimaleato de pixantrona).
Sin embargo, se debe ajustar la dosis antes del inicio de cada ciclo basándose en los recuentos hematológicos mínimos o en la toxicidad máxima del ciclo de tratamiento anterior. La cantidad de Pixuvri en miligramos que se debe administrar a un paciente se debe determinar sobre la base de la superficie corporal (SC) del paciente. La SC se debe determinar mediante la norma institucional para el cálculo de la SC y se debe utilizar un peso medido el día 1 de cada ciclo.
Se recomienda cierta precaución en pacientes obesos ya que los datos sobre la posología basada en la SC son muy limitados para este grupo.
Pautas para modificar la dosis
La modificación de la dosis y el horario de las dosis subsiguientes se deben determinar basándose en el criterio clínico en función del grado y la duración de la mielodepresión. Para los ciclos posteriores, por lo general se puede repetir la dosis si los recuentos de leucocitos y plaquetas han retornado a niveles aceptables.
Si el día 1 de cualquier ciclo la cifra absoluta de neutrófilos (CAN) es <1,0 x 109/l o el recuento de plaquetas es <75 x 109/l, se recomienda retrasar el tratamiento hasta que la CAN se recupere hasta >1,0 x 109/l y el recuento de plaquetas hasta >75 x 109/l.
La tabla 1 y la tabla 2 se recomiendan como guía para ajustar la dosis para los días 8 y 15 de los ciclos de 28 días.
|
Mor |
Tabla 1 ificaciones de la dosis por toxicidad hematológica los días 8 y 15 de cualquier ciclo | ||
|
Grado |
Recuento de plaquetas |
CAN |
Modificación de la dosis |
|
1-2 |
LIN* - 50 x 109/l |
LIN - 1,0 x 109/l |
Sin cambios en la dosis o pauta. |
|
3 |
<50 - 25 x 109/l |
<1,0 - 0,5 x 109/l |
Retrasar el tratamiento hasta la recuperación de un recuento de plaquetas >50 x 109/l y CAN** >1,0 x 109/l. |
|
4 |
<25 x 109/l |
<0,5 x 109/l |
Retrasar el tratamiento hasta la recuperación de un recuento de plaquetas >50 x 109/l y CAN** >1,0 x 109/l. Reducir la dosis en un 20 %. |
|
* LIN: Límite inferior del intervalo normal. ** CAN: Cifra absoluta de neutrófilos. | |||
|
Tabla 2 Modificaciones del tratamiento para toxicidades no hematológicas | |
|
Toxicidad |
Modificación |
|
Cualquier toxicidad no cardíaca de grado 3 o 4 relacionada con el fármaco salvo náuseas o vómitos. |
Retrasar el tratamiento hasta la recuperación del grado 1. Reducir la dosis en un 20 %. |
|
Cualquier toxicidad cardiovascular de grado 3 o 4 de la NYHA* o reducción persistente de la FEVI* *. |
Retrasar el tratamiento y vigilar hasta la recuperación. Contemplar la interrupción por una reducción persistente de la FEVI** >15 % del valor basal. |
|
* NYHA: New York Heart Association (Asociación de Cardiología de Nueva Y ork). ** FEVI: fracción de eyección del ventrículo izquierdo. | |
Poblaciones especiales Población pediátrica
No se ha establecido todavía la seguridad y eficacia de Pixuvri en niños menores de 18 años. No se dispone de datos.
Pacientes de edad avanzada
No es necesario un ajuste específico de la dosis en pacientes de edad avanzada (>65 años).
Insuficiencia renal
No se ha establecido la seguridad y eficacia de Pixuvri en pacientes con insuficiencia renal. Los pacientes con creatinina sérica >1,5 x límite superior del intervalo normal (LSN) se excluyeron del estudio aleatorizado. Por lo tanto, Pixuvri se debe utilizar con precaución en pacientes con insuficiencia renal.
Pacientes con insuficiencia hepática
No se ha establecido la seguridad y eficacia de Pixuvri en pacientes con insuficiencia hepática. Pixuvri se debe utilizar con precaución en pacientes con insuficiencia hepática leve o moderada. No se recomienda su uso en pacientes con insuficiencia hepática excretora grave (ver sección 4.3).
Pacientes con mal estado funcional
Actualmente no hay información sobre la seguridad y eficacia de los pacientes con mal estado funcional (ECOG > 2). Hay que tener precaución al tratar a estos pacientes.
Forma de administración
Pixuvri es solo para uso intravenoso. No se ha establecido la seguridad del uso intratecal.
Pixuvri está indicado para su administración en perfusión intravenosa lenta utilizando un filtro en línea (durante un mínimo de 60 minutos) solo después de su reconstitución con 5 ml de cloruro sódico de 9 mg/ml (0,9 %) solución inyectable y tras su posterior dilución con cloruro sódico de 9 mg/ml (0,9 %) solución inyectable hasta un volumen final de 250 ml.
Para obtener instrucciones sobre la reconstitución y dilución del medicamento antes de su administración, ver sección 6.6.
4.3 Contraindicaciones
- Hipersensibilidad a la pixantrona dimaleato o a alguno de los excipientes incluidos en la sección
6.1.
- Inmunización con vacunas de virus vivos.
- Depresión profunda de la médula ósea.
- Alteración grave de la función hepática.
4.4 Advertencias y precauciones especiales de empleo
Todo tratamiento inicial con Pixuvri debe ir precedido de una cuidadosa evaluación basal de los recuentos sanguíneos, los niveles séricos de bilirrubina total, los niveles séricos de creatinina total y la función cardíaca, medida por la fracción de eyección del ventrículo izquierdo (FEVI).
Mielodepresión
Se puede producir una mielodepresión intensa. Los pacientes tratados con Pixuvri son propensos a experimentar mielodepresión (neutropenia, leucopenia, anemia, trombocitopenia y linfopenia), cuya manifestación predominante es la neutropenia. Con la dosis y la posología recomendadas, la neutropenia suele ser transitoria, alcanzando su punto más bajo en los días 15-22 después de la administración en los días 1, 8 y 15; la recuperación suele producirse el día 28.
Es necesario controlar cuidadosamente los recuentos sanguíneos, incluidos los leucocitos, eritrocitos, plaquetas y recuento absoluto de neutrófilos. Se pueden utilizar factores de crecimiento hematopoyético recombinantes de acuerdo con las directrices institucionales o de la Sociedad Europea de Oncología Médica (ESMO). Se debe contemplar la modificación de la dosis (ver sección 4.2).
Cardiotoxicidad
Se pueden producir cambios de la función cardíaca, entre ellos disminución de la FEVI o insuficiencia cardíaca congestiva (ICC) mortal, durante el tratamiento con Pixuvri o después de él.
La enfermedad cardiovascular activa o latente, el tratamiento previo con antraciclinas o antraquinonas, la radioterapia previa o concurrente en la zona del mediastino o el uso concurrente de otros medicamentos cardiotóxicos pueden incrementar el riesgo de toxicidad cardíaca. Se puede producir toxicidad cardíaca con Pixuvri independientemente de la presencia de factores de riesgo cardíaco.
Los pacientes con cardiopatía o con factores de riesgo como un valor basal de la FEVI <45 % por ventriculografía nuclear (MUGA), alteraciones cardiovasculares clínicamente significativas (de grado 3 o 4 de la New York Heart Association [NYHA]), infarto de miocardio en los últimos 6 meses, arritmia grave, hipertensión no controlada, angina de pecho no controlada, o dosis acumuladas anteriores de doxorrubicina o su equivalente que superen los 450 mg/m2 se deben someter a una consideración cuidadosa de la relación riesgo/beneficio antes de recibir tratamiento con Pixuvri.
Se debe controlar la función cardíaca antes de iniciar un tratamiento con Pixuvri, y después periódicamente. Si se demuestra toxicidad cardíaca durante el tratamiento, se debe evaluar la relación riesgo/beneficio del tratamiento continuado con Pixuvri.
Cáncer secundario
El desarrollo de un cáncer hematológico, como la leucemia mielógena aguda (LMA) secundaria o el síndrome mielodisplásico (SMD), es un riesgo reconocido asociado con el tratamiento con antraciclinas y otros inhibidores de la topoisomerasa II. La aparición de cánceres secundarios, incluyendo LMA y SMD, puede ocurrir durante o después del tratamiento con Pixuvri.
Infección
Durante los ensayos clínicos se han registrado infecciones, entre ellas neumonía, celulitis, bronquitis y sepsis (ver sección 4.8). Las infecciones se han asociado con hospitalización, choque séptico y la muerte. Los pacientes con neutropenia son más propensos a las infecciones, aunque en los estudios clínicos no hubo aumento de la incidencia de infecciones atípicas de difícil tratamiento, tales como las infecciones micóticas sistémicas o las infecciones por microorganismos oportunistas como Pneumocystis jiroveci.
No se debe administrar Pixuvri a pacientes con una infección activa grave ni a pacientes con antecedentes de infecciones recurrentes o crónicas o con afecciones subyacentes que puedan predisponerles aún más a una infección grave.
Síndrome de lisis tumoral
La pixantrona puede inducir hiperuricemia como consecuencia del extenso catabolismo de purinas que acompaña a la lisis rápida de las células neoplásicas (síndrome de lisis tumoral) inducida por el fármaco y puede provocar desequilibrios electrolíticos, que pueden ocasionar daños renales. Hay que evaluar los niveles sanguíneos de ácido úrico, potasio, fosfato de calcio y creatinina después del tratamiento en pacientes con alto riesgo de lisis tumoral (LDH elevada, alto volumen tumoral, altos niveles basales de ácido úrico o de fosfato sérico). La hidratación, la alcalinización de la orina y la profilaxis con alopurinol u otros agentes para prevenir la hiperuricemia pueden minimizar las posibles complicaciones del síndrome de lisis tumoral.
Inmunización
La inmunización puede ser ineficaz cuando se administra durante el tratamiento con Pixuvri. La inmunización con vacunas de virus vivos está contraindicada debido a la inmunodepresión asociada con el tratamiento con Pixuvri (ver sección 4.3).
Extravasación
Si se produce una extravasación, la administración debe interrumpirse inmediatamente y reiniciarse en otra vena. Las propiedades no vesicantes de Pixuvri reducen al mínimo el riesgo de una reacción local tras la extravasación.
Prevención de reacciones de fotosensibilidad
La fotosensibilidad es un posible riesgo basado en datos preclínicos in vitro e in vivo y no se han registrado casos confirmados en el programa de ensayos clínicos. Como medida de precaución, se debe recomendar a los pacientes que utilicen estrategias de protección solar, entre ellas la ropa protectora del sol y el uso de protector solar. Como la mayor parte de las reacciones de fotosensibilidad inducidas por medicamentos se deben a longitudes de onda del intervalo UV-A, se recomienda un protector solar con fuerte absorción de UV-A.
Pacientes que siguen una dieta con restricción de sodio
Este medicamento contiene aproximadamente 1.000 mg (43 mmol) de sodio por dosis, después de la dilución. Deberá tenerse en cuenta en el tratamiento de pacientes con dietas pobres en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han registrado interacciones medicamentosas en seres humanos y no se han realizado ensayos de interacción entre fármacos en seres humanos.
Estudios de inhibición in vitro
Los estudios in vitro con las isoformas más comunes del citocromo P450 humano (entre ellas CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 y 3A4) han mostrado una posible inhibición de tipo mixto del CYP1A2 y del CYP2C8 que podría tener relevancia clínica. No se observó ninguna otra interacción significativa clínicamente relevante con los CYP450.
Teofilina: cuando se coadministra este medicamento de estrecho índice terapéutico, que se metaboliza principalmente por el CYP1A2, existe la inquietud teórica de que pueda aumentar la concentración de este sustrato, ocasionando toxicidad teofilínica. Hay que controlar cuidadosamente los niveles de teofilina en las semanas inmediatamente posteriores a la iniciación del tratamiento concurrente con Pixuvri.
La warfarina se metaboliza parcialmente por el CYP1A2; por lo tanto, existe inquietud teórica con respecto a la coadministración de este medicamento y el efecto que la inhibición de su metabolismo pueda tener sobre su acción prevista. Hay que controlar los parámetros de la coagulación, especialmente el índice internacional normalizado (IIN), en los días inmediatamente posteriores a la iniciación del tratamiento concurrente con Pixuvri.
La amitriptilina, el haloperidol, la clozapina, el ondansetrón y el propranolol se metabolizan por el CYP1A2, y por lo tanto existe la inquietud teórica de que la coadministración de Pixuvri pueda incrementar los niveles sanguíneos de este medicamento.
Aunque no se ha podido establecer un riesgo de inhibición de la pixantrona hacia el CYP2C8, se debe tener precaución cuando se coadministren sustancias que se metabolizan principalmente a través del CYP2C8, como la repaglinida, la rosiglitazona o elpaclitaxel, p. ej. mediante una vigilancia cuidadosa de los efectos secundarios.
Basándose en los estudios in vitro, se halló que la pixantrona era un sustrato para las proteínas transportadoras de membrana P-gp/BRCP y OCT1, y los agentes que inhiben estos transportadores tienen potencial para disminuir la captación hepática y la eficacia de excreción de la pixantrona. Hay que controlar estrechamente los recuentos sanguíneos cuando se coadministra con agentes que inhiben a estos transportadores, como la ciclosporina A o el tacrolimus, comúnmente utilizados para controlar la enfermedad de injerto contra huésped crónica, y los agentes anti-VIH ritonavir, saquinavir o nelfinavir.
Además, hay que tener precaución cuando se coadministra pixantrona continuamente con inductores del transporte de flujo de salida, tales como la rifampicina, la carbamazepina y los glucocorticoides, ya que la excreción de la pixantrona podría aumentar, con la consiguiente disminución de la exposición sistémica.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Se debe recomendar a las mujeres en edad fértil y a sus parejas que eviten el embarazo.
Las mujeres y los hombres deben utilizar métodos anticonceptivos efectivos durante el tratamiento y hasta 6 meses después de finalizarlo.
Embarazo
No hay datos relativos al uso de pixantrona en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3).
No se recomienda utilizar Pixuvri durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Lactancia
Se desconoce si Pixuvri/metabolitos se excretan en la leche materna.
No se puede excluir el riesgo en recién nacidos/niños.
Debe interrumpirse la lactancia durante el tratamiento con Pixuvri.
Fertilidad
Después de administraciones repetidas de Pixuvri en dosis tan bajas como 0,1 mg/kg/día, se detectó una atrofia testicular dependiente de la dosis en perros. Este efecto no se ha evaluado en seres humanos. Al igual que con otros agentes de la clase general de los agentes perjudiciales para el ácido desoxirribonucleico (ADN), Pixuvri puede asociarse con alteraciones de la fertilidad. Aunque no se ha determinado su efecto sobre la fertilidad, se debe recomendar por precaución a los pacientes varones el uso de métodos anticonceptivos (preferiblemente de barrera) durante el tratamiento y durante un plazo de 6 meses después del tratamiento para permitir la maduración de nuevos espermatozoides. Para evitar el riesgo de infertilidad a largo plazo, debe contemplarse el almacenamiento del esperma.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Se desconoce la influencia de Pixuvri sobre la capacidad de conducir y utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
La seguridad de Pixuvri se evaluó en 407 pacientes.
La toxicidad más frecuente es la depresión de la médula ósea, especialmente de la línea de los neutrófilos. Aunque la incidencia de depresión medular grave con consecuencias clínicas es relativamente baja, se controló estrechamente a los pacientes tratados con Pixuvri mediante recuentos sanguíneos frecuentes, particularmente para descartar la neutropenia. La incidencia de infecciones graves fue baja y no se observaron infecciones oportunistas asociadas con la inmunodepresión.
Aunque la aparición de toxicidad cardíaca manifestada por ICC parece ser menor de lo que cabría esperar con medicamentos relacionados, tales como las antraciclinas, se recomienda el control de la FEVI, ya sea por exploraciones MUGA o ecográficas, para evaluar la cardiotoxicidad subclínica. La experiencia con pixantrona se limita a pacientes con FEVI > 45 %, y la mayoría de los pacientes tenían valores >50 %. La experiencia de la administración de Pixuvri a pacientes con un deterioro cardíaco más significativo es limitada, y solo debe llevarse a cabo en el contexto de un ensayo clínico. Otras reacciones adversas que se observan con más frecuencia, como náuseas, vómitos y diarrea, fueron en general poco frecuentes, leves, reversibles, manejables y esperadas en pacientes tratados con agentes citotóxicos. Los efectos sobre la función hepática o renal fueron mínimos o inexistentes.
Lista tabulada de reacciones adversas
Las reacciones adversas (RA) notificadas con Pixuvri proceden de los datos finales de todos los estudios finalizados. Las RA se enumeran en la tabla 3 a continuación, por el sistema MedDRA de
clasificación de órganos y por frecuencia: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000), muy raras (<1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Tabla 3
Reacciones adversas notificadas en relación con Pixuvri en los estudios finalizados de Pixuvri por frecuencia
|
Sistema de clasificación de órganos |
Frecuencia |
Reacción adversa |
|
Infecciones e infestaciones |
Frecuentes |
Infección neutropénica, infección de las vías respiratorias, infección. |
|
Poco frecuentes |
Bronquitis, candidiasis, celulitis, herpes zóster, meningitis, infección de las uñas, infección fúngica oral, herpes oral, neumonía, gastroenteritis por Salmonella, choque séptico. | |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Poco frecuentes |
Progresión de una neoplasia. Cáncer secundario (incluyendo informes de LMA y SMD) |
|
Trastornos de la sangre y del sistema linfático* |
Muy frecuentes |
Neutropenia, leucopenia, linfopenia, anemia, trombocitopenia. |
|
Frecuentes |
Neutropenia febril, trastorno de la sangre. | |
|
Poco frecuentes |
Insuficiencia de la médula ósea, eosinofilia. | |
|
Trastornos del sistema inmunológico |
Poco frecuentes |
Hipersensibilidad al medicamento. |
|
Trastornos del metabolismo y de la nutrición |
Frecuentes |
Anorexia, hipofosfatemia. |
|
Poco frecuentes |
Hiperuricemia, hipocalcemia, hiponatremia. | |
|
Trastornos psiquiátricos |
Poco frecuentes |
Ansiedad, insomnio, trastornos del sueño. |
|
Trastornos del sistema nervioso |
Frecuentes |
Alteraciones del gusto, parestesia, dolor de cabeza, somnolencia. |
|
Poco frecuentes |
Mareo, letargo. | |
|
Trastornos oculares |
Frecuentes |
Conjuntivitis. |
|
Poco frecuentes |
Ojo seco, queratitis. | |
|
Trastornos del oído y del laberinto |
Poco frecuentes |
Vértigo. |
|
Trastornos cardíacos* |
Frecuentes |
Disfunción ventricular izquierda, trastorno cardíaco, insuficiencia cardíaca congestiva, bloqueo de rama, taquicardia. |
|
Poco frecuentes |
Arritmia. | |
|
Trastornos vasculares |
Frecuentes |
Palidez, coloración venosa, hipotensión. |
|
Poco frecuentes |
Flebopatía. | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Frecuentes |
Disnea, tos. |
|
Poco frecuentes |
Derrame pleural, neumonitis, rinorrea. |
Tabla 3
Reacciones adversas notificadas en relación con Pixuvri en los estudios finalizados de Pixuvri por frecuencia
|
Muy frecuentes |
Náuseas, vómitos. | |
|
Trastornos gastrointestinales |
Frecuentes |
Estomatitis, diarrea, estreñimiento, dolor abdominal, sequedad de boca, dispepsia. |
|
Poco frecuentes |
Esofagitis, parestesia oral, hemorragia rectal. | |
|
Trastornos hepatobiliares |
Poco frecuentes |
Hiperbilirrubinemia. |
|
Muy frecuentes |
Coloración de la piel, alopecia. | |
|
Trastornos de la piel y del tejido subcutáneo* |
Frecuentes |
Eritema, alteraciones de las uñas, prurito. |
|
Poco frecuentes |
Sudoración nocturna, petequias, exantema macular, úlceras cutáneas. | |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Frecuentes |
Dolor óseo. |
|
Poco frecuentes |
Artralgia, artritis, dolor de espalda, debilidad muscular, dolor torácico musculoesquelético, rigidez musculoesquelética, dolor de cuello, dolor en las extremidades. | |
|
Muy frecuentes |
Cromaturia. | |
|
Trastornos renales y urinarios |
Frecuentes |
Proteinuria, hematuria. |
|
Poco frecuentes |
Oliguria. | |
|
Trastornos del aparato reproductor y de la mama |
Poco frecuentes |
Erección espontánea del pene. |
|
Trastornos generales y alteraciones en el lugar de administración |
Muy frecuentes |
Astenia. |
|
Frecuentes |
Fatiga, inflamación de las mucosas, pirexia, dolor torácico, edema. | |
|
Poco frecuentes |
Escalofríos, frialdad en el lugar de la inyección, reacción local. | |
|
Exploraciones complementarias |
Frecuentes |
Aumento de la alanina-aminotransferasa, aumento de la aspartato-aminotransferasa, aumento de la fosfatasa alcalina sanguínea, aumento de la creatinina sanguínea. |
|
Poco frecuentes |
Bilirrubina en la orina, aumento del fósforo sanguíneo, aumento de la urea sanguínea, aumento de la y-glutamiltransferasa, aumento del recuento de neutrófilos, pérdida de peso. |
* RA comentadas a continuación.
Descripción de ciertas reacciones adversas
Toxicidades hematológicas y complicaciones de la neutropenia
Las toxicidades hematológicas han sido la toxicidad observada con más frecuencia pero, en general, se han tratado fácilmente con inmunoestimulantes y apoyo de transfusión en caso necesario. Aunque se produjo neutropenia de grado 3-4 en el ensayo aleatorizado con más frecuencia entre los receptores de Pixuvri, en la mayoría de los casos cursó sin complicaciones, no fue acumulativa y se asoció con una baja incidencia de neutropenia febril o infecciones. Es importante destacar que no se requirió apoyo con factor de crecimiento de forma rutinaria y las transfusiones de glóbulos rojos y plaquetas fueron poco frecuentes. (Ver sección 4.4).
Toxicidad cardíaca
En el estudio PIX 301, se produjo una disminución de la fracción de eyección en 13 pacientes (19,1 %) del grupo tratado con Pixuvri. En 11 pacientes tratados con Pixuvri, estos episodios fueron de grado 1-2 y en 2 pacientes fueron de grado 3; estos episodios fueron transitorios y no tuvieron relación con la dosis de Pixuvri. Se produjeron episodios de insuficiencia cardíaca (términos de MedDRA: insuficiencia cardíaca e insuficiencia cardíaca congestiva) en 6 pacientes (8,8 %) tratados con Pixuvri (2 pacientes con grado 1-2, 1 paciente con grado 3 y 3 pacientes con grado 5). Tres pacientes tratados con Pixuvri (4,4 %) presentaron taquicardia, arritmia, taquicardia sinusal o bradicardia.
Se recomienda una evaluación cardíaca basal mediante MUGA o ecografía, especialmente en pacientes con factores de riesgo de mayor toxicidad cardíaca. Se debe contemplar la repetición de las determinaciones de la FEVI mediante MUGA o ecografía en pacientes con factores de riesgo como una alta exposición acumulativa a antraciclinas previas o enfermedad cardíaca significativa preexistente. (Ver sección 4.4).
Otras toxicidades frecuentes
La coloración de la piel y la cromaturia son efectos conocidos relacionados con la administración de Pixuvri debido al color del compuesto (azul). La coloración de la piel desaparece por lo general en unos días o semanas a medida que se depura el medicamento.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No ha habido informes de sobredosis con Pixuvri.
Se han administrado dosis únicas de pixantrona de hasta 158 mg/m2 en ensayos clínicos de aumento de la dosis sin indicios de toxicidad relacionada con la dosis.
En caso de sobredosis, se recomienda tratamiento de apoyo.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: agentes antineoplásicos, antraciclinas y sustancias relacionadas. Código ATC: L01DB11
Mecanismo de acción
El principio activo de Pixuvri es la pixantrona, un aza-antraquinona citotóxica.
A diferencia de las antraciclinas (doxorrubicina y otras) y antraquinonas (mitoxantrona) autorizadas, la pixantrona solo es un inhibidor débil de la topoisomerasa II. Por otra parte, a diferencia de las antraciclinas o antraquinonas, la pixantrona alquila directamente el ADN, formando aductos estables de ADN y roturas de la cadena doble. Además, como incorpora un heteroátomo de nitrógeno en la estructura anular y no tiene grupos cetónicos, la pixantrona tiene menos potencial para la generación de especies reactivas del oxígeno, fijación de hierro y formación de metabolitos alcohólicos que se consideran responsables de la toxicidad cardíaca de las antraciclinas. Gracias a esta singular estructura, la pixantrona produjo una cardiotoxicidad mínima en modelos animales en comparación con la doxorrubicina o la mitoxantrona.
Un análisis completo retrospectivo poblacional de la FC/FD de los ensayos de fase 1 y de los regímenes combinados (fase 1/2) demostró que la supervivencia libre de progresión y la neutropenia de grado 2-3 estaban relacionadas con la exposición a Pixuvri.
Eficacia clínica y seguridad
Se evaluó la seguridad y la eficacia de Pixuvri como tratamiento de agente único en un ensayo multicéntrico aleatorizado, con control activo, en pacientes con LNH agresivo, recidivante o resistente al tratamiento, después de haber recibido al menos dos tratamientos previos (PIX301). En este ensayo se asignó al azar a 140 pacientes (1:1) para el tratamiento con Pixuvri o con una quimioterapia de agente único elegida por el investigador en el grupo de comparación. Los datos personales de los pacientes y las características basales de su enfermedad estaban bien equilibrados entre los grupos de tratamiento y no se observaron diferencias estadísticamente significativas. Para el estudio general, el promedio de edad de los pacientes fue de 59 años, un 61 % eran varones, un 64 % eran de raza blanca, un 76 % padecían una enfermedad en estadio III/IV de Ann Arbor al inicio del estudio, el 74 % tenía una puntuación del índice pronóstico internacional (IPI) >2 y un 60 % había recibido >3 quimioterapias anteriores. En el ensayo fundamental no se incluyó a pacientes con linfoma del manto. Los pacientes del PIX 301 debían haber sido sensibles a un tratamiento anterior con antraciclinas (RC o RP confirmada o sin confirmar).
Existen datos limitados en pacientes tratados anteriormente con rituximab (38 pacientes en el grupo tratado con Pixuvri y 39 pacientes en el grupo de comparación).
La respuesta tumoral fue evaluada por un panel central independiente de revisión con enmascaramiento de acuerdo con el grupo de trabajo internacional para estandarizar los criterios de respuesta para el LNH. Los pacientes tratados con Pixuvri mostraron una tasa significativamente mayor de respuestas completas y respuestas completas sin confirmar (RC/RCsc) y un índice de respuesta objetiva (IRO) más elevado, con respecto al grupo de comparación (ver la tabla 4).
|
Tabla 4 Resumen de respuestas según el panel de evaluación independiente (población por IDT) | ||||||
|
Final del tratamiento |
Final del ensayo | |||||
|
Pixuvri (n = 70) |
Fármaco de comparación (n = 70) |
Valor de P |
Pixuvri (n = 70) |
Fármaco de comparación (n = 70) |
Valor de P | |
|
RC/RCsc |
14 (20,0 %) |
4 (5,7 %) |
0,021 |
17 (24,3 %) |
5 (7,1 %) |
0,009 |
|
RC |
8 (11,4 %) |
0 (0 %) |
11 (15,7 %) |
0 (0,0 %) | ||
|
RCsc |
6 (8,6 %) |
4 (5,7 %) |
6 (8,6 %) |
5 (7,1 %) | ||
|
IRO (RC, RCsc y RP) |
26 (37,1 %) |
10 (14,3 %) |
0,003 |
28 (40,0 %) |
10 (14,3 %) |
0,001 |
|
Se utilizó la prueba exacta de Fisher para comparar las proporciones en los grupos de quimioterapia con Pixuvri y con el fármaco de comparación. | ||||||
Los pacientes tratados con Pixuvri mostraron una mejora del 40 % en la supervivencia libre de progresión con respecto a los pacientes tratados con los agentes de comparación, con una mediana de la SLP 2,7 meses mayor (cociente de riesgos instantáneos [CRI] = 0,60, p = 0,005 de orden logarítmico; ver la figura 1 más abajo).
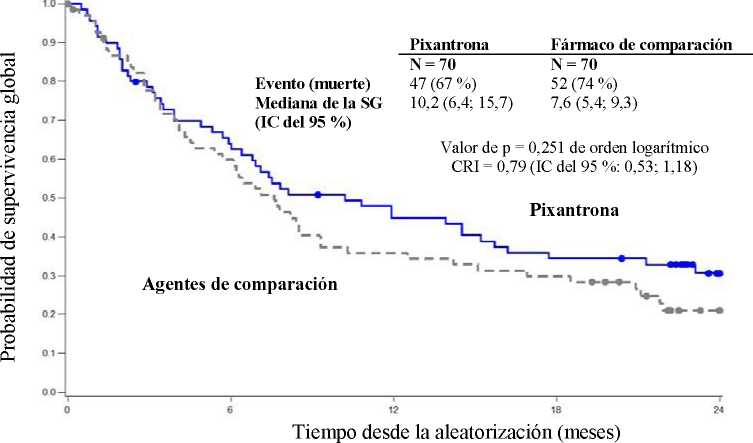
La mediana de la supervivencia global en pacientes tratados con Pixuvri fue 2,6 meses mayor que la de los pacientes tratados con el fármaco de comparación (CRI = 0,79, p = 0,25 de orden logarítmico; ver la figura 2 más abajo).
Figural
Supervivencia libre de progresión en PIX301, final del ensayo
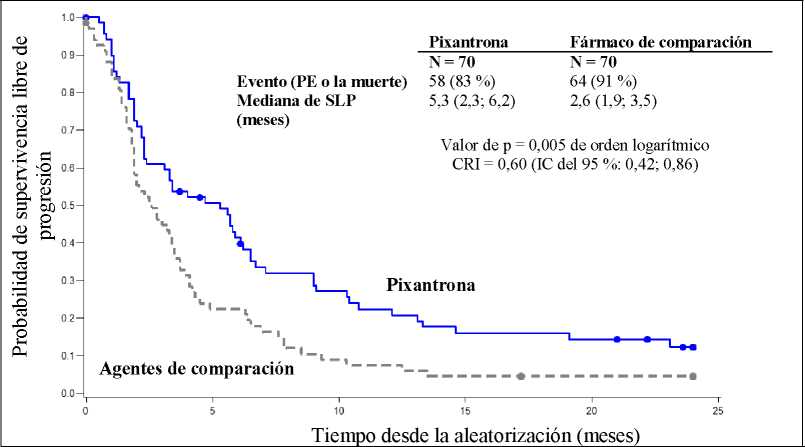
Figura 2
Supervivencia global en PIX 301, final del ensayo
Los resultados en los pacientes tratados previamente con rituximab todavía mostraban un beneficio del tratamiento superior con Pixuvri que con el fármaco de comparación en cuanto al índice de respuesta global (31,6 % con Pixuvri frente al 17,9 % con el fármaco de comparación) y a la mediana de supervivencia libre de progresión (3,3 meses con Pixuvri frente a 2,5 meses con el fármaco de comparación). Sin embargo, no se ha establecido el beneficio de Pixuvri cuando se utiliza como tratamiento de quinta línea o mayor en los pacientes refractarios al tratamiento anterior, y existen datos muy limitados en este grupo de pacientes.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido de la obligación de presentar los resultados de los ensayos con Pixuvri en los bebés desde el nacimiento hasta menos de 6 meses de edad, basándose en que el LNH no se produce en este subgrupo pediátrico específico.
La Agencia Europea de Medicamentos ha aplazado la obligación de presentar los resultados de los ensayos con Pixuvri en pacientes desde 6 meses hasta menos de 18 años de edad con LNH (ver sección 4.2 para obtener información sobre el uso pediátrico).
Este medicamento se ha autorizado con una “aprobación condicional”. Esta modalidad de aprobación significa que se espera obtener más información sobre este medicamento.
La Agencia Europea de Medicamentos revisará la información nueva de este medicamento al menos una vez al año, y esta Ficha Técnica o Resumen de las Características del Producto se actualizará cuando sea necesario.
5.2 Propiedades farmacocinéticas
Absorción
Tras la administración intravenosa, las concentraciones plasmáticas de pixantrona alcanzaron la concentración máxima al final de la perfusión y después disminuyeron de forma poliexponencial. La farmacocinética de Pixuvri fue independiente de la dosis en el intervalo de dosis de 3 mg/m2 a 105 mg/m2 y no se observaron grandes diferencias cuando el medicamento se administró como agente único o en estudios de combinación. La exposición media como agente único significó:
|
Dosis de Pixuvri (mg/m2) |
Número de pacientes |
ABC (0-24h) (ng.h/ml) |
|
33 |
3 |
982 ± 115 |
|
49 |
6 |
1727 ± 474 |
|
88 |
2 |
3811 |
A partir del análisis de los datos farmacocinéticos de la población, para una dosis diana registrada de 50 mg/m2 de pixantrona, la media de la exposición del ciclo de 28 días fue de 6.320 ng.h/ml (IC del 90 %, 5.990-6.800 ng.h/ml), para el ciclo de 3 dosis/4 semanas.
Distribución
Pixuvri tiene un gran volumen de distribución de 25,8 l y se fija aproximadamente en un 50 % a las proteínas plasmáticas.
Biotransformación
Los metabolitos acetilados son los principales productos de biotransformación de la pixantrona. Sin embargo, in vitro, la conversión de pixantrona en los metabolitos acetilados, ya sea por NAT1 o NAT2, fue muy limitada. En la orina humana, el compuesto se excretó principalmente sin cambios, y se hallaron cantidades muy pequeñas de metabolitos acetilados de fase I y II. Por lo tanto, el metabolismo no parece ser una vía de eliminación importante para la pixantrona. Los metabolitos acetilados eran farmacológicamente inactivos y metabólicamente estables.
Eliminación
La pixantrona tiene un aclaramiento plasmático total moderado o alto de 72,7 l/h y una excreción renal baja que constituye menos del 10 % de la dosis administrada en 0-24 horas. La semivida terminal varió desde 14,5 hasta 44,8 horas con una media de 23,3 ± 8,0 (n = 14, CV = 34 %) y una mediana de
21,2 horas. Debido a la limitada contribución del aclaramiento renal, el aclaramiento plasmático es principalmente no renal. Pixuvri puede metabolizarse en el hígado o excretarse en la bilis. Como el metabolismo parece ser limitado, la excreción biliar de pixantrona sin cambios puede ser la principal vía de eliminación. El aclaramiento hepático se aproxima al flujo de plasma hepático, lo que sugiere
un índice de extracción hepática elevado y, por tanto, la eliminación eficaz del principio activo matriz. La captación hepática de pixantrona está mediada posiblemente por transportadores OCT1 activos y la excreción biliar por P-gp y BCRP.
La pixantrona solo tuvo una capacidad débil o nula para inhibir la P-gp, BCRP, y el mecanismo de transporte de la BESB in vitro.
La pixantrona inhibió el transporte de metformina mediado por OCT1 in vitro, pero no se espera que inhiba el OTC1 in vivo a concentraciones clínicamente relevantes.
La pixantrona fue un inhibidor deficiente de los transportadores de captación de OATP1B1 y OATP1B3 in vitro.
Linealidad/no linealidad
La farmacocinética de la pixantrona resultó ser lineal en una amplia gama de dosis, desde 3 mg/m2 hasta 105 mg/m2.
Relación farmacocinética/farmacodinámica
Se ha observado una relación entre la exposición plasmática a la pixantrona y el recuento de neutrófilos.
5.3 Datos preclínicos sobre seguridad
Después de una administración intravenosa única de Pixuvri a 29 mg/kg y 38 mg/kg, se observaron muertes inmediatas en ratones (114 mg/m2, DL10). Se observaron disminuciones de los glóbulos blancos y rojos y alteraciones en la médula ósea, el bazo, los riñones y los testículos. Se registraron hallazgos similares en ratas y en perros a 116 mg/m2. En los perros, se produjo taquicardia y cambios electrocardiográficos (ECG) inmediatamente después del tratamiento.
En los estudios con dosis repetidas en ratones, ratas y perros, los principales hallazgos fueron mielotoxicidad, nefrotoxicidad (salvo en los perros) y daños testiculares.
En los perros, Pixuvri administrado entre 0,5 y 0,9 mg/kg durante seis ciclos no provocó mortalidad ni signos clínicos intensos, incluyendo cambios ECG o del peso corporal. Los machos eran más sensibles al tratamiento, en cuanto a la reducción de los recuentos de glóbulos blancos y plaquetas (reversible) y el agotamiento linfoide (bazo y timo), así como la marcada toxicidad para los órganos reproductores, como era de esperar en un agente citotóxico. A excepción de un aumento transitorio de la exposición en las hembras después del tercer ciclo, no hubo diferencias importantes en los parámetros farmacocinéticos. Los machos mostraron, sin embargo, una exposición ligeramente mayor que las hembras.
En los perros, el corazón no se vio afectado por el tratamiento, ya que no se observaron cambios ECG en diferentes momentos del tratamiento, ni se detectaron cambios cardíacos en la anatomía patológica macroscópica ni en la histopatología. De modo similar, tampoco la función ni la histología renal se vieron afectadas en los estudios de 4 y 26 semanas.
Se evaluó el potencial cardiotóxico de Pixuvri en comparación con dosis equiactivas de doxorrubicina y mitoxantrona en ratones sin tratamiento previo y pretratados con doxorrubicina. La pixantrona dimaleato hasta 27 mg/kg administrada dos veces por semana durante 4 semanas no indujo efectos cardiotóxicos, mientras que la mitoxantrona, como era de esperar, fue cardiotóxica a todas las dosis ensayadas (0,6, 1,6 y 1,5 mg/kg). Pixuvri indujo una nefropatía leve. Se demostró también la mínima cardiotoxicidad de Pixuvri con ciclos de tratamiento repetidos a las mismas dosis.
Los estudios de genotoxicidad confirmaron el potencial de efectos clastógenos en células de mamíferos in vitro e in vivo. Pixuvri fue mutagénico en la prueba de Ames, aumentó el número de aberraciones cromosómicas en linfocitos humanos y aumentó la frecuencia de micronúcleos in vivo.
Pixuvri provocó toxicidad materna y fetal en ratas y conejos, incluso a dosis tan bajas como 1,8 mg/kg administrados en los días 9-11 de gestación; las dosis mayores provocaron abortos y reabsorción embrionaria total. La embriotoxicidad se caracterizó por una reducción del peso medio del feto, malformaciones fetales y osificación fetal incompleta o retrasada. No se han llevado a cabo estudios a largo plazo en animales para establecer el potencial cancerígeno de Pixuvri. No se llevó a cabo ningún estudio de tolerancia local.
Pixuvri ha demostrado provocar efectos fototóxicos en linfocitos 3T3 in vitro.
En un estudio de unidades formadoras de colonias en ratones, la mielotoxicidad de Pixuvri y mitoxantrona administrados a su DL10 (pixantrona dimaleato 38 mg/kg y mitoxantrona 6,1 mg/kg) fue similar.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Cloruro sódico. Lactosa monohidrato.
Hidróxido sódico (para ajuste del pH).
Ácido clorhídrico (para ajuste del pH).
6.2 Incompatibilidades
Este medicamento no se debe mezclar con otros, excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez
Vial cerrado 5 años.
Solución reconstituida y diluida
Se ha demostrado la estabilidad química y física en uso durante 24 horas a temperatura ambiente (15 °C a 25 °C) y con exposición a la luz natural en bolsas de perfusión convencionales de polietileno (PE).
Desde el punto de vista microbiológico, el producto se debe utilizar inmediatamente. Si no se utiliza de inmediato, los tiempos de conservación en uso y las condiciones anteriores al uso son responsabilidad del usuario y normalmente no deberían superar las 24 horas entre 2 °C y 8 °C, a menos que la reconstitución y dilución se hayan realizado en condiciones asépticas controladas y validadas.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Para las condiciones de conservación tras la reconstitución y dilución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Vial de vidrio del tipo I con tapón de caucho de butilo gris con cierre de aluminio y tapa de plástico rojo que contiene 50 mg de pixantrona dimaleato equivalentes a 29 mg de pixantrona.
Envase de 1 vial.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Reconstitución y dilución
Antes de la reconstitución, comprobar visualmente el polvo liofilizado para descartar defectos anormales, como grietas, fusión o aspecto vítreo. Reconstituir asépticamente cada vial de 29 mg con 5 ml de cloruro sódico de 9 mg/ml (0,9 %) solución inyectable. El polvo liofilizado debe disolverse completamente en 60 segundos con agitación. Esto produce una solución azul oscuro con una concentración de pixantrona de 5,8 mg/ml.
Extraer de forma aséptica el volumen necesario para la dosis requerida (basada en la concentración de 5,8 mg/ml) y transferirlo a una bolsa de perfusión de 250 ml de cloruro sódico de 9 mg/ml (0,9 %) solución inyectable. La concentración final de pixantrona en la bolsa de perfusión debe ser inferior a 580 microgramos/ml basada en la aportación de medicamento reconstituido. No se ha determinado la compatibilidad con otros diluyentes. Después de la transferencia, mezclar bien el contenido de la bolsa de perfusión. La mezcla debe ser una solución transparente de color azul oscuro.
Se deben utilizar filtros en línea de polietersulfona de 0,2 micras de tamaño de poro durante la administración de la solución diluida de Pixuvri.
Pixuvri es un agente citotóxico. Evitar el contacto con los ojos y la piel. Utilizar guantes, máscaras y gafas protectoras al manipular Pixuvri y durante los procedimientos de descontaminación.
Precauciones especiales de eliminación
Pixuvri es para un solo uso. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, incluyendo los materiales utilizados para la reconstitución, dilución y administración, se realizará de acuerdo con la normativa local aplicable a los agentes citotóxicos.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
CTI Life Sciences Limited Highlands House Basingstoke Road Spencers Wood, Reading Berkshire RG7 1NT Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/764/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 10 mayo 2012 Fecha de la última renovación: 22 marzo 2016
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POST-AUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN CONDICIONAL
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes Catalent UK Packaging Limited
Lancaster Way, Wingates Industrial Estate Westhoughton, Bolton, Lancashire BL5 3XX Reino Unido
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos actualizados de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el plan de gestión de riesgos, especialmente como resultado de nueva
información disponible que pueda conllevar cambios relevantes en el perfil de beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POSTAUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN CONDICIONAL
Al ser esta una autorización de comercialización condicional y de según lo que establece el Artículo 14(7) del Reglamento (CE) 726/2004, el TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
Descripción |
Fecha límite |
|
Llevar a cabo un ensayo aleatorizado controlado de Fase 3 (PIX306) de pixantrona-rituximab frente a gemcitabina-rituximab en pacientes con LNH de linfocitos B agresivo, que no respondieron al tratamiento de primera línea con CHOP-R y no son elegibles para un autotrasplante de progenitores hematopoyéticos (ATPH, 2.a línea) o no respondieron al ATPH (3.a o 4.a línea). Se presentará un informe del ensayo clínico. |
31 de diciembre de 2018 |
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR ESTUCHE
1. NOMBRE DEL MEDICAMENTO
Pixuvri 29 mg polvo para concentrado para solución para perfusión pixantrona
2. PRINCIPIO(S) ACTIVO(S)
Cada vial contiene dimaleato de pixantrona equivalente a 29 mg de pixantrona. Después de la reconstitución, cada ml de concentrado contiene dimaleato de pixantrona equivalente a 5,8 mg de pixantrona.
3. LISTA DE EXCIPIENTES
Lactosa monohidrato, cloruro sódico, ácido clorhídrico, hidróxido sódico. Contiene sodio, para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para concentrado para solución para perfusión. Envase de 1 vial.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Reconstituir y diluir antes de su uso.
Leer el prospecto antes de utilizar este medicamento. Para uso intravenoso.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Citotóxico: manejar con precaución.
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera (entre 2 °C y 8 °C).
Conservar el vial en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
CTI Life Sciences Limited Highlands House Basingstoke Road Spencers Wood, Reading Berkshire RG7 1NT Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/764/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille.
INFORMACIÓN QUE DEBE FIGURAR EN EL ACONDICIONAMIENTO PRIMARIO VIAL
1. NOMBRE DEL MEDICAMENTO
Pixuvri 29 mg polvo para concentrado para solución para perfusión pixantrona
2. PRINCIPIO(S) ACTIVO(S)
Cada vial contiene dimaleato de pixantrona equivalente a 29 mg de pixantrona. Después de la reconstitución, cada ml de concentrado contiene dimaleato de pixantrona equivalente a 5,8 mg de pixantrona.
3. LISTA DE EXCIPIENTES
Lactosa monohidrato, cloruro sódico, ácido clorhídrico, hidróxido sódico. Contiene sodio, para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para concentrado para solución para perfusión.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Reconstituir y diluir antes de su uso.
Leer el prospecto antes de utilizar este medicamento. Para uso intravenoso.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA ADVERTENCIA ESPECIAL, SI ES NECESARIO
Citotóxico: manejar con precaución.
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera (entre 2 °C y 8 °C).
Conservar el vial en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
CTI Life Sciences Limited Highlands House Basingstoke Road Spencers Wood, Reading Berkshire RG7 1NT Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/764/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille.
B. PROSPECTO
Prospecto: información para el usuario
Pixuvri 29 mg polvo para concentrado para solución para perfusión
Pixantrona
"VEste medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Pixuvri y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Pixuvri
3. Cómo usar Pixuvri
4. Posibles efectos adversos
5. Conservación de Pixuvri
6. Contenido del envase e información adicional
1. Qué es Pixuvri y para qué se utiliza
Pixuvri pertenece a un grupo farmacoterapéutico de medicamentos conocidos como «agentes antineoplásicos». Se utilizan para tratar el cáncer.
Pixuvri se utiliza para el tratamiento de pacientes adultos con linfomas no hodgkinianos agresivos, multirrecidivantes o resistentes al tratamiento. Pixuvri mata las células cancerosas mediante su unión al ADN, ocasionando la muerte celular. Se utiliza para los pacientes cuyo cáncer no responde o ha reaparecido después de haber recibido otros tratamientos de quimioterapia.
2. Qué necesita saber antes de empezar a usar Pixuvri No use Pixuvri:
- Si es alérgico a la pixantrona o a cualquiera de los demás componentes de este medicamento
- (incluidos en la sección 6).
- Si ha recibido recientemente una vacuna.
- Si se le ha indicado que tiene un recuento bajo y persistente a largo plazo de glóbulos rojos, glóbulos blancos y plaquetas.
- Si padece problemas muy graves del hígado.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar Pixuvri:
- Si se le ha indicado que su recuento de glóbulos blancos es muy bajo.
- Si padece una enfermedad del corazón o tensión arterial alta no controlada, especialmente si se
le ha indicado alguna vez que padecía insuficiencia cardíaca o si ha sufrido un ataque al corazón en los últimos seis meses.
- Si sufre una infección.
- Si alguna vez ha recibido tratamiento contra el cáncer.
- Si sigue una dieta específica con restricción de sodio.
- Si está tomando otros medicamentos que pudieran interaccionar con Pixuvri (ver «Uso de
- Pixuvri con otros medicamentos» más adelante).
Sensibilidad de la piel a la luz solar
Durante el tratamiento con pixantrona, se debe minimizar o evitar la exposición a la luz solar natural o artificial (cámaras de bronceado o tratamiento con rayos UVA/B). Si va a estar expuesto a la luz solar, debe usar ropa protectora del sol y crema de protección solar que absorba fuertemente la radiación UV-A.
Niños y adolescentes
No administre este medicamento a niños y adolescentes menores de 18 años porque no hay información disponible sobre el tratamiento con Pixuvri en este grupo de pacientes.
Toma de Pixuvri con otros medicamentos
Informe a su médico si está tomando, ha tomado recientemente o podría tomar cualquier otro medicamento. Esto es extremadamente importante ya que el uso de más de un medicamento al mismo tiempo puede fortalecer o debilitar su efecto. Pixuvri no se debe utilizar con otros medicamentos a menos que su médico le haya indicado que es seguro hacerlo.
En particular, no deje de informar a su médico si está utilizando, o ha utilizado recientemente, alguno de los siguientes medicamentos:
Informe a su médico si toma medicamentos tales como:
- Warfarina para prevenir la formación de coágulos de sangre.
- Teofilina para el tratamiento de enfermedades pulmonares como el enfisema o el asma.
- Amitriptilina para tratar la depresión.
- Olanzapina, clozapina para el tratamiento de la esquizofrenia o del trastorno bipolar.
- Haloperidol para tratar la ansiedad y el insomnio.
- Ondansetrón para prevenir las náuseas y los vómitos durante la quimioterapia.
- Propranolol para tratar la tensión arterial elevada.
Toma de Pixuvri con alimentos y bebidas
No tiene que cambiar su dieta después del tratamiento con Pixuvri a menos que lo indique su médico. Embarazo, lactancia y fertilidad
Pixuvri no se debe administrar a mujeres embarazadas ya que puede causar daño a los fetos. Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
Hay que tomar precauciones anticonceptivas adecuadas cuando se recibe Pixuvri y hasta 6 meses después del tratamiento. Esto se aplica a las mujeres en edad fértil y los hombres que reciben Pixuvri y tienen capacidad para engendrar un hijo.
No amamante mientras esté en tratamiento con Pixuvri.
Conducción y uso de máquinas
Se desconoce la influencia de Pixuvri sobre la capacidad de conducir y utilizar máquinas
Información para los pacientes con una dieta baja en sal
Los pacientes con dietas pobres en sodio deben tener en cuenta que este medicamento contiene aproximadamente 1.000 mg (43 mmol) de sodio por dosis, después de la dilución.
3. Cómo usar Pixuvri Cuánto Pixuvri se administra
Las cantidades (dosis) de Pixuvri que se administrarán dependerán de su superficie corporal en metros cuadrados (m2). Esto está determinado por su estatura y peso. También se tendrán en cuenta los resultados de los análisis de sangre y su enfermedad. La dosis recomendada es de 50 mg/m2. Si es necesario, su médico le ajustará la dosis durante el tratamiento.
Su médico le realizará algunas pruebas antes de administrarle Pixuvri.
Con qué frecuencia se administra Pixuvri
Pixuvri se administra los días 1, 8 y 15 de cada ciclo de 28 días hasta un máximo de 6 ciclos.
Antes de administrar la perfusión se le pueden dar medicamentos para prevenir o reducir las posibles reacciones a Pixuvri, como los medicamentos para prevenir las náuseas.
Cómo se administra Pixuvri
Pixuvri se administra a través de un goteo en una vena (por perfusión intravenosa). Esto lo hará una enfermera o un médico.
Cuánto durará la perfusión
Durará aproximadamente una hora si no se indica lo contrario.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Reacciones a la perfusión
Se puede producir raramente dolor/enrojecimiento en el lugar de la inyección durante la perfusión de Pixuvri. Informe de inmediato a la persona que le administra la perfusión si siente dolor o si el lugar de la inyección enrojece. Tal vez haya que reducir la velocidad de perfusión o detenerla. Cuando desaparezcan o mejoren estos síntomas, la perfusión puede continuar.
Pixuvri tiene un color azul intenso y durante varios días después de recibir Pixuvri, la piel y los ojos pueden adquirir una coloración azulada, y la orina puede tener una coloración azulada. La coloración de la piel desaparece por lo general en pocos días o semanas a medida que se depura el medicamento.
Infecciones
Informe a su médico si presenta cualquier síntoma de una infección (por ejemplo, fiebre, escalofríos, dificultad para respirar, tos, úlceras en la boca, dificultad para tragar o diarrea intensa) después del tratamiento con Pixuvri. Puede contraer infecciones más fácilmente después de la administración de Pixuvri.
Corazón
Existe la posibilidad de que la función de bombeo del corazón pueda disminuir como consecuencia del tratamiento o incluso se podría presentar una enfermedad grave llamada insuficiencia cardíaca, sobre todo si la función del corazón ya estaba deteriorada al principio del tratamiento con Pixuvri. Su médico vigilará el funcionamiento del corazón si hay algún signo o síntoma de que se vea afectado.
Informe a su médico de inmediato si advierte alguno de estos efectos adversos
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
- Náuseas, vómitos.
- Coloración de la piel.
- Adelgazamiento o pérdida de pelo.
- Coloración anormal de la orina.
- Debilidad física.
- Bajo número de glóbulos blancos, bajo número de glóbulos rojos (anemia) y bajo número de plaquetas en la sangre (puede requerir transfusión).
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas):
- Infecciones, tales como infección pulmonar, infecciones de la piel, infecciones con recuentos bajos de glóbulos blancos, candidiasis oral.
- Fiebre.
- Alteraciones del gusto.
- Sensaciones anormales de la piel tales como entumecimiento, hormigueo, pinchazos
- (parestesia).
- Dolor de cabeza.
- Somnolencia.
- Cansancio.
- Inflamación de los ojos (conjuntivitis).
- Diarrea.
- Dolor en el abdomen.
- Inflamación o ulceración de la garganta y la boca.
- Boca seca, estreñimiento, indigestión, pérdida del apetito.
- Cambios en la piel como enrojecimiento y picor de la piel, cambios en las uñas.
- Daño en el corazón, disminución de la capacidad del corazón para bombear sangre, bloqueo de las señales eléctricas en el corazón, ritmo cardíaco irregular o rápido.
- Baja tensión arterial.
- Coloración de las venas, piel pálida.
- Falta de aliento, tos.
- Sangre en la orina.
- Exceso de proteínas en la orina.
- Hinchazón de piernas o tobillos u otras partes del cuerpo.
- Dolor de huesos.
- Dolor torácico.
- Bajos niveles de fosfato en la sangre.
- Análisis de sangre anormales de la función hepática o renal.
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 1.000 personas):
- Infecciones graves como choque séptico, bronquitis, neumonía, candidiasis, celulitis, meningitis, gastroenteritis.
- Infecciones víricas como el herpes zóster o reactivación de otros virus como el herpes de la boca.
- Nerviosismo, insomnio.
- Pérdida de energía.
- Mareo, vértigo.
- Sequedad del ojo.
- Entumecimiento de la boca.
- Infección de la córnea.
- Alergia al medicamento.
- Disminución del nivel de calcio y sodio en la sangre, aumento del nivel de ácido úrico en la sangre.
- Inflamación o acumulación de líquido alrededor de los pulmones.
- Moqueo nasal.
- Hemorragias, tales como hemorragia intestinal, manchas moradas en el cuerpo debidas a la rotura de vasos sanguíneos.
- Irritación de las venas.
- Sudores nocturnos.
- Latido irregular del corazón.
- Erecciones espontáneas.
- Erupciones o ulceración de la piel.
- Dolor, hinchazón, debilidad, rigidez de las articulaciones o músculos.
- Disminución del caudal urinario.
- Pérdida de peso.
- Aumento de bilirrubina en la sangre o en la orina.
- Inflamación del esófago.
- Dolor de cuello, espalda, extremidades.
- Infección de las uñas.
- Progresión de una neoplasia (tumor).
- Nuevos tipos de cáncer de la médula ósea o de la sangre, como la leucemia mielógena aguda (LMA) o el síndrome mielodisplásico (SMD)
- Insuficiencia de la médula ósea.
- Aumento de los eosinófilos en la sangre.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Pixuvri
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta del vial y en la caja después de «CAD». La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2 °C y 8 °C).
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Pixuvri no contiene nada para evitar el crecimiento de bacterias y se recomienda, por lo tanto, utilizarlo inmediatamente después de su reconstitución. Si no se utiliza de inmediato, los tiempos de conservación en uso y las condiciones anteriores al uso son responsabilidad del usuario y normalmente no deben superar las 24 horas entre 2 °C y 8 °C.
La solución de pixantrona reconstituida es estable durante un máximo de 24 horas a temperatura ambiente (15°C a 25°C) en bolsas de perfusión convencionales.
Pixuvri es para un solo uso. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, incluyendo los materiales utilizados para la reconstitución, dilución y administración, se realizará de acuerdo con la normativa local.
6. Contenido del envase e información adicional Composición de Pixuvri
- El principio activo es la pixantrona. Cada vial contiene 50 mg de pixantrona dimaleato (equivalentes a 29 mg de pixantrona). Los demás componentes son lactosa monohidrato, hidróxido sódico, ácido clorhídrico y cloruro sódico.
Aspecto del producto y contenido del envase
Pixuvri es un polvo para concentrado para solución para perfusión. Se presenta como un polvo de color azul oscuro envasado en viales que contienen 29 mg de pixantrona. Tamaño del envase: 1 vial.
Titular de la autorización de comercialización
CTI Life Sciences Limited Highlands House Basingstoke Road
Spencers Wood, Reading Berkshire RG7 1NT Reino Unido
Responsable de la fabricación
Catalent UK Packaging Limited Lancaster Way,Wingates Industrial Estate Westhoughton, Bolton,
Lancashire BL5 3XX Reino Unido
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien S.A. Servier Benelux N.V. Tél/Tel: +32 (0)2 529 43 11 |
Lietuva UAB “SERVIER PHARMA” Tel: +370 (5) 2 63 86 28 |
|
Etarapna CepBue MeguKaa EOOfl Tea.: +359 2 921 57 00 |
Luxembourg/Luxemburg S.A. Servier Benelux N.V. Tel: +32 (0)2 529 43 11 |
|
Ceská republika Servier s.r.o. Tel: +420 222 118 111 |
Magyarország Servier Hungaria Kft. Tel: +36 1 238 7799 |
|
Danmark CTI Life Sciences Limited Tlf: + 45 36 92 77 96 |
Malta GALEPHARMA Ltd Tel: +(356) 21 247 082 |
|
Deutschland CTI Life Sciences Limited Tel.: + 49 (0)6922 223384 |
Nederland Servier Nederland Farma B.V. Tel: + 31 (0)71 5246700 |
|
Eesti Servier Laboratories OÜ Tel:+ 372 664 5040 |
Norge CTI Life Sciences Limited Tlf: + 47 21 03 39 98 |
|
EXláóa IEPBIE EAAAI OAPMAKEYTIKH EnE TpA30 210 939 1000 |
Osterreich CTI Life Sciences Limited Tel: + 43 (0)19 287 896 |
|
España Laboratorios Servier S.L. Tel: + 34 91 748 96 30 |
Polska Servier Polska Sp. z o.o. Tel: + 48 (0) 22 594 90 00 |
|
France Les Laboratoires Servier Tél: + 33 (0)1 55 72 60 00 |
Portugal Servier Portugal, Lda Tel: + 351 21 312 20 00 |
|
Hrvatska Servier Pharma, d. o. o. Tel: + 385 (0)1 3016 222 |
Romania Servier Pharma SRL Tel: + 4 021 528 52 80 |
Slovenija
Servier Pharma d. o. o. Tel: + 386 (0)1 563 48 11
Ireland
Servier Laboratories (Ireland) Ltd. Tel: + 353 (0)1 663 8110
|
Ísland Servier Laboratories c/o Icepharma hf Sími: + 354 540 8000 |
Slovenská republika Servier Slovensko spol. s r.o. Tel: + 421 2 5920 41 11 |
|
Italia Servier Italia S.p.A. Tel: + 39 (06) 669081 |
Suomi/Finland CTI Life Sciences Limited Puh/Tel: + 358 9 23 195 429 |
|
Kúnpoq CA Papaellinas Ltd. Tr(k: + 357 22 741 741 |
Sverige CTI Life Sciences Limited Tel: + 46 (0) 850 33 48 19 |
|
Latvija SIA Servier Latvia Tel: + 371 67502039 |
United Kingdom CTI Life Sciences Limited Tel: + 44 (0)800 083 4014 |
Fecha de la última revisión de este prospecto: <mes AAAA>
Este medicamento se ha autorizado con una «aprobación condicional».
Esta modalidad de aprobación significa que se espera obtener más información de este medicamento. La Agencia Europea de Medicamentos revisará la información nueva de este medicamento al menos una vez al año y este prospecto se actualizará cuando sea necesario.
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
En la página web de la Agencia Europea de Medicamentos puede encontrarse este prospecto en todas las lenguas de la Unión Europea/Espacio Económico Europeo.
Esta información está destinada únicamente a profesionales del sector sanitario:
Instrucciones detalladas para los usuarios
LEA LAS INSTRUCCIONES COMPLETAS DE PREPARACIÓN ANTES DE LA RECONSTITUCIÓN
Precauciones especiales de empleo
Pixuvri es un medicamento contra el cáncer perjudicial para las células; hay que tener precaución en su manejo. Evitar el contacto con los ojos y la piel. Utilizar guantes, máscaras y gafas protectoras al manipularlo y durante los procedimientos de descontaminación. Si Pixuvri (polvo liofilizado o solución líquida reconstituida) entra en contacto con la piel, lavar la piel inmediatamente y enjuagar las membranas con abundante agua.
Reconstitución/preparación para administración intravenosa
Cada vial de un solo uso de Pixuvri contiene pixantrona dimaleato equivalente a 29 mg de pixantrona. Después de la reconstitución con 5 ml de cloruro sódico de 9 mg/ml (0,9 %) solución inyectable, cada ml de concentrado contiene dimaleato de pixantrona equivalente a 5,8 mg de pixantrona.
Antes de la reconstitución, comprobar visualmente el polvo liofilizado para descartar defectos anormales, como grietas, fusión o aspecto vítreo. Utilizando procedimientos estériles, reconstituir asépticamente cada vial de 29 mg con 5 ml de cloruro sódico de 9 mg/ml (0,9 %) solución inyectable.
El polvo debe disolverse completamente en 60 segundos con agitación. Esto produce una solución azul oscuro con una concentración de pixantrona de 5,8 mg/ml.
Utilizando procedimientos estériles, extraer el volumen necesario para la dosis requerida (basada en la concentración de 5,8 mg/ml) y transferirlo a una bolsa de perfusión de 250 ml de cloruro sódico de 9 mg/ml (0,9 %) solución inyectable. No se ha determinado la compatibilidad con otros diluyentes. Después de la transferencia, mezclar bien el contenido de la bolsa de perfusión. La mezcla debe ser una solución de color azul oscuro.
Se deben utilizar filtros en línea de polietersulfona de 0,2 micras de tamaño de poro durante la administración de la solución diluida de Pixuvri.
Condiciones de conservación en uso
Pixuvri no contiene nada para evitar el crecimiento de bacterias y se recomienda, por lo tanto, utilizarlo inmediatamente después de su reconstitución. Si no se utiliza de inmediato, los tiempos de conservación en uso y las condiciones anteriores al uso son responsabilidad del usuario y normalmente no deben superar las 24 horas entre 2°C y 8 °C.
La solución reconstituida y diluida es estable durante un máximo de 24 horas a temperatura ambiente (15°C a 25°C) y con exposición a la luz natural en bolsas de perfusión convencionales de polietileno (PE).
Precauciones especiales de eliminación y manipulación
Pixuvri es un agente citotóxico. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Los dispositivos y superficies contaminados accidentalmente con Pixuvri debe tratarse con una solución de hipoclorito sódico (100 pl de agua y 20 pl de hipoclorito sódico [7 ± 2 % de cloro disponible] para 0,58 mg de Pixuvri).
Los equipos tales como viales, jeringas y agujas utilizados para la administración de Pixuvri deben manejarse como residuos tóxicos.
34