Otezla 30Mg Comprimidos Recubiertos Con Pelicula
Información obsoleta, busque otroANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Otezla 10 mg comprimidos recubiertos con película Otezla 20 mg comprimidos recubiertos con película Otezla 30 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 10 mg de apremilast.
Cada comprimido recubierto con película contiene 20 mg de apremilast.
Cada comprimido recubierto con película contiene 30 mg de apremilast.
Excipiente con efecto conocido:
Cada comprimido recubierto con película contiene 57 mg de lactosa (como lactosa monohidrato). Cada comprimido recubierto con película contiene 114 mg de lactosa (como lactosa monohidrato). Cada comprimido recubierto con película contiene 171 mg de lactosa (como lactosa monohidrato).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
color rosa, de 8 mm de largo, color marrón, de 10 mm de color beige, de 12 mm de
Comprimido recubierto con película de 10 mg, con forma de rombo, de con “APR” grabado en una cara y “10” en la otra cara.
Comprimido recubierto con película de 20 mg, con forma de rombo, de largo, con “APR” grabado en una cara y “20” en la otra cara.
Comprimido recubierto con película de 30 mg, con forma de rombo, de largo, con “APR” grabado en una cara y “30” en la otra cara.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Artritis psoriásica
Otezla, solo o en combinación con Fármacos Antirreumáticos Modificadores de la Enfermedad (FAMEs), está indicado para el tratamiento de la artritis psoriásica (APs) activa en pacientes adultos que han tenido una respuesta inadecuada, o han presentado intolerancia al tratamiento previo con un FAME (ver sección 5.1).
Psoriasis
Otezla está indicado en el tratamiento de la psoriasis en placas crónica de moderada a grave en pacientes adultos que no han respondido o tienen contraindicado o no toleran otro tratamiento sistémico, incluyendo ciclosporina, metotrexato o psoraleno y luz ultravioleta A (PUVA).
4.2 Posología y forma de administración
El tratamiento con Otezla se debe iniciar por un médico especialista con experiencia en el diagnóstico y tratamiento de la psoriasis o de la artritis psoriásica.
Posología
La dosis recomendada de Otezla es de 30 mg dos veces al día por vía oral, por la mañana y por la noche, cada 12 horas aproximadamente, sin restricciones de alimentos. Es necesario un programa inicial de escalado de dosis como se muestra en la Tabla 1. Después del escalado inicial de la dosis, no es necesario un re-escalado de la dosis.
Tabla 1: Programa de escalado de dosis
|
Día 1 |
Día 2 |
Día 3 |
Día 4 |
Día 5 |
Día 6 y siguientes | |||||
|
a. m. |
a. m. |
p. m. |
a. m. |
p. m. |
a. m. |
p. m. |
a. m. |
p. m. |
a. m. |
p. m. |
|
10 mg |
10 mg |
10 mg |
10 mg |
20 mg |
20 mg |
20 mg |
20 mg |
30 mg |
30 mg |
30 mg |
Si el paciente se olvida una dosis, se debe tomar la siguiente dosis lo antes posible. Si está cerca de la hora de la siguiente dosis, no se debe tomar la dosis olvidada sino que se tomará la siguiente dosis a su hora habitual.
Durante los ensayos pivotales se observó la máxima mejoría en las primeras 24 semanas de tratamiento. Si un paciente no muestra indicios de beneficio terapéutico después de 24 semanas, se debe reconsiderar el tratamiento. Se debe evaluar la respuesta del paciente al tratamiento de forma periódica. No se dispone de experiencia clínica pasadas las 52 semanas (ver sección 5.1).
Poblaciones especiales
Pacientes de edad avanzada
No se requiere un ajuste de la dosis en esta población de pacientes (ver secciones 4.8 y 5.2).
Pacientes con insuficiencia renal
No es necesario un ajuste de la dosis en pacientes con insuficiencia renal leve y moderada. La dosis de apremilast se debe reducir a 30 mg una vez al día en pacientes con insuficiencia renal grave (aclaramiento de la creatinina (CLcr) menor de 30 ml por minuto, estimado mediante la fórmula de Cockcroft-Gault). Para el escalado inicial de la dosis en este grupo, se recomienda tomar únicamente las dosis de Otezla de la mañana del programa de la Tabla 1 y saltarse las dosis de la noche (ver sección 5.2).
Pacientes con insuficiencia hepática
No es necesario un ajuste de la dosis en pacientes con insuficiencia hepática (ver sección 5.2).
Población _ pediátrica
No se ha establecido la seguridad y eficacia de apremilast en niños de 0 a 17 años. No se dispone de datos.
Forma de administración
Otezla se administra por vía oral. Los comprimidos recubiertos con película se deben tragar enteros y se pueden tomar con o sin alimentos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Embarazo (ver sección 4.6)
4.4 Advertencias y precauciones especiales de empleo
Los pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia de lactasa de Lapp o malabsorción de glucosa-galactosa no deben tomar este medicamento.
La dosis de Otezla se debe reducir a 30 mg una vez al día en pacientes con insuficiencia renal grave (ver secciones 4.2 y 5.2).
Se debe monitorizar periódicamente el peso de los pacientes que, al comienzo del tratamiento, tengan un peso inferior al normal. En caso de una pérdida de peso clínicamente significativa y de causa desconocida, el médico general debe evaluar a estos pacientes y se debe considerar la interrupción del tratamiento.
4.5 Interacción con otros medicamentos y otras formas de interacción
La administración concomitante con el inductor enzimático potente del citocromo P450 3A4 (CYP3A4), rifampicina, produjo una reducción de la exposición sistémica de apremilast, lo que puede producir una pérdida de la eficacia de apremilast. Por lo tanto, no se recomienda usar inductores enzimáticos potentes del citocromo CYP3A4 (p. ej., rifampicina, fenobarbital, carbamazepina, fenitoína y hierba de San Juan) junto con apremilast. La administración concomitante de apremilast con dosis múltiples de rifampicina produjo una disminución en el área bajo la curva de concentración plasmática (AUC) de apremilast y de la concentración sérica máxima (Cmáx) aproximadamente del 72 % y del 43 %, respectivamente. La exposición de apremilast se reduce cuando se administra de forma concomitante con inductores potentes de CYP3A4 (p. ej., rifampicina) y puede dar lugar a una respuesta clínica reducida.
En los estudios clínicos se ha administrado apremilast de forma concomitante con tratamiento tópico (incluidos corticoesteroides, champú de alquitrán de hulla y preparados de ácido salicílico para el cuero cabelludo) y fototerapia UVB.
No hubo interacciones medicamentosas clínicamente significativas entre ketoconazol y apremilast. Se puede administrar apremilast de forma concomitante con un inhibidor potente de CYP3A4 como ketoconazol.
No hubo interacciones medicamentosas farmacocinéticas entre apremilast y metotrexato en pacientes con artritis psoriásica. Se puede administrar apremilast de forma concomitante con metotrexato.
No hubo interacciones medicamentosas farmacocinéticas entre apremilast y los anticonceptivos orales que contienen etinilestradiol y norgestimato. Se puede administrar apremilast de forma concomitante con anticonceptivos orales.
4.6 Fertilidad, embarazo y lactancia
Mujeres con capacidad de gestación
Se debe descartar el embarazo antes de poder iniciar el tratamiento. Las mujeres con capacidad de gestación deben utilizar un método anticonceptivo efectivo para prevenir el embarazo durante el tratamiento.
Embarazo
Los datos relativos al uso de apremilast en mujeres embarazadas son limitados.
Apremilast está contraindicado durante el embarazo. Los efectos de apremilast sobre el embarazo incluyeron pérdida embriofetal en ratones y monos, disminución del peso fetal y retraso en la osificación en ratones a dosis superiores a la dosis máxima humana actualmente recomendada. No se observaron dichos efectos cuando la exposición en los animales fue a dosis 1,3 veces la exposición clínica (ver sección 5.3).
Lactancia
Se ha detectado apremilast en la leche de ratones hembra en periodo de lactancia (ver sección 5.3). Se desconoce si apremilast, o sus metabolitos, se excretan en la leche materna humana. No puede descartarse un riesgo para el lactante; por lo tanto, apremilast no se debe utilizar durante la lactancia.
Fertilidad
No hay datos de fertilidad disponibles en seres humanos. En los estudios con ratones no se observaron efectos adversos sobre la fertilidad de los machos expuestos a niveles de 3 veces la exposición clínica ni de las hembras expuestas a niveles de 1 vez la exposición clínica. Para los datos de fertilidad de los estudios preclínicos ver sección 5.3.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de apremilast sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas Resumen del perfil de seguridad
Las reacciones adversas notificadas con mayor frecuencia en los estudios clínicos de fase III fueron trastornos gastrointestinales (GI) que incluyen diarrea (15,7 %) y náuseas (13,9 %). Estas reacciones adversas GI fueron, en su mayoría, de intensidad leve a moderada, con un 0,3 % de los casos de diarrea y un 0,3 % de los casos de náuseas notificados como severos. Estas reacciones adversas ocurrieron por lo general en las 2 primeras semanas de tratamiento y normalmente remitieron en 4 semanas. Las otras reacciones adversas notificadas con mayor frecuencia incluyeron infecciones del tracto respiratorio superior (8,4 %), cefalea (7,9 %) y cefalea tensional (7,2 %). En general, la mayoría de las reacciones adversas se consideraron de intensidad leve o moderada.
Las reacciones adversas más frecuentes que dieron lugar a la interrupción durante las primeras 16 semanas de tratamiento fueron diarrea (1,7 %) y náuseas (1,5 %). La incidencia global de reacciones adversas graves fue baja y no indicó afectación de ningún órgano ni sistema específico.
En los estudios clínicos de apremilast se observaron con poca frecuencia reacciones de hipersensibilidad (ver sección 4.3).
Tabla de reacciones adversas
Las reacciones adversas observadas en los pacientes tratados con apremilast se incluyen a continuación según el Sistema de Clasificación por de Órganos y sistemasl (SOC por sus siglas en inglés, System Organ Class) y la frecuencia de todas las reacciones adversas. Dentro de cada SOC y grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Las reacciones adversas al medicamento se determinaron basándose en los datos del programa de desarrollo clínico de apremilast. Las frecuencias de las reacciones adversas al medicamento son las notificadas en los grupos de apremilast de los cuatro estudios de fase III de artritis psoriásica (n = 1945) o de los dos estudios de fase III de psoriasis (n = 1184) (en la tabla 2 se representa la frecuencia más alta de los dos grupos de datos).
Las frecuencias se definen como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000).
Tabla 2. Resumen de las reacciones adversas en los estudios clínicos de fase III de artritis psoriásica (APs) y/o psoriasis (PSOR)
|
Sistema de Clasificación de Órganos |
Frecuencia |
Reacción adversa |
|
Infecciones e infestaciones |
Frecuentes |
Bronquitis |
|
Infección del tracto respiratorio superior | ||
|
Nasofaringitis* | ||
|
Trastornos del sistema inmunológico |
Poco frecuentes |
Hipersensibilidad |
|
Trastornos del metabolismo y de la nutrición |
Frecuentes |
Disminución del apetito* |
|
Trastornos psiquiátricos |
Frecuentes |
Insomnio |
|
Trastornos del sistema nervioso |
Frecuentes |
Migraña* |
|
Cefalea tensional* | ||
|
Cefalea* | ||
|
Trastornos respiratorios, torácicos y mediastínicos |
Frecuentes |
Tos |
|
Trastornos gastrointestinales |
Muy frecuentes |
Diarrea* |
|
Náuseas* | ||
|
Frecuentes |
Vómitos* | |
|
Dispepsia | ||
|
Movimientos intestinales frecuentes | ||
|
Dolor abdominal superior* | ||
|
Enfermedad por reflujo gastroesofágico | ||
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
Erupción |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Frecuentes |
Dolor de espalda* |
|
Trastornos generales y alteraciones en el lugar de administración |
Frecuentes |
Fatiga |
|
Exploraciones complementarias |
Poco frecuentes |
Pérdida de peso |
*Al menos una de estas reacciones adversas fue notificada como grave
Descripción de reacciones adversas seleccionadas
Pérdida de _peso
El peso de los pacientes se determinó de forma rutinaria en los estudios clínicos. La pérdida de peso media observada en los pacientes tratados hasta 52 semanas con apremilast fue de 1,99 kg. Se observó una pérdida de peso del 5 al 10 % en un total del 14,3 % de los pacientes tratados con apremilast, mientras que en el 5,7 % de los pacientes tratados con apremilast se observó una pérdida de peso mayor del 10 %. Ninguno de estos pacientes presentó consecuencias clínicas evidentes debido a la pérdida de peso. Un total del 0,1 % de los pacientes tratados con apremilast interrumpió el tratamiento debido a la reacción adversa de pérdida de peso.
Ver la advertencia adicional en la sección 4.4 para los pacientes con un peso inferior al normal al inicio del tratamiento.
Depresión
Durante el periodo controlado con placebo de los ensayos clínicos de fase III de psoriasis, el 1,2 % (14/1184) de los pacientes tratados con apremilast notificó depresión, en comparación con el 0,5 % (2/418) de los pacientes tratados con placebo. Ninguno de estos casos de depresión fue grave ni dio lugar a la retirada del estudio.
Poblaciones especiales
Pacientes de edad avanzada
No se observaron diferencias globales entre el perfil de seguridad de los pacientes de edad avanzada (>65 años) y el de los pacientes adultos más jóvenes (<65 años) en los estudios clínicos.
Pacientes con insuficiencia hepática
No se evaluó la seguridad de apremilast en los pacientes con artritis psoriásica o con psoriasis y con insuficiencia hepática.
Pacientes con insuficiencia renal
En los estudios clínicos de artritis psoriásica o de psoriasis, el perfil de seguridad observado en los pacientes con insuficiencia renal leve fue comparable al de los pacientes con función renal normal. No se evaluó la seguridad de apremilast en los pacientes con artritis psoriásica o con psoriasis y con insuficiencia renal moderada o grave en los estudios clínicos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Se estudió apremilast en sujetos sanos a una dosis máxima diaria total de 100 mg (administrada como 50 mg dos veces al día) durante 4,5 días y no se observaron indicios de toxicidad limitante de la dosis. En caso de sobredosis, se recomienda monitorizar al paciente para detectar cualquier signo o síntoma de efectos adversos e instaurar el tratamiento sintomático adecuado. En caso de sobredosis, se recomienda un cuidado sintomático y de soporte.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inmunosupresores, inmunosupresores selectivos, código ATC: L04AA32 Mecanismo de acción
Apremilast, una molécula pequeña que se administra por vía oral y que inhibe la fosfodiesterasa 4 (PDE4), actúa dentro de la célula modulando una red de mediadores proinflamatorios y antiinflamatorios. PDE4 es una fosfodiesterasa (PDE) específica del adenosín monofosfato cíclico (AMPc) y la PDE dominante en las células inflamatorias. La inhibición de PDE4 eleva los niveles intracelulares de AMPc, que a su vez regula disminuyendo la respuesta inflamatoria mediante modulación de la expresión de TNF-a, IL-23, IL-17 y otras citocinas inflamatorias. El AMP cíclico modula también los niveles de citocinas antiinflamatorias como IL-10. Estos mediadores proinflamatorios y antiinflamatorios están implicados en la artritis psoriásica y en la psoriasis.
Efectos _ farmacodinámicos
En los estudios clínicos en pacientes con artritis psoriásica, apremilast moduló de manera significativa, aunque sin inhibir por completo, los niveles de proteínas plasmáticas de IL-1a, IL-6, IL-8, MCP-1, MIP-ip, MMP-3 y TNF-a. Tras 40 semanas de tratamiento con apremilast, se observó una disminución en los niveles de proteínas plasmáticas de IL-17 e IL-23, y un aumento de IL-10. En los ensayos clínicos en pacientes con psoriasis, apremilast disminuyó el grosor epidérmico de la piel lesionada, la infiltración celular inflamatoria y la expresión de los genes proinflamatorios, incluidos aquellos que codifican para el óxido nítrico sintasa inducible (iNOS), IL-12/IL-23p40, IL-17A, IL-22 e IL-8.
Apremilast administrado a dosis de hasta 50 mg dos veces al día no prolongó el intervalo QT en sujetos sanos.
Experiencia en ensayos clínicos Artritis psoriásica
Se evaluaron la seguridad y la eficacia de apremilast en 3 estudios multicéntricos, aleatorizados, controlados con placebo y doble ciego (estudios PALACE 1, PALACE 2 y PALACE 3) de diseño similar en pacientes adultos con artritis psoriásica activa (>3 articulaciones inflamadas y >3 articulaciones dolorosas) a pesar del tratamiento previo con FAMEs de molécula pequeña o biológicos. Un total de 1493 pacientes fueron aleatorizados y tratados con placebo, 20 mg de apremilast o 30 mg de apremilast, administrado por vía oral dos veces al día (2 v/d).
Los pacientes de estos estudios tenían un diagnóstico de artritis psoriásica desde al menos 6 meses. Asimismo, en el estudio PALACE 3 era necesario tener una lesión psoriásica en la piel valorable (por lo menos de 2 cm de diámetro). Apremilast se utilizó en monoterapia (34,8 %) o en combinación con dosis estables de FAMEs de molécula pequeña (65,2 %). Los pacientes recibieron apremilast en combinación con uno o más de los siguientes: metotrexato (MTX, <25 mg/semana, 54,5 %), sulfasalazina (SSZ, <2 g/día, 9,0 %) y leflunomida (LEF; <20 mg/día, 7,4 %). No estaba permitido el tratamiento concomitante con FAMEs biológicos, incluidos los bloqueadores de TNF. Se incluyeron en los 3 estudios pacientes con cada subtipo de artritis psoriásica, incluidas poliartritis simétrica (62,0 %), oligoartritis asimétrica (26,9 %), artritis en las articulaciones interfalángeas distales (IFD) (6,2 %), artritis mutilante (2,7 %) y espondilitis predominante (2,1 %). Se incluyó a pacientes con entesopatía preexistente (63 %) o dactilitis preexistente (42 %). El 76,4 % de los pacientes habían sido tratados previamente solo con FAMEs de molécula pequeña y el 22,4 % de los pacientes habían sido tratados previamente con FAMEs biológicos, lo que incluye el 7,8 % que no había respondido al tratamiento previo con un FAME biológico. La mediana de duración de la artritis psoriásica fue de 5 años.
Basándose en el diseño del estudio, los pacientes cuyos recuentos de articulaciones dolorosas e inflamadas no habían mejorado como mínimo en un 20 % fueron considerados no respondedores en la semana 16. Los pacientes tratados con placebo que fueron considerados no respondedores fueron reasignados aleatoriamente en una proporción 1:1, de forma enmascarada, a 20 mg o a 30 mg de apremilast dos veces al día. En la semana 24, todos los demás pacientes tratados con placebo pasaron a recibir tratamiento con 20 mg o con 30 mg de apremilast dos veces al día.
El criterio principal de valoración fue el porcentaje de pacientes que obtuvieron una respuesta ACR20 en la semana 16 conforme a los criterios del Colegio Americano de Reumatología (ACR por sus siglas en inglés, American College of Rheumatology).
El tratamiento con apremilast produjo mejorías significativas en los signos y síntomas de la artritis psoriásica, como determinaron los criterios de respuesta ACR20, en comparación con el placebo, en la semana 16. La proporción de pacientes con respuestas ACR20/50/70 en los estudios PALACE 1,
PALACE 2 y PALACE 3 y los datos agrupados de los estudios PALACE 1, PALACE 2 y PALACE 3, con 30 mg de apremilast dos veces al día en la semana 16, se muestran en la tabla 3. Las respuestas ACR20/50/70 se mantenían en la semana 24.
Entre los pacientes que inicialmente fueron aleatorizados al tratamiento con 30 mg de apremilast dos veces al día, las tasas de respuesta ACR20/50/70 se mantuvieron hasta la semana 52 en los estudios agrupados PALACE 1, PALACE 2 y PALACE 3 (Figura 1).
Tabla 3. Proporción de pacientes con respuestas ACR en los estudios PALACE 1, PALACE 2 y PALACE 3 y en los estudios agrupados en la semana 16
|
PALACE1 |
PALACE 2 |
PALACE 3 |
AGRUPADOS | |||||
|
Placebo ± |
Apremilast |
Placebo ± |
Apremilast |
Placebo ± |
Apremilast |
Placebo ± |
Apremilast | |
|
FAMEs |
30 mg 2 v/d ± FAMEs |
FAMEs |
30 mg 2 v/d ± FAMEs |
FAMEs |
30 mg 2 v/d ± FAMEs |
FAMEs |
30 mg 2 v/d ± FAMEs | |
|
Na |
N = 168 |
N = 168 |
N = 159 |
N = 162 |
N = 169 |
N = 167 |
N = 496 |
N = 497 |
|
ACR20a | ||||||||
|
Semana 16 |
19,0 % |
38,1 %** |
18,9 % |
32,1 %* |
18,3 % |
40,7 %** |
18,8 % |
37,0 %** |
|
ACR50 | ||||||||
|
Semana 16 |
6,0 % |
16,1 %* |
5,0 % |
10,5 % |
8,3 % |
15,0 % |
6,5 % |
13,9 %** |
|
ACR70 | ||||||||
|
Semana 16 |
1,2 % |
4,2 % |
0,6 % |
1,2 % |
2,4 % |
3,6 % |
1,4 % |
3,0 % |
*p <0,01 para apremilast frente a placebo.
**p <0,001 para apremilast frente a placebo. a N es el número de pacientes aleatorizados y tratados.
Figura 1 Proporción de respondedores ACR20/50/70 hasta la semana 52 en el análisis agrupado de los estudios PALACE 1, PALACE 2 y PALACE 3 (NRI*)
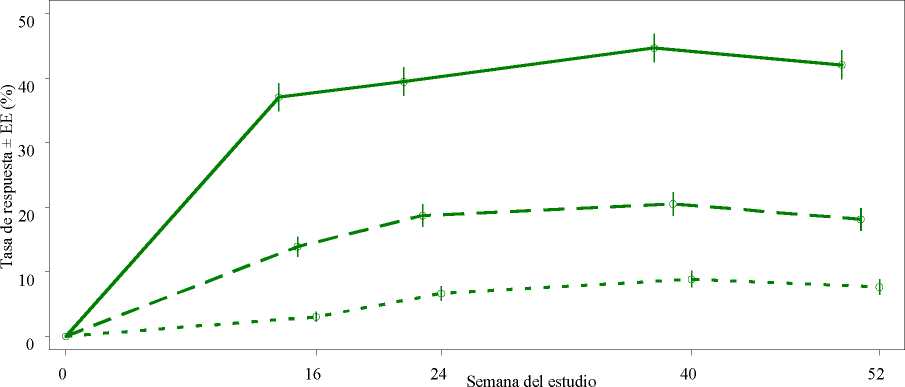
Criterio de valoración n/m (%) n/m (%)
ACR 20 184/497 (37,0) 196/497 (39,4)
ACR 50 69/497 (13,9) 93/497 (18,7)
ACR 70 15/497 ( 3,0) 33/497 ( 6,6)
n/m (%) 222/497 (44,7) 102/497 (20,5) 44/497 ( 8,9)
n/m (%) 209/497 (42,1) 90/497 (18,1) 38/497 ( 7,6)
Criterio de valoración o o o acr 20 acr 50 e « -o acr 70
*NRI (Por sus siglas en inglés, Non-responder imputation): imputación de los no respondedores. Los sujetos que abandonaron antes del punto temporal y los sujetos que no tenían datos suficientes para una determinación definitiva del estado de la respuesta en el punto temporal se cuentan como no respondedores.
Entre los 497 pacientes inicialmente aleatorizados a 30 mg de apremilast dos veces al día, 375 (75 %) pacientes seguían con este tratamiento en la semana 52. En estos pacientes, las respuestas ACR20/50/70 en la semana 52 fueron del 57 %, 25 % y 11 %, respectivamente.
Las respuestas observadas en el grupo tratado con apremilast fueron similares entre los pacientes que recibieron y los que no recibieron FAMEs de forma concomitante, incluido MTX.
Los pacientes tratados previamente con FAMEs o con tratamientos biológicos que recibieron apremilast alcanzaron una mayor respuesta ACR20 en la semana 16, en comparación con los pacientes que recibieron placebo.
Se observaron respuestas ACR similares en los pacientes con diferentes subtipos de artritis psoriásica, incluida la artritis en las articulaciones IFD. El número de pacientes con los subtipos artritis mutilante y espondilitis predominante era demasiado pequeño para permitir una evaluación significativa.
En los estudios PALACE 1, PALACE 2 y PALACE 3 las mejorías en la escala de actividad de la enfermedad (DAS28 por sus siglas en inglés, Disease Activity Score) utilizando la proteína C reactiva (PCR) y en la proporción de pacientes que alcanzó los criterios de respuesta de la artritis psoriásica (PsARC por sus siglas en inglés, Psoriatic Arthritis response criteria) modificados fueron mayores en elgrupo de apremilast, en comparación con placebo en la semana 16 (valor p nominal p<0,0004, valor p <0,0017, respectivamente). Estas mejorías se mantenían en la semana 24. Entre los pacientes que seguían con el tratamiento con apremilast al que fueron aleatorizados al comienzo del estudio, la puntuación DAS28 (PCR) y la respuesta en PsARC se mantuvieron hasta la semana 52.
Se observaron mejorías en las semanas 16 y 24 en los parámetros de la actividad periférica característica de la artritis psoriásica (p. ej., número de articulaciones inflamadas, número de articulaciones dolorosas/doloridas, dactilitis y entesitis) y en las manifestaciones cutáneas de psoriasis en los pacientes tratados con apremilast. Entre los pacientes que seguían con el tratamiento con apremilast al que fueron aleatorizados al comienzo del estudio, estas mejorías se mantuvieron hasta la semana 52.
Función física y calidad de vida relacionada con la salud
Los pacientes tratados con apremilast demostraron una mejoría estadísticamente significativa en la función física, como determinó el cambio con respecto a la situación basal en el índice de discapacidad del cuestionario de evaluación de la salud (HAQ-DI por sus siglas en inglés, Health Assessment Questionnaire Disability Index), en comparación con placebo a las 16 semanas en los estudios PALACE 1, PALACE 2 y PALACE 3 y en los estudios agrupados. La mejoría en las puntuaciones en HAQ-DI se mantuvo en la semana 24.
Entre los pacientes que fueron inicialmente aleatorizados al tratamiento con 30 mg de apremilast dos veces al día, el cambio con respecto a la situación basal en la puntuación en HAQ-DI en la semana 52 fue de -0,333 en el grupo de 30 mg de apremilast dos veces al día en un análisis agrupado de la fase abierta de los estudios PALACE 1, PALACE 2 y PALACE 3.
En los estudios PALACE 1, PALACE 2 y PALACE 3 se demostraron mejorías significativas en la calidad de vida relacionada con la salud, como determinaron los cambios con respecto a la situación basal en las puntuaciones del dominio de función física del cuestionario de la salud abreviado versión 2 (SF-36v2) y en las puntuaciones de la evaluación funcional del tratamiento de las enfermedades crónicas - fatiga (FACIT-fatiga) en los pacientes tratados con apremilast en comparación con placebo en las semanas 16 y 24. Entre los pacientes que seguían con el tratamiento con apremilast al que fueron aleatorizados al comienzo del estudio, la mejoría en la función física y en FACIT-fatiga se mantuvo hasta la semana 52.
Psoriasis
Se evaluaron la seguridad y la eficacia de apremilast en 2 estudios multicéntricos, aleatorizados, controlados con placebo y doble ciego (estudios ESTEEM 1 y ESTEEM 2) en los que participaron un total de 1257 pacientes con psoriasis en placas de moderada a grave que tenían un área de superficie corporal (BSA por sus siglas en inglés, Body surface area,) afectada >10 %, una puntuación en el índice de gravedad y área de la psoriasis (PASI por sus siglas en inglés, Psoriasis Area and Severity Index) de >12, una evaluación global estática del médico (sPGA por sus siglas en inglés, Static Physician Global Assessment) >3 (moderada o grave), y que eran candidatos a fototerapia o tratamiento sistémico.
El diseño de los estudios era similar hasta la semana 32. En ambos estudios, los pacientes fueron aleatotorizados en una proporción 2:1 a 30 mg de apremilast dos veces al día o a placebo durante 16 semanas (fase controlada con placebo) y desde la semana 16 hasta la 32 todos los pacientes recibieron 30 mg de apremilast dos veces al día (fase de mantenimiento). Durante la fase de retirada del tratamiento aleatorizado (semanas 32-52), los pacientes originalmente aleatorizados a apremilast que alcanzaron al menos una disminución del 75 % en la puntuación del PASI (PASI-75) (ESTEEM 1) o una disminución del 50 % en la puntuación (PASI-50) (ESTEEM 2) fueron reasignados aleatoriamente en la semana 32 a placebo o a 30 mg de apremilast dos veces al día. Los pacientes que fueron reasignados a placebo y que perdieron la respuesta PASI-75 (ESTEEM 1) o que perdieron el 50 % de la mejoría en el PASI en la semana 32 en comparación con la situación basal (ESTEEM 2) volvieron a recibir tratamiento con 30 mg de apremilast dos veces al día. Los pacientes que no alcanzaron la respuesta designada en el PASI en la semana 32, o que fueron inicialmente aleatorizados a placebo, siguieron con apremilast hasta la semana 52. Se permitió el uso de corticoesteroides tópicos de baja potencia en la cara, axilas e ingles, y el uso de champú de alquitrán de hulla y/o preparados de ácido salicílico para el cuero cabelludo durante los estudios. Además, en la semana 32, los sujetos que no alcanzaron una respuesta PASI-75 en ESTEEM 1 o una respuesta PASI-50 en ESTEEM 2 pudieron utilizar tratamientos tópicos para la psoriasis y/o fototerapia, además del tratamiento con 30 mg de apremilast dos veces al día.
En ambos estudios, el criterio principal de valoración fue la proporción de pacientes que alcanzó una respuesta PASI-75 en la semana 16. El principal criterio de valoración secundario fue la proporción de pacientes que alcanzó una puntuación en la sPGA de blanqueada (0) o casi blanqueada (1) en la semana 16.
La puntuación media en el PASI basal fue de 19,07 (mediana 16,80) y la proporción de pacientes con una puntuación en la sPGA basal de 3 (moderada) y 4 (grave) fue del 70,0 % y del 29,8 %, respectivamente, con una afectación media del BSA basal del 25,19 % (mediana 21,0 %). Aproximadamente el 30 % de todos los pacientes habían recibido fototerapia previa y el 54 % había recibido tratamiento sistémico convencional y/o biológico previo para el tratamiento de la psoriasis (incluidos los que no respondieron al tratamiento), de los que un 37 % había recibido tratamiento sistémico convencional previo y un 30 % tratamiento biológico previo. Aproximadamente un tercio de los pacientes no había recibido fototerapia previa ni tratamiento sistémico convencional o biológico previo. Un total del 18 % de los pacientes tenía historia de artritis psoriásica.
La proporción de pacientes que alcanzó respuestas PASI-50, PASI-75 y PASI-90, y una puntuación en la sPGA de blanqueada (0) o casi blanqueada (1), se presenta en la Tabla 4. El tratamiento con apremilast produjo una mejoría significativa de la psoriasis en placas de moderada a grave, como demostró la proporción de pacientes con respuesta PASI-75 en la semana 16, en comparación con placebo. La mejoría clínica determinada por las respuestas en sPGA, PASI-50 y PASI-90 se demostró también en la semana 16. Además, se demostró el beneficio del tratamiento con apremilast en múltiples manifestaciones de la psoriasis, incluyendo prurito, enfermedad ungueal, afectación del cuero cabelludo y medidas de calidad de vida.
Tabla 4. Respuesta clínica en la semana 16 en los estudios ESTEEM 1 y ESTEEM 2 (FASa LOCFb)
|
ESTEEM1 |
ESTEEM 2 | |||
|
Placebo |
Apremilast 30 mg 2 v/d * |
Placebo |
Apremilast 30 mg 2 v/d * | |
|
N |
282 |
562 |
137 |
274 |
|
PASIc-75, n (%) |
15 (5,3) |
186 (33,1) |
8 (5,8) |
79 (28,8) |
|
Blanqueada o casi blanqueada en sPGAd, n (%) |
11 (3,9) |
122 (21,7) |
6 (4,4) |
56 (20,4) |
|
PASI-50, n (%) |
48 (17,0) |
330 (58,7) |
27 (19,7) |
152 (55,5) |
|
PASI-90, n (%) |
1 (0,4) |
55 (9,8) |
2 (1,5) |
24 (8,8) |
|
Cambio en el BSAe (%) media ± Desviación Estándar (DE) |
-6,9 |
-47,8 |
-6,1 |
-48,4 |
|
±38,95 |
±38,48 |
±47,57 |
±40,78 | |
|
Cambio en el prurito en |
-7,3 |
-31,5 |
-12,2 |
-33,5 |
|
EVAf (mm), media ± DE |
±27,08 |
±32,43 |
±30,94 |
±35,46 |
|
Cambio en el DLQIg, |
-2,1 |
-6,6 |
-2,8 |
-6,7 |
|
media ± DE |
±5,69 |
±6,66 |
±7,22 |
±6,95 |
|
Cambio en el SF-36 MCS h, |
-1,02 |
2,39 |
0,00 |
2,58 |
|
media ± DE |
±9,161 |
±9,504 |
±10,498 |
±10,129 |
* p <0,0001 para apremilast frente a placebo, excepto en ESTEEM 2 PASI-90 y cambio en el SF-36 MCS donde p = 0,0042 y p = 0,0078, respectivamente.
a FAS (Full Analysis Set) = Conjunto de análisis completo
b LOCF (Last Observation Carried Forward)= Última observación disponible
c PASI = Índice de gravedad y área de la psoriasis
d sPGA = Evaluación global estática del médico
e BSC = Área de superficie corporal
f EVA = Escala visual analógica; 0 = nada, 100 = mucho
g DLQI (Dermatology Life Quality Index) = Índice de calidad de vida en dermatología; 0 = no impacto, 30 = máximo impacto h SF-36 MCS = Cuestionario de salud abreviado de 36 ítems para el estudio de los resultados médicos, resumen del componente mental
El beneficio clínico de apremilast se demostró en múltiples subgrupos definidos por las características demográficas basales y las características clínicas basales de la enfermedad (incluidos la duración de la enfermedad psoriásica y los pacientes con historia de artritis psoriásica). También se demostró el beneficio clínico de apremilast al margen del uso previo de medicamentos para la psoriasis y de la respuesta a los tratamientos previos para la psoriasis. Se observaron tasas de respuesta similares en todos los intervalos de peso.
La respuesta a apremilast fue rápida, con mejorías significativamente mayores en los signos y síntomas de la psoriasis, incluido el PASI, malestar/dolor en la piel y prurito, en comparación con placebo en la semana 2. En general, se alcanzaron las respuestas en el PASI en la semana 16 y se mantuvieron hasta la semana 32.
En ambos estudios, la mejoría porcentual media en el PASI con respecto al basal permaneció estable durante la fase de retirada del tratamiento aleatorizado para los pacientes reasignados aleatoriamente a apremilast en la semana 32 (Tabla 5).
Tabla 5. Persistencia del efecto entre los sujetos aleatorizados a 30 mg de apremilast dos
veces al día en la semana 0 y reasignados aleatoriamente a 30 mg de apremilast dos veces al día en la semana 32 hasta la semana 52
|
Punto temporal |
ESTEEM 1 |
ESTEEM 2 | |
|
Pacientes que alcanzaron una respuesta PASI-75 en la semana 32 |
Pacientes que alcanzaron una respuesta PASI-50 en la semana 32 | ||
|
Cambio porcentual en el PASI con respecto al basal, media (%) ± DEa |
Semana 16 |
-77,7 ± 20,30 |
-69,7 ± 24,23 |
|
Semana 32 |
-88 ± 8,30 |
-76,7 ± 13,42 | |
|
Semana 52 |
-80,5 ± 12,60 |
-74,4 ± 18,91 | |
|
Cambio en el DLQI con respecto al basal, media ± DEa |
Semana 16 |
-8,3 ± 6,26 |
-7,8 ± 6,41 |
|
Semana 32 |
-8,9 ± 6,68 |
-7,7 ± 5,92 | |
|
Semana 52 |
-7,8 ± 5,75 |
-7,5 ± 6,27 | |
|
Proporción de sujetos con psoriasis del cuero cabelludo y PGA de 0 o 1, n/N (%)b |
Semana 16 |
40/48 (83,3) |
21/37 (56,8) |
|
Semana 32 |
39/48 (81,3) |
27/37 (73,0) | |
|
Semana 52 |
35/48 (72,9) |
20/37 (54,1) |
a Incluye a los sujetos reasignados aleatoriamente a 30 mg de apremilast dos veces al día en la semana 32 con un valor basal y post-basal en la semana de evaluación del estudio.
bN se basa en el número de sujetos con psoriasis basal en el cuero cabelludo moderada o superior que fueron reasignados aleatoriamente a 30 mg de apremilast dos veces al día en la semana 32. Los sujetos sin datos contaron como no respondedores.
En el estudio ESTEEM 1, aproximadamente el 61 % de los pacientes reasignados aleatoriamente a apremilast en la semana 32 tenían una respuesta PASI-75 en la semana 52. De los pacientes con al menos una respuesta PASI-75 que fueron reasignados aleatoriamente a placebo en la semana 32 durante la fase de retirada del tratamiento aleatorizado, el 11,7 % mantenía una respuesta PASI-75 en la semana 52. La mediana de tiempo hasta la pérdida de la respuesta PASI-75 entre los pacientes reasignados aleatoriamente a placebo fue de 5,1 semanas.
En el estudio ESTEEM 2, aproximadamente el 80,3 % de los pacientes reasignados aleatoriamente a apremilast en la semana 32 tenían una respuesta PASI-50 en la semana 52. De los pacientes con al menos una respuesta PASI-50 que fueron reasignados aleatoriamente a placebo en la semana 32, el
24.2 % mantenía una respuesta PASI-50 en la semana 52. La mediana de tiempo hasta la pérdida del 50 % de la mejoría en PASI de la semana 32 fue de 12,4 semanas.
Después de la retirada aleatorizada del tratamiento en la semana 32, aproximadamente el 70 % de los pacientes del estudio ESTEEM 1 y el 65,6 % de los pacientes del estudio ESTEEM 2 volvieron a alcanzar una respuesta PASI-75 (ESTEEM 1) o una respuesta PASI-50 (ESTEEM 2) después de reiniciar el tratamiento con apremilast. Debido al diseño del estudio, la duración del retratamiento fue variable, y osciló desde 2,6 hasta 22,1 semanas.
En el estudio ESTEEM 1, los pacientes aleatorizados a apremilast al comienzo del estudio que no alcanzaron una respuesta PASI-75 en la semana 32 pudieron utilizar tratamientos tópicos y/o fototerapia UVB concomitantemente entre las semanas 32 y 52. De estos pacientes, el 12 % alcanzó una respuesta PASI-75 en la semana 52 con apremilast más un tratamiento tópico y/o fototerapia.
En los estudios ESTEEM 1 y ESTEEM 2, se observaron mejorías significativas (reducciones) en la psoriasis ungueal, como determinó el cambio porcentual medio en el índice de gravedad de la psoriasis ungueal (NAPSI por sus siglas en inglés, Nail Psoriasis Severity Index) con respecto al basal en los pacientes tratados con apremilast, en comparación con los tratados con placebo en la semana 16 (p <0,0001 y p = 0,0052, respectivamente). En la semana 32 se observaron mejorías adicionales en la psoriasis ungueal en los pacientes tratados continuamente con apremilast.
En los estudios ESTEEM 1 y ESTEEM 2, se observaron mejorías significativas en la psoriasis del cuero cabelludo de intensidad al menos moderada (>3), como determinó la proporción de pacientes que alcanzó la respuesta de blanqueada (0) o mínima (1) en la evaluación global del médico de la psoriasis del cuero cabelludo (ScPGA por sus siglas en inglés, Scalp Psoriasis Physician’s Global Assessment) en la semana 16, en los pacientes tratados con apremilast en comparación con los tratados con placebo (p <0,0001 en ambos estudios). Las mejorías, en general, se mantuvieron en los sujetos que fueron reasignados aleatoriamente a Otezla en la semana 32 hasta la semana 52 (Tabla 5).
En los estudios ESTEEM 1 y ESTEEM 2 se demostraron mejorías significativas en la calidad de vida, como determinaron el índice de calidad de vida en dermatología (DLQI) y el SF-36v2MCS, en los pacientes tratados con apremilast en comparación con los tratados con placebo (Tabla 4). Las mejorías en el DLQI se mantuvieron hasta la semana 52 en los sujetos reasignados aleatoriamente a apremilast en la semana 32 (Tabla 5). Además, en el estudio ESTEEM 1 se alcanzó una mejoría significativa en el cuestionario de limitaciones laborales (WLQ-25 por sus siglas en inglés, Work Limitations Questionnaire) en los pacientes tratados con apremilast, en comparación con los tratados con placebo.
5.2 Propiedades farmacocinéticas
Absorción
Apremilast se absorbe bien, con una biodisponibilidad oral absoluta del 73 % aproximadamente y concentraciones plasmáticas máximas (Cmáx) que se alcanzan en una mediana de tiempo (tmáx) de 2,5 horas aproximadamente. La farmacocinética de apremilast es lineal, con un aumento proporcional a la dosis en la exposición sistémica en el intervalo de dosis de 10 a 100 mg al día. La acumulación es mínima cuando apremilast se administra una vez al día y aproximadamente del 53 % en sujetos sanos y del 68 % en pacientes con psoriasis cuando se administra dos veces al día. La administración con alimentos no altera la biodisponibilidad, por lo tanto, apremilast se puede administrar con o sin alimentos.
Distribución
La unión de apremilast a las proteínas plasmáticas humanas es aproximadamente del 68 %. El volumen de distribución (Vd) aparente medio es de 87 l, lo que indica distribución extravascular.
Biotransformación
Apremilast se metaboliza extensamente por las vías mediadas por CYP y no mediadas por CYP, incluidas las vías de oxidación, hidrólisis y conjugación, lo que sugiere que no es probable que la inhibición de una única vía de aclaramiento cause una interacción medicamentosa destacable. El metabolismo oxidativo de apremilast está mediado principalmente por CYP3A4, con alguna contribución menor de CYP1A2 y CYP2A6. Apremilast es el principal componente circulante tras la administración oral. Apremilast se somete a un metabolismo extenso y solo el 3 % y el 7 % del compuesto original administrado se recuperan en orina y en heces, respectivamente. El principal metabolito inactivo circulante es el conjugado glucurónido de apremilast O-desmetilado (M12).
Debido a que apremilast es un sustrato de CYP3A4, la exposición de apremilast disminuye cuando se administra concomitantemente con rifampicina, un inductor potente de CYP3A4.
In vitro, apremilast no es un inhibidor ni un inductor de las enzimas del citocromo P450. Por lo tanto, es poco probable que la administración concomitante de apremilast con sustratos de las enzimas de CYP afecte al aclaramiento y a la exposición de los principios activos que se metabolizan por las enzimas de CYP.
In vitro, apremilast es un sustrato y un inhibidor débil de la glicoproteína P (P-gp) (CI50 >50 pM); sin embargo, no se espera que ocurran interacciones medicamentosas clínicamente relevantes mediadas por la P-gp.
In vitro, apremilast tiene escaso o ningún efecto inhibidor (CI50 >10 pM) en el transportador de aniones orgánicos (OAT por sus siglas en inglés, Organic Anion Transporter) 1 y OAT3, el transportador de cationes orgánicos (OCT por sus siglas en inglés, Organic Cation Transporter) 2, el polipéptido transportador de aniones orgánicos (OATP) 1B1 y OATP1B3, o en la proteína resistente al cáncer de mama (BCRP por sus siglas en inglés, Breast Cancer Resistance Protein), y no es un sustrato de estos transportadores. Por lo tanto, es poco probable que ocurran interacciones medicamentosas clínicamente relevantes cuando apremilast se administre concomitantemente con medicamentos que son sustratos o inhibidores de estos transportadores.
Eliminación
El aclaramiento plasmático medio de apremilast es de unos 10 l/h en sujetos sanos, con una semivida de eliminación terminal de 9 horas aproximadamente. Tras la administración oral de apremilast con radio marcaje, alrededor del 58 % y del 39 % de la radiactividad se recupera en la orina y en las heces, respectivamente, con alrededor del 3 % y del 7 % de la dosis radiactiva recuperada como apremilast en la orina y en las heces, respectivamente.
Pacientes de edad avanzada
Se estudió apremilast en sujetos sanos jóvenes y de edad avanzada. La exposición en los sujetos de edad avanzada (de 65 a 85 años) es alrededor de un 13 % mayor en el AUC y alrededor de un 6 % mayor en la Cmáx para apremilast que en los sujetos jóvenes (de 18 a 55 años). Los datos farmacocinéticos son limitados en sujetos mayores de 75 años en los ensayos clínicos. No es necesario un ajuste de la dosis en pacientes de edad avanzada.
Insuficiencia renal
No hay una diferencia significativa en la farmacocinética de apremilast entre los pacientes con insuficiencia renal leve o moderada y los sujetos sanos con las mismas características (N = 8 de cada). Los resultados respaldan que no es necesario ajustar la dosis en pacientes con insuficiencia renal leve o moderada. Se debe reducir la dosis de apremilast a 30 mg una vez al día en pacientes con insuficiencia renal grave (Filtración Glomerular estimada (FGe) menor de 30 ml/min/1,73 m2 o CLcr <30 ml/min). En 8 pacientes con insuficiencia renal grave que recibieron una dosis de 30 mg de apremilast, el AUC y la Cmáx de apremilast aumentaron en aproximadamente un 89 % y un 42 %, respectivamente.
Insuficiencia hepática
La farmacocinética de apremilast y la de su principal metabolito M12 no se ven afectadas por la insuficiencia hepática moderada o grave. No es necesario un ajuste de la dosis en pacientes con insuficiencia hepática.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad y toxicidad a dosis repetidas. No hay indicios de potencial inmunotóxico, de irritación dérmica o fototóxico.
Fertilidad y desarrollo embrionario temprano
En un estudio de fertilidad con ratones macho, la administración oral de apremilast a dosis de 1, 10, 25 y 50 mg/kg/día no produjo efectos en la fertilidad de los machos; el nivel sin efecto adverso observado (NOAEL por sus siglas en inglés, No Observed Adverse EffectLevel) para la fertilidad de los machos fue mayor de 50 mg/kg/día (3 veces la exposición clínica).
En un estudio combinado de toxicidad en el desarrollo embriofetal y de fertilidad en ratones hembra con dosis orales de 10, 20, 40 y 80 mg/kg/día se observaron una prolongación de los ciclos estrales y un mayor tiempo hasta el apareamiento con las dosis de 20 mg/kg/día y superiores. A pesar de esto, todos los ratones se aparearon y las tasas de gestación no se vieron afectadas. El nivel sin efecto observado (NOEL por sus siglas en inglés, No Observed Effect Level)para la fertilidad de las hembras fue de 10 mg/kg/día (1,0 vez la exposición clínica).
Desarrollo embriofetal
En un estudio combinado de toxicidad en el desarrollo embriofetal y de fertilidad en ratones hembra con dosis orales de 10, 20, 40 y 80 mg/kg/día, los pesos absolutos y/o relativos de los corazones de las madres aumentaron con las dosis de 20, 40 y 80 mg/kg/día. Se observaron un aumento del número de resorciones tempranas y una disminución del número de tarsos osificados con las dosis de 20, 40 y 80 mg/kg/día. Se observó una reducción de los pesos fetales y una osificación retardada del hueso supraoccipital del cráneo con las dosis de 40 y 80 mg/kg/día. En los ratones, el NOEL en el desarrollo y en la madre fue de 10 mg/kg/día (1,3 veces la exposición clínica).
En un estudio de toxicidad en el desarrollo embriofetal en monos, dosis orales de 20, 50, 200 y 1000 mg/kg/día dieron lugar a un aumento, relacionado con la dosis, de pérdidas prenatales (abortos) con las dosis de 50 mg/kg/día y superiores; no se observó ningún efecto relacionado con el medicamento del ensayo en las pérdidas prenatales a la dosis de 20 mg/kg/día (1,4 veces la exposición clínica).
Desarrollo prenatal y postnatal
En un estudio prenatal y postnatal se administró apremilast por vía oral a ratones hembra preñados a dosis de 10, 80 y 300 mg/kg/día desde el día 6 de gestación hasta el día 20 de lactancia. Se observaron reducciones en el peso corporal y en la ganancia de peso de las madres, así como un caso de muerte asociada a dificultad en el parto con la dosis de 300 mg/kg/día. También se observaron signos físicos de toxicidad materna asociados al parto en un ratón con la dosis de 80 mg/kg/día y con la dosis de 300 mg/kg/día. Se observó un aumento del número de muertes perinatales y postnatales de las crías y una reducción en la ganancia de peso de las crías durante la primera semana de lactancia con dosis >80 mg/kg/día (>4,0 veces la exposición clínica). No hubo efectos asociados a apremilast en la duración de la gestación, el número de ratones preñados al final del periodo de gestación, el número de ratones que parieron una camada, ni ningún efecto en el desarrollo de las crías pasado el día 7 de vida. Es probable que los efectos en el desarrollo de las crías observados durante la primera semana del periodo postnatal estuvieran asociados a la toxicidad de apremilast en las crías (peso y viabilidad de las crías reducidos) y/o a la falta de cuidados maternos (mayor incidencia de ausencia de leche en los estómagos de las crías). Todos los efectos en el desarrollo se observaron durante la primera semana del periodo postnatal; no se observaron efectos relacionados con apremilast durante el resto de los periodos antes o después del destete, incluidos los parámetros de maduración sexual, conductuales, apareamiento, fertilidad y uterinos. El NOEL en el ratón en cuanto a la toxicidad de las madres y a la primera generación (F1) fue de 10 mg/kg/día (1,3 veces el AUC clínica).
Estudios de carcinogenicidad
Los estudios de carcinogenicidad en ratones y ratas no mostraron indicios de carcinogenicidad asociada al tratamiento con apremilast.
Estudios de genotoxicidad
Apremilast no es genotóxico. Apremilast no indujo mutaciones en el ensayo de Ames ni aberraciones cromosómicas en cultivos de linfocitos de sangre periférica humana en presencia o ausencia de activación metabólica. Apremilast no fue clastogénico en un ensayo de micronúcleos en ratón in vivo con dosis de hasta 2000 mg/kg/día.
Otros estudios
No hay indicios de potencial inmunotóxico, de irritación dérmica o fototóxico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido Celulosa microcristalina Lactosa monohidrato Croscarmelosa sódica Estearato de magnesio
Cubierta pelicular Alcohol de polivinilo Dióxido de titanio (E171)
Macrogol 3350 Talco
Óxido de hierro rojo (E172)
Los comprimidos de 20 mg contienen también óxido de hierro amarillo (E172).
Los comprimidos de 30 mg contienen también óxido de hierro amarillo (E172) y óxido de hierro negro (E172).
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
21 meses.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30°C.
6.5 Naturaleza y contenido del envase
El pack de inicio de tratamiento contiene 27 comprimidos recubiertos con película (4 de 10 mg, 4 de 20 mg y 19 de 30 mg).
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd.
1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
8. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/981/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Otezla 30 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 30 mg de apremilast.
Excipiente con efecto conocido:
Cada comprimido recubierto con película contiene 171 mg de lactosa (como lactosa monohidrato). Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
Comprimido recubierto con película de 30 mg, con forma de rombo, de color beige, de 12 mm de largo, con “APR” grabado en una cara y “30” en la otra cara.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Artritis psoriásica
Otezla, solo o en combinación con Fármacos Antirreumáticos Modificadores de la Enfermedad (FAMEs), está indicado para el tratamiento de la artritis psoriásica (APs) activa en pacientes adultos que han tenido una respuesta inadecuada, o han presentado intolerancia al tratamiento previo con un FAME (ver sección 5.1).
Psoriasis
Otezla está indicado en el tratamiento de la psoriasis en placas crónica de moderada a grave en pacientes adultos que no han respondido o tienen contraindicado o no toleran otro tratamiento sistémico, incluyendo ciclosporina, metotrexato o psoraleno y luz ultravioleta A (PUVA).
4.2 Posología y forma de administración
El tratamiento con Otezla se debe iniciar por un médico especialista con experiencia en el diagnóstico y tratamiento de la psoriasis o de la artritis psoriásica.
Posología
La dosis recomendada de Otezla es de 30 mg dos veces al día por vía oral, por la mañana y por la noche, cada 12 horas aproximadamente, sin restricciones de alimentos. Es necesario un programa inicial de escalado de dosis como se muestra en la Tabla 1. Después del escalado inicial de la dosis, no es necesario un re-escalado de la dosis.
Tabla 1: Programa de escalado de dosis
|
Día 1 |
Día 2 |
Día 3 |
Día 4 |
Día 5 |
Día 6 y siguientes | |||||
|
a. m. |
a. m. |
p. m. |
a. m. |
p. m. |
a. m. |
p. m. |
a. m. |
p. m. |
a. m. |
p. m. |
|
10 mg |
10 mg |
10 mg |
10 mg |
20 mg |
20 mg |
20 mg |
20 mg |
30 mg |
30 mg |
30 mg |
Si el paciente se olvida una dosis, se debe tomar la siguiente dosis lo antes posible. Si está cerca de la hora de la siguiente dosis, no se debe tomar la dosis olvidada sino que se tomará la siguiente dosis a su hora habitual.
Durante los ensayos pivotales se observó la máxima mejoría en las primeras 24 semanas de tratamiento. Si un paciente no muestra indicios de beneficio terapéutico después de 24 semanas, se debe reconsiderar el tratamiento. Se debe evaluar la respuesta del paciente al tratamiento de forma periódica. No se dispone de experiencia clínica pasadas las 52 semanas (ver sección 5.1).
Poblaciones especiales
Pacientes de edad avanzada
No se requiere un ajuste de la dosis en esta población de pacientes (ver secciones 4.8 y 5.2).
Pacientes con insuficiencia renal
No es necesario un ajuste de la dosis en pacientes con insuficiencia renal leve y moderada. La dosis de apremilast se debe reducir a 30 mg una vez al día en pacientes con insuficiencia renal grave (aclaramiento de la creatinina (CLcr) menor de 30 ml por minuto, estimado mediante la fórmula de Cockcroft-Gault). Para el escalado inicial de la dosis en este grupo, se recomienda tomar únicamente las dosis de Otezla de la mañana del programa de la Tabla 1 y saltarse las dosis de la noche (ver sección 5.2).
Pacientes con insuficiencia hepática
No es necesario un ajuste de la dosis en pacientes con insuficiencia hepática (ver sección 5.2).
Población _ pediátrica
No se ha establecido la seguridad y eficacia de apremilast en niños de 0 a 17 años. No se dispone de datos.
Forma de administración
Otezla se administra por vía oral. Los comprimidos recubiertos con película se deben tragar enteros y se pueden tomar con o sin alimentos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Embarazo (ver sección 4.6)
4.4 Advertencias y precauciones especiales de empleo
Los pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia de lactasa de Lapp o malabsorción de glucosa-galactosa no deben tomar este medicamento.
La dosis de Otezla se debe reducir a 30 mg una vez al día en pacientes con insuficiencia renal grave (ver secciones 4.2 y 5.2).
Se debe monitorizar periódicamente el peso de los pacientes que, al comienzo del tratamiento, tengan un peso inferior al normal. En caso de una pérdida de peso clínicamente significativa y de causa desconocida, el médico general debe evaluar a estos pacientes y se debe considerar la interrupción del tratamiento.
4.5 Interacción con otros medicamentos y otras formas de interacción
La administración concomitante con el inductor enzimático potente del citocromo P450 3A4 (CYP3A4), rifampicina, produjo una reducción de la exposición sistémica de apremilast, lo que puede producir una pérdida de la eficacia de apremilast. Por lo tanto, no se recomienda usar inductores enzimáticos potentes del citocromo CYP3A4 (p. ej., rifampicina, fenobarbital, carbamazepina, fenitoína y hierba de San Juan) junto con apremilast. La administración concomitante de apremilast con dosis múltiples de rifampicina produjo una disminución en el área bajo la curva de concentración plasmática (AUC) de apremilast y de la concentración sérica máxima (Cmáx) aproximadamente del 72 % y del 43 %, respectivamente. La exposición de apremilast se reduce cuando se administra de forma concomitante con inductores potentes de CYP3A4 (p. ej., rifampicina) y puede dar lugar a una respuesta clínica reducida.
En los estudios clínicos se ha administrado apremilast de forma concomitante con tratamiento tópico (incluidos corticoesteroides, champú de alquitrán de hulla y preparados de ácido salicílico para el cuero cabelludo) y fototerapia UVB.
No hubo interacciones medicamentosas clínicamente significativas entre ketoconazol y apremilast. Se puede administrar apremilast de forma concomitante con un inhibidor potente de CYP3A4 como ketoconazol.
No hubo interacciones medicamentosas farmacocinéticas entre apremilast y metotrexato en pacientes con artritis psoriásica. Se puede administrar apremilast de forma concomitante con metotrexato.
No hubo interacciones medicamentosas farmacocinéticas entre apremilast y los anticonceptivos orales que contienen etinilestradiol y norgestimato. Se puede administrar apremilast de forma concomitante con anticonceptivos orales.
4.6 Fertilidad, embarazo y lactancia
Mujeres con capacidad de gestación
Se debe descartar el embarazo antes de poder iniciar el tratamiento. Las mujeres con capacidad de gestación deben utilizar un método anticonceptivo efectivo para prevenir el embarazo durante el tratamiento.
Embarazo
Los datos relativos al uso de apremilast en mujeres embarazadas son limitados.
Apremilast está contraindicado durante el embarazo. Los efectos de apremilast sobre el embarazo incluyeron pérdida embriofetal en ratones y monos, disminución del peso fetal y retraso en la osificación en ratones a dosis superiores a la dosis máxima humana actualmente recomendada. No se observaron dichos efectos cuando la exposición en los animales fue a dosis 1,3 veces la exposición clínica (ver sección 5.3).
Lactancia
Se ha detectado apremilast en la leche de ratones hembra en periodo de lactancia (ver sección 5.3). Se desconoce si apremilast, o sus metabolitos, se excretan en la leche materna humana. No puede descartarse un riesgo para el lactante; por lo tanto, apremilast no se debe utilizar durante la lactancia.
Fertilidad
No hay datos de fertilidad disponibles en seres humanos. En los estudios con ratones no se observaron efectos adversos sobre la fertilidad de los machos expuestos a niveles de 3 veces la exposición clínica ni de las hembras expuestas a niveles de 1 vez la exposición clínica. Para los datos de fertilidad de los estudios preclínicos ver sección 5.3.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de apremilast sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas Resumen del perfil de seguridad
Las reacciones adversas notificadas con mayor frecuencia en los estudios clínicos de fase III fueron trastornos gastrointestinales (GI) que incluyen diarrea (15,7 %) y náuseas (13,9 %). Estas reacciones adversas GI fueron, en su mayoría, de intensidad leve a moderada, con un 0,3 % de los casos de diarrea y un 0,3 % de los casos de náuseas notificados como severos. Estas reacciones adversas ocurrieron por lo general en las 2 primeras semanas de tratamiento y normalmente remitieron en 4 semanas. Las otras reacciones adversas notificadas con mayor frecuencia incluyeron infecciones del tracto respiratorio superior (8,4 %), cefalea (7,9 %) y cefalea tensional (7,2 %). En general, la mayoría de las reacciones adversas se consideraron de intensidad leve o moderada.
Las reacciones adversas más frecuentes que dieron lugar a la interrupción durante las primeras 16 semanas de tratamiento fueron diarrea (1,7 %) y náuseas (1,5 %). La incidencia global de reacciones adversas graves fue baja y no indicó afectación de ningún órgano ni sistema específico.
En los estudios clínicos de apremilast se observaron con poca frecuencia reacciones de hipersensibilidad (ver sección 4.3).
Tabla de reacciones adversas
Las reacciones adversas observadas en los pacientes tratados con apremilast se incluyen a continuación según el Sistema de Clasificación por de Órganos y sistemasl (SOC por sus siglas en inglés, System Organ Class) y la frecuencia de todas las reacciones adversas. Dentro de cada SOC y grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Las reacciones adversas al medicamento se determinaron basándose en los datos del programa de desarrollo clínico de apremilast. Las frecuencias de las reacciones adversas al medicamento son las notificadas en los grupos de apremilast de los cuatro estudios de fase III de artritis psoriásica (n = 1945) o de los dos estudios de fase III de psoriasis (n = 1184) (en la tabla 2 se representa la frecuencia más alta de los dos grupos de datos).
Las frecuencias se definen como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000).
Tabla 2. Resumen de las reacciones adversas en los estudios clínicos de fase III de artritis psoriásica (APs) y/o psoriasis (PSOR)
|
Sistema de Clasificación de Órganos |
Frecuencia |
Reacción adversa |
|
Infecciones e infestaciones |
Frecuentes |
Bronquitis |
|
Infección del tracto respiratorio superior | ||
|
Nasofaringitis* | ||
|
Trastornos del sistema inmunológico |
Poco frecuentes |
Hipersensibilidad |
|
Trastornos del metabolismo y de la nutrición |
Frecuentes |
Disminución del apetito* |
|
Trastornos psiquiátricos |
Frecuentes |
Insomnio |
|
Sistema de Clasificación de Órganos |
Frecuencia |
Reacción adversa |
|
Trastornos del sistema nervioso |
Frecuentes |
Migraña* |
|
Cefalea tensional* | ||
|
Cefalea* | ||
|
Trastornos respiratorios, torácicos y mediastínicos |
Frecuentes |
Tos |
|
Trastornos gastrointestinales |
Muy frecuentes |
Diarrea* |
|
Náuseas* | ||
|
Frecuentes |
Vómitos* | |
|
Dispepsia | ||
|
Movimientos intestinales frecuentes | ||
|
Dolor abdominal superior* | ||
|
Enfermedad por reflujo gastroesofágico | ||
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
Erupción |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Frecuentes |
Dolor de espalda* |
|
Trastornos generales y alteraciones en el lugar de administración |
Frecuentes |
Fatiga |
|
Exploraciones complementarias |
Poco frecuentes |
Pérdida de peso |
*Al menos una de estas reacciones adversas fue notificada como grave
Descripción de reacciones adversas seleccionadas
Pérdida de _peso
El peso de los pacientes se determinó de forma rutinaria en los estudios clínicos. La pérdida de peso media observada en los pacientes tratados hasta 52 semanas con apremilast fue de 1,99 kg. Se observó una pérdida de peso del 5 al 10 % en un total del 14,3 % de los pacientes tratados con apremilast, mientras que en el 5,7 % de los pacientes tratados con apremilast se observó una pérdida de peso mayor del 10 %. Ninguno de estos pacientes presentó consecuencias clínicas evidentes debido a la pérdida de peso. Un total del 0,1 % de los pacientes tratados con apremilast interrumpió el tratamiento debido a la reacción adversa de pérdida de peso.
Ver la advertencia adicional en la sección 4.4 para los pacientes con un peso inferior al normal al inicio del tratamiento.
Depresión
Durante el periodo controlado con placebo de los ensayos clínicos de fase III de psoriasis, el 1,2 % (14/1184) de los pacientes tratados con apremilast notificó depresión, en comparación con el 0,5 % (2/418) de los pacientes tratados con placebo. Ninguno de estos casos de depresión fue grave ni dio lugar a la retirada del estudio.
Poblaciones especiales
Pacientes de edad avanzada
No se observaron diferencias globales entre el perfil de seguridad de los pacientes de edad avanzada (>65 años) y el de los pacientes adultos más jóvenes (<65 años) en los estudios clínicos.
Pacientes con insuficiencia hepática
No se evaluó la seguridad de apremilast en los pacientes con artritis psoriásica o con psoriasis y con insuficiencia hepática.
Pacientes con insuficiencia renal
En los estudios clínicos de artritis psoriásica o de psoriasis, el perfil de seguridad observado en los pacientes con insuficiencia renal leve fue comparable al de los pacientes con función renal normal. No se evaluó la seguridad de apremilast en los pacientes con artritis psoriásica o con psoriasis y con insuficiencia renal moderada o grave en los estudios clínicos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Se estudió apremilast en sujetos sanos a una dosis máxima diaria total de 100 mg (administrada como 50 mg dos veces al día) durante 4,5 días y no se observaron indicios de toxicidad limitante de la dosis. En caso de sobredosis, se recomienda monitorizar al paciente para detectar cualquier signo o síntoma de efectos adversos e instaurar el tratamiento sintomático adecuado. En caso de sobredosis, se recomienda un cuidado sintomático y de soporte.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inmunosupresores, inmunosupresores selectivos, código ATC: L04AA32 Mecanismo de acción
Apremilast, una molécula pequeña que se administra por vía oral y que inhibe la fosfodiesterasa 4 (PDE4), actúa dentro de la célula modulando una red de mediadores proinflamatorios y antiinflamatorios. PDE4 es una fosfodiesterasa (PDE) específica del adenosín monofosfato cíclico (AMPc) y la PDE dominante en las células inflamatorias. La inhibición de PDE4 eleva los niveles intracelulares de AMPc, que a su vez regula disminuyendo la respuesta inflamatoria mediante modulación de la expresión de TNF-a, IL-23, IL-17 y otras citocinas inflamatorias. El AMP cíclico modula también los niveles de citocinas antiinflamatorias como IL-10. Estos mediadores proinflamatorios y antiinflamatorios están implicados en la artritis psoriásica y en la psoriasis.
Efectos farmacodinámicos
En los estudios clínicos en pacientes con artritis psoriásica, apremilast moduló de manera significativa, aunque sin inhibir por completo, los niveles de proteínas plasmáticas de IL-1a, IL-6, IL-8, MCP-1, MIP-ip, MMP-3 y TNF-a. Tras 40 semanas de tratamiento con apremilast, se observó una disminución en los niveles de proteínas plasmáticas de IL-17 e IL-23, y un aumento de IL-10. En los ensayos clínicos en pacientes con psoriasis, apremilast disminuyó el grosor epidérmico de la piel lesionada, la infiltración celular inflamatoria y la expresión de los genes proinflamatorios, incluidos aquellos que codifican para el óxido nítrico sintasa inducible (iNOS), IL-12/IL-23p40, IL-17A, IL-22 e IL-8.
Apremilast administrado a dosis de hasta 50 mg dos veces al día no prolongó el intervalo QT en sujetos sanos.
Experiencia en ensayos clínicos Artritis psoriásica
Se evaluaron la seguridad y la eficacia de apremilast en 3 estudios multicéntricos, aleatorizados, controlados con placebo y doble ciego (estudios PALACE 1, PALACE 2 y PALACE 3) de diseño similar en pacientes adultos con artritis psoriásica activa (>3 articulaciones inflamadas y >3 articulaciones dolorosas) a pesar del tratamiento previo con FAMEs de molécula pequeña o biológicos. Un total de 1493 pacientes fueron aleatorizados y tratados con placebo, 20 mg de apremilast o 30 mg de apremilast, administrado por vía oral dos veces al día (2 v/d).
Los pacientes de estos estudios tenían un diagnóstico de artritis psoriásica desde al menos 6 meses. Asimismo, en el estudio PALACE 3 era necesario tener una lesión psoriásica en la piel valorable (por lo menos de 2 cm de diámetro). Apremilast se utilizó en monoterapia (34,8 %) o en combinación con dosis estables de FAMEs de molécula pequeña (65,2 %). Los pacientes recibieron apremilast en combinación con uno o más de los siguientes: metotrexato (MTX, <25 mg/semana, 54,5 %), sulfasalazina (SSZ, <2 g/día, 9,0 %) y leflunomida (LEF; <20 mg/día, 7,4 %). No estaba permitido el tratamiento concomitante con FAMEs biológicos, incluidos los bloqueadores de TNF. Se incluyeron en los 3 estudios pacientes con cada subtipo de artritis psoriásica, incluidas poliartritis simétrica (62,0 %), oligoartritis asimétrica (26,9 %), artritis en las articulaciones interfalángeas distales (IFD) (6,2 %), artritis mutilante (2,7 %) y espondilitis predominante (2,1 %). Se incluyó a pacientes con entesopatía preexistente (63 %) o dactilitis preexistente (42 %). El 76,4 % de los pacientes habían sido tratados previamente solo con FAMEs de molécula pequeña y el 22,4 % de los pacientes habían sido tratados previamente con FAMEs biológicos, lo que incluye el 7,8 % que no había respondido al tratamiento previo con un FAME biológico. La mediana de duración de la artritis psoriásica fue de 5 años.
Basándose en el diseño del estudio, los pacientes cuyos recuentos de articulaciones dolorosas e inflamadas no habían mejorado como mínimo en un 20 % fueron considerados no respondedores en la semana 16. Los pacientes tratados con placebo que fueron considerados no respondedores fueron reasignados aleatoriamente en una proporción 1:1, de forma enmascarada, a 20 mg o a 30 mg de apremilast dos veces al día. En la semana 24, todos los demás pacientes tratados con placebo pasaron a recibir tratamiento con 20 mg o con 30 mg de apremilast dos veces al día.
El criterio principal de valoración fue el porcentaje de pacientes que obtuvieron una respuesta ACR20 en la semana 16 conforme a los criterios del Colegio Americano de Reumatología (ACR por sus siglas en inglés, American College of Rheumatology).
El tratamiento con apremilast produjo mejorías significativas en los signos y síntomas de la artritis psoriásica, como determinaron los criterios de respuesta ACR20, en comparación con el placebo, en la semana 16. La proporción de pacientes con respuestas ACR20/50/70 en los estudios PALACE 1, PALACE 2 y PALACE 3 y los datos agrupados de los estudios PALACE 1, PALACE 2 y PALACE 3, con 30 mg de apremilast dos veces al día en la semana 16, se muestran en la tabla 3. Las respuestas ACR20/50/70 se mantenían en la semana 24.
Entre los pacientes que inicialmente fueron aleatorizados al tratamiento con 30 mg de apremilast dos veces al día, las tasas de respuesta ACR20/50/70 se mantuvieron hasta la semana 52 en los estudios agrupados PALACE 1, PALACE 2 y PALACE 3 (Figura 1).
Tabla 3. Proporción de pacientes con respuestas ACR en los estudios PALACE 1, PALACE 2 y PALACE 3 y en los estudios agrupados en la semana 16
|
PALACE1 |
PALACE 2 |
PALACE 3 |
AGRUPADOS |
|
Placebo ± |
Apremilast |
Placebo ± |
Apremilast |
Placebo ± |
Apremilast |
Placebo ± |
Apremilast | |
|
FAMEs |
30 mg 2 v/d ± FAMEs |
FAMEs |
30 mg 2 v/d ± FAMEs |
FAMEs |
30 mg 2 v/d ± FAMEs |
FAMEs |
30 mg 2 v/d ± FAMEs | |
|
Na |
N = 168 |
N = 168 |
N = 159 |
N = 162 |
N = 169 |
N = 167 |
N = 496 |
N = 497 |
|
ACR20a | ||||||||
|
Semana 16 |
19,0 % |
38,1 %** |
18,9 % |
32,1 %* |
18,3 % |
40,7 %** |
18,8 % |
37,0 %** |
|
ACR50 | ||||||||
|
Semana 16 |
6,0 % |
16,1 %* |
5,0 % |
10,5 % |
8,3 % |
15,0 % |
6,5 % |
13,9 %** |
|
ACR70 | ||||||||
|
Semana 16 |
1,2 % |
4,2 % |
0,6 % |
1,2 % |
2,4 % |
3,6 % |
1,4 % |
3,0 % |
*p <0,01 para apremilast frente a placebo.
**p <0,001 para apremilast frente a placebo. a N es el número de pacientes aleatorizados y tratados.
Figura 1 Proporción de respondedores ACR20/50/70 hasta la semana 52 en el análisis agrupado de los estudios PALACE 1, PALACE 2 y PALACE 3 (NRI*)
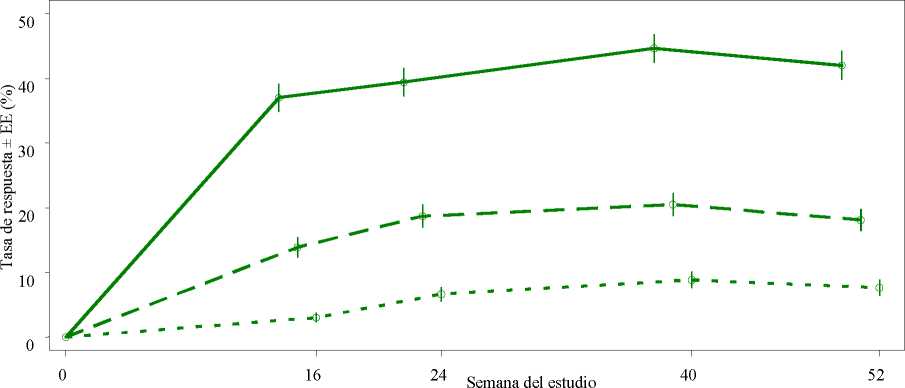
Criterici de varado!! n/m (%) n/m (%)
ACR 20 184/497 (37,0) 196/497 (39,4)
ACR 50 69/497 (13,9) 93/497 (18,7)
ACR 70 15/497 ( 3,0) 33/497 ( 6,6)
n/m (%) 222/497 (44,7) 102/497 (20,5) 44/497 ( 8,9)
n/m (%) 209/497 (42,1) 90/497 (18,1) 38/497 ( 7,6)
Criterio de valoración o o o acr 20 acr 50 ® ® o ACR 70
*NRI (Por sus siglas en inglés, Non-responder imputation): imputación de los no respondedores. Los sujetos que abandonaron antes del punto temporal y los sujetos que no tenían datos suficientes para una determinación definitiva del estado de la respuesta en el punto temporal se cuentan como no respondedores.
Entre los 497 pacientes inicialmente aleatorizados a 30 mg de apremilast dos veces al día, 375 (75 %) pacientes seguían con este tratamiento en la semana 52. En estos pacientes, las respuestas ACR20/50/70 en la semana 52 fueron del 57 %, 25 % y 11 %, respectivamente.
Las respuestas observadas en el grupo tratado con apremilast fueron similares entre los pacientes que recibieron y los que no recibieron FAMEs de forma concomitante, incluido MTX.
Los pacientes tratados previamente con FAMEs o con tratamientos biológicos que recibieron apremilast alcanzaron una mayor respuesta ACR20 en la semana 16, en comparación con los pacientes que recibieron placebo.
Se observaron respuestas ACR similares en los pacientes con diferentes subtipos de artritis psoriásica, incluida la artritis en las articulaciones IFD. El número de pacientes con los subtipos artritis mutilante y espondilitis predominante era demasiado pequeño para permitir una evaluación significativa.
En los estudios PALACE 1, PALACE 2 y PALACE 3 las mejorías en la escala de actividad de la enfermedad (DAS28 por sus siglas en inglés, Disease Activity Score) utilizando la proteína C reactiva (PCR) y en la proporción de pacientes que alcanzó los criterios de respuesta de la artritis psoriásica (PsARC por sus siglas en inglés, Psoriatic Arthritis response criteria) modificados fueron mayores en elgrupo de apremilast, en comparación con placebo en la semana 16 (valor p nominal p<0,0004, valor p <0,0017, respectivamente). Estas mejorías se mantenían en la semana 24. Entre los pacientes que seguían con el tratamiento con apremilast al que fueron aleatorizados al comienzo del estudio, la puntuación DAS28 (PCR) y la respuesta en PsARC se mantuvieron hasta la semana 52.
Se observaron mejorías en las semanas 16 y 24 en los parámetros de la actividad periférica característica de la artritis psoriásica (p. ej., número de articulaciones inflamadas, número de articulaciones dolorosas/doloridas, dactilitis y entesitis) y en las manifestaciones cutáneas de psoriasis en los pacientes tratados con apremilast. Entre los pacientes que seguían con el tratamiento con apremilast al que fueron aleatorizados al comienzo del estudio, estas mejorías se mantuvieron hasta la semana 52.
Función física y calidad de vida relacionada con la salud
Los pacientes tratados con apremilast demostraron una mejoría estadísticamente significativa en la función física, como determinó el cambio con respecto a la situación basal en el índice de discapacidad del cuestionario de evaluación de la salud (HAQ-DI por sus siglas en inglés, Health Assessment Questionnaire Disability Index), en comparación con placebo a las 16 semanas en los estudios PALACE 1, PALACE 2 y PALACE 3 y en los estudios agrupados. La mejoría en las puntuaciones en HAQ-DI se mantuvo en la semana 24.
Entre los pacientes que fueron inicialmente aleatorizados al tratamiento con 30 mg de apremilast dos veces al día, el cambio con respecto a la situación basal en la puntuación en HAQ-DI en la semana 52 fue de -0,333 en el grupo de 30 mg de apremilast dos veces al día en un análisis agrupado de la fase abierta de los estudios PALACE 1, PALACE 2 y PALACE 3.
En los estudios PALACE 1, PALACE 2 y PALACE 3 se demostraron mejorías significativas en la calidad de vida relacionada con la salud, como determinaron los cambios con respecto a la situación basal en las puntuaciones del dominio de función física del cuestionario de la salud abreviado versión 2 (SF-36v2) y en las puntuaciones de la evaluación funcional del tratamiento de las enfermedades crónicas - fatiga (FACIT-fatiga) en los pacientes tratados con apremilast en comparación con placebo en las semanas 16 y 24. Entre los pacientes que seguían con el tratamiento con apremilast al que fueron aleatorizados al comienzo del estudio, la mejoría en la función física y en FACIT-fatiga se mantuvo hasta la semana 52.
Psoriasis
Se evaluaron la seguridad y la eficacia de apremilast en 2 estudios multicéntricos, aleatorizados, controlados con placebo y doble ciego (estudios ESTEEM 1 y ESTEEM 2) en los que participaron un total de 1257 pacientes con psoriasis en placas de moderada a grave que tenían un área de superficie corporal (BSA por sus siglas en inglés, Body surface area,) afectada >10 %, una puntuación en el índice de gravedad y área de la psoriasis (PASI por sus siglas en inglés, Psoriasis Area and Severity Index) de >12, una evaluación global estática del médico (sPGA por sus siglas en inglés, Static
Physician Global Assessment) >3 (moderada o grave), y que eran candidatos a fototerapia o tratamiento sistémico.
El diseño de los estudios era similar hasta la semana 32. En ambos estudios, los pacientes fueron aleatotorizados en una proporción 2:1 a 30 mg de apremilast dos veces al día o a placebo durante 16 semanas (fase controlada con placebo) y desde la semana 16 hasta la 32 todos los pacientes recibieron 30 mg de apremilast dos veces al día (fase de mantenimiento). Durante la fase de retirada del tratamiento aleatorizado (semanas 32-52), los pacientes originalmente aleatorizados a apremilast que alcanzaron al menos una disminución del 75 % en la puntuación del PASI (PASI-75) (ESTEEM 1) o una disminución del 50 % en la puntuación (PASI-50) (ESTEEM 2) fueron reasignados aleatoriamente en la semana 32 a placebo o a 30 mg de apremilast dos veces al día. Los pacientes que fueron reasignados a placebo y que perdieron la respuesta PASI-75 (ESTEEM 1) o que perdieron el 50 % de la mejoría en el PASI en la semana 32 en comparación con la situación basal (ESTEEM 2) volvieron a recibir tratamiento con 30 mg de apremilast dos veces al día. Los pacientes que no alcanzaron la respuesta designada en el PASI en la semana 32, o que fueron inicialmente aleatorizados a placebo, siguieron con apremilast hasta la semana 52. Se permitió el uso de corticoesteroides tópicos de baja potencia en la cara, axilas e ingles, y el uso de champú de alquitrán de hulla y/o preparados de ácido salicílico para el cuero cabelludo durante los estudios. Además, en la semana 32, los sujetos que no alcanzaron una respuesta PASI-75 en ESTEEM 1 o una respuesta PASI-50 en ESTEEM 2 pudieron utilizar tratamientos tópicos para la psoriasis y/o fototerapia, además del tratamiento con 30 mg de apremilast dos veces al día.
En ambos estudios, el criterio principal de valoración fue la proporción de pacientes que alcanzó una respuesta PASI-75 en la semana 16. El principal criterio de valoración secundario fue la proporción de pacientes que alcanzó una puntuación en la sPGA de blanqueada (0) o casi blanqueada (1) en la semana 16.
La puntuación media en el PASI basal fue de 19,07 (mediana 16,80) y la proporción de pacientes con una puntuación en la sPGA basal de 3 (moderada) y 4 (grave) fue del 70,0 % y del 29,8 %, respectivamente, con una afectación media del BSA basal del 25,19 % (mediana 21,0 %). Aproximadamente el 30 % de todos los pacientes habían recibido fototerapia previa y el 54 % había recibido tratamiento sistémico convencional y/o biológico previo para el tratamiento de la psoriasis (incluidos los que no respondieron al tratamiento), de los que un 37 % había recibido tratamiento sistémico convencional previo y un 30 % tratamiento biológico previo. Aproximadamente un tercio de los pacientes no había recibido fototerapia previa ni tratamiento sistémico convencional o biológico previo. Un total del 18 % de los pacientes tenía historia de artritis psoriásica.
La proporción de pacientes que alcanzó respuestas PASI-50, PASI-75 y PASI-90, y una puntuación en la sPGA de blanqueada (0) o casi blanqueada (1), se presenta en la Tabla 4. El tratamiento con apremilast produjo una mejoría significativa de la psoriasis en placas de moderada a grave, como demostró la proporción de pacientes con respuesta PASI-75 en la semana 16, en comparación con placebo. La mejoría clínica determinada por las respuestas en sPGA, PASI-50 y PASI-90 se demostró también en la semana 16. Además, se demostró el beneficio del tratamiento con apremilast en múltiples manifestaciones de la psoriasis, incluyendo prurito, enfermedad ungueal, afectación del cuero cabelludo y medidas de calidad de vida.
Tabla 4. Respuesta clínica en la semana 16 en los estudios ESTEEM 1 y ESTEEM 2 (FASa LOCFb)
|
ESTEEM1 |
ESTEEM 2 | |||
|
Placebo |
Apremilast 30 mg 2 v/d * |
Placebo |
Apremilast 30 mg 2 v/d * | |
|
N |
282 |
562 |
137 |
274 |
|
PASIc-75, n (%) |
15 (5,3) |
186 (33,1) |
8 (5,8) |
79 (28,8) |
|
Blanqueada o casi blanqueada en sPGAd, n (%) |
11 (3,9) |
122 (21,7) |
6 (4,4) |
56 (20,4) |
|
PASI-50, n (%) |
48 (17,0) |
330 (58,7) |
27 (19,7) |
152 (55,5) |
|
PASI-90, n (%) |
1 (0,4) |
55 (9,8) |
2 (1,5) |
24 (8,8) |
|
Cambio en el BSAe (%) media ± Desviación Estándar (DE) |
-6,9 |
-47,8 |
-6,1 |
-48,4 |
|
±38,95 |
±38,48 |
±47,57 |
±40,78 | |
|
Cambio en el prurito en |
-7,3 |
-31,5 |
-12,2 |
-33,5 |
|
EVAf (mm), media ± DE |
±27,08 |
±32,43 |
±30,94 |
±35,46 |
|
Cambio en el DLQIg, |
-2,1 |
-6,6 |
-2,8 |
-6,7 |
|
media ± DE |
±5,69 |
±6,66 |
±7,22 |
±6,95 |
|
Cambio en el SF-36 MCS h, |
-1,02 |
2,39 |
0,00 |
2,58 |
|
media ± DE |
±9,161 |
±9,504 |
±10,498 |
±10,129 |
* p <0,0001 para apremilast frente a placebo, excepto en ESTEEM 2 PASI-90 y cambio en el SF-36 MCS donde p = 0,0042 y p = 0,0078, respectivamente.
a FAS (Full Analysis Set) = Conjunto de análisis completo
b LOCF (Last Observation Carried Forward)= Última observación disponible
c PASI = Índice de gravedad y área de la psoriasis
d sPGA = Evaluación global estática del médico
e BSC = Área de superficie corporal
f EVA = Escala visual analógica; 0 = nada, 100 = mucho
8 DLQI (Dermatology Life Quality Index) = Índice de calidad de vida en dermatología; 0 = no impacto, 30 = máximo impacto h SF-36 MCS = Cuestionario de salud abreviado de 36 ítems para el estudio de los resultados médicos, resumen del componente mental
El beneficio clínico de apremilast se demostró en múltiples subgrupos definidos por las características demográficas basales y las características clínicas basales de la enfermedad (incluidos la duración de la enfermedad psoriásica y los pacientes con historia de artritis psoriásica). También se demostró el beneficio clínico de apremilast al margen del uso previo de medicamentos para la psoriasis y de la respuesta a los tratamientos previos para la psoriasis. Se observaron tasas de respuesta similares en todos los intervalos de peso.
La respuesta a apremilast fue rápida, con mejorías significativamente mayores en los signos y síntomas de la psoriasis, incluido el PASI, malestar/dolor en la piel y prurito, en comparación con placebo en la semana 2. En general, se alcanzaron las respuestas en el PASI en la semana 16 y se mantuvieron hasta la semana 32.
En ambos estudios, la mejoría porcentual media en el PASI con respecto al basal permaneció estable durante la fase de retirada del tratamiento aleatorizado para los pacientes reasignados aleatoriamente a apremilast en la semana 32 (Tabla 5).
Tabla 5. Persistencia del efecto entre los sujetos aleatorizados a 30 mg de apremilast dos
veces al día en la semana 0 y reasignados aleatoriamente a 30 mg de apremilast dos veces al día en la semana 32 hasta la semana 52
|
ESTEEM 1 |
ESTEEM 2 |
|
Punto temporal |
Pacientes que alcanzaron una respuesta PASI-75 en la semana 32 |
Pacientes que alcanzaron una respuesta PASI-50 en la semana 32 | |
|
Cambio porcentual en el PASI con respecto al basal, media (%) ± DEa |
Semana 16 |
-77,7 ± 20,30 |
-69,7 ± 24,23 |
|
Semana 32 |
-88 ± 8,30 |
-76,7 ± 13,42 | |
|
Semana 52 |
-80,5 ± 12,60 |
-74,4 ± 18,91 | |
|
Cambio en el DLQI con respecto al basal, media ± DEa |
Semana 16 |
-8,3 ± 6,26 |
-7,8 ± 6,41 |
|
Semana 32 |
-8,9 ± 6,68 |
-7,7 ± 5,92 | |
|
Semana 52 |
-7,8 ± 5,75 |
-7,5 ± 6,27 | |
|
Proporción de sujetos con psoriasis del cuero cabelludo y PGA de 0 o 1, n/N (%)b |
Semana 16 |
40/48 (83,3) |
21/37 (56,8) |
|
Semana 32 |
39/48 (81,3) |
27/37 (73,0) | |
|
Semana 52 |
35/48 (72,9) |
20/37 (54,1) |
a Incluye a los sujetos reasignados aleatoriamente a 30 mg de apremilast dos veces al día en la semana 32 con un valor basal y post-basal en la semana de evaluación del estudio.
bN se basa en el número de sujetos con psoriasis basal en el cuero cabelludo moderada o superior que fueron reasignados aleatoriamente a 30 mg de apremilast dos veces al día en la semana 32. Los sujetos sin datos contaron como no respondedores.
En el estudio ESTEEM 1, aproximadamente el 61 % de los pacientes reasignados aleatoriamente a apremilast en la semana 32 tenían una respuesta PASI-75 en la semana 52. De los pacientes con al menos una respuesta PASI-75 que fueron reasignados aleatoriamente a placebo en la semana 32 durante la fase de retirada del tratamiento aleatorizado, el 11,7 % mantenía una respuesta PASI-75 en la semana 52. La mediana de tiempo hasta la pérdida de la respuesta PASI-75 entre los pacientes reasignados aleatoriamente a placebo fue de 5,1 semanas.
En el estudio ESTEEM 2, aproximadamente el 80,3 % de los pacientes reasignados aleatoriamente a apremilast en la semana 32 tenían una respuesta PASI-50 en la semana 52. De los pacientes con al menos una respuesta PASI-50 que fueron reasignados aleatoriamente a placebo en la semana 32, el 24,2 % mantenía una respuesta PASI-50 en la semana 52. La mediana de tiempo hasta la pérdida del 50 % de la mejoría en PASI de la semana 32 fue de 12,4 semanas.
Después de la retirada aleatorizada del tratamiento en la semana 32, aproximadamente el 70 % de los pacientes del estudio ESTEEM 1 y el 65,6 % de los pacientes del estudio ESTEEM 2 volvieron a alcanzar una respuesta PASI-75 (ESTEEM 1) o una respuesta PASI-50 (ESTEEM 2) después de reiniciar el tratamiento con apremilast. Debido al diseño del estudio, la duración del retratamiento fue variable, y osciló desde 2,6 hasta 22,1 semanas.
En el estudio ESTEEM 1, los pacientes aleatorizados a apremilast al comienzo del estudio que no alcanzaron una respuesta PASI-75 en la semana 32 pudieron utilizar tratamientos tópicos y/o fototerapia UVB concomitantemente entre las semanas 32 y 52. De estos pacientes, el 12 % alcanzó una respuesta PASI-75 en la semana 52 con apremilast más un tratamiento tópico y/o fototerapia.
En los estudios ESTEEM 1 y ESTEEM 2, se observaron mejorías significativas (reducciones) en la psoriasis ungueal, como determinó el cambio porcentual medio en el índice de gravedad de la psoriasis ungueal (NAPSI por sus siglas en inglés, Nail Psoriasis Severity Index) con respecto al basal en los pacientes tratados con apremilast, en comparación con los tratados con placebo en la semana 16 (p <0,0001 y p = 0,0052, respectivamente). En la semana 32 se observaron mejorías adicionales en la psoriasis ungueal en los pacientes tratados continuamente con apremilast.
En los estudios ESTEEM 1 y ESTEEM 2, se observaron mejorías significativas en la psoriasis del cuero cabelludo de intensidad al menos moderada (>3), como determinó la proporción de pacientes que alcanzó la respuesta de blanqueada (0) o mínima (1) en la evaluación global del médico de la psoriasis del cuero cabelludo (ScPGA por sus siglas en inglés, Scalp Psoriasis Physician’s Global Assessment) en la semana 16, en los pacientes tratados con apremilast en comparación con los tratados con placebo (p <0,0001 en ambos estudios). Las mejorías, en general, se mantuvieron en los sujetos que fueron reasignados aleatoriamente a Otezla en la semana 32 hasta la semana 52 (Tabla 5).
En los estudios ESTEEM 1 y ESTEEM 2 se demostraron mejorías significativas en la calidad de vida, como determinaron el índice de calidad de vida en dermatología (DLQI) y el SF-36v2MCS, en los pacientes tratados con apremilast en comparación con los tratados con placebo (Tabla 4). Las mejorías en el DLQI se mantuvieron hasta la semana 52 en los sujetos reasignados aleatoriamente a apremilast en la semana 32 (Tabla 5). Además, en el estudio ESTEEM 1 se alcanzó una mejoría significativa en el cuestionario de limitaciones laborales (WLQ-25 por sus siglas en inglés, Work Limitations Questionnaire) en los pacientes tratados con apremilast, en comparación con los tratados con placebo.
5.2 Propiedades farmacocinéticas
Absorción
Apremilast se absorbe bien, con una biodisponibilidad oral absoluta del 73 % aproximadamente y concentraciones plasmáticas máximas (Cmáx) que se alcanzan en una mediana de tiempo (tmáx) de
2,5 horas aproximadamente. La farmacocinética de apremilast es lineal, con un aumento proporcional a la dosis en la exposición sistémica en el intervalo de dosis de 10 a 100 mg al día. La acumulación es mínima cuando apremilast se administra una vez al día y aproximadamente del 53 % en sujetos sanos y del 68 % en pacientes con psoriasis cuando se administra dos veces al día. La administración con alimentos no altera la biodisponibilidad, por lo tanto, apremilast se puede administrar con o sin alimentos.
Distribución
La unión de apremilast a las proteínas plasmáticas humanas es aproximadamente del 68 %. El volumen de distribución (Vd) aparente medio es de 87 l, lo que indica distribución extravascular.
Biotransformación
Apremilast se metaboliza extensamente por las vías mediadas por CYP y no mediadas por CYP, incluidas las vías de oxidación, hidrólisis y conjugación, lo que sugiere que no es probable que la inhibición de una única vía de aclaramiento cause una interacción medicamentosa destacable. El metabolismo oxidativo de apremilast está mediado principalmente por CYP3A4, con alguna contribución menor de CYP1A2 y CYP2A6. Apremilast es el principal componente circulante tras la administración oral. Apremilast se somete a un metabolismo extenso y solo el 3 % y el 7 % del compuesto original administrado se recuperan en orina y en heces, respectivamente. El principal metabolito inactivo circulante es el conjugado glucurónido de apremilast O-desmetilado (M12).
Debido a que apremilast es un sustrato de CYP3A4, la exposición de apremilast disminuye cuando se administra concomitantemente con rifampicina, un inductor potente de CYP3A4.
In vitro, apremilast no es un inhibidor ni un inductor de las enzimas del citocromo P450. Por lo tanto, es poco probable que la administración concomitante de apremilast con sustratos de las enzimas de CYP afecte al aclaramiento y a la exposición de los principios activos que se metabolizan por las enzimas de CYP.
In vitro, apremilast es un sustrato y un inhibidor débil de la glicoproteína P (P-gp) (CI50 >50 pM); sin embargo, no se espera que ocurran interacciones medicamentosas clínicamente relevantes mediadas por la P-gp.
In vitro, apremilast tiene escaso o ningún efecto inhibidor (CI50 >10 pM) en el transportador de aniones orgánicos (OAT por sus siglas en inglés, Organic Anion Transporter) 1 y OAT3, el transportador de cationes orgánicos (OCT por sus siglas en inglés, Organic Cation Transporter) 2, el polipéptido transportador de aniones orgánicos (OATP) 1B1 y OATP1B3, o en la proteína resistente al cáncer de mama (BCRP por sus siglas en inglés, Breast Cancer Resistance Protein), y no es un sustrato de estos transportadores. Por lo tanto, es poco probable que ocurran interacciones medicamentosas clínicamente relevantes cuando apremilast se administre concomitantemente con medicamentos que son sustratos o inhibidores de estos transportadores.
Eliminación
El aclaramiento plasmático medio de apremilast es de unos 10 l/h en sujetos sanos, con una semivida de eliminación terminal de 9 horas aproximadamente. Tras la administración oral de apremilast con radio marcaje, alrededor del 58 % y del 39 % de la radiactividad se recupera en la orina y en las heces, respectivamente, con alrededor del 3 % y del 7 % de la dosis radiactiva recuperada como apremilast en la orina y en las heces, respectivamente.
Pacientes de edad avanzada
Se estudió apremilast en sujetos sanos jóvenes y de edad avanzada. La exposición en los sujetos de edad avanzada (de 65 a 85 años) es alrededor de un 13 % mayor en el AUC y alrededor de un 6 % mayor en la Cmáx para apremilast que en los sujetos jóvenes (de 18 a 55 años). Los datos farmacocinéticos son limitados en sujetos mayores de 75 años en los ensayos clínicos. No es necesario un ajuste de la dosis en pacientes de edad avanzada.
Insuficiencia renal
No hay una diferencia significativa en la farmacocinética de apremilast entre los pacientes con insuficiencia renal leve o moderada y los sujetos sanos con las mismas características (N = 8 de cada). Los resultados respaldan que no es necesario ajustar la dosis en pacientes con insuficiencia renal leve o moderada. Se debe reducir la dosis de apremilast a 30 mg una vez al día en pacientes con insuficiencia renal grave (Filtración Glomerular estimada (FGe) menor de 30 ml/min/1,73 m2 o CLcr <30 ml/min). En 8 pacientes con insuficiencia renal grave que recibieron una dosis de 30 mg de apremilast, el AUC y la Cmáx de apremilast aumentaron en aproximadamente un 89 % y un 42 %, respectivamente.
Insuficiencia hepática
La farmacocinética de apremilast y la de su principal metabolito M12 no se ven afectadas por la insuficiencia hepática moderada o grave. No es necesario un ajuste de la dosis en pacientes con insuficiencia hepática.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad y toxicidad a dosis repetidas. No hay indicios de potencial inmunotóxico, de irritación dérmica o fototóxico.
Fertilidad y desarrollo embrionario temprano
En un estudio de fertilidad con ratones macho, la administración oral de apremilast a dosis de 1, 10, 25 y 50 mg/kg/día no produjo efectos en la fertilidad de los machos; el nivel sin efecto adverso observado (NOAEL por sus siglas en inglés, No Observed Adverse EffectLevel) para la fertilidad de los machos fue mayor de 50 mg/kg/día (3 veces la exposición clínica).
En un estudio combinado de toxicidad en el desarrollo embriofetal y de fertilidad en ratones hembra con dosis orales de 10, 20, 40 y 80 mg/kg/día se observaron una prolongación de los ciclos estrales y un mayor tiempo hasta el apareamiento con las dosis de 20 mg/kg/día y superiores. A pesar de esto, todos los ratones se aparearon y las tasas de gestación no se vieron afectadas. El nivel sin efecto observado (NOEL por sus siglas en inglés, No Observed Effect Level)para la fertilidad de las hembras fue de 10 mg/kg/día (1,0 vez la exposición clínica).
Desarrollo embriofetal
En un estudio combinado de toxicidad en el desarrollo embriofetal y de fertilidad en ratones hembra con dosis orales de 10, 20, 40 y 80 mg/kg/día, los pesos absolutos y/o relativos de los corazones de las madres aumentaron con las dosis de 20, 40 y 80 mg/kg/día. Se observaron un aumento del número de resorciones tempranas y una disminución del número de tarsos osificados con las dosis de 20, 40 y 80 mg/kg/día. Se observó una reducción de los pesos fetales y una osificación retardada del hueso supraoccipital del cráneo con las dosis de 40 y 80 mg/kg/día. En los ratones, el NOEL en el desarrollo y en la madre fue de 10 mg/kg/día (1,3 veces la exposición clínica).
En un estudio de toxicidad en el desarrollo embriofetal en monos, dosis orales de 20, 50, 200 y 1000 mg/kg/día dieron lugar a un aumento, relacionado con la dosis, de pérdidas prenatales (abortos) con las dosis de 50 mg/kg/día y superiores; no se observó ningún efecto relacionado con el medicamento del ensayo en las pérdidas prenatales a la dosis de 20 mg/kg/día (1,4 veces la exposición clínica).
Desarrollo prenatal y postnatal
En un estudio prenatal y postnatal se administró apremilast por vía oral a ratones hembra preñados a dosis de 10, 80 y 300 mg/kg/día desde el día 6 de gestación hasta el día 20 de lactancia. Se observaron reducciones en el peso corporal y en la ganancia de peso de las madres, así como un caso de muerte asociada a dificultad en el parto con la dosis de 300 mg/kg/día. También se observaron signos físicos de toxicidad materna asociados al parto en un ratón con la dosis de 80 mg/kg/día y con la dosis de 300 mg/kg/día. Se observó un aumento del número de muertes perinatales y postnatales de las crías y una reducción en la ganancia de peso de las crías durante la primera semana de lactancia con dosis >80 mg/kg/día (>4,0 veces la exposición clínica). No hubo efectos asociados a apremilast en la duración de la gestación, el número de ratones preñados al final del periodo de gestación, el número de ratones que parieron una camada, ni ningún efecto en el desarrollo de las crías pasado el día 7 de vida. Es probable que los efectos en el desarrollo de las crías observados durante la primera semana del periodo postnatal estuvieran asociados a la toxicidad de apremilast en las crías (peso y viabilidad de las crías reducidos) y/o a la falta de cuidados maternos (mayor incidencia de ausencia de leche en los estómagos de las crías). Todos los efectos en el desarrollo se observaron durante la primera semana del periodo postnatal; no se observaron efectos relacionados con apremilast durante el resto de los periodos antes o después del destete, incluidos los parámetros de maduración sexual, conductuales, apareamiento, fertilidad y uterinos. El NOEL en el ratón en cuanto a la toxicidad de las madres y a la primera generación (F1) fue de 10 mg/kg/día (1,3 veces el AUC clínica).
Estudios de carcinogenicidad
Los estudios de carcinogenicidad en ratones y ratas no mostraron indicios de carcinogenicidad asociada al tratamiento con apremilast.
Estudios de genotoxicidad
Apremilast no es genotóxico. Apremilast no indujo mutaciones en el ensayo de Ames ni aberraciones cromosómicas en cultivos de linfocitos de sangre periférica humana en presencia o ausencia de activación metabólica. Apremilast no fue clastogénico en un ensayo de micronúcleos en ratón in vivo con dosis de hasta 2000 mg/kg/día.
Otros estudios
No hay indicios de potencial inmunotóxico, de irritación dérmica o fototóxico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido Celulosa microcristalina Lactosa monohidrato Croscarmelosa sódica
Estearato de magnesio
Cubierta pelicular Alcohol de polivinilo Dióxido de titanio (E171)
Macrogol 3350 Talco
Óxido de hierro rojo (E172)
Los comprimidos de 30 mg contienen también óxido de hierro amarillo (E172) y óxido de hierro negro (E172).
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
21 meses.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30°C.
6.5 Naturaleza y contenido del envase
Blísters de PVC/lámina de aluminio que contienen 14 comprimidos recubiertos con película, en tamaños de envase de 56 comprimidos (30 mg) y 168 comprimidos (30 mg).
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd.
1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
8. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/981/002
EU/1/14/981/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE
COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN
SEGURA Y EFICAZ DEL MEDICAMENTO
Nombre y dirección del fabricante responsable de la liberación de los lotes
Celgene Europe Limited
1 Longwalk Road
Stockley Park
Uxbridge
Middlesex
UB11 1DB
Reino Unido
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (Ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará el primer informe periódico de seguridad para este medicamento en un plazo de 6 meses después de la autorización. Posteriormente, el TAC presentará informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR Tarjeta tipo estuche que contiene un pack de inicio de tratamiento para 2 semanas
1. NOMBRE DEL MEDICAMENTO
Otezla 10 mg comprimidos recubiertos con película Otezla 20 mg comprimidos recubiertos con película Otezla 30 mg comprimidos recubiertos con película apremilast
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 10 mg, 20 mg o 30 mg de apremilast.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto .
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Comprimidos recubiertos con película Pack de inicio de tratamiento 4 comprimidos recubiertos con película de 10 mg 4 comprimidos recubiertos con película de 20 mg 19 comprimidos recubiertos con película de 30 mg
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30°C.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd. 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/981/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
MEDICAMENTO SUJETO A PRESCRIPCIÓN MÉDICA.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Otezla 10 mg Otezla 20 mg Otezla 30 mg
Blíster (Información impresa directamente en la tarjeta tipo estuche con el blíster en blanco sellado en el interior).
1. NOMBRE DEL MEDICAMENTO
Otezla 10 mg comprimidos Otezla 20 mg comprimidos Otezla 30 mg comprimidos
apremilast
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
Caja_
1. NOMBRE DEL MEDICAMENTO
Otezla 30 mg comprimidos recubiertos con película apremilast
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 30 mg de apremilast.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto .
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Comprimidos recubiertos con película 56 comprimidos recubiertos con película 168 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30°C.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd. 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/981/002
EU/1/14/981/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
MEDICAMENTO SUJETO A PRESCRIPCIÓN MÉDICA.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Otezla 30 mg
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLÍSTER
1. NOMBRE DEL MEDICAMENTO
Otezla 30 mg comprimidos apremilast
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
B. PROSPECTO
Prospecto: información para el paciente
Otezla 10 mg comprimidos recubiertos con película Otezla 20 mg comprimidos recubiertos con película Otezla 30 mg comprimidos recubiertos con película
Apremilast
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Otezla y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Otezla
3. Cómo tomar Otezla
4. Posibles efectos adversos
5. Conservación de Otezla
6. Contenido del envase e información adicional
1. Qué es Otezla y para qué se utiliza Qué es Otezla
Otezla contiene el principio activo “apremilast”. Pertenece a un grupo de medicamentos llamados inhibidores de la fosfodiesterasa 4, que ayudan a reducir la inflamación.
Para qué se utiliza Otezla
Otezla se utiliza para tratar a adultos con las siguientes enfermedades:
• Artritis psoriásica: si no puede utilizar otro tipo de medicamentos llamados “Fármacos Antirreumáticos Modificadores de la Enfermedad” (FAMEs) o cuando ya ha probado uno de estos medicamentos y no ha funcionado.
• Psoriasis en placas de moderada a grave: si no puede utilizar uno de los siguientes tratamientos o cuando ya ha probado uno de estos tratamientos y no ha funcionado:
- fototerapia: un tratamiento en el que ciertas zonas de la piel se exponen a luz ultravioleta
- tratamiento sistémico: un tratamiento que actúa en todo el cuerpo en vez de a una zona
localizada, como la “ciclosporina” o el “metotrexato”.
Qué es la artritis psoriásica
La artritis psoriásica es una enfermedad inflamatoria de las articulaciones, generalmente va acompañada de psoriasis, una enfermedad inflamatoria de la piel.
Qué es la psoriasis en placas
La psoriasis es una enfermedad inflamatoria de la piel, que puede producir lesiones rojas, descamativas, engrosadas, con picor o dolorosas sobre la piel, y también puede afectar al cuero cabelludo y a las uñas.
Cómo actúa Otezla
La artritis psoriásica y la psoriasis son por lo general enfermedades crónicas que actualmente no tienen cura. Otezla actúa reduciendo la actividad de una enzima del organismo que se llama “fosfodiesterasa 4”, que está involucrada en el proceso inflamatorio. Al reducir la actividad de esta enzima, Otezla puede ayudar a controlar la inflamación asociada a la artritis psoriásica y a la psoriasis y, de este modo, reducir los signos y los síntomas de estas enfermedades.
En la artritis psoriásica, el tratamiento con Otezla produce una mejoría en las articulaciones inflamadas y dolorosas y puede mejorar su función física general.
En la psoriasis, el tratamiento con Otezla reduce las placas de psoriasis en la piel y otros signos y síntomas de la enfermedad.
Otezla también ha mostrado que mejora la calidad de vida de los pacientes con psoriasis o con artritis psoriásica. Esto significa que el impacto de su enfermedad en las actividades cotidianas, en las relaciones y en otros factores debe ser menor que antes.
2. Qué necesita saber antes de empezar a tomar Otezla No tome Otezla:
- si es alérgico a apremilast o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si está embarazada o cree que pudiera estarlo
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar Otezla.
Si su médico considera que tiene un peso inferior al normal y usted observa una pérdida involuntaria de peso mientras está en tratamiento con Otezla, debe informar a su médico.
Si tiene problemas de riñón graves, entonces la dosis recomendada de Otezla es de 30 mg una vez al día (dosis de la mañana). Su médico le indicará cómo aumentar la dosis cuando empiece a tomar Otezla por primera vez.
Niños y adolescentes
Otezla no se ha estudiado en niños y adolescentes; por lo tanto, no se recomienda utilizarlo en niños y adolescentes de 17 años o menores.
Uso de Otezla con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento. Esto incluye los medicamentos obtenidos sin receta y los medicamentos a base de plantas. Esto se debe a que Otezla puede afectar a la forma de actuar de otros medicamentos. Además, algunos medicamentos pueden afectar a la forma de actuar de Otezla.
En concreto, informe a su médico o farmacéutico antes de empezar a tomar Otezla si está tomando alguno de los siguientes medicamentos:
• rifampicina: un antibiótico que se utiliza para la tuberculosis;
• fenitoína, fenobarbital y carbamazepina: medicamentos que se utilizan en el tratamientos de las crisis convulsivas o de la epilepsia;
• hierba de San Juan: un medicamento a base de plantas que se utiliza para la ansiedad y la depresión leves.
Embarazo y lactancia
Hay poca información relativa a los efectos de Otezla durante el embarazo. No se debe quedar embarazada mientras toma este medicamento y debe utilizar métodos anticonceptivos efectivos durante el tratamiento con Otezla. Se desconoce si este medicamento pasa a la leche materna. Otezla no se debe utilizar mientras se esté dando el pecho .
Si cree que podría estar embarazada o tiene intención de quedarse embarazada o si está en periodo de lactancia o tiene intención de hacerlo, informe a su médico.
Conducción y uso de máquinas
La influencia de Otezla sobre la capacidad para conducir y utilizar máquinas es nula.
Otezla contiene lactosa
Otezla contiene lactosa (un tipo de azúcar). Si su médico le ha indicado que padece una intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento.
3. Cómo tomar Otezla
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico.
En caso de duda, consulte de nuevo a su médico o farmacéutico.
Cuánto tomar
• Cuando empiece a tomar Otezla por primera vez, recibirá un “pack de inicio de tratamiento” que contiene todas las dosis de la forma descrita en la tabla siguiente.
• El “pack de inicio de tratamiento” está claramente etiquetado para estar seguros de que toma la dosis correcta a la hora correcta.
• Su tratamiento comenzará a una dosis más baja e irá aumentando paulatinamente durante los primeros 6 días de tratamiento.
• El “pack de inicio de tratamiento” también contendrá el número suficiente de comprimidos para otros 8 días a la dosis recomendada (días del 7 al 14).
• La dosis recomendada de Otezla es de 30 mg dos veces al día después de completar la fase de escalado, una dosis de 30 mg por la mañana y una dosis de 30 mg por la noche, cada 12 horas aproximadamente, con o sin alimentos.
• Esto hace una dosis diaria total de 60 mg. Al final del día 6 ya habrá alcanzado esta dosis recomendada.
• Una vez alcanzada la dosis recomendada, los envases recetados contendrán únicamente los comprimidos de 30 mg. Solo tendrá que pasar por este proceso de ir aumentando la dosis paulatinamente una vez, aunque tenga que reiniciar el tratamiento.
|
Dosis diaria total | ||||
|
Día |
Dosis de la mañana |
Dosis de la noche | ||
|
Día 1 |
1 |
10 mg (rosa) |
No tome la dosis |
10 mg |
|
Día 2 |
10 mg (rosa) |
10 mg (rosa) |
20 mg | |
|
Día 3 |
10 mg (rosa) |
20 mg (marrón) |
30 mg | |
|
Día 4 |
20 mg (marrón) |
20 mg (marrón) |
40 mg | |
|
Día 5 |
20 mg (marrón) |
30 mg (beige) |
50 mg | |
|
Día 6 en adelante |
30 mg (beige) |
30 mg (beige) |
60 mg | |
Personas con problemas de riñón
Si tiene problemas de riñón graves, entonces la dosis recomendada de Otezla es de 30 mg una vez al día (dosis de la mañana). Su médico le indicará cómo aumentar la dosis cuando empiece a tomar Otezla por primera vez.
Cómo y cuándo tomar Otezla
• Trague los comprimidos enteros, preferiblemente con agua.
Puede tomar los comprimidos con o sin alimentos.
• Tome Otezla aproximadamente a la misma hora cada día, un comprimido por la mañana y un comprimido por la noche.
• Si no mejora su enfermedad después de seis meses de tratamiento, consulte a su médico.
Si toma más Otezla del que debe
Si toma más Otezla del que debe, consulte a un médico o vaya a un hospital inmediatamente. Lleve el envase del medicamento y el prospecto con usted.
Si olvidó tomar Otezla
• Si se salta una dosis de Otezla, tómela lo antes posible. Si está cerca de la hora de la siguiente dosis, sáltese la dosis olvidada. Tómese la siguiente dosis a su hora habitual.
• No tome dos dosis al mismo tiempo.
Si interrumpe el tratamiento con Otezla
• Debe continuar tomando Otezla hasta que su médico le indique que lo deje.
• No deje de tomar Otezla sin consultar antes a su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas)
• diarrea
• náuseas
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas)
• tos
• dolor de espalda
• vómitos
• cansancio
• dolor de estómago
• pérdida de apetito
• deposiciones frecuentes
• dificultad para dormir (insomnio)
• indigestión o ardor de estómago
• dolores de cabeza, migrañas o dolores de cabeza de tensión
• infecciones del tracto respiratorio superior tales como resfriado, moqueo, infección de los senos paranasales (sinusitis)
• inflamación e hinchazón de las vías que van a los pulmones (bronquitis)
• resfriado común (nasofaringitis)
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas)
• erupción
• pérdida de peso
• reacción alérgica
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Otezla
• Mantener este medicamento fuera de la vista y del alcance de los niños.
• No utilice este medicamento después de la fecha de caducidad que aparece en el blíster y la caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
• No conservar a temperatura superior a 30°C.
• No utilice este medicamento si observa algún deterioro o indicios de manipulación del envase del medicamento.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de Otezla
• El principio activo es apremilast.
• Cada comprimido recubierto con película contiene 10 mg de apremilast.
• Cada comprimido recubierto con película contiene 20 mg de apremilast.
• Cada comprimido recubierto con película contiene 30 mg de apremilast.
• Los demás componentes del núcleo del comprimido son celulosa microcristalina, lactosa monohidrato, croscarmelosa sódica y estearato de magnesio.
• El recubrimiento contiene alcohol de polivinilo, dióxido de titanio (E171), macrogol, talco, óxido de hierro rojo (E172).
• El comprimido recubierto con película de 20 mg contiene también óxido de hierro amarillo (E172).
• El comprimido recubierto con película de 30 mg contiene también óxido de hierro amarillo (E172) y óxido de hierro negro (E172).
Aspecto del producto y contenido del envase
Otezla 10 mg comprimido recubierto con película es un comprimido recubierto con película con forma de rombo, de color rosa, con “APR” grabado en una cara y “10” en la otra cara.
Otezla 20 mg comprimido recubierto con película es un comprimido recubierto con película con forma de rombo, de color marrón con “APR” grabado en una cara y “20” en la otra cara.
Otezla 30 mg comprimido recubierto con película es un comprimido recubierto con película con forma de rombo, de color beige, con “APR” grabado en una cara y “30” en la otra cara.
Tamaños de envase
• El pack de inicio de tratamiento es un estuche desplegable que contiene 27 comprimidos:
4 comprimidos de 10 mg, 4 comprimidos de 20 mg y 19 comprimidos de 30 mg.
• El envase estándar para un mes contiene 56 comprimidos de 30 mg.
• El envase estándar para tres meses contiene 168 comprimidos de 30 mg.
Titular de la autorización de comercialización y responsable de la fabricación
Celgene Europe Ltd.
1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
52