Opdivo 10 Mg/Ml Concentrado Para Solucion Para Perfusion
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
OPDIVO 10 mg/ml concentrado para solución para perfusión.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada ml de concentrado contiene 10 mg de nivolumab.
Un vial de 4 ml contiene 40 mg de nivolumab.
Un vial de 10 ml contiene 100 mg de nivolumab.
Nivolumab se produce en células de ovario de hámster chino mediante tecnología de ADN recombinante.
Excipiente con efecto conocido
Cada ml de este concentrado contiene 0,1 mmol (ó 2,5 mg) de sodio.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Concentrado para solución para perfusión (concentrado estéril).
Líquido, de incoloro a amarillo pálido, de transparente a opalescente que puede contener algunas (pocas) partículas. La solución tiene un pH de aproximadamente 6,0 y una osmolalidad de aproximadamente 340 mOsm/kg.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Melanoma
OPDIVO en monoterapia o en combinación con ipilimumab está indicado para el tratamiento del melanoma avanzado (irresecable o metastásico) en adultos.
En comparación con nivolumab en monoterapia se ha establecido un aumento de la supervivencia libre de progresión (SLP) para la combinación de nivolumab con ipilimumab, solamente en los pacientes con baja expresión de PD-L1 en el tumor (ver las secciones 4.4 y 5.1).
Cáncer de pulmón no microcítico (CPNM)
OPDIVO está indicado para el tratamiento de cáncer de pulmón no microcítico (CPNM), localmente avanzado o metastásico después de quimioterapia previa, en adultos.
Carcinoma de Células Renales (CCR)
OPDIVO en monoterapia está indicado para el tratamiento de carcinoma de células renales avanzado después de tratamiento previo, en adultos.
El tratamiento debe ser iniciado y supervisado por médicos con experiencia en el tratamiento del cáncer.
Posología
OPDIVO en monoterapia
La dosis recomendada de OPDIVO es 3 mg/kg de nivolumab administrado por vía intravenosa durante 60 minutos cada 2 semanas.
OPDIVO en combinación con ipilimumab
La dosis recomendada es 1 mg/kg de nivolumab, administrado por vía intravenosa durante 60 minutos cada 3 semanas para las primeras 4 dosis en combinación con 3 mg/kg de ipilimumab administrados por vía intravenosa durante 90 minutos.
Después se continúa con una segunda fase en la que se administran 3 mg/kg de nivolumab por vía intravenosa durante 60 minutos cada 2 semanas.
El tratamiento con OPDIVO, ya sea en monoterapia o en combinación con ipilimumab, se debe prolongar mientras se observe beneficio clínico o hasta que el paciente ya no tolere el tratamiento.
No se recomienda la escalada de la dosis ni su reducción. Puede que sea necesario el retraso o la suspensión de la dosificación de acuerdo con la seguridad y tolerabilidad individual. Las directrices para la interrupción permanente o la suspensión de alguna dosis se describen en la Tabla 1. Las directrices detalladas para el manejo de las reacciones adversas inmunorrelacionadas se describen en la sección 4.4.
Tabla 1: Modificaciones del tratamiento recomendadas para OPDIVO u OPDIVO
|
en combinación con ipilimumab | ||
|
Reacción adversa inmunorrelacionada |
Gravedad |
Modificación del tratamiento |
|
Neumonitis inmunorrelacionada |
Neumonitis de Grado 2 |
Suspender la(s) dosis hasta que los síntomas se resuelvan y mejoren las anomalías radiográficas y el tratamiento con corticosteroides, haya finalizado |
|
Neumonitis de Grado 3 ó 4 |
Suspender de forma permanente el tratamiento | |
|
Colitis Inmunorrelacionada |
Diarrea o colitis de Grado 2 Diarrea o colitis de Grado 3 - OPDIVO en monoterapia |
Suspender la(s) dosis hasta que los síntomas se resuelvan y el tratamiento con corticosteroides, si fuese necesario, haya finalizado Suspender la(s) dosis hasta que los síntomas se resuelvan y el tratamiento con corticosteroides, si fuese necesario, haya finalizado |
|
- OPDIVO+ipilimumab |
Suspender de forma permanente el tratamiento | |
|
Diarrea o colitis de Grado 4 |
Suspender de forma permanente el tratamiento | |
|
Hepatitis inmunorrelacionada |
Elevación de la aspartato aminotransferasa (AST), alanina aminotransferasa (ALT) de Grado 2 o bilirrubina total |
Suspender la(s) dosis hasta que los valores de laboratorio disminuyan hasta el valor basal y el tratamiento con corticosteroides, si fuese necesario, haya finalizado |
|
Elevación de AST, ALT de Grado 3 ó 4 o bilirrubina total |
Suspender de forma permanente el tratamiento | |
|
Nefritis inmunorrelacionada e insuficiencia renal |
Elevación de creatinina de Grado 2 ó 3 |
Suspender la(s) dosis hasta que el valor de creatinina disminuya hasta el valor basal y el tratamiento con corticosteroides haya finalizado |
|
Elevación de creatinina de Grado 4 |
Suspender de forma permanente el tratamiento | |
|
Endocrinopatías inmunorrelacionadas |
Hipotiroidismo, hipertiroidismo, hipofisitis sintomáticos de Grado 2 ó 3 Insuficiencia suprarrenal de Grado 2 Diabetes de Grado 3 |
Suspender la(s) dosis hasta que los síntomas se resuelvan y el tratamiento con corticosteroides (si fuese necesario para los síntomas de inflamación aguda) haya finalizado El tratamiento debe continuarse en presencia de tratamientoa hormonal de sustitución hasta que los síntomas desaparezcan |
|
Hipotiroidismo de Grado 4 Hipertiroidismo de Grado 4 Hipofisitis de Grado 4 Insuficiencia suprarrenal de Grado 3 ó 4 Diabetes de Grado 4 |
Suspender de forma permanente el tratamiento | |
|
Erupción cutánea inmunorrelacionada |
Erupción cutánea de Grado 3 Erupción cutánea de Grado 4 |
Suspender la(s) dosis hasta que los síntomas se resuelvan y el tratamiento con corticosteroides haya finalizado Suspender de forma permanente el tratamiento |
Tabla 1: Modificaciones del tratamiento recomendadas para OPDIVO u OPDIVO
en combinación con ipilimumab
Suspender la(s) dosis
Grado 3 (la primera vez que ocurre)
Grado 4 o Grado 3 recurrente;
Otreas reacciones Grado 2 ó 3 persistente a pesar de la
Suspender de forma permanente el tratamiento
adversas modificación del tratamiento;
imposibilidad de reducir la dosis de corticosteroides hasta 10 mg de _prednisona o equivalente por día
Nota: los grados de toxicidad se determinan de acuerdo con los Criterios de terminología común para acontecimientos adversos del National Cáncer Institute, versión 4.0 (NC-CTCAE v4). a La recomendación para el uso de tratamiento hormonal de sustitución se incluye en la sección 4.4.
Los pacientes en tratamiento con OPDIVO deben recibir la tarjeta de información para el paciente y deben ser informados sobre los riesgos de OPDIVO (ver también el prospecto de este medicamento).
Cuando se administra OPDIVO en combinación con ipilimumab, si se interrumpe la administración de uno de ellos, el otro también se debe interrumpir. Si se decide continuar el tratamiento después de haberse interrumpido, tanto el tratamiento de combinación como la monoterapia con OPDIVO deben continuarse en base a la evaluación individual de cada paciente.
Poblaciones especiales
Población pediátrica
No se ha establecido la seguridad y eficacia de OPDIVO en niños menores de 18 años. No se dispone de datos.
Pacientes de edad avanzada
No es necesario el ajuste de la dosis en pacientes de edad avanzada (> 65 años) (ver las secciones 5.1 y 5.2).
Cáncer de pulmón no microcítico
Los datos de los pacientes con 75 años de edad o mayores son demasiado limitados para sacar conclusiones sobre esta población.
Insuficiencia renal
De acuerdo con los resultados de la farmacocinética (FC) poblacional, no es necesario el ajuste de la dosis en pacientes con insuficiencia renal leve o moderada (ver sección 5.2). Los datos de pacientes con insuficiencia renal grave son demasiado limitados para sacar conclusiones en esta población.
Insuficiencia hepática
De acuerdo con los resultados de la FC poblacional, no es necesario el ajuste de la dosis en pacientes con insuficiencia hepática leve (ver sección 5.2). Los datos de pacientes con insuficiencia hepática moderada o grave son demasiado limitados para sacar conclusiones en estas poblaciones. OPDIVO se debe administrar con precaución en pacientes con insuficiencia hepática moderada (bilirrubina total > 1,5 x a 3 x el límite superior de normalidad [LSN] y cualquier valor de AST) o grave (bilirrubina total > 3 x LSN y cualquier valor de AST).
Cáncer de pulmón no microcítico
Los pacientes con estado funcional según el Eastern Cooperative Oncology Group (ECOG, por sus siglas en inglés) > 2 fueron excluidos de los ensayos clínicos de CPNM (ver las secciones 4.4 y 5.1).
OPDIVO se utiliza sólo por vía intravenosa. Se debe administrar en forma de perfusión intravenosa durante un periodo de tiempo de 60 minutos. La perfusión se debe administrar a través de un filtro en línea estéril, no pirógeno y de baja unión a proteínas, con un tamaño de poro de 0,2-1,2 pm.
OPDIVO no se debe administrar como inyección en bolo intravenoso.
La dosis total necesaria de OPDIVO se puede perfundir directamente como una solución de 10 mg/ml o se puede diluir hasta una concentración tan baja como 1 mg/ml con una solución de cloruro sódico para preparaciones inyectables a una concentración de 9 mg/ml (0,9%) o una solución de glucosa para preparaciones inyectables a una concentración de 50 mg/ml (5%).
Cuando se administre el tratamiento en combinación con ipilimumab, OPDIVO se debe administrar en primer lugar seguido de ipilimumab en el mismo día. Utilizar bolsas de perfusión y filtros distintos para cada perfusión.
Para consultar las instrucciones sobre la dilución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Cuando se administre nivolumab en combinación con ipilimumab, consultar la Ficha Técnica de ipilimumab antes de iniciar el tratamiento. Se han producido reacciones adversas inmunorrelacionadas con mayor frecuencia cuando se utiliza nivolumab en combinación con ipilimumab que cuando se utiliza nivolumab en monoterapia. La mayoría de las reacciones adversas inmunorrelacionadas mejoran o se resuelven con un manejo adecuado, incluyendo la iniciación del tratamiento con corticosteroides y las modificaciones del tratamiento (ver sección 4.2).
También se han notificado reacciones adversas cardíacas y embolismo pulmonar con el tratamiento de combinación. Los pacientes deben monitorizarse continuamente para detectar reacciones adversas cardíacas y pulmonares así como signos clínicos, síntomas, alteraciones de los valores de laboratorio que indican alteraciones de electrolitos, deshidratación previa y periódicamente durante el tratamiento. Nivolumab en combinación con ipilimumab debe interrumpirse si se producen reacciones adversas cardíacas y pulmonares graves recurrentes o que sean potencialmente mortales.
Los pacientes deben ser monitorizados continuamente (como mínimo hasta 5 meses después de la última dosis) ya que se puede producir una reacción adversa con nivolumab o con nivolumab en combinación con ipilimumab en cualquier momento durante o después de la suspensión del tratamiento.
Para sospechas de reacciones adversas inmunorrelacionadas, se debe realizar una evaluación adecuada para confirmar esta etiología o excluir otra causa. De acuerdo a la gravedad de las reacciones adversas, se debe suspender el tratamiento con nivolumab o nivolumab en combinación con ipilimumab y se deben administrar corticosteroides. Si se emplea inmunosupresión con corticosteroides para tratar una reacción adversa, se debe iniciar una reducción progresiva de la dosis de al menos 1 mes de duración en cuanto se observe mejoría. Una disminución rápida de la dosis puede provocar un empeoramiento o recurrencia de la reacción adversa. Se debe añadir tratamiento inmunosupresor sin corticosteroides si se observa un empeoramiento o no se produce una mejoría.
Nivolumab o nivolumab en combinación con ipilimumab no se deben reanudar mientras el paciente esté recibiendo dosis inmunosupresoras de corticosteroides u otro tratamiento inmunosupresor. Se deben utilizar antibióticos profilácticos para prevenir la aparición de infecciones oportunistas en pacientes que reciben tratamiento inmunosupresor.
Nivolumab o nivolumab en combinación con ipilimumab se deben suspender de forma permanente si se produce cualquier reacción adversa inmunorrelacionada grave, recurrente y ante cualquier reacción adversa inmunorrelacionada que pueda ser potencialmente mortal.
Uso de nivolumab en pacientes con melanoma con una rápida progresión de la enfermedad Los médicos prescriptores deben considerar que el efecto de nivolumab puede aparecer con cierto retraso antes de iniciar el tratamiento en pacientes cuya enfermedad progresa rápidamente (ver sección 5.1).
Uso de nivolumab en CPNM de histología no escamosa
Los médicos prescriptores deben tener en cuenta que el efecto de nivolumab puede aparecer con cierto retraso antes de iniciar el tratamiento en pacientes con características de peor pronóstico y/o enfermedad agresiva. En CPNM de histología no escamosa, se ha observado un mayor número de muertes durante los 3 primeros meses en el brazo de nivolumab comparado con el de docetaxel. Los factores asociados a muertes tempranas eran factores de peor pronóstico y/o enfermedad más agresiva combinada con una baja o ninguna expresión de PD-L1 (ver sección 5.1).
Neumonitis inmunorrelacionada
Se ha observado neumonitis grave o enfermedad pulmonar intersticial, incluyendo casos mortales, con nivolumab en monoterapia o nivolumab en combinación con ipilimumab (ver sección 4.8). Los pacientes se deben monitorizar en cuanto a los signos y síntomas de neumonitis, tales como cambios radiográficos (p. ej., opacidades focales vitrales, en la base, infiltrados en parches), disnea e hipoxia. Se deben descartar etiologías infecciosas o relacionadas con la enfermedad.
En el caso de neumonitis de Grado 3 ó 4, nivolumab o nivolumab en combinación con ipilimumab se deben suspender de forma permanente y se debe iniciar tratamiento con corticosteroides a una dosis equivalente de 2 a 4 mg/kg/día de metilprednisolona.
Para neumonitis de Grado 2 (sintomática), nivolumab o nivolumab en combinación con ipilimumab se deben interrumpir e iniciar tratamiento con corticosteroides a una dosis equivalente de 1 mg/kg/día de metilprednisolona. Una vez que se produzca la mejoría, nivolumab o nivolumab en combinación con ipilimumab, se deben reanudar tras la disminución gradual de la dosis de corticosteroides. Si se produjese un empeoramiento o no se observase una mejoría a pesar del inicio del tratamiento con corticosteroides, la dosis equivalente de corticosteroides debe aumentarse hasta 2 a 4 mg/kg/día de metilprednisolona y suspender nivolumab o nivolumab en combinación con ipilimumab de forma permanente.
Colitis inmunorrelacionada
Se ha observado diarrea o colitis grave asociada a nivolumab en monoterapia o nivolumab en combinación con ipilimumab (ver sección 4.8). Los pacientes se deben monitorizar en relación a su diarrea y a otros síntomas relacionados con la colitis, como dolor abdominal y presencia de moco o sangre en las heces. Se deben descartar etiologías infecciosas o relacionadas con la enfermedad.
Para diarrea o colitis de Grado 4, se debe suspender permanentemente nivolumab o nivolumab en combinación con ipilimumab, e iniciar tratamiento con corticosteroides a una dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona.
Nivolumab en monoterapia, se debe suspender en casos de diarrea o colitis de Grado 3, e iniciar tratamiento con corticosteroides a una dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona. Una vez que se observe una mejoría, reiniciar nivolumab en monoterapia tras la disminución gradual de la dosis de corticosteroides. Si se produjese un empeoramiento o no se observase una mejoría a pesar del inicio del tratamiento con corticosteroides, se debe suspender nivolumab en monoterapia de forma permanente. La diarrea o colitis de Grado 3 observadas con nivolumab en combinación con ipilimumab requiere también la interrupción permanente del tratamiento y la iniciación de tratamiento con corticosteroides a dosis equivalentes de 1 a 2 mg/kg/día de metilprednisolona.
En el caso de diarrea o colitis de Grado 2, suspender nivolumab o nivolumab en combinación con ipilimumab. Si persiste la diarrea o la colitis, se debe manejar con corticosteroides a una dosis equivalente de 0,5 a 1 mg/kg/día de metilprednisolona. Si se produce mejoría, reanudar el tratamiento
7
con nivolumab, o nivolumab en combinación con ipilimumab tras la disminución gradual de la dosis de corticosteroides si fuese necesario. Si se produjese un empeoramiento o no se observase una mejoría a pesar del inicio del tratamiento con corticosteroides, aumentar la dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona y suspender nivolumab o nivolumab en combinación con ipilimumab de forma permanente.
Hepatitis inmunorrelacionada
Se ha observado hepatitis grave con nivolumab en monoterapia o nivolumab en combinación con ipilimumab (ver sección 4.8). Los pacientes se deben monitorizar para la detección de signos y síntomas de hepatitis, tales como elevaciones de transaminasas y bilirrubina total. Se deben descartar etiologías infecciosas o relacionadas con la enfermedad.
En el caso de elevaciones de transaminasas o bilirrubina total de Grado 3 ó 4, nivolumab o nivolumab en combinación con ipilimumab se deben suspender definitivamente e iniciar el tratamiento con corticosteroides a una dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona.
En el caso de elevaciones de transaminasas o bilirrubina total de Grado 2, nivolumab o nivolumab en combinación con ipilimumab, se deben suspender. Si las elevaciones en estos parámetros de laboratorio persisten, se deben manejar con corticosteroides a una dosis de 0,5 a 1 mg/kg/día de metilprednisolona o equivalente. Una vez que se produzca la mejoría, se debe reiniciar nivolumab o nivolumab en combinación con ipilimumab tras la disminución gradual de la dosis de corticosteroides, si fuesen necesarios. Si se produce un empeoramiento o no se observa una mejoría a pesar del inicio del tratamiento con corticosteroides, aumentar la dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona y suspender nivolumab o nivolumab en combinación con ipilimumab de forma permanente.
Nefritis inmunorrelacionada e insuficiencia renal
Se han observado nefritis grave e insuficiencia renal, con el tratamiento con nivolumab en monoterapia o nivolumab en combinación con ipilimumab (ver sección 4.8). Los pacientes se deben monitorizar para la detección de signos y síntomas de nefritis o insuficiencia renal. La mayoría de los pacientes presentan un aumento asintomático de la creatinina sérica. Se deben descartar etiologías infecciosas o relacionadas con la enfermedad.
En el caso del aumento de la creatinina sérica de Grado 4, suspender permanentemente nivolumab o nivolumab en combinación con ipilimumab e iniciar el tratamiento con corticosteroides a una dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona.
En el caso del aumento de la creatinina sérica de Grado 2 ó 3, se deben suspender nivolumab o nivolumab en combinación con ipilimumab e iniciar tratamiento con corticosteroides a una dosis equivalente de 0,5 a 1 mg/kg/día de metilprednisolona. Una vez que se produzca la mejoría, se debe reiniciar nivolumab o nivolumab en combinación con ipilimumab tras la disminución gradual de la dosis de corticosteroides. Si se produjese un empeoramiento o no se observase una mejoría a pesar del inicio del tratamiento con corticosteroides, aumentar la dosis equivalente de 1 a 2 mg/kg/día de metilprednisolona y suspender nivolumab o nivolumab en combinación con ipilimumab de forma permanente.
Endocrinopatías inmunorrelacionadas
Se han observado endocrinopatías graves, incluyendo hipotiroidismo, hipertiroidismo, insuficiencia suprarrenal, hipofisitis, diabetes mellitus, y cetoacidosis diabética asociadas a nivolumab en monoterapia o nivolumab en combinación con ipilimumab (ver sección 4.8).
Los pacientes se deben monitorizar para la detección de signos y síntomas de endocrinopatías e hiperglucemias y para evaluar los cambios en la función tiroidea (al comienzo del tratamiento, periódicamente durante el tratamiento y como se ha indicado en base a su evaluación clínica). Los pacientes pueden presentar fatiga, cefalea, cambios en el estado mental, dolor abdominal, hábitos intestinales inusuales e hipotensión o síntomas no específicos que pueden parecerse a otras causas como metástasis cerebrales u otra enfermedad subyacente. A menos que otra etiología alternativa se haya identificado, los signos y síntomas de endocrinopatías se deben considera inmunorrelacionados.
Para hipotiroidismo sintomático, nivolumab o nivolumab en combinación con ipilimumab se deben suspender e iniciar tratamiento de sustitución con hormona tiroidea si fuese necesario. Para hipertiroidismo sintomático, nivolumab o nivolumab en combinación con ipilimumab se deben suspender e iniciar tratamiento con medicación antitiroidea si fuese necesario. También podría considerarse el tratamiento con corticosteroides a una dosis equivalente entre 1 y 2 mg/kg/día de metilprednisolona, si se sospechase inflamación aguda del tiroides. Una vez que se produzca una mejoría, se debe reiniciar nivolumab o nivolumab en combinación con ipilimumab tras la disminución gradual de la dosis de corticosteroides, si fuese necesario. La monitorización de la función tiroidea debe continuar para asegurar que se ha utilizado el tratamiento de sustitución hormonal adecuado. Nivolumab o nivolumab en combinación con ipilimumab se deben suspender de forma permanente por hipertiroidismo o hipotiroidismo que puedan resultar potencialmente mortales.
Para insuficiencia suprarrenal sintomática de Grado 2, nivolumab o nivolumab en combinación con ipilimumab se deben suspender e iniciar tratamiento fisiológico de sustitución con corticosteroides si fuese necesario. Nivolumab o nivolumab en combinación con ipilimumab se deben suspender de forma permanente por insuficiencia suprarrenal grave (Grado 3) o por insuficiencia suprarrenal (Grado°4) que pueda resultar potencialmente mortal. La monitorización de la función suprarrenal y los niveles hormonales deben continuar para asegurar que se ha utilizado el tratamiento de sustitución con corticosteroides adecuado.
Para hipofisitis sintomática de Grado 2 ó 3, nivolumab o nivolumab en combinación con ipilimumab se deben suspender e iniciar tratamiento de sustitución hormonal si fuese necesario. También se podría considerar el tratamiento con corticosteroides a una dosis equivalente entre 1 y 2 mg/kg/día de metilprednisolona si se sospecha inflamación aguda de la glándula pituitaria. Una vez que se produzca una mejoría, se debe reiniciar nivolumab o nivolumab en combinación con ipilimumab tras la disminución gradual de la dosis de corticosteroides, si fuese necesario. Nivolumab o nivolumab en combinación con ipilimumab se deben suspender de forma permanente por hipofisitis (Grado 4) que pueda resultar potencialmente mortal. La monitorización de la función de la hipófisis y de los niveles hormonales deben continuar para asegurar que se ha utilizado el tratamiento de sustitución hormonal adecuado.
Para diabetes sintomática nivolumab o nivolumab en combinación con ipilimumab se deben suspender e iniciar tratamiento de sustitución con insulina si fuese necesario. La monitorización de la glucosa en sangre debe continuar para asegurar que se ha utilizado el tratamiento de sustitución de insulina adecuado. Nivolumab o nivolumab en combinación con ipilimumab se deben suspender de forma permanente por diabetes que pueda resultar potencialmente mortal.
Erupción cutánea inmunorrelacionada
Se ha observado erupción cutánea grave con el tratamiento con nivolumab en combinación con ipilimumab y con menor frecuencia con nivolumab en monoterapia (ver sección 4.8). Nivolumab o nivolumab en combinación con ipilimumab se deben suspender si se producen erupciones cutáneas de Grado 3 y suspenderse definitivamente con erupciones cutáneas de Grado 4. Las erupciones cutáneas graves se deben manejar con dosis altas de corticosteroides equivalentes a dosis de 1 a 2 mg/kg/día de metilprednisolona.
Se han observado casos raros de necrólisis epidérmica tóxica (NET) algunos de ellos con desenlace mortal. Si aparecen síntomas o signos del Síndrome de Stevens-Johnson (SSJ) o NET, el tratamiento con nivolumab se debe interrumpir y el paciente debe ser derivado a una unidad especializada para evaluación y tratamiento. Si el paciente ha desarrollado SSJ o NET con el uso de nivolumab, se recomienda la interrupción permanente de nivolumab.
Se debe actuar con precaución, cuando se considere el uso de nivolumab en un paciente que haya experimentado previamente reacciones adversa cutáneas graves o que pueda resultar potencialmente mortal para la vida en un tratamiento previo con otros medicamentos anticancerígenos inmunoestimuladores.
Otras reacciones adversas inmunorrelacionadas
Las siguientes reacciones adversas inmunorrelacionadas se notificaron en menos del 1% de los pacientes tratados con nivolumab en monoterapia en ensayos clínicos en diversos tipos de tumores:
9
pancreatitis, uveítis, desmielinización, neuropatía autoinmune (incluyendo parálisis de los nervios faciales y del nervio abducente), síndrome de Guillain-Barré, hipopituitarismo y síndrome miasténico.
En los ensayos clínicos de nivolumab en combinación con ipilimumab, se notificaron las siguientes reacciones adversas inmunorrelacionadas adicionales, clínicamente significativas en menos del 1% de pacientes: gastritis, sarcoidosis y duodenitis.
Para sospecha de reacciones adversas inmunorrelacionadas, se debe realizar una evaluación adecuada para confirmar su etiología y excluir otras causas. De acuerdo a la gravedad de las reacciones adversas, nivolumab o nivolumab en combinación con ipilimumab se deben suspender e iniciar tratamiento con corticosteroides. Una vez que se produzca una mejoría, se deben reiniciar nivolumab o nivolumab en combinación con ipilimumab tras la disminución gradual de la dosis de corticosteroides. Nivolumab o nivolumab en combinación con ipilimumab se deben interrumpir de forma permanente por cualquier reacción adversa grave inmunorrelacionada recurrente y por cualquier reacción adversa inmunorrelacionada que suponga una amenaza para la vida.
Reacciones a la perfusión
Se han notificado reacciones graves a la perfusión en los ensayos clínicos de nivolumab o nivolumab en combinación con ipilimumab (ver sección 4.8). En caso de una reacción a la perfusión grave o que pueda resultar potencialmente mortal, nivolumab o nivolumab en combinación con ipilimumab se deben interrumpir y se debe administrar tratamiento médico adecuado. Los pacientes con reacciones a la perfusión leves o moderadas pueden recibir nivolumab o nivolumab en combinación con ipilimumab con una estrecha monitorización y uso de premedicación de acuerdo a las guías locales de tratamiento profiláctico de las reacciones a la perfusión.
Precauciones específicas de la enfermedad
Melanoma
Se excluyeron de los ensayos clínicos de nivolumab o nivolumab en combinación con ipilimumab, los pacientes con un estado funcional basal > 2, metástasis cerebrales activas o enfermedad autoinmune, y pacientes que hayan recibido inmunosupresores sistémicos antes de su entrada en un estudio. Se excluyeron de los ensayos clínicos de melanoma, lo pacientes con melanoma ocular/uveal. Además, se excluyeron pacientes del ensayo CA209037, que habían tenido una reacción adversa Grado 4 relacionada con el tratamiento anti-CTLA-4 (ver sección 5.1). Ante la ausencia de datos, nivolumab se debe utilizar con precaución en estas poblaciones, después de valorar cuidadosamente el potencial riesgo/beneficio de forma individual en cada paciente.
En comparación con nivolumab en monoterapia se ha establecido un aumento de la supervivencia libre de progresión (SLP) para la combinación de nivolumab con ipilimumab, solamente en los pacientes con baja expresión de PD-L1 en el tumor. Antes de iniciar el tratamiento con la combinación, se aconseja a los prescriptores que evalúen cuidadosamente tanto a los pacientes individualmente como las características del tumor, teniendo en cuenta los beneficios observados y la toxicidad de la combinación con respecto a nivolumab en monoterapia (ver las secciones 4.8 y 5.1).
Cáncer de pulmón no microcítico
Se excluyeron de los ensayos clínicos de cáncer de pulmón no microcítico (CPNM) los pacientes con un estado funcional basal > 2, con metástasis cerebrales activas o con enfermedad autoinmune o enfermedad intersticial pulmonar sintomática, y pacientes que hubieran recibido inmunosupresores sistémicos antes de su entrada en el estudio (ver las secciones 4.5 y 5.1). Ante la ausencia de datos, nivolumab se debe utilizar con precaución en estas poblaciones, después de valorar cuidadosamente el potencial riesgo/beneficio de forma individual en cada paciente.
Carcinoma de Células Renales
Se excluyeron del ensayo pivotal de CCR los pacientes con antecedentes previos o actuales de metástasis cerebrales, enfermedad autoinmune activa, o situación clínica que requiriera
10
inmunosupresión sistémica (ver las secciones 4.5 y 5.1). Ante la ausencia de datos, nivolumab se debe utilizar con precaución en estas poblaciones, después de valorar cuidadosamente el potencial riesgo/beneficio de forma individual en cada paciente.
Pacientes con dietas bajas en sodio
Cada ml de este medicamento contiene 0,1 mmol (ó 2,5 mg) de sodio. Lo que se debe tener en cuenta a la hora de tratar a pacientes con dietas pobres en sodio.
Tarjeta de Información para el Paciente
Todos los prescriptores de OPDIVO, deben estar familiarizados con la información dirigida al Profesional Sanitario y las Guías de manejo. El prescriptor debe discutir el riesgo del tratamiento con OPDIVO con el paciente. Al paciente se le suministrará la Tarjeta de Información para el Paciente con cada prescripción.
4.5 Interacción con otros medicamentos y otras formas de interacción
Nivolumab es un anticuerpo monoclonal humano, y por tanto no se han realizado estudios de interacciones farmacocinéticas. Dado que los anticuerpos monoclonales no se metabolizan por las enzimas del citocromo P450 (CYP) u otras enzimas metabolizadoras de medicamentos, no se prevé que ni la inhibición ni la inducción de estas enzimas por medicamentos administrados de forma conjunta afecte a la farmacocinética de nivolumab.
Otras formas de interacción
Inmunosupresión sistémica
Se debe evitar la utilización de corticosteroides sistémicos y otros inmunosupresores, antes de comenzar tratamiento con nivolumab, debido a su interferencia potencial con la actividad farmacodinámica. Sin embargo, se pueden usar corticosteroides sistémicos y otros inmunosupresores después de comenzar el tratamiento con nivolumab para tratar las reacciones adversas inmunorrelacionadas. Los resultados preliminares muestran que la inmunosupresión sistémica después del comienzo del tratamiento con nivolumab no parece excluir la respuesta de nivolumab.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos relativos al uso de nivolumab en mujeres embarazadas. Los estudios en animales han mostrado toxicidad embriofetal (ver sección 5.3). Se sabe que la IgG4 humana atraviesa la barrera placentaria, y nivolumab es una IgG4; por tanto nivolumab tiene el potencial de transmitirse de la madre al feto en desarrollo. No se recomienda el uso de nivolumab durante el embarazo ni en las mujeres en edad fértil que no estén utilizando métodos anticonceptivos eficaces, a menos que el beneficio clínico supere el posible riesgo. Se deben utilizar métodos anticonceptivos efectivos al menos hasta los 5 meses siguientes a la última dosis de nivolumab.
Lactancia
Se desconoce si nivolumab se excreta en la leche materna. Puesto que muchos medicamentos, incluidos sus metabolitos, se excretan a través de la leche materna, no se puede excluir el riesgo para los recién nacidos/lactantes. Se debe tomar una decisión acerca de interrumpir la lactancia o interrumpir el tratamiento con nivolumab, teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Fertilidad
No se han realizado estudios que evalúen el efecto de nivolumab sobre la fertilidad. Por tanto, se desconoce el efecto de nivolumab sobre la fertilidad masculina y femenina.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
De acuerdo con sus propiedades farmacodinámicas, es poco probable que nivolumab afecte a la capacidad para conducir y utilizar máquinas. Debido a las posibles reacciones adversas como fatiga (ver sección 4.8), se debe advertir a los pacientes que tengan precaución al conducir o utilizar maquinaria hasta que estén seguros de que nivolumab no les afecta de forma negativa.
4.8 Reacciones adversas
Resumen del perfil de seguridad
En los datos agrupados de nivolumab 3 mg/kg en monoterapia en los diferentes tumores (CA209066, CA209037, CA209067 (cohorte de monoterapia), CA209017, CA209057, CA209063 y CA209025), las reacciones adversas más frecuentes (> 10%) fueron fatiga (34%), erupción cutánea (19%), prurito (14%), diarrea (13%), náuseas (13%) y disminución del apetito (10%). La mayoría de las reacciones adversas fueron de leves a moderadas (Grado 1 ó 2).
En los datos agrupados de nivolumab en combinación con ipilimumab en melanoma (CA209067 (cohorte de la combinación), CA209069 y CA209004-cohorte 8), las reacciones adversas más frecuentes (> 10%) fueron erupción cutánea (51%), fatiga (43%), diarrea (42%), prurito (35%), nausea (25%), pirexia (19%), disminución del apetito (15%), hipotiroidismo (15%), vómitos (14%), colitis (14%), dolor abdominal (13%), artralgia (11%), y cefalea (11%). La mayoría de las reacciones adversas fueron de leves a moderadas (Grado 1 ó 2).
Entre los pacientes tratados con nivolumab en combinación con ipilimumab en el ensayo CA209067, 151/313 (48%) aparecieron las primeras reacciones adversas de Grado 3 o 4 durante la fase de combinación inicial. Entre los 147 pacientes en este grupo que continuaron tratamiento con nivolumab en monoterapia, 37 (25%) experimentaron como mínimo una reacción adversa de Grado 3 ó 4 durante la fase de monoterapia.
Tabla de reacciones adversas
Las reacciones adversas notificadas en los datos agrupados de los pacientes tratados con nivolumab en monoterapia (n = 1728) y de los pacientes tratados con nivolumab en combinación con ipilimumab (n = 448) se presentan en la Tabla 2. Estas reacciones se presentan según el sistema de clasificación de órganos y por frecuencias. Las frecuencias se definen como: muy frecuentes (> 1/10); frecuentes (de > 1/100 a < 1/10); poco frecuentes (de > 1/1.000 a < 1/100); raras (de > 1/10.000 a < 1/1.000); muy raras (< 1/10.000). Dentro de cada intervalo de frecuencias, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 2: Reacciones adversas en ensayos clínicos
|
Nivolumab en monoterapia |
Nivolumab en combinación con ipilimumab | |
|
Infeciones e infestaciones | ||
|
Frecuentes |
infección del tracto respiratorio superior |
neumoniaa, infección del tracto respiratorio superior |
|
Poco frecuentes |
neumoniaa, bronquitis |
bronquitis |
|
Neoplasias benignas, malignas y no especificadas (incluyendo quistes y pólipos) | ||
|
Raras |
linfoadenitis histocítica necrotizante (linfoadenitis Kikuchi) | |
|
Trastornos de la sangre y del sistema linfático | ||
|
Frecuentes |
eosinofilia | |
|
Poco frecuentes |
eosinofilia | |
|
Trastornos del sistema inmunológico | ||
|
Frecuentes |
reacción relacionada con la perfusiónb, hipersensibilidad |
reacción relacionada con la perfusiónb, hipersensibilidad |
|
Poco frecuentes |
reacción anafilácticab |
sarcoidosis |
|
Trastornos endocrinos | ||
|
Muy frecuentes |
hipotiroidismo | |
|
Frecuentes |
hipotiroidismo, hipertiroidismo, hiperglucemiab |
insuficiencia suprarrenal, hipopituitarismo, hipofisitis, hipertiroidismo, tiroiditis, hiperglucemiab |
|
Poco frecuentes |
insuficiencia suprarrenal, hipopituitarismo, hipofisitis, tiroiditis, cetoacidosis diabética |
cetoacidosis diabética, diabetes mellitus |
|
Raras |
diabetes mellitus | |
|
Trastornos del metabolismo y la nutrición | ||
|
Muy frecuentes |
disminución del apetito |
disminución del apetito |
|
Frecuentes |
deshidratación | |
|
Poco frecuentes |
deshidratación, acidosis metabólica | |
|
Trastornos hepatobiliares | ||
|
Frecuentes |
hepatitisb | |
|
Poco frecuentes |
hepatitisb, hiperbilirrubinemia | |
|
Raras |
colestasis | |
|
Trastornos del sistema nervioso | ||
|
Muy frecuentes |
cefalea | |
|
Frecuentes |
neuropatía periférica, cefalea, mareo |
neuropatía periférica, mareo |
|
Poco frecuentes |
polineuropatía |
síndrome Guillain-Barré, polineuropatía, neuritis, parálisis del nervio peroneal, neuropatía autoinmune (incluyendo parálisis de los nervios faciales y del nervio abducente) |
|
Raras |
síndrome Guillain-Barré, desmielinización, síndrome miasténico, neuropatía autoinmune (incluyendo parálisis de los nervios faciales y del nervio abducente) | |
|
Trastornos oculares | ||
|
Frecuentes |
visión borrosa, ojo seco |
uveítis, visión borrosa |
|
Poco frecuentes |
uveitis | |
|
Trastornos cardíacos | ||
|
Frecuentes |
taquicardia | |
|
Poco frecuentes |
taquicardia |
arritmia (incluyendo arritmia ventricular)c, fibrilación auricular |
|
Raras |
arritmia (incluyendo arritmia ventricular)c, fibrilación auricular | |
|
Trastornos vasculares | ||
|
Frecuentes |
hipertensión |
hipertensión |
|
Poco frecuentes |
vasculitis | |
|
Trastornos respiratorios. torácicos y mediastínicos | ||
|
Frecuentes |
neumonitisa,b, disnea, tos |
neumonitisa,b, embolismo pulmonara, disnea, tos |
|
Poco frecuentes |
derrame pleural |
derrame pleural |
|
Raras |
infiltración pulmonar | |
|
Trastornos gastrointestinales | ||
|
Muy frecuentes |
diarrea, nausea |
colitis, diarrea, vómitos, nausea, dolor abdominal |
|
Frecuentes |
colitis, estomatitis, vómitos, dolor abdominal, estreñimiento, sequedad bucal |
estomatitis, gastritis, estreñimiento, sequedad bucal |
|
Poco frecuentes |
pancreatitis |
pancreatitis, perforación intestinal, duodenitis |
|
Raras |
gastritis, úlcera duodenal | |
|
Trastornos de la piel y del tejido subcutáneo | ||
|
Muy frecuentes |
erupción cutánead, prurito |
erupción cutánead, prurito |
|
Frecuentes |
vitiligo, piel seca, eritema, alopecia |
vitiligo, piel seca, eritema, alopecia, urticaria |
|
Poco frecuentes |
eritema multiforme, psoriasis, rosácea, urticaria |
psoriasis |
|
Raras |
necrólisis epidérmica tóxicaa,e |
necrólisis epidérmica tóxicaa,e |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | ||
|
Muy frecuentes |
artralgia | |
|
Frecuentes |
dolor musculoesqueléticof, artralgia |
dolor musculoesqueléticof |
|
Poco frecuentes |
polimialgia reumática, artritis |
espondiloartropatía, síndrome de Sjogren, artritis, miopatía |
|
Raras |
miopatía | |
|
Trastornos renales y urinarios | ||
|
Frecuentes |
fallo renalab | |
|
Poco frecuentes |
nefritis tubulointersticial, fallo renala,b |
nefritis tubulointersticial |
|
Trastornos generales y alteraciones en el lugar de la administración | ||
|
Muy frecuentes |
fatiga |
fatiga, pirexia |
|
Frecuentes |
pirexia, edema (incluyendo edema periférico) |
edema (incluyendo edema periférico), dolor |
|
Poco frecuentes |
dolor, dolor torácico |
dolor torácico |
|
Exploraciones complementariasg | ||
|
Muy frecuentes |
elevación de la AST, elevación de la ALT, elevación de la fosfatasa alcalina, elevación de la lipasa, elevación de la amilasa, hipocalcemia, elevación de la creatinina, linfopenia, leucopenia, trombocitopenia, anemia, hipercalcemia, hiperpotasemia , hipopotasemia, hipomagnesemia, hiponatremia |
elevación de la AST, elevación de la ALT, elevación de la bilirrubina total, elevación de la fosfatasa alcalina, elevación de la lipasa, elevación de la amilasa, elevación de la creatinina, linfopenia, leucopenia, neutropenia, trombocitopenia, anemia, hipocalcemia, hiperpotasemia, hipopotasemia, hipomagnesemia, hiponatremia |
|
Frecuentes |
elevación de la bilirubina total, neutropenia, hipermagnesemia, hipernatremia, disminución de peso |
hipercalcemia, hipermagnesemia, hipernatremia, disminución de peso |
a
b
c
d
e
f
g
Los casos mortales se han notificado en estudios finalizados o en curso.
Los casos potencialmente mortales se han notificado en estudios finalizados o en curso.
La frecuencia de acontecimientos adversos relacionados con trastornos cardíacos según el sistema de clasificación de órganos de acontecimientos adversos con independencia de la causalidad, fue más alta en el grupo de nivolumab que en el grupo de quimioterapia en la población con melanoma metastásico previamente tratada con CTLA4/Inhibidor BRAF. El porcentaje de las tasas de incidencia personas/años en exposición fue 9,3 % vs 0%; se notificaron acontecimientos adversos en un 4,9% de pacientes en el grupo de nivolumab vs 0% en el grupo de tratamiento elegido por el investigador. La frecuencia de acontecimientos adversos cardíacos fue más baja en el grupo de nivolumab que en el grupo de dacarbacina en la población con melanoma metastásico sin tratamiento previo. Todos fueron considerados no relacionados con nivolumab por los investigadores excepto arritmia (fibrilación auricular, taquicardia, y arritmia ventricular).
Erupción cutánea es un término compuesto que incluye erupción maculopaular, erupción eritematosa, erupción pruriginosa, erupción folicular, erupción macular, erupción morbiliforme, erupción papular, erupción pustular, erupción papuloescamosa, erupción vesicular, erupción generalizada, dermatitis, dermatitis acneiforme, dermatitis alérgica, dermatitis atópica, dermatitis bullosa, dermatitis exfoliativa, dermatitis psoriasiforme y erupción medicamentosa.
Notificado en estudios fuera del conjunto de datos agrupados. La frecuencia se basa en la exposición durante todo el programa.
El dolor musculoesquelético es un término compuesto que incluye dolor de espalda, dolor óseo, dolor musculoesquelético torácico, molestias musculoesqueléticas, mialgia, dolor de cuello, dolor en las extremidades y dolor en la columna vertebral.
Las frecuencias de las exploraciones complementarias reflejan la proporción de pacientes que experimentaron un empeoramiento de los valores basales de laboratorio. Ver “Descripción de las reacciones adversas seleccionadas; anomalías de laboratorio” a continuación.
Descripción de reacciones adversas seleccionadas
Nivolumab o nivolumab en combinación con ipilimumab se asocia con reacciones adversas inmunorrelacionadas. Con tratamiento médico adecuado, las reacciones adversas inmunorrelacionadas se resolvieron en la mayoría de los casos. La interrupción permanente del tratamiento fue necesaria en una mayor proporción de pacientes que recibieron nivolumab en combinación con ipilimumab que en aquellos que recibieron nivolumab en monoterapia para colitis inmunorrelacionada (16% y 0,7% respectivamente), hepatitis inmunorrelacionada (9% y 0,9%) y endocrinopatías inmunorrelacionadas (2,5% y 0,1%). Entre los pacientes que experimentaron un acontecimiento, requirieron en mayor proporción dosis altas de corticosteroides (al menos 40 mg de prednisona o equivalente) los pacientes que recibieron el régimen de combinación, que los pacientes que recibieron nivolumab en monoterapia para el manejo de la colitis inmunorrelacionada (47% y 14% respectivamente) y hepatitis inmunorrelacionada (46% y 16%). Las directrices de manejo para estas reacciones adversas se describen en la sección 4.4.
Neumonitis inmunorrelacionada
En pacientes tratados con nivolumab en monoterapia, la incidencia de neumonitis, incluida la enfermedad pulmonar intersticial, fue 3,2% (56/1728). La mayoría de los casos fueron de Grado 1 ó 2 de gravedad notificados en un 0,7% (12/1728) y 1,7 % (29/1728) de los pacientes respectivamente. Se notificaron casos de Grado 3 y 4 en 0,8% (14/1728) y <0,1% (1/1728) de los pacientes respectivamente. No se notificaron casos de Grado 5 en estos ensayos. La mediana hasta su aparición fue de 3,6 meses (rango: 0,4-19,6). La resolución se produjo en 47 pacientes (84%) con una mediana de tiempo hasta la resolución de 5,3 semanas (rango: 0,6-53,1+); + indica una observación censurada.
En pacientes tratados con nivolumab en combinación con ipilimumab, la incidencia de neumonitis incluyendo infiltración pulmonar, fue 7,4% (33/448). Se notificaron casos de Grado 2, Grado 3, y Grado 4 en 4,5% (20/448); 1,1% (5/448); y 0,2% (1/448) de pacientes, respectivamente. Una de las neumonitis de Grado 3 empeoró durante 11 días con desenlace mortal. La mediana del tiempo hasta su aparición fue 2,3 meses (rango: 0,7-6,7). La resolución se produjo en 29 pacientes (87,9%) con una mediana de tiempo hasta su resolución de 6,1 semanas (rango: 0,3-46,9+).
Colitis inmunorrelacionada
En pacientes tratados con nivolumab en monoterapia, la incidencia de diarrea o colitis fue 13,6% (235/1728). La mayoría de los casos fueron de Grado 1 ó 2 de gravedad notificados en un 9,0% (156/1728) y 3,0% (52/1728) de los pacientes respectivamente. Se notificaron casos de Grado 3 en 1,6% (27/1728) de los pacientes. No se notificaron casos de Grado 4 ó 5 en estos ensayos. La mediana
hasta su aparición fue de 1,8 meses (rango: 0,0-20,9). La resolución se produjo en 207 pacientes (89%) con una mediana de tiempo hasta la resolución de 2,1 semanas (rango: 0,1-88,3+).
En pacientes tratados con nivolumab en combinación con ipilimumab, la incidencia de diarrea o colitis fue 45,5% (204/448). Se notificaron casos de Grado 2, Grado 3, y Grado 4 en 13,2% (59/448),
15,4% (69/448), y 0,4% (2/448) de los pacientes, respectivamente. No se notificaron casos de Grado 5. La mediana del tiempo hasta su aparición fue 1,1 meses (rango: 0,0-10,4). La resolución se produjo en 184 pacientes (90,6%) con una mediana de tiempo hasta su resolución de 3,0 semanas (rango: 0,1-78,7+).
Hepatitis inmunorrelacionada
En pacientes tratados con nivolumab en monoterapia, la incidencia de anormalidades en las pruebas de función hepática fue 7,0% (121/1728). La mayoría de los casos fueron de Grado 1 ó 2 de gravedad notificados en un 3,9% (68/1728) y 1,3 % (22/1728) de los pacientes respectivamente. Se notificaron casos de Grado 3 y 4 en 1,4% (25/1728) y 0,3% (6/1728) de los pacientes respectivamente. No se notificaron casos de Grado 5 en estos ensayos. La mediana hasta su aparición fue de 1,9 meses (rango: 0,0-18,7). La resolución se produjo en 95 pacientes (79%) con una mediana del tiempo hasta la resolución de 5,1 semanas (rango: 0,1-82,6+).
En pacientes tratados con nivolumab en combinación con ipilimumab, la incidencia de anormalidades en las pruebas de función hepática fue 27,9% (125/448). Se notificaron casos de Grado 2, Grado 3, y Grado 4 en 6,3% (28/448), 15,0% (67/448) y 1,8% (8/448) de los pacientes, respectivamente. No se notificaron casos de Grado 5. La mediana del tiempo hasta su aparición fue 1,4 meses (rango: 0,0-11,0). La resolución se produjo en 116 pacientes (92,8%) con una mediana de tiempo hasta su resolución de 5,0 semanas (rango: 0,1-53,1).
Nefritis inmunorrelacionada e insuficiencia renal
En pacientes tratados con nivolumab en monoterapia, la incidencia de nefritis o insuficiencia renal fue 3,2% (55/1728). La mayoría de los casos fueron de Grado 1 ó 2 de gravedad notificados en un 1,9% (32/1728) y 0,8 % (14/1728) de los pacientes respectivamente. Se notificaron casos de Grado 3 y 4 en un 0,5% (8/1728) y <0,1 % (1/1728) de los pacientes respectivamente. No se notificaron casos de nefritis o insuficiencia renal de Grado 5 en estos ensayos. La mediana hasta su aparición fue de 2,3 meses (rango: 0,0-18,2). La resolución se produjo en 33 pacientes (62%) con una mediana de tiempo hasta la resolución de 11,1 semanas (rango: 0,1-77,1+).
En pacientes tratados con nivolumab en combinación con ipilimumab, la incidencia de nefritis e insuficiencia renal fue 4,2% (19/448). Se notificaron casos de Grado 2, Grado 3 y Grado 4 en 1,1% (5/448), 0,9% (4/448) y 0,7% (3/448) de los pacientes, respectivamente. No se notificaron casos de Grado 5. La mediana de tiempo hasta su aparición fue de 2,6 meses (rango: 0,5-14,7). La resolución se produjo en 17 pacientes (89,5%) con una mediana de tiempo hasta su resolución de 1,9 semanas (rango: 0,4-42,6+).
Endocrinopatías inmunorrelacionadas
En pacientes tratados con nivolumab en monoterapia, la incidencia de trastornos en la función tiroidea, incluyendo hipotiroidismo o hipertiroidismo, fue 8,6% (149/1728). La mayoría de los casos fueron de Grado 1 ó 2 de gravedad notificados en un 3,6% (62/1728) y 4,9% (85/1728) de los pacientes respectivamente. Se notificaron casos de Grado 3 de alteraciones de la función tiroidea en 0,1% (2/1728) de los pacientes. Se notificaron hipofisitis (1 caso de Grado 1; 1 caso de Grado 2, y 3 casos de Grado 3), insuficiencia suprarrenal (1 caso de Grado 1; 5 casos de Grado 2, y 4 casos de Grado 3), diabetes mellitus (1 caso de Grado 2) y cetoacidosis diabética (2 casos de Grado 3). No se notificaron casos de Grado 4 ó 5 en estos ensayos. La mediana hasta la aparición de estas endocrinopatías fue de
2,8 meses (rango: 0,4-14,0). La resolución se produjo en 74 pacientes (45%) con una mediana de tiempo hasta la resolución de 66,6 semanas (rango: 0,4-96,1+).
En pacientes tratados con nivolumab en combinación con ipilimumab, la incidencia de trastornos tiroideos fue 23,7% (106/448). Se notificaron casos de Grado 2 y Grado 3 de trastornos tiroideos en 13,4% (60/448) y 1,6% (7/448) de los pacientes, respectivamente. Hipofisitis de Grado 2 y Grado 3 se produjo en 6,0% (27/448) y 1, 8% (8/448) de los pacientes respectivamente. Se produjo insuficiencia suprarrenal de Grado 2 y Grado 3 en 1,1% (5/448), en cada caso e insuficiencia suprarrenal de Grado 4
16
en 0,2% (1/448) de los pacientes. No se notificaron endocrinopatías de Grado 5. La mediana de tiempo hasta la aparición de estas endocrinopatías fue 1,5 meses (rango: 0,0-10,1). La resolución se produjo en 59 pacientes (45,0%). El tiempo hasta la resolución va de 0,4 hasta 74,4+ semanas.
Erupción cutánea inmunorrelacionada
En pacientes tratados con nivolumab en monoterapia, la incidencia de erupción cutánea fue 28,0% (484/1728). La mayoría de los casos fueron de Grado 1 de gravedad notificados en un 21,9% (378/1728) de los pacientes. Se notificaron casos de Grado 2 y Grado 3 en 5,2% (89/1728) y 1,0% (17/1728) de los pacientes respectivamente. No se notificó ningún caso de Grado 4 ó 5 en estos ensayos. La mediana hasta su aparición fue de 1,4 meses (rango: 0,0-17,2). La resolución se produjo en 295 pacientes (62%) con una mediana de tiempo hasta la resolución de 18,1 semanas (rango: 0,1-113,7+).
En pacientes tratados con nivolumab en combinación con ipilimumab, la incidencia de erupción cutánea fue 63,4% (284/448). Se notificaron casos de Grado 2 y Grado 3 en 19,2% (86/448) y 7,4% (33/448) de los pacientes respectivamente. No se notificaron casos de Grado 4 ó 5. La mediana de tiempo hasta su aparición fue 0,5 meses (rango: 0,0-9,7). La resolución se produjo en 192 pacientes (67,6%) con una mediana de tiempo hasta la resolución de 10,4 semanas (rango: 0,1-74,0+).
Reacciones a la perfusión
En pacientes tratados con nivolumab en monoterapia, la incidencia de hipersensibilidad/reacciones a la perfusión fue 4,1% (71/1728), incluyendo 3 casos de Grado 3 y 2 casos de Grado 4.
En pacientes tratados con nivolumab en combinación con ipilimumab, la incidencia de reacciones de hipersensibilidad/reacciones a la perfusión fue 3,8% (17/448); todos los casos fueron de Grado 1 ó 2 de gravedad. Se notificaron casos de Grado 2 en 2,2% (10/448) de los pacientes. No se notificaron casos de Grado 3-5.
Anomalías de laboratorio
En pacientes tratados con nivolumab en monoterapia, el porcentaje de pacientes que experimentó un cambio desde el momento basal hasta una anomalía de laboratorio de Grado 3 ó 4 fue el siguiente: 4,4% para anemia (todas de Grado 3), 0,4% para trombocitopenia, 7,7% para linfopenia, 0,5% para neutropenia, 1,7% para el aumento de la fosfatasa alcalina, 2,7% para el aumento de la AST, 2,4% para el aumento de la ALT, 1,0% para el aumento de la bilirrubina total, 0,8% para el aumento de la creatinina, 2,4% para el aumento de la amilasa, 8,0% para el aumento de la lipasa, 5,9% para hiponatremia, 2,1% para hiperpotasemia, 1,5% para hipopotasemia, 1,3% para hipercalcemia, 0,8% para hipermagnesemia, 0,5% para hipomagnesemia, 0,6% para hipocalcaemia, 0,5% para leucopenia y 0,1% para hipernatremia.
En pacientes tratados con nivolumab en combinación con ipilimumab, la proporción de pacientes que experimentaron un empeoramiento desde el nivel basal hasta anomalías de laboratorio de Grado 3 ó 4 fue la siguiente: 2,8% para anemia (todas de Grado 3), 1,2% para trombocitopenia, 0,5% para leucopenia, 6,4% para linfopenia, 0,7% para neutropenia, 4,1% para elevación de la fosfatasa alcalina, 11,9% para elevaciones de la AST, 14,6% para elevaciones de la ALT, 0,9% para elevaciones de la bilirrubina total, 2,4% para elevaciones de la creatinina, 8,5% para elevaciones de la amilasa, 18,2% para elevaciones de la lipasa, 1,3% para hipocalcemia, 0,3% para cada una de hipercalcemia, hiperpotasemia, hipermagnesemia e hipernatremia, 4,5% para hipopotasemia y 9,2% para hiponatremia.
Inmunogenicidad
De los 1.408 pacientes que se trataron con nivolumab 3 mg/kg, cada 2 semanas, en monoterapia y evaluables para la presencia de anticuerpos-anti-medicamento, 155 pacientes (11,0%), fueron positivos al tratamiento por anticuerpos emergentes anti-medicamento y nueve pacientes (0,6%) tuvieron anticuerpos neutralizantes.
De los 394 pacientes tratados con nivolumab en combinación con ipilimumab y evaluables para la presencia de anticuerpos anti-nivolumab, 149 pacientes (37,8%) fueron positivos al tratamiento por anticuerpos emergentes anti-nivolumab y 18 pacientes (4,6%) tuvieron anticuerpos anti-neutralizantes.
Aunque el aclaramiento de nivolumab aumentó en un 25% cuando estaban presentes anticuerpos anti-nivolumab, la presencia de anticuerpos no se asoció a una pérdida de eficacia o alteración del perfil de toxicidad en presencia de anticuerpos anti-nivolumab de acuerdo a los análisis farmacocinéticos y de respuesta a la exposición de ambos, monoterapia y combinación.
Pacientes de edad avanzada
No se han notificado diferencias en la seguridad entre pacientes de edad avanzada (> 65 años) y pacientes más jóvenes (< 65 años). Los datos de CPNM de los pacientes de 75 años de edad o mayores son demasiado limitados para sacar conclusiones en esta población (ver sección 5.1).
Insuficiencia hepática o renal
En el ensayo de CPNM de histología no escamosa (CA209057), el perfil de seguridad en pacientes con insuficiencia renal o hepática basales fue comparable al de la población general. Estos resultados deben ser interpretados con precaución debido al pequeño tamaño de la muestra dentro de los subgrupos.
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación incluido en el Apéndice V.
4.9 Sobredosis
No se notificaron casos de sobredosis en los ensayos clínicos. En caso de sobredosis, los pacientes deben monitorizarse estrechamente para signos y síntomas de reacciones adversas, y debe instaurarse tratamiento sintomático adecuado inmediatamente.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes antineoplásicos, anticuerpos monoclonales.
Código ATC: L01XC17.
Mecanismo de acción
Nivolumab es un anticuerpo monoclonal humano de tipo inmunoglobulina G4 (IgG4) (HuMAb) que se une al receptor de muerte programada 1 (PD-1) y bloquea su interacción con PD-L1 y PD-L2. El receptor PD-1 es un regulador negativo de la actividad de los linfocitos-T, que se ha visto que está implicado en el control de la respuesta inmunitaria de los linfocitos-T. El acoplamiento de PD-1 con los ligandos PD-L1 y PD-L2, que se expresan en las células presentadoras de antígenos, podría ser expresado por tumores u otras células en el microambiente tumoral, produce la inhibición de la proliferación de los linfocitos-T y la secreción de citoquinas. Nivolumab potencia las respuestas de los linfocitos-T incluyendo respuestas antitumorales, por medio del bloqueo de PD-1, evitando su unión a los ligandos PD-L1 y PD-L2. En modelos sinérgicos de ratón, el bloqueo de la actividad de PD-1 produce una disminución del crecimiento del tumor.
La combinación de nivolumab (anti-PD-1) e ipilimumab (anti-CTLA-4) produjo una mejora en la respuesta antitumoral en melanoma metastásico. En modelos de tumores sinérgicos murinos, el doble bloqueo de PD-1 y CTLA-4 dio como resultado una actividad antitumoral sinérgica.
Eficacia clínica y seguridad
Melanoma
Ensayo de fase 3 aleatorizado frente a dacarbacina (CA209066)
La seguridad y eficacia de nivolumab 3 mg/kg para el tratamiento del melanoma avanzado (irresecable o metastásico) se evaluaron en un ensayo de fase 3, aleatorizado y abierto (CA209066). El ensayo
18
incluyó pacientes adultos con melanoma previamente no tratados (con 18 años o mayores),
Estadio III o IV confirmado, BRAF no mutado, y un estado funcional de acuerdo a la escala ECOG de 0 ó 1. Se excluyeron del ensayo los pacientes con enfermedad autoinmune activa, melanoma ocular, o metástasis cerebrales o leptomeníngeas activas.
Un total de 418 pacientes fueron aleatorizados en una proporción 1:1 para recibir nivolumab (n = 210), administrado por vía intravenosa durante 60 minutos a una concentración de 3 mg/kg cada 2 semanas o dacarbacina (n = 208) a una concentración de 1000 mg/m2 cada 3 semanas. La aleatorización se estratificó por expresión de PD-L1 en el tumor y estadio M (M0/M1a/M1b frente a M1c). El tratamiento continuó mientras se observó beneficio clínico o hasta tolerabilidad inaceptable. El tratamiento después de la progresión de la enfermedad se permitió para pacientes que habían obtenido beneficio clínico y no habían sufrido efectos adversos importantes relacionados con el medicamento del ensayo de acuerdo al criterio del investigador. Las evaluaciones del tumor según los " Criterios de Evaluación de Respuesta en Tumores Sólidos" (RECIST), versión 1.1, se realizaron 9 semanas después de la aleatorización y se siguieron efectuando cada 6 semanas durante el primer año y posteriormente cada 12 semanas. La variable principal fue Supervivencia Global (SG) y las variables secundarias evaluadas por el investigador fueron SLP y Tasa de Respuesta Objetiva (TRO).
Las características basales de los pacientes fueron similares en los dos grupos de pacientes. La mediana de edad fue de 65 años (rango: 18-87). El 59% de los pacientes fueron varones y el 99,5% de raza blanca. La mayoría de pacientes tenían un estado funcional ECOG de 0 (64%) o 1 (34%). El sesenta y uno por ciento de los pacientes presentaban un estadio M1c de la enfermedad en el momento de incorporarse al estudio. El setenta y cuatro por ciento de los pacientes tenía melanoma cutáneo y el 11% melanoma mucoso; el 35% de los pacientes presentaban melanoma con expresión de PD-L1 positiva (> 5% de expresión del tumor en células de membrana). Dieciséis por ciento de pacientes habían recibido tratamiento adyuvante previo; el tratamiento adyuvante más común fue interferón (9%). El cuatro por ciento de los pacientes tenía historia de metástasis cerebrales y el 37% de los pacientes tenían un nivel de LDH basal superior al LSN en el momento de incorporarse al estudio.
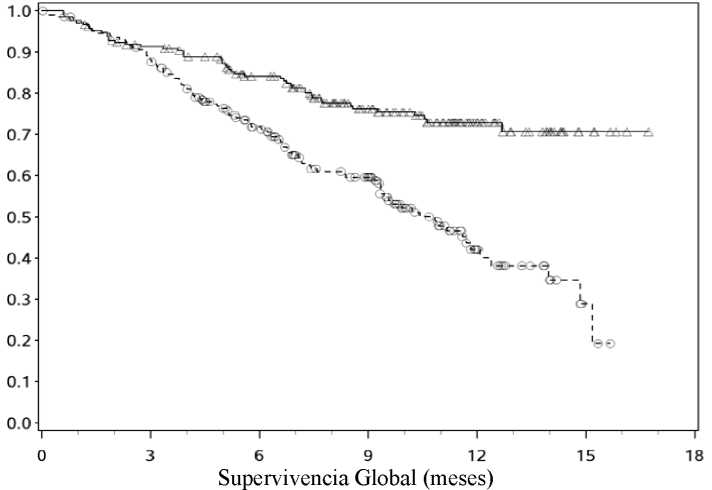
Las curvas Kaplan-Meier para la SG se muestran en la Figura 1.
Número de Sujetos en Riesgo Nivolumab
|
210 Dacarbacina |
185 |
150 |
105 |
45 |
8 |
0 |
|
208 |
177 |
123 |
82 |
22 |
3 |
0 |
-A-Nivolumab (eventos: 50/210), mediana e IC 95%: N.A.
—O— Dacarbacina (eventos: 96/208), mediana e IC 95%: 10,84 (9,33; 12,09)
El beneficio en SG observado fue consistente con el demostrado a través de los subgrupos de pacientes incluyendo estado funcional ECOG, estadio M, historia de metástasis cerebrales y nivel de LDH basal. Se observó beneficio en la supervivencia independientemente de que los pacientes se hubiesen clasificado como PD-L1 positivo o PD-L1 negativo, (la expresión de PD-L1 en la membrana del tumor, estará por encima del límite de 5% o 10%).
Los datos disponibles muestran que la aparición del efecto de nivolumab se retrasa de modo que el beneficio de nivolumab frente a la quimioterapia puede tardar 2-3 meses.
Los resultados de eficacia se muestran en la Tabla 3.
|
nivolumab (n = 210) |
dacarbacina (n = 208) | ||
|
Supervivencia global Eventos Hazard ratio IC 99,79% IC 95% valor-p |
50 (23,8) |
0,42 (0,25; 0,73) (0,30; 0,60) < 0,0001 |
96 (46,2) |
|
Mediana (IC 95%) Tasa (IC 95%) A 6 meses A 12 meses Supervivencia libre de progresión Eventos Hazard ratio IC 95% valor-p |
No alcanzada 84,1 (78,3; 88,5) 72,9 (65,5; 78,9) 108 (51,4) |
0,43 (0,34; 0,56) < 0,0001 |
10.8 (9,33; 12,09) 71.8 (64,9; 77,6) 42,1 (33,0; 50,9) 163 (78,4) |
|
Mediana (IC 95%) Tasa (IC 95%) A 6 meses A 12 meses |
5,1 (3,48; 10,81) 48,0 (40,8; 54,9) 41,8 (34,0; 49,3) |
2,2 (2,10; 2,40) 18,5 (13,1; 24,6) NA | |
|
Respuesta objetiva (IC 95%) Odds ratio (IC 95%) valor-p |
84 (40,0%) (33,3; 47,0) 4,06 (2,52;6,54) < 0,0001 |
29 (13,9%) (9,5; 19,4) | |
|
Respuesta completa (RC) Respuesta parcial (RP) Enfermedad estable (EE) |
16 (7,6%) 68 (32,4%) 35 (16,7%) |
2 (1,0%) 27 (13,0%) 46 (22,1%) | |
|
Mediana de la duración de la respuesta Meses (rango) |
No alcanzado (0+ -12,5+) |
6,0 (1,1-10,0+) | |
|
Mediana del tiempo hasta respuesta Meses (rango) |
2,1 (1,2-7,6) |
2,1 (1,8-3,6) | |
+” indica una observación censurada
Ensayo de fase 3 aleatorizado frente a quimioterapia (CA209037)
La seguridad y eficacia de nivolumab 3 mg/kg para el tratamiento del melanoma avanzado (irresecable o metastásico) se evaluó en un ensayo de fase 3, aleatorizado y abierto (CA209037). El ensayo incluyó pacientes adultos que habían progresado antes o después a ipilimumab y aquellos que presentan la mutación BRAF V600 positiva y también habían progresado durante o después de otro tratamiento con un inhibidor de BRAF. Se excluyeron del ensayo los pacientes con enfermedad autoinmune activa, melanoma ocular o historia conocida de reacciones adversas relacionadas con ipilimumab de grado elevado (Grado 4 según los CTCAE v4.0), salvo en el caso de náuseas, fatiga, reacciones a la perfusión o endocrinopatías que se habían resuelto.
Un total de 405 pacientes fueron aleatorizados para recibir nivolumab (n = 272) administrado por vía intravenosa durante 60 minutos con una concentración de 3 mg/kg cada 2 semanas o quimioterapia (n = 133) que consistía en la elección del investigador o dacarbacina (1000 mg/m2 cada 3 semanas) o carboplatino (AUC 6 cada 3 semanas) y paclitaxel (175 mg/m2 cada 3 semanas). La aleatorización se estratificó por estado mutacional BRAF, expresión de PD-L1 en el tumor y la mejor respuesta previa a ipilimumab.
Las variables principales de la eficacia fueron TRO confirmada, en los primeros 120 pacientes tratados con nivolumab, medida por un comité independiente de revisión radiológica (IRRC) utilizando (RECIST, versión 1.1), y la comparación de la SG entre nivolumab y la quimioterapia. Las variables adicionales incluyeron la duración y el tiempo de la respuesta.
La mediana de la edad fue 60 años (rango: 23-88). Sesenta y cuatro por ciento de los pacientes fueron varones y el 98% de raza blanca. El estado funcional ECOG fue de 0 para el 61% de los pacientes y de 1 para el 39% de los pacientes. La mayoría (75%) de los pacientes presentaba un estadio M1c de la enfermedad en el momento de incorporarse al estudio. El setenta y tres por ciento de los pacientes tenía melanoma cutáneo y el 10% melanoma mucoso. El número de tratamientos sistémicos previos que habían recibido fue 1 para un 27% de los pacientes, 2 para un 51% de los pacientes y > 2 para un 21% de los pacientes. Un veintidós por ciento de los pacientes que tenían tumor presentaban la mutación BRAF positiva y un 50% de los pacientes que tenían tumor fueron considerados PD-L1 positivo. El sesenta y cuatro por ciento de los pacientes no tuvieron beneficio clínico previo (RC/RP o EE) con ipilimumab. Las características basales fueron similares para ambos grupos de tratamiento excepto para la proporción de pacientes con antecedentes de metástasis cerebrales (19% y 13% en el grupo de nivolumab y en el grupo de quimioterapia respectivamente) y pacientes con un nivel de LDH basal superior al LSN (51% y 35%, respectivamente).
En el momento del análisis final de TRO, los resultados obtenidos de 120 pacientes tratados con nivolumab y 47 tratados con quimioterapia, durante un mínimo de 6 meses de seguimiento fueron analizados. Los resultados de eficacia se muestran en la Tabla 4.
Tabla 4: Mejor respuesta global, tiempo y duración de la respuesta (CA209037)
|
nivolumab (n = 120) |
quimioterapia (n = 47) | |
|
Respuesta objetiva confirmada (ROC) (IC 95%) |
38 (31,7%) (23,5; 40,8) |
5 (10,6%) (3,5; 23,1) |
|
Respuesta completa (RC) Respuesta parcial (RP) Enfermedad estable (EE) |
4 (3,3%) 34 (28,3%) 28 (23,3%) |
0 5 (10,6%) 16 (34,0%) |
|
Mediana de la Duración de la Respuesta Meses (rango) |
No alcanzada |
3,6 (No disponible) |
|
Mediana del Tiempo hasta la Respuesta Meses (rango) |
2,1 (1,6-7,4) |
3,5 (2,1-6,1) |
Se observaron respuestas objetivas a nivolumab (de acuerdo a la definición de las variables principales) en pacientes con melanoma con o sin mutación BRAF positiva. De los pacientes que recibieron nivolumab, la TRO en el subgrupo BRAF mutación positiva (n = 26) fue 23% (IC 95%:9,0; 43,6), y 34% (IC 95%:24,6; 44,5) en pacientes cuyos tumores eran BRAF no mutado (n = 94). Se observaron respuestas objetivas a nivolumab, independientemente de que los pacientes se hubiesen clasificado como PD-L1 positivo o PD-L1 negativo, (la expresión de PD-L1 en la membrana del tumor, estará por encima del límite de 5% o 10%). Sin embargo, el papel de este biomarcador (expresión de PD-L1 en el tumor) no ha sido completamente aclarado.
Los datos de SG eran inmaduros en el momento del análisis de SLP. No hubo diferencias estadísticamente significativas entre nivolumab y quimioterapia en el análisis preliminar de SG que no fue ajustado a los efectos potenciales de confusión del tratamiento posterior. Hay que destacar que 42 (31,6%) de los pacientes en el brazo de quimioterapia y posteriormente tratados con un anti-PD1.
Los datos disponibles muestran que la aparición del efecto de nivolumab se retrasa de modo que el beneficio de nivolumab frente a la quimioterapia puede tardar 2-3 meses.
La evaluación del investigador confirmó que la TRO en todos los pacientes tratados fue de 25,7%
[IC 95%: 20,6; 31,4] en el grupo de nivolumab (n = 268) vs 10,8% [IC 95%: 5,5; 18,5] en el grupo de quimioterapia, (n = 102) con una diferencia en la TRO de 15,0% [IC 95%: 6,0; 22,2]. La evaluación del investigador confirmó que la TRO en los pacientes con la mutación BRAF positiva (n = 79) fue del
19,3% [IC 95%: 10,0; 31,9] vs 13,6% [IC 95%: 2,9; 34,9], respectivamente, y en los pacientes sin mutación BRAF (wild-type) (n = 291) fue del 27,5% [IC 95%: 21,6; 34,0] vs 10,0% [IC 95%: 24,4; 18,8] respectivamente.
La SLP numéricamente favoreció al grupo de nivolumab vs el grupo de quimioterapia en todos los pacientes aleatorizados, los pacientes con mutación BRAF positiva y los pacientes sin mutación BRAF (BRAF WT) (HRs 0,74 [IC 95%: 0,57; 0,97], 0,98 [IC 95%: 0,56; 1,70] y 0,63 [IC 95%: 0,47; 0,85] respectivamente.
Ensayo de fase 1 abierto, con escalada de la dosis (MDX1106-03)
La seguridad y tolerabilidad de nivolumab se investigaron en un ensayo de fase 1, abierto, de escalada de la dosis en varios tipos de tumor, incluido el melanoma maligno. De los 306 pacientes previamente tratados, reclutados en el estudio, 107 tenían melanoma y recibieron nivolumab a dosis de 0,1 mg/kg, 0,3 mg/kg, 1 mg/kg, 3 mg/kg o 10 mg/kg, durante un máximo de 2 años. En esta población la respuesta objetiva se notificó en 33 pacientes (31%), con una mediana de la duración de la respuesta de 22,9 meses (IC 95%: 17,0, NR). La mediana de la SLP fue 3,7 meses (IC 95%: 1,9; 9,3). La mediana de la SG fue 17,3 meses (IC 95%: 12,5; 36,7) y las tasas estimadas de la SG fueron el 63% (IC 95%: 53,71) a 1 año, el 48% (IC 95%: 38, 57) a 2 años y el 41%
(IC 95%: 31,51) a 3 años.
Ensayo de fase 3 aleatorizado de nivolumab en combinación con ipilimumab o nivolumab en monoterapia frente a ipilimumab en monoterapia (CA209067)
La seguridad y eficacia de nivolumab en combinación con ipilimumab o nivolumab frente a ipilimumab en monoterapia para el tratamiento de melanoma avanzado (irresecable o metastásico) se evaluaron en un ensayo de fase 3, aleatorizado y doble ciego (CA209067). Se evaluaron las diferencias descriptivamente entre los dos grupos que contienen nivolumab. El ensayo incluyó pacientes adultos con melanoma irresecable confirmado en Estadio III o Estadio IV. El estado funcional ECOG de los pacientes fue de 0 ó 1. Fueron elegibles los pacientes que no habían recibido tratamiento anticanceroso sistémico previo para melanoma irresecable o metastásico. Se permitió el tratamiento adyuvante o neoadyuvante si se completaba al menos 6 semanas antes de la aleatorización. Los pacientes con enfermedad autoinmune activa, melanoma ocular/uveal, o metástasis cerebral o leptomeníngea activas fueron excluidos de este ensayo.
Un total de 945 pacientes fueron aleatorizados para recibir nivolumab en combinación con ipilimumab (n = 314), nivolumab en monoterapia (n = 316), o ipilimumab en monoterapia (n = 315). Los pacientes en el brazo de combinación recibieron nivolumab 1 mg/kg durante 60 minutos e ipilimumab 3 mg/kg durante 90 minutos administrados de forma intravenosa cada 3 semanas durante las primeras 4 dosis, seguido de nivolumab 3 mg/kg en monoterapia cada 2 semanas. Los pacientes en el brazo de nivolumab en monoterapia recibieron nivolumab 3 mg/kg cada 2 semanas. Los pacientes en el brazo comparador recibieron ipilimumab 3 mg/kg y placebo de nivolumab administrados de forma intravenosa cada 3 semanas, durante 4 dosis seguido de placebo cada 2 semanas. La aleatorización se estratificó según la expresión de PD-L1 (>5% vs. <5% de expresión en la membrana celular del tumor), el estado BRAF y el estadio M según el sistema de clasificación del American Joint Committee on Cancer (AJCCpor sus siglas en inglés). El tratamiento continuó mientras se observó beneficio clínico o hasta que el tratamiento no fue tolerado. Las evaluaciones del tumor se realizaron 12 semanas después de la aleatorización y se siguieron efectuando cada 6 semanas durante el primer año y cada 12 semanas posteriormente. Los objetivos medidos fueron la supervivencia libre de progresión y la SG. La TRO y la duración de la respuesta también se evaluaron.
Las características basales de los pacientes fueron similares en los tres grupos de tratamiento. La mediana de edad fue 61 años (rango: 18 a 90 años), el 65% de los pacientes fueron varones y el 97% eran de raza blanca. El nivel basal del estado funcional ECOG fue 0 (73%) o 1 (27%). La mayoría de los pacientes tenían un Estadio IV de la enfermedad según el AJCC (93%); un 58% tenían la enfermedad en estadio M1c en el momento de incorporarse al estudio. El veintidós por ciento de los pacientes había recibido tratamiento adyuvante previo. El treinta y dos por ciento de los pacientes tenía melanoma con mutación BRAF positiva; el 26,5% de los pacientes tenía >5% de expresión de PD-L1 en la membrana celular del tumor. El cuatro por ciento de los pacientes tenía historia de metástasis cerebral y el 36% de los pacientes tenía un nivel de LDH basal superior al LSN en el momento de incorporarse al estudio. Entre los pacientes con expresión de PD-L1 cuantificable en el
23
tumor, la distribución de pacientes fue similar en los tres grupos de tratamiento. La expresión de PD-L1 en el tumor se determinó usando el test PD-L1 IHC 28-8 pharmDx.
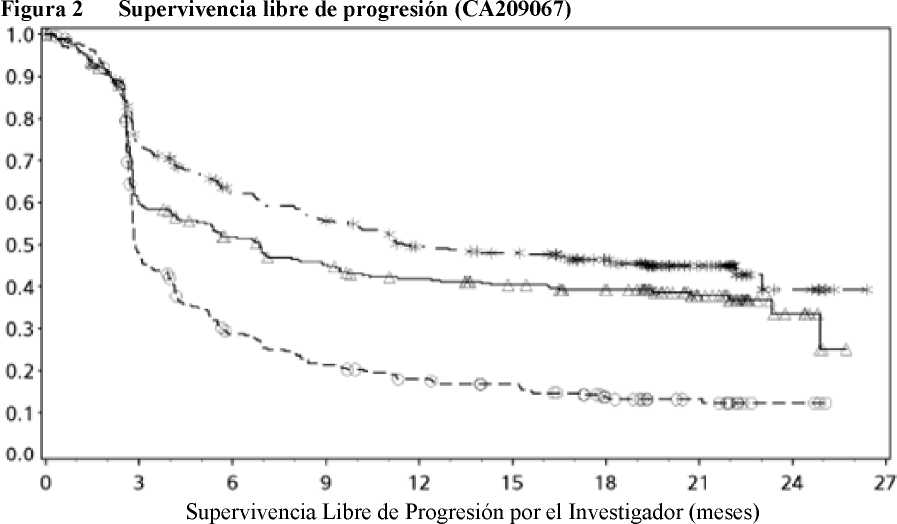
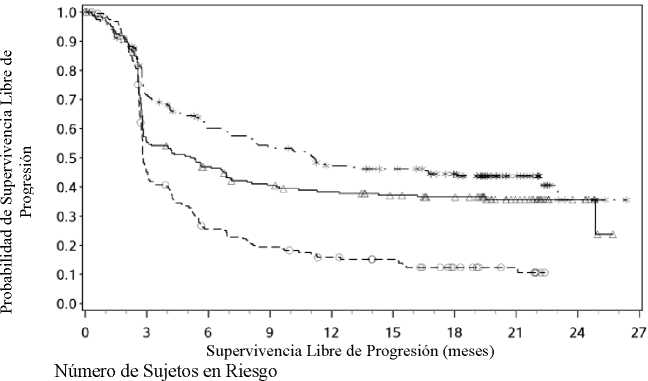
El seguimiento mínimo fue de 18 meses. La supervivencia global no estaba madura en el momento de este análisis. Los resultados de SLP se muestran en la Figura 2 (toda la población aleatorizada),
Figura 3 (cut-off del 5% de PD-L1 en el tumor) y Figura 4 (cut-off del 1% de PD-L1 en el tumor). Las respuestas se resumen en la Tabla 5.
Probabilidad de Supervivencia Libre de Progresión
Número de Sujetos en Riesgo Nivolumab + Ipilimumab
|
314 219 Nivolumab |
174 |
156 |
133 |
126 |
103 |
48 |
8 |
0 |
|
316 177 Ipilimumab |
148 |
127 |
114 |
104 |
94 |
46 |
8 |
0 |
|
315 137 |
78 |
58 |
46 |
40 |
25 |
15 |
3 |
0 |
---*---- Nivolumab+ipilimumab (eventos: 161/314), mediana e IC 95%: 11,50 (8,90; 22,18).
Tasa de SLP a los 12 meses e IC 95%: 49% (44; 55)
-A- Nivolumab (eventos: 183/316), mediana e IC 95%: 6,87 (4,34; 9,46).
Tasa de SLP a los 12 meses e IC 95%: 42% (36; 47)
---O— Ipilimumab (eventos: 245/315), mediana e IC 95%: 2,89 (2,79; 3,42).
Tasa de SLP a los 12 meses e IC 95%: 18% (14; 23)
Nivolumab+ipilimumab vs ipilimumab (análisis primario) - HR (IC 99,5%): 0,42 (0,32; 0,56); valor-p: <0,0001 Nivolumab vs ipilimumab (análisis primario) - HR (IC 99,5%): 0,55 (0,42; 0,73); valor-p: <0,0001 Nivolumab+ipilimumab vs nivolumab (análisis descriptivo) - HR (IC 95%): 0,76 (0,62; 0,95)
Expresión de PD-L1 < 5%
Nivolumab + Ipilimumab
|
210 142 Nivolumab |
113 |
101 |
86 |
81 |
69 |
31 |
5 |
0 |
|
208 108 Ipilimumab |
89 |
75 |
69 |
62 |
55 |
29 |
7 |
0 |
|
202 82 |
45 |
34 |
26 |
22 |
12 |
7 |
0 |
0 |
---*---- Nivolumab+Ipilimumab (eventos: 111/210), mediana e IC 95%: 11,10 (7,98; 22,18)
—A- Nivolumab (eventos: 125/208), mediana e IC 95%: 5,32 (2,83; 7,06)
---O--- Ipilimumab (eventos: 159/202), mediana e IC 95%: 2,83 (2,76; 3,09)
Nivolumab+Ipilimumab vs. Ipilimumab - hazard ratio: 0,42 (0,33; 0,54)
Nivolumab vs. Ipilimumab - hazard ratio: 0,57 (0,45; 0,72)
Nivolumab+Ipilimumab vs. Nivolumab - hazard ratio: 0,74 (0,58; 0,96)
Nivolumab + Ipilimumab
|
68 53 Nivolumab |
44 |
39 |
33 |
31 |
22 |
13 |
3 |
0 |
|
80 57 Ipilimumab |
51 |
45 |
39 |
37 |
36 |
16 |
1 |
0 |
|
75 40 |
21 |
17 |
14 |
12 |
8 |
6 |
2 |
0 |
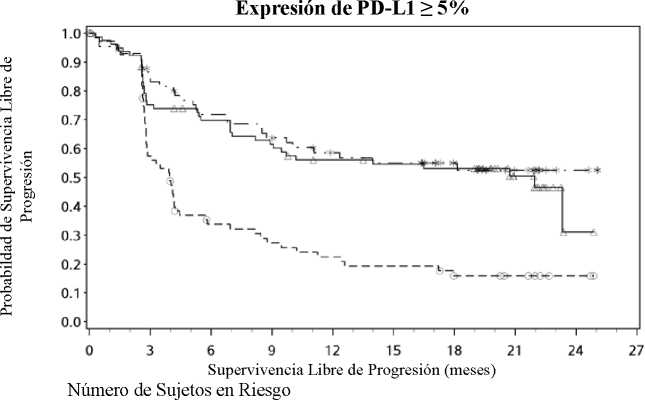
---*---- Nivolumab+Ipilimumab (eventos: 29/68), mediana e IC 95%: N.A. (9,72; N.A.)
—A- Nivolumab (eventos: 38/80), mediana e IC 95%: 21,95 (8,90; N.A.)
---O--- Ipilimumab (eventos: 57/75), mediana e IC 95%: 3,94 (2,79; 4,21)
Nivolumab+Ipilimumab vs. Ipilimumab - hazard ratio: 0,35 (0,22; 0,55) Nivolumab vs. Ipilimumab - hazard ratio: 0,41 (0,27; 0,62) Nivolumab+Ipilimumab vs. Nivolumab - hazard ratio: 0,87 (0,54; 1,41)
Expresión de PD-L1 < 1%
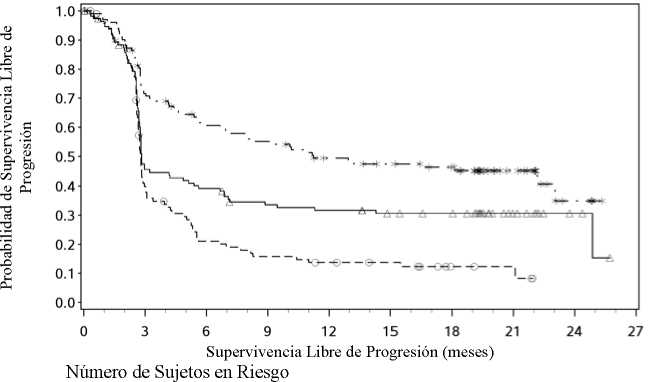
Nivolumab + Ipilimumab
|
123 82 Nivolumab |
65 |
59 |
50 |
46 |
41 |
18 |
4 |
0 |
|
117 50 Ipilimumab |
43 |
35 |
33 |
29 |
27 |
11 |
3 |
0 |
|
113 39 |
20 |
15 |
12 |
10 |
4 |
3 |
0 |
0 |
---*---- Nivolumab+Ipilimumab (eventos: 63/123), mediana e IC 95%: 11,24 (6,93; 23,03)
—A- Nivolumab (eventos: 77/117), mediana e IC 95%: 2,83 (2,76; 5,13)
---O--- Ipilimumab (eventos: 87/113), mediana e IC 95%: 2,79 (2,66; 2,96)
Nivolumab+Ipilimumab vs. Ipilimumab - hazard ratio: 0,39 (0,28; 0,54) Nivolumab vs. Ipilimumab - hazard ratio: 0,65 (0,48; 0,88) Nivolumab+Ipilimumab vs. Nivolumab - hazard ratio: 0,60 (0,43; 0,84)
Expresión de PD-L1 > 1%
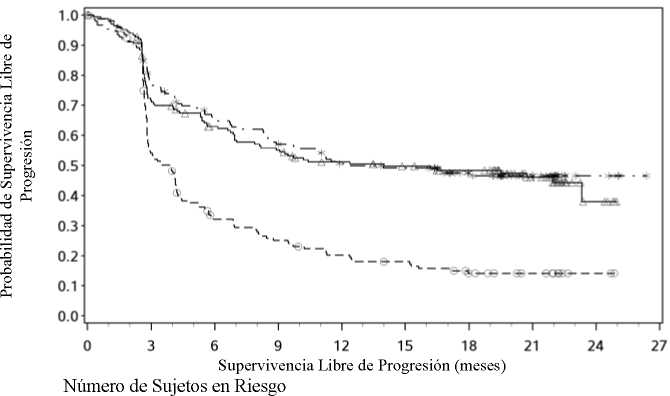
Nivolumab + Ipilimumab
|
155 113 Nivolumab |
92 |
81 |
69 |
66 |
50 |
26 |
4 |
0 |
|
1 71 1 1 5 Ipilimumab |
97 |
85 |
75 |
70 |
64 |
34 |
5 |
0 |
|
164 83 |
46 |
36 |
28 |
24 |
16 |
10 |
2 |
0 |
---*---- Nivolumab+Ipilimumab (eventos: 77/155), mediana e IC 95%: 12,35 (8,74; N.A.)
—A- Nivolumab (eventos: 86/171), mediana e IC 95%: 14,00 (7,03; N.A.)
---O--- Ipilimumab (eventos: 129/164), mediana e IC 95%: 3,91 (2,83; 4,17)
Nivolumab+Ipilimumab vs. Ipilimumab - hazard ratio: 0,42 (0,31; 0,55) Nivolumab vs. Ipilimumab - hazard ratio: 0,44 (0,34; 0,58) Nivolumab+Ipilimumab vs. Nivolumab - hazard ratio: 0,94 (0,69; 1,28)
Tabla 5: Respuesta objetiva (CA209067)
|
nivolumab + ipilimumab (n=314) |
nivolumab (n=316) |
ipilimumab (n=315) | |
|
Respuesta objetiva |
181 (58%) |
138 (44%) |
60 (19%) |
|
(IC 95%) |
(52,0; 63,2) |
(38,1; 49,3) |
(14,9; 23,8) |
|
Odds ratio (vs. ipilimumab) |
6,09 |
3,40 | |
|
(IC 99,5%) |
(3,59; 10,33) |
(2,02; 5,72) | |
|
valor-p |
p<0,0001 |
p<0,0001 | |
|
Respuesta completa (RC) |
38 (12%) |
31 (10%) |
7 (2%) |
|
Respuesta parcial (RP) |
143 (46%) |
107 (34%) |
53 (17%) |
|
Enfermedad estable (EE) |
41 (13%) |
33 (10%) |
69 (22%) |
|
Mediana de la duración de la respuesta | |||
|
Meses (rango) |
No alcanzado (0+ - 24+) |
22,3 (0+ - 23+) |
14,4 (1,4 - 22,3+) |
|
TRO (IC 95%) según el nivel de expresión de PD-L1 en el tumor | |||
|
<5% |
55% (47,8; 61,6) n=210 |
41% (34,6; 48,4) n=208 |
18% (12,8; 23,8) n=202 |
|
>5% |
72% (59,9; 82,3) n=68 |
58% (45,9; 68,5) n=80 |
21% (12,7; 32,3) n=75 |
|
<1% |
52% (42,8; 61,1) n=123 |
33% (24,9; 42,6) n=117 |
19% (11,9; 27,0) n=113 |
|
>1% |
65% (56,4; 72,0) n=155 |
54% (46,6; 62,0) n=171 |
19% (13,2; 25,7) n=164 |
Ambos brazos que contienen nivolumab demostraron beneficio significativo en SLP y mayor TRO comparado con ipilimumab solo y los resultados observados de SLP y TRO a los 12 meses de seguimiento fueron consistentemente demostrados en todos los subgrupos de pacientes, incluyendo el nivel basal del estado funcional ECOG, estado BRAF, estadio M, edad, historia de metástasis cerebral y LDH en el nivel basal.
Entre los 128 pacientes que interrumpieron nivolumab en combinación con ipilimumab debido a una reacción adversa, la mediana de SLP fue de 16,7 meses (IC 95%: 10,2; NA), y la de TRO fue de 69% (88/128) con 15% (19/128) alcanzando una respuesta completa.
Ambos brazos que contienen nivolumab demostraron mayores tasas de respuesta objetiva que ipilimumab, independientemente de los niveles de expresión de PD-L1. Las TRO fueron mayores para la combinación de nivolumab e ipilimumab con respecto a nivolumab en monoterapia en todos los niveles de expresión de PD-L1 en el tumor (Tabla 5). La mediana de la duración de la respuesta para pacientes con expresión de PD-L1 en el tumor >5% no se alcanzó (rango: 0+-22,3+) en el brazo de la combinación, fue de 20,8 meses (rango: 2,8-20,8) en el brazo de nivolumab en monoterapia y no se alcanzó en el brazo de ipilimumab (rango: 1,4-19,9+). Cuando la expresión de PD-L1 en el tumor fue <5%, la mediana de la duración de la respuesta no se alcanzó (rango: 0+-24+) en el brazo de la combinación, fue de 22,3 meses (rango: 0+-23+) en el brazo de nivolumab en monoterapia y de 18,2 meses (rango: 1,4-19,8+) en el brazo de ipilimumab en monoterapia.
No se puede establecer un límite claro de la expresión de PD-L1 a la hora de considerar los objetivos relevantes de respuesta tumoral y SLP. Los resultados del análisis post-hoc exploratorio multivariante indican que otras características tanto del paciente como del tumor (p. ej. estado funcional ECOG, estadio M, estadio AJCC, género, región y LDH en estado basal) podrían contribuir al resultado clínico.
Eficacia según el estado BRAF: los pacientes con mutación BRAF[V600] positiva y BRAF wild-type
aleatorizados a nivolumab en combinación con ipilimumab tuvieron una mediana de SLP de
15,5 meses (IC 95%: 8,0; NA) y 11,3 meses (IC 95%: 8,3; 22,2), y de TRO de 66,7% (IC 95%: 56,6;
75,7; n=102) y 53,3% (IC 95%: 46,3; 60,2; n=212) respectivamente, mientras que los pacientes aleatorizados a nivolumab en monoterapia tuvieron una mediana de SLP de 5,6 meses (IC 95%: 2,8; 9,3) y 7,1 meses (IC 95%: 4,9; 14,3), y de TRO de 36,7% (IC 95%: 27,2; 47,1; n=98) y 46,8%
(IC 95%: 40,0; 53,6; n=218), respectivamente.
Ensayo de _ fase 2 aleatorizado de nivolumab en combinación con ipilimumab e ipilimumab (CA209069)
El estudio CA209069 fue un estudio de Fase 2, aleatorizado, doble ciego qué comparó la combinación de nivolumab e ipilimumab con ipilimumab solo, en 142 pacientes con melanoma avanzado (irresecable o mestastásico) con criterios de inclusión similares al estudio CA209067 y el análisis primario en pacientes con melanoma BRAF wild-type (77% de los pacientes). La TRO evaluada por el investigador fue de 61% (IC 95%: 48,9; 72,4) en el brazo de combinación (n=72) versus 11% (IC 95%: 3,0; 25,4) en el brazo de ipilimumab (n=37). Las tasas de SG estimadas a los 12 y 18 meses fueron del 79% (IC 95%: 67; 87) y del 73% (IC 95%: 61; 82) respectivamente para la combinación y del 62% (IC 95%: 44; 75) y del 56% (IC 95%: 39; 70) respectivamente para ipilimumab.
Cáncer de Pulmón No Microcítico CPNM de histología escamosa
Ensayo de fase 3 aleatorizado frente a docetaxel (CA209017)
La seguridad y eficacia de nivolumab 3 mg/kg para el tratamiento del CPNM de histología escamosa avanzado se evaluaron en un ensayo de fase 3, aleatorizado y abierto (CA209017). El ensayo incluyó pacientes (con 18 años de edad o mayores) que habían experimentado una progresión de la enfermedad durante o después de un régimen de quimioterapia basado en un doblete de platino y un estado funcional ECOG de 0 a 1. Los pacientes se incluyeron con independencia de la expresión de PD-L1 en el tumor. Los pacientes con enfermedad autoinmune, enfermedad pulmonar intersticial sintomática, o metástasis cerebrales sin tratar se excluyeron de este estudio. Los pacientes con metástasis cerebrales tratadas fueron elegibles si neurológicamente habían vuelto al valor basal como mínimo 2 semanas antes del reclutamiento, y si habían terminado el tratamiento con corticosteroides o si recibían tratamiento con una dosis estable o descendiendo a una dosis equivalente a < 10 mg diarios de prednisona.
Un total de 272 pacientes fueron aleatorizados para recibir nivolumab 3 mg/kg; (n = 135) administrado de forma intravenosa durante 60 minutos cada 2 semanas o docetaxel (n = 137) 75 mg/m2 cada 3 semanas. El tratamiento continuó mientras se observó beneficio clínico o hasta que el tratamiento no fue tolerado. Las evaluaciones del tumor según los RECIST, versión 1.1, se realizaron 9 semanas después de la aleatorización y se siguieron efectuando cada 6 semanas posteriormente. La variable principal fue la SG y las variables secundarias evaluadas por el investigador fueron la TRO y la SLP. Además la mejoría en los síntomas y el estatus de salud global se evaluaron utilizando la media de la Escala de Síntomas de Cáncer de Pulmón (ESCP) el índice de carga sintomática media y el EQ-5D la escala visual análoga (EQ-VAS), respectivamente.
Las características basales de los pacientes fueron similares en los dos grupos de pacientes. La mediana de edad fue 63 años (rango: 39-85) con 44% >65 años de edad y 11% >75 años de edad. La mayoría de los pacientes fueron de raza blanca (93%) y varones (76%). El treinta y uno por ciento tenían enfermedad progresiva notificada como respuesta mejor a su régimen de tratamiento previo más reciente y un 45% recibió nivolumab dentro de los 3 meses después de completar su régimen de tratamiento previo más reciente. El nivel basal del estado funcional ECOG fue 0 (24%) o 1 (76%).
Las curvas de Kaplan-Meier para la SG se muestran en la Figura 5.
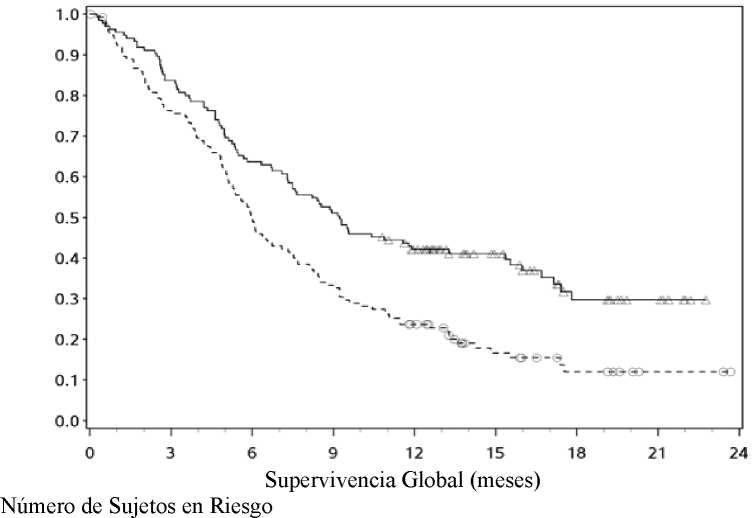
Nivolumab 3 mg/kg
|
135 |
113 |
86 |
69 |
52 |
31 |
15 |
7 |
0 |
|
Docetaxel 137 |
103 |
68 |
45 |
30 |
14 |
7 |
2 |
0 |
-A-Nivolumab 3 mg/kg (eventos: 86/135), mediana e IC 95%: 9,23 (7,33; 13,27)
—O— Docetaxel (eventos: 113/137), mediana e IC 95%: 6,01 (5,13; 7,33)
El beneficio en SG observado fue consistente con el demostrado a través de los subgrupos de pacientes. Se observó beneficio en la supervivencia independientemente de que los pacientes se hubiesen clasificado como PD-L1 positivo o PD-L1 negativo, (la expresión de PD-L1 en la membrana del tumor, estará por encima del límite de 1%, 5% o 10%). Sin embargo, el papel de este biomarcador, (expresión de PD-L1 en el tumor) no se ha aclarado completamente.
El ensayo CA209017 incluyó un número limitado de pacientes > 75 años de edad (11 en el grupo de nivolumab y 18 en el grupo de docetaxel). Nivolumab demostró numéricamente menos efecto sobre la SG (HR = 1,85; IC 95%: 0,76; 4,51), SLP (HR = 1,76; IC 95%: 0,77; 4,05) y TRO (9,1% vs. 16,7%). Debido al pequeño tamaño de muestra, no pueden sacarse conclusiones definitivas de estos datos.
Los resultados de eficacia se muestran en la Tabla 6.
|
Tabla 6: Resultados de eficacia (CA209017) | |||
|
nivolumab (n = 135) |
docetaxel (n = 137) | ||
|
Supervivencia global | |||
|
Eventos Hazard ratio IC 96,85% valor-p |
86 (63,7) |
0,59 (0,43; 0,81) 0,0002 |
113 (82,5) |
|
Mediana (IC 95%) meses |
9,23 (7,33; 13,27) |
6,01 (5,13; 7,33) | |
|
Tasa (IC 95%) a 12 meses |
42,1 (33,7; 50,3) |
23,7 (16,9; 31,1) | |
|
Respuesta objetiva confirmada |
27 (20,0%) |
12 (8,8%) | |
|
(IC 95%) |
(13,6; 27,7) |
(4,6; 14,8) | |
|
Odds ratio (IC 95%) valor-p |
2,64 (1,27; 5,49) 0,0083 | ||
|
Respuesta completa (RC) |
1 (0,7%) |
0 | |
|
Respuesta parcial (RP) |
26 (19,3%) |
12 (8,8%) | |
|
Enfermedad estable (EE) |
39 (28,9%) |
47 (34,3%) | |
|
Mediana de la duración de la respuesta | |||
|
Meses (rango) |
No alcanzada (2,9 - 20,5+) |
8,4 (1,4+ - 15,2+) | |
|
Mediana del tiempo hasta respuesta | |||
|
Meses (rango) |
2,2 (1,6 - 11,8) |
2,1 (1,8 - 9,5) | |
|
Supervivencia libre de progresión | |||
|
Eventos Hazard ratio IC 95% valor-p |
105 (77,8) |
0,62 (0,47; 0,81) < 0,0004 |
122 (89,1) |
|
Mediana (IC 95%) (meses) |
3,48 (2,14; 4,86) |
2,83 (2,10; 3,52) | |
|
Tasa (IC 95%) a 12 meses |
20,8 (14,0; 28,4) |
6,4 (2,9; 11,8) | |
La mejora de la tasa que relaciona síntomas y enfermedad, medida por ESCP, fue similar entre el grupo de nivolumab (18,5%) y el grupo de docetaxel (21,2%). La media de EQ-VAS aumentó en el tiempo para ambos grupos de tratamiento, indicando un mejor estatus de salud global para los pacientes que permanecen en tratamiento.
Ensayo de brazo único de fase 2 (CA209063)
El ensayo CA209063 fue un ensayo de un solo brazo, abierto, realizado en 117 pacientes con CPNM de histología escamosa, localmente avanzado o metastásico después de dos o más líneas de tratamiento; aunque se aplicaron criterios de inclusión similares a los del ensayo CA209017. Nivolumab 3 mg/kg demostró una tasa de respuesta global de 14,5% (IC 95%: 8,7-22,2%), una mediana de SG de 8,21 meses (IC 95%: 6,05-10,9) y una mediana de SLP de 1,87 meses (IC 95%: 1,77-3,15). La SLP se midió por RECIST, versión 1.1. La tasa estimada de supervivencia a 1 año fue 41%.
CPNM de histología no escamosa
Ensayo de fase 3 aleatorizado frente a docetaxel (CA209057)
La seguridad y eficacia de nivolumab 3 mg/kg para el tratamiento del CPNM de histología no escamosa avanzado o metastásico se evaluaron en un ensayo fase 3, aleatorizado y abierto (CA209057). El ensayo incluyó pacientes (con 18 años o mayores) que habían experimentado una progresión de la enfermedad durante o después de un tratamiento de quimioterapia basado en un doblete de platino, que podría estar incluido en el tratamiento de mantenimiento y que tuvieran un
estado funcional ECOG de 0 ó 1. Se permitió una línea adicional de tratamiento con ITK para los pacientes con mutación conocida de EGFR o translocación de ALK. Los pacientes se incluyeron con independencia de la expresión de PD-L1 en el tumor. Los pacientes con enfermedad autoinmune activa, enfermedad pulmonar intersticial sintomática, o metástasis cerebrales sin tratar se excluyeron de este estudio. Los pacientes con metástasis cerebrales tratadas fueron elegibles si neurológicamente habían recuperado su valor basal como mínimo 2 semanas antes del reclutamiento, y si habían terminado el tratamiento con corticosteroides o si recibían tratamiento con una dosis estable o en pauta descendente a una dosis equivalente a < 10 mg diarios de prednisona.
Un total de 582 pacientes fueron aleatorizados para recibir nivolumab 3 mg/kg administrado de forma intravenosa durante 60 minutos cada 2 semanas (n = 292) o docetaxel 75 mg/m2 cada 3 semanas (n = 290). El tratamiento continuó mientras se observó beneficio clínico o hasta que el tratamiento no fue tolerado. Las evaluaciones del tumor se realizaron según los criterios RECIST. La variable principal fue la SG. Las variables secundarias evaluadas por el investigador fueron la TRO y la SLP.
Se realizaron análisis adicionales de subgrupos preespecificados para evaluar la eficacia de la expresión de PD-L1 en el tumor a niveles predefinidos de 1%, 5% y 10%. La evaluación de acuerdo a intervalos discretos de expresión de PD-L1 no fueron incluidos en los análisis preespecificados debido al pequeño tamaño de la muestra dentro de los intervalos.
Antes del estudio y antes de la aleatorización, se recogieron muestras de forma sistemática de tejido tumoral con el fin de realizar análisis de eficacia planificados con anterioridad según la expresión de PD-L1 en el tumor. La expresión de PD-L1 en el tumor se midió usando el test PD-L1 IHC 28-8 pharmDx.
La mediana de edad fue 62 años (rango: 21 a 85) con un 34% >65 años y un 7% >75 años. La mayoría de los pacientes fueron de raza blanca (92%) y varones (55%). El nivel basal del estado funcional ECOG fue 0 (31%) o 1 (69%). Setenta y nueve por ciento de los pacientes eran exfumadores/fumadores.
Las curvas de Kaplan-Meier para la SG se muestran en la Figura 6.
Figura 6: Curvas de Kaplan-Meier de SG (CA209057)
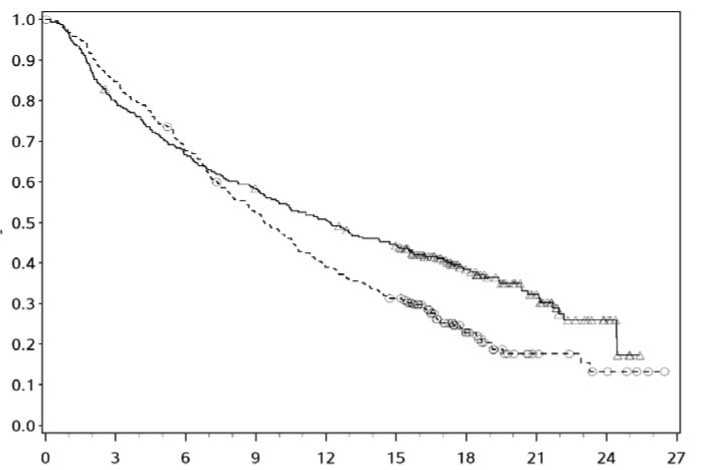
.2
’o
tí
<D
£
<D
tí
O
Ü
"O
Supervivencia global (meses) Número de Sujetos en Riesgo Nivolumab 3 mg/kg
|
292 Docetaxel |
232 |
194 |
169 |
146 |
123 |
62 |
32 |
9 |
0 |
|
290 |
244 |
194 |
150 |
111 |
88 |
34 |
10 |
5 |
0 |
-A-Nivolumab 3 mg/kg (eventos: 190/292); mediana e IC 95%: 12,19 (9,66; 14,98)
—O— Docetaxel (eventos: 223/290); mediana e IC 95%: 9,36 (8,05; 10,68)
31
El ensayo demostró aumento estadísticamente significativo en la SG en los pacientes aleatorizados en el brazo de nivolumab comparado con el de docetaxel en el análisis intermedio preespecificado cuando se observaron 413 eventos (93% de los eventos planificados para el análisis final).
Los resultados de eficacia se muestan en la Tabla 7.
Tabla 7: Resultados de eficacia (CA209057)
|
nivolumab (n = 292) |
docetaxel (n = 290) | ||
|
Análisis intermedio preespecificado Supervivencia global | |||
|
Eventos (%) |
190 (65,1%) |
223 (76,9%) | |
|
Hazard ratioa (IC 95,92%) valor-pb Mediana (IC 95%) meses |
12,19 (9,66; 14,98) |
0,73 (0,59; 0,89) 0,0015 |
9,36 (8,05; 10,68) |
|
Tasa (IC 95%) a los 12 meses |
50,5 (44,6; 56,1) |
39,0 (33,3; 44,6) | |
|
Respuesta objetiva confirmada |
56 (19,2%) |
36 (12,4%) | |
|
(IC 95%) |
(14,8; 24,2) |
(8,8; 16,8) | |
|
Odds ratio (IC 95%) |
1,68 (1,07; 2,64) | ||
|
valor-p Respuesta completa (RC) |
4 (1,4%) |
0,0246 |
1 (0,3%) |
|
Respuesta parcial (RP) |
52 (17,8%) |
35 (12,1%) | |
|
Enfermedad estable (EE) |
74 (25,3%) |
122 (42,1%) | |
|
Mediana de la duración de la respuesta | |||
|
Meses (rango) |
17,15 (1,8; 22,6+) |
5,55 (1,2+; 15,2+) | |
|
Mediana del tiempo de la respuesta | |||
|
Meses (rango) |
2,10 (1,2; 8,6) |
2,61 (1,4; 6,3) | |
|
Supervivencia libre de progresión | |||
|
Eventos |
234 (80,1%) |
245 (84,5%) | |
|
Hazard ratio IC 95% valor-p Mediana (IC 95%) (meses) |
2,33 (2,17; 3,32) |
0,92 (0,77; 1,11) 0,3932 |
4,21 (3,45; 4,86) |
|
Tasa (IC 95%) a los 12 meses |
18,5 (14,1; 23,4) |
8,1 (5,1; 12,0) | |
Derivado de un modelo de riesgo proporcional estratificado.
Valor-p se obtiene a partir de una prueba de log-rank estratificada por tratamiento de mantenimiento previo y la línea del tratamiento; el correspondiente nivel O'Brien-Fleming de significación que limita la eficacia es 0,0408.
Indica una observación censurada.
a
b
Se midió la expresión cuantificable de PD-L1 en el tumor en el 79% de los pacientes del grupo de OPDIVO y en el 77% de los pacientes del grupo de docetaxel. Los niveles de la expresión de PD-L1 en el tumor fueron equilibrados entre los dos grupos de tratamiento (nivolumab vs. docetaxel) en cada nivel de expresión de PD-L1 tumoral predefinido, en >1% (53% vs. 55%), >5% (41% vs. 38%) o >10% (37% vs. 35%).
Los pacientes con expresión de PD-L1 en el tumor según todos los niveles de expresión predefinidos en el grupo de OPDIVO demostraron una mayor probabilidad de aumento de la supervivencia en comparación con docetaxel, mientras que la supervivencia fue similar a la de docetaxel en pacientes con bajo o ninguna expresión de PD-L1 en el tumor. Respecto a la TRO, el aumento de la expresión de PD-L1 se asoció con una mayor TRO. En compración con la población general, la mediana de la duración de la respuesta fue superior con nivolumab vs. docetaxel en pacientes sin expresión de PD-L1 (18,3 meses vs. 5,6 meses) y en pacientes con expresión de PD-L1 (16,0 meses vs. 5,6 meses).
La tabla 8 resume los resultados de TRO y SG según la expresión de PD-L1 en el tumor.
|
Expresión de PD-L1 |
nivolumab |
docetaxel | |
|
TRO según la expresión de PD-L1 en el tumor | |||
|
Odds Ratio (IC 95%) | |||
|
<1% |
10/108 (9,3%) IC 95%: 4,5; 16,4 |
15/101 (14,9%) IC 95%: 8,6; 23,3 |
0,59 (0,22; 1,48) |
|
>1% |
38/123 (30,9%) IC 95%: 22,9; 39,9 |
15/123 (12,2%) IC 95%: 7,0; 19,3 |
3,22 (1,60; 6,71) |
|
>1% a <10%a |
6/37 (16,2%) IC 95%: 6,2; 32,0 |
5/ 44 (11,4%) IC 95%: 3,8; 24,6 |
1,51 (0,35; 6,85) |
|
>10% a <50%a |
5/20 (25,0%) IC 95%: 8,7; 49,1 |
7/33 (21,2%) IC 95%: 9,0; 38,9 |
1,24 (0,26; 5,48) |
|
>50%a |
27/66 (40,9%) IC 95%: 29,0; 53,7 |
3/46 (6,5%) IC 95%: 1,4; 17,9 |
9,92 (2,68; 54,09) |
|
SG según la expresión de PD-L1 en el tumor Número de eventos (número de pacientes) |
Hazard Ratio no estratificado (IC 95%) | ||
|
<1% |
77 (108) |
75 (101) |
0,90 (0,66; 1,24) |
|
>1% |
68 (123) |
93 (123) |
0,59 (0,43; 0,82) |
|
>1% a <10%a |
27 (37) |
30 (44) |
1,33 (0,79; 2,24) |
|
>10% a <50%a |
11 (20) |
26 (33) |
0,61 (0,30; 1,23) |
|
>50%a |
30 (66) |
37 (46) |
0,32 (0,20; 0,53) |
a Los resultados del análisis post-hoc deben ser interpretados con precaución debido al pequeño tamaño de la muestra de los subgrupos y a que en el momento del análisis, el test PD-L1 IHC 28-8 pharmDx no estaba validado analíticamente para los niveles de expresión del 10% o 50%
Una mayor proporción de pacientes fallecieron dentro de los 3 primeros meses en el brazo de nivolumab (59/292; 20,2%) en comparación con el grupo de docetaxel (44/290; 15,2%). Los resultados de un análisis multivariante indicaron que los pacientes tratados con nivolumab con características de peor pronóstico y/o enfermedad agresiva cuando se combinó con baja (p. ej. < 50%) o ninguna expresión de PD-L1 en el tumor pueden estar en mayor riesgo de muerte dentro de los primeros 3 meses de tratamiento.
En los análisis de subgrupos, no se muestra beneficio en la supervivencia en comparación con docetaxel para los pacientes que nunca habían sido fumadores o cuyos tumores aportaban mutaciones activadoras del EGFR; sin embargo, debido al pequeño número de pacientes, no se pueden extraer conclusiones definitivas de estos datos.
Carcinoma de Células Renales
La seguridad y eficacia de nivolumab 3 mg/kg para el tratamiento del CCR avanzado con componente de células claras se evaluaron en un ensayo de Fase 3, aleatorizado, y abierto (CA209025). El ensayo incluyó pacientes (con 18 años o mayores) que habían experimentado una progresión de la enfermedad durante o después de 1 ó 2 regímenes de tratamiento antiangiogénico y no más de 3 tratamientos sistémicos previos. Los pacientes tenían que tener una puntuación de >70% del Karnofsky Performance Score (KPS). Este ensayo incluyó pacientes independientemente de la expresion de PD-L1 en el tumor. Pacientes con antecedentes o presencia de metástasis cerebrales, tratados previamente con un inhibidor de la diana de rapamicina en células de mamífero (mTOR, por sus siglas en inglés, mammalian Target of Rapamycin), cualquier enfermedad autoinmune activa, o condición médica que requiriera tratamiento sistémico de inmunosupresión fueron excluidos del estudio.
Un total de 821 pacientes fueron aleatorizados para recibir nivolumab (n=410) administrado por vía intravenosa durante 60 minutos con una concentración de 3 mg/kg cada 2 semanas o everolimus (n=411) 10 mg al día, administrado por vía oral. El tratamiento continuó mientras se observó beneficio clínico o hasta que el tratamiento no fuera tolerado. Las primeras evaluaciones del tumor se realizaron 8 semanas después de la aleatorización y se siguieron efectuando cada 8 semanas durantel el primer año, y después cada 12 semanas hasta alcanzar progresión o suspensión del tratamiento, lo que ocurriera más tarde. Las evaluaciones del tumor se continuaron después de la suspensión de tratamiento en pacientes que lo habían discontinuado por razones que no fueran progresión. El tratamiento más allá de la progresión, establecida por el investigador según criterios RECIST, versión 1.1, se permitió si el paciente presentaba beneficio clínico y toleraba el medicamento del ensayo según lo determinado por el investigador. La variable principal de eficacia fue la SG y las variables secundarias evaluadas por el investigador fueron TRO y SLP.
Las características basales fueron generalmente similares para ambos grupos. La mediana de edad fue 62 años (rango: 18-88) con 40% >65 años y 9% >75 años. La mayoría de los pacientes eran varones (75%) y de raza blanca (88%), estaban representados todos los grupos de riesgo del Memorial Sloan Kettering Cáncer Center (MSKCC), y el 34% y 66% de los pacientes tenían puntuación del estado funcional basal de Karnofsky (KPS) de 70 a 80% y de 90 a 100%, respectivamente. La mayoría de los pacientes (72%) fueron tratados después de un régímen de tratamiento antiangiogénico. La mediana de duración de tiempo desde el diagnóstico inicial hasta la aleatorización fue de 2,6 años en ambos grupos, el de nivolumab y el de everolimus. La mediana de duración de tratamiento fue de 5.5 meses (rango: 0-29,6+ meses) en pacientes tratados con nivolumab y fue de 3,7 meses (rango: 6 días-25,7+ meses) en pacientes tratados con everolimus.
El tratamiento con Nivolumab se continuó después de la progresión en un 44% de los pacientes.
Las curvas Kaplan-Meier para la SG se muestran en la Figura 7.
Número de Sujetos en Riesgo
|
Nivolumab 410 389 |
359 |
337 |
305 |
275 |
213 |
139 |
73 |
29 |
3 |
0 |
|
Everolimus 411 366 |
324 |
287 |
265 |
241 |
187 |
115 |
61 |
20 |
2 |
0 |
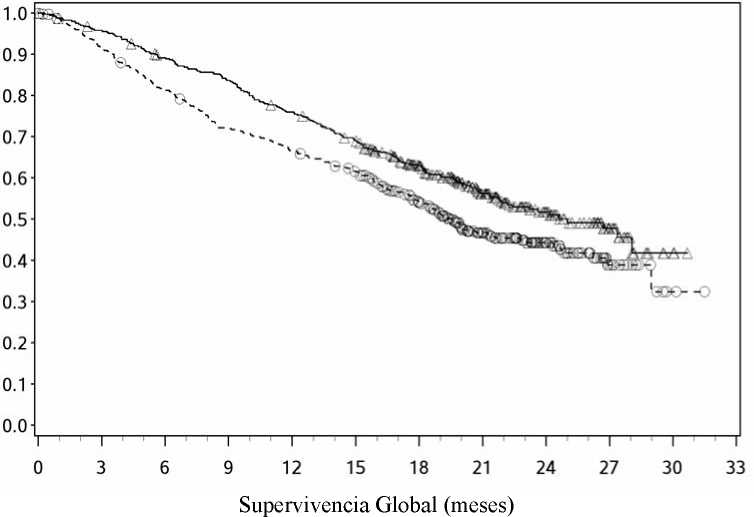
Nivolumab 3 mg/kg (eventos: 183/410), mediana e IC 95%: 25,00 (21,75; N.A.)
■ Everolimus 10 mg (eventos: 215/411), mediana e IC 95%: 19,55 (17,64; 23,06)
El ensayo demostró mejora estadísticamente significativa en la SG en los pacientes aleatorizados en el brazo de nivolumab comparado con el de everolimus en el análisis intermedio preespecificado cuando se observaron 398 eventos (70% de los eventos planificados para el análisis final). (Tabla 9 y Figura 7). Se observó beneficio en la SG independientemente del nivel de expresión de PD-L1 en el tumor.
Los resultados de eficacia se muestan en la Tabla 9.
|
nivolumab (n = 410) |
everolimus (n = 411) | ||
|
Supervivencia global | |||
|
Eventos Hazard ratio IC 98,52% valor-p |
183 (45) |
0,73 (0,57; 0,93) 0,0018 |
215 (52) |
|
Mediana (IC 95%) |
25,0 (21,7; NE) |
19,6 (17,6; 23,1) | |
|
Tasa (IC 95%) | |||
|
A los 6 meses |
89,2 (85,7, 91,8) |
81,2 (77,0; 84,7) | |
|
A los 12 meses |
76,0 (71,5; 79,9) |
66,7 (61,8; 71,0) | |
|
Respuesta objetiva |
103 (25,1%) |
22 (5,4%) | |
|
(IC 95%) Odds ratio (IC 95%) valor-p |
(21,0; 29,6) |
5,98 (3,68; 9,72) < 0,0001 |
(3,4; 8,0) |
|
Respuesta completa (RC) |
4 (1,0%) |
2 (0,5%) | |
|
Respuesta parcial (RP) |
99 (24,1%) |
20 (4,9%) | |
|
Enfermedad estable (EE) |
141 (34,4%) |
227 (55,2%) | |
|
Mediana de la duración de la respuesta | |||
|
Meses (rango) |
11,99 (0,0-27,6+) |
11,99 (0,0+-22,2+) | |
|
Mediana del tiempo hasta respuesta | |||
|
Meses (rango) |
3,5 (1,4-24,8) |
3,7 (1,5-11,2) | |
|
Supervivencia libre de progresión | |||
|
Eventos Hazard ratio IC 95% valor-p |
318 (77,6) |
0,88 (0,75; 1,03) 0,1135 |
322 (78,3) |
|
Mediana (IC 95%) |
4,6 (3,71; 5,39) |
4,4 (3,71; 5,52) | |
“+” indica una observación censurada.
La mediana de tiempo hasta la aparición de respuesta objetiva fUe de 3,5 meses (rango: 1,4-24,8 meses) tras empezar un tratamiento de nivolumab. Cuarenta y nueve (47,6%) de los pacientes que respondieron se mantenían en respuesta en el momento del análisis del estudio con un rango de duración de entre 0,0 y 27,6+ meses.
La supervivencia global podría estar acompañada de una mejora en el tiempo de los síntomas relacionados con la enfermedad y la calidad de vida no específica de la enfermedad tal y como se evaluó usando las escalas validadas de la Functional Assessment of Cáncer Therapy-Kidney Symptom Index-Disease Related Symptoms (FKSI-DRS) y el EuroQoL EQ-5D. Síntoma aparentemente significativo de la mejoría (MID=2 puntos de cambio en la puntuación FKSI-DRS; p<0,001) y el tiempo para la mejora (HR= 1,66 (1,33; 2,08), p<0,001) fueron significativamente mejores para pacienes en el brazo de nivolumab. Aunque ambos brazos del estudio recibieron terapia activa, los datos de calidad de vida se deben interpretar en el contexto de un estudio abierto y por tanto se deben tomar con precaución.
Seguridad y eficacia en pacientes de edad avanzada
No se han notificado diferencias generales en la seguridad o eficacia entre pacientes de edad avanzada (> 65 años) y pacientes más jóvenes (< 65 años). Los datos de CPNM de los pacientes de 75 años o mayores son demasiado limitados para sacar conclusiones en esta población.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con nivolumab en los diferentes grupos de la población pediátrica en el tratamiento de tumores sólidos malignos (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
La farmacocinética (FC) de nivolumab es lineal en el intervalo de dosis de 0,1 a 10 mg/kg. Las medias geométricas del aclaramiento (CL), la semivida terminal de nivolumab y la exposición media en el estado estable a 3mg/kg cada 2 semanas fueron 9,5 ml/h y 26,7 días, y 75,3 pg/ml respectivamente, de acuerdo con un análisis FC poblacional.
El CL de nivolumab se vio incrementado con el aumento del peso corporal. La dosificación normalizada en función del peso corporal generó una concentración mínima aproximadamente uniforme en el estado estacionario a lo largo de un amplio rango de pesos (34-162 kg).
No se ha caracterizado la ruta metabólica de nivolumab. Nivolumab se espera que se degrade en pequeños péptidos y aminoácidos a través de vías catabólicas de la misma manera que las IgG endógenas.
OPDIVO en combinación con ipilimumab: la media geométrica de CL, Vee, y la semivida terminal de nivolumab fueron 9,83 ml/h, 7,62 l, y 24,1 días, respectivamente. Cuando se administró en combinación, el CL de nivolumab aumentó en un 35%, mientras que no hubo efecto sobre el CL de ipilimumab.
Cuando se administró en combinación, el CL de nivolumab aumentó en un 25% en presencia de anticuerpos anti-nivolumab. No hubo efecto de los anticuerpos anti-ipilimumab sobre el CL de ipilimumab.
Poblaciones especiales
Un análisis FC poblacional sugirió que no había diferencias en el CL de nivolumab en función de la edad, el sexo, la raza, el tipo de tumor, el tamaño del tumor y la insuficiencia hepática. Aunque el estado ECOG, la tasa de filtración glomerular (TFG) basal, la albúmina, el peso corporal y la insuficiencia hepática leve, tuvieron un efecto sobre el CL de nivolumab, el efecto no fue clínicamente significativo.
Insuficiencia renal
El efecto de la insuficiencia renal sobre el CL de nivolumab se evaluó en pacientes con insuficiencia renal leve (TFG < 90 y > 60 ml/min/1,73 m2; n = 379), moderada (TFG < 60 y > 30 ml/min/1,73 m2; n = 179) o grave (TFG < 30 y > 15 ml/min/1,73 m2; n = 2) en comparación con los pacientes con una función renal normal (TFG > 90 ml/min/1,73 m2; n = 342) en análisis FC poblacionales. No se encontraron diferencias clínicamente importantes en el CL de nivolumab entre pacientes con insuficiencias renales leves o moderadas y pacientes con una función renal normal. Los datos de pacientes con insuficiencia renal grave son demasiado limitados para sacar conclusiones en esta población (ver sección 4.2).
Insuficiencia hepática
El efecto de la insuficiencia hepática sobre el CL de nivolumab se evaluó en pacientes con insuficiencia renal leve (bilirrubina total de 1,0 * a 1,5 * LSN o AST > LSN según la definición de los criterios del National Cancer Institute para la insuficiencia hepática; n = 92) comparado con los pacientes con una función hepática normal (bilirrubina total y AST < LSN; n = 804) en los análisis FC poblacionales. No se encontraron diferencias clínicamente relevantes en el CL de nivolumab entre
37
pacientes con insuficiencia hepática leve y con una función hepática normal. Nivolumab no se ha estudiado en pacientes con insuficiencia hepática moderada (bilirrubina total > de 1,5 * a 3 * LSN y cualquier valor de AST) o grave (bilirrubina total > 3 * LSN y cualquier valor de AST) (ver sección 4.2).
5.3 Datos preclínicos sobre seguridad
El bloqueo de la señalización PD-L1 se ha demostrado en modelos murinos de embarazos que interrumpe la tolerancia al feto e incrementa la pérdida del feto. Los efectos de nivolumab sobre el desarrollo prenatal y postnatal se evaluaron en monas que recibieron nivolumab dos veces a la semana desde el inicio de la organogénesis en el primer trimestre hasta el parto, a niveles de exposición 8 ó 35 veces superiores a los observados a la dosis clínica de 3 mg/kg de nivolumab (de acuerdo al AUC). Hubo un aumento dosis-dependiente en pérdidas de fetos y un incremento de la mortalidad neonatal, al inicio del tercer trimestre.
La progenie restante de las hembras tratadas con nivolumab sobrevivió a la terminación programada, sin signos clínicos, alteraciones respecto al desarrollo normal, efectos en el peso de los órganos o cambios patológicos macroscópicos o microscópicos asociados al tratamiento. Los resultados de los índices de crecimiento, así como de los parámetros teratogénicos, neuroconductuales, inmunológicos y de patología clínica a lo largo de un periodo postnatal de 6 meses fueron comparables a los del grupo control. Sin embargo, de acuerdo a este mecanismo de acción, la exposición del feto a nivolumab puede aumentar el riesgo de desarrollar alteraciones inmunorrelacionadas o alteraciones de la respuesta inmunitaria normal y se han notificado alteraciones inmunorrelacionadas en ratones machos PD-1.
No se han realizado estudios de fertilidad con nivolumab.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Citrato sódico di hidratado Cloruro sódico Manitol (E421)
Ácido pentético (ácido dietilentriaminopentaacético)
Polisorbato 80
Hidróxido sódico (para el ajuste del pH)
Ácido clorhídrico (para el ajuste del pH)
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no se debe mezclar con otros. OPDIVO no se debe perfundir simultáneamente a través de la misma vía intravenosa con otros medicamentos.
6.3 Periodo de validez
Vial sin abrir 2 años.
Una vez abierto
Desde un punto de vista microbiológico, una vez abierto, el medicamento se debe perfundir o diluir y perfundir inmediatamente.
Tras la preparación de la perfusión
Desde un punto de vista microbiológico, el medicamento se debe utilizar inmediatamente.
Si no se utiliza inmediatamente, se ha demostrado la estabilidad física y química de OPDIVO durante 24 horas entre 2°C y 8°C, protegido de la luz y un máximo de 4 horas a 20°C-25°C y expuesta a la luz
38
ambiental; (este periodo máximo de 4 horas del total de 24 horas se debe incluir en el periodo de administración del producto).
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el envase original para protegerlo de la luz.
Para las condiciones de conservación tras la preparación de la perfusión, ver sección 6.3.
6.5 Naturaleza y contenido del envase
4 ml de concentrado estéril en un vial de 10 ml (vidrio de Tipo I) con un tapón (caucho de butilo revestido) y un sello de tipo flip-off de color azul oscuro (aluminio). Envase con 1 vial.
10 ml de concentrado estéril en un vial de 10 ml (vidrio de Tipo I) con un tapón (caucho de butilo revestido) y un sello de tipo flip-off de color gris (aluminio). Envase con 1 vial.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
La preparación la debe realizar personal formado de acuerdo con la normas de buenas prácticas, especialmente en lo que respecta a la asepsia.
Preparación y administración
Cálculo de la dosis
La dosis prescrita para el paciente se administra en mg/kg. De acuerdo con esta dosis prescrita, calcular la dosis total a administrar. Puede ser necesario más de un vial de OPDIVO concentrado para suministrar la dosis total del paciente.
■ La dosis total de nivolumab en mg = el peso del paciente en kg * la dosis prescrita en mg/kg.
■ El volumen de OPDIVO concentrado para preparar la dosis (ml) = la dosis total en mg, dividida entre 10 (la concentración del concentrado de OPDIVO es 10 mg/ml).
Preparación de la perfusión
Hay que garantizar una manipulación aséptica al preparar la perfusión. La perfusión se debe preparar en una campana de flujo laminar o una cabina de seguridad empleando las precauciones habituales para la manipulación segura de los agentes intravenosos.
OPDIVO se puede utilizar para administración intravenosa:
■ sin dilución, después de transferir a un recipiente para perfusión utilizando una jeringa estéril adecuada; o
■ tras su dilución a concentraciones tan bajas como 1 mg/ml. La concentración final de la perfusión debe oscilar entre 1 y 10 mg/ml. El concentrado de OPDIVO se puede diluir con:
■ solución de cloruro sódico para preparaciones inyectables a una concentración de 9 mg/ml (0,9%); o
■ solución de glucosa para preparaciones inyectables a una concentración de 50 mg/ml (5%). PASO 1
■ Inspeccionar el concentrado de OPDIVO para la detección de partículas o cambios de color. No agitar el vial. El concentrado de OPDIVO es un líquido de transparente a opalescente, de incoloro a amarillo pálido. Deseche el vial si la solución está turbia, descolorida o si contiene otras partículas que no sean unas pocas de translúcidas a blancas.
■ Extraer el volumen necesario de concentrado de OPDIVO empleando una jeringa estéril adecuada.
■ Transferir el concentrado a un frasco de vidrio estéril evacuado o envase para solución IV (PVC o poliolefina).
■ Si procede, diluir con el volumen preciso de solución de cloruro sódico para preparaciones inyectables a una concentración de 9 mg/ml (0,9%) o una solución de glucosa para preparaciones inyectables a una concentración de 50 mg/ml (5%). Mezclar suavemente la perfusión por rotación manual. No agitar.
Administración
La perfusión de OPDIVO no se debe administrar en forma de inyección en bolo intravenoso. Administrar la perfusión de OPDIVO por vía intravenosa durante un periodo de tiempo de 60 minutos. La perfusión de OPDIVO no se debe perfundir simultáneamente a través de la misma vía intravenosa con otros medicamentos. Usar una vía de perfusión aparte para la perfusión.
Usar un equipo de perfusión y un filtro en línea estéril, no pirógeno y de baja unión a proteínas (tamaño de poro de 0,2 pm a 1,2 pm).
La perfusión de OPDIVO es compatible con contenedores de PVC y poliolefina, frascos de vidrio, equipos de perfusión de PVC y filtro en línea con membranas de poliétersulfona con tamaños de poro de 0,2 pm a 1,2 pm.
Tras la administración de la dosis, de nivolumab purgar la vía con una solución de cloruro sódico para preparaciones inyectables a una concentración de 9 mg/ml (0,9%) o una solución de glucosa para preparaciones inyectables a una concentración de 50 mg/ml (5%).
Eliminación
No almacenar la solución para perfusión no utilizada para su reutilización. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bristol-Myers Squibb Pharma EEIG Uxbridge Business Park Sanderson Road Uxbridge UB8 1DH Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/15/1014/001-002
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 19/junio/2015
10. FECHA DE LA REVISIÓN DEL TEXTO
DD/MM/YYYY
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema. europa.eu
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de los fabricantes del principio activo biológico.
Bristol-Myers Squibb Company 6000 Thompson Road East Syracuse, New York 13057 Estados Unidos
Lonza Biologics, Inc.
101 International Drive Portsmouth, New Hampshire 03801 Estados Unidos
Samsung Biologics Co. Ltd.
125, Cheomdan-daero Yeonsu-gu, Incheon, 21987 Corea
Nombre y dirección del fabricante responsable de la liberación de los lotes
Bristol-Myers Squibb S.r.l.
Loc. Fontana del Ceraso 03012 Anagni (FR)
Italia
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad actualizados (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y las siguientes actualizaciones publicadas en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Medidas adicionales de minimización de riesgos
Antes del lanzamiento de OPDIVO en cada Estdo Miembro el Titular de la Autorización de Comercialización (TAC) debe acordar con la Autoridad Nacional Competente el contenido y formato de los materiales informativos sobre seguridad, incluyendo medios de comunicación, modalidades de distribución y cualquier otro aspecto de estas medidas.
Los materiales informativos sobre seguridad, tienen como objetivo incrementar el conocimiento sobre las reacciones adversas potenciales relacionadas con el sistema inmunitario asociados al uso de OPDIVO y proporcionar una guía sobre cómo manejarlas y cómo aumentar el conocimiento de los pacientes o de sus cuidadores sobre los signos y síntomas relevantes de aquellos efectos adversos tempranos.
El TAC debe acordar en cada Estado Miembro donde OPDIVO se comercialice, que todos los profesionales sanitarios y pacientes/cuidadores que se espera prescriban y utilicen OPDIVO tengan acceso a/se les faciliten los siguientes materiales informativos sobre seguridad:
• Información de Seguridad para el Profesional Sanitario
• Tarjeta de Información para el Paciente
La información de seguridad para el profesional sanitario contendrá:
• El Resumen de Características del Producto
• La Guía para el Manejo de las Reacciones Adversas
La Guía para el manejo de las reacciones adversas contendrá los siguientes elementos clave:
• Información relevante (por ejemplo: severidad, gravedad, frecuencia, tiempo de aparición, reversibilidad de los acontecimientos adversos según proceda) para las siguientes cuestiones de seguridad:
o Neumonitis inmunorrelacionada o Colitis inmunorrelacionada o Hepatitis inmunorrelacionada o Nefritis e insuficiencia renal inmunorrelacionadas o Endocrinopatías inmunorrelacionadas o Erupción cutánea inmunorrelacionada o Otras reacciones adversas inmunorrelacionadas
• Detalles de cómo minimizar las cuestiones de seguridad a través de la monitorización y el manejo apropiados
• La tarjeta de información para el paciente contendrá los siguientes mensajes clave:
o Neumonitis inmunorrelacionada o Colitis inmunorrelacionada o Hepatitis inmunorrelacionada o Nefritis e insuficiencia renal inmunorrelacionadas o Endocrinopatías inmunorrelacionadas o Erupción cutánea inmunorrelacionada o Otras reacciones adversas inmunorrelacionadas
• Signos y síntomas de las cuestiones de seguridad y cómo buscar la atención de un profesional sanitario
• Detalles para contactar con el prescriptor de OPDIVO
• Obligación de llevar a cabo medidas posautorización
El TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
1. Estudio de eficacia posautorización (EPA): el TAC deberá presentar el informe final del ensayo CA209037: ensayo de fase 3, aleatorizado, abierto, de nivolumab frente al tratamiento elegido por el investigador en pacientes con melanoma avanzado (irresecable o metastásico) en progresión después de tratamiento con anti-CTLA-4. |
El informe final del ensayo deberá presentarse antes del 31 de octubre de 2016 |
|
2. Estudio de eficacia posautorización (EPA): el TAC debe presentar el informe final del Estudio para el ensayo CA209067: ensayo aleatorizado y doble ciego en sujetos tratados con nivolumab en monoterapia, ipilimumab en monoterapia y nivolumab combinado con ipilimumab. |
El informe final del ensayo clínico se debe presentar antes del 31 de marzo de 2017 |
|
3. El valor de los biomarcadores para predecir la eficacia de nivolumab y/o la combinación de nivolumab + ipilimumab debe ser investigado especificamente: 1. Continuar investigando el corte óptimo para la positividad de PD-L1 de acuerdo al método de ensayo vigente utilizado para posteriores dilucidaciones sobre su valor como predictivo de la eficacia de nivolumab. Estos análisis se realizarán en toda la población del ensayo CA209037 en pacientes con melanoma avanzado. Estos análisis se pueden presentar dentro del informe final del Estudio para el ensayo CA209037 como adenda. |
31 de octubre de 2016 |
|
2. Continuar investigando el valor de otros biomarcadores distintos de la expresión de PD-L1 en la membrana celular del tumor por IHQ (p. ej., otros métodos/ensayos basados en la genómica, y cut-offs asociados, que podrían demostrar mayor sensibilidad y especificidad en la predicción de la respuesta al tratamiento según PD-L1, PD-L2, linfocitos infiltrantes en el tumor con medidas de CD8+T densidad, firma ARN, expresión de componentes de los complejos de presentación de antígenos y/u otros receptores/ligandos inhibidores de puntos de control dentro del tumor etc.) como predictivo de la eficacia de nivolumab y/o de la combinación de nivolumab + ipilimumab. Esto será proporcionado para todas las indicaciones aprobadas: - Melanoma en monoterapia: ensayos CA209-038 y CA209-066 - Melanoma en combinación (con ipilimumab): ensayos CA209-038, CA209-067 y CA209-069 - CPNM: ensayos CA209-017, CA209-057, CA209-026 - CCR: ensayos CA209-025 y CA209-009 Además, los niveles de células supresoras de origen mieloide en circulación se estudiarán en el ensayo CA209038. |
30 septiembre 2017 31 marzo 2019 31 marzo 2018 31 marzo 2018 |
|
3. Continuar investigando la relación entre la expresión de PDL-1 y PDL-2 en los estudios de Fase 1 (CA209009, CA209038 y CA209064). |
31 marzo 2017 |
|
4. Continuar investigando el análisis asociativo entre la expresión de PDL-1 y PDL-2 realizado en los ensayos CA209-066, CA209-057 y CA209-025. |
30 junio 2018 |
|
5. Continuar investigando el cambio posible en la expresión de PD-L1 en el tumor durante el tratamiento y/o progresión del tumor en los ensayos CA209-009, CA209-038 y CA209-064. |
30 septiembre 2017 |
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CARTONAJE EXTERIOR
1. NOMBRE DEL MEDICAMENTO
OPDIVO 10 mg/ml concentrado para solución para perfusión nivolumab
2. PRINCIPIO(S) ACTIVO(S)
Cada ml de concentrado contiene 10 mg de nivolumab. Cada vial de 4 ml contiene 40 mg de nivolumab.
Cada vial de 10 ml contiene 100 mg de nivolumab.
3. LISTA DE EXCIPIENTES_
Excipientes: citrato sódico di hidratado, cloruro sódico, manitol (E421), ácido pentético, polisorbato 80, hidróxido sódico, ácido clorhídrico, agua para preparaciones inyectables.
Para mayor información, consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE_
Concentrado para solución para perfusión.
40 mg/4 ml 100 mg/10 ml
1 vial
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía intravenosa.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Para un solo uso.
8. FECHA DE CADUCIDAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera.
No congelar.
Conservar en el envase original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bristol-Myers Squibb Pharma EEIG Uxbridge Business Park Sanderson Road Uxbridge UB8 1DH Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/15/1014/001 vial de 40 mg EU/1/15/1014/002 vial de 100 mg
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
INFORMACIÓN MÍNIMA A INCLUIR EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETA DEL VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
OPDIVO 10 mg/ml concentrado estéril
nivolumab
Vía IV
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento.
3. FECHA DE CADUCIDAD
EXP
4. NUMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
40 mg/4 ml 100 mg/10 ml
6. OTROS
Para un solo uso.
B. PROSPECTO
OPDIVO 10 mg/ml concentrado para solución para perfusión
nivolumab
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Es importante que tenga siempre con usted, la "Tarjeta de Información para el Paciente" durante el tratamiento.
- Si tiene alguna duda, consulte a su médico.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es OPDIVO y para qué se utiliza
2. Qué necesita saber antes de empezar a usar OPDIVO
3. Cómo usar OPDIVO
4. Posibles efectos adversos
5. Conservación de OPDIVO
6. Contenido del envase e información adicional
1. Qué es OPDIVO y para qué se utiliza
OPDIVO es un medicamento que se utiliza para tratar:
■ melanoma avanzado (un tipo de cáncer de piel) en adultos
■ cáncer de pulmón no microcítico (CPNM) avanzado (un tipo de cáncer de pulmón) en adultos
■ carcinoma avanzado de células renales (cáncer de riñón avanzado) en adultos.
Contiene nivolumab como principio activo, que es un anticuerpo monoclonal, un tipo de proteína diseñada para reconocer y atacar una substancia diana específica que se encuentra en el cuerpo.
Nivolumab ataca y bloquea una proteína diana llamada receptor de muerte programada-1 (PD-1), que puede inhibir la actividad de las células T (un tipo de glóbulos blancos de la sangre que forman parte del sistema inmunitario, la defensa natural del organismo). Atacando a PD-1, nivolumab bloquea su acción y previene de la inhibición de sus células T. Esto ayuda a aumentar su actividad frente a las células del melanoma, del cáncer de pulmón o del cáncer de riñón.
OPDIVO se puede administrar en combinación con ipilimumab. Es importante que también lea el prospecto de este medicamento. Si tiene alguna duda sobre ipilimumab, por favor consulte a su médico.
2. Qué necesita saber antes de empezar a usar OPDIVO No use OPDIVO:
■ si es alérgico a nivolumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6 "Contenido del envase e información adicional"). En caso de duda consulte a su médico.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar OPDIVO, ya que puede producir:
■ Problemas en sus pulmones como dificultad para respirar o tos. Estos pueden ser signos de inflamación de los pulmones (neumonitis o enfermedad pulmonar intersticial).
■ Diarrea (heces acuosas, sueltas o blandas) o cualquier síntoma de inflamación de los intestinos (colitis), tales como dolor de estómago o presencia de moco o sangre en las heces.
■ Inflamación del hígado (hepatitis). Los signos y síntomas de hepatitis pueden incluir pruebas de función hepática anormales, amarilleamiento de los ojos o la piel (ictericia), dolor en la parte derecha del área del estómago o cansancio.
■ Problemas con sus riñones. Los signos y síntomas pueden incluir pruebas de función renal anormales, disminución del volumen de orina.
■ Problemas con sus glándulas productoras de hormonas (incluyendo la glándula pituitaria, glándulas tiroideas y suprarrenales) que pueden afectar a cómo funcionan estas glándulas. Los signos y síntomas de que estas glándulas no están funcionando adecuadamente, pueden incluir fatiga (cansancio extremo), cambios de peso, o dolor de cabeza y alteraciones visuales.
■ Diabetes (los síntomas incluyen sed excesiva, el paso de una gran cantidad incrementada de orina, aumento del apetito con pérdida de peso, sentirse cansado, somnolencia, debilidad, depresión, irritabilidad y en general no sentirse bien) o cetoacidosis diabética (ácido en la sangre producido por la diabetes).
■ Inflamación de la piel que puede conducir a erupción cutánea y picor.
Informe a su médico inmediatamente si presenta alguno de estos signos o síntomas, o si éstos empeoraran. No intente tratar sus síntomas con otros medicamentos por su cuenta. Su médico puede
■ administrarle otros medicamentos para evitar complicaciones y reducir sus síntomas,
■ suspender la siguiente dosis de OPDIVO,
■ interrumpir completamente su tratamiento con OPDIVO.
Tenga en cuenta que estos signos y síntomas a veces se producen de forma tardía, y podrían aparecer semanas o meses después de recibir su última dosis. Antes del tratamiento, su médico comprobará su estado de salud general. También se le realizarán análisis de sangre durante su tratamiento.
Compruebe con su médico antes de recibir OPDIVO si:
■ tiene una enfermedad autoinmune (un trastorno en el que el organismo ataca a sus propias células);
■ tiene melanoma ocular;
■ le han administrado previamente ipilimumab, otro medicamento para tratar el melanoma, y ha experimentado efectos adversos graves a consecuencia de ese medicamento;
■ le han dicho que su cáncer se ha extendido a su cerebro;
■ tiene antecedentes de inflamación de los pulmones;
■ ha estado tomando medicamentos que deprimen su sistema inmunitario.
Niños y adolescentes
OPDIVO no se debe usar en niños y adolescentes menores de 18 años.
Uso de OPDIVO con otros medicamentos
Antes de recibir OPDIVO informe a su médico si está tomando algún medicamento que suprima su sistema inmunitario, como corticosteroides, puesto que estos medicamentos pueden interferir con el efecto de OPDIVO. Sin embargo, una vez en tratamiento con OPDIVO, su médico, puede administrarle corticosteroides para reducir cualquier efecto adverso posible que pudiese tener durante su tratamiento y esto no impactará en el efecto del medicamento.
Informe a su médico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento. No tome ningún otro medicamento durante su tratamiento sin consultarlo primero con su médico.
Embarazo y lactancia
Si está embarazada o en período de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico.
No use OPDIVO si está embarazada, a menos que su médico se lo diga específicamente. Se desconocen los efectos de OPDIVO en mujeres embarazadas, pero es posible que el principio activo, nivolumab, pueda ocasionar daños fetales.
■ Debe utilizar un método anticonceptivo eficaz mientras recibe tratamiento con OPDIVO, y como mínimo hasta los 5 meses siguientes a la última dosis de OPDIVO, en caso de que sea una mujer que se podría quedar embarazada.
■ Si se queda embarazada mientras usa OPDIVO informe a su médico.
Se desconoce si nivolumab pasa a la leche materna. No se puede excluir el riesgo para el lactante. Pregunte a su médico si puede dar el pecho durante o después del tratamiento con OPDIVO.
Conducción y uso de máquinas
No se han llevado a cabo estudios acerca de la capacidad para conducir y utilizar máquinas. Es poco probable que nivolumab afecte a su capacidad para conducir y utilizar máquinas; no obstante, tenga precaución al realizar estas actividades, hasta que esté seguro de que nivolumab no le afecta de forma negativa.
OPDIVO contiene sodio
Informe a su médico si está siguiendo una dieta baja en sodio (baja en sal) antes de recibir OPDIVO. Este medicamento contiene 2,5 mg de sodio por ml de concentrado.
Usted también encontrará esta información en la "Tarjeta de Información para el Paciente", que le dará su médico. Es importante que tenga siempre con usted esta Tarjeta de Información y se la muestre a su pareja o cuidadores.
3. Cómo usar OPDIVO Cuánto OPDIVO se administra
La cantidad de OPDIVO que vaya a recibir se calculará en función de su peso corporal. La dosis recomendada es 3 mg de nivolumab por kilogramo de peso.
Dependiendo de su dosis, la cantidad adecuada de OPDIVO podría diluirse antes de su uso con una solución de cloruro sódico para preparaciones inyectables a una concentración de 9 mg/ml (0,9%) o una solución de glucosa para preparaciones inyectables a una concentración de 50 mg/ml (5%). Es posible que sea necesario más de un vial de OPDIVO para obtener la dosis necesaria.
Cuando se administre OPDIVO en combinación con ipilimumab, la dosis recomendada de OPDIVO es de 1 mg de nivolumab por kilogramo de su peso corporal durante las primeras 4 dosis (fase de combinación). Posteriormente, la dosis recomendada de OPDIVO es de 3 mg de nivolumab por kilogramo de su peso corporal (fase como agente único).
Cómo se administra OPDIVO
Usted recibirá tratamiento con OPDIVO en un hospital o una clínica, bajo la supervisión de un médico experimentado.
OPDIVO se le administrará en forma de perfusión (un gotero) en una vena (por vía intravenosa) durante un periodo de tiempo de 60 minutos, cada 2 semanas. Su médico seguirá administrándole OPDIVO mientras se beneficie de él o hasta que ya no tolere el tratamiento.
Cuando se administre OPDIVO en combinación con ipilimumab, se le administrará una perfusión durante un periodo de tiempo de 60 minutos, cada 3 semanas durante las primeras 4 dosis (fase de combinación). Posteriormente, se le administrará en forma de perfusión durante un periodo de tiempo de 60 minutos, cada 2 semanas (fase como agente único).
Si olvidó usar OPDIVO
Es muy importante que acuda a todas sus citas para recibir OPDIVO. Si falta a alguna de ellas, pregunte a su médico cuándo se puede programar su siguiente dosis.
Si interrumpe el tratamiento con OPDIVO
La interrupción de su tratamiento puede detener el efecto del medicamento. No interrumpa el tratamiento con OPDIVO a menos que lo haya comentado con su médico.
Si tiene cualquier otra duda sobre su tratamiento o el uso de este medicamento, pregunte a su médico.
Cuando se administre OPDIVO en combinación con ipilimumab, primero se le administrará OPDIVO seguido de ipilimumab.
Por favor, consulte el prospecto de ipilimumab para entender el uso de este medicamento. Si tiene alguna duda sobre este medicamento, por favor consulte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Su médico los comentará con usted y le explicará los riesgos y beneficios de su tratamiento.
Sea consciente de la importancia de los síntomas de inflamación. OPDIVO actúa sobre su sistema inmunitario y podría causar inflamación en alguna parte de su cuerpo. La inflamación podría causar un daño grave a su cuerpo y algunas condiciones inflamatorias podrían suponer una amenaza para la vida y necesitará tratamiento o la retirada de nivolumab.
Se han notificado los siguientes efectos adversos en los ensayos clínicos con nivolumab solo:
Muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes)
■ Disminución del apetito
■ Diarrea (heces acuosas, sueltas o blandas), náuseas
■ Erupción cutánea, picor
■ Sensación de cansancio o debilidad
Frecuentes (pueden afectar hasta 1 de cada 10 pacientes)
■ Infecciones del tracto respiratorio superior
■ Reacción alérgica, reacciones relacionadas con la perfusión del medicamento
■ Disminución de la función de la glándula tiroides, lo que puede provocar cansancio o ganancia de peso, aumento de la función de la glándula tiroides, que puede producir un aumento de la frecuencia cardíaca, sudoración y pérdida de peso
■ Niveles altos de azúcar en sangre (hiperglucemia)
■ Inflamación de los nervios que causa entumecimiento, debilidad, hormigueo o quemazón de los brazos y las piernas; cefalea, mareo
■ Visión borrosa, ojos secos
■ Elevación de la tensión sanguínea (hipertensión)
■ Inflamación de los pulmones (neumonitis), caracterizada por tos y dificultad para respirar, falta de aliento (disnea), tos
■ Inflamación de los intestinos (colitis), úlceras en la boca y herpes (estomatitis), vómitos, dolor de estómago, estreñimiento, sequedad bucal
■ Cambio del color de la piel en parches (vitíligo), piel seca, enrojecimiento de la piel, pérdida inusual o debilitamiento del cabello
■ Dolor en los músculos, huesos y articulaciones
■ Fiebre, edema (hinchazón)
Poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes)
■ Infección pulmonar grave (neumonía), bronquitis
■ Elevación de algunas células de la serie blanca de la sangre
■ Disminución de la secreción de hormonas producidas por las glándulas suprarrenales (glándulas situadas encima de los riñones), baja actividad de funcionamiento (hipopituitarismo) o inflamación (hipofisitis) de la glándula pituitaria situada en la base del cerebro, hinchazón de la glándula tiroides en el cuello, ácido en la sangre producido por diabetes (cetoacidosis diabética)
54
■ Deshidratación, aumento del nivel de ácido en la sangre
■ Inflamación del hígado (hepatitis)
■ Amarilleamiento de piel y/u ojos (ictericia)
■ Nervios dañados causando entumecimiento y debilidad (polineuropatía)
■ Inflamación de los ojos, que produce dolor y enrojecimiento, problemas visuales o visión borrosa
■ Latido cardíaco rápido
■ Inflamación de los vasos sanguíneos
■ Líquido alrededor de los pulmones
■ Inflamación del páncreas
■ Condición grave de la piel que causa, enrojecimiento, con frecuencia manchas que pican, similares a la erupción del sarampión que comienza en las extremidades y a veces en la cara y en el resto del cuerpo (eritema multiforme), enfermedad cutánea con zonas abultadas de piel roja, con frecuencia con escamas plateadas (psoriasis), condición de la piel de la cara dónde la nariz y las mejillas enrojecidas de forma inusual (rosácea), urticaria (picor, erupción abultada)
■ Inflamación de los músculos que causa dolor o rigidez, dolor en las articulaciones
■ Inflamación del riñón, insuficiencia renal
■ Dolor, dolor en el pecho
Raros (pueden afectar hasta 1 de cada 1.000 pacientes)
■ Una enfermedad que causa inflamación o aumento de los nódulos linfáticos (linfoadenitis de Kikuchi)
■ Diabetes
■ Obstrucción de los conductos biliares
■ Inflamación temporal de los nervios que causa dolor, debilidad y parálisis en las extremidades (síndrome de Guillain-Barré); pérdida de la vaina protectora de los nervios (desmielinización); una condición en la que los músculos se debilitan y se cansan fácilmente (síndrome miasténico), inflamación de los nervios causada por el ataque al cuerpo en sí, causando insensibilidad, debilidad, hormigueo o quemazón
■ Cambios en el ritmo o la frecuencia del latido cardíaco, latido cardíaco rápido, ritmo cardíaco anormal
■ Líquido en los pulmones
■ Gastritis (inflamación del estómago), úlceras en el intestino delgado
■ Descamación de la piel grave y posiblemente mortal (necrólisis epidérmica tóxica)
■ Miopatía (dolor en los músculos, sensibilidad o debilidad muscular, no causadas por el ejercicio)
Si experimenta alguno de los efectos adversos descritos más arriba, informe a su médico
inmediatamente. No intente tratar estos síntomas con otros medicamentos por su cuenta.
Cambios en los resultados de los análisis de laboratorio
OPDIVO puede provocar cambios en los resultados de los análisis de laboratorio efectuados por su
médico, entre otros:
■ Pruebas de función hepática anormales (aumento de la concentración en sangre de las enzimas hepáticas aspartato aminotransferasa, alanina aminotransferasa o fosfatasa alcalina, aumento de los niveles sanguíneos del producto de desecho, bilirrubina)
■ Pruebas de función renal anormales (aumento de la concentración de creatinina en sangre)
■ Disminución del recuento de glóbulos rojos (que transportan oxígeno), glóbulos blancos (importantes para combatir las infecciones) o plaquetas (las células que ayudan a la coagulación sanguínea)
■ Aumento del nivel de la enzima que rompe las grasas y de la enzima que rompe el almidón
■ Aumento o disminución de la cantidad de calcio o potasio
■ Aumento o disminución de los niveles de magnesio o sodio en su sangre
■ Disminución del peso corporal
Se han comunicado los siguientes efectos adversos en los ensayos clínicos con nivolumab en
combinación con ipilimumab:
Muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes)
■ Disminución de la función de la glándula tiroidea, que puede causar cansancio o aumento de peso
■ Disminución del apetito
■ Dolor de cabeza
■ Inflamación de los intestinos (colitis), diarrea (heces acuosas, sueltas o blandas), vómitos, náuseas, dolor de estómago
■ Erupción cutánea, picor
■ Dolor en las articulaciones
■ Sensación de cansancio o debilidad, fiebre
Frecuentes (pueden afectar hasta 1 de cada 10 pacientes)
■ Infecciones del tracto respiratorio superior, infección pulmonar grave (neumonía)
■ Elevación de algunas células de la serie blanca de la sangre
■ Reacción alérgica, reacciones relacionadas con la perfusión del medicamento
■ Disminución de la secreción de hormonas producidas por las glándulas suprarrenales (glándulas situadas encima de los riñones); baja actividad de funcionamiento (hipopituitarismo) o inflamación (hipofisitis) de la glándula pituitaria situada en la base del cerebro; aumento de la actividad de la glándula tiroides en el cuello, que puede producir un aumento de la frecuencia cardíaca, sudoración y pérdida de peso; inflamación de la glándula tiroides; hinchazón de la glándula tiroides, niveles altos de azúcar en sangre (hiperglucemia)
■ Deshidratación
■ Inflamación del hígado
■ Inflamación de los nervios que causa entumecimiento, debilidad, hormigueo o quemazón de los brazos y las piernas; mareo
■ Inflamación de los ojos, que produce dolor y enrojecimiento, problemas visuales o visión borrosa
■ Latido cardíaco rápido
■ Elevación de la tensión sanguínea (hipertensión)
■ Inflamación de los pulmones (neumonitis), caracterizada por tos y dificultad para respirar, coágulos sanguíneos, falta de aliento (disnea), tos
■ Úlceras en la boca y herpes (estomatitis), gastritis (inflamación del estómago), estreñimiento, sequedad bucal
■ Cambio del color de la piel en parches (vitíligo), piel seca, enrojecimiento de la piel, pérdida inusual o debilitamiento del cabello, urticaria (erupción cutánea con picor)
■ Dolor en los músculos y huesos
■ Insuficiencia renal
■ Edema (hinchazón), dolor
Poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes)
■ Bronquitis
■ Enfermedades crónicas asociadas con la acumulación de células inflamatorias en varios órganos y tejidos, más comúnmente en los pulmones (sarcoidosis)
■ Ácido en la sangre producido por diabetes (cetoacidosis diabética), diabetes
■ Inflamación temporal de los nervios que causa dolor, debilidad y parálisis en las extremidades (síndrome de Guillain-Barré); nervios dañados causando entumecimiento y debilidad (polineuropatía); inflamación de los nervios; pie caído (parálisis del nervio peroneo); inflamación de los nervios causada por el ataque al cuerpo en sí, causando insensibilidad y debilidad, hormigueo o quemazón
■ Cambios en el ritmo o la frecuencia del latido cardíaco, ritmo cardíaco anormal
■ Líquido en los pulmones
■ Inflamación del páncreas, perforación intestinal, inflamación del duodeno
■ Enfermedad cutánea con zonas abultadas de piel roja, con frecuencia con escamas plateadas (psoriasis)
■ Enfermedad crónica de las articulaciones (espondiloartropatía); enfermedad en la cual el sistema inmunitario ataca las glándulas que producen la hidratación del cuerpo, tales como lágrimas y saliva (síndrome de Sjogren), dolor en las articulaciones; miopatía (dolor en los músculos, sensibilidad o debilidad muscular, no causadas por el ejercicio)
■ Inflamación del riñón
Dolor en el pecho
Raros (pueden afectar hasta 1 de cada 1.000 pacientes)
■ Descamación de la piel grave y posiblemente mortal (necrólisis epidérmica tóxica)
Si experimenta alguno de los efectos adversos descritos más arriba, informe a su médico inmediatamente. No intente tratar estos síntomas con otros medicamentos por su cuenta.
Cambios en los resultados de los análisis de laboratorio
OPDIVO en combinación con ipilimumab puede provocar cambios en los resultados de los análisis de laboratorio efectuados por su médico, entre otros:
■ Pruebas de función hepática anormales (aumento de la concentración en sangre de las enzimas hepáticas aspartato aminotransferasa, alanina aminotransferasa o fosfatasa alcalina, aumento de los niveles sanguíneos del producto de desecho, bilirrubina)
■ Pruebas de función renal anormales (aumento de la concentración de creatinina en sangre)
■ Disminución del recuento de glóbulos rojos (que transportan oxígeno), glóbulos blancos (importantes para combatir las infecciones) o plaquetas (las células que ayudan a la coagulación sanguínea)
■ Aumento del nivel de la enzima que rompe las grasas y de la enzima que rompe el almidón
■ Aumento o disminución de la cantidad de calcio o potasio
■ Aumento o disminución de los niveles de magnesio o sodio en su sangre
■ Disminución del peso corporal
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de OPDIVO
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y la etiqueta del vial después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar este medicamento en el embalaje original para protegerlo de la luz.
No conserve la solución de perfusión no utilizada para un nuevo uso. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
6. Contenido del envase e información adicional
Composición de OPDIVO
■ El principio activo es nivolumab.
Cada ml de concentrado para solución para perfusión contiene 10 mg de nivolumab. Cada vial contiene 40 mg (en 4 ml) o 100 mg (en 10 ml) de nivolumab.
■ Los demás componentes son citrato sódico di hidratado, cloruro sódico (ver sección 2 "OPDIVO contiene sodio"), manitol (E421), ácido pentético, polisorbato 80, hidróxido sódico, ácido clorhídrico y agua para preparaciones inyectables.
Aspecto de OPDIVO y contenido del envase
OPDIVO concentrado para solución para perfusión (concentrado estéril) es un líquido de transparente a opalescente, de incoloro a amarillo pálido que puede contener algunas (pocas) partículas.
Está disponible en envases que contienen 1 vial de 4 ml ó 1 vial de 10 ml.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Bristol-Myers Squibb Pharma EEIG Uxbridge Business Park Sanderson Road Uxbridge UB8 1DH Reino Unido
Fabricante
Bristol-Myers Squibb S.r.l.
Loc. Fontana del Ceraso 03012 Anagni (FR)
Italia
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización.
Belgique/Belgie/Belgien Lietuva
N.V. Bristol-Myers Squibb Belgium S.A.
Tél/Tel: + 32 2 352 76 11
Btnrapnn
Bristol-Myers Squibb Gyógyszerkereskedelmi Kft Ten.: + 359 800 12 400
Ceská republika
Bristol-Myers Squibb spol. s r.o.
Tel: + 420 221 016 111
Danmark
Bristol-Myers Squibb Tlf: + 45 45 93 05 06
Deutschland
Bristol-Myers Squibb GmbH & Co. KGaA Tel: + 49 89 121 42-0
Eesti
Bristol-Myers Squibb Gyógyszerkereskedelmi Kft Tel: + 372 6827 400
EXXáóa
Bristol-Myers Squibb A.E.
Tr(k: + 30 210 6074300
España
BRISTOL-MYERS SQUIBB, S.A.
Tel: + 34 91 456 53 00
Bristol-Myers Squibb Gyógyszerkereskedelmi Kft. Tel: + 370 5 2790 762
Luxembourg/Luxemburg
.N.V. Bristol-Myers Squibb Belgium S.A.
Tél/Tel: + 32 2 352 76 11
Magyarország
Bristol-Myers Squibb Gyógyszerkereskedelmi Kft. Tel.: + 36 1 301 9700
Malta
Bristol-Myers Squibb S.r.l.
Tel: + 39 06 50 39 61
Nederland
Bristol-Myers Squibb B.V.
Tel: + 31 (0)30 300 2222
Norge
. Bristol-Myers Squibb Norway Ltd Tlf: + 47 67 55 53 50
Osterreich
Bristol-Myers Squibb GesmbH Tel: + 43 1 60 14 30
Polska
Bristol-Myers Squibb Polska Sp. z o.o.
Tel.: + 48 22 5796666
S.A.
Tel: + 351 21 440 70 00
Hrvatska Romanía
Bristol-Myers Squibb spol. s r.o. Bristol-Myers Squibb Gyógyszerkereskedelmi Kft.
TEL: +385 (1) 6311-833 Tel: + 40 (0)21 272 16 00
Ireland Sloveníja
Bristol-Myers Squibb Pharmaceuticals Ltd Bristol-Myers Squibb spol. s r.o.
Tel: + 353 (1 800) 749 749 Tel: + 386 1 236 47 00
Slovenská republika
Bristol-Myers Squibb spol. s r.o.
Tel: + 421 2 59298411
Italía
Bristol-Myers Squibb S.r.l.
Tel: + 39 06 50 39 61
Kónpoq
Bristol-Myers Squibb A.E.
Tr(k: + 357 800 92666
Latvija
Bristol-Myers Squibb Gyógyszerkereskedelmi Tel: + 371 67 50 21 85
Suomi/Finland
Oy Bristol-Myers Squibb (Finland) Ab Puh/Tel: + 358 9 251 21 230
Sverige
Bristol-Myers Squibb AB Tel: + 46 8 704 71 00
United Kingdom
Kft. Bristol-Myers Squibb Pharmaceuticals Ltd Tel: + 44 (0800) 731 1736
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
Esta información está destinada unicamente a profesionales del sector sanitario:
Preparación y administración de OPDIVO
La preparación la debe realizar personal formado de acuerdo con la normas de buenas prácticas, especialmente en lo que respecta a la asepsia.
Cálculo de la dosis
La dosis prescrita para el paciente se administra en mg/kg. De acuerdo con esta dosis prescrita, calcular la dosis total a administrar. Puede ser necesario más de un vial de concentrado de OPDIVO para suministrar la dosis total del paciente.
■ La dosis total de nivolumab en mg = el peso del paciente en kg * la dosis prescrita en mg/kg.
■ El volumen de concentrado de OPDIVO para preparar la dosis (ml) = la dosis total en mg, dividida entre 10 (la concentración del concentrado de OPDIVO es 10 mg/ml).
Preparación de la perfusión
Hay que garantizar una manipulación aséptica al preparar la perfusión. Ésta se debe preparar en una campana de flujo laminar o cabina de seguridad empleando las precauciones habituales para la manipulación segura de los agentes intravenosos.
OPDIVO se puede utilizar para la administración intravenosa:
■ sin dilución, después de transferir a un recipiente para perfusión utilizando una jeringa estéril adecuada;
o
■ tras su dilución a concentraciones tan bajas como 1 mg/ml. La concentración final de la
perfusión debe oscilar entre 1 y 10 mg/ml. OPDIVO concentrado se puede diluir con:
■ solución de cloruro sódico para preparaciones inyectables a una concentración de 9 mg/ml (0,9%); o
■ solución de glucosa para preparaciones inyectables a una concentración de 50 mg/ml (5%).
PASO 1
■ Inspeccionar el concentrado de OPDIVO para la detección de partículas o cambios de color. No agitar el vial. El concentrado de OPDIVO es un líquido de transparente a opalescente, de incoloro a amarillo pálido. Deseche el vial si la solución está turbia, descolorida o si contiene otras partículas que no sean unas pocas de translúcidas a blancas.
■ Extraer el volumen necesario de concentrado de OPDIVO empleando una jeringa estéril adecuada.
PASO 2
■ Transferir el concentrado a un frasco de vidrio estéril evacuado o envase para solución intravenosa (PVC o poliolefina).
■ Si procede, diluir con el volumen preciso de solución de cloruro sódico para preparaciones inyectables a una concentración de 9 mg/ml (0,9%) o una solución de glucosa para preparaciones inyectables a una concentración de 50 mg/ml (5%). Mezclar suavemente la perfusión por rotación manual. No agitar.
Administración
La perfusión de OPDIVO no se debe administrar en forma de inyección en bolo intravenoso. Administrar la perfusión de OPDIVO por vía intravenosa durante un período de tiempo de 60 minutos.
La perfusión de OPDIVO no se debe perfundir simultáneamente a través de la misma vía intravenosa con otros medicamentos. Usar una vía de perfusión aparte para la perfusión.
Usar un equipo de perfusión y un filtro en línea estéril, no pirógeno y de baja unión a proteínas (tamaño de poro de 0,2 pm a 1,2 pm).
La perfusión de OPDIVO es compatible con:
■ Contenedores de PVC
■ Contenedores de poliolefina
■ Frascos de vidrio
■ Equipos de perfusión de PVC
■ Filtro en línea con membranas de poliétersulfona con tamaños de poro de 0,2 pm a 1,2 pm.
Tras la administración de la dosis, purgar la vía con una solución de cloruro sódico para preparaciones inyectables a una concentración de 9 mg/ml (0,9%) o una solución de glucosa para preparaciones inyectables a una concentración de 50 mg/ml (5%).
Condiciones de conservación y periodo de validez Vial sin abrir
OPDIVO se debe conservar en nevera (2°C-8°C). Los viales se deben mantener en el embalaje original para protegerlos de la luz. OPDIVO no se debe congelar.
No utilice OPDIVO después de la fecha de caducidad que aparece en el envase y la etiqueta del vial después de CAD. La fecha de caducidad es el último día del mes que se indica.
Perfusión de OPDIVO
La perfusión de OPDIVO se debe completar en un plazo de 24 horas a partir de su preparación. Si no se utiliza inmediatamente, la solución se puede conservar en nevera (2°C-8°C) y protegida de la luz hasta 24 horas [un máximo de 4 horas del total de 24 horas se puede mantener a temperatura ambiente (20°C-25°C) y expuesta a la luz ambiental]. Otros tiempos y condiciones de conservación durante su uso son responsabilidad del usuario.
Eliminación
No conservar la solución de perfusión no utilizada para un nuevo uso. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
61