Omnitrope 15 Mg/1,5 Ml Solucion Inyectable (1 Cartucho)
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Omnitrope 1,3 mg/ml polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Tras la reconstitución, un vial contiene 1,3 mg de somatropina* (que corresponde a 4 UI) por ml. * Producida en Escherichia coli mediante tecnología de DNA recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable. El polvo es blanco.
El disolvente es límpido e incoloro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lactantes, niños y adolescentes:
- Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento (deficiencia de la hormona del crecimiento, DGH).
- Trastorno del crecimiento asociado al síndrome de Turner.
- Trastorno del crecimiento asociado a insuficiencia renal crónica.
- Trastorno del crecimiento (puntuación de la desviación estándar actual de la talla (SDS) < -2,5 y
SDS de la talla < -1 ajustada para los padres) en los niños y los adolescentes nacidos con baja
talla para su edad gestacional (PEG), con un peso al nacer y/o longitud por debajo de -
2 desviaciones estándar (DE), que no alcanzaron el estirón de crecimiento (velocidad de crecimiento (VC) SDS < 0 durante el último año) a los 4 años de edad o más.
- El síndrome de Prader-Willi (SPW) para la mejoría del crecimiento y composición corporal. El diagnóstico de SPW debe ser confirmado por pruebas genéticas apropiadas.
Adultos
- Terapia de sustitución en los adultos con deficiencia pronunciada de la hormona del crecimiento.
- Inicio en la edad adulta: Pacientes que padecen una deficiencia grave de la hormona del crecimiento, asociada a múltiples deficiencias hormonales a consecuencia de una patología hipotalámica o hipofisaria conocida y que padecen, por lo menos, una deficiencia conocida de una hormona hipofisaria que no sea la prolactina. Estos pacientes deben someterse a una prueba dinámica adecuada, para así diagnosticar o excluir una deficiencia de la hormona del crecimiento.
- Inicio en la niñez: Pacientes que padecían una deficiencia de la hormona del crecimiento en la niñez a consecuencia de causas congénitas, genéticas, adquiridas o idiopáticas. En los pacientes con DGH de inicio en la niñez se debe reevaluar la capacidad secretora de la hormona del crecimiento después de concluir su crecimiento longitudinal. En los pacientes con una probabilidad alta de DGH persistente, es decir, una causa congénita o una DGH secundaria a una enfermedad o lesión hipotálamo-hipofisaria, un SDS < -2 del factor de crecimiento de tipo insulínico I (IGF-I) cuando no reciben tratamiento con hormona de crecimiento por lo menos durante cuatro semanas debe considerarse una prueba suficiente de DGH profunda.
Todos los demás pacientes requerirán un análisis del IGF-I y una prueba de estimulación con la
hormona del crecimiento.
El diagnóstico y el tratamiento con somatropina deben iniciarse y monitorizarse por médicos que tengan la capacitación y la experiencia adecuadas en el diagnóstico y el tratamiento de los pacientes con trastornos del crecimiento.
Posología
Población _ pediátrica
La posología y la pauta de dosificación deben individualizarse.
Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento en los pacientes pediátricos:
En términos generales, se recomienda una dosis de 0,025 a 0,035 mg/kg de peso corporal por día o de 0,7 a 1,0 mg/m2 de superficie corporal por día. Incluso dosis más altas han sido utilizadas.
Cuando la DGH de inicio en la niñez persiste hasta la adolescencia, se debe continuar el tratamiento a fin de alcanzar un desarrollo somático completo (p. ej., composición corporal, masa ósea). Para la monitorización, el logro de una masa ósea máxima normal, definida como una puntuación T > -1 (es decir, normalizado hasta la masa ósea máxima promedio en el adulto, determinada mediante absorciometría de rayos X de doble energía, teniendo en cuenta el sexo y el origen étnico del paciente) es uno de los objetivos terapéuticos durante el período de transición. Para la orientación en cuanto a la posología, ver el apartado sobre los adultos, a continuación.
Síndrome de Prader-Willi, para la mejoría del crecimiento y composición corporal en los pacientes pediátricos
En general, se recomienda una dosis de 0,035 mg/kg de peso corporal por día ó 1,0 mg/m2 de superficie corporal por día. No se excederán las dosis diarias de 2,7 mg. El tratamiento no se utilizará en los pacientes pediátricos con una velocidad de crecimiento inferior a 1 cm al año y cerca del cierre de las epífisis.
Trastorno del crecimiento debido al síndrome de Turner
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal al día ó 1,4 mg/m2 de superficie corporal.
Trastorno del crecimiento en la insuficiencia renal crónica
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal y día (de 1,4 mg/m2 de superficie corporal y día). Pueden precisarse dosis más altas si la velocidad de crecimiento es muy lenta. Puede ser necesario corregir la dosis después de seis meses de tratamiento (ver sección 4.4).
Trastorno del crecimiento en los niños y los adolescentes nacidos con baja talla para su edad gestacional (PEG)
Se recomienda una dosis de 0,035 mg/kg de peso corporal al día (1 mg/m2 de superficie corporal al día) hasta que se alcance la talla final (ver sección 5.1). El tratamiento se debe interrumpir después del primer año de tratamiento, si la SDS de la velocidad de crecimiento es inferior a + 1. El tratamiento se interrumpirá si la velocidad de crecimiento es < 2 cm/año y, si se requiere confirmación, la edad ósea es > 14 años (niñas) o > 16 años (niños), correspondiente con el cierre de las placas de crecimiento epifisario.
|
Indicación |
Dosis diaria en mg/kg de peso corporal |
Dosis diaria en mg/m2 de superficie corporal |
|
Deficiencia de la hormona del crecimiento |
0,025 - 0,035 |
0,7 - 1,0 |
|
Síndrome de Prader-Willi |
0,035 |
1,0 |
|
Síndrome de Turner |
0,045 - 0,050 |
1,4 |
|
Insuficiencia renal crónica |
0,045 - 0,050 |
1,4 |
|
Niños y adolescentes nacidos con talla baja para su edad gestacional (PEG) |
0,035 |
1,0 |
Pacientes adultos con deficiencia de la hormona del crecimiento
La dosis recomendada para reanudar el tratamiento en aquellos pacientes que continúen con hormona de crecimiento tras un déficit de GH en la infancia, es de 0,2-0,5 mg por día. La dosis se debe ir incrementando o disminuyendo gradualmente de acuerdo a las necesidades individuales del paciente, determinadas de acuerdo a la concentración del IGF-I.
En los pacientes con DGH de inicio en la edad adulta, el tratamiento debe iniciarse con una dosis baja, de 0,15 a 0,3 mg al día. La dosis se aumentará gradualmente según las necesidades de cada paciente y según lo determine la concentración de IGF-I.
En ambos casos, el objetivo del tratamiento debe ser alcanzar concentraciones de factor de crecimiento de tipo insulina (IGF-I) dentro de 2 SDS de la media corregida por la edad. A los pacientes con concentraciones IGF-I normales al inicio del tratamiento se les debe administrar la hormona del crecimiento hasta una concentración de IGF-I dentro de los límites superiores normales, sin sobrepasar 2 SDS. La respuesta clínica y los efectos adversos pueden también usarse como guía para el ajuste de la dosis. Se conoce que hay pacientes con DGH que no llegan a normalizar los niveles de IGF-I a pesar de tener una buena respuesta clínica, y por tanto, no requieren ajuste de dosis. La dosis de mantenimiento raramente excede 1,0 mg al día. Las mujeres pueden necesitar dosis más altas que los hombres, mientras que los varones muestran una sensibilidad mayor al aumento de la IGF-I con el tiempo. Esto significa que hay un riesgo de que las mujeres, en especial aquellas en sustitución con estrógenos orales, estén infra-tratadas mientras que los hombres estén sobre-tratados. La exactitud de la dosis de la hormona del crecimiento debe, por lo tanto, controlarse cada 6 meses. Habida cuenta de que la producción fisiológica de la hormona del crecimiento desciende con la edad, pueden reducirse las necesidades de la dosis.
Poblaciones especiales
Personas de edad avanzada
En los pacientes mayores de 60 años, el tratamiento debe comenzar con una dosis de 0,1 a 0,2 mg diarios, y debe aumentarse gradualmente, según las necesidades individuales del paciente. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento en estos pacientes rara vez es superior a 0,5 mg diarios.
Forma de administración
Se debe administrar la inyección por vía subcutánea, cambiando el lugar de inyección para evitar la lipoatrofia.
Para consultar las instrucciones de uso y manipulación, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes.
La somatropina no debe usarse cuando exista evidencia de actividad de un tumor. Los tumores intracraneales deben ser inactivos y el tratamiento antitumoral se debe completar antes de iniciar el tratamiento con GH. El tratamiento se debe suspender si hay pruebas de crecimiento tumoral.
La somatropina no debe usarse en la promoción del crecimiento de niños con epífisis cerradas.
Los pacientes con enfermedad crítica aguda que presenten complicaciones después de cirugía cardiaca, abdominal, traumatismo múltiple por accidente, insuficiencia respiratoria aguda o enfermedades similares, no deben ser tratados con somatropina (en cuanto a los pacientes sometidos a tratamiento de sustitución, ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
No se debe superar la dosis diaria máxima recomendada (ver sección 4.2).
Sensibilidad a la insulina
La somatropina puede reducir la sensibilidad a la insulina. En pacientes con diabetes mellitus, puede ser necesario ajustar la dosis de insulina tras la instauración del tratamiento con somatropina. Durante el tratamiento con somatropina, se deben monitorizar cuidadosamente los pacientes con diabetes, intolerancia a la glucosa o con factores de riesgo adicionales de sufrir diabetes.
Función tiroidea
La hormona de crecimiento aumenta la conversión extratiroidea de T4 a T3, lo que puede causar una reducción de la concentración T4 y un aumento de la T3 en el suero. Si bien las concentraciones de la hormona tiroidea periférica han permanecido dentro de los límites de referencia en la mayoría de sujetos sanos, teóricamente, se puede desarrollar hipotiroidismo en los sujetos con hipotiroidismo subclínico. En consecuencia, se debe llevar a cabo una monitorización de la función tiroidea en todos los pacientes. El potencial efecto del tratamiento con hormona de crecimiento sobre la función tiroidea se debe monitorizar cuidadosamente en pacientes con hipopituitarismo tratados con terapia sustitutiva estándar.
En la deficiencia de la hormona del crecimiento, secundaria al tratamiento de una neoplasia maligna, se recomienda prestar atención a los signos de recaída de la malignidad. Entre los supervivientes de cáncer pediátrico se ha referido un mayor riesgo de una segunda neoplasia en los pacientes tratados con somatropina después de su primera neoplasia. Las más frecuentes de estas segundas neoplasias en los pacientes tratados con radioterapia en la cabeza para su primera neoplasia fueron los tumores intracraneales, en concreto los meningiomas.
En los pacientes con trastornos endocrinos, incluida la deficiencia de la hormona del crecimiento, el deslizamiento de la epífisis de la cadera puede producirse con más frecuencia que en la población general. Los pacientes que cojean durante el tratamiento con somatropina deben ser controlados clínicamente.
Hipertensión intracraneal benigna
En casos de cefalea recurrente o grave, problemas visuales, náuseas y/o vómitos, se recomienda practicar una fundoscopia para descartar un edema de papila. Si se confirma el edema de papila, debe considerarse una hipertensión intracraneal benigna y, si fuese apropiado, debe de retirarse el tratamiento con la hormona del crecimiento. Si se reinicia el tratamiento con hormona del crecimiento, se debe instaurar un control cuidadoso para, si fuese necesario, detectar la hipertensión intracraneal.
Leucemia
Se ha notificado leucemia en un pequeño número de pacientes con deficiencia de la hormona de crecimiento, algunos de los cuales han sido tratados con somatropina. Sin embargo, no existe evidencia de que haya un aumento en la incidencia de la leucemia en pacientes tratados con hormona del crecimiento sin factores de predisposición.
Anticuerpos
En un pequeño porcentaje de pacientes pueden aparecer anticuerpos dirigidos contra Omnitrope. Omnitrope ha dado lugar a la formación de anticuerpos aproximadamente en el 1% de los pacientes. La capacidad de fijación de estos anticuerpos es baja y no hay ningún efecto sobre la velocidad de crecimiento. En cualquier paciente con una falta de respuesta por lo demás no explicada se deben hacer análisis de anticuerpos contra la somatropina.
Pacientes de edad avanzada
La experiencia con los pacientes mayores de 80 años es limitada. Los pacientes de edad avanzada pueden ser más sensibles a la acción de Omnitrope y, por lo tanto, pueden ser más propensos a presentar reacciones adversas.
Enfermedad agudas graves
Los efectos de la somatropina sobre la recuperación de pacientes adultos críticos se han evaluado en dos ensayos controlados con placebo en 522 pacientes adultos con complicaciones posteriores a cirugía a corazón abierto, cirugía abdominal, traumatismo accidental múltiple o insuficiencia respiratoria aguda. La mortalidad fue superior en los pacientes tratados diariamente con 5,3 u 8 mg de somatropina, en comparación con los pacientes que recibieron placebo, 42% vs. 19%. En base a esta información, este tipo de pacientes no deben ser tratados con somatropina. Dado que no existe información disponible sobre la seguridad del tratamiento sustitutivo con hormona de crecimiento en pacientes críticos, los beneficios del tratamiento continuado en esta situación deben considerarse sobre la base de los riesgos potenciales.
En todos los pacientes que desarrollen algún otro tipo de enfermedad aguda en fase crítica o similar, el posible beneficio del tratamiento con somatropina deberá ser sopesado en relación con el riesgo potencial que implica.
Población pediátrica
Pancreatitis
A pesar de que la pancreatitis es rara, debe ser considerada en niños tratados con somatropina que presenten dolor abdominal.
Síndrome de Prader-Willi
En los pacientes con SPW, el tratamiento debe ser siempre en combinación con una dieta baja en calorías.
Existen informes de muertes asociadas al uso de la hormona del crecimiento en los pacientes pediátricos con SPW que presentaron uno o más factores de riesgo: obesidad grave (aquellos pacientes con un peso/altura superior al 200%), antecedentes de insuficiencia respiratoria o apnea del sueño o de infección respiratoria no identificada. Los pacientes con SPW y uno o más de estos factores de riesgo podrían estar en mayor riesgo.
Antes de iniciar el tratamiento con somatropina los pacientes con SPW serán valorados respecto de la obstrucción de las vías aéreas altas, apnea del sueño o infección respiratoria.
Si, durante la evaluación de la obstrucción de las vías respiratorias altas, se observan resultados anatomopatológicos, se debe derivar al niño al otorrinolaringólogo (ENT) para el tratamiento y la resolución del trastorno respiratorio antes de iniciar el tratamiento con hormona de crecimiento.
La apnea del sueño debe evaluarse antes del inicio del tratamiento con hormona de crecimiento por métodos reconocidos como la polisomnografía o la oximetría, durante la noche, y los pacientes deben controlarse, si se sospecha apnea del sueño.
Si, durante el tratamiento con somatropina, los pacientes muestran signos de obstrucción de las vías respiratorias altas (incluido el inicio o el aumento de ronquidos), el tratamiento debe interrumpirse y debe efectuarse una nueva evaluación de ENT.
Todos los pacientes con SPW deben valorarse en cuanto a la apnea y ser controlados si se sospecha la presencia de apnea del sueño. Los pacientes deben controlarse para detectar signos de infecciones respiratorias, que deben ser diagnosticadas lo más pronto posible y tratadas de forma contundente.
Todos los pacientes con SPW deben someterse a un control eficaz del peso antes y, también, durante el tratamiento con hormona del crecimiento.
La escoliosis es muy frecuente en los pacientes con SPW. La escoliosis puede progresar en cualquier niño durante el crecimiento rápido. Deben controlarse los signos de escoliosis durante el tratamiento.
La experiencia con el tratamiento prolongado en los adultos y en los pacientes con SPW es limitada.
Nacidos pequeños para su edad gestacional
En los niños y los adolescentes PEG nacidos bajos antes de iniciar el tratamiento se deben descartar otras razones médicas u otros tratamientos que pudieran explicar la perturbación del crecimiento antes de iniciar el tratamiento.
En los niños y los adolescentes PEG se recomienda medir la insulina y la glucosa sanguínea en ayunas antes de iniciar el tratamiento y, posteriormente, en períodos anuales. En los pacientes con un aumento del riesgo de diabetes mellitus (p. ej., antecedentes familiares de diabetes, obesidad, resistencia grave a la insulina, acantosis nigricans) debe realizarse la prueba de tolerancia a la glucosa (OGTT). Si aparece una diabetes clara, no se debe administrar hormona del crecimiento.
En los niños y los adolescentes PEG se recomienda medir la concentración de IGF-I antes de iniciar el tratamiento y después, dos veces al año. Si en mediciones repetidas las concentraciones de IGF-I exceden en +2 DS comparado con la edad de referencia y estado puberal, la proporción IGF-I/IGFBP-3 debe tenerse en cuenta para considerar un ajuste de la dosis.
La experiencia al iniciar el tratamiento en los pacientes PEG cerca del comienzo de la pubertad es limitada. Por tanto, no se recomienda iniciar el tratamiento cerca de la pubertad. La experiencia en los pacientes con síndrome de Silver-Rusell es limitada.
Parte del aumento de estatura obtenido al tratar a los niños y los adolescentes nacidos PEG y bajos con hormona del crecimiento puede perderse si se interrumpe el tratamiento antes de que se alcance la estatura final.
Insuficiencia renal crónica
En la insuficiencia renal crónica, la función renal debe ser un 50% de la normal antes de iniciar el tratamiento. Para verificar la perturbación del crecimiento, se debe hacer un seguimiento del crecimiento durante un año antes de iniciar el tratamiento. Durante este periodo, se debe iniciar un tratamiento conservador para la insuficiencia renal (incluido el control de la acidosis, el hiperparatiroidismo y el estado nutricional), y debe mantenerse durante todo el tratamiento.
El tratamiento se debe interrumpir en casos de trasplante renal.
Hasta la fecha, no se disponen de datos de la estatura final de los pacientes con insuficiencia renal crónica tratados con Omnitrope.
4.5 Interacción con otros medicamentos y otras formas de interacción
El tratamiento concomitante con glucocorticoides puede inhibir los efectos de promoción del crecimiento de los productos que contienen somatropina. Por tanto, los pacientes tratados con glucocorticoides deben tener su crecimiento cuidadosamente monitorizado con el fin de valorar el potencial impacto del tratamiento con glucocorticoides sobre el crecimiento.
Los datos de un estudio de interacción, realizado en los adultos con deficiencia de la hormona del crecimiento, sugieren que la administración de somatropina puede aumentar la eliminación de compuestos que son metabolizados por las isoenzimas del citocromo P450. El metabolismo de los compuestos metabolizados por el citocromo P450 3A4 (p. ej.: esteroides sexuales, corticoesteroides, anticonvulsivantes y ciclosporinas) puede aumentar, dando como resultado unas concentraciones plasmáticas más bajas de estos compuestos. Se desconoce la significación clínica de esto.
Ver también en la sección 4.4 la información relacionada con la diabetes mellitus y los trastornos tiroideos, y en la sección 4.2 la información sobre la terapia de sustitución de estrógenos orales.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos o éstos son limitados relativos al uso de somatropina en mujeres embarazadas. Los estudios realizados en animales son insuficientes en términos de toxicidad para la reproducción (ver sección 5.3). No se recomienda utilizar somatropina durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Lactancia
No se han llevado a cabo estudios clínicos con medicamentos que contienen somatropina en mujeres en periodo de lactancia. Se desconoce si la somatropina se excreta en la leche materna, pero es poco probable la absorción de proteína intacta del tubo digestivo del niño. Por tanto, se debe tener precaución cuando se administre Omnitrope a mujeres en periodo de lactancia.
Fertilidad
No se han realizado estudios de fertilidad con Omnitrope.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Omnitrope sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los pacientes que padecen una deficiencia de la hormona de crecimiento se caracterizan por un déficit del volumen extracelular. Al iniciar el tratamiento con somatropina, este déficit se corrige rápidamente. En los pacientes adultos, son frecuentes las reacciones adversas relacionadas con la retención de líquidos, como el edema periférico, la rigidez musculoesquelética, la artralgia, la mialgia y las parestesias. En general, estas reacciones son leves a moderadas, se producen en los primeros meses de tratamiento y remiten espontáneamente o al reducir la dosis.
La incidencia de estas reacciones adversas está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de hormona de crecimiento. En los niños, tales reacciones adversas son poco frecuentes.
Omnitrope ha dado lugar a la formación de anticuerpos en aproximadamente el 1% de los pacientes. La capacidad de fijación de estos anticuerpos ha sido baja y no hay cambios clínicos asociados a su formación (ver sección 4.4).
Tabla de reacciones adversas
Las tablas 1-6 muestran las reacciones adversas clasificadas según los apartados del sistema de clasificación de órganos y las frecuencias, utilizando la convención siguiente: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); no conocida (no puede estimarse a partir de los datos disponibles) para cada una de las afecciones indicadas.
Ensayos clínicos en niños con DGH
|
Tabla 1 Tratamiento a largo plazo de niños con alteración del crecimiento debida a una secreción insuficiente de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias1 Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia1 |
Mialgia1 Rigidez musculoesquelética1 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico1 | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre1 | |||||
Ensayos clínicos en niños con síndrome de Turner
|
Tabla 2 Tratamiento a largo plazo de niños con alteración del crecimiento debida al síndrome de Turner | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico* Reacción en el lugar de la inyección$ | |||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* | |||||
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con insuficiencia renal crónica
|
Tabla 3 | ||||||
|
Tratamiento a largo plazo de niños con alteración del crecimiento debida a una insuficiencia renal crónica | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas |
Leucemiaf | |||||
|
y no especificadas (incl. quistes y pólipos) | ||||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* Rigidez musculoesquelética* | |||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con PEG
|
Tabla 4 Tratamiento a largo plazo de niños con alteración del crecimiento debida al nacimiento con baja talla para su edad gestacional | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión | |||||
|
intracraneal benigna | ||||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con SPW
|
Tabla 5 Tratamiento a largo debida al síndrome ( |
plazo y mejora de la composición corporal en niños con alteración del crecimiento e Prader-Willi | |||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* |
Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de |
Edema periférico* |
Reacción en el lugar de la inyección$ | ||||
|
administración | ||||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre2 |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en adultos con DGH
|
Tabla 6 Terapia de sustitución en adultos con deficiencia de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo 2 | |||||
|
Trastornos del sistema nervioso |
Parestesias * Síndrome del túnel carpiano |
Hipertensión intracraneal benigna | ||||
|
Trastornos musculoesquelét icos y del tejido conjuntivo |
Artralgia2 |
Mialgia2 Rigidez musculoesq uelética2 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico2 |
Reacción en el lugar de la inyección$ | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre2 | |||||
Descripción de reacciones adversas seleccionadas Niveles reducidos de cortisol sérico
Se ha notificado que la somatropina reduce las concentraciones séricas de cortisol, posiblemente al afectar a las proteínas transportadoras o mediante un aumento de la depuración hepática. La relevancia clínica de estas observaciones puede ser limitada. No obstante, antes de iniciar el tratamiento, debe optimizarse el tratamiento de reemplazo con corticoesteroides.
Síndrome de Prader-Willi
Se han notificado casos raros de muerte súbita en pacientes con síndrome de Prader-Willi tratados con somatropina en la experiencia post-comercialización, aunque no se ha demostrado que exista relación causal.
Leucemia
Se han notificado casos (raros o muy raros) de leucemia en los niños que padecen una deficiencia de la hormona de crecimiento y son tratados con somatropina e incluidos en la experiencia post-comercialización. Sin embargo, no existen indicios de un aumento del riesgo de leucemia sin factores predisponentes, tales como la radioterapia cerebral o de la cabeza.
Epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes
Se han notificado epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes en niños tratados con la hormona del crecimiento. La epifisiólisis de la cabeza femoral se produce con mayor frecuencia en caso de trastornos endocrinos y la enfermedad de Legg-Calvé-Perthes es más frecuente en caso de baja estatura. No obstante, se desconoce si estas dos afecciones aparecen o no con mayor frecuencia durante el tratamiento con somatropina. Debe tenerse en cuenta la posibilidad de su diagnóstico en niños que presenten molestias o dolor en la cadera o la rodilla.
Otras reacciones adversas al medicamento
Otras reacciones adversas al medicamento pueden considerarse efectos de clase de la somatropina, tales como una posible hiperglucemia causada por la disminución de la sensibilidad a la insulina, la disminución de la concentración de tiroxina libre y a una hipertensión intracraneal benigna.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Síntomas:
La sobredosis aguda puede conducir inicialmente a hipoglucemia y posteriormente a hiperglucemia.
La sobredosis prolongada puede causar signos y síntomas compatibles con los efectos conocidos de exceso de la hormona del crecimiento.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: hormonas del lóbulo anterior de la hipófisis y sus análogos, código ATC: H01AC01.
Omnitrope es un medicamento biosimilar. La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Mecanismo de acción
La somatropina es una potente hormona metabólica, importante en el metabolismo de lípidos, carbohidratos y proteínas. En los niños con hormona del crecimiento endógena insuficiente, la somatropina estimula el crecimiento lineal y aumenta la tasa de crecimiento. En los adultos y también en los niños, la somatropina mantiene una composición corporal normal aumentando la retención de nitrógeno y la estimulación del crecimiento del músculo esquelético y la movilización de la grasa corporal. El tejido adiposo visceral responde en particular a la somatropina. Además, para mejorar la lipólisis, la somatropina reduce la captación de triglicéridos de los depósitos de grasa corporal. Las concentraciones séricas de IGF-I (factor de crecimiento de tipo insulina I) y la IGFBP3 (proteína de unión del factor de crecimiento de tipo insulina 3) son aumentadas por la somatropina. Además, se han demostrado las siguientes acciones.
Efectos farmacodinámicos
Metabolismo lipídico
La somatropina induce los receptores de colesterol LDL hepáticos y afecta el perfil de lípidos séricos y las lipoproteínas. En general, la administración de somatropina a pacientes deficitarios de la hormona del crecimiento resulta en una reducción del DLD sérico y de la apoliproteína B. También se puede observar una reducción del colesterol total.
Metabolismo de los carbohidratos
La somatropina aumenta la insulina, pero la glucosa sanguínea en ayunas permanece, por lo general, sin cambios. Los niños con hipopituitarismo experimentan hipoglucemia en ayunas. Esta condición es corregida por la somatropina.
Metabolismo del agua y de los minerales
La deficiencia de la hormona del crecimiento se asocia a una reducción de los volúmenes plasmático y extracelular. Ambos aumentan rápidamente tras el tratamiento con somatropina. La somatropina induce la retención de sodio, potasio y fósforo.
Metabolismo óseo
La somatropina estimula el recambio del hueso esquelético. La administración a largo plazo de somatropina a pacientes con osteopenia con deficiencia de la hormona del crecimiento resulta en un aumento del contenido mineral y de la densidad ósea en los lugares portadores de peso.
Capacidad _ física
Mejoría de la potencia muscular y del ejercicio físico tras un tratamiento a largo plazo con somatropina. La somatropina también aumenta la frecuencia cardíaca, pero el mecanismo de acción no ha sido aún aclarado. Puede que una reducción de la resistencia vascular periférica contribuya a este efecto.
Eficacia clínica y seguridad
En ensayos clínicos en los niños y los adolescentes PEG y de talla baja se utilizaron dosis de 0,033 y 0,067 mg/kg de peso corporal, por día hasta alcanzar la altura final. En 56 pacientes tratados de forma continuada y que alcanzaron (casi) la altura final, el cambio principal de talla al inicio del tratamiento fue de +1,90 SDS (0,033 mg/kg de peso corporal por día) y +2,19 SDS (0,067 mg/kg de peso corporal por día). Los datos publicados de niños y adolescentes PEG no tratados sin estirón espontáneo y temprano sugieren un crecimiento tardío, de la SDS de 0,5. Los datos de seguridad a largo plazo son todavía limitados.
5.2 Propiedades farmacocinéticas
Absorción
La biodisponibilidad de la somatropina, administrada subcutáneamente, es aproximadamente del 80% en ambos casos, en las personas sanas y en los pacientes con deficiencia de la hormona de crecimiento. Una dosis subcutánea de 5 mg de Omnitrope polvo y disolvente para solución inyectable en los adultos sanos produce unos valores de Cmax en el plasma de 71 ± 24 pig/l (media ± DE) y una mediana del valor de tmax de 4 horas (límites, 2 y 8 horas), respectivamente.
Eliminación
La media de la vida media terminal de somatropina después de la administración intravenosa en los pacientes adultos con deficiencia de hormona de crecimiento, es alrededor de 0,4 horas. Sin embargo, después de la administración subcutánea de Omnitrope polvo y disolvente para solución inyectable, se alcanza una vida media de 3 horas. Es probable que la diferencia observada se deba a la lenta absorción de la inyección después de la administración subcutánea.
Poblaciones especiales
La biodisponibilidad absoluta de somatropina parece ser similar tanto en los hombres como en las mujeres después de la administración subcutánea.
La información sobre las propiedades farmacocinéticas de somatropina en geriatría y pediatría, en diferentes razas y en los pacientes con insuficiencia renal, hepática o cardiaca es o bien escasa o incompleta.
5.3 Datos preclínicos sobre seguridad
En los estudios con Omnitrope referentes a toxicidad subaguda y tolerancia local no se han observado casos con efectos clínicamente relevantes.
En otros estudios con somatropina referente a toxicidad general, tolerancia local y reproducción de la toxicidad no se han observado efectos clínicamente relevantes.
Con somatropina, los estudios de genotoxicidad in vitro e in vivo o de mutaciones genéticas e inducción de aberraciones cromosómicas han sido negativos.
Se ha observado un aumento de fragilidad cromosómica en un estudio in vitro con linfocitos, tomados de pacientes en tratamiento prolongado con somatropina y bajo la adición de bleomicina, un medicamento radiomimético. El significado clínico de esta observación no está claro.
En otro estudio con somatropina, no se encontró incremento de anomalías cromosómicas en linfocitos de pacientes que recibieron terapia con somatropina durante largo tiempo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Glicina
Fosfato de hidrógeno disódico heptahidrato Fosfato dihidrógeno sódico dihidrato
Disolvente:
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
2 años.
Periodo de validez tras la reconstitución
Tras la reconstitución, desde un punto de vista microbiológico, se recomienda su uso inmediato. No obstante, se ha demostrado la estabilidad hasta un periodo de 24 horas, entre 2°C y 8°C, en el embalaje original. Conservar y transportar refrigerado (entre 2°C y 8°C). No congelar. Conservar en el embalaje original para protegerlo de la luz.
6.4 Precauciones especiales de conservación
Cartucho sin abrir
Conservar y transportar refrigerado (entre 2°C y 8°C). No congelar. Conservar en el embalaje original para protegerlo de la luz.
Para las condiciones de conservación del medicamento en uso, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Polvo en un vial (de vidrio tipo I) con un tapón (goma de butilo laminado y capa de resina-flúor) y un tapón (tapa “flip-off’ de polipropileno violeta), y 1 ml de disolvente en un vial (de vidrio de tipo 1) con un tapón (de elastómero clorobutilo laminado con resina de flúor), una cinta (de aluminio laqueado), y una cápsula de cierre (blanca, de polipropileno, de tipo “flip-off’ de polipropileno blanco).
Paquete de 1 cartucho.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Omnitrope 1,3 mg/ml se presenta en viales que contienen el principio activo en polvo para solución inyectable y el disolvente, de uso único. Cada vial debe ser reconstituido solamente en el solvente. La solución reconstituida deberá administrase utilizando una aguja para inyección estéril y una jeringa para inyección desechables.
Lo que sigue es una descripción general del proceso de reconstitución y administración. La reconstitución debe efectuarse de conformidad con las normas de buena práctica, sobre todo en lo que respecta a la asepsia.
1. Las manos deben lavarse.
2. Retirar los tapones protectores de plástico de los viales.
3. La parte superior del vial deberá desinfectarse con una solución antiséptica para evitar la contaminación del contenido.
4. Utilizar una jeringa para inyección desechable, estéril (p. ej.: una jeringa para inyección de 2 ml) y una aguja para inyección, también desechable (p. ej.: de 0,33 mm x 12,7 mm) para retirar todo el disolvente del vial.
5. Coger el vial de polvo para solución inyectable, e introducir la aguja para inyección a través del tapón de goma e inyectar el disolvente lentamente en el vial, enfocando el chorro de líquido contra la pared de cristal y así evitar la producción de espuma.
6. Girar suavemente el vial unas cuantas veces, hasta que el contenido esté completamente disuelto. No agitar, ya que podría causar la desnaturalización del principio activo.
7. Si la solución inyectable se vuelve turbia o contiene partículas, no deberá utilizarse. El contenido deberá ser claro e incoloro tras la reconstitución.
8. Invertir el vial y utilizando otra jeringa para inyección desechable, estéril, de tamaño apropiado (p. ej.: jeringa para inyección de 1 ml) y una aguja para inyección (p. ej.: de 0,25 mm x 8 mm) retirar un poco más de la dosis necesaria. Eliminar las burbujas de aire de la jeringa para inyección. Medir la dosis correcta necesaria dentro de la jeringa para inyección.
9. Limpiar el lugar de la inyección con una torunda de algodón y administrar Omnitrope por vía subcutánea.
La solución inyectable es para un solo uso. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Austria
8. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 12 de abril de 2006 Fecha de la última renovación: 12 de abril de 2011
10. FECHA DE LA REVISIÓN DEL TEXTO
<{MM/AAAA}>
1. NOMBRE DEL MEDICAMENTO
Omnitrope 5 mg/ml polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Tras la reconstitución, 1 vial contiene 5 mg de somatropina* (que corresponde a 15 UI) por ml. * Producida en Escherichia coli mediante tecnología de DNA recombinante.
Excipiente(s) con efecto conocido:
Tras la reconstitución, un ml contiene 15 mg de alcohol bencílico.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable. El polvo es blanco.
El disolvente es límpido e incoloro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lactantes, niños y adolescentes:
- Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento (deficiencia de la hormona del crecimiento, DGH).
- Trastorno del crecimiento asociado al síndrome de Turner.
- Trastorno del crecimiento asociado a insuficiencia renal crónica.
- Trastorno del crecimiento (puntuación de la desviación estándar actual de la talla (SDS) < -2,5 y
SDS de la talla < -1 ajustada para los padres) en los niños y los adolescentes nacidos con baja talla para su edad gestacional (PEG), con un peso al nacer y/o longitud por debajo de -
2 desviaciones estándar (DE), que no alcanzaron el estirón de crecimiento (velocidad de crecimiento (VC) SDS < 0 durante el último año) a los 4 años de edad o más.
- El síndrome de Prader-Willi (SPW) para la mejoría del crecimiento y composición corporal. El diagnóstico de SPW debe ser confirmado por pruebas genéticas apropiadas.
Adultos
- Terapia de sustitución en los adultos con deficiencia pronunciada de la hormona del crecimiento.
- Inicio en la edad adulta: Pacientes que padecen una deficiencia grave de la hormona del crecimiento, asociada a múltiples deficiencias hormonales a consecuencia de una patología hipotalámica o hipofisaria conocida y que padecen, por lo menos, una deficiencia conocida de una hormona hipofisaria que no sea la prolactina. Estos pacientes deben someterse a una prueba dinámica adecuada, para así diagnosticar o excluir una deficiencia de la hormona del crecimiento.
- Inicio en la niñez: Pacientes que padecían una deficiencia de la hormona del crecimiento en la niñez a consecuencia de causas congénitas, genéticas, adquiridas o idiopáticas. En los pacientes con DGH de inicio en la niñez se debe reevaluar la capacidad secretora de la hormona del crecimiento después de concluir su crecimiento longitudinal. En los pacientes con una probabilidad alta de DGH persistente, es decir, una causa congénita o una DGH secundaria a una enfermedad o lesión hipotálamo-hipofisaria, un SDS < -2 del factor de crecimiento de tipo insulínico I (IGF-I) cuando no reciben tratamiento con hormona de crecimiento por lo menos durante cuatro semanas debe considerarse una prueba suficiente de DGH profunda.
Todos los demás pacientes requerirán un análisis del IGF-I y una prueba de estimulación con la hormona del crecimiento.
4.2 Posología y forma de administración
El diagnóstico y el tratamiento con somatropina deben iniciarse y monitorizarse por médicos que tengan la capacitación y la experiencia adecuadas en el diagnóstico y el tratamiento de los pacientes con trastornos del crecimiento.
Posología
Población _ pediátrica
La posología y la pauta de dosificación deben individualizarse.
Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento en los pacientes pediátricos:
En términos generales, se recomienda una dosis de 0,025 a 0,035 mg/kg de peso corporal por día o de 0,7 a 1,0 mg/m2 de superficie corporal por día. Incluso dosis más altas han sido utilizadas.
Cuando la DGH de inicio en la niñez persiste hasta la adolescencia, se debe continuar el tratamiento a fin de alcanzar un desarrollo somático completo (p. ej., composición corporal, masa ósea). Para la monitorización, el logro de una masa ósea máxima normal, definida como una puntuación T > -1 (es decir, normalizado hasta la masa ósea máxima promedio en el adulto, determinada mediante absorciometría de rayos X de doble energía, teniendo en cuenta el sexo y el origen étnico del paciente) es uno de los objetivos terapéuticos durante el período de transición. Para la orientación en cuanto a la posología, ver el apartado sobre los adultos, a continuación.
Síndrome de Prader-Willi, para la mejoría del crecimiento y composición corporal en los pacientes pediátricos
En general, se recomienda una dosis de 0,035 mg/kg de peso corporal por día ó 1,0 mg/m2 de superficie corporal por día. No se excederán las dosis diarias de 2,7 mg. El tratamiento no se utilizará en los pacientes pediátricos con una velocidad de crecimiento inferior a 1 cm al año y cerca del cierre de las epífisis.
Trastorno del crecimiento debido al síndrome de Turner
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal al día ó 1,4 mg/m2 de superficie corporal.
Trastorno del crecimiento en la insuficiencia renal crónica
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal y día (de 1,4 mg/m2 de superficie corporal y día). Pueden precisarse dosis más altas si la velocidad de crecimiento es muy lenta. Puede ser necesario corregir la dosis después de seis meses de tratamiento (ver sección 4.4).
Trastorno del crecimiento en los niños y los adolescentes nacidos con baja talla para su edad gestacional (PEG)
Se recomienda una dosis de 0,035 mg/kg de peso corporal al día (1 mg/m2 de superficie corporal al día) hasta que se alcance la talla final (ver sección 5.1). El tratamiento se debe interrumpir después del primer año de tratamiento, si la SDS de la velocidad de crecimiento es inferior a + 1. El tratamiento se interrumpirá si la velocidad de crecimiento es < 2 cm/año y, si se requiere confirmación, la edad ósea es > 14 años (niñas) o > 16 años (niños), correspondiente con el cierre de las placas de crecimiento epifisario.
Posología recomendada en pacientes pediátricos
|
Indicación |
Dosis diaria en mg/kg de peso corporal |
Dosis diaria en mg/m2 de superficie corporal |
|
Deficiencia de la hormona del crecimiento |
0,025 - 0,035 |
0,7 - 1,0 |
|
Síndrome de Prader-Willi |
0,035 |
1,0 |
|
Síndrome de Turner |
0,045 - 0,050 |
1,4 |
|
Insuficiencia renal crónica |
0,045 - 0,050 |
1,4 |
|
Niños y adolescentes nacidos con talla baja para su edad gestacional (PEG) |
0,035 |
1,0 |
Pacientes adultos con deficiencia de la hormona del crecimiento
La dosis recomendada para reanudar el tratamiento en aquellos pacientes que continúen con hormona de crecimiento tras un déficit de GH en la infancia, es de 0,2-0,5 mg por día. La dosis se debe ir incrementando o disminuyendo gradualmente de acuerdo a las necesidades individuales del paciente, determinadas de acuerdo a la concentración del IGF-I.
En los pacientes con DGH de inicio en la edad adulta, el tratamiento debe iniciarse con una dosis baja, de 0,15 a 0,3 mg al día. La dosis se aumentará gradualmente según las necesidades de cada paciente y según lo determine la concentración de IGF-I.
En ambos casos, el objetivo del tratamiento debe ser alcanzar concentraciones de factor de crecimiento de tipo insulina (IGF-I) dentro de 2 SDS de la media corregida por la edad. A los pacientes con concentraciones IGF-I normales al inicio del tratamiento se les debe administrar la hormona del crecimiento hasta una concentración de IGF-I dentro de los límites superiores normales, sin sobrepasar 2 SDS. La respuesta clínica y los efectos adversos pueden también usarse como guía para el ajuste de la dosis. Se conoce que hay pacientes con DGH que no llegan a normalizar los niveles de IGF-I a pesar de tener una buena respuesta clínica, y por tanto, no requieren ajuste de dosis. La dosis de mantenimiento raramente excede 1,0 mg al día. Las mujeres pueden necesitar dosis más altas que los hombres, mientras que los varones muestran una sensibilidad mayor al aumento de la IGF-I con el tiempo. Esto significa que hay un riesgo de que las mujeres, en especial aquellas en sustitución con estrógenos orales, estén infra-tratadas mientras que los hombres estén sobre-tratados. La exactitud de la dosis de la hormona del crecimiento debe, por lo tanto, controlarse cada 6 meses. Habida cuenta de que la producción fisiológica de la hormona del crecimiento desciende con la edad, pueden reducirse las necesidades de la dosis.
Poblaciones especiales
Personas de edad avanzada
En los pacientes mayores de 60 años, el tratamiento debe comenzar con una dosis de 0,1 a 0,2 mg diarios, y debe aumentarse gradualmente, según las necesidades individuales del paciente. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento en estos pacientes rara vez es superior a 0,5 mg diarios.
Forma de administración
Se debe administrar la inyección por vía subcutánea, cambiando el lugar de inyección para evitar la lipoatrofia.
Para consultar las instrucciones de uso y manipulación, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes.
La somatropina no debe usarse cuando exista evidencia de actividad de un tumor. Los tumores intracraneales deben ser inactivos y el tratamiento antitumoral se debe completar antes de iniciar el tratamiento con GH. El tratamiento se debe suspender si hay pruebas de crecimiento tumoral.
La somatropina no debe usarse en la promoción del crecimiento de niños con epífisis cerradas.
Los pacientes con enfermedad crítica aguda que presenten complicaciones después de cirugía cardiaca, abdominal, traumatismo múltiple por accidente, insuficiencia respiratoria aguda o enfermedades similares, no deben ser tratados con somatropina (en cuanto a los pacientes sometidos a tratamiento de sustitución, ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
No se debe superar la dosis diaria máxima recomendada (ver sección 4.2).
Sensibilidad a la insulina
La somatropina puede reducir la sensibilidad a la insulina. En pacientes con diabetes mellitus, puede ser necesario ajustar la dosis de insulina tras la instauración del tratamiento con somatropina. Durante el tratamiento con somatropina, se deben monitorizar cuidadosamente los pacientes con diabetes, intolerancia a la glucosa o con factores de riesgo adicionales de sufrir diabetes.
Función tiroidea
La hormona de crecimiento aumenta la conversión extratiroidea de T4 a T3, lo que puede causar una reducción de la concentración T4 y un aumento de la T3 en el suero. Si bien las concentraciones de la hormona tiroidea periférica han permanecido dentro de los límites de referencia en la mayoría de sujetos sanos, teóricamente, se puede desarrollar hipotiroidismo en los sujetos con hipotiroidismo subclínico. En consecuencia, se debe llevar a cabo una monitorización de la función tiroidea en todos los pacientes. El potencial efecto del tratamiento con hormona de crecimiento sobre la función tiroidea se debe monitorizar cuidadosamente en pacientes con hipopituitarismo tratados con terapia sustitutiva estándar.
En la deficiencia de la hormona del crecimiento, secundaria al tratamiento de una neoplasia maligna, se recomienda prestar atención a los signos de recaída de la malignidad. Entre los supervivientes de cáncer pediátrico se ha referido un mayor riesgo de una segunda neoplasia en los pacientes tratados con somatropina después de su primera neoplasia. Las más frecuentes de estas segundas neoplasias en los pacientes tratados con radioterapia en la cabeza para su primera neoplasia fueron los tumores intracraneales, en concreto los meningiomas.
En los pacientes con trastornos endocrinos, incluida la deficiencia de la hormona del crecimiento, el deslizamiento de la epífisis de la cadera puede producirse con más frecuencia que en la población general. Los pacientes que cojean durante el tratamiento con somatropina deben ser controlados clínicamente.
Hipertensión intracraneal benigna
En casos de cefalea recurrente o grave, problemas visuales, náuseas y/o vómitos, se recomienda practicar una fundoscopia para descartar un edema de papila. Si se confirma el edema de papila, debe considerarse una hipertensión intracraneal benigna y, si fuese apropiado, debe de retirarse el tratamiento con la hormona del crecimiento. Si se reinicia el tratamiento con hormona del crecimiento, se debe instaurar un control cuidadoso para, si fuese necesario, detectar la hipertensión intracraneal.
Leucemia
Se ha notificado leucemia en un pequeño número de pacientes con deficiencia de la hormona de crecimiento, algunos de los cuales han sido tratados con somatropina. Sin embargo, no existe evidencia de que haya un aumento en la incidencia de la leucemia en pacientes tratados con hormona del crecimiento sin factores de predisposición.
Anticuerpos
En un pequeño porcentaje de pacientes pueden aparecer anticuerpos dirigidos contra Omnitrope. Omnitrope ha dado lugar a la formación de anticuerpos aproximadamente en el 1% de los pacientes. La capacidad de fijación de estos anticuerpos es baja y no hay ningún efecto sobre la velocidad de crecimiento. En cualquier paciente con una falta de respuesta por lo demás no explicada se deben hacer análisis de anticuerpos contra la somatropina.
Pacientes de edad avanzada
La experiencia con los pacientes mayores de 80 años es limitada. Los pacientes de edad avanzada pueden ser más sensibles a la acción de Omnitrope y, por lo tanto, pueden ser más propensos a presentar reacciones adversas.
Enfermedad agudas graves
Los efectos de la somatropina sobre la recuperación de pacientes adultos críticos se han evaluado en dos ensayos controlados con placebo en 522 pacientes adultos con complicaciones posteriores a cirugía a corazón abierto, cirugía abdominal, traumatismo accidental múltiple o insuficiencia respiratoria aguda. La mortalidad fue superior en los pacientes tratados diariamente con 5,3 u 8 mg de somatropina, en comparación con los pacientes que recibieron placebo, 42% vs. 19%. En base a esta información, este tipo de pacientes no deben ser tratados con somatropina. Dado que no existe información disponible sobre la seguridad del tratamiento sustitutivo con hormona de crecimiento en pacientes críticos, los beneficios del tratamiento continuado en esta situación deben considerarse sobre la base de los riesgos potenciales.
En todos los pacientes que desarrollen algún otro tipo de enfermedad aguda en fase crítica o similar, el posible beneficio del tratamiento con somatropina deberá ser sopesado en relación con el riesgo potencial que implica.
Población pediátrica
Pancreatitis
A pesar de que la pancreatitis es rara, debe ser considerada en niños tratados con somatropina que presenten dolor abdominal.
Síndrome de Prader-Willi
En los pacientes con SPW, el tratamiento debe ser siempre en combinación con una dieta baja en calorías.
Existen informes de muertes asociadas al uso de la hormona del crecimiento en los pacientes pediátricos con SPW que presentaron uno o más factores de riesgo: obesidad grave (aquellos pacientes con un peso/altura superior al 200%), antecedentes de insuficiencia respiratoria o apnea del sueño o de infección respiratoria no identificada. Los pacientes con SPW y uno o más de estos factores de riesgo podrían estar en mayor riesgo.
Antes de iniciar el tratamiento con somatropina los pacientes con SPW serán valorados respecto de la obstrucción de las vías aéreas altas, apnea del sueño o infección respiratoria.
Si, durante la evaluación de la obstrucción de las vías respiratorias altas, se observan resultados anatomopatológicos, se debe derivar al niño al otorrinolaringólogo (ENT) para el tratamiento y la resolución del trastorno respiratorio antes de iniciar el tratamiento con hormona de crecimiento.
La apnea del sueño debe evaluarse antes del inicio del tratamiento con hormona de crecimiento por métodos reconocidos como la polisomnografía o la oximetría, durante la noche, y los pacientes deben controlarse, si se sospecha apnea del sueño.
Si, durante el tratamiento con somatropina, los pacientes muestran signos de obstrucción de las vías respiratorias altas (incluido el inicio o el aumento de ronquidos), el tratamiento debe interrumpirse y debe efectuarse una nueva evaluación de ENT.
Todos los pacientes con SPW deben valorarse en cuanto a la apnea y ser controlados si se sospecha la presencia de apnea del sueño. Los pacientes deben controlarse para detectar signos de infecciones respiratorias, que deben ser diagnosticadas lo más pronto posible y tratadas de forma contundente.
Todos los pacientes con SPW deben someterse a un control eficaz del peso antes y, también, durante el tratamiento con hormona del crecimiento.
La escoliosis es muy frecuente en los pacientes con SPW. La escoliosis puede progresar en cualquier niño durante el crecimiento rápido. Deben controlarse los signos de escoliosis durante el tratamiento.
La experiencia con el tratamiento prolongado en los adultos y en los pacientes con SPW es limitada.
Nacidos pequeños para su edad gestacional
En los niños y los adolescentes PEG nacidos bajos antes de iniciar el tratamiento se deben descartar otras razones médicas u otros tratamientos que pudieran explicar la perturbación del crecimiento antes de iniciar el tratamiento.
En los niños y los adolescentes PEG se recomienda medir la insulina y la glucosa sanguínea en ayunas antes de iniciar el tratamiento y, posteriormente, en períodos anuales. En los pacientes con un aumento del riesgo de diabetes mellitus (p. ej., antecedentes familiares de diabetes, obesidad, resistencia grave a la insulina, acantosis nigricans) debe realizarse la prueba de tolerancia a la glucosa (OGTT). Si aparece una diabetes clara, no se debe administrar hormona del crecimiento.
En los niños y los adolescentes PEG se recomienda medir la concentración de IGF-I antes de iniciar el tratamiento y después, dos veces al año. Si en mediciones repetidas las concentraciones de IGF-I exceden en +2 DS comparado con la edad de referencia y estado puberal, la proporción IGF-I/IGFBP-3 debe tenerse en cuenta para considerar un ajuste de la dosis.
La experiencia al iniciar el tratamiento en los pacientes PEG cerca del comienzo de la pubertad es limitada. Por tanto, no se recomienda iniciar el tratamiento cerca de la pubertad. La experiencia en los pacientes con síndrome de Silver-Rusell es limitada.
Parte del aumento de estatura obtenido al tratar a los niños y los adolescentes nacidos PEG y bajos con hormona del crecimiento puede perderse si se interrumpe el tratamiento antes de que se alcance la estatura final.
Insuficiencia renal crónica
En la insuficiencia renal crónica, la función renal debe ser un 50% de la normal antes de iniciar el tratamiento. Para verificar la perturbación del crecimiento, se debe hacer un seguimiento del crecimiento durante un año antes de iniciar el tratamiento. Durante este periodo, se debe iniciar un tratamiento conservador para la insuficiencia renal (incluido el control de la acidosis, el hiperparatiroidismo y el estado nutricional), y debe mantenerse durante todo el tratamiento.
El tratamiento se debe interrumpir en casos de trasplante renal.
Hasta la fecha, no se disponen de datos de la estatura final de los pacientes con insuficiencia renal crónica tratados con Omnitrope.
Debido a la presencia de alcohol bencílico, el medicamento no debe administrarse a recién nacidos prematuros ni a neonatos. Puede causar reacciones tóxicas y reacciones anafilactoides en los lactantes y en los niños de hasta tres años.
4.5 Interacción con otros medicamentos y otras formas de interacción
El tratamiento concomitante con glucocorticoides puede inhibir los efectos de promoción del crecimiento de los productos que contienen somatropina. Por tanto, los pacientes tratados con glucocorticoides deben tener su crecimiento cuidadosamente monitorizado con el fin de valorar el potencial impacto del tratamiento con glucocorticoides sobre el crecimiento.
Los datos de un estudio de interacción, realizado en los adultos con deficiencia de la hormona del crecimiento, sugieren que la administración de somatropina puede aumentar la eliminación de compuestos que son metabolizados por las isoenzimas del citocromo P450. El metabolismo de los compuestos metabolizados por el citocromo P450 3A4 (p. ej.: esteroides sexuales, corticoesteroides, anticonvulsivantes y ciclosporinas) puede aumentar, dando como resultado unas concentraciones plasmáticas más bajas de estos compuestos. Se desconoce la significación clínica de esto.
Ver también en la sección 4.4 la información relacionada con la diabetes mellitus y los trastornos tiroideos, y en la sección 4.2 la información sobre la terapia de sustitución de estrógenos orales.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos o éstos son limitados relativos al uso de somatropina en mujeres embarazadas. Los estudios realizados en animales son insuficientes en términos de toxicidad para la reproducción (ver sección 5.3). No se recomienda utilizar somatropina durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Lactancia
No se han llevado a cabo estudios clínicos con medicamentos que contienen somatropina en mujeres en periodo de lactancia. Se desconoce si la somatropina se excreta en la leche materna, pero es poco probable la absorción de proteína intacta del tubo digestivo del niño. Por tanto, se debe tener precaución cuando se administre Omnitrope a mujeres en periodo de lactancia.
Fertilidad
No se han realizado estudios de fertilidad con Omnitrope.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Omnitrope sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los pacientes que padecen una deficiencia de la hormona de crecimiento se caracterizan por un déficit del volumen extracelular. Al iniciar el tratamiento con somatropina, este déficit se corrige rápidamente. En los pacientes adultos, son frecuentes las reacciones adversas relacionadas con la retención de líquidos, como el edema periférico, la rigidez musculoesquelética, la artralgia, la mialgia y las parestesias. En general, estas reacciones son leves a moderadas, se producen en los primeros meses de tratamiento y remiten espontáneamente o al reducir la dosis.
La incidencia de estas reacciones adversas está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de hormona de crecimiento. En los niños, tales reacciones adversas son poco frecuentes.
Omnitrope ha dado lugar a la formación de anticuerpos en aproximadamente el 1% de los pacientes. La capacidad de fijación de estos anticuerpos ha sido baja y no hay cambios clínicos asociados a su formación (ver sección 4.4).
Tabla de reacciones adversas
Las tablas 1-6 muestran las reacciones adversas clasificadas según los apartados del sistema de clasificación de órganos y las frecuencias, utilizando la convención siguiente: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); no conocida (no puede estimarse a partir de los datos disponibles) para cada una de las afecciones indicadas.
Ensayos clínicos en niños con DGH
|
Tabla 1 Tratamiento a largo plazo de niños con alteración del crecimiento debida a una secreción insuficiente de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias3 Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia3 |
Mialgia3 Rigidez musculoesquelética3 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico3 | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre3 | |||||
Ensayos clínicos en niños con síndrome de Turner
|
Tabla 2 Tratamiento a largo plazo de niños con alteración del crecimiento debida al síndrome de Turner | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico* Reacción en el lugar de la inyección$ | |||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* | |||||
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con insuficiencia renal crónica
|
Tabla 3 | ||||||
|
Tratamiento a largo plazo de niños con alteración del crecimiento debida a una insuficiencia renal crónica | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas |
Leucemiaf | |||||
|
y no especificadas (incl. quistes y pólipos) | ||||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* Rigidez musculoesquelética* | |||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con PEG
|
Tabla 4 Tratamiento a largo plazo de niños con alteración del crecimiento debida al nacimiento con baja talla para su edad gestacional | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión | |||||
|
intracraneal benigna | ||||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con SPW
|
Tabla 5 Tratamiento a largo debida al síndrome ( |
plazo y mejora de la composición corporal en niños con alteración del crecimiento e Prader-Willi | |||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* |
Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de |
Edema periférico* |
Reacción en el lugar de la inyección$ | ||||
|
administración | ||||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre4 |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en adultos con DGH
|
Tabla 6 Terapia de sustitución en adultos con deficiencia de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo 2 | |||||
|
Trastornos del sistema nervioso |
Parestesias * Síndrome del túnel carpiano |
Hipertensión intracraneal benigna | ||||
|
Trastornos musculoesquelét icos y del tejido conjuntivo |
Artralgia4 |
Mialgia4 Rigidez musculoesq uelética4 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico4 |
Reacción en el lugar de la inyección$ | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre4 | |||||
Descripción de reacciones adversas seleccionadas Niveles reducidos de cortisol sérico
Se ha notificado que la somatropina reduce las concentraciones séricas de cortisol, posiblemente al afectar a las proteínas transportadoras o mediante un aumento de la depuración hepática. La relevancia clínica de estas observaciones puede ser limitada. No obstante, antes de iniciar el tratamiento, debe optimizarse el tratamiento de reemplazo con corticoesteroides.
Síndrome de Prader-Willi
Se han notificado casos raros de muerte súbita en pacientes con síndrome de Prader-Willi tratados con somatropina en la experiencia post-comercialización, aunque no se ha demostrado que exista relación causal.
Leucemia
Se han notificado casos (raros o muy raros) de leucemia en los niños que padecen una deficiencia de la hormona de crecimiento y son tratados con somatropina e incluidos en la experiencia post-comercialización. Sin embargo, no existen indicios de un aumento del riesgo de leucemia sin factores predisponentes, tales como la radioterapia cerebral o de la cabeza.
Epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes
Se han notificado epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes en niños tratados con la hormona del crecimiento. La epifisiólisis de la cabeza femoral se produce con mayor frecuencia en caso de trastornos endocrinos y la enfermedad de Legg-Calvé-Perthes es más frecuente en caso de baja estatura. No obstante, se desconoce si estas dos afecciones aparecen o no con mayor frecuencia durante el tratamiento con somatropina. Debe tenerse en cuenta la posibilidad de su diagnóstico en niños que presenten molestias o dolor en la cadera o la rodilla.
Otras reacciones adversas al medicamento
Otras reacciones adversas al medicamento pueden considerarse efectos de clase de la somatropina, tales como una posible hiperglucemia causada por la disminución de la sensibilidad a la insulina, la disminución de la concentración de tiroxina libre y a una hipertensión intracraneal benigna.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Síntomas:
La sobredosis aguda puede conducir inicialmente a hipoglucemia y posteriormente a hiperglucemia.
La sobredosis prolongada puede causar signos y síntomas compatibles con los efectos conocidos de exceso de la hormona del crecimiento.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: hormonas del lóbulo anterior de la hipófisis y sus análogos, código ATC: H01AC01.
Omnitrope es un medicamento biosimilar. La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Mecanismo de acción
La somatropina es una potente hormona metabólica, importante en el metabolismo de lípidos, carbohidratos y proteínas. En los niños con hormona del crecimiento endógena insuficiente, la somatropina estimula el crecimiento lineal y aumenta la tasa de crecimiento. En los adultos y también en los niños, la somatropina mantiene una composición corporal normal aumentando la retención de nitrógeno y la estimulación del crecimiento del músculo esquelético y la movilización de la grasa corporal. El tejido adiposo visceral responde en particular a la somatropina. Además, para mejorar la lipólisis, la somatropina reduce la captación de triglicéridos de los depósitos de grasa corporal. Las concentraciones séricas de IGF-I (factor de crecimiento de tipo insulina I) y la IGFBP3 (proteína de unión del factor de crecimiento de tipo insulina 3) son aumentadas por la somatropina. Además, se han demostrado las siguientes acciones.
Efectos farmacodinámicos
Metabolismo lipídico
La somatropina induce los receptores de colesterol LDL hepáticos y afecta el perfil de lípidos séricos y las lipoproteínas. En general, la administración de somatropina a pacientes deficitarios de la hormona del crecimiento resulta en una reducción del DLD sérico y de la apoliproteína B. También se puede observar una reducción del colesterol total.
Metabolismo de los carbohidratos
La somatropina aumenta la insulina, pero la glucosa sanguínea en ayunas permanece, por lo general, sin cambios. Los niños con hipopituitarismo experimentan hipoglucemia en ayunas. Esta condición es corregida por la somatropina.
Metabolismo del agua y de los minerales
La deficiencia de la hormona del crecimiento se asocia a una reducción de los volúmenes plasmático y extracelular. Ambos aumentan rápidamente tras el tratamiento con somatropina. La somatropina induce la retención de sodio, potasio y fósforo.
Metabolismo óseo
La somatropina estimula el recambio del hueso esquelético. La administración a largo plazo de somatropina a pacientes con osteopenia con deficiencia de la hormona del crecimiento resulta en un aumento del contenido mineral y de la densidad ósea en los lugares portadores de peso.
Capacidad _ física
Mejoría de la potencia muscular y del ejercicio físico tras un tratamiento a largo plazo con somatropina. La somatropina también aumenta la frecuencia cardíaca, pero el mecanismo de acción no ha sido aún aclarado. Puede que una reducción de la resistencia vascular periférica contribuya a este efecto.
Eficacia clínica y seguridad
En ensayos clínicos en los niños y los adolescentes PEG y de talla baja se utilizaron dosis de 0,033 y 0,067 mg/kg de peso corporal, por día hasta alcanzar la altura final. En 56 pacientes tratados de forma continuada y que alcanzaron (casi) la altura final, el cambio principal de talla al inicio del tratamiento fue de +1,90 SDS (0,033 mg/kg de peso corporal por día) y +2,19 SDS (0,067 mg/kg de peso corporal por día). Los datos publicados de niños y adolescentes PEG no tratados sin estirón espontáneo y temprano sugieren un crecimiento tardío, de la SDS de 0,5. Los datos de seguridad a largo plazo son todavía limitados.
5.2 Propiedades farmacocinéticas
Absorción
La biodisponibilidad de la somatropina, administrada subcutáneamente, es aproximadamente del 80% en ambos casos, en las personas sanas y en los pacientes con deficiencia de la hormona de crecimiento. Una dosis subcutánea de 5 mg de Omnitrope polvo y disolvente para solución inyectable en los adultos sanos produce unos valores de Cmax en el plasma de 71 ± 24 pig/l (media ± DE) y una mediana del valor de tmax de 4 horas (límites, 2 y 8 horas), respectivamente.
Eliminación
La media de la vida media terminal de somatropina después de la administración intravenosa en los pacientes adultos con deficiencia de hormona de crecimiento, es alrededor de 0,4 horas. Sin embargo, después de la administración subcutánea de Omnitrope 5 mg/ml polvo y disolvente para solución inyectable, se alcanza una vida media de 3 horas. Es probable que la diferencia observada se deba a la lenta absorción de la inyección después de la administración subcutánea.
Poblaciones especiales
La biodisponibilidad absoluta de somatropina parece ser similar tanto en los hombres como en las mujeres después de la administración subcutánea.
La información sobre las propiedades farmacocinéticas de somatropina en geriatría y pediatría, en diferentes razas y en los pacientes con insuficiencia renal, hepática o cardiaca es o bien escasa o incompleta.
5.3 Datos preclínicos sobre seguridad
En los estudios con Omnitrope referentes a toxicidad subaguda y tolerancia local no se han observado casos con efectos clínicamente relevantes.
En otros estudios con somatropina referente a toxicidad general, tolerancia local y reproducción de la toxicidad no se han observado efectos clínicamente relevantes.
Con somatropina, los estudios de genotoxicidad in vitro e in vivo o de mutaciones genéticas e inducción de aberraciones cromosómicas han sido negativos.
Se ha observado un aumento de fragilidad cromosómica en un estudio in vitro con linfocitos, tomados de pacientes en tratamiento prolongado con somatropina y bajo la adición de bleomicina, un medicamento radiomimético. El significado clínico de esta observación no está claro.
En otro estudio con somatropina, no se encontró incremento de anomalías cromosómicas en linfocitos de pacientes que recibieron terapia con somatropina durante largo tiempo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Glicina
Fosfato de hidrógeno disódico heptahidrato Fosfato dihidrógeno sódico dihidrato
Disolvente:
Agua para preparaciones inyectables Alcohol bencílico
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 3 años.
Periodo de validez tras la reconstitución
Tras la reconstitución y del primer uso, el cartucho deberá permanecer en la pluma y debe conservarse en una nevera (entre 2°C y 8°C) durante un máximo de 21 días. Conservar y transportar refrigerado (entre 2°C y 8°C). No congelar. Conservar en la pluma original para protegerlo de la luz.
6.4 Precauciones especiales de conservación
Cartucho sin abrir
Conservar y transportar refrigerado (entre 2°C y 8°C). No congelar. Conservar en el embalaje original para protegerlo de la luz.
Para las condiciones de conservación del medicamento en uso, ver sección 6.3
6.5 Naturaleza y contenido del recipiente
Polvo en un vial (de vidrio tipo 1), con tapón (de goma de butilo laminada de resina de flúor), una cinta (de aluminio) y una cápsula de cierre (verde, de propileno, de tipo “flip-off’) y 1 ml de disolvente en un cartucho (de vidrio de tipo 1) con un tapón (de elastómero de clorobutilo laminado con resina de flúor), una cinta (de aluminio laqueado) y una cápsula de cierre (blanca, de polipropileno, de tipo “flip-off’).
Paquetes de 1 y 5 cartuchos.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Omnitrope 5 mg/ml se presenta en un vial que contiene el principio activo en forma de polvo con el disolvente dentro de un cartucho. Debe reconstituirse mediante un set de transferencia según se recomienda en la información facilitada conjuntamente con el set de transferencia.
Esta presentación está pensada para uso múltiple. Sólo debería ser administrada con Omnitrope Pen L, un dispositivo de inyección específicamente desarrollado para usarse con Omnitrope 5 mg/ml reconstituido para inyección. Se administrará utilizando agujas para inyección de pluma estériles y desechables. Los pacientes y cuidadores han de recibir del médico o de otro profesional de la salud, entrenamiento e instrucciones adecuadas sobre el uso correcto de los viales de Omnitrope, los cartuchos de disolvente, el set de transferencia y la pluma.
Lo que sigue es una descripción de los procesos generales de reconstitución y administración. Para la reconstitución, la carga del cartucho, el montaje de la aguja para inyección y la administración deberán seguirse las instrucciones del fabricante que aparecen con el juego individual de transferencia de Omnitrope 5 mg/ml polvo para solución inyectable.
1. Las manos deben lavarse.
2. Retirar la tapa protectora del vial. La parte superior protectora del cartucho se limpiará con una solución antiséptica para evitar la contaminación del contenido.
3. Utilizar el set de transferencia para transferir el disolvente del cartucho al vial
4. Girar suavemente el vial unas cuantas veces, hasta que el contenido esté completamente disuelto. No agitar, ya que podría provocar la desnaturalización del principio activo.
5. No se debe usar si la solución inyectable está turbia o contiene partículas. Tras la reconstitución, el contenido deberá estar claro y ser incoloro.
6. Mediante el set de transferencia, transferir la solución inyectable al cartucho.
7. Montar la pluma según las instrucciones de uso.
8. Si fuese necesario, eliminar las burbujas de aire.
9. Limpiar el lugar de aplicación de la inyección con una torunda empapada en alcohol.
10. Administrar por vía subcutánea la dosis apropiada, usando una aguja para inyección de pluma estéril. Retirar la aguja para inyección de pluma y desecharla según las normativas locales.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Austria
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/002
EU/1/06/332/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 12 de abril de 2006 Fecha de la última renovación: 12 de abril de 2011
10. FECHA DE LA REVISIÓN DEL TEXTO
<{MM/AAAA}>
1. NOMBRE DEL MEDICAMENTO
Omnitrope 5 mg/1,5 ml solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada ml de solución contiene 3,3 mg de somatropina* (que corresponde a 10 UI). Un cartucho contiene 1,5 ml, que corresponde a 5 mg de somatropina* (15 UI).
* Producida en Escherichia coli mediante tecnología de DNA recombinante.
Excipiente(s) con efecto conocido:
Un ml contiene 9 mg de alcohol bencílico.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable La solución es límpida e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lactantes, niños y adolescentes:
- Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento (deficiencia de la hormona del crecimiento, DGH).
- Trastorno del crecimiento asociado al síndrome de Turner.
- Trastorno del crecimiento asociado a insuficiencia renal crónica.
- Trastorno del crecimiento (puntuación de la desviación estándar actual de la talla (SDS) < -2,5 y
SDS de la talla < -1 ajustada para los padres) en los niños y los adolescentes nacidos con baja
talla para su edad gestacional (PEG), con un peso al nacer y/o longitud por debajo de -
2 desviaciones estándar (DE), que no alcanzaron el estirón de crecimiento (velocidad de crecimiento (VC) SDS < 0 durante el último año) a los 4 años de edad o más.
- El síndrome de Prader-Willi (SPW) para la mejoría del crecimiento y composición corporal. El diagnóstico de SPW debe ser confirmado por pruebas genéticas apropiadas.
Adultos
- Terapia de sustitución en los adultos con deficiencia pronunciada de la hormona del crecimiento.
- Inicio en la edad adulta: Pacientes que padecen una deficiencia grave de la hormona del crecimiento, asociada a múltiples deficiencias hormonales a consecuencia de una patología hipotalámica o hipofisaria conocida y que padecen, por lo menos, una deficiencia conocida de una hormona hipofisaria que no sea la prolactina. Estos pacientes deben someterse a una prueba dinámica adecuada, para así diagnosticar o excluir una deficiencia de la hormona del crecimiento.
- Inicio en la niñez: Pacientes que padecían una deficiencia de la hormona del crecimiento en la niñez a consecuencia de causas congénitas, genéticas, adquiridas o idiopáticas. En los pacientes con DGH de inicio en la niñez se debe reevaluar la capacidad secretora de la hormona del crecimiento después de concluir su crecimiento longitudinal. En los pacientes con una probabilidad alta de DGH persistente, es decir, una causa congénita o una DGH secundaria a una enfermedad o lesión hipotálamo-hipofisaria, un SDS < -2 del factor de crecimiento de tipo insulínico I (IGF-I) cuando no reciben tratamiento con hormona de crecimiento por lo menos durante cuatro semanas debe considerarse una prueba suficiente de DGH profunda.
Todos los demás pacientes requerirán un análisis del IGF-I y una prueba de estimulación con la hormona del crecimiento.
4.2 Posología y forma de administración
El diagnóstico y el tratamiento con somatropina deben iniciarse y monitorizarse por médicos que tengan la capacitación y la experiencia adecuadas en el diagnóstico y el tratamiento de los pacientes con trastornos del crecimiento.
Posología
Población _ pediátrica
La posología y la pauta de dosificación deben individualizarse.
Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento en los pacientes pediátricos:
En términos generales, se recomienda una dosis de 0,025 a 0,035 mg/kg de peso corporal por día o de 0,7 a 1,0 mg/m2 de superficie corporal por día. Incluso dosis más altas han sido utilizadas.
Cuando la DGH de inicio en la niñez persiste hasta la adolescencia, se debe continuar el tratamiento a fin de alcanzar un desarrollo somático completo (p. ej., composición corporal, masa ósea). Para la monitorización, el logro de una masa ósea máxima normal, definida como una puntuación T > -1 (es decir, normalizado hasta la masa ósea máxima promedio en el adulto, determinada mediante absorciometría de rayos X de doble energía, teniendo en cuenta el sexo y el origen étnico del paciente) es uno de los objetivos terapéuticos durante el período de transición. Para la orientación en cuanto a la posología, ver el apartado sobre los adultos, a continuación.
Síndrome de Prader-Willi, para la mejoría del crecimiento y composición corporal en los pacientes pediátricos
En general, se recomienda una dosis de 0,035 mg/kg de peso corporal por día ó 1,0 mg/m2 de superficie corporal por día. No se excederán las dosis diarias de 2,7 mg. El tratamiento no se utilizará en los pacientes pediátricos con una velocidad de crecimiento inferior a 1 cm al año y cerca del cierre de las epífisis.
Trastorno del crecimiento debido al síndrome de Turner
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal al día ó 1,4 mg/m2 de superficie corporal.
Trastorno del crecimiento en la insuficiencia renal crónica
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal y día (de 1,4 mg/m2 de superficie corporal y día). Pueden precisarse dosis más altas si la velocidad de crecimiento es muy lenta. Puede ser necesario corregir la dosis después de seis meses de tratamiento (ver sección 4.4).
Trastorno del crecimiento en los niños y los adolescentes nacidos con baja talla para su edad gestacional (PEG)
Se recomienda una dosis de 0,035 mg/kg de peso corporal al día (1 mg/m2 de superficie corporal al día) hasta que se alcance la talla final (ver sección 5.1). El tratamiento se debe interrumpir después del primer año de tratamiento, si la SDS de la velocidad de crecimiento es inferior a + 1. El tratamiento se interrumpirá si la velocidad de crecimiento es < 2 cm/año y, si se requiere confirmación, la edad ósea es > 14 años (niñas) o > 16 años (niños), correspondiente con el cierre de las placas de crecimiento epifisario.
Posología recomendada en pacientes pediátricos
|
Indicación |
Dosis diaria en mg/kg de peso corporal |
Dosis diaria en mg/m2 de superficie corporal |
|
Deficiencia de la hormona del crecimiento |
0,025 - 0,035 |
0,7 - 1,0 |
|
Síndrome de Prader-Willi |
0,035 |
1,0 |
|
Síndrome de Turner |
0,045 - 0,050 |
1,4 |
|
Insuficiencia renal crónica |
0,045 - 0,050 |
1,4 |
|
Niños y adolescentes nacidos con talla baja para su edad gestacional (PEG) |
0,035 |
1,0 |
Pacientes adultos con deficiencia de la hormona del crecimiento
La dosis recomendada para reanudar el tratamiento en aquellos pacientes que continúen con hormona de crecimiento tras un déficit de GH en la infancia, es de 0,2-0,5 mg por día. La dosis se debe ir incrementando o disminuyendo gradualmente de acuerdo a las necesidades individuales del paciente, determinadas de acuerdo a la concentración del IGF-I.
En los pacientes con DGH de inicio en la edad adulta, el tratamiento debe iniciarse con una dosis baja, de 0,15 a 0,3 mg al día. La dosis se aumentará gradualmente según las necesidades de cada paciente y según lo determine la concentración de IGF-I.
En ambos casos, el objetivo del tratamiento debe ser alcanzar concentraciones de factor de crecimiento de tipo insulina (IGF-I) dentro de 2 SDS de la media corregida por la edad. A los pacientes con concentraciones IGF-I normales al inicio del tratamiento se les debe administrar la hormona del crecimiento hasta una concentración de IGF-I dentro de los límites superiores normales, sin sobrepasar 2 SDS. La respuesta clínica y los efectos adversos pueden también usarse como guía para el ajuste de la dosis. Se conoce que hay pacientes con DGH que no llegan a normalizar los niveles de IGF-I a pesar de tener una buena respuesta clínica, y por tanto, no requieren ajuste de dosis. La dosis de mantenimiento raramente excede 1,0 mg al día. Las mujeres pueden necesitar dosis más altas que los hombres, mientras que los varones muestran una sensibilidad mayor al aumento de la IGF-I con el tiempo. Esto significa que hay un riesgo de que las mujeres, en especial aquellas en sustitución con estrógenos orales, estén infra-tratadas mientras que los hombres estén sobre-tratados. La exactitud de la dosis de la hormona del crecimiento debe, por lo tanto, controlarse cada 6 meses. Habida cuenta de que la producción fisiológica de la hormona del crecimiento desciende con la edad, pueden reducirse las necesidades de la dosis.
Poblaciones especiales
Personas de edad avanzada
En los pacientes mayores de 60 años, el tratamiento debe comenzar con una dosis de 0,1 a 0,2 mg diarios, y debe aumentarse gradualmente, según las necesidades individuales del paciente. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento en estos pacientes rara vez es superior a 0,5 mg diarios.
Forma de administración
Se debe administrar la inyección por vía subcutánea, cambiando el lugar de inyección para evitar la lipoatrofia.
Para consultar las instrucciones de uso y manipulación, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes.
La somatropina no debe usarse cuando exista evidencia de actividad de un tumor. Los tumores intracraneales deben ser inactivos y el tratamiento antitumoral se debe completar antes de iniciar el tratamiento con GH. El tratamiento se debe suspender si hay pruebas de crecimiento tumoral.
La somatropina no debe usarse en la promoción del crecimiento de niños con epífisis cerradas.
Los pacientes con enfermedad crítica aguda que presenten complicaciones después de cirugía cardiaca, abdominal, traumatismo múltiple por accidente, insuficiencia respiratoria aguda o enfermedades similares, no deben ser tratados con somatropina (en cuanto a los pacientes sometidos a tratamiento de sustitución, ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
No se debe superar la dosis diaria máxima recomendada (ver sección 4.2).
Sensibilidad a la insulina
La somatropina puede reducir la sensibilidad a la insulina. En pacientes con diabetes mellitus, puede ser necesario ajustar la dosis de insulina tras la instauración del tratamiento con somatropina. Durante el tratamiento con somatropina, se deben monitorizar cuidadosamente los pacientes con diabetes, intolerancia a la glucosa o con factores de riesgo adicionales de sufrir diabetes.
Función tiroidea
La hormona de crecimiento aumenta la conversión extratiroidea de T4 a T3, lo que puede causar una reducción de la concentración T4 y un aumento de la T3 en el suero. Si bien las concentraciones de la hormona tiroidea periférica han permanecido dentro de los límites de referencia en la mayoría de sujetos sanos, teóricamente, se puede desarrollar hipotiroidismo en los sujetos con hipotiroidismo subclínico. En consecuencia, se debe llevar a cabo una monitorización de la función tiroidea en todos los pacientes. El potencial efecto del tratamiento con hormona de crecimiento sobre la función tiroidea se debe monitorizar cuidadosamente en pacientes con hipopituitarismo tratados con terapia sustitutiva estándar.
En la deficiencia de la hormona del crecimiento, secundaria al tratamiento de una neoplasia maligna, se recomienda prestar atención a los signos de recaída de la malignidad. Entre los supervivientes de cáncer pediátrico se ha referido un mayor riesgo de una segunda neoplasia en los pacientes tratados con somatropina después de su primera neoplasia. Las más frecuentes de estas segundas neoplasias en los pacientes tratados con radioterapia en la cabeza para su primera neoplasia fueron los tumores intracraneales, en concreto los meningiomas.
En los pacientes con trastornos endocrinos, incluida la deficiencia de la hormona del crecimiento, el deslizamiento de la epífisis de la cadera puede producirse con más frecuencia que en la población general. Los pacientes que cojean durante el tratamiento con somatropina deben ser controlados clínicamente.
Hipertensión intracraneal benigna
En casos de cefalea recurrente o grave, problemas visuales, náuseas y/o vómitos, se recomienda practicar una fundoscopia para descartar un edema de papila. Si se confirma el edema de papila, debe considerarse una hipertensión intracraneal benigna y, si fuese apropiado, debe de retirarse el tratamiento con la hormona del crecimiento. Si se reinicia el tratamiento con hormona del crecimiento, se debe instaurar un control cuidadoso para, si fuese necesario, detectar la hipertensión intracraneal.
Leucemia
Se ha notificado leucemia en un pequeño número de pacientes con deficiencia de la hormona de crecimiento, algunos de los cuales han sido tratados con somatropina. Sin embargo, no existe evidencia de que haya un aumento en la incidencia de la leucemia en pacientes tratados con hormona del crecimiento sin factores de predisposición.
Anticuerpos
En un pequeño porcentaje de pacientes pueden aparecer anticuerpos dirigidos contra Omnitrope. Omnitrope ha dado lugar a la formación de anticuerpos aproximadamente en el 1% de los pacientes. La capacidad de fijación de estos anticuerpos es baja y no hay ningún efecto sobre la velocidad de crecimiento. En cualquier paciente con una falta de respuesta por lo demás no explicada se deben hacer análisis de anticuerpos contra la somatropina.
Pacientes de edad avanzada
La experiencia con los pacientes mayores de 80 años es limitada. Los pacientes de edad avanzada pueden ser más sensibles a la acción de Omnitrope y, por lo tanto, pueden ser más propensos a presentar reacciones adversas.
Enfermedad agudas graves
Los efectos de la somatropina sobre la recuperación de pacientes adultos críticos se han evaluado en dos ensayos controlados con placebo en 522 pacientes adultos con complicaciones posteriores a cirugía a corazón abierto, cirugía abdominal, traumatismo accidental múltiple o insuficiencia respiratoria aguda. La mortalidad fue superior en los pacientes tratados diariamente con 5,3 u 8 mg de somatropina, en comparación con los pacientes que recibieron placebo, 42% vs. 19%. En base a esta información, este tipo de pacientes no deben ser tratados con somatropina. Dado que no existe información disponible sobre la seguridad del tratamiento sustitutivo con hormona de crecimiento en pacientes críticos, los beneficios del tratamiento continuado en esta situación deben considerarse sobre la base de los riesgos potenciales.
En todos los pacientes que desarrollen algún otro tipo de enfermedad aguda en fase crítica o similar, el posible beneficio del tratamiento con somatropina deberá ser sopesado en relación con el riesgo potencial que implica.
Población pediátrica
Pancreatitis
A pesar de que la pancreatitis es rara, debe ser considerada en niños tratados con somatropina que presenten dolor abdominal.
Síndrome de Prader-Willi
En los pacientes con SPW, el tratamiento debe ser siempre en combinación con una dieta baja en calorías.
Existen informes de muertes asociadas al uso de la hormona del crecimiento en los pacientes pediátricos con SPW que presentaron uno o más factores de riesgo: obesidad grave (aquellos pacientes con un peso/altura superior al 200%), antecedentes de insuficiencia respiratoria o apnea del sueño o de infección respiratoria no identificada. Los pacientes con SPW y uno o más de estos factores de riesgo podrían estar en mayor riesgo.
Antes de iniciar el tratamiento con somatropina los pacientes con SPW serán valorados respecto de la obstrucción de las vías aéreas altas, apnea del sueño o infección respiratoria.
Si, durante la evaluación de la obstrucción de las vías respiratorias altas, se observan resultados anatomopatológicos, se debe derivar al niño al otorrinolaringólogo (ENT) para el tratamiento y la resolución del trastorno respiratorio antes de iniciar el tratamiento con hormona de crecimiento.
La apnea del sueño debe evaluarse antes del inicio del tratamiento con hormona de crecimiento por métodos reconocidos como la polisomnografía o la oximetría, durante la noche, y los pacientes deben controlarse, si se sospecha apnea del sueño.
Si, durante el tratamiento con somatropina, los pacientes muestran signos de obstrucción de las vías respiratorias altas (incluido el inicio o el aumento de ronquidos), el tratamiento debe interrumpirse y debe efectuarse una nueva evaluación de ENT.
Todos los pacientes con SPW deben valorarse en cuanto a la apnea y ser controlados si se sospecha la presencia de apnea del sueño. Los pacientes deben controlarse para detectar signos de infecciones respiratorias, que deben ser diagnosticadas lo más pronto posible y tratadas de forma contundente.
Todos los pacientes con SPW deben someterse a un control eficaz del peso antes y, también, durante el tratamiento con hormona del crecimiento.
La escoliosis es muy frecuente en los pacientes con SPW. La escoliosis puede progresar en cualquier niño durante el crecimiento rápido. Deben controlarse los signos de escoliosis durante el tratamiento.
La experiencia con el tratamiento prolongado en los adultos y en los pacientes con SPW es limitada.
Nacidos pequeños para su edad gestacional
En los niños y los adolescentes PEG nacidos bajos antes de iniciar el tratamiento se deben descartar otras razones médicas u otros tratamientos que pudieran explicar la perturbación del crecimiento antes de iniciar el tratamiento.
En los niños y los adolescentes PEG se recomienda medir la insulina y la glucosa sanguínea en ayunas antes de iniciar el tratamiento y, posteriormente, en períodos anuales. En los pacientes con un aumento del riesgo de diabetes mellitus (p. ej., antecedentes familiares de diabetes, obesidad, resistencia grave a la insulina, acantosis nigricans) debe realizarse la prueba de tolerancia a la glucosa (OGTT). Si aparece una diabetes clara, no se debe administrar hormona del crecimiento.
En los niños y los adolescentes PEG se recomienda medir la concentración de IGF-I antes de iniciar el tratamiento y después, dos veces al año. Si en mediciones repetidas las concentraciones de IGF-I exceden en +2 DS comparado con la edad de referencia y estado puberal, la proporción IGF-I/IGFBP-3 debe tenerse en cuenta para considerar un ajuste de la dosis.
La experiencia al iniciar el tratamiento en los pacientes PEG cerca del comienzo de la pubertad es limitada. Por tanto, no se recomienda iniciar el tratamiento cerca de la pubertad. La experiencia en los pacientes con síndrome de Silver-Rusell es limitada.
Parte del aumento de estatura obtenido al tratar a los niños y los adolescentes nacidos PEG y bajos con hormona del crecimiento puede perderse si se interrumpe el tratamiento antes de que se alcance la estatura final.
Insuficiencia renal crónica
En la insuficiencia renal crónica, la función renal debe ser un 50% de la normal antes de iniciar el tratamiento. Para verificar la perturbación del crecimiento, se debe hacer un seguimiento del crecimiento durante un año antes de iniciar el tratamiento. Durante este periodo, se debe iniciar un tratamiento conservador para la insuficiencia renal (incluido el control de la acidosis, el hiperparatiroidismo y el estado nutricional), y debe mantenerse durante todo el tratamiento.
El tratamiento se debe interrumpir en casos de trasplante renal.
Hasta la fecha, no se disponen de datos de la estatura final de los pacientes con insuficiencia renal crónica tratados con Omnitrope.
Debido a la presencia de alcohol bencílico, el medicamento no debe administrarse a recién nacidos prematuros ni a neonatos. Puede causar reacciones tóxicas y reacciones anafilactoides en los lactantes y en los niños de hasta tres años.
4.5 Interacción con otros medicamentos y otras formas de interacción
El tratamiento concomitante con glucocorticoides puede inhibir los efectos de promoción del crecimiento de los productos que contienen somatropina. Por tanto, los pacientes tratados con glucocorticoides deben tener su crecimiento cuidadosamente monitorizado con el fin de valorar el potencial impacto del tratamiento con glucocorticoides sobre el crecimiento.
Los datos de un estudio de interacción, realizado en los adultos con deficiencia de la hormona del crecimiento, sugieren que la administración de somatropina puede aumentar la eliminación de compuestos que son metabolizados por las isoenzimas del citocromo P450. El metabolismo de los compuestos metabolizados por el citocromo P450 3A4 (p. ej.: esteroides sexuales, corticoesteroides, anticonvulsivantes y ciclosporinas) puede aumentar, dando como resultado unas concentraciones plasmáticas más bajas de estos compuestos. Se desconoce la significación clínica de esto.
Ver también en la sección 4.4 la información relacionada con la diabetes mellitus y los trastornos tiroideos, y en la sección 4.2 la información sobre la terapia de sustitución de estrógenos orales.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos o éstos son limitados relativos al uso de somatropina en mujeres embarazadas. Los estudios realizados en animales son insuficientes en términos de toxicidad para la reproducción (ver sección 5.3). No se recomienda utilizar somatropina durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Lactancia
No se han llevado a cabo estudios clínicos con medicamentos que contienen somatropina en mujeres en periodo de lactancia. Se desconoce si la somatropina se excreta en la leche materna, pero es poco probable la absorción de proteína intacta del tubo digestivo del niño. Por tanto, se debe tener precaución cuando se administre Omnitrope a mujeres en periodo de lactancia.
Fertilidad
No se han realizado estudios de fertilidad con Omnitrope.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Omnitrope sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los pacientes que padecen una deficiencia de la hormona de crecimiento se caracterizan por un déficit del volumen extracelular. Al iniciar el tratamiento con somatropina, este déficit se corrige rápidamente. En los pacientes adultos, son frecuentes las reacciones adversas relacionadas con la retención de líquidos, como el edema periférico, la rigidez musculoesquelética, la artralgia, la mialgia y las parestesias. En general, estas reacciones son leves a moderadas, se producen en los primeros meses de tratamiento y remiten espontáneamente o al reducir la dosis.
La incidencia de estas reacciones adversas está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de hormona de crecimiento. En los niños, tales reacciones adversas son poco frecuentes.
Omnitrope ha dado lugar a la formación de anticuerpos en aproximadamente el 1% de los pacientes. La capacidad de fijación de estos anticuerpos ha sido baja y no hay cambios clínicos asociados a su formación (ver sección 4.4).
Tabla de reacciones adversas
Las tablas 1-6 muestran las reacciones adversas clasificadas según los apartados del sistema de clasificación de órganos y las frecuencias, utilizando la convención siguiente: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); no conocida (no puede estimarse a partir de los datos disponibles) para cada una de las afecciones indicadas.
Ensayos clínicos en niños con DGH
|
Tabla 1 Tratamiento a largo plazo de niños con alteración del crecimiento debida a una secreción insuficiente de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias5 Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia5 |
Mialgia5 Rigidez musculoesquelética5 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico5 | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre5 | |||||
Ensayos clínicos en niños con síndrome de Turner
|
Tabla 2 Tratamiento a largo plazo de niños con alteración del crecimiento debida al síndrome de Turner | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico* Reacción en el lugar de la inyección$ | |||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* | |||||
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con insuficiencia renal crónica
|
Tabla 3 | ||||||
|
Tratamiento a largo plazo de niños con alteración del crecimiento debida a una insuficiencia renal crónica | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas |
Leucemiaf | |||||
|
y no especificadas (incl. quistes y pólipos) | ||||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* Rigidez musculoesquelética* | |||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con PEG
|
Tabla 4 Tratamiento a largo plazo de niños con alteración del crecimiento debida al nacimiento con baja talla para su edad gestacional | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión | |||||
|
intracraneal benigna | ||||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección$ |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangrej |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con SPW
|
Tabla 5 Tratamiento a largo debida al síndrome ( |
plazo y mejora de la composición corporal en niños con alteración del crecimiento e Prader-Willi | |||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* |
Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de |
Edema periférico* |
Reacción en el lugar de la inyección$ | ||||
|
administración | ||||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre6 |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en adultos con DGH
|
Tabla 6 Terapia de sustitución en adultos con deficiencia de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo 2 | |||||
|
Trastornos del sistema nervioso |
Parestesias * Síndrome del túnel carpiano |
Hipertensión intracraneal benigna | ||||
|
Trastornos musculoesquelét icos y del tejido conjuntivo |
Artralgia6 |
Mialgia6 Rigidez musculoesq uelética6 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico6 |
Reacción en el lugar de la inyección$ | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre6 | |||||
Descripción de reacciones adversas seleccionadas Niveles reducidos de cortisol sérico
Se ha notificado que la somatropina reduce las concentraciones séricas de cortisol, posiblemente al afectar a las proteínas transportadoras o mediante un aumento de la depuración hepática. La relevancia clínica de estas observaciones puede ser limitada. No obstante, antes de iniciar el tratamiento, debe optimizarse el tratamiento de reemplazo con corticoesteroides.
Síndrome de Prader-Willi
Se han notificado casos raros de muerte súbita en pacientes con síndrome de Prader-Willi tratados con somatropina en la experiencia post-comercialización, aunque no se ha demostrado que exista relación causal.
Leucemia
Se han notificado casos (raros o muy raros) de leucemia en los niños que padecen una deficiencia de la hormona de crecimiento y son tratados con somatropina e incluidos en la experiencia post-comercialización. Sin embargo, no existen indicios de un aumento del riesgo de leucemia sin factores predisponentes, tales como la radioterapia cerebral o de la cabeza.
Epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes
Se han notificado epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes en niños tratados con la hormona del crecimiento. La epifisiólisis de la cabeza femoral se produce con mayor frecuencia en caso de trastornos endocrinos y la enfermedad de Legg-Calvé-Perthes es más frecuente en caso de baja estatura. No obstante, se desconoce si estas dos afecciones aparecen o no con mayor frecuencia durante el tratamiento con somatropina. Debe tenerse en cuenta la posibilidad de su diagnóstico en niños que presenten molestias o dolor en la cadera o la rodilla.
Otras reacciones adversas al medicamento
Otras reacciones adversas al medicamento pueden considerarse efectos de clase de la somatropina, tales como una posible hiperglucemia causada por la disminución de la sensibilidad a la insulina, la disminución de la concentración de tiroxina libre y a una hipertensión intracraneal benigna.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Síntomas:
La sobredosis aguda puede conducir inicialmente a hipoglucemia y posteriormente a hiperglucemia.
La sobredosis prolongada puede causar signos y síntomas compatibles con los efectos conocidos de exceso de la hormona del crecimiento.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: hormonas del lóbulo anterior de la hipófisis y sus análogos, código ATC: H01AC01.
Omnitrope es un medicamento biosimilar. La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Mecanismo de acción
La somatropina es una potente hormona metabólica, importante en el metabolismo de lípidos, carbohidratos y proteínas. En los niños con hormona del crecimiento endógena insuficiente, la somatropina estimula el crecimiento lineal y aumenta la tasa de crecimiento. En los adultos y también en los niños, la somatropina mantiene una composición corporal normal aumentando la retención de nitrógeno y la estimulación del crecimiento del músculo esquelético y la movilización de la grasa corporal. El tejido adiposo visceral responde en particular a la somatropina. Además, para mejorar la lipólisis, la somatropina reduce la captación de triglicéridos de los depósitos de grasa corporal. Las concentraciones séricas de IGF-I (factor de crecimiento de tipo insulina I) y la IGFBP3 (proteína de unión del factor de crecimiento de tipo insulina 3) son aumentadas por la somatropina. Además, se han demostrado las siguientes acciones.
Efectos farmacodinámicos
Metabolismo lipídico
La somatropina induce los receptores de colesterol LDL hepáticos y afecta el perfil de lípidos séricos y las lipoproteínas. En general, la administración de somatropina a pacientes deficitarios de la hormona del crecimiento resulta en una reducción del DLD sérico y de la apoliproteína B. También se puede observar una reducción del colesterol total.
Metabolismo de los carbohidratos
La somatropina aumenta la insulina, pero la glucosa sanguínea en ayunas permanece, por lo general, sin cambios. Los niños con hipopituitarismo experimentan hipoglucemia en ayunas. Esta condición es corregida por la somatropina.
Metabolismo del agua y de los minerales
La deficiencia de la hormona del crecimiento se asocia a una reducción de los volúmenes plasmático y extracelular. Ambos aumentan rápidamente tras el tratamiento con somatropina. La somatropina induce la retención de sodio, potasio y fósforo.
Metabolismo óseo
La somatropina estimula el recambio del hueso esquelético. La administración a largo plazo de somatropina a pacientes con osteopenia con deficiencia de la hormona del crecimiento resulta en un aumento del contenido mineral y de la densidad ósea en los lugares portadores de peso.
Capacidad _ física
Mejoría de la potencia muscular y del ejercicio físico tras un tratamiento a largo plazo con somatropina. La somatropina también aumenta la frecuencia cardíaca, pero el mecanismo de acción no ha sido aún aclarado. Puede que una reducción de la resistencia vascular periférica contribuya a este efecto.
Eficacia clínica y seguridad
En ensayos clínicos en los niños y los adolescentes PEG y de talla baja se utilizaron dosis de 0,033 y 0,067 mg/kg de peso corporal, por día hasta alcanzar la altura final. En 56 pacientes tratados de forma continuada y que alcanzaron (casi) la altura final, el cambio principal de talla al inicio del tratamiento fue de +1,90 SDS (0,033 mg/kg de peso corporal por día) y +2,19 SDS (0,067 mg/kg de peso corporal por día). Los datos publicados de niños y adolescentes PEG no tratados sin estirón espontáneo y temprano sugieren un crecimiento tardío, de la SDS de 0,5. Los datos de seguridad a largo plazo son todavía limitados.
5.2 Propiedades farmacocinéticas
Absorción
La biodisponibilidad de la somatropina, administrada subcutáneamente, es aproximadamente del 80% en ambos casos, en las personas sanas y en los pacientes con deficiencia de la hormona de crecimiento. Una dosis subcutánea de 5 mg de Omnitrope 5 mg/1,5 ml solución inyectable en los adultos sanos lleva a unos valores plasmáticos de Cmax de 72 ± 28 microgramos/l, y de tmax, de
4,0 ± 2,0 horas.
Eliminación
La media de la vida media terminal de la somatropina después de la administración intravenosa en los pacientes adultos con deficiencia de la hormona de crecimiento es de aproximadamente 0,4 horas. Sin embargo, después de la administración subcutánea de Omnitrope 5 mg/1,5 ml solución inyectable, se alcanza una vida media de 3 horas. Es probable que la diferencia observada se deba a la lenta absorción de la inyección después de la administración subcutánea.
Poblaciones especiales
La biodisponibilidad absoluta de somatropina parece ser similar tanto en los hombres como en las mujeres después de la administración subcutánea.
La información sobre las propiedades farmacocinéticas de somatropina en geriatría y pediatría, en diferentes razas y en los pacientes con insuficiencia renal, hepática o cardiaca es o bien escasa o incompleta.
5.3 Datos preclínicos sobre seguridad
En los estudios con Omnitrope referentes a toxicidad subaguda y tolerancia local no se han observado casos con efectos clínicamente relevantes.
En otros estudios con somatropina referente a toxicidad general, tolerancia local y reproducción de la toxicidad no se han observado efectos clínicamente relevantes.
Con somatropina, los estudios de genotoxicidad in vitro e in vivo o de mutaciones genéticas e inducción de aberraciones cromosómicas han sido negativos.
Se ha observado un aumento de fragilidad cromosómica en un estudio in vitro con linfocitos, tomados de pacientes en tratamiento prolongado con somatropina y bajo la adición de bleomicina, un medicamento radiomimético. El significado clínico de esta observación no está claro.
En otro estudio con somatropina, no se encontró incremento de anomalías cromosómicas en linfocitos de pacientes que recibieron terapia con somatropina durante largo tiempo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Fosfato hidrógeno disódico heptahidrato Fosfato dihidrógeno sódico dihidrato Manitol
Poloxámero 188 Alcohol bencílico
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
Periodo de validez después del primer uso
Después del primer uso, el cartucho debe permanecer en la pluma y debe conservarse en una nevera (entre 2°C y 8°C) durante un máximo de 28 días. Conservar y transportar refrigerado (entre 2°C y 8°C). No congelar. Conservar en la pluma original para protegerlo de la luz.
6.4 Precauciones especiales de conservación
Cartucho sin abrir
Conservar y transportar refrigerado (entre 2°C y 8°C). No congelar. Conservar en el embalaje original para protegerlo de la luz.
Para las condiciones de conservación del medicamento en uso, ver sección 6.3.
6.5 Naturaleza y contenido del envase
1.5 ml de solución en un cartucho (de vidrio de tipo I), con émbolo en un lado (de bromobutilo siliconizado), un disco (de bromobutilo) y una cápsula de cierre (de aluminio) en el otro lado.
Paquetes de 1, 5 y 10 cartuchos.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Omnitrope 5 mg/1,5 ml solución inyectable es una solución estéril y lista para usar, para inyección subcutánea, cargada en un cartucho de vidrio.
Esta presentación está pensada para varios usos. Sólo debería administrarse con Omnitrope Pen 5, un dispositivo de inyección específicamente desarrollado para usarse con Omnitrope 5 mg/1,5 ml solución inyectable. Se administrará utilizando agujas para inyección de pluma estériles y desechables. Los pacientes y los cuidadores han de recibir del médico o de otro profesional de la salud formación e instrucciones adecuadas sobre el uso correcto de los cartuchos y de la pluma Omnitrope.
Lo que sigue es una descripción general del proceso de administración. Deben seguirse las instrucciones del fabricante para cargar el cartucho, conectar la aguja para inyección y para la administración.
1. Las manos deben lavarse.
2. Si la solución está turbia o si contiene material en partículas, no debe utilizarse. El contenido debe ser cristalino e incoloro.
3. Desinfectar la membrana de goma del cartucho con una torunda para limpiar.
4. Introducir el cartucho en la pluma Omnitrope Pen 5, según las instrucciones de uso
suministradas con la pluma.
5. Limpiar el lugar de aplicación de la inyección con una torunda empapada en alcohol.
6. Administrar la dosis adecuada mediante inyección subcutánea, utilizando la aguja de pluma estéril. Extraer la aguja de la pluma y eliminarla con arreglo a los requisitos locales.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Austria
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/004
EU/1/06/332/005
EU/1/06/332/006
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 12 de abril de 2006 Fecha de la última renovación: 12 de abril de 2011
10. FECHA DE LA REVISIÓN DEL TEXTO
<{MM/AAAA}>
1. NOMBRE DEL MEDICAMENTO
Omnitrope 10 mg/1,5 ml solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada ml de solución contiene 6,7 mg de somatropina* (que corresponden a 20 UI). Un cartucho contiene 1,5 ml, que corresponde a 10 mg de somatropina* (30 UI).
* Producida en Escherichia coli mediante tecnología de DNA recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable La solución es límpida e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lactantes, niños y adolescentes:
- Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento (deficiencia de la hormona del crecimiento, DGH).
- Trastorno del crecimiento asociado al síndrome de Turner.
- Trastorno del crecimiento asociado a insuficiencia renal crónica.
- Trastorno del crecimiento (puntuación de la desviación estándar actual de la talla (SDS) < -2,5 y
SDS de la talla < -1 ajustada para los padres) en los niños y los adolescentes nacidos con baja
talla para su edad gestacional (PEG), con un peso al nacer y/o longitud por debajo de -
2 desviaciones estándar (DE), que no alcanzaron el estirón de crecimiento (velocidad de crecimiento (VC) SDS < 0 durante el último año) a los 4 años de edad o más.
- El síndrome de Prader-Willi (SPW) para la mejoría del crecimiento y composición corporal. El diagnóstico de SPW debe ser confirmado por pruebas genéticas apropiadas.
Adultos
- Terapia de sustitución en los adultos con deficiencia pronunciada de la hormona del crecimiento.
- Inicio en la edad adulta: Pacientes que padecen una deficiencia grave de la hormona del crecimiento, asociada a múltiples deficiencias hormonales a consecuencia de una patología hipotalámica o hipofisaria conocida y que padecen, por lo menos, una deficiencia conocida de una hormona hipofisaria que no sea la prolactina. Estos pacientes deben someterse a una prueba dinámica adecuada, para así diagnosticar o excluir una deficiencia de la hormona del crecimiento.
- Inicio en la niñez: Pacientes que padecían una deficiencia de la hormona del crecimiento en la niñez a consecuencia de causas congénitas, genéticas, adquiridas o idiopáticas. En los pacientes con DGH de inicio en la niñez se debe reevaluar la capacidad secretora de la hormona del crecimiento después de concluir su crecimiento longitudinal. En los pacientes con una probabilidad alta de DGH persistente, es decir, una causa congénita o una DGH secundaria a una enfermedad o lesión hipotálamo-hipofisaria, un SDS < -2 del factor de crecimiento de tipo insulínico I (IGF-I) cuando no reciben tratamiento con hormona de crecimiento por lo menos durante cuatro semanas debe considerarse una prueba suficiente de DGH profunda.
Todos los demás pacientes requerirán un análisis del IGF-I y una prueba de estimulación con la hormona del crecimiento.
4.2 Posología y forma de administración
El diagnóstico y el tratamiento con somatropina deben iniciarse y monitorizarse por médicos que tengan la capacitación y la experiencia adecuadas en el diagnóstico y el tratamiento de los pacientes con trastornos del crecimiento.
Posología
Población pediátrica
La posología y la pauta de dosificación deben individualizarse.
Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento en los pacientes pediátricos:
En términos generales, se recomienda una dosis de 0,025 a 0,035 mg/kg de peso corporal por día o de 0,7 a 1,0 mg/m2 de superficie corporal por día. Incluso dosis más altas han sido utilizadas.
Cuando la DGH de inicio en la niñez persiste hasta la adolescencia, se debe continuar el tratamiento a fin de alcanzar un desarrollo somático completo (p. ej., composición corporal, masa ósea). Para la monitorización, el logro de una masa ósea máxima normal, definida como una puntuación T > -1 (es decir, normalizado hasta la masa ósea máxima promedio en el adulto, determinada mediante absorciometría de rayos X de doble energía, teniendo en cuenta el sexo y el origen étnico del paciente) es uno de los objetivos terapéuticos durante el período de transición. Para la orientación en cuanto a la posología, ver el apartado sobre los adultos, a continuación.
Síndrome de Prader-Willi, para la mejoría del crecimiento y composición corporal en los pacientes pediátricos
En general, se recomienda una dosis de 0,035 mg/kg de peso corporal por día ó 1,0 mg/m2 de superficie corporal por día. No se excederán las dosis diarias de 2,7 mg. El tratamiento no se utilizará en los pacientes pediátricos con una velocidad de crecimiento inferior a 1 cm al año y cerca del cierre de las epífisis.
Trastorno del crecimiento debido al síndrome de Turner
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal al día ó 1,4 mg/m2 de superficie corporal.
Trastorno del crecimiento en la insuficiencia renal crónica
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal y día (de 1,4 mg/m2 de superficie corporal y día). Pueden precisarse dosis más altas si la velocidad de crecimiento es muy lenta. Puede ser necesario corregir la dosis después de seis meses de tratamiento (ver sección 4.4).
Trastorno del crecimiento en los niños y los adolescentes nacidos con baja talla para su edad gestacional (PEG)
Se recomienda una dosis de 0,035 mg/kg de peso corporal al día (1 mg/m2 de superficie corporal al día) hasta que se alcance la talla final (ver sección 5.1). El tratamiento se debe interrumpir después del primer año de tratamiento, si la SDS de la velocidad de crecimiento es inferior a + 1. El tratamiento se interrumpirá si la velocidad de crecimiento es < 2 cm/año y, si se requiere confirmación, la edad ósea es > 14 años (niñas) o > 16 años (niños), correspondiente con el cierre de las placas de crecimiento epifisario.
Posología recomendada en pacientes pediátricos
|
Indicación |
Dosis diaria en mg/kg de peso corporal |
Dosis diaria en mg/m2 de superficie corporal |
|
Deficiencia de la hormona del crecimiento |
0,025 - 0,035 |
0,7 - 1,0 |
|
Síndrome de Prader-Willi |
0,035 |
1,0 |
|
Síndrome de Turner |
0,045 - 0,050 |
1,4 |
|
Insuficiencia renal crónica |
0,045 - 0,050 |
1,4 |
|
Niños y adolescentes nacidos con talla baja para su edad gestacional (PEG) |
0,035 |
1,0 |
Pacientes adultos con deficiencia de la hormona del crecimiento
La dosis recomendada para reanudar el tratamiento en aquellos pacientes que continúen con hormona de crecimiento tras un déficit de GH en la infancia, es de 0,2-0,5 mg por día. La dosis se debe ir incrementando o disminuyendo gradualmente de acuerdo a las necesidades individuales del paciente, determinadas de acuerdo a la concentración del IGF-I.
En los pacientes con DGH de inicio en la edad adulta, el tratamiento debe iniciarse con una dosis baja, de 0,15 a 0,3 mg al día. La dosis se aumentará gradualmente según las necesidades de cada paciente y según lo determine la concentración de IGF-I.
En ambos casos, el objetivo del tratamiento debe ser alcanzar concentraciones de factor de crecimiento de tipo insulina (IGF-I) dentro de 2 SDS de la media corregida por la edad. A los pacientes con concentraciones IGF-I normales al inicio del tratamiento se les debe administrar la hormona del crecimiento hasta una concentración de IGF-I dentro de los límites superiores normales, sin sobrepasar 2 SDS. La respuesta clínica y los efectos adversos pueden también usarse como guía para el ajuste de la dosis. Se conoce que hay pacientes con DGH que no llegan a normalizar los niveles de IGF-I a pesar de tener una buena respuesta clínica, y por tanto, no requieren ajuste de dosis. La dosis de mantenimiento raramente excede 1,0 mg al día. Las mujeres pueden necesitar dosis más altas que los hombres, mientras que los varones muestran una sensibilidad mayor al aumento de la IGF-I con el tiempo. Esto significa que hay un riesgo de que las mujeres, en especial aquellas en sustitución con estrógenos orales, estén infra-tratadas mientras que los hombres estén sobre-tratados. La exactitud de la dosis de la hormona del crecimiento debe, por lo tanto, controlarse cada 6 meses. Habida cuenta de que la producción fisiológica de la hormona del crecimiento desciende con la edad, pueden reducirse las necesidades de la dosis.
Poblaciones especiales
Personas de edad avanzada
En los pacientes mayores de 60 años, el tratamiento debe comenzar con una dosis de 0,1 a 0,2 mg diarios, y debe aumentarse gradualmente, según las necesidades individuales del paciente. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento en estos pacientes rara vez es superior a 0,5 mg diarios.
Forma de administración
Se debe administrar la inyección por vía subcutánea, cambiando el lugar de inyección para evitar la lipoatrofia.
Para consultar las instrucciones de uso y manipulación, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes.
La somatropina no debe usarse cuando exista evidencia de actividad de un tumor. Los tumores intracraneales deben ser inactivos y el tratamiento antitumoral se debe completar antes de iniciar el tratamiento con GH. El tratamiento se debe suspender si hay pruebas de crecimiento tumoral.
La somatropina no debe usarse en la promoción del crecimiento de niños con epífisis cerradas.
Los pacientes con enfermedad crítica aguda que presenten complicaciones después de cirugía cardiaca, abdominal, traumatismo múltiple por accidente, insuficiencia respiratoria aguda o enfermedades similares, no deben ser tratados con somatropina (en cuanto a los pacientes sometidos a tratamiento de sustitución, ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
No se debe superar la dosis diaria máxima recomendada (ver sección 4.2).
Sensibilidad a la insulina
La somatropina puede reducir la sensibilidad a la insulina. En pacientes con diabetes mellitus, puede ser necesario ajustar la dosis de insulina tras la instauración del tratamiento con somatropina. Durante el tratamiento con somatropina, se deben monitorizar cuidadosamente los pacientes con diabetes, intolerancia a la glucosa o con factores de riesgo adicionales de sufrir diabetes.
Función tiroidea
La hormona de crecimiento aumenta la conversión extratiroidea de T4 a T3, lo que puede causar una reducción de la concentración T4 y un aumento de la T3 en el suero. Si bien las concentraciones de la hormona tiroidea periférica han permanecido dentro de los límites de referencia en la mayoría de sujetos sanos, teóricamente, se puede desarrollar hipotiroidismo en los sujetos con hipotiroidismo subclínico. En consecuencia, se debe llevar a cabo una monitorización de la función tiroidea en todos los pacientes. El potencial efecto del tratamiento con hormona de crecimiento sobre la función tiroidea se debe monitorizar cuidadosamente en pacientes con hipopituitarismo tratados con terapia sustitutiva estándar.
En la deficiencia de la hormona del crecimiento, secundaria al tratamiento de una neoplasia maligna, se recomienda prestar atención a los signos de recaída de la malignidad. Entre los supervivientes de cáncer pediátrico se ha referido un mayor riesgo de una segunda neoplasia en los pacientes tratados con somatropina después de su primera neoplasia. Las más frecuentes de estas segundas neoplasias en los pacientes tratados con radioterapia en la cabeza para su primera neoplasia fueron los tumores intracraneales, en concreto los meningiomas.
En los pacientes con trastornos endocrinos, incluida la deficiencia de la hormona del crecimiento, el deslizamiento de la epífisis de la cadera puede producirse con más frecuencia que en la población general. Los pacientes que cojean durante el tratamiento con somatropina deben ser controlados clínicamente.
Hipertensión intracraneal benigna
En casos de cefalea recurrente o grave, problemas visuales, náuseas y/o vómitos, se recomienda practicar una fundoscopia para descartar un edema de papila. Si se confirma el edema de papila, debe considerarse una hipertensión intracraneal benigna y, si fuese apropiado, debe de retirarse el tratamiento con la hormona del crecimiento. Si se reinicia el tratamiento con hormona del crecimiento, se debe instaurar un control cuidadoso para, si fuese necesario, detectar la hipertensión intracraneal.
Leucemia
Se ha notificado leucemia en un pequeño número de pacientes con deficiencia de la hormona de crecimiento, algunos de los cuales han sido tratados con somatropina. Sin embargo, no existe evidencia de que haya un aumento en la incidencia de la leucemia en pacientes tratados con hormona del crecimiento sin factores de predisposición.
Anticuerpos
En un pequeño porcentaje de pacientes pueden aparecer anticuerpos dirigidos contra Omnitrope. Omnitrope ha dado lugar a la formación de anticuerpos aproximadamente en el 1% de los pacientes. La capacidad de fijación de estos anticuerpos es baja y no hay ningún efecto sobre la velocidad de crecimiento. En cualquier paciente con una falta de respuesta por lo demás no explicada se deben hacer análisis de anticuerpos contra la somatropina.
Pacientes de edad avanzada
La experiencia con los pacientes mayores de 80 años es limitada. Los pacientes de edad avanzada pueden ser más sensibles a la acción de Omnitrope y, por lo tanto, pueden ser más propensos a presentar reacciones adversas.
Enfermedad agudas graves
Los efectos de la somatropina sobre la recuperación de pacientes adultos críticos se han evaluado en dos ensayos controlados con placebo en 522 pacientes adultos con complicaciones posteriores a cirugía a corazón abierto, cirugía abdominal, traumatismo accidental múltiple o insuficiencia respiratoria aguda. La mortalidad fue superior en los pacientes tratados diariamente con 5,3 u 8 mg de somatropina, en comparación con los pacientes que recibieron placebo, 42% vs. 19%. En base a esta información, este tipo de pacientes no deben ser tratados con somatropina. Dado que no existe información disponible sobre la seguridad del tratamiento sustitutivo con hormona de crecimiento en pacientes críticos, los beneficios del tratamiento continuado en esta situación deben considerarse sobre la base de los riesgos potenciales.
En todos los pacientes que desarrollen algún otro tipo de enfermedad aguda en fase crítica o similar, el posible beneficio del tratamiento con somatropina deberá ser sopesado en relación con el riesgo potencial que implica.
Población pediátrica
Pancreatitis
A pesar de que la pancreatitis es rara, debe ser considerada en niños tratados con somatropina que presenten dolor abdominal.
Síndrome de Prader-Willi
En los pacientes con SPW, el tratamiento debe ser siempre en combinación con una dieta baja en calorías.
Existen informes de muertes asociadas al uso de la hormona del crecimiento en los pacientes pediátricos con SPW que presentaron uno o más factores de riesgo: obesidad grave (aquellos pacientes con un peso/altura superior al 200%), antecedentes de insuficiencia respiratoria o apnea del sueño o de infección respiratoria no identificada. Los pacientes con SPW y uno o más de estos factores de riesgo podrían estar en mayor riesgo.
Antes de iniciar el tratamiento con somatropina los pacientes con SPW serán valorados respecto de la obstrucción de las vías aéreas altas, apnea del sueño o infección respiratoria.
Si, durante la evaluación de la obstrucción de las vías respiratorias altas, se observan resultados anatomopatológicos, se debe derivar al niño al otorrinolaringólogo (ENT) para el tratamiento y la resolución del trastorno respiratorio antes de iniciar el tratamiento con hormona de crecimiento.
La apnea del sueño debe evaluarse antes del inicio del tratamiento con hormona de crecimiento por métodos reconocidos como la polisomnografía o la oximetría, durante la noche, y los pacientes deben controlarse, si se sospecha apnea del sueño.
Si, durante el tratamiento con somatropina, los pacientes muestran signos de obstrucción de las vías respiratorias altas (incluido el inicio o el aumento de ronquidos), el tratamiento debe interrumpirse y debe efectuarse una nueva evaluación de ENT.
Todos los pacientes con SPW deben valorarse en cuanto a la apnea y ser controlados si se sospecha la presencia de apnea del sueño. Los pacientes deben controlarse para detectar signos de infecciones respiratorias, que deben ser diagnosticadas lo más pronto posible y tratadas de forma contundente.
Todos los pacientes con SPW deben someterse a un control eficaz del peso antes y, también, durante el tratamiento con hormona del crecimiento.
La escoliosis es muy frecuente en los pacientes con SPW. La escoliosis puede progresar en cualquier niño durante el crecimiento rápido. Deben controlarse los signos de escoliosis durante el tratamiento.
La experiencia con el tratamiento prolongado en los adultos y en los pacientes con SPW es limitada.
Nacidos pequeños para su edad gestacional
En los niños y los adolescentes PEG nacidos bajos antes de iniciar el tratamiento se deben descartar otras razones médicas u otros tratamientos que pudieran explicar la perturbación del crecimiento antes de iniciar el tratamiento.
En los niños y los adolescentes PEG se recomienda medir la insulina y la glucosa sanguínea en ayunas antes de iniciar el tratamiento y, posteriormente, en períodos anuales. En los pacientes con un aumento del riesgo de diabetes mellitus (p. ej., antecedentes familiares de diabetes, obesidad, resistencia grave a la insulina, acantosis nigricans) debe realizarse la prueba de tolerancia a la glucosa (OGTT). Si aparece una diabetes clara, no se debe administrar hormona del crecimiento.
En los niños y los adolescentes PEG se recomienda medir la concentración de IGF-I antes de iniciar el tratamiento y después, dos veces al año. Si en mediciones repetidas las concentraciones de IGF-I exceden en +2 DS comparado con la edad de referencia y estado puberal, la proporción IGF-I/IGFBP-3 debe tenerse en cuenta para considerar un ajuste de la dosis.
La experiencia al iniciar el tratamiento en los pacientes PEG cerca del comienzo de la pubertad es limitada. Por tanto, no se recomienda iniciar el tratamiento cerca de la pubertad. La experiencia en los pacientes con síndrome de Silver-Rusell es limitada.
Parte del aumento de estatura obtenido al tratar a los niños y los adolescentes nacidos PEG y bajos con hormona del crecimiento puede perderse si se interrumpe el tratamiento antes de que se alcance la estatura final.
Insuficiencia renal crónica
En la insuficiencia renal crónica, la función renal debe ser un 50% de la normal antes de iniciar el tratamiento. Para verificar la perturbación del crecimiento, se debe hacer un seguimiento del crecimiento durante un año antes de iniciar el tratamiento. Durante este periodo, se debe iniciar un tratamiento conservador para la insuficiencia renal (incluido el control de la acidosis, el hiperparatiroidismo y el estado nutricional), y debe mantenerse durante todo el tratamiento.
El tratamiento se debe interrumpir en casos de trasplante renal.
Hasta la fecha, no se disponen de datos de la estatura final de los pacientes con insuficiencia renal crónica tratados con Omnitrope.
4.5 Interacción con otros medicamentos y otras formas de interacción
El tratamiento concomitante con glucocorticoides puede inhibir los efectos de promoción del crecimiento de los productos que contienen somatropina. Por tanto, los pacientes tratados con glucocorticoides deben tener su crecimiento cuidadosamente monitorizado con el fin de valorar el potencial impacto del tratamiento con glucocorticoides sobre el crecimiento.
Los datos de un estudio de interacción, realizado en los adultos con deficiencia de la hormona del crecimiento, sugieren que la administración de somatropina puede aumentar la eliminación de compuestos que son metabolizados por las isoenzimas del citocromo P450. El metabolismo de los compuestos metabolizados por el citocromo P450 3A4 (p. ej.: esteroides sexuales, corticoesteroides, anticonvulsivantes y ciclosporinas) puede aumentar, dando como resultado unas concentraciones plasmáticas más bajas de estos compuestos. Se desconoce la significación clínica de esto.
Ver también en la sección 4.4 la información relacionada con la diabetes mellitus y los trastornos tiroideos, y en la sección 4.2 la información sobre la terapia de sustitución de estrógenos orales.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos o éstos son limitados relativos al uso de somatropina en mujeres embarazadas. Los estudios realizados en animales son insuficientes en términos de toxicidad para la reproducción (ver sección 5.3). No se recomienda utilizar somatropina durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Lactancia
No se han llevado a cabo estudios clínicos con medicamentos que contienen somatropina en mujeres en periodo de lactancia. Se desconoce si la somatropina se excreta en la leche materna, pero es poco probable la absorción de proteína intacta del tubo digestivo del niño. Por tanto, se debe tener precaución cuando se administre Omnitrope a mujeres en periodo de lactancia.
Fertilidad
No se han realizado estudios de fertilidad con Omnitrope.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Omnitrope sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los pacientes que padecen una deficiencia de la hormona de crecimiento se caracterizan por un déficit del volumen extracelular. Al iniciar el tratamiento con somatropina, este déficit se corrige rápidamente. En los pacientes adultos, son frecuentes las reacciones adversas relacionadas con la retención de líquidos, como el edema periférico, la rigidez musculoesquelética, la artralgia, la mialgia y las parestesias. En general, estas reacciones son leves a moderadas, se producen en los primeros meses de tratamiento y remiten espontáneamente o al reducir la dosis.
La incidencia de estas reacciones adversas está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de hormona de crecimiento. En los niños, tales reacciones adversas son poco frecuentes.
Omnitrope ha dado lugar a la formación de anticuerpos en aproximadamente el 1% de los pacientes. La capacidad de fijación de estos anticuerpos ha sido baja y no hay cambios clínicos asociados a su formación (ver sección 4.4).
Tabla de reacciones adversas
Las tablas 1-6 muestran las reacciones adversas clasificadas según los apartados del sistema de clasificación de órganos y las frecuencias, utilizando la convención siguiente: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); no conocida (no puede estimarse a partir de los datos disponibles) para cada una de las afecciones indicadas.
Ensayos clínicos en niños con DGH
|
Tabla 1 Tratamiento a largo plazo de niños con alteración del crecimiento debida a una secreción insuficiente de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias7 Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia7 |
Mialgia7 Rigidez musculoesquelética7 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico7 | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre7 | |||||
Ensayos clínicos en niños con síndrome de Turner
|
Tabla 2 Tratamiento a largo plazo de niños con alteración del crecimiento debida al síndrome de Turner | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico* Reacción en el lugar de la inyección$ | |||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* | |||||
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con insuficiencia renal crónica
|
Tabla 3 | ||||||
|
Tratamiento a largo plazo de niños con alteración del crecimiento debida a una insuficiencia renal crónica | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas |
Leucemiaf | |||||
|
y no especificadas (incl. quistes y pólipos) | ||||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* Rigidez musculoesquelética* | |||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con PEG
|
Tabla 4 Tratamiento a largo plazo de niños con alteración del crecimiento debida al nacimiento con baja talla para su edad gestacional | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión | |||||
|
intracraneal benigna | ||||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con SPW
|
Tabla 5 Tratamiento a largo debida al síndrome ( |
plazo y mejora de la composición corporal en niños con alteración del crecimiento e Prader-Willi | |||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* |
Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de |
Edema periférico* |
Reacción en el lugar de la inyección$ | ||||
|
administración | ||||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre8 |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en adultos con DGH
|
Tabla 6 Terapia de sustitución en adultos con deficiencia de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo 2 | |||||
|
Trastornos del sistema nervioso |
Parestesias * Síndrome del túnel carpiano |
Hipertensión intracraneal benigna | ||||
|
Trastornos musculoesquelét icos y del tejido conjuntivo |
Artralgia8 |
Mialgia8 Rigidez musculoesq uelética8 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico8 |
Reacción en el lugar de la inyección$ | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre8 | |||||
Descripción de reacciones adversas seleccionadas Niveles reducidos de cortisol sérico
Se ha notificado que la somatropina reduce las concentraciones séricas de cortisol, posiblemente al afectar a las proteínas transportadoras o mediante un aumento de la depuración hepática. La relevancia clínica de estas observaciones puede ser limitada. No obstante, antes de iniciar el tratamiento, debe optimizarse el tratamiento de reemplazo con corticoesteroides.
Síndrome de Prader-Willi
Se han notificado casos raros de muerte súbita en pacientes con síndrome de Prader-Willi tratados con somatropina en la experiencia post-comercialización, aunque no se ha demostrado que exista relación causal.
Leucemia
Se han notificado casos (raros o muy raros) de leucemia en los niños que padecen una deficiencia de la hormona de crecimiento y son tratados con somatropina e incluidos en la experiencia post-comercialización. Sin embargo, no existen indicios de un aumento del riesgo de leucemia sin factores predisponentes, tales como la radioterapia cerebral o de la cabeza.
Epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes
Se han notificado epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes en niños tratados con la hormona del crecimiento. La epifisiólisis de la cabeza femoral se produce con mayor frecuencia en caso de trastornos endocrinos y la enfermedad de Legg-Calvé-Perthes es más frecuente en caso de baja estatura. No obstante, se desconoce si estas dos afecciones aparecen o no con mayor frecuencia durante el tratamiento con somatropina. Debe tenerse en cuenta la posibilidad de su diagnóstico en niños que presenten molestias o dolor en la cadera o la rodilla.
Otras reacciones adversas al medicamento
Otras reacciones adversas al medicamento pueden considerarse efectos de clase de la somatropina, tales como una posible hiperglucemia causada por la disminución de la sensibilidad a la insulina, la disminución de la concentración de tiroxina libre y a una hipertensión intracraneal benigna.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Síntomas:
La sobredosis aguda puede conducir inicialmente a hipoglucemia y posteriormente a hiperglucemia.
La sobredosis prolongada puede causar signos y síntomas compatibles con los efectos conocidos de exceso de la hormona del crecimiento.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: hormonas del lóbulo anterior de la hipófisis y sus análogos, código ATC: H01AC01.
Omnitrope es un medicamento biosimilar. La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Mecanismo de acción
La somatropina es una potente hormona metabólica, importante en el metabolismo de lípidos, carbohidratos y proteínas. En los niños con hormona del crecimiento endógena insuficiente, la somatropina estimula el crecimiento lineal y aumenta la tasa de crecimiento. En los adultos y también en los niños, la somatropina mantiene una composición corporal normal aumentando la retención de nitrógeno y la estimulación del crecimiento del músculo esquelético y la movilización de la grasa corporal. El tejido adiposo visceral responde en particular a la somatropina. Además, para mejorar la lipólisis, la somatropina reduce la captación de triglicéridos de los depósitos de grasa corporal. Las concentraciones séricas de IGF-I (factor de crecimiento de tipo insulina I) y la IGFBP3 (proteína de unión del factor de crecimiento de tipo insulina 3) son aumentadas por la somatropina. Además, se han demostrado las siguientes acciones.
Efectos farmacodinámicos
Metabolismo lipídico
La somatropina induce los receptores de colesterol LDL hepáticos y afecta el perfil de lípidos séricos y las lipoproteínas. En general, la administración de somatropina a pacientes deficitarios de la hormona del crecimiento resulta en una reducción del DLD sérico y de la apoliproteína B. También se puede observar una reducción del colesterol total.
Metabolismo de los carbohidratos
La somatropina aumenta la insulina, pero la glucosa sanguínea en ayunas permanece, por lo general, sin cambios. Los niños con hipopituitarismo experimentan hipoglucemia en ayunas. Esta condición es corregida por la somatropina.
Metabolismo del agua y de los minerales
La deficiencia de la hormona del crecimiento se asocia a una reducción de los volúmenes plasmático y extracelular. Ambos aumentan rápidamente tras el tratamiento con somatropina. La somatropina induce la retención de sodio, potasio y fósforo.
Metabolismo óseo
La somatropina estimula el recambio del hueso esquelético. La administración a largo plazo de somatropina a pacientes con osteopenia con deficiencia de la hormona del crecimiento resulta en un aumento del contenido mineral y de la densidad ósea en los lugares portadores de peso.
Capacidad _ física
Mejoría de la potencia muscular y del ejercicio físico tras un tratamiento a largo plazo con somatropina. La somatropina también aumenta la frecuencia cardíaca, pero el mecanismo de acción no ha sido aún aclarado. Puede que una reducción de la resistencia vascular periférica contribuya a este efecto.
Eficacia clínica y seguridad
En ensayos clínicos en los niños y los adolescentes PEG y de talla baja se utilizaron dosis de 0,033 y 0,067 mg/kg de peso corporal, por día hasta alcanzar la altura final. En 56 pacientes tratados de forma continuada y que alcanzaron (casi) la altura final, el cambio principal de talla al inicio del tratamiento fue de +1,90 SDS (0,033 mg/kg de peso corporal por día) y +2,19 SDS (0,067 mg/kg de peso corporal por día). Los datos publicados de niños y adolescentes PEG no tratados sin estirón espontáneo y temprano sugieren un crecimiento tardío, de la SDS de 0,5. Los datos de seguridad a largo plazo son todavía limitados.
5.2 Propiedades farmacocinéticas
Absorción
La biodisponibilidad de la somatropina, administrada subcutáneamente, es aproximadamente del 80% en ambos casos, en las personas sanas y en los pacientes con deficiencia de la hormona de crecimiento. Una dosis subcutánea de 5 mg de Omnitrope 10 mg/1,5 ml solución inyectable en los adultos sanos produce unos valores plasmáticos de Cmax de 74 ± 22 microgramos/l, y de tmax, de
3,9 ± 1,2 horas.
Eliminación
La media de la vida media terminal de somatropina después de la administración intravenosa en los pacientes adultos con deficiencia de la hormona de crecimiento es de aproximadamente 0,4 horas. Sin embargo, después de la administración subcutánea de Omnitrope 10 mg/1,5 ml solución inyectable se alcanza una vida media de 3 horas. Es probable que la diferencia observada se deba a la lenta absorción de la inyección después de la administración subcutánea.
Poblaciones especiales
La biodisponibilidad absoluta de somatropina parece ser similar tanto en los hombres como en las mujeres después de la administración subcutánea.
La información sobre las propiedades farmacocinéticas de la somatropina en geriatría y pediatría, en diferentes razas y en los pacientes con insuficiencia renal, hepática o cardiaca es o bien escasa o incompleta.
5.3 Datos preclínicos sobre seguridad
En los estudios con Omnitrope referentes a toxicidad subaguda y tolerancia local no se han observado casos con efectos clínicamente relevantes.
En otros estudios con somatropina referente a toxicidad general, tolerancia local y reproducción de la toxicidad no se han observado efectos clínicamente relevantes.
Con somatropina, los estudios de genotoxicidad in vitro e in vivo o de mutaciones genéticas e inducción de aberraciones cromosómicas han sido negativos.
Se ha observado un aumento de fragilidad cromosómica en un estudio in vitro con linfocitos, tomados de pacientes en tratamiento prolongado con somatropina y bajo la adición de bleomicina, un medicamento radiomimético. El significado clínico de esta observación no está claro.
En otro estudio con somatropina, no se encontró incremento de anomalías cromosómicas en linfocitos de pacientes que recibieron terapia con somatropina durante largo tiempo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Fosfato hidrógeno disódico heptahidrato Fosfato dihidrógeno sódico dihidrato Glicina
Poloxámero 188 Fenol
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 18 meses.
Periodo de validez después del primer uso
Después del primer uso, el cartucho debe permanecer en la pluma y debe conservarse en una nevera (entre 2°C y 8°C) durante un máximo de 28 días. Conservar y transportar refrigerado (entre 2°C y 8°C).No congelar. Conservar en la pluma original para protegerlo de la luz.
6.4 Precauciones especiales de conservación
Cartucho sin abrir
Conservar y transportar refrigerado (entre 2°C y 8°C). No congelar. Conservar en el embalaje original para protegerlo de la luz.
Para las condiciones de conservación del medicamento en uso, ver sección 6.3.
6.5 Naturaleza y contenido del envase
1.5 ml de solución en un cartucho (de vidrio incoloro tipo I), con un émbolo en un lado (de bromobutilo siliconizado) un disco (de bromobutilo) y una cápsula de cierre (de aluminio) en el otro. Envases de 1, 5 y 10 cartuchos.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Omnitrope 10 mg/1,5 ml solución inyectable es una solución estéril y lista para usar que se presenta en un cartucho de vidrio destinado para inyección subcutánea,
Este cartucho está previsto para varios usos. Sólo debe administrarse con Omnitrope Pen 10, un dispositivo de inyección específicamente desarrollado para la administración de Omnitrope 10 mg/1,5 ml solución inyectable. Se administrará utilizando agujas para inyección de pluma estériles y desechables. Los pacientes y los cuidadores deben recibir la formación e instrucciones adecuadas sobre el correcto uso de los cartuchos y de la pluma Omnitrope del médico o de otro profesional de la salud.
Lo que sigue es una descripción general del proceso de administración. Deben seguirse las instrucciones del fabricante para cargar el cartucho, conectar la aguja para inyección y para la administración.
1. Las manos deben lavarse.
2. Si la solución está turbia o si contiene material en partículas, no debe utilizarse. El contenido debe ser cristalino e incoloro.
3. Desinfectar la membrana de goma del cartucho con una torunda para limpiar.
4. Introducir el cartucho en la Omnitrope Pen 10, según las instrucciones de uso
suministradas con la pluma.
5. Limpiar el lugar de aplicación de la inyección con una torunda empapada en alcohol.
6. Administrar la dosis adecuada mediante inyección subcutánea, utilizando la aguja de pluma estéril. Extraer la aguja de la pluma y eliminarla con arreglo a los requisitos locales.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl
Austria
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/007
EU/1/06/332/008
EU/1/06/332/009
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA
AUTORIZACIÓN
Fecha de la primera autorización: 12 de abril de 2006 Fecha de la última renovación: 12 de abril de 2011
10. FECHA DE LA REVISIÓN DEL TEXTO
<{MM/AAAA}>
1. NOMBRE DEL MEDICAMENTO
Omnitrope 5 mg/1,5 ml solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada ml de solución contiene 3,3 mg de somatropina* (que corresponde a 10 UI). Un cartucho contiene 1,5 ml, que corresponde a 5 mg de somatropina* (15 UI).
* Producida en Escherichia coli mediante tecnología de DNA recombinante.
Excipiente(s) con efecto conocido:
Un ml contiene 9 mg de alcohol bencílico.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable en un cartucho para SurePal 5. La solución es límpida e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lactantes, niños y adolescentes:
- Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento (deficiencia de la hormona del crecimiento, DGH).
- Trastorno del crecimiento asociado al síndrome de Turner.
- Trastorno del crecimiento asociado a insuficiencia renal crónica.
- Trastorno del crecimiento (puntuación de la desviación estándar actual de la talla (SDS) < -2,5 y
SDS de la talla < -1 ajustada para los padres) en los niños y los adolescentes nacidos con baja talla para su edad gestacional (PEG), con un peso al nacer y/o longitud por debajo de -
2 desviaciones estándar (DE), que no alcanzaron el estirón de crecimiento (velocidad de crecimiento (VC) SDS < 0 durante el último año) a los 4 años de edad o más.
- El síndrome de Prader-Willi (SPW) para la mejoría del crecimiento y composición corporal. El diagnóstico de SPW debe ser confirmado por pruebas genéticas apropiadas.
Adultos
- Terapia de sustitución en los adultos con deficiencia pronunciada de la hormona del crecimiento.
- Inicio en la edad adulta: Pacientes que padecen una deficiencia grave de la hormona del crecimiento, asociada a múltiples deficiencias hormonales a consecuencia de una patología hipotalámica o hipofisaria conocida y que padecen, por lo menos, una deficiencia conocida de una hormona hipofisaria que no sea la prolactina. Estos pacientes deben someterse a una prueba dinámica adecuada, para así diagnosticar o excluir una deficiencia de la hormona del crecimiento.
- Inicio en la niñez: Pacientes que padecían una deficiencia de la hormona del crecimiento en la niñez a consecuencia de causas congénitas, genéticas, adquiridas o idiopáticas. En los pacientes con DGH de inicio en la niñez se debe reevaluar la capacidad secretora de la hormona del crecimiento después de concluir su crecimiento longitudinal. En los pacientes con una probabilidad alta de DGH persistente, es decir, una causa congénita o una DGH secundaria a una enfermedad o lesión hipotálamo-hipofisaria, un SDS < -2 del factor de crecimiento de tipo insulínico I (IGF-I) cuando no reciben tratamiento con hormona de crecimiento por lo menos durante cuatro semanas debe considerarse una prueba suficiente de DGH profunda.
Todos los demás pacientes requerirán un análisis del IGF-I y una prueba de estimulación con la hormona del crecimiento.
4.2 Posología y forma de administración
El diagnóstico y el tratamiento con somatropina deben iniciarse y monitorizarse por médicos que tengan la capacitación y la experiencia adecuadas en el diagnóstico y el tratamiento de los pacientes con trastornos del crecimiento.
Posología
Población _ pediátrica
La posología y la pauta de dosificación deben individualizarse.
Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento en los pacientes pediátricos:
En términos generales, se recomienda una dosis de 0,025 a 0,035 mg/kg de peso corporal por día o de 0,7 a 1,0 mg/m2 de superficie corporal por día. Incluso dosis más altas han sido utilizadas.
Cuando la DGH de inicio en la niñez persiste hasta la adolescencia, se debe continuar el tratamiento a fin de alcanzar un desarrollo somático completo (p. ej., composición corporal, masa ósea). Para la monitorización, el logro de una masa ósea máxima normal, definida como una puntuación T > -1 (es decir, normalizado hasta la masa ósea máxima promedio en el adulto, determinada mediante absorciometría de rayos X de doble energía, teniendo en cuenta el sexo y el origen étnico del paciente) es uno de los objetivos terapéuticos durante el período de transición. Para la orientación en cuanto a la posología, ver el apartado sobre los adultos, a continuación.
Síndrome de Prader-Willi, para la mejoría del crecimiento y composición corporal en los pacientes pediátricos
En general, se recomienda una dosis de 0,035 mg/kg de peso corporal por día ó 1,0 mg/m2 de superficie corporal por día. No se excederán las dosis diarias de 2,7 mg. El tratamiento no se utilizará en los pacientes pediátricos con una velocidad de crecimiento inferior a 1 cm al año y cerca del cierre de las epífisis.
Trastorno del crecimiento debido al síndrome de Turner
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal al día ó 1,4 mg/m2 de superficie corporal.
Trastorno del crecimiento en la insuficiencia renal crónica
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal y día (de 1,4 mg/m2 de superficie corporal y día). Pueden precisarse dosis más altas si la velocidad de crecimiento es muy lenta. Puede ser necesario corregir la dosis después de seis meses de tratamiento (ver sección 4.4).
Trastorno del crecimiento en los niños y los adolescentes nacidos con baja talla para su edad gestacional (PEG)
Se recomienda una dosis de 0,035 mg/kg de peso corporal al día (1 mg/m2 de superficie corporal al día) hasta que se alcance la talla final (ver sección 5.1). El tratamiento se debe interrumpir después del primer año de tratamiento, si la SDS de la velocidad de crecimiento es inferior a + 1. El tratamiento se interrumpirá si la velocidad de crecimiento es < 2 cm/año y, si se requiere confirmación, la edad ósea es > 14 años (niñas) o > 16 años (niños), correspondiente con el cierre de las placas de crecimiento epifisario.
Posología recomendada en pacientes pediátricos
|
Indicación |
Dosis diaria en mg/kg de peso corporal |
Dosis diaria en mg/m2 de superficie corporal |
|
Deficiencia de la hormona del crecimiento |
0,025 - 0,035 |
0,7 - 1,0 |
|
Síndrome de Prader-Willi |
0,035 |
1,0 |
|
Síndrome de Turner |
0,045 - 0,050 |
1,4 |
|
Insuficiencia renal crónica |
0,045 - 0,050 |
1,4 |
|
Niños y adolescentes nacidos con talla baja para su edad gestacional (PEG) |
0,035 |
1,0 |
Pacientes adultos con deficiencia de la hormona del crecimiento
La dosis recomendada para reanudar el tratamiento en aquellos pacientes que continúen con hormona de crecimiento tras un déficit de GH en la infancia, es de 0,2-0,5 mg por día. La dosis se debe ir incrementando o disminuyendo gradualmente de acuerdo a las necesidades individuales del paciente, determinadas de acuerdo a la concentración del IGF-I.
En los pacientes con DGH de inicio en la edad adulta, el tratamiento debe iniciarse con una dosis baja, de 0,15 a 0,3 mg al día. La dosis se aumentará gradualmente según las necesidades de cada paciente y según lo determine la concentración de IGF-I.
En ambos casos, el objetivo del tratamiento debe ser alcanzar concentraciones de factor de crecimiento de tipo insulina (IGF-I) dentro de 2 SDS de la media corregida por la edad. A los pacientes con concentraciones IGF-I normales al inicio del tratamiento se les debe administrar la hormona del crecimiento hasta una concentración de IGF-I dentro de los límites superiores normales, sin sobrepasar 2 SDS. La respuesta clínica y los efectos adversos pueden también usarse como guía para el ajuste de la dosis. Se conoce que hay pacientes con DGH que no llegan a normalizar los niveles de IGF-I a pesar de tener una buena respuesta clínica, y por tanto, no requieren ajuste de dosis. La dosis de mantenimiento raramente excede 1,0 mg al día. Las mujeres pueden necesitar dosis más altas que los hombres, mientras que los varones muestran una sensibilidad mayor al aumento de la IGF-I con el tiempo. Esto significa que hay un riesgo de que las mujeres, en especial aquellas en sustitución con estrógenos orales, estén infra-tratadas mientras que los hombres estén sobre-tratados. La exactitud de la dosis de la hormona del crecimiento debe, por lo tanto, controlarse cada 6 meses. Habida cuenta de que la producción fisiológica de la hormona del crecimiento desciende con la edad, pueden reducirse las necesidades de la dosis.
Poblaciones especiales
Personas de edad avanzada
En los pacientes mayores de 60 años, el tratamiento debe comenzar con una dosis de 0,1 a 0,2 mg diarios, y debe aumentarse gradualmente, según las necesidades individuales del paciente. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento en estos pacientes rara vez es superior a 0,5 mg diarios.
Forma de administración
Se debe administrar la inyección por vía subcutánea, cambiando el lugar de inyección para evitar la lipoatrofia.
Para consultar las instrucciones de uso y manipulación, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes.
La somatropina no debe usarse cuando exista evidencia de actividad de un tumor. Los tumores intracraneales deben ser inactivos y el tratamiento antitumoral se debe completar antes de iniciar el tratamiento con GH. El tratamiento se debe suspender si hay pruebas de crecimiento tumoral.
La somatropina no debe usarse en la promoción del crecimiento de niños con epífisis cerradas.
Los pacientes con enfermedad crítica aguda que presenten complicaciones después de cirugía cardiaca, abdominal, traumatismo múltiple por accidente, insuficiencia respiratoria aguda o enfermedades similares, no deben ser tratados con somatropina (en cuanto a los pacientes sometidos a tratamiento de sustitución, ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
No se debe superar la dosis diaria máxima recomendada (ver sección 4.2).
Sensibilidad a la insulina
La somatropina puede reducir la sensibilidad a la insulina. En pacientes con diabetes mellitus, puede ser necesario ajustar la dosis de insulina tras la instauración del tratamiento con somatropina. Durante el tratamiento con somatropina, se deben monitorizar cuidadosamente los pacientes con diabetes, intolerancia a la glucosa o con factores de riesgo adicionales de sufrir diabetes.
Función tiroidea
La hormona de crecimiento aumenta la conversión extratiroidea de T4 a T3, lo que puede causar una reducción de la concentración T4 y un aumento de la T3 en el suero. Si bien las concentraciones de la hormona tiroidea periférica han permanecido dentro de los límites de referencia en la mayoría de sujetos sanos, teóricamente, se puede desarrollar hipotiroidismo en los sujetos con hipotiroidismo subclínico. En consecuencia, se debe llevar a cabo una monitorización de la función tiroidea en todos los pacientes. El potencial efecto del tratamiento con hormona de crecimiento sobre la función tiroidea se debe monitorizar cuidadosamente en pacientes con hipopituitarismo tratados con terapia sustitutiva estándar.
En la deficiencia de la hormona del crecimiento, secundaria al tratamiento de una neoplasia maligna, se recomienda prestar atención a los signos de recaída de la malignidad. Entre los supervivientes de cáncer pediátrico se ha referido un mayor riesgo de una segunda neoplasia en los pacientes tratados con somatropina después de su primera neoplasia. Las más frecuentes de estas segundas neoplasias en los pacientes tratados con radioterapia en la cabeza para su primera neoplasia fueron los tumores intracraneales, en concreto los meningiomas.
En los pacientes con trastornos endocrinos, incluida la deficiencia de la hormona del crecimiento, el deslizamiento de la epífisis de la cadera puede producirse con más frecuencia que en la población general. Los pacientes que cojean durante el tratamiento con somatropina deben ser controlados clínicamente.
Hipertensión intracraneal benigna
En casos de cefalea recurrente o grave, problemas visuales, náuseas y/o vómitos, se recomienda practicar una fundoscopia para descartar un edema de papila. Si se confirma el edema de papila, debe considerarse una hipertensión intracraneal benigna y, si fuese apropiado, debe de retirarse el tratamiento con la hormona del crecimiento. Si se reinicia el tratamiento con hormona del crecimiento, se debe instaurar un control cuidadoso para, si fuese necesario, detectar la hipertensión intracraneal.
Leucemia
Se ha notificado leucemia en un pequeño número de pacientes con deficiencia de la hormona de crecimiento, algunos de los cuales han sido tratados con somatropina. Sin embargo, no existe evidencia de que haya un aumento en la incidencia de la leucemia en pacientes tratados con hormona del crecimiento sin factores de predisposición.
Anticuerpos
En un pequeño porcentaje de pacientes pueden aparecer anticuerpos dirigidos contra Omnitrope. Omnitrope ha dado lugar a la formación de anticuerpos aproximadamente en el 1% de los pacientes. La capacidad de fijación de estos anticuerpos es baja y no hay ningún efecto sobre la velocidad de crecimiento. En cualquier paciente con una falta de respuesta por lo demás no explicada se deben hacer análisis de anticuerpos contra la somatropina.
Pacientes de edad avanzada
La experiencia con los pacientes mayores de 80 años es limitada. Los pacientes de edad avanzada pueden ser más sensibles a la acción de Omnitrope y, por lo tanto, pueden ser más propensos a presentar reacciones adversas.
Enfermedad agudas graves
Los efectos de la somatropina sobre la recuperación de pacientes adultos críticos se han evaluado en dos ensayos controlados con placebo en 522 pacientes adultos con complicaciones posteriores a cirugía a corazón abierto, cirugía abdominal, traumatismo accidental múltiple o insuficiencia respiratoria aguda. La mortalidad fue superior en los pacientes tratados diariamente con 5,3 u 8 mg de somatropina, en comparación con los pacientes que recibieron placebo, 42% vs. 19%. En base a esta información, este tipo de pacientes no deben ser tratados con somatropina. Dado que no existe información disponible sobre la seguridad del tratamiento sustitutivo con hormona de crecimiento en pacientes críticos, los beneficios del tratamiento continuado en esta situación deben considerarse sobre la base de los riesgos potenciales.
En todos los pacientes que desarrollen algún otro tipo de enfermedad aguda en fase crítica o similar, el posible beneficio del tratamiento con somatropina deberá ser sopesado en relación con el riesgo potencial que implica.
Población pediátrica
Pancreatitis
A pesar de que la pancreatitis es rara, debe ser considerada en niños tratados con somatropina que presenten dolor abdominal.
Síndrome de Prader-Willi
En los pacientes con SPW, el tratamiento debe ser siempre en combinación con una dieta baja en calorías.
Existen informes de muertes asociadas al uso de la hormona del crecimiento en los pacientes pediátricos con SPW que presentaron uno o más factores de riesgo: obesidad grave (aquellos pacientes con un peso/altura superior al 200%), antecedentes de insuficiencia respiratoria o apnea del sueño o de infección respiratoria no identificada. Los pacientes con SPW y uno o más de estos factores de riesgo podrían estar en mayor riesgo.
Antes de iniciar el tratamiento con somatropina los pacientes con SPW serán valorados respecto de la obstrucción de las vías aéreas altas, apnea del sueño o infección respiratoria.
Si, durante la evaluación de la obstrucción de las vías respiratorias altas, se observan resultados anatomopatológicos, se debe derivar al niño al otorrinolaringólogo (ENT) para el tratamiento y la resolución del trastorno respiratorio antes de iniciar el tratamiento con hormona de crecimiento.
La apnea del sueño debe evaluarse antes del inicio del tratamiento con hormona de crecimiento por métodos reconocidos como la polisomnografía o la oximetría, durante la noche, y los pacientes deben controlarse, si se sospecha apnea del sueño.
Si, durante el tratamiento con somatropina, los pacientes muestran signos de obstrucción de las vías respiratorias altas (incluido el inicio o el aumento de ronquidos), el tratamiento debe interrumpirse y debe efectuarse una nueva evaluación de ENT.
Todos los pacientes con SPW deben valorarse en cuanto a la apnea y ser controlados si se sospecha la presencia de apnea del sueño. Los pacientes deben controlarse para detectar signos de infecciones respiratorias, que deben ser diagnosticadas lo más pronto posible y tratadas de forma contundente.
Todos los pacientes con SPW deben someterse a un control eficaz del peso antes y, también, durante el tratamiento con hormona del crecimiento.
La escoliosis es muy frecuente en los pacientes con SPW. La escoliosis puede progresar en cualquier niño durante el crecimiento rápido. Deben controlarse los signos de escoliosis durante el tratamiento.
La experiencia con el tratamiento prolongado en los adultos y en los pacientes con SPW es limitada.
Nacidos pequeños para su edad gestacional
En los niños y los adolescentes PEG nacidos bajos antes de iniciar el tratamiento se deben descartar otras razones médicas u otros tratamientos que pudieran explicar la perturbación del crecimiento antes de iniciar el tratamiento.
En los niños y los adolescentes PEG se recomienda medir la insulina y la glucosa sanguínea en ayunas antes de iniciar el tratamiento y, posteriormente, en períodos anuales. En los pacientes con un aumento del riesgo de diabetes mellitus (p. ej., antecedentes familiares de diabetes, obesidad, resistencia grave a la insulina, acantosis nigricans) debe realizarse la prueba de tolerancia a la glucosa (OGTT). Si aparece una diabetes clara, no se debe administrar hormona del crecimiento.
En los niños y los adolescentes PEG se recomienda medir la concentración de IGF-I antes de iniciar el tratamiento y después, dos veces al año. Si en mediciones repetidas las concentraciones de IGF-I exceden en +2 DS comparado con la edad de referencia y estado puberal, la proporción IGF-I/IGFBP-3 debe tenerse en cuenta para considerar un ajuste de la dosis.
La experiencia al iniciar el tratamiento en los pacientes PEG cerca del comienzo de la pubertad es limitada. Por tanto, no se recomienda iniciar el tratamiento cerca de la pubertad. La experiencia en los pacientes con síndrome de Silver-Rusell es limitada.
Parte del aumento de estatura obtenido al tratar a los niños y los adolescentes nacidos PEG y bajos con hormona del crecimiento puede perderse si se interrumpe el tratamiento antes de que se alcance la estatura final.
Insuficiencia renal crónica
En la insuficiencia renal crónica, la función renal debe ser un 50% de la normal antes de iniciar el tratamiento. Para verificar la perturbación del crecimiento, se debe hacer un seguimiento del crecimiento durante un año antes de iniciar el tratamiento. Durante este periodo, se debe iniciar un tratamiento conservador para la insuficiencia renal (incluido el control de la acidosis, el hiperparatiroidismo y el estado nutricional), y debe mantenerse durante todo el tratamiento.
El tratamiento se debe interrumpir en casos de trasplante renal.
Hasta la fecha, no se disponen de datos de la estatura final de los pacientes con insuficiencia renal crónica tratados con Omnitrope.
Debido a la presencia de alcohol bencílico, el medicamento no debe administrarse a recién nacidos prematuros ni a neonatos. Puede causar reacciones tóxicas y reacciones anafilactoides en los lactantes y en los niños de hasta tres años.
4.5 Interacción con otros medicamentos y otras formas de interacción
El tratamiento concomitante con glucocorticoides puede inhibir los efectos de promoción del crecimiento de los productos que contienen somatropina. Por tanto, los pacientes tratados con glucocorticoides deben tener su crecimiento cuidadosamente monitorizado con el fin de valorar el potencial impacto del tratamiento con glucocorticoides sobre el crecimiento.
Los datos de un estudio de interacción, realizado en los adultos con deficiencia de la hormona del crecimiento, sugieren que la administración de somatropina puede aumentar la eliminación de compuestos que son metabolizados por las isoenzimas del citocromo P450. El metabolismo de los compuestos metabolizados por el citocromo P450 3A4 (p. ej.: esteroides sexuales, corticoesteroides, anticonvulsivantes y ciclosporinas) puede aumentar, dando como resultado unas concentraciones plasmáticas más bajas de estos compuestos. Se desconoce la significación clínica de esto.
Ver también en la sección 4.4 la información relacionada con la diabetes mellitus y los trastornos tiroideos, y en la sección 4.2 la información sobre la terapia de sustitución de estrógenos orales.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos o éstos son limitados relativos al uso de somatropina en mujeres embarazadas. Los estudios realizados en animales son insuficientes en términos de toxicidad para la reproducción (ver sección 5.3). No se recomienda utilizar somatropina durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Lactancia
No se han llevado a cabo estudios clínicos con medicamentos que contienen somatropina en mujeres en periodo de lactancia. Se desconoce si la somatropina se excreta en la leche materna, pero es poco probable la absorción de proteína intacta del tubo digestivo del niño. Por tanto, se debe tener precaución cuando se administre Omnitrope a mujeres en periodo de lactancia.
Fertilidad
No se han realizado estudios de fertilidad con Omnitrope.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Omnitrope sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los pacientes que padecen una deficiencia de la hormona de crecimiento se caracterizan por un déficit del volumen extracelular. Al iniciar el tratamiento con somatropina, este déficit se corrige rápidamente. En los pacientes adultos, son frecuentes las reacciones adversas relacionadas con la retención de líquidos, como el edema periférico, la rigidez musculoesquelética, la artralgia, la mialgia y las parestesias. En general, estas reacciones son leves a moderadas, se producen en los primeros meses de tratamiento y remiten espontáneamente o al reducir la dosis.
La incidencia de estas reacciones adversas está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de hormona de crecimiento. En los niños, tales reacciones adversas son poco frecuentes.
Omnitrope ha dado lugar a la formación de anticuerpos en aproximadamente el 1% de los pacientes. La capacidad de fijación de estos anticuerpos ha sido baja y no hay cambios clínicos asociados a su formación (ver sección 4.4).
Tabla de reacciones adversas
Las tablas 1-6 muestran las reacciones adversas clasificadas según los apartados del sistema de clasificación de órganos y las frecuencias, utilizando la convención siguiente: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); no conocida (no puede estimarse a partir de los datos disponibles) para cada una de las afecciones indicadas.
Ensayos clínicos en niños con DGH
|
Tabla 1 Tratamiento a largo plazo de niños con alteración del crecimiento debida a una secreción insuficiente de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias9 Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia9 |
Mialgia9 Rigidez musculoesquelética9 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico9 | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre9 | |||||
Ensayos clínicos en niños con síndrome de Turner
|
Tabla 2 Tratamiento a largo plazo de niños con alteración del crecimiento debida al síndrome de Turner | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico* Reacción en el lugar de la inyección$ | |||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* | |||||
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con insuficiencia renal crónica
|
Tabla 3 | ||||||
|
Tratamiento a largo plazo de niños con alteración del crecimiento debida a una insuficiencia renal crónica | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas |
Leucemiaf | |||||
|
y no especificadas (incl. quistes y pólipos) | ||||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* Rigidez musculoesquelética* | |||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con PEG
|
Tabla 4 Tratamiento a largo plazo de niños con alteración del crecimiento debida al nacimiento con baja talla para su edad gestacional | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión | |||||
|
intracraneal benigna | ||||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con SPW
|
Tabla 5 Tratamiento a largo debida al síndrome ( |
plazo y mejora de la composición corporal en niños con alteración del crecimiento e Prader-Willi | |||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* |
Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de |
Edema periférico* |
Reacción en el lugar de la inyección$ | ||||
|
administración | ||||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre10 |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en adultos con DGH
|
Tabla 6 Terapia de sustitución en adultos con deficiencia de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo 2 | |||||
|
Trastornos del sistema nervioso |
Parestesias * Síndrome del túnel carpiano |
Hipertensión intracraneal benigna | ||||
|
Trastornos musculoesquelét icos y del tejido conjuntivo |
Artralgia10 |
Mialgia10 Rigidez musculoesq uelética10 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico10 |
Reacción en el lugar de la inyección$ | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre10 | |||||
Descripción de reacciones adversas seleccionadas Niveles reducidos de cortisol sérico
Se ha notificado que la somatropina reduce las concentraciones séricas de cortisol, posiblemente al afectar a las proteínas transportadoras o mediante un aumento de la depuración hepática. La relevancia clínica de estas observaciones puede ser limitada. No obstante, antes de iniciar el tratamiento, debe optimizarse el tratamiento de reemplazo con corticoesteroides.
Síndrome de Prader-Willi
Se han notificado casos raros de muerte súbita en pacientes con síndrome de Prader-Willi tratados con somatropina en la experiencia post-comercialización, aunque no se ha demostrado que exista relación causal.
Leucemia
Se han notificado casos (raros o muy raros) de leucemia en los niños que padecen una deficiencia de la hormona de crecimiento y son tratados con somatropina e incluidos en la experiencia post-comercialización. Sin embargo, no existen indicios de un aumento del riesgo de leucemia sin factores predisponentes, tales como la radioterapia cerebral o de la cabeza.
Epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes
Se han notificado epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes en niños tratados con la hormona del crecimiento. La epifisiólisis de la cabeza femoral se produce con mayor frecuencia en caso de trastornos endocrinos y la enfermedad de Legg-Calvé-Perthes es más frecuente en caso de baja estatura. No obstante, se desconoce si estas dos afecciones aparecen o no con mayor frecuencia durante el tratamiento con somatropina. Debe tenerse en cuenta la posibilidad de su diagnóstico en niños que presenten molestias o dolor en la cadera o la rodilla.
Otras reacciones adversas al medicamento
Otras reacciones adversas al medicamento pueden considerarse efectos de clase de la somatropina, tales como una posible hiperglucemia causada por la disminución de la sensibilidad a la insulina, la disminución de la concentración de tiroxina libre y a una hipertensión intracraneal benigna.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Síntomas:
La sobredosis aguda puede conducir inicialmente a hipoglucemia y posteriormente a hiperglucemia.
La sobredosis prolongada puede causar signos y síntomas compatibles con los efectos conocidos de exceso de la hormona del crecimiento.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: hormonas del lóbulo anterior de la hipófisis y sus análogos, código ATC: H01AC01.
Omnitrope es un medicamento biosimilar. La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Mecanismo de acción
La somatropina es una potente hormona metabólica, importante en el metabolismo de lípidos, carbohidratos y proteínas. En los niños con hormona del crecimiento endógena insuficiente, la somatropina estimula el crecimiento lineal y aumenta la tasa de crecimiento. En los adultos y también en los niños, la somatropina mantiene una composición corporal normal aumentando la retención de nitrógeno y la estimulación del crecimiento del músculo esquelético y la movilización de la grasa corporal. El tejido adiposo visceral responde en particular a la somatropina. Además, para mejorar la lipólisis, la somatropina reduce la captación de triglicéridos de los depósitos de grasa corporal. Las concentraciones séricas de IGF-I (factor de crecimiento de tipo insulina I) y la IGFBP3 (proteína de unión del factor de crecimiento de tipo insulina 3) son aumentadas por la somatropina. Además, se han demostrado las siguientes acciones.
Efectos farmacodinámicos
Metabolismo lipídico
La somatropina induce los receptores de colesterol LDL hepáticos y afecta el perfil de lípidos séricos y las lipoproteínas. En general, la administración de somatropina a pacientes deficitarios de la hormona del crecimiento resulta en una reducción del DLD sérico y de la apoliproteína B. También se puede observar una reducción del colesterol total.
Metabolismo de los carbohidratos
La somatropina aumenta la insulina, pero la glucosa sanguínea en ayunas permanece, por lo general, sin cambios. Los niños con hipopituitarismo experimentan hipoglucemia en ayunas. Esta condición es corregida por la somatropina.
Metabolismo del agua y de los minerales
La deficiencia de la hormona del crecimiento se asocia a una reducción de los volúmenes plasmático y extracelular. Ambos aumentan rápidamente tras el tratamiento con somatropina. La somatropina induce la retención de sodio, potasio y fósforo.
Metabolismo óseo
La somatropina estimula el recambio del hueso esquelético. La administración a largo plazo de somatropina a pacientes con osteopenia con deficiencia de la hormona del crecimiento resulta en un aumento del contenido mineral y de la densidad ósea en los lugares portadores de peso.
Capacidad _ física
Mejoría de la potencia muscular y del ejercicio físico tras un tratamiento a largo plazo con somatropina. La somatropina también aumenta la frecuencia cardíaca, pero el mecanismo de acción no ha sido aún aclarado. Puede que una reducción de la resistencia vascular periférica contribuya a este efecto.
Eficacia clínica y seguridad
En ensayos clínicos en los niños y los adolescentes PEG y de talla baja se utilizaron dosis de 0,033 y 0,067 mg/kg de peso corporal, por día hasta alcanzar la altura final. En 56 pacientes tratados de forma continuada y que alcanzaron (casi) la altura final, el cambio principal de talla al inicio del tratamiento fue de +1,90 SDS (0,033 mg/kg de peso corporal por día) y +2,19 SDS (0,067 mg/kg de peso corporal por día). Los datos publicados de niños y adolescentes PEG no tratados sin estirón espontáneo y temprano sugieren un crecimiento tardío, de la SDS de 0,5. Los datos de seguridad a largo plazo son todavía limitados.
5.2 Propiedades farmacocinéticas
Absorción
La biodisponibilidad de la somatropina, administrada subcutáneamente, es aproximadamente del 80% en ambos casos, en las personas sanas y en los pacientes con deficiencia de la hormona de crecimiento. Una dosis subcutánea de 5 mg de Omnitrope 5 mg/1,5 ml solución inyectable en los adultos sanos lleva a unos valores plasmáticos de Cmax de 72 ± 28 microgramos/l, y de tmax, de
4,0 ± 2,0 horas.
Eliminación
La media de la vida media terminal de la somatropina después de la administración intravenosa en los pacientes adultos con deficiencia de la hormona de crecimiento es de aproximadamente 0,4 horas. Sin embargo, después de la administración subcutánea de Omnitrope 5 mg/1,5 ml solución inyectable, se alcanza una vida media de 3 horas. Es probable que la diferencia observada se deba a la lenta absorción de la inyección después de la administración subcutánea.
Poblaciones especiales
La biodisponibilidad absoluta de somatropina parece ser similar tanto en los hombres como en las mujeres después de la administración subcutánea.
La información sobre las propiedades farmacocinéticas de somatropina en geriatría y pediatría, en diferentes razas y en los pacientes con insuficiencia renal, hepática o cardiaca es o bien escasa o incompleta.
5.3 Datos preclínicos sobre seguridad
En los estudios con Omnitrope referentes a toxicidad subaguda y tolerancia local no se han observado casos con efectos clínicamente relevantes.
En otros estudios con somatropina referente a toxicidad general, tolerancia local y reproducción de la toxicidad no se han observado efectos clínicamente relevantes.
Con somatropina, los estudios de genotoxicidad in vitro e in vivo o de mutaciones genéticas e inducción de aberraciones cromosómicas han sido negativos.
Se ha observado un aumento de fragilidad cromosómica en un estudio in vitro con linfocitos, tomados de pacientes en tratamiento prolongado con somatropina y bajo la adición de bleomicina, un medicamento radiomimético. El significado clínico de esta observación no está claro.
En otro estudio con somatropina, no se encontró incremento de anomalías cromosómicas en linfocitos de pacientes que recibieron terapia con somatropina durante largo tiempo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Fosfato hidrógeno disódico heptahidrato Fosfato dihidrógeno sódico dihidrato Manitol
Poloxámero 188 Alcohol bencílico
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
Periodo de validez después del primer uso
Después del primer uso, el cartucho debe permanecer en la pluma y debe conservarse en una nevera (entre 2°C y 8°C) durante un máximo de 28 días. Conservar y transportar refrigerado (entre 2°C y 8°C). No congelar. Conservar en la pluma original para protegerlo de la luz.
6.4 Precauciones especiales de conservación
Cartucho sin abrir
Conservar y transportar refrigerado (entre 2°C y 8°C). No congelar. Conservar en el embalaje original para protegerlo de la luz.
Para las condiciones de conservación del medicamento en uso, ver sección 6.3.
6.5 Naturaleza y contenido del envase
1.5 ml de solución en un cartucho (de vidrio de tipo I), con émbolo en un lado (de bromobutilo siliconizado), un disco (de bromobutilo) y una cápsula de cierre (de aluminio) en el otro lado. El cartucho de vidrio está integrado irreversiblemente en un recipiente transparente y está montado en un mecanismo de plástico con una barra roscada en una extremidad.
Paquetes de 1, 5 y 10 cartuchos.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Omnitrope 5 mg/1,5 ml solución inyectable es una solución estéril y lista para usar, para inyección subcutánea, cargada en un cartucho de vidrio.
Esta presentación está pensada para varios usos. Sólo debería administrarse con SurePal 5, un dispositivo de inyección específicamente desarrollado para usarse con Omnitrope 5 mg/1,5 ml solución inyectable. Se administrará utilizando agujas para inyección de pluma estériles y desechables. Los pacientes y los cuidadores han de recibir del médico o de otro profesional de la salud formación e instrucciones adecuadas sobre el uso correcto de los cartuchos y de la pluma Omnitrope.
Lo que sigue es una descripción general del proceso de administración. Deben seguirse las instrucciones del fabricante para cargar el cartucho, conectar la aguja para inyección y para la administración.
1. Las manos deben lavarse.
2. Si la solución está turbia o si contiene material en partículas, no debe utilizarse. El contenido debe ser cristalino e incoloro.
3. Desinfectar la membrana de goma del cartucho con una torunda para limpiar.
4. Introducir el cartucho en la pluma SurePal 5, según las instrucciones de uso
suministradas con la pluma.
5. Limpiar el lugar de aplicación de la inyección con una torunda empapada en alcohol.
6. Administrar la dosis adecuada mediante inyección subcutánea, utilizando la aguja de pluma estéril. Extraer la aguja de la pluma y eliminarla con arreglo a los requisitos locales.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestrasse 10
A-6250 Kundl Austria
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/013
EU/1/06/332/014
EU/1/06/332/015
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 12 de abril de 2006 Fecha de la última renovación: 12 de abril de 2011
10. FECHA DE LA REVISIÓN DEL TEXTO
<{MM/AAAA}>
1. NOMBRE DEL MEDICAMENTO
Omnitrope 10 mg/1,5 ml solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada ml de solución contiene 6,7 mg de somatropina* (que corresponden a 20 UI). Un cartucho contiene 1,5 ml, que corresponde a 10 mg de somatropina* (30 UI).
* Producida en Escherichia coli mediante tecnología de DNA recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable en un cartucho para SurePal 10. La solución es límpida e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lactantes, niños y adolescentes:
- Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento (deficiencia de la hormona del crecimiento, DGH).
- Trastorno del crecimiento asociado al síndrome de Turner.
- Trastorno del crecimiento asociado a insuficiencia renal crónica.
- Trastorno del crecimiento (puntuación de la desviación estándar actual de la talla (SDS) < -2,5 y
SDS de la talla < -1 ajustada para los padres) en los niños y los adolescentes nacidos con baja
talla para su edad gestacional (PEG), con un peso al nacer y/o longitud por debajo de -
2 desviaciones estándar (DE), que no alcanzaron el estirón de crecimiento (velocidad de crecimiento (VC) SDS < 0 durante el último año) a los 4 años de edad o más.
- El síndrome de Prader-Willi (SPW) para la mejoría del crecimiento y composición corporal. El diagnóstico de SPW debe ser confirmado por pruebas genéticas apropiadas.
Adultos
- Terapia de sustitución en los adultos con deficiencia pronunciada de la hormona del crecimiento.
- Inicio en la edad adulta: Pacientes que padecen una deficiencia grave de la hormona del crecimiento, asociada a múltiples deficiencias hormonales a consecuencia de una patología hipotalámica o hipofisaria conocida y que padecen, por lo menos, una deficiencia conocida de una hormona hipofisaria que no sea la prolactina. Estos pacientes deben someterse a una prueba dinámica adecuada, para así diagnosticar o excluir una deficiencia de la hormona del crecimiento.
- Inicio en la niñez: Pacientes que padecían una deficiencia de la hormona del crecimiento en la niñez a consecuencia de causas congénitas, genéticas, adquiridas o idiopáticas. En los pacientes con DGH de inicio en la niñez se debe reevaluar la capacidad secretora de la hormona del crecimiento después de concluir su crecimiento longitudinal. En los pacientes con una probabilidad alta de DGH persistente, es decir, una causa congénita o una DGH secundaria a una enfermedad o lesión hipotálamo-hipofisaria, un SDS < -2 del factor de crecimiento de tipo insulínico I (IGF-I) cuando no reciben tratamiento con hormona de crecimiento por lo menos durante cuatro semanas debe considerarse una prueba suficiente de DGH profunda.
Todos los demás pacientes requerirán un análisis del IGF-I y una prueba de estimulación con la hormona del crecimiento.
4.2 Posología y forma de administración
El diagnóstico y el tratamiento con somatropina deben iniciarse y monitorizarse por médicos que tengan la capacitación y la experiencia adecuadas en el diagnóstico y el tratamiento de los pacientes con trastornos del crecimiento.
Posología
Población pediátrica
La posología y la pauta de dosificación deben individualizarse.
Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento en los pacientes pediátricos:
En términos generales, se recomienda una dosis de 0,025 a 0,035 mg/kg de peso corporal por día o de 0,7 a 1,0 mg/m2 de superficie corporal por día. Incluso dosis más altas han sido utilizadas.
Cuando la DGH de inicio en la niñez persiste hasta la adolescencia, se debe continuar el tratamiento a fin de alcanzar un desarrollo somático completo (p. ej., composición corporal, masa ósea). Para la monitorización, el logro de una masa ósea máxima normal, definida como una puntuación T > -1 (es decir, normalizado hasta la masa ósea máxima promedio en el adulto, determinada mediante absorciometría de rayos X de doble energía, teniendo en cuenta el sexo y el origen étnico del paciente) es uno de los objetivos terapéuticos durante el período de transición. Para la orientación en cuanto a la posología, ver el apartado sobre los adultos, a continuación.
Síndrome de Prader-Willi, para la mejoría del crecimiento y composición corporal en los pacientes pediátricos
En general, se recomienda una dosis de 0,035 mg/kg de peso corporal por día ó 1,0 mg/m2 de superficie corporal por día. No se excederán las dosis diarias de 2,7 mg. El tratamiento no se utilizará en los pacientes pediátricos con una velocidad de crecimiento inferior a 1 cm al año y cerca del cierre de las epífisis.
Trastorno del crecimiento debido al síndrome de Turner
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal al día ó 1,4 mg/m2 de superficie corporal.
Trastorno del crecimiento en la insuficiencia renal crónica
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal y día (de 1,4 mg/m2 de superficie corporal y día). Pueden precisarse dosis más altas si la velocidad de crecimiento es muy lenta. Puede ser necesario corregir la dosis después de seis meses de tratamiento (ver sección 4.4).
Trastorno del crecimiento en los niños y los adolescentes nacidos con baja talla para su edad gestacional (PEG)
Se recomienda una dosis de 0,035 mg/kg de peso corporal al día (1 mg/m2 de superficie corporal al día) hasta que se alcance la talla final (ver sección 5.1). El tratamiento se debe interrumpir después del primer año de tratamiento, si la SDS de la velocidad de crecimiento es inferior a + 1. El tratamiento se interrumpirá si la velocidad de crecimiento es < 2 cm/año y, si se requiere confirmación, la edad ósea es > 14 años (niñas) o > 16 años (niños), correspondiente con el cierre de las placas de crecimiento epifisario.
Posología recomendada en pacientes pediátricos
|
Indicación |
Dosis diaria en mg/kg de peso corporal |
Dosis diaria en mg/m2 de superficie corporal |
|
Deficiencia de la hormona del crecimiento |
0,025 - 0,035 |
0,7 - 1,0 |
|
Síndrome de Prader-Willi |
0,035 |
1,0 |
|
Síndrome de Turner |
0,045 - 0,050 |
1,4 |
|
Insuficiencia renal crónica |
0,045 - 0,050 |
1,4 |
|
Niños y adolescentes nacidos con talla baja para su edad gestacional (PEG) |
0,035 |
1,0 |
Pacientes adultos con deficiencia de la hormona del crecimiento
La dosis recomendada para reanudar el tratamiento en aquellos pacientes que continúen con hormona de crecimiento tras un déficit de GH en la infancia, es de 0,2-0,5 mg por día. La dosis se debe ir incrementando o disminuyendo gradualmente de acuerdo a las necesidades individuales del paciente, determinadas de acuerdo a la concentración del IGF-I.
En los pacientes con DGH de inicio en la edad adulta, el tratamiento debe iniciarse con una dosis baja, de 0,15 a 0,3 mg al día. La dosis se aumentará gradualmente según las necesidades de cada paciente y según lo determine la concentración de IGF-I.
En ambos casos, el objetivo del tratamiento debe ser alcanzar concentraciones de factor de crecimiento de tipo insulina (IGF-I) dentro de 2 SDS de la media corregida por la edad. A los pacientes con concentraciones IGF-I normales al inicio del tratamiento se les debe administrar la hormona del crecimiento hasta una concentración de IGF-I dentro de los límites superiores normales, sin sobrepasar 2 SDS. La respuesta clínica y los efectos adversos pueden también usarse como guía para el ajuste de la dosis. Se conoce que hay pacientes con DGH que no llegan a normalizar los niveles de IGF-I a pesar de tener una buena respuesta clínica, y por tanto, no requieren ajuste de dosis. La dosis de mantenimiento raramente excede 1,0 mg al día. Las mujeres pueden necesitar dosis más altas que los hombres, mientras que los varones muestran una sensibilidad mayor al aumento de la IGF-I con el tiempo. Esto significa que hay un riesgo de que las mujeres, en especial aquellas en sustitución con estrógenos orales, estén infra-tratadas mientras que los hombres estén sobre-tratados. La exactitud de la dosis de la hormona del crecimiento debe, por lo tanto, controlarse cada 6 meses. Habida cuenta de que la producción fisiológica de la hormona del crecimiento desciende con la edad, pueden reducirse las necesidades de la dosis.
Poblaciones especiales
Personas de edad avanzada
En los pacientes mayores de 60 años, el tratamiento debe comenzar con una dosis de 0,1 a 0,2 mg diarios, y debe aumentarse gradualmente, según las necesidades individuales del paciente. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento en estos pacientes rara vez es superior a 0,5 mg diarios.
Forma de administración
Se debe administrar la inyección por vía subcutánea, cambiando el lugar de inyección para evitar la lipoatrofia.
Para consultar las instrucciones de uso y manipulación, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes.
La somatropina no debe usarse cuando exista evidencia de actividad de un tumor. Los tumores intracraneales deben ser inactivos y el tratamiento antitumoral se debe completar antes de iniciar el tratamiento con GH. El tratamiento se debe suspender si hay pruebas de crecimiento tumoral.
La somatropina no debe usarse en la promoción del crecimiento de niños con epífisis cerradas.
Los pacientes con enfermedad crítica aguda que presenten complicaciones después de cirugía cardiaca, abdominal, traumatismo múltiple por accidente, insuficiencia respiratoria aguda o enfermedades similares, no deben ser tratados con somatropina (en cuanto a los pacientes sometidos a tratamiento de sustitución, ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
No se debe superar la dosis diaria máxima recomendada (ver sección 4.2).
Sensibilidad a la insulina
La somatropina puede reducir la sensibilidad a la insulina. En pacientes con diabetes mellitus, puede ser necesario ajustar la dosis de insulina tras la instauración del tratamiento con somatropina. Durante el tratamiento con somatropina, se deben monitorizar cuidadosamente los pacientes con diabetes, intolerancia a la glucosa o con factores de riesgo adicionales de sufrir diabetes.
Función tiroidea
La hormona de crecimiento aumenta la conversión extratiroidea de T4 a T3, lo que puede causar una reducción de la concentración T4 y un aumento de la T3 en el suero. Si bien las concentraciones de la hormona tiroidea periférica han permanecido dentro de los límites de referencia en la mayoría de sujetos sanos, teóricamente, se puede desarrollar hipotiroidismo en los sujetos con hipotiroidismo subclínico. En consecuencia, se debe llevar a cabo una monitorización de la función tiroidea en todos los pacientes. El potencial efecto del tratamiento con hormona de crecimiento sobre la función tiroidea se debe monitorizar cuidadosamente en pacientes con hipopituitarismo tratados con terapia sustitutiva estándar.
En la deficiencia de la hormona del crecimiento, secundaria al tratamiento de una neoplasia maligna, se recomienda prestar atención a los signos de recaída de la malignidad. Entre los supervivientes de cáncer pediátrico se ha referido un mayor riesgo de una segunda neoplasia en los pacientes tratados con somatropina después de su primera neoplasia. Las más frecuentes de estas segundas neoplasias en los pacientes tratados con radioterapia en la cabeza para su primera neoplasia fueron los tumores intracraneales, en concreto los meningiomas.
En los pacientes con trastornos endocrinos, incluida la deficiencia de la hormona del crecimiento, el deslizamiento de la epífisis de la cadera puede producirse con más frecuencia que en la población general. Los pacientes que cojean durante el tratamiento con somatropina deben ser controlados clínicamente.
Hipertensión intracraneal benigna
En casos de cefalea recurrente o grave, problemas visuales, náuseas y/o vómitos, se recomienda practicar una fundoscopia para descartar un edema de papila. Si se confirma el edema de papila, debe considerarse una hipertensión intracraneal benigna y, si fuese apropiado, debe de retirarse el tratamiento con la hormona del crecimiento. Si se reinicia el tratamiento con hormona del crecimiento, se debe instaurar un control cuidadoso para, si fuese necesario, detectar la hipertensión intracraneal.
Leucemia
Se ha notificado leucemia en un pequeño número de pacientes con deficiencia de la hormona de crecimiento, algunos de los cuales han sido tratados con somatropina. Sin embargo, no existe evidencia de que haya un aumento en la incidencia de la leucemia en pacientes tratados con hormona del crecimiento sin factores de predisposición.
Anticuerpos
En un pequeño porcentaje de pacientes pueden aparecer anticuerpos dirigidos contra Omnitrope. Omnitrope ha dado lugar a la formación de anticuerpos aproximadamente en el 1% de los pacientes. La capacidad de fijación de estos anticuerpos es baja y no hay ningún efecto sobre la velocidad de crecimiento. En cualquier paciente con una falta de respuesta por lo demás no explicada se deben hacer análisis de anticuerpos contra la somatropina.
Pacientes de edad avanzada
La experiencia con los pacientes mayores de 80 años es limitada. Los pacientes de edad avanzada pueden ser más sensibles a la acción de Omnitrope y, por lo tanto, pueden ser más propensos a presentar reacciones adversas.
Enfermedad agudas graves
Los efectos de la somatropina sobre la recuperación de pacientes adultos críticos se han evaluado en dos ensayos controlados con placebo en 522 pacientes adultos con complicaciones posteriores a cirugía a corazón abierto, cirugía abdominal, traumatismo accidental múltiple o insuficiencia respiratoria aguda. La mortalidad fue superior en los pacientes tratados diariamente con 5,3 u 8 mg de somatropina, en comparación con los pacientes que recibieron placebo, 42% vs. 19%. En base a esta información, este tipo de pacientes no deben ser tratados con somatropina. Dado que no existe información disponible sobre la seguridad del tratamiento sustitutivo con hormona de crecimiento en pacientes críticos, los beneficios del tratamiento continuado en esta situación deben considerarse sobre la base de los riesgos potenciales.
En todos los pacientes que desarrollen algún otro tipo de enfermedad aguda en fase crítica o similar, el posible beneficio del tratamiento con somatropina deberá ser sopesado en relación con el riesgo potencial que implica.
Población pediátrica
Pancreatitis
A pesar de que la pancreatitis es rara, debe ser considerada en niños tratados con somatropina que presenten dolor abdominal.
Síndrome de Prader-Willi
En los pacientes con SPW, el tratamiento debe ser siempre en combinación con una dieta baja en calorías.
Existen informes de muertes asociadas al uso de la hormona del crecimiento en los pacientes pediátricos con SPW que presentaron uno o más factores de riesgo: obesidad grave (aquellos pacientes con un peso/altura superior al 200%), antecedentes de insuficiencia respiratoria o apnea del sueño o de infección respiratoria no identificada. Los pacientes con SPW y uno o más de estos factores de riesgo podrían estar en mayor riesgo.
Antes de iniciar el tratamiento con somatropina los pacientes con SPW serán valorados respecto de la obstrucción de las vías aéreas altas, apnea del sueño o infección respiratoria.
Si, durante la evaluación de la obstrucción de las vías respiratorias altas, se observan resultados anatomopatológicos, se debe derivar al niño al otorrinolaringólogo (ENT) para el tratamiento y la resolución del trastorno respiratorio antes de iniciar el tratamiento con hormona de crecimiento.
La apnea del sueño debe evaluarse antes del inicio del tratamiento con hormona de crecimiento por métodos reconocidos como la polisomnografía o la oximetría, durante la noche, y los pacientes deben controlarse, si se sospecha apnea del sueño.
Si, durante el tratamiento con somatropina, los pacientes muestran signos de obstrucción de las vías respiratorias altas (incluido el inicio o el aumento de ronquidos), el tratamiento debe interrumpirse y debe efectuarse una nueva evaluación de ENT.
Todos los pacientes con SPW deben valorarse en cuanto a la apnea y ser controlados si se sospecha la presencia de apnea del sueño. Los pacientes deben controlarse para detectar signos de infecciones respiratorias, que deben ser diagnosticadas lo más pronto posible y tratadas de forma contundente.
Todos los pacientes con SPW deben someterse a un control eficaz del peso antes y, también, durante el tratamiento con hormona del crecimiento.
La escoliosis es muy frecuente en los pacientes con SPW. La escoliosis puede progresar en cualquier niño durante el crecimiento rápido. Deben controlarse los signos de escoliosis durante el tratamiento.
La experiencia con el tratamiento prolongado en los adultos y en los pacientes con SPW es limitada.
Nacidos pequeños para su edad gestacional
En los niños y los adolescentes PEG nacidos bajos antes de iniciar el tratamiento se deben descartar otras razones médicas u otros tratamientos que pudieran explicar la perturbación del crecimiento antes de iniciar el tratamiento.
En los niños y los adolescentes PEG se recomienda medir la insulina y la glucosa sanguínea en ayunas antes de iniciar el tratamiento y, posteriormente, en períodos anuales. En los pacientes con un aumento del riesgo de diabetes mellitus (p. ej., antecedentes familiares de diabetes, obesidad, resistencia grave a la insulina, acantosis nigricans) debe realizarse la prueba de tolerancia a la glucosa (OGTT). Si aparece una diabetes clara, no se debe administrar hormona del crecimiento.
En los niños y los adolescentes PEG se recomienda medir la concentración de IGF-I antes de iniciar el tratamiento y después, dos veces al año. Si en mediciones repetidas las concentraciones de IGF-I exceden en +2 DS comparado con la edad de referencia y estado puberal, la proporción IGF-I/IGFBP-3 debe tenerse en cuenta para considerar un ajuste de la dosis.
La experiencia al iniciar el tratamiento en los pacientes PEG cerca del comienzo de la pubertad es limitada. Por tanto, no se recomienda iniciar el tratamiento cerca de la pubertad. La experiencia en los pacientes con síndrome de Silver-Rusell es limitada.
Parte del aumento de estatura obtenido al tratar a los niños y los adolescentes nacidos PEG y bajos con hormona del crecimiento puede perderse si se interrumpe el tratamiento antes de que se alcance la estatura final.
Insuficiencia renal crónica
En la insuficiencia renal crónica, la función renal debe ser un 50% de la normal antes de iniciar el tratamiento. Para verificar la perturbación del crecimiento, se debe hacer un seguimiento del crecimiento durante un año antes de iniciar el tratamiento. Durante este periodo, se debe iniciar un tratamiento conservador para la insuficiencia renal (incluido el control de la acidosis, el hiperparatiroidismo y el estado nutricional), y debe mantenerse durante todo el tratamiento.
El tratamiento se debe interrumpir en casos de trasplante renal.
Hasta la fecha, no se disponen de datos de la estatura final de los pacientes con insuficiencia renal crónica tratados con Omnitrope.
4.5 Interacción con otros medicamentos y otras formas de interacción
El tratamiento concomitante con glucocorticoides puede inhibir los efectos de promoción del crecimiento de los productos que contienen somatropina. Por tanto, los pacientes tratados con glucocorticoides deben tener su crecimiento cuidadosamente monitorizado con el fin de valorar el potencial impacto del tratamiento con glucocorticoides sobre el crecimiento.
Los datos de un estudio de interacción, realizado en los adultos con deficiencia de la hormona del crecimiento, sugieren que la administración de somatropina puede aumentar la eliminación de compuestos que son metabolizados por las isoenzimas del citocromo P450. El metabolismo de los compuestos metabolizados por el citocromo P450 3A4 (p. ej.: esteroides sexuales, corticoesteroides, anticonvulsivantes y ciclosporinas) puede aumentar, dando como resultado unas concentraciones plasmáticas más bajas de estos compuestos. Se desconoce la significación clínica de esto.
Ver también en la sección 4.4 la información relacionada con la diabetes mellitus y los trastornos tiroideos, y en la sección 4.2 la información sobre la terapia de sustitución de estrógenos orales.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos o éstos son limitados relativos al uso de somatropina en mujeres embarazadas. Los estudios realizados en animales son insuficientes en términos de toxicidad para la reproducción (ver sección 5.3). No se recomienda utilizar somatropina durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Lactancia
No se han llevado a cabo estudios clínicos con medicamentos que contienen somatropina en mujeres en periodo de lactancia. Se desconoce si la somatropina se excreta en la leche materna, pero es poco probable la absorción de proteína intacta del tubo digestivo del niño. Por tanto, se debe tener precaución cuando se administre Omnitrope a mujeres en periodo de lactancia.
Fertilidad
No se han realizado estudios de fertilidad con Omnitrope.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Omnitrope sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los pacientes que padecen una deficiencia de la hormona de crecimiento se caracterizan por un déficit del volumen extracelular. Al iniciar el tratamiento con somatropina, este déficit se corrige rápidamente. En los pacientes adultos, son frecuentes las reacciones adversas relacionadas con la retención de líquidos, como el edema periférico, la rigidez musculoesquelética, la artralgia, la mialgia y las parestesias. En general, estas reacciones son leves a moderadas, se producen en los primeros meses de tratamiento y remiten espontáneamente o al reducir la dosis.
La incidencia de estas reacciones adversas está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de hormona de crecimiento. En los niños, tales reacciones adversas son poco frecuentes.
Omnitrope ha dado lugar a la formación de anticuerpos en aproximadamente el 1% de los pacientes. La capacidad de fijación de estos anticuerpos ha sido baja y no hay cambios clínicos asociados a su formación (ver sección 4.4).
Tabla de reacciones adversas
Las tablas 1-6 muestran las reacciones adversas clasificadas según los apartados del sistema de clasificación de órganos y las frecuencias, utilizando la convención siguiente: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); no conocida (no puede estimarse a partir de los datos disponibles) para cada una de las afecciones indicadas.
Ensayos clínicos en niños con DGH
|
Tabla 1 Tratamiento a largo plazo de niños con alteración del crecimiento debida a una secreción insuficiente de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias11 Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia11 |
Mialgia11 Rigidez musculoesquelética11 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico11 | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre11 | |||||
Ensayos clínicos en niños con síndrome de Turner
|
Tabla 2 Tratamiento a largo plazo de niños con alteración del crecimiento debida al síndrome de Turner | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico* Reacción en el lugar de la inyección$ | |||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* | |||||
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con insuficiencia renal crónica
|
Tabla 3 | ||||||
|
Tratamiento a largo plazo de niños con alteración del crecimiento debida a una insuficiencia renal crónica | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas |
Leucemiaf | |||||
|
y no especificadas (incl. quistes y pólipos) | ||||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* Rigidez musculoesquelética* | |||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con PEG
|
Tabla 4 Tratamiento a largo plazo de niños con alteración del crecimiento debida al nacimiento con baja talla para su edad gestacional | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión | |||||
|
intracraneal benigna | ||||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con SPW
|
Tabla 5 Tratamiento a largo debida al síndrome ( |
plazo y mejora de la composición corporal en niños con alteración del crecimiento e Prader-Willi | |||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* |
Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de |
Edema periférico* |
Reacción en el lugar de la inyección$ | ||||
|
administración | ||||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre12 |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en adultos con DGH
|
Tabla 6 Terapia de sustitución en adultos con deficiencia de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo 2 | |||||
|
Trastornos del sistema nervioso |
Parestesias * Síndrome del túnel carpiano |
Hipertensión intracraneal benigna | ||||
|
Trastornos musculoesquelét icos y del tejido conjuntivo |
Artralgia12 |
Mialgia12 Rigidez musculoesq uelética12 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico12 |
Reacción en el lugar de la inyección$ | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre12 | |||||
Descripción de reacciones adversas seleccionadas Niveles reducidos de cortisol sérico
Se ha notificado que la somatropina reduce las concentraciones séricas de cortisol, posiblemente al afectar a las proteínas transportadoras o mediante un aumento de la depuración hepática. La relevancia clínica de estas observaciones puede ser limitada. No obstante, antes de iniciar el tratamiento, debe optimizarse el tratamiento de reemplazo con corticoesteroides.
Síndrome de Prader-Willi
Se han notificado casos raros de muerte súbita en pacientes con síndrome de Prader-Willi tratados con somatropina en la experiencia post-comercialización, aunque no se ha demostrado que exista relación causal.
Leucemia
Se han notificado casos (raros o muy raros) de leucemia en los niños que padecen una deficiencia de la hormona de crecimiento y son tratados con somatropina e incluidos en la experiencia post-comercialización. Sin embargo, no existen indicios de un aumento del riesgo de leucemia sin factores predisponentes, tales como la radioterapia cerebral o de la cabeza.
Epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes
Se han notificado epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes en niños tratados con la hormona del crecimiento. La epifisiólisis de la cabeza femoral se produce con mayor frecuencia en caso de trastornos endocrinos y la enfermedad de Legg-Calvé-Perthes es más frecuente en caso de baja estatura. No obstante, se desconoce si estas dos afecciones aparecen o no con mayor frecuencia durante el tratamiento con somatropina. Debe tenerse en cuenta la posibilidad de su diagnóstico en niños que presenten molestias o dolor en la cadera o la rodilla.
Otras reacciones adversas al medicamento
Otras reacciones adversas al medicamento pueden considerarse efectos de clase de la somatropina, tales como una posible hiperglucemia causada por la disminución de la sensibilidad a la insulina, la disminución de la concentración de tiroxina libre y a una hipertensión intracraneal benigna.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Síntomas:
La sobredosis aguda puede conducir inicialmente a hipoglucemia y posteriormente a hiperglucemia.
La sobredosis prolongada puede causar signos y síntomas compatibles con los efectos conocidos de exceso de la hormona del crecimiento.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: hormonas del lóbulo anterior de la hipófisis y sus análogos, código ATC: H01AC01.
Omnitrope es un medicamento biosimilar. La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Mecanismo de acción
La somatropina es una potente hormona metabólica, importante en el metabolismo de lípidos, carbohidratos y proteínas. En los niños con hormona del crecimiento endógena insuficiente, la somatropina estimula el crecimiento lineal y aumenta la tasa de crecimiento. En los adultos y también en los niños, la somatropina mantiene una composición corporal normal aumentando la retención de nitrógeno y la estimulación del crecimiento del músculo esquelético y la movilización de la grasa corporal. El tejido adiposo visceral responde en particular a la somatropina. Además, para mejorar la lipólisis, la somatropina reduce la captación de triglicéridos de los depósitos de grasa corporal. Las concentraciones séricas de IGF-I (factor de crecimiento de tipo insulina I) y la IGFBP3 (proteína de unión del factor de crecimiento de tipo insulina 3) son aumentadas por la somatropina. Además, se han demostrado las siguientes acciones.
Efectos farmacodinámicos
Metabolismo lipídico
La somatropina induce los receptores de colesterol LDL hepáticos y afecta el perfil de lípidos séricos y las lipoproteínas. En general, la administración de somatropina a pacientes deficitarios de la hormona del crecimiento resulta en una reducción del DLD sérico y de la apoliproteína B. También se puede observar una reducción del colesterol total.
Metabolismo de los carbohidratos
La somatropina aumenta la insulina, pero la glucosa sanguínea en ayunas permanece, por lo general, sin cambios. Los niños con hipopituitarismo experimentan hipoglucemia en ayunas. Esta condición es corregida por la somatropina.
Metabolismo del agua y de los minerales
La deficiencia de la hormona del crecimiento se asocia a una reducción de los volúmenes plasmático y extracelular. Ambos aumentan rápidamente tras el tratamiento con somatropina. La somatropina induce la retención de sodio, potasio y fósforo.
Metabolismo óseo
La somatropina estimula el recambio del hueso esquelético. La administración a largo plazo de somatropina a pacientes con osteopenia con deficiencia de la hormona del crecimiento resulta en un aumento del contenido mineral y de la densidad ósea en los lugares portadores de peso.
Capacidad _ física
Mejoría de la potencia muscular y del ejercicio físico tras un tratamiento a largo plazo con somatropina. La somatropina también aumenta la frecuencia cardíaca, pero el mecanismo de acción no ha sido aún aclarado. Puede que una reducción de la resistencia vascular periférica contribuya a este efecto.
Eficacia clínica y seguridad
En ensayos clínicos en los niños y los adolescentes PEG y de talla baja se utilizaron dosis de 0,033 y 0,067 mg/kg de peso corporal, por día hasta alcanzar la altura final. En 56 pacientes tratados de forma continuada y que alcanzaron (casi) la altura final, el cambio principal de talla al inicio del tratamiento fue de +1,90 SDS (0,033 mg/kg de peso corporal por día) y +2,19 SDS (0,067 mg/kg de peso corporal por día). Los datos publicados de niños y adolescentes PEG no tratados sin estirón espontáneo y temprano sugieren un crecimiento tardío, de la SDS de 0,5. Los datos de seguridad a largo plazo son todavía limitados.
5.2 Propiedades farmacocinéticas
Absorción
La biodisponibilidad de la somatropina, administrada subcutáneamente, es aproximadamente del 80% en ambos casos, en las personas sanas y en los pacientes con deficiencia de la hormona de crecimiento. Una dosis subcutánea de 5 mg de Omnitrope 10 mg/1,5 ml solución inyectable en los adultos sanos produce unos valores plasmáticos de Cmax de 74 ± 22 microgramos/l, y de tmax, de
3,9 ± 1,2 horas.
Eliminación
La media de la vida media terminal de somatropina después de la administración intravenosa en los pacientes adultos con deficiencia de la hormona de crecimiento es de aproximadamente 0,4 horas. Sin embargo, después de la administración subcutánea de Omnitrope 10 mg/1,5 ml solución inyectable se alcanza una vida media de 3 horas. Es probable que la diferencia observada se deba a la lenta absorción de la inyección después de la administración subcutánea.
Poblaciones especiales
La biodisponibilidad absoluta de somatropina parece ser similar tanto en los hombres como en las mujeres después de la administración subcutánea.
La información sobre las propiedades farmacocinéticas de la somatropina en geriatría y pediatría, en diferentes razas y en los pacientes con insuficiencia renal, hepática o cardiaca es o bien escasa o incompleta.
5.3 Datos preclínicos sobre seguridad
En los estudios con Omnitrope referentes a toxicidad subaguda y tolerancia local no se han observado casos con efectos clínicamente relevantes.
En otros estudios con somatropina referente a toxicidad general, tolerancia local y reproducción de la toxicidad no se han observado efectos clínicamente relevantes.
Con somatropina, los estudios de genotoxicidad in vitro e in vivo o de mutaciones genéticas e inducción de aberraciones cromosómicas han sido negativos.
Se ha observado un aumento de fragilidad cromosómica en un estudio in vitro con linfocitos, tomados de pacientes en tratamiento prolongado con somatropina y bajo la adición de bleomicina, un medicamento radiomimético. El significado clínico de esta observación no está claro.
En otro estudio con somatropina, no se encontró incremento de anomalías cromosómicas en linfocitos de pacientes que recibieron terapia con somatropina durante largo tiempo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Fosfato hidrógeno disódico heptahidrato Fosfato dihidrógeno sódico dihidrato Glicina
Poloxámero 188 Fenol
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 18 meses.
Periodo de validez después del primer uso
Después del primer uso, el cartucho debe permanecer en la pluma y debe conservarse en una nevera (entre 2°C y 8°C) durante un máximo de 28 días. Conservar y transportar refrigerado (entre 2°C y 8°C).No congelar. Conservar en la pluma original para protegerlo de la luz.
6.4 Precauciones especiales de conservación
Cartucho sin abrir
Conservar y transportar refrigerado (entre 2°C y 8°C). No congelar. Conservar en el embalaje original para protegerlo de la luz.
Para las condiciones de conservación del medicamento en uso, ver sección 6.3.
6.5 Naturaleza y contenido del envase
1.5 ml de solución en un cartucho (de vidrio incoloro tipo I), con un émbolo en un lado (de bromobutilo siliconizado) un disco (de bromobutilo) y una cápsula de cierre (de aluminio) en el otro. El cartucho de vidrio está integrado irreversiblemente en un recipiente transparente y está montado en un mecanismo de plástico con una barra roscada en una extremidad.
Envases de 1, 5 y 10 cartuchos.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Omnitrope 10 mg/1,5 ml solución inyectable es una solución estéril y lista para usar que se presenta en un cartucho de vidrio destinado para inyección subcutánea,
Este cartucho está previsto para varios usos. Sólo debe administrarse con SurePal 10, un dispositivo de inyección específicamente desarrollado para la administración de Omnitrope 10 mg/1,5 ml solución inyectable. Se administrará utilizando agujas para inyección de pluma estériles y desechables. Los pacientes y los cuidadores deben recibir la formación e instrucciones adecuadas sobre el correcto uso de los cartuchos y de la pluma Omnitrope del médico o de otro profesional de la salud.
Lo que sigue es una descripción general del proceso de administración. Deben seguirse las instrucciones del fabricante para cargar el cartucho, conectar la aguja para inyección y para la administración.
1. Las manos deben lavarse.
2. Si la solución está turbia o si contiene material en partículas, no debe utilizarse. El contenido debe ser cristalino e incoloro.
3. Desinfectar la membrana de goma del cartucho con una torunda para limpiar.
4. Introducir el cartucho en la SurePal 10, según las instrucciones de uso suministradas con la pluma.
5. Limpiar el lugar de aplicación de la inyección con una torunda empapada en alcohol.
6. Administrar la dosis adecuada mediante inyección subcutánea, utilizando la aguja de pluma estéril. Extraer la aguja de la pluma y eliminarla con arreglo a los requisitos locales.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestrasse 10
A-6250 Kundl Austria
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/016
EU/1/06/332/017
EU/1/06/332/018
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 12 de abril de 2006 Fecha de la última renovación: 12 de abril de 2011
10. FECHA DE LA REVISIÓN DEL TEXTO
<{MM/AAAA}>
1. NOMBRE DEL MEDICAMENTO
Omnitrope 15 mg/1,5 ml solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada ml de solución contiene 10 mg de somatropina* (que corresponden a 30 UI). Un cartucho contiene 1,5 ml, que corresponde a 15 mg de somatropina* (45 UI).
* Producida en Escherichia coli mediante tecnología de DNA recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable en un cartucho para SurePal 15 La solución es límpida e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lactantes, niños y adolescentes:
- Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento (deficiencia de la hormona del crecimiento, DGH).
- Trastorno del crecimiento asociado al síndrome de Turner.
- Trastorno del crecimiento asociado a insuficiencia renal crónica.
- Trastorno del crecimiento (puntuación de la desviación estándar actual de la talla (SDS) < -2,5 y
SDS de la talla < -1 ajustada para los padres) en los niños y los adolescentes nacidos con baja
talla para su edad gestacional (PEG), con un peso al nacer y/o longitud por debajo de -
2 desviaciones estándar (DE), que no alcanzaron el estirón de crecimiento (velocidad de crecimiento (VC) SDS < 0 durante el último año) a los 4 años de edad o más.
- El síndrome de Prader-Willi (SPW) para la mejoría del crecimiento y composición corporal. El diagnóstico de SPW debe ser confirmado por pruebas genéticas apropiadas.
Adultos
- Terapia de sustitución en los adultos con deficiencia pronunciada de la hormona del crecimiento.
- Inicio en la edad adulta: Pacientes que padecen una deficiencia grave de la hormona del crecimiento, asociada a múltiples deficiencias hormonales a consecuencia de una patología hipotalámica o hipofisaria conocida y que padecen, por lo menos, una deficiencia conocida de una hormona hipofisaria que no sea la prolactina. Estos pacientes deben someterse a una prueba dinámica adecuada, para así diagnosticar o excluir una deficiencia de la hormona del crecimiento.
- Inicio en la niñez: Pacientes que padecían una deficiencia de la hormona del crecimiento en la niñez a consecuencia de causas congénitas, genéticas, adquiridas o idiopáticas. En los pacientes con DGH de inicio en la niñez se debe reevaluar la capacidad secretora de la hormona del crecimiento después de concluir su crecimiento longitudinal. En los pacientes con una probabilidad alta de DGH persistente, es decir, una causa congénita o una DGH secundaria a una enfermedad o lesión hipotálamo-hipofisaria, un SDS < -2 del factor de crecimiento de tipo insulínico I (IGF-I) cuando no reciben tratamiento con hormona de crecimiento por lo menos durante cuatro semanas debe considerarse una prueba suficiente de DGH profunda.
Todos los demás pacientes requerirán un análisis del IGF-I y una prueba de estimulación con la hormona del crecimiento.
4.2 Posología y forma de administración
El diagnóstico y el tratamiento con somatropina deben iniciarse y monitorizarse por médicos que tengan la capacitación y la experiencia adecuadas en el diagnóstico y el tratamiento de los pacientes con trastornos del crecimiento.
Posología
Población pediátrica
La posología y la pauta de dosificación deben individualizarse.
Trastorno del crecimiento debido a secreción insuficiente de la hormona del crecimiento en los pacientes pediátricos:
En términos generales, se recomienda una dosis de 0,025 a 0,035 mg/kg de peso corporal por día o de 0,7 a 1,0 mg/m2 de superficie corporal por día. Incluso dosis más altas han sido utilizadas.
Cuando la DGH de inicio en la niñez persiste hasta la adolescencia, se debe continuar el tratamiento a fin de alcanzar un desarrollo somático completo (p. ej., composición corporal, masa ósea). Para la monitorización, el logro de una masa ósea máxima normal, definida como una puntuación T > -1 (es decir, normalizado hasta la masa ósea máxima promedio en el adulto, determinada mediante absorciometría de rayos X de doble energía, teniendo en cuenta el sexo y el origen étnico del paciente) es uno de los objetivos terapéuticos durante el período de transición. Para la orientación en cuanto a la posología, ver el apartado sobre los adultos, a continuación.
Síndrome de Prader-Willi, para la mejoría del crecimiento y composición corporal en los pacientes pediátricos
En general, se recomienda una dosis de 0,035 mg/kg de peso corporal por día ó 1,0 mg/m2 de superficie corporal por día. No se excederán las dosis diarias de 2,7 mg. El tratamiento no se utilizará en los pacientes pediátricos con una velocidad de crecimiento inferior a 1 cm al año y cerca del cierre de las epífisis.
Trastorno del crecimiento debido al síndrome de Turner
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal al día ó 1,4 mg/m2 de superficie corporal.
Trastorno del crecimiento en la insuficiencia renal crónica
Se recomienda una dosis de 0,045 a 0,050 mg/kg de peso corporal y día (de 1,4 mg/m2 de superficie corporal y día). Pueden precisarse dosis más altas si la velocidad de crecimiento es muy lenta. Puede ser necesario corregir la dosis después de seis meses de tratamiento (ver sección 4.4).
Trastorno del crecimiento en los niños y los adolescentes nacidos con baja talla para su edad gestacional (PEG)
Se recomienda una dosis de 0,035 mg/kg de peso corporal al día (1 mg/m2 de superficie corporal al día) hasta que se alcance la talla final (ver sección 5.1). El tratamiento se debe interrumpir después del primer año de tratamiento, si la SDS de la velocidad de crecimiento es inferior a + 1. El tratamiento se interrumpirá si la velocidad de crecimiento es < 2 cm/año y, si se requiere confirmación, la edad ósea es > 14 años (niñas) o > 16 años (niños), correspondiente con el cierre de las placas de crecimiento epifisario.
Posología recomendada en pacientes pediátricos
|
Indicación |
Dosis diaria en mg/kg de peso corporal |
Dosis diaria en mg/m2 de superficie corporal |
|
Deficiencia de la hormona del crecimiento |
0,025 - 0,035 |
0,7 - 1,0 |
|
Síndrome de Prader-Willi |
0,035 |
1,0 |
|
Síndrome de Turner |
0,045 - 0,050 |
1,4 |
|
Insuficiencia renal crónica |
0,045 - 0,050 |
1,4 |
|
Niños y adolescentes nacidos con talla baja para su edad gestacional (PEG) |
0,035 |
1,0 |
Pacientes adultos con deficiencia de la hormona del crecimiento
La dosis recomendada para reanudar el tratamiento en aquellos pacientes que continúen con hormona de crecimiento tras un déficit de GH en la infancia, es de 0,2-0,5 mg por día. La dosis se debe ir incrementando o disminuyendo gradualmente de acuerdo a las necesidades individuales del paciente, determinadas de acuerdo a la concentración del IGF-I.
En los pacientes con DGH de inicio en la edad adulta, el tratamiento debe iniciarse con una dosis baja, de 0,15 a 0,3 mg al día. La dosis se aumentará gradualmente según las necesidades de cada paciente y según lo determine la concentración de IGF-I.
En ambos casos, el objetivo del tratamiento debe ser alcanzar concentraciones de factor de crecimiento de tipo insulina (IGF-I) dentro de 2 SDS de la media corregida por la edad. A los pacientes con concentraciones IGF-I normales al inicio del tratamiento se les debe administrar la hormona del crecimiento hasta una concentración de IGF-I dentro de los límites superiores normales, sin sobrepasar 2 SDS. La respuesta clínica y los efectos adversos pueden también usarse como guía para el ajuste de la dosis. Se conoce que hay pacientes con DGH que no llegan a normalizar los niveles de IGF-I a pesar de tener una buena respuesta clínica, y por tanto, no requieren ajuste de dosis. La dosis de mantenimiento raramente excede 1,0 mg al día. Las mujeres pueden necesitar dosis más altas que los hombres, mientras que los varones muestran una sensibilidad mayor al aumento de la IGF-I con el tiempo. Esto significa que hay un riesgo de que las mujeres, en especial aquellas en sustitución con estrógenos orales, estén infra-tratadas mientras que los hombres estén sobre-tratados. La exactitud de la dosis de la hormona del crecimiento debe, por lo tanto, controlarse cada 6 meses. Habida cuenta de que la producción fisiológica de la hormona del crecimiento desciende con la edad, pueden reducirse las necesidades de la dosis.
Poblaciones especiales
Personas de edad avanzada
En los pacientes mayores de 60 años, el tratamiento debe comenzar con una dosis de 0,1 a 0,2 mg diarios, y debe aumentarse gradualmente, según las necesidades individuales del paciente. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento en estos pacientes rara vez es superior a 0,5 mg diarios.
Forma de administración
Se debe administrar la inyección por vía subcutánea, cambiando el lugar de inyección para evitar la lipoatrofia.
Para consultar las instrucciones de uso y manipulación, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes.
La somatropina no debe usarse cuando exista evidencia de actividad de un tumor. Los tumores intracraneales deben ser inactivos y el tratamiento antitumoral se debe completar antes de iniciar el tratamiento con GH. El tratamiento se debe suspender si hay pruebas de crecimiento tumoral.
La somatropina no debe usarse en la promoción del crecimiento de niños con epífisis cerradas.
Los pacientes con enfermedad crítica aguda que presenten complicaciones después de cirugía cardiaca, abdominal, traumatismo múltiple por accidente, insuficiencia respiratoria aguda o enfermedades similares, no deben ser tratados con somatropina (en cuanto a los pacientes sometidos a tratamiento de sustitución, ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
No se debe superar la dosis diaria máxima recomendada (ver sección 4.2).
Sensibilidad a la insulina
La somatropina puede reducir la sensibilidad a la insulina. En pacientes con diabetes mellitus, puede ser necesario ajustar la dosis de insulina tras la instauración del tratamiento con somatropina. Durante el tratamiento con somatropina, se deben monitorizar cuidadosamente los pacientes con diabetes, intolerancia a la glucosa o con factores de riesgo adicionales de sufrir diabetes.
Función tiroidea
La hormona de crecimiento aumenta la conversión extratiroidea de T4 a T3, lo que puede causar una reducción de la concentración T4 y un aumento de la T3 en el suero. Si bien las concentraciones de la hormona tiroidea periférica han permanecido dentro de los límites de referencia en la mayoría de sujetos sanos, teóricamente, se puede desarrollar hipotiroidismo en los sujetos con hipotiroidismo subclínico. En consecuencia, se debe llevar a cabo una monitorización de la función tiroidea en todos los pacientes. El potencial efecto del tratamiento con hormona de crecimiento sobre la función tiroidea se debe monitorizar cuidadosamente en pacientes con hipopituitarismo tratados con terapia sustitutiva estándar.
En la deficiencia de la hormona del crecimiento, secundaria al tratamiento de una neoplasia maligna, se recomienda prestar atención a los signos de recaída de la malignidad. Entre los supervivientes de cáncer pediátrico se ha referido un mayor riesgo de una segunda neoplasia en los pacientes tratados con somatropina después de su primera neoplasia. Las más frecuentes de estas segundas neoplasias en los pacientes tratados con radioterapia en la cabeza para su primera neoplasia fueron los tumores intracraneales, en concreto los meningiomas.
En los pacientes con trastornos endocrinos, incluida la deficiencia de la hormona del crecimiento, el deslizamiento de la epífisis de la cadera puede producirse con más frecuencia que en la población general. Los pacientes que cojean durante el tratamiento con somatropina deben ser controlados clínicamente.
Hipertensión intracraneal benigna
En casos de cefalea recurrente o grave, problemas visuales, náuseas y/o vómitos, se recomienda practicar una fundoscopia para descartar un edema de papila. Si se confirma el edema de papila, debe considerarse una hipertensión intracraneal benigna y, si fuese apropiado, debe de retirarse el tratamiento con la hormona del crecimiento. Si se reinicia el tratamiento con hormona del crecimiento, se debe instaurar un control cuidadoso para, si fuese necesario, detectar la hipertensión intracraneal.
Leucemia
Se ha notificado leucemia en un pequeño número de pacientes con deficiencia de la hormona de crecimiento, algunos de los cuales han sido tratados con somatropina. Sin embargo, no existe evidencia de que haya un aumento en la incidencia de la leucemia en pacientes tratados con hormona del crecimiento sin factores de predisposición.
Anticuerpos
En un pequeño porcentaje de pacientes pueden aparecer anticuerpos dirigidos contra Omnitrope. Omnitrope ha dado lugar a la formación de anticuerpos aproximadamente en el 1% de los pacientes. La capacidad de fijación de estos anticuerpos es baja y no hay ningún efecto sobre la velocidad de crecimiento. En cualquier paciente con una falta de respuesta por lo demás no explicada se deben hacer análisis de anticuerpos contra la somatropina.
Pacientes de edad avanzada
La experiencia con los pacientes mayores de 80 años es limitada. Los pacientes de edad avanzada pueden ser más sensibles a la acción de Omnitrope y, por lo tanto, pueden ser más propensos a presentar reacciones adversas.
Enfermedad agudas graves
Los efectos de la somatropina sobre la recuperación de pacientes adultos críticos se han evaluado en dos ensayos controlados con placebo en 522 pacientes adultos con complicaciones posteriores a cirugía a corazón abierto, cirugía abdominal, traumatismo accidental múltiple o insuficiencia respiratoria aguda. La mortalidad fue superior en los pacientes tratados diariamente con 5,3 u 8 mg de somatropina, en comparación con los pacientes que recibieron placebo, 42% vs. 19%. En base a esta información, este tipo de pacientes no deben ser tratados con somatropina. Dado que no existe información disponible sobre la seguridad del tratamiento sustitutivo con hormona de crecimiento en pacientes críticos, los beneficios del tratamiento continuado en esta situación deben considerarse sobre la base de los riesgos potenciales.
En todos los pacientes que desarrollen algún otro tipo de enfermedad aguda en fase crítica o similar, el posible beneficio del tratamiento con somatropina deberá ser sopesado en relación con el riesgo potencial que implica.
Población pediátrica
Pancreatitis
A pesar de que la pancreatitis es rara, debe ser considerada en niños tratados con somatropina que presenten dolor abdominal.
Síndrome de Prader-Willi
En los pacientes con SPW, el tratamiento debe ser siempre en combinación con una dieta baja en calorías.
Existen informes de muertes asociadas al uso de la hormona del crecimiento en los pacientes pediátricos con SPW que presentaron uno o más factores de riesgo: obesidad grave (aquellos pacientes con un peso/altura superior al 200%), antecedentes de insuficiencia respiratoria o apnea del sueño o de infección respiratoria no identificada. Los pacientes con SPW y uno o más de estos factores de riesgo podrían estar en mayor riesgo.
Antes de iniciar el tratamiento con somatropina los pacientes con SPW serán valorados respecto de la obstrucción de las vías aéreas altas, apnea del sueño o infección respiratoria.
Si, durante la evaluación de la obstrucción de las vías respiratorias altas, se observan resultados anatomopatológicos, se debe derivar al niño al otorrinolaringólogo (ENT) para el tratamiento y la resolución del trastorno respiratorio antes de iniciar el tratamiento con hormona de crecimiento.
La apnea del sueño debe evaluarse antes del inicio del tratamiento con hormona de crecimiento por métodos reconocidos como la polisomnografía o la oximetría, durante la noche, y los pacientes deben controlarse, si se sospecha apnea del sueño.
Si, durante el tratamiento con somatropina, los pacientes muestran signos de obstrucción de las vías respiratorias altas (incluido el inicio o el aumento de ronquidos), el tratamiento debe interrumpirse y debe efectuarse una nueva evaluación de ENT.
Todos los pacientes con SPW deben valorarse en cuanto a la apnea y ser controlados si se sospecha la presencia de apnea del sueño. Los pacientes deben controlarse para detectar signos de infecciones respiratorias, que deben ser diagnosticadas lo más pronto posible y tratadas de forma contundente.
Todos los pacientes con SPW deben someterse a un control eficaz del peso antes y, también, durante el tratamiento con hormona del crecimiento.
La escoliosis es muy frecuente en los pacientes con SPW. La escoliosis puede progresar en cualquier niño durante el crecimiento rápido. Deben controlarse los signos de escoliosis durante el tratamiento.
La experiencia con el tratamiento prolongado en los adultos y en los pacientes con SPW es limitada.
Nacidos pequeños para su edad gestacional
En los niños y los adolescentes PEG nacidos bajos antes de iniciar el tratamiento se deben descartar otras razones médicas u otros tratamientos que pudieran explicar la perturbación del crecimiento antes de iniciar el tratamiento.
En los niños y los adolescentes PEG se recomienda medir la insulina y la glucosa sanguínea en ayunas antes de iniciar el tratamiento y, posteriormente, en períodos anuales. En los pacientes con un aumento del riesgo de diabetes mellitus (p. ej., antecedentes familiares de diabetes, obesidad, resistencia grave a la insulina, acantosis nigricans) debe realizarse la prueba de tolerancia a la glucosa (OGTT). Si aparece una diabetes clara, no se debe administrar hormona del crecimiento.
En los niños y los adolescentes PEG se recomienda medir la concentración de IGF-I antes de iniciar el tratamiento y después, dos veces al año. Si en mediciones repetidas las concentraciones de IGF-I exceden en +2 DS comparado con la edad de referencia y estado puberal, la proporción IGF-I/IGFBP-3 debe tenerse en cuenta para considerar un ajuste de la dosis.
La experiencia al iniciar el tratamiento en los pacientes PEG cerca del comienzo de la pubertad es limitada. Por tanto, no se recomienda iniciar el tratamiento cerca de la pubertad. La experiencia en los pacientes con síndrome de Silver-Rusell es limitada.
Parte del aumento de estatura obtenido al tratar a los niños y los adolescentes nacidos PEG y bajos con hormona del crecimiento puede perderse si se interrumpe el tratamiento antes de que se alcance la estatura final.
Insuficiencia renal crónica
En la insuficiencia renal crónica, la función renal debe ser un 50% de la normal antes de iniciar el tratamiento. Para verificar la perturbación del crecimiento, se debe hacer un seguimiento del crecimiento durante un año antes de iniciar el tratamiento. Durante este periodo, se debe iniciar un tratamiento conservador para la insuficiencia renal (incluido el control de la acidosis, el hiperparatiroidismo y el estado nutricional), y debe mantenerse durante todo el tratamiento.
El tratamiento se debe interrumpir en casos de trasplante renal.
Hasta la fecha, no se disponen de datos de la estatura final de los pacientes con insuficiencia renal crónica tratados con Omnitrope.
4.5 Interacción con otros medicamentos y otras formas de interacción
El tratamiento concomitante con glucocorticoides puede inhibir los efectos de promoción del crecimiento de los productos que contienen somatropina. Por tanto, los pacientes tratados con glucocorticoides deben tener su crecimiento cuidadosamente monitorizado con el fin de valorar el potencial impacto del tratamiento con glucocorticoides sobre el crecimiento.
Los datos de un estudio de interacción, realizado en los adultos con deficiencia de la hormona del crecimiento, sugieren que la administración de somatropina puede aumentar la eliminación de compuestos que son metabolizados por las isoenzimas del citocromo P450. El metabolismo de los compuestos metabolizados por el citocromo P450 3A4 (p. ej.: esteroides sexuales, corticoesteroides, anticonvulsivantes y ciclosporinas) puede aumentar, dando como resultado unas concentraciones plasmáticas más bajas de estos compuestos. Se desconoce la significación clínica de esto.
Ver también en la sección 4.4 la información relacionada con la diabetes mellitus y los trastornos tiroideos, y en la sección 4.2 la información sobre la terapia de sustitución de estrógenos orales.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos o éstos son limitados relativos al uso de somatropina en mujeres embarazadas. Los estudios realizados en animales son insuficientes en términos de toxicidad para la reproducción (ver sección 5.3). No se recomienda utilizar somatropina durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Lactancia
No se han llevado a cabo estudios clínicos con medicamentos que contienen somatropina en mujeres en periodo de lactancia. Se desconoce si la somatropina se excreta en la leche materna, pero es poco probable la absorción de proteína intacta del tubo digestivo del niño. Por tanto, se debe tener precaución cuando se administre Omnitrope a mujeres en periodo de lactancia.
Fertilidad
No se han realizado estudios de fertilidad con Omnitrope.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Omnitrope sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los pacientes que padecen una deficiencia de la hormona de crecimiento se caracterizan por un déficit del volumen extracelular. Al iniciar el tratamiento con somatropina, este déficit se corrige rápidamente. En los pacientes adultos, son frecuentes las reacciones adversas relacionadas con la retención de líquidos, como el edema periférico, la rigidez musculoesquelética, la artralgia, la mialgia y las parestesias. En general, estas reacciones son leves a moderadas, se producen en los primeros meses de tratamiento y remiten espontáneamente o al reducir la dosis.
La incidencia de estas reacciones adversas está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de hormona de crecimiento. En los niños, tales reacciones adversas son poco frecuentes.
Omnitrope ha dado lugar a la formación de anticuerpos en aproximadamente el 1% de los pacientes. La capacidad de fijación de estos anticuerpos ha sido baja y no hay cambios clínicos asociados a su formación (ver sección 4.4).
Tabla de reacciones adversas
Las tablas 1-6 muestran las reacciones adversas clasificadas según los apartados del sistema de clasificación de órganos y las frecuencias, utilizando la convención siguiente: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); no conocida (no puede estimarse a partir de los datos disponibles) para cada una de las afecciones indicadas.
Ensayos clínicos en niños con DGH
|
Tabla 1 Tratamiento a largo plazo de niños con alteración del crecimiento debida a una secreción insuficiente de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias13 Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia13 |
Mialgia13 Rigidez musculoesquelética13 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico13 | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre13 | |||||
Ensayos clínicos en niños con síndrome de Turner
|
Tabla 2 Tratamiento a largo plazo de niños con alteración del crecimiento debida al síndrome de Turner | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico* Reacción en el lugar de la inyección$ | |||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* | |||||
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con insuficiencia renal crónica
|
Tabla 3 | ||||||
|
Tratamiento a largo plazo de niños con alteración del crecimiento debida a una insuficiencia renal crónica | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas |
Leucemiaf | |||||
|
y no especificadas (incl. quistes y pólipos) | ||||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* Rigidez musculoesquelética* | |||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con PEG
|
Tabla 4 Tratamiento a largo plazo de niños con alteración del crecimiento debida al nacimiento con baja talla para su edad gestacional | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión | |||||
|
intracraneal benigna | ||||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* |
Mialgia* Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección |
Edema periférico* | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre* |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en niños con SPW
|
Tabla 5 Tratamiento a largo debida al síndrome ( |
plazo y mejora de la composición corporal en niños con alteración del crecimiento e Prader-Willi | |||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Leucemiaf | |||||
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo II | |||||
|
Trastornos del sistema nervioso |
Parestesias* Hipertensión intracraneal benigna | |||||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia* Mialgia* |
Rigidez musculoesquelética* | ||||
|
Trastornos generales y alteraciones en el lugar de |
Edema periférico* |
Reacción en el lugar de la inyección$ | ||||
|
administración | ||||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre14 |
*En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
Ensayos clínicos en adultos con DGH
|
Tabla 6 Terapia de sustitución en adultos con deficiencia de la hormona del crecimiento | ||||||
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes >1/100 a <1/10 |
Poco frecuentes >1/1.000 a <1/100 |
Raras >1/10.000 a <1/1.000 |
Muy raras <1/10.000 |
No conocida (no puede estimarse a partir de los datos disponibles) |
|
Trastornos del metabolismo y de la nutrición |
Diabetes mellitus de tipo 2 | |||||
|
Trastornos del sistema nervioso |
Parestesias * Síndrome del túnel carpiano |
Hipertensión intracraneal benigna | ||||
|
Trastornos musculoesquelét icos y del tejido conjuntivo |
Artralgia14 |
Mialgia14 Rigidez musculoesq uelética14 | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema periférico14 |
Reacción en el lugar de la inyección$ | ||||
|
Exploraciones complementarias |
Disminución del cortisol en sangre14 | |||||
Descripción de reacciones adversas seleccionadas Niveles reducidos de cortisol sérico
Se ha notificado que la somatropina reduce las concentraciones séricas de cortisol, posiblemente al afectar a las proteínas transportadoras o mediante un aumento de la depuración hepática. La relevancia clínica de estas observaciones puede ser limitada. No obstante, antes de iniciar el tratamiento, debe optimizarse el tratamiento de reemplazo con corticoesteroides.
Síndrome de Prader-Willi
Se han notificado casos raros de muerte súbita en pacientes con síndrome de Prader-Willi tratados con somatropina en la experiencia post-comercialización, aunque no se ha demostrado que exista relación causal.
Leucemia
Se han notificado casos (raros o muy raros) de leucemia en los niños que padecen una deficiencia de la hormona de crecimiento y son tratados con somatropina e incluidos en la experiencia post-comercialización. Sin embargo, no existen indicios de un aumento del riesgo de leucemia sin factores predisponentes, tales como la radioterapia cerebral o de la cabeza.
Epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes
Se han notificado epifisiólisis de la cabeza femoral y enfermedad de Legg-Calvé-Perthes en niños tratados con la hormona del crecimiento. La epifisiólisis de la cabeza femoral se produce con mayor frecuencia en caso de trastornos endocrinos y la enfermedad de Legg-Calvé-Perthes es más frecuente en caso de baja estatura. No obstante, se desconoce si estas dos afecciones aparecen o no con mayor frecuencia durante el tratamiento con somatropina. Debe tenerse en cuenta la posibilidad de su diagnóstico en niños que presenten molestias o dolor en la cadera o la rodilla.
Otras reacciones adversas al medicamento
Otras reacciones adversas al medicamento pueden considerarse efectos de clase de la somatropina, tales como una posible hiperglucemia causada por la disminución de la sensibilidad a la insulina, la disminución de la concentración de tiroxina libre y a una hipertensión intracraneal benigna.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Síntomas:
La sobredosis aguda puede conducir inicialmente a hipoglucemia y posteriormente a hiperglucemia.
La sobredosis prolongada puede causar signos y síntomas compatibles con los efectos conocidos de exceso de la hormona del crecimiento.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: hormonas del lóbulo anterior de la hipófisis y sus análogos, código ATC: H01AC01.
Omnitrope es un medicamento biosimilar. La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Mecanismo de acción
La somatropina es una potente hormona metabólica, importante en el metabolismo de lípidos, carbohidratos y proteínas. En los niños con hormona del crecimiento endógena insuficiente, la somatropina estimula el crecimiento lineal y aumenta la tasa de crecimiento. En los adultos y también en los niños, la somatropina mantiene una composición corporal normal aumentando la retención de nitrógeno y la estimulación del crecimiento del músculo esquelético y la movilización de la grasa corporal. El tejido adiposo visceral responde en particular a la somatropina. Además, para mejorar la lipólisis, la somatropina reduce la captación de triglicéridos de los depósitos de grasa corporal. Las concentraciones séricas de IGF-I (factor de crecimiento de tipo insulina I) y la IGFBP3 (proteína de unión del factor de crecimiento de tipo insulina 3) son aumentadas por la somatropina. Además, se han demostrado las siguientes acciones.
Efectos farmacodinámicos
Metabolismo lipídico
La somatropina induce los receptores de colesterol LDL hepáticos y afecta el perfil de lípidos séricos y las lipoproteínas. En general, la administración de somatropina a pacientes deficitarios de la hormona del crecimiento resulta en una reducción del DLD sérico y de la apoliproteína B. También se puede observar una reducción del colesterol total.
Metabolismo de los carbohidratos
La somatropina aumenta la insulina, pero la glucosa sanguínea en ayunas permanece, por lo general, sin cambios. Los niños con hipopituitarismo experimentan hipoglucemia en ayunas. Esta condición es corregida por la somatropina.
Metabolismo del agua y de los minerales
La deficiencia de la hormona del crecimiento se asocia a una reducción de los volúmenes plasmático y extracelular. Ambos aumentan rápidamente tras el tratamiento con somatropina. La somatropina induce la retención de sodio, potasio y fósforo.
Metabolismo óseo
La somatropina estimula el recambio del hueso esquelético. La administración a largo plazo de somatropina a pacientes con osteopenia con deficiencia de la hormona del crecimiento resulta en un aumento del contenido mineral y de la densidad ósea en los lugares portadores de peso.
Capacidad _ física
Mejoría de la potencia muscular y del ejercicio físico tras un tratamiento a largo plazo con somatropina. La somatropina también aumenta la frecuencia cardíaca, pero el mecanismo de acción no ha sido aún aclarado. Puede que una reducción de la resistencia vascular periférica contribuya a este efecto.
Eficacia clínica y seguridad
En ensayos clínicos en los niños y los adolescentes PEG y de talla baja se utilizaron dosis de 0,033 y 0,067 mg/kg de peso corporal, por día hasta alcanzar la altura final. En 56 pacientes tratados de forma continuada y que alcanzaron (casi) la altura final, el cambio principal de talla al inicio del tratamiento fue de +1,90 SDS (0,033 mg/kg de peso corporal por día) y +2,19 SDS (0,067 mg/kg de peso corporal por día). Los datos publicados de niños y adolescentes PEG no tratados sin estirón espontáneo y temprano sugieren un crecimiento tardío, de la SDS de 0,5. Los datos de seguridad a largo plazo son todavía limitados.
5.2 Propiedades farmacocinéticas
Absorción
La biodisponibilidad de la somatropina, administrada subcutáneamente, es aproximadamente del 80% en ambos casos, en las personas sanas y en los pacientes con deficiencia de la hormona de crecimiento. Una dosis subcutánea de 5 mg de Omnitrope 15 mg/1,5 ml solución inyectable en los adultos sanos produce unos valores plasmáticos de Cmax de 52 ± 19 microgramos/l, y de tmax, de
3,7 ± 1,2 horas.
Eliminación
La media de la vida media terminal de la somatropina después de la administración intravenosa en los pacientes adultos con deficiencia de la hormona de crecimiento es de aproximadamente 0,4 horas. Sin embargo, después de la administración subcutánea de Omnitrope 15 mg/1,5 ml solución inyectable se alcanza una vida media de 2,76 horas. Es probable que la diferencia observada se deba a la lenta absorción de la inyección después de la administración subcutánea.
Poblaciones especiales
La biodisponibilidad absoluta de somatropina parece ser similar tanto en los hombres como en las mujeres después de la administración subcutánea.
La información sobre las propiedades farmacocinéticas de somatropina en geriatría y pediatría, en diferentes razas y en los pacientes con insuficiencia renal, hepática o cardiaca es o bien escasa o incompleta.
5.3 Datos preclínicos sobre seguridad
En los estudios con Omnitrope referentes a toxicidad subaguda y tolerancia local no se han observado casos con efectos clínicamente relevantes.
En otros estudios con somatropina referente a toxicidad general, tolerancia local y reproducción de la toxicidad no se han observado efectos clínicamente relevantes.
Con somatropina, los estudios de genotoxicidad in vitro e in vivo o de mutaciones genéticas e inducción de aberraciones cromosómicas han sido negativos.
Se ha observado un aumento de fragilidad cromosómica en un estudio in vitro con linfocitos, tomados de pacientes en tratamiento prolongado con somatropina y bajo la adición de bleomicina, un medicamento radiomimético. El significado clínico de esta observación no está claro.
En otro estudio con somatropina, no se encontró incremento de anomalías cromosómicas en linfocitos de pacientes que recibieron terapia con somatropina durante largo tiempo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Fosfato hidrógeno disódico heptahidrato Fosfato dihidrógeno sódico dihidrato Cloruro de sodio Poloxámero 188 Fenol
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
Periodo de validez después del primer uso
Después del primer uso, el cartucho debe permanecer en la pluma y debe conservarse en una nevera (entre 2°C y 8°C) durante un máximo de 28 días. Conservar y transportar refrigerado (entre 2°C y 8°C).No congelar. Conservar en la pluma original para protegerlo de la luz.
6.4 Precauciones especiales de conservación
Cartucho sin abrir
Conservar y transportar refrigerado (entre 2°C y 8°C). No congelar. Conservar en el embalaje original para protegerlo de la luz.
Para las condiciones de conservación del medicamento en uso, ver sección 6.3.
6.5 Naturaleza y contenido del envase
1.5 ml de solución en un cartucho (de vidrio incoloro tipo I), con un émbolo y un anillo azul en un lado (de bromobutilo siliconizado) un disco (de bromobutilo) y una cápsula de cierre (de aluminio) en el otro. El cartucho de vidrio está integrado irreversiblemente en un recipiente transparente y está montado en un mecanismo de plástico con una barra roscada en una extremidad.
Envases de 1, 5 y 10 cartuchos.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Omnitrope 15 mg/1,5 ml solución inyectable es una solución estéril y lista para usar que se presenta en un cartucho de vidrio destinado para inyección subcutánea,
Este cartucho está previsto para varios usos. Sólo debe administrarse con SurePal 15, un dispositivo de inyección específicamente desarrollado para la administración de Omnitrope 15 mg/1,5 ml solución inyectable. Se administrará utilizando agujas para inyección de pluma estériles y desechables. Los pacientes y los cuidadores deben recibir la formación e instrucciones adecuadas sobre el correcto uso de los cartuchos y de la pluma Omnitrope del médico o de otro profesional de la salud.
Lo que sigue es una descripción general del proceso de administración. Deben seguirse las instrucciones del fabricante para cargar el cartucho, conectar la aguja para inyección y para la administración.
1. Las manos deben lavarse.
2. Si la solución está turbia o si contiene material en partículas, no debe utilizarse. El contenido debe ser cristalino e incoloro.
3. Desinfectar la membrana de goma del cartucho con una torunda para limpiar.
4. Introducir el cartucho en SurePal 15, según las instrucciones de uso suministradas con la
pluma.
5. Limpiar el lugar de aplicación de la inyección con una torunda empapada en alcohol.
6. Administrar la dosis adecuada mediante inyección subcutánea, utilizando la aguja de pluma estéril. Extraer la aguja de la pluma y eliminarla con arreglo a los requisitos locales.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestrasse 10
A-6250 Kundl Austria
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/010 EU/1/06/332/011 EU/1/06/332/012
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 12 de abril de 2006 Fecha de la última renovación: 12 de abril de 2011
10. FECHA DE LA REVISIÓN DEL TEXTO
<{MM/AAAA}>
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES RELATIVAS AL USO SEGURO Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE DEL PRINCIPO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante del principio activo biológico.
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Austria
Nombre y dirección del fabricante responsable de la liberación de los lotes
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Austria
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
1. NOMBRE DEL MEDICAMENTO
Omnitrope, 1,3 mg/ml (4 UI) polvo y disolvente para solución inyectable Somatropina
2. PRINCIPIO ACTIVO
Somatropina 1,3 mg (4 UI/ml) en un vial. Tras la reconstitución, un vial contiene 1,3 mg de somatropina (que corresponde a 4 UI) por ml.
3. LISTA DE EXCIPIENTES
Otros componentes:
Polvo: glicina, fosfato de hidrógeno disódico heptahidrato, fosfato dihidrógeno sódico dihidrato. Disolvente: agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 vial de 1,3 mg de polvo 1 vial de 1 ml de disolvente Envase de 1 unidad.
5. FORMA Y VÍA DE ADMINISTRACIÓN
Para un solo uso. Utilizar solo la solución cristalina. Leer el prospecto antes de utilizar este medicamento. Para uso por vía subcutánea después de la reconstitución
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRAS ADVERTENCIA ESPECIALE, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar y transportar refrigerado (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestr. 10 A-6250 Kundl Austria
12. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Omnitrope 1,3 mg/ml
ETIQUETA DEL VIAL DE OMNITROPE
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Omnitrope 1,3 mg/ml polvo para solución inyectable Somatropina
SC
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Disolvente para Omnitrope (agua para preparaciones inyectables) Vía subcutánea
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
1. NOMBRE DEL MEDICAMENTO
Omnitrope 5 mg/ml polvo y disolvente para solución inyectable Somatropina
2. PRINCIPIO ACTIVO
Somatropina 5 mg (15 UI)/ml en un vial. Tras la reconstitución, un cartucho contiene 5 mg de somatropina (que corresponde a 15 UI) por ml.
3. LISTA DE EXCIPIENTES
Otros componentes:
Polvo: glicina, fosfato de hidrógeno disódico heptahidrato, fosfato dihidrógeno sódico dihidrato. Disolvente: alcohol bencílico, agua para preparaciones inyectables Contiene alcohol bencílico. Ver información adicional en el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 vial de 5 mg de polvo 1 cartucho de 1 ml de disolvente 5 viales de 5 mg de polvo 5 cartuchos de 1 ml de disolvente
5. FORMA Y VÍA DE ADMINISTRACIÓN
Utilizar solo la solución cristalina. Utilizar solo con Omnitrope Pen L. Leer el prospecto antes de utilizar este medicamento.
Para uso por vía subcutánea tras la reconstitución
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA ADVERTENCIA ESPECIAL, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar y transportar refrigerado (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestr. 10 A-6250 Kundl Austria
12. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/002
EU/1/06/332/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE LA DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Omnitrope 5 mg/ml
ETIQUETA DEL VIAL DE OMNITROPE
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Omnitrope 5 mg/ml polvo para solución inyectable
Somatropina
SC
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
ETIQUETA DEL CARTUCHO DE DISOLVENTE
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Disolvente para Omnitrope (agua para preparaciones inyectables con alcohol bencílico al 1,5%) Vía subcutánea
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
1. NOMBRE DEL MEDICAMENTO
Omnitrope 5 mg/1,5 ml solución inyectable Somatropina
2. PRINCIPIO ACTIVO
Somatropina 3,3 mg (10 UI)/ml en un cartucho.
Un cartucho contiene 1,5 ml, que corresponde a 5 mg de somatropina (15 UI).
3. LISTA DE EXCIPIENTES
Otros componentes: fosfato hidrógeno disódico heptahidrato, fosfato dihidrógeno sódico dihidrato, manitol, poloxámero 188, alcohol bencílico, agua para preparaciones inyectables.
Contiene alcohol bencílico. Ver información adicional en el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable 1 cartucho 5 cartuchos 10 cartuchos
5. FORMA Y VÍA DE ADMINISTRACIÓN
Utilizar sólo la solución cristalina. Utilizar sólo con Omnitrope Pen 5. Leer el prospecto antes de utilizar este medicamento.
Vía subcutánea
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA ADVERTENCIA ESPECIAL, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar y transportar refrigerado (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestr. 10 A-6250 Kundl Austria
12. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/004
EU/1/06/332/005
EU/1/06/332/006
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
ETIQUETA DEL CARTUCHO DE OMNITROPE
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Omnitrope 5 mg/1,5 ml Inyectable Somatropina
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
1. NOMBRE DEL MEDICAMENTO
Omnitrope 10 mg/1,5 ml solución inyectable Somatropina
2. PRINCIPIO ACTIVO
Somatropina 6,7 mg (20 UI)/ml.
Un cartucho contiene 1,5 ml, que corresponde a 10 mg de somatropina (30 UI).
3. LISTA DE EXCIPIENTES
Otros componentes: fosfato hidrógeno disódico heptahidrato, fosfato dihidrógeno sódico dihidrato, glicina, poloxámero 188, fenol, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable 1 cartucho 5 cartuchos 10 cartuchos
5. FORMA Y VÍA DE ADMINISTRACIÓN
Utilizar sólo la solución cristalina. Utilizar sólo con Omnitrope Pen 10. Leer el prospecto antes de utilizar este medicamento.
Vía subcutánea
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA ADVERTENCIA ESPECIAL, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar y transportar refrigerado (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestr. 10 A-6250 Kundl Austria
12. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/007
EU/1/06/332/008
EU/1/06/332/009
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
ETIQUETA DEL CARTUCHO DE OMNITROPE
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Omnitrope 10 mg/1,5 ml Inyectable Somatropina
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
1. NOMBRE DEL MEDICAMENTO
Omnitrope 5 mg/1,5 ml solución inyectable Somatropina
2. PRINCIPIO ACTIVO
Somatropina 3,3 mg (10 UI)/ml en un cartucho.
Un cartucho contiene 1,5 ml, que corresponde a 5 mg de somatropina (15 UI).
3. LISTA DE EXCIPIENTES
Otros componentes: fosfato hidrógeno disódico heptahidrato, fosfato dihidrógeno sódico dihidrato, manitol, poloxámero 188, alcohol bencílico, agua para preparaciones inyectables.
Contiene alcohol bencílico. Ver información adicional en el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable 1 cartucho para SurePal 5 5 cartuchos para SurePal 5 10 cartuchos para SurePal 5
5. FORMA Y VÍA DE ADMINISTRACIÓN
Utilizar sólo la solución cristalina. Utilizar sólo con SurePal 5. Leer el prospecto antes de utilizar este medicamento.
Vía subcutánea
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA ADVERTENCIA ESPECIAL, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar y transportar refrigerado (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestr. 10 A-6250 Kundl Austria
12. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/013
EU/1/06/332/014
EU/1/06/332/015
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
ETIQUETA DEL CARTUCHO DE OMNITROPE
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Omnitrope 5 mg/1,5 ml Inyectable Somatropina
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
1. NOMBRE DEL MEDICAMENTO
Omnitrope 10 mg/1,5 ml solución inyectable Somatropina
2. PRINCIPIO ACTIVO
Somatropina 6,7 mg (20 UI)/ml.
Un cartucho contiene 1,5 ml, que corresponde a 10 mg de somatropina (30 UI).
3. LISTA DE EXCIPIENTES
Otros componentes: fosfato hidrógeno disódico heptahidrato, fosfato dihidrógeno sódico dihidrato, glicina, poloxámero 188, fenol, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable 1 cartucho para SurePal 10 5 cartuchos para SurePal 10 10 cartuchos para SurePal 10
5. FORMA Y VÍA DE ADMINISTRACIÓN
Utilizar sólo la solución cristalina. Utilizar sólo con SurePal 10. Leer el prospecto antes de utilizar este medicamento.
Vía subcutánea
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA ADVERTENCIA ESPECIAL, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar y transportar refrigerado (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestr. 10 A-6250 Kundl Austria
12. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/016
EU/1/06/332/017
EU/1/06/332/018
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
ETIQUETA DEL CARTUCHO DE OMNITROPE
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Omnitrope 10 mg/1,5 ml Inyectable Somatropina
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
1. NOMBRE DEL MEDICAMENTO
Omnitrope 15 mg/1,5 ml solución inyectable Somatropina
2. PRINCIPIO ACTIVO
Somatropina 10 mg (30 UI)/ml.
Un cartucho contiene 10 mg en 1,5 ml, que corresponde a 15 mg de somatropina (45 UI).
3. LISTA DE EXCIPIENTES
Otros componentes: fosfato hidrógeno disódico heptahidrato, fosfato dihidrógeno sódico dihidrato, cloruro de sodio, poloxámero 188, fenol, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable.
1 cartucho para SurePal 15 5 cartuchos para SurePal 15 10 cartuchos para SurePal 15
5. FORMA Y VÍA DE ADMINISTRACIÓN
Utilizar sólo la solución cristalina. Utilizar sólo con SurePal 15. Leer el prospecto antes de utilizar este medicamento.
Vía subcutánea
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA ADVERTENCIA ESPECIAL, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar y transportar refrigerado (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandoz GmbH Biochemiestr. 10 A-6250 Kundl Austria
12. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/332/010
EU/1/06/332/011
EU/1/06/332/012
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
ETIQUETA DEL CARTUCHO DE OMNITROPE
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Omnitrope 15 mg/1,5 ml Inyectable Somatropina
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
B. PROSPECTO
Prospecto: Información para el usuario Omnitrope 1,3 mg/ml polvo y disolvente para solución inyectable
Somatropina
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Omnitrope y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Omnitrope
3. Cómo usar Omnitrope
4. Posibles efectos adversos
5. Conservación de Omnitrope
6. Contenido del envase e información adicional
1. Qué es Omnitrope y para qué se utiliza
Omnitrope es una hormona del crecimiento humana recombinante (también llamada somatropina). Tiene la misma estructura que la hormona del crecimiento humana natural, que es necesaria para que los huesos y los músculos crezcan. También ayuda a que los tejidos grasos y musculares se desarrollen en las cantidades correctas. Es recombinante, lo que significa que no se elabora a partir de tejido humano o animal.
En los niños, Omnitrope se usa para tratar los siguientes trastornos del crecimiento:
• Si no creces adecuadamente y no tienes suficiente hormona del crecimiento propia.
• Si padeces un síndrome de Turner, que es un trastorno genético en las niñas que puede afectar al crecimiento; el médico te lo habrá dicho si padeces este trastorno.
• Si padeces una insuficiencia renal crónica. A medida que los riñones pierden su capacidad para funcionar normalmente, esto puede afectar al crecimiento.
• Si eras demasiado pequeño o demasiado ligero al nacer. La hormona de crecimiento puede ayudar a que crezcas más si no has podido tener un estirón o mantener un crecimiento normal a los cuatro años de edad o en adelante.
• Si padeces un síndrome de Prader-Willi (un trastorno cromosómico). La hormona de crecimiento puede ayudar a que crezca más si sigue creciendo y también mejorará la composición de tu organismo. El exceso de grasa se reducirá y la masa muscular disminuida mejorará.
En los adultos, Omnitrope se usa para
• tratar a las personas con una deficiencia pronunciada de la hormona del crecimiento. Esta puede empezar durante la edad adulta o puede continuar desde la niñez.
Si usted ha sido tratado con Omnitrope por una deficiencia de la hormona de crecimiento durante la niñez, se volverá a examinar el estado de la hormona de crecimiento después de finalizar el crecimiento. Si se confirma una deficiencia grave de la hormona, el médico propondrá la continuación del tratamiento con Omnitrope.
Sólo debe recibir este medicamento de un médico que tenga experiencia con la hormona de crecimiento y que haya confirmado su diagnóstico.
2. Qué necesita saber antes de empezar a usar Omnitrope
No use Omnitrope
• si es alérgico (hipersensible) a la somatropina o a cualquiera de los demás componentes de Omnitrope.
• e informe a su médico si usted padece un tumor activo (cáncer). Los tumores deben ser inactivos y usted debe haber terminado su tratamiento antitumoral antes de empezar su tratamiento con Omnitrope.
• e informe a su médico si se le ha prescrito Omitrope para estimular el crecimiento pero usted ya ha dejado de crecer (epífisis cerradas).
• si está gravemente enfermo (por ejemplo, complicaciones post-quirúrgicas a corazón abierto, cirugía abdominal, traumatismo accidental, insuficiencia respiratoria aguda o afecciones similares). Si a usted le van a practicar o le han practicado una operación mayor, o si va al hospital por cualquier motivo, infórmele a su médico y recuérdeles a los otros médicos a los que ve que usted usa la hormona de crecimiento.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar Omnitrope.
• Si tiene riesgo de presentar diabetes, el médico deberá controlar regularmente la concentración de la glucosa en la sangre durante el tratamiento con somatropina.
• Si padece diabetes mellitus, deberá vigilar atentamente la concentración de glucosa en la sangre durante el tratamiento con somatropina y hablar con el médico acerca de los resultados, a fin de decidir si tiene que cambiar la dosis de sus medicamentos para tratar la diabetes.
• Después de comenzar el tratamiento con somatropina, algunos pacientes pueden tener que comenzar un reemplazo con hormona tiroidea.
• Si recibe tratamiento con hormonas tiroideas, puede ser necesario ajustar la dosis de hormona tiroidea.
• Si usted tiene un aumento de la presión intracraneal (que causa síntomas, tales como dolor de cabeza intenso, alteraciones visuales o vómitos) deberá informar al médico acerca de ello.
• Si camina cojeando o si empieza a cojear durante el tratamiento con hormona de crecimiento, deberá informar al médico.
• Si está recibiendo somatropina para una deficiencia de la hormona de crecimiento después de un tumor previo (cáncer), deberán examinarlo regularmente para detectar la recurrencia del tumor o cualquier otro cáncer.
• Si experimenta un dolor abdominal que empeora, debe informar a su médico.
• La experiencia en pacientes de más de 80 años es limitada. Las personas de edad avanzada
pueden ser más sensibles a la acción de la somatropina y, por lo tanto, pueden ser más propensas a presentar reacciones adversas.
Niños con insuficiencia renal crónica
• El médico deberá examinar la función de los riñones y la velocidad de crecimiento antes de empezar el tratamiento con somatropina. El tratamiento médico de los riñones debe continuarse. El tratamiento con somatropina debe interrumpirse en caso de trasplante renal.
Niños con síndrome de Prader-Willi
• El médico le dará restricciones en la dieta que debe seguir para controlar su peso.
• El médico evaluará los signos de obstrucción de las vías respiratorias altas, apnea del sueño (en
que la respiración se interrumpe durante el sueño) o infección respiratoria antes de comenzar el tratamiento con somatropina.
• Durante el tratamiento con somatropina, informe al médico si presenta signos de obstrucción de las vías respiratorias altas (incluso comenzar a roncar o un empeoramiento de los ronquidos).
Tal vez el médico tenga que examinarle y puede interrumpir el tratamiento con somatropina.
• Durante el tratamiento, el médico le examinará para ver si hay signos de escoliosis, un tipo de deformidad vertebral.
• Durante el tratamiento, si presenta una infección pulmonar, informe al médico para que pueda tratar la infección.
Niños nacidos demasiado pequeños o bajos de peso
• Si eras demasiado pequeño o demasiado ligero al nacer y tienes de 9 a 12 años, consulta al médico específicamente en relación con la pubertad y el tratamiento con este medicamento.
• El tratamiento debe continuar hasta que hayas dejado de crecer.
• El médico examinará las concentraciones de glucosa e insulina antes de comenzar el tratamiento y cada año durante el tratamiento.
Uso de Omnitrope con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Debe informar al médico si está utilizando:
• medicamentos para tratar la diabetes;
• hormonas tiroideas;
• medicamentos para controlar la epilepsia (anticonvulsivantes);
• ciclosporina (un medicamento que debilita el sistema inmunitario después de los trasplantes);
• hormonas sexuales (por ejemplo, estrógenos);
• hormonas suprarrenales sintéticas (corticoesteroides).
Tal vez el médico tenga que ajustar la dosis de estos medicamentos o la dosis de somatropina. Embarazo y lactancia
No debe usar Omnitrope si está embarazada o tratando de quedarse embarazada.
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Información importante sobre algunos de los componentes de Omnitrope
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por ml; esto es, esencialmente “exento de sodio”.
3. Cómo usar Omnitrope
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero. La dosis depende de su tamaño, de la afección para la que recibe tratamiento y de lo bien que funcione la hormona de crecimiento en usted. Todas las personas son diferentes. El médico le aconsejará acerca de su dosis individualizada de Omnitrope en miligramos (mg) a partir de su peso corporal en kilogramos (kg) o por su superficie corporal, calculada a partir de su estatura y peso en metros cuadrados (m2), así como su pauta de tratamiento. No cambie la dosificación y la pauta de tratamiento sin consultarle al médico.
La dosis recomendada es para:
Niños con deficiencia de la hormona de crecimiento:
0,025 a 0,035 mg/kg de peso corporal al día o 0,7 a 1,0 mg/m2 de superficie corporal al día. Pueden utilizarse dosis más altas. Cuando la deficiencia de hormona de crecimiento continúa durante la adolescencia, Omnitrope debe continuarse hasta finalizar el desarrollo físico.
Niñas con síndrome de Turner:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día.
Niños con insuficiencia renal crónica:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día. Pueden ser necesarias dosis más altas si la velocidad de crecimiento es demasiado baja. Puede ser necesario un ajuste de la dosis después de seis meses de tratamiento.
Niños con síndrome de Prader-Willi:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. La dosificación diaria no debe ser superior a 2,7 mg. El tratamiento no debe utilizarse en los niños que casi han dejado de crecer después de la pubertad.
Niños nacidos más pequeños o con peso más bajo que lo esperado y con un trastorno del crecimiento:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. Es importante continuar el tratamiento hasta que se alcance la estatura final. El tratamiento debe suspenderse después del primer año si no responde, o si ha alcanzado la estatura final y dejado de crecer.
Adultos con deficiencia de la hormona de crecimiento:
Si continúa utilizando Omnitrope después del tratamiento durante la niñez, debe comenzar con 0,2 a 0,5 mg al día.
Esta dosificación se debe aumentar o reducir gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios.
Si la deficiencia de la hormona de crecimiento comienza durante la vida adulta, debe comenzar con 0,15 a 0,3 mg al día. Esta dosificación debe aumentarse gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios. La dosis de mantenimiento diaria rara vez es superior a 1,0 mg diarios. Las mujeres pueden necesitar dosis más altas que los hombres. La dosificación debe vigilarse cada seis meses. Las personas de más de 60 años deben comenzar con una dosis de 0,1 a 0,2 mg diarios que debe aumentarse lentamente según las necesidades individuales. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento rara vez supera 0,5 mg al día. Siga las instrucciones que le haya dado el médico.
Inyección de Omnitrope
Inyéctese la hormona del crecimiento más o menos a la misma hora cada día. La hora de acostarse es un buen momento porque es fácil de recordar. Además, también es natural tener una concentración más elevada de hormona del crecimiento por la noche.
Omnitrope está indicado para su empleo por vía subcutánea. Esto significa que se inyecta por medio de una pequeña aguja para inyección en el tejido adiposo, por debajo de la piel. La mayor parte de las personas se inyectan en el muslo o en las nalgas. Póngase la inyección en el sitio que le haya enseñado su médico. El tejido adiposo de la piel puede verse reducido en el lugar de la inyección. Para evitar esto, utilice cada vez un sitio ligeramente diferente para inyectarse. Esto proporciona a la piel y a la zona por debajo de ella tiempo para recuperarse de una inyección antes de recibir otra en el mismo sitio.
El médico debe haberle enseñado ya cómo utilizar Omnitrope. Inyéctese siempre Omnitrope tal como el médico le ha dicho. Si no está seguro, compruebe con su médico o farmacéutico.
Cómo inyectar Omnitrope 1,3 mg/ml
Las siguientes instrucciones explican cómo inyectarse Omnitrope 1,3 mg/ml usted mismo. Lea detenidamente las instrucciones y sígalas paso a paso. Su médico o la enfermera le enseñará cómo inyectarse Omnitrope. No intente inyectarse a menos que esté seguro de que entiende el procedimiento y lo que conlleva la inyección.
- Tras la reconstitución, Omnitrope se administra como una inyección debajo de la piel (vía subcutánea).
- Inspeccione cuidadosamente la solución inyectable antes de inyectarla y úsela sólo si es clara e incolora.
Cambie el lugar de la inyección para minimizar el riesgo de lipoatrofia local (reducción local del tejido adiposo debajo de la piel).
Preparación
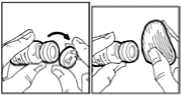
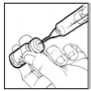
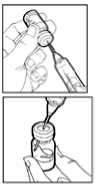
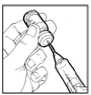
Antes de empezar, debe tener todo lo necesario:
- Vial con Omnitrope 1,3 mg/ml polvo para solución inyectable
- Vial con disolvente (líquido) para Omnitrope 1,3 mg/ml.
- Jeringa para inyección estéril desechable (p. ej.: jeringa para inyección de 2 ml) y aguja para inyección (p. ej.: 0,33 mm x
12,7 mm) para retirar el disolvente del vial (no suministrada con el envase).
- Jeringa para inyección estéril, desechable, de tamaño apropiado (p. ej.: jeringa para inyección de 1 ml) y aguja para inyección (p. ej.: 0,25 mm x 8 mm) para inyección subcutánea (no suministrada con el envase).
- 2 torundas para la limpieza (no suministradas con el envase).
Lávese las manos antes de continuar con los siguiente pasos.
Reconstitución de Omnitrope
- Retire las tapas protectoras de los dos viales de la caja. Con una torunda limpia, desinfecte las membranas de goma del vial que contiene el polvo para solución inyectable y del vial con el disolvente.
- Coja el vial con el disolvente y la jeringa para inyección estéril, desechable (p. ej.: jeringa para inyección de 2 ml) y la aguja para inyección (p. ej.: de 0,33 mm x 12,7 mm). Presione la aguja para inyección ya colocada en la jeringa para inyección a través de la membrana de goma.
- Gire el vial con el disolvente de arriba abajo y extraiga el disolvente del vial.
Coja el vial que contiene el polvo para solución inyectable y empujar la aguja para inyección a través de la membrana de goma del vial. Inyecte lentamente el disolvente. Procure dirigir el chorro del líquido hacia la pared de vidrio y así evitar la formación de espuma. Retire la jeringa para inyección y la aguja para inyección. Con suavidad, gire el vial reconstituido hasta que el contenido se haya disuelto por completo. No lo agite..
Si la solución inyectable está turbia (y la turbidez no desaparece en unos diez minutos) o si contiene partículas, no deberá utilizarse. El contenido deberá ser claro e incoloro.
Use la solución inyectable de inmediato.
Midiendo la dosis de Omnitrope a ser inyectada
- Tomar la jeringa para inyección estéril, desechable, de tamaño apropiado (p. ej.: una jeringa para inyección de 1 ml) y la aguja para inyección (p. ej.: de 0,.25 mm x 8 mm).
- Empujar la aguja para inyección a través del tapón de goma del vial que contiene la solución inyectable reconstituida.
Con una mano, invertir el vial y la jeringa para inyección de arriba abajo.
Asegúrese de que la punta de la jeringa para inyección esté dentro de la solución inyectable reconstituida de Omnitrope. Con su otra mano podrá mover el émbolo.
Retire lentamente el émbolo y extraiga justo un poco más de la dosis prescrita por su médico.
Sostenga hacia arriba la jeringa para inyección y la aguja para inyección que están en el vial, y retire la jeringa para inyección del vial.
Verifique que la jeringa para inyección no tiene burbujas de aire. Si observa que hay burbujas, retire un poco el émbolo hacia atrás, golpee suavemente la jeringa para inyección, con la aguja para inyección hacia arriba, hasta que desaparezcan las burbujas. Empujar el émbolo lentamente hasta alcanzar la dosis correcta. Antes de la administración inspeccionar visualmente la solución inyectable reconstituida. No utilizarla si la solución inyectable está turbia o contiene partículas. Ahora puede inyectar la dosis.
Inyección de Omnitrope
- Seleccionar el lugar de la inyección. Los mejores lugares para la inyección son los tejidos con una capa de grasa entre la piel y el músculo, tal como el muslo o el vientre (pero no el ombligo y la cintura).
- Asegúrese de inyectarse, como mínimo, a 1 cm de distancia del lugar utilizado para la administración de la inyección anterior y de cambiar los lugares donde se inyecte, tal y como se le ha enseñado.
- Antes de administrar la inyección, limpie bien la piel con una torunda empapada en alcohol. Esperar a que la zona se seque.
- Con la otra mano, pinzar un pliegue de piel suelta. Con la mano libre, sostenga la jeringa para inyección tal y como lo haría con un lápiz. Introduzca la aguja para inyección dentro del pliegue cutáneo a un ángulo de 45° a 90°. Una vez introducida la aguja para inyección, retire la mano que utilizó para pinzar la piel y utilícela para sostener el cuerpo de la jeringa para inyección. Si aparece sangre en la jeringa para inyección, significa que la aguja para inyección ha penetrado un vaso sanguíneo. No se inyecte en este lugar; retire la aguja para inyección que ha penetrado el vaso sanguíneo y repita este paso. Empujando el émbolo en su totalidad, inyectar la solución inyectable lentamente.
- Retirar la aguja para inyección de la piel.
Después de la inyección
- Después de administrarse la inyección, presionar durante unos segundos el lugar de la inyección con un pequeño vendaje de gasa estéril. No frote el lugar de la inyección.
- La solución inyectable residual, viales y materiales empleados en la administración de la inyección están pensados para un solo uso y deberán ser desechados. Deseche de la jeringa para inyección en un contenedor cerrado y seguro.
Si usa más Omnitrope del que debe
Si se inyecta mucho más de lo que debiera, consulte lo antes posible a su médico o farmacéutico. Su concentración de azúcar en la sangre podría descender demasiado y después aumentar demasiado. Tal vez se sienta con temblores, sudoroso, somnoliento o “como si no fuera usted mismo”, y podría desmayarse.
Si olvidó usar Omnitrope
No use una dosis doble para compensar las dosis olvidadas. Lo mejor es usar la hormona de crecimiento con regularidad. Si se olvida de usar una dosis, póngase la siguiente inyección a la hora habitual, al día siguiente. Tome nota de las inyecciones olvidadas e infórmele al médico en el siguiente control.
Si interrumpe el tratamiento con Omnitrope
Consulte a su médico antes de dejar de usar Omnitrope.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Los efectos adversos muy frecuentes y frecuentes en los adultos pueden comenzar en los primeros meses de tratamiento y pueden detenerse espontáneamente o si se reduce la dosis.
Los efectos adversos muy frecuentes (que probablemente se produzcan en más de 1 de cada 10 pacientes) incluyen los siguientes:
En los adultos
• Dolor articular
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos)
Los efectos adversos frecuentes (que probablemente afecten a menos de 1 de cada 10 pacientes) incluyen los siguientes:
En los niños
• Enrojecimiento, picor o dolor temporal en el lugar de la inyección
• Dolor articular
En los adultos
• Adormecimiento, hormigueos
• Rigidez de los brazos y las piernas, dolor muscular
• Dolor o sensación de escozor en las manos o los antebrazos (conocido como síndrome del túnel carpiano)
Los efectos adversos poco frecuentes (que probablemente afecten a menos de 1 de cada 100 pacientes) incluyen los siguientes:
En los niños
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos, durante un tiempo corto al comienzo del tratamiento)
Los efectos adversos raros (que probablemente afecten a menos de 1 de cada 1.000 pacientes) incluyen los siguientes:
En los niños
• Adormecimiento, hormigueos
• Leucemia (se ha observado en un pequeño número de pacientes con deficiencia de la hormona del crecimiento, algunos de cuales habían sido tratados con somatropina. Sin embargo, no hay ningún indicio de que la incidencia de leucemia sea mayor en receptores de la hormona del crecimiento sin factores predisponentes).
• Aumento de la presión intracraneal (que causa síntomas, como cefalea intensa, alteraciones visuales o vómitos)
• Dolor muscular
Frecuencia no conocida: no puede estimarse a partir de los datos disponibles
• Diabetes de tipo 2
• Disminución de los niveles de la hormona cortisol en la sangre
En los niños
• Rigidez en los brazos y las piernas
En los adultos
• Aumento de la presión intracraneal (que causa síntomas, como dolor de cabeza intenso, alteraciones visuales o vómitos)
• Enrojecimiento, picor o dolor en el lugar de la inyección
Formación de anticuerpos contra la hormona del crecimiento inyectada, pero estos no parecen hacer que la hormona del crecimiento deje de funcionar.
La piel alrededor de la zona de inyección puede ponerse irregular o con bultos, pero esto no deberá ocurrir si se inyecta en un lugar diferente cada vez.
Ha habido casos raros de muerte súbita en pacientes con síndrome de Prader-Willi. No obstante, estos casos no se han relacionado con el tratamiento con Omnitrope.
Su médico puede considerar una epifisiólisis de la cabeza femoral o una enfermedad de Legg-Calvé-Perthes si experimenta molestias o dolor en la cadera o la rodilla mientras está siendo tratado con Omnitrope.
Otros posibles efectos adversos relacionados con su tratamiento con la hormona del crecimiento pueden incluir los siguientes:
Usted (o su hijo) puede tener niveles elevados de azúcar en sangre o niveles reducidos de la hormona tiroidea. Esto lo puede analizar su médico y, si es necesario, su médico le recetará el tratamiento adecuado. En casos raros se ha observado inflamación del páncreas en pacientes tratados con la hormona del crecimiento.
Si considera que alguno de los efectos adversos que sufre es grave o si aprecia cualquier efecto adverso no mencionado en este prospecto, informe a su médico o farmacéutico.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Omnitrope
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y en la caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
• Conservar y transportar refrigerado (entre 2°C y 8°C).
• No congelar.
• Conservar en el embalaje original para protegerlo de la luz.
• Tras la reconstitución, desde un punto de vista microbiológico, el producto debe utilizarse inmediatamente. Sin embargo, se ha demostrado la estabilidad durante el uso durante un periodo de hasta 24 horas, a una temperatura entre 2 y 8°C, en el embalaje original.
• Este producto es para un solo uso.
No utilice Omnitrope si se observa que la solución está turbia.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Omnitrope
El principio activo de Omnitrope es somatropina.
Un vial contiene 1,3 mg (que corresponde a 4 UI) de somatropina tras su reconstitución con 1 ml de disolvente.
Los demás componentes son:
Polvo:
glicina
fosfato de hidrógeno disódico heptahidrato fosfato dihidrógeno sódico dihidrato
Disolvente:
agua para preparaciones inyectables Aspecto del producto y contenido del envase
Polvo y disolvente para solución inyectable (polvo en un vial (1,3 mg), disolvente en un vial (1 ml)). Envase de una unidad.
El polvo es blanco y el disolvente es una solución transparente e incolora.
Titular de la autorización de comercialización y responsable de la fabricación
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Austria
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Lietuva
Sandoz Pharmaceuticals d.d. filialas Tel.: +370 5 2636 037
Luxembourg/Luxemburg
Sandoz GmbH Tel.: +43 5338 2000
Magyarország
Sandoz Hungária Kft.
Tel.: +36 1 430 2890
Malta
Sandoz GmbH Tel.: +43 5338 2000
Belgie/Belgique/Belgien
Sandoz nv-sa Tél/Tel.: +32 2 722 97 97
Btnrapun
TtproBCKO npegcTaBHTencTBO CaHgo3 g.g.
Tea.: +359 2 970 47 47
Ceská republika
Sandoz s.r.o.
Tel.: +420 221 421 611
Danmark/Norge/Ísland/Sverige
Sandoz A/S Tlf.: +45 6395 1000
Deutschland
Hexal AG
Tel.: +49 8024 908 0 Eesti
Sandoz d.d. Eesti filiaal Tel.: +372 665 2400
EXláóa Sandoz dd
T^: +30 216 600 500 0 España
Sandoz Farmacéutica, S.A.
Tel.: +34 900 456 856
France
Sandoz SAS
Tél.: +33 1 49 64 48 00
Hrvatska
Sandoz d.o.o.
Tel: +385 1 23 53 111
Ireland
Rowex Ltd Tel.: +353 27 50077
Italia
Sandoz S.p.A.
Tel.: +39 02 96541
Kúrcpog
P.T.Hadjigeorgiou co ltd Tel.: +357 25372425
Latvija
Sandoz d.d. Parstavnieciba Latvija Tel.: +371 67 892 006
Nederland
Sandoz B.V.
Tel.: +31 36 52 41 600
Osterreich
Sandoz GmbH Tel.: +43 5338 2000
Polska
Sandoz Polska Sp. z o.o.
Tel.: +48 22 209 70 00
Portugal
Sandoz Farmacéutica Lda.
Tel.: +351 21 924 19 11
Romania
S.C. Sandoz S.R.L.
Tel.: +40 265 208 120
Slovenija
Lek farmacevtska druzba d.d. Tel.: +386 1 580 21 11
Slovenská republika
Sandoz d.d. - organizacná zlozka Tel.: +421 2 48 200 600
Suomi/Finland
Sandoz A/S
Puh/Tel.: +358 10 6133 415
United Kingdom
Sandoz Ltd
Tel.: + 44 1276 69 8020
Fecha de la última revisión de este prospecto: {MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
Prospecto: Información para el usuario Omnitrope 5 mg/ml polvo y disolvente para solución inyectable
Somatropina
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Omnitrope y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Omnitrope
3. Cómo usar Omnitrope
4. Posibles efectos adversos
5. Conservación de Omnitrope
6. Contenido del envase e información adicional
1. Qué es Omnitrope y para qué se utiliza
Omnitrope es una hormona del crecimiento humana recombinante (también llamada somatropina). Tiene la misma estructura que la hormona del crecimiento humana natural, que es necesaria para que los huesos y los músculos crezcan. También ayuda a que los tejidos grasos y musculares se desarrollen en las cantidades correctas. Es recombinante, lo que significa que no se elabora a partir de tejido humano o animal.
En los niños, Omnitrope se usa para tratar los siguientes trastornos del crecimiento:
• Si no creces adecuadamente y no tienes suficiente hormona del crecimiento propia.
• Si padeces un síndrome de Turner, que es un trastorno genético en las niñas que puede afectar al crecimiento; el médico te lo habrá dicho si padeces este trastorno.
• Si padeces una insuficiencia renal crónica. A medida que los riñones pierden su capacidad para funcionar normalmente, esto puede afectar al crecimiento.
• Si eras demasiado pequeño o demasiado ligero al nacer. La hormona de crecimiento puede ayudar a que crezcas más si no has podido tener un estirón o mantener un crecimiento normal a los cuatro años de edad o en adelante.
• Si padeces un síndrome de Prader-Willi (un trastorno cromosómico). La hormona de crecimiento puede ayudar a que crezca más si sigue creciendo y también mejorará la composición de tu organismo. El exceso de grasa se reducirá y la masa muscular disminuida mejorará.
En los adultos, Omnitrope se usa para
• tratar a las personas con una deficiencia pronunciada de la hormona del crecimiento. Esta puede empezar durante la edad adulta o puede continuar desde la niñez.
Si usted ha sido tratado con Omnitrope por una deficiencia de la hormona de crecimiento durante la niñez, se volverá a examinar el estado de la hormona de crecimiento después de finalizar el crecimiento. Si se confirma una deficiencia grave de la hormona, el médico propondrá la continuación del tratamiento con Omnitrope.
Sólo debe recibir este medicamento de un médico que tenga experiencia con la hormona de crecimiento y que haya confirmado su diagnóstico.
2. Qué necesita saber antes de empezar a usar Omnitrope
No use Omnitrope
• si es alérgico (hipersensible) a la somatropina o a cualquiera de los demás componentes de Omnitrope.
• e informe a su médico si usted padece un tumor activo (cáncer). Los tumores deben ser inactivos y usted debe haber terminado su tratamiento antitumoral antes de empezar su tratamiento con Omnitrope.
• e informe a su médico si se le ha prescrito Omitrope para estimular el crecimiento pero usted ya ha dejado de crecer (epífisis cerradas).
• si está gravemente enfermo (por ejemplo, complicaciones post-quirúrgicas a corazón abierto, cirugía abdominal, traumatismo accidental, insuficiencia respiratoria aguda o afecciones similares). Si a usted le van a practicar o le han practicado una operación mayor, o si va al hospital por cualquier motivo, infórmele a su médico y recuérdeles a los otros médicos a los que ve que usted usa la hormona de crecimiento.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar Omnitrope.
• Si tiene riesgo de presentar diabetes, el médico deberá controlar regularmente la concentración de la glucosa en la sangre durante el tratamiento con somatropina.
• Si padece diabetes mellitus, deberá vigilar atentamente la concentración de glucosa en la sangre durante el tratamiento con somatropina y hablar con el médico acerca de los resultados, a fin de decidir si tiene que cambiar la dosis de sus medicamentos para tratar la diabetes.
• Después de comenzar el tratamiento con somatropina, algunos pacientes pueden tener que comenzar un reemplazo con hormona tiroidea.
• Si recibe tratamiento con hormonas tiroideas, puede ser necesario ajustar la dosis de hormona tiroidea.
• Si usted tiene un aumento de la presión intracraneal (que causa síntomas, tales como dolor de cabeza intenso, alteraciones visuales o vómitos) deberá informar al médico acerca de ello.
• Si camina cojeando o si empieza a cojear durante el tratamiento con hormona de crecimiento, deberá informar al médico.
• Si está recibiendo somatropina para una deficiencia de la hormona de crecimiento después de un tumor previo (cáncer), deberán examinarlo regularmente para detectar la recurrencia del tumor o cualquier otro cáncer.
• Si experimenta un dolor abdominal que empeora, debe informar a su médico.
• La experiencia en pacientes de más de 80 años es limitada. Las personas de edad avanzada
pueden ser más sensibles a la acción de la somatropina y, por lo tanto, pueden ser más propensas a presentar reacciones adversas.
Niños con insuficiencia renal crónica
• El médico deberá examinar la función de los riñones y la velocidad de crecimiento antes de empezar el tratamiento con somatropina. El tratamiento médico de los riñones debe continuarse. El tratamiento con somatropina debe interrumpirse en caso de trasplante renal.
Niños con síndrome de Prader-Willi
• El médico le dará restricciones en la dieta que debe seguir para controlar su peso.
• El médico evaluará los signos de obstrucción de las vías respiratorias altas, apnea del sueño (en
que la respiración se interrumpe durante el sueño) o infección respiratoria antes de comenzar el tratamiento con somatropina.
• Durante el tratamiento con somatropina, informe al médico si presenta signos de obstrucción de las vías respiratorias altas (incluso comenzar a roncar o un empeoramiento de los ronquidos).
Tal vez el médico tenga que examinarle y puede interrumpir el tratamiento con somatropina.
• Durante el tratamiento, el médico le examinará para ver si hay signos de escoliosis, un tipo de deformidad vertebral.
• Durante el tratamiento, si presenta una infección pulmonar, informe al médico para que pueda tratar la infección.
Niños nacidos demasiado pequeños o bajos de peso
• Si eras demasiado pequeño o demasiado ligero al nacer y tienes de 9 a 12 años, consulta al médico específicamente en relación con la pubertad y el tratamiento con este medicamento.
• El tratamiento debe continuar hasta que hayas dejado de crecer.
• El médico examinará las concentraciones de glucosa e insulina antes de comenzar el tratamiento y cada año durante el tratamiento.
Uso de Omnitrope con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Debe informar al médico si está utilizando:
• medicamentos para tratar la diabetes;
• hormonas tiroideas;
• medicamentos para controlar la epilepsia (anticonvulsivantes);
• ciclosporina (un medicamento que debilita el sistema inmunitario después de los trasplantes);
• hormonas sexuales (por ejemplo, estrógenos);
• hormonas suprarrenales sintéticas (corticoesteroides).
Tal vez el médico tenga que ajustar la dosis de estos medicamentos o la dosis de somatropina. Embarazo y lactancia
No debe usar Omnitrope si está embarazada o tratando de quedarse embarazada.
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Información importante sobre algunos de los componentes de Omnitrope
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por ml; esto es, esencialmente “exento de sodio”.
Tras la reconstitución, un ml contiene 15 mg de alcohol bencílico.
Debido a la presencia de alcohol bencílico, el medicamento no se debe administrar a niños prematuros ni recién nacidos. Puede provocar reacciones tóxicas y reacciones alérgicas en niños de hasta 3 años de edad.
3. Cómo usar Omnitrope
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero. La dosis depende de su tamaño, de la afección para la que recibe tratamiento y de lo bien que funcione la hormona de crecimiento en usted. Todas las personas son diferentes. El médico le aconsejará acerca de su dosis individualizada de Omnitrope en miligramos (mg) a partir de su peso corporal en kilogramos (kg) o por su superficie corporal, calculada a partir de su estatura y peso en metros cuadrados (m2), así como su pauta de tratamiento. No cambie la dosificación y la pauta de tratamiento sin consultarle al médico.
La dosis recomendada es para:
Niños con deficiencia de la hormona de crecimiento:
0,025 a 0,035 mg/kg de peso corporal al día o 0,7 a 1,0 mg/m2 de superficie corporal al día. Pueden utilizarse dosis más altas. Cuando la deficiencia de hormona de crecimiento continúa durante la adolescencia, Omnitrope debe continuarse hasta finalizar el desarrollo físico.
Niñas con síndrome de Turner:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día.
Niños con insuficiencia renal crónica:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día. Pueden ser necesarias dosis más altas si la velocidad de crecimiento es demasiado baja. Puede ser necesario un ajuste de la dosis después de seis meses de tratamiento.
Niños con síndrome de Prader-Willi:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. La dosificación diaria no debe ser superior a 2,7 mg. El tratamiento no debe utilizarse en los niños que casi han dejado de crecer después de la pubertad.
Niños nacidos más pequeños o con peso más bajo que lo esperado y con un trastorno del crecimiento:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. Es importante continuar el tratamiento hasta que se alcance la estatura final. El tratamiento debe suspenderse después del primer año si no responde, o si ha alcanzado la estatura final y dejado de crecer.
Adultos con deficiencia de la hormona de crecimiento:
Si continúa utilizando Omnitrope después del tratamiento durante la niñez, debe comenzar con 0,2 a 0,5 mg al día.
Esta dosificación se debe aumentar o reducir gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios.
Si la deficiencia de la hormona de crecimiento comienza durante la vida adulta, debe comenzar con 0,15 a 0,3 mg al día. Esta dosificación debe aumentarse gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios. La dosis de mantenimiento diaria rara vez es superior a 1,0 mg diarios. Las mujeres pueden necesitar dosis más altas que los hombres. La dosificación debe vigilarse cada seis meses. Las personas de más de 60 años deben comenzar con una dosis de 0,1 a 0,2 mg diarios que debe aumentarse lentamente según las necesidades individuales. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento rara vez supera 0,5 mg al día. Siga las instrucciones que le haya dado el médico.
Inyección de Omnitrope
Inyéctese la hormona del crecimiento más o menos a la misma hora cada día. La hora de acostarse es un buen momento porque es fácil de recordar. Además, también es natural tener una concentración más elevada de hormona del crecimiento por la noche.
Omnitrope 5 mg/ml está indicado para varios usos. Sólo deberá administrarse con el Omnitrope Pen L, un dispositivo para inyección que se ha desarrollado específicamente para su uso con Omnitrope 5 mg/ml polvo y disolvente para solución inyectable.
Omnitrope está indicado para su empleo por vía subcutánea. Esto significa que se inyecta por medio de una pequeña aguja para inyección en el tejido adiposo, por debajo de la piel. La mayor parte de las personas se inyectan en el muslo o en las nalgas. Póngase la inyección en el sitio que le haya enseñado su médico. El tejido adiposo de la piel puede verse reducido en el lugar de la inyección. Para evitar esto, utilice cada vez un sitio ligeramente diferente para inyectarse. Esto proporciona a la piel y a la zona por debajo de ella tiempo para recuperarse de una inyección antes de recibir otra en el mismo sitio.
El médico debe haberle enseñado ya cómo utilizar Omnitrope. Inyéctese siempre Omnitrope tal como el médico le ha dicho. Si no está seguro, compruebe con su médico o farmacéutico.
Cómo inyectar Omnitrope 5 mg/ml
Las siguientes instrucciones explican cómo inyectarse Omnitrope 5 mg/ml usted mismo. Lea detenidamente las instrucciones y sígalas paso a paso. Su médico o la enfermera le enseñará cómo inyectarse Omnitrope. No intente inyectarse a menos que esté seguro de que entiende el procedimiento y lo que conlleva la inyección.
- Tras la reconstitución, Omnitrope se administra como una inyección debajo de la piel (vía subcutánea).
- Inspeccione cuidadosamente la solución antes de inyectarla y úsela sólo si es clara e incolora.
- Cambie el lugar de la inyección para así minimizar el riesgo de lipoatrofia local (reducción local del tejido adiposo debajo de la piel).
Preparación

Antes de empezar, debe tener todo lo necesario:
- Vial con Omnitrope 5 mg/ml polvo para solución inyectable
- Cartucho con disolvente para solución inyectable para Omnitrope 5 mg/ml.
- Juego de transferencia para el mezclado y transferido de la solución inyectable al cartucho (véase Instrucciones de Uso del inyector de pluma).
- Omnitrope Pen L, un dispositivo de inyección específicamente desarrollado para usarse con Omnitrope 5 mg/ml reconstituido para inyección. (no suministrado en el envase, ver Instrucciones de Uso del juego de transferencia del aparato de inyección).
- Pluma para inyección subcutánea.
- 2 torundas para la limpieza (no suministradas con el envase)
Lávese las manos antes de continuar con los siguiente pasos.
Reconstitución de Omnitrope
- Retire el protector del vial. Con una torunda limpia, desinfectar tanto la membrana de goma del vial con polvo para solución inyectable, como la membrana de goma del cartucho que contiene el disolvente.
- Utilice el juego de transferencia para transferir todo el disolvente del cartucho al vial. Seguir las instrucciones que vienen en el juego de transferencia.
- Suavemente, gire el vial reconstituido hasta que el contenido esté completamente disuelto. No agitar.
- Si la solución aparece turbia (y la turbidez no desaparece en unos diez minutos) o contiene partículas, no deberá utilizarse. El contenido deberá ser claro e incoloro.
- Transferir toda la solución disuelta al cartucho, usando para ello el juego de transferencia.
Inyección de Omnitrope
- Coloque el cartucho con Omnitrope diluido en la pluma de inyección. Siga las Instrucciones de Uso del inyector de pluma.
Para ajustar la pluma, marque la dosis.
- Elimine las burbujas.
- Seleccione el lugar de la inyección. Los mejores lugares para la administración de la inyección son los tejidos con una capa de grasa entre la piel y el músculo, como el muslo o el vientre
(excepto el ombligo o la cintura).

Asegúrese de que se inyecta a una distancia mínima de 1 cm respecto del lugar de la última inyección y de que cambia los lugares en donde se inyecte, tal y como se le habrá enseñado. Antes de administrar la inyección, limpie la bien piel con una torunda de alcohol. Espere a que la zona se seque.
Introduzca la aguja para inyección en la piel en la forma que le ha enseñado el médico.
Después de inyectarse
- Después de la inyección, presione el lugar de la inyección con un pequeño vendaje o gasa estéril durante unos segundos. No masajee el lugar de la inyección.
- Retire la aguja para inyección de la pluma utilizando la tapa exterior y elimine la misma. Esto mantendrá Omnitrope estéril e impedirá que gotee. También impedirá que el aire se introduzca en la pluma, taponando la aguja. No comparta las agujas. No comparta la pluma.
- Deje el cartucho en la pluma, reponga el capuchón de la pluma y guárdela en una nevera.
- Después de retirar de la nevera, la solución deberá ser clara. No la utilice si la solución está turbia o si contiene partículas.
Si usa más Omnitrope del que debe
Si se inyecta mucho más de lo que debiera, consulte lo antes posible a su médico o farmacéutico. Su concentración de azúcar en la sangre podría descender demasiado y después aumentar demasiado. Tal vez se sienta con temblores, sudoroso, somnoliento o “como si no fuera usted mismo”, y podría desmayarse.
Si olvidó usar Omnitrope
No use una dosis doble para compensar las dosis olvidadas. Lo mejor es usar la hormona de crecimiento con regularidad. Si se olvida de usar una dosis, póngase la siguiente inyección a la hora habitual, al día siguiente. Tome nota de las inyecciones olvidadas e infórmele al médico en el siguiente control.
Si interrumpe el tratamiento con Omnitrope
Consulte a su médico antes de dejar de usar Omnitrope.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Los efectos adversos muy frecuentes y frecuentes en los adultos pueden comenzar en los primeros meses de tratamiento y pueden detenerse espontáneamente o si se reduce la dosis.
Los efectos adversos muy frecuentes (que probablemente se produzcan en más de 1 de cada 10 pacientes) incluyen los siguientes:
En los adultos
• Dolor articular
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos)
Los efectos adversos frecuentes (que probablemente afecten a menos de 1 de cada 10 pacientes) incluyen los siguientes:
En los niños
• Enrojecimiento, picor o dolor temporal en el lugar de la inyección
• Dolor articular
En los adultos
• Adormecimiento, hormigueos
• Rigidez de los brazos y las piernas, dolor muscular
• Dolor o sensación de escozor en las manos o los antebrazos (conocido como síndrome del túnel
carpiano)
Los efectos adversos poco frecuentes (que probablemente afecten a menos de 1 de cada 100 pacientes) incluyen los siguientes:
En los niños
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos, durante un tiempo corto al comienzo del tratamiento)
Los efectos adversos raros (que probablemente afecten a menos de 1 de cada 1.000 pacientes) incluyen los siguientes:
En los niños
• Adormecimiento, hormigueos
• Leucemia (se ha observado en un pequeño número de pacientes con deficiencia de la hormona del crecimiento, algunos de cuales habían sido tratados con somatropina. Sin embargo, no hay ningún indicio de que la incidencia de leucemia sea mayor en receptores de la hormona del crecimiento sin factores predisponentes).
• Aumento de la presión intracraneal (que causa síntomas, como cefalea intensa, alteraciones visuales o vómitos)
• Dolor muscular
Frecuencia no conocida: no puede estimarse a partir de los datos disponibles
• Diabetes de tipo 2
• Disminución de los niveles de la hormona cortisol en la sangre En los niños
• Rigidez en los brazos y las piernas
En los adultos
• Aumento de la presión intracraneal (que causa síntomas, como dolor de cabeza intenso, alteraciones visuales o vómitos)
• Enrojecimiento, picor o dolor en el lugar de la inyección
Formación de anticuerpos contra la hormona del crecimiento inyectada, pero estos no parecen hacer que la hormona del crecimiento deje de funcionar.
La piel alrededor de la zona de inyección puede ponerse irregular o con bultos, pero esto no deberá ocurrir si se inyecta en un lugar diferente cada vez.
Ha habido casos raros de muerte súbita en pacientes con síndrome de Prader-Willi. No obstante, estos casos no se han relacionado con el tratamiento con Omnitrope.
Su médico puede considerar una epifisiólisis de la cabeza femoral o una enfermedad de Legg-Calvé-Perthes si experimenta molestias o dolor en la cadera o la rodilla mientras está siendo tratado con Omnitrope.
Otros posibles efectos adversos relacionados con su tratamiento con la hormona del crecimiento pueden incluir los siguientes:
Usted (o su hijo) puede tener niveles elevados de azúcar en sangre o niveles reducidos de la hormona tiroidea. Esto lo puede analizar su médico y, si es necesario, su médico le recetará el tratamiento adecuado. En casos raros se ha observado inflamación del páncreas en pacientes tratados con la hormona del crecimiento.
Si considera que alguno de los efectos adversos que sufre es grave o si aprecia cualquier efecto adverso no mencionado en este prospecto, informe a su médico o farmacéutico.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Omnitrope
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y en la caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
• Conservar y transportar refrigerado (entre 2°C y 8°C).
• No congelar.
• Conservar en el embalaje original para protegerlo de la luz.
• Después de la administración de la primera inyección, el cartucho deberá permanecer en el inyector de pluma y debe conservarse en una nevera (entre 2°C y 8°C), y debe utilizarse en un máximo de 21 días.
No utilice Omnitrope si se observa que la solución está turbia.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Omnitrope
El principio activo de Omnitrope es somatropina.
Un cartucho contiene 5 mg (que corresponde a 15 UI) de somatropina tras su reconstitución con 1 ml de disolvente.
Los demás componentes son:
Polvo:
glicina
fosfato de hidrógeno disódico heptahidrato fosfato dihidrógeno sódico dihidrato
Disolvente:
agua para preparaciones inyectables alcohol bencílico
Aspecto del producto y contenido del envase
Polvo y disolvente para solución inyectable (polvo en un vial (5 mg), disolvente en un cartucho (1 ml)).
Envases de 1 y 5 cartuchos.
El polvo es blanco y el disolvente es una solución transparente e incolora.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Austria
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Lietuva
Sandoz Pharmaceuticals d.d. filialas Tel.: +370 5 2636 037
Luxembourg/Luxemburg
Sandoz GmbH Tel.: +43 5338 2000
Magyarország
Sandoz Hungária Kft.
Tel.: +36 1 430 2890
Malta
Sandoz GmbH Tel.: +43 5338 2000
Nederland
Sandoz B.V.
Tel.: +31 36 52 41 600
Osterreich
Sandoz GmbH Tel.: +43 5338 2000
Polska
Sandoz Polska Sp. z o.o.
Tel.: +48 22 209 70 00
Portugal
Sandoz Farmacéutica Lda.
Tel.: +351 21 924 19 11
Romania
S.C. Sandoz S.R.L.
Tel.: +40 265 208 120
Slovenija
Lek farmacevtska druzba d.d.
Tel.: +386 1 580 21 11
Belgie/Belgique/Belgien
Sandoz nv-sa Tél/Tel.: +32 2 722 97 97
Etarapna
TtproBCKO npegcTaBHTencTBO CaHgo3 g.g. Tea.: +359 2 970 47 47
Ceská republika
Sandoz s.r.o.
Tel.: +420 221 421 611
Danmark/Norge/Ísland/Sverige
Sandoz A/S Tlf.: +45 6395 1000
Deutschland
Hexal AG
Tel.: +49 8024 908 0 Eesti
Sandoz d.d. Eesti filiaal Tel.: +372 665 2400
EXláóa Sandoz dd
Tq^: +30 216 600 500 0 España
Sandoz Farmacéutica, S.A.
Tel.: +34 900 456 856
France
Sandoz SAS
Tél.: +33 1 49 64 48 00
Hrvatska
Sandoz d.o.o.
Tel: +385 1 23 53 111
Ireland
Rowex Ltd Tel.: +353 27 50077
Italia
Sandoz S.p.A.
Tel.: +39 02 96541
Kúrcpog
P.T.Hadjigeorgiou co ltd Tel.: +357 25372425
Latvija
Sandoz d.d. Párstávnieciba Latvija Tel.: +371 67 892 006
Slovenská republika
Sandoz d.d. - organizacná zlozka Tel.: +421 2 48 200 600
Suomi/Finland
Sandoz A/S
Puh/Tel.: +358 10 6133 415
United Kingdom
Sandoz Ltd
Tel.: + 44 1276 69 8020
Fecha de la última revisión de este prospecto: {MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
Prospecto: Información para el usuario Omnitrope 5 mg/1,5 ml solución inyectable
Somatropina
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Omnitrope y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Omnitrope
3. Cómo usar Omnitrope
4. Posibles efectos adversos
5. Conservación de Omnitrope
6. Contenido del envase e información adicional
1. Qué es Omnitrope y para qué se utiliza
Omnitrope es una hormona del crecimiento humana recombinante (también llamada somatropina). Tiene la misma estructura que la hormona del crecimiento humana natural, que es necesaria para que los huesos y los músculos crezcan. También ayuda a que los tejidos grasos y musculares se desarrollen en las cantidades correctas. Es recombinante, lo que significa que no se elabora a partir de tejido humano o animal.
En los niños, Omnitrope se usa para tratar los siguientes trastornos del crecimiento:
• Si no creces adecuadamente y no tienes suficiente hormona del crecimiento propia.
• Si padeces un síndrome de Turner, que es un trastorno genético en las niñas que puede afectar al crecimiento; el médico te lo habrá dicho si padeces este trastorno.
• Si padeces una insuficiencia renal crónica. A medida que los riñones pierden su capacidad para funcionar normalmente, esto puede afectar al crecimiento.
• Si eras demasiado pequeño o demasiado ligero al nacer. La hormona de crecimiento puede ayudar a que crezcas más si no has podido tener un estirón o mantener un crecimiento normal a los cuatro años de edad o en adelante.
• Si padeces un síndrome de Prader-Willi (un trastorno cromosómico). La hormona de crecimiento puede ayudar a que crezca más si sigue creciendo y también mejorará la composición de tu organismo. El exceso de grasa se reducirá y la masa muscular disminuida mejorará.
En los adultos, Omnitrope se usa para
• tratar a las personas con una deficiencia pronunciada de la hormona del crecimiento. Esta puede empezar durante la edad adulta o puede continuar desde la niñez.
Si usted ha sido tratado con Omnitrope por una deficiencia de la hormona de crecimiento durante la niñez, se volverá a examinar el estado de la hormona de crecimiento después de finalizar el crecimiento. Si se confirma una deficiencia grave de la hormona, el médico propondrá la continuación del tratamiento con Omnitrope.
Sólo debe recibir este medicamento de un médico que tenga experiencia con la hormona de crecimiento y que haya confirmado su diagnóstico.
2. Qué necesita saber antes de empezar a usar Omnitrope
No use Omnitrope
• si es alérgico (hipersensible) a la somatropina o a cualquiera de los demás componentes de Omnitrope.
• e informe a su médico si usted padece un tumor activo (cáncer). Los tumores deben ser inactivos y usted debe haber terminado su tratamiento antitumoral antes de empezar su tratamiento con Omnitrope.
• e informe a su médico si se le ha prescrito Omitrope para estimular el crecimiento pero usted ya ha dejado de crecer (epífisis cerradas).
• si está gravemente enfermo (por ejemplo, complicaciones post-quirúrgicas a corazón abierto, cirugía abdominal, traumatismo accidental, insuficiencia respiratoria aguda o afecciones similares). Si a usted le van a practicar o le han practicado una operación mayor, o si va al hospital por cualquier motivo, infórmele a su médico y recuérdeles a los otros médicos a los que ve que usted usa la hormona de crecimiento.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar Omnitrope.
• Si tiene riesgo de presentar diabetes, el médico deberá controlar regularmente la concentración de la glucosa en la sangre durante el tratamiento con somatropina.
• Si padece diabetes mellitus, deberá vigilar atentamente la concentración de glucosa en la sangre durante el tratamiento con somatropina y hablar con el médico acerca de los resultados, a fin de decidir si tiene que cambiar la dosis de sus medicamentos para tratar la diabetes.
• Después de comenzar el tratamiento con somatropina, algunos pacientes pueden tener que comenzar un reemplazo con hormona tiroidea.
• Si recibe tratamiento con hormonas tiroideas, puede ser necesario ajustar la dosis de hormona tiroidea.
• Si usted tiene un aumento de la presión intracraneal (que causa síntomas, tales como dolor de cabeza intenso, alteraciones visuales o vómitos) deberá informar al médico acerca de ello.
• Si camina cojeando o si empieza a cojear durante el tratamiento con hormona de crecimiento, deberá informar al médico.
• Si está recibiendo somatropina para una deficiencia de la hormona de crecimiento después de un tumor previo (cáncer), deberán examinarlo regularmente para detectar la recurrencia del tumor o cualquier otro cáncer.
• Si experimenta un dolor abdominal que empeora, debe informar a su médico.
• La experiencia en pacientes de más de 80 años es limitada. Las personas de edad avanzada
pueden ser más sensibles a la acción de la somatropina y, por lo tanto, pueden ser más propensas a presentar reacciones adversas.
Niños con insuficiencia renal crónica
• El médico deberá examinar la función de los riñones y la velocidad de crecimiento antes de empezar el tratamiento con somatropina. El tratamiento médico de los riñones debe continuarse. El tratamiento con somatropina debe interrumpirse en caso de trasplante renal.
Niños con síndrome de Prader-Willi
• El médico le dará restricciones en la dieta que debe seguir para controlar su peso.
• El médico evaluará los signos de obstrucción de las vías respiratorias altas, apnea del sueño (en
que la respiración se interrumpe durante el sueño) o infección respiratoria antes de comenzar el tratamiento con somatropina.
• Durante el tratamiento con somatropina, informe al médico si presenta signos de obstrucción de las vías respiratorias altas (incluso comenzar a roncar o un empeoramiento de los ronquidos).
Tal vez el médico tenga que examinarle y puede interrumpir el tratamiento con somatropina.
• Durante el tratamiento, el médico le examinará para ver si hay signos de escoliosis, un tipo de deformidad vertebral.
• Durante el tratamiento, si presenta una infección pulmonar, informe al médico para que pueda tratar la infección.
Niños nacidos demasiado pequeños o bajos de peso
• Si eras demasiado pequeño o demasiado ligero al nacer y tienes de 9 a 12 años, consulta al médico específicamente en relación con la pubertad y el tratamiento con este medicamento.
• El tratamiento debe continuar hasta que hayas dejado de crecer.
• El médico examinará las concentraciones de glucosa e insulina antes de comenzar el tratamiento y cada año durante el tratamiento.
Uso de Omnitrope con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Debe informar al médico si está utilizando:
• medicamentos para tratar la diabetes;
• hormonas tiroideas;
• medicamentos para controlar la epilepsia (anticonvulsivantes);
• ciclosporina (un medicamento que debilita el sistema inmunitario después de los trasplantes);
• hormonas sexuales (por ejemplo, estrógenos);
• hormonas suprarrenales sintéticas (corticoesteroides).
Tal vez el médico tenga que ajustar la dosis de estos medicamentos o la dosis de somatropina. Embarazo y lactancia
No debe usar Omnitrope si está embarazada o tratando de quedarse embarazada.
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Información importante sobre algunos de los componentes de Omnitrope
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por ml; esto es, esencialmente “exento de sodio”.
Un ml contiene 9 mg de alcohol bencílico.
Debido a la presencia de alcohol bencílico, el medicamento no se debe administrar a niños prematuros ni recién nacidos. Puede provocar reacciones tóxicas y reacciones alérgicas en niños de hasta 3 años de edad.
3. Cómo usar Omnitrope
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero. La dosis depende de su tamaño, de la afección para la que recibe tratamiento y de lo bien que funcione la hormona de crecimiento en usted. Todas las personas son diferentes. El médico le aconsejará acerca de su dosis individualizada de Omnitrope en miligramos (mg) a partir de su peso corporal en kilogramos (kg) o por su superficie corporal, calculada a partir de su estatura y peso en metros cuadrados (m2), así como su pauta de tratamiento. No cambie la dosificación y la pauta de tratamiento sin consultarle al médico.
La dosis recomendada es para:
Niños con deficiencia de la hormona de crecimiento:
0,025 a 0,035 mg/kg de peso corporal al día o 0,7 a 1,0 mg/m2 de superficie corporal al día. Pueden utilizarse dosis más altas. Cuando la deficiencia de hormona de crecimiento continúa durante la adolescencia, Omnitrope debe continuarse hasta finalizar el desarrollo físico.
Niñas con síndrome de Turner:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día.
Niños con insuficiencia renal crónica:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día. Pueden ser necesarias dosis más altas si la velocidad de crecimiento es demasiado baja. Puede ser necesario un ajuste de la dosis después de seis meses de tratamiento.
Niños con síndrome de Prader-Willi:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. La dosificación diaria no debe ser superior a 2,7 mg. El tratamiento no debe utilizarse en los niños que casi han dejado de crecer después de la pubertad.
Niños nacidos más pequeños o con peso más bajo que lo esperado y con un trastorno del crecimiento:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. Es importante continuar el tratamiento hasta que se alcance la estatura final. El tratamiento debe suspenderse después del primer año si no responde, o si ha alcanzado la estatura final y dejado de crecer.
Adultos con deficiencia de la hormona de crecimiento:
Si continúa utilizando Omnitrope después del tratamiento durante la niñez, debe comenzar con 0,2 a 0,5 mg al día.
Esta dosificación se debe aumentar o reducir gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios.
Si la deficiencia de la hormona de crecimiento comienza durante la vida adulta, debe comenzar con 0,15 a 0,3 mg al día. Esta dosificación debe aumentarse gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios. La dosis de mantenimiento diaria rara vez es superior a 1,0 mg diarios. Las mujeres pueden necesitar dosis más altas que los hombres. La dosificación debe vigilarse cada seis meses. Las personas de más de 60 años deben comenzar con una dosis de 0,1 a 0,2 mg diarios que debe aumentarse lentamente según las necesidades individuales. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento rara vez supera 0,5 mg al día. Siga las instrucciones que le haya dado el médico.
Inyección de Omnitrope
Inyéctese la hormona del crecimiento más o menos a la misma hora cada día. La hora de acostarse es un buen momento porque es fácil de recordar. Además, también es natural tener una concentración más elevada de hormona del crecimiento por la noche.
Omnitrope 5 mg/1,5 ml está pensado para varios usos. Sólo debe administrarse con la pluma Omnitrope Pen 5, un dispositivo inyectable desarrollado específicamente para su uso con Omnitrope 5 mg/1,5 ml solución inyectable.
Omnitrope está indicado para su empleo por vía subcutánea. Esto significa que se inyecta por medio de una pequeña aguja para inyección en el tejido adiposo, por debajo de la piel. La mayor parte de las personas se inyectan en el muslo o en las nalgas. Póngase la inyección en el sitio que le haya enseñado su médico. El tejido adiposo de la piel puede verse reducido en el lugar de la inyección. Para evitar esto, utilice cada vez un sitio ligeramente diferente para inyectarse. Esto proporciona a la piel y a la zona por debajo de ella tiempo para recuperarse de una inyección antes de recibir otra en el mismo sitio.
El médico debe haberle enseñado ya cómo utilizar Omnitrope. Inyéctese siempre Omnitrope tal como el médico le ha dicho. Si no está seguro, compruebe con su médico o farmacéutico.
Cómo inyectar Omnitrope 5 mg/1,5 ml
Las siguientes instrucciones explican cómo inyectarse Omnitrope 5 mg/1,5 ml usted mismo. Lea detenidamente las instrucciones y sígalas paso a paso. Su médico o la enfermera le enseñará cómo inyectarse Omnitrope. No intente inyectarse a menos que esté seguro de que entiende el procedimiento y lo que conlleva la inyección.
- Omnitrope se administra como una inyección debajo de la piel.
- Inspeccione cuidadosamente la solución antes de inyectarla y úsela sólo si es clara e incolora.
- Cambie el lugar de la inyección para así minimizar el riesgo de lipoatrofia local (reducción local del tejido adiposo debajo de la piel).
Preparación
Antes de empezar, debe tener todo lo necesario:
- Un cartucho con Omnitrope 5 mg/1,5 ml solución inyectable.
El Omnitrope Pen 5, un dispositivo para inyección desarrollado específicamente para usarlo junto con Omnitrope 5 mg/1,5 ml solución inyectable (este dispositivo no se suministra en este envase; deben consultarse las Instrucciones de uso proporcionadas junto con Omnitrope Pen 5).
Una aguja de pluma para inyección subcutánea.
2 gasas de limpieza (no suministradas en el envase).
Lávese las manos antes continuar con los siguiente pasos.
Inyección de Omnitrope
- Con una gasa, desinfecte la membrana de goma del cartucho.
- El contenido del cartucho debe ser transparente e incoloro .
Introduzca el cartucho en la pluma inyectable. Siga las Instrucciones de uso de la pluma para inyección (Pen). Para ajustar la pluma, dosifique la cantidad necesaria.
Seleccione el lugar de la inyección. Los mejores lugares para la administración de la inyección son los tejidos con una capa de grasa entre la piel y el músculo, como el muslo o el vientre (excepto el ombligo o la cintura).
Asegúrese de que se inyecta al menos a 1 cm de distancia respecto del lugar de la última inyección y de que cambia los lugares de inyección, tal y como le habrán enseñado.
Antes de administrar la inyección, limpie bien la piel con una gasa empapada en alcohol. Espere a que la zona se seque.
Introduzca la aguja en la piel de la forma que le haya enseñado el médico.
Después de inyectarse
- Después de la inyección, presione el lugar de la inyección con un pequeño vendaje o gasa estéril durante unos segundos. No masajee el lugar de la inyección.
- Retire la aguja de la pluma utilizando para ello la tapa exterior y elimínela. Esto mantendrá la solución de Omnitrope estéril e impedirá que gotee. También impedirá que el aire se introduzca en la pluma, taponando la aguja. No comparta las agujas. No comparta la pluma.
- Deje el cartucho en la pluma, reponga el capuchón en la pluma y guárdela en una nevera.
- La solución debe ser transparente cuando se saque de la nevera. No se debe utilizar si la solución está turbia o si contiene partículas.
Si usa más Omnitrope del que debe
Si se inyecta mucho más de lo que debiera, consulte lo antes posible a su médico o farmacéutico. Su concentración de azúcar en la sangre podría descender demasiado y después aumentar demasiado. Tal vez se sienta con temblores, sudoroso, somnoliento o “como si no fuera usted mismo”, y podría desmayarse.
Si olvidó usar Omnitrope
No use una dosis doble para compensar las dosis olvidadas. Lo mejor es usar la hormona de crecimiento con regularidad. Si se olvida de usar una dosis, póngase la siguiente inyección a la hora habitual, al día siguiente. Tome nota de las inyecciones olvidadas e infórmele al médico en el siguiente control.
Si interrumpe el tratamiento con Omnitrope
Consulte a su médico antes de dejar de usar Omnitrope.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Los efectos adversos muy frecuentes y frecuentes en los adultos pueden comenzar en los primeros meses de tratamiento y pueden detenerse espontáneamente o si se reduce la dosis.
Los efectos adversos muy frecuentes (que probablemente se produzcan en más de 1 de cada 10 pacientes) incluyen los siguientes:
En los adultos
• Dolor articular
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos)
Los efectos adversos frecuentes (que probablemente afecten a menos de 1 de cada 10 pacientes) incluyen los siguientes:
En los niños
• Enrojecimiento, picor o dolor temporal en el lugar de la inyección
• Dolor articular
En los adultos
• Adormecimiento, hormigueos
• Rigidez de los brazos y las piernas, dolor muscular
• Dolor o sensación de escozor en las manos o los antebrazos (conocido como síndrome del túnel carpiano)
Los efectos adversos poco frecuentes (que probablemente afecten a menos de 1 de cada 100 pacientes) incluyen los siguientes:
En los niños
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos, durante un tiempo corto al comienzo del tratamiento)
Los efectos adversos raros (que probablemente afecten a menos de 1 de cada 1.000 pacientes) incluyen los siguientes:
En los niños
• Adormecimiento, hormigueos
• Leucemia (se ha observado en un pequeño número de pacientes con deficiencia de la hormona del crecimiento, algunos de cuales habían sido tratados con somatropina. Sin embargo, no hay ningún indicio de que la incidencia de leucemia sea mayor en receptores de la hormona del crecimiento sin factores predisponentes).
• Aumento de la presión intracraneal (que causa síntomas, como cefalea intensa, alteraciones visuales o vómitos)
• Dolor muscular
Frecuencia no conocida: no puede estimarse a partir de los datos disponibles
• Diabetes de tipo 2
• Disminución de los niveles de la hormona cortisol en la sangre En los niños
• Rigidez en los brazos y las piernas En los adultos
• Aumento de la presión intracraneal (que causa síntomas, como dolor de cabeza intenso, alteraciones visuales o vómitos)
• Enrojecimiento, picor o dolor en el lugar de la inyección
Formación de anticuerpos contra la hormona del crecimiento inyectada, pero estos no parecen hacer que la hormona del crecimiento deje de funcionar.
La piel alrededor de la zona de inyección puede ponerse irregular o con bultos, pero esto no deberá ocurrir si se inyecta en un lugar diferente cada vez.
Ha habido casos raros de muerte súbita en pacientes con síndrome de Prader-Willi. No obstante, estos casos no se han relacionado con el tratamiento con Omnitrope.
Su médico puede considerar una epifisiólisis de la cabeza femoral o una enfermedad de Legg-Calvé-Perthes si experimenta molestias o dolor en la cadera o la rodilla mientras está siendo tratado con Omnitrope.
Otros posibles efectos adversos relacionados con su tratamiento con la hormona del crecimiento pueden incluir los siguientes:
Usted (o su hijo) puede tener niveles elevados de azúcar en sangre o niveles reducidos de la hormona tiroidea. Esto lo puede analizar su médico y, si es necesario, su médico le recetará el tratamiento adecuado. En casos raros se ha observado inflamación del páncreas en pacientes tratados con la hormona del crecimiento.
Si considera que alguno de los efectos adversos que sufre es grave o si aprecia cualquier efecto adverso no mencionado en este prospecto, informe a su médico o farmacéutico.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Omnitrope
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y en la caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
• Conservar y transportar refrigerado (entre 2°C y 8°C).
• No congelar.
• Conservar en el embalaje original para protegerlo de la luz.
• Después de la administración de la primera inyección, el cartucho debe permanecer en el inyector de pluma y debe conservarse en una nevera, a una temperatura de 2 a 8 °C, y sólo debe usarse durante un máximo de 28 días.
No utilice Omnitrope si se observa que la solución está turbia.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Omnitrope
- El principio activo de Omnitrope es somatropina.
Cada ml de solución contiene 3,3 mg de somatropina (que corresponde a 10 UI).
Un cartucho contiene 5,0 mg (que corresponde a 15 UI) de somatropina en 1,5 ml.
- Los demás componentes son:
Fosfato hidrógeno disódico heptahidrato Fosfato dihidrógeno sódico dihidrato Manitol
Poloxámero 188 Alcohol bencílico
Agua para preparaciones inyectables
Aspecto del producto y contenido del envase
Omnitrope es una solución inyectable transparente e incolora.
Envases con 1, 5 ó 10 cartuchos.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Austria
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Lietuva
Belgie/Belgique/Belgien
Sandoz nv-sa Tél/Tel.: +32 2 722 97 97
Sandoz Pharmaceuticals d.d. filíalas Tel.: +370 5 2636 037
|
Etarapna TtproBCKO npegcTaBHTeacTBO CaHgo3 g.g. Tea.: +359 2 970 47 47 |
Luxembourg/Luxemburg Sandoz GmbH Tel.: +43 5338 2000 |
|
Ceská republika Sandoz s.r.o. Tel.: +420 221 421 611 |
Magyarország Sandoz Hungária Kft. Tel.: +36 1 430 2890 |
|
Danmark/Norge/Ísland/Sverige Sandoz A/S Tlf.: +45 6395 1000 |
Malta Sandoz GmbH Tel.: +43 5338 2000 |
|
Deutschland Hexal AG Tel.: +49 8024 908 0 |
Nederland Sandoz B.V. Tel.: +31 36 52 41 600 |
|
Eesti Sandoz d.d. Eesti filiaal Tel.: +372 665 2400 |
Osterreich Sandoz GmbH Tel.: +43 5338 2000 |
|
EXláda Sandoz dd T^: +30 216 600 500 0 |
Polska Sandoz Polska Sp. z o.o. Tel.: +48 22 209 70 00 |
|
España Sandoz Farmacéutica, S.A. Tel.: +34 900 456 856 |
Portugal Sandoz Farmacéutica Lda. Tel.: +351 21 924 19 11 |
|
France Sandoz SAS Tél.: +33 1 49 64 48 00 |
Romania S.C. Sandoz S.R.L. Tel.: +40 265 208 120 |
|
Hrvatska Sandoz d.o.o. |
Slovenija Lek farmacevtska druzba d.d. |
|
Tel: +385 1 23 53 111 |
Tel.: +386 1 580 21 11 |
|
Ireland Rowex Ltd Tel.: +353 27 50077 |
Slovenská republika Sandoz d.d. - organizacná zlozka Tel.: +421 2 48 200 600 |
|
Italia Sandoz S.p.A. Tel.: +39 02 96541 |
Suomi/Finland Sandoz A/S Puh/Tel.: +358 10 6133 415 |
|
Kúnpoq P.T.Hadjigeorgiou co ltd Tel.: +357 25372425 |
United Kingdom Sandoz Ltd Tel.: + 44 1276 69 8020 |
Latvija
Sandoz d.d. Párstávniedba Latvija Tel.: +371 67 892 006
Fecha de la última revisión de este prospecto: {MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
Prospecto: Información para el usuario Omnitrope 10 mg/1,5 ml solución inyectable
Somatropina
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Omnitrope y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Omnitrope
3. Cómo usar Omnitrope
4. Posibles efectos adversos
5. Conservación de Omnitrope
6. Contenido del envase e información adicional
1. Qué es Omnitrope y para qué se utiliza
Omnitrope es una hormona del crecimiento humana recombinante (también llamada somatropina). Tiene la misma estructura que la hormona del crecimiento humana natural, que es necesaria para que los huesos y los músculos crezcan. También ayuda a que los tejidos grasos y musculares se desarrollen en las cantidades correctas. Es recombinante, lo que significa que no se elabora a partir de tejido humano o animal.
En los niños, Omnitrope se usa para tratar los siguientes trastornos del crecimiento:
• Si no creces adecuadamente y no tienes suficiente hormona del crecimiento propia.
• Si padeces un síndrome de Turner, que es un trastorno genético en las niñas que puede afectar al crecimiento; el médico te lo habrá dicho si padeces este trastorno.
• Si padeces una insuficiencia renal crónica. A medida que los riñones pierden su capacidad para funcionar normalmente, esto puede afectar al crecimiento.
• Si eras demasiado pequeño o demasiado ligero al nacer. La hormona de crecimiento puede ayudar a que crezcas más si no has podido tener un estirón o mantener un crecimiento normal a los cuatro años de edad o en adelante.
• Si padeces un síndrome de Prader-Willi (un trastorno cromosómico). La hormona de crecimiento puede ayudar a que crezca más si sigue creciendo y también mejorará la composición de tu organismo. El exceso de grasa se reducirá y la masa muscular disminuida mejorará.
En los adultos, Omnitrope se usa para
• tratar a las personas con una deficiencia pronunciada de la hormona del crecimiento. Esta puede empezar durante la edad adulta o puede continuar desde la niñez.
Si usted ha sido tratado con Omnitrope por una deficiencia de la hormona de crecimiento durante la niñez, se volverá a examinar el estado de la hormona de crecimiento después de finalizar el crecimiento. Si se confirma una deficiencia grave de la hormona, el médico propondrá la continuación del tratamiento con Omnitrope.
Sólo debe recibir este medicamento de un médico que tenga experiencia con la hormona de crecimiento y que haya confirmado su diagnóstico.
2. Qué necesita saber antes de empezar a usar Omnitrope
No use Omnitrope
• si es alérgico (hipersensible) a la somatropina o a cualquiera de los demás componentes de Omnitrope.
• e informe a su médico si usted padece un tumor activo (cáncer). Los tumores deben ser inactivos y usted debe haber terminado su tratamiento antitumoral antes de empezar su tratamiento con Omnitrope.
• e informe a su médico si se le ha prescrito Omitrope para estimular el crecimiento pero usted ya ha dejado de crecer (epífisis cerradas).
• si está gravemente enfermo (por ejemplo, complicaciones post-quirúrgicas a corazón abierto, cirugía abdominal, traumatismo accidental, insuficiencia respiratoria aguda o afecciones similares). Si a usted le van a practicar o le han practicado una operación mayor, o si va al hospital por cualquier motivo, infórmele a su médico y recuérdeles a los otros médicos a los que ve que usted usa la hormona de crecimiento.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar Omnitrope.
• Si tiene riesgo de presentar diabetes, el médico deberá controlar regularmente la concentración de la glucosa en la sangre durante el tratamiento con somatropina.
• Si padece diabetes mellitus, deberá vigilar atentamente la concentración de glucosa en la sangre durante el tratamiento con somatropina y hablar con el médico acerca de los resultados, a fin de decidir si tiene que cambiar la dosis de sus medicamentos para tratar la diabetes.
• Después de comenzar el tratamiento con somatropina, algunos pacientes pueden tener que comenzar un reemplazo con hormona tiroidea.
• Si recibe tratamiento con hormonas tiroideas, puede ser necesario ajustar la dosis de hormona tiroidea.
• Si usted tiene un aumento de la presión intracraneal (que causa síntomas, tales como dolor de cabeza intenso, alteraciones visuales o vómitos) deberá informar al médico acerca de ello.
• Si camina cojeando o si empieza a cojear durante el tratamiento con hormona de crecimiento, deberá informar al médico.
• Si está recibiendo somatropina para una deficiencia de la hormona de crecimiento después de un tumor previo (cáncer), deberán examinarlo regularmente para detectar la recurrencia del tumor o cualquier otro cáncer.
• Si experimenta un dolor abdominal que empeora, debe informar a su médico.
• La experiencia en pacientes de más de 80 años es limitada. Las personas de edad avanzada
pueden ser más sensibles a la acción de la somatropina y, por lo tanto, pueden ser más propensas a presentar reacciones adversas.
Niños con insuficiencia renal crónica
• El médico deberá examinar la función de los riñones y la velocidad de crecimiento antes de empezar el tratamiento con somatropina. El tratamiento médico de los riñones debe continuarse. El tratamiento con somatropina debe interrumpirse en caso de trasplante renal.
Niños con síndrome de Prader-Willi
• El médico le dará restricciones en la dieta que debe seguir para controlar su peso.
• El médico evaluará los signos de obstrucción de las vías respiratorias altas, apnea del sueño (en
que la respiración se interrumpe durante el sueño) o infección respiratoria antes de comenzar el tratamiento con somatropina.
• Durante el tratamiento con somatropina, informe al médico si presenta signos de obstrucción de las vías respiratorias altas (incluso comenzar a roncar o un empeoramiento de los ronquidos).
Tal vez el médico tenga que examinarle y puede interrumpir el tratamiento con somatropina.
• Durante el tratamiento, el médico le examinará para ver si hay signos de escoliosis, un tipo de deformidad vertebral.
• Durante el tratamiento, si presenta una infección pulmonar, informe al médico para que pueda tratar la infección.
Niños nacidos demasiado pequeños o bajos de peso
• Si eras demasiado pequeño o demasiado ligero al nacer y tienes de 9 a 12 años, consulta al médico específicamente en relación con la pubertad y el tratamiento con este medicamento.
• El tratamiento debe continuar hasta que hayas dejado de crecer.
• El médico examinará las concentraciones de glucosa e insulina antes de comenzar el tratamiento y cada año durante el tratamiento.
Uso de Omnitrope con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Debe informar al médico si está utilizando:
• medicamentos para tratar la diabetes;
• hormonas tiroideas;
• medicamentos para controlar la epilepsia (anticonvulsivantes);
• ciclosporina (un medicamento que debilita el sistema inmunitario después de los trasplantes);
• hormonas sexuales (por ejemplo, estrógenos);
• hormonas suprarrenales sintéticas (corticoesteroides).
Tal vez el médico tenga que ajustar la dosis de estos medicamentos o la dosis de somatropina. Embarazo y lactancia
No debe usar Omnitrope si está embarazada o tratando de quedarse embarazada.
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Información importante sobre algunos de los componentes de Omnitrope
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por ml; esto es, esencialmente “exento de sodio”.
3. Cómo usar Omnitrope
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero. La dosis depende de su tamaño, de la afección para la que recibe tratamiento y de lo bien que funcione la hormona de crecimiento en usted. Todas las personas son diferentes. El médico le aconsejará acerca de su dosis individualizada de Omnitrope en miligramos (mg) a partir de su peso corporal en kilogramos (kg) o por su superficie corporal, calculada a partir de su estatura y peso en metros cuadrados (m2), así como su pauta de tratamiento. No cambie la dosificación y la pauta de tratamiento sin consultarle al médico.
La dosis recomendada es para:
Niños con deficiencia de la hormona de crecimiento:
0,025 a 0,035 mg/kg de peso corporal al día o 0,7 a 1,0 mg/m2 de superficie corporal al día. Pueden utilizarse dosis más altas. Cuando la deficiencia de hormona de crecimiento continúa durante la adolescencia, Omnitrope debe continuarse hasta finalizar el desarrollo físico.
Niñas con síndrome de Turner:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día.
Niños con insuficiencia renal crónica:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día. Pueden ser necesarias dosis más altas si la velocidad de crecimiento es demasiado baja. Puede ser necesario un ajuste de la dosis después de seis meses de tratamiento.
Niños con síndrome de Prader-Willi:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. La dosificación diaria no debe ser superior a 2,7 mg. El tratamiento no debe utilizarse en los niños que casi han dejado de crecer después de la pubertad.
Niños nacidos más pequeños o con peso más bajo que lo esperado y con un trastorno del crecimiento:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. Es importante continuar el tratamiento hasta que se alcance la estatura final. El tratamiento debe suspenderse después del primer año si no responde, o si ha alcanzado la estatura final y dejado de crecer.
Adultos con deficiencia de la hormona de crecimiento:
Si continúa utilizando Omnitrope después del tratamiento durante la niñez, debe comenzar con 0,2 a 0,5 mg al día.
Esta dosificación se debe aumentar o reducir gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios.
Si la deficiencia de la hormona de crecimiento comienza durante la vida adulta, debe comenzar con 0,15 a 0,3 mg al día. Esta dosificación debe aumentarse gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios. La dosis de mantenimiento diaria rara vez es superior a 1,0 mg diarios. Las mujeres pueden necesitar dosis más altas que los hombres. La dosificación debe vigilarse cada seis meses. Las personas de más de 60 años deben comenzar con una dosis de 0,1 a 0,2 mg diarios que debe aumentarse lentamente según las necesidades individuales. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento rara vez supera 0,5 mg al día. Siga las instrucciones que le haya dado el médico.
Inyección de Omnitrope
Inyéctese la hormona del crecimiento más o menos a la misma hora cada día. La hora de acostarse es un buen momento porque es fácil de recordar. Además, también es natural tener una concentración más elevada de hormona del crecimiento por la noche.
Omnitrope 10 mg/1,5 ml está previsto para varios usos. Sólo debe administrarse con la pluma Omnitrope Pen 10, un dispositivo de inyección desarrollado específicamente para la administración de Omnitrope 10 mg/1,5 ml solución inyectable.
Omnitrope está indicado para su empleo por vía subcutánea. Esto significa que se inyecta por medio de una pequeña aguja para inyección en el tejido adiposo, por debajo de la piel. La mayor parte de las personas se inyectan en el muslo o en las nalgas. Póngase la inyección en el sitio que le haya enseñado su médico. El tejido adiposo de la piel puede verse reducido en el lugar de la inyección. Para evitar esto, utilice cada vez un sitio ligeramente diferente para inyectarse. Esto proporciona a la piel y a la zona por debajo de ella tiempo para recuperarse de una inyección antes de recibir otra en el mismo sitio.
El médico debe haberle enseñado ya cómo utilizar Omnitrope. Inyéctese siempre Omnitrope tal como el médico le ha dicho. Si no está seguro, compruebe con su médico o farmacéutico.
Cómo inyectar Omnitrope 10 mg/1,5 ml
Las siguientes instrucciones explican cómo inyectarse Omnitrope 10 mg/1,5 ml usted mismo. Lea detenidamente las instrucciones y sígalas paso a paso. Su médico o la enfermera le enseñará cómo inyectarse Omnitrope. No intente inyectarse a menos que esté seguro de que entiende el procedimiento y lo que conlleva la inyección.
- Omnitrope se administra con una inyección debajo de la piel.
- Inspeccione cuidadosamente la solución antes de inyectarla y úsela sólo si es clara e incolora.
- Cambie el lugar de la inyección para así minimizar el riesgo de lipoatrofia local (reducción local del tejido adiposo debajo de la piel).
Preparación
Antes de empezar, debe tener todo lo necesario:
- Un cartucho con Omnitrope 10 mg/1,5 ml solución inyectable.
- El Omnitrope Pen 10, un dispositivo para inyecciói desarrollado específicamente para usarlo junto con Omnitrope 10 mg/1,5 ml solución inyectable (este dispositivo no se suministra en este envase; deben consultarse las Instrucciones de uso proporcionadas junto con Omnitrope Pen 10).
- Una aguja de pluma para inyección subcutánea.
- 2 gasas de limpieza (no suministradas en el envase).
Lávese las manos antes de continuar con los siguiente pasos.
Inyección de Omnitrope
- Con una gasa, desinfecte la membrana de goma del cartucho.
- El contenido del cartucho debe ser transparente e incoloro.
Introduzca el cartucho en la pluma inyectable. Siga las Instrucciones de uso de la pluma para inyección (Pen). Para ajustar la pluma, dosifique la cantidad necesaria.
Seleccione el lugar de la inyección. Los mejores lugares para la administración de la inyección son los tejidos con una capa de grasa entre la piel y el músculo, como el muslo o el vientre (excepto el ombligo o la cintura).
Asegúrese de que se inyecta al menos a 1 cm de distancia respecto del lugar de la última inyección y de que cambia los lugares de inyección, tal y como le habrán enseñado.
Antes de administrar la inyección, limpie bien la piel con una gasa empapada en alcohol. Espere a que la zona se seque.
Introduzca la aguja en la piel de la forma que le haya enseñado el médico.
Después de inyectarse
- Después de la inyección, presione el lugar de la inyección con un pequeño vendaje o gasa estéril durante unos segundos. No masajee el lugar de la inyección.
- Retire la aguja de la pluma utilizando para ello la tapa exterior y elimínela. Esto mantendrá la solución de Omnitrope estéril e impedirá que gotee. También impedirá que el aire se introduzca en la pluma , taponando la aguja. No comparta las agujas. No comparta la pluma.
- Deje el cartucho en la pluma, reponga el capuchón en la pluma y guárdela en una nevera.
- La solución debe ser transparente cuando se saque de la nevera . No se debe utilizar si la solución está turbia o si contiene partículas.
Si se inyecta mucho más de lo que debiera, consulte lo antes posible a su médico o farmacéutico. Su concentración de azúcar en la sangre podría descender demasiado y después aumentar demasiado. Tal vez se sienta con temblores, sudoroso, somnoliento o “como si no fuera usted mismo”, y podría desmayarse.
Si olvidó usar Omnitrope
No use una dosis doble para compensar las dosis olvidadas. Lo mejor es usar la hormona de crecimiento con regularidad. Si se olvida de usar una dosis, póngase la siguiente inyección a la hora habitual, al día siguiente. Tome nota de las inyecciones olvidadas e infórmele al médico en el siguiente control.
Si interrumpe el tratamiento con Omnitrope
Consulte a su médico antes de dejar de usar Omnitrope.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Los efectos adversos muy frecuentes y frecuentes en los adultos pueden comenzar en los primeros meses de tratamiento y pueden detenerse espontáneamente o si se reduce la dosis.
Los efectos adversos muy frecuentes (que probablemente se produzcan en más de 1 de cada 10 pacientes) incluyen los siguientes:
En los adultos
• Dolor articular
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos)
Los efectos adversos frecuentes (que probablemente afecten a menos de 1 de cada 10 pacientes) incluyen los siguientes:
En los niños
• Enrojecimiento, picor o dolor temporal en el lugar de la inyección
• Dolor articular
En los adultos
• Adormecimiento, hormigueos
• Rigidez de los brazos y las piernas, dolor muscular
• Dolor o sensación de escozor en las manos o los antebrazos (conocido como síndrome del túnel carpiano)
Los efectos adversos poco frecuentes (que probablemente afecten a menos de 1 de cada 100 pacientes) incluyen los siguientes:
En los niños
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos, durante un tiempo corto al comienzo del tratamiento)
Los efectos adversos raros (que probablemente afecten a menos de 1 de cada 1.000 pacientes) incluyen los siguientes:
En los niños
• Adormecimiento, hormigueos
• Leucemia (se ha observado en un pequeño número de pacientes con deficiencia de la hormona del crecimiento, algunos de cuales habían sido tratados con somatropina. Sin embargo, no hay ningún indicio de que la incidencia de leucemia sea mayor en receptores de la hormona del crecimiento sin factores predisponentes).
• Aumento de la presión intracraneal (que causa síntomas, como cefalea intensa, alteraciones visuales o vómitos)
• Dolor muscular
Frecuencia no conocida: no puede estimarse a partir de los datos disponibles
• Diabetes de tipo 2
• Disminución de los niveles de la hormona cortisol en la sangre En los niños
• Rigidez en los brazos y las piernas
En los adultos
• Aumento de la presión intracraneal (que causa síntomas, como dolor de cabeza intenso, alteraciones visuales o vómitos)
• Enrojecimiento, picor o dolor en el lugar de la inyección
Formación de anticuerpos contra la hormona del crecimiento inyectada, pero estos no parecen hacer que la hormona del crecimiento deje de funcionar.
La piel alrededor de la zona de inyección puede ponerse irregular o con bultos, pero esto no deberá ocurrir si se inyecta en un lugar diferente cada vez.
Ha habido casos raros de muerte súbita en pacientes con síndrome de Prader-Willi. No obstante, estos casos no se han relacionado con el tratamiento con Omnitrope.
Su médico puede considerar una epifisiólisis de la cabeza femoral o una enfermedad de Legg-Calvé-Perthes si experimenta molestias o dolor en la cadera o la rodilla mientras está siendo tratado con Omnitrope.
Otros posibles efectos adversos relacionados con su tratamiento con la hormona del crecimiento pueden incluir los siguientes:
Usted (o su hijo) puede tener niveles elevados de azúcar en sangre o niveles reducidos de la hormona tiroidea. Esto lo puede analizar su médico y, si es necesario, su médico le recetará el tratamiento adecuado. En casos raros se ha observado inflamación del páncreas en pacientes tratados con la hormona del crecimiento.
Si considera que alguno de los efectos adversos que sufre es grave o si aprecia cualquier efecto adverso no mencionado en este prospecto, informe a su médico o farmacéutico.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y en la caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
• Conservar y transportar refrigerado (entre 2°C y 8°C).
• No congelar.
• Conservar en el embalaje original para protegerlo de la luz.
• Después de la administración de la primera inyección, el cartucho debe permanecer en el inyector de pluma y debe conservarse en una nevera, a una temperatura de 2 a 8 °C, y sólo debe usarse durante un máximo de 28 días.
No utilice Omnitrope si se observa que la solución está turbia.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Omnitrope
- El principio activo de Omnitrope es somatropina.
Cada ml de solución contiene 6,7 mg de somatropina (que corresponde a 20 UI).
Un cartucho contiene 10,0 mg (que corresponden a 30 UI) de somatropina en 1,5 ml.
- Los demás componentes son:
Fosfato hidrógeno disódico heptahidrato Fosfato dihidrógeno sódico dihidrato Glicina
Poloxámero 188 Fenol
Agua para preparaciones inyectables
Aspecto del producto y contenido del envase
Omnitrope es una solución inyectable transparente e incolora.
Envases de 1, 5 ó 10 cartuchos.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Austria
Pueden solicitar más información respecto a este titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Sandoz nv-sa Tél/Tel.: +32 2 722 97 97
Etarapna
TtproBCKO npegcTaBHTeacTBO CaHgo3 g.g.
Tea.: +359 2 970 47 47
medicamento dirigiéndose al representante local del
Lietuva
Sandoz Pharmaceuticals d.d. filialas Tel.: +370 5 2636 037
Luxembourg/Luxemburg
Sandoz GmbH Tel.: +43 5338 2000
Ceská republika
Sandoz s.r.o.
Tel.: +420 221 421 611
Magyarország
Sandoz Hungária Kft. Tel.: +36 1 430 2890
|
Danmark/Norge/Ísland/Sverige Sandoz A/S Tlf.: +45 6395 1000 |
Malta Sandoz GmbH Tel.: +43 5338 2000 |
|
Deutschland Hexal AG Tel.: +49 8024 908 0 |
Nederland Sandoz B.V. Tel.: +31 36 52 41 600 |
|
Eesti Sandoz d.d. Eesti filiaal Tel.: +372 665 2400 |
Osterreich Sandoz GmbH Tel.: +43 5338 2000 |
|
EXláóa Sandoz dd T^: +30 216 600 500 0 |
Polska Sandoz Polska Sp. z o.o. Tel.: +48 22 209 70 00 |
|
España Sandoz Farmacéutica, S.A. Tel.: +34 900 456 856 |
Portugal Sandoz Farmacéutica Lda. Tel.: +351 21 924 19 11 |
|
France Sandoz SAS Tél.: +33 1 49 64 48 00 |
Romania S.C. Sandoz S.R.L. Tel.: +40 265 208 120 |
|
Hrvatska Sandoz d.o.o. Tel: +385 1 23 53 111 |
Slovenija Lek farmacevtska druzba d.d. Tel.: +386 1 580 21 11 |
|
Ireland Rowex Ltd Tel.: +353 27 50077 |
Slovenská republika Sandoz d.d. - organizacná zlozka Tel.: +421 2 48 200 600 |
|
Italia Sandoz S.p.A. Tel.: +39 02 96541 |
Suomi/Finland Sandoz A/S Puh/Tel.: +358 10 6133 415 |
|
Kúnpoq P.T.Hadjigeorgiou co ltd Tel.: +357 25372425 Latvija Sandoz d.d. Parstavnieciba Latvija Tel.: +371 67 892 006 |
United Kingdom Sandoz Ltd Tel.: + 44 1276 69 8020 |
Fecha de la última revisión de este prospecto: {MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
Prospecto: Información para el usuario Omnitrope 5 mg/1,5 ml solución inyectable
Somatropina
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Omnitrope y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Omnitrope
3. Cómo usar Omnitrope
4. Posibles efectos adversos
5. Conservación de Omnitrope
6. Contenido del envase e información adicional
1. Qué es Omnitrope y para qué se utiliza
Omnitrope es una hormona del crecimiento humana recombinante (también llamada somatropina). Tiene la misma estructura que la hormona del crecimiento humana natural, que es necesaria para que los huesos y los músculos crezcan. También ayuda a que los tejidos grasos y musculares se desarrollen en las cantidades correctas. Es recombinante, lo que significa que no se elabora a partir de tejido humano o animal.
En los niños, Omnitrope se usa para tratar los siguientes trastornos del crecimiento:
• Si no creces adecuadamente y no tienes suficiente hormona del crecimiento propia.
• Si padeces un síndrome de Turner, que es un trastorno genético en las niñas que puede afectar al crecimiento; el médico te lo habrá dicho si padeces este trastorno.
• Si padeces una insuficiencia renal crónica. A medida que los riñones pierden su capacidad para funcionar normalmente, esto puede afectar al crecimiento.
• Si eras demasiado pequeño o demasiado ligero al nacer. La hormona de crecimiento puede ayudar a que crezcas más si no has podido tener un estirón o mantener un crecimiento normal a los cuatro años de edad o en adelante.
• Si padeces un síndrome de Prader-Willi (un trastorno cromosómico). La hormona de crecimiento puede ayudar a que crezca más si sigue creciendo y también mejorará la composición de tu organismo. El exceso de grasa se reducirá y la masa muscular disminuida mejorará.
En los adultos, Omnitrope se usa para
• tratar a las personas con una deficiencia pronunciada de la hormona del crecimiento. Esta puede empezar durante la edad adulta o puede continuar desde la niñez.
Si usted ha sido tratado con Omnitrope por una deficiencia de la hormona de crecimiento durante la niñez, se volverá a examinar el estado de la hormona de crecimiento después de finalizar el crecimiento. Si se confirma una deficiencia grave de la hormona, el médico propondrá la continuación del tratamiento con Omnitrope.
Sólo debe recibir este medicamento de un médico que tenga experiencia con la hormona de crecimiento y que haya confirmado su diagnóstico.
2. Qué necesita saber antes de empezar a usar Omnitrope
No use Omnitrope
• si es alérgico (hipersensible) a la somatropina o a cualquiera de los demás componentes de Omnitrope.
• e informe a su médico si usted padece un tumor activo (cáncer). Los tumores deben ser inactivos y usted debe haber terminado su tratamiento antitumoral antes de empezar su tratamiento con Omnitrope.
• e informe a su médico si se le ha prescrito Omitrope para estimular el crecimiento pero usted ya ha dejado de crecer (epífisis cerradas).
• si está gravemente enfermo (por ejemplo, complicaciones post-quirúrgicas a corazón abierto, cirugía abdominal, traumatismo accidental, insuficiencia respiratoria aguda o afecciones similares). Si a usted le van a practicar o le han practicado una operación mayor, o si va al hospital por cualquier motivo, infórmele a su médico y recuérdeles a los otros médicos a los que ve que usted usa la hormona de crecimiento.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar Omnitrope.
• Si tiene riesgo de presentar diabetes, el médico deberá controlar regularmente la concentración de la glucosa en la sangre durante el tratamiento con somatropina.
• Si padece diabetes mellitus, deberá vigilar atentamente la concentración de glucosa en la sangre durante el tratamiento con somatropina y hablar con el médico acerca de los resultados, a fin de decidir si tiene que cambiar la dosis de sus medicamentos para tratar la diabetes.
• Después de comenzar el tratamiento con somatropina, algunos pacientes pueden tener que comenzar un reemplazo con hormona tiroidea.
• Si recibe tratamiento con hormonas tiroideas, puede ser necesario ajustar la dosis de hormona tiroidea.
• Si usted tiene un aumento de la presión intracraneal (que causa síntomas, tales como dolor de cabeza intenso, alteraciones visuales o vómitos) deberá informar al médico acerca de ello.
• Si camina cojeando o si empieza a cojear durante el tratamiento con hormona de crecimiento, deberá informar al médico.
• Si está recibiendo somatropina para una deficiencia de la hormona de crecimiento después de un tumor previo (cáncer), deberán examinarlo regularmente para detectar la recurrencia del tumor o cualquier otro cáncer.
• Si experimenta un dolor abdominal que empeora, debe informar a su médico.
• La experiencia en pacientes de más de 80 años es limitada. Las personas de edad avanzada
pueden ser más sensibles a la acción de la somatropina y, por lo tanto, pueden ser más propensas a presentar reacciones adversas.
Niños con insuficiencia renal crónica
• El médico deberá examinar la función de los riñones y la velocidad de crecimiento antes de empezar el tratamiento con somatropina. El tratamiento médico de los riñones debe continuarse. El tratamiento con somatropina debe interrumpirse en caso de trasplante renal.
Niños con síndrome de Prader-Willi
• El médico le dará restricciones en la dieta que debe seguir para controlar su peso.
• El médico evaluará los signos de obstrucción de las vías respiratorias altas, apnea del sueño (en
que la respiración se interrumpe durante el sueño) o infección respiratoria antes de comenzar el tratamiento con somatropina.
• Durante el tratamiento con somatropina, informe al médico si presenta signos de obstrucción de las vías respiratorias altas (incluso comenzar a roncar o un empeoramiento de los ronquidos).
Tal vez el médico tenga que examinarle y puede interrumpir el tratamiento con somatropina.
• Durante el tratamiento, el médico le examinará para ver si hay signos de escoliosis, un tipo de deformidad vertebral.
• Durante el tratamiento, si presenta una infección pulmonar, informe al médico para que pueda tratar la infección.
Niños nacidos demasiado pequeños o bajos de peso
• Si eras demasiado pequeño o demasiado ligero al nacer y tienes de 9 a 12 años, consulta al médico específicamente en relación con la pubertad y el tratamiento con este medicamento.
• El tratamiento debe continuar hasta que hayas dejado de crecer.
• El médico examinará las concentraciones de glucosa e insulina antes de comenzar el tratamiento y cada año durante el tratamiento.
Uso de Omnitrope con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Debe informar al médico si está utilizando:
• medicamentos para tratar la diabetes;
• hormonas tiroideas;
• medicamentos para controlar la epilepsia (anticonvulsivantes);
• ciclosporina (un medicamento que debilita el sistema inmunitario después de los trasplantes);
• hormonas sexuales (por ejemplo, estrógenos);
• hormonas suprarrenales sintéticas (corticoesteroides).
Tal vez el médico tenga que ajustar la dosis de estos medicamentos o la dosis de somatropina. Embarazo y lactancia
No debe usar Omnitrope si está embarazada o tratando de quedarse embarazada.
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Información importante sobre algunos de los componentes de Omnitrope
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por ml; esto es, esencialmente “exento de sodio”.
Un ml contiene 9 mg de alcohol bencílico.
Debido a la presencia de alcohol bencílico, el medicamento no se debe administrar a niños prematuros ni recién nacidos. Puede provocar reacciones tóxicas y reacciones alérgicas en niños de hasta 3 años de edad.
3. Cómo usar Omnitrope
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero. La dosis depende de su tamaño, de la afección para la que recibe tratamiento y de lo bien que funcione la hormona de crecimiento en usted. Todas las personas son diferentes. El médico le aconsejará acerca de su dosis individualizada de Omnitrope en miligramos (mg) a partir de su peso corporal en kilogramos (kg) o por su superficie corporal, calculada a partir de su estatura y peso en metros cuadrados (m2), así como su pauta de tratamiento. No cambie la dosificación y la pauta de tratamiento sin consultarle al médico.
La dosis recomendada es para:
Niños con deficiencia de la hormona de crecimiento:
0,025 a 0,035 mg/kg de peso corporal al día o 0,7 a 1,0 mg/m2 de superficie corporal al día. Pueden utilizarse dosis más altas. Cuando la deficiencia de hormona de crecimiento continúa durante la adolescencia, Omnitrope debe continuarse hasta finalizar el desarrollo físico.
Niñas con síndrome de Turner:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día.
Niños con insuficiencia renal crónica:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día. Pueden ser necesarias dosis más altas si la velocidad de crecimiento es demasiado baja. Puede ser necesario un ajuste de la dosis después de seis meses de tratamiento.
Niños con síndrome de Prader-Willi:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. La dosificación diaria no debe ser superior a 2,7 mg. El tratamiento no debe utilizarse en los niños que casi han dejado de crecer después de la pubertad.
Niños nacidos más pequeños o con peso más bajo que lo esperado y con un trastorno del crecimiento:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. Es importante continuar el tratamiento hasta que se alcance la estatura final. El tratamiento debe suspenderse después del primer año si no responde, o si ha alcanzado la estatura final y dejado de crecer.
Adultos con deficiencia de la hormona de crecimiento:
Si continúa utilizando Omnitrope después del tratamiento durante la niñez, debe comenzar con 0,2 a 0,5 mg al día.
Esta dosificación se debe aumentar o reducir gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios.
Si la deficiencia de la hormona de crecimiento comienza durante la vida adulta, debe comenzar con 0,15 a 0,3 mg al día. Esta dosificación debe aumentarse gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios. La dosis de mantenimiento diaria rara vez es superior a 1,0 mg diarios. Las mujeres pueden necesitar dosis más altas que los hombres. La dosificación debe vigilarse cada seis meses. Las personas de más de 60 años deben comenzar con una dosis de 0,1 a 0,2 mg diarios que debe aumentarse lentamente según las necesidades individuales. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento rara vez supera 0,5 mg al día. Siga las instrucciones que le haya dado el médico.
Inyección de Omnitrope
Inyéctese la hormona del crecimiento más o menos a la misma hora cada día. La hora de acostarse es un buen momento porque es fácil de recordar. Además, también es natural tener una concentración más elevada de hormona del crecimiento por la noche.
Omnitrope 5 mg/1,5 ml en un cartucho para SurePal 5 está pensado para varios usos. Sólo debe administrarse con SurePal 5, un dispositivo inyectable desarrollado específicamente para su uso con Omnitrope 5 mg/1,5 ml solución inyectable.
Omnitrope está indicado para su empleo por vía subcutánea. Esto significa que se inyecta por medio de una pequeña aguja para inyección en el tejido adiposo, por debajo de la piel. La mayor parte de las personas se inyectan en el muslo o en las nalgas. Póngase la inyección en el sitio que le haya enseñado su médico. El tejido adiposo de la piel puede verse reducido en el lugar de la inyección. Para evitar esto, utilice cada vez un sitio ligeramente diferente para inyectarse. Esto proporciona a la piel y a la zona por debajo de ella tiempo para recuperarse de una inyección antes de recibir otra en el mismo sitio.
El médico debe haberle enseñado ya cómo utilizar Omnitrope. Inyéctese siempre Omnitrope tal como el médico le ha dicho. Si no está seguro, compruebe con su médico o farmacéutico.
Cómo inyectar Omnitrope 5 mg/1,5 ml
Las siguientes instrucciones explican cómo inyectarse Omnitrope 5 mg/1,5 ml usted mismo. Lea detenidamente las instrucciones y sígalas paso a paso. Su médico o la enfermera le enseñará cómo inyectarse Omnitrope. No intente inyectarse a menos que esté seguro de que entiende el procedimiento y lo que conlleva la inyección.
- Omnitrope se administra como una inyección debajo de la piel.
- Inspeccione cuidadosamente la solución antes de inyectarla y úsela sólo si es clara e incolora.
- Cambie el lugar de la inyección para así minimizar el riesgo de lipoatrofia local (reducción local del tejido adiposo debajo de la piel).
Preparación
Antes de empezar, debe tener todo lo necesario:
- Un cartucho con Omnitrope 5 mg/1,5 ml solución inyectable.
- SurePal 5, un dispositivo para inyección desarrollado específicamente para usarlo junto con Omnitrope
5 mg/1,5 ml solución inyectable (este dispositivo no se suministra en este envase; deben consultarse las Instrucciones de uso proporcionadas junto con SurePal 5).
- Una aguja de pluma para inyección subcutánea.
- 2 gasas de limpieza (no suministradas en el envase).
Lávese las manos antes continuar con los siguiente pasos.
Inyección de Omnitrope
- Con una gasa, desinfecte la membrana de goma del cartucho.
- El contenido del cartucho debe ser transparente e incoloro .
Introduzca el cartucho en la pluma inyectable. Siga las Instrucciones de uso de la pluma para inyección (Pen). Para ajustar la pluma, dosifique la cantidad necesaria.
Seleccione el lugar de la inyección. Los mejores lugares para la administración de la inyección son los tejidos con una capa de grasa entre la piel y el músculo, como el muslo o el vientre (excepto el ombligo o la cintura).
Asegúrese de que se inyecta al menos a 1 cm de distancia respecto del lugar de la última inyección y de que cambia los lugares de inyección, tal y como le habrán enseñado.
Antes de administrar la inyección, limpie bien la piel con una gasa empapada en alcohol. Espere a que la zona se seque.
Introduzca la aguja en la piel de la forma que le haya enseñado el médico.
Después de inyectarse
- Después de la inyección, presione el lugar de la inyección con un pequeño vendaje o gasa estéril durante unos segundos. No masajee el lugar de la inyección.
- Retire la aguja de la pluma utilizando para ello la tapa
exterior y elimínela. Esto mantendrá la solución de Omnitrope estéril e impedirá que gotee. También impedirá que el aire se introduzca en la pluma, taponando la aguja. No comparta las agujas. No comparta la pluma.
- Deje el cartucho en la pluma, reponga el capuchón en la pluma y guárdela en una nevera.
- La solución debe ser transparente cuando se saque de la nevera. No se debe utilizar si la solución está turbia o si contiene partículas.
Si usa más Omnitrope del que debe
Si se inyecta mucho más de lo que debiera, consulte lo antes posible a su médico o farmacéutico. Su concentración de azúcar en la sangre podría descender demasiado y después aumentar demasiado. Tal vez se sienta con temblores, sudoroso, somnoliento o “como si no fuera usted mismo”, y podría desmayarse.
Si olvidó usar Omnitrope
No use una dosis doble para compensar las dosis olvidadas. Lo mejor es usar la hormona de crecimiento con regularidad. Si se olvida de usar una dosis, póngase la siguiente inyección a la hora habitual, al día siguiente. Tome nota de las inyecciones olvidadas e infórmele al médico en el siguiente control.
Si interrumpe el tratamiento con Omnitrope
Consulte a su médico antes de dejar de usar Omnitrope.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Los efectos adversos muy frecuentes y frecuentes en los adultos pueden comenzar en los primeros meses de tratamiento y pueden detenerse espontáneamente o si se reduce la dosis.
Los efectos adversos muy frecuentes (que probablemente se produzcan en más de 1 de cada 10 pacientes) incluyen los siguientes:
En los adultos
• Dolor articular
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos)
Los efectos adversos frecuentes (que probablemente afecten a menos de 1 de cada 10 pacientes) incluyen los siguientes:
En los niños
• Enrojecimiento, picor o dolor temporal en el lugar de la inyección
• Dolor articular
En los adultos
• Adormecimiento, hormigueos
• Rigidez de los brazos y las piernas, dolor muscular
• Dolor o sensación de escozor en las manos o los antebrazos (conocido como síndrome del túnel carpiano)
Los efectos adversos poco frecuentes (que probablemente afecten a menos de 1 de cada 100 pacientes) incluyen los siguientes:
En los niños
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos, durante un tiempo corto al comienzo del tratamiento)
Los efectos adversos raros (que probablemente afecten a menos de 1 de cada 1.000 pacientes) incluyen los siguientes:
En los niños
• Adormecimiento, hormigueos
• Leucemia (se ha observado en un pequeño número de pacientes con deficiencia de la hormona del crecimiento, algunos de cuales habían sido tratados con somatropina. Sin embargo, no hay ningún indicio de que la incidencia de leucemia sea mayor en receptores de la hormona del crecimiento sin factores predisponentes).
• Aumento de la presión intracraneal (que causa síntomas, como cefalea intensa, alteraciones visuales o vómitos)
• Dolor muscular
Frecuencia no conocida: no puede estimarse a partir de los datos disponibles
• Diabetes de tipo 2
• Disminución de los niveles de la hormona cortisol en la sangre En los niños
• Rigidez en los brazos y las piernas En los adultos
• Aumento de la presión intracraneal (que causa síntomas, como dolor de cabeza intenso, alteraciones visuales o vómitos)
• Enrojecimiento, picor o dolor en el lugar de la inyección
Formación de anticuerpos contra la hormona del crecimiento inyectada, pero estos no parecen hacer que la hormona del crecimiento deje de funcionar.
La piel alrededor de la zona de inyección puede ponerse irregular o con bultos, pero esto no deberá ocurrir si se inyecta en un lugar diferente cada vez.
Ha habido casos raros de muerte súbita en pacientes con síndrome de Prader-Willi. No obstante, estos casos no se han relacionado con el tratamiento con Omnitrope.
Su médico puede considerar una epifisiólisis de la cabeza femoral o una enfermedad de Legg-Calvé-Perthes si experimenta molestias o dolor en la cadera o la rodilla mientras está siendo tratado con Omnitrope.
Otros posibles efectos adversos relacionados con su tratamiento con la hormona del crecimiento pueden incluir los siguientes:
Usted (o su hijo) puede tener niveles elevados de azúcar en sangre o niveles reducidos de la hormona tiroidea. Esto lo puede analizar su médico y, si es necesario, su médico le recetará el tratamiento adecuado. En casos raros se ha observado inflamación del páncreas en pacientes tratados con la hormona del crecimiento.
Si considera que alguno de los efectos adversos que sufre es grave o si aprecia cualquier efecto adverso no mencionado en este prospecto, informe a su médico o farmacéutico.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Omnitrope
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y en la caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
• Conservar y transportar refrigerado (entre 2°C y 8°C).
• No congelar.
• Conservar en el embalaje original para protegerlo de la luz.
• Después de la administración de la primera inyección, el cartucho debe permanecer en el inyector de pluma y debe conservarse en una nevera, a una temperatura de 2 a 8 °C, y sólo debe usarse durante un máximo de 28 días.
No utilice Omnitrope si se observa que la solución está turbia.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Omnitrope
- El principio activo de Omnitrope es somatropina.
Cada ml de solución contiene 3,3 mg de somatropina (que corresponde a 10 UI). Un cartucho contiene 5,0 mg (que corresponde a 15 UI) de somatropina en 1,5 ml.
- Los demás componentes son:
Fosfato hidrógeno disódico heptahidrato Fosfato dihidrógeno sódico dihidrato Manitol
Poloxámero 188 Alcohol bencílico
Agua para preparaciones inyectables
Aspecto del producto y contenido del envase
Omnitrope es una solución inyectable transparente e incolora.
Omnitrope 5 mg/1,5 ml solución para inyección es para su uso sólo en SurePal 5. Envases con 1, 5 ó 10 cartuchos.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Austria
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Sandoz nv-sa Tél/Tel.: +32 2 722 97 97 |
Lietuva Sandoz Pharmaceuticals d.d. filialas Tel.: +370 5 2636 037 |
|
Etarapun TuproBCKO npegcTaBHTencTBO CaHgo3 g.g. Tea.: +359 2 970 47 47 |
Luxembourg/Luxemburg Sandoz GmbH Tel.: +43 5338 2000 |
|
Ceská republika Sandoz s.r.o. Tel.: +420 221 421 611 |
Magyarország Sandoz Hungária Kft. Tel.: +36 1 430 2890 |
|
Danmark/Norge/Ísland/Sverige Sandoz A/S Tlf.: +45 6395 1000 |
Malta Sandoz GmbH Tel.: +43 5338 2000 |
|
Deutschland Hexal AG Tel.: +49 8024 908 0 |
Nederland Sandoz B.V. Tel.: +31 36 52 41 600 |
|
Eesti Sandoz d.d. Eesti filiaal Tel.: +372 665 2400 |
Osterreich Sandoz GmbH Tel.: +43 5338 2000 |
|
EXláda Sandoz dd Tp^: +30 216 600 500 0 |
Polska Sandoz Polska Sp. z o.o. Tel.: +48 22 209 70 00 |
|
España Sandoz Farmacéutica, S.A. Tel.: +34 900 456 856 |
Portugal Sandoz Farmacéutica Lda. Tel.: +351 21 924 19 11 |
|
France Sandoz SAS Tél.: +33 1 49 64 48 00 |
Romania S.C. Sandoz S.R.L. Tel.: +40 265 208 120 |
|
Hrvatska Sandoz d.o.o. Tel: +385 1 23 53 111 |
Slovenija Lek farmacevtska druzba d.d. Tel.: +386 1 580 21 11 |
|
Ireland Rowex Ltd Tel.: +353 27 50077 |
Slovenská republika Sandoz d.d. - organizacná zlozka Tel.: +421 2 48 200 600 |
|
Italia Sandoz S.p.A. Tel.: +39 02 96541 |
Suomi/Finland Sandoz A/S Puh/Tel.: +358 10 6133 415 |
|
Kúnpoq P.T.Hadjigeorgiou co ltd Tel.: +357 25372425 |
United Kingdom Sandoz Ltd Tel.: + 44 1276 69 8020 |
Latvija
Sandoz d.d. Párstávniedba Latvija Tel.: +371 67 892 006
Fecha de la última revisión de este prospecto: {MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Prospecto: Información para el usuario Omnitrope 10 mg/1,5 ml solución inyectable
Somatropina
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Omnitrope y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Omnitrope
3. Cómo usar Omnitrope
4. Posibles efectos adversos
5. Conservación de Omnitrope
6. Contenido del envase e información adicional
1. Qué es Omnitrope y para qué se utiliza
Omnitrope es una hormona del crecimiento humana recombinante (también llamada somatropina). Tiene la misma estructura que la hormona del crecimiento humana natural, que es necesaria para que los huesos y los músculos crezcan. También ayuda a que los tejidos grasos y musculares se desarrollen en las cantidades correctas. Es recombinante, lo que significa que no se elabora a partir de tejido humano o animal.
En los niños, Omnitrope se usa para tratar los siguientes trastornos del crecimiento:
• Si no creces adecuadamente y no tienes suficiente hormona del crecimiento propia.
• Si padeces un síndrome de Turner, que es un trastorno genético en las niñas que puede afectar al crecimiento; el médico te lo habrá dicho si padeces este trastorno.
• Si padeces una insuficiencia renal crónica. A medida que los riñones pierden su capacidad para funcionar normalmente, esto puede afectar al crecimiento.
• Si eras demasiado pequeño o demasiado ligero al nacer. La hormona de crecimiento puede ayudar a que crezcas más si no has podido tener un estirón o mantener un crecimiento normal a los cuatro años de edad o en adelante.
• Si padeces un síndrome de Prader-Willi (un trastorno cromosómico). La hormona de crecimiento puede ayudar a que crezca más si sigue creciendo y también mejorará la composición de tu organismo. El exceso de grasa se reducirá y la masa muscular disminuida mejorará.
En los adultos, Omnitrope se usa para
• tratar a las personas con una deficiencia pronunciada de la hormona del crecimiento. Esta puede empezar durante la edad adulta o puede continuar desde la niñez.
Si usted ha sido tratado con Omnitrope por una deficiencia de la hormona de crecimiento durante la niñez, se volverá a examinar el estado de la hormona de crecimiento después de finalizar el crecimiento. Si se confirma una deficiencia grave de la hormona, el médico propondrá la continuación del tratamiento con Omnitrope.
Sólo debe recibir este medicamento de un médico que tenga experiencia con la hormona de crecimiento y que haya confirmado su diagnóstico.
2. Qué necesita saber antes de empezar a usar Omnitrope
No use Omnitrope
• si es alérgico (hipersensible) a la somatropina o a cualquiera de los demás componentes de Omnitrope.
• e informe a su médico si usted padece un tumor activo (cáncer). Los tumores deben ser inactivos y usted debe haber terminado su tratamiento antitumoral antes de empezar su tratamiento con Omnitrope.
• e informe a su médico si se le ha prescrito Omitrope para estimular el crecimiento pero usted ya ha dejado de crecer (epífisis cerradas).
• si está gravemente enfermo (por ejemplo, complicaciones post-quirúrgicas a corazón abierto, cirugía abdominal, traumatismo accidental, insuficiencia respiratoria aguda o afecciones similares). Si a usted le van a practicar o le han practicado una operación mayor, o si va al hospital por cualquier motivo, infórmele a su médico y recuérdeles a los otros médicos a los que ve que usted usa la hormona de crecimiento.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar Omnitrope.
• Si tiene riesgo de presentar diabetes, el médico deberá controlar regularmente la concentración de la glucosa en la sangre durante el tratamiento con somatropina.
• Si padece diabetes mellitus, deberá vigilar atentamente la concentración de glucosa en la sangre durante el tratamiento con somatropina y hablar con el médico acerca de los resultados, a fin de decidir si tiene que cambiar la dosis de sus medicamentos para tratar la diabetes.
• Después de comenzar el tratamiento con somatropina, algunos pacientes pueden tener que comenzar un reemplazo con hormona tiroidea.
• Si recibe tratamiento con hormonas tiroideas, puede ser necesario ajustar la dosis de hormona tiroidea.
• Si usted tiene un aumento de la presión intracraneal (que causa síntomas, tales como dolor de cabeza intenso, alteraciones visuales o vómitos) deberá informar al médico acerca de ello.
• Si camina cojeando o si empieza a cojear durante el tratamiento con hormona de crecimiento, deberá informar al médico.
• Si está recibiendo somatropina para una deficiencia de la hormona de crecimiento después de un tumor previo (cáncer), deberán examinarlo regularmente para detectar la recurrencia del tumor o cualquier otro cáncer.
• Si experimenta un dolor abdominal que empeora, debe informar a su médico.
• La experiencia en pacientes de más de 80 años es limitada. Las personas de edad avanzada
pueden ser más sensibles a la acción de la somatropina y, por lo tanto, pueden ser más propensas a presentar reacciones adversas.
Niños con insuficiencia renal crónica
• El médico deberá examinar la función de los riñones y la velocidad de crecimiento antes de empezar el tratamiento con somatropina. El tratamiento médico de los riñones debe continuarse. El tratamiento con somatropina debe interrumpirse en caso de trasplante renal.
Niños con síndrome de Prader-Willi
• El médico le dará restricciones en la dieta que debe seguir para controlar su peso.
• El médico evaluará los signos de obstrucción de las vías respiratorias altas, apnea del sueño (en
que la respiración se interrumpe durante el sueño) o infección respiratoria antes de comenzar el tratamiento con somatropina.
• Durante el tratamiento con somatropina, informe al médico si presenta signos de obstrucción de las vías respiratorias altas (incluso comenzar a roncar o un empeoramiento de los ronquidos).
Tal vez el médico tenga que examinarle y puede interrumpir el tratamiento con somatropina.
• Durante el tratamiento, el médico le examinará para ver si hay signos de escoliosis, un tipo de deformidad vertebral.
• Durante el tratamiento, si presenta una infección pulmonar, informe al médico para que pueda tratar la infección.
Niños nacidos demasiado pequeños o bajos de peso
• Si eras demasiado pequeño o demasiado ligero al nacer y tienes de 9 a 12 años, consulta al médico específicamente en relación con la pubertad y el tratamiento con este medicamento.
• El tratamiento debe continuar hasta que hayas dejado de crecer.
• El médico examinará las concentraciones de glucosa e insulina antes de comenzar el tratamiento y cada año durante el tratamiento.
Uso de Omnitrope con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Debe informar al médico si está utilizando:
• medicamentos para tratar la diabetes;
• hormonas tiroideas;
• medicamentos para controlar la epilepsia (anticonvulsivantes);
• ciclosporina (un medicamento que debilita el sistema inmunitario después de los trasplantes);
• hormonas sexuales (por ejemplo, estrógenos);
• hormonas suprarrenales sintéticas (corticoesteroides).
Tal vez el médico tenga que ajustar la dosis de estos medicamentos o la dosis de somatropina. Embarazo y lactancia
No debe usar Omnitrope si está embarazada o tratando de quedarse embarazada.
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Información importante sobre algunos de los componentes de Omnitrope
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por ml; esto es, esencialmente “exento de sodio”.
3. Cómo usar Omnitrope
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero. La dosis depende de su tamaño, de la afección para la que recibe tratamiento y de lo bien que funcione la hormona de crecimiento en usted. Todas las personas son diferentes. El médico le aconsejará acerca de su dosis individualizada de Omnitrope en miligramos (mg) a partir de su peso corporal en kilogramos (kg) o por su superficie corporal, calculada a partir de su estatura y peso en metros cuadrados (m2), así como su pauta de tratamiento. No cambie la dosificación y la pauta de tratamiento sin consultarle al médico.
La dosis recomendada es para:
Niños con deficiencia de la hormona de crecimiento:
0,025 a 0,035 mg/kg de peso corporal al día o 0,7 a 1,0 mg/m2 de superficie corporal al día. Pueden utilizarse dosis más altas. Cuando la deficiencia de hormona de crecimiento continúa durante la adolescencia, Omnitrope debe continuarse hasta finalizar el desarrollo físico.
Niñas con síndrome de Turner:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día.
Niños con insuficiencia renal crónica:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día. Pueden ser necesarias dosis más altas si la velocidad de crecimiento es demasiado baja. Puede ser necesario un ajuste de la dosis después de seis meses de tratamiento.
Niños con síndrome de Prader-Willi:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. La dosificación diaria no debe ser superior a 2,7 mg. El tratamiento no debe utilizarse en los niños que casi han dejado de crecer después de la pubertad.
Niños nacidos más pequeños o con peso más bajo que lo esperado y con un trastorno del crecimiento:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. Es importante continuar el tratamiento hasta que se alcance la estatura final. El tratamiento debe suspenderse después del primer año si no responde, o si ha alcanzado la estatura final y dejado de crecer.
Adultos con deficiencia de la hormona de crecimiento:
Si continúa utilizando Omnitrope después del tratamiento durante la niñez, debe comenzar con 0,2 a 0,5 mg al día.
Esta dosificación se debe aumentar o reducir gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios.
Si la deficiencia de la hormona de crecimiento comienza durante la vida adulta, debe comenzar con 0,15 a 0,3 mg al día. Esta dosificación debe aumentarse gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios. La dosis de mantenimiento diaria rara vez es superior a 1,0 mg diarios. Las mujeres pueden necesitar dosis más altas que los hombres. La dosificación debe vigilarse cada seis meses. Las personas de más de 60 años deben comenzar con una dosis de 0,1 a 0,2 mg diarios que debe aumentarse lentamente según las necesidades individuales. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento rara vez supera 0,5 mg al día. Siga las instrucciones que le haya dado el médico.
Inyección de Omnitrope
Inyéctese la hormona del crecimiento más o menos a la misma hora cada día. La hora de acostarse es un buen momento porque es fácil de recordar. Además, también es natural tener una concentración más elevada de hormona del crecimiento por la noche.
Omnitrope 10 mg/1,5 ml en un cartucho para SurePal 10 está previsto para varios usos. Sólo debe administrarse con SurePal 10, un dispositivo de inyección desarrollado específicamente para la administración de Omnitrope 10 mg/1,5 ml solución inyectable.
Omnitrope está indicado para su empleo por vía subcutánea. Esto significa que se inyecta por medio de una pequeña aguja para inyección en el tejido adiposo, por debajo de la piel. La mayor parte de las personas se inyectan en el muslo o en las nalgas. Póngase la inyección en el sitio que le haya enseñado su médico. El tejido adiposo de la piel puede verse reducido en el lugar de la inyección. Para evitar esto, utilice cada vez un sitio ligeramente diferente para inyectarse. Esto proporciona a la piel y a la zona por debajo de ella tiempo para recuperarse de una inyección antes de recibir otra en el mismo sitio.
El médico debe haberle enseñado ya cómo utilizar Omnitrope. Inyéctese siempre Omnitrope tal como el médico le ha dicho. Si no está seguro, compruebe con su médico o farmacéutico.
Cómo inyectar Omnitrope 10 mg/1,5 ml
Las siguientes instrucciones explican cómo inyectarse Omnitrope 10 mg/1,5 ml usted mismo. Lea detenidamente las instrucciones y sígalas paso a paso. Su médico o la enfermera le enseñará cómo inyectarse Omnitrope. No intente inyectarse a menos que esté seguro de que entiende el procedimiento y lo que conlleva la inyección.
- Omnitrope se administra con una inyección debajo de la piel.
Inspeccione cuidadosamente la solución antes de inyectarla y úsela sólo si es clara e incolora. Cambie el lugar de la inyección para así minimizar el riesgo de lipoatrofia local (reducción local del tejido adiposo debajo de la piel).
Preparación
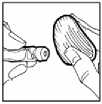
Antes de empezar, debe tener todo lo necesario:
- Un cartucho con Omnitrope 10 mg/1,5 ml solución inyectable.
- SurePal 10, un dispositivo para inyección desarrollado específicamente para usarlo junto con Omnitrope
10 mg/1,5 ml solución inyectable (este dispositivo no se suministra en este envase; deben consultarse las Instrucciones de uso proporcionadas junto con SurePal 10).
- Una aguja de pluma para inyección subcutánea.
- 2 gasas de limpieza (no suministradas en el envase).
Lávese las manos antes de continuar con los siguiente pasos.
Inyección de Omnitrope
- Con una gasa, desinfecte la membrana de goma del cartucho.
- El contenido del cartucho debe ser transparente e incoloro.
Introduzca el cartucho en la pluma inyectable. Siga las Instrucciones de uso de la pluma para inyección (Pen). Para ajustar la pluma, dosifique la cantidad necesaria.
Seleccione el lugar de la inyección. Los mejores lugares para la administración de la inyección son los tejidos con una capa de grasa entre la piel y el músculo, como el muslo o el vientre (excepto el ombligo o la cintura).
Asegúrese de que se inyecta al menos a 1 cm de distancia respecto del lugar de la última inyección y de que cambia los lugares de inyección, tal y como le habrán enseñado.
Antes de administrar la inyección, limpie bien la piel con una gasa empapada en alcohol. Espere a que la zona se seque.
Introduzca la aguja en la piel de la forma que le haya enseñado el médico.
Después de inyectarse
- Después de la inyección, presione el lugar de la inyección con un pequeño vendaje o gasa estéril durante unos segundos. No masajee el lugar de la inyección.
- Retire la aguja de la pluma utilizando para ello la tapa exterior y elimínela. Esto mantendrá la solución de Omnitrope estéril e impedirá que gotee. También impedirá que el aire se introduzca en la pluma , taponando la aguja. No comparta las agujas. No comparta la pluma.
- Deje el cartucho en la pluma, reponga el capuchón en la pluma y guárdela en una nevera.
- La solución debe ser transparente cuando se saque de la nevera . No se debe utilizar si la solución está turbia o si contiene partículas.
Si se inyecta mucho más de lo que debiera, consulte lo antes posible a su médico o farmacéutico. Su concentración de azúcar en la sangre podría descender demasiado y después aumentar demasiado. Tal vez se sienta con temblores, sudoroso, somnoliento o “como si no fuera usted mismo”, y podría desmayarse.
Si olvidó usar Omnitrope
No use una dosis doble para compensar las dosis olvidadas. Lo mejor es usar la hormona de crecimiento con regularidad. Si se olvida de usar una dosis, póngase la siguiente inyección a la hora habitual, al día siguiente. Tome nota de las inyecciones olvidadas e infórmele al médico en el siguiente control.
Si interrumpe el tratamiento con Omnitrope
Consulte a su médico antes de dejar de usar Omnitrope.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Los efectos adversos muy frecuentes y frecuentes en los adultos pueden comenzar en los primeros meses de tratamiento y pueden detenerse espontáneamente o si se reduce la dosis.
Los efectos adversos muy frecuentes (que probablemente se produzcan en más de 1 de cada 10 pacientes) incluyen los siguientes:
En los adultos
• Dolor articular
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos)
Los efectos adversos frecuentes (que probablemente afecten a menos de 1 de cada 10 pacientes) incluyen los siguientes:
En los niños
• Enrojecimiento, picor o dolor temporal en el lugar de la inyección
• Dolor articular
En los adultos
• Adormecimiento, hormigueos
• Rigidez de los brazos y las piernas, dolor muscular
• Dolor o sensación de escozor en las manos o los antebrazos (conocido como síndrome del túnel carpiano)
Los efectos adversos poco frecuentes (que probablemente afecten a menos de 1 de cada 100 pacientes) incluyen los siguientes:
En los niños
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos, durante un tiempo corto al comienzo del tratamiento)
Los efectos adversos raros (que probablemente afecten a menos de 1 de cada 1.000 pacientes) incluyen los siguientes:
En los niños
• Adormecimiento, hormigueos
• Leucemia (se ha observado en un pequeño número de pacientes con deficiencia de la hormona del crecimiento, algunos de cuales habían sido tratados con somatropina. Sin embargo, no hay ningún indicio de que la incidencia de leucemia sea mayor en receptores de la hormona del crecimiento sin factores predisponentes).
• Aumento de la presión intracraneal (que causa síntomas, como cefalea intensa, alteraciones visuales o vómitos)
• Dolor muscular
Frecuencia no conocida: no puede estimarse a partir de los datos disponibles
• Diabetes de tipo 2
• Disminución de los niveles de la hormona cortisol en la sangre En los niños
• Rigidez en los brazos y las piernas
En los adultos
• Aumento de la presión intracraneal (que causa síntomas, como dolor de cabeza intenso, alteraciones visuales o vómitos)
• Enrojecimiento, picor o dolor en el lugar de la inyección
Formación de anticuerpos contra la hormona del crecimiento inyectada, pero estos no parecen hacer que la hormona del crecimiento deje de funcionar.
La piel alrededor de la zona de inyección puede ponerse irregular o con bultos, pero esto no deberá ocurrir si se inyecta en un lugar diferente cada vez.
Ha habido casos raros de muerte súbita en pacientes con síndrome de Prader-Willi. No obstante, estos casos no se han relacionado con el tratamiento con Omnitrope.
Su médico puede considerar una epifisiólisis de la cabeza femoral o una enfermedad de Legg-Calvé-Perthes si experimenta molestias o dolor en la cadera o la rodilla mientras está siendo tratado con Omnitrope.
Otros posibles efectos adversos relacionados con su tratamiento con la hormona del crecimiento pueden incluir los siguientes:
Usted (o su hijo) puede tener niveles elevados de azúcar en sangre o niveles reducidos de la hormona tiroidea. Esto lo puede analizar su médico y, si es necesario, su médico le recetará el tratamiento adecuado. En casos raros se ha observado inflamación del páncreas en pacientes tratados con la hormona del crecimiento.
Si considera que alguno de los efectos adversos que sufre es grave o si aprecia cualquier efecto adverso no mencionado en este prospecto, informe a su médico o farmacéutico.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y en la caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
• Conservar y transportar refrigerado (entre 2°C y 8°C).
• No congelar.
• Conservar en el embalaje original para protegerlo de la luz.
• Después de la administración de la primera inyección, el cartucho debe permanecer en el inyector de pluma y debe conservarse en una nevera, a una temperatura de 2 a 8 °C, y sólo debe usarse durante un máximo de 28 días.
No utilice Omnitrope si se observa que la solución está turbia.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Omnitrope
- El principio activo de Omnitrope es somatropina.
Cada ml de solución contiene 6,7 mg de somatropina (que corresponde a 20 UI).
Un cartucho contiene 10,0 mg (que corresponden a 30 UI) de somatropina en 1,5 ml.
- Los demás componentes son:
Fosfato hidrógeno disódico heptahidrato Fosfato dihidrógeno sódico dihidrato Glicina
Poloxámero 188 Fenol
Agua para preparaciones inyectables
Aspecto del producto y contenido del envase
Omnitrope es una solución inyectable transparente e incolora.
Omnitrope 10 mg/1,5 ml solución para inyección es para su uso sólo en SurePal 10. Envases de 1, 5 ó 10 cartuchos.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Austria
Pueden solicitar más información respecto a este titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Sandoz nv-sa Tél/Tel.: +32 2 722 97 97
Etarapna
TtproBCKO npegcTaBHTeacTBO CaHgo3 g.g.
Tea.: +359 2 970 47 47
medicamento dirigiéndose al representante local del
Lietuva
Sandoz Pharmaceuticals d.d. filialas Tel.: +370 5 2636 037
Luxembourg/Luxemburg
Sandoz GmbH Tel.: +43 5338 2000
|
Ceská republika Sandoz s.r.o. Tel.: +420 221 421 611 |
Magyarország Sandoz Hungária Kft. Tel.: +36 1 430 2890 |
|
Danmark/Norge/Ísland/Sverige Sandoz A/S Tlf.: +45 6395 1000 |
Malta Sandoz GmbH Tel.: +43 5338 2000 |
|
Deutschland Hexal AG Tel.: +49 8024 908 0 |
Nederland Sandoz B.V. Tel.: +31 36 52 41 600 |
|
Eesti Sandoz d.d. Eesti filiaal Tel.: +372 665 2400 |
Osterreich Sandoz GmbH Tel.: +43 5338 2000 |
|
ELLáóa Sandoz dd T^: +30 216 600 500 0 |
Polska Sandoz Polska Sp. z o.o. Tel.: +48 22 209 70 00 |
|
España Sandoz Farmacéutica, S.A. Tel.: +34 900 456 856 |
Portugal Sandoz Farmacéutica Lda. Tel.: +351 21 924 19 11 |
|
France Sandoz SAS Tél.: +33 1 49 64 48 00 |
Romania S.C. Sandoz S.R.L. Tel.: +40 265 208 120 |
|
Hrvatska Sandoz d.o.o. |
Slovenija Lek farmacevtska druzba d.d. |
|
Tel: +385 1 23 53 111 |
Tel.: +386 1 580 21 11 |
|
Ireland Rowex Ltd Tel.: +353 27 50077 |
Slovenská republika Sandoz d.d. - organizacná zlozka Tel.: +421 2 48 200 600 |
|
Italia Sandoz S.p.A. Tel.: +39 02 96541 |
Suomi/Finland Sandoz A/S Puh/Tel.: +358 10 6133 415 |
|
Kúnpoq P.T.Hadjigeorgiou co ltd Tel.: +357 25372425 |
United Kingdom Sandoz Ltd Tel.: + 44 1276 69 8020 |
|
Latvija Sandoz d.d. Párstávnieciba Latvija Tel.: +371 67 892 006 |
Fecha de la última revisión de este prospecto: {MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
Prospecto: Información para el usuario Omnitrope 15 mg/1,5 ml solución inyectable
Somatropina
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Omnitrope y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Omnitrope
3. Cómo usar Omnitrope
4. Posibles efectos adversos
5. Conservación de Omnitrope
6. Contenido del envase e información adicional
1. Qué es Omnitrope y para qué se utiliza
Omnitrope es una hormona del crecimiento humana recombinante (también llamada somatropina). Tiene la misma estructura que la hormona del crecimiento humana natural, que es necesaria para que los huesos y los músculos crezcan. También ayuda a que los tejidos grasos y musculares se desarrollen en las cantidades correctas. Es recombinante, lo que significa que no se elabora a partir de tejido humano o animal.
En los niños, Omnitrope se usa para tratar los siguientes trastornos del crecimiento:
• Si no creces adecuadamente y no tienes suficiente hormona del crecimiento propia.
• Si padeces un síndrome de Turner, que es un trastorno genético en las niñas que puede afectar al crecimiento; el médico te lo habrá dicho si padeces este trastorno.
• Si padeces una insuficiencia renal crónica. A medida que los riñones pierden su capacidad para funcionar normalmente, esto puede afectar al crecimiento.
• Si eras demasiado pequeño o demasiado ligero al nacer. La hormona de crecimiento puede ayudar a que crezcas más si no has podido tener un estirón o mantener un crecimiento normal a los cuatro años de edad o en adelante.
• Si padeces un síndrome de Prader-Willi (un trastorno cromosómico). La hormona de crecimiento puede ayudar a que crezca más si sigue creciendo y también mejorará la composición de tu organismo. El exceso de grasa se reducirá y la masa muscular disminuida mejorará.
En los adultos, Omnitrope se usa para
• tratar a las personas con una deficiencia pronunciada de la hormona del crecimiento. Esta puede empezar durante la edad adulta o puede continuar desde la niñez.
Si usted ha sido tratado con Omnitrope por una deficiencia de la hormona de crecimiento durante la niñez, se volverá a examinar el estado de la hormona de crecimiento después de finalizar el crecimiento. Si se confirma una deficiencia grave de la hormona, el médico propondrá la continuación del tratamiento con Omnitrope.
Sólo debe recibir este medicamento de un médico que tenga experiencia con la hormona de crecimiento y que haya confirmado su diagnóstico.
2. Qué necesita saber antes de empezar a usar Omnitrope
No use Omnitrope
• si es alérgico (hipersensible) a la somatropina o a cualquiera de los demás componentes de Omnitrope.
• e informe a su médico si usted padece un tumor activo (cáncer). Los tumores deben ser inactivos y usted debe haber terminado su tratamiento antitumoral antes de empezar su tratamiento con Omnitrope.
• e informe a su médico si se le ha prescrito Omitrope para estimular el crecimiento pero usted ya ha dejado de crecer (epífisis cerradas).
• si está gravemente enfermo (por ejemplo, complicaciones post-quirúrgicas a corazón abierto, cirugía abdominal, traumatismo accidental, insuficiencia respiratoria aguda o afecciones similares). Si a usted le van a practicar o le han practicado una operación mayor, o si va al hospital por cualquier motivo, infórmele a su médico y recuérdeles a los otros médicos a los que ve que usted usa la hormona de crecimiento.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar Omnitrope.
• Si tiene riesgo de presentar diabetes, el médico deberá controlar regularmente la concentración de la glucosa en la sangre durante el tratamiento con somatropina.
• Si padece diabetes mellitus, deberá vigilar atentamente la concentración de glucosa en la sangre durante el tratamiento con somatropina y hablar con el médico acerca de los resultados, a fin de decidir si tiene que cambiar la dosis de sus medicamentos para tratar la diabetes.
• Después de comenzar el tratamiento con somatropina, algunos pacientes pueden tener que comenzar un reemplazo con hormona tiroidea.
• Si recibe tratamiento con hormonas tiroideas, puede ser necesario ajustar la dosis de hormona tiroidea.
• Si usted tiene un aumento de la presión intracraneal (que causa síntomas, tales como dolor de cabeza intenso, alteraciones visuales o vómitos) deberá informar al médico acerca de ello.
• Si camina cojeando o si empieza a cojear durante el tratamiento con hormona de crecimiento, deberá informar al médico.
• Si está recibiendo somatropina para una deficiencia de la hormona de crecimiento después de un tumor previo (cáncer), deberán examinarlo regularmente para detectar la recurrencia del tumor o cualquier otro cáncer.
• Si experimenta un dolor abdominal que empeora, debe informar a su médico.
• La experiencia en pacientes de más de 80 años es limitada. Las personas de edad avanzada
pueden ser más sensibles a la acción de la somatropina y, por lo tanto, pueden ser más propensas a presentar reacciones adversas.
Niños con insuficiencia renal crónica
• El médico deberá examinar la función de los riñones y la velocidad de crecimiento antes de empezar el tratamiento con somatropina. El tratamiento médico de los riñones debe continuarse. El tratamiento con somatropina debe interrumpirse en caso de trasplante renal.
Niños con síndrome de Prader-Willi
• El médico le dará restricciones en la dieta que debe seguir para controlar su peso.
• El médico evaluará los signos de obstrucción de las vías respiratorias altas, apnea del sueño (en
que la respiración se interrumpe durante el sueño) o infección respiratoria antes de comenzar el tratamiento con somatropina.
• Durante el tratamiento con somatropina, informe al médico si presenta signos de obstrucción de las vías respiratorias altas (incluso comenzar a roncar o un empeoramiento de los ronquidos).
Tal vez el médico tenga que examinarle y puede interrumpir el tratamiento con somatropina.
• Durante el tratamiento, el médico le examinará para ver si hay signos de escoliosis, un tipo de deformidad vertebral.
• Durante el tratamiento, si presenta una infección pulmonar, informe al médico para que pueda tratar la infección.
Niños nacidos demasiado pequeños o bajos de peso
• Si eras demasiado pequeño o demasiado ligero al nacer y tienes de 9 a 12 años, consulta al médico específicamente en relación con la pubertad y el tratamiento con este medicamento.
• El tratamiento debe continuar hasta que hayas dejado de crecer.
• El médico examinará las concentraciones de glucosa e insulina antes de comenzar el tratamiento y cada año durante el tratamiento.
Uso de Omnitrope con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Debe informar al médico si está utilizando:
• medicamentos para tratar la diabetes;
• hormonas tiroideas;
• medicamentos para controlar la epilepsia (anticonvulsivantes);
• ciclosporina (un medicamento que debilita el sistema inmunitario después de los trasplantes);
• hormonas sexuales (por ejemplo, estrógenos);
• hormonas suprarrenales sintéticas (corticoesteroides).
Tal vez el médico tenga que ajustar la dosis de estos medicamentos o la dosis de somatropina. Embarazo y lactancia
No debe usar Omnitrope si está embarazada o tratando de quedarse embarazada.
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Información importante sobre algunos de los componentes de Omnitrope
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por ml; esto es, esencialmente “exento de sodio”.
3. Cómo usar Omnitrope
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero. La dosis depende de su tamaño, de la afección para la que recibe tratamiento y de lo bien que funcione la hormona de crecimiento en usted. Todas las personas son diferentes. El médico le aconsejará acerca de su dosis individualizada de Omnitrope en miligramos (mg) a partir de su peso corporal en kilogramos (kg) o por su superficie corporal, calculada a partir de su estatura y peso en metros cuadrados (m2), así como su pauta de tratamiento. No cambie la dosificación y la pauta de tratamiento sin consultarle al médico.
La dosis recomendada es para:
Niños con deficiencia de la hormona de crecimiento:
0,025 a 0,035 mg/kg de peso corporal al día o 0,7 a 1,0 mg/m2 de superficie corporal al día. Pueden utilizarse dosis más altas. Cuando la deficiencia de hormona de crecimiento continúa durante la adolescencia, Omnitrope debe continuarse hasta finalizar el desarrollo físico.
Niñas con síndrome de Turner:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día.
Niños con insuficiencia renal crónica:
0,045 a 0,050 mg/kg de peso corporal al día o 1,4 mg/m2 de superficie corporal al día. Pueden ser necesarias dosis más altas si la velocidad de crecimiento es demasiado baja. Puede ser necesario un ajuste de la dosis después de seis meses de tratamiento.
Niños con síndrome de Prader-Willi:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. La dosificación diaria no debe ser superior a 2,7 mg. El tratamiento no debe utilizarse en los niños que casi han dejado de crecer después de la pubertad.
Niños nacidos más pequeños o con peso más bajo que lo esperado y con un trastorno del crecimiento:
0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal al día. Es importante continuar el tratamiento hasta que se alcance la estatura final. El tratamiento debe suspenderse después del primer año si no responde, o si ha alcanzado la estatura final y dejado de crecer.
Adultos con deficiencia de la hormona de crecimiento:
Si continúa utilizando Omnitrope después del tratamiento durante la niñez, debe comenzar con 0,2 a 0,5 mg al día.
Esta dosificación se debe aumentar o reducir gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios.
Si la deficiencia de la hormona de crecimiento comienza durante la vida adulta, debe comenzar con 0,15 a 0,3 mg al día. Esta dosificación debe aumentarse gradualmente según los resultados de los análisis de sangre, así como la respuesta clínica y los efectos secundarios. La dosis de mantenimiento diaria rara vez es superior a 1,0 mg diarios. Las mujeres pueden necesitar dosis más altas que los hombres. La dosificación debe vigilarse cada seis meses. Las personas de más de 60 años deben comenzar con una dosis de 0,1 a 0,2 mg diarios que debe aumentarse lentamente según las necesidades individuales. Debe utilizarse la dosis mínima eficaz. La dosis de mantenimiento rara vez supera 0,5 mg al día. Siga las instrucciones que le haya dado el médico.
Inyección de Omnitrope
Inyéctese la hormona del crecimiento más o menos a la misma hora cada día. La hora de acostarse es un buen momento porque es fácil de recordar. Además, también es natural tener una concentración más elevada de hormona del crecimiento por la noche.
Omnitrope 15 mg/1,5 ml en un cartucho para SurePal 15 está previsto para varios usos. Sólo debe administrarse con SurePal 15, un dispositivo de inyección desarrollado específicamente para la administración de Omnitrope 15 mg/1.5 ml solución inyectable.
Omnitrope está indicado para su empleo por vía subcutánea. Esto significa que se inyecta por medio de una pequeña aguja para inyección en el tejido adiposo, por debajo de la piel. La mayor parte de las personas se inyectan en el muslo o en las nalgas. Póngase la inyección en el sitio que le haya enseñado su médico. El tejido adiposo de la piel puede verse reducido en el lugar de la inyección. Para evitar esto, utilice cada vez un sitio ligeramente diferente para inyectarse. Esto proporciona a la piel y a la zona por debajo de ella tiempo para recuperarse de una inyección antes de recibir otra en el mismo sitio.
El médico debe haberle enseñado ya cómo utilizar Omnitrope. Inyéctese siempre Omnitrope tal como el médico le ha dicho. Si no está seguro, compruebe con su médico o farmacéutico.
Cómo inyectar Omnitrope 15 mg/1,5 ml
Las siguientes instrucciones explican cómo inyectarse Omnitrope 15 mg/1,5 ml usted mismo. Lea detenidamente las instrucciones y sígalas paso a paso. Su médico o la enfermera le enseñará cómo inyectarse Omnitrope. No intente inyectarse a menos que esté seguro de que entiende el procedimiento y lo que conlleva la inyección.
- Omnitrope se administra con una inyección debajo de la piel.
Inspeccione cuidadosamente la solución antes de inyectarla y úsela sólo si es clara e incolora. Cambie el lugar de la inyección para así minimizar el riesgo de lipoatrofia local (reducción local del tejido adiposo debajo de la piel).
Preparación
Antes de empezar, debe tener todo lo necesario:
- Un cartucho especial con Omnitrope 15 mg/1,5 ml solución inyectable.
- SurePal 15, un dispositivo para inyección desarrollado específicamente para usarlo junto con Omnitrope
15 mg/1,5 ml solución inyectable (este dispositivo no se suministra en este envase; deben consultarse las Instrucciones de uso proporcionadas junto con SurePal 15).
- Una aguja de pluma para inyección subcutánea.
- 2 gasas de limpieza (no suministradas en el envase).
Lávese las manos antes de continuar con los siguiente pasos.
Inyección de Omnitrope
- Con una gasa, desinfecte la membrana de goma del cartucho.
- El contenido del cartucho debe ser transparente e incoloro.
Introduzca el cartucho en la pluma inyectable. Siga las Instrucciones de uso de la pluma para inyección (Pen). Para ajustar la pluma, dosifique la cantidad necesaria.
Seleccione el lugar de la inyección. Los mejores lugares para la administración de la inyección son los tejidos con una capa de grasa entre la piel y el músculo, como el muslo o el vientre (excepto el ombligo o la cintura).
Asegúrese de que se inyecta al menos a 1 cm de distancia respecto del lugar de la última inyección y de que cambia los lugares de inyección, tal y como le habrán enseñado.
Antes de administrar la inyección, limpie bien la piel con una gasa empapada en alcohol. Espere a que la zona se seque.
Introduzca la aguja en la piel de la forma que le haya enseñado el médico.
Después de inyectarse
- Después de la inyección, presione el lugar de la inyección con un pequeño vendaje o gasa estéril durante unos segundos. No masajee el lugar de la inyección.
- Retire la aguja de la pluma utilizando para ello la tapa exterior y elimínela. Esto mantendrá la solución de Omnitrope estéril e impedirá que gotee. También impedirá que el aire se introduzca en la pluma , taponando la aguja. No comparta las agujas. No comparta la pluma.
- Deje el cartucho en la pluma, reponga el capuchón en la pluma y guárdela en una nevera.
- La solución debe ser transparente cuando se saque de la nevera . No se debe utilizar si la solución está turbia o si contiene partículas.
Si se inyecta mucho más de lo que debiera, consulte lo antes posible a su médico o farmacéutico. Su concentración de azúcar en la sangre podría descender demasiado y después aumentar demasiado. Tal vez se sienta con temblores, sudoroso, somnoliento o “como si no fuera usted mismo”, y podría desmayarse.
Si olvidó usar Omnitrope
No use una dosis doble para compensar las dosis olvidadas. Lo mejor es usar la hormona de crecimiento con regularidad. Si se olvida de usar una dosis, póngase la siguiente inyección a la hora habitual, al día siguiente. Tome nota de las inyecciones olvidadas e infórmele al médico en el siguiente control.
Si interrumpe el tratamiento con Omnitrope
Consulte a su médico antes de dejar de usar Omnitrope.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Los efectos adversos muy frecuentes y frecuentes en los adultos pueden comenzar en los primeros meses de tratamiento y pueden detenerse espontáneamente o si se reduce la dosis.
Los efectos adversos muy frecuentes (que probablemente se produzcan en más de 1 de cada 10 pacientes) incluyen los siguientes:
En los adultos
• Dolor articular
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos)
Los efectos adversos frecuentes (que probablemente afecten a menos de 1 de cada 10 pacientes) incluyen los siguientes:
En los niños
• Enrojecimiento, picor o dolor temporal en el lugar de la inyección
• Dolor articular
En los adultos
• Adormecimiento, hormigueos
• Rigidez de los brazos y las piernas, dolor muscular
• Dolor o sensación de escozor en las manos o los antebrazos (conocido como síndrome del túnel carpiano)
Los efectos adversos poco frecuentes (que probablemente afecten a menos de 1 de cada 100 pacientes) incluyen los siguientes:
En los niños
• Retención de agua (que se presenta como hinchazón de los dedos o los tobillos, durante un tiempo corto al comienzo del tratamiento)
Los efectos adversos raros (que probablemente afecten a menos de 1 de cada 1.000 pacientes) incluyen los siguientes:
En los niños
• Adormecimiento, hormigueos
• Leucemia (se ha observado en un pequeño número de pacientes con deficiencia de la hormona del crecimiento, algunos de cuales habían sido tratados con somatropina. Sin embargo, no hay ningún indicio de que la incidencia de leucemia sea mayor en receptores de la hormona del crecimiento sin factores predisponentes).
• Aumento de la presión intracraneal (que causa síntomas, como cefalea intensa, alteraciones visuales o vómitos)
• Dolor muscular
Frecuencia no conocida: no puede estimarse a partir de los datos disponibles
• Diabetes de tipo 2
• Disminución de los niveles de la hormona cortisol en la sangre En los niños
• Rigidez en los brazos y las piernas
En los adultos
• Aumento de la presión intracraneal (que causa síntomas, como dolor de cabeza intenso, alteraciones visuales o vómitos)
• Enrojecimiento, picor o dolor en el lugar de la inyección
Formación de anticuerpos contra la hormona del crecimiento inyectada, pero estos no parecen hacer que la hormona del crecimiento deje de funcionar.
La piel alrededor de la zona de inyección puede ponerse irregular o con bultos, pero esto no deberá ocurrir si se inyecta en un lugar diferente cada vez.
Ha habido casos raros de muerte súbita en pacientes con síndrome de Prader-Willi. No obstante, estos casos no se han relacionado con el tratamiento con Omnitrope.
Su médico puede considerar una epifisiólisis de la cabeza femoral o una enfermedad de Legg-Calvé-Perthes si experimenta molestias o dolor en la cadera o la rodilla mientras está siendo tratado con Omnitrope.
Otros posibles efectos adversos relacionados con su tratamiento con la hormona del crecimiento pueden incluir los siguientes:
Usted (o su hijo) puede tener niveles elevados de azúcar en sangre o niveles reducidos de la hormona tiroidea. Esto lo puede analizar su médico y, si es necesario, su médico le recetará el tratamiento adecuado. En casos raros se ha observado inflamación del páncreas en pacientes tratados con la hormona del crecimiento.
Si considera que alguno de los efectos adversos que sufre es grave o si aprecia cualquier efecto adverso no mencionado en este prospecto, informe a su médico o farmacéutico.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y en la caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
• Conservar y transportar refrigerado (entre 2°C y 8°C).
• No congelar.
• Conservar en el embalaje original para protegerlo de la luz.
• Después de la administración de la primera inyección, el cartucho debe permanecer en el inyector de pluma y debe conservarse en una nevera, a una temperatura de 2 a 8 °C, y sólo debe usarse durante un máximo de 28 días.
No utilice Omnitrope si se observa que la solución está turbia.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Omnitrope
- El principio activo de Omnitrope es somatropina.
Cada ml de solución contiene 10 mg de somatropina (que corresponde a 30 UI).
Un cartucho contiene 15 mg (que corresponden a 45 UI) de somatropina en 1,5 ml.
- Los demás componentes son:
Fosfato hidrógeno disódico heptahidrato Fosfato dihidrógeno sódico dihidrato Cloruro de sodio Poloxámero 188 Fenol
Agua para preparaciones inyectables
Aspecto del producto y contenido del envase
Omnitrope es una solución inyectable transparente e incolora.
Omnitrope 15 mg/1,5 ml solución para inyección es para su uso sólo en SurePal 15. Envases de 1, 5 ó 10 cartuchos.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Sandoz GmbH Biochemiestrasse 10 A-6250 Kundl Austria
Pueden solicitar más información respecto a este titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Sandoz nv-sa Tél/Tel.: +32 2 722 97 97
Etarapna
TtproBCKO npegcTaBHTeacTBO CaHgo3 g.g.
Tea.: +359 2 970 47 47
medicamento dirigiéndose al representante local del
Lietuva
Sandoz Pharmaceuticals d.d. filialas Tel.: +370 5 2636 037
Luxembourg/Luxemburg
Sandoz GmbH Tel.: +43 5338 2000
|
Ceská republika Sandoz s.r.o. Tel.: +420 221 421 611 |
Magyarország Sandoz Hungária Kft. Tel.: +36 1 430 2890 |
|
Danmark/Norge/Ísland/Sverige Sandoz A/S Tlf.: +45 6395 1000 |
Malta Sandoz GmbH Tel.: +43 5338 2000 |
|
Deutschland Hexal AG Tel.: +49 8024 908 0 |
Nederland Sandoz B.V. Tel.: +31 36 52 41 600 |
|
Eesti Sandoz d.d. Eesti filiaal Tel.: +372 665 2400 |
Osterreich Sandoz GmbH Tel.: +43 5338 2000 |
|
ELLáóa Sandoz dd T^: +30 216 600 500 0 |
Polska Sandoz Polska Sp. z o.o. Tel.: +48 22 209 70 00 |
|
España Sandoz Farmacéutica, S.A. Tel.: +34 900 456 856 |
Portugal Sandoz Farmacéutica Lda. Tel.: +351 21 924 19 11 |
|
France Sandoz SAS Tél.: +33 1 49 64 48 00 |
Romania S.C. Sandoz S.R.L. Tel.: +40 265 208 120 |
|
Hrvatska Sandoz d.o.o. |
Slovenija Lek farmacevtska druzba d.d. |
|
Tel: +385 1 23 53 111 |
Tel.: +386 1 580 21 11 |
|
Ireland Rowex Ltd Tel.: +353 27 50077 |
Slovenská republika Sandoz d.d. - organizacná zlozka Tel.: +421 2 48 200 600 |
|
Italia Sandoz S.p.A. Tel.: +39 02 96541 |
Suomi/Finland Sandoz A/S Puh/Tel.: +358 10 6133 415 |
|
Kúnpoq P.T.Hadjigeorgiou co ltd Tel.: +357 25372425 |
United Kingdom Sandoz Ltd Tel.: + 44 1276 69 8020 |
|
Latvija Sandoz d.d. Párstávnieciba Latvija Tel.: +371 67 892 006 |
Fecha de la última revisión de este prospecto: {MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
215
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.
f Notificada en niños que padecen deficiencia de la hormona del crecimiento tratados con somatropina; sin embargo, se aprecia que la incidencia es parecida a la de los niños sin esta deficiencia.
En general, estos efectos adversos son leves o moderados, aparecen durante los primeros meses de tratamiento, y remiten espontáneamente o al reducir la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y, posiblemente, está relacionada inversamente con la edad de los pacientes al inicio de la deficiencia de la hormona del crecimiento.
$ Se han notificado reacciones transitorias en el lugar de la inyección en niños.
{ Se desconoce su importancia clínica.