Novoeight 1000Ui Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
^Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
NovoEight 250 UI polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial de polvo contiene 250 UI de factor VIII de coagulación humano (ADNr), turoctocog alfa.
NovoEight contiene aproximadamente 62,5 UI/ml de factor VIII de coagulación humano (ADNr), turoctocog alfa, después de la reconstitución.
La potencia (UI) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de NovoEight es aproximadamente de 8.300 UI/mg de proteína.
Turoctocog alfa (factor VIII de coagulación humano (ADNr)) es una proteína purificada que contiene 1.445 aminoácidos con una masa molecular aproximada de 166 kDA. Se produce mediante tecnología de ADN recombinante en células ováricas de hámster chino (CHO) y se prepara sin añadir ninguna proteína humana ni derivada de animal durante el proceso de cultivo de las células, la purificación o la formulación final.
T uroctocog alfa es un factor VIII de coagulación humano recombinante truncado de dominio B (el dominio B consiste en 21 aminoácidos del dominio silvestre de tipo B) sin ninguna otra modificación de la secuencia de aminoácidos.
Excipiente con efecto conocido:
0,31 mmol de sodio (7 mg) por ml de solución reconstituida.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo o masa friable de color blanco o ligeramente amarillo. Solución inyectable transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
T ratamiento y profilaxis de hemorragias en pacientes con hemofilia A (deficiencia congénita del factor VIII). NovoEight se puede usar en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de hemofilia.
Pacientes no tratados previamente
No se ha establecido todavía la seguridad y la eficacia de NovoEight en pacientes no tratados previamente.
No se dispone de datos.
Posología
La dosis y duración de la terapia de sustitución dependen de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que se corresponden con el estándar actual de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa como porcentaje (relativo al nivel en plasma humano normal) o en Unidades Internacionales (relativas al estándar internacional para el factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII presente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor VIII se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad plasmática de factor VIII en 2 UI/dl. La dosis requerida se determina utilizando la fórmula siguiente:
UI requeridas = peso corporal (kg) * aumento deseado de factor VIII (%) (UI/dl) x 0,5 (UI/kg por UI/dl).
La dosis y la frecuencia de la administración deberá individualizarse según la eficacia clínica en cada caso en particular.
En el caso de los eventos hemorrágicos siguientes, la actividad del factor VIII no deberá bajar del nivel de actividad plasmática indicado (en % del normal o UI/dl) durante el periodo correspondiente. La tabla siguiente se puede usar como guía para determinar la dosis en episodios hemorrágicos y cirugía:
T abla 1 Guía para determinar la dosis en episodios hemorrágicos y cirugía
Grado de la hemorragia/Tipo de Nivel de factor VIII Frecuencia de dosificación
procedimiento quirúrgico necesario (%) (UI/dl) (horas)/Duración del tratamiento
(días)
Hemorragia
Hemartrosis precoz, sangrado muscular 20-40 Repetir cada 12 - 24 horas al menos 1
o sangrado de la cavidad oral día hasta que el episodio hemorrágico
se haya resuelto, en función del dolor, o hasta la cicatrización de la herida.
Hemartrosis más extensa, sangrado 30-60 muscular o hematoma
Hemorragias con riesgo vital 60-100
Repetir la perfusión cada 12 - 24 horas durante 3 - 4 días o hasta que el dolor y la discapacidad aguda se hayan resuelto.
Repetir la perfusión cada 8 - 24 horas hasta que el riesgo desaparezca.
Cirugía
Cirugía menor, incluidas las 30-60 Cada 24 horas, al menos 1 día, hasta
extracciones dentales la cicatrización.
Grado de la hemorragia/Tipo de Nivel de factor VIII Frecuencia de dosificación
procedimiento quirúrgico necesario (%) (UI/dl) (horas)/Duración del tratamiento
(días)
Cirugía mayor 80-100 Repetir la perfusión cada 8-24 horas
(pre- y postoperatorio) hasta que se consiga una cicatrización
adecuada. A continuación, tratamiento durante por lo menos 7 días más para mantener una actividad del factor VIII del 30 % al 60 % (UI/dl).
Profilaxis
En la profilaxis a largo plazo para prevenir hemorragias en pacientes con hemofilia A grave. Las dosis habituales recomendadas son de 20 a 40 UI de factor VIII por kg de peso corporal a días alternos o de 20 a 50 UI de factor VIII por kg de peso corporal 3 veces por semana. En algunos casos, especialmente en los pacientes más jóvenes, puede ser necesario un intervalo de dosis más corto o una dosis mayor.
Supervisión del tratamiento
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis a administrar y la frecuencia de las inyecciones repetidas. Especialmente en el caso de las intervenciones de cirugía mayor, es indispensable controlar con precisión la terapia de sustitución mediante pruebas de coagulación (actividad plasmática del factor VIII). La respuesta individual de cada paciente frente al factor VIII puede variar y alcanzar distintos niveles de recuperación in vivo, y presentar semividas diferentes.
Cirugía
No existe experiencia en cirugía en pacientes pediátricos.
Pacientes de edad avanzada
No existe experiencia en pacientes de más de 65 años.
Población pediátrica
En la profilaxis a largo plazo para prevenir hemorragias en pacientes de menos de 12 años, las dosis recomendadas son de 25 a 50 UI de factor VIII por kg de peso corporal a días alternos o de 25 a 60 UI de factor VIII por kg de peso corporal 3 veces por semana. En pacientes pediátricos de más de 12 años, la dosis recomendada es la misma que para los adultos.
Forma de administración Vía intravenosa.
La velocidad de perfusión recomendada para NovoEight es de 1 - 2 ml/min. La velocidad se deberá determinar de acuerdo con el nivel de confort del paciente.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Reacciones alérgicas conocidas a las proteínas de hámster.
4.4 Advertencias y precauciones especiales de empleo Hipersensibilidad
Es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico a NovoEight. El producto contiene trazas de proteínas de hámster, las cuales pueden provocar reacciones alérgicas en algunos
4
pacientes. Si se produjesen síntomas de hipersensibilidad, se deberá instruir a los pacientes que deben interrumpir el uso del medicamento y ponerse inmediatamente en contacto con su médico. Se debe informar a los pacientes de los síntomas iniciales de las reacciones de hipersensibilidad, como urticaria localizada o generalizada, opresión en el pecho, respiración sibilante, hipotensión y anafilaxia.
En caso de shock, se seguirán las pautas médicas habituales para su tratamiento.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, y se cuantifican en Unidades Bethesda (UB) por ml de plasma usando el ensayo modificado. El riesgo de desarrollo de inhibidores está correlacionado con la exposición al factor VIII, y el riesgo mayor se produce durante los primeros 20 días de exposición. Los inhibidores raramente se forman después de los primeros 100 días de tratamiento.
Se han observado casos recurrentes de inhibidores (con título bajo), después de cambiar de un medicamento con factor VIII a otro a pacientes que habían recibido un tratamiento previo de más de 100 días de exposición y que tienen antecedentes de desarrollo de inhibidores. Por consiguiente se recomienda controlar a todos los pacientes atentamente para determinar la presencia de inhibidores después de cualquier cambio de medicamento.
En general, se deberá controlar atentamente a todos los pacientes tratados con medicamentos con factor VIII para determinar el desarrollo de inhibidores mediante la observación clínica y las pruebas de laboratorio adecuadas. Si no se obtienen los niveles plasmáticos de actividad del factor VIII esperados, o si no se controla la hemorragia con una dosis adecuada, se deberán realizar análisis para detectar la presencia de inhibidores del factor VIII. En pacientes con niveles elevados de inhibidores, es posible que el tratamiento con factor VIII no sea eficaz y se deberán considerar otras opciones terapéuticas. El tratamiento de estos pacientes debe hacerse bajo la dirección de un médico con experiencia en el cuidado de la hemofilia y los inhibidores del factor VIII.
Se recomienda encarecidamente registrar el nombre y el número de lote del medicamento cada vez que se administre NovoEight a un paciente con el fin de mantener un vínculo entre el paciente y el lote del medicamento.
Consideraciones relacionadas con los excipientes
Después de la reconstitución, este medicamento contiene 0,31 mmol de sodio (7 mg) por ml de solución reconstituida. Esto deberá tenerse en cuenta en pacientes que sigan una dieta controlada en sodio.
Complicaciones relacionadas con el catéter
Si se requiere un dispositivo de acceso venoso central (DAVC), se debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, como por ejemplo, infecciones locales, bacteriemia y trombosis en el lugar de inserción del catéter.
Población pediátrica
Las advertencias y precauciones indicadas son aplicables a adultos y niños.
4.5 Interacción con otros medicamentos y otras formas de interacción No se han realizado estudios de interacciones con NovoEight.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción animal con NovoEight. Teniendo en cuenta la escasa ocurrencia de hemofilia A en mujeres, no existe experiencia en relación con el uso de factor VIII durante el embarazo y la lactancia. Por consiguiente, solo se deberá usar factor VIII durante el embarazo y la lactancia si está estrictamente indicado.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NovoEight sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Resumen del perfil de seguridad
Hipersensibilidad o reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el punto de perfusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habones, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, cosquilleo, vómitos, sibilancias) se han observado raramente y, en algunos casos, puede progresar hasta una anafilaxia grave (incluido shock).
En muy raras ocasiones se ha observado el desarrollo de anticuerpos frente a la proteína de hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizadores (inhibidores) del factor VIII. Si se generan inhibidores de este tipo, la situación se pondrá de manifiesto por una respuesta clínica insuficiente. En tales casos, se recomienda ponerse en contacto con un centro especializado en hemofilia.
T abla de reacciones adversas
La tabla que se muestra a continuación sigue la clasificación de sistemas de órganos de MedDRA (clasificación por órganos y sistemas (SOC) y nivel de término preferente).
Las frecuencias se han evaluado conforme a la convención siguiente: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); raras (> 1/10.000 a < 1/1.000); muy raras (< 1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
En cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
T abla 2 Frecuencia de los reacciones adversas en ensayos clínicos
|
Clasificación por órganos y sistemas |
Frecuencia* |
Reacción adversa |
|
Trastornos psiquiátricos |
Poco frecuentes |
Insomnio |
|
Trastornos del sistema nervioso |
Poco frecuentes |
Cefalea, mareo |
|
Trastornos cardiacos |
Poco frecuentes |
Taquicardia sinusal |
|
Trastornos vasculares |
Poco frecuentes |
Hipertensión, linfoedema |
|
Trastornos hepatobiliares |
Frecuentes |
Enzimas hepáticas elevadas** |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
Erupción |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Poco frecuentes |
Rigidez musculoesquelética, artropatía, dolor en las extremidades, dolor musculoesquelético |
|
Trastornos generales y alteraciones en el lugar de administración |
Frecuentes |
Reacciones en la zona de inyección*** |
|
Poco frecuentes |
Fatiga, sensación de calor, edema periférico, pirexia | |
|
Exploraciones complementarias |
Poco frecuentes |
Frecuencia cardiaca aumentada |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Poco frecuentes |
Contusión |
Cálculo basado en el número total de pacientes distintos en todos los estudios clínicos (214).
Entre las enzimas hepáticas elevadas se encuentran la aminotransferasa, aspartato aminotransferasa, gamma glutamiltransferasa y bilirrubina.
*** Entre las reacciones en la zona de inyección se encuentran eritema, extravasación y prurito en la zona de inyección.
Descripción de las reacciones adversas señaladas
Durante todos los estudios clínicos con NovoEight, se han notificado un total de 30 reacciones adversas en
19 de 214 pacientes expuestos a NovoEight. Las reacciones adversas de las que se informó con mayor frecuencia fueron reacciones en la zona de inyección y enzimas hepáticas elevadas. De las 30 reacciones adversas, 2 de ellas se notificaron en 1 de 31 pacientes de menos de 6 años de edad, ninguna entre los pacientes de 6 a 18 años de edad y 28 en 18 de 127 adultos.
Población pediátrica
En estudios clínicos con 63 pacientes pediátricos de entre 0 y 12 años de edad y 24 adolescentes entre 12 y 18 años de edad con hemofilia A grave, no se observó ninguna diferencia en el perfil de seguridad de NovoEight entre pacientes pediátricos y adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han observado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos, factor VIII de la coagulación sanguínea, código ATC: B02BD02.
Mecanismo de acción
NovoEight contiene turoctocog alfa, un factor VIII de coagulación humano (ADNr) con un dominio B truncado. Esta glicoproteína tiene la misma estructura que el factor VIII cuando se activa, y modificaciones postranslacionales similares a las de la molécula derivada del plasma. Se ha observado que el punto de sulfatación de tirosina presente en Tyr1680 (longitud completa nativa), que es importante para el enlace con el factor de von Willebrand, está totalmente sulfatado en la molécula de turoctocog alfa. Cuando se administra mediante perfusión en un paciente con hemofilia, el factor VIII se enlaza con el factor de von Willebrand endógeno en la circulación del paciente. El complejo factor VIII/factor von Willebrand consiste en dos moléculas (factor VIII y factor de von Willebrand) con funciones fisiológicas distintas. El factor VIII activado actúa como cofactor del factor IX activado, y acelera la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo. La hemofilia es una alteración hereditaria vinculada al sexo de la coagulación de la sangre debida a niveles reducidos de factor VIII:C y da como resultado hemorragias difusas en articulaciones, músculos u órganos internos, ya sea de forma espontánea como tras un accidente o un trauma quirúrgico. La terapia de sustitución, aumenta los niveles plasmáticos de factor VIII, lo cual permite corregir temporalmente la deficiencia del factor y la tendencia al sangrado.
Eficacia clínica
Se han realizado tres ensayos multicéntricos, abiertos y no controlados para evaluar la seguridad y la eficacia de NovoEight para la prevención y el tratamiento de sangrado en pacientes tratados previamente con hemofilia A grave (actividad de FVEI < 1 %). Los ensayos incluyeron a 213 pacientes expuestos; 150 pacientes adolescentes o adultos sin inhibidores desde la edad de 12 años (> 150 días de exposición) y 63 pacientes pediátricos sin inhibidores de menos de 12 años de edad (> 50 días de exposición). 187 de 213 pacientes continuaron en el ensayo ampliado de seguridad. Se observó que el tratamiento con NovoEight es seguro y tiene el efecto hemostático y preventivo deseado. Durante una exposición acumulada de 54.000 días (correspondientes a 342 años paciente), no se observó el desarrollo de inhibidores del factor VIII en los ensayos clínicos en fase 3a con pacientes tratados previamente. De los 1.377 sangrados observados en 177 de los 213 pacientes, 1.244 (90,3 %) de los sangrados se resolvieron con 1 o 2 perfusiones de NovoEight.
|
Tabla 3 Consumo de turoctocog alfa y tasas de éxito glo |
bales | ||||
|
Niños pequeños (0 - <6 años) |
Niños mayores (6 - <12 años) |
Adolescentes (12 -<18 años) |
Adultos (>18 años) |
Total | |
|
Número de pacientes |
31 |
32 |
24 |
126 |
213 |
|
Dosis empleada como prevención por paciente (UI/kg PC) Valor medio (DE) Mín; Máx |
40,1 (8,5) 26,5 ; 57,3 |
36,6 (9,0) 24,9 ; 57,9 |
27,0 (7,6) 20,5 ; 46,9 |
26,9 (6,9) 20,0 ; 50,8 |
30,3 (9,2) 20,0 ; 57,9 |
|
Dosis utilizada para el tratamiento del sangrado (UI/kg BW) Valor medio (DE) Mín; Máx |
44,4 (17,9) 25,9 ; 193,8 |
40,0 (10,4) 25,5 ; 65,5 |
28,2 (10,2) 12,4 ; 76,8 |
33,8 (11,9) 9,3 ; 104,0 |
34,5 (12,6) 9,3 ; 193,8 |
|
Número de infusiones para el tratamiento del sangrado Valor medio (DE) Mín; Máx |
1,2 (0,6) 1 ; 6 |
1,5 (1,1) 1 ; 8 |
1,7 (1,3) 1 ; 9 |
1,5 (2,4) 1 ; 49 |
1,5 (2,1) 1 ; 49 |
|
T asa de éxito* % |
92,9 % |
88,9 % |
79,7 % |
85,6 % |
85,9 % |
PC: Peso corporal, DE: Desviación estándar *El éxito se define como “Excelente” o “Bueno”.
En total se realizaron 14 intervenciones quirúrgicas en 14 pacientes, de las cuales 13 fueron de cirugía mayor y 1 de cirugía menor. La hemostasis fue correcta en todas las intervenciones quirúrgicas y no se observó ningún fallo de tratamiento.
5.2 Propiedades farmacocinéticas
Todos los estudios de farmacocinética con turoctocog alfa se realizaron en pacientes tratados previamente con hemofilia A grave (FVHI < 1 %). El análisis de las muestras de plasma se realizó mediante análisis de coagulación de una fase y análisis cromogénico.
En un estudio internacional que implicó a 36 laboratorios, se evaluó el rendimiento de NovoEight en ensayos de FVHI:C y se comparó con un medicamento comercializado de FVIII recombinante de longitud completa. El estudio reveló que se obtienen resultados comparables y coherentes con ambos productos y que NovoEight se puede medir en plasma de forma fiable sin necesidad de usar un estándar aparte para NovoEight.
Los parámetros farmacocinéticos de NovoEight en una sola dosis se indican en la T abla 4 para el análisis de coagulación y en la Tabla 5 para el análisis cromogénico.
Tabla 4 Farmacocinética de una sola dosis de turoctocog alfa en pacientes con hemofilia A grave (FVIII < 1 %), análisis de coagulación___
|
Parámetro |
0 - <6 años |
6 - <12 años |
>12 años |
|
n = 14 |
n = 14 |
n = 33 | |
|
Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) | |
|
Recuperación incremental (UI/ml)/(UI/kg) |
0,018 (0,007) |
0,020 (0,004) |
0,022 (0,004) |
|
AUC ((UI*h)/ml) |
9,92 (4,11) |
11,09 (3,74) |
15,26 (5,77) |
|
CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
|
t/ (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
|
Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
|
Cmax (UI/ml) |
1,00 (0,58) |
1,07 (0,35) |
1,226 (0,41) |
|
Tiempo medio de residencia (h) |
9,63 (2,50) |
9,91 (2,57) |
14,19 (5,08) |
Tabla 5 Farmacocinética de una sola dosis de turoctocog alfa en pacientes con hemofilia A grave (FVIII < 1 %), análisis cromogénico___
|
Parámetro |
0 - <6 años |
6 - <12 años |
>12 años |
|
n = 14 |
n = 14 |
n = 33 | |
|
Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) | |
|
Recuperación incremental (UI/ml)/(UI/kg) |
0,022 (0,006) |
0,025 (0,006) |
0,029 (0,006) |
|
AUC ((UI*h)/ml) |
12,23 (4,36) |
14,37 (3,48) |
19,63 (7,73) |
|
CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
|
t/ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
|
Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
|
Cmax (UI/ml) |
1,12 (0,31) |
1,25 (0,27) |
1,63 (0,50) |
|
Tiempo medio de residencia (h) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
Los parámetros farmacocinéticos fueron comparables entre pacientes pediátricos de menos de 6 años de edad y pacientes pediátricos mayores de 6 y menores de 12 años de edad. Se observó alguna variación entre los parámetros farmacocinéticos de NovoEight en pacientes pediátricos y adultos. Los valores mayores de CL y menores de t./2 en pacientes pediátricos en comparación con pacientes adultos con hemofilia A se pueden deber en parte al hecho conocido de que el volumen de plasma por kilogramo de peso corporal es mayor en los pacientes más jóvenes.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad y toxicidad a dosis repetidas.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Cloruro de sodio L-histidina Sacarosa Polisorbato 80 L-metionina
Cloruro de calcio dihidratado Hidróxido de sodio Ácido clorhídrico
Disolvente:
Cloruro de sodio
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
Sin abrir:
30 meses
Durante el periodo de validez, el medicamento se puede almacenar a temperatura ambiente (<30°C) durante un solo periodo de hasta 9 meses. Una vez que el producto se saca de la nevera, no se debe volver a refrigerar. Registrar la fecha de inicio del almacenamiento a temperatura ambiente en el embalaje del producto.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Después de la reconstitución:
Se ha demostrado la estabilidad química y física en uso durante 24 horas almacenado entre 2°C y 8°C y 4 horas almacenado a temperatura ambiente (<30°C).
Desde el punto de vista microbiológico, el medicamento se debería usar inmediatamente después de la reconstitución. Si no se utiliza de inmediato, los tiempos y las condiciones de almacenamiento en uso antes del uso son responsabilidad del usuario y no deberían superarse las 4 horas a temperatura ambiente (<30°C) o 24 horas entre 2°C y 8°C, a menos que la reconstitución se haya realizado en condiciones asépticas controladas y validadas.
El medicamento no utilizado almacenado a temperatura ambiente durante más de 4 horas se deberá desechar.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C). No congelar.
Para consultar la información de almacenamiento a temperatura ambiente y las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Cada envase de NovoEight 250 UI polvo y disolvente para solución inyectable contiene:
- 1 vial de vidrio (tipo I) con polvo y un tapón de goma de clorobutilo
- 1 adaptador de vial estéril para la reconstitución
- 1 jeringa precargada con 4 ml de disolvente con mecanismo de protección (polipropileno), un émbolo
de goma (bromobutilo) y un capuchón de aguja con tapón (bromobutilo)
- 1 varilla del émbolo (polipropileno).
6.6 Precauciones especiales de eliminación y otras manipulaciones
NovoEight se debe administrar por vía intravenosa después de la reconstitución del polvo con el disolvente suministrado en la jeringa. Después de la reconstitución, la solución es transparente o ligeramente opalescente. No utilice la solución si tiene aspecto turbio o presenta depósitos.
También necesitará un conjunto de perfusión (tubos y aguja mariposa), toallitas estériles con alcohol, gasas y tiritas. Estos dispositivos no se incluyen en el paquete NovoEight.
Utilice siempre una técnica aséptica.
Reconstitución
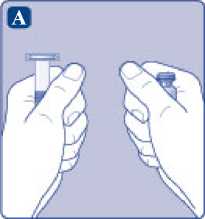
A)
Saque el vial, el adaptador del vial y la jeringa precargada del envase. Deje la varilla del émbolo sin tocar en el envase. Lleve el vial y la jeringa precargada a temperatura ambiente. Puede hacerlo manteniendo ambos en las manos hasta que sienta que están a la misma temperatura que sus manos. No utilice ningún otro sistema para calentar el vial y la jeringa precargada.


B)
Quite el capuchón de plástico del vial. Si el capuchón de plástico está flojo o falta, no utilice el vial. Limpie el tapón de goma del vial con una toallita estéril con alcohol y deje que se seque al aire unos segundos antes de usarlo.
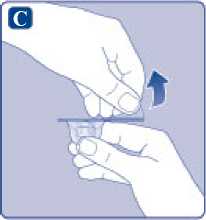
C)
Quite el precinto de papel del adaptador del vial. Si el precinto de papel no está totalmente sellado o si está roto, no utilice el adaptador del vial. No saque el adaptador del vial del capuchón protector con los dedos.
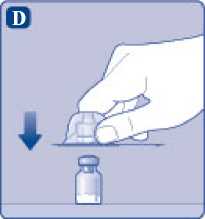
D)
Ponga el capuchón protector del adaptador del vial boca abajo y coloque el adaptador del vial a presión sobre el vial. Una vez unido, no retire del vial el adaptador del vial.
E)
Comprima ligeramente el capuchón protector entre los dedos pulgar e índice tal como se muestra. Quite el capuchón protector del adaptador del vial.
|
0/ Xj Vví ♦ir |
L | |
|
; i |
L_ | |
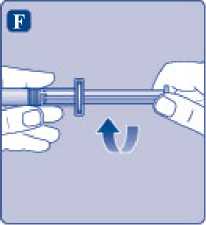
F)
Sujete la varilla del émbolo por el extremo ancho e inmediatamente conéctelo a la jeringa girándolo hacia la derecha dentro del émbolo en el interior de la jeringa precargada hasta que se sienta resistencia.
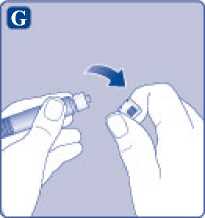
G)
Quite el tapón de la jeringa precargada doblándolo hacia abajo hasta que se rompa la perforación. No toque la punta de la jeringa debajo del tapón de la jeringa.
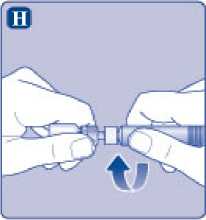
H)
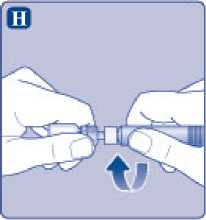
Enrosque la jeringa precargada con firmeza en el adaptador del vial hasta que se sienta resistencia.
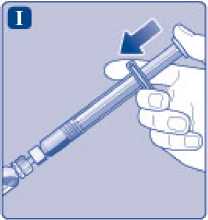
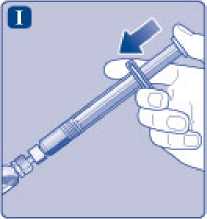
I)
Sujete la jeringa precargada ligeramente inclinada con el vial apuntando hacia abajo. Presione la varilla del émbolo para inyectar todo el disolvente en el vial.
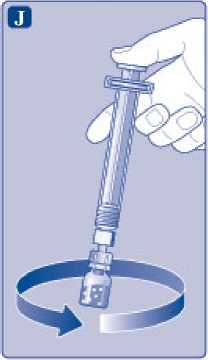
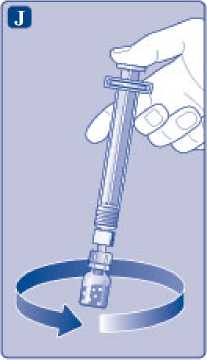
J)
Mantenga la varilla del vial presionada y remueva suavemente el vial hasta que el polvo se haya disuelto. No agite el vial, ya que esto produciría espuma.
Se recomienda utilizar NovoEight inmediatamente después de la reconstitución. Para consultar las condiciones de conservación del medicamento reconstituido, ver sección 6.3.
Si se necesita una dosis mayor, repita los pasos A a J con más viales, adaptadores de viales y jeringas precargadas.
Administración de la solución reconstituida
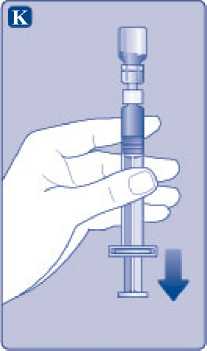
K)
Mantenga la varilla del émbolo totalmente empujada hacia el interior. Gire la jeringa con el vial boca abajo. Deje de empujar la varilla del émbolo y deje que retroceda por sí misma mientras la solución reconstituida llena la jeringa. Tire de la varilla del émbolo ligeramente hacia abajo para hacer pasar la solución reconstituida hacia la jeringa.
Si solo necesita una parte del vial, utilice la escala de la jeringa para ver la cantidad de solución reconstituida que ha retirado, tal como le haya indicado su médico o su enfermera.
Mientras sujeta el vial cabeza abajo, golpee suavemente la jeringa para que las posibles burbujas suban hasta la parte superior. Empuje la varilla del émbolo lentamente hasta que se hayan eliminado las burbujas.
L)
Desenrosque el adaptador del vial con el vial.
Ahora NovoEight está listo para ser inyectado.
Busque un lugar adecuado e inyecte lentamente NovoEight en la vena en un periodo de 2 a 5 minutos.
Eliminación
Después de la inyección, deseche con seguridad toda la solución NovoEight no utilizada, la jeringa con el juego de perfusión, el vial con el adaptador del vial y los demás residuos siguiendo las indicaciones de su farmacéutico.
No los deseche como residuos domésticos ordinarios.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dinamarca
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/888/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 13 Noviembre 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
^Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
NovoEight 500 UI polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial de polvo contiene 500 UI de factor VIII de coagulación humano (ADNr), turoctocog alfa.
NovoEight contiene aproximadamente 125 UI/ml de factor VIII de coagulación humano (ADNr), turoctocog alfa, después de la reconstitución.
La potencia (UI) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de NovoEight es aproximadamente de 8.300 UI/mg de proteína.
Turoctocog alfa (factor VIII de coagulación humano (ADNr)) es una proteína purificada que contiene 1.445 aminoácidos con una masa molecular aproximada de 166 kDA. Se produce mediante tecnología de ADN recombinante en células ováricas de hámster chino (CHO) y se prepara sin añadir ninguna proteína humana ni derivada de animal durante el proceso de cultivo de las células, la purificación o la formulación final.
Turoctocog alfa es un factor VIII de coagulación humano recombinante truncado de dominio B (el dominio B consiste en 21 aminoácidos del dominio silvestre de tipo B) sin ninguna otra modificación de la secuencia de aminoácidos.
Excipiente con efecto conocido:
0,31 mmol de sodio (7 mg) por ml de solución reconstituida.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo o masa friable de color blanco o ligeramente amarillo. Solución inyectable transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
T ratamiento y profilaxis de hemorragias en pacientes con hemofilia A (deficiencia congénita del factor VIII). NovoEight se puede usar en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de hemofilia.
Pacientes no tratados previamente
No se ha establecido todavía la seguridad y la eficacia de NovoEight en pacientes no tratados previamente.
No se dispone de datos.
Posología
La dosis y duración de la terapia de sustitución dependen de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que se corresponden con el estándar actual de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa como porcentaje (relativo al nivel en plasma humano normal) o en Unidades Internacionales (relativas al estándar internacional para el factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII presente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor VIII se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad plasmática de factor VIII en 2 UI/dl. La dosis requerida se determina utilizando la fórmula siguiente:
UI requeridas = peso corporal (kg) * aumento deseado de factor VIII (%) (UI/dl) x 0,5 (UI/kg por UI/dl).
La dosis y la frecuencia de la administración deberá individualizarse según la eficacia clínica en cada caso en particular.
En el caso de los eventos hemorrágicos siguientes, la actividad del factor VIII no deberá bajar del nivel de actividad plasmática indicado (en % del normal o UI/dl) durante el periodo correspondiente. La tabla siguiente se puede usar como guía para determinar la dosis en episodios hemorrágicos y cirugía:
T abla 1 Guía para determinar la dosis en episodios hemorrágicos y cirugía
Grado de la hemorragia/Tipo de Nivel de factor VIII Frecuencia de dosificación
procedimiento quirúrgico necesario (%) (UI/dl) (horas)/Duración del tratamiento
(días)
Hemorragia
Hemartrosis precoz, sangrado muscular 20-40 Repetir cada 12 - 24 horas al menos 1
o sangrado de la cavidad oral día hasta que el episodio hemorrágico
se haya resuelto, en función del dolor, o hasta la cicatrización de la herida.
Hemartrosis más extensa, sangrado 30-60 muscular o hematoma
Hemorragias con riesgo vital 60-100
Repetir la perfusión cada 12 - 24 horas durante 3 - 4 días o hasta que el dolor y la discapacidad aguda se hayan resuelto.
Repetir la perfusión cada 8 - 24 horas hasta que el riesgo desaparezca.
Cirugía
Cirugía menor, incluidas las 30-60 Cada 24 horas, al menos 1 día, hasta
extracciones dentales la cicatrización.
Grado de la hemorragia/Tipo de Nivel de factor VIII Frecuencia de dosificación
procedimiento quirúrgico necesario (%) (UI/dl) (horas)/Duración del tratamiento
(días)
Cirugía mayor 80-100 Repetir la perfusión cada 8-24 horas
(pre- y postoperatorio) hasta que se consiga una cicatrización
adecuada. A continuación, tratamiento durante por lo menos 7 días más para mantener una actividad del factor VIII del 30 % al 60 % (UI/dl).
Profilaxis
En la profilaxis a largo plazo para prevenir hemorragias en pacientes con hemofilia A grave. Las dosis habituales recomendadas son de 20 a 40 UI de factor VIII por kg de peso corporal a días alternos o de 20 a 50 UI de factor VIII por kg de peso corporal 3 veces por semana. En algunos casos, especialmente en los pacientes más jóvenes, puede ser necesario un intervalo de dosis más corto o una dosis mayor.
Supervisión del tratamiento
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis a administrar y la frecuencia de las inyecciones repetidas. Especialmente en el caso de las intervenciones de cirugía mayor, es indispensable controlar con precisión la terapia de sustitución mediante pruebas de coagulación (actividad plasmática del factor VIII). La respuesta individual de cada paciente frente al factor VIII puede variar y alcanzar distintos niveles de recuperación in vivo, y presentar semividas diferentes.
Cirugía
No existe experiencia en cirugía en pacientes pediátricos.
Pacientes de edad avanzada
No existe experiencia en pacientes de más de 65 años.
Población pediátrica
En la profilaxis a largo plazo para prevenir hemorragias en pacientes de menos de 12 años, las dosis recomendadas son de 25 a 50 UI de factor VIII por kg de peso corporal a días alternos o de 25 a 60 UI de factor VIII por kg de peso corporal 3 veces por semana. En pacientes pediátricos de más de 12 años, la dosis recomendada es la misma que para los adultos.
Forma de administración Vía intravenosa.
La velocidad de perfusión recomendada para NovoEight es de 1 - 2 ml/min. La velocidad se deberá determinar de acuerdo con el nivel de confort del paciente.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Reacciones alérgicas conocidas a las proteínas de hámster.
4.4 Advertencias y precauciones especiales de empleo Hipersensibilidad
Es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico a NovoEight. El producto contiene trazas de proteínas de hámster, las cuales pueden provocar reacciones alérgicas en algunos
17
pacientes. Si se produjesen síntomas de hipersensibilidad, se deberá instruir a los pacientes que deben interrumpir el uso del medicamento y ponerse inmediatamente en contacto con su médico. Se debe informar a los pacientes de los síntomas iniciales de las reacciones de hipersensibilidad, como urticaria localizada o generalizada, opresión en el pecho, respiración sibilante, hipotensión y anafilaxia.
En caso de shock, se seguirán las pautas médicas habituales para su tratamiento.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, y se cuantifican en Unidades Bethesda (UB) por ml de plasma usando el ensayo modificado. El riesgo de desarrollo de inhibidores está correlacionado con la exposición al factor VIII, y el riesgo mayor se produce durante los primeros 20 días de exposición. Los inhibidores raramente se forman después de los primeros 100 días de tratamiento.
Se han observado casos recurrentes de inhibidores (con título bajo), después de cambiar de un medicamento con factor VIII a otro a pacientes que habían recibido un tratamiento previo de más de 100 días de exposición y que tienen antecedentes de desarrollo de inhibidores. Por consiguiente se recomienda controlar a todos los pacientes atentamente para determinar la presencia de inhibidores después de cualquier cambio de medicamento.
En general, se deberá controlar atentamente a todos los pacientes tratados con medicamentos con factor VIII para determinar el desarrollo de inhibidores mediante la observación clínica y las pruebas de laboratorio adecuadas. Si no se obtienen los niveles plasmáticos de actividad del factor VIII esperados, o si no se controla la hemorragia con una dosis adecuada, se deberán realizar análisis para detectar la presencia de inhibidores del factor VIII. En pacientes con niveles elevados de inhibidores, es posible que el tratamiento con factor VIII no sea eficaz y se deberán considerar otras opciones terapéuticas. El tratamiento de estos pacientes debe hacerse bajo la dirección de un médico con experiencia en el cuidado de la hemofilia y los inhibidores del factor VIII.
Se recomienda encarecidamente registrar el nombre y el número de lote del medicamento cada vez que se administre NovoEight a un paciente con el fin de mantener un vínculo entre el paciente y el lote del medicamento.
Consideraciones relacionadas con los excipientes
Después de la reconstitución, este medicamento contiene 0,31 mmol de sodio (7 mg) por ml de solución reconstituida. Esto deberá tenerse en cuenta en pacientes que sigan una dieta controlada en sodio.
Complicaciones relacionadas con el catéter
Si se requiere un dispositivo de acceso venoso central (DAVC), se debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, como por ejemplo, infecciones locales, bacteriemia y trombosis en el lugar de inserción del catéter.
Población pediátrica
Las advertencias y precauciones indicadas son aplicables a adultos y niños.
4.5 Interacción con otros medicamentos y otras formas de interacción No se han realizado estudios de interacciones con NovoEight.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción animal con NovoEight. Teniendo en cuenta la escasa ocurrencia de hemofilia A en mujeres, no existe experiencia en relación con el uso de factor VIII durante el embarazo y la lactancia. Por consiguiente, solo se deberá usar factor VIII durante el embarazo y la lactancia si está estrictamente indicado.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NovoEight sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Resumen del perfil de seguridad
Hipersensibilidad o reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el punto de perfusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habones, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, cosquilleo, vómitos, sibilancias) se han observado raramente y, en algunos casos, puede progresar hasta una anafilaxia grave (incluido shock).
En muy raras ocasiones se ha observado el desarrollo de anticuerpos frente a la proteína de hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizadores (inhibidores) del factor VIII. Si se generan inhibidores de este tipo, la situación se pondrá de manifiesto por una respuesta clínica insuficiente. En tales casos, se recomienda ponerse en contacto con un centro especializado en hemofilia.
T abla de reacciones adversas
La tabla que se muestra a continuación sigue la clasificación de sistemas de órganos de MedDRA (clasificación por órganos y sistemas (SOC) y nivel de término preferente).
Las frecuencias se han evaluado conforme a la convención siguiente: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); raras (> 1/10.000 a < 1/1.000); muy raras (< 1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
En cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
T abla 2 Frecuencia de los reacciones adversas en ensayos clínicos
|
Clasificación por órganos y sistemas |
Frecuencia* |
Reacción adversa |
|
Trastornos psiquiátricos |
Poco frecuentes |
Insomnio |
|
Trastornos del sistema nervioso |
Poco frecuentes |
Cefalea, mareo |
|
Trastornos cardiacos |
Poco frecuentes |
Taquicardia sinusal |
|
Trastornos vasculares |
Poco frecuentes |
Hipertensión, linfoedema |
|
Trastornos hepatobiliares |
Frecuentes |
Enzimas hepáticas elevadas** |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
Erupción |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Poco frecuentes |
Rigidez musculoesquelética, artropatía, dolor en las extremidades, dolor musculoesquelético |
|
Trastornos generales y alteraciones en el lugar de administración |
Frecuentes |
Reacciones en la zona de inyección*** |
|
Poco frecuentes |
Fatiga, sensación de calor, edema periférico, pirexia | |
|
Exploraciones complementarias |
Poco frecuentes |
Frecuencia cardiaca aumentada |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Poco frecuentes |
Contusión |
Cálculo basado en el número total de pacientes distintos en todos los estudios clínicos (214).
Entre las enzimas hepáticas elevadas se encuentran la aminotransferasa, aspartato aminotransferasa, gamma glutamiltransferasa y bilirrubina.
*** Entre las reacciones en la zona de inyección se encuentran eritema, extravasación y prurito en la zona de inyección.
Descripción de las reacciones adversas señaladas
Durante todos los estudios clínicos con NovoEight, se han notificado un total de 30 reacciones adversas en
19 de 214 pacientes expuestos a NovoEight. Las reacciones adversas de las que se informó con mayor frecuencia fueron reacciones en la zona de inyección y enzimas hepáticas elevadas. De las 30 reacciones adversas, 2 de ellas se notificaron en 1 de 31 pacientes de menos de 6 años de edad, ninguna entre los pacientes de 6 a 18 años de edad y 28 en 18 de 127 adultos.
Población pediátrica
En estudios clínicos con 63 pacientes pediátricos de entre 0 y 12 años de edad y 24 adolescentes entre 12 y 18 años de edad con hemofilia A grave, no se observó ninguna diferencia en el perfil de seguridad de NovoEight entre pacientes pediátricos y adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación
incluido en el Anexo V.
4.9 Sobredosis
No se han observado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos, factor VIII de la coagulación sanguínea, código ATC: B02BD02.
Mecanismo de acción
NovoEight contiene turoctocog alfa, un factor VIII de coagulación humano (ADNr) con un dominio B truncado. Esta glicoproteína tiene la misma estructura que el factor VIII cuando se activa, y modificaciones postranslacionales similares a las de la molécula derivada del plasma. Se ha observado que el punto de sulfatación de tirosina presente en Tyr1680 (longitud completa nativa), que es importante para el enlace con el factor de von Willebrand, está totalmente sulfatado en la molécula de turoctocog alfa. Cuando se administra mediante perfusión en un paciente con hemofilia, el factor VIII se enlaza con el factor de von Willebrand endógeno en la circulación del paciente. El complejo factor VIII/factor von Willebrand consiste en dos moléculas (factor VIII y factor de von Willebrand) con funciones fisiológicas distintas. El factor VIII activado actúa como cofactor del factor IX activado, y acelera la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo. La hemofilia es una alteración hereditaria vinculada al sexo de la coagulación de la sangre debida a niveles reducidos de factor VIII:C y da como resultado hemorragias difusas en articulaciones, músculos u órganos internos, ya sea de forma espontánea como tras un accidente o un trauma quirúrgico. La terapia de sustitución, aumenta los niveles plasmáticos de factor VIII, lo cual permite corregir temporalmente la deficiencia del factor y la tendencia al sangrado.
Eficacia clínica
Se han realizado tres ensayos multicéntricos, abiertos y no controlados para evaluar la seguridad y la eficacia de NovoEight para la prevención y el tratamiento de sangrado en pacientes tratados previamente con hemofilia A grave (actividad de FVEI < 1 %). Los ensayos incluyeron a 213 pacientes expuestos; 150 pacientes adolescentes o adultos sin inhibidores desde la edad de 12 años (> 150 días de exposición) y 63 pacientes pediátricos sin inhibidores de menos de 12 años de edad (> 50 días de exposición). 187 de 213 pacientes continuaron en el ensayo ampliado de seguridad. Se observó que el tratamiento con NovoEight es seguro y tiene el efecto hemostático y preventivo deseado. Durante una exposición acumulada de 54.000 días (correspondientes a 342 años paciente), no se observó el desarrollo de inhibidores del factor VIII en los ensayos clínicos en fase 3a con pacientes tratados previamente. De los 1.377 sangrados observados en 177 de los 213 pacientes, 1.244 (90,3 %) de los sangrados se resolvieron con 1 o 2 perfusiones de NovoEight.
|
Tabla 3 Consumo de turoctocog alfa y tasas de éxito glo |
bales | ||||
|
Niños pequeños (0 - <6 años) |
Niños mayores (6 - <12 años) |
Adolescentes (12 -<18 años) |
Adultos (>18 años) |
Total | |
|
Número de pacientes |
31 |
32 |
24 |
126 |
213 |
|
Dosis empleada como prevención por paciente (UI/kg PC) Valor medio (DE) Mín; Máx |
40,1 (8,5) 26,5 ; 57,3 |
36,6 (9,0) 24,9 ; 57,9 |
27,0 (7,6) 20,5 ; 46,9 |
26,9 (6,9) 20,0 ; 50,8 |
30,3 (9,2) 20,0 ; 57,9 |
|
Dosis utilizada para el tratamiento del sangrado (UI/kg BW) Valor medio (DE) Mín; Máx |
44,4 (17,9) 25,9 ; 193,8 |
40,0 (10,4) 25,5 ; 65,5 |
28,2 (10,2) 12,4 ; 76,8 |
33,8 (11,9) 9,3 ; 104,0 |
34,5 (12,6) 9,3 ; 193,8 |
|
Número de infusiones para el tratamiento del sangrado Valor medio (DE) Mín; Máx |
1,2 (0,6) 1 ; 6 |
1,5 (1,1) 1 ; 8 |
1,7 (1,3) 1 ; 9 |
1,5 (2,4) 1 ; 49 |
1,5 (2,1) 1 ; 49 |
|
T asa de éxito* % |
92,9 % |
88,9 % |
79,7 % |
85,6 % |
85,9 % |
PC: Peso corporal, DE: Desviación estándar *El éxito se define como “Excelente” o “Bueno”.
En total se realizaron 14 intervenciones quirúrgicas en 14 pacientes, de las cuales 13 fueron de cirugía mayor y 1 de cirugía menor. La hemostasis fue correcta en todas las intervenciones quirúrgicas y no se observó ningún fallo de tratamiento.
5.2 Propiedades farmacocinéticas
T odos los estudios de farmacocinética con turoctocog alfa se realizaron en pacientes tratados previamente con hemofilia A grave (FVHI < 1 %). El análisis de las muestras de plasma se realizó mediante análisis de coagulación de una fase y análisis cromogénico.
En un estudio internacional que implicó a 36 laboratorios, se evaluó el rendimiento de NovoEight en ensayos de FVHI:C y se comparó con un medicamento comercializado de FVIII recombinante de longitud completa. El estudio reveló que se obtienen resultados comparables y coherentes con ambos productos y que NovoEight se puede medir en plasma de forma fiable sin necesidad de usar un estándar aparte para NovoEight.
Los parámetros farmacocinéticos de NovoEight en una sola dosis se indican en la T abla 4 para el análisis de coagulación y en la Tabla 5 para el análisis cromogénico.
Tabla 4 Farmacocinética de una sola dosis de turoctocog alfa en pacientes con hemofilia A grave (FVIII < 1 %), análisis de coagulación___
|
Parámetro |
0 - <6 años |
6 - <12 años |
>12 años |
|
n = 14 |
n = 14 |
n = 33 | |
|
Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) | |
|
Recuperación incremental (UI/ml)/(UI/kg) |
0,018 (0,007) |
0,020 (0,004) |
0,022 (0,004) |
|
AUC ((UI*h)/ml) |
9,92 (4,11) |
11,09 (3,74) |
15,26 (5,77) |
|
CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
|
t/ (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
|
Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
|
Cmax (UI/ml) |
1,00 (0,58) |
1,07 (0,35) |
1,226 (0,41) |
|
Tiempo medio de residencia (h) |
9,63 (2,50) |
9,91 (2,57) |
14,19 (5,08) |
Tabla 5 Farmacocinética de una sola dosis de turoctocog alfa en pacientes con hemofilia A grave (FVIII < 1 %), análisis cromogénico___
|
Parámetro |
0 - <6 años |
6 - <12 años |
>12 años |
|
n = 14 |
n = 14 |
n = 33 | |
|
Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) | |
|
Recuperación incremental (UI/ml)/(UI/kg) |
0,022 (0,006) |
0,025 (0,006) |
0,029 (0,006) |
|
AUC ((UI*h)/ml) |
12,23 (4,36) |
14,37 (3,48) |
19,63 (7,73) |
|
CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
|
t/ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
|
Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
|
Cmax (UI/ml) |
1,12 (0,31) |
1,25 (0,27) |
1,63 (0,50) |
|
Tiempo medio de residencia (h) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
Los parámetros farmacocinéticos fueron comparables entre pacientes pediátricos de menos de 6 años de edad y pacientes pediátricos mayores de 6 y menores de 12 años de edad. Se observó alguna variación entre los parámetros farmacocinéticos de NovoEight en pacientes pediátricos y adultos. Los valores mayores de CL y menores de t./2 en pacientes pediátricos en comparación con pacientes adultos con hemofilia A se pueden deber en parte al hecho conocido de que el volumen de plasma por kilogramo de peso corporal es mayor en los pacientes más jóvenes.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad y toxicidad a dosis repetidas.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Cloruro de sodio L-histidina Sacarosa Polisorbato 80 L-metionina
Cloruro de calcio dihidratado Hidróxido de sodio Ácido clorhídrico
Disolvente:
Cloruro de sodio
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
Sin abrir:
30 meses
Durante el periodo de validez, el medicamento se puede almacenar a temperatura ambiente (<30°C) durante un solo periodo de hasta 9 meses. Una vez que el producto se saca de la nevera, no se debe volver a refrigerar. Registrar la fecha de inicio del almacenamiento a temperatura ambiente en el embalaje del producto.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Después de la reconstitución:
Se ha demostrado la estabilidad química y física en uso durante 24 horas almacenado entre 2°C y 8°C y 4 horas almacenado a temperatura ambiente (<30°C).
Desde el punto de vista microbiológico, el medicamento se debería usar inmediatamente después de la reconstitución. Si no se utiliza de inmediato, los tiempos y las condiciones de almacenamiento en uso antes del uso son responsabilidad del usuario y no deberían superarse las 4 horas a temperatura ambiente (<30°C) o 24 horas entre 2°C y 8°C, a menos que la reconstitución se haya realizado en condiciones asépticas controladas y validadas.
El medicamento no utilizado almacenado a temperatura ambiente durante más de 4 horas se deberá desechar.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C). No congelar.
Para consultar la información de almacenamiento a temperatura ambiente y las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Cada envase de NovoEight 500 UI polvo y disolvente para solución inyectable contiene:
- 1 vial de vidrio (tipo I) con polvo y un tapón de goma de clorobutilo
- 1 adaptador de vial estéril para la reconstitución
- 1 jeringa precargada con 4 ml de disolvente con mecanismo de protección (polipropileno), un émbolo
de goma (bromobutilo) y un capuchón de aguja con tapón (bromobutilo)
- 1 varilla del émbolo (polipropileno).
6.6 Precauciones especiales de eliminación y otras manipulaciones
NovoEight se debe administrar por vía intravenosa después de la reconstitución del polvo con el disolvente suministrado en la jeringa. Después de la reconstitución, la solución es transparente o ligeramente opalescente. No utilice la solución si tiene aspecto turbio o presenta depósitos.
También necesitará un conjunto de perfusión (tubos y aguja mariposa), toallitas estériles con alcohol, gasas y tiritas. Estos dispositivos no se incluyen en el paquete NovoEight.
Utilice siempre una técnica aséptica.
Reconstitución
A)
Saque el vial, el adaptador del vial y la jeringa precargada del envase. Deje la varilla del émbolo sin tocar en el envase. Lleve el vial y la jeringa precargada a temperatura ambiente. Puede hacerlo manteniendo ambos en las manos hasta que sienta que están a la misma temperatura que sus manos. No utilice ningún otro sistema para calentar el vial y la jeringa precargada.
|
0/ tv |
“1 l\ .”-J ■ \^J | ||
|
3 | |||
|
ñ | |||
B)
Quite el capuchón de plástico del vial. Si el capuchón de plástico está flojo o falta, no utilice el vial. Limpie el tapón de goma del vial con una toallita estéril con alcohol y deje que se seque al aire unos segundos antes de usarlo.
C)
Quite el precinto de papel del adaptador del vial. Si el precinto de papel no está totalmente sellado o si está roto, no utilice el adaptador del vial. No saque el adaptador del vial del capuchón protector con los dedos.
D)
Ponga el capuchón protector del adaptador del vial boca abajo y coloque el adaptador del vial a presión sobre el vial. Una vez unido, no retire del vial el adaptador del vial.
E)
Comprima ligeramente el capuchón protector entre los dedos pulgar e índice tal como se muestra. Quite el capuchón protector del adaptador del vial.
F)
Sujete la varilla del émbolo por el extremo ancho e inmediatamente conéctelo a la jeringa girándolo hacia la derecha dentro del émbolo en el interior de la jeringa precargada hasta que se sienta resistencia.

G)
Quite el tapón de la jeringa precargada doblándolo hacia abajo hasta que se rompa la perforación. No toque la punta de la jeringa debajo del tapón de la jeringa.
H)
Enrosque la jeringa precargada con firmeza en el adaptador del vial hasta que se sienta resistencia.
I)
Sujete la jeringa precargada ligeramente inclinada con el vial apuntando hacia abajo. Presione la varilla del émbolo para inyectar todo el disolvente en el vial.
J)
Mantenga la varilla del vial presionada y remueva suavemente el vial hasta que el polvo se haya disuelto. No agite el vial, ya que esto produciría espuma.
Se recomienda utilizar NovoEight inmediatamente después de la reconstitución. Para consultar las condiciones de conservación del medicamento reconstituido, ver sección 6.3.
Si se necesita una dosis mayor, repita los pasos A a J con más viales, adaptadores de viales y jeringas precargadas.
Administración de la solución reconstituida
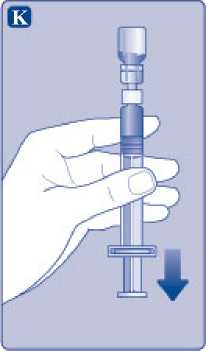
K)
Mantenga la varilla del émbolo totalmente empujada hacia el interior. Gire la jeringa con el vial boca abajo. Deje de empujar la varilla del émbolo y deje que retroceda por sí misma mientras la solución reconstituida llena la jeringa. T ire de la varilla del émbolo ligeramente hacia abajo para hacer pasar la solución reconstituida hacia la jeringa.
Si solo necesita una parte del vial, utilice la escala de la jeringa para ver la cantidad de solución reconstituida que ha retirado, tal como le haya indicado su médico o su enfermera.
Mientras sujeta el vial cabeza abajo, golpee suavemente la jeringa para que las posibles burbujas suban hasta la parte superior. Empuje la varilla del émbolo lentamente hasta que se hayan eliminado las burbujas.
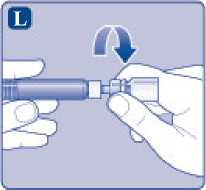
L)
Desenrosque el adaptador del vial con el vial.
Ahora NovoEight está listo para ser inyectado. Busque un lugar adecuado e inyecte lentamente NovoEight en la vena en un periodo de 2 a 5 minutos.
Eliminación
Después de la inyección, deseche con seguridad toda la solución NovoEight no utilizada, la jeringa con el juego de perfusión, el vial con el adaptador del vial y los demás residuos siguiendo las indicaciones de su farmacéutico.
No los deseche como residuos domésticos ordinarios.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novo Nordisk A/S
Novo Allé DK-2880 Bagsv^rd Dinamarca
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/888/002
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 13 Noviembre 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
^Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
NovoEight 1.000 UI polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial de polvo contiene 1.000 UI de factor VIII de coagulación humano (ADNr), turoctocog alfa.
NovoEight contiene aproximadamente 250 UI/ml de factor VIII de coagulación humano (ADNr), turoctocog alfa, después de la reconstitución.
La potencia (UI) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de NovoEight es aproximadamente de 8.300 UI/mg de proteína.
Turoctocog alfa (factor VIII de coagulación humano (ADNr)) es una proteína purificada que contiene 1.445 aminoácidos con una masa molecular aproximada de 166 kDA. Se produce mediante tecnología de ADN recombinante en células ováricas de hámster chino (CHO) y se prepara sin añadir ninguna proteína humana ni derivada de animal durante el proceso de cultivo de las células, la purificación o la formulación final.
Turoctocog alfa es un factor VIII de coagulación humano recombinante truncado de dominio B (el dominio B consiste en 21 aminoácidos del dominio silvestre de tipo B) sin ninguna otra modificación de la secuencia de aminoácidos.
Excipiente con efecto conocido:
0,31 mmol de sodio (7 mg) por ml de solución reconstituida.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo o masa friable de color blanco o ligeramente amarillo. Solución inyectable transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
T ratamiento y profilaxis de hemorragias en pacientes con hemofilia A (deficiencia congénita del factor VIII). NovoEight se puede usar en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de hemofilia.
Pacientes no tratados previamente
No se ha establecido todavía la seguridad y la eficacia de NovoEight en pacientes no tratados previamente.
No se dispone de datos.
Posología
La dosis y duración de la terapia de sustitución dependen de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que se corresponden con el estándar actual de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa como porcentaje (relativo al nivel en plasma humano normal) o en Unidades Internacionales (relativas al estándar internacional para el factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII presente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor VIII se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad plasmática de factor VIII en 2 UI/dl. La dosis requerida se determina utilizando la fórmula siguiente:
UI requeridas = peso corporal (kg) * aumento deseado de factor VIII (%) (UI/dl) x 0,5 (UI/kg por UI/dl).
La dosis y la frecuencia de la administración deberá individualizarse según la eficacia clínica en cada caso en particular.
En el caso de los eventos hemorrágicos siguientes, la actividad del factor VIII no deberá bajar del nivel de actividad plasmática indicado (en % del normal o UI/dl) durante el periodo correspondiente. La tabla siguiente se puede usar como guía para determinar la dosis en episodios hemorrágicos y cirugía:
T abla 1 Guía para determinar la dosis en episodios hemorrágicos y cirugía
Grado de la hemorragia/Tipo de Nivel de factor VIII Frecuencia de dosificación
procedimiento quirúrgico necesario (%) (UI/dl) (horas)/Duración del tratamiento
(días)
Hemorragia
Hemartrosis precoz, sangrado muscular 20-40 Repetir cada 12 - 24 horas al menos 1
o sangrado de la cavidad oral día hasta que el episodio hemorrágico
se haya resuelto, en función del dolor, o hasta la cicatrización de la herida.
Hemartrosis más extensa, sangrado 30-60 muscular o hematoma
Hemorragias con riesgo vital 60-100
Repetir la perfusión cada 12 - 24 horas durante 3 - 4 días o hasta que el dolor y la discapacidad aguda se hayan resuelto.
Repetir la perfusión cada 8 - 24 horas hasta que el riesgo desaparezca.
Cirugía
Cirugía menor, incluidas las 30-60 Cada 24 horas, al menos 1 día, hasta
extracciones dentales la cicatrización.
Grado de la hemorragia/Tipo de Nivel de factor VIII Frecuencia de dosificación
procedimiento quirúrgico necesario (%) (UI/dl) (horas)/Duración del tratamiento
(días)
Cirugía mayor 80-100 Repetir la perfusión cada 8-24 horas
(pre- y postoperatorio) hasta que se consiga una cicatrización
adecuada. A continuación, tratamiento durante por lo menos 7 días más para mantener una actividad del factor VIII del 30 % al 60 % (UI/dl).
Profilaxis
En la profilaxis a largo plazo para prevenir hemorragias en pacientes con hemofilia A grave. Las dosis habituales recomendadas son de 20 a 40 UI de factor VIII por kg de peso corporal a días alternos o de 20 a 50 UI de factor VIII por kg de peso corporal 3 veces por semana. En algunos casos, especialmente en los pacientes más jóvenes, puede ser necesario un intervalo de dosis más corto o una dosis mayor.
Supervisión del tratamiento
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis a administrar y la frecuencia de las inyecciones repetidas. Especialmente en el caso de las intervenciones de cirugía mayor, es indispensable controlar con precisión la terapia de sustitución mediante pruebas de coagulación (actividad plasmática del factor VIII). La respuesta individual de cada paciente frente al factor VIII puede variar y alcanzar distintos niveles de recuperación in vivo, y presentar semividas diferentes.
Cirugía
No existe experiencia en cirugía en pacientes pediátricos.
Pacientes de edad avanzada
No existe experiencia en pacientes de más de 65 años.
Población pediátrica
En la profilaxis a largo plazo para prevenir hemorragias en pacientes de menos de 12 años, las dosis recomendadas son de 25 a 50 UI de factor VIII por kg de peso corporal a días alternos o de 25 a 60 UI de factor VIII por kg de peso corporal 3 veces por semana. En pacientes pediátricos de más de 12 años, la dosis recomendada es la misma que para los adultos.
Forma de administración Vía intravenosa.
La velocidad de perfusión recomendada para NovoEight es de 1 - 2 ml/min. La velocidad se deberá determinar de acuerdo con el nivel de confort del paciente.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Reacciones alérgicas conocidas a las proteínas de hámster.
4.4 Advertencias y precauciones especiales de empleo Hipersensibilidad
Es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico a NovoEight. El producto contiene trazas de proteínas de hámster, las cuales pueden provocar reacciones alérgicas en algunos
30
pacientes. Si se produjesen síntomas de hipersensibilidad, se deberá instruir a los pacientes que deben interrumpir el uso del medicamento y ponerse inmediatamente en contacto con su médico. Se debe informar a los pacientes de los síntomas iniciales de las reacciones de hipersensibilidad, como urticaria localizada o generalizada, opresión en el pecho, respiración sibilante, hipotensión y anafilaxia.
En caso de shock, se seguirán las pautas médicas habituales para su tratamiento.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, y se cuantifican en Unidades Bethesda (UB) por ml de plasma usando el ensayo modificado. El riesgo de desarrollo de inhibidores está correlacionado con la exposición al factor VIII, y el riesgo mayor se produce durante los primeros 20 días de exposición. Los inhibidores raramente se forman después de los primeros 100 días de tratamiento.
Se han observado casos recurrentes de inhibidores (con título bajo), después de cambiar de un medicamento con factor VIII a otro a pacientes que habían recibido un tratamiento previo de más de 100 días de exposición y que tienen antecedentes de desarrollo de inhibidores. Por consiguiente se recomienda controlar a todos los pacientes atentamente para determinar la presencia de inhibidores después de cualquier cambio de medicamento.
En general, se deberá controlar atentamente a todos los pacientes tratados con medicamentos con factor VIII para determinar el desarrollo de inhibidores mediante la observación clínica y las pruebas de laboratorio adecuadas. Si no se obtienen los niveles plasmáticos de actividad del factor VIII esperados, o si no se controla la hemorragia con una dosis adecuada, se deberán realizar análisis para detectar la presencia de inhibidores del factor VIII. En pacientes con niveles elevados de inhibidores, es posible que el tratamiento con factor VIII no sea eficaz y se deberán considerar otras opciones terapéuticas. El tratamiento de estos pacientes debe hacerse bajo la dirección de un médico con experiencia en el cuidado de la hemofilia y los inhibidores del factor VIII.
Se recomienda encarecidamente registrar el nombre y el número de lote del medicamento cada vez que se administre NovoEight a un paciente con el fin de mantener un vínculo entre el paciente y el lote del medicamento.
Consideraciones relacionadas con los excipientes
Después de la reconstitución, este medicamento contiene 0,31 mmol de sodio (7 mg) por ml de solución reconstituida. Esto deberá tenerse en cuenta en pacientes que sigan una dieta controlada en sodio.
Complicaciones relacionadas con el catéter
Si se requiere un dispositivo de acceso venoso central (DAVC), se debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, como por ejemplo, infecciones locales, bacteriemia y trombosis en el lugar de inserción del catéter.
Población pediátrica
Las advertencias y precauciones indicadas son aplicables a adultos y niños.
4.5 Interacción con otros medicamentos y otras formas de interacción No se han realizado estudios de interacciones con NovoEight.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción animal con NovoEight. Teniendo en cuenta la escasa ocurrencia de hemofilia A en mujeres, no existe experiencia en relación con el uso de factor VIII durante el embarazo y la lactancia. Por consiguiente, solo se deberá usar factor VIII durante el embarazo y la lactancia si está estrictamente indicado.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NovoEight sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Resumen del perfil de seguridad
Hipersensibilidad o reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el punto de perfusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habones, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, cosquilleo, vómitos, sibilancias) se han observado raramente y, en algunos casos, puede progresar hasta una anafilaxia grave (incluido shock).
En muy raras ocasiones se ha observado el desarrollo de anticuerpos frente a la proteína de hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizadores (inhibidores) del factor VIII. Si se generan inhibidores de este tipo, la situación se pondrá de manifiesto por una respuesta clínica insuficiente. En tales casos, se recomienda ponerse en contacto con un centro especializado en hemofilia.
T abla de reacciones adversas
La tabla que se muestra a continuación sigue la clasificación de sistemas de órganos de MedDRA (clasificación por órganos y sistemas (SOC) y nivel de término preferente).
Las frecuencias se han evaluado conforme a la convención siguiente: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); raras (> 1/10.000 a < 1/1.000); muy raras (< 1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
En cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
T abla 2 Frecuencia de los reacciones adversas en ensayos clínicos
|
Clasificación por órganos y sistemas |
Frecuencia* |
Reacción adversa |
|
Trastornos psiquiátricos |
Poco frecuentes |
Insomnio |
|
Trastornos del sistema nervioso |
Poco frecuentes |
Cefalea, mareo |
|
Trastornos cardiacos |
Poco frecuentes |
Taquicardia sinusal |
|
Trastornos vasculares |
Poco frecuentes |
Hipertensión, linfoedema |
|
Trastornos hepatobiliares |
Frecuentes |
Enzimas hepáticas elevadas** |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
Erupción |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Poco frecuentes |
Rigidez musculoesquelética, artropatía, dolor en las extremidades, dolor musculoesquelético |
|
Trastornos generales y alteraciones en el lugar de administración |
Frecuentes |
Reacciones en la zona de inyección*** |
|
Poco frecuentes |
Fatiga, sensación de calor, edema periférico, pirexia | |
|
Exploraciones complementarias |
Poco frecuentes |
Frecuencia cardiaca aumentada |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Poco frecuentes |
Contusión |
Cálculo basado en el número total de pacientes distintos en todos los estudios clínicos (214).
Entre las enzimas hepáticas elevadas se encuentran la aminotransferasa, aspartato aminotransferasa, gamma glutamiltransferasa y bilirrubina.
*** Entre las reacciones en la zona de inyección se encuentran eritema, extravasación y prurito en la zona de inyección.
Descripción de las reacciones adversas señaladas
Durante todos los estudios clínicos con NovoEight, se han notificado un total de 30 reacciones adversas en
19 de 214 pacientes expuestos a NovoEight. Las reacciones adversas de las que se informó con mayor frecuencia fueron reacciones en la zona de inyección y enzimas hepáticas elevadas. De las 30 reacciones adversas, 2 de ellas se notificaron en 1 de 31 pacientes de menos de 6 años de edad, ninguna entre los pacientes de 6 a 18 años de edad y 28 en 18 de 127 adultos.
Población pediátrica
En estudios clínicos con 63 pacientes pediátricos de entre 0 y 12 años de edad y 24 adolescentes entre 12 y 18 años de edad con hemofilia A grave, no se observó ninguna diferencia en el perfil de seguridad de NovoEight entre pacientes pediátricos y adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han observado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos, factor VIII de la coagulación sanguínea, código ATC: B02BD02.
Mecanismo de acción
NovoEight contiene turoctocog alfa, un factor VIII de coagulación humano (ADNr) con un dominio B truncado. Esta glicoproteína tiene la misma estructura que el factor VIII cuando se activa, y modificaciones postranslacionales similares a las de la molécula derivada del plasma. Se ha observado que el punto de sulfatación de tirosina presente en Tyr1680 (longitud completa nativa), que es importante para el enlace con el factor de von Willebrand, está totalmente sulfatado en la molécula de turoctocog alfa. Cuando se administra mediante perfusión en un paciente con hemofilia, el factor VIII se enlaza con el factor de von Willebrand endógeno en la circulación del paciente. El complejo factor VIII/factor von Willebrand consiste en dos moléculas (factor VIII y factor de von Willebrand) con funciones fisiológicas distintas. El factor VIII activado actúa como cofactor del factor IX activado, y acelera la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo. La hemofilia es una alteración hereditaria vinculada al sexo de la coagulación de la sangre debida a niveles reducidos de factor VIII:C y da como resultado hemorragias difusas en articulaciones, músculos u órganos internos, ya sea de forma espontánea como tras un accidente o un trauma quirúrgico. La terapia de sustitución, aumenta los niveles plasmáticos de factor VIII, lo cual permite corregir temporalmente la deficiencia del factor y la tendencia al sangrado.
Eficacia clínica
Se han realizado tres ensayos multicéntricos, abiertos y no controlados para evaluar la seguridad y la eficacia de NovoEight para la prevención y el tratamiento de sangrado en pacientes tratados previamente con hemofilia A grave (actividad de FVEI < 1 %). Los ensayos incluyeron a 213 pacientes expuestos; 150 pacientes adolescentes o adultos sin inhibidores desde la edad de 12 años (> 150 días de exposición) y 63 pacientes pediátricos sin inhibidores de menos de 12 años de edad (> 50 días de exposición). 187 de 213 pacientes continuaron en el ensayo ampliado de seguridad. Se observó que el tratamiento con NovoEight es seguro y tiene el efecto hemostático y preventivo deseado. Durante una exposición acumulada de 54.000 días (correspondientes a 342 años paciente), no se observó el desarrollo de inhibidores del factor VIII en los ensayos clínicos en fase 3a con pacientes tratados previamente. De los 1.377 sangrados observados en 177 de los 213 pacientes, 1.244 (90,3 %) de los sangrados se resolvieron con 1 o 2 perfusiones de NovoEight.
|
Tabla 3 Consumo de turoctocog alfa y tasas de éxito glo |
bales | ||||
|
Niños pequeños (0 - <6 años) |
Niños mayores (6 - <12 años) |
Adolescentes (12 -<18 años) |
Adultos (>18 años) |
Total | |
|
Número de pacientes |
31 |
32 |
24 |
126 |
213 |
|
Dosis empleada como prevención por paciente (UI/kg PC) Valor medio (DE) Mín; Máx |
40,1 (8,5) 26,5 ; 57,3 |
36,6 (9,0) 24,9 ; 57,9 |
27,0 (7,6) 20,5 ; 46,9 |
26,9 (6,9) 20,0 ; 50,8 |
30,3 (9,2) 20,0 ; 57,9 |
|
Dosis utilizada para el tratamiento del sangrado (UI/kg BW) Valor medio (DE) Mín; Máx |
44,4 (17,9) 25,9 ; 193,8 |
40,0 (10,4) 25,5 ; 65,5 |
28,2 (10,2) 12,4 ; 76,8 |
33,8 (11,9) 9,3 ; 104,0 |
34,5 (12,6) 9,3 ; 193,8 |
|
Número de infusiones para el tratamiento del sangrado Valor medio (DE) Mín; Máx |
1,2 (0,6) 1 ; 6 |
1,5 (1,1) 1 ; 8 |
1,7 (1,3) 1 ; 9 |
1,5 (2,4) 1 ; 49 |
1,5 (2,1) 1 ; 49 |
|
T asa de éxito* % |
92,9 % |
88,9 % |
79,7 % |
85,6 % |
85,9 % |
PC: Peso corporal, DE: Desviación estándar *El éxito se define como “Excelente” o “Bueno”.
En total se realizaron 14 intervenciones quirúrgicas en 14 pacientes, de las cuales 13 fueron de cirugía mayor y 1 de cirugía menor. La hemostasis fue correcta en todas las intervenciones quirúrgicas y no se observó ningún fallo de tratamiento.
5.2 Propiedades farmacocinéticas
T odos los estudios de farmacocinética con turoctocog alfa se realizaron en pacientes tratados previamente con hemofilia A grave (FVHI < 1 %). El análisis de las muestras de plasma se realizó mediante análisis de coagulación de una fase y análisis cromogénico.
En un estudio internacional que implicó a 36 laboratorios, se evaluó el rendimiento de NovoEight en ensayos de FVHI:C y se comparó con un medicamento comercializado de FVIII recombinante de longitud completa. El estudio reveló que se obtienen resultados comparables y coherentes con ambos productos y que NovoEight se puede medir en plasma de forma fiable sin necesidad de usar un estándar aparte para NovoEight.
Los parámetros farmacocinéticos de NovoEight en una sola dosis se indican en la T abla 4 para el análisis de coagulación y en la Tabla 5 para el análisis cromogénico.
Tabla 4 Farmacocinética de una sola dosis de turoctocog alfa en pacientes con hemofilia A grave (FVIII < 1 %), análisis de coagulación___
|
Parámetro |
0 - <6 años |
6 - <12 años |
>12 años |
|
n = 14 |
n = 14 |
n = 33 | |
|
Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) | |
|
Recuperación incremental (UI/ml)/(UI/kg) |
0,018 (0,007) |
0,020 (0,004) |
0,022 (0,004) |
|
AUC ((UI*h)/ml) |
9,92 (4,11) |
11,09 (3,74) |
15,26 (5,77) |
|
CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
|
t/ (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
|
Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
|
Cmax (UI/ml) |
1,00 (0,58) |
1,07 (0,35) |
1,226 (0,41) |
|
Tiempo medio de residencia (h) |
9,63 (2,50) |
9,91 (2,57) |
14,19 (5,08) |
Tabla 5 Farmacocinética de una sola dosis de turoctocog alfa en pacientes con hemofilia A grave (FVIII < 1 %), análisis cromogénico___
|
Parámetro |
0 - <6 años |
6 - <12 años |
>12 años |
|
n = 14 |
n = 14 |
n = 33 | |
|
Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) | |
|
Recuperación incremental (UI/ml)/(UI/kg) |
0,022 (0,006) |
0,025 (0,006) |
0,029 (0,006) |
|
AUC ((UI*h)/ml) |
12,23 (4,36) |
14,37 (3,48) |
19,63 (7,73) |
|
CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
|
t/ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
|
Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
|
Cmax (UI/ml) |
1,12 (0,31) |
1,25 (0,27) |
1,63 (0,50) |
|
Tiempo medio de residencia (h) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
Los parámetros farmacocinéticos fueron comparables entre pacientes pediátricos de menos de 6 años de edad y pacientes pediátricos mayores de 6 y menores de 12 años de edad. Se observó alguna variación entre los parámetros farmacocinéticos de NovoEight en pacientes pediátricos y adultos. Los valores mayores de CL y menores de t./2 en pacientes pediátricos en comparación con pacientes adultos con hemofilia A se pueden deber en parte al hecho conocido de que el volumen de plasma por kilogramo de peso corporal es mayor en los pacientes más jóvenes.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad y toxicidad a dosis repetidas.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Cloruro de sodio L-histidina Sacarosa Polisorbato 80 L-metionina
Cloruro de calcio dihidratado Hidróxido de sodio Ácido clorhídrico
Disolvente:
Cloruro de sodio
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
Sin abrir:
30 meses
Durante el periodo de validez, el medicamento se puede almacenar a temperatura ambiente (<30°C) durante un solo periodo de hasta 9 meses. Una vez que el producto se saca de la nevera, no se debe volver a refrigerar. Registrar la fecha de inicio del almacenamiento a temperatura ambiente en el embalaje del producto.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Después de la reconstitución:
Se ha demostrado la estabilidad química y física en uso durante 24 horas almacenado entre 2°C y 8°C y 4 horas almacenado a temperatura ambiente (<30°C).
Desde el punto de vista microbiológico, el medicamento se debería usar inmediatamente después de la reconstitución. Si no se utiliza de inmediato, los tiempos y las condiciones de almacenamiento en uso antes del uso son responsabilidad del usuario y no deberían superarse las 4 horas a temperatura ambiente (<30°C) o 24 horas entre 2°C y 8°C, a menos que la reconstitución se haya realizado en condiciones asépticas controladas y validadas.
El medicamento no utilizado almacenado a temperatura ambiente durante más de 4 horas se deberá desechar.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C). No congelar.
Para consultar la información de almacenamiento a temperatura ambiente y las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Cada envase de NovoEight 1.000 UI polvo y disolvente para solución inyectable contiene:
- 1 vial de vidrio (tipo I) con polvo y un tapón de goma de clorobutilo
- 1 adaptador de vial estéril para la reconstitución
- 1 jeringa precargada con 4 ml de disolvente con mecanismo de protección (polipropileno), un émbolo
de goma (bromobutilo) y un capuchón de aguja con tapón (bromobutilo)
- 1 varilla del émbolo (polipropileno).
6.6 Precauciones especiales de eliminación y otras manipulaciones
NovoEight se debe administrar por vía intravenosa después de la reconstitución del polvo con el disolvente suministrado en la jeringa. Después de la reconstitución, la solución es transparente o ligeramente opalescente. No utilice la solución si tiene aspecto turbio o presenta depósitos.
También necesitará un conjunto de perfusión (tubos y aguja mariposa), toallitas estériles con alcohol, gasas y tiritas. Estos dispositivos no se incluyen en el paquete NovoEight.
Utilice siempre una técnica aséptica.
Reconstitución
A)
Saque el vial, el adaptador del vial y la jeringa precargada del envase. Deje la varilla del émbolo sin tocar en el envase. Lleve el vial y la jeringa precargada a temperatura ambiente. Puede hacerlo manteniendo ambos en las manos hasta que sienta que están a la misma temperatura que sus manos. No utilice ningún otro sistema para calentar el vial y la jeringa precargada.
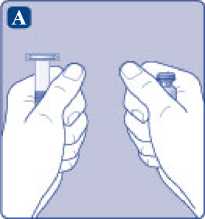

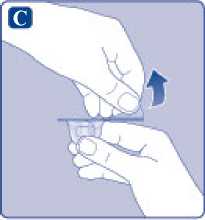

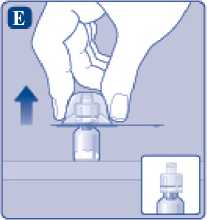
B)
Quite el capuchón de plástico del vial. Si el capuchón de plástico está flojo o falta, no utilice el vial. Limpie el tapón de goma del vial con una toallita estéril con alcohol y deje que se seque al aire unos segundos antes de usarlo.
C)
Quite el precinto de papel del adaptador del vial. Si el precinto de papel no está totalmente sellado o si está roto, no utilice el adaptador del vial. No saque el adaptador del vial del capuchón protector con los dedos.
D)
Ponga el capuchón protector del adaptador del vial boca abajo y coloque el adaptador del vial a presión sobre el vial. Una vez unido, no retire del vial el adaptador del vial.
E)
Comprima ligeramente el capuchón protector entre los dedos pulgar e índice tal como se muestra. Quite el capuchón protector del adaptador del vial.
F)
Sujete la varilla del émbolo por el extremo ancho e inmediatamente conéctelo a la jeringa
girándolo hacia la derecha dentro del émbolo en el interior de la jeringa precargada hasta que se sienta resistencia.
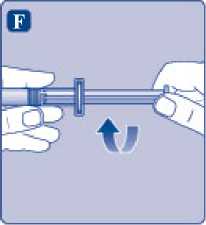
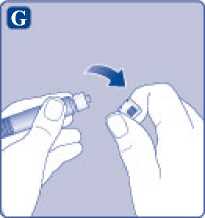
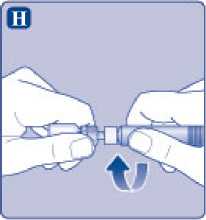
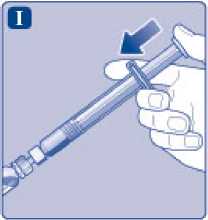
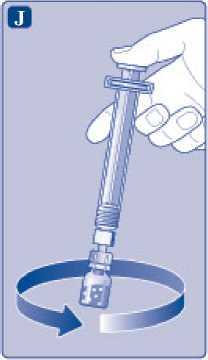
G)
Quite el tapón de la jeringa precargada doblándolo hacia abajo hasta que se rompa la perforación. No toque la punta de la jeringa debajo del tapón de la jeringa.
H)
Enrosque la jeringa precargada con firmeza en el adaptador del vial hasta que se sienta resistencia.
I)
Sujete la jeringa precargada ligeramente inclinada con el vial apuntando hacia abajo. Presione la varilla del émbolo para inyectar todo el disolvente en el vial.
J)
Mantenga la varilla del vial presionada y remueva suavemente el vial hasta que el polvo se haya disuelto. No agite el vial, ya que esto produciría espuma.
Se recomienda utilizar NovoEight inmediatamente después de la reconstitución. Para consultar las condiciones de conservación del medicamento reconstituido, ver sección 6.3.
Si se necesita una dosis mayor, repita los pasos A a J con más viales, adaptadores de viales y jeringas precargadas.
Administración de la solución reconstituida
K)
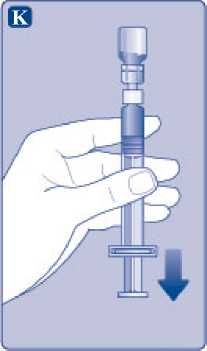
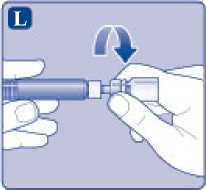
Mantenga la varilla del émbolo totalmente empujada hacia el interior. Gire la jeringa con el vial boca abajo. Deje de empujar la varilla del émbolo y deje que retroceda por sí misma mientras la solución reconstituida llena la jeringa. T ire de la varilla del émbolo ligeramente hacia abajo para hacer pasar la solución reconstituida hacia la jeringa.
Si solo necesita una parte del vial, utilice la escala de la jeringa para ver la cantidad de solución reconstituida que ha retirado, tal como le haya indicado su médico o su enfermera.
Mientras sujeta el vial cabeza abajo, golpee suavemente la jeringa para que las posibles burbujas suban hasta la parte superior. Empuje la varilla del émbolo lentamente hasta que se hayan eliminado las burbujas.
L)
Desenrosque el adaptador del vial con el vial.
Ahora NovoEight está listo para ser inyectado. Busque un lugar adecuado e inyecte lentamente NovoEight en la vena en un periodo de 2 a 5 minutos.
Eliminación
Después de la inyección, deseche con seguridad toda la solución NovoEight no utilizada, la jeringa con el juego de perfusión, el vial con el adaptador del vial y los demás residuos siguiendo las indicaciones de su farmacéutico.
No los deseche como residuos domésticos ordinarios.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novo Nordisk A/S
Novo Allé DK-2880 Bagsv^rd Dinamarca
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/888/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 13 Noviembre 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
^Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
NovoEight 1.500 UI polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial de polvo contiene 1.500 UI de factor VIII de coagulación humano (ADNr), turoctocog alfa.
NovoEight contiene aproximadamente 375 UI/ml de factor VIII de coagulación humano (ADNr), turoctocog alfa, después de la reconstitución.
La potencia (UI) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de NovoEight es aproximadamente de 8.300 UI/mg de proteína.
Turoctocog alfa (factor VIII de coagulación humano (ADNr)) es una proteína purificada que contiene 1.445 aminoácidos con una masa molecular aproximada de 166 kDA. Se produce mediante tecnología de ADN recombinante en células ováricas de hámster chino (CHO) y se prepara sin añadir ninguna proteína humana ni derivada de animal durante el proceso de cultivo de las células, la purificación o la formulación final.
Turoctocog alfa es un factor VIII de coagulación humano recombinante truncado de dominio B (el dominio B consiste en 21 aminoácidos del dominio silvestre de tipo B) sin ninguna otra modificación de la secuencia de aminoácidos.
Excipiente con efecto conocido:
0,31 mmol de sodio (7 mg) por ml de solución reconstituida.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo o masa friable de color blanco o ligeramente amarillo. Solución inyectable transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
T ratamiento y profilaxis de hemorragias en pacientes con hemofilia A (deficiencia congénita del factor VIII). NovoEight se puede usar en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de hemofilia.
Pacientes no tratados previamente
No se ha establecido todavía la seguridad y la eficacia de NovoEight en pacientes no tratados previamente.
No se dispone de datos.
Posología
La dosis y duración de la terapia de sustitución dependen de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que se corresponden con el estándar actual de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa como porcentaje (relativo al nivel en plasma humano normal) o en Unidades Internacionales (relativas al estándar internacional para el factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII presente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor VIII se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad plasmática de factor VIII en 2 UI/dl. La dosis requerida se determina utilizando la fórmula siguiente:
UI requeridas = peso corporal (kg) * aumento deseado de factor VIII (%) (UI/dl) x 0,5 (UI/kg por UI/dl).
La dosis y la frecuencia de la administración deberá individualizarse según la eficacia clínica en cada caso en particular.
En el caso de los eventos hemorrágicos siguientes, la actividad del factor VIII no deberá bajar del nivel de actividad plasmática indicado (en % del normal o UI/dl) durante el periodo correspondiente. La tabla siguiente se puede usar como guía para determinar la dosis en episodios hemorrágicos y cirugía:
T abla 1 Guía para determinar la dosis en episodios hemorrágicos y cirugía
Grado de la hemorragia/Tipo de Nivel de factor VIII Frecuencia de dosificación
procedimiento quirúrgico necesario (%) (UI/dl) (horas)/Duración del tratamiento
(días)
Hemorragia
Hemartrosis precoz, sangrado muscular 20-40 Repetir cada 12 - 24 horas al menos 1
o sangrado de la cavidad oral día hasta que el episodio hemorrágico
se haya resuelto, en función del dolor, o hasta la cicatrización de la herida.
Hemartrosis más extensa, sangrado 30-60 muscular o hematoma
Hemorragias con riesgo vital 60-100
Repetir la perfusión cada 12 - 24 horas durante 3 - 4 días o hasta que el dolor y la discapacidad aguda se hayan resuelto.
Repetir la perfusión cada 8 - 24 horas hasta que el riesgo desaparezca.
Cirugía
Cirugía menor, incluidas las 30-60 Cada 24 horas, al menos 1 día, hasta
extracciones dentales la cicatrización.
Grado de la hemorragia/Tipo de Nivel de factor VIII Frecuencia de dosificación
procedimiento quirúrgico necesario (%) (UI/dl) (horas)/Duración del tratamiento
(días)
Cirugía mayor 80-100 Repetir la perfusión cada 8-24 horas
(pre- y postoperatorio) hasta que se consiga una cicatrización
adecuada. A continuación, tratamiento durante por lo menos 7 días más para mantener una actividad del factor VIII del 30 % al 60 % (UI/dl).
Profilaxis
En la profilaxis a largo plazo para prevenir hemorragias en pacientes con hemofilia A grave. Las dosis habituales recomendadas son de 20 a 40 UI de factor VIII por kg de peso corporal a días alternos o de 20 a 50 UI de factor VIII por kg de peso corporal 3 veces por semana. En algunos casos, especialmente en los pacientes más jóvenes, puede ser necesario un intervalo de dosis más corto o una dosis mayor.
Supervisión del tratamiento
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis a administrar y la frecuencia de las inyecciones repetidas. Especialmente en el caso de las intervenciones de cirugía mayor, es indispensable controlar con precisión la terapia de sustitución mediante pruebas de coagulación (actividad plasmática del factor VIII). La respuesta individual de cada paciente frente al factor VIII puede variar y alcanzar distintos niveles de recuperación in vivo, y presentar semividas diferentes.
Cirugía
No existe experiencia en cirugía en pacientes pediátricos.
Pacientes de edad avanzada
No existe experiencia en pacientes de más de 65 años.
Población pediátrica
En la profilaxis a largo plazo para prevenir hemorragias en pacientes de menos de 12 años, las dosis recomendadas son de 25 a 50 UI de factor VIII por kg de peso corporal a días alternos o de 25 a 60 UI de factor VIII por kg de peso corporal 3 veces por semana. En pacientes pediátricos de más de 12 años, la dosis recomendada es la misma que para los adultos.
Forma de administración Vía intravenosa.
La velocidad de perfusión recomendada para NovoEight es de 1 - 2 ml/min. La velocidad se deberá determinar de acuerdo con el nivel de confort del paciente.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Reacciones alérgicas conocidas a las proteínas de hámster.
4.4 Advertencias y precauciones especiales de empleo Hipersensibilidad
Es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico a NovoEight. El producto contiene trazas de proteínas de hámster, las cuales pueden provocar reacciones alérgicas en algunos
43
pacientes. Si se produjesen síntomas de hipersensibilidad, se deberá instruir a los pacientes que deben interrumpir el uso del medicamento y ponerse inmediatamente en contacto con su médico. Se debe informar a los pacientes de los síntomas iniciales de las reacciones de hipersensibilidad, como urticaria localizada o generalizada, opresión en el pecho, respiración sibilante, hipotensión y anafilaxia.
En caso de shock, se seguirán las pautas médicas habituales para su tratamiento.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, y se cuantifican en Unidades Bethesda (UB) por ml de plasma usando el ensayo modificado. El riesgo de desarrollo de inhibidores está correlacionado con la exposición al factor VIII, y el riesgo mayor se produce durante los primeros 20 días de exposición. Los inhibidores raramente se forman después de los primeros 100 días de tratamiento.
Se han observado casos recurrentes de inhibidores (con título bajo), después de cambiar de un medicamento con factor VIII a otro a pacientes que habían recibido un tratamiento previo de más de 100 días de exposición y que tienen antecedentes de desarrollo de inhibidores. Por consiguiente se recomienda controlar a todos los pacientes atentamente para determinar la presencia de inhibidores después de cualquier cambio de medicamento.
En general, se deberá controlar atentamente a todos los pacientes tratados con medicamentos con factor VIII para determinar el desarrollo de inhibidores mediante la observación clínica y las pruebas de laboratorio adecuadas. Si no se obtienen los niveles plasmáticos de actividad del factor VIII esperados, o si no se controla la hemorragia con una dosis adecuada, se deberán realizar análisis para detectar la presencia de inhibidores del factor VIII. En pacientes con niveles elevados de inhibidores, es posible que el tratamiento con factor VIII no sea eficaz y se deberán considerar otras opciones terapéuticas. El tratamiento de estos pacientes debe hacerse bajo la dirección de un médico con experiencia en el cuidado de la hemofilia y los inhibidores del factor VIII.
Se recomienda encarecidamente registrar el nombre y el número de lote del medicamento cada vez que se administre NovoEight a un paciente con el fin de mantener un vínculo entre el paciente y el lote del medicamento.
Consideraciones relacionadas con los excipientes
Después de la reconstitución, este medicamento contiene 0,31 mmol de sodio (7 mg) por ml de solución reconstituida. Esto deberá tenerse en cuenta en pacientes que sigan una dieta controlada en sodio.
Complicaciones relacionadas con el catéter
Si se requiere un dispositivo de acceso venoso central (DAVC), se debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, como por ejemplo, infecciones locales, bacteriemia y trombosis en el lugar de inserción del catéter.
Población pediátrica
Las advertencias y precauciones indicadas son aplicables a adultos y niños.
4.5 Interacción con otros medicamentos y otras formas de interacción No se han realizado estudios de interacciones con NovoEight.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción animal con NovoEight. Teniendo en cuenta la escasa ocurrencia de hemofilia A en mujeres, no existe experiencia en relación con el uso de factor VIII durante el embarazo y la lactancia. Por consiguiente, solo se deberá usar factor VIII durante el embarazo y la lactancia si está estrictamente indicado.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NovoEight sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Resumen del perfil de seguridad
Hipersensibilidad o reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el punto de perfusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habones, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, cosquilleo, vómitos, sibilancias) se han observado raramente y, en algunos casos, puede progresar hasta una anafilaxia grave (incluido shock).
En muy raras ocasiones se ha observado el desarrollo de anticuerpos frente a la proteína de hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizadores (inhibidores) del factor VIII. Si se generan inhibidores de este tipo, la situación se pondrá de manifiesto por una respuesta clínica insuficiente. En tales casos, se recomienda ponerse en contacto con un centro especializado en hemofilia.
T abla de reacciones adversas
La tabla que se muestra a continuación sigue la clasificación de sistemas de órganos de MedDRA (clasificación por órganos y sistemas (SOC) y nivel de término preferente).
Las frecuencias se han evaluado conforme a la convención siguiente: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); raras (> 1/10.000 a < 1/1.000); muy raras (< 1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
En cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
T abla 2 Frecuencia de los reacciones adversas en ensayos clínicos
|
Clasificación por órganos y sistemas |
Frecuencia* |
Reacción adversa |
|
Trastornos psiquiátricos |
Poco frecuentes |
Insomnio |
|
Trastornos del sistema nervioso |
Poco frecuentes |
Cefalea, mareo |
|
Trastornos cardiacos |
Poco frecuentes |
Taquicardia sinusal |
|
Trastornos vasculares |
Poco frecuentes |
Hipertensión, linfoedema |
|
Trastornos hepatobiliares |
Frecuentes |
Enzimas hepáticas elevadas** |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
Erupción |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Poco frecuentes |
Rigidez musculoesquelética, artropatía, dolor en las extremidades, dolor musculoesquelético |
|
Trastornos generales y alteraciones en el lugar de administración |
Frecuentes |
Reacciones en la zona de inyección*** |
|
Poco frecuentes |
Fatiga, sensación de calor, edema periférico, pirexia | |
|
Exploraciones complementarias |
Poco frecuentes |
Frecuencia cardiaca aumentada |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Poco frecuentes |
Contusión |
Cálculo basado en el número total de pacientes distintos en todos los estudios clínicos (214).
Entre las enzimas hepáticas elevadas se encuentran la aminotransferasa, aspartato aminotransferasa, gamma glutamiltransferasa y bilirrubina.
*** Entre las reacciones en la zona de inyección se encuentran eritema, extravasación y prurito en la zona de inyección.
Descripción de las reacciones adversas señaladas
Durante todos los estudios clínicos con NovoEight, se han notificado un total de 30 reacciones adversas en
19 de 214 pacientes expuestos a NovoEight. Las reacciones adversas de las que se informó con mayor frecuencia fueron reacciones en la zona de inyección y enzimas hepáticas elevadas. De las 30 reacciones adversas, 2 de ellas se notificaron en 1 de 31 pacientes de menos de 6 años de edad, ninguna entre los pacientes de 6 a 18 años de edad y 28 en 18 de 127 adultos.
Población pediátrica
En estudios clínicos con 63 pacientes pediátricos de entre 0 y 12 años de edad y 24 adolescentes entre 12 y 18 años de edad con hemofilia A grave, no se observó ninguna diferencia en el perfil de seguridad de NovoEight entre pacientes pediátricos y adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han observado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos, factor VIII de la coagulación sanguínea, código ATC: B02BD02.
Mecanismo de acción
NovoEight contiene turoctocog alfa, un factor VIII de coagulación humano (ADNr) con un dominio B truncado. Esta glicoproteína tiene la misma estructura que el factor VIII cuando se activa, y modificaciones postranslacionales similares a las de la molécula derivada del plasma. Se ha observado que el punto de sulfatación de tirosina presente en Tyr1680 (longitud completa nativa), que es importante para el enlace con el factor de von Willebrand, está totalmente sulfatado en la molécula de turoctocog alfa. Cuando se administra mediante perfusión en un paciente con hemofilia, el factor VIII se enlaza con el factor de von Willebrand endógeno en la circulación del paciente. El complejo factor VIII/factor von Willebrand consiste en dos moléculas (factor VIII y factor de von Willebrand) con funciones fisiológicas distintas. El factor VIII activado actúa como cofactor del factor IX activado, y acelera la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo. La hemofilia es una alteración hereditaria vinculada al sexo de la coagulación de la sangre debida a niveles reducidos de factor VIII:C y da como resultado hemorragias difusas en articulaciones, músculos u órganos internos, ya sea de forma espontánea como tras un accidente o un trauma quirúrgico. La terapia de sustitución, aumenta los niveles plasmáticos de factor VIII, lo cual permite corregir temporalmente la deficiencia del factor y la tendencia al sangrado.
Eficacia clínica
Se han realizado tres ensayos multicéntricos, abiertos y no controlados para evaluar la seguridad y la eficacia de NovoEight para la prevención y el tratamiento de sangrado en pacientes tratados previamente con hemofilia A grave (actividad de FVEI < 1 %). Los ensayos incluyeron a 213 pacientes expuestos; 150 pacientes adolescentes o adultos sin inhibidores desde la edad de 12 años (> 150 días de exposición) y 63 pacientes pediátricos sin inhibidores de menos de 12 años de edad (> 50 días de exposición). 187 de 213 pacientes continuaron en el ensayo ampliado de seguridad. Se observó que el tratamiento con NovoEight es seguro y tiene el efecto hemostático y preventivo deseado. Durante una exposición acumulada de 54.000 días (correspondientes a 342 años paciente), no se observó el desarrollo de inhibidores del factor VIII en los ensayos clínicos en fase 3a con pacientes tratados previamente. De los 1.377 sangrados observados en 177 de los 213 pacientes, 1.244 (90,3 %) de los sangrados se resolvieron con 1 o 2 perfusiones de NovoEight.
|
Tabla 3 Consumo de turoctocog alfa y tasas de éxito glo |
bales | ||||
|
Niños pequeños (0 - <6 años) |
Niños mayores (6 - <12 años) |
Adolescentes (12 -<18 años) |
Adultos (>18 años) |
Total | |
|
Número de pacientes |
31 |
32 |
24 |
126 |
213 |
|
Dosis empleada como prevención por paciente (UI/kg PC) Valor medio (DE) Mín; Máx |
40,1 (8,5) 26,5 ; 57,3 |
36,6 (9,0) 24,9 ; 57,9 |
27,0 (7,6) 20,5 ; 46,9 |
26,9 (6,9) 20,0 ; 50,8 |
30,3 (9,2) 20,0 ; 57,9 |
|
Dosis utilizada para el tratamiento del sangrado (UI/kg BW) Valor medio (DE) Mín; Máx |
44,4 (17,9) 25,9 ; 193,8 |
40,0 (10,4) 25,5 ; 65,5 |
28,2 (10,2) 12,4 ; 76,8 |
33,8 (11,9) 9,3 ; 104,0 |
34,5 (12,6) 9,3 ; 193,8 |
|
Número de infusiones para el tratamiento del sangrado Valor medio (DE) Mín; Máx |
1,2 (0,6) 1 ; 6 |
1,5 (1,1) 1 ; 8 |
1,7 (1,3) 1 ; 9 |
1,5 (2,4) 1 ; 49 |
1,5 (2,1) 1 ; 49 |
|
T asa de éxito* % |
92,9 % |
88,9 % |
79,7 % |
85,6 % |
85,9 % |
PC: Peso corporal, DE: Desviación estándar *El éxito se define como “Excelente” o “Bueno”.
En total se realizaron 14 intervenciones quirúrgicas en 14 pacientes, de las cuales 13 fueron de cirugía mayor y 1 de cirugía menor. La hemostasis fue correcta en todas las intervenciones quirúrgicas y no se observó ningún fallo de tratamiento.
5.2 Propiedades farmacocinéticas
T odos los estudios de farmacocinética con turoctocog alfa se realizaron en pacientes tratados previamente con hemofilia A grave (FVHI < 1 %). El análisis de las muestras de plasma se realizó mediante análisis de coagulación de una fase y análisis cromogénico.
En un estudio internacional que implicó a 36 laboratorios, se evaluó el rendimiento de NovoEight en ensayos de FVHI:C y se comparó con un medicamento comercializado de FVIII recombinante de longitud completa. El estudio reveló que se obtienen resultados comparables y coherentes con ambos productos y que NovoEight se puede medir en plasma de forma fiable sin necesidad de usar un estándar aparte para NovoEight.
Los parámetros farmacocinéticos de NovoEight en una sola dosis se indican en la T abla 4 para el análisis de coagulación y en la Tabla 5 para el análisis cromogénico.
Tabla 4 Farmacocinética de una sola dosis de turoctocog alfa en pacientes con hemofilia A grave (FVIII < 1 %), análisis de coagulación___
|
Parámetro |
0 - <6 años |
6 - <12 años |
>12 años |
|
n = 14 |
n = 14 |
n = 33 | |
|
Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) | |
|
Recuperación incremental (UI/ml)/(UI/kg) |
0,018 (0,007) |
0,020 (0,004) |
0,022 (0,004) |
|
AUC ((UI*h)/ml) |
9,92 (4,11) |
11,09 (3,74) |
15,26 (5,77) |
|
CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
|
t/ (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
|
Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
|
Cmax (UI/ml) |
1,00 (0,58) |
1,07 (0,35) |
1,226 (0,41) |
|
Tiempo medio de residencia (h) |
9,63 (2,50) |
9,91 (2,57) |
14,19 (5,08) |
Tabla 5 Farmacocinética de una sola dosis de turoctocog alfa en pacientes con hemofilia A grave (FVIII < 1 %), análisis cromogénico___
|
Parámetro |
0 - <6 años |
6 - <12 años |
>12 años |
|
n = 14 |
n = 14 |
n = 33 | |
|
Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) | |
|
Recuperación incremental (UI/ml)/(UI/kg) |
0,022 (0,006) |
0,025 (0,006) |
0,029 (0,006) |
|
AUC ((UI*h)/ml) |
12,23 (4,36) |
14,37 (3,48) |
19,63 (7,73) |
|
CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
|
t/ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
|
Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
|
Cmax (UI/ml) |
1,12 (0,31) |
1,25 (0,27) |
1,63 (0,50) |
|
Tiempo medio de residencia (h) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
Los parámetros farmacocinéticos fueron comparables entre pacientes pediátricos de menos de 6 años de edad y pacientes pediátricos mayores de 6 y menores de 12 años de edad. Se observó alguna variación entre los parámetros farmacocinéticos de NovoEight en pacientes pediátricos y adultos. Los valores mayores de CL y menores de t./2 en pacientes pediátricos en comparación con pacientes adultos con hemofilia A se pueden deber en parte al hecho conocido de que el volumen de plasma por kilogramo de peso corporal es mayor en los pacientes más jóvenes.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad y toxicidad a dosis repetidas.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Cloruro de sodio L-histidina Sacarosa Polisorbato 80 L-metionina
Cloruro de calcio dihidratado Hidróxido de sodio Ácido clorhídrico
Disolvente:
Cloruro de sodio
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
Sin abrir:
30 meses
Durante el periodo de validez, el medicamento se puede almacenar a temperatura ambiente (<30°C) durante un solo periodo de hasta 9 meses. Una vez que el producto se saca de la nevera, no se debe volver a refrigerar. Registrar la fecha de inicio del almacenamiento a temperatura ambiente en el embalaje del producto.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Después de la reconstitución:
Se ha demostrado la estabilidad química y física en uso durante 24 horas almacenado entre 2°C y 8°C y 4 horas almacenado a temperatura ambiente (<30°C).
Desde el punto de vista microbiológico, el medicamento se debería usar inmediatamente después de la reconstitución. Si no se utiliza de inmediato, los tiempos y las condiciones de almacenamiento en uso antes del uso son responsabilidad del usuario y no deberían superarse las 4 horas a temperatura ambiente (<30°C) o 24 horas entre 2°C y 8°C, a menos que la reconstitución se haya realizado en condiciones asépticas controladas y validadas.
El medicamento no utilizado almacenado a temperatura ambiente durante más de 4 horas se deberá desechar.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C). No congelar.
Para consultar la información de almacenamiento a temperatura ambiente y las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Cada envase de NovoEight 1.500 UI polvo y disolvente para solución inyectable contiene:
- 1 vial de vidrio (tipo I) con polvo y un tapón de goma de clorobutilo
- 1 adaptador de vial estéril para la reconstitución
- 1 jeringa precargada con 4 ml de disolvente con mecanismo de protección (polipropileno), un émbolo
de goma (bromobutilo) y un capuchón de aguja con tapón (bromobutilo)
- 1 varilla del émbolo (polipropileno).
6.6 Precauciones especiales de eliminación y otras manipulaciones
NovoEight se debe administrar por vía intravenosa después de la reconstitución del polvo con el disolvente suministrado en la jeringa. Después de la reconstitución, la solución es transparente o ligeramente opalescente. No utilice la solución si tiene aspecto turbio o presenta depósitos.
También necesitará un conjunto de perfusión (tubos y aguja mariposa), toallitas estériles con alcohol, gasas y tiritas. Estos dispositivos no se incluyen en el paquete NovoEight.
Utilice siempre una técnica aséptica.
Reconstitución
A)
Saque el vial, el adaptador del vial y la jeringa
precargada del envase. Deje la varilla del émbolo sin tocar en el envase. Lleve el vial y la jeringa precargada a temperatura ambiente. Puede hacerlo manteniendo ambos en las manos hasta que sienta que están a la misma temperatura que sus manos. No utilice ningún otro sistema para calentar el vial y la jeringa precargada.
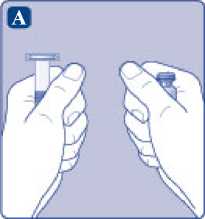

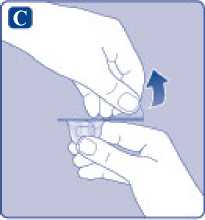

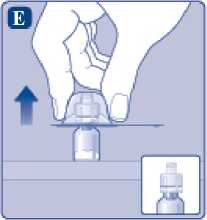
B)
Quite el capuchón de plástico del vial. Si el capuchón de plástico está flojo o falta, no utilice el vial. Limpie el tapón de goma del vial con una toallita estéril con alcohol y deje que se seque al aire unos segundos antes de usarlo.
C)
Quite el precinto de papel del adaptador del vial. Si el precinto de papel no está totalmente sellado o si está roto, no utilice el adaptador del vial. No saque el adaptador del vial del capuchón protector con los dedos.
D)
Ponga el capuchón protector del adaptador del vial boca abajo y coloque el adaptador del vial a presión sobre el vial. Una vez unido, no retire del vial el adaptador del vial.
E)
Comprima ligeramente el capuchón protector entre los dedos pulgar e índice tal como se muestra. Quite el capuchón protector del adaptador del vial.
F)
Sujete la varilla del émbolo por el extremo ancho e inmediatamente conéctelo a la jeringa
girándolo hacia la derecha dentro del émbolo en el interior de la jeringa precargada hasta que se sienta resistencia.
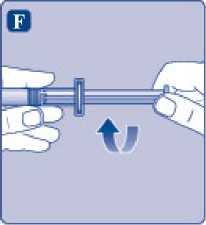
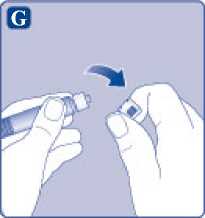
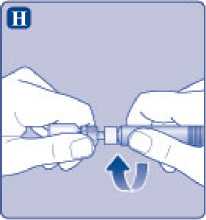
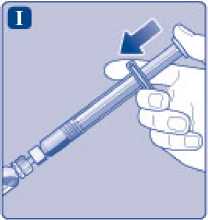
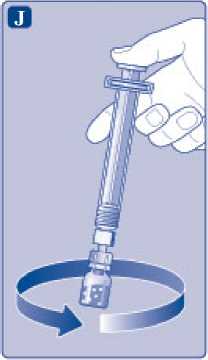
G)
Quite el tapón de la jeringa precargada doblándolo hacia abajo hasta que se rompa la perforación. No toque la punta de la jeringa debajo del tapón de la jeringa.
H)
Enrosque la jeringa precargada con firmeza en el adaptador del vial hasta que se sienta resistencia.
I)
Sujete la jeringa precargada ligeramente inclinada con el vial apuntando hacia abajo. Presione la varilla del émbolo para inyectar todo el disolvente en el vial.
J)
Mantenga la varilla del vial presionada y remueva suavemente el vial hasta que el polvo se haya disuelto. No agite el vial, ya que esto produciría espuma.
Se recomienda utilizar NovoEight inmediatamente después de la reconstitución. Para consultar las condiciones de conservación del medicamento reconstituido, ver sección 6.3.
Si se necesita una dosis mayor, repita los pasos A a J con más viales, adaptadores de viales y jeringas precargadas.
Administración de la solución reconstituida
K)
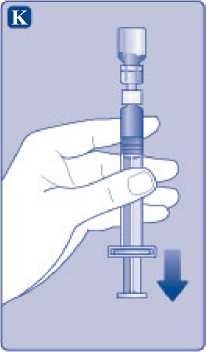
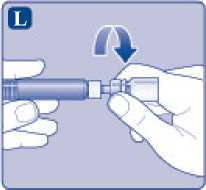
Mantenga la varilla del émbolo totalmente empujada hacia el interior. Gire la jeringa con el vial boca abajo. Deje de empujar la varilla del émbolo y deje que retroceda por sí misma mientras la solución reconstituida llena la jeringa. T ire de la varilla del émbolo ligeramente hacia abajo para hacer pasar la solución reconstituida hacia la jeringa.
Si solo necesita una parte del vial, utilice la escala de la jeringa para ver la cantidad de solución reconstituida que ha retirado, tal como le haya indicado su médico o su enfermera.
Mientras sujeta el vial cabeza abajo, golpee suavemente la jeringa para que las posibles burbujas suban hasta la parte superior. Empuje la varilla del émbolo lentamente hasta que se hayan eliminado las burbujas.
L)
Desenrosque el adaptador del vial con el vial.
Ahora NovoEight está listo para ser inyectado. Busque un lugar adecuado e inyecte lentamente NovoEight en la vena en un periodo de 2 a 5 minutos.
Eliminación
Después de la inyección, deseche con seguridad toda la solución NovoEight no utilizada, la jeringa con el juego de perfusión, el vial con el adaptador del vial y los demás residuos siguiendo las indicaciones de su farmacéutico.
No los deseche como residuos domésticos ordinarios.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novo Nordisk A/S Novo Allé
DK-2880 Bagsv^rd Dinamarca
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/888/004
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 13 Noviembre 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
^Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
NovoEight 2.000 UI polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial de polvo contiene 2.000 UI de factor VIII de coagulación humano (ADNr), turoctocog alfa.
NovoEight contiene aproximadamente 500 UI/ml de factor VIII de coagulación humano (ADNr), turoctocog alfa, después de la reconstitución.
La potencia (UI) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de NovoEight es aproximadamente de 8.300 UI/mg de proteína.
Turoctocog alfa (factor VIII de coagulación humano (ADNr)) es una proteína purificada que contiene 1.445 aminoácidos con una masa molecular aproximada de 166 kDA. Se produce mediante tecnología de ADN recombinante en células ováricas de hámster chino (CHO) y se prepara sin añadir ninguna proteína humana ni derivada de animal durante el proceso de cultivo de las células, la purificación o la formulación final.
Turoctocog alfa es un factor VIII de coagulación humano recombinante truncado de dominio B (el dominio B consiste en 21 aminoácidos del dominio silvestre de tipo B) sin ninguna otra modificación de la secuencia de aminoácidos.
Excipiente con efecto conocido:
0,31 mmol de sodio (7 mg) por ml de solución reconstituida.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo o masa friable de color blanco o ligeramente amarillo. Solución inyectable transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
T ratamiento y profilaxis de hemorragias en pacientes con hemofilia A (deficiencia congénita del factor VIII). NovoEight se puede usar en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de hemofilia.
Pacientes no tratados previamente
No se ha establecido todavía la seguridad y la eficacia de NovoEight en pacientes no tratados previamente.
No se dispone de datos.
Posología
La dosis y duración de la terapia de sustitución dependen de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que se corresponden con el estándar actual de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa como porcentaje (relativo al nivel en plasma humano normal) o en Unidades Internacionales (relativas al estándar internacional para el factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII presente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor VIII se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad plasmática de factor VIII en 2 UI/dl. La dosis requerida se determina utilizando la fórmula siguiente:
UI requeridas = peso corporal (kg) * aumento deseado de factor VIII (%) (UI/dl) x 0,5 (UI/kg por UI/dl).
La dosis y la frecuencia de la administración deberá individualizarse según la eficacia clínica en cada caso en particular.
En el caso de los eventos hemorrágicos siguientes, la actividad del factor VIII no deberá bajar del nivel de actividad plasmática indicado (en % del normal o UI/dl) durante el periodo correspondiente. La tabla siguiente se puede usar como guía para determinar la dosis en episodios hemorrágicos y cirugía:
T abla 1 Guía para determinar la dosis en episodios hemorrágicos y cirugía
Grado de la hemorragia/Tipo de Nivel de factor VIII Frecuencia de dosificación
procedimiento quirúrgico necesario (%) (UI/dl) (horas)/Duración del tratamiento
(días)
Hemorragia
Hemartrosis precoz, sangrado muscular 20-40 Repetir cada 12 - 24 horas al menos 1
o sangrado de la cavidad oral día hasta que el episodio hemorrágico
se haya resuelto, en función del dolor, o hasta la cicatrización de la herida.
Hemartrosis más extensa, sangrado 30-60 muscular o hematoma
Hemorragias con riesgo vital 60-100
Repetir la perfusión cada 12 - 24 horas durante 3 - 4 días o hasta que el dolor y la discapacidad aguda se hayan resuelto.
Repetir la perfusión cada 8 - 24 horas hasta que el riesgo desaparezca.
Cirugía
Cirugía menor, incluidas las 30-60 Cada 24 horas, al menos 1 día, hasta
extracciones dentales la cicatrización.
Grado de la hemorragia/Tipo de Nivel de factor VIII Frecuencia de dosificación
procedimiento quirúrgico necesario (%) (UI/dl) (horas)/Duración del tratamiento
(días)
Cirugía mayor 80-100 Repetir la perfusión cada 8-24 horas
(pre- y postoperatorio) hasta que se consiga una cicatrización
adecuada. A continuación, tratamiento durante por lo menos 7 días más para mantener una actividad del factor VIII del 30 % al 60 % (UI/dl).
Profilaxis
En la profilaxis a largo plazo para prevenir hemorragias en pacientes con hemofilia A grave. Las dosis habituales recomendadas son de 20 a 40 UI de factor VIII por kg de peso corporal a días alternos o de 20 a 50 UI de factor VIII por kg de peso corporal 3 veces por semana. En algunos casos, especialmente en los pacientes más jóvenes, puede ser necesario un intervalo de dosis más corto o una dosis mayor.
Supervisión del tratamiento
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis a administrar y la frecuencia de las inyecciones repetidas. Especialmente en el caso de las intervenciones de cirugía mayor, es indispensable controlar con precisión la terapia de sustitución mediante pruebas de coagulación (actividad plasmática del factor VIII). La respuesta individual de cada paciente frente al factor VIII puede variar y alcanzar distintos niveles de recuperación in vivo, y presentar semividas diferentes.
Cirugía
No existe experiencia en cirugía en pacientes pediátricos.
Pacientes de edad avanzada
No existe experiencia en pacientes de más de 65 años.
Población pediátrica
En la profilaxis a largo plazo para prevenir hemorragias en pacientes de menos de 12 años, las dosis recomendadas son de 25 a 50 UI de factor VIII por kg de peso corporal a días alternos o de 25 a 60 UI de factor VIII por kg de peso corporal 3 veces por semana. En pacientes pediátricos de más de 12 años, la dosis recomendada es la misma que para los adultos.
Forma de administración Vía intravenosa.
La velocidad de perfusión recomendada para NovoEight es de 1 - 2 ml/min. La velocidad se deberá determinar de acuerdo con el nivel de confort del paciente.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Reacciones alérgicas conocidas a las proteínas de hámster.
4.4 Advertencias y precauciones especiales de empleo Hipersensibilidad
Es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico a NovoEight. El producto contiene trazas de proteínas de hámster, las cuales pueden provocar reacciones alérgicas en algunos
56
pacientes. Si se produjesen síntomas de hipersensibilidad, se deberá instruir a los pacientes que deben interrumpir el uso del medicamento y ponerse inmediatamente en contacto con su médico. Se debe informar a los pacientes de los síntomas iniciales de las reacciones de hipersensibilidad, como urticaria localizada o generalizada, opresión en el pecho, respiración sibilante, hipotensión y anafilaxia.
En caso de shock, se seguirán las pautas médicas habituales para su tratamiento.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, y se cuantifican en Unidades Bethesda (UB) por ml de plasma usando el ensayo modificado. El riesgo de desarrollo de inhibidores está correlacionado con la exposición al factor VIII, y el riesgo mayor se produce durante los primeros 20 días de exposición. Los inhibidores raramente se forman después de los primeros 100 días de tratamiento.
Se han observado casos recurrentes de inhibidores (con título bajo), después de cambiar de un medicamento con factor VIII a otro a pacientes que habían recibido un tratamiento previo de más de 100 días de exposición y que tienen antecedentes de desarrollo de inhibidores. Por consiguiente se recomienda controlar a todos los pacientes atentamente para determinar la presencia de inhibidores después de cualquier cambio de medicamento.
En general, se deberá controlar atentamente a todos los pacientes tratados con medicamentos con factor VIII para determinar el desarrollo de inhibidores mediante la observación clínica y las pruebas de laboratorio adecuadas. Si no se obtienen los niveles plasmáticos de actividad del factor VIII esperados, o si no se controla la hemorragia con una dosis adecuada, se deberán realizar análisis para detectar la presencia de inhibidores del factor VIII. En pacientes con niveles elevados de inhibidores, es posible que el tratamiento con factor VIII no sea eficaz y se deberán considerar otras opciones terapéuticas. El tratamiento de estos pacientes debe hacerse bajo la dirección de un médico con experiencia en el cuidado de la hemofilia y los inhibidores del factor VIII.
Se recomienda encarecidamente registrar el nombre y el número de lote del medicamento cada vez que se administre NovoEight a un paciente con el fin de mantener un vínculo entre el paciente y el lote del medicamento.
Consideraciones relacionadas con los excipientes
Después de la reconstitución, este medicamento contiene 0,31 mmol de sodio (7 mg) por ml de solución reconstituida. Esto deberá tenerse en cuenta en pacientes que sigan una dieta controlada en sodio.
Complicaciones relacionadas con el catéter
Si se requiere un dispositivo de acceso venoso central (DAVC), se debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, como por ejemplo, infecciones locales, bacteriemia y trombosis en el lugar de inserción del catéter.
Población pediátrica
Las advertencias y precauciones indicadas son aplicables a adultos y niños.
4.5 Interacción con otros medicamentos y otras formas de interacción No se han realizado estudios de interacciones con NovoEight.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción animal con NovoEight. Teniendo en cuenta la escasa ocurrencia de hemofilia A en mujeres, no existe experiencia en relación con el uso de factor VIII durante el embarazo y la lactancia. Por consiguiente, solo se deberá usar factor VIII durante el embarazo y la lactancia si está estrictamente indicado.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NovoEight sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Resumen del perfil de seguridad
Hipersensibilidad o reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el punto de perfusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habones, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, cosquilleo, vómitos, sibilancias) se han observado raramente y, en algunos casos, puede progresar hasta una anafilaxia grave (incluido shock).
En muy raras ocasiones se ha observado el desarrollo de anticuerpos frente a la proteína de hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizadores (inhibidores) del factor VIII. Si se generan inhibidores de este tipo, la situación se pondrá de manifiesto por una respuesta clínica insuficiente. En tales casos, se recomienda ponerse en contacto con un centro especializado en hemofilia.
T abla de reacciones adversas
La tabla que se muestra a continuación sigue la clasificación de sistemas de órganos de MedDRA (clasificación por órganos y sistemas (SOC) y nivel de término preferente).
Las frecuencias se han evaluado conforme a la convención siguiente: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); raras (> 1/10.000 a < 1/1.000); muy raras (< 1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
En cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
T abla 2 Frecuencia de los reacciones adversas en ensayos clínicos
|
Clasificación por órganos y sistemas |
Frecuencia* |
Reacción adversa |
|
Trastornos psiquiátricos |
Poco frecuentes |
Insomnio |
|
Trastornos del sistema nervioso |
Poco frecuentes |
Cefalea, mareo |
|
Trastornos cardiacos |
Poco frecuentes |
Taquicardia sinusal |
|
Trastornos vasculares |
Poco frecuentes |
Hipertensión, linfoedema |
|
Trastornos hepatobiliares |
Frecuentes |
Enzimas hepáticas elevadas** |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
Erupción |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Poco frecuentes |
Rigidez musculoesquelética, artropatía, dolor en las extremidades, dolor musculoesquelético |
|
Trastornos generales y alteraciones en el lugar de administración |
Frecuentes |
Reacciones en la zona de inyección*** |
|
Poco frecuentes |
Fatiga, sensación de calor, edema periférico, pirexia | |
|
Exploraciones complementarias |
Poco frecuentes |
Frecuencia cardiaca aumentada |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Poco frecuentes |
Contusión |
Cálculo basado en el número total de pacientes distintos en todos los estudios clínicos (214).
Entre las enzimas hepáticas elevadas se encuentran la aminotransferasa, aspartato aminotransferasa, gamma glutamiltransferasa y bilirrubina.
*** Entre las reacciones en la zona de inyección se encuentran eritema, extravasación y prurito en la zona de inyección.
Descripción de las reacciones adversas señaladas
Durante todos los estudios clínicos con NovoEight, se han notificado un total de 30 reacciones adversas en
19 de 214 pacientes expuestos a NovoEight. Las reacciones adversas de las que se informó con mayor frecuencia fueron reacciones en la zona de inyección y enzimas hepáticas elevadas. De las 30 reacciones adversas, 2 de ellas se notificaron en 1 de 31 pacientes de menos de 6 años de edad, ninguna entre los pacientes de 6 a 18 años de edad y 28 en 18 de 127 adultos.
Población pediátrica
En estudios clínicos con 63 pacientes pediátricos de entre 0 y 12 años de edad y 24 adolescentes entre 12 y 18 años de edad con hemofilia A grave, no se observó ninguna diferencia en el perfil de seguridad de NovoEight entre pacientes pediátricos y adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han observado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos, factor VIII de la coagulación sanguínea, código ATC: B02BD02.
Mecanismo de acción
NovoEight contiene turoctocog alfa, un factor VIII de coagulación humano (ADNr) con un dominio B truncado. Esta glicoproteína tiene la misma estructura que el factor VIII cuando se activa, y modificaciones postranslacionales similares a las de la molécula derivada del plasma. Se ha observado que el punto de sulfatación de tirosina presente en Tyr1680 (longitud completa nativa), que es importante para el enlace con el factor de von Willebrand, está totalmente sulfatado en la molécula de turoctocog alfa. Cuando se administra mediante perfusión en un paciente con hemofilia, el factor VIII se enlaza con el factor de von Willebrand endógeno en la circulación del paciente. El complejo factor VIII/factor von Willebrand consiste en dos moléculas (factor VIII y factor de von Willebrand) con funciones fisiológicas distintas. El factor VIII activado actúa como cofactor del factor IX activado, y acelera la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo. La hemofilia es una alteración hereditaria vinculada al sexo de la coagulación de la sangre debida a niveles reducidos de factor VIII:C y da como resultado hemorragias difusas en articulaciones, músculos u órganos internos, ya sea de forma espontánea como tras un accidente o un trauma quirúrgico. La terapia de sustitución, aumenta los niveles plasmáticos de factor VIII, lo cual permite corregir temporalmente la deficiencia del factor y la tendencia al sangrado.
Eficacia clínica
Se han realizado tres ensayos multicéntricos, abiertos y no controlados para evaluar la seguridad y la eficacia de NovoEight para la prevención y el tratamiento de sangrado en pacientes tratados previamente con hemofilia A grave (actividad de FVEI < 1 %). Los ensayos incluyeron a 213 pacientes expuestos; 150 pacientes adolescentes o adultos sin inhibidores desde la edad de 12 años (> 150 días de exposición) y 63 pacientes pediátricos sin inhibidores de menos de 12 años de edad (> 50 días de exposición). 187 de 213 pacientes continuaron en el ensayo ampliado de seguridad. Se observó que el tratamiento con NovoEight es seguro y tiene el efecto hemostático y preventivo deseado. Durante una exposición acumulada de 54.000 días (correspondientes a 342 años paciente), no se observó el desarrollo de inhibidores del factor VIII en los ensayos clínicos en fase 3a con pacientes tratados previamente. De los 1.377 sangrados observados en 177 de los 213 pacientes, 1.244 (90,3 %) de los sangrados se resolvieron con 1 o 2 perfusiones de NovoEight.
|
Tabla 3 Consumo de turoctocog alfa y tasas de éxito glo |
bales | ||||
|
Niños pequeños (0 - <6 años) |
Niños mayores (6 - <12 años) |
Adolescentes (12 -<18 años) |
Adultos (>18 años) |
Total | |
|
Número de pacientes |
31 |
32 |
24 |
126 |
213 |
|
Dosis empleada como prevención por paciente (UI/kg PC) Valor medio (DE) Mín; Máx |
40,1 (8,5) 26,5 ; 57,3 |
36,6 (9,0) 24,9 ; 57,9 |
27,0 (7,6) 20,5 ; 46,9 |
26,9 (6,9) 20,0 ; 50,8 |
30,3 (9,2) 20,0 ; 57,9 |
|
Dosis utilizada para el tratamiento del sangrado (UI/kg BW) Valor medio (DE) Mín; Máx |
44,4 (17,9) 25,9 ; 193,8 |
40,0 (10,4) 25,5 ; 65,5 |
28,2 (10,2) 12,4 ; 76,8 |
33,8 (11,9) 9,3 ; 104,0 |
34,5 (12,6) 9,3 ; 193,8 |
|
Número de infusiones para el tratamiento del sangrado Valor medio (DE) Mín; Máx |
1,2 (0,6) 1 ; 6 |
1,5 (1,1) 1 ; 8 |
1,7 (1,3) 1 ; 9 |
1,5 (2,4) 1 ; 49 |
1,5 (2,1) 1 ; 49 |
|
T asa de éxito* % |
92,9 % |
88,9 % |
79,7 % |
85,6 % |
85,9 % |
PC: Peso corporal, DE: Desviación estándar *El éxito se define como “Excelente” o “Bueno”.
En total se realizaron 14 intervenciones quirúrgicas en 14 pacientes, de las cuales 13 fueron de cirugía mayor y 1 de cirugía menor. La hemostasis fue correcta en todas las intervenciones quirúrgicas y no se observó ningún fallo de tratamiento.
5.2 Propiedades farmacocinéticas
T odos los estudios de farmacocinética con turoctocog alfa se realizaron en pacientes tratados previamente con hemofilia A grave (FVHI < 1 %). El análisis de las muestras de plasma se realizó mediante análisis de coagulación de una fase y análisis cromogénico.
En un estudio internacional que implicó a 36 laboratorios, se evaluó el rendimiento de NovoEight en ensayos de FVHI:C y se comparó con un medicamento comercializado de FVIII recombinante de longitud completa. El estudio reveló que se obtienen resultados comparables y coherentes con ambos productos y que NovoEight se puede medir en plasma de forma fiable sin necesidad de usar un estándar aparte para NovoEight.
Los parámetros farmacocinéticos de NovoEight en una sola dosis se indican en la T abla 4 para el análisis de coagulación y en la Tabla 5 para el análisis cromogénico.
Tabla 4 Farmacocinética de una sola dosis de turoctocog alfa en pacientes con hemofilia A grave (FVIII < 1 %), análisis de coagulación___
|
Parámetro |
0 - <6 años |
6 - <12 años |
>12 años |
|
n = 14 |
n = 14 |
n = 33 | |
|
Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) | |
|
Recuperación incremental (UI/ml)/(UI/kg) |
0,018 (0,007) |
0,020 (0,004) |
0,022 (0,004) |
|
AUC ((UI*h)/ml) |
9,92 (4,11) |
11,09 (3,74) |
15,26 (5,77) |
|
CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
|
t/ (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
|
Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
|
Cmax (UI/ml) |
1,00 (0,58) |
1,07 (0,35) |
1,226 (0,41) |
|
Tiempo medio de residencia (h) |
9,63 (2,50) |
9,91 (2,57) |
14,19 (5,08) |
Tabla 5 Farmacocinética de una sola dosis de turoctocog alfa en pacientes con hemofilia A grave (FVIII < 1 %), análisis cromogénico___
|
Parámetro |
0 - <6 años |
6 - <12 años |
>12 años |
|
n = 14 |
n = 14 |
n = 33 | |
|
Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) | |
|
Recuperación incremental (UI/ml)/(UI/kg) |
0,022 (0,006) |
0,025 (0,006) |
0,029 (0,006) |
|
AUC ((UI*h)/ml) |
12,23 (4,36) |
14,37 (3,48) |
19,63 (7,73) |
|
CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
|
t/ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
|
Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
|
Cmax (UI/ml) |
1,12 (0,31) |
1,25 (0,27) |
1,63 (0,50) |
|
Tiempo medio de residencia (h) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
Los parámetros farmacocinéticos fueron comparables entre pacientes pediátricos de menos de 6 años de edad y pacientes pediátricos mayores de 6 y menores de 12 años de edad. Se observó alguna variación entre los parámetros farmacocinéticos de NovoEight en pacientes pediátricos y adultos. Los valores mayores de CL y menores de t./2 en pacientes pediátricos en comparación con pacientes adultos con hemofilia A se pueden deber en parte al hecho conocido de que el volumen de plasma por kilogramo de peso corporal es mayor en los pacientes más jóvenes.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad y toxicidad a dosis repetidas.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Cloruro de sodio L-histidina Sacarosa Polisorbato 80 L-metionina
Cloruro de calcio dihidratado Hidróxido de sodio Ácido clorhídrico
Disolvente:
Cloruro de sodio
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
Sin abrir:
30 meses
Durante el periodo de validez, el medicamento se puede almacenar a temperatura ambiente (<30°C) durante un solo periodo de hasta 9 meses. Una vez que el producto se saca de la nevera, no se debe volver a refrigerar. Registrar la fecha de inicio del almacenamiento a temperatura ambiente en el embalaje del producto.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Después de la reconstitución:
Se ha demostrado la estabilidad química y física en uso durante 24 horas almacenado entre 2°C y 8°C y 4 horas almacenado a temperatura ambiente (<30°C).
Desde el punto de vista microbiológico, el medicamento se debería usar inmediatamente después de la reconstitución. Si no se utiliza de inmediato, los tiempos y las condiciones de almacenamiento en uso antes del uso son responsabilidad del usuario y no deberían superarse las 4 horas a temperatura ambiente (<30°C) o 24 horas entre 2°C y 8°C, a menos que la reconstitución se haya realizado en condiciones asépticas controladas y validadas.
El medicamento no utilizado almacenado a temperatura ambiente durante más de 4 horas se deberá desechar.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C). No congelar.
Para consultar la información de almacenamiento a temperatura ambiente y las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Cada envase de NovoEight 2.000 UI polvo y disolvente para solución inyectable contiene:
- 1 vial de vidrio (tipo I) con polvo y un tapón de goma de clorobutilo
- 1 adaptador de vial estéril para la reconstitución
- 1 jeringa precargada con 4 ml de disolvente con mecanismo de protección (polipropileno), un émbolo
de goma (bromobutilo) y un capuchón de aguja con tapón (bromobutilo)
- 1 varilla del émbolo (polipropileno).
6.6 Precauciones especiales de eliminación y otras manipulaciones
NovoEight se debe administrar por vía intravenosa después de la reconstitución del polvo con el disolvente suministrado en la jeringa. Después de la reconstitución, la solución es transparente o ligeramente opalescente. No utilice la solución si tiene aspecto turbio o presenta depósitos.
También necesitará un conjunto de perfusión (tubos y aguja mariposa), toallitas estériles con alcohol, gasas y tiritas. Estos dispositivos no se incluyen en el paquete NovoEight.
Utilice siempre una técnica aséptica.
Reconstitución
Saque el vial, el adaptador del vial y la jeringa precargada del envase. Deje la varilla del émbolo sin tocar en el envase. Lleve el vial y la jeringa precargada a temperatura ambiente. Puede hacerlo manteniendo ambos en las manos hasta que sienta que están a la misma temperatura que sus manos. No utilice ningún otro sistema para calentar el vial y la jeringa precargada.




|
0/ |
L | |
|
; | ||
B)
Quite el capuchón de plástico del vial. Si el capuchón de plástico está flojo o falta, no utilice el vial. Limpie el tapón de goma del vial con una toallita estéril con alcohol y deje que se seque al aire unos segundos antes de usarlo.
C)
Quite el precinto de papel del adaptador del vial. Si el precinto de papel no está totalmente sellado o si está roto, no utilice el adaptador del vial. No saque el adaptador del vial del capuchón protector con los dedos.
D)
Ponga el capuchón protector del adaptador del vial boca abajo y coloque el adaptador del vial a presión sobre el vial. Una vez unido, no retire del vial el adaptador del vial.
E)
Comprima ligeramente el capuchón protector entre los dedos pulgar e índice tal como se muestra. Quite el capuchón protector del adaptador del vial.
F)
Sujete la varilla del émbolo por el extremo ancho e inmediatamente conéctelo a la jeringa girándolo hacia la derecha dentro del émbolo en el interior de la jeringa precargada hasta que se sienta resistencia.





G)
Quite el tapón de la jeringa precargada doblándolo hacia abajo hasta que se rompa la perforación. No toque la punta de la jeringa debajo del tapón de la jeringa.
H)
Enrosque la jeringa precargada con firmeza en el adaptador del vial hasta que se sienta resistencia.
I)
Sujete la jeringa precargada ligeramente inclinada con el vial apuntando hacia abajo. Presione la varilla del émbolo para inyectar todo el disolvente en el vial.
J)
Mantenga la varilla del vial presionada y remueva suavemente el vial hasta que el polvo se haya disuelto. No agite el vial, ya que esto produciría espuma.
Se recomienda utilizar NovoEight inmediatamente después de la reconstitución. Para consultar las condiciones de conservación del medicamento reconstituido, ver sección 6.3.
Si se necesita una dosis mayor, repita los pasos A a J con más viales, adaptadores de viales y jeringas precargadas.
Administración de la solución reconstituida
K)


Mantenga la varilla del émbolo totalmente empujada hacia el interior. Gire la jeringa con el vial boca abajo. Deje de empujar la varilla del émbolo y deje que retroceda por sí misma mientras la solución reconstituida llena la jeringa. T ire de la varilla del émbolo ligeramente hacia abajo para hacer pasar la solución reconstituida hacia la jeringa.
Si solo necesita una parte del vial, utilice la escala de la jeringa para ver la cantidad de solución reconstituida que ha retirado, tal como le haya indicado su médico o su enfermera.
Mientras sujeta el vial cabeza abajo, golpee suavemente la jeringa para que las posibles burbujas suban hasta la parte superior. Empuje la varilla del émbolo lentamente hasta que se hayan eliminado las burbujas.
L)
Desenrosque el adaptador del vial con el vial.
Ahora NovoEight está listo para ser inyectado. Busque un lugar adecuado e inyecte lentamente NovoEight en la vena en un periodo de 2 a 5 minutos.
Eliminación
Después de la inyección, deseche con seguridad toda la solución NovoEight no utilizada, la jeringa con el juego de perfusión, el vial con el adaptador del vial y los demás residuos siguiendo las indicaciones de su farmacéutico.
No los deseche como residuos domésticos ordinarios.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novo Nordisk A/S Novo Allé
DK-2880 Bagsv^rd Dinamarca
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/888/005
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 13 Noviembre 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
^Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
NovoEight 3.000 UI polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial de polvo contiene 3.000 UI de factor VIII de coagulación humano (ADNr), turoctocog alfa.
NovoEight contiene aproximadamente 750 UI/ml de factor VIII de coagulación humano (ADNr), turoctocog alfa, después de la reconstitución.
La potencia (UI) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de NovoEight es aproximadamente de 8.300 UI/mg de proteína.
Turoctocog alfa (factor VIII de coagulación humano (ADNr)) es una proteína purificada que contiene 1.445 aminoácidos con una masa molecular aproximada de 166 kDA. Se produce mediante tecnología de ADN recombinante en células ováricas de hámster chino (CHO) y se prepara sin añadir ninguna proteína humana ni derivada de animal durante el proceso de cultivo de las células, la purificación o la formulación final.
Turoctocog alfa es un factor VIII de coagulación humano recombinante truncado de dominio B (el dominio B consiste en 21 aminoácidos del dominio silvestre de tipo B) sin ninguna otra modificación de la secuencia de aminoácidos.
Excipiente con efecto conocido:
0,31 mmol de sodio (7 mg) por ml de solución reconstituida.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo o masa friable de color blanco o ligeramente amarillo. Solución inyectable transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
T ratamiento y profilaxis de hemorragias en pacientes con hemofilia A (deficiencia congénita del factor VIII). NovoEight se puede usar en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de hemofilia.
Pacientes no tratados previamente
No se ha establecido todavía la seguridad y la eficacia de NovoEight en pacientes no tratados previamente.
No se dispone de datos.
Posología
La dosis y duración de la terapia de sustitución dependen de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que se corresponden con el estándar actual de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa como porcentaje (relativo al nivel en plasma humano normal) o en Unidades Internacionales (relativas al estándar internacional para el factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII presente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor VIII se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad plasmática de factor VIII en 2 UI/dl. La dosis requerida se determina utilizando la fórmula siguiente:
UI requeridas = peso corporal (kg) * aumento deseado de factor VIII (%) (UI/dl) x 0,5 (UI/kg por UI/dl).
La dosis y la frecuencia de la administración deberá individualizarse según la eficacia clínica en cada caso en particular.
En el caso de los eventos hemorrágicos siguientes, la actividad del factor VIII no deberá bajar del nivel de actividad plasmática indicado (en % del normal o UI/dl) durante el periodo correspondiente. La tabla siguiente se puede usar como guía para determinar la dosis en episodios hemorrágicos y cirugía:
T abla 1 Guía para determinar la dosis en episodios hemorrágicos y cirugía
Grado de la hemorragia/Tipo de Nivel de factor VIII Frecuencia de dosificación
procedimiento quirúrgico necesario (%) (UI/dl) (horas)/Duración del tratamiento
(días)
Hemorragia
Hemartrosis precoz, sangrado muscular 20-40 Repetir cada 12 - 24 horas al menos 1
o sangrado de la cavidad oral día hasta que el episodio hemorrágico
se haya resuelto, en función del dolor, o hasta la cicatrización de la herida.
Hemartrosis más extensa, sangrado 30-60 muscular o hematoma
Hemorragias con riesgo vital 60-100
Repetir la perfusión cada 12 - 24 horas durante 3 - 4 días o hasta que el dolor y la discapacidad aguda se hayan resuelto.
Repetir la perfusión cada 8 - 24 horas hasta que el riesgo desaparezca.
Cirugía
Cirugía menor, incluidas las 30-60 Cada 24 horas, al menos 1 día, hasta
extracciones dentales la cicatrización.
Grado de la hemorragia/Tipo de Nivel de factor VIII Frecuencia de dosificación
procedimiento quirúrgico necesario (%) (UI/dl) (horas)/Duración del tratamiento
(días)
Cirugía mayor 80-100 Repetir la perfusión cada 8-24 horas
(pre- y postoperatorio) hasta que se consiga una cicatrización
adecuada. A continuación, tratamiento durante por lo menos 7 días más para mantener una actividad del factor VIII del 30 % al 60 % (UI/dl).
Profilaxis
En la profilaxis a largo plazo para prevenir hemorragias en pacientes con hemofilia A grave. Las dosis habituales recomendadas son de 20 a 40 UI de factor VIII por kg de peso corporal a días alternos o de 20 a 50 UI de factor VIII por kg de peso corporal 3 veces por semana. En algunos casos, especialmente en los pacientes más jóvenes, puede ser necesario un intervalo de dosis más corto o una dosis mayor.
Supervisión del tratamiento
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis a administrar y la frecuencia de las inyecciones repetidas. Especialmente en el caso de las intervenciones de cirugía mayor, es indispensable controlar con precisión la terapia de sustitución mediante pruebas de coagulación (actividad plasmática del factor VIII). La respuesta individual de cada paciente frente al factor VIII puede variar y alcanzar distintos niveles de recuperación in vivo, y presentar semividas diferentes.
Cirugía
No existe experiencia en cirugía en pacientes pediátricos.
Pacientes de edad avanzada
No existe experiencia en pacientes de más de 65 años.
Población pediátrica
En la profilaxis a largo plazo para prevenir hemorragias en pacientes de menos de 12 años, las dosis recomendadas son de 25 a 50 UI de factor VIII por kg de peso corporal a días alternos o de 25 a 60 UI de factor VIII por kg de peso corporal 3 veces por semana. En pacientes pediátricos de más de 12 años, la dosis recomendada es la misma que para los adultos.
Forma de administración Vía intravenosa.
La velocidad de perfusión recomendada para NovoEight es de 1 - 2 ml/min. La velocidad se deberá determinar de acuerdo con el nivel de confort del paciente.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Reacciones alérgicas conocidas a las proteínas de hámster.
4.4 Advertencias y precauciones especiales de empleo Hipersensibilidad
Es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico a NovoEight. El producto contiene trazas de proteínas de hámster, las cuales pueden provocar reacciones alérgicas en algunos
69
pacientes. Si se produjesen síntomas de hipersensibilidad, se deberá instruir a los pacientes que deben interrumpir el uso del medicamento y ponerse inmediatamente en contacto con su médico. Se debe informar a los pacientes de los síntomas iniciales de las reacciones de hipersensibilidad, como urticaria localizada o generalizada, opresión en el pecho, respiración sibilante, hipotensión y anafilaxia.
En caso de shock, se seguirán las pautas médicas habituales para su tratamiento.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, y se cuantifican en Unidades Bethesda (UB) por ml de plasma usando el ensayo modificado. El riesgo de desarrollo de inhibidores está correlacionado con la exposición al factor VIII, y el riesgo mayor se produce durante los primeros 20 días de exposición. Los inhibidores raramente se forman después de los primeros 100 días de tratamiento.
Se han observado casos recurrentes de inhibidores (con título bajo), después de cambiar de un medicamento con factor VIII a otro a pacientes que habían recibido un tratamiento previo de más de 100 días de exposición y que tienen antecedentes de desarrollo de inhibidores. Por consiguiente se recomienda controlar a todos los pacientes atentamente para determinar la presencia de inhibidores después de cualquier cambio de medicamento.
En general, se deberá controlar atentamente a todos los pacientes tratados con medicamentos con factor VIII para determinar el desarrollo de inhibidores mediante la observación clínica y las pruebas de laboratorio adecuadas. Si no se obtienen los niveles plasmáticos de actividad del factor VIII esperados, o si no se controla la hemorragia con una dosis adecuada, se deberán realizar análisis para detectar la presencia de inhibidores del factor VIII. En pacientes con niveles elevados de inhibidores, es posible que el tratamiento con factor VIII no sea eficaz y se deberán considerar otras opciones terapéuticas. El tratamiento de estos pacientes debe hacerse bajo la dirección de un médico con experiencia en el cuidado de la hemofilia y los inhibidores del factor VIII.
Se recomienda encarecidamente registrar el nombre y el número de lote del medicamento cada vez que se administre NovoEight a un paciente con el fin de mantener un vínculo entre el paciente y el lote del medicamento.
Consideraciones relacionadas con los excipientes
Después de la reconstitución, este medicamento contiene 0,31 mmol de sodio (7 mg) por ml de solución reconstituida. Esto deberá tenerse en cuenta en pacientes que sigan una dieta controlada en sodio.
Complicaciones relacionadas con el catéter
Si se requiere un dispositivo de acceso venoso central (DAVC), se debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, como por ejemplo, infecciones locales, bacteriemia y trombosis en el lugar de inserción del catéter.
Población pediátrica
Las advertencias y precauciones indicadas son aplicables a adultos y niños.
4.5 Interacción con otros medicamentos y otras formas de interacción No se han realizado estudios de interacciones con NovoEight.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción animal con NovoEight. Teniendo en cuenta la escasa ocurrencia de hemofilia A en mujeres, no existe experiencia en relación con el uso de factor VIII durante el embarazo y la lactancia. Por consiguiente, solo se deberá usar factor VIII durante el embarazo y la lactancia si está estrictamente indicado.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NovoEight sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Resumen del perfil de seguridad
Hipersensibilidad o reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el punto de perfusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habones, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, cosquilleo, vómitos, sibilancias) se han observado raramente y, en algunos casos, puede progresar hasta una anafilaxia grave (incluido shock).
En muy raras ocasiones se ha observado el desarrollo de anticuerpos frente a la proteína de hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizadores (inhibidores) del factor VIII. Si se generan inhibidores de este tipo, la situación se pondrá de manifiesto por una respuesta clínica insuficiente. En tales casos, se recomienda ponerse en contacto con un centro especializado en hemofilia.
T abla de reacciones adversas
La tabla que se muestra a continuación sigue la clasificación de sistemas de órganos de MedDRA (clasificación por órganos y sistemas (SOC) y nivel de término preferente).
Las frecuencias se han evaluado conforme a la convención siguiente: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); raras (> 1/10.000 a < 1/1.000); muy raras (< 1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
En cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
T abla 2 Frecuencia de los reacciones adversas en ensayos clínicos
|
Clasificación por órganos y sistemas |
Frecuencia* |
Reacción adversa |
|
Trastornos psiquiátricos |
Poco frecuentes |
Insomnio |
|
Trastornos del sistema nervioso |
Poco frecuentes |
Cefalea, mareo |
|
Trastornos cardiacos |
Poco frecuentes |
Taquicardia sinusal |
|
Trastornos vasculares |
Poco frecuentes |
Hipertensión, linfoedema |
|
Trastornos hepatobiliares |
Frecuentes |
Enzimas hepáticas elevadas** |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
Erupción |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Poco frecuentes |
Rigidez musculoesquelética, artropatía, dolor en las extremidades, dolor musculoesquelético |
|
Trastornos generales y alteraciones en el lugar de administración |
Frecuentes |
Reacciones en la zona de inyección*** |
|
Poco frecuentes |
Fatiga, sensación de calor, edema periférico, pirexia | |
|
Exploraciones complementarias |
Poco frecuentes |
Frecuencia cardiaca aumentada |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Poco frecuentes |
Contusión |
Cálculo basado en el número total de pacientes distintos en todos los estudios clínicos (214).
Entre las enzimas hepáticas elevadas se encuentran la aminotransferasa, aspartato aminotransferasa, gamma glutamiltransferasa y bilirrubina.
*** Entre las reacciones en la zona de inyección se encuentran eritema, extravasación y prurito en la zona de inyección.
Descripción de las reacciones adversas señaladas
Durante todos los estudios clínicos con NovoEight, se han notificado un total de 30 reacciones adversas en
19 de 214 pacientes expuestos a NovoEight. Las reacciones adversas de las que se informó con mayor frecuencia fueron reacciones en la zona de inyección y enzimas hepáticas elevadas. De las 30 reacciones adversas, 2 de ellas se notificaron en 1 de 31 pacientes de menos de 6 años de edad, ninguna entre los pacientes de 6 a 18 años de edad y 28 en 18 de 127 adultos.
Población pediátrica
En estudios clínicos con 63 pacientes pediátricos de entre 0 y 12 años de edad y 24 adolescentes entre 12 y 18 años de edad con hemofilia A grave, no se observó ninguna diferencia en el perfil de seguridad de NovoEight entre pacientes pediátricos y adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han observado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos, factor VIII de la coagulación sanguínea, código ATC: B02BD02.
Mecanismo de acción
NovoEight contiene turoctocog alfa, un factor VIII de coagulación humano (ADNr) con un dominio B truncado. Esta glicoproteína tiene la misma estructura que el factor VIII cuando se activa, y modificaciones postranslacionales similares a las de la molécula derivada del plasma. Se ha observado que el punto de sulfatación de tirosina presente en Tyr1680 (longitud completa nativa), que es importante para el enlace con el factor de von Willebrand, está totalmente sulfatado en la molécula de turoctocog alfa. Cuando se administra mediante perfusión en un paciente con hemofilia, el factor VIII se enlaza con el factor de von Willebrand endógeno en la circulación del paciente. El complejo factor VIII/factor von Willebrand consiste en dos moléculas (factor VIII y factor de von Willebrand) con funciones fisiológicas distintas. El factor VIII activado actúa como cofactor del factor IX activado, y acelera la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo. La hemofilia es una alteración hereditaria vinculada al sexo de la coagulación de la sangre debida a niveles reducidos de factor VIII:C y da como resultado hemorragias difusas en articulaciones, músculos u órganos internos, ya sea de forma espontánea como tras un accidente o un trauma quirúrgico. La terapia de sustitución, aumenta los niveles plasmáticos de factor VIII, lo cual permite corregir temporalmente la deficiencia del factor y la tendencia al sangrado.
Eficacia clínica
Se han realizado tres ensayos multicéntricos, abiertos y no controlados para evaluar la seguridad y la eficacia de NovoEight para la prevención y el tratamiento de sangrado en pacientes tratados previamente con hemofilia A grave (actividad de FVEI < 1 %). Los ensayos incluyeron a 213 pacientes expuestos; 150 pacientes adolescentes o adultos sin inhibidores desde la edad de 12 años (> 150 días de exposición) y 63 pacientes pediátricos sin inhibidores de menos de 12 años de edad (> 50 días de exposición). 187 de 213 pacientes continuaron en el ensayo ampliado de seguridad. Se observó que el tratamiento con NovoEight es seguro y tiene el efecto hemostático y preventivo deseado. Durante una exposición acumulada de 54.000 días (correspondientes a 342 años paciente), no se observó el desarrollo de inhibidores del factor VIII en los ensayos clínicos en fase 3a con pacientes tratados previamente. De los 1.377 sangrados observados en 177 de los 213 pacientes, 1.244 (90,3 %) de los sangrados se resolvieron con 1 o 2 perfusiones de NovoEight.
|
Tabla 3 Consumo de turoctocog alfa y tasas de éxito glo |
bales | ||||
|
Niños pequeños (0 - <6 años) |
Niños mayores (6 - <12 años) |
Adolescentes (12 -<18 años) |
Adultos (>18 años) |
Total | |
|
Número de pacientes |
31 |
32 |
24 |
126 |
213 |
|
Dosis empleada como prevención por paciente (UI/kg PC) Valor medio (DE) Mín; Máx |
40,1 (8,5) 26,5 ; 57,3 |
36,6 (9,0) 24,9 ; 57,9 |
27,0 (7,6) 20,5 ; 46,9 |
26,9 (6,9) 20,0 ; 50,8 |
30,3 (9,2) 20,0 ; 57,9 |
|
Dosis utilizada para el tratamiento del sangrado (UI/kg BW) Valor medio (DE) Mín; Máx |
44,4 (17,9) 25,9 ; 193,8 |
40,0 (10,4) 25,5 ; 65,5 |
28,2 (10,2) 12,4 ; 76,8 |
33,8 (11,9) 9,3 ; 104,0 |
34,5 (12,6) 9,3 ; 193,8 |
|
Número de infusiones para el tratamiento del sangrado Valor medio (DE) Mín; Máx |
1,2 (0,6) 1 ; 6 |
1,5 (1,1) 1 ; 8 |
1,7 (1,3) 1 ; 9 |
1,5 (2,4) 1 ; 49 |
1,5 (2,1) 1 ; 49 |
|
T asa de éxito* % |
92,9 % |
88,9 % |
79,7 % |
85,6 % |
85,9 % |
PC: Peso corporal, DE: Desviación estándar *El éxito se define como “Excelente” o “Bueno”.
En total se realizaron 14 intervenciones quirúrgicas en 14 pacientes, de las cuales 13 fueron de cirugía mayor y 1 de cirugía menor. La hemostasis fue correcta en todas las intervenciones quirúrgicas y no se observó ningún fallo de tratamiento.
5.2 Propiedades farmacocinéticas
T odos los estudios de farmacocinética con turoctocog alfa se realizaron en pacientes tratados previamente con hemofilia A grave (FVHI < 1 %). El análisis de las muestras de plasma se realizó mediante análisis de coagulación de una fase y análisis cromogénico.
En un estudio internacional que implicó a 36 laboratorios, se evaluó el rendimiento de NovoEight en ensayos de FVHI:C y se comparó con un medicamento comercializado de FVIII recombinante de longitud completa. El estudio reveló que se obtienen resultados comparables y coherentes con ambos productos y que NovoEight se puede medir en plasma de forma fiable sin necesidad de usar un estándar aparte para NovoEight.
Los parámetros farmacocinéticos de NovoEight en una sola dosis se indican en la T abla 4 para el análisis de coagulación y en la Tabla 5 para el análisis cromogénico.
Tabla 4 Farmacocinética de una sola dosis de turoctocog alfa en pacientes con hemofilia A grave (FVIII < 1 %), análisis de coagulación___
|
Parámetro |
0 - <6 años |
6 - <12 años |
>12 años |
|
n = 14 |
n = 14 |
n = 33 | |
|
Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) | |
|
Recuperación incremental (UI/ml)/(UI/kg) |
0,018 (0,007) |
0,020 (0,004) |
0,022 (0,004) |
|
AUC ((UI*h)/ml) |
9,92 (4,11) |
11,09 (3,74) |
15,26 (5,77) |
|
CL (ml/h/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
|
t/ (h) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
|
Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
|
Cmax (UI/ml) |
1,00 (0,58) |
1,07 (0,35) |
1,226 (0,41) |
|
Tiempo medio de residencia (h) |
9,63 (2,50) |
9,91 (2,57) |
14,19 (5,08) |
Tabla 5 Farmacocinética de una sola dosis de turoctocog alfa en pacientes con hemofilia A grave (FVIII < 1 %), análisis cromogénico___
|
Parámetro |
0 - <6 años |
6 - <12 años |
>12 años |
|
n = 14 |
n = 14 |
n = 33 | |
|
Valor medio (DE) |
Valor medio (DE) |
Valor medio (DE) | |
|
Recuperación incremental (UI/ml)/(UI/kg) |
0,022 (0,006) |
0,025 (0,006) |
0,029 (0,006) |
|
AUC ((UI*h)/ml) |
12,23 (4,36) |
14,37 (3,48) |
19,63 (7,73) |
|
CL (ml/h/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
|
t/ (h) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
|
Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
|
Cmax (UI/ml) |
1,12 (0,31) |
1,25 (0,27) |
1,63 (0,50) |
|
Tiempo medio de residencia (h) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
Los parámetros farmacocinéticos fueron comparables entre pacientes pediátricos de menos de 6 años de edad y pacientes pediátricos mayores de 6 y menores de 12 años de edad. Se observó alguna variación entre los parámetros farmacocinéticos de NovoEight en pacientes pediátricos y adultos. Los valores mayores de CL y menores de t./2 en pacientes pediátricos en comparación con pacientes adultos con hemofilia A se pueden deber en parte al hecho conocido de que el volumen de plasma por kilogramo de peso corporal es mayor en los pacientes más jóvenes.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad y toxicidad a dosis repetidas.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Cloruro de sodio L-histidina Sacarosa Polisorbato 80 L-metionina
Cloruro de calcio dihidratado Hidróxido de sodio Ácido clorhídrico
Disolvente:
Cloruro de sodio
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
Sin abrir:
30 meses
Durante el periodo de validez, el medicamento se puede almacenar a temperatura ambiente (<30°C) durante un solo periodo de hasta 9 meses. Una vez que el producto se saca de la nevera, no se debe volver a refrigerar. Registrar la fecha de inicio del almacenamiento a temperatura ambiente en el embalaje del producto.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Después de la reconstitución:
Se ha demostrado la estabilidad química y física en uso durante 24 horas almacenado entre 2°C y 8°C y 4 horas almacenado a temperatura ambiente (<30°C).
Desde el punto de vista microbiológico, el medicamento se debería usar inmediatamente después de la reconstitución. Si no se utiliza de inmediato, los tiempos y las condiciones de almacenamiento en uso antes del uso son responsabilidad del usuario y no deberían superarse las 4 horas a temperatura ambiente (<30°C) o 24 horas entre 2°C y 8°C, a menos que la reconstitución se haya realizado en condiciones asépticas controladas y validadas.
El medicamento no utilizado almacenado a temperatura ambiente durante más de 4 horas se deberá desechar.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C). No congelar.
Para consultar la información de almacenamiento a temperatura ambiente y las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Cada envase de NovoEight 3.000 UI polvo y disolvente para solución inyectable contiene:
- 1 vial de vidrio (tipo I) con polvo y un tapón de goma de clorobutilo
- 1 adaptador de vial estéril para la reconstitución
- 1 jeringa precargada con 4 ml de disolvente con mecanismo de protección (polipropileno), un émbolo
de goma (bromobutilo) y un capuchón de aguja con tapón (bromobutilo)
- 1 varilla del émbolo (polipropileno).
6.6 Precauciones especiales de eliminación y otras manipulaciones
NovoEight se debe administrar por vía intravenosa después de la reconstitución del polvo con el disolvente suministrado en la jeringa. Después de la reconstitución, la solución es transparente o ligeramente opalescente. No utilice la solución si tiene aspecto turbio o presenta depósitos.
También necesitará un conjunto de perfusión (tubos y aguja mariposa), toallitas estériles con alcohol, gasas y tiritas. Estos dispositivos no se incluyen en el paquete NovoEight.
Utilice siempre una técnica aséptica.
Reconstitución
Saque el vial, el adaptador del vial y la jeringa precargada del envase. Deje la varilla del émbolo sin tocar en el envase. Lleve el vial y la jeringa precargada a temperatura ambiente. Puede hacerlo manteniendo ambos en las manos hasta que sienta que están a la misma temperatura que sus manos. No utilice ningún otro sistema para calentar el vial y la jeringa precargada.




|
0/ Xj Vví ♦ir |
L | |
|
; i |
L_ | |
B)
Quite el capuchón de plástico del vial. Si el capuchón de plástico está flojo o falta, no utilice el vial. Limpie el tapón de goma del vial con una toallita estéril con alcohol y deje que se seque al aire unos segundos antes de usarlo.
C)
Quite el precinto de papel del adaptador del vial.
Si el precinto de papel no está totalmente sellado o si está roto, no utilice el adaptador del vial. No saque el adaptador del vial del capuchón protector con los dedos.
D)
Ponga el capuchón protector del adaptador del vial boca abajo y coloque el adaptador del vial a presión sobre el vial. Una vez unido, no retire del vial el adaptador del vial.
E)
Comprima ligeramente el capuchón protector entre los dedos pulgar e índice tal como se muestra. Quite el capuchón protector del adaptador del vial.
F)
Sujete la varilla del émbolo por el extremo ancho e inmediatamente conéctelo a la jeringa girándolo hacia la derecha dentro del émbolo en el interior
de la jeringa precargada hasta que se sienta resistencia.





G)
Quite el tapón de la jeringa precargada doblándolo hacia abajo hasta que se rompa la perforación. No toque la punta de la jeringa debajo del tapón de la jeringa.
H)
Enrosque la jeringa precargada con firmeza en el adaptador del vial hasta que se sienta resistencia.
I)
Sujete la jeringa precargada ligeramente inclinada con el vial apuntando hacia abajo. Presione la varilla del émbolo para inyectar todo el disolvente en el vial.
J)
Mantenga la varilla del vial presionada y remueva suavemente el vial hasta que el polvo se haya disuelto. No agite el vial, ya que esto produciría espuma.
Se recomienda utilizar NovoEight inmediatamente después de la reconstitución. Para consultar las condiciones de conservación del medicamento reconstituido, ver sección 6.3.
Si se necesita una dosis mayor, repita los pasos A a J con más viales, adaptadores de viales y jeringas precargadas.
Administración de la solución reconstituida
K)


Mantenga la varilla del émbolo totalmente empujada hacia el interior. Gire la jeringa con el vial boca abajo. Deje de empujar la varilla del émbolo y deje que retroceda por sí misma mientras la solución reconstituida llena la jeringa. T ire de la varilla del émbolo ligeramente hacia abajo para hacer pasar la solución reconstituida hacia la jeringa.
Si solo necesita una parte del vial, utilice la escala de la jeringa para ver la cantidad de solución reconstituida que ha retirado, tal como le haya indicado su médico o su enfermera.
Mientras sujeta el vial cabeza abajo, golpee suavemente la jeringa para que las posibles burbujas suban hasta la parte superior. Empuje la varilla del émbolo lentamente hasta que se hayan eliminado las burbujas.
L)
Desenrosque el adaptador del vial con el vial.
Ahora NovoEight está listo para ser inyectado. Busque un lugar adecuado e inyecte lentamente NovoEight en la vena en un periodo de 2 a 5 minutos.
Eliminación
Después de la inyección, deseche con seguridad toda la solución NovoEight no utilizada, la jeringa con el juego de perfusión, el vial con el adaptador del vial y los demás residuos siguiendo las indicaciones de su farmacéutico.
No los deseche como residuos domésticos ordinarios.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novo Nordisk A/S Novo Allé
DK-2880 Bagsv^rd Dinamarca
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/888/006
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 13 Noviembre 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de los fabricantes del principio activo biológico
BioReliance Ltd
Todd Campus, West of Scotland Science Park,
Glasgow, G20 0XA Reino Unido
Novo Nordisk A/S Brennum Park DK-3400 Hillerod Dinamarca
Nombre y dirección del fabricante responsable de la liberación de los lotes
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dinamarca
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará el primer informe periódico de seguridad para este medicamento en un plazo de 8 meses después de la autorización. Posteriormente, el titular de la autorización de comercialización presentará informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, (párrafo 7), de la Directiva 2001/83/CE y publicados en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
NovoEight 250 UI polvo y disolvente para solución inyectable turoctocog alfa (factor VIII de coagulación humano (ADNr))
2. PRINCIPIO ACTIVO
Un ml de NovoEight contiene aproximadamente 62,5 UI de factor VIII de coagulación humano (ADNr), turoctocog alfa, después de la reconstitución
3. LISTA DE EXCIPIENTES
Polvo: cloruro sódico, L-histidina, sacarosa, polisorbato 80, L-metionina, cloruro de calcio dihidratado, hidróxido de sodio, ácido clorhídrico Disolvente: solución inyectable de cloruro sódico
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
El envase contiene: 1 vial de polvo, 4 ml de disolvente en una jeringa precargada, varilla del émbolo y adaptador del vial
5. FORMA Y VÍA DE ADMINISTRACIÓN_
Vía intravenosa
Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA
DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS_
Mantener fuera de la vista y del alcance de los niños
7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en el embalaje original para protegerlo de la luz
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dinamarca
EU/1/13/888/001
Lote
Medicamento sujeto a prescripción médica
NovoEight 250 UI
Vial
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
NovoEight 250 UI polvo para solución inyectable turoctocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
250 UI
6. OTROS
NovoEight 500 UI polvo y disolvente para solución inyectable turoctocog alfa (factor VIII de coagulación humano (ADNr))
2. PRINCIPIO ACTIVO
Un ml de NovoEight contiene aproximadamente 125 UI de factor VIII de coagulación humano (ADNr), turoctocog alfa, después de la reconstitución
3. LISTA DE EXCIPIENTES
Polvo: cloruro sódico, L-histidina, sacarosa, polisorbato 80, L-metionina, cloruro de calcio dihidratado, hidróxido de sodio, ácido clorhídrico Disolvente: solución inyectable de cloruro sódico
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
El envase contiene: 1 vial de polvo, 4 ml de disolvente en una jeringa precargada, varilla del émbolo y adaptador del vial
5. FORMA Y VÍA DE ADMINISTRACIÓN_
Vía intravenosa
Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA
DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS_
Mantener fuera de la vista y del alcance de los niños
7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en el embalaje original para protegerlo de la luz
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dinamarca
EU/1/13/888/002
Lote
Medicamento sujeto a prescripción médica
NovoEight 500 UI
Vial
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
NovoEight 500 UI polvo para solución inyectable turoctocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
500 UI
6. OTROS
NovoEight 1.000 UI polvo y disolvente para solución inyectable turoctocog alfa (factor VIII de coagulación humano (ADNr))
2. PRINCIPIO ACTIVO
Un ml de NovoEight contiene aproximadamente 250 UI de factor VIII de coagulación humano (ADNr), turoctocog alfa, después de la reconstitución
3. LISTA DE EXCIPIENTES
Polvo: cloruro sódico, L-histidina, sacarosa, polisorbato 80, L-metionina, cloruro de calcio dihidratado, hidróxido de sodio, ácido clorhídrico Disolvente: solución inyectable de cloruro sódico
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
El envase contiene: 1 vial de polvo, 4 ml de disolvente en una jeringa precargada, varilla del émbolo y adaptador del vial
5. FORMA Y VÍA DE ADMINISTRACIÓN_
Vía intravenosa
Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA
DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS_
Mantener fuera de la vista y del alcance de los niños
7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en el embalaje original para protegerlo de la luz
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dinamarca
EU/1/13/888/003
Lote
Medicamento sujeto a prescripción médica
NovoEight 1.000 UI
Vial
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
NovoEight 1.000 UI polvo para solución inyectable turoctocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
1.000 UI
6. OTROS
NovoEight 1.500 UI polvo y disolvente para solución inyectable turoctocog alfa (factor VIII de coagulación humano (ADNr))
2. PRINCIPIO ACTIVO
Un ml de NovoEight contiene aproximadamente 375 UI de factor VIII de coagulación humano (ADNr), turoctocog alfa, después de la reconstitución
3. LISTA DE EXCIPIENTES
Polvo: cloruro sódico, L-histidina, sacarosa, polisorbato 80, L-metionina, cloruro de calcio dihidratado, hidróxido de sodio, ácido clorhídrico Disolvente: solución inyectable de cloruro sódico
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
El envase contiene: 1 vial de polvo, 4 ml de disolvente en una jeringa precargada, varilla del émbolo y adaptador del vial
5. FORMA Y VÍA DE ADMINISTRACIÓN_
Vía intravenosa
Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA
DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS_
Mantener fuera de la vista y del alcance de los niños
7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en el embalaje original para protegerlo de la luz
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dinamarca
EU/1/13/888/004
Lote
Medicamento sujeto a prescripción médica
NovoEight 1.500 UI
Vial
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
NovoEight 1.500 UI polvo para solución inyectable turoctocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
1.500 UI
6. OTROS
NovoEight 2.000 UI polvo y disolvente para solución inyectable turoctocog alfa (factor VIII de coagulación humano (ADNr))
Un ml de NovoEight contiene aproximadamente 500 UI de factor VIII de coagulación humano (ADNr), turoctocog alfa, después de la reconstitución
Polvo: cloruro sódico, L-histidina, sacarosa, polisorbato 80, L-metionina, cloruro de calcio dihidratado, hidróxido de sodio, ácido clorhídrico Disolvente: solución inyectable de cloruro sódico
Polvo y disolvente para solución inyectable
El envase contiene: 1 vial de polvo, 4 ml de disolvente en una jeringa precargada, varilla del émbolo y adaptador del vial
5. FORMA Y VÍA DE ADMINISTRACIÓN
Vía intravenosa
Leer el prospecto antes de utilizar este medicamento
|
6. |
ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS |
|
Mantener fuera de la vista y del alcance de los niños | |
|
7. |
OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO |
|
8. |
FECHA DE CADUCIDAD |
CAD
Conservar en el embalaje original para protegerlo de la luz
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dinamarca
EU/1/13/888/005
Lote
Medicamento sujeto a prescripción médica
NovoEight 2.000 UI
Vial
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
NovoEight 2.000 UI polvo para solución inyectable turoctocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
2.000 UI
6. OTROS
NovoEight 3.000 UI polvo y disolvente para solución inyectable turoctocog alfa (factor VIII de coagulación humano (ADNr))
2. PRINCIPIO ACTIVO
Un ml de NovoEight contiene aproximadamente 750 UI de factor VIII de coagulación humano (ADNr), turoctocog alfa, después de la reconstitución
3. LISTA DE EXCIPIENTES
Polvo: cloruro sódico, L-histidina, sacarosa, polisorbato 80, L-metionina, cloruro de calcio dihidratado, hidróxido de sodio, ácido clorhídrico Disolvente: solución inyectable de cloruro sódico
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
El envase contiene: 1 vial de polvo, 4 ml de disolvente en una jeringa precargada, varilla del émbolo y adaptador del vial
5. FORMA Y VÍA DE ADMINISTRACIÓN_
Vía intravenosa
Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA
DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS_
Mantener fuera de la vista y del alcance de los niños
7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en el embalaje original para protegerlo de la luz
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Novo Nordisk A/S Novo Allé DK-2880 Bagsv^rd Dinamarca
EU/1/13/888/006
Lote
Medicamento sujeto a prescripción médica
NovoEight 3.000 UI
Vial
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
NovoEight 3.000 UI polvo para solución inyectable turoctocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
3.000 UI
6. OTROS
Jeringa precargada_
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Disolvente para NovoEight Cloruro de sodio 9 mg/ml
|
2. |
FORMA DE ADMINISTRACIÓN |
|
3. |
FECHA DE CADUCIDAD |
|
CAD | |
|
4. |
NÚMERO DE LOTE |
|
Lote | |
|
5. |
CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES |
|
4 ml | |
|
6. |
OTROS |
Novo Nordisk A/S
B. PROSPECTO
Prospecto: información para el usuario
NovoEight 250 UI polvo y disolvente para solución inyectable NovoEight 500 UI polvo y disolvente para solución inyectable NovoEight 1.000 UI polvo y disolvente para solución inyectable NovoEight 1.500 UI polvo y disolvente para solución inyectable NovoEight 2.000 UI polvo y disolvente para solución inyectable NovoEight 3.000 UI polvo y disolvente para solución inyectable
turoctocog alfa (factor VIII de coagulación humano (ADNr))
'VEste medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es NovoEight y para qué se utiliza
2. Qué necesita saber antes de empezar a usar NovoEight
3. Cómo usar NovoEight
4. Posibles efectos adversos
5. Conservación de NovoEight
6. Contenido del envase e información adicional 1. Qué es NovoEight y para qué se utiliza
NovoEight contiene la sustancia activa turoctocog alfa, factor VIII de coagulación humano. El factor VIII es una proteína que se encuentra de forma natural en la sangre y la ayuda a coagular..
NovoEight se utiliza para tratar y prevenir episodios de sangrado en pacientes con hemofilia A (deficiencia congénita de factor VIII) y se puede usar en todos los grupos de edad.
En los pacientes con hemofilia A, el factor VIII falta o bien no funciona correctamente. NovoEight sustituye este “factor VIII” que falta o no funciona correctamente y ayuda a que la sangre forme coágulos en el lugar del sangrado.
2. Qué necesita saber antes de empezar a usar NovoEight No use NovoEight:
• si es alérgico al principio activo o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6)
• si es alérgico a las proteínas de hámster.
No use NovoEight en cualquiera de los casos anteriores. Si no está seguro, hable con su médico antes de empezar a usar este medicamento.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar NovoEight.
Existe una probabilidad muy pequeña de que se produzca una reacción anafiláctica (una reacción alérgica repentina y grave) a NovoEight. Esté atento a los primeros signos de reacción alérgica como son erupción, habones, ronchas, picor generalizado, hinchazón de los labios y la lengua, dificultad para respirar, sibilancias, opresión en el pecho, sensación general de malestar y mareo.
Si se produce cualquiera de estos síntomas, detenga la inyección de inmediato y póngase en contacto con su médico.
Hable con su médico si cree que el sangrado no queda controlado con la dosis que se le administra, ya que puede haber distintos motivos para ello. Algunas personas que usan este medicamento pueden desarrollar anticuerpos frente al factor VIII (también conocidos como inhibidores del factor VIII). Los inhibidores del factor VIII reducen la eficacia de NovoEight para evitar o controlar el sangrado. Si esto sucede, es posible que necesite una dosis mayor de NovoEight u otro medicamento para controlar el sangrado. No aumente la dosis total de NovoEight para controlar el sangrado sin hablar con su médico. Debe comunicar a su médico si ha sido tratado previamente con medicamentos con factor VIII, especialmente si ha desarrollado inhibidores, puesto que el riesgo de que esto vuelva a suceder es mayor.
Uso de NovoEight con otros medicamentos
Informe a su médico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
Conducción y uso de máquinas
La influencia de NovoEight sobre la capacidad para conducir y utilizar máquinas es nula.
NovoEight contiene sodio
Este medicamento contiene 28 mg de sodio (7 mg/ml) después de su reconstitución.
Si sigue una dieta controlada en sodio, hable con su médico.
3. Cómo usar NovoEight
Un médico con experiencia en el tratamiento de pacientes con hemofilia A iniciará al tratamiento con NovoEight. Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico.
Su médico calculará la dosis para usted en función de su peso y de para qué se utilice el medicamento. Prevención del sangrado
La dosis habitual de NovoEight es de 20 a 50 unidades internacionales (UI) por kg de peso corporal. La inyección se administra cada 2 o 3 días. En algunos casos, especialmente en pacientes más jóvenes, pueden ser necesarias inyecciones más frecuentes o dosis mayores.
Tratamiento del sangrado
La dosis de NovoEight se calcula en función del peso corporal y los niveles de factor VIII que se desee alcanzar. El valor deseado de niveles de factor VIII depende de la gravedad y la localización del sangrado.
Uso en niños y adolescentes
NovoEight se puede usar en niños de todas las edades. En niños de menos de 12 años pueden ser necesarias
106
dosis mayores o inyecciones más frecuentes. Los niños de más de 12 años y los adolescentes pueden usar la misma dosis que los adultos.
Cómo se administra NovoEight
NovoEight debe inyectarse en una vena. Ver “Instrucciones de uso de NovoEight” para obtener más información.
Si usa más NovoEight del que debe
Si usa más NovoEight del que debería, póngase en contacto con su médico o acuda a un hospital directamente.
Si olvidó usar NovoEight
Si se ha saltado una dosis y no sabe cómo compensarlo, póngase en contacto con su médico.
Si interrumpe el tratamiento con NovoEight
Si interrumpe el tratamiento con NovoEight dejará de estar protegido frente al sangrado o es posible que un sangrado ya existente no se detenga. No interrumpa el tratamiento con NovoEight sin hablar antes con su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Con este medicamento se pueden producir los efectos adversos siguientes.
Si se producen reacciones alérgicas (reacciones anafilácticas) graves y repentinas (muy raras), es necesario detener la inyección inmediatamente. Si sufre alguno de los siguientes síntomas tempranos de reacciones alérgicas (de hipersensibilidad) (poco frecuentes), póngase en contacto con su médico inmediatamente:
• dificultad para respirar, respiración difícil o sibilancia
• opresión en el pecho
• hinchazón de los labios y la lengua
• erupciones, habones, ronchas o picor generalizado
• sensación de mareo o pérdida de conciencia
• presión arterial baja (piel pálida y fría, palpitaciones).
Los síntomas graves, incluida la dificultad al tragar o respirar y el enrojecimiento y la hinchazón de la cara o las manos, exigen tratamiento urgente de inmediato.
Si sufre una reacción alérgica grave, es posible que su médico cambie su medicamento.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas)
• análisis de sangre que indiquen cambios en el funcionamiento del hígado
• reacciones (rojez y picores) en el lugar donde se ha inyectado el medicamento
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas)
• sensación de cansancio
• cefalea
• sensación de mareo
• dificultad para dormir (insomnio)
• palpitaciones
• mayor presión sanguínea
• erupción
• fiebre
• sensación de calor
• rigidez muscular
• dolor muscular
• dolor en brazos y piernas
• hinchazón de brazos y piernas
• enfermedad articular
• cardenales.
Otros efectos adversos en niños y adolescentes
Los efectos adversos observados en niños y adolescentes son los mismos que se observan en adultos. Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de NovoEight
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en las etiquetas del vial y de la jeringa precargada después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en el embalaje original para protegerlo de la luz.
Conservar en nevera (entre 2°C y 8°C). No congelar.
Antes de reconstituirlo, el polvo de NovoEight se puede conservar a temperatura ambiente (hasta 30°C) durante un periodo inferior a 9 meses. Anote la fecha en la que empiece a conservar NovoEight a temperatura ambiente en el envase del producto. Una vez que se haya conservado a temperatura ambiente, no vuelva a conservar NovoEight en la nevera.
Una vez que se ha reconstituido, NovoEight se debe usar inmediatamente. Si no puede usar la solución de NovoEight reconstituida inmediatamente, deberá usarla en un plazo de 4 horas si está conservada a temperatura ambiente (hasta 30°C) y en un plazo de 24 horas si está conservada entre 2°C y 8°C. Si no va a utilizar el medicamento reconstituido inmediatamente, consérvelo en el vial. De lo contrario, dejará de ser estéril y podría provocar una infección. No conserve la solución a menos que siga los consejos de su médico.
El polvo del vial es un polvo blanco o ligeramente amarillo. Si el color del polvo ha cambiado, no lo utilice.
La solución reconstituida será entre transparente y ligeramente opalescente. No utilice este medicamento si observa que está turbio o contiene partículas visibles.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de NovoEight
- El principio activo es turoctocog alfa (factor VIII de coagulación humano (ADNr)). Cada vial de NovoEight contiene 250, 500, 1.000, 1.500, 2.000 o 3.000 UI de turoctocog alfa.
- Los demás componentes son L-histidina, sacarosa, polisorbato 80, cloruro de sodio, L-metionina, cloruro de calcio dihidratado, hidróxido de sodio y ácido clorhídrico.
- El componente del disolvente es cloruro de sodio 9 mg/ml.
Después de la reconstitución con el disolvente suministrado (solución inyectable con cloruro sódico 9 mg/ml (0,9 %)), la solución inyectable preparada contiene 62,5, 125, 250, 375, 500 o 750 UI de turoctocog alfa por ml, respectivamente (basado en la concentración de turoctocog alfa, eso es, 250, 500, 1.000, 1.500, 2.000 o 3.000 UI).
Aspecto del producto y contenido del envase
NovoEight está disponible en envases de 250, 500, 1.000, 1.500, 2.000 o 3.000 UI.
Cada envase de NovoEight contiene un vial con polvo blanco o ligeramente amarillo, una jeringa precargada con 4 ml de una solución incolora y transparente, una varilla de émbolo y un adaptador de vial.
Titular de la autorización de comercialización y responsable de la fabricación
Novo Nordisk A/S Novo Allé
DK-2880 Bagsv^rd, Dinamarca
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
LEA ATENTAMENTE ESTAS INSTRUCCIONES ANTES DE USAR NOVOEIGHT.
NovoEight se suministra en forma de polvo. Antes de la inyección (administración) se debe reconstituir con el disolvente suministrado en la jeringa. El disolvente es una solución de cloruro de sodio 9 mg/ml (0,9%). NovoEight reconstituido se debe inyectar en una vena (inyección intravenosa). Los elementos de este envase están diseñados para reconstituir e inyectar NovoEight.
También necesitará un conjunto de perfusión (tubos y aguja mariposa), toallitas estériles con alcohol, gasas y tiritas. Estos dispositivos no se incluyen en el paquete NovoEight.
No utilice el equipo sin haber recibido la formación adecuada de su médico o enfermero.
Lávese siempre las manos y asegúrese siempre de que el área a su alrededor esté limpia.
Al preparar e inyectar el medicamento directamente en las venas, es importante usar una técnica limpia y sin gérmenes (aséptica). Una técnica inadecuada puede introducir gérmenes capaces de infectar la sangre.
No abra el equipo hasta que esté listo para usarlo.
No utilice el equipo si se ha caído o se ha dañado. Utilice un envase nuevo en su lugar.
No use el equipo si ha caducado. Utilice un envase nuevo en su lugar. La fecha de caducidad está impresa en el embalaje exterior, el vial, el adaptador del vial y la jeringa precargada después de CAD.
No use el equipo si sospecha que está contaminado. Utilice un envase nuevo en su lugar.
No deseche ningún elemento hasta que se haya inyectado la solución reconstituida.
El equipo es de un solo uso.
Co ntenido
El envase contiene:
• 1 vial con polvo NovoEight
• 1 adaptador del vial
• 1 jeringa precargada con disolvente
• 1 varilla del émbolo (colocada debajo de la jeringa)
Aspecto general
Vial con polvo NovoEight
Capuchón
de plástico Tapón de goma
(debajo del capuchón de plástico)

Adaptador del vial
Cubierta protectora

Jeringa precargada con disolvente
Varilla del émbolo
Punta de la jeringa (debajo del tapón
Émbolo
|
ñ | |||
|
El |
A__i |
U ^3 | |
Tapón de la jeringa
Rosca
Extremo
ancho
1. Prepare el vial y la jeringa
• Prepare el número de envases de NovoEight que necesite.
• Compruebe la fecha de caducidad.
• Compruebe el nombre, la concentración y el color del envase para comprobar que contenga el producto correcto.
• Lávese las manos y séquelas correctamente con una toalla limpia o al aire.
• Saque el vial, el adaptador del vial y la jeringa precargada del envase. Deje la varilla del émbolo sin tocar en el envase.
• Lleve el vial y la jeringa precargada a temperatura ambiente. Puede hacerlo manteniendo ambos en las manos hasta que sienta que están a la
_misma temperatura que sus manos.

No utilice ningún otro sistema para calentar el vial y la jeringa precargada.
Quite el capuchón de plástico del vial. Si el capuchón de plástico está flojo o falta, no utilice el vial.
Limpie el tapón de goma con una toallita estéril con alcohol y deje que se seque al aire unos segundos antes de usarlo para asegurarse de que esté tan libre de gérmenes como sea posible.
No toque el tapón de goma con los dedos, ya que esto puede transferir gérmenes.

2. Coloque el adaptador del vial
• Quite el precinto de papel del
adaptador del vial.
Si el precinto de papel no está totalmente sellado o si está roto, no utilice el adaptador del vial.
No saque el adaptador del vial del capuchón protector con los dedos. Si
toca la espiga del adaptador del vial, puede transferirle gérmenes de sus dedos.

Ponga el vial sobre una superficie plana y dura.
Ponga el capuchón protector del adaptador del vial boca abajo y
coloque el adaptador del vial a presión sobre el vial.
Una vez unido, no retire del vial el adaptador del vial.

Comprima ligeramente el capuchón protector entre los dedos pulgar e índice tal como se muestra.
Quite el capuchón protector del
adaptador del vial.
No quite el adaptador del vial al
quitar el capuchón protector .

3. Monte la varilla del émbolo y la jeringa
Sujete la varilla del émbolo por el extremo ancho y sáquela del envase.
No toque los lados ni la rosca de la varilla del émbolo. Si toca los lados o la rosca, les puede transferir gérmenes de sus dedos.
Conecte inmediatamente la varilla del émbolo con la jeringa girándola hacia la derecha dentro del émbolo en el interior de la jeringa precargada hasta que note resistencia.

Quite el tapón de la jeringa de la
jeringa precargada doblándolo hacia abajo hasta que se rompa la perforación.
No toque la punta de la jeringa debajo del tapón de la jeringa. Si
toca la punta de la jeringa, puede le transferir gérmenes de sus dedos.
Si el capuchón de la jeringa está flojo o falta, no utilice la jeringa precargada.
Enrosque la jeringa precargada con firmeza en el adaptador del vial hasta que se sienta resistencia.
4. Reconstituya el polvo con el disolvente
• Sujete la jeringa precargada ligeramente inclinada con el vial apuntando hacia abajo.
• Presione la varilla del émbolo para inyectar todo el disolvente en el vial.


Mantenga la varilla del émbolo presionada y remueva suavemente el vial hasta que el polvo se haya disuelto.
No agite el vial, ya que esto produciría espuma.
Compruebe la solución reconstituida. Debe ser transparente o ligeramente opalescente (un poco menos transparente). Si se ve turbia o contiene partículas visibles, no la utilice. Utilice un envase nuevo en su lugar.

Se recomienda usar NovoEight inmediatamente después de reconstituirlo, ya que, si se deja, el medicamento puede dejar de ser estéril y podría causar infecciones.
Si no puede usar la solución de NovoEight reconstituida inmediatamente, deberá usarla en un plazo de 4 horas si está conservada a temperatura ambiente (hasta 30°C) y en un plazo de 24 horas si está conservada entre 2°C y 8°C. Conserve el producto reconstituido en el vial.
No congelar la solución de NovoEight reconstituida ni la guarde en jeringas.
No guardar la solución sin el consejo de su médico.
Guardar la solución de NovoEight reconstituida alejada de la luz directa.
®
Si su dosis requiere más de un vial, repita los pasos A a J con más viales, adaptadores de vial y jeringas precargadas hasta alcanzar la dosis necesaria.
Mantenga la varilla del émbolo empujada totalmente hacia dentro.
Ponga la jeringa con el vial cabeza abajo.
Deje de empujar la varilla del émbolo y deje que retroceda por sí
misma mientras la solución reconstituida llena la jeringa.
Tire de la varilla del émbolo ligeramente hacia abajo para hacer pasar la solución reconstituida hacia la jeringa.
Si solo necesita una parte del vial, utilice la escala de la jeringa para ver la cantidad de solución reconstituida que ha retirado, tal como le haya indicado su médico o su enfermera.

Si en algún momento hay demasiado aire en la jeringa, inyecte el aire de nuevo en el vial.
Mientras sujeta el vial cabeza abajo, golpee suavemente la jeringa para que las posibles burbujas suban hasta la parte superior.
Empuje la varilla del émbolo
lentamente hasta que se hayan eliminado las burbujas.
Desenrosque el adaptador del vial
con el vial.
No toque la punta de la jeringa. Si
toca la punta de la jeringa, puede le transferir gérmenes de sus dedos.

5. Inyecte la solución reconstituida
Ahora NovoEight está listo para ser inyectado en una vena.
• Inyecte la solución reconstituida siguiendo las instrucciones de su médico o enfermero.
• Inyecte lentamente durante de 2 a 5 minutos.
• No mezcle NovoEight con ninguna otra perfusión ni medicamento por vía intravenosa.
Inyección de NovoEight a través de conectores sin aguja para catéteres intravenosos (IV)
Precaución: la jeringa precargada es de cristal y está diseñada para ser compatible con conexiones luer-lock estándar. Algunos conectores sin aguja que presentan una espiga interna, son incompatibles con la jeringa precargada. Esta incompatibilidad puede impedir la administración del medicamento y/o originar un daño del conector sin aguja.
Inyección de la solución a través de un dispositivo de acceso venoso central (DAVC) como un catéter venoso central o un puerto subcutáneo:
• Utilice una técnica limpia y libre de gérmenes (aséptica). Siga las instrucciones para el uso adecuado de su conector y DAVC con el asesoramiento de su médico o enfermero.
• La inyección en un DAVC puede requerir utilizar una jeringa de plástico estéril de 10 ml para extraer la solución reconstituida. Esto debe llevarse a cabo justo después del paso J.
• Si es necesario enjuagar la línea DAVC antes o después de la inyección de NovoEight, utilice solución inyectable de cloruro de sodio 9 mg/ml.
Eliminación
• Después de la inyección, deseche con seguridad toda la solución NovoEight no utilizada, la jeringa con el juego de _perfusión, el vial con el adaptador del
|
vial y los demás residuos siguiendo las indicaciones de su farmacéutico. No los deseche como residuos domésticos ordinarios. |
(33 /% * & | ||
|
No desmonte el equipo antes de eliminarlo. No reutilice el equipo. | |||
116
i