Mekinist 2 Mg 30 Comprimidos
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Mekinist 0,5 mg comprimidos recubiertos con película Mekinist 2 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Mekinist 0,5 mg comprimidos recubiertos con película
Cada comprimido recubierto con película contiene trametinib dimetil sulfóxido, equivalente a 0,5 mg de trametinib.
Mekinist 2 mg comprimidos recubiertos con película
Cada comprimido recubierto con película contiene trametinib dimetil sulfóxido, equivalente a 2 mg de trametinib.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película
Mekinist 0,5 mg comprimidos recubiertos con película
Los comprimidos son de color amarillo, ovalados, biconvexos, recubiertos con película, de 4,8 x 8,9 mm aproximadamente, marcados con ‘GS’ en una cara y ‘TFC’ en la cara opuesta.
Mekinist 2 mg comprimidos recubiertos con película
Los comprimidos son de color rosa, redondos, biconvexos, recubiertos con película, de 7,5 mm aproximadamente, marcados con ‘GS’ en una cara y ‘HMJ’ en la cara opuesta.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Trametinib en monoterapia o en combinación con dabrafenib está indicado para el tratamiento de pacientes adultos con melanoma no resecable o metastásico con mutación BRAF V600 (ver sección 4.4 y 5.1).
Trametinib en monoterapia no ha demostrado actividad clínica en pacientes que han progresado a un tratamiento previo con un inhibidor BRAF (ver sección 5.1).
4.2 Posología y forma de administración
El tratamiento con trametinib solo se debe iniciar y supervisar por un médico especializado en el uso de medicamentos anticancerígenos.
Antes de comenzar el tratamiento con trametinib, los pacientes deben tener un diagnóstico de mutación BRAF V600 positiva en el tumor, confirmado por un test validado.
Posología
La dosis recomendada de trametinib, tanto en monoterapia como en combinación con dabrafenib, es de 2 mg una vez al día. La dosis recomendada de dabrafenib, cuando se utiliza en combinación con trametinib, es de 150 mg dos veces al día.
Dosis olvidadas
Si olvida tomar una dosis de trametinib, tome la dosis olvidada solo en caso de que falten más de 12 horas hasta la siguiente dosis.
Si olvida tomar una dosis de dabrafenib, cuando se utiliza en combinación con trametinib, tome la dosis olvidada de dabrafenib solo en caso de que falten más de 6 horas hasta la siguiente dosis.
Duración del tratamiento
Se recomienda que los pacientes continúen el tratamiento con trametinib hasta que no obtengan un beneficio clínico o cuando se desarrolle una toxicidad intolerable.
Modificaciones de dosis
El manejo de las reacciones adversas puede requerir reducciones de dosis, interrupción o suspensión del tratamiento (ver Tablas 1 y 2).
No se recomiendan modificaciones de la dosis en el caso de reacciones adversas de carcinoma de células escamosas cutáneo (CCE) o por un nuevo melanoma primario (ver Ficha Técnica de dabrafenib para más detalles)
Tabla 1 Reducciones de dosis recomendadas
|
Nivel de dosis |
Dosis de Trametinib Utilizado en monoterapia o en combinación con dabrafenib |
Dosis de Dabrafenib* Unicamente cuando se utiliza en combinación con trametinib |
|
Dosis de inicio |
2 mg una vez al día |
150 mg dos veces al día |
|
1a reducción de dosis |
1,5 mg una vez al día |
100 mg dos veces al día |
|
2a reducción de dosis |
1 mg una vez al día |
75 mg dos veces al día |
|
3a reducción de dosis (únicamente en combinación) |
1 mg una vez al día |
50 mg dos veces al día |
|
No se recomienda realizar modificaciones de dosis por debajo de 1 mg de trametinib una vez al día, cuando se utiliza como monoterapia o en combinación con dabrafenib. No se recomienda realizar modificaciones de dosis por debajo de 50 mg de dabrafenib dos veces al día, cuando se utiliza en combinación con trametinib. | ||
|
*Por favor, para instrucciones de dosis en monoterapia con dabrafenib, consultar la sección “Posología y forma de administración” de la Ficha Técnica de dabrafenib. | ||
Esquema de modificaciones de dosis basadas en el grado de cualquier Acontecimiento Adverso (AA)
|
Grado (CTC-AA)* |
Modificaciones de dosis recomendadas para trametinib Utilizado en monoterapia o en combinación con dabrafenib |
|
Grado 1 o Grado 2 (Tolerable) |
Continuar el tratamiento y monitorizar a los pacientes en función de la clínica. |
|
Grado 2 (Intolerable) o Grado 3 |
Interrumpir el tratamiento hasta que la toxicidad sea de Grado 0 a 1, y reducir la dosis un nivel cuando se reinicie el tratamiento. |
|
Grado 4 |
Suspender permanentemente o interrumpir el tratamiento hasta que la toxicidad sea de Grado 0 a 1, y reducir la dosis un nivel cuando se reinicie el tratamiento. |
|
* Grado de intensidad de acontecimientos adversos clínicos según los criterios de Common Terminology Criteria for Adverse Events (CTC-AE) v4.0. | |
Tabla 2
Cuando una reacción adversa individual se maneja de manera efectiva, se puede considerar realizar un re-escalado de dosis, siguiendo las mismas pautas posológicas empleadas para las reducciones de dosis. La dosis de trametinib no debe exceder de 2 mg una vez al día.
Si apareciera toxicidad relacionada con el tratamiento cuando se utiliza en combinación con dabrafenib se debe suspender o interrumpir o reducir la dosis de los dos tratamientos simultáneamente. Solo en los casos de pirexia, de uveítis, de cáncer no cutáneo con mutación RAS positiva y prolongación del intervalo QT (principalmente relacionado con dabrafenib), de reducción en la fracción de eyección del ventrículo izquierdo (FEVI), de oclusión de las venas retinianas (OCV), de desprendimiento del epitelio pigmentario retiniano (DEPR) y de enfermedad pulmonar intersticial (EPI) / Pneumonitis (principalmente relacionado con trametinib), podría ser necesario que solo se modificara la dosis de uno de los dos tratamientos.
Excepciones de modificación de dosis (cuando se reduce la dosis de uno de los dos tratamientos) Pirexia
Cuando se utiliza trametinib junto con dabrafenib y la temperatura del paciente es >38,5°C, se debe consultar la Ficha Técnica de dabrafenib (sección 4.2.) para modificar la dosis de dabrafenib. No es necesario modificar la dosis de trametinib cuando se toma en combinación con dabrafenib.
Uveitis
Si el tratamiento local puede controlar la inflamación ocular, no es necesario hacer ningún ajuste de dosis para la uveítis. En el caso que no respondiera al tratamiento local ocular, se debe suspender dabrafenib hasta que se resuelva la inflamación ocular y se debe reiniciar con dabrafenib reducido en un nivel de dosis. No es necesario modificar la dosis de trametinib cuando se toma en combinación con dabrafenib (ver sección 4.4).
Cáncer no-cutáneo con mutación-RAS-positiva
En pacientes con un cáncer no cutáneo con mutación RAS positivo sopesar los beneficios y riesgos antes de continuar con el tratamiento con dabrafenib. No es necesario modificar la dosis de trametinib cuando se toma en combinación con dabrafenib.
Prolongación del intervalo QT
Si durante el tratamiento el intervalo QTc sobrepasa 500 msec, se debe consultar la Ficha Técnica de dabrafenib (sección 4.2) para modificar la dosis de dabrafenib. No es necesario modificar la dosis de trametinib cuando se toma en combinación con dabrafenib.
Reducción en la fracción de eyección del ventrículo izquierdo (FEVI)/Disfunción del ventrículo izquierdo
El tratamiento con trametinib se debe interrumpir en pacientes que han tenido de manera asintomática una reducción absoluta de >10% de la FEVI en comparación con la situación basal, y que está por debajo del límite inferior normal establecido (ver sección 4.4). No es necesario modificar la dosis de dabrafenib cuando se toma trametinib en combinación con dabratenib. Si se recuperan los valores de la FEVI, se puede reiniciar el tratamiento con trametinib, pero la dosis empleada se debe reducir un nivel y realizar una monitorización cuidadosa de los pacientes (ver sección 4.4).
Si la disfunción del ventrículo izquierdo es de Grado 3 o 4, o si la FEVI no recupera los valores normales, el tratamiento con trametinib se debe suspender permanentemente (ver sección 4.4).
Oclusión de las venas retinianas (OCV) y desprendimiento del epitelio pigmentario retiniano (DEPR) Si durante el tratamiento con trametinib los pacientes notifican nuevas alteraciones en la visión, como una disminución de la visión central, visión borrosa o pérdida de visión, se recomienda realizar de inmediato una evaluación oftalmológica. En pacientes diagnosticados de oclusión de las venas retinianas, el tratamiento con trametinib, ya sea en monoterapia o en combinación con dabrafenib, se debe suspender permanentemente. No es necesario modificar la dosis de dabrafenib cuando se toma trametinib en combinación con dabrafenib. En pacientes a los que se les diagnostica desprendimiento del epitelio pigmentario retiniano, se deben seguir los ajustes de dosis indicados en la Tabla 3 (ver sección 4.4).
Tabla 3 Modificaciones de dosis de trametinib recomendadas para pacientes con DEPR
|
DEPR de Grado 1 |
Continuar el tratamiento con trametinib realizando una revisión de retina mensual hasta que se resuelva. Si el desprendimiento del epitelio pigmentario retiniano empeora, seguir las instrucciones que aparecen a continuación y retirar el tratamiento con trametinib durante 3 semanas. |
|
DEPR de Grado 2-3 |
Retirar el tratamiento con trametinib durante 3 semanas. |
|
DEPR de Grado 2-3 que mejora a Grado 0-1 en el plazo de 3 semanas |
Reiniciar el tratamiento con trametinib a una dosis inferior (reducida en 0,5 mg), o suspender el tratamiento en aquellos pacientes que estén tomando 1 mg de trametinib una vez al día. |
|
DEPR Grado 2-3 que no mejora como mínimo a Grado 1 en el plazo de 3 semanas |
Suspender permanentemente el tratamiento con trametinib. |
Enfermedad pulmonar intersticial (EPI)/Pneumonitis
En espera de tener una confirmación clínica, se debe retirar el tratamiento con trametinib en pacientes con sospechas de padecer EPI o pneumonitis, incluyendo pacientes que presenten síntomas pulmonares nuevos o progresivos y signos de tos, disnea, hipoxia, derrame pleural o infiltrados. Se debe suspender el tratamiento con trametinib en pacientes diagnosticados con EPI o pneumonitis relacionada con el tratamiento. No es necesario modificar la dosis de dabrafenib cuando se toma trametinib en combinación con dabrafenib en los casos de enfermedad pulmonar intersticial o pneumonitis.
Insuficiencia renal
No se requieren ajustes de dosis en pacientes con insuficiencia renal leve o moderada (ver sección 5.2). No se dispone de datos del uso de trametinib en pacientes con insuficiencia renal grave, y por lo tanto, no se puede determinar la posible necesidad de ajuste inicial de dosis. Trametinib se debe utilizar con precaución en pacientes con insuficiencia renal grave cuando se administre como monoterapia o en combinación con dabrafenib.
Insuficiencia hepática
No se requieren ajustes de dosis en pacientes con insuficiencia hepática leve (ver sección 5.2). No existen datos clínicos en pacientes con insuficiencia hepática moderada o grave, y por lo tanto, no se ha podido determinar la posible necesidad de ajuste inicial de dosis. Trametinib se debe utilizar con precaución en pacientes con insuficiencia hepática moderada o grave cuando se administre como monoterapia o en combinación con dabrafenib.
Pacientes no caucásicos
No se ha establecido la eficacia y seguridad de trametinib en pacientes no caucásicos. No se dispone de datos.
Pacientes de edad avanzada
No se requieren ajustes de la dosis inicial en pacientes >65 años de edad. Los pacientes >65 años pueden necesitar ajustes de dosis (ver Tablas 1 y 2) con mayor frecuencia (ver sección 4.8).
Población pediátrica
No se ha establecido la eficacia y seguridad de trametinib en niños y adolescentes (<18 años de edad). No se dispone de datos. Los estudios en animales jóvenes han mostrado efectos adversos de trametinib que no se observaron en los animales adultos (ver sección 5.3).
Forma de administración
Los comprimidos de trametinb se deben tomar por vía oral con un vaso lleno de agua. Los comprimidos de trametinib no se deben masticar ni machacar. Los comprimidos de trametinib se deben de tomar sin alimentos, al menos 1 hora antes o 2 horas después de una comida.
Se recomienda que la dosis de trametinib se tome a la misma hora del día, todos los días. Cuando se tome trametinib en combinación con dabrafenib, la dosis diaria de trametinib se debe tomar a la vez que la dosis matutina o la dosis vespertina de dabrafenib.
Si el paciente vomita después de tomar trametinib, no debe volver a tomar la dosis, y debe esperar a la siguiente toma.
Por favor, consulte la Ficha Técnica de dabrafenib para información sobre la forma de administración cuando se toma en combinación con trametinib.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Cuando se da trametinib en combinación con dabrafenib, se debe consultar la Ficha Técnica de dabrafenib antes de comenzar el tratamiento. Por favor, consultar la Ficha Técnica de dabrafenib para información adicional sobre las advertencias y precauciones asociados al tratamiento con dabrafenib.
Test mutación BRAF V600
No se ha evaluado la eficacia y seguridad de trametinib en pacientes con resultado negativo en el test diagnóstico de la mutación BRAF V600.
No hay estudios clínicos en pacientes con mutación positiva de BRAF V600 con melanoma irresecable o metastásico, en los que se compare el tratamiento de trametinib en monoterapia frente a un inhibidor BRAF. En base a las comparaciones de los estudios de eficacia entre trametinib y los inhibidores de BRAF, los datos de supervivencia global y de supervivencia libre de progresión parecen ser similares, sin embargo, las tasas de respuesta global notificadas en pacientes tratados con trametinib fueron más bajas que en pacientes tratados con inhibidores de BRAF.
Trametinib en combinación con dabrafenib en pacientes que han progresado con un inhibidor de BRAF
Existen pocos datos de pacientes en combinación de trametinib con dabrafenib que han progresado a un tratamiento previo con un inhibidor de BRAF. Estos datos muestran que la eficacia de la combinación es menor en estos pacientes (ver sección 5.1). Por tanto se deben considerar otras opciones terapéuticas antes de tratar con la combinación a esta población tratada previamente con un inhibidor de BRAF. No se ha establecido la secuencia de tratamientos tras progresión con un tratamiento inhibidor de BRAF.
Trametinib en combinación con dabrafenib en pacientes con metástasis en el cerebro
La seguridad y eficacia de la combinación de trametinib y dabrafenib no se ha evaluado en pacientes con melanoma BRAF v600 positivo con metástasis en el cerebro.
Nuevo cáncer
Pueden aparecer nuevos cánceres, cutáneos y no cutáneos, cuando trametinib se utiliza en combinación con dabrafenib.
Carcinoma cutáneo de células escamosas (CCE)
Se han notificado casos de CCE (incluido queratoacantoma) en pacientes que estaban en tratamiento con trametinib en combinación con dabrafenib. Los casos de CCE se pueden controlar por extirpación sin necesidad de modificar el tratamiento. Por favor, consulte la Ficha Técnica de dabrafenib (sección 4.4).
Nuevo melanoma primario
Se han notificado nuevos casos de melanoma primario en pacientes que estaban en tratamiento con trametinib en combinación con dabrafenib. Los nuevos casos de melanoma primario se pueden controlar por escisión sin necesidad de modificar el tratamiento. Por favor, consulte la Ficha Técnica de dabrafenib (sección 4.4).
Cáncer no cutaneo
En base a su mecanismo de acción, dabrafenib puede aumentar el riesgo de cánceres no cutaneos cuando la mutación RAS está presente. Cuando trametinib se utiliza en combinación con dabrafenib, por favor, consulte la Ficha Técnica de dabrafenib (ver sección 4.4). No es necesario modificar la dosis de trametinib con cánceres RAS positivos cuando se toma en combinación con dabrafenib.
Hemorragias
Se han dado casos de hemorragias, incluidas hemorragias graves y mortales, en pacientes en tratamiento con trametinib en monoterapia y en combinación con dabrafenib (ver sección 4.8). La mayoría de los sangrados fueron moderados. Las hemorragias intracraneales mortales sucedieron en el 1% (3/209) de los pacientes tratados con trametinib en combinación con dabrafenib en el estudio MEK115306 y en <1% (3/350) de los pacientes en el estudio MEK116513. En estos ensayos clínicos de combinación de trametinib y dabrafenib, el tiempo medio de aparición de la primera hemorragia fue 94 días en ambos estudios de combinación de trametinib y dabrafenib. No se ha establecido la posibilidad de que se produzcan estos eventos en pacientes con metástasis cerebrales o con niveles bajos de plaquetas (<75.000), debido a que estos pacientes fueron excluidos de los ensayos clínicos. El riesgo de hemorragia se puede incrementar con el uso concomitante de tratamientos anticoagulantes o antiplaquetarios. Si se produce una hemorragia, se debe tratar a los pacientes según la práctica clínica adecuada.
Reducción de la FEVI/Disfunción del ventrículo izquierdo
Se ha notificado que trametinib disminuye la FEVI, cuando se utiliza en monoterapia o en combinación con dabrafenib (ver sección 4.8). En los ensayos clínicos, el tiempo medio de aparición de la primera disfunción del ventrículo izquierdo, fallo cardiaco y disminución de la FEVI fue entre 2 y 5 meses.
Trametinib se debe usar con precaución en pacientes en los que la función del ventriculo izquierdo este alterada. Los pacientes con disfunción del ventrículo izquierdo, fallo cardiaco de Clase II, III o IV según la New York Heart Association, síndrome coronario agudo durante los últimos 6 meses, aritmias clínicamente significativas no controladas e hipertensión no controlada, fueron excluidos de los ensayos clínicos y por lo tanto el uso seguro en esta población es desconocido. Se debe evaluar la FEVI a todos los pacientes, antes de iniciar el tratamiento con trametinib, un mes después de iniciar el tratamiento, y posteriormente en intervalos de aproximadamente 3 meses durante el tratamiento (ver sección 4.2 en relación a las modificaciones de dosis).
Ocasionalmente se han notificado casos de disfunción aguda, grave del ventrículo izquierdo debida a miocarditis en pacientes que han recibido trametinib en combinación con dabrafenib. Se observó una recuperación total cuando se interrumpió el tratamiento. Los médicos deben estar alerta sobre la posibilidad de que se produzca miocarditis en pacientes que han desarrollado nuevos signos o síntomas cardiacos o si éstos han empeorado.
Pirexia
En los ensayos clínicos con trametinib en monoterapia y en combinación con dabrafenib (ver sección 4.8) se ha notificado fiebre. La incidencia y gravedad de la pirexia aumenta con el tratamiento combinado (ver sección 4.4 de la Ficha Técnica de dabrafenib). En pacientes que reciben trametinib en combinación con dabrafenib, la pirexia podría ir acompañada de deshidratación e hipotensión grave y en algunos casos, podría provocar una insuficiencia renal aguda.
Cuando trametinib se utilice en combinación con dabrafenib y la temperatura del paciente es >38,5°C, por favor consulte la Ficha Técnica de dabrafenib (sección 4.2) para modificaciones de la dosis de dabrafenib. No es necesario modificar la dosis de trametinib cuando se toma en combinación con dabrafenib.
Hipertensión
Se han notificado elevaciones de la presión arterial asociadas al uso de trametinib en monoterapia y en combinación con dabrafenib, en pacientes con y sin hipertensión preexistente (ver sección 4.8). Se debe medir la presión arterial al inicio del tratamiento, llevar a cabo una monitorización durante el tratamiento con trametinib, y controlar la hipertensión con un tratamiento estándar apropiado.
Enfermedad pulmonar intersticial (EPI)/Pneumonitis
En un estudio clínico fase III, el 2,4% (5/211) de los pacientes tratados con trametinib en monoterapia desarrolló EPI o pneumonitis, y los 5 pacientes necesitaron ser hospitalizados. La mediana del tiempo hasta la primera aparición de EPI o pneumonitis fue de 160 días (rango: de 60 a 172 días). En los estudios MEK115306 y MEK116513, <1% (2/209) y el 1% (4/350) respectivamente, de los pacientes tratados con trametinib en combinación con dabrafemib desarrollaron neumonitis o EPI (ver sección 4.8).
En espera de tener una confirmación clínica, se debe retirar el tratamiento con trametinib en pacientes con sospecha de padecer EPI o pneumonitis, incluyendo pacientes que presenten síntomas pulmonares nuevos o progresivos y signos de tos, disnea, hipoxia, derrame pleural o infiltrados. Se debe suspender permanentemente el tratamiento con trametinib en pacientes diagnosticados con EPI o pneumonitis relacionada con el tratamiento (ver sección 4.2). Si trametinib se utilizara en combinación con dabrafenib, podría continuar con el tratamiento con dabrafenib a la misma dosis.
Alteraciones visuales
En pacientes tratados con trametinib en monoterapia como en combinación con dabrafenib, podrían aparecerles alteraciones visuales incluyendo oclusión de las venas retinianas y desprendimiento del epitelio pigmentario retiniano. En los ensayos clínicos con trametinib se han notificado síntomas de visión borrosa, disminución de la agudeza visual, y otros fenómenos visuales (ver sección 4.8). En los ensayos clínicos con trametinib en combinación con dabrafenib se han notificado uveítis e iriociclitis.
Trametinib no está recomendado en pacientes con historial de oclusión de las venas retinianas. No se ha establecido la seguridad de trametinib en sujetos con factores que predispongan a padecer oclusión de las venas retinianas, incluyendo glaucoma no controlado o hipertensión ocular, hipertensión no controlada, diabetes mellitus no controlada, o un historial de hiperviscosidad o sindromes de hipercoagulabilidad.
Si durante el tratamiento con trametinib los pacientes notifican nuevas alteraciones en la visión, como disminución de la visión central, visión borrosa o pérdida de visión, se recomienda realizar de inmediato una evaluación oftalmológica. En pacientes diagnosticados de desprendimiento del epitelio pigmentario retiniano, se deben seguir los ajustes de dosis indicados en la Tabla 3 (ver sección 4.2); si se diagnosticara uveítis, por favor refiérase a la sección 4.4. de la ficha técnica de dabrafenib. Se debe suspender permanentemente el tratamiento con trametinib, en pacientes diagnosticados de oclusión de las venas retinianas. Tras el diagnóstico de OCV o DEPR, no es necesario modificar la dosis de dabrafenib cuando se toma en combinación con trametinib. Tras el diagnóstico de uveitis, no es necesario modificar la dosis de trametinib cuando se toma en combinación con dabrafenib.
Erupción
Se han observado erupciones aproximadamente en el 60% de los pacientes de los estudios con trametinib en monoterapia y en un 25% de los pacientes de los estudios con trametinib y dabrafenib combinado, MEK115306 y MEK116513 (ver sección 4.8). La mayoría de estos casos fueron de Grado 1 o 2 y no requirieron interrupciones de tratamiento ni reducciones de dosis.
Rabdomiolisis
En pacientes tratados con trametinib en monoterapia o en combinación con dabrafenib (ver sección 4.8) se han notificado casos de rabdomiolisis. En algunos casos, los pacientes fueron capaces de continuar el tratamiento. En los casos más graves se requirió hospitalización, interrupción o suspensión permanente del tratamiento. Ante signos o síntomas de rabdomiolisis, se debe garantizar una evaluación clínica y recomendar el tratamiento adecuado.
Fallo renal
En los ensayos clínicos se ha identificado fallo renal en pacientes tratados con trametinib en combinación con dabrafenib. Por favor consulte la Ficha Técnica de dabrafenib (sección 4.4).
Pancreatitis
En los ensayos clínicos se ha notificado pancreatitis en pacientes tratados con trametinib en combinación con dabrafenib. Por favor consulte la Ficha Técnica de dabrafenib (sección 4.4).
Prolongación del intervalo QT
Si durante el tratamiento el intervalo QT sobrepasa 500 msec, por favor consulte la sección 4.4. de la Ficha Técnica de dabrafenib.
Acontecimientos hepáticos
En ensayos clínicos con trametinib en monoterapia como en combinación, se han notificado acontecimientos adversos hepáticos (ver sección 4.8). Se recomienda realizar una monitorización de la función hepática cada cuatro semanas durante 6 meses tras iniciar el tratamiento con trametinib en monoterapia o en combinación con dabrafenib. A partir ahí, la monitorización hepática puede continuar según indique la práctica clínica.
Insuficiencia hepática
Debido a que el metabolismo hepático y la secreción biliar son las principales rutas de eliminación de trametinib, la administración de trametinib se debe llevar a cabo con precaución en pacientes con insuficiencia hepática moderada o grave (ver secciones 4.2 y 5.2).
Trombosis venosa profunda/Embolismo pulmonar
Cuando trametinib se utiliza en monoterapia o en combinación con dabrafenib, puede aparecer embolismo pulmonar o trombosis venosa profunda. Si el paciente desarrolla síntomas de embolismo pulmonar o trombosis venosa profunda tales como respiración entrecortada, dolor en el pecho, hinchazón de brazos o piernas, debe buscar atención médica urgente. Interrumpir trametinib y dabrafenib de manera permanente por riesgo de muerte por embolismo pulmonar.
4.5 Interacción con otros medicamentos y otras formas de interacción
Efecto de otros medicamentos sobre trametinib
Debido a que trametinib se metaboliza principalmente vía deacetilación mediada por enzimas hidrolíticas (p. ej. carboxil-esterasas), es poco problable que su farmacocinética se vea afectada por otros agentes a traves de interacciones metabólicas (ver sección 5.2). No se puede descartar la interacción entre fármacos por la vía de estas enzimas hidrolíticas, la cual podría influenciar a la exposición a trametinib.
Trametinib, in vitro, es un sustrato del flujo de transportadores P-gp. Como no se puede excluir que una fuerte inhibición hepática de P-gp pudiera elevar los niveles de trametinib, es por lo que se recomienda precaución cuando se administre trametinib con medicamentos que sean inhibidores potentes de P-gp (p.ej. verapamilo, ciclosporina, ritonavir, quinidina, itraconazol).
En base a los datos obtenidos in vitro e in vivo, es poco probable que trametinib afecte de forma significativa a la farmacocinética de otros medicamentos por la vía de interacción con enzimas CYP o transportadores (ver sección 5.2). Trametinib puede producir una inhibición transitoria de los sustratos de BCRP en el intestino (por ejemplo, pitavastatina), que puede minimizarse con una escalada de dosis (diferencia de dos horas) de estos medicamentos y trametinib.
Combinación con dabrafenib
Cuando trametinib se utiliza en combinación con dabrafenib ver las interacciones en las secciones 4.4 y 4.5 de la Ficha Técnica de dabrafenib.
Efecto de los alimentos sobre trametinib
Debido al efecto de los alimentos sobre la absorción de trametinib, los pacientes deben tomar trametinib en monoterapia o en combinación con dabrafenib, al menos una hora antes o dos horas después de las comidas (ver sección 4.2 y 5.2).
Trastornos gastrointestinales
En pacientes tratados con trametinib en monoterapia o en combinación con dabrafenib (ver sección 4.8) se han notificado colitis y perforación gastrointestinal, incluyendo desenlace mortal. El tratamiento con trametinib en monoterapia o en combinación con dabrafenib debe ser usado con precaución en pacientes que presenten factores de riesgo de perforación gastrointestinal, incluyendo antecedentes de diverticulitis, metástasis en el tracto gastrointestinal y uso concomitante de medicamentos con riesgo conocido de perforación gastrointestinal.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil/Anticoncepción en mujeres
Se debe advertir a las pacientes femeninas en edad fértil sobre la utilización de un método anticonceptivo altamente eficaz durante el tratamiento con trametinib y durante los 4 meses siguientes a la finalización del tratamiento.
Actualmente se desconoce si los anticonceptivos hormonales se ven afectados por el uso de trametinib. Para evitar el embarazo, se debe aconsejar a las pacientes femeninas que utilicen anticonceptivos hormonales que utilicen algún otro método adicional o alternativo durante el tratamiento y durante los 4 meses siguientes a la suspensión del tratamiento con trametinib.
El uso de dabrafenib puede disminuir la eficacia de los anticonceptivos hormonales por lo que cuando trametinib se tome en combinación con dabrafenib se debe utilizar otro método anticonceptivo, tales como los métodos de barrera. Para más información consulte la Ficha Técnica de dabrafenib.
Embarazo
No hay estudios adecuados y bien controlados de trametinib en mujeres embarazadas. Los estudios en animales han mostrado toxicidad en la reproducción (ver sección 5.3). Trametinib, no se debe administrar a mujeres embarazadas o madres lactantes. Si se usa trametinib durante el embarazo o si la paciente se queda embarazada durante el tratamiento con trametinib, la paciente debe ser informada sobre los posibles riesgos para el feto.
Lactancia
Se desconoce si trametinib se excreta en la leche materna. Debido a que muchos medicamentos se excretan en la leche materna, no se puede descartar la existencia de riesgo para los lactantes. Se debe decidir si es necesario suspender la lactancia o suspender el tratamiento con trametinib, tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Fertilidad
No hay datos de trametinib en monoterapia ni en combinación con dabrafenib en seres humanos. En animales, no se han llevado a cabo estudios de fertilidad, pero se han observado efectos adversos sobre los organos reproductores femeninos (ver sección 5.3). Trametinib puede afectar a la fertilidad de los seres humanos.
Hombres que toman trametinib en combinación con dabrafenib
En animales a los que se admistró dabrafenib se ha observado efectos sobre la espermatogénesis. Se ha de advertir a los pacientes varones que tomen trametinib en combinación con dabrafenib del posible riesgo de deterioro de la espermatogénesis, que puede ser irreversible. Para más información consulte la Ficha Técnica de dabrafenib.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de trametinib sobre la capacidad para conducir y utilizar máquinas es pequeña. A la hora de considerar la capacidad del paciente para realizar tareas que requieran juicio, habilidades motoras o cognitivas, se deben tener en cuenta tanto el estado clínico del paciente como el perfil de reacciones adversas de trametinib. Los pacientes deben ser conscientes de la posibilidad de padecer fatiga, mareos o problemas oculares que pueden afectar a estas actividades.
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad de trametinib en monoterapia se ha evaluado en la población de seguridad integrada de 329 pacientes con melanoma metastásico tratados con 2 mg de trametinib una vez al día. De estos pacientes, 211 fueron tratados con trametinib en un estudio fase III abierto, aleatorizado en pacientes con melanoma con mutación BRAF V600 (ver sección 5.1). Las reacciones adversas más frecuentes (>20%) con trametinib incluyeron, erupción, diarrea, fatiga, edema periférico, náusea y dermatitis acneiforme.
La seguridad de trametinib en combinación con dabrafenib se ha evaluado en 2 estudios fase III,
MEK115306 y MEK116513, donde se ha realizado un análisis de la seguridad de trametinib en combinación con dabrafenib en 209 y 350 pacientes, respectivamente, con melanoma no resecable o metastásico con mutación BRAF V600 que recibieron tratamiento combinado (ver tratamiento combinado en la sección 5.1) de trametinib (2 mg una vez al día) con dabrafenib (150 mg dos veces al día). Las reacciones adversas más frecuentes (>20%) del tratamiento combinado de trametinib y dabrafenib son pirexia, fatiga, naúseas, cefalea, escalofríos, diarrea, prurito, artralgia, hipertensión, vómitos y tos.
Tabla de reacciones adversas
Las reacciones adversas que fueron notificadas se incluyen bajo la clasificación de órganos del sistema MedDRA, por frecuencia y por nivel de gravedad.
La siguiente clasificación se ha utilizado para ordenarlas por frecuencia:
Muy frecuentes: Frecuentes:
Poco frecuentes: Raras:
No conocida:
>1/10
>1/100 a <1/10 >1/1.000 a <1/100 >1/10.000 a <1/1.000
(no se pueden estimar a partir de los datos disponibles)
Las categorías han sido asignadas en base a las frecuencias absolutas procedentes de los datos de los ensayos clínicos. Dentro de cada frecuencia, las reacciones adversas se enumeran en orden decreciente de gravedad.
Trametinib en monoterapia
Tabla 4 Reacciones adversas que aparecieron en pacientes tratados con trametinib en la población de seguridad integrada (n=329)
|
Sistema de Clasificación de Órganos |
Frecuencia (todos los grados) |
Reacciones adversas |
|
Trastornos de la sangre y del sistema linfático |
Frecuentes |
Anemia |
|
Trastornos del sistema inmunológico |
Frecuentes |
Hipersensibilidada |
|
Trastornos del metabolismo y la nutrición |
Frecuentes |
Deshidratación |
|
Trastornos oculares |
Frecuentes |
Visión borrosa |
|
Edema periorbital | ||
|
Alteración visual | ||
|
Poco frecuentes |
Corioretinopatía | |
|
Papiledema | ||
|
Desprendimiento de retina | ||
|
Oclusión venosa retiniana | ||
|
Trastornos cardiacos |
Frecuentes |
Disfunción del ventrículo izquierdo |
|
Disminución de la fracción de eyección | ||
|
Bradicardia | ||
|
Poco frecuentes |
Fallo cardiaco | |
|
Trastornos vasculares |
Muy frecuentes |
Hipertensión |
|
Hemorragiab | ||
|
Frecuentes |
Linfoedema | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy frecuentes |
Tos |
|
Disnea | ||
|
Frecuentes |
Pneumonitis | |
|
Poco frecuentes |
Enfermedad pulmonar intersticial | |
|
Trastornos gastrointestinales |
Muy frecuentes |
Diarrea |
|
Náusea | ||
|
Vómitos | ||
|
Estreñimiento | ||
|
Dolor abdominal | ||
|
Boca seca | ||
|
Frecuentes |
Estomatitis | |
|
Poco frecuentes |
Perforación gastrointestinal | |
|
Colitis |
|
Trastornos de la piel y del tejido subcutáneo |
Muy frecuentes |
Erupción |
|
Dermatitis acneiforme | ||
|
Piel seca | ||
|
Prurito | ||
|
Alopecia | ||
|
Frecuentes |
Eritema | |
|
Síndrome de eritrodisestesia palmoplantar | ||
|
Fisuras de la piel | ||
|
Piel agrietada | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Poco frecuentes |
Rabdomiolisis |
|
Trastornos generales y alteraciones en el lugar de administración |
Muy frecuentes |
Fatiga |
|
Edema periférico | ||
|
Pirexia | ||
|
Frecuentes |
Edema facial | |
|
Inflamación de la mucosa | ||
|
Astenia | ||
|
Infecciones e infestaciones |
Frecuentes |
Foliculitis |
|
Paroniquia | ||
|
Celulitis | ||
|
Erupción pustular | ||
|
Exploraciones complementarias |
Muy frecuentes |
Aspartato aminotransferasa elevada |
|
Frecuentes |
Alanina aminotransferasa elevada | |
|
Fosfatasa alcalina en sangre elevada | ||
|
Creatina fosfoquinasa en sangre elevada | ||
|
Disminución del ritmo cardiaco | ||
|
a Se puede presentar con síntomas como fiebre, erupción, aumento de transaminasas hepáticas y alteraciones visuales. b Estos eventos incluyen, aunque no limitados a: epistaxis, hematoquecia, sangrado gingival, hematuria y hemorragia rectal, hemorroidal, gástrica, vaginal, conjuntival y hemorragia intracraneal y postoperatoria. | ||
Tratamiento combinado de trametinib y dabrafenib
Tabla 5 Reacciones adversas que aparecieron en los estudios fase III randomizados de la combinación MEK115306 (n = 209) y MEK116513a (n = 350)
|
Sistema de Clasificación de Órganos |
Frecuencia (todos los grados) |
Reacciones adversas |
|
Infecciones e infestaciones |
Muy frecuentes |
Infecciones del tracto urinario |
|
Nasofaringitis | ||
|
Frecuentes |
Celulitis | |
|
Foliculitis | ||
|
Paroniquia | ||
|
Erupción pustular | ||
|
Neoplasias benignas, malignas y no especificadas (incl quistes y pólipos) |
Frecuentes |
Carcinoma de células escamosasb |
|
Papilomac | ||
|
Queratosis seborreica | ||
|
Acrocordón (marcas en la piel) | ||
|
Poco frecuentes |
Nuevo melanoma primario | |
|
Trastornos de la sangre y del sistema linfático |
Muy frecuentes |
Neutropenia |
|
Frecuentes |
Anemia | |
|
Trombocitopenia | ||
|
Leucopenia |
|
Trastornos del sistema inmunológico |
Poco frecuentes |
Hipersensibilidad a medicamentos |
|
Trastornos del metabolismo y la nutrición |
Muy frecuentes |
Disminución del apetito |
|
Frecuentes |
Deshidratación | |
|
Hiponatremia | ||
|
Hipofosfatemia | ||
|
Hiperglucemia | ||
|
Trastornos del sistema nervioso |
Muy frecuentes |
Cefalea |
|
Mareo | ||
|
Trastornos oculares |
Frecuentes |
Visión borrosa |
|
Alteraciones visuales | ||
|
Poco frecuentes |
Coriorretinopatía | |
|
Uveitis | ||
|
Desprendimiento de retina | ||
|
Edema periorbital | ||
|
Trastornos cardiacos |
Frecuentes |
Disminución de la fracción de eyección |
|
Bradicardia | ||
|
No conocida |
Miocarditis | |
|
Trastornos vasculares |
Muy frecuentes |
Hipertensión |
|
Hemorragiad | ||
|
Frecuentes |
Hipotensión | |
|
Poco frecuentes |
Limfoedemaa | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy frecuentes |
Tos |
|
Frecuentes |
Disnea | |
|
Poco frecuentes |
Pneumonitis | |
|
Trastornos gastrointestinales |
Muy frecuentes |
Dolor abdominal |
|
Estreñimiento | ||
|
Diarrea | ||
|
Nausea | ||
|
Vómitos | ||
|
Frecuentes |
Sequedad de boca | |
|
Estomatitis | ||
|
Poco frecuentes |
Pancreatitis | |
|
Perforación gastrointestinal | ||
|
Colitis | ||
|
Trastornos hepatobiliares |
Muy frecuentes |
Alanina aminotransferasa elevada |
|
Aspartato aminotransferase elevada | ||
|
Frecuentes |
Fosfatasa alcalina en sangre elevada | |
|
Gamma-glutamiltransferasa elevada |
|
Trastornos de la piel y del tejido subcutáneo |
Muy frecuentes |
Piel seca |
|
Prurito | ||
|
Erupción | ||
|
Dermatitis acneiforme | ||
|
Frecuentes |
Eritema | |
|
Queratosis actinica | ||
|
Sudores nocturnos | ||
|
Hiperqueratosis | ||
|
Alopecia | ||
|
Síndrome de eritrodisestesia palmoplantar | ||
|
Lesiones de piel | ||
|
Hiperhidrosis | ||
|
Paniculitis | ||
|
Fisuras de la piel | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Muy frecuentes |
Artralgia |
|
Mialgia | ||
|
Dolor en las extremidades | ||
|
Frecuentes |
Espasmos musculares3 | |
|
Creatina fosfoquinasa en sangre elevada | ||
|
Trastornos renales y urinarios |
Poco frecuentes |
Fallo renala |
|
Nefritis | ||
|
Trastornos generales y alteraciones en el lugar de administración |
Muy frecuentes |
Fatiga |
|
Escalofríos | ||
|
Astenia | ||
|
Edema periférico | ||
|
Pirexia | ||
|
Frecuentes |
Inflamación de la mucosa | |
|
Síntomas gripales | ||
|
Edema facial | ||
|
Exploraciones complementarias |
Frecuentes |
Disminución del ritmo caridiaco |
|
a El perfil de seguridad de MEK116513 es en general similar al de MEK115306 con las siguientes excepciones: 1) Las siguientes reacciones adversas se presentan en mayor frecuencia en comparación con MEK115306: espasmos musculares (muy frecuentes); fallo renal y linfoedema (frecuentes); fallo renal agudo (poco frecuente); 2) Las siguientes reacciones adversas aparecieron en MEK116513 pero no en MEK115306: fallo cardiaco, disfunción del ventrículo izquierdo, enfermedad pulmonar intersticial, rabdomiolisis (poco frecuente). b CCE: CEE de la piel, CCE in situ (enfermedad de Bowen) y queratoacantoma c Papiloma, papiloma de piel d Sangrado de varios sitios, incluido hemorragia intracranial y sangrado grave y mortal | ||
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
Descripción de reacciones adversas seleccionadas
Nuevos cánceres
Pueden aparece nuevos cánceres, cutáneos y no cutáneos, cuando trametinib se utiliza en combinación con dabrafenib. Por favor, consulte la Ficha Técnica de dabrafenib.
Se han dado casos de hemorragias, incluidas hemorragias graves y mortales, en pacientes en tratamiento con trametinib en monoterapia y en combinación con dabrafenib. La mayoría de los sangrados fueron moderados. Las hemorragias intracraneales graves - mortales sucedieron en el 1% (3/209) de los pacientes tratados con trametinib en combinación con dabrafenib en el estudio MEK115306 y en <1% (3/350) de los pacientes en el estudio MEK116513. El riesgo de hemorragia se puede incrementar con el uso concomitante de tratamientos anticoagulantes o antiplaquetarios. Si se produce una hemorragia, se debe tratar a los pacientes según la práctica clínica adecuada (ver sección 4.4).
Reducción de la FEVI/Disfunción del ventrículo izquierdo
Se ha notificado que trametinib disminuye la FEVI, cuando se utiliza en monoterapia o en combinación con dabrafenib. En ensayos clínicos, el tiempo medio de aparición de la primera disfunción del ventrículo izquierdo, fallo cardiaco y disminución de la FEVI fue entre 2 y 5 meses. En 2 ensayos clínicos fase III se notificó que trametinib disminuye la FEVI en el 6 al 8% de los pacientes tratados con trametinib en combinación con dabrafenib, en la mayoría de los casos asintomáticos y reversibles. Los pacientes con FEVI menor que el límite inferior normal aceptado, no se incluyeron en los ensayos clínicos con trametinib.Trametinib se debe usar con precaución en pacientes con afecciones que puedan alterar la función del ventrículo izquierdo (ver secciones 4.2 y 4.4).
Pirexia
En los ensayos clínicos con trametinib en monoterapia y en combinación con dabrafenib se ha notificado fiebre; sin embargo, la incidencia y gravedad de la pirexia aumenta con el tratamiento combinado. Por favor, consulte las secciones 4.4 y 4.8 de la Ficha Técnica de dabrafenib.
Acontecimientos hepáticos
En ensayos clínicos con trametinib en monoterapia y en combinación con dabrafenib, se han notificado acontecimientos adversos hepáticos. De los eventos hepáticos, los más frecuentes fueron elevación de ALT y AST, y la mayoría fueron de Grado 1 o 2. Más del 90% de estos eventos hepáticos, sucedieron dentro de los primeros 6 meses de tratamiento con trametinib en monoterapia. Los eventos hepáticos fueron detectados en los ensayos clínicos mediante la monitorización realizada cada cuatro semanas. Se recomienda realizar una monitorización de la función hepática cada cuatro semanas durante 6 meses a los pacientes tratados con trametinib en monoterapia o en combinación con dabrafenib. A partir de ahí, la monitorización hepática puede continuar según indique la práctica clínica (ver sección 4.4).
Hipertensión
Se han notificado elevaciones de la presión arterial asociadas al uso de trametinib en monoterapia o en combinación con dabrafenib, en pacientes con y sin hipertensión preexistente. Se debe medir la presión arterial al inicio del tratamiento, llevar a cabo una monitorización durante el tratamiento con trametinib, y cuando proceda controlar la hipertensión con un tratamiento estándar (ver sección 4.4).
Enfermedad pulmonar intersticial (EPI)/Pneumonitis
Los pacientes en tratamiento con trametinib o en combinación con dabrafenib pueden desarrollar EPI o pneumonitis. En espera de tener una confirmación clínica, se debe retirar el tratamiento con trametinib en pacientes con sospecha de padecer EPI o pneumonitis, incluyendo pacientes que presenten síntomas pulmonares nuevos o progresivos y signos como tos, disnea, hipoxia, derrame pleural o infiltrados. Se debe suspender el tratamiento con trametinib en pacientes diagnosticados con EPI o pneumonitis relacionada con el tratamiento (ver secciones 4.2 y 4.4).
Alteraciones visuales
En pacientes tratados con trametinib se han observado alteraciones visuales, incluyendo oclusión de las venas retinianas y desprendimiento del epitelio pigmentario retiniano. En los ensayos clínicos con trametinib se han notificado síntomas de visión borrosa, disminución de la agudeza visual, y otras alteraciones visuales (ver secciones 4.2 y 4.4).
Erupción
Se han observado erupciones aproximadamente en el 60% de los pacientes de los estudios con trametinib en monoterapia y un 25% de los pacientes de los estudios con trametinib y dabrafenib combinado, MEK115306 y MEK116513. La mayoría de estos casos fueron de Grado 1 o 2 y no requirieron interrupciones del tratamiento ni reducciones de dosis (ver secciones 4.2 y 4.4).
Rabdomiolisis
En pacientes tratados solo con trametinib o en combinación con dabrafenib se han notificado casos de rabdomiolisis. Ante signos o síntomas de rabdomiolisis, se debe garantizar una evaluación clínica y recomendar el tratamiento adecuado (ver sección 4.4).
Pancreatitis
Se ha notificado pancreatitis con dabrafenib en combinación con trametinib. Por favor consulte la Ficha Técnica de dabrafenib.
Fallo renal
Se ha notificado fallo renal con dabrafenib en combinación con trametinib. Por favor consulte la Ficha Técnica de dabrafenib.
Poblaciones especiales
Pacientes de edad avanzada
En el estudio fase III en el que pacientes con melanoma irresecable o metastásico fueron tratados con trametinib (N = 211), 49 pacientes (23%) tenían >65 años, y 9 pacientes (4%) tenían >75 años. El porcentaje de sujetos que experimentaron acontecimientos adversos y acontecimientos adversos graves, fue similar en sujetos de <65 años y en sujetos >65 años de edad. Los pacientes >65 años presentaron una mayor probabilidad de experimentar acontecimientos adversos que conducían a la interrupción permanente del tratamiento, reducciones de dosis e interrupciones de dosis, en comparación con pacientes <65 años.
Los pacientes >65 años que participaron en los estudios de fase III, MEK115306 (n = 209) y MEK116513 (n = 350) con trametinib en combinación con dabrafenib en pacientes con melanoma no resecable o metastásico, fueron 56 pacientes (27%) y 77 pacientes (22%), respectivamente; los pacientes >75 años fueron 11 pacientes (5%) y 21 pacientes (6%), respectivamente. En ambos estudios, la proporción de pacientes que experimentaron acontecimientos adversos fue similar para los <65 años que para los >65 años. Sin embargo los pacientes >65 años fueron más propensos a sufrir efectos adversos, incluso tuvieron que interrumpir el tratamiento de forma permanente, reducir o interrumpir su dosis debido a los efectos adversos, que los pacientes <65 años.
Insuficiencia renal
No se requiere ajuste de dosis en pacientes con insuficiencia renal leve o moderada (ver sección 5.2). Trametinib se debe utilizar con precaución en pacientes con insuficiencia renal grave (ver secciones 4.2 y 4.4).
No se requiere ajuste de dosis en pacientes con insuficiencia hepática leve (ver sección 5.2). Trametinib se debe utilizar con precaución en pacientes con insuficiencia hepática moderada o grave (ver secciones 4.2 y 4.4).
4.9 Sobredosis
En los ensayos clínicos con trametinib en monoterapia se notificó un caso de sobredosis accidental, correspondiente a una única dosis de 4 mg. No se notificaron acontecimientos adversos como consecuencia de esta sobredosis de trametinib. En los ensayos clínicos con la combinación de trametinib y dabrafenib, se notificaron 11 pacientes con sobredosis de trametinib (4 mg); no se notificaron acontecimientos adversos graves. No existe un tratamiento específico para tratar la sobredosis de trametinib. Si se produce una sobredosis, el paciente debe ser tratado con medidas complementarias y llevar a cabo una monitorización adecuada según sea necesario.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agente antineoplásico, inhibidor de la proteína quinasa, código ATC: L01XE25
Mecanismo de acción
Trametinib es un inhibidor alostérico, reversible, altamente selectivo, de la señal extracelular activada por mitógenos que regula la activación y la actividad quinasa, de las quinasas MEK1 y MEK2. Las proteínas MEK con componentes de la vía de señalización extracelular relacionada con quinasas (ERK). En el melanoma y en otros tipos de cáncer, esta vía está activada frecuentemente por isoformas mutadas de BRAF que activan MEK. Trametinib inhíbe la activación de MEK por BRAF e inhíbe la actividad quinasa de MEK. Trametinib inhíbe el crecimiento de líneas celulares de melanoma con mutación BRAF V600, y ha demostrado efectos anti tumorales en modelos animales, en melanoma con mutación BRAF V600.
Combinación con dabrafenib
Dabrafenib es un inhibidor de quinasas RAF. Las mutaciones oncogénicas en BRAF conducen a la activación constitutiva de la vía RAS/RAF/MEK/ERK. Por tanto, trametinib y dabrafenib inhiben dos quinasas de la misma vía, MEK y RAF, por lo que la combinación proporciona una doble inhibición de esta vía. La combinación de trametinib con dabrafenib ha demostado actividad antitumoral in vitro en las lineas celulares con melanoma con la mutación BRAF V600 y ha retrasado la aparición de resistencias in vivo en injertos con melanoma con la mutación BRAF V600.
Determinación del estado de la mutación BRAF
Antes de empezar el tratamiento con trametinib o en combinación con dabrafenib, se debe confirmar que los pacientes tienen tumores con mutación positiva BRAF V600, mediante la realización de un test validado.
En los ensayos clínicos, la determinación de la mutación BRAF V600 se realizó de manera centralizada, y se utilizó un ensayo de mutación BRAF que se llevó a cabo con las muestras más recientes de tumor disponibles. Los tumores primarios o tumores procedentes de lugares donde se habían producido metástasis, fueron analizados mediante un ensayo validado de la reacción de la cadena de la polimerasa (PCR), desarrollado por Response Genetics Inc. Este ensayo fue específicamente diseñado para diferenciar las mutaciones V600E y V600K. Solamente aquellos pacientes con tumores con mutación positiva BRAF V600E o V600K fueron candidatos a participar en el estudio.
Posteriormente, todas las muestras de los pacientes fueron analizadas de nuevo utilizando el ensayo validado bioMerieux (bMx) THxID BRAF, que posee marcado CE. El ensayo bMx THxID BRAF es un ensayo de identificación de un alelo específico de PCR realizado sobre ADN extraído de tejido tumoral fijado con formalina y embebido en parafina (FFPE). Este ensayo se diseñó para detectar con alta sensibilidad las mutaciones BRAF V600E y V600K (menos de un 5% de secuencias V600E y V600K, sobre un panel de secuencias de tipo nativo a partir de ADN extraído de un tejido tumoral en FFPE). Los análisis de secuenciación retrospectivos y bidireccionales, realizados por el método Sanger en estudios clínicos y preclínicos, han demostrado que el ensayo también detecta las mutaciones menos frecuentes BRAF V600D y V600E/K601E con menor sensibilidad. De las muestras procedentes de estudios preclínicos y clínicos (n = 876) que presentaron mutación positiva por el test THxID BRAF y que posteriormente fueron secuenciadas utilizando el método de referencia, la especificidad del ensayo fue del 94%.
Efectos farmacodinámicos
Trametinib suprime los niveles de ERK fosforilado en líneas celulares de melanoma con mutación BRAF y en modelos xenográficos de melanoma.
En pacientes con melanoma con mutacion positiva BRAF y NRAS, la administración de trametinib provocó cambios de manera dosis dependiente en los biomarcadores tumorales, incluyendo la inhibición de ERK fosforilado, inhibición de Ki67 (un marcador de proliferación celular), e incrementos en p27 (un marcador de apoptosis). Tras la administración de dosis repetidas de 2 mg de trametinib una vez al día, la media de las concentraciones observadas sobrepasó la concentración preclínica diana durante el intervalo de dosis de 24 horas, y por lo tanto proporcionó una inhibición sostenida de la vía de señalización de MEK.
Eficacia clínica y seguridad
En los estudios clínicos solamente se estudiaron los pacientes con melanoma cutáneo. No se ha evaluado la eficacia en pacientes con melanoma ocular o melanoma en mucosas.
Trametinib en combinación con dabrafenib
Pacientes que no habían recibido un tratamiento previo
La seguridad y eficacia de trametinib (2 mg una vez al día) con dabrafenib (150 mg dos veces al día) en pacientes adultos con melanoma no resecable o metastásico con mutación BRAF V600 se ha estudiado en dos estudios fase III y un estudio fase I/II de soporte.
MEK115306 (COMBI-d):
MEK115306 es un estudio fase III, aleatorizado, doble ciego que compara la combinación de dabrafenib y trametinib con dabrafenib y placebo en primera línea para pacientes con melanoma no resecable (estadío IIIC) o mestatásico (estadío IV) con mutación BRAF V600. La variable primaria del estudio fue la supervivencia libre de progresión (SLP) y la variable secundaria fue la supervivencia global (SG). Los pacientes se clasificaron por niveles de lactato de deshidrogenasa (LDH) (>vs < del límite superior normal) y por tipo de mutación BRAF (V600E vs V600K).
Se aleatorizaron 423 pacientes en una relación 1:1, N = 211, en el grupo de la combinación y N = 212 en el grupo de dabrafenib. La mayoría de los sujetos fueron de raza caucásica (>99%) y varones (53%), con una edad media de 56 años (28% fueron >65 años). La mayoría se encontraban en un estadío IVM1c (67%) de la enfermedad. Al inicio, la mayoría tenían un LDH < del límite superior normal (65%), un estado de desarrollo ECOG de 0 (72%) y con enfermedad visceral (73%). La mayoría tenían mutación BRAF V600E (85%). Los sujetos con metástasis cerebrales no se incluyeron en el ensayo.
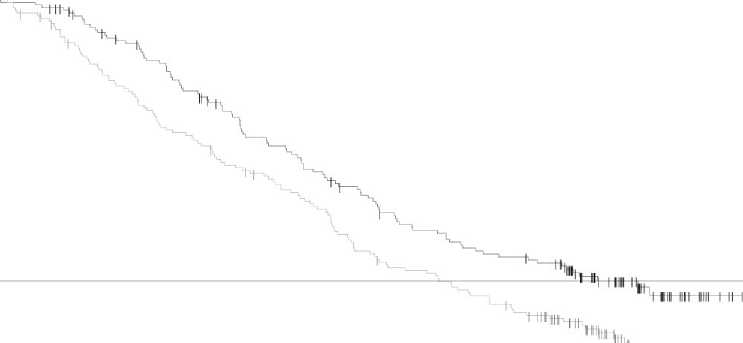
El análisis final de la supervivencia global (a 12 de enero de 2015) demostró una mejoría estadísticamente significativa de la SG con la combinación en comparación con dabrafenib en monoterapia (Figura 1). Se estima una mayor SG para el brazo de la combinación (74% al año y 51% a los 2 años) que en dabrafenib en monoterapia (68% y 42%, respectivamente).
Figura 1 Curvas Kaplan-Meier de supervivencia global del estudio MEK115306 (Población por intención de tratar (ITT)
Page 1 of 1
Dabrafenib + Trametinib Dabrafenib + Placebo
mi i til - 11 t -
1.0
0.8 0.7
0.4
0.3
Dabrafenib + Trametinib
Dabrafenib + Placebo
Number at risk 211 208
212 206
200
191
187
175
174
159
159
147
10
144
138
135
127
124
111
112
104
106
95
12 14 16 18 20
Time from Randomization (Months)
103
88
i
22
88
70
24
53
26
21
10
28
3
30
32
0.2
|
Dabrafenib + trametinib (N = 211) |
Dabrafenib + placebo (N = 212) | |||||
|
Supervivencia Global 12 Enero 2015 | ||||||
|
Número de eventos (%) |
99 (47%) |
123 (58%) | ||||
|
SG media (meses) |
25,1 |
18,7 | ||||
|
Hazard Ratio ajustado (95% CI) |
0,71 (0,55, 0,92) | |||||
|
Valor P Rango-log estratificado |
0,011 | |||||
Se observaron mejorías estadísticamente significativas en la variable principal, SLP y en la secundaria, tasa de respuesta global (TRG). Se observó también una mayor duración de la respuesta (Tabla 6)
|
Objetivo |
Dabrafenib + Trametinib (N = 211) |
Dabrafenib + Placebo (N = 212) |
Dabrafenib + Trametinib (N = 211) |
Dabrafenib + Placebo (N = 212) |
|
Fecha de corte de datos |
26 Agosto 2013 |
12 Enero 2015 | ||
|
SLPa | ||||
|
Progresión de la enfermedad o muerte, n (%) |
102 (48) |
109 (51) |
139 (66) |
162 (76) |
|
SLP medio (meses) (95% IC) |
9,3 (7,7, 11,1) |
8,8 (5,9, 10,9) |
11.0 (8,0, 13,9) |
8.8 (5,9, 9,3) |
|
HR (Hazard Ratio) (95% IC) |
0,75 (0,57, 0,99) |
0,67 (0,53, 0,84) | ||
|
valor P |
0,035 |
<0,001 | ||
|
TRGb (95% IC) |
67 (59,9, 73,0) |
51 (44,5, 58,4) |
69 (61,8,74,8) |
53 (46,3, 60,2) |
|
Diferencia TRG (95% IC) |
15e (5,9, 24,5) |
15e (6,0, 24,5) | ||
|
valor P |
0,0015 |
0,0014 | ||
|
DdRc medio (meses) (95% CI) |
9,2d (7,4, NA) |
10.2d (7,5, NA) |
12.9 (9.4,19,5) |
10.6 (9,1, 13,8) |
|
a - Supervivencia libre de progresión (valorado por el investigador) b - Tasa de respuesta global (TRG) = Respuesta completa + Respuesta parcial c - Duración de la respuesta (DdR) d - Estaba en marcha en el momento de recogida de la mayoría de las respuestas evaluadas por el investigador (>59%) e - diferencia de TRG calculada a partir de los resultados de TRG sin redondear NA = No alcanzado | ||||
MEK116513 (COMBI-v):
MEK116513 es un estudio abierto fase III, aleatorizado, que compara la combinación de dabrafenib y trametinib con vemurafenib en monoterapia en pacientes con melanoma metastásico con mutación BRAF V600. La variable primaria del estudio fue la supervivencia global con SLP, como variable secundaria. Los pacientes se clasificaron por niveles de lactato de deshidrogenasa (LDH) (>vs < del límite superior normal) y por tipo de mutación BRAF (V600E vs V600K).
Se aleatorizaron 704 pacientes en una relación 1:1 tanto en el grupo de la combinación como en el grupo de vemurafenib. La mayoría de los sujetos fueron de raza caucásica (>96%) y varones (55%), con una edad media de 55 años (24% fueron >65 años). La mayoría se encontraban en un estadío IVM1c (61%) de la enfermedad. Al inicio, la mayoría tenían un LDH < del límite superior normal (67%), un estado de desarrollo ECOG de 0 (70%) y con enfermedad visceral (78%). La mayoría, 54% de los pacientes, teían <3 enfermedades al inicio. La mayoría tenían mutación BRAF V600E (89%). Los sujetos con metástasis cerebrales no se incluyeron en el ensayo.
El análisis actualizado de SG (13 de marzo de 2015) demostró una mejoría estadísticamente significativa de la SG en la combinación en comparación con vemurafenib en monoterapia (Figura 2). A los 12 meses, se estima una SG del 72% para el tratamiento de la combinación y de un 65% para vemurafenib.
Page 1 of1
Vemurafenib Dabrafenib + Trametinib

mui i i i
0.2
0.1
Vemurafenib
Dabrafenib + Trametinib
|
Dabrafenib + trametinib (N = 352) |
Vemurafenib (N = 352) | ||||||
|
Supervivencia Global 13 Marzo2015 | |||||||
|
Número de eventos (%) |
155 (44%) |
195 (55%) | |||||
|
SG media (meses) |
25,6 |
18,0 | |||||
|
Hazard Ratio ajustado (95% CI) |
0,66 (0,53, 0,81) | ||||||
|
Valor P Rango-log estratificado |
<0,001 | ||||||
Number at risk 352 341
352 342
315
336
286
311
252
286
231
260
201
245
187
230
166
217
152
198
129
173
88
128
46
68
28
38
7
16
12 14 16 18 20
Time from Randomization (Months)
Se observaron mejorías significativas en la variable secundaria SLP y en la TRG. Se observó también una mayor da duración de la respuesta (Tabla 7).
|
Objetivo |
Dabrafenib + Trametinib (N=352) |
Vemurafenib (N=352) |
|
SLP | ||
|
Progresión de la enfermedad o |
166 (47) |
217 (62) |
|
muerte, n (%) | ||
|
SLP medio (meses) (95% IC) |
11,4 |
7,3 |
|
(9,9, 14,9) |
(5,8, 7,8) | |
|
HR (Hazard Ratio) |
0,56 | |
|
(95% IC) |
(0,46, 0,69) | |
|
valor P |
<0,001 | |
|
TRG |
226 (64) |
180 (51) |
|
(95% IC) |
(59,1, 69,4) |
(46,1, 56,8) |
|
Diferencia TRG |
13 | |
|
(95% IC) |
(5,7, 20,2) | |
|
valor P |
0,0005 | |
|
DdRc medio (meses) | ||
|
(95% IC) |
13,8 |
7,5 |
|
(11,0, NR) |
(7,3, 9,3) | |
Tratamiento previo con un inhibidor BRAF
Hay pocos datos de pacientes que tomaran la combinación de trametinib con dabrafenib que hayan progresado con un inhibidor BRAF previo.
La Parte B del estudio BRF113220 (incluida la cohorte de 26 pacientes que progresaron con un inhibidor BRAF). La combinación de 2 mg de trametibib una vez al día y 150 mg de dabrafenib dos veces al día demostró actividad clínica limitada en pacientes que habían progresado con un inhibidor BRAF (ver sección 4.4). La evaluación del investigador confirmó la tasa de respuesta del 15% (95% IC: 4,4, 34,9) y la SLP fue de 3,6 meses (95% IC: 1,9, 5,2). Se vieron resultados similares en 45 pacientes que pasaron de dabrafenib en monoterapia a la combinación 2 mg de trametinib una vez al día y 150 mg de dabrafenib dos veces al día en la Parte C del estudio. En estos pacientes un 13% (95% IC: 5,0, 27,0) confirmó la tasa de respuesta con un SLP media de 3,6 meses (95% IC: 2, 4).
Trametinib en monoterapia
Pacientes que no habían recibido un tratamiento previo
La eficacia y seguridad de trametinib en pacientes con melanoma con mutación BRAF (V600E y V600K) positiva, se evaluó en un estudio fase III abierto, aleatorizado (MEK114267). Este estudio requeria, que a todos los pacientes se les diagnosticara la mutación BRAF V600.
Los pacientes que no habían recibido tratamiento previo (N = 322), o que hubiesen recibido un tratamiento previo con quimioterapia en el entorno metastásico [Población por intención de tratar ITT], fueron randomizados 2:1 para recibir 2 mg de trametinib una vez al día o quimioterapia (1.000 mg/m2 de dacarbazina cada 3 semanas o 175 mg/m2 de paclitaxel cada 3 semanas). Todos los pacientes continuaron el tratamiento hasta progresión de la enfermedad, muerte o retirada del estudio.
La variable primaria del estudio fue evaluar la eficacia de trametinib frente a la quimioterapia en relación a la supervivencia libre de progresión (SLP), en pacientes con melanoma avanzado/metastásico con mutación BRAF V600E positiva, que no presentaban antecedentes previos de metástasis cerebrales (N = 273), que se consideró la población primaria para determinar la eficacia. Las variables secundarias fueron la supervivencia libre de progresión en la población por intención de tratar (ITT), la supervivencia global (SG), la tasa de respuesta global (ORR), y la duración de la respuesta en la población primaria de eficacia y en la población por intención de tratar (ITT). A los pacientes del brazo de quimioterapia se les permitía pasar a recibir tratamiento con trametinib tras confirmación de progresión de enfermedad por un comité independiente. Un total de 51 (47%) pacientes del brazo de quimioterapia, recibieron tratamiento con trametinib tras confirmar que presentaban progresión de enfermedad.
Las características basales de ambos grupos de tratamiento estaban equilibradas, tanto en la población primaria de eficacia como en la población ITT. En la población ITT, el 54% de los pacientes eran hombres y todos ellos caucásicos. La mediana de la edad fue de 54 años (el 22% eran >65 años), todos los pacientes presentaban puntuación 0 ó 1 en la escala ECOG, y el 3% presentaba un historial de metástasis cerebrales. La mayoría de los pacientes (87%) de la población ITT presentaban mutación BRAF V600E, y el 12% de los pacientes presentaban mutación BRAF V600K. La mayoría de los pacientes (66%) no habian recibido tratamiento previo con quimioterapia para la enfermedad avanzada o metastásica.
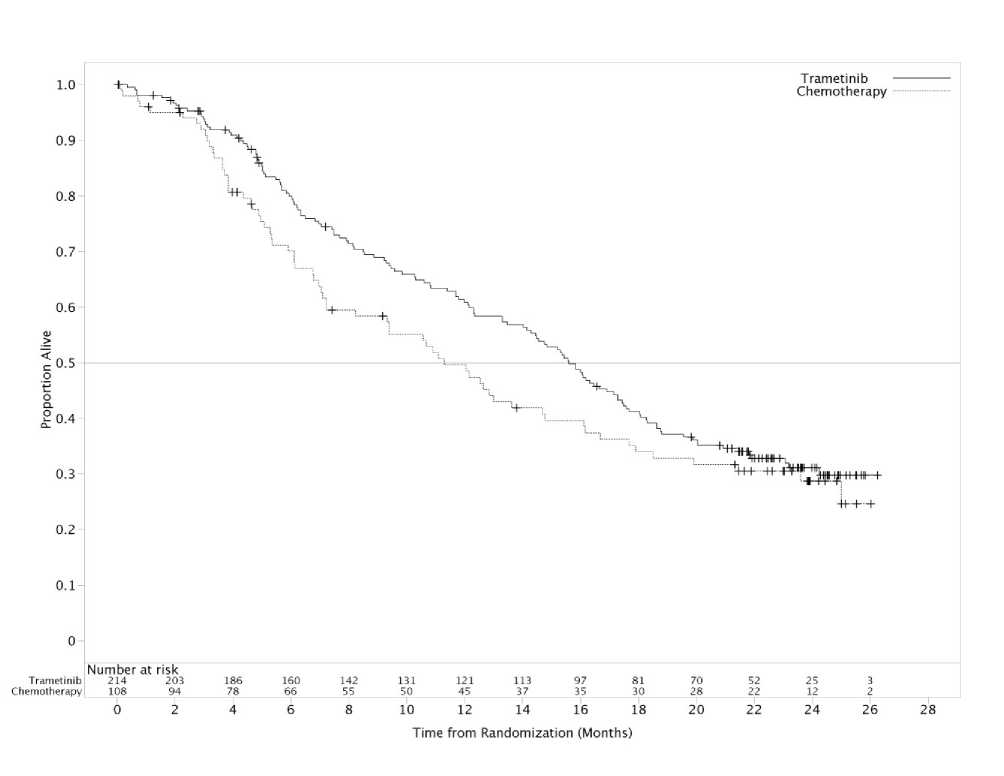
Los resultados de eficacia en la población primaria de eficacia fueron consistentes con los obtenidos en la población por ITT, y por lo tanto, en la Tabla 8 solo se presentan los datos de eficacia de la población por ITT. En la Figura 3 se presentan las curvas Kaplan-Meier para la supervivencia global evaluadas por el investigador (análisis post-hoc realizado el 20 de Mayo de 2013).
Tabla 8 Resultados de eficacia en la población por ITT evaluados por el investigador
|
Variable |
Trametinib |
Quimioterapia3 |
|
Supervivencia libre de progresión |
(N=214) |
(N=108) |
|
Mediana de la SLP (meses) |
4,8 |
1,5 |
|
(IC 95%) |
(4,3; 4,9) |
(1,4; 2,7) |
|
Hazard Ratio |
0,45 | |
|
(IC 95%) |
(0,33; 0,63) | |
|
Valor de p |
<0,0001 | |
|
Tasa de respuesta global (ORR) (%) |
22 |
8 |
|
ITT = intención de tratar; SLP = supervivencia libre de progresión; IC = intervalo de confianza. | ||
|
a Quimioterapia: se incluyeron pacientes en tratamiento con 1000 mg/m2 de dacarbazina (DTIC) cada | ||
|
3 semanas o pacientes en tratamiento con 175 mg/m2 de paclitaxel cada 3 semanas. | ||
El resultado de la SLP fue consistente en el subgrupo de pacientes con melanoma y mutación positiva V600K (HR = 0,50; [IC 95%: 0,18; 1,35], valor de p =0,0788).
Se llevó acabo un análisis adicional de supervivencia global en base a los datos de corte obtenidos el 20 de mayo de 2013. En la Tabla 9, de octubre de 2011, el 47% de los sujetos habían cruzado al tratamiento con trametinib, mientras en mayo de 2013, el 65% de los sujetos habían cruzado de tratamiento.
|
Fechas de corte |
Tratamiento |
Número de muertes (%) |
Mediana de meses SG (IC 95%) |
Hazard ratio (IC 95%) |
Porcentaje de Supervivencia a los 12 meses (IC 95%) |
|
26 de octubre de 2011 |
Quimioterapia (N=108) |
29 (27) |
NA |
0,54 (0,32; 0,92) |
NA |
|
Trametinib (N=214) |
35 (16) |
NA |
NA | ||
|
20 de mayo de 2013 |
Quimioterapia (N=108) |
67 (62) |
11,3 (7,2; 14,8) |
0,78 (0,57; 1,06) |
50 (39,59) |
|
Trametinib (N=214) |
137 (64) |
15,6 (14,0; 17,4) |
61(54, 67) | ||
|
NA=no alcanzado | |||||
Figura 3 Curvas Kaplan-Meier de supervivencia global (análisis post-hoc realizado el 20 de Mayo de 2013)
Tratamiento previo con BRAF
En un estudio fase II (MEK113583) de un solo brazo, diseñado para evaluar la tasa de repuesta objetiva, la seguridad y la farmacocinética del tratamiento con 2 mg de trametinib una vez al día, en pacientes con melanoma metastásico y mutación positiva BRAF V600E, V600K o V600 D, se reclutaron dos tipos de cohortes: la Cohorte A que incluía pacientes que habían recibido tratamiento previo con un inhibidor BRAF y que podian haber recibido otro tratamiento o no, y la Cohorte B que incluía pacientes que habían recibido al menos un tratamiento previo con quimioterapia o inmunoterapia y que no habían recibido tratamiento previo con un inhibidor BRAF.
En la Cohorte A de este estudio, trametinib no demostro actividad clínica en pacientes que habían progresado a un tratamiento previo con un inhibidor BRAF.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con trametinib en los diferentes grupos de la población pediátrica en melanoma (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Trametinib se absorbe por vía oral con una mediana del tiempo hasta alcanzar el pico de concentración de 1,5 horas desde la administración de la dosis. La biodisponibilidad absoluta media, de una dosis única de un comprimido de 2 mg es del 72%, en relación con una microdosis intravenosa (IV). Tras la administración de dosis repetidas, el incremento en la exposición (Cmáx y el AUC) fue proporcional a la dosis. Tras la administración de 2 mg diarios, la media geométrica de la Cmáx, el AUC(0-X) y la concentración previa en estado estacionario, fueron de 22,2 ng/ml, 370 ng*hr/ml y 12,1 ng/ml respectivamente, con un pico mínimo:ratio (1,8). La variabilidad entre los sujetos en el estado estacionario fue baja (<28%).
Con dosis repetidas de 2 mg de trametinib una vez al día, el ratio medio de acumulación es de 6,0. El estado estacionario se alcanzó el día 15.
La administración de una dosis única de trametinib con una comida de alto contenido en grasas y calorías, provocó una reducción del 70% y del 10% en la Cmáx y el AUC respectivamente, en comparación con condiciones de ayuno (ver secciones 4.2 y 4.5).
Distribución
La unión de trametinib a las proteínas plasmáticas humanas es del 97,4%. El volumen de distribución de trametinib es aproximadamente de 1.200 L, determinado tras la administración intravenosa de una microdosis de 5 pg.
Biotransformación
Estudios in vitro mostraron que trametinib se metaboliza principalmente por la vía deacetilzación sola o con mono oxigenación, o en combinación con vías de biotransformación por glucuronidación. La oxidación por el CYP3A4 se considera una vía metabólica menor. La deacetilación está mediada por las carboxil esterasas (p. ej. carboxil esterasa 1b/c y 2) y puede que también estén mediadas por otras enzimas hidrolíticas. Sin embargo, todavía se desconoce qué enzima(s) están involucradas en el metabolismo de trametinib.
Tras la administración de una única dosis y de dosis repetidas, trametinib como molécula inalterada, es el principal componente circulante en el plasma.
Tras la administración de una única dosis, la media de la semivida de eliminación es de 127 horas (5,3 días). El aclaramiento plasmático de trametinib IV es de 3,21 l/hora.
Tras la administración de una única dosis oral de trametinib radiomarcado en forma de solución, la recuperación total de la dosis después de 10 días de periodo de recogida, fue baja (<50%) debido a la prolongada semivida de eliminación. Tras la administración oral de trametinib-[14C], la principal ruta de eliminación es la excreción fecal con >80% de radioactividad excretada recuperada, mientras que la excreción urinaria fue de <19% de la radioactividad excretada recuperada. Menos de un 0,1% de la dosis excretada en orina fue recuperada como trametinib inalterado.
Poblaciones especiales
Insuficiencia hepática
Un análisis farmacocinético poblacional indicó que una leve elevación de bilirrubina y/o de los niveles de AST (en base a la clasificación del Instituto Nacional del Cáncer [NCI]) no afectó de manera significativa al aclaramiento de trametinib administrado por vía oral. No hay datos disponibles en pacientes con insuficiencia hepática moderada o grave. Dado que el metabolismo y la excreción biliar son las principales rutas de eliminación de trametinib, la administración de trametinib se debe realizar con precaución en pacientes con insuficiencia hepática de moderada a grave (ver sección 4.2).
Insuficiencia renal
Es poco probable que la insuficiencia renal tenga un efecto clínicamente relevante en la farmacocinética de trametinib debido a su baja excreción renal. La farmacocinética de trametinib se caracterizó mediante un análisis farmacocinético poblacional realizado en 223 pacientes con insuficiencia renal leve, y 35 pacientes con insuficiencia renal moderada, que participaron en los ensayos clínicos con trametinib. La insuficiencia renal leve y moderada no tuvo efecto sobre la exposición a trametinib (<6% en cada uno de los grupos). No hay datos disponibles en pacientes con insuficiencia renal grave (ver sección 4.2).
Pacientes de edad avanzada
En base a un análisis farmacocinético poblacional (rango de edad de 19 a 92 años de edad), la edad no presentó un efecto clínico relevante sobre la farmacocinética de trametinib. Los datos de seguridad en pacientes >75 años son limitados (ver sección 4.8).
Raza
Los datos que hay son insuficientes para evaluar el posible efecto de la raza en la farmacocinética de trametinib, dado que la experiencia clínica está limitada a pacientes de raza caucásica.
Población pediátrica
No se han realizado estudios para investigar la farmacocinética de trametinb en pacientes pediátricos.
Sexo/Peso corporal
En base al análisis farmacocinético poblacional, se determinó que el sexo y el peso corporal influyen en el aclaramiento de trametinib administrado por vía oral. Aunque es predecible que las mujeres de menor tamaño presenten una mayor exposición que los hombres de gran tamaño, es poco problable que estas diferencias sean clínicamente relevantes y por lo tanto no es necesario realizar ajustes de dosis.
Efectos de trametinib sobre enzimas metabolizadoras de fármacos y transportadores: los datos in vivo e in vitro sugieren que es poco probable que trametinib afecte a la farmacocinética de otros medicamentos. En base a los estudios in vitro, trametinib no es un inhibidor de CYP1A2, CYP2A6, CYP2B6, CYP2D6 y CYP3A4. In vitro, trametinib resultó ser un inhibidor de CYP2C8, CYP2C9 y CYP2C19, un inductor de CYP3A4 y un inhibidor de los transportadores OAT1, OAT3, OCT2, MATE1, OATP1B1, OATP1B3, Pgp y BCRP. Sin embargo, en base a la baja dosis y la baja exposición sistémica clínica, relacionada con valores de potencia de inhibición o de inducción in vitro, no se considera que trametinib sea un inhibidor o inductor in vivo de estas enzimas o transportadores, aunque puede producirse una inhibición transitoria de los sustratos de BCRP en el intestino (ver sección 4.5).
Efectos de otros medicamentos sobre trametinib: los datos in vivo e in vitro sugieren que es poco probable que la farmacocinética de trametinib se vea afectada por otros medicamentos. Trametinib no es un sustrato de enzimas CYP, ni de los transportadores BCRP, OATP1B1, OATP1B3, OATP2B1, OCT1, MRP2, y MATE1. Trametinib, in vitro, es sustrato de BSEP y del flujo de transportadores P-gp. Aunque parece improbable que la exposición de trametinib se vea afectada por la inhibición de BSEP, no se puede excluir un aumento de lo niveles de trametinib por una potente inhibición de la P-gp hepática (ver sección 4.5).
5.3 Datos preclínicos sobre seguridad
No se han realizado estudios de carcinogenicidad con trametinib. Trametinib no fue genotóxico en el ensayo de mutación inversa en bacterias, ni en estudios que evaluaron aberraciones cromosómicas en células de mamíferos, ni en estudios con micronúcleos en la médula ósea de ratas.
Trametinib puede afectar a la fertilidad femenina en humanos. En estudios a dosis repetidas en ratas hembras, se observaron incrementos en los folículos císticos y disminución del cuerpo lúteo, a exposiciones de trametinib inferiores a la exposición clínica en humanos en base al AUC.
Además, en ratas jóvenes a las que se le dio trametinib se observó disminución del peso de los ovarios, ligeros retrasos en signos de la maduración sexual femenina (abertura vaginal y aumento de la incidencia de brotes prominentes extremos terminales dentro de la glándula mamaria) y una ligera hipertrofia de la superficie del epitelio uterino. Todos estos efectos fueron reversibles tras un periodo sin tratamiento y atribuibles a la farmacología. Sin embargo, en estudios de toxicidad en ratas y perros de hasta 13 semanas de duración, no se observaron efectos relacionados con el tratamiento sobre el tejido reproductor masculino.
En estudios de toxicidad reproductiva en ratas y conejos, trametinib provocó toxicidad maternal y toxicidad en el desarrollo. En ratas, a exposiciones por debajo o ligeramente por encima de la exposición clínica en base al AUC, se observó una disminución del peso fetal y un incremento de pérdidas tras la implantación. En conejas preñadas a exposiciones sub-clínicas en base al AUC, se observó disminución del peso corporal del feto, aumento de los abortos, incremento en la incidencia de osificación incompleta y malformaciones esqueléticas.
En estudios a dosis repetidas, los efectos observados tras la exposición a trametinib fueron principalmente en la piel, tracto gastrointestinal, sistema hematológico, huesos e hígado. La mayoría de los hallazgos son reversibles tras un periodo de recuperación libre de fármaco. En ratas, se observó necrosis hepatocelular y elevación de transaminasas después de 8 semanas a dosis >0,062 mg/Kg/día (aproximadamente 0,8 veces la exposición clínica en humanos en base al AUC).
En ratones sin histopatología cardiaca, se observó una disminución de la frecuencia cardiaca, del peso del corazón y de la función del ventrículo izquierdo, tras recibir durante 3 semanas dosis de trametinib >0,25 mg/Kg/día (aproximadamente 3 veces la exposición clínica en humanos en base al AUC). En ratas adultas, la mineralización de múltiples órganos estuvo asociada con un incremento del fósforo sérico y estrechamente relacionada con necrosis en el corazón, hígado y riñones y hemorragias en los pulmones, a exposiciones comparables a la exposición clínica en humanos. En ratas, se observó hipertrofia epifisiaria y un incremento del recambio óseo, pero no es de esperar que la hipertrofia epifisiaria sea clinicamente relevante para adultos humanos. En ratas y perros a los que se les adminitró trametinib a dosis clínicas o por debajo de la exposición clínica, se observó necrosis en la médula ósea, atrofía linfoide en el timo y en el tejido linfoide asociado a las mucosas, y necrosis linfoide en los nódulos linfáticos, bazo y timo, lo que hace posible que afecte a la función inmunológica. En ratas jóvenes, a dosis de 0,35 mg/kg/día (aproximadamente 2 veces la exposición clínica en humanos adultos en base al AUC) se observó un aumento del peso del corazón sin histopatologías.
En un ensayo in vitro de captación del rojo neutro (Neutral Red Uptake) realizado en fibroblastos 3T3 de ratón, trametinib fue fototóxico a concentraciones significativamente mayores que las exposiciones clínicas (IC50 a 2,92 pg/ml, >130 veces la exposición clínica en base a la Cmax), lo cual indica que el riesgo de fototoxicidad en pacientes tratados con trametinib, es bajo.
Combinación con dabrafenib
En un estudio en perros a los que se les dio trametinib y dabrafenib en combinación durante 4 semanas se observó signos de toxicidad gastrointestinasl y disminución de las células linfoides del timo a niveles inferiores que los perros con solo trametinib. Por lo contrario, se observaron misma toxicidad es que los estudios en monoterapia.
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Mekinist 0,5 mg comprimidos recubiertos con película Núcleo del comprimido Manitol (E421)
Celulosa microcristalina (E460)
Hipromelosa (E464)
Croscarmelosa sódica (E468)
Estearato de magnesio (E470b)
Laurilsulfato sódico
Dióxido de sílice coloidal (E551)
Película de recubrimiento del comprimido Hipromelosa (E464)
Dióxido de titanio (E171)
Polietilenglicol
Óxido de hierro amarillo (E172)
Mekinist 2 mg comprimidos recubiertos con película Núcleo del comprimido Manitol (E421)
Celulosa microcristalina (E460)
Hipromelosa (E464)
Croscarmelosa sódica (E468)
Estearato de magnesio (E470b)
Laurilsulfato sódico
Dióxido de sílice coloidal (E551)
Película de recubrimiento del comprimido Hipromelosa (E464)
Dióxido de titanio (E171)
Polietilenglicol Polisorbato 80 (E433)
Óxido de hierro rojo (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
Frasco no abierto: 18 meses.
Frasco abierto: 30 días.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C)
Conservar en el embalaje original para protegerlo de la luz y la humedad.
Mantener el frasco perfectamente cerrado.
Una vez abierto, el frasco se puede conservar durante 30 días a una temperatura por debajo de 30°C.
6.5 Naturaleza y contenido del envase
Frascos de polietileno de alta densidad (PEAD) con tapón de polipropileno resistente a niños. El frasco contiene un desecante.
Tamaño de los envases: los frascos pueden contener 7 ó 30 comprimidos.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
Mekinist 0,5 mg comprimidos recubiertos con película
EU/1/14/931/001
EU/1/14/931/002
Mekinist 2 mg comprimidos recubiertos con película
EU/1/14/931/005
EU/1/14/931/006
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
30 de Junio de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de los fabricantes responsables de la liberación de los lotes
Glaxo Wellcome, S.A.
Avda. Extremadura, 3 09400, Aranda de Duero Burgos España
Novartis Pharmaceuticals UK Limited
Frimley Business Park
Frimley
Camberley, Surrey GU16 7SR Reino Unido
Novartis Pharma GmbH RoonstraBe 25 D-90429 Nuremberg Alemania
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 quater, apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo
acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de
Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Mekinist 0,5 mg comprimidos recubiertos con película trametinib
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene trametinib dimetil sulfóxido equivalente a 0,5 mg de trametinib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Comprimidos recubiertos con película
7 comprimidos recubiertos con película 30 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Contiene un desecante que no se debe retirar ni tragar.
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera (entre 2°C y 8°C)
Conservar en el embalaje original para protegerlo de la luz y la humedad. Mantener el frasco perfectamente cerrado.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/931/001 7 comprimidos recubiertos con película
EU/1/14/931/002 30 comprimidos recubiertos con película
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
mekinist 0,5 mg
1. NOMBRE DEL MEDICAMENTO
Mekinist 0,5 mg comprimidos recubiertos con película trametinib
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene trametinib dimetil sulfóxido equivalente a 0,5 mg de trametinib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Comprimidos recubiertos con película
7 comprimidos 30 comprimidos
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera (entre 2°C y 8°C). Mantener el frasco perfectamente cerrado.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/931/001 7 comprimidos recubiertos con película
EU/1/14/931/002 30 comprimidos recubiertos con película
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
1. NOMBRE DEL MEDICAMENTO
Mekinist 2 mg comprimidos recubiertos con película trametinib
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene trametinib dimetil sulfóxido equivalente a 2 mg de trametinib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Comprimidos recubiertos con película
7 comprimidos recubiertos con película 30 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Contiene un desecante que no se debe retirar ni tragar.
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera (entre 2°C y 8°C)
Conservar en el embalaje original para protegerlo de la luz y la humedad. Mantener el frasco perfectamente cerrado.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/931/005 7 comprimidos recubiertos con película
EU/1/14/931/006 30 comprimidos recubiertos con película
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
mekinist 2 mg
1. NOMBRE DEL MEDICAMENTO
Mekinist 2 mg comprimidos recubiertos con película trametinib
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene trametinib dimetil sulfóxido equivalente a 2 mg de trametinib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Comprimidos recubiertos con película
7 comprimidos 30 comprimidos
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera (entre 2°C y 8°C). Mantener el frasco perfectamente cerrado.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/931/005 7 comprimidos recubiertos con película
EU/1/14/931/006 30 comprimidos recubiertos con película
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
B. PROSPECTO
Prospecto: información para el paciente
Mekinist 0,5 mg comprimidos recubiertos con película Mekinist 2 mg comprimidos recubiertos con película
trametinib
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto.Ver sección 4.
Contenido del prospecto
1. Qué es Mekinist y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Mekinist
3. Cómo tomar Mekinist
4. Posibles efectos adversos
5. Conservación de Mekinist
6. Contenido del envase e información adicional
1. Qué es Mekinist y para qué se utiliza
Mekinist es un medicamento que contiene el principio activo trametinib. Se utiliza solo o en combinación con otro medicamento que contiene dabrafenib para tratar un tipo de cáncer de piel llamado melanoma,
• que tiene un cambio concreto (mutación) en un gen llamado BRAF, y
• que se ha extendido a otras partes del cuerpo, o no puede ser eliminado mediante cirugía. Esta mutación en el gen, puede haber causado el desarrollo del melanoma. Este medicamento actúa sobre las proteínas producidas por el gen modificado y ralentiza o detiene el desarrollo del cáncer.
2. Qué necesita saber antes de empezar a tomar Mekinist
Mekinist sólo se puede utilizar para tratar melanomas que presenten mutación BRAF. Por lo tanto, antes de iniciar el tratamiento, su médico comprobará si presenta esta mutación.
Si su médico decidiera que tome el tratamiento combinado de Mekinist y dabrafenib, lea detenidamente el prospecto de dabrafenib así como este prospecto.
Si tiene preguntas adicionales sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
No tome Mekinist:
• si es alérgico a trametinib o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
Consulte con su médico si piensa que esto puede sucederle.
Advertencias y precauciones
Consulte a su médico antes de empezar a tomar este medicamento. Su médico necesita saber:
• si tiene problemas de hígado. Mientras esté tomando este medicamento, su médico puede tomar muestras de sangre para comprobar el funcionamiento de su hígado.
• si tiene o ha tenido algúna vez problemas de riñón.
• si tiene o ha tenido algúna vez problemas de respiración o pulmonares
• si tiene problemas de corazón como fallo cardiaco o problemas con la forma en que late el corazón
• si tiene problemas oculares, como bloqueo de las venas que irrigan el ojo (oclusión de las venas de la retina) o hinchazón del ojo que se pueda deber a un bloqueo de fluido (coriorretinopatía).
Antes de tomar Mekinist en combinación con dabrafenib, su médico debe saber:
• si ha tenido otro tipo de cáncer distinto al melanoma, ya que podría tener mayor riesgo de desarrollar otros cánceres que no sean de piel mientras esté tomando Mekinist.
Consulte con su médico si piensa que alguna de las circunstancias anteriores le puede afectar.
Afecciones a las que debe prestar atención
Algunas personas que toman Mekinist desarrollan otras afecciones que pueden ser graves. Usted necesita saber a qué síntomas debe prestar atención mientras esté tomando este medicamento.
Sangrados
Tomar Mekinist o Mekinist en combinación con dabrafenib puede causar sangrados graves en el cerebro, sistema digestivo (tales como estómago, recto o intestino), pulmones y otros órganos, que pueden provocarle la muerte. Los síntomas pueden ser:
• dolor de cabeza, mareo o sentirse sin fuerzas
• que pase sangre a las heces o heces negras
• que pase sangre a la orina
• dolor de estómago
• tos / vómitos de sangre
Infome lo antes posible a su médico si siente estos síntomas Alteraciones del corazón
Mekinist puede causar problemas en el corazón, o puede hacer que problemas existentes empeoren (ver “Afecciones del corazón” en la sección 4 de este prospecto).
Informe a su médico si presenta algún problema en el corazón. Antes de empezar el tratamiento y durante el tratamiento con este medicamento, su médico le realizará pruebas para comprobar si su corazón funciona adecuadamente. Informe a su médico inmediatamente si siente que: su corazón late con fuerza, se le acelera el corazón o late a un ritmo irregular, si presenta mareos, se siente cansado, aturdido, le falta el aire o se le hinchan las piernas. Si es necesario, su médico puede decidir interrumpir el tratamiento o suspenderlo.
Fiebre (temperatura alta)
Tomar Mekinist o Mekinist en combinación con dabrafenib puede causar fiebre, aunque es más probable si toma la combinación. En algunos casos hay gente con fiebre que desarrolla tensión sanguinea baja, mareo y otros síntomas.
Infome inmediatamente a su médico si su temperatura es más de 38,5°C mientras esté tomando este medicamento.
Cambios en la piel que pueden indicar un nuevo cáncer de piel
Tomar Mekinist o Mekinist en combinación con dabrafenib puede causar diferentes tipos de cáncer de piel llamados cáncer de piel de células escamosas. Normalmente estas lesiones se quedan a nivel local y pueden quitarse con cirugía y puede continuar con el tratamiento.
Algunas personas que toman Mekinist en combinación con dabrafenib pueden también notar que tienen nuevos melanomas. Estos melanomas son eliminados normalmente mediante cirugía y continuar con el tratamiento sin necesidad de interrumpirlo.
Su médico le examinará la piel antes de empezar el tratamiento, y después, de forma periódica. Informe a su médico inmediatamente si notara algún cambio en la piel mientras esté en tratamiento con este medicamento o tras el tratamiento (vea también la sección 4).
Problemas de hígado
en su hígado que puede ser mortal. Su médico le adecuadamente son:
Mekinist, o la combinación con dabrafenib, puede causar problemas desarrollar enfermedades como hepatitis o fallo hepático, que puede controlará periodicamente. Los signos de que su hígado no funciona
• pérdida de apetito
• encontrarse mal (náuseas)
• sentirse enfermo (vómitos)
• dolor en el estómago (abdomen)
• amarilleo de su piel o del blanco de los ojos (ictericia)
• orina de color oscuro
• picores de piel
Infome lo antes posible a su médico si siente estos síntomas.
Problemas en los ojos
Su médico debe examinar sus ojos mientras esté tomando este medicamento. Mekinst puede causar problemas en los ojos, incluso ceguera. Mekinist no está recomendado si alguna vez ha tenido un bloqueo en las venas que drenan los ojos (oclusión de las venas retinianas). Informe a su médico inmediatamente si durante el tratamiento presenta los siguientes síntomas relacionados con problemas en los ojos: visión borrosa, pérdida de visión u otros cambios en la visión, si ve puntos de colores o halos (visión borrosa alrededor de objetos). Si es necesario, su médico puede decidir interrumpir el tratamiento o suspenderlo.
Problemas de respiración o pulmonares
Informe a su médico si tiene problemas de respiración o pulmonares, incluyendo dificultad para respirar frecuentes acopañados de tos seca, respiración entrecortada y fatiga. Su médico puede pedirle que le revisen la función pulmonar antes de empezar a tomar este medicamento.
Dolor muscular
Mekinist puede provocarle deterioro de los músculos (rabdomiolisis). Informe a su médico lo antes posible si nota alguno de estos síntomas:
• dolor muscular
• orina oscura debido al daño en riñones
Si fuera necesario, su médico podría decidir interrumpir su tratamiento o detenerlo por completo.
Lea la información ‘Posibles efectos adversos graves’ en la sección 4 de este prospecto. Perforación en el estómago o en el intestino
Tomar Mekinist o Mekinist en combinación con dabrafenib puede aumentar el riesgo de desarrollar perforación en la pared del intestino. Si usted siente dolor abdominal grave dígaselo a su médico lo antes posible.
Niños y adolescentes
Mekinist no está recomendado en niños y adolescentes debido a que no se conocen los efectos de Mekinist en personas menores de 18 años de edad.
Uso de Mekinist con otros medicamentos
Antes de empezar el tratamiento, informe a su médico, farmacéutico o enfermero si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento. Esto incluye medicamentos adquiridos sin receta médica.
Haga una lista de los medicamentos que esté tomando, y enséñesela a su médico, farmacéutico o enfermero si empieza a tomar un nuevo medicamento.
Uso de Mekinist con alimentos y bebidas
Es importante que tome Mekinist con el estómago vacio debido a que los alimentos pueden afectar a la absorción de este medicamento (ver sección 3).
Embarazo, lactancia y fertilidad
El uso de Mekinist no está recomendado durante el embarazo.
• Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento. Mekinist puede provocar daños en el feto.
• Si usted es una mujer en edad fértil, debe utilizar un método anticonceptivo fiable mientras esté tomando Mekinist, y durante los 4 meses siguientes a la finalización del tratamiento.
• Los métodos anticonceptivos a base de hormonas (pildora anticonceptiva, inyecciones o parches anticonceptivos) puede que no sean efectivos durante el tratamiento con Mekinist o con el tratamiento combinado (Mekinist así como dabrafenib). Se recomienda usar otro método anticonceptivo adicional tal como un método de barrera (p.ej. un preservativo) para no quedarse embarazada durante el tratamiento con este medicamento. Consulte a su médico, farmacéutico o enfermero para que le aconseje al respecto.
• Si se queda embarazada durante el tratamiento con Mekinist, informe a su médico inmediatamente.
No se recomienda utilizar Mekinist durante la lactancia
Se desconoce si los componentes de este medicamento pueden pasar a la leche materna.
Si está en periodo de lactancia o planea dar el pecho, debe indicárselo a su médico. Se recomienda no dar el pecho mientras esté tomando Mekinist. Usted y su médico decidirán si toma este medicamento, o si decide dar el pecho.
Fertilidad de hombres y mujeres
Mekinist puede alterar la fertilidad de hombres y mujeres.
Si toma Mekinist con dabrafenib: Dabrafenib puede reducir permanentemente la fertilidad de los hombres. Así mismo, los hombres que toman dabrafenib puede que se les reduzca el recuento de esperma y puede que su esperma no vuelva a los niveles normales hasta que haya dejado de tomar este medicamento.
Antes de empezar el tratamiento con dabrafenib, hable con su médico sobre las opciones de poder tener hijos en el futuro.
Si tiene preguntas adicionales sobre los efectos de este medicamento en la fertilidad, pregunte a su médico, farmacéutico o enfermero.
Conducción y uso de máquinas
Mekinist puede provocar efectos adversos que afecten a la capacidad de conducir o utilizar máquinas. Evite conducir o utilizar máquinas si se siente cansado o débil, si tiene problemas de visión o si se siente falto de energía.
La descripción de estos efectos adversos se puede encontrar en otras secciones de este prospecto (ver secciónes 2 y 4.).
Lea toda la información contenida en este prospecto.
Si no está seguro, hable con su médico, farmacéutico o enfermero. Su capacidad para conducir y utilizar máquinas se puede ver afectada incluso por la propia enfermedad, los síntomas o el tratamiento.
3. Cómo tomar Mekinist
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero.
Cuánto tomar
La dosis recomendada de Mekinist, tanto se utilice solo o en combinación con dabrafenib, es de 1 comprimido de 2 mg una vez al día. La dosis recomendada de dabrafenib, cuando se utilice en combinación con Mekinist es de 150 mg dos veces al día.
Su médico decidirá si es necesario disminuir la dosis en función de los efectos adversos que tenga.
No tome más Mekinist del que le ha recomendado su médico.
Cómo tomarlo
Trague los comprimidos enteros con la ayuda de un vaso lleno de agua.
Tome Mekinist una vez al día, con el estómago vacío (al menos 1 hora antes de la comida o 2 horas después de la comida). Es decir:
• una vez que haya tomado Mekinist, debe esperar por lo menos 1 hora antes de comer, o
• si ya ha comido, debe esperar por lo menos 2 horas antes de tomar Mekinist.
Tome Mekinist a la misma hora del día, todos los días.
Si toma más Mekinist del que debe
Si toma demasiados comprimidos de Mekinist, contacte con su médico, farmacéutico o enfermero. Si le es posible, enséñeles en envase de Mekinist junto con este prospecto.
Si olvidó tomar Mekinist
Si han transcurrido menos de 12 horas desde la hora habitual a la que debería haber tomado Mekinist, tómelo tan pronto como se acuerde.
Si han transcurrido más de 12 horas desde la hora habitual a la que debería haber tomado Mekinist, sáltese esta dosis y tome la siguiente a su hora habitual. Luego, continúe tomando los comprimidos a su hora habitual.
No tome una dosis doble para compensar las dosis olvidadas.
Si interrumpe el tratamiento con Mekinist
Tome Mekinist durante el tiempo que le haya recomendado su médico. No deje de tomar este medicamento a menos que su médico se lo recomiende.
Si tiene cualquier otra duda sobre cómo tomar Mekinist, pregunte a su médico, farmacéutico o enfermero.
Cómo debe tomar Mekinist en combinación con dabrafenib
• Tome Mekinist en combinación con dabrafenib exactamente como le ha dicho su médico, enfermera o farmacéutico. No cambie la dosis o deje de tomar Mekinist o dabrafenib a menos que su médico, enfermera o farmacéutico se lo indique.
• Tome Mekinist una vez al día y tome dabrafenib dos veces al día. Sería bueno para usted que tuviera el hábito de tomarse los dos medicamentos a la vez cada día. Debe tomar Mekinist tanto con la dosis matutina de dabrafenib o con la dosis nocturna de dabrafenib. Las dosis de dabrafenib se deben tomar con una diferencia de 12 horas.
• Tome Mekinist y dabrafenib con el estómago vacío, al menos una hora antes o dos horas depués de una comida. Tómeselos enteros con un vaso lleno de agua.
• Si se le olvida una dosis de Mekinist o de dabrafenib, tómesela tan pronto se acuerde. No compense la dosis olvidada y tómese la siguiente dosis cuando le tocara:
o Si quedan menos de 12 horas hasta la siguiente dosis de Mekinist, que se toma una al día
o Si quedan menos de 6 horas hasta la siguiente dosis de dabrafenib, que se toma dos veces
al día
• Si toma más Mekinist o dabrafenib, contacte inmediatamente con su médico, enfermero o farmacéutico. Llévese los comprimidos de Mekinist y las cápsulas de dabrafenib si puede. Si fuera posible, enseñe el envase de Mekinist y dabrafenib con el prospecto.
• Si presenta efectos adversos su médico puede decidir darle una dosis inferior de Mekinist y de dabrafenib. Tóme la dosis de Mekinist exactamente como le ha dicho su médico, enfermera o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Posibles efectos adversos graves
Afecciones del corazón
Mekinist puede afectar al funcionamiento de su corazón. Es más probable que afecte a personas con un problema de corazón existente. Durante el tratamiento con Mekinist le realizaran pruebas de corazón. Entre los signos y síntomas relacionados con problemas de corazón se incluyen:
• palpitaciones, aumento del ritmo del corazón o latidos irregulares
• mareos
• cansancio
• sentirse aturdido
• falta de aire
• piernas hinchadas
Si experimenta cualquiera de estos síntomas, informe a su médico tan pronto como sea posible, tanto si los experimenta por primera vez como si considera que están empeorando.
Elevación de la presión arterial
Mekinist puede aumentar la presión arterial (hipertensión) o empeorarla. Su médico o enfermero deben comprobar su presión arterial durante el tratamiento con Mekinist. Contacte inmediatamente con su médico o enfermero si presenta presión arterial elevada, si la presión arterial empeora, o si presenta fuertes dolores de cabeza, se siente aturdido o mareado.
Problemas de sangrado
Mekinist puede causar problemas de sangrado graves, especialmente en el cerebro o en el estómago. Contacte con su médico o enfermero, y obtenga atención médica inmediata si presenta signos inusuales de sangrado, entre los que se incluyen:
• dolor de cabeza, mareo o debilidad
• tos con sangre o tos con coágulos de sangre
• si vomita sangre o si el vómito tiene aspecto de “granos de café”
• si presenta heces de color rojo, o de color negro con aspecto de alquitrán.
Problemas en los ojos (visión)
Mekinist puede causar problemas en los ojos. No se recomienda tomar Mekinist si alguna vez ha tenido una obstrucción en las venas que drenan los ojos (oclusión de las venas retinianas). Su médico le aconsejará que se realice un chequeo de los ojos antes de empezar el tratamiento con Mekinist y durante el tratamiento. Su médico puede pedirle que deje de tomar Mekinist o derivarle a un especialista, si experimenta signos y síntomas en la visión, entre los que se incluyen:
• pérdida de visión
• enrojecimiento e irritación en los ojos
• si ve puntos de colores
• si ve un halo (visión borrosa alrededor de objetos)
• visión borrosa.
Problemas en la piel
Si nota cualquier cambio en su piel mientras está tomando este medicamento, consulte con su médico, farmacéutico o enfermero tan pronto como le sea posible.
Hasta 3 de cada 100 personas que toman Mekinist en combinación con dabrafenib pueden desarrollar un tipo diferente de cáncer de piel llamado Carcinoma Cutáneo de Células Escamosas(CCE). Otros, pueden desarrollar un tipo de cáncer llamado Carcinoma Basocelular (CBC). Normalmente, estos cambios sólo afectan a la piel de forma local y pueden ser eliminados con cirugía y continuar el tratamiento con Mekinist y dabrafenib sin necesidad de interrumpirlo.
Algunas personas que toman Mekinist en combinación con dabrafenib pueden también notar que tienen nuevos melanomas. Estos melanomas son eliminados normalmente mediante cirugía y continuar el tratamiento con Mekinist y dabrafenib sin necesidad de interrumpirlo.
Su médico examinará su piel antes de empezar el tratamiento con dabrafenib, después mensualmente durante todo el tratamiento y durante los 6 meses posteriores a la finalización del tratamiento. El motivo de estas revisiones es buscar lesiones cancerígenas nuevas.
Su médico también examinará su cabeza, cuello, boca y ganglios linfáticos y le realizarán regularmente un escáner (Tomografía Computerizada) de pecho y de la zona abdominal. También puede que le realicen análisis de sangre. Estas revisiones sirven para detectar si ha desarrollado otros cánceres, incluyendo carcinoma de células escamosas. Se recomienda que tanto al principio como al final del tratamiento se realice un examen pélvico (en mujeres) y anal.
Revise regularmente su piel mientras toma dabrafenib
Si nota cualquiera de los siguientes:
• verrugas nuevas
• llagas en la piel o bultos enrojecidos que sangran o no se curan
• cambios en el tamaño o color de los lunares
Consulte con su médico, farmacéutico o enfermero tan pronto como le sea posible
tanto si los síntomas aparecen por primera vez como si empeoran.
Mekinist tanto en monoterapia como en combinación con dabrafenib puede provocar erupción o erupción similar al acné. Siga las instrucciones de su médico para saber qué debe hacer para prevenir la aparición de una erupción. Si experimenta cualquiera de estos síntomas por primera vez o si estos empeoran, informe a su médico o enfermero tan pronto como le sea posible.
Contacte con su médico inmediatamente si experimenta erupción cutánea grave con alguno de los siguientes síntomas: ampollas en la piel, ampollas o llagas en la boca, descamación de la piel, fiebre, enrojecimiento o hinchazón en la cara, ampollas en las plantas de los pies.
Si presenta erupción en la piel o si esta empeora, informe a su médico o enfermero tan pronto como sea posible.
Dolor muscular
Mekinist puede provocar daños en los músculos (rabdomiolisis). Informe a su médico o enfermera si siente un nuevo síntoma o si alguno de los siguientes síntomas empeora:
• dolor muscular
• orina de color oscuro, debido a daños en el riñón.
Problemas respiratorios o en los pulmones
Mekinist puede provocar inflamación de los pulmones (pneumonitis o enfermedad pulmonar intersticial). Informe a su médico o su enfermero, si presenta nuevos síntomas o un empeoramiento de los síntomas asociados a problemas para respirar o problemas en los pulmones, incluyendo:
• falta de aire
• tos
• fatiga
Otros efectos adversos que se pueden observar mientras toma solo Mekinist:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
• erupción cutánea, erupción similar al acné, enrojecimiento en la cara, piel seca o picor en la piel (ver también “Problemas en la piel” en la sección 4).
• diarrea
• náuseas, vómitos
• estreñimiento
• dolor de estómago
• boca seca
• falta de energía, debilidad, cansancio
• hinchazón de las manos o de los pies
• pérdida inusual del cabello o cabello fino
• presión arterial alta (hipertensión)
• sangrado en diferentes partes del cuerpo, que puede ser leve o grave
• fiebre (temperatura elevada)
• tos
• falta de aire.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas):
• inflamación de los folículos pilosos de la piel
• erupción cutánea con ampollas llenas de pus (ver también “Problemas en la piel”, al principio de la sección 4)
• enrojecimiento, grietas o hendiduras en la piel
• infección de la piel (celulitis)
• trastornos en las uñas de tipo alteraciones en las cuencas de las uñas, dolor en las uñas, infección e hinchazón de las cutículas.
• enrojecimiento, dolor en manos y pies
• sangrado de nariz
• deshidratación (niveles bajos de agua o fluidos)
• úlceras o llagas en la boca, inflamación de las membranas de las mucosas
• inflamación en los pulmones (pneumonitis o enfermedad pulmonar intersticial)
• hinchazón en la cara, con tejido localizado inflamado
• hinchazón alrededor de los ojos
• visión borrosa
• problemas de visión (ver también “Problemas en los ojos (visión)”, al principio de la sección 4 de este prospecto)
• cambios en los latidos del corazón (disfunción del ventrículo izquierdo) (ver también “Afecciones del corazón”, al principio de la sección 4 de este prospecto).
• El ritmo del corazón es inferior de lo normal y/o existe una disminución del ritmo del corazón.
• análisis de sangre con alteraciones de los resultados relacionados con el hígado, disminución de glóbulos rojo (anemia), resultados anormales de creatina fosfoquinasa, una enzima que se encuentra principalmente en el corazón, cerebro y músculo esquelético.
• reacción alérgica (hipersensibilidad).
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
• obstrucción de las venas que drenan los ojos (oclusión de las venas retinianas) (ver también “Problemas en los ojos (visión)”, al principio de la sección 4 de este prospecto).
• hinchazón en los ojos causada por pérdida de líquido (corioretinopatía) (ver también “Problemas en los ojos (visión)”, al principio de la sección 4 de este prospecto).
• daño en los músculos que puede causar dolor muscular y daño en los riñones (rabdomiolisis).
• inflamación de los nervios de la parte posterior del ojo (papiloedema) (ver también “Problemas en los ojos (visión)”, al principio de la sección 4 de este prospecto).
• separación de la membrana sensible a la luz de la parte posterior del ojo (desprendimiento de retina) (ver también “Problemas en los ojos (visión)”, al principio de la sección 4 de este prospecto).
• el corazón bombea la sangre con menor eficiencia causando dificultad para respirar, cansancio extremo e inflamación en los tobillos y en las piernas (fallo cardiaco)
• perforación de la pared del estómago o de los intestinos
• inflamación de los intestinos (colitis).
Si experimenta cualquiera de estos efectos adversos, informe a su médico, farmacéutico o enfermero. También se debe comunicar cualquier otro efecto adverso que no aparezca en este prospecto.
Efectos adversos cuando Mekinist se toma junto con dabrafenib
Cuando Mekinist se toma junto con dabrafenib puede sentir cualquiera de los efectos adversos incluido antes, aunque su frecuencia puede cambiar (aumentar o disminuir)
También podría aparecerle nuevos efectos adversos debido a tomar dabrafenib a la vez con
Mekinist, que se incluyen a continuación.
Informe a su médico tan pronto como le sea posible si nota tanto que los síntomas aparecen por primera vez como si empeoran.
Lea el prospecto de dabrafenib para más detalles sobre los efectos adversos o que podrían aparecerle mientras toma este medicamento.
Los efectos adversos que puede verse mientras tome Mekinist en combinación con dabrafenib son los siguientes:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
• mareo
• escalofríos
• temperatura elevada
• erupción, piel seca, picor, problemas similares al acné
• disminución del apetito
• dolor de cabeza
• tensión sanguínea elevada
• tos
• dolor de estómago
• sentirse mal (náusea), estar mal (vómitos)
• diarrea
• estreñimiento
• dolor en las articulaciones, dolor muscular o dolor en las manos o pies
• falta de energía, sentirse débil
• manos y pies hinchados
• inflamación de la nariz y de la garganta
• sangrados (hemorragia)
• infección del sistema urinario
Efectos adversos muy frecuentes que pueden aparecer en los análisis de sangre:
• niveles bajos de glóbulos blancos
• resultados anormales en sangre de las pruebas de hígado
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
• tensión sanguínea baja
• sudoración excesiva
• efectos en la piel incluyendo parches de piel rugosos y con escamas, erupciones en la piel con ampollas de pus, engrosamiento de la piel de color marrón o amarillento, marcas en la piel, grietas, crecimiento de verrugas o enrojecimiento en las palmas de las manos y pies, cáncer de piel de células escamosas (un tipo de cáncer de piel), inflamación de la capa grasa de la piel, papiloma (un tipo de tumor que normalmente no es dañino), infección de la piel (celulitis), inflamación de los folículos pilosos
• alteraciones en las uñas, como los cambios en el lecho de las uñas, dolor en las uñas, infección e inflamación de las cutículas
• pérdida inusual de pelo o de volumen
• visión borrosa, problemas de la vista
• respiración entrecortada
• úlceras o dolor en la boca, inflamación de la mucosa
• sequedad de boca
• síntomas gripales
• espasmos musculares
• hinchazón de la cara
• sudores nocturnos
• bombeo del corazón menos eficiente
• el ritmo del corazón es inferior de lo normal y/o existe una disminución en el ritmo cardiaco
Efectos adversos frecuentes que pueden aparecer en los análisis de sangre:
• disminución de las plaquetas en sangre (células que ayudan a la sangre a coagular)
• disminución de glóbulos rojos (Anemia) y un tipo de células blancas (leucopenia)
• niveles bajos de sal en sangre
• aumento de algunas sustancias (enzimas) producidas por el hígado
aumento de la creatinina fosfoquinasa, una enzima que se encuentra principalmente en el corazón, cerebro y músculo esquelético
• aumento de los niveles de azúcar en sangre
• niveles bajos de fosfato en sangre
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
• reacciones alérgicas
• cambios en el ojo, como hinchazón debido al acumulación de líquido (coriorretinopatía), inflamación del ojo (uveítis), separación de la membrana sensible a la luz en la parte posterior del ojo (la retina) de sus capas de soporte (desprendimiento de retina) e hinchazón alrededor de los ojos
• hinchazón de la cara, localizado hinchazón de los tejidos
• inflamación del páncreas
• fallo renal, inflamación de los riñones
• inflamación del pulmón
• nuevo melanoma primario
• perforación de la pared del estómago o de los intestinos
• inflamación de los intestinos (colitis)
No conocida (no se puede estimar la frecuencia a partir de los datos disponibles):
• Inflamación del músculo del corazón (miocarditis) que puede producir dificultad para respirar, fiebre, palpitaciones y dolor en el pecho.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Mekinist
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta del frasco y en el envase después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2°C y 8°C).
Conservar en el embalaje original para protegerlo de la luz y la humedad.
Mantener el envase perfectamente cerrado. El frasco contiene un desecante en un contenedor de forma cilindrica. No debe retirar ni tragar el desecante.
El frasco no debe permanecer fuera de la nevera durante más de 30 días.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
Contenido del envase e información adicional
6.
Composición de Mekinist
- El principio activo es trametinib. Cada comprimido recubierto con película contiene trametinib dimetil sulfóxido equivante a 0,5 mg o 2 mg de trametinib.
- Los demás componentes son:
- Comprimido: manitol (E421), celulosa microcristalina (E460), hipromelosa (E464), croscarmelosa sódica (E468), estearato de magnesio (E470b), laurilsulfato de sodio y dióxido de sílice coloidal (E551).
- Cubierta pelicular: hipromelosa (E464), dióxido de titanio (E171), polietilenglicol, óxido de hierro amarillo (E172) (para los comprimidos de 0,5 mg), polisorbato 80 (E433) and óxido de hierro rojo (E172) (para los comprimidos de 2 mg).
Aspecto del producto y contenido del envase
Mekinist 0,5 mg: los comprimidos son recubiertos con película, de color amarillo, ovalados, biconvexos, marcados con ‘GS’ en una de las caras y ‘TFC’ en la cara opuesta.
Mekinist 2 mg: los comprimidos son recubiertos con película, de color rosa, redondos, biconvexos, marcados con ‘GS’ en una de las caras y ‘HMJ’ en la cara opuesta.
Los comprimidos recubiertos con película se suministran en frascos de plástico blancos opacos con tapones de rosca de plástico.
Los frascos incluyen un desecante de silica gel en un pequeño contenedor de forma cilíndrica. El desecante debe mantenerse dentro del frasco y no se debe ingerir.
Los frascos contienen 7 o 30 comprimidos.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Glaxo Wellcome, S.A., Avd. Extremadura, 3, 09400 Aranda de Duero, Burgos, España
|
Novartis Pharmaceuticals UK Limited, Frimley Business Park, Frimley, Camberley, Surrey GU16 | ||
|
7SR, Reino Unido | ||
|
Novartis Pharma GmbH, RoonstraBe 25, D-90429 Nuremberg, Alemania | ||
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Bt^rapnn Novartis Pharma Services Inc. Ten: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Ceská republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 555 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXláda Novartis (Hellas) A.E.B.E. Tpk: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
En la página web de la Agencia Europea de Medicamentos puede encontrarse este prospecto en todas las lenguas de la Unión Europea/Espacio Económico Europeo.
ANEXO IV
CONCLUSIONES CIENTÍFICAS Y MOTIVOS PARA LA MODIFICACIÓN DE LAS CONDICIONES DE LAS AUTORIZACIONES DE COMERCIALIZACIÓN
Conclusiones científicas
Teniendo en cuenta lo dispuesto en el Informe de Evaluación del Comité para la Evaluación de Riesgos en Farmacovigilancia (PRAC) sobre los informes periódicos de seguridad (IPS) para trametinib, las conclusiones científicas del Comité de Medicamentos de Uso Humano (CHMP) son las siguientes:
Se han notificado de forma acumulada tres casos de miocarditis en pacientes que recibieron trametinib en combinación con dabrafenib. En los tres casos se ha notificado relación de causalidad asociada a la combinación de trametinib/dabrafenib y se clasificaron como serios.Un caso fue mortal y en los otros dos casos que no fueron mortales se produjo la recuperación después de la interrupción del tratamiento con la combinación de trametinib/dabrafenib. Basándose en el evidencia presentada, el PRAC consideró que la relación causal entre miocarditis y la combinación trametinib/dabrafenib es probable en los dos casos no mortales en base a una relación temporal, una retirada de medicación positiva y la ausencia de otras causas alternativas convincentes.
Además, en el contexto de miocarditis como acontecimiento potencialmente mortal que puede diagnosticarse erroneamente, el PRAC recomendó que la advertencia existente sobre la reducción de la fracción de eyección del ventrículo izquierdo (FEVI)/Disfunción del ventrículo izquierdo se debe actualizar para incluir una referencia específica a estos casos de miocarditis con el fin de concienciar a los prescriptores sobre la posibilidad de que ocurra esta reacción adversa y señalar que la interrupción del tratamiento resolvió la miocarditis.
Por tanto, en vista de los datos presentados en el PSUR revisado, el PRAC consideró que los cambios en la información del producto de los medicamentos conteniendo trametinib estaban justificados.
El CHMP está de acuerdo con las conclusiones científicas del PRAC.
Motivos para la modificación de las condiciones de la(s) autorización(es) de comercialización
De acuerdo con las conclusiones científicas para trametimib, el CHMP considera que el balance beneficio-riesgo del medicamento o medicamentos que contienen trametinib no se modifica sujeto a los cambios propuestos en la información del producto.
El CHMP recomienda que se modifiquen las condiciones de la(s) autorización(es) de comercialización.
62