Magnograf 0,5 Mmol/Ml Solucion Inyectable En Jeringa Precargada
Información obsoleta, busque otroDE SANIDAD, POLÍTICA SOCIAL E IGUALDAD
agencia española de medicamentos y productos sanitarios
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Magnograf 0,5 mmol/ml solución inyectable.
Magnograf 0,5 mmol/ml solución inyectable en jeringa precargada.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
1 ml de solución acuosa contiene 469 mg (0,5 mmol) de gadopentetato de dimeglumina.
|
Concentración del medio de contraste |
(mg/ml) |
469,01 |
|
(mol/l) |
0,5 | |
|
Contenido en medio de contraste por |
100 ml |
46,900 g |
|
30 ml |
14,070 g | |
|
20 ml |
9,380 g | |
|
15 ml |
7,035 g | |
|
10 ml |
4,690 g | |
|
5 ml |
2,345 g | |
|
Osmolaridad a 37°C (osm/l solución) |
1,44 | |
|
Osmolalidad a 37°C (osm/kg H2O) |
1,96 | |
|
Presión osmótica a 37°C (atm) |
49,8 | |
|
(mPa) |
5,06 | |
|
Densidad a 20°C (kg/l) |
1,210 | |
|
a 37°C (Kg/l) |
1,195 |
|
Viscosidad (mPa.s ó cP) a 20°C |
4,9 |
|
a 37°C |
2,9 |
pH 6,5-8,0
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA Solución para inyección intravenosa.
Correo electrónicoI
C / CAMPEZO, 1 - EDIFICIO 8 28022 MADRID
4.
4.1. Indicaciones terapéuticas
Este medicamento es únicamente para uso diagnóstico.
• Resonancia magnética (RM) craneal y espinal
En especial para la detección de tumores y el ulterior diagnóstico diferencial en los casos de sospecha de meningiomas, neurinomas (del acústico), tumores invasivos (por ejemplo, gliomas) y metástasis; para la demostración de la existencia de tumores pequeños y/o isointensos; en la sospecha de recurrencia después de cirugía o radioterapia; para la representación diferencial de neoplasias raras, como hemangioblastomas, ependimomas y adenomas pequeños de la hipófisis; para la mejor evaluación de la extensión de tumores que no sean de origen cerebral.
Adicionalmente en la RM de la médula espinal: diferenciación de tumores intra- y extramedulares; demostración de zonas tumorales sólidas en siringomielia establecida; determinación de la extensión tumoral intramedular.
• RM de otras regiones corporales
Incluyen la región otorrinofaríngea, cavidad torácica y abdominal, mamas, pelvis y el sistema músculoesquelético.
En particular, Magnograf suministra información diagnóstica para:
- la demostración o exclusión de tumores, inflamaciones y lesiones vasculares, la determinación de su extensión y demarcación, y la diferenciación de su estructura interna
- la evaluación del flujo sanguíneo en tejidos normales y patológicos
- la diferenciación entre tumor y tejido cicatricial después de terapia
- el diagnóstico del prolapso recurrente del disco intervertebral después de cirugía
- la evaluación semicuantitativa de la función renal en combinación con el diagnóstico anatómico del órgano.
4.2. Posología y forma de administración
- En caso de función renal comprometida ver secciones 4.3 y 4.4.
• RM craneal y espinal
Las siguientes directrices sobre dosificación son aplicables a adultos y niños (incluidos neonatos y bebés).
Generalmente, la administración de 0,2 ml (0,1 mmol) de Magnograf/kg de peso corporal es suficiente para obtener una buena opacificación que permita aclarar el problema diagnóstico planteado. Si a pesar de una RM con contraste normal subsistiera una fuerte sospecha clínica de existencia de lesión, puede incrementarse el valor diagnóstico de la exploración volviendo a administrar otra inyección de la misma dosis original en el intervalo de 30 minutos y repitiendo a continuación la RM.
• RM de otras regiones corporales
Las siguientes directrices sobre dosificación son aplicables a adultos y niños. La experiencia en la indicación de “RM de otras regiones corporales” en niños menores de dos años de edad es limitada.
Generalmente la administración de 0,2 ml (0,1 mmol) de Magnograf/kg de peso corporal es suficiente para obtener una buena opacificación que permita aclarar el problema diagnóstico planteado.
En casos especiales, por ej. en lesiones con escasa vascularización y/o poco espacio extracelular, puede ser necesario administrar 0,4 ml (0,2 mmol) Magnograf/kg de peso corporal para obtener un contraste adecuado, en particular si se emplean secuencias relativamente poco potenciadas en T1.
Forma de empleo
El paciente debe estar en ayunas desde dos horas antes de la exploración.
Han de observarse las normas de seguridad habituales en resonancia magnética, p.ej., exclusión de marcapasos cardíacos e implantes ferromagnéticos.
La dosis necesaria se administra por inyección intravenosa; si se desea, en bolo. La RM con intensificación de contraste puede empezar inmediatamente después. Generalmente se obtiene un contraste óptimo aproximadamente en un período de 45 minutos tras la inyección de Magnograf.
Las secuencias de imagen potenciadas en T1 son especialmente idóneas para las exploraciones con contraste.
Las recomendaciones para el empleo de Magnograf son aplicables entre 0,14 Tesla y 1,5 Tesla. Dentro de este rango, éstas son independientes de la intensidad del campo magnético empleado.
Siempre que sea posible, la administración intravascular del medio de contraste debe realizarse con el paciente en decúbito; después de la inyección, el paciente debe permanecer bajo observación, como mínimo, durante media hora.
• Presentación en vial:
No debe extraerse Magnograf con la jeringa hasta inmediatamente antes de la administración.
En ningún caso debe perforarse más de una vez el tapón de goma.
Cualquier resto del medio de contraste no empleado en una exploración debe desecharse.
• Presentación en jeringa precargada:
La jeringa precargada se deberá extraer del envase y preparar para la inyección inmediatamente antes de la administración. Inmediatamente antes del uso, se debe separar la tapa del extremo (capuchón) de la jeringa precargada.
Cualquier resto del medio de contraste no empleado en una exploración debe desecharse.
• Presentación en frasco de 100 ml:
La administración del medio de contraste deberá realizarse mediante inyector, cuyo uso está contraindicado en neonatos y bebés (ver sección 4.9). Los elementos del inyector en contacto con el paciente deberán ser sustituidos después de cada exploración, al estar contaminados con sangre. Los restos de solución del medio de contraste que permanezcan en el frasco, así como los tubos de conexión y los elementos fungibles del sistema de inyección, se desecharán al final del día de la exploración. Es imprescindible seguir las instrucciones adicionales facilitadas por los fabricantes del inyector.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes.
Pacientes con insuficiencia renal grave (Tasa de filtración glomerular-TFG <30 ml/min/1,73m2).
4.4 Advertencias y precauciones especiales de empleo
En pacientes con tendencia a reacciones alérgicas, la decisión de realizar una exploración con Magnograf debe tomarse valorando cuidadosamente la relación beneficio - riesgo, ya que, según muestra la experiencia, en estos pacientes se presentan con mayor frecuencia reacciones de hipersensibilidad.
•Reacciones de hipersensibilidad:
Como ocurre con otros medios de contraste, Magnograf puede asociarse a reacciones de hipersensibilidad/anafilácticas o a otras reacciones idiosincrásicas caracterizadas por manifestaciones cardiovasculares, respiratorias o cutáneas.
La mayoría de estas reacciones ocurren como mínimo a la media hora tras la administración.
El riesgo de reacciones de hipersensibilidad es mayor en los siguientes casos:
-reacción previa a medios de contraste -antecedentes de asma bronquial -antecedentes de trastornos alérgicos
•Insuficiencia renal:
Se han notificado casos de fibrosis sistémica nefrogénica (FSN) asociada al uso de Magnograf y otros agentes de contraste que contienen gadolinio en pacientes con insuficiencia renal grave (TFG <30ml/min/1,73m2). Por ello, Magnograf no se debe administrar en estos pacientes (ver sección 4.3).
También se han notificado casos de FSN en pacientes con insuficiencia renal aguda de cualquier intensidad debido al síndrome hepato-renal o en el periodo preoperatorio de un transplante de hígado, asociados a la utilización de algunos agentes de contraste que contienen gadolinio. Por consiguiente, Magnevist debe ser utilizado en estos pacientes solamente tras una evaluación cuidadosa del riesgo/beneficio, incluyendo la consideración de otros métodos de diagnóstico por la imagen alternativos, y no a dosis mayores de 0,1 mmol/kg de peso corporal (= 0,2 ml/kg de peso corporal).
Se debe asegurar un periodo de tiempo suficiente para la eliminación del agente de contraste del cuerpo, antes de cualquier otra re-administración.
El riesgo de desarrollar FSN en pacientes con insuficiencia renal moderada es desconocido. Por lo tanto, Magnograf se debe utilizar con precaución en pacientes con insuficiencia renal moderada (TFG 30-59 ml/min/1,73m2).
Todos los pacientes, sobre todo aquellos mayores de 65 años, deberán someterse a una revisión para detectar una posible disfunción renal a partir de su historia clínica o mediante pruebas de laboratorio.
La hemodiálisis poco después de la administración de Magnograf en pacientes ya en tratamiento con hemodiálisis puede resultar útil para la eliminación de Magnograf del organismo. No hay evidencia que apoye el inicio de la hemodiálisis para la prevención o tratamiento de la FSN en pacientes que todavía no están sometidos a hemodialisis.
Tras la administración de Magnograf se han observado aisladamente aumentos leves y asintomáticos de los valores de hierro y bilirrubina séricos que, sin embargo, regresaron por lo general a los valores iniciales dentro de las 24 horas siguientes a su administración.
A causa del DTPA (ácido dietilentriaminopentaacético = ácido pentético) libre contenido en la solución de medio de contraste, la determinación del hierro en suero por métodos complexométricos (p.ej., batofenantrolina) puede resultar demasiado baja hasta 24 horas después de la exploración con Magnograf.
•Trastornos convulsivos:
Los pacientes con trastornos convulsivos o lesiones intracraneales pueden tener un mayor riesgo de actividad convulsiva ya que en ocasiones raras se ha informado de casos de este tipo con la administración de Magnograf.
• Uso en neonatos y niños:
En neonatos y niños hasta 1 año de edad, Magnograf sólo debería usarse tras una evaluación cuidadosa debido a la inmadurez de su función renal. (Ver sección 4.2)
• Uso en ancianos:
Dosificación igual que en adultos.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se conocen interacciones con otros medicamentos.
Interacción con otras pruebas diagnósticas:
A causa del DTPA (ácido dietilentriaminopentaacético = ácido pentético) libre contenido en la solución de medio de contraste, la determinación del hierro en suero por métodos complexométricos (p.ej., batofenantrolina) puede resultar demasiado baja hasta 24 horas después de la exploración con Magnograf.
4.6 Embarazo y lactancia
Aún no se ha demostrado la inocuidad de Magnograf durante la gestación.
En la experimentación animal se ha observado el paso de gadopentetato de dimeglumina a la leche materna en cantidades mínimas (un máximo del 0,04% de la dosis administrada).
Por tanto, antes de emplear Magnograf durante el embarazo y la lactancia, debe valorarse cuidadosamente la indicación y el riesgo.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No procede debido al perfil farmacológico del producto
4.8 Reacciones adversas
Las reacciones adversas asociadas con el uso de Magnograf son de naturaleza de leve a moderada y transitorias. Las reacciones adversas notificadas más frecuentemente son náuseas, vómitos, cefaleas, mareos y reacciones en el lugar de inyección (p.ej. dolor, enfriamento, calor).
Se han notificado reacciones graves y muy graves así como también muertes.
En ocasiones raras, se han producido reacciones retardadas de los medios de contraste (Ver sección 4.4 Advertencias y precauciones especiales de empleo).
Frecuencia de las reacciones adversas a partir de los datos de los estudios clínicos:
Ninguna de las reacciones adversas alcanzó una frecuencia superior a poco frecuente.
En base a la experiencia en más de 11.000 pacientes, se han observado las siguientes reacciones adversas y los investigadores las han clasificado como relacionadas con el fármaco.
En la tabla siguiente se enumeran las reacciones adversas según la clasificación MedDRA de órganos del sistema (MedDRA SOCs). Para describir una determinada reacción y sus sinónimos y trastornos relacionados se utiliza el término MedDRA más adecuado.
|
Poco frecuentes > 1/1000 a <1 /100 |
Raras >1/10000 a <1/1000 | |
|
Trastornos psiquiátricos |
Desorientación | |
|
Trastornos del sistema nervioso |
Mareos Cefaleas Disgeusia |
Convulsiones Parestesia Sensación de ardor Temblores |
|
Trastornos oculares |
Conjuntivitis | |
|
Trastornos del sistema cardiovascular |
Taquicardia Arritmia | |
|
Trastornos vasculares |
Tromboflebitis Rubores Vasodilatación | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea Irritación de la garganta / estrechamiento de la garganta Dolor faringolaríngeo / molestias de faringe Tos Estornudos Sibilanzias | |
|
Trastornos gastrointestinales |
Vómitos |
Dolor abdominal |
|
Poco frecuentes > 1/1000 a <1 /100 |
Raras >1/10000 a <1/1000 | |
|
Náuseas |
Molestias estomacales Diarrea Dolor de muelas Sequedad de boca Dolor en los tejidos blandos bucales y parestesia | |
|
Trastornos de la piel y el tejido subcutáneo |
Urticaria Prurito Erupción Eritema | |
|
Trastornos musculoesqueléticos y del tejido conjuntivo y de los huesos |
Dolor en las extremidades | |
|
Trastornos generales y alteraciones del lugar de administración |
Dolor Sensación de calor Sensación de frío Varios tipos de reacciones en el lugar de inyección* |
Edema facial Dolor del tórax Pirexia Edema periférico Malestar general Fatiga Sed Astenia |
* Varios tipos de reacciones en el lugar de inyección (enfriamiento del lugar de inyección, parestesia del lugar de inyección, hinchazón del lugar de inyección, calor en el lugar de inyección, dolor en el lugar de inyección, edema en el lugar de inyección, irritación en el lugar de inyección, hemorragia en el lugar de inyección, molestias en el lugar de inyección).
Reacciones adversas adicionales según los datos post-comercialización (notificación espontanea):
|
Raras (<1 /1000) | |
|
Trastornos de la sangre y del sistema linfático |
Niveles de hierro en sangre elevados |
|
Trastornos del sistema inmunológico |
Choque anafiláctico/reacciones anafilactoides Reacciones de hipersensibilidad. |
|
Trastornos psiquiátricos |
Agitación Confusión |
|
Trastornos del sistema nervioso |
Coma Pérdida de la conciencia Somnolencia Trastornos del habla Parosmia |
|
Trastornos oculares |
Trastornos visuales Dolor ocular Lagrimación |
|
Trastornos del oído y del laberinto |
Deterioro de la audición Dolor de oido |
|
Trastornos del sistema cardiovascular |
Parada cardíaca Disminución del latido del corazón Taquicardia refleja |
|
Trastornos vasculares |
Shock Síncope Reacción vasovagal Hipotensión Aumento de la presión sanguínea |
|
Raras (<1 /1000) | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Parada respiratoria Dolor respiratorio Aumento /disminución de la velocidad de respiración Broncospasmo Laringeospasmo Edema laríngeo Edema faríngeo Edema pulmonar Cianosis Rinitis |
|
Trastornos gastrointestinales |
Salivación |
|
Trastornos hepatobiliares |
Niveles elevados de bilirrubina en sangre Niveles elevados de enzimas hepáticos en sangre |
|
Trastornos de la piel y el tejido subcutáneo |
Angioedema |
|
Trastornos musculoesqueléticos y del tejido conjuntivo y de los huesos |
Dolor de espalda Artralgia |
|
Trastornos renales y urinarios |
Insuficiencia renal grave* Niveles elevados de creatinina en sangre* Incontinencia urinaria Urgencia urinaria |
|
Trastornos generales y alteraciones del lugar de administración |
Escalofríos Sudoración Temperatura corporal elevada/disminuida Sensación de calor Varios tipos de reacciones en el lugar de inyección** |
* En pacientes con insuficiencia renal pre-existente
** Varios tipos de reacciones en el lugar de inyección (necrosis del lugar de inyección, tromboflebitis del lugar de inyección, flebitis del lugar de inyección, inflamación del lugar de inyección, extravasación del lugar de inyección).
En pacientes con insuficiencia renal diálisis-dependientes tratados con Magnograf, se han observado comúnmente reacciones retardadas y transitorias del tipo inflamatorio tales como fiebre, escalofríos y aumento de proteína C-reactiva. En estos pacientes se realizó una Resonancia Magnética un día antes a la hemodiálisis.
Se ha informado de casos de fibrosis sistémica nefrogénica (FSN) con Magnograf.
4.9 Sobredosis
Hasta el momento no se han observado ni se ha informado de signos de intoxicación debidos a sobredosificación durante el empleo en clínica. Sobre la base de los resultados de los estudios de toxicidad aguda, es altamente improbable el riesgo de intoxicación aguda por el empleo de Magnograf. Esta afirmación es también cierta para neonatos y bebés únicamente si la dosis de Magnograf especificada para este grupo de pacientes se inyecta manualmente, sin el empleo de un inyector.
No obstante, la sobredosificación accidental de Magnograf, a causa de la hiperosmolalidad de la solución podría dar lugar (particularmente en niños pequeños y en dependencia del peso corporal) a los siguientes efectos:
- Sistémicos: Aumento de la presión arterial pulmonar, hipervolemia, diuresis osmótica,
deshidratación.
- Locales:
Dolor vascular

Teniendo en cuenta el pequeño volumen que se administra y la bajísima tasa de absorción gastrointestinal de Magnograf (< 1%), es extremadamente improbable una intoxicación por ingestión oral inadvertida de este medio de contraste.
En el caso de sobredosificación accidental, o en pacientes con función renal gravemente restringida, Magnograf puede ser eliminado del organismo mediante hemodiálisis extra-corpórea.
En pacientes con insuficiencia renal debe monitorizarse la función renal.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Magnograf es un medio de contraste paramagnético para empleo en resonancia magnética. El efecto intensificador de contraste es producido por la sal di-N-metilglucamínica del ácido gadopentético (Gd-DTPA; gadopentetato), complejo de gadolinio del ácido pentético (dietilentriaminopentaacético = DTPA). Utilizando una secuencia adecuada (p.ej., técnica espín-eco potenciada en T1) en la resonancia magnética de protones, el acortamiento del tiempo de relajación espín-red de los núcleos atómicos excitados, inducido por el gadolinio, da lugar a un aumento de la intensidad de señal y, por tanto, a un incremento del contraste de la imagen de ciertos tejidos.
El gadopentetato es un compuesto fuertemente paramagnético que reduce notablemente el tiempo de relajación, incluso a bajas concentraciones. La efectividad paramagnética, conocida como relaxividad, determinada por la influencia sobre el tiempo de relajación espín-red de los protones en agua, es de aproximadamente 3,8 l/mmol/seg, a pH 7 y a temperatura de 39°C, y depende sólo débilmente de la intensidad del campo magnético.
El ion paramagnético gadolinio forma con DTPA un complejo extraordinariamente estable tanto in vivo como in vitro (log k = 22-23). El gadopentetato de dimeglumina es un compuesto extremadamente hidrofílico, muy soluble en agua, cuyo coeficiente de reparto entre n-butanol y tampón a pH = 7,6 es de aprox. 0,0001. La sustancia no se une a proteínas en cantidad apreciable ni muestra interacción inhibitoria con enzimas tales como las ATP-asas de Na+ y K+ del miocardio. Magnograf tampoco activa el sistema de complemento y, por tanto, tiene muy escaso riesgo de provocar una reacción anafilactoide. No se ha observado deterioro de la función renal.
In vitro, la morfología eritrocitaria sólo se afectó, en grado ligero, tras incubación durante tiempo prolongado a altas concentraciones de gadopentetato. Tras la administración i.v. de Magnograf en humanos, el proceso reversible puede causar una leve hemólisis intravascular, lo que explicaría el ligero incremento de bilirrubina y hierro séricos que se ha observado, ocasionalmente, en las primeras horas postadministración.
Los resultados de los ensayos clínicos no suministraron indicio alguno de deterioro del estado general ni de las funciones hepática, renal o cardiovascular.
5.2 Propiedades farmacocinéticas
El gadopentetato se comporta en el organismo como otros compuestos altamente hidrófilos, biológicamente inertes (p.ej., manitol o inulina).
Después de la administración i.v., el compuesto difunde rápidamente en el espacio extracelular y se elimina inalterado en la orina, por filtración glomerular. La porción eliminada extrarrenalmente es extremadamente pequeña. Siete días después de la administración i.v. de gadopentetato marcado radioactivamente, se hallaron en el cuerpo de la rata y del perro cantidades muy inferiores al 1% de la dosis administrada. Las concentraciones relativamente más altas se encontraron en los riñones, en forma de Gd-DTPA inalterado. El compuesto no atraviesa la barrera hematoencefálica intacta ni la hemato-seminal. La escasa cantidad que atraviesa la barrera placentaria es rápidamente eliminada por el feto.
Los parámetros farmacocinéticos observados en el hombre fueron independientes de la dosis. Hasta 0,25 mmol de gadopentetato /kg de peso corporal (= 0,5 ml Magnograf/ kg p.c.), los niveles plasmáticos, después de una primera fase de distribución de pocos minutos de duración, declinaron con una vida media de aproximadamente 90 minutos, idéntica a la velocidad de eliminación renal. Con una dosis de 0,1 mmol de gadopentetato/ Kg de peso corporal (=0,2 ml Magnograf/ kg p.c.) se hallaron 0,6 mmol de gadopentetato/ l de plasma a los 3 min post-inyección y 0,24 mmol de gadopentetato/l de plasma a los 60 min p.i. Una media del 83% de la dosis se elimina por vía renal a las 6 horas, y aproximadamente el 91% de la dosis se recupera en orina dentro de las primeras 24 horas. Al 5° día después de la inyección, la porción de dosis eliminada por heces fue menor del 1%. No se detectó ruptura ni degradación metabólica del complejo. El aclaramiento renal del gadopentetato, referido a 1,73 m2 fue de aproximadamente 120 ml/min y es, por tanto, comparable al de inulina o 51Cr-EDTA.
El gadopentetato se elimina casi completamente por vía renal, aún en presencia de función renal alterada (aclaramiento de creatinina > 20 ml/min); la vida media en plasma aumenta en relación con el grado de insuficiencia renal, pero no se observa incremento de la eliminación extrarrenal.
Puesto que la vida media plasmática en el suero se prolonga (hasta 30 horas) en presencia de función renal severamente restringida (aclaramiento de creatinina < 20 ml/min), el gadopentetato debería eliminarse mediante hemodiálisis extracorpórea.
Se ha observado un incremento de la semivida de eliminación en neonatos debido a la inmadurez de la función renal.
5.3 Datos preclínicos sobre seguridad
De acuerdo con los resultados de toxicidad por dosis única, el riesgo de intoxicación aguda con el empleo de Magnograf es extremadamente improbable.
Los estudios de tolerancia sistémica tras administración intravenosa repetida diaria, no revelan ningún hallazgo que desaconseje la administración diagnóstica única de Magnograf en el ser humano.
Los estudios de embriotoxicidad no indicaron potencial teratogénico o embriotóxico alguno tras la administración de Magnograf durante la gestación.
Los estudios de tolerancia local después de la administración intravenosa única y repetida, así como tras la administración intraarterial única, no indicaron que se puedan producir efectos adversos locales en los vasos sanguíneos.
Los estudios de tolerancia local tras administración única paravenosa, subcutánea, así como aplicación intramuscular, indicaron que se podrían manifestar ligeras reacciones de intolerancia local en el lugar de la administración, después de la inyección accidental paravenosa.
En los estudios sobre genotoxicidad no se halló ningún efecto mutagénico, ni in vi tro ni in vivo. Estos resultados y la ausencia de indicios de efectos tóxicos sobre los tejidos de crecimiento rápido en los estudios de administración repetida, así como la farmacocinética del producto y su frecuencia de administración diagnóstica, normalmente única, llevan a considerar como muy improbable el riesgo de efecto tumorígeno en humanos.
Los estudios de sensibilización por contacto en animales de experimentación no indicaron la existencia de potencial sensibilizante alguno con Magnograf.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Meglumina
Ácido dietilentriaminopentaacético (DTPA)
Agua para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
6.3 Período de validez
Magnograf 0,5 mmol/ml solución inyectable: 5 años.
Magnograf 0,5 mmol/ml solución inyectable en jeringa precargada: 3 años.
• Conservación una vez abierto el envase:
Después de abrir el vial/frasco o preparar la jeringa precargada, Magnograf permanece estable durante el día de la exploración. Este tiempo de validez no se refiere a estabilidad físico-química, sino a la posibilidad de contaminación microbiana.
6.4 Precauciones especiales de conservación
Magnograf debe conservarse en lugar fresco y seco, y protegido de la luz.
6.5 Naturaleza y contenido del envase
Magnograf se presenta en:
- viales de 5 ml, 10 ml, 15 ml y 30 ml
- jeringas precargadas de 10 ml, 15ml y 20 ml
- frascos de 100 ml para empleo con inyector.

6.6 Precauciones especiales de manipulación y otras manipulaciones
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
• Presentación en vial/jeringa precargada
La solución se extrae del vial o se prepara la jeringa precargada inmediatamente antes de su administración. El resto del medio de contraste no empleado en una exploración debe desecharse (ver sección 4.2).
Para el montaje de la jeringa precargada, pueden verse las Figuras 1 a 6 que se presentan a continuación:
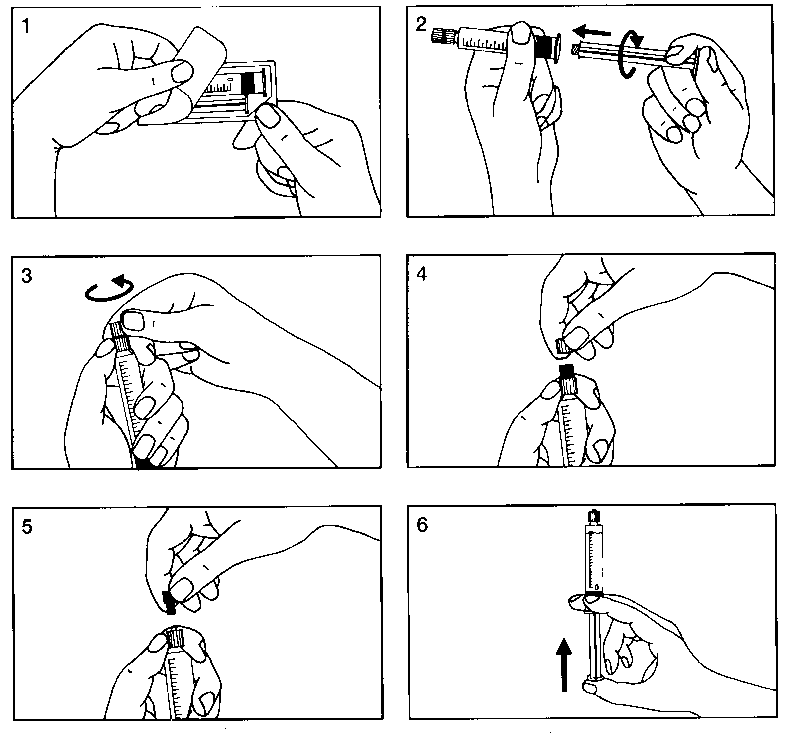
• Presentación en frasco de 100 ml:
La administración del medio de contraste deberá realizarse mediante inyector, cuyo uso está contraindicado en neonatos y bebés (ver sección 4.9). Los elementos del inyector en contacto con el paciente deberán ser sustituidos después de cada exploración, al estar contaminados con sangre. Los restos de solución del medio de contraste que permanezcan en el frasco, así como los tubos de conexión y
los elementos fungibles del sistema de inyección, se desecharán al final del día de la exploración. Es imprescindible seguir las instrucciones adicionales facilitadas por los fabricantes del inyector.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Hispania, S.L.
Av. Baix Llobregat, 3-5
08970 Sant Joan Despí (Barcelona)
España
8. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
Magnograf 0,5 mmol/ml solución inyectable: 59.380.
Magnograf 0,5 mmol/ml solución inyectable en jeringa precargada: 62.020.
9. FECHA DE PRIMERA AUTORIZACIÓN/REVALIDACIÓN DE LA AUTORIZACIÓN
Magnograf 0,5 mmol/ml solución inyectable: 02/03/93.
Magnograf 0,5 mmol/ml solución inyectable en jeringa precargada: 13/10/98.
10. FECHA DE REVISIÓN DEL TEXTO
Mayo 2010.
Agencia española de
medicamentos y
productos sanitarios