Macugen 0,3 Mg Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Macugen 0,3 mg solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una jeringa precargada proporciona una cantidad para un solo uso de 90 microlitros de pegaptanib de sodio, correspondientes a 0,3 mg de la forma del ácido libre del oligonucleótido.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable (inyectable).
La solución es transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Macugen está indicado para el tratamiento de la degeneración macular neovascular (exudativa) asociada a la edad (DMAE) en adultos (ver sección 5.1).
4.2 Posología y forma de administración
Macugen sólo debe ser administrado por oftalmólogos que tengan experiencia en inyecciones intravítreas.
Posología
Antes de llevar a cabo el procedimiento de inyección intravítrea, se debe evaluar detalladamente la historia clínica del paciente en cuanto a reacciones de hipersensibilidad (ver sección 4.4).
La dosis recomendada es de 0,3 mg de pegaptanib, equivalente a 90 microlitros, administrada una vez cada seis semanas (9 inyecciones por año) mediante una inyección intravítrea en el ojo afectado.
Después de la inyección, se han observado casos de elevación transitoria de la presión intraocular en pacientes tratados con Macugen. Por ello, debe realizarse un seguimiento de la perfusión de la cabeza del nervio óptico y de la presión intraocular. Asimismo, se debe realizar un estrecho seguimiento de los pacientes por si hubiera hemorragia del vítreo y endoftalmitis en las dos semanas posteriores a la inyección. Se debe instruir a los pacientes en la necesidad de comunicar inmediatamente cualquier síntoma que sugiera alguna de estas condiciones (ver sección 4.4).
Después de dos inyecciones consecutivas de Macugen, si el paciente no muestra un beneficio con el tratamiento (pérdida de menos de 15 letras de agudeza visual) en la visita de las 12 semanas, se podría considerar el aplazamiento o la suspensión de la terapia con Macugen.
Poblaciones especiales
Pacientes de edad avanzada
No es necesaria ninguna consideración especial.
Insuficiencia hepática
Macugen no ha sido estudiado en pacientes con insuficiencia hepática.
No obstante, no es necesaria ninguna consideración especial en esta población (ver sección 5.2).
Insuficiencia renal
Macugen no ha sido suficientemente estudiado en pacientes con alteración renal grave. No se recomienda un ajuste de dosis en pacientes con alteración renal leve o moderada (ver sección 5.2).
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de Macugen en niños menores de 18 años. No hay datos disponibles.
Forma de administración
Para vía intravítrea exclusivamente.
Antes de su administración, Macugen debe ser inspeccionado visualmente para comprobar si hay partículas en suspensión y decoloración (ver sección 6.6).
La inyección debe realizarse en condiciones asépticas que incluyen el lavado quirúrgico de las manos, el uso de guantes estériles, un campo estéril, un blefarostato estéril para los párpados (o equivalente) y una paracentesis estéril (en caso necesario). Antes de la inyección se debe administrar una anestesia adecuada y un microbicida tópico de amplio espectro.
La jeringa precargada se suministra con un excedente de volumen de producto. Inyectar todo el volumen de la jeringa precargada puede suponer una sobredosis (ver secciones 4.8 y 4.9). Ver sección 6.6 antes de la inyección, para las instrucciones de eliminación del excedente de volumen.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Infección ocular o periocular activa o sospecha de éstas.
4.4 Advertencias y precauciones especiales de empleo
Endoftalmitis
Los procedimientos de inyección intravítrea se asocian con un riesgo de endoftalmitis; en los ensayos clínicos de Macugen, la incidencia de endoftalmitis fue de 0,1 % por inyección (ver sección 4.2)
Incremento de la presión intraocular
Tal y como se puede esperar de la administración de inyecciones intravítreas, se pueden observar aumentos transitorios en la presión intraocular. Por lo tanto, se debe verificar la perfusión de la cabeza del nervio óptico, debiéndose monitorizar el aumento de la presión intraocular adecuadamente después de cada inyección.
En un estudio observacional post-comercialización también se ha notificado un pequeño riesgo de que se produzca un aumento lento y sostenido de la presión intraocular (ver sección 4.8).
Hemorragias intravítreas
Tras la administración de las inyecciones de pegaptanib, se pueden producir hemorragias intravítreas tanto de forma inmediata (el día de la inyección) como con posterioridad (ver sección 4.2).
Reacciones de hipersensibilidad
Durante la etapa post-comercialización se han observado casos de anafilaxia/reacciones anafilactoides, incluyendo angioedema, en las horas siguientes al procedimiento de administración intravítrea con pegaptanib. En ninguno de estos casos se ha establecido una relación directa con Macugen, con otros tratamientos administrados como parte del procedimiento de preparación de la inyección, o con otros factores.
Efectos sistémicos
Se han notificado reacciones adversas sistémicas incluyendo hemorragias no oculares y eventos tromboembólicos arteriales tras la inyección intravitrea de inhibidores de VEGF (factor de crecimiento endotelial vascular) y existe un riesgo teórico de que pueda estar relacionado con la inhibición de VEGF. Existen datos limitados sobre la seguridad en pacientes con antecedentes previos de accidentes cerebrovasculares o ataques isquémicos transitorios. Se deben tomar precauciones al tratar a estos pacientes (ver sección 4.8, título “Reacciones adversas relacionadas con la clase de medicamento”).
Volumen de sobrellenado
La inyección del volumen completo de la jeringa precargada puede dar lugar a reacciones adversas graves; por ello, el excedente de volumen debe ser eliminado antes de la inyección (ver secciones 4.8 y 6.6).
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis, esto es, esencialmente “exento de sodio”.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacción de Macugen con otros medicamentos. Pegaptanib se metaboliza mediante nucleasas, por lo que no son previsibles interacciones farmacológicas mediadas por el citocromo P450.
En dos ensayos clínicos exploratorios realizados en pacientes que recibieron Macugen solo o en combinación con Terapia Fotodinámica (TFD) no se observó ninguna diferencia significativa en la farmacocinética plasmática de pegaptanib.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No existen estudios de pegaptanib en mujeres embarazadas. Los estudios en animales son insuficientes, si bien han mostrado toxicidad reproductiva a niveles de exposición sistémicos elevados (ver sección 5.3). Se desconoce el riesgo potencial en seres humanos. Se estima que la exposición sistémica a pegaptanib es muy baja después de la administración ocular. No obstante, Macugen debe usarse durante el embarazo únicamente si el beneficio potencial para la madre justifica el riesgo potencial para el feto.
Lactancia
Se desconoce si Macugen se excreta por la leche materna. No se recomienda el uso de Macugen durante el periodo de lactancia.
Fertilidad
No hay datos disponibles en humanos del efecto de Macugen sobre la fertilidad. En estudios con animales no se observaron efectos en la fertilidad de machos y hembras de ratones. Ver sección 5.3.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Macugen tiene una influencia menor sobre la capacidad de conducir y el uso de máquinas debido a la posible visión borrosa que aparece de forma transitoria tras la administración de Macugen mediante inyección intravítrea. Los pacientes no deben conducir ni utilizar maquinaria hasta que estos síntomas hayan desaparecido.
4.8 Reacciones adversas
Resumen del perfil de seguridad
La mayoría de las reacciones adversas reportadas de Macugen están relacionadas con el procedimiento de inyección intravítrea.
Las reacciones adversas oculares tras la inyección de Macugen reportadas con mayor frecuencia en los ensayos clínicos son: inflamación de la cámara anterior, dolor de ojo, incremento de la presión intraocular,queratitis punteada, cuerpos flotantes y opacificidades vítreas.
Se reportaron menos frecuentemente reacciones adversas oculares graves incluyendo endoftalmitis, hemorragia retiniana, hemorragia del vítreo y desprendimiento de retina.
Tabla de reacciones adversas
Los datos de seguridad que se describen a continuación resumen las reacciones adversas y las debidas al procedimiento de inyección de los 295 pacientes del grupo de tratamiento que recibió 0,3 mg. Las reacciones adversas se enumeran según clasificación de órganos y frecuencia: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), y frecuencia no conocida (no puede estimarse a partir de los datos disponibles)].
Las notificaciones procedentes de la experiencia post-comercialización se incluyen en cursiva.
Clasificación de órganos del sistema Reacción adversa
MedDRA
Trastornos del sistema inmunológico
Frecuencia no conocida reacción anafiláctica*
Trastornos psiquiátricos
Poco frecuentes pesadillas, depresión
Trastornos del sistema nervioso
Frecuentes Cefalea
Trastornos oculares
Muy frecuentes inflamación de la cámara anterior, dolor ocular, aumento de
la presión intraocular, queratitis puntiforme, partículas flotantes en el vítreo y opacidades del vítreo
Frecuentes sensación anormal en el ojo, catarata, hemorragia
conjuntival, hiperemia conjuntival, edema conjuntival, conjuntivitis, distrofia corneal, defecto del epitelio corneal, trastorno del epitelio de la córnea, edema corneal, ojo seco, endoftalmitis, secreción ocular, inflamación ocular, irritación ocular, prurito ocular, ojo rojo, hinchazón ocular, edema palpebral, aumento del lagrimeo, degeneración macular, midriasis, molestia ocular, hipertensión ocular, hematoma periorbital, fotofobia, fotopsia, hemorragia retiniana, visión borrosa, disminución de la agudeza visual, deterioro visual, desprendimiento del cuerpo vítreo y trastorno del cuerpo vítreo
|
Poco frecuentes |
astenopia, blefaritis, conjuntivitis alérgica, depósitos corneales, hemorragia ocular, prurito palpebral, queratitis, hemorragia del vítreo, reflejo pupilar alterado, abrasión corneal, exudados retinianos, ptosis palpebral, cicatriz retiniana, chalazión, erosión corneal, disminución de la presión intraocular, reacción en la zona de inyección, vesículas en la zona de inyección, desprendimiento de retina, alteración corneal, oclusión arterial retiniana, desgarro retiniano, ectropión, trastorno de los movimientos oculares, irritación palpebral, hifema, trastorno pupilar, trastorno del iris, ictericia ocular, uveítis anterior, depósito en el ojo, iritis, excavación del nervio óptico, deformidad pupilar, oclusión venosa retiniana y prolapso del vítreo |
|
Trastornos del oído y del laberinto Poco frecuentes |
sordera, enfermedad de Méniere agravada, vértigo |
|
Trastornos cardiacos Poco frecuentes |
palpitaciones |
|
Trastornos vasculares Poco frecuentes |
hipertensión, aneurisma aórtico |
|
Trastornos respiratorios, torácicos y mediastínicos Frecuentes Poco frecuentes |
rinorrea nasofaringitis |
|
Trastornos gastrointestinales Poco frecuentes |
vómitos, dispepsia |
|
Trastornos de la piel y del tejido subcutáneo Poco frecuentes Frecuencia no conocida |
dermatitis de contacto, eczema, cambios de color del pelo, erupción cutánea, prurito, sudoración nocturna angioedema* |
|
Trastornos musculoesqueléticos y del tejido conjuntivo Poco frecuentes |
Dolor de espalda |
|
Trastornos generales y alteraciones en el lugar de administración Poco frecuentes |
fatiga, escalofríos, dolor a la palpación, dolor de pecho, enfermedad de tipo gripal |
|
Exploraciones complementarias Poco frecuentes |
incremento de la actividad de la gamma-glutamiltransferasa |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos Poco frecuentes |
Abrasión |
*Experiencia post-comercialización; ver “Descripción de reacciones adversas seleccionadas”
Descripción de reacciones adversas seleccionadas
Casos de anafilaxia/reacciones anafilactoides, incluyendo angioedema, en las horas siguientes a la administración de pegaptanib junto con otros medicamentos que forman parte del procedimiento de preparación de la inyección (ver secciones 4.2 y 4.4).
Se han comunicado casos graves de aumento de la presión intraocular cuando no se eliminaba el excedente de volumen de la jeringa pre-cargada antes de la inyección.
En un estudio observacional post-comercialización, también se han notificado pequeños aumentos sostenidos de la presión intraocular (PIO) después de la administración intravítrea de dosis repetidas. La probabilidad de un incremento de la PIO aumentó según un factor de 1,128 por cada inyección adicional (p= 0,0003). No se encontró ninguna diferencia estadística en la incidencia de PIO elevada entre pacientes con antecedentes de PIO alta o glaucoma respecto a los pacientes sin estos antecedentes.
Reacciones adversas relacionadas con la clase de medicamento
En el estudio clínico, la frecuencia general de las hemorragias no oculares, un potencial efecto adverso relacionado con la inhibición sistémica de VEGF (factor del crecimiento endotelial vascular), aumentó ligeramente en los pacientes tratados con inhibidores de VEGF intravítreo. Sin embargo, no existe un patrón consistente entre las diferentes hemorragias. Los eventos tromboembólicos arteriales (ATEs) son efectos adversos potencialmente relacionados con la inhibición de VEGF. Existe un riesgo teórico de sufrir eventos tromboembólicos arteriales, incluyendo accidentes cerebrovasculares e infarto de miocardio, después del uso intravítreo con inhibidores de VEGF.
En los ensayos clínicos con pegaptanib en pacientes con DMAE o edema macular diabético, se observaron algunos casos de eventos arteriales tromboembólicos, y no hubo diferencias importantes entre los grupos tratados con pegaptanib comprados con el grupo control.
Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V
4.9 Sobredosis
No se han notificado casos de sobredosis con Macugen en los ensayos clínicos.
Por tanto, no se dispone de datos relativos a síntomas agudos, signos o secuelas asociados a sobredosis.
La sobredosis por volumen de inyección superior (p.e., cuando el exceso de volumen en la jeringa precargada no ha sido expulsado antes de la inyección) puede elevar la presión intraocular (ver sección 4.8). El médico debe expulsar el exceso de volumen de la solución siempre, de acuerdo con las instrucciones indicadas en la sección 6.6.
Por tanto, en caso de sobredosis, se debe monitorizar la presión intraocular y si el médico lo considera necesario, adecuar el tratamiento.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Oftalmológicos, Agentes contra desórdenes vasculares oculares, código ATC S01LA03.
Mecanismo de acción
Pegaptanib es un oligonucleótido modificado y pegilado que liga con una especificidad y afinidad altas al Factor de Crecimiento del Endotelio Vascular (VEGF165) extracelular, inhibiendo su actividad. El VEGF es una proteína secretada que induce la angiogénesis, la permeabilidad vascular y la inflamación; se cree que todos estos procesos contribuyen a la progresión de la forma exudativa (húmeda) de la degeneración macular asociada a la edad (DMAE).
Efectos farmacodinámicos
El VEGF165 es la isoforma de VEGF que está implicada de forma preferencial en la neovascularización ocular patológica. En animales, la inhibición selectiva con pegaptanib se mostró tan eficaz en suprimir la neovascularización patológica como en la inhibición de todas las isoformas del VEGF. No obstante, pegaptanib no afectó la vasculatura normal mientras que la inhibición de todas las isoformas del VEGF sí que lo hizo.
Se ha demostrado que se reduce el crecimiento del tamaño medio de lesión total, el tamaño de la neovascularización coroidea (tamaño NVC) y el tamaño del exudado de fluoresceína, en los pacientes con DMAE que fueron tratados con Macugen.
Eficacia clínica y seguridad
Pegaptanib fue estudiado en dos ensayos aleatorizados, controlados, doble ciego y de idéntico diseño (EOP1003; EOP1004) en pacientes con DMAE neovascular. Se trató un total de 1.190 pacientes [892 con pegaptanib, 298 con tratamiento simulado o sham (control)], con una edad media de 77 años. Los pacientes recibieron una media de entre 8,4-8,6 tratamientos de un posible total de 9 en todos los brazos de tratamiento durante el primer año.
Los pacientes fueron aleatorizados para recibir bien el tratamiento simulado o bien dosis de 0,3 mg, 1 mg, o 3 mg de pegaptanib, administrados como inyecciones intravítreas cada 6 semanas durante un periodo de 48 semanas. Se permitió el uso de terapia fotodinámica con verteporfina (TFD) en pacientes con lesiones predominantemente clásicas a criterio de los investigadores.
En los dos ensayos se reclutaron pacientes con todos los subtipos de lesiones de DMAE neovascular (25% con predominantemente clásica, 39% con oculta no clásica y 36% con mínimamente clásica), con tamaños de lesiones de hasta 12 áreas de discos, de las cuales hasta un 50% pudieron estar constituidas por una hemorragia subretiniana y/o hasta un 25% por una cicatriz fibrótica o daño atrófico. A los pacientes se les había administrado hasta una TFD previa, y presentaban una agudeza visual inicial en el ojo del estudio de entre 20/40 y 20/320.
Al cabo de 1 año, se obtuvo un beneficio estadísticamente significativo con 0,3 mg de pegaptanib para la variable primaria de eficacia, es decir, la proporción de pacientes que perdieron menos de 15 letras de agudeza visual (según un análisis combinado pre-especificado: 0,3mg de pegaptanib, 70% vs. tratamiento simulado, 55%, p= 0,0001; EOP1003: 0,3mg de pegaptanib, 73% vs. tratamiento simulado, 59%, p= 0,0105; EOP1004: 0,3mg de pegaptanib, 67% vs. tratamiento simulado, 52%, p= 0,0031).
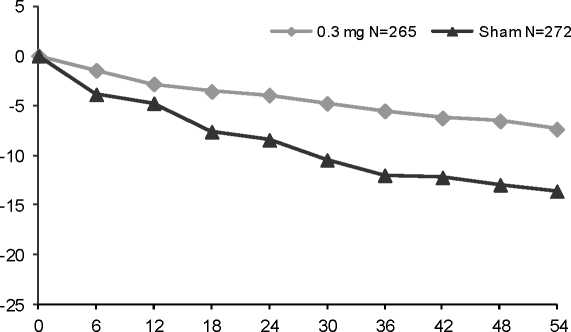
Semanas
N: número de pacientes reclutados
Se demostró un beneficio con 0,3 mg de pegaptanib, independientemente del subtipo de lesión inicial, del tamaño de lesión inicial y de la agudeza visual inicial, así como de la edad, del sexo, de la pigmentación del iris y de la utilización de TFD previa y/o al inicio del ensayo.
Al final del primer año (semana 54), 1.053 pacientes volvieron a ser aleatorizados, bien para que continuasen, bien para que interrumpiesen el tratamiento hasta la semana 102.
De forma general, se mantuvo el beneficio del tratamiento a lo largo de las 102 semanas, conservándose la agudeza visual de forma continuada en aquellos pacientes que volvieron a ser aleatorizados para continuar con pegaptanib. Los pacientes que tras la nueva aleatorización interrumpieron el tratamiento con pegaptanib después de un año, perdieron agudeza visual durante el segundo año.
|
EOP 1003 |
EOP 1004 | |||||
|
Tratamiento |
Tratamiento (tto) simulado - | |||||
|
0,3- interrupción |
(tto) simulado - tto. simulado/ tto. simulado + interrupción |
0,3- | ||||
|
0,3-0,3 |
0,3-0,3 |
interrupció n |
tto. simulado / tto. simulado + interrupción | |||
|
N |
67 |
66 |
54 |
66 |
66 |
53 |
|
Cambio medio en la AV en la |
-1,9 |
-0,0 |
-4,4 |
-1,9 |
-2,0 |
-3,4 |
|
semana 6 Cambio medio en la AV en la |
-4,3 |
-2,0 |
-4,8 |
-2,8 |
-2,2 |
-4,7 |
|
semana 12 Cambio medio en la AV en la |
-9,6 |
-4,3 |
-11,7 |
-8,0 |
-7,6 |
-15,6 |
|
semana 54 Cambio medio en la AV en la semana 102 |
-10,8 |
-9,7 |
-13,1 |
-8,0 |
-12,7 |
-21,1 |
Los datos obtenidos durante el periodo de dos años indican que el tratamiento con Macugen debe iniciarse tan pronto como sea posible. En los estadios avanzados de la enfermedad, debe considerarse el inicio y la continuación del tratamiento con Macugen con respecto al potencial de visión útil en el ojo.
El tratamiento con Macugen administrado en los dos ojos de forma concurrente no ha sido estudiado. No se ha demostrado la seguridad y eficacia de Macugen más allá de los 2 años.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Macugen en los diferentes grupos de la población pediátrica en degeneración macular asociada a la edad. Ver sección 4.2 para consultar la información sobre el uso en población pediátrica.
5.2 Propiedades farmacocinéticas
Absorción:
En animales, pegaptanib se absorbe lentamente hacia la circulación sistémica desde el ojo después de una administración intravitrea. La velocidad de absorción desde el ojo es la fase limitante para la disponibilidad de pegaptanib en animales y es probable que también sea asi en los seres humanos. En los humanos, la media ± la desviación estándar de la semivida plasmática aparente de pegaptanib después de una dosis mono-ocular de 3 mg (10 veces la dosis recomendada) es de 10 ± 4 días.
Tras un periodo de 1 a 4 dias después de una dosis mono-ocular de 3 mg en seres humanos, la concentración plasmática máxima media es de unos 80 ng/ml. La media del área bajo la curva (AUC) de concentración plasmática frente al tiempo es aproximadamente 25 microgramos-h/ml a esta dosis. Pegaptanib no se acumula en el plasma cuando se administra de forma intravitrea cada 6 semanas. A dosis inferiores a 0,5 mg/ojo, las concentraciones plasmáticas de pegaptanib probablemente no superan 10 ng/ml.
No se ha valorado la biodisponibilidad absoluta de pegaptanib después de la administración intravítrea en seres humanos, siendo de aproximadamente 70-100% en conejos, perros y monos.
En los animales que recibieron dosis de pegaptanib de hasta 0,5 mg/ojo en ambos ojos, las concentraciones plasmáticas fueron 0,03% a 0,15% de las alcanzadas en el humor vítreo.
Distribución, biotransformación y eliminación:
En ratones, ratas, conejos, perros y monos, después de una administración intravenosa, pegaptanib se distribuye principalmente en el plasma y no llega a los tejidos periféricos en cantidades apreciables. A las veinticuatro horas de una administración intravítrea de una dosis radiomarcada de pegaptanib en ambos ojos de conejos, la radioactividad fue principalmente distribuida en el humor vítreo, la retina y el humor acuoso. Tanto después de la administración intravítrea como de la intravenosa de pegaptanib radiomarcado a conejos, las concentraciones más altas de radioactividad fueron obtenidas en el riñón (excluyendo las obtenidas en el ojo tras una dosis intravítrea). En conejos, el componente nucleótido 2’-fluorouridina se encuentra en plasma y orina después de dosis únicas intravenosas e intravítreas de pegaptanib radiomarcado. Pegaptanib se metaboliza por endo- y exonucleasas. En conejos, pegaptanib es eliminado tanto en su forma inalterada como sus metabolitos, principalmente en la orina.
Poblaciones especiales:
La farmacocinética de pegaptanib es similar tanto en pacientes mujeres y hombres, como dentro del rango de edad comprendido entre 50 y 90 años.
No se ha estudiado suficientemente el uso de pegaptanib de sodio en pacientes con aclaramiento de creatinina por debajo de 20 ml/min. Una disminución en el aclaramiento de creatinina por debajo de 20 ml/min puede asociarse con un incremento de hasta 2,3 veces el AUC de pegaptanib. No es necesaria ninguna consideración especial en pacientes con un aclaramiento de creatinina superior a 20 ml/min y que sean tratados con la dosis recomendada de 0,3 mg de pegaptanib de sodio.
No se ha estudiado la farmacocinética de pegaptanib en pacientes con insuficiencia hepática. Se estima que la exposición sistémica esté dentro de un rango bien tolerado en pacientes con insuficiencia hepática, dado que se toleró bien una dosis 10 veces más alta (3 mg/ojo).
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas y genotoxicidad. No existe ningún estudio sobre el potencial carcinogénico de pegaptanib.
Pegaptanib no produjo toxicidad materna ni evidencias de teratogenicidad o mortalidad fetal en ratones con dosis intravenosas de 1 a 40 mg/kg/día. Se observó una reducción del peso de las crías (del 5%) y un ligero retraso en la osificación de las falanges de las patas delanteras; esto sólo tuvo lugar a unos niveles de exposición (calculados en base al AUC) 300 veces superiores a los que se estiman en el ser humano. Por lo tanto, se considera que estos hallazgos tienen una relevancia clínica limitada. En el grupo que recibió 40 mg/kg/día, las concentraciones de pegaptanib en el líquido amniótico fueron un 0,05% de las concentraciones plasmáticas maternas. No hay estudios de toxicidad reproductiva en conejos.
No hay datos disponibles para evaluar los índices de fertilidad o de apareamiento entre machos y hembras.
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Cloruro de sodio
Fosfato monobásico de sodio monohidratado Fosfato dibásico de sodio heptahidratado Hidróxido de sodio (para ajuste de pH)
Ácido clorhídrico (para ajuste de pH)
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8oC). No congelar.
La solución para inyección debe alcanzar la temperatura ambiente (por debajo de 25°C) antes de la inyección.
Este medicamento se debe descartar si se ha mantenido a temperatura ambiente más de dos semanas. Para prevenir de la contaminación, la jeringa no debe sacarse de la bolsa hasta que el paciente esté preparado para la inyección.
6.5 Naturaleza y contenido del envase
Cada envase contiene una bolsa en un estuche, con una jeringa precargada de vidrio Tipo I, de 1 ml sellada con una junta de estanqueidad elastomérica (goma de bromobutilo) y con un vástago prefijado, todo ello sostenido por un soporte de sujección de plástico. La jeringa tiene un adaptador prefijado de policarbonato plástico denominado luer lock cuyo extremo cónico se cierra con un protector elastomérico (isopreno sintético de bromobutilo).
Cada jeringa precargada contiene aproximadamente 0,25 - 0, 27 ml de solución.
Cada caja contiene una jeringa precargada en un estuche (embalaje monodosis).
El envase se suministra sin aguja.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Macugen es para un solo uso. No debe utilizarse Macugen si la solución está turbia, si se observan partículas en suspensión, si existiera cualquier evidencia de daño en la jeringa, si falta o no está unido el soporte de sujección de plástico.
Antes de la administración, la jeringa se debe separar del soporte de sujección de plástico y debe quitarse el protector. En el adaptador luer lock se ha de insertar una aguja de 27 ó 30 G x ‘A pulgada, de forma que permita la administración del medicamento (Ver Figura 1 abajo).
ADVERTENCIA: Como la jeringa precargada contiene un volumen de medicamento superior (250-270 microlitros) al recomendado (90 microlitros), se debe descartar una parte del volumen contenido en la jeringa antes de la administración. Siga las instrucciones descritas a continuación para la eliminación del volumen excedente antes de la inyección.
Figura 1. Antes de eliminar la burbuja de aire y el exceso de medicamento
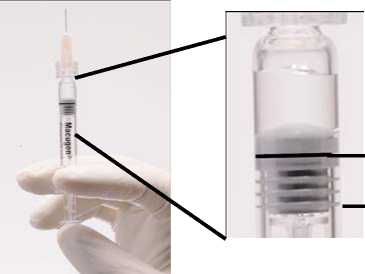
Línea de dosificación
3" estría (borde superior)
(La formación de la burbuja de aire puede variar)
Se debe comprobar que la jeringa con la aguja ya insertada, y colocada en posición vertical, no tiene burbujas en el interior. Si las tuviera, deben darse unos golpecitos suaves con un dedo a la jeringa hasta que las burbujas suban a la parte superior de la misma.
Empujar LENTAMENTE el émbolo para eliminar todas las burbujas y eliminar el excedente de medicamento de manera que el borde superior de la 3a estría de la junta de estaqueidad, se alinee con la línea de dosificación negra pre-impresa (ver figura 2, debajo). La junta de estanqueidad no debe retirarse.
Figura 2. Después de eliminar la burbuja de aire y el excedente de medicamento

Línea de dosificación y borde superior de la 3a estría alineadas
En este momento, el contenido que queda en la jeringa debe ser inyectado.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
PharmaSwiss Ceská republika s.r.o.
Jankovcova 1569/2c 170 00 Praha 7 República Checa
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/325/002
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 31/01/2006 Fecha de la última renovación: 19/11/2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema. europa.eu/
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes
Pfizer Manufacturing Belgium NV,
Rijksweg 12 B-2870 Puurs Belgium
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (Ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
• Medidas adicionales de minimización de riesgos
Previo al lanzamiento en cada uno de los Estados Miembros, el Titular de la Autorización de Comercialización (TAC) debe acordar los materiales educacionales finales con las autoridades nacionales competentes.
El TAC debe garantizar que, tras la discusión y el acuerdo con las autoridades nacionales competentes de cada uno de los Estados Miembros en los que Macugen está comercializado, durante el lanzamiento y tras el lanzamiento, todas las clínicas oftalmológicas donde se pueda utilizar Macugen tengan materiales educacionales actualizados que incluyan los siguientes elementos:
• Ficha técnica.
• Manual de seguridad para el médico.
• Vídeo sobre el procedimiento de inyección intravítrea.
• Pictograma sobre el procedimiento de inyección intravítrea.
• Información para el paciente.
El manual de seguridad para el médico debe contener los siguientes elementos clave:
a) El procedimiento de inyección intravítrea según se realizó en los ensayos clínicos pivotales junto con todas las mejoras técnicas.
b) La utilización de povidona iodada.
c) La limpieza de los párpados.
d) La utilización de anestesia para garantizar el confort de los pacientes.
e) Las técnicas estériles para minimizar el riesgo de infección.
f) La utilización de antibióticos.
g) Las técnicas de inyección intravítrea.
h) Signos y síntomas clave de reacciones adversas relacionadas con la inyección intravítrea, entre otros, endoftalmitis, aumento de la presión intraocular, daño en la retina, hemorragia intraocular, catarata traumática, hipersensibilidad e inyección del volumen excedente.
i) El control y seguimiento de la presión intraocular.
j) El control y seguimiento de la endoftalmitis.
k) El entendimiento de los factores de riesgo involucrados en el desarrollo de endoftalmitis.
l) La notificación de acontecimientos adversos graves (tarjeta de alerta).
La información al paciente debe contener los siguientes elementos clave:
m) Signos y síntomas clave de los acontecimientos adversos graves asociados con el procedimiento de inyección intravítrea, entre otros, endoftalmitis, aumento de la presión intraocular, daño en la retina, hemorragia intraocular, catarata traumática, hipersensibilidad e inyección del volumen excedente.
n) Cuándo debe requerir atención médica urgente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR Estuche
1. NOMBRE DEL MEDICAMENTO
Macugen 0,3 mg solución inyectable pegaptanib
Una jeringa precargada proporciona una dosis única de 90 microlitros que contiene pegaptanib de sodio, que corresponde a 0,3 mg del ácido libre del oligonucleótido.
3. LISTA DE EXCIPIENTES
Cloruro de sodio, fosfato monobásico de sodio monohidratado, fosfato dibásico de sodio heptahidratado, hidróxido de sodio y ácido clorhídrico (para ajustar pH), agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable.
Proporciona un dosis única de 0,3 mg en 90 microlitros.
El envase contiene una jeringa precargada, una junta de estanqueidad y un vástago prefijado. Se suministra sin aguja.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Para un único uso.
Leer el prospecto antes de utilizar este medicamento. Vía intravítrea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
ADVERTENCIA: Eliminar el excedente de volumen antes de inyectar.
Alinear la 3a estría de la junta de estanqueidad con la línea de dosificación negra pre-impresa.
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera. No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
PharmaSwiss Ceská republika s.r.o. Jankovcova 1569/2c 170 00 Praha 7 República Checa
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/325/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
Jeringa precargada
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Macugen 0,3 mg inyectable pegaptanib
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
3. FECHA DE CADUCIDAD
4. NÚMERO DE LOTE
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
Única dosis: 0,3 mg / 90 pl
6. OTROS
Bolsa conteniendo la jeringa precargada, una junta de estanqueidad y un vástago prefijado
1. NOMBRE DEL MEDICAMENTO
Macugen 0,3 mg solución inyectable
pegaptanib
Vía intravítrea
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
Única dosis: 0,3 mg / 90 pl
6. OTROS
No debe abrirse la bolsa hasta que el paciente haya sido preparado para recibir la inyección.
ADVERTENCIA: Eliminar el excedente de volumen antes de inyectar.
Alinear la 3a estría de la junta de estanqueidad con la línea de dosificación negra pre-impresa.
B. PROSPECTO
Prospecto: información para el paciente
Macugen 0,3 mg solución inyectable
Pegaptanib
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Si experimenta efectos adversos consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Macugen y para qué se utiliza
2. Qué necesita saber antes de que le administrenr Macugen
3. Cómo le administrarán Macugen
4. Posibles efectos adversos
5 Conservación de Macugen
6. Contenido del envase e información adicional
1. Qué es Macugen y para qué se utiliza
Macugen es una solución que se inyecta en el ojo. Pegaptanib, el principio activo de este medicamento, inhibe la actividad del factor implicado en la formación anormal de nuevos vasos sanguíneos en el ojo, conocido como Factor de Crecimiento Endotelial Vascular (VEGF).
Macugen se utiliza para el tratamiento de la forma exudativa de degeneración macular asociada a la edad (DMAE). Esta enfermedad provoca una pérdida de visión como resultado del daño en la parte central de la retina (llamada mácula), localizada en la zona posterior del ojo. La mácula permite que el ojo tenga una visión central fina, la cual es necesaria para conducir un coche, leer letra pequeña y otras tareas similares.
La forma exudativa de DMAE es causada por el crecimiento anómalo de vasos sanguíneos debajo de la retina y de la mácula. Estos nuevos vasos sanguíneos pueden tener fugas de sangre o líquido que provocan que la mácula aumente de grosor y se levante, y en consecuencia, se distorsione o se destruya la visión central. En estas circunstancias, la pérdida de visión puede ser rápida y grave. Este medicamento actúa mediante la inhibición del crecimiento de estos vasos sanguíneos anómalos y deteniendo el sangrado o fugas. Macugen es utilizado para el tratamiento de todos los tipos de crecimiento de vasos sanguíneos anómalos en pacientes con DMAE.
2. Qué necesita saber antes de que le administren Macugen No deben administrarle Macugen
Si es alérgico a pegaptanib o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Si tiene una infección del ojo o alrededor del mismo.
Advertencias y precauciones
Consulte a su médico antes de que le administren Macugen.
Ocasionalmente, puede producirse una infección o un sangrado en el interior del ojo tras la inyección con Macugen (en las dos semanas posteriores). Es importante identificar y tratar este tipo de
situaciones lo más rápidamente posible. Consulte a su médico inmediatamente si notase cualquiera de los siguientes síntomas: dolor ocular o aumento de la molestia ocular, aumento del enrojecimiento del ojo, visión borrosa o disminución de la visión, incremento de la sensibilidad a la luz e incremento del número de pequeñas manchas en la visión. Si no puede contactar con su médico, contacte inmediatamente con otro médico.
En algunos pacientes, la presión dentro del ojo tratado puede verse incrementada durante un corto periodo de tiempo inmediatamente después de la inyección. Su médico puede realizarle un seguimiento después de cada inyección.
Pueden producirse reacciones alérgicas graves poco después de la inyección. Los síntomas que puede experimentar y las instrucciones sobre que debe hacer en estos casos están descritas en la sección 4 de este prospecto.
Niños y adolescentes
Macugen no debe utilizarse en niños ni adolescentes menores de 18 años de edad.
Uso de Macugen con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes del tratamiento con Macugen.
• No se ha utilizado Macugen en mujeres embarazadas; por tanto, se desconocen los riesgos potenciales.
No se recomienda el uso de Macugen durante la lactancia ya que se desconoce si Macugen pasa a la leche materna. Consulte a su médico o farmacéutico antes de recibir tratamiento con Macugen.
Conducción y uso de máquinas
Puede experimentar visión borrosa temporalmente después de recibir Macugen. Si esto le ocurriera, no conduzca ni utilice máquinas hasta que desaparezcan estos síntomas.
Información importante sobre algunos componentes de Macugen
Este medicamento contiene menos de 23 mg (1 mmol) de sodio por dosis de 90 microlitros, por lo que se considera esencialmente “exento de sodio” (ver sección 6).
3. Cómo le administrarán Macugen
Su médico le administrará todas las inyecciones de Macugen.
Macugen es administrado como una única inyección (0,3 mg) en el ojo a intervalos de 6 semanas (es decir, 9 veces al año). La inyección es administrada en el vítreo del ojo, que es una sustancia de tipo gelatinoso que hay dentro del ojo. Su médico deberá realizar un seguimiento de su enfermedad y determinará la duración del tratamiento con Macugen.
Antes de administrar el tratamiento, su médico puede pedirle que use antibiótico en gotas oftálmicas, o que se lave los ojos cuidadosamente. Antes de la inyección, su médico le administrará anestesia local (medicamento desensibilizante). Este permitirá reducir o aliviar el posible dolor que pueda sufrir con la inyección.
Por favor no olvide informar a su médico si sabe que usted es alérgico a algún medicamento.
Después de cada inyección puede que se le recomiende que utilice un antibiótico en gotas oftálmicas (o algún otro tipo de tratamiento antibiótico) con el fin de evitar la aparición de una infección ocular.
Si se le ha administrado más Macugen del que se debe
En caso de que se haya inyectado un volumen de Macugen en exceso, podría producirse un aumento de la presión intraocular grave.
Siempre que experimente visión borrosa, dolor/incomodidad en el ojo, ojos rojos o náuseas y vómitos, acuda inmediatamente al médico.
Si tiene cualquier otra duda sobre el uso de este producto, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Se han comunicado algunos casos de reacción alérgica grave, incluyendo reacción anafiláctica y angioedema, cuyos síntomas se describen abajo, poco tiempo después de la inyección. Por favor solicite asistencia sanitaria inmediatamente si experimenta cualquiera de los siguientes síntomas tras la inyección: dificultad respiratoria repentina o respiración con pitos, hinchazón de la boca, de la cara, de las manos o de los pies, picor de la piel, desvanecimiento, pulso acelerado, calambres en el estómago, náuseas, vómitos o diarrea. La frecuencia de estos efectos adversos no puede estimarse con los datos disponibles.
Ocasionalmente, puede desarrollarse una infección en la parte interna del ojo tras dos semanas de tratamiento con Macugen. Los síntomas que pueda experimentar están descritos en el apartado 2 de este prospecto (“Advertencias y precauciones”). Por favor, lea ese apartado donde se le indica qué tiene que hacer en el caso de presentar uno de estos síntomas.
Otros posibles efectos adversos son los siguientes:
Muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes)
Estos efectos adversos probablemente están más relacionados con el procedimiento de inyección que con el medicamento, e incluyen los siguientes:
• inflamación del ojo
• dolor ocular
• incremento de la presión dentro del ojo
• pequeñas marcas en la superficie del ojo (queratitis puntiforme)
• pequeñas manchas o puntos en la visión (como ‘moscas flotantes’ o manchas oscuras).
Frecuentes (pueden afectar hasta 1 de cada 10 pacientes)
Otros efectos adversos oculares comunicados frecuentemente y que pueden estar causados posiblemente por el medicamento o por el procedimiento de inyección, son los siguientes:
• visión borrosa
• alteración visual
• molestia ocular
• disminución de la visión
• aumento de la sensibilidad a la luz, aparición de destellos
• hemorragia alrededor del ojo (hemorragia periorbital)
• hemorragia en el ojo (hemorragia subconjuntival)
• alteración de la parte gelatinosa que hay dentro del ojo (alteración del cuerpo vítreo), así como el desprendimiento o desgarro (desprendimiento del cuerpo vítreo)
• opacificación del cristalino (catarata)
• alteración de la superficie del ojo (córnea)
• hinchazón o inflamación del párpado, hinchazón del área interior del párpado o de la superficie externa del ojo (conjuntiva)
• inflamación del ojo, lagrimeo, inflamación de la conjuntiva (conjuntivitis), sequedad del ojo, secreción ocular, irritación del ojo, picor en el ojo, enrojecimiento ocular o agrandamiento de la pupila.
Otros efectos adversos no oculares comunicados frecuentemente y que pueden estar causados posiblemente por el medicamento o por el procedimiento de inyección, son los siguientes:
• dolor de cabeza
• secreción nasal.
Poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes)
Otros efectos adversos oculares comunicados poco frecuentemente y que pueden estar causados posiblemente por el medicamento o por el procedimiento de inyección, son los siguientes:
• inflamación del ojo o de la superficie del mismo
• hemorragia en el ojo o en la parte interna del ojo (vítreo)
• tensión ocular
• inflamación de la parte central de la superficie del ojo (queratitis)
• pequeños depósitos en el ojo o en la superficie del ojo (córnea), depósitos en el fondo del ojo
• picor de párpados
• alteración de la reacción de los ojos a la luz (reflejo pupilar alterado)
• pequeña erosión en la parte central de la superficie del ojo (córnea)
• párpados caídos
• cicatriz dentro del ojo (cicatriz retiniana)
• pequeño bulto en el párpado debido a un proceso inflamatorio (chalazión)
• descenso de la presión dentro del ojo
• reacción en el lugar de la inyección, vesículas en el lugar de la inyección
• desplazamiento o desgarro de la capa situada en el fondo del ojo (retina)
• alteración de la pupila, alteración del área coloreada del ojo (iris)
• oclusión de la arteria de la retina
• párpado vuelto hacia fuera, alteración del movimiento del ojo, irritación del párpado
• hematoma en el ojo, decoloración del ojo, depósito en el ojo
• inflamación del ojo (iritis)
• daño en la cabeza del nervio óptico
• deformación de la pupila
• oclusión de la vena situada en el fondo del ojo
• secreción de la sustancia gelatinosa del ojo.
Otros efectos adversos no oculares comunicados poco frecuentemente y que pueden estar causados posiblemente por el medicamento o por el procedimiento de inyección, son los siguientes:
• pesadillas, depresión, sordera, vértigo
• palpitaciones, presión arterial alta, dilatación de la aorta (vaso sanguíneo principal)
• inflamación del tracto respiratorio superior, vómitos, indigestión
• irritación e inflamación de la piel, cambios en el color de pelo, erupción de la piel, picor
• sudoración nocturna, dolor de espalda, cansancio, escalofríos, dolor al tacto, dolor de pecho, fiebre repentina y síntomas similares a un catarro (dolor generalizado)
• elevación de las enzimas hepáticas y abrasión.
Existe un pequeño riesgo de que se produzca un ligero aumento duradero de la presión en el interior del ojo después de que se realicen varias inyecciones en el ojo.
Comunicación de efectos adversos:
Si experimenta cualquier tipo de efecto adverso, consulte con su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V.
. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Macugen
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y en el envase después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Se debe desechar este medicamento si se ha mantenido a temperatura ambiente durante un periodo superior a dos semanas.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Macugen
- El principio activo es pegaptanib. Con cada jeringa monodosis, precargada, se administra una dosis de 0,3 mg de pegaptanib en 90 microlitros.
- Los demás componentes son cloruro de sodio, fosfato monobásico de sodio monohidratado, fosfato dibásico de sodio heptahidratado, hidróxido de sodio y ácido clorhídrico (para ajustar el pH) y agua para preparaciones inyectables. Para más información sobre el contenido de sodio de Macugen, ver sección 2.
Aspecto de Macugen y contenido del envase
Macugen solución inyectable se presenta en un envase de un solo uso.
Cada envase contiene una bolsa con una jeringa precargada de vidrio Tipo I, que contiene 0,25 - 0,27 ml de solución, sellada con una junta de estanqueidad elastomérica y con un vástago prefijado, todo ello sostenido por un soporte de sujeción de plástico. La jeringa tiene un adaptador prefijado de policarbonato plástico denominado luer lock cuyo extremo cónico se cierra con un protector elastomérico.
El envase se suministra sin aguja.
Titular de la autorización de comercialización
PharmaSwiss Ceská republika s.r.o.
Jankovcova 1569/2c 170 00 Praha 7 República Checa
Responsable de la fabricación:
Pfizer Manufacturing Belgium NV,
Rijksweg 12 B-2870 Puurs Belgium
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Bausch & Lomb Pharma nv/sa, Belgium Tél/Tel: + 32 (0)3 280 82 84 |
Lietuva PharmaSwiss UAB Latvia Tel. + 370 5 279 0762 |
|
Etarapna PharmaSwiss EOOD Tea.: + 359 2 89 52 110 |
Luxembourg/Luxemburg Bausch & Lomb Pharma nv/sa, Belgium Tél/Tel: + 32 (0)3 280 82 84 |
|
Ceská republika PharmaSwiss Ceská republika s.r.o. Tel: + 420 234 719 600 |
Magyarország Valeant Pharma Magyarország Kft. Tel. +36 1 345 5900 |
|
Danmark Bausch & Lomb Nordic AB Tlf: 80 88 82 68 Tel (fra udlandet): +46 8 616 95 85 |
Malta Laboratoire Chauvin, France Tél: + 33 (0)4 67 12 30 30 |
|
Deutschland Bausch & Lomb GmbH Tel: + 49 (0)30 33093 0 |
Nederland Bausch & Lomb Pharma nv/sa, Belgium Tel: + 32 (0)3 280 82 84 |
|
Eesti PharmaSwiss Eesti OÜ Tel: +372 6 827 400 |
Norge Bausch & Lomb Nordic AB Tlf: 800 19 841 Fra utlandet Tlf: +46 8 616 95 85 |
|
EXláóa Pharmaswiss Hellas A.E. Tq^: +30 210 8108 460 |
Osterreich Bausch & Lomb GmbH Tel: + 49 (0)30 33093 0 |
|
España Bausch & Lomb, S.A. Tel: + 34 91 657 63 00 |
Polska Valeant sp. z o.o. sp. j. Tel.: +48 17 865 51 00 |
|
France Laboratoire Chauvin SAS Tél: + 33 (0)4 67 12 30 30 |
Portugal Bausch & Lomb, S.A. (Sucursal Portugal) Tel: + 351 21 424 15 10 |
|
Hrvatska PharmaSwiss d.o.o. Tel: +385 1 6311 833 |
Romania Valeant Pharma S.R.L. Tel: +40 374 102 600 |
|
Ireland Bausch & Lomb UK Ltd. Tel: +44 (0) 1748 828864 |
Slovenija PharmaSwiss d.o.o. Tel: + 386 1 2364 700 |
Ísland
Bausch & Lomb UK Ltd.
Sími frá útlondum: +44 (0) 1748 828864
Italia
Bausch & Lomb-IOM S.p.A.
Tel: + 39 (0)2 27407300
Slovenská republika
Valeant Slovakia s.r.o.
Tel: +421 2 3233 4900
Suomi/Finland
Bausch & Lomb Nordic AB
Puh./Tel: 0800 773 851
Ulkomailta/Fran utmlands: +46 8 616 95 85
Sverige
Bausch & Lomb Nordic AB
Tel: 020 088 3496
Fran utomlands: +46 8 616 95 85
United Kingdom Bausch & Lomb UK Ltd.
Tel: +44 (0) 1748 828864
Fecha de la última revisión de este prospecto
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema. europa.eu/
La siguiente información está destinada unicamente a profesionales sanitarios:
ADVERTENCIA: Como la jeringa precargada se suministra con un volumen de producto (250270 microlitros) superior a la dosis recomendada (90 microlitros), se debe descartar una parte del volumen contenido en la jeringa antes de la administración. Siga las instrucciones descritas a continuación para expulsar el exceso de volumen antes de la inyección.
Figura 1. Antes de eliminar la burbuja de aire y el exceso de medicamento
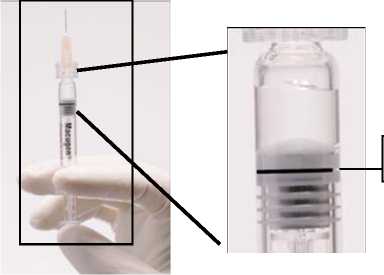
Línea de dosificación
3a estría (borde superior)
(La formación de la burbuja de aire puede variar)
Se debe comprobar que la jeringa con la aguja ya insertada, y colocada en posición vertical, no tiene burbujas en el interior. Si las tuviera, deben darse unos golpecitos suaves con un dedo a la jeringa hasta que las burbujas suban a la parte superior de la misma.
Empujar LENTAMENTE el émbolo para eliminar todas las burbujas y expulsar el excedente de medicamento de manera que el borde superior de la 3a estría de la junta de estanqueidad, se alinee con la línea de dosificación negra pre-impresa (ver figura 2, debajo). La junta de estanqueidad no debe retirarse.
Figura 2. Después de eliminar la burbuja de aire y el excedente de medicamento
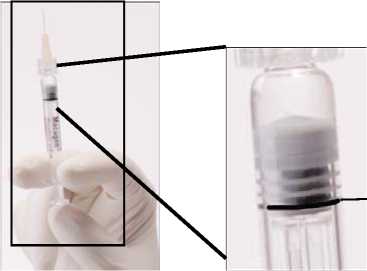
Línea de dosificación y borde superior de la 3a estría alineadas.
En este momento, el contenido que queda en la jeringa debe ser inyectado.
32