Lucentis 10 Mg/Ml Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Lucentis 10 mg/ml solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un ml contiene 10 mg de ranibizumab*. Cada vial contiene 2,3 mg de ranibizumab en 0,23 ml de solución.
*Ranibizumab es un fragmento de anticuerpo monoclonal humanizado producido en células de Escherichia coli mediante tecnología de ADN recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución acuosa transparente, de incolora a amarillo pálido.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lucentis está indicado en adultos para:
• El tratamiento de la degeneración macular asociada a la edad (DMAE) neovascular (exudativa)
• El tratamiento de la alteración visual debida al edema macular diabético (EMD)
• El tratamiento de la alteración visual debida al edema macular secundario a la oclusión de la
vena retiniana (OVR) (oclusión de la rama venosa retiniana u oclusión de la vena central retiniana)
• El tratamiento de la alteración visual debida a la neovascularización coroidea (NVC) secundaria a la miopía patológica (MP)
4.2 Posología y forma de administración
Lucentis debe ser administrado por un oftalmólogo que tenga experiencia en la administración de inyecciones intravítreas.
La dosis recomendada de Lucentis es 0,5 mg administrada en forma de inyección intravítrea única. Esto corresponde a un volumen de inyección de 0,05 ml. El intervalo entre dos dosis inyectadas en el mismo ojo debe ser como mínimo de cuatro semanas.
El tratamiento se inicia con una inyección al mes hasta alcanzar la agudeza visual máxima y/o no haya signos de actividad de la enfermedad, es decir ningún cambio en la agudeza visual ni en otros signos y síntomas de la enfermedad bajo tratamiento continuado. En pacientes con DMAE exudativa, EMD y OVR inicialmente pueden ser necesarias tres o más inyecciones consecutivas administradas mensualmente.
A partir de ese momento, los intervalos de monitorización y tratamiento se deben determinar según criterio médico y en base a la actividad de la enfermedad, valorada mediante la agudeza visual y/o parámetros anatómicos.
Se debe interrumpir el tratamiento con Lucentis si bajo criterio del médico, los parámetros visuales y anatómicos indican que el paciente no se está beneficiando del tratamiento continuado.
La monitorización para determinar la actividad de la enfermedad puede incluir examen clínico, control funcional o técnicas de imagen (p. ej. tomografía de coherencia óptica o angiografía con fluoresceína).
Si se está tratando a los pacientes de acuerdo a un régimen de tratar y extender, una vez se ha alcanzado la agudeza visual máxima y/o no hay signos de actividad de la enfermedad, los intervalos de tratamiento se pueden espaciar de forma gradual hasta que vuelvan a aparecer signos de actividad de la enfermedad o alteración visual. En el caso de la DMAE exudativa el intervalo de tratamiento no debe espaciarse en más de dos semanas cada vez y en el caso del EMD se puede espaciar hasta un mes cada vez. Para la OVR, los intervalos de tratamiento también pueden espaciarse de forma gradual, sin embargo los datos que hay no son suficientes para determinar la duración de estos intervalos. Si vuelve a aparecer actividad de la enfermedad, se debe acortar el intervalo de tratamiento de manera consecuente.
En el tratamiento de la alteración visual debida a la NVC secundaria a MP, muchos pacientes pueden necesitar únicamente una o dos inyecciones durante el primer año, mientras que algunos pacientes pueden necesitar tratamiento con mayor frecuencia (ver sección 5.1).
Lucentis y fotocoagulación con láser en EMD y en edema macular secundario a oclusión de la rama venosa retiniana (ORVR)
Existe alguna experiencia con Lucentis administrado concomitantemente con fotocoagulación con láser (ver sección 5.1). Cuando se administren en el mismo día, Lucentis se debe administrar como mínimo 30 minutos después de la fotocoagulación con láser. Lucentis puede administrarse en pacientes que han recibido fotocoagulación con láser previamente.
Lucentis y la terapia fotodinámica con Visudyne en la NVC secundaria a MP No hay experiencia en la administración concomitante de Lucentis y Visudyne.
Poblaciones especiales
Insuficiencia hepática
Lucentis no ha sido estudiado en pacientes con insuficiencia hepática. Sin embargo, no es necesaria ninguna consideración especial en esta población.
Insuficiencia renal
No es necesario ningún ajuste de la dosis en pacientes con insuficiencia renal (ver sección 5.2).
Pacientes de edad avanzada
No se requiere ningún ajuste de la dosis en pacientes de edad avanzada. Existe experiencia limitada en pacientes con EMD mayores de 75 años.
Población pediátrica
No se ha establecido la seguridad y eficacia de Lucentis en niños y adolescentes menores de 18 años de edad. No se dispone de datos.
Forma de administración
Vial para un solo uso. Únicamente para vía intravítrea.
Antes de la administración de Lucentis se debe comprobar visualmente la ausencia de partículas y decoloración.
El procedimiento de inyección deberá llevarse a cabo bajo condiciones asépticas, que incluyen el lavado quirúrgico de las manos, el uso de guantes estériles, un campo estéril, un blefarostato estéril para los párpados (o equivalente) y la disponibilidad de una paracentesis estéril (en caso necesario).
Antes de realizar el procedimiento de inyección intravítrea, se deberá evaluar detalladamente la historia clínica del paciente en cuanto a reacciones de hipersensibilidad (ver sección 4.4). Antes de la inyección se debe administrar una anestesia adecuada y un microbicida tópico de amplio espectro para desinfectar la piel de la zona periocular, párpado y superficie ocular, de acuerdo con la práctica local.
Para la información relativa a la preparación de Lucentis, ver sección 6.6.
La aguja para inyección se deberá introducir 3,5-4,0 mm por detrás del limbo en la cavidad vítrea, evitando el meridiano horizontal y en dirección al centro del globo. Seguidamente debe liberarse el volumen de inyección de 0,05 ml; las inyecciones siguientes deberán aplicarse cada vez en un punto escleral distinto.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Pacientes con infecciones oculares o perioculares o con sospecha de éstas.
Pacientes con inflamación intraocular grave.
4.4 Advertencias y precauciones especiales de empleo Reacciones relacionadas con la inyección intravítrea
Las inyecciones intravítreas, incluidas las de Lucentis, se han asociado a endoftalmitis, inflamación intraocular, desprendimiento de retina regmatógeno, desgarro retiniano y catarata traumática iatrogénica (ver sección 4.8). Siempre que se administre Lucentis se deben emplear técnicas de inyección asépticas adecuadas. Además, deberá realizarse un seguimiento de los pacientes durante la semana posterior a la inyección para poder administrar tratamiento temprano en caso de infección. Se deberá instruir a los pacientes sobre la necesidad de comunicar inmediatamente cualquier síntoma que sugiera endoftalmitis o cualquiera de los acontecimientos mencionados anteriormente.
Aumento de la presión intraocular
Se han observado aumentos transitorios de la presión intraocular (PIO) en los 60 minutos siguientes a la inyección de Lucentis. También se han identificado aumentos sostenidos de la PIO (ver sección 4.8). Tanto la presión intraocular como la perfusión de la cabeza del nervio óptico, se deben monitorizar y tratar adecuadamente.
Tratamiento bilateral
Los escasos datos existentes sobre el tratamiento bilateral con Lucentis (incluyendo la administración en el mismo día) no sugieren un riesgo incrementado de efectos adversos sistémicos en comparación con el tratamiento unilateral.
Inmunogenicidad
Hay un potencial de inmunogenicidad con Lucentis. Dado que en sujetos con EMD existe un potencial para una exposición sistémica incrementada, no puede excluirse un mayor riesgo para desarrollar hipersensibilidad en esta población de pacientes. También se deberá instruir a los pacientes sobre la necesidad de notificar si la inflamación intraocular incrementa en su gravedad, lo que puede ser un signo clínico atribuible a la formación de anticuerpos intraoculares.
Uso concomitante con otros anti-VEGF (factor de crecimiento endotelial vascular)
Lucentis no se deberá administrar de forma concurrente con otros medicamentos anti-VEGF (sistémicos u oculares).
Aplazamiento del tratamiento con Lucentis
La administración se deberá aplazar y el tratamiento no deberá reanudarse antes del siguiente tratamiento programado en caso de:
• una disminución en la agudeza visual mejor corregida (AVMC) de >30 letras comparado con la última evaluación de la agudeza visual;
• una presión intraocular de >30 mmHg;
• una rotura retiniana;
• una hemorragia subretiniana que afecte al centro de la fóvea, o, si el tamaño de la hemorragia es >50% del área total de la lesión;
• cirugía intraocular realizada en los 28 días previos o prevista durante los 28 días posteriores. Desgarro del epitelio pigmentario de la retina
Los factores de riesgo asociados con el desarrollo de un desgarro del epitelio pigmentario de la retina tras la terapia con anti-VEGF para la DMAE exudativa, incluyen un desprendimiento del epitelio pigmentario de la retina extenso y/o elevado. Cuando se inicie la terapia con Lucentis se debe tener precaución en pacientes con estos factores de riesgo de desarrollar desgarros del epitelio pigmentario de la retina.
Desprendimiento de retina regmatógeno o agujeros maculares
El tratamiento se debe interrumpir en sujetos con desprendimiento de retina regmatógeno o agujeros maculares en estadíos 3 ó 4.
Poblaciones con datos limitados
Sólo existe experiencia limitada en el tratamiento de sujetos con EMD debido a diabetes tipo I. Lucentis no ha sido estudiado en pacientes que hayan recibido previamente inyecciones intravítreas, en pacientes con infecciones sistémicas activas, retinopatía diabética proliferativa, ni en pacientes con enfermedades oculares simultáneas tales como desprendimiento de retina o agujero macular.
Tampoco existe experiencia en el tratamiento con Lucentis en pacientes diabéticos con un HbA1c por encima del 12% e hipertensión no controlada. El médico debe tener en cuenta esta falta de información al tratar a tales pacientes.
No hay datos suficientes que permitan establecer una conclusión acerca del efecto de Lucentis en pacientes con OVR que presentan pérdida irreversible de la función visual isquémica.
En pacientes con MP, hay datos limitados del efecto de Lucentis en pacientes que han sido sometidos previamente a un tratamiento de terapia fotodinámica con verteporfina (TFDv) sin exito. Además, mientras que en sujetos con lesiones subfoveales y yuxtafoveales se observó un efecto consistente, no hay datos suficientes para establecer conclusiones sobre el efecto de Lucentis en sujetos con MP y lesiones extrafoveales.
Efectos sistémicos tras el uso intravítreo
Se han notificado acontecimientos adversos sistémicos, incluyendo hemorragias no oculares y acontecimientos tromboembólicos arteriales tras la inyección intravítrea de inhibidores del VEGF.
Existen datos limitados sobre seguridad en el tratamiento de pacientes con EMD, edema macular debido a OVR y NVC secundaria a MP que tengan antecedentes de accidente cerebrovascular o ataques isquémicos transitorios. Se debe tener precaución cuando se traten tales pacientes (ver sección 4.8).
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones formales.
Para el uso conjunto de terapia fotodinámica (TFD) con verteporfina y Lucentis en la DMAE exudativa y en la MP, ver sección 5.1.
Para el uso conjunto de fotocoagulación con láser y Lucentis en EMD y ORVR, ver secciones 4.2 y 5.1.
En ensayos clínicos para el tratamiento de la alteración visual debida al EMD, el tratamiento concomitante con tiazolidinedionas en pacientes tratados con Lucentis, no afectó el resultado en relación a la agudeza visual o al grosor del subcampo central de la retina (GSCR).
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil/anticoncepción en mujeres
Las mujeres en edad fértil deben utilizar métodos anticonceptivos efectivos durante el tratamiento. Embarazo
No se dispone de datos clínicos de exposición a ranibizumab en embarazos. Los estudios en monos cinomolgos no sugieren efectos perjudiciales directos ni indirectos en términos de embarazo o desarrollo embrional/fetal (ver sección 5.3). La exposición sistémica a ranibizumab tras la administración ocular es baja, pero debido a su mecanismo de acción, ranibizumab debe considerarse como potencialmente teratogénico y embrio-/fetotóxico. Por ello ranibizumab no se deberá usar durante el embarazo salvo que el beneficio esperado supere el riesgo potencial para el feto. Para mujeres que deseen quedarse embarazadas y hayan sido tratadas con ranibizumab, se recomienda esperar como mínimo 3 meses tras la última dosis de ranibizumab antes de concebir un hijo.
Lactancia
Se desconoce si Lucentis se excreta en la leche materna. No se recomienda la lactancia durante el uso de Lucentis.
Fertilidad
No hay datos disponibles sobre fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
El procedimiento de tratamiento con Lucentis puede producir alteraciones visuales transitorias, que pueden afectar la capacidad para conducir o utilizar máquinas (ver sección 4.8). Los pacientes que experimenten estos signos no deben conducir ni utilizar máquinas hasta que dichas alteraciones visuales transitorias remitan.
4.8 Reacciones adversas
Resumen del perfil de seguridad
La mayoría de las reacciones adversas notificadas tras la administración de Lucentis están relacionadas con el procedimiento de inyección intravítrea.
Las reacciones adversas oculares tras la inyección de Lucentis notificadas más frecuentemente son: dolor ocular, hiperemia ocular, aumento de la presión intraocular, vitritis, desprendimiento de vítreo, hemorragia retiniana, alteración visual, partículas flotantes en el vítreo, hemorragia conjuntival, irritación ocular, sensación de cuerpo extraño en los ojos, aumento del lagrimeo, blefaritis, ojo seco, y prurito ocular.
Las reacciones adversas no oculares notificadas más frecuentemente son cefalea, nasofaringitis y artralgia.
Las reacciones adversas notificadas con menor frecuencia, pero de mayor gravedad, incluyen endoftalmitis, ceguera, desprendimiento de retina, desgarro retiniano y catarata traumática iatrogénica (ver sección 4.4).
Se debe informar a los pacientes de los síntomas de estas reacciones adversas potenciales e instruirlos para que informen a su médico en caso de aparición de signos tales como dolor ocular o aumento del malestar en el ojo, empeoramiento del enrojecimiento del ojo, visión borrosa o disminución de la visión, aumento del número de pequeñas manchas en su visión o aumento de la sensibilidad a la luz.
En la siguiente tabla se resumen las reacciones adversas ocurridas tras la administración de Lucentis en los ensayos clínicos.
Tabla de reacciones adversas#
Las reacciones adversas se listan con un sistema de clasificación de órganos y frecuencia usando el siguiente criterio: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000), muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo frecuencia.
Infecciones e infestaciones
Muy frecuentes Nasofaringitis
Frecuentes Infección de las vías urinarias*
Trastornos de la sangre y del sistema linfático
Frecuentes Anemia
Trastornos del sistema inmunológico
Frecuentes Hipersensibilidad
Trastornos psiquiátricos
Frecuentes Ansiedad
Trastornos del sistema nervioso
Muy frecuentes Cefalea
Trastornos oculares
Muy frecuentes
Vitritis, desprendimiento de vitreo, hemorragia retiniana, alteración visual, dolor ocular, partículas flotantes en el vítreo, hemorragia conjuntival, irritación ocular, sensación de cuerpo extraño en los ojos, aumento del lagrimeo, blefaritis, ojo seco, hiperemia ocular, prurito ocular.
Frecuentes
Degeneración retiniana, trastorno retiniano, desprendimiento de retina, desgarro retiniano, desprendimiento del epitelio pigmentario retiniano, desgarro del epitelio pigmentario retiniano, agudeza visual reducida, hemorragia vítrea, trastorno del cuerpo vítreo, uveítis, iritis, iridociclitis, catarata, catarata subcapsular, opacificación de la cápsula posterior, queratitis punctata, abrasión corneal, células flotantes en la cámara anterior, visión borrosa, hemorragia en el lugar de inyección, hemorragia ocular, conjuntivitis, conjuntivitis alérgica, secreción ocular, fotopsia, fotofobia, molestia ocular, edema palpebral, dolor palpebral, hiperemia conjuntival.
Poco frecuentes
Ceguera, endoftalmitis, hipopion, hipema, queratopatía, adhesión del iris, depósitos corneales, edema corneal, estrías corneales, dolor en el lugar de inyección, irritación en el lugar de inyección, sensación anormal en el ojo, irritación palpebral.
Trastornos respiratorios, torácicos y mediastínicos
Frecuentes Tos
Trastornos gastrointestinales
Frecuentes Náuseas
Trastornos de la piel y del tejido subcutáneo
Frecuentes Reacciones alérgicas (erupción, urticaria, prurito, eritema)
Trastornos musculoesqueléticos y del tejido conjuntivo Muy frecuentes Artralgia
Exploraciones complementarias
Muy frecuentes Aumento de la presión intraocular
#-Las reacciones adversas se definieron como acontecimientos adversos (en al menos 0,5 puntos porcentuales de pacientes) que ocurrieron con una frecuencia superior (como mínimo 2 puntos porcentuales) en los pacientes que recibieron el tratamiento con Lucentis 0,5 mg respecto a los que recibieron el tratamiento control (tratamiento simulado (sham) o TFD con verteporfina).
* observado sólo en población con EMD
Reacciones adversas de clase terapéutica
En los ensayos fase III en DMAE exudativa, la frecuencia global de hemorragias no oculares, un efecto adverso potencialmente relacionado con la inhibición sistémica del VEGF (factor de crecimiento endotelial vascular), fue ligeramente superior en los pacientes tratados con ranibizumab. Sin embargo, no hubo un patrón consistente entre las distintas hemorragias. Tras el uso intravítreo de inhibidores del VEGF existe un riesgo teórico de acontecimientos tromboembólicos arteriales, incluyendo accidente cerebrovascular e infarto de miocardio. En los ensayos clínicos con Lucentis se observó una incidencia baja de acontecimientos tromboembólicos arteriales en pacientes con DMAE, EMD, OVR y MP y no hubo ninguna diferencia destacable entre los grupos tratados con ranibizumab comparado con el control.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Se han notificado casos de sobredosis accidental procedentes de los ensayos clínicos en DMAE exudativa y de los datos post-comercialización. Las reacciones adversas que se asociaron a estos casos notificados fueron aumento de la presión intraocular, ceguera transitoria, agudeza visual reducida, edema corneal, dolor corneal y dolor ocular. En caso de sobredosis, se deberá realizar un seguimiento y tratamiento de la presión intraocular, si el médico lo considera necesario.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Oftalmológicos, agentes antineovascularización, código ATC: S01LA04
Ranibizumab es un fragmento de anticuerpo monoclonal recombinante humanizado dirigido contra el factor de crecimiento endotelial vascular A (VEGF-A) humano. Se une con alta afinidad a las isoformas del VEGF-A (p. ej. VEGFn0, VEGFm y VEGFJ65), impidiendo, por tanto, la unión del VEGF-A a sus receptores VEGFR-1 y VEGFR-2. La unión del VEGF-A a sus receptores conduce a la proliferación de las células endoteliales y la neovascularización, así como a la exudación vascular, todo lo cual se cree que contribuye a la progresión de la forma neovascular de la degeneración macular asociada a la edad, la miopía patológica o a la alteración visual causada por el edema macular diabético o por el edema macular secundario a OVR.
Tratamiento de la DMAE exudativa
En la DMAE exudativa, la eficacia y seguridad clínicas de Lucentis se han evaluado en tres ensayos aleatorizados, doble ciego, controlados con tratamiento simulado (sham) o con tratamiento activo de 24 meses de duración, en pacientes con DMAE neovascular. En estos ensayos fueron reclutados un total de 1.323 pacientes (879 con tratamiento activo y 444 con control).
En el ensayo FVF2598g (MARINA), se aleatorizaron 716 pacientes con lesiones mínimamente clásicas u ocultas sin componente clásico en un ratio 1:1:1 para recibir inyecciones de Lucentis 0,3 mg, Lucentis 0,5 mg o tratamiento simulado una vez al mes.
En el ensayo FVF2587g (ANCHOR), se aleatorizaron 423 pacientes con lesiones de NVC predominantemente clásicas en un ratio 1:1:1 para recibir Lucentis 0,3 mg una vez al mes, Lucentis 0,5 mg una vez al mes o TFD con verteporfina (al inicio y posteriormente cada 3 meses si la angiografía fluoresceínica mostraba persistencia o recurrencia de la exudación vascular).
En la Tabla 1 y en la Figura 1 se resumen los resultados clave.
Tabla 1 Resultados al Mes 12 y al Mes 24 en el ensayo FVF2598g (MARINA) y FVF2587g (ANCHOR)
|
FVF2598g (MARINA) |
FVF2587g (ANCHOR) | ||||
|
Medida del resultado |
Mes |
Tratamient o simulado o sham (n=238) |
Lucentis 0,5 mg (n=240) |
TFD con verteporfina (n=143) |
Lucentis 0,5 mg (n=140) |
|
Pérdida de <15 letras de agudeza visual (%)a (mantenimiento de la visión, variable primaria) |
Mes 12 |
62% |
95% |
64% |
96% |
|
Mes 24 |
53% |
90% |
66% |
90% | |
|
Ganancia de >15 letras de agudeza visual (%)a |
Mes 12 |
5% |
34% |
6% |
40% |
|
Mes 24 |
4% |
33% |
6% |
41% | |
|
Cambio medio en la agudeza visual (letras) (desviación estándar)a |
Mes 12 |
-10,5 (16,6) |
+7,2 (14,4) |
-9,5 (16,4) |
+11,3 (14,6) |
|
Mes 24 |
-14,9 (18,7) |
+6,6 (16,5) |
-9,8 (17,6) |
+10,7 (16,5) | |
a p<0,01
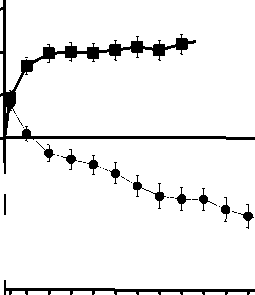
Figura 1 Cambio medio en la agudeza visual desde el inicio hasta el Mes 24 en el ensayo FVF2598g (MARINA) y el ensayo FVF2587g (ANCHOR)
C/5
>
co
N
CD
T3
CD
O
T3
CD
E
O
!q
E
co
O
Ensayo FVF2598g (MARINA)
+6,6 ^
15
10
5
0
— <D C
<L>
O
<L>
E
O
E
Cü
O
~-5
-10
-15
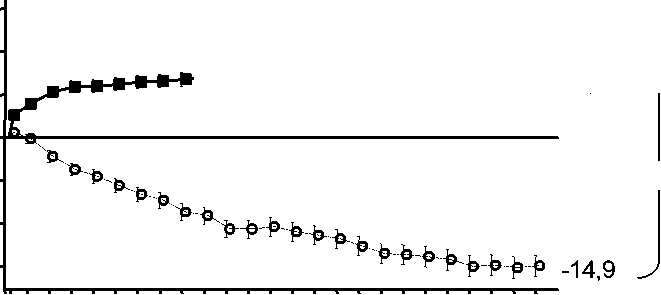
V +21,5
0 2 4 6 8 10 12 14 16 18 20 22 24
Mes
15
10
5
0
co "S
"-5 ■
-10 ■ -15 ■
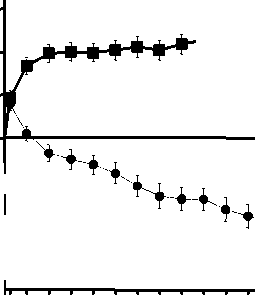
Ensayo FVF2587g (ANCHOR)
frf-j +10,7
r +20,5
-9'8
0 2 4 6 8 10 12 14 16 18 20 22 24
Mes
MARINA
LUCENTIS 0,5 mg (n=240)
& Tratamiento simulado (n=238)
ANCHOR
LUCENTIS 0,5 mg (n=140)
• TFD con verteporfina (n=143)
Los resultados de ambos ensayos indicaron que el tratamiento continuado con ranibizumab también puede ser beneficioso en pacientes que perdieron >15 letras de agudeza visual mejor corregida (AVMC) en el primer año de tratamiento.
En ambos ensayos MARINA y ANCHOR se observaron beneficios en la función visual notificados por el mismo paciente estadísticamente significativos en el tratamiento con ranibizumab respecto al grupo control, medidos según el NEI VFQ-25.
En el ensayo FVF3192g (PIER), se aleatorizaron 184 pacientes que presentaban todas las formas de DMAE neovascular en un ratio 1:1:1 para recibir Lucentis 0,3 mg, Lucentis 0,5 mg o inyecciones simuladas (sham) una vez al mes en 3 dosis consecutivas, seguido de la administración de una dosis una vez cada 3 meses. A partir del Mes 14 del ensayo, se permitió a los pacientes tratados con inyecciones simuladas (sham) recibir ranibizumab, y a partir del Mes 19 fueron posibles tratamientos más frecuentes. Los pacientes tratados con Lucentis en el ensayo PIER recibieron un promedio de 10 tratamientos.
En general, tras un incremento inicial en la agudeza visual (después de la dosificación mensual), la agudeza visual de los pacientes disminuyó con la dosis trimestral, volviendo al valor basal en el Mes 12 y este efecto se mantuvo al Mes 24 en la mayoría de los pacientes tratados con ranibizumab (82%). Datos limitados procedentes de sujetos que recibieron tratamiento simulado y que posteriormente fueron tratados con ranibizumab, sugirieron que el inicio temprano del tratamiento puede asociarse a una mejor conservación de la agudeza visual.
Los datos de dos ensayos (MONT BLANC, BPD952A2308 y DENALI, BPD952A2309) realizados tras la autorización de comercialización, confirmaron la eficacia de Lucentis pero no demostraron un efecto adicional en la administración combinada de verteporfina (Visudyne TFD) y Lucentis comparado con Lucentis en monoterapia.
Tratamiento de la alteración visual debida a EMD
La eficacia y seguridad de Lucentis se han evaluado en tres ensayos aleatorizados, controlados de al menos 12 meses de duración. En estos ensayos fueron reclutados un total de 868 pacientes (708 con tratamiento activo y 160 con control).
En el ensayo de fase II D2201 (RESOLVE), 151 pacientes fueron tratados con ranibizumab (6 mg/ml, n=51, 10 mg/ml, n=51) o con tratamiento simulado (n=49) mediante inyecciones intravítreas mensuales. El cambio medio en la AVMC desde el Mes 1 al Mes 12 comparado con el inicio fue de +7,8 (±7,72) letras en los pacientes tratados con ranibizumab combinados (n=102), comparado con -0,1 (±9,77) letras para pacientes con tratamiento simulado; y el cambio medio en la AVMC en el Mes 12 desde el valor inicial fue de 10,3 (±9,1) letras comparado con -1,4 (±14,2) letras respectivamente (p<0,0001 para la diferencia de tratamiento).
En el ensayo de fase III D2301 (RESTORE), se aleatorizaron 345 pacientes en un ratio 1:1:1 para recibir ranibizumab 0,5 mg en monoterapia y fotocoagulación con láser simulada, o ranibizumab 0,5 mg y fotocoagulación con láser combinados, o inyección simulada y fotocoagulación con láser. En un ensayo de extensión de 24 meses multicéntrico y abierto (Extension del ensayo RESTORE) fueron reclutados 240 pacientes, que previamente habían completado el ensayo RESTORE de 12 meses. Los pacientes fueron tratados con ranibizumab 0,5 mg pro re nata (PRN) en el mismo ojo que en el ensayo principal (D2301 RESTORE).
Los resultados clave se resumen en la Tabla 2 (RESTORE y Extensión) y en la Figura 2 (RESTORE).
Figura 2
Cambio medio en la agudeza visual en el tiempo desde el inicio, en el estudio D2301 (RESTORE)
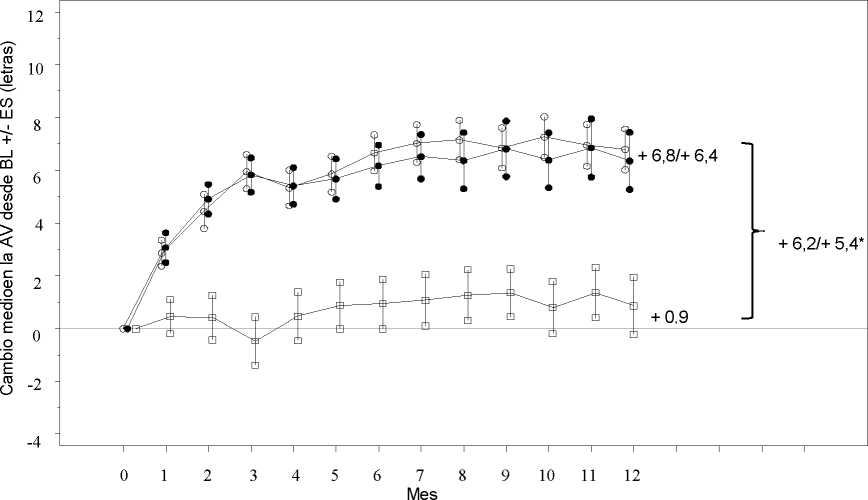
Ranibizumab 0,5 mg (n=115)
• • • Ranibizumab 0,5 mg + Láser (n=118) Láser (n=110)
Grupo de tratamiento
BL=valor inicial; ES= error estándar de la media
* Diferencia de medias de mínimos cuadrados, p<0,0001/0,0004 basado en el test bilateral de Cochran-Mantel-Haenszel
El efecto a los 12 meses fue consistente en la mayoría de subgrupos. Sin embargo, sujetos con una AVMC al inicio >73 letras y con un edema macular con un grosor de la retina central <300 pm, no parecían beneficiarse del tratamiento con ranibizumab comparado con la fotocoagulación con láser.
Tabla 2 Resultados al Mes 12 en el estudio D2301 (RESTORE) y al Mes 36 en el estudio D2301-E1 (Extensión del estudio RESTORE)
|
Medidas del resultado al Mes 12 comparado con el valor inicial en el estudio D2301 (RESTORE) |
Ranibizumab 0,5 mg n=115 |
Ranibizumab 0,5 mg + Láser n=118 |
Láser n=110 |
|
Cambio medio en la AVMC desde el Mes 1 al Mes 12a (±desviación estándar) |
6,1 (6,4)a |
5,9 (7,9)a |
0,8 (8,6) |
|
Cambio medio en la AVMC al Mes 12 (±desviación estándar) |
6,8 (8,3)a |
6,4 (11,8)a |
0,9 (11,4) |
|
Ganancia de >15 letras o AVMC >84 letras al Mes 12 (%) |
22,6 |
22,9 |
8,2 |
|
Número medio de inyecciones (Meses 011) |
7,0 |
6,8 |
7,3 (tratamient o simulado) |
|
Medida del resultado al Mes 36 comparado con el valor inicial del estudio D2301 (RESTORE) en el estudio D2301-E1 (Extension del estudio RESTORE) |
Ranibizumab 0,5 mg previo n=83 |
Ranibizumab 0,5 mg + láser previos n=83 |
Láser previo n=74 |
|
Cambio medio en la AVMC al Mes 24 (desviación estándar) |
7,9 (9,0) |
6,7 (7,9) |
5,4 (9,0) |
|
Cambio medio en la AVMC al Mes 36 (desviación estándar) |
8,0 (10,1) |
6,7 (9,6) |
6,0 (9,4) |
|
Ganancia de >15 letras o AVMC >84 letras al Mes 36 (%) |
27,7 |
30,1 |
21,6 |
|
Número medio de inyecciones (Meses 1235)* |
6,8 |
6,0 |
6,5 |
ap<0,0001 para comparaciones de los grupos de ranibizumab frente al grupo de láser. n en D2301-E1 (Extension del estudio RESTORE) es el número de pacientes con un valor tanto en el valor inicial del estudio D2301 (RESTORE) (Mes 0) como en la visita del Mes 36.
• Las proporciones de pacientes que no necesitaron ningún tratamiento con ranibizumab durante la fase de extensión fueron del 19%, 25% y 20% en los grupos ranibizumab previo, ranibizumab + láser previos y láser previo, respectivamente.
En el tratamiento con ranibizumab (con o sin láser) se observaron beneficios notificados por el mismo paciente estadísticamente significativos para la mayoría de funciones relacionadas con la visión respecto al grupo control, medidos según el NEI VFQ-25. Para las otras subescalas de este cuestionario no pudieron establecerse diferencias ligadas al tratamiento.
El perfil de seguridad a largo plazo de ranibizumab observado en el ensayo de extensión de 24 meses es consistente con el perfil de seguridad de Lucentis conocido.
En el ensayo de fase IIIb D2304 (RETAIN), se aleatorizaron 372 pacientes en un ratio 1:1:1 para recibir:
• ranibizumab 0,5 mg con fotocoagulación con láser concomitante en un régimen de tratar y extender (TE), o bien
• ranibizumab 0,5 mg en monoterapia en un régimen TE, o bien
• ranibizumab 0,5 mg en monoterapia en un régimen PRN.
En todos los grupos, ranibizumab se administró mensualmente hasta que la AVMC era estable durante al menos tres controles mensuales consecutivos. En el régimen TE ranibizumab se administró a intervalos de tratamiento de 2-3 meses. En todos los grupos, el tratamiento mensual se reiniciaba en cuanto había una disminución de la AVMC debida a la progresión del EMD y continuaba hasta que se alcanzaba nuevamente una AVMC estable.
Después de las 3 inyecciones iniciales, el número de visitas de tratamiento programadas fueron de 13 y 20 para los regímenes TE y PRN respectivamente. Con ambos regímenes TE, más del 70% de los pacientes mantuvieron su AVMC con una frecuencia media de visitas de >2 meses.
Los resultados clave se resumen en la Tabla 3.
Tabla 3 Resultados en el estudio D2304 (RETAIN)
|
Medida del resultado comparado con el valor inicial |
Ranibizumab 0,5 mg + láser en TE n=117 |
Ranibizumab 0,5 mg solo en TE n=125 |
Ranibizumab 0,5 mg en PRN n=117 |
|
Cambio medio en la AVMC desde el Mes 1 al Mes 12 (desviación estándar) |
5,9 (5,5)a |
6,1 (5,7)a |
6,2 (6,0) |
|
Cambio medio en la AVMC desde el Mes 1 al Mes 24 (desviación estándar) |
6,8 (6,0) |
6,6 (7,1) |
7,0 (6,4) |
|
Cambio medio en la AVMC al Mes 24 (desviación estándar) |
8,3 (8,1) |
6,5 (10,9) |
8,1 (8,5) |
|
Ganancia de >15 letras o AVMC >84 letras al Mes 24(%) |
25,6 |
28,0 |
30,8 |
|
Número medio de inyecciones (meses 023) |
12,4 |
12,8 |
10,7 |
“p<0,0001 para la valoración de no inferioridad a PRN
En los ensayos en EMD, la mejora en la AVMC se acompañó de una reducción en el tiempo del GSCR medio en todos los grupos de tratamiento.
Tratamiento de la alteración visual debida al edema macular secundario a OVR La seguridad y eficacia clínicas de Lucentis en pacientes con alteración visual debida al edema macular secundario a OVR se han evaluado en los ensayos BRAVO y CRUISE, controlados, aleatorizados, doble ciego que reclutaron sujetos con ORVR (n=397) y con OVCR (n=392), respectivamente. En ambos ensayos, los sujetos recibieron o bien ranibizumab 0,3 mg o 0,5 mg o inyecciones simuladas. Después de 6 meses, los pacientes en los grupos control con inyección simulada cambiaron a ranibizumab 0,5 mg.
|
BRAVO |
CRUISE | |||
|
Tratamiento simulado/Lucenti s 0,5 mg (n=132) |
Lucentis 0,5 mg (n=131) |
Tratamiento simulado/Lucenti s 0,5 mg (n=130) |
Lucentis 0,5 mg (n=130) | |
|
Cambio medio en la agudeza visual al Mes 6a (letras) (desviación estándar) (variable primaria) |
7,3 (13,0) |
18,3 (13,2) |
0,8 (16,2) |
14,9 (13,2) |
|
Cambio medio en la AVMC al Mes 12 (letras) (desviación estándar) |
12,1 (14,4) |
18,3 (14,6) |
7,3 (15,9) |
13,9 (14,2) |
|
Ganancia de >15 letras en agudeza visual al Mes 6a (%) |
28,8 |
61,1 |
16,9 |
47,7 |
|
Ganancia de >15 letras en agudeza visual al Mes 12 (%) |
43,9 |
60,3 |
33,1 |
50,8 |
|
Proporción (%) que recibió rescate con láser en 12 meses |
61,4 |
34,4 |
NA |
NA |
a p<0,0001 para ambos ensayos
Cambio medio en la AVMC desde el inicio en el tiempo hasta el Mes 6 y el Mes 12 (BRAVO)
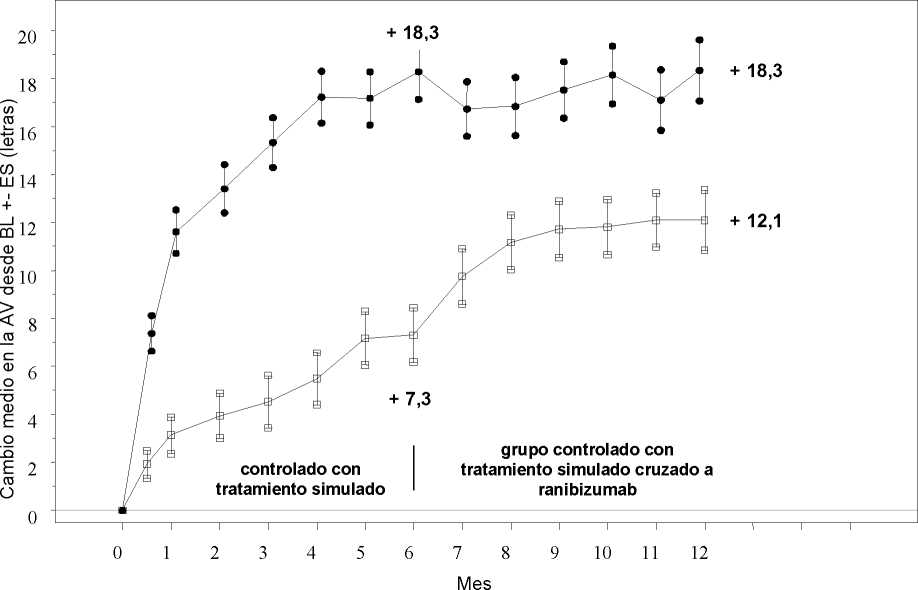
Grupo de tratamiento Tratamiento simulado/Ranibizumab 0,5 mg (n=132)
* * * Ranibizumab 0,5 mg (n=131)
BL=valor inicial; ES=error estándar de la media
Cambio medio en la AVMC desde el inicio en el tiempo hasta el Mes 6 y el Mes 12 (CRUISE)
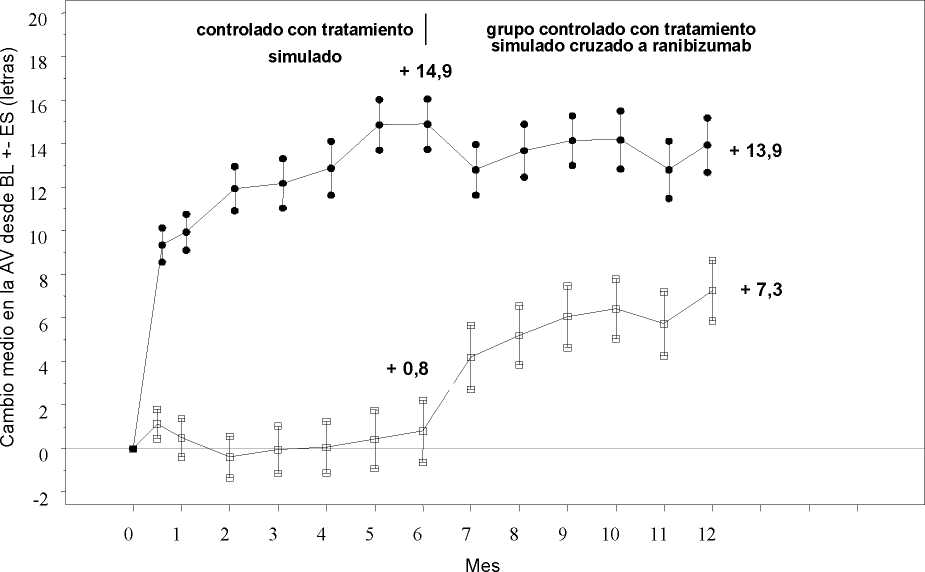
Grupo de tratamiento Tratamiento simulado/Ranibizumab 0,5 mg (n=130)
* * * Ranibizumab 0,5 mg (n=130)
BL=valor inicial; ES=error estándar de la media
En ambos estudios, la mejora de la visión se acompañó de una reducción continua y significativa del edema macular medido según el grosor central de la retina.
En pacientes con OVCR (CRUISE y ensayo de extensión HORIZON): Los sujetos tratados con tratamiento simulado en los primeros 6 meses que posteriormente recibieron ranibizumab, no alcanzaron ganancias comparables en AV en el Mes 24 (~6 letras) en comparación con los sujetos tratados con ranibizumab desde el inicio del ensayo (~12 letras).
En el tratamiento con ranibizumab se observaron beneficios notificados por el mismo paciente estadísticamente significativos en las subescalas relativas a la actividad de cerca y de lejos respecto al grupo control, medidos según el NEI VFQ-25.
La seguridad y eficacia clínicas de Lucentis a largo plazo (24 meses) en pacientes con alteración visual debida al edema macular secundario a OVR se evaluaron en los ensayos BRIGHTER (BRVO) y CRYSTAL (CRVO). En ambos ensayos, los sujetos recibieron ranibizumab 0,5 mg en un regimen de dosificación PRN que obedece a criterios de estabilización individualizados. BRIGHTER era un ensayo aleatorizado con 3 grupos controlado con tratamiento activo que comparaba ranibizumab 0,5 mg administrado en monoterapia o en combinación con fotocoagulación con láser adjunta frente a fotocoagulación con láser sola. Después de 6 meses, los sujetos en el grupo del láser podían recibir ranibizumab 0,5 mg. CRYSTAL era un ensayo de un grupo único con ranibizumab 0,5 mg en monoterapia.
|
BRIGHTER |
CRYSTAL | |||
|
Lucentis 0,5 mg N=180 |
Lucentis 0,5 mg + Láser N=178 |
Láser* N=90 |
Lucentis 0,5 mg N=356 | |
|
Cambio medio en la AVMC al Mes 6a (letras) (desviación estándar) |
+14,8 (10,7) |
+14,8 (11,13) |
+6,0 (14,27) |
+12,0 (13,95) |
|
Cambio medio en la AVMC al Mes 24b (letras) (desviación estándar) |
+15,5 (13,91) |
+17,3 (12,61) |
+11,6 (16,09) |
+12,1 (18,60) |
|
Ganancia de >15 letras en AVMC al Mes 24 (%) |
52,8 |
59,6 |
43,3 |
49,2 |
|
Número medio de inyecciones (desviación estándar) (Meses 0-23) |
11,4 (5,81) |
11,3 (6,02) |
NA |
13,1 (6,39) |
|
a p<0,0001 para ambas comparaciones en el ensayo BRIGHTER al Mes 6: Lucentis 0,5 mg frente a Láser y Lucentis 0,5 mg + Láser frente a Láser. b p<0,0001 para la hipótesis nula en el ensayo CRYSTAL en que el cambio medio al Mes 24 desde el valor inicial es cero. * A partir del Mes 6 se permitió el tratamiento con ranibizumab 0,5 mg (24 pacientes fueron tratados solo con láser). | ||||
En el ensayo BRIGHTER, ranibizumab 0,5 mg con terapia con láser adjunta demostró no inferioridad comparado con ranibizumab en monoterapia desde el valor inicial hasta el Mes 24 (95% IC -2,8, 1,4).
En ambos ensayos, se observó en el Mes 1 una reducción rápida y estadísticamente significativa del grosor del subcampo central de la retina desde el valor inicial. Este efecto se mantuvo hasta el Mes 24.
El efecto del tratamiento con ranibizumab fue similar de forma independiente de la presencia de isquemia retiniana. En el ensayo BRIGHTER, los pacientes que presentaban isquemia (N=46) o los que no la presentaban (N=133) y que fueron tratados con ranibizumab en monoterapia, tuvieron un cambio medio desde el valor inicial de +15,3 y +15,6 letras respectivamente, en el Mes 24. En el ensayo CRYSTAL, los pacientes que presentaban isquemia (N=53) o los que no la presentaban (N=300) y que fueron tratados con ranibizumab en monoterapia, tuvieron un cambio medio desde el valor inicial de +15,0 y +11,5 letras respectivamente.
En todos los pacientes tratados con ranibizumab 0,5 mg en monoterapia se observó el efecto en términos de mejora visual en ambos ensayos BRIGHTER y CRYSTAL de forma independiente de la duración de la enfermedad. En pacientes con una duración de la enfermedad <3 meses se observó un aumento en la agudeza visual de 13,3 y 10,0 letras en el Mes 1; y 17,7 y 13,2 letras en el Mes 24 en BRIGHTER y CRYSTAL respectivamente. En pacientes con una duración de la enfermedad >12 meses, la ganancia de agudeza visual correspondiente fue de 8,6 y 8,4 letras en los respectivos ensayos. Se debe considerar el inicio del tratamiento en el momento del diagnóstico.
El perfil de seguridad a largo plazo de ranibizumab observado en los ensayos de 24 meses es consistente con el perfil de seguridad de Lucentis conocido.
Tratamiento de la alteración visual debida a la NVC secundaria a MP
La seguridad y eficacia clínicas de Lucentis en pacientes con alteración visual debida a la NVC secundaria a MP se han evaluado de acuerdo a los datos de 12 meses del ensayo pivotal F2301 (RADIANCE) controlado, doble ciego. En este ensayo 277 pacientes fueron aleatorizados en un ratio 2:2:1 para los siguientes grupos:
• Grupo I (ranibizumab 0,5 mg, régimen de tratamiento basado en criterios de “estabilidad” definida como ningún cambio en la AVMC en comparación con las dos evaluaciones mensuales previas).
• Grupo II (ranibizumab 0,5 mg, régimen de tratamiento basado en criterios de “actividad de la enfermedad” definida como alteración de la visión atribuible a fluido intra o subretiniano o exudación activa debido a la lesión de NVC y evaluada mediante OCT y/o AF).
• Grupo III (TFDv - se permitió a los pacientes recibir tratamiento con ranibizumab a partir del Mes 3).
En el Grupo II, que se corresponde con la posología recomendada (ver sección 4.2), el 50,9% de los pacientes requirió 1 ó 2 inyecciones, el 34,5% requirió de 3 a 5 inyecciones y el 14,7% requirió de 6 a 12 inyecciones durante el periodo de 12 meses del ensayo. El 62,9% de los pacientes del Grupo II no requirió inyecciones en el segundo semestre del ensayo.
|
Grupo I |
Grupo II |
Grupo III | |
|
Ranibizumab |
Ranibizumab |
TFDvb | |
|
0,5 mg |
0,5 mg | ||
|
“estabilidad de |
“actividad de la | ||
|
la visión” |
enfermedad” | ||
|
(n=105) |
(n=116) |
(n=55) | |
|
Mes 3 Cambio medio en la AVMC desde el Mes 1 |
+10,5 |
+10,6 |
+2,2 |
|
al Mes 3 comparado con el inicioa (letras) Proporción de pacientes que ganaron: >15 letras, o alcanzaron >84 letras en la AVMC |
38,1% |
43,1% |
14,5% |
|
Mes 12 Número de inyecciones hasta el Mes 12: Media |
4,6 |
3,5 |
N/A |
|
Mediana |
4,0 |
2,5 |
N/A |
|
Cambio medio en la AVMC desde el Mes 1 |
+12,8 |
+12,5 |
N/A |
|
al Mes 12 comparado con el inicio (letras) Proporción de pacientes que ganaron: >15 letras, o alcanzaron >84 letras en la AVMC |
53,3% |
51,7% |
N/A |
a p<0,00001 comparación con el control de TFDv
b Control comparativo hasta el Mes 3. A los pacientes aleatorizados a TFDv se les permitió recibir tratamiento con ranibizumab a partir del Mes 3 (en el Grupo III, 38 pacientes recibieron ranibizumab a partir del Mes 3)
Figura 5 Cambio medio en la AVMC desde el inicio en el tiempo hasta el Mes 12 (RADIANCE)
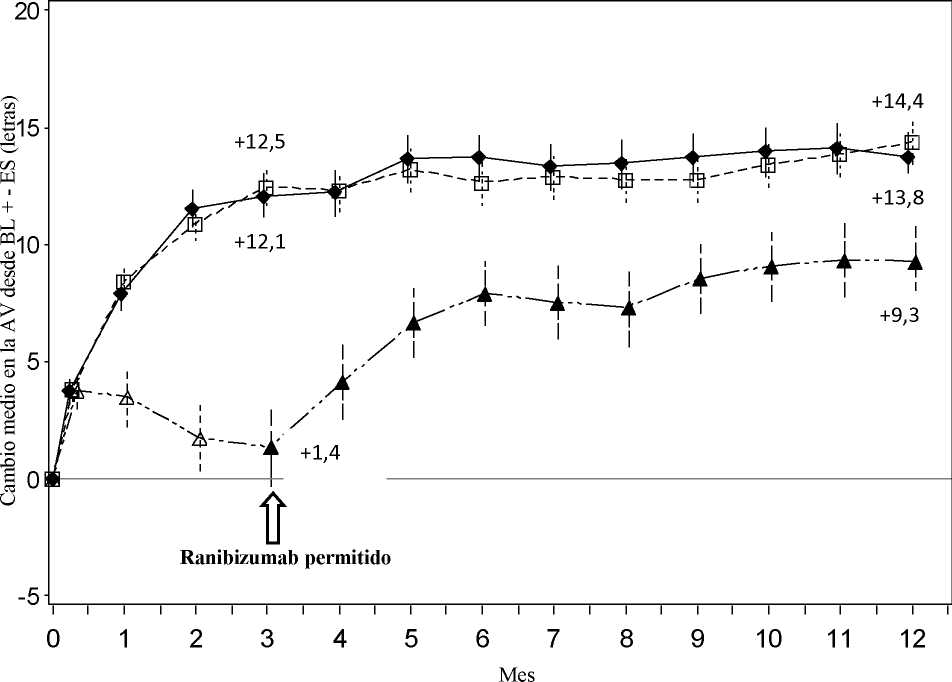
Ranibizumab 0,5 mg Grupo I por estabilización (n=105)
Ranibizumab 0,5 mg Grupo II
por actividad de la enfermedad (n=116)
Ranibizumab 0,5 mg/TFD con verteporfina Grupo III desde el Mes 3 en adelante (n=55)
A-a-a TFD con verteporfina Grupo III (n=55)
La mejora de la visión se acompañó de una reducción del grosor central de la retina.
Los beneficios comunicados por el paciente se observaron más en los grupos de tratamiento con ranibizumab que en la TFDv (valor p<0,05) en términos de mejora en la puntuación compuesta y varias subescalas (visión general, actividades de cerca, salud mental y dependencia) del NEI VFQ-25.
Población pediátrica
La seguridad y eficacia de ranibizumab aún no han sido estudiadas en pacientes pediátricos.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Lucentis en los diferentes grupos de la población pediátrica en la DMAE neovascular, la alteración visual debida al EMD, la alteración visual debida al edema macular secundario a la OVR y la alteración visual debida a la NVC secundaria a MP (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
Tras la administración intravítrea mensual de Lucentis a pacientes con DMAE neovascular, las concentraciones séricas de ranibizumab fueron en general bajas, con niveles máximos (Cmax) por debajo de la concentración de ranibizumab necesaria para inhibir la actividad biológica del VEGF en un 50% (11-27 ng/ml, valorado en un ensayo de proliferación celular in vitro). La Cmax fue proporcional a la dosis, en el rango de dosis de 0,05 a 1,0 mg/ojo. Las concentraciones séricas en un número limitado de pacientes con EMD indican que no puede excluirse una exposición sistémica ligeramente superior en comparación con la observada en pacientes con DMAE neovascular. Las concentraciones séricas de ranibizumab en pacientes con OVR fueron similares o ligeramente superiores en comparación con las observadas en pacientes con DMAE neovascular.
En base al análisis farmacocinético poblacional y a la desaparición sérica de ranibizumab en pacientes con DMAE neovascular tratados con la dosis de 0,5 mg, el promedio de la vida media de eliminación vítrea de ranibizumab es de 9 días aproximadamente. Tras la administración intravítrea mensual de Lucentis 0,5 mg/ojo, se prevé que la Cmax de ranibizumab sérica alcanzada aproximadamente 1 día después de la administración, varíe en general en un rango de entre 0,79 y 2,90 ng/ml, y que la Cmin varíe en general en un rango de entre 0,07 y 0,49 ng/ml. Se prevé que las concentraciones séricas de ranibizumab sean aproximadamente 90.000 veces inferiores a las concentraciones vítreas de ranibizumab.
Pacientes con insuficiencia renal: No se han realizado estudios formales para investigar la farmacocinética de Lucentis en pacientes con insuficiencia renal. En un análisis farmacocinético poblacional de pacientes con DMAE neovascular, el 68% (136 de 200) de los pacientes tenían insuficiencia renal (leve en un 46,5% [50-80 ml/min], moderada en un 20% [30-50 ml/min] y grave en un 1,5% [<30 ml/min]). En el caso de pacientes con OVR, el 48,2% (253 de 525) tenían insuficiencia renal (leve en un 36,4%, moderada en un 9,5% y grave en un 2,3%). El aclaramiento sistémico fue ligeramente inferior, pero esto no fue clínicamente significativo.
Insuficiencia hepática: No se han realizado estudios formales para investigar la farmacocinética de Lucentis en pacientes con insuficiencia hepática.
5.3 Datos preclínicos sobre seguridad
La administración intravítrea bilateral de ranibizumab a macacos, a dosis de entre 0,25 mg/ojo y
2,0 mg/ojo una vez cada 2 semanas durante 26 semanas, ocasionó efectos oculares dosis-dependientes.
Intraocularmente, se observaron incrementos dosis-dependientes de exudados y de células en la cámara anterior, con un máximo a los 2 días después de la inyección. Por lo general, la gravedad de la respuesta inflamatoria disminuyó con las inyecciones posteriores o durante el periodo de recuperación. En el segmento posterior, hubo infiltración de células en la cámara vítrea y partículas flotantes, que tendieron también a ser dosis-dependientes y que, en general, persistieron hasta el final del periodo de tratamiento. En el ensayo a 26 semanas, la gravedad de la inflamación vítrea aumentó con el número de inyecciones. Sin embargo, se observó evidencia de reversibilidad tras el periodo de recuperación. La naturaleza y cronología de la inflamación en el segmento posterior sugiere una respuesta inmunitaria mediada por anticuerpos, que puede ser clínicamente irrelevante. En algunos animales se observó la formación de cataratas tras un periodo relativamente largo de inflamación intensa, lo cual sugiere que las alteraciones en el cristalino fueron secundarias a la inflamación grave. Tras las inyecciones intravítreas se observó un aumentó transitorio de la presión intraocular independiente de la dosis.
Los cambios oculares microscópicos fueron relacionados con la inflamación y no eran indicativos de procesos degenerativos. Se observaron cambios inflamatorios granulomatosos en el disco óptico de algunos ojos. Estas alteraciones en el segmento posterior disminuyeron, y en algunos casos se resolvieron, durante el periodo de recuperación.
Tras la administración intravítrea, no se detectaron signos de toxicidad sistémica. En un subgrupo de animales tratados se detectaron anticuerpos séricos y vítreos contra ranibizumab.
No se dispone de datos de carcinogenicidad ni de mutagenicidad.
En hembras de mono preñadas, el tratamiento con ranibizumab intravítreo resultando en exposiciones sistémicas máximas 0,9-7 veces la peor exposición clínica, no provocó toxicidad en el desarrollo ni teratogenicidad y no tuvo ningún efecto sobre el peso o la estructura de la placenta, aunque en base a su efecto farmacológico, ranibizumab debe considerarse potencialmente teratogénico y embrio/fetotóxico.
La ausencia de efectos mediados por ranibizumab sobre el desarrollo embrio-fetal está plausiblemente relacionado principalmente con la incapacidad del fragmento Fab de atravesar la placenta. Sin embargo, se describió un caso de niveles séricos de ranibizumab maternos elevados y presencia de ranibizumab en el suero fetal lo que sugiere que el anticuerpo contra ranibizumab actuó como proteína transportadora (conteniendo la región Fc) para ranibizumab, disminuyendo de ese modo su aclaramiento sérico materno y permitiendo su paso a la placenta. Dado que las investigaciones en el desarrollo embrio-fetal se llevaron a cabo en animales preñados sanos y las enfermedades (tales como la diabetes) pueden modificar la permeabilidad de la placenta para el fragmento Fab, el estudio debe interpretarse con cautela.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
a,a-trehalosa dihidrato
Hidrocloruro de histidina monohidrato
Histidina
Polisorbato 20
Agua para inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
3 años
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Antes de usar, el vial sin abrir se puede conservar a temperatura ambiente (25°C) durante un máximo de 24 horas.
6.5 Naturaleza y contenido del envase
0. 23.ml de solución estéril en un vial (vidrio tipo I) con un tapón (goma de clorobutilo), 1 aguja roma con filtro (18G x 1V", 1,2 mm x 40 mm, 5 pm), 1 aguja para inyección (30G x V", 0,3 mm x 13 mm) y 1 jeringa (polipropileno) (1 ml). El envase contiene 1 vial.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El vial, la aguja para inyección, la aguja con filtro y la jeringa son para un solo uso. La reutilización puede dar lugar a una infección u otra enfermedad/lesión. Todos los componentes son estériles. No debe utilizarse ningún componente cuyo envase muestre signos de deterioro o manipulación. La esterilidad sólo se puede garantizar si el sellado del envase de los componentes se mantiene intacto.
Para la preparación de Lucentis para administración intravítrea, siga las siguientes instrucciones:
1. Antes de extraer la solución, se debe desinfectar la parte exterior del tapón de goma del vial.
2. Incorporar la aguja con filtro de 5 pm (18G x 1V", 1,2 mm x 40 mm, suministrada) a la jeringa de 1 ml (suministrada) usando técnicas asépticas. Insertar la aguja roma con filtro en el centro del tapón del vial hasta que la aguja toque el extremo inferior del vial.
3. Extraer todo el líquido del vial, manteniendo el vial en posición vertical, ligeramente inclinado para facilitar la extracción completa.
4. Al vaciar el vial, asegurar que el émbolo se retira hacia atrás lo suficiente de forma que se vacíe por completo la aguja con filtro.
5. Dejar la aguja roma con filtro en el vial y desconectarla de la jeringa. La aguja con filtro se debe desechar tras extraer el contenido del vial, y no se debe utilizar para la inyección intravítrea.
6. Incorporar la aguja para inyección (30G x V", 0,3 mm x 13 mm, suministrada) a la jeringa con firmeza y de forma aséptica.
7. Quitar la cápsula de cierre de la aguja para inyección cuidadosamente sin desconectar la aguja para inyección de la jeringa.
Nota: Sujetar la aguja para inyección por el cono amarillo mientras se retira la cápsula de cierre.
8. Expulsar el aire de la jeringa y ajustar la dosis hasta la marca de 0,05 ml en la jeringa cuidadosamente. La jeringa está lista para la inyección.
Nota: No secar la aguja para inyección. No tirar del émbolo hacia atrás.
Tras la inyección, no tapar la aguja con la cápsula de cierre ni separarla de la jeringa. Eliminar la jeringa usada junto con la aguja en un contenedor para objetos punzantes o eliminar de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/374/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 22 Enero 2007 Fecha de la última renovación: 24 Enero 2012
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
1. NOMBRE DEL MEDICAMENTO
Lucentis 10 mg/ml solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un ml contiene 10 mg de ranibizumab*. Cada vial contiene 2,3 mg de ranibizumab en 0,23 ml de solución.
*Ranibizumab es un fragmento de anticuerpo monoclonal humanizado producido en células de Escherichia coli mediante tecnología de ADN recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución acuosa transparente, de incolora a amarillo pálido.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lucentis está indicado en adultos para:
• El tratamiento de la degeneración macular asociada a la edad (DMAE) neovascular (exudativa)
• El tratamiento de la alteración visual debida al edema macular diabético (EMD)
• El tratamiento de la alteración visual debida al edema macular secundario a la oclusión de la
vena retiniana (OVR) (oclusión de la rama venosa retiniana u oclusión de la vena central retiniana)
• El tratamiento de la alteración visual debida a la neovascularización coroidea (NVC) secundaria a la miopía patológica (MP)
4.2 Posología y forma de administración
Lucentis debe ser administrado por un oftalmólogo que tenga experiencia en la administración de inyecciones intravítreas.
La dosis recomendada de Lucentis es 0,5 mg administrada en forma de inyección intravítrea única. Esto corresponde a un volumen de inyección de 0,05 ml. El intervalo entre dos dosis inyectadas en el mismo ojo debe ser como mínimo de cuatro semanas.
El tratamiento se inicia con una inyección al mes hasta alcanzar la agudeza visual máxima y/o no haya signos de actividad de la enfermedad, es decir ningún cambio en la agudeza visual ni en otros signos y síntomas de la enfermedad bajo tratamiento continuado. En pacientes con DMAE exudativa, EMD y OVR inicialmente pueden ser necesarias tres o más inyecciones consecutivas administradas mensualmente.
A partir de ese momento, los intervalos de monitorización y tratamiento se deben determinar según criterio médico y en base a la actividad de la enfermedad, valorada mediante la agudeza visual y/o parámetros anatómicos.
Se debe interrumpir el tratamiento con Lucentis si bajo criterio del médico, los parámetros visuales y anatómicos indican que el paciente no se está beneficiando del tratamiento continuado.
La monitorización para determinar la actividad de la enfermedad puede incluir examen clínico, control funcional o técnicas de imagen (p. ej. tomografía de coherencia óptica o angiografía con fluoresceína).
Si se está tratando a los pacientes de acuerdo a un régimen de tratar y extender, una vez se ha alcanzado la agudeza visual máxima y/o no hay signos de actividad de la enfermedad, los intervalos de tratamiento se pueden espaciar de forma gradual hasta que vuelvan a aparecer signos de actividad de la enfermedad o alteración visual. En el caso de la DMAE exudativa el intervalo de tratamiento no debe espaciarse en más de dos semanas cada vez y en el caso del EMD se puede espaciar hasta un mes cada vez. Para la OVR, los intervalos de tratamiento también pueden espaciarse de forma gradual, sin embargo los datos que hay no son suficientes para determinar la duración de estos intervalos. Si vuelve a aparecer actividad de la enfermedad, se debe acortar el intervalo de tratamiento de manera consecuente.
En el tratamiento de la alteración visual debida a la NVC secundaria a MP, muchos pacientes pueden necesitar únicamente una o dos inyecciones durante el primer año, mientras que algunos pacientes pueden necesitar tratamiento con mayor frecuencia (ver sección 5.1).
Lucentis y fotocoagulación con láser en EMD y en edema macular secundario a oclusión de la rama venosa retiniana (ORVR)
Existe alguna experiencia con Lucentis administrado concomitantemente con fotocoagulación con láser (ver sección 5.1). Cuando se administren en el mismo día, Lucentis se debe administrar como mínimo 30 minutos después de la fotocoagulación con láser. Lucentis puede administrarse en pacientes que han recibido fotocoagulación con láser previamente.
Lucentis y la terapia fotodinámica con Visudyne en la NVC secundaria a MP No hay experiencia en la administración concomitante de Lucentis y Visudyne.
Poblaciones especiales
Insuficiencia hepática
Lucentis no ha sido estudiado en pacientes con insuficiencia hepática. Sin embargo, no es necesaria ninguna consideración especial en esta población.
Insuficiencia renal
No es necesario ningún ajuste de la dosis en pacientes con insuficiencia renal (ver sección 5.2).
Pacientes de edad avanzada
No se requiere ningún ajuste de la dosis en pacientes de edad avanzada. Existe experiencia limitada en pacientes con EMD mayores de 75 años.
Población pediátrica
No se ha establecido la seguridad y eficacia de Lucentis en niños y adolescentes menores de 18 años de edad. No se dispone de datos.
Forma de administración
Vial para un solo uso. Únicamente para vía intravítrea.
Antes de la administración de Lucentis se debe comprobar visualmente la ausencia de partículas y decoloración.
El procedimiento de inyección deberá llevarse a cabo bajo condiciones asépticas, que incluyen el lavado quirúrgico de las manos, el uso de guantes estériles, un campo estéril, un blefarostato estéril para los párpados (o equivalente) y la disponibilidad de una paracentesis estéril (en caso necesario). Antes de realizar el procedimiento de inyección intravítrea, se deberá evaluar detalladamente la historia clínica del paciente en cuanto a reacciones de hipersensibilidad (ver sección 4.4). Antes de la inyección se debe administrar una anestesia adecuada y un microbicida tópico de amplio espectro para desinfectar la piel de la zona periocular, párpado y superficie ocular, de acuerdo con la práctica local.
Para la información relativa a la preparación de Lucentis, ver sección 6.6.
La aguja para inyección se deberá introducir 3,5-4,0 mm por detrás del limbo en la cavidad vítrea, evitando el meridiano horizontal y en dirección al centro del globo. Seguidamente debe liberarse el volumen de inyección de 0,05 ml; las inyecciones siguientes deberán aplicarse cada vez en un punto escleral distinto.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Pacientes con infecciones oculares o perioculares o con sospecha de éstas.
Pacientes con inflamación intraocular grave.
4.4 Advertencias y precauciones especiales de empleo Reacciones relacionadas con la inyección intravítrea
Las inyecciones intravítreas, incluidas las de Lucentis, se han asociado a endoftalmitis, inflamación intraocular, desprendimiento de retina regmatógeno, desgarro retiniano y catarata traumática iatrogénica (ver sección 4.8). Siempre que se administre Lucentis se deben emplear técnicas de inyección asépticas adecuadas. Además, deberá realizarse un seguimiento de los pacientes durante la semana posterior a la inyección para poder administrar tratamiento temprano en caso de infección. Se deberá instruir a los pacientes sobre la necesidad de comunicar inmediatamente cualquier síntoma que sugiera endoftalmitis o cualquiera de los acontecimientos mencionados anteriormente.
Aumento de la presión intraocular
Se han observado aumentos transitorios de la presión intraocular (PIO) en los 60 minutos siguientes a la inyección de Lucentis. También se han identificado aumentos sostenidos de la PIO (ver sección 4.8). Tanto la presión intraocular como la perfusión de la cabeza del nervio óptico, se deben monitorizar y tratar adecuadamente.
Tratamiento bilateral
Los escasos datos existentes sobre el tratamiento bilateral con Lucentis (incluyendo la administración en el mismo día) no sugieren un riesgo incrementado de efectos adversos sistémicos en comparación con el tratamiento unilateral.
Inmunogenicidad
Hay un potencial de inmunogenicidad con Lucentis. Dado que en sujetos con EMD existe un potencial para una exposición sistémica incrementada, no puede excluirse un mayor riesgo para desarrollar hipersensibilidad en esta población de pacientes. También se deberá instruir a los pacientes sobre la necesidad de notificar si la inflamación intraocular incrementa en su gravedad, lo que puede ser un signo clínico atribuible a la formación de anticuerpos intraoculares.
Uso concomitante con otros anti-VEGF (factor de crecimiento endotelial vascular)
Lucentis no se deberá administrar de forma concurrente con otros medicamentos anti-VEGF (sistémicos u oculares).
Aplazamiento del tratamiento con Lucentis
La administración se deberá aplazar y el tratamiento no deberá reanudarse antes del siguiente tratamiento programado en caso de:
• una disminución en la agudeza visual mejor corregida (AVMC) de >30 letras comparado con la última evaluación de la agudeza visual;
• una presión intraocular de >30 mmHg;
• una rotura retiniana;
• una hemorragia subretiniana que afecte al centro de la fóvea, o, si el tamaño de la hemorragia es >50% del área total de la lesión;
• cirugía intraocular realizada en los 28 días previos o prevista durante los 28 días posteriores. Desgarro del epitelio pigmentario de la retina
Los factores de riesgo asociados con el desarrollo de un desgarro del epitelio pigmentario de la retina tras la terapia con anti-VEGF para la DMAE exudativa, incluyen un desprendimiento del epitelio pigmentario de la retina extenso y/o elevado. Cuando se inicie la terapia con Lucentis se debe tener precaución en pacientes con estos factores de riesgo de desarrollar desgarros del epitelio pigmentario de la retina.
Desprendimiento de retina regmatógeno o agujeros maculares
El tratamiento se debe interrumpir en sujetos con desprendimiento de retina regmatógeno o agujeros maculares en estadíos 3 ó 4.
Poblaciones con datos limitados
Sólo existe experiencia limitada en el tratamiento de sujetos con EMD debido a diabetes tipo I. Lucentis no ha sido estudiado en pacientes que hayan recibido previamente inyecciones intravítreas, en pacientes con infecciones sistémicas activas, retinopatía diabética proliferativa, ni en pacientes con enfermedades oculares simultáneas tales como desprendimiento de retina o agujero macular.
Tampoco existe experiencia en el tratamiento con Lucentis en pacientes diabéticos con un HbA1c por encima del 12% e hipertensión no controlada. El médico debe tener en cuenta esta falta de información al tratar a tales pacientes.
No hay datos suficientes que permitan establecer una conclusión acerca del efecto de Lucentis en pacientes con OVR que presentan pérdida irreversible de la función visual isquémica.
En pacientes con MP, hay datos limitados del efecto de Lucentis en pacientes que han sido sometidos previamente a un tratamiento de terapia fotodinámica con verteporfina (TFDv) sin exito. Además, mientras que en sujetos con lesiones subfoveales y yuxtafoveales se observó un efecto consistente, no hay datos suficientes para establecer conclusiones sobre el efecto de Lucentis en sujetos con MP y lesiones extrafoveales.
Efectos sistémicos tras el uso intravítreo
Se han notificado acontecimientos adversos sistémicos, incluyendo hemorragias no oculares y acontecimientos tromboembólicos arteriales tras la inyección intravítrea de inhibidores del VEGF.
Existen datos limitados sobre seguridad en el tratamiento de pacientes con EMD, edema macular debido a OVR y NVC secundaria a MP que tengan antecedentes de accidente cerebrovascular o ataques isquémicos transitorios. Se debe tener precaución cuando se traten tales pacientes (ver sección 4.8).
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones formales.
Para el uso conjunto de terapia fotodinámica (TFD) con verteporfina y Lucentis en la DMAE exudativa y en la MP, ver sección 5.1.
Para el uso conjunto de fotocoagulación con láser y Lucentis en EMD y ORVR, ver secciones 4.2 y 5.1.
En ensayos clínicos para el tratamiento de la alteración visual debida al EMD, el tratamiento concomitante con tiazolidinedionas en pacientes tratados con Lucentis, no afectó el resultado en relación a la agudeza visual o al grosor del subcampo central de la retina (GSCR).
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil/anticoncepción en mujeres
Las mujeres en edad fértil deben utilizar métodos anticonceptivos efectivos durante el tratamiento. Embarazo
No se dispone de datos clínicos de exposición a ranibizumab en embarazos. Los estudios en monos cinomolgos no sugieren efectos perjudiciales directos ni indirectos en términos de embarazo o desarrollo embrional/fetal (ver sección 5.3). La exposición sistémica a ranibizumab tras la administración ocular es baja, pero debido a su mecanismo de acción, ranibizumab debe considerarse como potencialmente teratogénico y embrio-/fetotóxico. Por ello ranibizumab no se deberá usar durante el embarazo salvo que el beneficio esperado supere el riesgo potencial para el feto. Para mujeres que deseen quedarse embarazadas y hayan sido tratadas con ranibizumab, se recomienda esperar como mínimo 3 meses tras la última dosis de ranibizumab antes de concebir un hijo.
Lactancia
Se desconoce si Lucentis se excreta en la leche materna. No se recomienda la lactancia durante el uso de Lucentis.
Fertilidad
No hay datos disponibles sobre fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
El procedimiento de tratamiento con Lucentis puede producir alteraciones visuales transitorias, que pueden afectar la capacidad para conducir o utilizar máquinas (ver sección 4.8). Los pacientes que experimenten estos signos no deben conducir ni utilizar máquinas hasta que dichas alteraciones visuales transitorias remitan.
4.8 Reacciones adversas
Resumen del perfil de seguridad
La mayoría de las reacciones adversas notificadas tras la administración de Lucentis están relacionadas con el procedimiento de inyección intravítrea.
Las reacciones adversas oculares tras la inyección de Lucentis notificadas más frecuentemente son: dolor ocular, hiperemia ocular, aumento de la presión intraocular, vitritis, desprendimiento de vítreo, hemorragia retiniana, alteración visual, partículas flotantes en el vítreo, hemorragia conjuntival, irritación ocular, sensación de cuerpo extraño en los ojos, aumento del lagrimeo, blefaritis, ojo seco, y prurito ocular.
Las reacciones adversas no oculares notificadas más frecuentemente son cefalea, nasofaringitis y artralgia.
Las reacciones adversas notificadas con menor frecuencia, pero de mayor gravedad, incluyen endoftalmitis, ceguera, desprendimiento de retina, desgarro retiniano y catarata traumática iatrogénica (ver sección 4.4).
Se debe informar a los pacientes de los síntomas de estas reacciones adversas potenciales e instruirlos para que informen a su médico en caso de aparición de signos tales como dolor ocular o aumento del malestar en el ojo, empeoramiento del enrojecimiento del ojo, visión borrosa o disminución de la visión, aumento del número de pequeñas manchas en su visión o aumento de la sensibilidad a la luz.
En la siguiente tabla se resumen las reacciones adversas ocurridas tras la administración de Lucentis en los ensayos clínicos.
Tabla de reacciones adversas#
Las reacciones adversas se listan con un sistema de clasificación de órganos y frecuencia usando el siguiente criterio: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000), muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo frecuencia.
Infecciones e infestaciones
Muy frecuentes Nasofaringitis
Frecuentes Infección de las vías urinarias*
Trastornos de la sangre y del sistema linfático
Frecuentes Anemia
Trastornos del sistema inmunológico
Frecuentes Hipersensibilidad
Trastornos psiquiátricos
Frecuentes Ansiedad
Trastornos del sistema nervioso
Muy frecuentes Cefalea
Trastornos oculares
Muy frecuentes
Vitritis, desprendimiento de vitreo, hemorragia retiniana, alteración visual, dolor ocular, partículas flotantes en el vítreo, hemorragia conjuntival, irritación ocular, sensación de cuerpo extraño en los ojos, aumento del lagrimeo, blefaritis, ojo seco, hiperemia ocular, prurito ocular.
Frecuentes
Degeneración retiniana, trastorno retiniano, desprendimiento de retina, desgarro retiniano, desprendimiento del epitelio pigmentario retiniano, desgarro del epitelio pigmentario retiniano, agudeza visual reducida, hemorragia vítrea, trastorno del cuerpo vítreo, uveítis, iritis, iridociclitis, catarata, catarata subcapsular, opacificación de la cápsula posterior, queratitis punctata, abrasión corneal, células flotantes en la cámara anterior, visión borrosa, hemorragia en el lugar de inyección, hemorragia ocular, conjuntivitis, conjuntivitis alérgica, secreción ocular, fotopsia, fotofobia, molestia ocular, edema palpebral, dolor palpebral, hiperemia conjuntival.
Poco frecuentes
Ceguera, endoftalmitis, hipopion, hipema, queratopatía, adhesión del iris, depósitos corneales, edema corneal, estrías corneales, dolor en el lugar de inyección, irritación en el lugar de inyección, sensación anormal en el ojo, irritación palpebral.
Trastornos respiratorios, torácicos y mediastínicos
Frecuentes Tos
Trastornos gastrointestinales
Frecuentes Náuseas
Trastornos de la piel y del tejido subcutáneo
Frecuentes Reacciones alérgicas (erupción, urticaria, prurito, eritema)
Trastornos musculoesqueléticos y del tejido conjuntivo Muy frecuentes Artralgia
Exploraciones complementarias
Muy frecuentes Aumento de la presión intraocular
#-Las reacciones adversas se definieron como acontecimientos adversos (en al menos 0,5 puntos porcentuales de pacientes) que ocurrieron con una frecuencia superior (como mínimo 2 puntos porcentuales) en los pacientes que recibieron el tratamiento con Lucentis 0,5 mg respecto a los que recibieron el tratamiento control (tratamiento simulado (sham) o TFD con verteporfina).
* observado sólo en población con EMD
Reacciones adversas de clase terapéutica
En los ensayos fase III en DMAE exudativa, la frecuencia global de hemorragias no oculares, un efecto adverso potencialmente relacionado con la inhibición sistémica del VEGF (factor de crecimiento endotelial vascular), fue ligeramente superior en los pacientes tratados con ranibizumab. Sin embargo, no hubo un patrón consistente entre las distintas hemorragias. Tras el uso intravítreo de inhibidores del VEGF existe un riesgo teórico de acontecimientos tromboembólicos arteriales, incluyendo accidente cerebrovascular e infarto de miocardio. En los ensayos clínicos con Lucentis se observó una incidencia baja de acontecimientos tromboembólicos arteriales en pacientes con DMAE, EMD, OVR y MP y no hubo ninguna diferencia destacable entre los grupos tratados con ranibizumab comparado con el control.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Se han notificado casos de sobredosis accidental procedentes de los ensayos clínicos en DMAE exudativa y de los datos post-comercialización. Las reacciones adversas que se asociaron a estos casos notificados fueron aumento de la presión intraocular, ceguera transitoria, agudeza visual reducida, edema corneal, dolor corneal y dolor ocular. En caso de sobredosis, se deberá realizar un seguimiento y tratamiento de la presión intraocular, si el médico lo considera necesario.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Oftalmológicos, agentes antineovascularización, código ATC: S01LA04
Ranibizumab es un fragmento de anticuerpo monoclonal recombinante humanizado dirigido contra el factor de crecimiento endotelial vascular A (VEGF-A) humano. Se une con alta afinidad a las isoformas del VEGF-A (p. ej. VEGFn0, VEGFm y VEGFJ65), impidiendo, por tanto, la unión del VEGF-A a sus receptores VEGFR-1 y VEGFR-2. La unión del VEGF-A a sus receptores conduce a la proliferación de las células endoteliales y la neovascularización, así como a la exudación vascular, todo lo cual se cree que contribuye a la progresión de la forma neovascular de la degeneración macular asociada a la edad, la miopía patológica o a la alteración visual causada por el edema macular diabético o por el edema macular secundario a OVR.
Tratamiento de la DMAE exudativa
En la DMAE exudativa, la eficacia y seguridad clínicas de Lucentis se han evaluado en tres ensayos aleatorizados, doble ciego, controlados con tratamiento simulado (sham) o con tratamiento activo de 24 meses de duración, en pacientes con DMAE neovascular. En estos ensayos fueron reclutados un total de 1.323 pacientes (879 con tratamiento activo y 444 con control).
En el ensayo FVF2598g (MARINA), se aleatorizaron 716 pacientes con lesiones mínimamente clásicas u ocultas sin componente clásico en un ratio 1:1:1 para recibir inyecciones de Lucentis 0,3 mg, Lucentis 0,5 mg o tratamiento simulado una vez al mes.
En el ensayo FVF2587g (ANCHOR), se aleatorizaron 423 pacientes con lesiones de NVC predominantemente clásicas en un ratio 1:1:1 para recibir Lucentis 0,3 mg una vez al mes, Lucentis 0,5 mg una vez al mes o TFD con verteporfina (al inicio y posteriormente cada 3 meses si la angiografía fluoresceínica mostraba persistencia o recurrencia de la exudación vascular).
Tabla 1 Resultados al Mes 12 y al Mes 24 en el ensayo FVF2598g (MARINA) y FVF2587g (ANCHOR)
|
FVF2598g (MARINA) |
FVF2587g (ANCHOR) | ||||
|
Medida del resultado |
Mes |
Tratamient o simulado o sham (n=238) |
Lucentis 0,5 mg (n=240) |
TFD con verteporfina (n=143) |
Lucentis 0,5 mg (n=140) |
|
Pérdida de <15 letras de agudeza visual (%)a (mantenimiento de la visión, variable primaria) |
Mes 12 |
62% |
95% |
64% |
96% |
|
Mes 24 |
53% |
90% |
66% |
90% | |
|
Ganancia de >15 letras de agudeza visual (%)a |
Mes 12 |
5% |
34% |
6% |
40% |
|
Mes 24 |
4% |
33% |
6% |
41% | |
|
Cambio medio en la agudeza visual (letras) (desviación estándar)a |
Mes 12 |
-10,5 (16,6) |
+7,2 (14,4) |
-9,5 (16,4) |
+11,3 (14,6) |
|
Mes 24 |
-14,9 (18,7) |
+6,6 (16,5) |
-9,8 (17,6) |
+10,7 (16,5) | |
a p<0,01
Figura 1 Cambio medio en la agudeza visual desde el inicio hasta el Mes 24 en el ensayo FVF2598g (MARINA) y el ensayo FVF2587g (ANCHOR)
C/5
>
co
N
CD
T3
CD
O
T3
CD
E
O
!q
E
co
O
Ensayo FVF2598g (MARINA)
+6,6 ^
15
10
5
0
— <D C
<L>
O
<L>
E
O
E
Cü
O
~-5
-10
-15
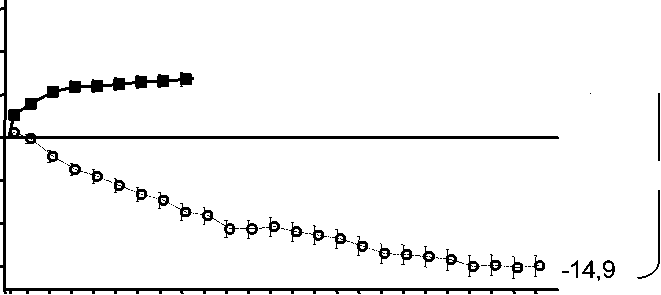
V +21,5
0 2 4 6 8 10 12 14 16 18 20 22 24
Mes
15
10
5
0
co "S
"-5 ■
-10 ■ -15 ■
Ensayo FVF2587g (ANCHOR)
frf-j +10,7
r +20,5
-9'8
0 2 4 6 8 10 12 14 16 18 20 22 24
Mes
MARINA
LUCENTIS 0,5 mg (n=240)
& Tratamiento simulado (n=238)
ANCHOR
LUCENTIS 0,5 mg (n=140)
• TFD con verteporfina (n=143)
Los resultados de ambos ensayos indicaron que el tratamiento continuado con ranibizumab también puede ser beneficioso en pacientes que perdieron >15 letras de agudeza visual mejor corregida (AVMC) en el primer año de tratamiento.
En ambos ensayos MARINA y ANCHOR se observaron beneficios en la función visual notificados por el mismo paciente estadísticamente significativos en el tratamiento con ranibizumab respecto al grupo control, medidos según el NEI VFQ-25.
En el ensayo FVF3192g (PIER), se aleatorizaron 184 pacientes que presentaban todas las formas de DMAE neovascular en un ratio 1:1:1 para recibir Lucentis 0,3 mg, Lucentis 0,5 mg o inyecciones simuladas (sham) una vez al mes en 3 dosis consecutivas, seguido de la administración de una dosis una vez cada 3 meses. A partir del Mes 14 del ensayo, se permitió a los pacientes tratados con inyecciones simuladas (sham) recibir ranibizumab, y a partir del Mes 19 fueron posibles tratamientos más frecuentes. Los pacientes tratados con Lucentis en el ensayo PIER recibieron un promedio de 10 tratamientos.
En general, tras un incremento inicial en la agudeza visual (después de la dosificación mensual), la agudeza visual de los pacientes disminuyó con la dosis trimestral, volviendo al valor basal en el Mes 12 y este efecto se mantuvo al Mes 24 en la mayoría de los pacientes tratados con ranibizumab (82%). Datos limitados procedentes de sujetos que recibieron tratamiento simulado y que posteriormente fueron tratados con ranibizumab, sugirieron que el inicio temprano del tratamiento puede asociarse a una mejor conservación de la agudeza visual.
Los datos de dos ensayos (MONT BLANC, BPD952A2308 y DENALI, BPD952A2309) realizados tras la autorización de comercialización, confirmaron la eficacia de Lucentis pero no demostraron un efecto adicional en la administración combinada de verteporfina (Visudyne TFD) y Lucentis comparado con Lucentis en monoterapia.
Tratamiento de la alteración visual debida a EMD
La eficacia y seguridad de Lucentis se han evaluado en tres ensayos aleatorizados, controlados de al menos 12 meses de duración. En estos ensayos fueron reclutados un total de 868 pacientes (708 con tratamiento activo y 160 con control).
En el ensayo de fase II D2201 (RESOLVE), 151 pacientes fueron tratados con ranibizumab (6 mg/ml, n=51, 10 mg/ml, n=51) o con tratamiento simulado (n=49) mediante inyecciones intravítreas mensuales. El cambio medio en la AVMC desde el Mes 1 al Mes 12 comparado con el inicio fue de +7,8 (±7,72) letras en los pacientes tratados con ranibizumab combinados (n=102), comparado con -0,1 (±9,77) letras para pacientes con tratamiento simulado; y el cambio medio en la AVMC en el Mes 12 desde el valor inicial fue de 10,3 (±9,1) letras comparado con -1,4 (±14,2) letras respectivamente (p<0,0001 para la diferencia de tratamiento).
En el ensayo de fase III D2301 (RESTORE), se aleatorizaron 345 pacientes en un ratio 1:1:1 para recibir ranibizumab 0,5 mg en monoterapia y fotocoagulación con láser simulada, o ranibizumab 0,5 mg y fotocoagulación con láser combinados, o inyección simulada y fotocoagulación con láser. En un ensayo de extensión de 24 meses multicéntrico y abierto (Extension del ensayo RESTORE) fueron reclutados 240 pacientes, que previamente habían completado el ensayo RESTORE de 12 meses. Los pacientes fueron tratados con ranibizumab 0,5 mg pro re nata (PRN) en el mismo ojo que en el ensayo principal (D2301 RESTORE).
Los resultados clave se resumen en la Tabla 2 (RESTORE y Extensión) y en la Figura 2 (RESTORE).
Figura 2
Cambio medio en la agudeza visual en el tiempo desde el inicio, en el estudio D2301 (RESTORE)
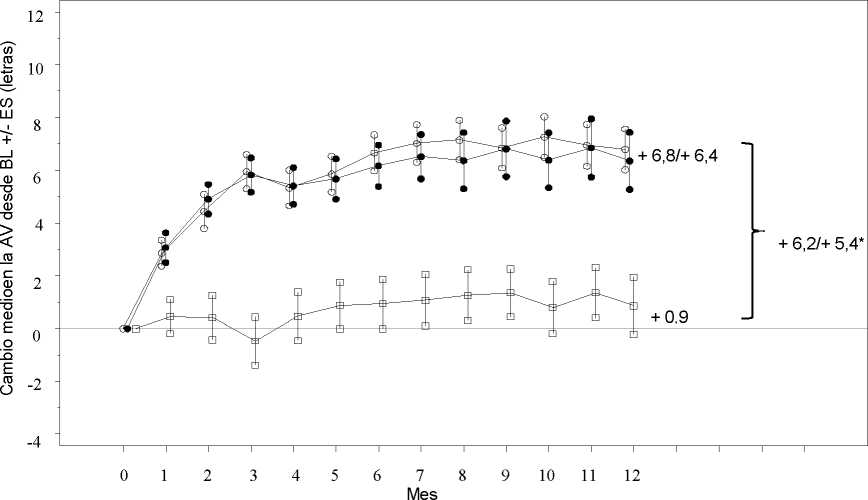
Ranibizumab 0,5 mg (n=115)
• • • Ranibizumab 0,5 mg + Láser (n=118) Láser (n=110)
Grupo de tratamiento
BL=valor inicial; ES= error estándar de la media
* Diferencia de medias de mínimos cuadrados, p<0,0001/0,0004 basado en el test bilateral de Cochran-Mantel-Haenszel
El efecto a los 12 meses fue consistente en la mayoría de subgrupos. Sin embargo, sujetos con una AVMC al inicio >73 letras y con un edema macular con un grosor de la retina central <300 pm, no parecían beneficiarse del tratamiento con ranibizumab comparado con la fotocoagulación con láser.
Tabla 2 Resultados al Mes 12 en el estudio D2301 (RESTORE) y al Mes 36 en el estudio D2301-E1 (Extensión del estudio RESTORE)
|
Medidas del resultado al Mes 12 comparado con el valor inicial en el estudio D2301 (RESTORE) |
Ranibizumab 0,5 mg n=115 |
Ranibizumab 0,5 mg + Láser n=118 |
Láser n=110 |
|
Cambio medio en la AVMC desde el Mes 1 al Mes 12a (±desviación estándar) |
6,1 (6,4)a |
5,9 (7,9)a |
0,8 (8,6) |
|
Cambio medio en la AVMC al Mes 12 (±desviación estándar) |
6,8 (8,3)a |
6,4 (11,8)a |
0,9 (11,4) |
|
Ganancia de >15 letras o AVMC >84 letras al Mes 12 (%) |
22,6 |
22,9 |
8,2 |
|
Número medio de inyecciones (Meses 011) |
7,0 |
6,8 |
7,3 (tratamient o simulado) |
|
Medida del resultado al Mes 36 comparado con el valor inicial del estudio D2301 (RESTORE) en el estudio D2301-E1 (Extension del estudio RESTORE) |
Ranibizumab 0,5 mg previo n=83 |
Ranibizumab 0,5 mg + láser previos n=83 |
Láser previo n=74 |
|
Cambio medio en la AVMC al Mes 24 (desviación estándar) |
7,9 (9,0) |
6,7 (7,9) |
5,4 (9,0) |
|
Cambio medio en la AVMC al Mes 36 (desviación estándar) |
8,0 (10,1) |
6,7 (9,6) |
6,0 (9,4) |
|
Ganancia de >15 letras o AVMC >84 letras al Mes 36 (%) |
27,7 |
30,1 |
21,6 |
|
Número medio de inyecciones (Meses 1235)* |
6,8 |
6,0 |
6,5 |
ap<0,0001 para comparaciones de los grupos de ranibizumab frente al grupo de láser. n en D2301-E1 (Extension del estudio RESTORE) es el número de pacientes con un valor tanto en el valor inicial del estudio D2301 (RESTORE) (Mes 0) como en la visita del Mes 36.
• Las proporciones de pacientes que no necesitaron ningún tratamiento con ranibizumab durante la fase de extensión fueron del 19%, 25% y 20% en los grupos ranibizumab previo, ranibizumab + láser previos y láser previo, respectivamente.
En el tratamiento con ranibizumab (con o sin láser) se observaron beneficios notificados por el mismo paciente estadísticamente significativos para la mayoría de funciones relacionadas con la visión respecto al grupo control, medidos según el NEI VFQ-25. Para las otras subescalas de este cuestionario no pudieron establecerse diferencias ligadas al tratamiento.
El perfil de seguridad a largo plazo de ranibizumab observado en el ensayo de extensión de 24 meses es consistente con el perfil de seguridad de Lucentis conocido.
En el ensayo de fase IIIb D2304 (RETAIN), se aleatorizaron 372 pacientes en un ratio 1:1:1 para recibir:
• ranibizumab 0,5 mg con fotocoagulación con láser concomitante en un régimen de tratar y extender (TE), o bien
• ranibizumab 0,5 mg en monoterapia en un régimen TE, o bien
• ranibizumab 0,5 mg en monoterapia en un régimen PRN.
En todos los grupos, ranibizumab se administró mensualmente hasta que la AVMC era estable durante al menos tres controles mensuales consecutivos. En el régimen TE ranibizumab se administró a intervalos de tratamiento de 2-3 meses. En todos los grupos, el tratamiento mensual se reiniciaba en cuanto había una disminución de la AVMC debida a la progresión del EMD y continuaba hasta que se alcanzaba nuevamente una AVMC estable.
Después de las 3 inyecciones iniciales, el número de visitas de tratamiento programadas fueron de 13 y 20 para los regímenes TE y PRN respectivamente. Con ambos regímenes TE, más del 70% de los pacientes mantuvieron su AVMC con una frecuencia media de visitas de >2 meses.
Los resultados clave se resumen en la Tabla 3.
Tabla 3 Resultados en el estudio D2304 (RETAIN)
|
Medida del resultado comparado con el valor inicial |
Ranibizumab 0,5 mg + láser en TE n=117 |
Ranibizumab 0,5 mg solo en TE n=125 |
Ranibizumab 0,5 mg en PRN n=117 |
|
Cambio medio en la AVMC desde el Mes 1 al Mes 12 (desviación estándar) |
5,9 (5,5)a |
6,1 (5,7)a |
6,2 (6,0) |
|
Cambio medio en la AVMC desde el Mes 1 al Mes 24 (desviación estándar) |
6,8 (6,0) |
6,6 (7,1) |
7,0 (6,4) |
|
Cambio medio en la AVMC al Mes 24 (desviación estándar) |
8,3 (8,1) |
6,5 (10,9) |
8,1 (8,5) |
|
Ganancia de >15 letras o AVMC >84 letras al Mes 24(%) |
25,6 |
28,0 |
30,8 |
|
Número medio de inyecciones (meses 023) |
12,4 |
12,8 |
10,7 |
“p<0,0001 para la valoración de no inferioridad a PRN
En los ensayos en EMD, la mejora en la AVMC se acompañó de una reducción en el tiempo del GSCR medio en todos los grupos de tratamiento.
Tratamiento de la alteración visual debida al edema macular secundario a OVR La seguridad y eficacia clínicas de Lucentis en pacientes con alteración visual debida al edema macular secundario a OVR se han evaluado en los ensayos BRAVO y CRUISE, controlados, aleatorizados, doble ciego que reclutaron sujetos con ORVR (n=397) y con OVCR (n=392), respectivamente. En ambos ensayos, los sujetos recibieron o bien ranibizumab 0,3 mg o 0,5 mg o inyecciones simuladas. Después de 6 meses, los pacientes en los grupos control con inyección simulada cambiaron a ranibizumab 0,5 mg.
|
BRAVO |
CRUISE | |||
|
Tratamiento simulado/Lucenti s 0,5 mg (n=132) |
Lucentis 0,5 mg (n=131) |
Tratamiento simulado/Lucenti s 0,5 mg (n=130) |
Lucentis 0,5 mg (n=130) | |
|
Cambio medio en la agudeza visual al Mes 6a (letras) (desviación estándar) (variable primaria) |
7,3 (13,0) |
18,3 (13,2) |
0,8 (16,2) |
14,9 (13,2) |
|
Cambio medio en la AVMC al Mes 12 (letras) (desviación estándar) |
12,1 (14,4) |
18,3 (14,6) |
7,3 (15,9) |
13,9 (14,2) |
|
Ganancia de >15 letras en agudeza visual al Mes 6a (%) |
28,8 |
61,1 |
16,9 |
47,7 |
|
Ganancia de >15 letras en agudeza visual al Mes 12 (%) |
43,9 |
60,3 |
33,1 |
50,8 |
|
Proporción (%) que recibió rescate con láser en 12 meses |
61,4 |
34,4 |
NA |
NA |
a p<0,0001 para ambos ensayos
Cambio medio en la AVMC desde el inicio en el tiempo hasta el Mes 6 y el Mes 12 (BRAVO)
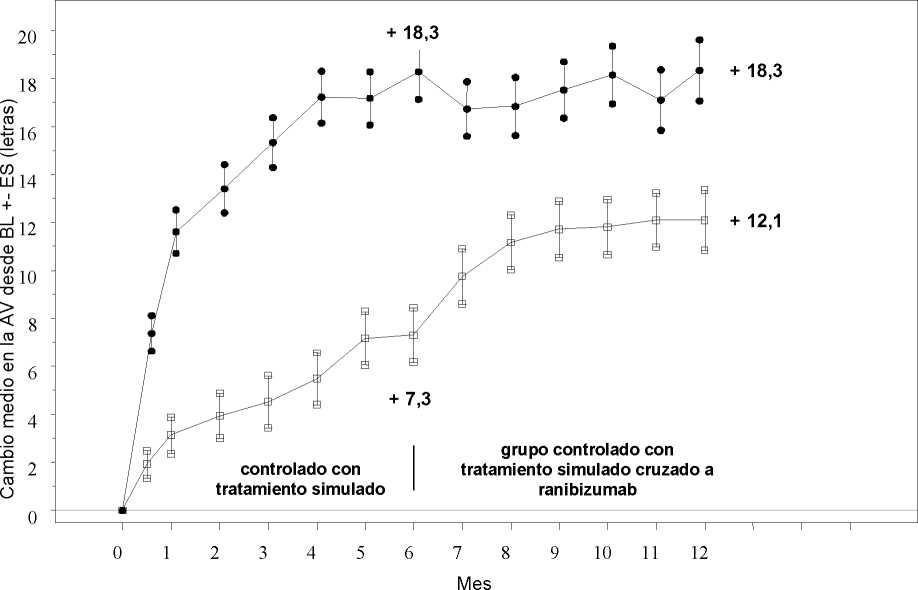
Grupo de tratamiento Tratamiento simulado/Ranibizumab 0,5 mg (n=132)
* * * Ranibizumab 0,5 mg (n=131)
BL=valor inicial; ES=error estándar de la media
Cambio medio en la AVMC desde el inicio en el tiempo hasta el Mes 6 y el Mes 12 (CRUISE)
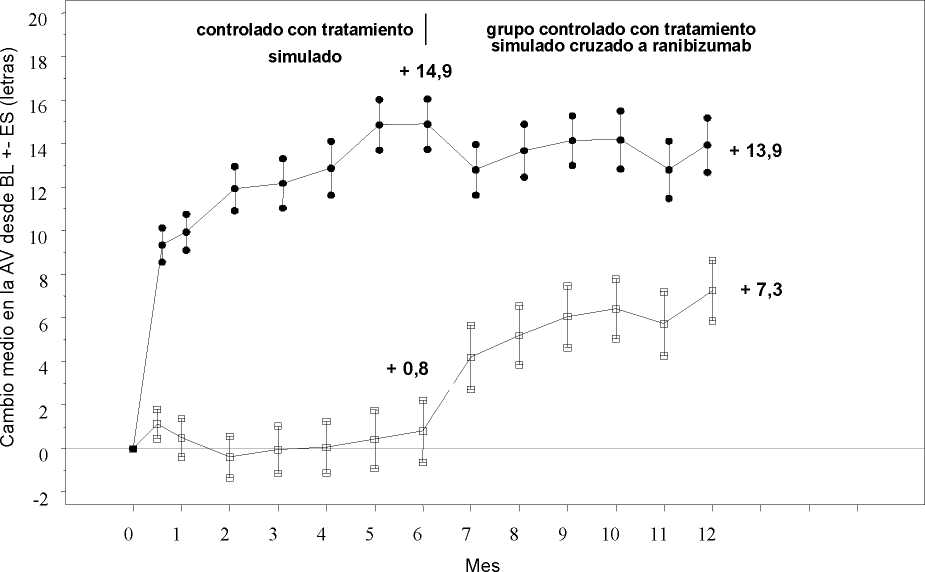
Grupo de tratamiento Tratamiento simulado/Ranibizumab 0,5 mg (n=130)
* * * Ranibizumab 0,5 mg (n=130)
BL=valor inicial; ES=error estándar de la media
En ambos estudios, la mejora de la visión se acompañó de una reducción continua y significativa del edema macular medido según el grosor central de la retina.
En pacientes con OVCR (CRUISE y ensayo de extensión HORIZON): Los sujetos tratados con tratamiento simulado en los primeros 6 meses que posteriormente recibieron ranibizumab, no alcanzaron ganancias comparables en AV en el Mes 24 (~6 letras) en comparación con los sujetos tratados con ranibizumab desde el inicio del ensayo (~12 letras).
En el tratamiento con ranibizumab se observaron beneficios notificados por el mismo paciente estadísticamente significativos en las subescalas relativas a la actividad de cerca y de lejos respecto al grupo control, medidos según el NEI VFQ-25.
La seguridad y eficacia clínicas de Lucentis a largo plazo (24 meses) en pacientes con alteración visual debida al edema macular secundario a OVR se evaluaron en los ensayos BRIGHTER (BRVO) y CRYSTAL (CRVO). En ambos ensayos, los sujetos recibieron ranibizumab 0,5 mg en un regimen de dosificación PRN que obedece a criterios de estabilización individualizados. BRIGHTER era un ensayo aleatorizado con 3 grupos controlado con tratamiento activo que comparaba ranibizumab 0,5 mg administrado en monoterapia o en combinación con fotocoagulación con láser adjunta frente a fotocoagulación con láser sola. Después de 6 meses, los sujetos en el grupo del láser podían recibir ranibizumab 0,5 mg. CRYSTAL era un ensayo de un grupo único con ranibizumab 0,5 mg en monoterapia.
|
BRIGHTER |
CRYSTAL | |||
|
Lucentis 0,5 mg N=180 |
Lucentis 0,5 mg + Láser N=178 |
Láser* N=90 |
Lucentis 0,5 mg N=356 | |
|
Cambio medio en la AVMC al Mes 6a (letras) (desviación estándar) |
+14,8 (10,7) |
+14,8 (11,13) |
+6,0 (14,27) |
+12,0 (13,95) |
|
Cambio medio en la AVMC al Mes 24b (letras) (desviación estándar) |
+15,5 (13,91) |
+17,3 (12,61) |
+11,6 (16,09) |
+12,1 (18,60) |
|
Ganancia de >15 letras en AVMC al Mes 24 (%) |
52,8 |
59,6 |
43,3 |
49,2 |
|
Número medio de inyecciones (desviación estándar) (Meses 0-23) |
11,4 (5,81) |
11,3 (6,02) |
NA |
13,1 (6,39) |
|
a p<0,0001 para ambas comparaciones en el ensayo BRIGHTER al Mes 6: Lucentis 0,5 mg frente a Láser y Lucentis 0,5 mg + Láser frente a Láser. b p<0,0001 para la hipótesis nula en el ensayo CRYSTAL en que el cambio medio al Mes 24 desde el valor inicial es cero. * A partir del Mes 6 se permitió el tratamiento con ranibizumab 0,5 mg (24 pacientes fueron tratados solo con láser). | ||||
En el ensayo BRIGHTER, ranibizumab 0,5 mg con terapia con láser adjunta demostró no inferioridad comparado con ranibizumab en monoterapia desde el valor inicial hasta el Mes 24 (95% IC -2,8, 1,4).
En ambos ensayos, se observó en el Mes 1 una reducción rápida y estadísticamente significativa del grosor del subcampo central de la retina desde el valor inicial. Este efecto se mantuvo hasta el Mes 24.
El efecto del tratamiento con ranibizumab fue similar de forma independiente de la presencia de isquemia retiniana. En el ensayo BRIGHTER, los pacientes que presentaban isquemia (N=46) o los que no la presentaban (N=133) y que fueron tratados con ranibizumab en monoterapia, tuvieron un cambio medio desde el valor inicial de +15,3 y +15,6 letras respectivamente, en el Mes 24. En el ensayo CRYSTAL, los pacientes que presentaban isquemia (N=53) o los que no la presentaban (N=300) y que fueron tratados con ranibizumab en monoterapia, tuvieron un cambio medio desde el valor inicial de +15,0 y +11,5 letras respectivamente.
En todos los pacientes tratados con ranibizumab 0,5 mg en monoterapia se observó el efecto en términos de mejora visual en ambos ensayos BRIGHTER y CRYSTAL de forma independiente de la duración de la enfermedad. En pacientes con una duración de la enfermedad <3 meses se observó un aumento en la agudeza visual de 13,3 y 10,0 letras en el Mes 1; y 17,7 y 13,2 letras en el Mes 24 en BRIGHTER y CRYSTAL respectivamente. En pacientes con una duración de la enfermedad >12 meses, la ganancia de agudeza visual correspondiente fue de 8,6 y 8,4 letras en los respectivos ensayos. Se debe considerar el inicio del tratamiento en el momento del diagnóstico.
El perfil de seguridad a largo plazo de ranibizumab observado en los ensayos de 24 meses es consistente con el perfil de seguridad de Lucentis conocido.
Tratamiento de la alteración visual debida a la NVC secundaria a MP
La seguridad y eficacia clínicas de Lucentis en pacientes con alteración visual debida a la NVC secundaria a MP se han evaluado de acuerdo a los datos de 12 meses del ensayo pivotal F2301 (RADIANCE) controlado, doble ciego. En este ensayo 277 pacientes fueron aleatorizados en un ratio 2:2:1 para los siguientes grupos:
• Grupo I (ranibizumab 0,5 mg, régimen de tratamiento basado en criterios de “estabilidad” definida como ningún cambio en la AVMC en comparación con las dos evaluaciones mensuales previas).
• Grupo II (ranibizumab 0,5 mg, régimen de tratamiento basado en criterios de “actividad de la enfermedad” definida como alteración de la visión atribuible a fluido intra o subretiniano o exudación activa debido a la lesión de NVC y evaluada mediante OCT y/o AF).
• Grupo III (TFDv - se permitió a los pacientes recibir tratamiento con ranibizumab a partir del Mes 3).
En el Grupo II, que se corresponde con la posología recomendada (ver sección 4.2), el 50,9% de los pacientes requirió 1 ó 2 inyecciones, el 34,5% requirió de 3 a 5 inyecciones y el 14,7% requirió de 6 a 12 inyecciones durante el periodo de 12 meses del ensayo. El 62,9% de los pacientes del Grupo II no requirió inyecciones en el segundo semestre del ensayo.
|
Grupo I |
Grupo II |
Grupo III | |
|
Ranibizumab |
Ranibizumab |
TFDvb | |
|
0,5 mg |
0,5 mg | ||
|
“estabilidad de |
“actividad de la | ||
|
la visión” |
enfermedad” | ||
|
(n=105) |
(n=116) |
(n=55) | |
|
Mes 3 Cambio medio en la AVMC desde el Mes 1 |
+10,5 |
+10,6 |
+2,2 |
|
al Mes 3 comparado con el inicioa (letras) Proporción de pacientes que ganaron: >15 letras, o alcanzaron >84 letras en la AVMC |
38,1% |
43,1% |
14,5% |
|
Mes 12 Número de inyecciones hasta el Mes 12: Media |
4,6 |
3,5 |
N/A |
|
Mediana |
4,0 |
2,5 |
N/A |
|
Cambio medio en la AVMC desde el Mes 1 |
+12,8 |
+12,5 |
N/A |
|
al Mes 12 comparado con el inicio (letras) Proporción de pacientes que ganaron: >15 letras, o alcanzaron >84 letras en la AVMC |
53,3% |
51,7% |
N/A |
a p<0,00001 comparación con el control de TFDv
b Control comparativo hasta el Mes 3. A los pacientes aleatorizados a TFDv se les permitió recibir tratamiento con ranibizumab a partir del Mes 3 (en el Grupo III, 38 pacientes recibieron ranibizumab a partir del Mes 3)
Figura 5 Cambio medio en la AVMC desde el inicio en el tiempo hasta el Mes 12 (RADIANCE)
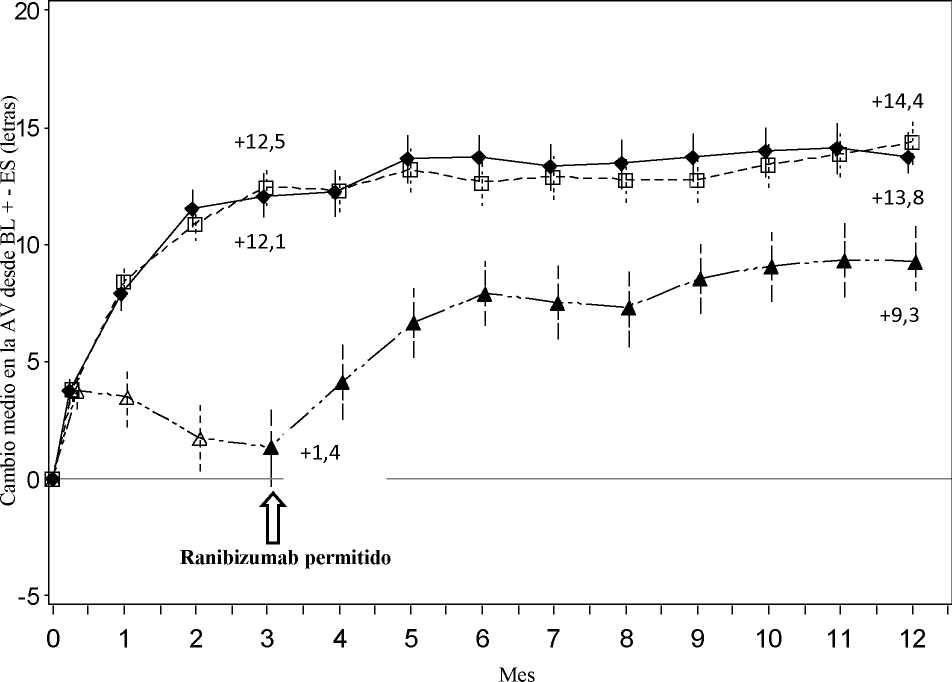
Ranibizumab 0,5 mg Grupo I por estabilización (n=105)
Ranibizumab 0,5 mg Grupo II
por actividad de la enfermedad (n=116)
Ranibizumab 0,5 mg/TFD con verteporfina Grupo III desde el Mes 3 en adelante (n=55)
A-a-a TFD con verteporfina Grupo III (n=55)
La mejora de la visión se acompañó de una reducción del grosor central de la retina.
Los beneficios comunicados por el paciente se observaron más en los grupos de tratamiento con ranibizumab que en la TFDv (valor p<0,05) en términos de mejora en la puntuación compuesta y varias subescalas (visión general, actividades de cerca, salud mental y dependencia) del NEI VFQ-25.
Población pediátrica
La seguridad y eficacia de ranibizumab aún no han sido estudiadas en pacientes pediátricos.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Lucentis en los diferentes grupos de la población pediátrica en la DMAE neovascular, la alteración visual debida al EMD, la alteración visual debida al edema macular secundario a la OVR y la alteración visual debida a la NVC secundaria a MP (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
Tras la administración intravítrea mensual de Lucentis a pacientes con DMAE neovascular, las concentraciones séricas de ranibizumab fueron en general bajas, con niveles máximos (Cmax) por debajo de la concentración de ranibizumab necesaria para inhibir la actividad biológica del VEGF en un 50% (11-27 ng/ml, valorado en un ensayo de proliferación celular in vitro). La Cmax fue proporcional a la dosis, en el rango de dosis de 0,05 a 1,0 mg/ojo. Las concentraciones séricas en un número limitado de pacientes con EMD indican que no puede excluirse una exposición sistémica ligeramente superior en comparación con la observada en pacientes con DMAE neovascular. Las concentraciones séricas de ranibizumab en pacientes con OVR fueron similares o ligeramente superiores en comparación con las observadas en pacientes con DMAE neovascular.
En base al análisis farmacocinético poblacional y a la desaparición sérica de ranibizumab en pacientes con DMAE neovascular tratados con la dosis de 0,5 mg, el promedio de la vida media de eliminación vítrea de ranibizumab es de 9 días aproximadamente. Tras la administración intravítrea mensual de Lucentis 0,5 mg/ojo, se prevé que la Cmax de ranibizumab sérica alcanzada aproximadamente 1 día después de la administración, varíe en general en un rango de entre 0,79 y 2,90 ng/ml, y que la Cmin varíe en general en un rango de entre 0,07 y 0,49 ng/ml. Se prevé que las concentraciones séricas de ranibizumab sean aproximadamente 90.000 veces inferiores a las concentraciones vítreas de ranibizumab.
Pacientes con insuficiencia renal: No se han realizado estudios formales para investigar la farmacocinética de Lucentis en pacientes con insuficiencia renal. En un análisis farmacocinético poblacional de pacientes con DMAE neovascular, el 68% (136 de 200) de los pacientes tenían insuficiencia renal (leve en un 46,5% [50-80 ml/min], moderada en un 20% [30-50 ml/min] y grave en un 1,5% [<30 ml/min]). En el caso de pacientes con OVR, el 48,2% (253 de 525) tenían insuficiencia renal (leve en un 36,4%, moderada en un 9,5% y grave en un 2,3%). El aclaramiento sistémico fue ligeramente inferior, pero esto no fue clínicamente significativo.
Insuficiencia hepática: No se han realizado estudios formales para investigar la farmacocinética de Lucentis en pacientes con insuficiencia hepática.
5.3 Datos preclínicos sobre seguridad
La administración intravítrea bilateral de ranibizumab a macacos, a dosis de entre 0,25 mg/ojo y
2,0 mg/ojo una vez cada 2 semanas durante 26 semanas, ocasionó efectos oculares dosis-dependientes.
Intraocularmente, se observaron incrementos dosis-dependientes de exudados y de células en la cámara anterior, con un máximo a los 2 días después de la inyección. Por lo general, la gravedad de la respuesta inflamatoria disminuyó con las inyecciones posteriores o durante el periodo de recuperación. En el segmento posterior, hubo infiltración de células en la cámara vítrea y partículas flotantes, que tendieron también a ser dosis-dependientes y que, en general, persistieron hasta el final del periodo de tratamiento. En el ensayo a 26 semanas, la gravedad de la inflamación vítrea aumentó con el número de inyecciones. Sin embargo, se observó evidencia de reversibilidad tras el periodo de recuperación. La naturaleza y cronología de la inflamación en el segmento posterior sugiere una respuesta inmunitaria mediada por anticuerpos, que puede ser clínicamente irrelevante. En algunos animales se observó la formación de cataratas tras un periodo relativamente largo de inflamación intensa, lo cual sugiere que las alteraciones en el cristalino fueron secundarias a la inflamación grave. Tras las inyecciones intravítreas se observó un aumentó transitorio de la presión intraocular independiente de la dosis.
Los cambios oculares microscópicos fueron relacionados con la inflamación y no eran indicativos de procesos degenerativos. Se observaron cambios inflamatorios granulomatosos en el disco óptico de algunos ojos. Estas alteraciones en el segmento posterior disminuyeron, y en algunos casos se resolvieron, durante el periodo de recuperación.
Tras la administración intravítrea, no se detectaron signos de toxicidad sistémica. En un subgrupo de animales tratados se detectaron anticuerpos séricos y vítreos contra ranibizumab.
No se dispone de datos de carcinogenicidad ni de mutagenicidad.
En hembras de mono preñadas, el tratamiento con ranibizumab intravítreo resultando en exposiciones sistémicas máximas 0,9-7 veces la peor exposición clínica, no provocó toxicidad en el desarrollo ni teratogenicidad y no tuvo ningún efecto sobre el peso o la estructura de la placenta, aunque en base a su efecto farmacológico, ranibizumab debe considerarse potencialmente teratogénico y embrio/fetotóxico.
La ausencia de efectos mediados por ranibizumab sobre el desarrollo embrio-fetal está plausiblemente relacionado principalmente con la incapacidad del fragmento Fab de atravesar la placenta. Sin embargo, se describió un caso de niveles séricos de ranibizumab maternos elevados y presencia de ranibizumab en el suero fetal lo que sugiere que el anticuerpo contra ranibizumab actuó como proteína transportadora (conteniendo la región Fc) para ranibizumab, disminuyendo de ese modo su aclaramiento sérico materno y permitiendo su paso a la placenta. Dado que las investigaciones en el desarrollo embrio-fetal se llevaron a cabo en animales preñados sanos y las enfermedades (tales como la diabetes) pueden modificar la permeabilidad de la placenta para el fragmento Fab, el estudio debe interpretarse con cautela.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
a,a-trehalosa dihidrato
Hidrocloruro de histidina monohidrato
Histidina
Polisorbato 20
Agua para inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
3 años
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Antes de usar, el vial sin abrir se puede conservar a temperatura ambiente (25°C) durante un máximo de 24 horas.
6.5 Naturaleza y contenido del envase
0,23 ml de solución estéril en un vial (vidrio tipo I) con un tapón (goma de clorobutilo). El envase contiene 1 vial.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El vial es para un solo uso. Tras la inyección se debe desechar cualquier sobrante de producto no utilizado. No debe utilizarse ningún vial que muestre signos de deterioro o manipulación. La esterilidad sólo se puede garantizar si el sellado del envase se mantiene intacto.
Para la preparación y la inyección intravítrea se necesitan los siguientes productos sanitarios de un solo uso:
- una aguja con filtro de 5 pm (18G)
- una jeringa estéril de 1 ml
- una aguja para inyección (30G x 'A").
Estos productos sanitarios no se incluyen en el envase de Lucentis.
Para la preparación de Lucentis para administración intravítrea, siga las siguientes instrucciones:
1. Antes de extraer la solución, se debe desinfectar la parte exterior del tapón de goma del vial.
2. Incorporar una aguja con filtro de 5 pm (18G) a una jeringa de 1 ml usando técnicas asépticas. Insertar la aguja con filtro en el centro del tapón del vial hasta que la aguja toque el extremo inferior del vial.
3. Extraer todo el líquido del vial, manteniendo el vial en posición vertical, ligeramente inclinado para facilitar la extracción completa.
4. Al vaciar el vial, asegurar que el émbolo se retira hacia atrás lo suficiente de forma que se vacíe por completo la aguja con filtro.
5. Dejar la aguja con filtro en el vial y desconectarla de la jeringa. La aguja con filtro se debe desechar tras extraer el contenido del vial, y no se debe utilizar para la inyección intravítrea.
6. Incorporar una aguja para inyección (30G x V") a la jeringa con firmeza y de forma aséptica.
7. Quitar la cápsula de cierre de la aguja para inyección cuidadosamente sin desconectar la aguja para inyección de la jeringa.
Nota: Sujetar la aguja para inyección por el cono mientras se retira la cápsula de cierre.
8. Expulsar el aire de la jeringa y ajustar la dosis hasta la marca de 0,05 ml en la jeringa cuidadosamente. La jeringa está lista para la inyección.
Nota: No secar la aguja para inyección. No tirar del émbolo hacia atrás.
Tras la inyección, no tapar la aguja con la cápsula de cierre ni separarla de la jeringa. Eliminar la jeringa usada junto con la aguja en un contenedor para objetos punzantes o eliminar de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/374/002
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 22 Enero 2007 Fecha de la última renovación: 24 Enero 2012
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
1. NOMBRE DEL MEDICAMENTO
Lucentis 10 mg/ml solución inyectable en jeringa precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un ml contiene 10 mg de ranibizumab*. Una jeringa precargada contiene 0,165 ml, equivalente a 1,65 mg de ranibizumab. El volumen extraíble de una jeringa precargada es 0,1 ml. Esto aporta una cantidad utilizable que proporciona una dosis única de 0,05 ml, que contiene 0,5 mg de ranibizumab. *Ranibizumab es un fragmento de anticuerpo monoclonal humanizado producido en células de Escherichia coli mediante tecnología de ADN recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución acuosa transparente, de incolora a amarillo pálido.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lucentis está indicado en adultos para:
• El tratamiento de la degeneración macular asociada a la edad (DMAE) neovascular (exudativa)
• El tratamiento de la alteración visual debida al edema macular diabético (EMD)
• El tratamiento de la alteración visual debida al edema macular secundario a la oclusión de la
vena retiniana (OVR) (oclusión de la rama venosa retiniana u oclusión de la vena central retiniana)
• El tratamiento de la alteración visual debida a la neovascularización coroidea (NVC) secundaria a la miopía patológica (MP)
4.2 Posología y forma de administración
Lucentis debe ser administrado por un oftalmólogo que tenga experiencia en la administración de inyecciones intravítreas.
La dosis recomendada de Lucentis es 0,5 mg administrada en forma de inyección intravítrea única. Esto corresponde a un volumen de inyección de 0,05 ml. El intervalo entre dos dosis inyectadas en el mismo ojo debe ser como mínimo de cuatro semanas.
El tratamiento se inicia con una inyección al mes hasta alcanzar la agudeza visual máxima y/o no haya signos de actividad de la enfermedad, es decir ningún cambio en la agudeza visual ni en otros signos y síntomas de la enfermedad bajo tratamiento continuado. En pacientes con DMAE exudativa, EMD y OVR inicialmente pueden ser necesarias tres o más inyecciones consecutivas administradas mensualmente.
A partir de ese momento, los intervalos de monitorización y tratamiento se deben determinar según criterio médico y en base a la actividad de la enfermedad, valorada mediante la agudeza visual y/o parámetros anatómicos.
Se debe interrumpir el tratamiento con Lucentis si bajo criterio del médico, los parámetros visuales y anatómicos indican que el paciente no se está beneficiando del tratamiento continuado.
La monitorización para determinar la actividad de la enfermedad puede incluir examen clínico, control funcional o técnicas de imagen (p. ej. tomografía de coherencia óptica o angiografía con fluoresceína).
Si se está tratando a los pacientes de acuerdo a un régimen de tratar y extender, una vez se ha alcanzado la agudeza visual máxima y/o no hay signos de actividad de la enfermedad, los intervalos de tratamiento se pueden espaciar de forma gradual hasta que vuelvan a aparecer signos de actividad de la enfermedad o alteración visual. En el caso de la DMAE exudativa el intervalo de tratamiento no debe espaciarse en más de dos semanas cada vez y en el caso del EMD se puede espaciar hasta un mes cada vez. Para la OVR, los intervalos de tratamiento también pueden espaciarse de forma gradual, sin embargo los datos que hay no son suficientes para determinar la duración de estos intervalos. Si vuelve a aparecer actividad de la enfermedad, se debe acortar el intervalo de tratamiento de manera consecuente.
En el tratamiento de la alteración visual debida a la NVC secundaria a MP, muchos pacientes pueden necesitar únicamente una o dos inyecciones durante el primer año, mientras que algunos pacientes pueden necesitar tratamiento con mayor frecuencia (ver sección 5.1).
Lucentis y fotocoagulación con láser en EMD y en edema macular secundario a oclusión de la rama venosa retiniana (ORVR)
Existe alguna experiencia con Lucentis administrado concomitantemente con fotocoagulación con láser (ver sección 5.1). Cuando se administren en el mismo día, Lucentis se debe administrar como mínimo 30 minutos después de la fotocoagulación con láser. Lucentis puede administrarse en pacientes que han recibido fotocoagulación con láser previamente.
Lucentis y la terapia fotodinámica con Visudyne en la NVC secundaria a MP No hay experiencia en la administración concomitante de Lucentis y Visudyne.
Poblaciones especiales
Insuficiencia hepática
Lucentis no ha sido estudiado en pacientes con insuficiencia hepática. Sin embargo, no es necesaria ninguna consideración especial en esta población.
Insuficiencia renal
No es necesario ningún ajuste de la dosis en pacientes con insuficiencia renal (ver sección 5.2).
Pacientes de edad avanzada
No se requiere ningún ajuste de la dosis en pacientes de edad avanzada. Existe experiencia limitada en pacientes con EMD mayores de 75 años.
Población pediátrica
No se ha establecido la seguridad y eficacia de Lucentis en niños y adolescentes menores de 18 años de edad. No se dispone de datos.
Forma de administración
Jeringa precargada para un solo uso. Unicamente para vía intravítrea. La jeringa precargada contiene más cantidad que la dosis recomendada de 0,5 mg. El volumen extraíble de la jeringa precargada (0,1 ml) no se administrará en su totalidad. El exceso de volumen se debe expulsar antes de la inyección. Si se inyecta todo el volumen de la jeringa precargada puede dar lugar a una sobredosis. Para expulsar las burbujas de aire y el exceso de medicamento, presione lentamente el émbolo hasta que el borde inferior de la cúpula que forma el extremo del tapón de goma quede alineado con la línea negra de dosificación de la jeringa (equivalente a 0,05 ml, es decir, 0,5 mg de ranibizumab).
Antes de la administración de Lucentis se debe comprobar visualmente la ausencia de partículas y decoloración.
El procedimiento de inyección deberá llevarse a cabo bajo condiciones asépticas, que incluyen el lavado quirúrgico de las manos, el uso de guantes estériles, un campo estéril, un blefarostato estéril para los párpados (o equivalente) y la disponibilidad de una paracentesis estéril (en caso necesario). Antes de realizar el procedimiento de inyección intravítrea, se deberá evaluar detalladamente la historia clínica del paciente en cuanto a reacciones de hipersensibilidad (ver sección 4.4). Antes de la inyección se debe administrar una anestesia adecuada y un microbicida tópico de amplio espectro para desinfectar la piel de la zona periocular, párpado y superficie ocular, de acuerdo con la práctica local.
Para la información relativa a la preparación de Lucentis, ver sección 6.6.
La aguja para inyección se deberá introducir 3,5-4,0 mm por detrás del limbo en la cavidad vítrea, evitando el meridiano horizontal y en dirección al centro del globo. Seguidamente debe liberarse el volumen de inyección de 0,05 ml; las inyecciones siguientes deberán aplicarse cada vez en un punto escleral distinto. Cada jeringa precargada se debe usar exclusivamente para el tratamiento de un solo ojo.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Pacientes con infecciones oculares o perioculares o con sospecha de éstas.
Pacientes con inflamación intraocular grave.
4.4 Advertencias y precauciones especiales de empleo Reacciones relacionadas con la inyección intravítrea
Las inyecciones intravítreas, incluidas las de Lucentis, se han asociado a endoftalmitis, inflamación intraocular, desprendimiento de retina regmatógeno, desgarro retiniano y catarata traumática iatrogénica (ver sección 4.8). Siempre que se administre Lucentis se deben emplear técnicas de inyección asépticas adecuadas. Además, deberá realizarse un seguimiento de los pacientes durante la semana posterior a la inyección para poder administrar tratamiento temprano en caso de infección. Se deberá instruir a los pacientes sobre la necesidad de comunicar inmediatamente cualquier síntoma que sugiera endoftalmitis o cualquiera de los acontecimientos mencionados anteriormente.
Aumento de la presión intraocular
Se han observado aumentos transitorios de la presión intraocular (PIO) en los 60 minutos siguientes a la inyección de Lucentis. También se han identificado aumentos sostenidos de la PIO (ver sección 4.8). Tanto la presión intraocular como la perfusión de la cabeza del nervio óptico, se deben monitorizar y tratar adecuadamente.
Tratamiento bilateral
Los escasos datos existentes sobre el tratamiento bilateral con Lucentis (incluyendo la administración en el mismo día) no sugieren un riesgo incrementado de efectos adversos sistémicos en comparación con el tratamiento unilateral.
Inmunogenicidad
Hay un potencial de inmunogenicidad con Lucentis. Dado que en sujetos con EMD existe un potencial para una exposición sistémica incrementada, no puede excluirse un mayor riesgo para desarrollar hipersensibilidad en esta población de pacientes. También se deberá instruir a los pacientes sobre la necesidad de notificar si la inflamación intraocular incrementa en su gravedad, lo que puede ser un signo clínico atribuible a la formación de anticuerpos intraoculares.
Uso concomitante con otros anti-VEGF (factor de crecimiento endotelial vascular)
Lucentis no se deberá administrar de forma concurrente con otros medicamentos anti-VEGF (sistémicos u oculares).
Aplazamiento del tratamiento con Lucentis
La administración se deberá aplazar y el tratamiento no deberá reanudarse antes del siguiente tratamiento programado en caso de:
• una disminución en la agudeza visual mejor corregida (AVMC) de >30 letras comparado con la última evaluación de la agudeza visual;
• una presión intraocular de >30 mmHg;
• una rotura retiniana;
• una hemorragia subretiniana que afecte al centro de la fóvea, o, si el tamaño de la hemorragia es >50% del área total de la lesión;
• cirugía intraocular realizada en los 28 días previos o prevista durante los 28 días posteriores. Desgarro del epitelio pigmentario de la retina
Los factores de riesgo asociados con el desarrollo de un desgarro del epitelio pigmentario de la retina tras la terapia con anti-VEGF para la DMAE exudativa, incluyen un desprendimiento del epitelio pigmentario de la retina extenso y/o elevado. Cuando se inicie la terapia con Lucentis se debe tener precaución en pacientes con estos factores de riesgo de desarrollar desgarros del epitelio pigmentario de la retina.
Desprendimiento de retina regmatógeno o agujeros maculares
El tratamiento se debe interrumpir en sujetos con desprendimiento de retina regmatógeno o agujeros maculares en estadíos 3 ó 4.
Poblaciones con datos limitados
Sólo existe experiencia limitada en el tratamiento de sujetos con EMD debido a diabetes tipo I. Lucentis no ha sido estudiado en pacientes que hayan recibido previamente inyecciones intravítreas, en pacientes con infecciones sistémicas activas, retinopatía diabética proliferativa, ni en pacientes con enfermedades oculares simultáneas tales como desprendimiento de retina o agujero macular.
Tampoco existe experiencia en el tratamiento con Lucentis en pacientes diabéticos con un HbA1c por encima del 12% e hipertensión no controlada. El médico debe tener en cuenta esta falta de información al tratar a tales pacientes.
No hay datos suficientes que permitan establecer una conclusión acerca del efecto de Lucentis en pacientes con OVR que presentan pérdida irreversible de la función visual isquémica.
En pacientes con MP, hay datos limitados del efecto de Lucentis en pacientes que han sido sometidos previamente a un tratamiento de terapia fotodinámica con verteporfina (TFDv) sin exito. Además, mientras que en sujetos con lesiones subfoveales y yuxtafoveales se observó un efecto consistente, no hay datos suficientes para establecer conclusiones sobre el efecto de Lucentis en sujetos con MP y lesiones extrafoveales.
Efectos sistémicos tras el uso intravítreo
Se han notificado acontecimientos adversos sistémicos, incluyendo hemorragias no oculares y acontecimientos tromboembólicos arteriales tras la inyección intravítrea de inhibidores del VEGF.
Existen datos limitados sobre seguridad en el tratamiento de pacientes con EMD, edema macular debido a OVR y NVC secundaria a MP que tengan antecedentes de accidente cerebrovascular o ataques isquémicos transitorios. Se debe tener precaución cuando se traten tales pacientes (ver sección 4.8).
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones formales.
Para el uso conjunto de terapia fotodinámica (TFD) con verteporfina y Lucentis en la DMAE exudativa y en la MP, ver sección 5.1.
Para el uso conjunto de fotocoagulación con láser y Lucentis en EMD y ORVR, ver secciones 4.2 y 5.1.
En ensayos clínicos para el tratamiento de la alteración visual debida al EMD, el tratamiento concomitante con tiazolidinedionas en pacientes tratados con Lucentis, no afectó el resultado en relación a la agudeza visual o al grosor del subcampo central de la retina (GSCR).
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil/anticoncepción en mujeres
Las mujeres en edad fértil deben utilizar métodos anticonceptivos efectivos durante el tratamiento. Embarazo
No se dispone de datos clínicos de exposición a ranibizumab en embarazos. Los estudios en monos cinomolgos no sugieren efectos perjudiciales directos ni indirectos en términos de embarazo o desarrollo embrional/fetal (ver sección 5.3). La exposición sistémica a ranibizumab tras la administración ocular es baja, pero debido a su mecanismo de acción, ranibizumab debe considerarse como potencialmente teratogénico y embrio-/fetotóxico. Por ello ranibizumab no se deberá usar durante el embarazo salvo que el beneficio esperado supere el riesgo potencial para el feto. Para mujeres que deseen quedarse embarazadas y hayan sido tratadas con ranibizumab, se recomienda esperar como mínimo 3 meses tras la última dosis de ranibizumab antes de concebir un hijo.
Lactancia
Se desconoce si Lucentis se excreta en la leche materna. No se recomienda la lactancia durante el uso de Lucentis.
Fertilidad
No hay datos disponibles sobre fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
El procedimiento de tratamiento con Lucentis puede producir alteraciones visuales transitorias, que pueden afectar la capacidad para conducir o utilizar máquinas (ver sección 4.8). Los pacientes que experimenten estos signos no deben conducir ni utilizar máquinas hasta que dichas alteraciones visuales transitorias remitan.
4.8 Reacciones adversas
Resumen del perfil de seguridad
La mayoría de las reacciones adversas notificadas tras la administración de Lucentis están relacionadas con el procedimiento de inyección intravítrea.
Las reacciones adversas oculares tras la inyección de Lucentis notificadas más frecuentemente son: dolor ocular, hiperemia ocular, aumento de la presión intraocular, vitritis, desprendimiento de vítreo, hemorragia retiniana, alteración visual, partículas flotantes en el vítreo, hemorragia conjuntival, irritación ocular, sensación de cuerpo extraño en los ojos, aumento del lagrimeo, blefaritis, ojo seco, y prurito ocular.
Las reacciones adversas no oculares notificadas más frecuentemente son cefalea, nasofaringitis y artralgia.
Las reacciones adversas notificadas con menor frecuencia, pero de mayor gravedad, incluyen endoftalmitis, ceguera, desprendimiento de retina, desgarro retiniano y catarata traumática iatrogénica (ver sección 4.4).
Se debe informar a los pacientes de los síntomas de estas reacciones adversas potenciales e instruirlos para que informen a su médico en caso de aparición de signos tales como dolor ocular o aumento del malestar en el ojo, empeoramiento del enrojecimiento del ojo, visión borrosa o disminución de la visión, aumento del número de pequeñas manchas en su visión o aumento de la sensibilidad a la luz.
En la siguiente tabla se resumen las reacciones adversas ocurridas tras la administración de Lucentis en los ensayos clínicos.
Tabla de reacciones adversas#
Las reacciones adversas se listan con un sistema de clasificación de órganos y frecuencia usando el siguiente criterio: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000), muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo frecuencia.
Infecciones e infestaciones
Muy frecuentes Nasofaringitis
Frecuentes Infección de las vías urinarias*
Trastornos de la sangre y del sistema linfático
Frecuentes Anemia
Trastornos del sistema inmunológico
Frecuentes Hipersensibilidad
Trastornos psiquiátricos
Frecuentes Ansiedad
Trastornos del sistema nervioso
Muy frecuentes Cefalea
Trastornos oculares
Muy frecuentes
Vitritis, desprendimiento de vitreo, hemorragia retiniana, alteración visual, dolor ocular, partículas flotantes en el vítreo, hemorragia conjuntival, irritación ocular, sensación de cuerpo extraño en los ojos, aumento del lagrimeo, blefaritis, ojo seco, hiperemia ocular, prurito ocular.
Frecuentes
Degeneración retiniana, trastorno retiniano, desprendimiento de retina, desgarro retiniano, desprendimiento del epitelio pigmentario retiniano, desgarro del epitelio pigmentario retiniano, agudeza visual reducida, hemorragia vítrea, trastorno del cuerpo vítreo, uveítis, iritis, iridociclitis, catarata, catarata subcapsular, opacificación de la cápsula posterior, queratitis punctata, abrasión corneal, células flotantes en la cámara anterior, visión borrosa, hemorragia en el lugar de inyección, hemorragia ocular, conjuntivitis, conjuntivitis alérgica, secreción ocular, fotopsia, fotofobia, molestia ocular, edema palpebral, dolor palpebral, hiperemia conjuntival.
Poco frecuentes
Ceguera, endoftalmitis, hipopion, hipema, queratopatía, adhesión del iris, depósitos corneales, edema corneal, estrías corneales, dolor en el lugar de inyección, irritación en el lugar de inyección, sensación anormal en el ojo, irritación palpebral.
Trastornos respiratorios, torácicos y mediastínicos
Frecuentes Tos
Trastornos gastrointestinales
Frecuentes Náuseas
Trastornos de la piel y del tejido subcutáneo
Frecuentes Reacciones alérgicas (erupción, urticaria, prurito, eritema)
Trastornos musculoesqueléticos y del tejido conjuntivo Muy frecuentes Artralgia
Exploraciones complementarias
Muy frecuentes Aumento de la presión intraocular
#-Las reacciones adversas se definieron como acontecimientos adversos (en al menos 0,5 puntos porcentuales de pacientes) que ocurrieron con una frecuencia superior (como mínimo 2 puntos porcentuales) en los pacientes que recibieron el tratamiento con Lucentis 0,5 mg respecto a los que recibieron el tratamiento control (tratamiento simulado (sham) o TFD con verteporfina).
* observado sólo en población con EMD
Reacciones adversas de clase terapéutica
En los ensayos fase III en DMAE exudativa, la frecuencia global de hemorragias no oculares, un efecto adverso potencialmente relacionado con la inhibición sistémica del VEGF (factor de crecimiento endotelial vascular), fue ligeramente superior en los pacientes tratados con ranibizumab. Sin embargo, no hubo un patrón consistente entre las distintas hemorragias. Tras el uso intravítreo de inhibidores del VEGF existe un riesgo teórico de acontecimientos tromboembólicos arteriales, incluyendo accidente cerebrovascular e infarto de miocardio. En los ensayos clínicos con Lucentis se observó una incidencia baja de acontecimientos tromboembólicos arteriales en pacientes con DMAE, EMD, OVR y MP y no hubo ninguna diferencia destacable entre los grupos tratados con ranibizumab comparado con el control.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Se han notificado casos de sobredosis accidental procedentes de los ensayos clínicos en DMAE exudativa y de los datos post-comercialización. Las reacciones adversas que se asociaron a estos casos notificados fueron aumento de la presión intraocular, ceguera transitoria, agudeza visual reducida, edema corneal, dolor corneal y dolor ocular. En caso de sobredosis, se deberá realizar un seguimiento y tratamiento de la presión intraocular, si el médico lo considera necesario.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Oftalmológicos, agentes antineovascularización, código ATC: S01LA04
Ranibizumab es un fragmento de anticuerpo monoclonal recombinante humanizado dirigido contra el factor de crecimiento endotelial vascular A (VEGF-A) humano. Se une con alta afinidad a las isoformas del VEGF-A (p. ej. VEGFn0, VEGFm y VEGFJ65), impidiendo, por tanto, la unión del VEGF-A a sus receptores VEGFR-1 y VEGFR-2. La unión del VEGF-A a sus receptores conduce a la proliferación de las células endoteliales y la neovascularización, así como a la exudación vascular, todo lo cual se cree que contribuye a la progresión de la forma neovascular de la degeneración macular asociada a la edad, la miopía patológica o a la alteración visual causada por el edema macular diabético o por el edema macular secundario a OVR.
Tratamiento de la DMAE exudativa
En la DMAE exudativa, la eficacia y seguridad clínicas de Lucentis se han evaluado en tres ensayos aleatorizados, doble ciego, controlados con tratamiento simulado (sham) o con tratamiento activo de 24 meses de duración, en pacientes con DMAE neovascular. En estos ensayos fueron reclutados un total de 1.323 pacientes (879 con tratamiento activo y 444 con control).
En el ensayo FVF2598g (MARINA), se aleatorizaron 716 pacientes con lesiones mínimamente clásicas u ocultas sin componente clásico en un ratio 1:1:1 para recibir inyecciones de Lucentis 0,3 mg, Lucentis 0,5 mg o tratamiento simulado una vez al mes.
En el ensayo FVF2587g (ANCHOR), se aleatorizaron 423 pacientes con lesiones de NVC predominantemente clásicas en un ratio 1:1:1 para recibir Lucentis 0,3 mg una vez al mes, Lucentis 0,5 mg una vez al mes o TFD con verteporfina (al inicio y posteriormente cada 3 meses si la angiografía fluoresceínica mostraba persistencia o recurrencia de la exudación vascular).
En la Tabla 1 y en la Figura 1 se resumen los resultados clave.
Tabla 1 Resultados al Mes 12 y al Mes 24 en el ensayo FVF2598g (MARINA) y FVF2587g (ANCHOR)
|
FVF2598g (MARINA) |
FVF2587g (ANCHOR) | ||||
|
Medida del resultado |
Mes |
Tratamient o simulado o sham (n=238) |
Lucentis 0,5 mg (n=240) |
TFD con verteporfina (n=143) |
Lucentis 0,5 mg (n=140) |
|
Pérdida de <15 letras de agudeza visual (%)a (mantenimiento de la visión, variable primaria) |
Mes 12 |
62% |
95% |
64% |
96% |
|
Mes 24 |
53% |
90% |
66% |
90% | |
|
Ganancia de >15 letras de agudeza visual (%)a |
Mes 12 |
5% |
34% |
6% |
40% |
|
Mes 24 |
4% |
33% |
6% |
41% | |
|
Cambio medio en la agudeza visual (letras) (desviación estándar)a |
Mes 12 |
-10,5 (16,6) |
+7,2 (14,4) |
-9,5 (16,4) |
+11,3 (14,6) |
|
Mes 24 |
-14,9 (18,7) |
+6,6 (16,5) |
-9,8 (17,6) |
+10,7 (16,5) | |
a p<0,01
Figura 1 Cambio medio en la agudeza visual desde el inicio hasta el Mes 24 en el ensayo FVF2598g (MARINA) y el ensayo FVF2587g (ANCHOR)
C/5
>
co
N
CD
T3
CD
O
T3
CD
E
O
!q
E
co
O
Ensayo FVF2598g (MARINA)
+6,6 ^
15
10
5
0
— <D C
<L>
O
<L>
E
O
E
Cü
O
~-5
-10
-15
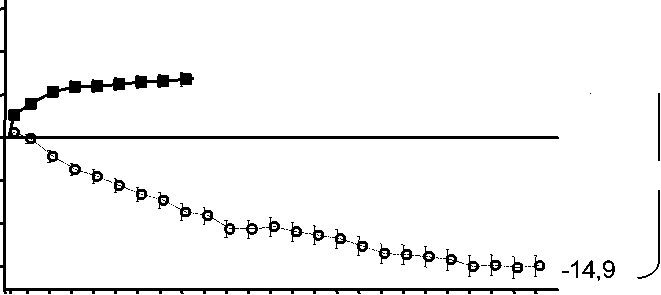
V +21,5
0 2 4 6 8 10 12 14 16 18 20 22 24
Mes
15
10
5
0
co "S
"-5 ■
-10 ■ -15 ■
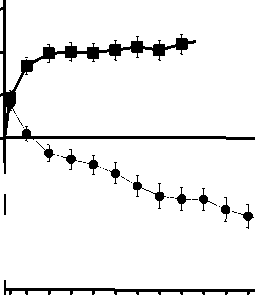
Ensayo FVF2587g (ANCHOR)
frf-j +10,7
r +20,5
-9'8
0 2 4 6 8 10 12 14 16 18 20 22 24
Mes
MARINA
LUCENTIS 0,5 mg (n=240)
& Tratamiento simulado (n=238)
ANCHOR
LUCENTIS 0,5 mg (n=140)
• TFD con verteporfina (n=143)
Los resultados de ambos ensayos indicaron que el tratamiento continuado con ranibizumab también puede ser beneficioso en pacientes que perdieron >15 letras de agudeza visual mejor corregida (AVMC) en el primer año de tratamiento.
En ambos ensayos MARINA y ANCHOR se observaron beneficios en la función visual notificados por el mismo paciente estadísticamente significativos en el tratamiento con ranibizumab respecto al grupo control, medidos según el NEI VFQ-25.
En el ensayo FVF3192g (PIER), se aleatorizaron 184 pacientes que presentaban todas las formas de DMAE neovascular en un ratio 1:1:1 para recibir Lucentis 0,3 mg, Lucentis 0,5 mg o inyecciones simuladas (sham) una vez al mes en 3 dosis consecutivas, seguido de la administración de una dosis una vez cada 3 meses. A partir del Mes 14 del ensayo, se permitió a los pacientes tratados con inyecciones simuladas (sham) recibir ranibizumab, y a partir del Mes 19 fueron posibles tratamientos más frecuentes. Los pacientes tratados con Lucentis en el ensayo PIER recibieron un promedio de 10 tratamientos.
En general, tras un incremento inicial en la agudeza visual (después de la dosificación mensual), la agudeza visual de los pacientes disminuyó con la dosis trimestral, volviendo al valor basal en el Mes 12 y este efecto se mantuvo al Mes 24 en la mayoría de los pacientes tratados con ranibizumab (82%). Datos limitados procedentes de sujetos que recibieron tratamiento simulado y que posteriormente fueron tratados con ranibizumab, sugirieron que el inicio temprano del tratamiento puede asociarse a una mejor conservación de la agudeza visual.
Los datos de dos ensayos (MONT BLANC, BPD952A2308 y DENALI, BPD952A2309) realizados tras la autorización de comercialización, confirmaron la eficacia de Lucentis pero no demostraron un efecto adicional en la administración combinada de verteporfina (Visudyne TFD) y Lucentis comparado con Lucentis en monoterapia.
Tratamiento de la alteración visual debida a EMD
La eficacia y seguridad de Lucentis se han evaluado en tres ensayos aleatorizados, controlados de al menos 12 meses de duración. En estos ensayos fueron reclutados un total de 868 pacientes (708 con tratamiento activo y 160 con control).
En el ensayo de fase II D2201 (RESOLVE), 151 pacientes fueron tratados con ranibizumab (6 mg/ml, n=51, 10 mg/ml, n=51) o con tratamiento simulado (n=49) mediante inyecciones intravítreas mensuales. El cambio medio en la AVMC desde el Mes 1 al Mes 12 comparado con el inicio fue de +7,8 (±7,72) letras en los pacientes tratados con ranibizumab combinados (n=102), comparado con -0,1 (±9,77) letras para pacientes con tratamiento simulado; y el cambio medio en la AVMC en el Mes 12 desde el valor inicial fue de 10,3 (±9,1) letras comparado con -1,4 (±14,2) letras respectivamente (p<0,0001 para la diferencia de tratamiento).
En el ensayo de fase III D2301 (RESTORE), se aleatorizaron 345 pacientes en un ratio 1:1:1 para recibir ranibizumab 0,5 mg en monoterapia y fotocoagulación con láser simulada, o ranibizumab 0,5 mg y fotocoagulación con láser combinados, o inyección simulada y fotocoagulación con láser. En un ensayo de extensión de 24 meses multicéntrico y abierto (Extension del ensayo RESTORE) fueron reclutados 240 pacientes, que previamente habían completado el ensayo RESTORE de 12 meses. Los pacientes fueron tratados con ranibizumab 0,5 mg pro re nata (PRN) en el mismo ojo que en el ensayo principal (D2301 RESTORE).
Los resultados clave se resumen en la Tabla 2 (RESTORE y Extensión) y en la Figura 2 (RESTORE).
Figura 2
Cambio medio en la agudeza visual en el tiempo desde el inicio, en el estudio D2301 (RESTORE)
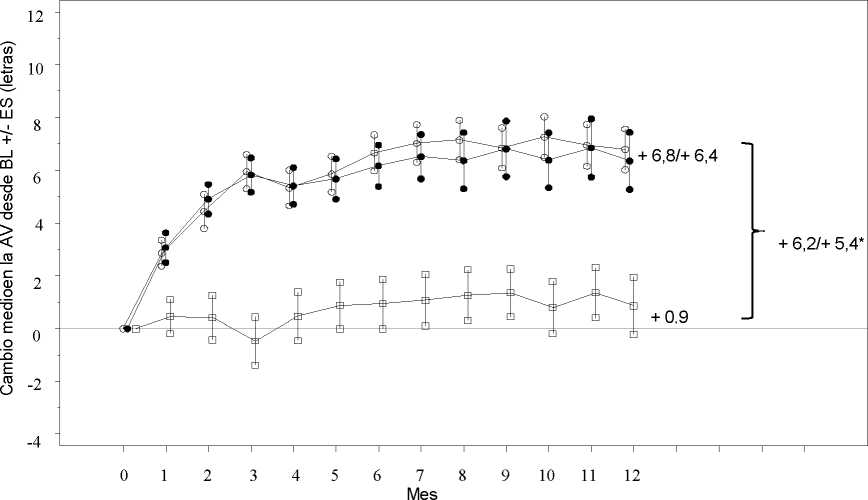
Ranibizumab 0,5 mg (n=115)
• • • Ranibizumab 0,5 mg + Láser (n=118) Láser (n=110)
Grupo de tratamiento
BL=valor inicial; ES= error estándar de la media
* Diferencia de medias de mínimos cuadrados, p<0,0001/0,0004 basado en el test bilateral de Cochran-Mantel-Haenszel
El efecto a los 12 meses fue consistente en la mayoría de subgrupos. Sin embargo, sujetos con una AVMC al inicio >73 letras y con un edema macular con un grosor de la retina central <300 pm, no parecían beneficiarse del tratamiento con ranibizumab comparado con la fotocoagulación con láser.
Tabla 2 Resultados al Mes 12 en el estudio D2301 (RESTORE) y al Mes 36 en el estudio D2301-E1 (Extensión del estudio RESTORE)
|
Medidas del resultado al Mes 12 comparado con el valor inicial en el estudio D2301 (RESTORE) |
Ranibizumab 0,5 mg n=115 |
Ranibizumab 0,5 mg + Láser n=118 |
Láser n=110 |
|
Cambio medio en la AVMC desde el Mes 1 al Mes 12a (±desviación estándar) |
6,1 (6,4)a |
5,9 (7,9)a |
0,8 (8,6) |
|
Cambio medio en la AVMC al Mes 12 (±desviación estándar) |
6,8 (8,3)a |
6,4 (11,8)a |
0,9 (11,4) |
|
Ganancia de >15 letras o AVMC >84 letras al Mes 12 (%) |
22,6 |
22,9 |
8,2 |
|
Número medio de inyecciones (Meses 011) |
7,0 |
6,8 |
7,3 (tratamient o simulado) |
|
Medida del resultado al Mes 36 comparado con el valor inicial del estudio D2301 (RESTORE) en el estudio D2301-E1 (Extension del estudio RESTORE) |
Ranibizumab 0,5 mg previo n=83 |
Ranibizumab 0,5 mg + láser previos n=83 |
Láser previo n=74 |
|
Cambio medio en la AVMC al Mes 24 (desviación estándar) |
7,9 (9,0) |
6,7 (7,9) |
5,4 (9,0) |
|
Cambio medio en la AVMC al Mes 36 (desviación estándar) |
8,0 (10,1) |
6,7 (9,6) |
6,0 (9,4) |
|
Ganancia de >15 letras o AVMC >84 letras al Mes 36 (%) |
27,7 |
30,1 |
21,6 |
|
Número medio de inyecciones (Meses 1235)* |
6,8 |
6,0 |
6,5 |
ap<0,0001 para comparaciones de los grupos de ranibizumab frente al grupo de láser. n en D2301-E1 (Extension del estudio RESTORE) es el número de pacientes con un valor tanto en el valor inicial del estudio D2301 (RESTORE) (Mes 0) como en la visita del Mes 36.
• Las proporciones de pacientes que no necesitaron ningún tratamiento con ranibizumab durante la fase de extensión fueron del 19%, 25% y 20% en los grupos ranibizumab previo, ranibizumab + láser previos y láser previo, respectivamente.
En el tratamiento con ranibizumab (con o sin láser) se observaron beneficios notificados por el mismo paciente estadísticamente significativos para la mayoría de funciones relacionadas con la visión respecto al grupo control, medidos según el NEI VFQ-25. Para las otras subescalas de este cuestionario no pudieron establecerse diferencias ligadas al tratamiento.
El perfil de seguridad a largo plazo de ranibizumab observado en el ensayo de extensión de 24 meses es consistente con el perfil de seguridad de Lucentis conocido.
En el ensayo de fase IIIb D2304 (RETAIN), se aleatorizaron 372 pacientes en un ratio 1:1:1 para recibir:
• ranibizumab 0,5 mg con fotocoagulación con láser concomitante en un régimen de tratar y extender (TE), o bien
• ranibizumab 0,5 mg en monoterapia en un régimen TE, o bien
• ranibizumab 0,5 mg en monoterapia en un régimen PRN.
En todos los grupos, ranibizumab se administró mensualmente hasta que la AVMC era estable durante al menos tres controles mensuales consecutivos. En el régimen TE ranibizumab se administró a intervalos de tratamiento de 2-3 meses. En todos los grupos, el tratamiento mensual se reiniciaba en cuanto había una disminución de la AVMC debida a la progresión del EMD y continuaba hasta que se alcanzaba nuevamente una AVMC estable.
Después de las 3 inyecciones iniciales, el número de visitas de tratamiento programadas fueron de 13 y 20 para los regímenes TE y PRN respectivamente. Con ambos regímenes TE, más del 70% de los pacientes mantuvieron su AVMC con una frecuencia media de visitas de >2 meses.
Los resultados clave se resumen en la Tabla 3.
Tabla 3 Resultados en el estudio D2304 (RETAIN)
|
Medida del resultado comparado con el valor inicial |
Ranibizumab 0,5 mg + láser en TE n=117 |
Ranibizumab 0,5 mg solo en TE n=125 |
Ranibizumab 0,5 mg en PRN n=117 |
|
Cambio medio en la AVMC desde el Mes 1 al Mes 12 (desviación estándar) |
5,9 (5,5)a |
6,1 (5,7)a |
6,2 (6,0) |
|
Cambio medio en la AVMC desde el Mes 1 al Mes 24 (desviación estándar) |
6,8 (6,0) |
6,6 (7,1) |
7,0 (6,4) |
|
Cambio medio en la AVMC al Mes 24 (desviación estándar) |
8,3 (8,1) |
6,5 (10,9) |
8,1 (8,5) |
|
Ganancia de >15 letras o AVMC >84 letras al Mes 24(%) |
25,6 |
28,0 |
30,8 |
|
Número medio de inyecciones (meses 023) |
12,4 |
12,8 |
10,7 |
“p<0,0001 para la valoración de no inferioridad a PRN
En los ensayos en EMD, la mejora en la AVMC se acompañó de una reducción en el tiempo del GSCR medio en todos los grupos de tratamiento.
Tratamiento de la alteración visual debida al edema macular secundario a OVR La seguridad y eficacia clínicas de Lucentis en pacientes con alteración visual debida al edema macular secundario a OVR se han evaluado en los ensayos BRAVO y CRUISE, controlados, aleatorizados, doble ciego que reclutaron sujetos con ORVR (n=397) y con OVCR (n=392), respectivamente. En ambos ensayos, los sujetos recibieron o bien ranibizumab 0,3 mg o 0,5 mg o inyecciones simuladas. Después de 6 meses, los pacientes en los grupos control con inyección simulada cambiaron a ranibizumab 0,5 mg.
|
BRAVO |
CRUISE | |||
|
Tratamiento simulado/Lucenti s 0,5 mg (n=132) |
Lucentis 0,5 mg (n=131) |
Tratamiento simulado/Lucenti s 0,5 mg (n=130) |
Lucentis 0,5 mg (n=130) | |
|
Cambio medio en la agudeza visual al Mes 6a (letras) (desviación estándar) (variable primaria) |
7,3 (13,0) |
18,3 (13,2) |
0,8 (16,2) |
14,9 (13,2) |
|
Cambio medio en la AVMC al Mes 12 (letras) (desviación estándar) |
12,1 (14,4) |
18,3 (14,6) |
7,3 (15,9) |
13,9 (14,2) |
|
Ganancia de >15 letras en agudeza visual al Mes 6a (%) |
28,8 |
61,1 |
16,9 |
47,7 |
|
Ganancia de >15 letras en agudeza visual al Mes 12 (%) |
43,9 |
60,3 |
33,1 |
50,8 |
|
Proporción (%) que recibió rescate con láser en 12 meses |
61,4 |
34,4 |
NA |
NA |
a p<0,0001 para ambos ensayos
Cambio medio en la AVMC desde el inicio en el tiempo hasta el Mes 6 y el Mes 12 (BRAVO)
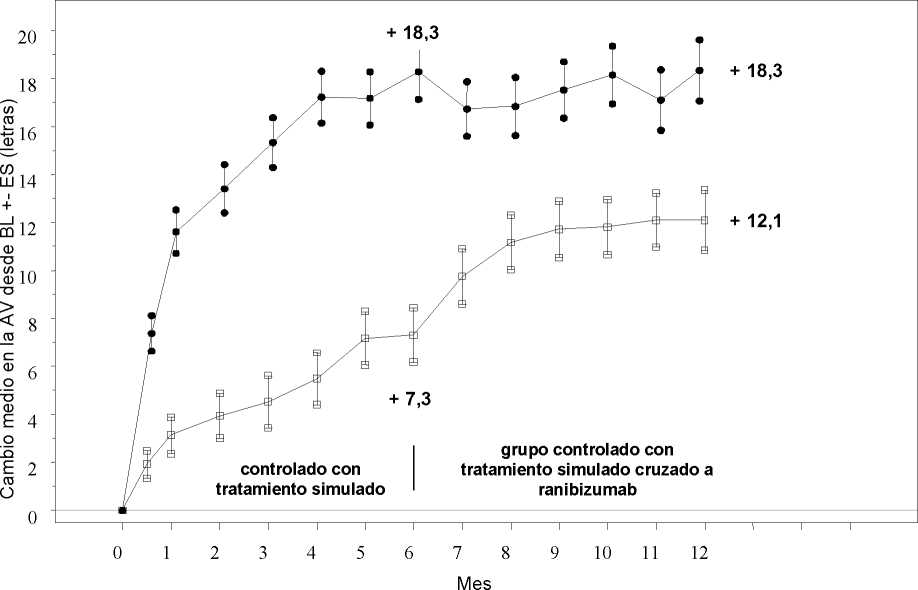
Grupo de tratamiento Tratamiento simulado/Ranibizumab 0,5 mg (n=132)
* * * Ranibizumab 0,5 mg (n=131)
BL=valor inicial; ES=error estándar de la media
Cambio medio en la AVMC desde el inicio en el tiempo hasta el Mes 6 y el Mes 12 (CRUISE)

Grupo de tratamiento Tratamiento simulado/Ranibizumab 0,5 mg (n=130)
* * * Ranibizumab 0,5 mg (n=130)
BL=valor inicial; ES=error estándar de la media
En ambos estudios, la mejora de la visión se acompañó de una reducción continua y significativa del edema macular medido según el grosor central de la retina.
En pacientes con OVCR (CRUISE y ensayo de extensión HORIZON): Los sujetos tratados con tratamiento simulado en los primeros 6 meses que posteriormente recibieron ranibizumab, no alcanzaron ganancias comparables en AV en el Mes 24 (~6 letras) en comparación con los sujetos tratados con ranibizumab desde el inicio del ensayo (~12 letras).
En el tratamiento con ranibizumab se observaron beneficios notificados por el mismo paciente estadísticamente significativos en las subescalas relativas a la actividad de cerca y de lejos respecto al grupo control, medidos según el NEI VFQ-25.
La seguridad y eficacia clínicas de Lucentis a largo plazo (24 meses) en pacientes con alteración visual debida al edema macular secundario a OVR se evaluaron en los ensayos BRIGHTER (BRVO) y CRYSTAL (CRVO). En ambos ensayos, los sujetos recibieron ranibizumab 0,5 mg en un regimen de dosificación PRN que obedece a criterios de estabilización individualizados. BRIGHTER era un ensayo aleatorizado con 3 grupos controlado con tratamiento activo que comparaba ranibizumab 0,5 mg administrado en monoterapia o en combinación con fotocoagulación con láser adjunta frente a fotocoagulación con láser sola. Después de 6 meses, los sujetos en el grupo del láser podían recibir ranibizumab 0,5 mg. CRYSTAL era un ensayo de un grupo único con ranibizumab 0,5 mg en monoterapia.
|
BRIGHTER |
CRYSTAL | |||
|
Lucentis 0,5 mg N=180 |
Lucentis 0,5 mg + Láser N=178 |
Láser* N=90 |
Lucentis 0,5 mg N=356 | |
|
Cambio medio en la AVMC al Mes 6a (letras) (desviación estándar) |
+14,8 (10,7) |
+14,8 (11,13) |
+6,0 (14,27) |
+12,0 (13,95) |
|
Cambio medio en la AVMC al Mes 24b (letras) (desviación estándar) |
+15,5 (13,91) |
+17,3 (12,61) |
+11,6 (16,09) |
+12,1 (18,60) |
|
Ganancia de >15 letras en AVMC al Mes 24 (%) |
52,8 |
59,6 |
43,3 |
49,2 |
|
Número medio de inyecciones (desviación estándar) (Meses 0-23) |
11,4 (5,81) |
11,3 (6,02) |
NA |
13,1 (6,39) |
|
a p<0,0001 para ambas comparaciones en el ensayo BRIGHTER al Mes 6: Lucentis 0,5 mg frente a Láser y Lucentis 0,5 mg + Láser frente a Láser. b p<0,0001 para la hipótesis nula en el ensayo CRYSTAL en que el cambio medio al Mes 24 desde el valor inicial es cero. * A partir del Mes 6 se permitió el tratamiento con ranibizumab 0,5 mg (24 pacientes fueron tratados solo con láser). | ||||
En el ensayo BRIGHTER, ranibizumab 0,5 mg con terapia con láser adjunta demostró no inferioridad comparado con ranibizumab en monoterapia desde el valor inicial hasta el Mes 24 (95% IC -2,8, 1,4).
En ambos ensayos, se observó en el Mes 1 una reducción rápida y estadísticamente significativa del grosor del subcampo central de la retina desde el valor inicial. Este efecto se mantuvo hasta el Mes 24.
El efecto del tratamiento con ranibizumab fue similar de forma independiente de la presencia de isquemia retiniana. En el ensayo BRIGHTER, los pacientes que presentaban isquemia (N=46) o los que no la presentaban (N=133) y que fueron tratados con ranibizumab en monoterapia, tuvieron un cambio medio desde el valor inicial de +15,3 y +15,6 letras respectivamente, en el Mes 24. En el ensayo CRYSTAL, los pacientes que presentaban isquemia (N=53) o los que no la presentaban (N=300) y que fueron tratados con ranibizumab en monoterapia, tuvieron un cambio medio desde el valor inicial de +15,0 y +11,5 letras respectivamente.
En todos los pacientes tratados con ranibizumab 0,5 mg en monoterapia se observó el efecto en términos de mejora visual en ambos ensayos BRIGHTER y CRYSTAL de forma independiente de la duración de la enfermedad. En pacientes con una duración de la enfermedad <3 meses se observó un aumento en la agudeza visual de 13,3 y 10,0 letras en el Mes 1; y 17,7 y 13,2 letras en el Mes 24 en BRIGHTER y CRYSTAL respectivamente. En pacientes con una duración de la enfermedad >12 meses, la ganancia de agudeza visual correspondiente fue de 8,6 y 8,4 letras en los respectivos ensayos. Se debe considerar el inicio del tratamiento en el momento del diagnóstico.
El perfil de seguridad a largo plazo de ranibizumab observado en los ensayos de 24 meses es consistente con el perfil de seguridad de Lucentis conocido.
Tratamiento de la alteración visual debida a la NVC secundaria a MP
La seguridad y eficacia clínicas de Lucentis en pacientes con alteración visual debida a la NVC secundaria a MP se han evaluado de acuerdo a los datos de 12 meses del ensayo pivotal F2301 (RADIANCE) controlado, doble ciego. En este ensayo 277 pacientes fueron aleatorizados en un ratio 2:2:1 para los siguientes grupos:
• Grupo I (ranibizumab 0,5 mg, régimen de tratamiento basado en criterios de “estabilidad” definida como ningún cambio en la AVMC en comparación con las dos evaluaciones mensuales previas).
• Grupo II (ranibizumab 0,5 mg, régimen de tratamiento basado en criterios de “actividad de la enfermedad” definida como alteración de la visión atribuible a fluido intra o subretiniano o exudación activa debido a la lesión de NVC y evaluada mediante OCT y/o AF).
• Grupo III (TFDv - se permitió a los pacientes recibir tratamiento con ranibizumab a partir del Mes 3).
En el Grupo II, que se corresponde con la posología recomendada (ver sección 4.2), el 50,9% de los pacientes requirió 1 ó 2 inyecciones, el 34,5% requirió de 3 a 5 inyecciones y el 14,7% requirió de 6 a 12 inyecciones durante el periodo de 12 meses del ensayo. El 62,9% de los pacientes del Grupo II no requirió inyecciones en el segundo semestre del ensayo.
|
Grupo I |
Grupo II |
Grupo III | |
|
Ranibizumab |
Ranibizumab |
TFDvb | |
|
0,5 mg |
0,5 mg | ||
|
“estabilidad de |
“actividad de la | ||
|
la visión” |
enfermedad” | ||
|
(n=105) |
(n=116) |
(n=55) | |
|
Mes 3 Cambio medio en la AVMC desde el Mes 1 |
+10,5 |
+10,6 |
+2,2 |
|
al Mes 3 comparado con el inicioa (letras) Proporción de pacientes que ganaron: >15 letras, o alcanzaron >84 letras en la AVMC |
38,1% |
43,1% |
14,5% |
|
Mes 12 Número de inyecciones hasta el Mes 12: Media |
4,6 |
3,5 |
N/A |
|
Mediana |
4,0 |
2,5 |
N/A |
|
Cambio medio en la AVMC desde el Mes 1 |
+12,8 |
+12,5 |
N/A |
|
al Mes 12 comparado con el inicio (letras) Proporción de pacientes que ganaron: >15 letras, o alcanzaron >84 letras en la AVMC |
53,3% |
51,7% |
N/A |
a p<0,00001 comparación con el control de TFDv
b Control comparativo hasta el Mes 3. A los pacientes aleatorizados a TFDv se les permitió recibir tratamiento con ranibizumab a partir del Mes 3 (en el Grupo III, 38 pacientes recibieron ranibizumab a partir del Mes 3)
Figura 5 Cambio medio en la AVMC desde el inicio en el tiempo hasta el Mes 12 (RADIANCE)
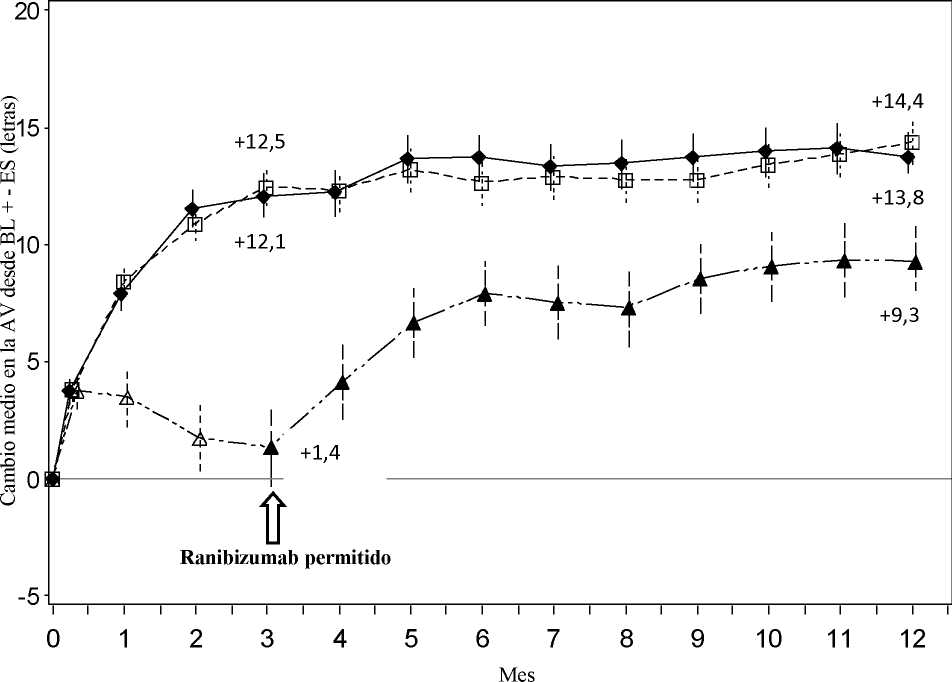
Ranibizumab 0,5 mg Grupo I por estabilización (n=105)
Ranibizumab 0,5 mg Grupo II
por actividad de la enfermedad (n=116)
Ranibizumab 0,5 mg/TFD con verteporfina Grupo III desde el Mes 3 en adelante (n=55)
A-a-a TFD con verteporfina Grupo III (n=55)
La mejora de la visión se acompañó de una reducción del grosor central de la retina.
Los beneficios comunicados por el paciente se observaron más en los grupos de tratamiento con ranibizumab que en la TFDv (valor p<0,05) en términos de mejora en la puntuación compuesta y varias subescalas (visión general, actividades de cerca, salud mental y dependencia) del NEI VFQ-25.
Población pediátrica
La seguridad y eficacia de ranibizumab aún no han sido estudiadas en pacientes pediátricos.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Lucentis en los diferentes grupos de la población pediátrica en la DMAE neovascular, la alteración visual debida al EMD, la alteración visual debida al edema macular secundario a la OVR y la alteración visual debida a la NVC secundaria a MP (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
Tras la administración intravítrea mensual de Lucentis a pacientes con DMAE neovascular, las concentraciones séricas de ranibizumab fueron en general bajas, con niveles máximos (Cmax) por debajo de la concentración de ranibizumab necesaria para inhibir la actividad biológica del VEGF en un 50% (11-27 ng/ml, valorado en un ensayo de proliferación celular in vitro). La Cmax fue proporcional a la dosis, en el rango de dosis de 0,05 a 1,0 mg/ojo. Las concentraciones séricas en un número limitado de pacientes con EMD indican que no puede excluirse una exposición sistémica ligeramente superior en comparación con la observada en pacientes con DMAE neovascular. Las concentraciones séricas de ranibizumab en pacientes con OVR fueron similares o ligeramente superiores en comparación con las observadas en pacientes con DMAE neovascular.
En base al análisis farmacocinético poblacional y a la desaparición sérica de ranibizumab en pacientes con DMAE neovascular tratados con la dosis de 0,5 mg, el promedio de la vida media de eliminación vítrea de ranibizumab es de 9 días aproximadamente. Tras la administración intravítrea mensual de Lucentis 0,5 mg/ojo, se prevé que la Cmax de ranibizumab sérica alcanzada aproximadamente 1 día después de la administración, varíe en general en un rango de entre 0,79 y 2,90 ng/ml, y que la Cmin varíe en general en un rango de entre 0,07 y 0,49 ng/ml. Se prevé que las concentraciones séricas de ranibizumab sean aproximadamente 90.000 veces inferiores a las concentraciones vítreas de ranibizumab.
Pacientes con insuficiencia renal: No se han realizado estudios formales para investigar la farmacocinética de Lucentis en pacientes con insuficiencia renal. En un análisis farmacocinético poblacional de pacientes con DMAE neovascular, el 68% (136 de 200) de los pacientes tenían insuficiencia renal (leve en un 46,5% [50-80 ml/min], moderada en un 20% [30-50 ml/min] y grave en un 1,5% [<30 ml/min]). En el caso de pacientes con OVR, el 48,2% (253 de 525) tenían insuficiencia renal (leve en un 36,4%, moderada en un 9,5% y grave en un 2,3%). El aclaramiento sistémico fue ligeramente inferior, pero esto no fue clínicamente significativo.
Insuficiencia hepática: No se han realizado estudios formales para investigar la farmacocinética de Lucentis en pacientes con insuficiencia hepática.
5.3 Datos preclínicos sobre seguridad
La administración intravítrea bilateral de ranibizumab a macacos, a dosis de entre 0,25 mg/ojo y
2,0 mg/ojo una vez cada 2 semanas durante 26 semanas, ocasionó efectos oculares dosis-dependientes.
Intraocularmente, se observaron incrementos dosis-dependientes de exudados y de células en la cámara anterior, con un máximo a los 2 días después de la inyección. Por lo general, la gravedad de la respuesta inflamatoria disminuyó con las inyecciones posteriores o durante el periodo de recuperación. En el segmento posterior, hubo infiltración de células en la cámara vítrea y partículas flotantes, que tendieron también a ser dosis-dependientes y que, en general, persistieron hasta el final del periodo de tratamiento. En el ensayo a 26 semanas, la gravedad de la inflamación vítrea aumentó con el número de inyecciones. Sin embargo, se observó evidencia de reversibilidad tras el periodo de recuperación. La naturaleza y cronología de la inflamación en el segmento posterior sugiere una respuesta inmunitaria mediada por anticuerpos, que puede ser clínicamente irrelevante. En algunos animales se observó la formación de cataratas tras un periodo relativamente largo de inflamación intensa, lo cual sugiere que las alteraciones en el cristalino fueron secundarias a la inflamación grave. Tras las inyecciones intravítreas se observó un aumentó transitorio de la presión intraocular independiente de la dosis.
Los cambios oculares microscópicos fueron relacionados con la inflamación y no eran indicativos de procesos degenerativos. Se observaron cambios inflamatorios granulomatosos en el disco óptico de algunos ojos. Estas alteraciones en el segmento posterior disminuyeron, y en algunos casos se resolvieron, durante el periodo de recuperación.
Tras la administración intravítrea, no se detectaron signos de toxicidad sistémica. En un subgrupo de animales tratados se detectaron anticuerpos séricos y vítreos contra ranibizumab.
No se dispone de datos de carcinogenicidad ni de mutagenicidad.
En hembras de mono preñadas, el tratamiento con ranibizumab intravítreo resultando en exposiciones sistémicas máximas 0,9-7 veces la peor exposición clínica, no provocó toxicidad en el desarrollo ni teratogenicidad y no tuvo ningún efecto sobre el peso o la estructura de la placenta, aunque en base a su efecto farmacológico, ranibizumab debe considerarse potencialmente teratogénico y embrio/fetotóxico.
La ausencia de efectos mediados por ranibizumab sobre el desarrollo embrio-fetal está plausiblemente relacionado principalmente con la incapacidad del fragmento Fab de atravesar la placenta. Sin embargo, se describió un caso de niveles séricos de ranibizumab maternos elevados y presencia de ranibizumab en el suero fetal lo que sugiere que el anticuerpo contra ranibizumab actuó como proteína transportadora (conteniendo la región Fc) para ranibizumab, disminuyendo de ese modo su aclaramiento sérico materno y permitiendo su paso a la placenta. Dado que las investigaciones en el desarrollo embrio-fetal se llevaron a cabo en animales preñados sanos y las enfermedades (tales como la diabetes) pueden modificar la permeabilidad de la placenta para el fragmento Fab, el estudio debe interpretarse con cautela.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
a,a-trehalosa dihidrato
Hidrocloruro de histidina monohidrato
Histidina
Polisorbato 20
Agua para inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
3 años
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar la jeringa precargada en su bandeja sellada en la caja para protegerlo de la luz.
Antes de usar, la bandeja sin abrir se puede conservar a temperatura ambiente (25°C) durante un máximo de 24 horas.
6.5 Naturaleza y contenido del envase
0,165 ml de solución estéril en una jeringa precargada (vidrio tipo I) con un tapón con émbolo de goma de bromobutilo y una cápsula de cierre de la jeringa que consiste en un cierre rígido blanco con precinto de seguridad con un tapón de goma de bromobutilo gris y que incluye un adaptador Luer Lock. La jeringa precargada tiene un émbolo y una aleta de sujeción, y se envasa en una bandeja contenedora sellada.
Tamaño de envase: una jeringa precargada.
6.6 Precauciones especiales de eliminación y otras manipulaciones
La jeringa precargada es para un solo uso. La jeringa precargada es estéril. No use el producto si el envase está deteriorado. La esterilidad de la jeringa precargada sólo se puede garantizar si la bandeja se mantiene sellada. No use la jeringa precargada si la solución ha cambiado de color, está turbia o contiene partículas.
La jeringa precargada contiene más cantidad que la dosis recomendada de 0,5 mg. El volumen extraíble de la jeringa precargada (0,1 ml) no se administrará en su totalidad. El exceso de volumen se debe expulsar antes de la inyección. Si se inyecta todo el volumen de la jeringa precargada puede dar lugar a una sobredosis. Para expulsar las burbujas de aire y el exceso de medicamento, presione lentamente el émbolo hasta que el borde inferior de la cúpula que forma el extremo del tapón de goma quede alineado con la línea negra de dosificación de la jeringa (equivalente a 0,05 ml, es decir, 0,5 mg de ranibizumab).
Para la inyección intravítrea, se debe usar una aguja para inyección estéril 30G x A".
Para la preparación de Lucentis para administración intravítrea, siga las instrucciones de uso:
Lea todas las instrucciones detenidamente antes de usar la jeringa precargada.
La jeringa precargada es para un solo uso. La jeringa precargada es estéril. No usar el producto si el envase está deteriorado. La apertura de la bandeja sellada y los siguientes pasos se deben realizar bajo condiciones asépticas.
Nota: La dosis a administrar se debe ajustar a 0,05 ml._
Cápsula de cierre de la jeringa
Marca de dosis de 0,05 ml
Aleta de sujeción
I
Luer lock Tapón de goma
Émbolo
1. Asegurarse que el envase contiene:
• una jeringa precargada estéril en una bandeja sellada.
2. Quitar la cubierta de la bandeja contenedora de la jeringa y, usando una
técnica aséptica, extraer la jeringa cuidadosamente._
|
Comprobar la |
3. |
|
jeringa |
• |
|
• | |
|
• | |
|
4. | |
|
Quitar la cápsula de cierre de la |
5. |
|
jeringa |
6. |
Comprobar que:
la cápsula de cierre de la jeringa no esté separado del Luer lock. la jeringa no esté deteriorada. la solución tenga un aspecto transparente, de incolora a amarillo pálido y no contenga ninguna partícula.
Si alguno de los puntos anteriores no es cierto, desechar la jeringa precargada y usar una nueva.
Tirar y desprender la cápsula de cierre de la jeringa (no lo gire ni lo retuerza) (ver Figura 2).
Desechar la cápsula de cierre de la jeringa (ver Figura 3).
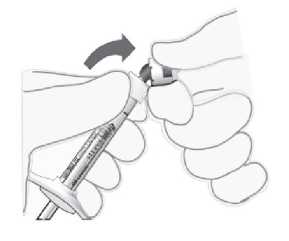
Figura 2
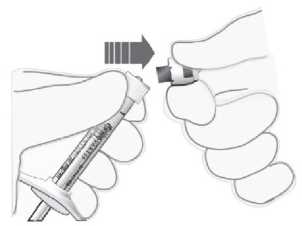
Figura 3
Conectar la aguja
7. Conectar con firmeza a la jeringa una aguja para inyección estéril 30G x A", enroscando bien la aguja en el Luer lock (ver Figura 4).
8. Quitar la cápsula de cierre de la aguja cuidadosamente, tirando directamente de él hacia fuera (ver Figura 5).
Nota: No secar la aguja en ningún momento.

Figura 4 Figura 5
|
Extraer las |
9. |
|
burbujas de |
c |
|
aire |
10. S s |
Mantener la jeringa en posición vertical con la aguja dirigida hacia arriba.
Si hay alguna burbuja de aire, golpear suavemente la jeringa con el dedo hasta que las burbujas asciendan a su parte superior (ver Figura 6).

Figura 6
Ajustar la dosis
11. Mantener la jeringa a la altura de los ojos y presionar cuidadosamente el émbolo hasta que el borde inferior de la cúpula que forma el extremo del tapón de goma quede alineado con la marca de dosis (ver Figura 7). Con esto se expulsará el aire y el exceso de solución y se ajustará la dosis a 0,05 ml.
Nota: El émbolo no está unido al tapón de goma - esto es para evitar la entrada de aire en la jeringa.

Figura 7
Inyección
El procedimiento de inyección debe llevarse a cabo bajo condiciones asépticas.
12. La aguja para inyección se debe introducir 3,5-4,0 mm por detrás del limbo en la cavidad vítrea, evitando el meridiano horizontal y en dirección al centro del globo.
13. Inyectar lentamente hasta que el tapón de goma llegue a la parte inferior de la jeringa, con el fin de administrar el volumen de 0,05 ml.
14. Las inyecciones siguientes se deben aplicar cada vez en un punto escleral distinto.
15. Tras la inyección, no tapar la aguja con la cápsula de cierre ni separarla de la
jeringa. Eliminar la jeringa usada junto con la aguja en un contenedor para objetos punzantes o eliminar de acuerdo con la normativa local._
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/374/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 22 Enero 2007 Fecha de la última renovación: 24 Enero 2012
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
1. NOMBRE DEL MEDICAMENTO
Lucentis 10 mg/ml solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un ml contiene 10 mg de ranibizumab*. Cada vial contiene 2,3 mg de ranibizumab en 0,23 ml de solución.
*Ranibizumab es un fragmento de anticuerpo monoclonal humanizado producido en células de Escherichia coli mediante tecnología de ADN recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución acuosa transparente, de incolora a amarillo pálido.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lucentis está indicado en adultos para:
• El tratamiento de la degeneración macular asociada a la edad (DMAE) neovascular (exudativa)
• El tratamiento de la alteración visual debida al edema macular diabético (EMD)
• El tratamiento de la alteración visual debida al edema macular secundario a la oclusión de la
vena retiniana (OVR) (oclusión de la rama venosa retiniana u oclusión de la vena central retiniana)
• El tratamiento de la alteración visual debida a la neovascularización coroidea (NVC) secundaria a la miopía patológica (MP)
4.2 Posología y forma de administración
Lucentis debe ser administrado por un oftalmólogo que tenga experiencia en la administración de inyecciones intravítreas.
La dosis recomendada de Lucentis es 0,5 mg administrada en forma de inyección intravítrea única. Esto corresponde a un volumen de inyección de 0,05 ml. El intervalo entre dos dosis inyectadas en el mismo ojo debe ser como mínimo de cuatro semanas.
El tratamiento se inicia con una inyección al mes hasta alcanzar la agudeza visual máxima y/o no haya signos de actividad de la enfermedad, es decir ningún cambio en la agudeza visual ni en otros signos y síntomas de la enfermedad bajo tratamiento continuado. En pacientes con DMAE exudativa, EMD y OVR inicialmente pueden ser necesarias tres o más inyecciones consecutivas administradas mensualmente.
A partir de ese momento, los intervalos de monitorización y tratamiento se deben determinar según criterio médico y en base a la actividad de la enfermedad, valorada mediante la agudeza visual y/o parámetros anatómicos.
Se debe interrumpir el tratamiento con Lucentis si bajo criterio del médico, los parámetros visuales y anatómicos indican que el paciente no se está beneficiando del tratamiento continuado.
La monitorización para determinar la actividad de la enfermedad puede incluir examen clínico, control funcional o técnicas de imagen (p. ej. tomografía de coherencia óptica o angiografía con fluoresceína).
Si se está tratando a los pacientes de acuerdo a un régimen de tratar y extender, una vez se ha alcanzado la agudeza visual máxima y/o no hay signos de actividad de la enfermedad, los intervalos de tratamiento se pueden espaciar de forma gradual hasta que vuelvan a aparecer signos de actividad de la enfermedad o alteración visual. En el caso de la DMAE exudativa el intervalo de tratamiento no debe espaciarse en más de dos semanas cada vez y en el caso del EMD se puede espaciar hasta un mes cada vez. Para la OVR, los intervalos de tratamiento también pueden espaciarse de forma gradual, sin embargo los datos que hay no son suficientes para determinar la duración de estos intervalos. Si vuelve a aparecer actividad de la enfermedad, se debe acortar el intervalo de tratamiento de manera consecuente.
En el tratamiento de la alteración visual debida a la NVC secundaria a MP, muchos pacientes pueden necesitar únicamente una o dos inyecciones durante el primer año, mientras que algunos pacientes pueden necesitar tratamiento con mayor frecuencia (ver sección 5.1).
Lucentis y fotocoagulación con láser en EMD y en edema macular secundario a oclusión de la rama venosa retiniana (ORVR)
Existe alguna experiencia con Lucentis administrado concomitantemente con fotocoagulación con láser (ver sección 5.1). Cuando se administren en el mismo día, Lucentis se debe administrar como mínimo 30 minutos después de la fotocoagulación con láser. Lucentis puede administrarse en pacientes que han recibido fotocoagulación con láser previamente.
Lucentis y la terapia fotodinámica con Visudyne en la NVC secundaria a MP No hay experiencia en la administración concomitante de Lucentis y Visudyne.
Poblaciones especiales
Insuficiencia hepática
Lucentis no ha sido estudiado en pacientes con insuficiencia hepática. Sin embargo, no es necesaria ninguna consideración especial en esta población.
Insuficiencia renal
No es necesario ningún ajuste de la dosis en pacientes con insuficiencia renal (ver sección 5.2).
Pacientes de edad avanzada
No se requiere ningún ajuste de la dosis en pacientes de edad avanzada. Existe experiencia limitada en pacientes con EMD mayores de 75 años.
Población pediátrica
No se ha establecido la seguridad y eficacia de Lucentis en niños y adolescentes menores de 18 años de edad. No se dispone de datos.
Forma de administración
Vial para un solo uso. Únicamente para vía intravítrea.
Antes de la administración de Lucentis se debe comprobar visualmente la ausencia de partículas y decoloración.
El procedimiento de inyección deberá llevarse a cabo bajo condiciones asépticas, que incluyen el lavado quirúrgico de las manos, el uso de guantes estériles, un campo estéril, un blefarostato estéril para los párpados (o equivalente) y la disponibilidad de una paracentesis estéril (en caso necesario). Antes de realizar el procedimiento de inyección intravítrea, se deberá evaluar detalladamente la historia clínica del paciente en cuanto a reacciones de hipersensibilidad (ver sección 4.4). Antes de la inyección se debe administrar una anestesia adecuada y un microbicida tópico de amplio espectro para desinfectar la piel de la zona periocular, párpado y superficie ocular, de acuerdo con la práctica local.
Para la información relativa a la preparación de Lucentis, ver sección 6.6.
La aguja para inyección se deberá introducir 3,5-4,0 mm por detrás del limbo en la cavidad vítrea, evitando el meridiano horizontal y en dirección al centro del globo. Seguidamente debe liberarse el volumen de inyección de 0,05 ml; las inyecciones siguientes deberán aplicarse cada vez en un punto escleral distinto.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Pacientes con infecciones oculares o perioculares o con sospecha de éstas.
Pacientes con inflamación intraocular grave.
4.4 Advertencias y precauciones especiales de empleo Reacciones relacionadas con la inyección intravítrea
Las inyecciones intravítreas, incluidas las de Lucentis, se han asociado a endoftalmitis, inflamación intraocular, desprendimiento de retina regmatógeno, desgarro retiniano y catarata traumática iatrogénica (ver sección 4.8). Siempre que se administre Lucentis se deben emplear técnicas de inyección asépticas adecuadas. Además, deberá realizarse un seguimiento de los pacientes durante la semana posterior a la inyección para poder administrar tratamiento temprano en caso de infección. Se deberá instruir a los pacientes sobre la necesidad de comunicar inmediatamente cualquier síntoma que sugiera endoftalmitis o cualquiera de los acontecimientos mencionados anteriormente.
Aumento de la presión intraocular
Se han observado aumentos transitorios de la presión intraocular (PIO) en los 60 minutos siguientes a la inyección de Lucentis. También se han identificado aumentos sostenidos de la PIO (ver sección 4.8). Tanto la presión intraocular como la perfusión de la cabeza del nervio óptico, se deben monitorizar y tratar adecuadamente.
Tratamiento bilateral
Los escasos datos existentes sobre el tratamiento bilateral con Lucentis (incluyendo la administración en el mismo día) no sugieren un riesgo incrementado de efectos adversos sistémicos en comparación con el tratamiento unilateral.
Inmunogenicidad
Hay un potencial de inmunogenicidad con Lucentis. Dado que en sujetos con EMD existe un potencial para una exposición sistémica incrementada, no puede excluirse un mayor riesgo para desarrollar hipersensibilidad en esta población de pacientes. También se deberá instruir a los pacientes sobre la necesidad de notificar si la inflamación intraocular incrementa en su gravedad, lo que puede ser un signo clínico atribuible a la formación de anticuerpos intraoculares.
Uso concomitante con otros anti-VEGF (factor de crecimiento endotelial vascular)
Lucentis no se deberá administrar de forma concurrente con otros medicamentos anti-VEGF (sistémicos u oculares).
Aplazamiento del tratamiento con Lucentis
La administración se deberá aplazar y el tratamiento no deberá reanudarse antes del siguiente tratamiento programado en caso de:
• una disminución en la agudeza visual mejor corregida (AVMC) de >30 letras comparado con la última evaluación de la agudeza visual;
• una presión intraocular de >30 mmHg;
• una rotura retiniana;
• una hemorragia subretiniana que afecte al centro de la fóvea, o, si el tamaño de la hemorragia es >50% del área total de la lesión;
• cirugía intraocular realizada en los 28 días previos o prevista durante los 28 días posteriores. Desgarro del epitelio pigmentario de la retina
Los factores de riesgo asociados con el desarrollo de un desgarro del epitelio pigmentario de la retina tras la terapia con anti-VEGF para la DMAE exudativa, incluyen un desprendimiento del epitelio pigmentario de la retina extenso y/o elevado. Cuando se inicie la terapia con Lucentis se debe tener precaución en pacientes con estos factores de riesgo de desarrollar desgarros del epitelio pigmentario de la retina.
Desprendimiento de retina regmatógeno o agujeros maculares
El tratamiento se debe interrumpir en sujetos con desprendimiento de retina regmatógeno o agujeros maculares en estadíos 3 ó 4.
Poblaciones con datos limitados
Sólo existe experiencia limitada en el tratamiento de sujetos con EMD debido a diabetes tipo I. Lucentis no ha sido estudiado en pacientes que hayan recibido previamente inyecciones intravítreas, en pacientes con infecciones sistémicas activas, retinopatía diabética proliferativa, ni en pacientes con enfermedades oculares simultáneas tales como desprendimiento de retina o agujero macular.
Tampoco existe experiencia en el tratamiento con Lucentis en pacientes diabéticos con un HbA1c por encima del 12% e hipertensión no controlada. El médico debe tener en cuenta esta falta de información al tratar a tales pacientes.
No hay datos suficientes que permitan establecer una conclusión acerca del efecto de Lucentis en pacientes con OVR que presentan pérdida irreversible de la función visual isquémica.
En pacientes con MP, hay datos limitados del efecto de Lucentis en pacientes que han sido sometidos previamente a un tratamiento de terapia fotodinámica con verteporfina (TFDv) sin exito. Además, mientras que en sujetos con lesiones subfoveales y yuxtafoveales se observó un efecto consistente, no hay datos suficientes para establecer conclusiones sobre el efecto de Lucentis en sujetos con MP y lesiones extrafoveales.
Efectos sistémicos tras el uso intravítreo
Se han notificado acontecimientos adversos sistémicos, incluyendo hemorragias no oculares y acontecimientos tromboembólicos arteriales tras la inyección intravítrea de inhibidores del VEGF.
Existen datos limitados sobre seguridad en el tratamiento de pacientes con EMD, edema macular debido a OVR y NVC secundaria a MP que tengan antecedentes de accidente cerebrovascular o ataques isquémicos transitorios. Se debe tener precaución cuando se traten tales pacientes (ver sección 4.8).
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones formales.
Para el uso conjunto de terapia fotodinámica (TFD) con verteporfina y Lucentis en la DMAE exudativa y en la MP, ver sección 5.1.
Para el uso conjunto de fotocoagulación con láser y Lucentis en EMD y ORVR, ver secciones 4.2 y 5.1.
En ensayos clínicos para el tratamiento de la alteración visual debida al EMD, el tratamiento concomitante con tiazolidinedionas en pacientes tratados con Lucentis, no afectó el resultado en relación a la agudeza visual o al grosor del subcampo central de la retina (GSCR).
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil/anticoncepción en mujeres
Las mujeres en edad fértil deben utilizar métodos anticonceptivos efectivos durante el tratamiento. Embarazo
No se dispone de datos clínicos de exposición a ranibizumab en embarazos. Los estudios en monos cinomolgos no sugieren efectos perjudiciales directos ni indirectos en términos de embarazo o desarrollo embrional/fetal (ver sección 5.3). La exposición sistémica a ranibizumab tras la administración ocular es baja, pero debido a su mecanismo de acción, ranibizumab debe considerarse como potencialmente teratogénico y embrio-/fetotóxico. Por ello ranibizumab no se deberá usar durante el embarazo salvo que el beneficio esperado supere el riesgo potencial para el feto. Para mujeres que deseen quedarse embarazadas y hayan sido tratadas con ranibizumab, se recomienda esperar como mínimo 3 meses tras la última dosis de ranibizumab antes de concebir un hijo.
Lactancia
Se desconoce si Lucentis se excreta en la leche materna. No se recomienda la lactancia durante el uso de Lucentis.
Fertilidad
No hay datos disponibles sobre fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
El procedimiento de tratamiento con Lucentis puede producir alteraciones visuales transitorias, que pueden afectar la capacidad para conducir o utilizar máquinas (ver sección 4.8). Los pacientes que experimenten estos signos no deben conducir ni utilizar máquinas hasta que dichas alteraciones visuales transitorias remitan.
4.8 Reacciones adversas
Resumen del perfil de seguridad
La mayoría de las reacciones adversas notificadas tras la administración de Lucentis están relacionadas con el procedimiento de inyección intravítrea.
Las reacciones adversas oculares tras la inyección de Lucentis notificadas más frecuentemente son: dolor ocular, hiperemia ocular, aumento de la presión intraocular, vitritis, desprendimiento de vítreo, hemorragia retiniana, alteración visual, partículas flotantes en el vítreo, hemorragia conjuntival, irritación ocular, sensación de cuerpo extraño en los ojos, aumento del lagrimeo, blefaritis, ojo seco, y prurito ocular.
Las reacciones adversas no oculares notificadas más frecuentemente son cefalea, nasofaringitis y artralgia.
Las reacciones adversas notificadas con menor frecuencia, pero de mayor gravedad, incluyen endoftalmitis, ceguera, desprendimiento de retina, desgarro retiniano y catarata traumática iatrogénica (ver sección 4.4).
Se debe informar a los pacientes de los síntomas de estas reacciones adversas potenciales e instruirlos para que informen a su médico en caso de aparición de signos tales como dolor ocular o aumento del malestar en el ojo, empeoramiento del enrojecimiento del ojo, visión borrosa o disminución de la visión, aumento del número de pequeñas manchas en su visión o aumento de la sensibilidad a la luz.
En la siguiente tabla se resumen las reacciones adversas ocurridas tras la administración de Lucentis en los ensayos clínicos.
Tabla de reacciones adversas#
Las reacciones adversas se listan con un sistema de clasificación de órganos y frecuencia usando el siguiente criterio: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000), muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo frecuencia.
Infecciones e infestaciones
Muy frecuentes Nasofaringitis
Frecuentes Infección de las vías urinarias*
Trastornos de la sangre y del sistema linfático
Frecuentes Anemia
Trastornos del sistema inmunológico
Frecuentes Hipersensibilidad
Trastornos psiquiátricos
Frecuentes Ansiedad
Trastornos del sistema nervioso
Muy frecuentes Cefalea
Trastornos oculares
Muy frecuentes Vitritis, desprendimiento de vítreo, hemorragia retiniana,
alteración visual, dolor ocular, partículas flotantes en el vítreo, hemorragia conjuntival, irritación ocular, sensación de cuerpo extraño en los ojos, aumento del lagrimeo, blefaritis, ojo seco, hiperemia ocular, prurito ocular.
Frecuentes Degeneración retiniana, trastorno retiniano, desprendimiento de
retina, desgarro retiniano, desprendimiento del epitelio pigmentario retiniano, desgarro del epitelio pigmentario retiniano, agudeza visual reducida, hemorragia vítrea, trastorno del cuerpo vítreo, uveítis, iritis, iridociclitis, catarata, catarata subcapsular, opacificación de la cápsula posterior, queratitis punctata, abrasión corneal, células flotantes en la cámara anterior, visión borrosa, hemorragia en el lugar de inyección, hemorragia ocular, conjuntivitis, conjuntivitis alérgica, secreción ocular, fotopsia, fotofobia, molestia ocular, edema palpebral, dolor palpebral, hiperemia conjuntival.
Poco frecuentes Ceguera, endoftalmitis, hipopion, hipema, queratopatía, adhesión
del iris, depósitos corneales, edema corneal, estrías corneales, dolor en el lugar de inyección, irritación en el lugar de inyección, sensación anormal en el ojo, irritación palpebral.
Trastornos respiratorios, torácicos y mediastínicos
Frecuentes Tos
Trastornos gastrointestinales
Frecuentes Náuseas
Trastornos de la piel y del tejido subcutáneo
Frecuentes Reacciones alérgicas (erupción, urticaria, prurito, eritema)
Trastornos musculoesqueléticos y del tejido conjuntivo Muy frecuentes Artralgia
Exploraciones complementarias
Muy frecuentes Aumento de la presión intraocular
#-Las reacciones adversas se definieron como acontecimientos adversos (en al menos 0,5 puntos porcentuales de pacientes) que ocurrieron con una frecuencia superior (como mínimo 2 puntos porcentuales) en los pacientes que recibieron el tratamiento con Lucentis 0,5 mg respecto a los que recibieron el tratamiento control (tratamiento simulado (sham) o TFD con verteporfina).
* observado sólo en población con EMD
Reacciones adversas de clase terapéutica
En los ensayos fase III en DMAE exudativa, la frecuencia global de hemorragias no oculares, un efecto adverso potencialmente relacionado con la inhibición sistémica del VEGF (factor de crecimiento endotelial vascular), fue ligeramente superior en los pacientes tratados con ranibizumab. Sin embargo, no hubo un patrón consistente entre las distintas hemorragias. Tras el uso intravítreo de inhibidores del VEGF existe un riesgo teórico de acontecimientos tromboembólicos arteriales, incluyendo accidente cerebrovascular e infarto de miocardio. En los ensayos clínicos con Lucentis se observó una incidencia baja de acontecimientos tromboembólicos arteriales en pacientes con DMAE, EMD, OVR y MP y no hubo ninguna diferencia destacable entre los grupos tratados con ranibizumab comparado con el control.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Se han notificado casos de sobredosis accidental procedentes de los ensayos clínicos en DMAE exudativa y de los datos post-comercialización. Las reacciones adversas que se asociaron a estos casos notificados fueron aumento de la presión intraocular, ceguera transitoria, agudeza visual reducida, edema corneal, dolor corneal y dolor ocular. En caso de sobredosis, se deberá realizar un seguimiento y tratamiento de la presión intraocular, si el médico lo considera necesario.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Oftalmológicos, agentes antineovascularización, código ATC: S01LA04
Ranibizumab es un fragmento de anticuerpo monoclonal recombinante humanizado dirigido contra el factor de crecimiento endotelial vascular A (VEGF-A) humano. Se une con alta afinidad a las isoformas del VEGF-A (p. ej. VEGF110, VEGF121 y VEGF165), impidiendo, por tanto, la unión del VEGF-A a sus receptores VEGFR-1 y VEGFR-2. La unión del VEGF-A a sus receptores conduce a la proliferación de las células endoteliales y la neovascularización, así como a la exudación vascular, todo lo cual se cree que contribuye a la progresión de la forma neovascular de la degeneración macular asociada a la edad, la miopía patológica o a la alteración visual causada por el edema macular diabético o por el edema macular secundario a OVR.
Tratamiento de la DMAE exudativa
En la DMAE exudativa, la eficacia y seguridad clínicas de Lucentis se han evaluado en tres ensayos aleatorizados, doble ciego, controlados con tratamiento simulado (sham) o con tratamiento activo de 24 meses de duración, en pacientes con DMAE neovascular. En estos ensayos fueron reclutados un total de 1.323 pacientes (879 con tratamiento activo y 444 con control).
En el ensayo FVF2598g (MARINA), se aleatorizaron 716 pacientes con lesiones mínimamente clásicas u ocultas sin componente clásico en un ratio 1:1:1 para recibir inyecciones de Lucentis 0,3 mg, Lucentis 0,5 mg o tratamiento simulado una vez al mes.
En el ensayo FVF2587g (ANCHOR), se aleatorizaron 423 pacientes con lesiones de NVC predominantemente clásicas en un ratio 1:1:1 para recibir Lucentis 0,3 mg una vez al mes, Lucentis 0,5 mg una vez al mes o TFD con verteporfina (al inicio y posteriormente cada 3 meses si la angiografía fluoresceínica mostraba persistencia o recurrencia de la exudación vascular).
En la Tabla 1 y en la Figura 1 se resumen los resultados clave.
Tabla 1 Resultados al Mes 12 y al Mes 24 en el ensayo FVF2598g (MARINA) y FVF2587g (ANCHOR)
|
FVF2598g (MARINA) |
FVF2587g (ANCHOR) | ||||
|
Medida del resultado |
Mes |
Tratamient o simulado o sham (n=238) |
Lucentis 0,5 mg (n=240) |
TFD con verteporfina (n=143) |
Lucentis 0,5 mg (n=140) |
|
Pérdida de <15 letras de agudeza visual (%)a (mantenimiento de la visión, variable primaria) |
Mes 12 |
62% |
95% |
64% |
96% |
|
Mes 24 |
53% |
90% |
66% |
90% | |
|
Ganancia de >15 letras de agudeza visual (%)a |
Mes 12 |
5% |
34% |
6% |
40% |
|
Mes 24 |
4% |
33% |
6% |
41% | |
|
Cambio medio en la agudeza visual (letras) (desviación estándar)a |
Mes 12 |
-10,5 (16,6) |
+7,2 (14,4) |
-9,5 (16,4) |
+11,3 (14,6) |
|
Mes 24 |
-14,9 (18,7) |
+6,6 (16,5) |
-9,8 (17,6) |
+10,7 (16,5) | |
a p<0,01
Figura 1 Cambio medio en la agudeza visual desde el inicio hasta el Mes 24 en el ensayo FVF2598g (MARINA) y el ensayo FVF2587g (ANCHOR)
C/5
>
co
N
CD
T3
CD
O
T3
CD
E
O
!q
E
co
O
Ensayo FVF2598g (MARINA)
+6,6 ^
15
10
5
0
— <D C
<L>
O
<L>
E
O
E
Cü
O
~-5
-10
-15

V +21,5
0 2 4 6 8 10 12 14 16 18 20 22 24
Mes
15
10
5
0
co "S
"-5 ■
-10 ■ -15 ■

Ensayo FVF2587g (ANCHOR)
frf-j +10,7
r +20,5
-9'8
0 2 4 6 8 10 12 14 16 18 20 22 24
Mes
MARINA
LUCENTIS 0,5 mg (n=240)
& Tratamiento simulado (n=238)
ANCHOR
LUCENTIS 0,5 mg (n=140)
• TFD con verteporfina (n=143)
Los resultados de ambos ensayos indicaron que el tratamiento continuado con ranibizumab también puede ser beneficioso en pacientes que perdieron >15 letras de agudeza visual mejor corregida (AVMC) en el primer año de tratamiento.
En ambos ensayos MARINA y ANCHOR se observaron beneficios en la función visual notificados por el mismo paciente estadísticamente significativos en el tratamiento con ranibizumab respecto al grupo control, medidos según el NEI VFQ-25.
En el ensayo FVF3192g (PIER), se aleatorizaron 184 pacientes que presentaban todas las formas de DMAE neovascular en un ratio 1:1:1 para recibir Lucentis 0,3 mg, Lucentis 0,5 mg o inyecciones simuladas (sham) una vez al mes en 3 dosis consecutivas, seguido de la administración de una dosis una vez cada 3 meses. A partir del Mes 14 del ensayo, se permitió a los pacientes tratados con inyecciones simuladas (sham) recibir ranibizumab, y a partir del Mes 19 fueron posibles tratamientos más frecuentes. Los pacientes tratados con Lucentis en el ensayo PIER recibieron un promedio de 10 tratamientos.
En general, tras un incremento inicial en la agudeza visual (después de la dosificación mensual), la agudeza visual de los pacientes disminuyó con la dosis trimestral, volviendo al valor basal en el Mes 12 y este efecto se mantuvo al Mes 24 en la mayoría de los pacientes tratados con ranibizumab (82%). Datos limitados procedentes de sujetos que recibieron tratamiento simulado y que posteriormente fueron tratados con ranibizumab, sugirieron que el inicio temprano del tratamiento puede asociarse a una mejor conservación de la agudeza visual.
Los datos de dos ensayos (MONT BLANC, BPD952A2308 y DENALI, BPD952A2309) realizados tras la autorización de comercialización, confirmaron la eficacia de Lucentis pero no demostraron un efecto adicional en la administración combinada de verteporfina (Visudyne TFD) y Lucentis comparado con Lucentis en monoterapia.
Tratamiento de la alteración visual debida a EMD
La eficacia y seguridad de Lucentis se han evaluado en tres ensayos aleatorizados, controlados de al menos 12 meses de duración. En estos ensayos fueron reclutados un total de 868 pacientes (708 con tratamiento activo y 160 con control).
En el ensayo de fase II D2201 (RESOLVE), 151 pacientes fueron tratados con ranibizumab (6 mg/ml, n=51, 10 mg/ml, n=51) o con tratamiento simulado (n=49) mediante inyecciones intravítreas mensuales. El cambio medio en la AVMC desde el Mes 1 al Mes 12 comparado con el inicio fue de +7,8 (±7,72) letras en los pacientes tratados con ranibizumab combinados (n=102), comparado con -0,1 (±9,77) letras para pacientes con tratamiento simulado; y el cambio medio en la AVMC en el Mes 12 desde el valor inicial fue de 10,3 (±9,1) letras comparado con -1,4 (±14,2) letras respectivamente (p<0,0001 para la diferencia de tratamiento).
En el ensayo de fase III D2301 (RESTORE), se aleatorizaron 345 pacientes en un ratio 1:1:1 para recibir ranibizumab 0,5 mg en monoterapia y fotocoagulación con láser simulada, o ranibizumab 0,5 mg y fotocoagulación con láser combinados, o inyección simulada y fotocoagulación con láser. En un ensayo de extensión de 24 meses multicéntrico y abierto (Extension del ensayo RESTORE) fueron reclutados 240 pacientes, que previamente habían completado el ensayo RESTORE de 12 meses. Los pacientes fueron tratados con ranibizumab 0,5 mg pro re nata (PRN) en el mismo ojo que en el ensayo principal (D2301 RESTORE).
Los resultados clave se resumen en la Tabla 2 (RESTORE y Extensión) y en la Figura 2 (RESTORE).
Figura 2
Cambio medio en la agudeza visual en el tiempo desde el inicio, en el estudio D2301 (RESTORE)

Ranibizumab 0,5 mg (n=115)
• • • Ranibizumab 0,5 mg + Láser (n=118) Láser (n=110)
Grupo de tratamiento
BL=valor inicial; ES= error estándar de la media
* Diferencia de medias de mínimos cuadrados, p<0,0001/0,0004 basado en el test bilateral de Cochran-Mantel-Haenszel
El efecto a los 12 meses fue consistente en la mayoría de subgrupos. Sin embargo, sujetos con una AVMC al inicio >73 letras y con un edema macular con un grosor de la retina central <300 pm, no parecían beneficiarse del tratamiento con ranibizumab comparado con la fotocoagulación con láser.
Tabla 2 Resultados al Mes 12 en el estudio D2301 (RESTORE) y al Mes 36 en el estudio D2301-E1 (Extensión del estudio RESTORE)
|
Medidas del resultado al Mes 12 comparado con el valor inicial en el estudio D2301 (RESTORE) |
Ranibizumab 0,5 mg n=115 |
Ranibizumab 0,5 mg + Láser n=118 |
Láser n=110 |
|
Cambio medio en la AVMC desde el Mes 1 al Mes 12a (±desviación estándar) |
6,1 (6,4)a |
5,9 (7,9)a |
0,8 (8,6) |
|
Cambio medio en la AVMC al Mes 12 (±desviación estándar) |
6,8 (8,3)a |
6,4 (11,8)a |
0,9 (11,4) |
|
Ganancia de >15 letras o AVMC >84 letras al Mes 12 (%) |
22,6 |
22,9 |
8,2 |
|
Número medio de inyecciones (Meses 011) |
7,0 |
6,8 |
7,3 (tratamient o simulado) |
|
Medida del resultado al Mes 36 comparado con el valor inicial del estudio D2301 (RESTORE) en el estudio D2301-E1 (Extension del estudio RESTORE) |
Ranibizumab 0,5 mg previo n=83 |
Ranibizumab 0,5 mg + láser previos n=83 |
Láser previo n=74 |
|
Cambio medio en la AVMC al Mes 24 (desviación estándar) |
7,9 (9,0) |
6,7 (7,9) |
5,4 (9,0) |
|
Cambio medio en la AVMC al Mes 36 (desviación estándar) |
8,0 (10,1) |
6,7 (9,6) |
6,0 (9,4) |
|
Ganancia de >15 letras o AVMC >84 letras al Mes 36 (%) |
27,7 |
30,1 |
21,6 |
|
Número medio de inyecciones (Meses 1235)* |
6,8 |
6,0 |
6,5 |
ap<0,0001 para comparaciones de los grupos de ranibizumab frente al grupo de láser. n en D2301-E1 (Extension del estudio RESTORE) es el número de pacientes con un valor tanto en el valor inicial del estudio D2301 (RESTORE) (Mes 0) como en la visita del Mes 36.
* Las proporciones de pacientes que no necesitaron ningún tratamiento con ranibizumab durante la fase de extensión fueron del 19%, 25% y 20% en los grupos ranibizumab previo, ranibizumab + láser previos y láser previo, respectivamente.
En el tratamiento con ranibizumab (con o sin láser) se observaron beneficios notificados por el mismo paciente estadísticamente significativos para la mayoría de funciones relacionadas con la visión respecto al grupo control, medidos según el NEI VFQ-25. Para las otras subescalas de este cuestionario no pudieron establecerse diferencias ligadas al tratamiento.
El perfil de seguridad a largo plazo de ranibizumab observado en el ensayo de extensión de 24 meses es consistente con el perfil de seguridad de Lucentis conocido.
En el ensayo de fase IlIb D2304 (RETAIN), se aleatorizaron 372 pacientes en un ratio 1:1:1 para recibir:
• ranibizumab 0,5 mg con fotocoagulación con láser concomitante en un régimen de tratar y extender (TE), o bien
• ranibizumab 0,5 mg en monoterapia en un régimen TE, o bien
• ranibizumab 0,5 mg en monoterapia en un régimen PRN.
En todos los grupos, ranibizumab se administró mensualmente hasta que la AVMC era estable durante al menos tres controles mensuales consecutivos. En el régimen TE ranibizumab se administró a intervalos de tratamiento de 2-3 meses. En todos los grupos, el tratamiento mensual se reiniciaba en cuanto había una disminución de la AVMC debida a la progresión del EMD y continuaba hasta que se alcanzaba nuevamente una AVMC estable.
Después de las 3 inyecciones iniciales, el número de visitas de tratamiento programadas fueron de 13 y 20 para los regímenes TE y PRN respectivamente. Con ambos regímenes TE, más del 70% de los pacientes mantuvieron su AVMC con una frecuencia media de visitas de >2 meses.
Los resultados clave se resumen en la Tabla 3.
Tabla 3 Resultados en el estudio D2304 (RETAIN)
|
Medida del resultado comparado con el valor inicial |
Ranibizumab 0,5 mg + láser en TE n=117 |
Ranibizumab 0,5 mg solo en TE n=125 |
Ranibizumab 0,5 mg en PRN n=117 |
|
Cambio medio en la AVMC desde el Mes 1 al Mes 12 (desviación estándar) |
5,9 (5,5)a |
6,1 (5,7)a |
6,2 (6,0) |
|
Cambio medio en la AVMC desde el Mes 1 al Mes 24 (desviación estándar) |
6,8 (6,0) |
6,6 (7,1) |
7,0 (6,4) |
|
Cambio medio en la AVMC al Mes 24 (desviación estándar) |
8,3 (8,1) |
6,5 (10,9) |
8,1 (8,5) |
|
Ganancia de >15 letras o AVMC >84 letras al Mes 24(%) |
25,6 |
28,0 |
30,8 |
|
Número medio de inyecciones (meses 023) |
12,4 |
12,8 |
10,7 |
“p<0,0001 para la valoración de no inferioridad a PRN
En los ensayos en EMD, la mejora en la AVMC se acompañó de una reducción en el tiempo del GSCR medio en todos los grupos de tratamiento.
Tratamiento de la alteración visual debida al edema macular secundario a OVR La seguridad y eficacia clínicas de Lucentis en pacientes con alteración visual debida al edema macular secundario a OVR se han evaluado en los ensayos BRAVO y CRUISE, controlados, aleatorizados, doble ciego que reclutaron sujetos con ORVR (n=397) y con OVCR (n=392), respectivamente. En ambos ensayos, los sujetos recibieron o bien ranibizumab 0,3 mg o 0,5 mg o inyecciones simuladas. Después de 6 meses, los pacientes en los grupos control con inyección simulada cambiaron a ranibizumab 0,5 mg.
En la Tabla 4 y las Figuras 3 y 4 se resumen los resultados clave de los ensayos BRAVO y CRUISE. Tabla 4 Resultados a los Meses 6 y 12 (BRAVO y CRUISE)
|
BRAVO |
CRUISE | |||
|
Tratamiento simulado/Lucenti s 0,5 mg (n=132) |
Lucentis 0,5 mg (n=131) |
Tratamiento simulado/Lucenti s 0,5 mg (n=130) |
Lucentis 0,5 mg (n=130) | |
|
Cambio medio en la agudeza visual al Mes 6a (letras) (desviación estándar) (variable primaria) |
7,3 (13,0) |
18,3 (13,2) |
0,8 (16,2) |
14,9 (13,2) |
|
Cambio medio en la AVMC al Mes 12 (letras) (desviación estándar) |
12,1 (14,4) |
18,3 (14,6) |
7,3 (15,9) |
13,9 (14,2) |
|
Ganancia de >15 letras en agudeza visual al Mes 6a (%) |
28,8 |
61,1 |
16,9 |
47,7 |
|
Ganancia de >15 letras en agudeza visual al Mes 12 (%) |
43,9 |
60,3 |
33,1 |
50,8 |
|
Proporción (%) que recibió rescate con láser en 12 meses |
61,4 |
34,4 |
NA |
NA |
a p<0,0001 para ambos ensayos
Cambio medio en la AVMC desde el inicio en el tiempo hasta el Mes 6 y el Mes 12 (BRAVO)

Grupo de tratamiento Tratamiento simulado/Ranibizumab 0,5 mg (n=132)
* * * Ranibizumab 0,5 mg (n=131)
BL=valor inicial; ES=error estándar de la media
Cambio medio en la AVMC desde el inicio en el tiempo hasta el Mes 6 y el Mes 12 (CRUISE)

Grupo de tratamiento Tratamiento simulado/Ranibizumab 0,5 mg (n=130)
* * * Ranibizumab 0,5 mg (n=130)
BL=valor inicial; ES=error estándar de la media
En ambos estudios, la mejora de la visión se acompañó de una reducción continua y significativa del edema macular medido según el grosor central de la retina.
En pacientes con OVCR (CRUISE y ensayo de extensión HORIZON): Los sujetos tratados con tratamiento simulado en los primeros 6 meses que posteriormente recibieron ranibizumab, no alcanzaron ganancias comparables en AV en el Mes 24 (~6 letras) en comparación con los sujetos tratados con ranibizumab desde el inicio del ensayo (~12 letras).
En el tratamiento con ranibizumab se observaron beneficios notificados por el mismo paciente estadísticamente significativos en las subescalas relativas a la actividad de cerca y de lejos respecto al grupo control, medidos según el NEI VFQ-25.
La seguridad y eficacia clínicas de Lucentis a largo plazo (24 meses) en pacientes con alteración visual debida al edema macular secundario a OVR se evaluaron en los ensayos BRIGHTER (BRVO) y CRYSTAL (CRVO). En ambos ensayos, los sujetos recibieron ranibizumab 0,5 mg en un regimen de dosificación PRN que obedece a criterios de estabilización individualizados. BRIGHTER era un ensayo aleatorizado con 3 grupos controlado con tratamiento activo que comparaba ranibizumab 0,5 mg administrado en monoterapia o en combinación con fotocoagulación con láser adjunta frente a fotocoagulación con láser sola. Después de 6 meses, los sujetos en el grupo del láser podían recibir ranibizumab 0,5 mg. CRYSTAL era un ensayo de un grupo único con ranibizumab 0,5 mg en monoterapia.
|
BRIGHTER |
CRYSTAL | |||
|
Lucentis 0,5 mg N=180 |
Lucentis 0,5 mg + Láser N=178 |
Láser* N=90 |
Lucentis 0,5 mg N=356 | |
|
Cambio medio en la AVMC al Mes 6a (letras) (desviación estándar) |
+14,8 (10,7) |
+14,8 (11,13) |
+6,0 (14,27) |
+12,0 (13,95) |
|
Cambio medio en la AVMC al Mes 24b (letras) (desviación estándar) |
+15,5 (13,91) |
+17,3 (12,61) |
+11,6 (16,09) |
+12,1 (18,60) |
|
Ganancia de >15 letras en AVMC al Mes 24 (%) |
52,8 |
59,6 |
43,3 |
49,2 |
|
Número medio de inyecciones (desviación estándar) (Meses 0-23) |
11,4 (5,81) |
11,3 (6,02) |
NA |
13,1 (6,39) |
|
a p<0,0001 para ambas comparaciones en el ensayo BRIGHTER al Mes 6: Lucentis 0,5 mg frente a Láser y Lucentis 0,5 mg + Láser frente a Láser. b p<0,0001 para la hipótesis nula en el ensayo CRYSTAL en que el cambio medio al Mes 24 desde el valor inicial es cero. * A partir del Mes 6 se permitió el tratamiento con ranibizumab 0,5 mg (24 pacientes fueron tratados solo con láser). | ||||
En el ensayo BRIGHTER, ranibizumab 0,5 mg con terapia con láser adjunta demostró no inferioridad comparado con ranibizumab en monoterapia desde el valor inicial hasta el Mes 24 (95% IC -2,8, 1,4).
En ambos ensayos, se observó en el Mes 1 una reducción rápida y estadísticamente significativa del grosor del subcampo central de la retina desde el valor inicial. Este efecto se mantuvo hasta el Mes 24.
El efecto del tratamiento con ranibizumab fue similar de forma independiente de la presencia de isquemia retiniana. En el ensayo BRIGHTER, los pacientes que presentaban isquemia (N=46) o los que no la presentaban (N=133) y que fueron tratados con ranibizumab en monoterapia, tuvieron un cambio medio desde el valor inicial de +15,3 y +15,6 letras respectivamente, en el Mes 24. En el ensayo CRYSTAL, los pacientes que presentaban isquemia (N=53) o los que no la presentaban (N=300) y que fueron tratados con ranibizumab en monoterapia, tuvieron un cambio medio desde el valor inicial de +15,0 y +11,5 letras respectivamente.
En todos los pacientes tratados con ranibizumab 0,5 mg en monoterapia se observó el efecto en términos de mejora visual en ambos ensayos BRIGHTER y CRYSTAL de forma independiente de la duración de la enfermedad. En pacientes con una duración de la enfermedad <3 meses se observó un aumento en la agudeza visual de 13,3 y 10,0 letras en el Mes 1; y 17,7 y 13,2 letras en el Mes 24 en BRIGHTER y CRYSTAL respectivamente. En pacientes con una duración de la enfermedad >12 meses, la ganancia de agudeza visual correspondiente fue de 8,6 y 8,4 letras en los respectivos ensayos. Se debe considerar el inicio del tratamiento en el momento del diagnóstico.
El perfil de seguridad a largo plazo de ranibizumab observado en los ensayos de 24 meses es consistente con el perfil de seguridad de Lucentis conocido.
Tratamiento de la alteración visual debida a la NVC secundaria a MP
La seguridad y eficacia clínicas de Lucentis en pacientes con alteración visual debida a la NVC secundaria a MP se han evaluado de acuerdo a los datos de 12 meses del ensayo pivotal F2301 (RADIANCE) controlado, doble ciego. En este ensayo 277 pacientes fueron aleatorizados en un ratio 2:2:1 para los siguientes grupos:
• Grupo I (ranibizumab 0,5 mg, régimen de tratamiento basado en criterios de “estabilidad” definida como ningún cambio en la AVMC en comparación con las dos evaluaciones mensuales previas).
• Grupo II (ranibizumab 0,5 mg, régimen de tratamiento basado en criterios de “actividad de la enfermedad” definida como alteración de la visión atribuible a fluido intra o subretiniano o exudación activa debido a la lesión de NVC y evaluada mediante OCT y/o AF).
• Grupo III (TFDv - se permitió a los pacientes recibir tratamiento con ranibizumab a partir del Mes 3).
En el Grupo II, que se corresponde con la posología recomendada (ver sección 4.2), el 50,9% de los pacientes requirió 1 ó 2 inyecciones, el 34,5% requirió de 3 a 5 inyecciones y el 14,7% requirió de 6 a 12 inyecciones durante el periodo de 12 meses del ensayo. El 62,9% de los pacientes del Grupo II no requirió inyecciones en el segundo semestre del ensayo.
|
Grupo I |
Grupo II |
Grupo III | |
|
Ranibizumab |
Ranibizumab |
TFDvb | |
|
0,5 mg |
0,5 mg | ||
|
“estabilidad de |
“actividad de la | ||
|
la visión” |
enfermedad” | ||
|
(n=105) |
(n=116) |
(n=55) | |
|
Mes 3 Cambio medio en la AVMC desde el Mes 1 |
+10,5 |
+10,6 |
+2,2 |
|
al Mes 3 comparado con el inicioa (letras) Proporción de pacientes que ganaron: >15 letras, o alcanzaron >84 letras en la AVMC |
38,1% |
43,1% |
14,5% |
|
Mes 12 Número de inyecciones hasta el Mes 12: Media |
4,6 |
3,5 |
N/A |
|
Mediana |
4,0 |
2,5 |
N/A |
|
Cambio medio en la AVMC desde el Mes 1 |
+12,8 |
+12,5 |
N/A |
|
al Mes 12 comparado con el inicio (letras) Proporción de pacientes que ganaron: >15 letras, o alcanzaron >84 letras en la AVMC |
53,3% |
51,7% |
N/A |
a p<0,00001 comparación con el control de TFDv
b Control comparativo hasta el Mes 3. A los pacientes aleatorizados a TFDv se les permitió recibir tratamiento con ranibizumab a partir del Mes 3 (en el Grupo III, 38 pacientes recibieron ranibizumab a partir del Mes 3)
Figura 5 Cambio medio en la AVMC desde el inicio en el tiempo hasta el Mes 12 (RADIANCE)

Ranibizumab 0,5 mg Grupo I por estabilización (n=105)
Ranibizumab 0,5 mg Grupo II
por actividad de la enfermedad (n=116)
Ranibizumab 0,5 mg/TFD con verteporfina Grupo III desde el Mes 3 en adelante (n=55)
A-a-a TFD con verteporfina Grupo III (n=55)
La mejora de la visión se acompañó de una reducción del grosor central de la retina.
Los beneficios comunicados por el paciente se observaron más en los grupos de tratamiento con ranibizumab que en la TFDv (valor p<0,05) en términos de mejora en la puntuación compuesta y varias subescalas (visión general, actividades de cerca, salud mental y dependencia) del NEI VFQ-25.
Población pediátrica
La seguridad y eficacia de ranibizumab aún no han sido estudiadas en pacientes pediátricos.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Lucentis en los diferentes grupos de la población pediátrica en la DMAE neovascular, la alteración visual debida al EMD, la alteración visual debida al edema macular secundario a la OVR y la alteración visual debida a la NVC secundaria a MP (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
Tras la administración intravítrea mensual de Lucentis a pacientes con DMAE neovascular, las concentraciones séricas de ranibizumab fueron en general bajas, con niveles máximos (Cmax) por debajo de la concentración de ranibizumab necesaria para inhibir la actividad biológica del VEGF en un 50% (11-27 ng/ml, valorado en un ensayo de proliferación celular in vitro). La Cmax fue proporcional a la dosis, en el rango de dosis de 0,05 a 1,0 mg/ojo. Las concentraciones séricas en un número limitado de pacientes con EMD indican que no puede excluirse una exposición sistémica ligeramente superior en comparación con la observada en pacientes con DMAE neovascular. Las concentraciones séricas de ranibizumab en pacientes con OVR fueron similares o ligeramente superiores en comparación con las observadas en pacientes con DMAE neovascular.
En base al análisis farmacocinético poblacional y a la desaparición sérica de ranibizumab en pacientes con DMAE neovascular tratados con la dosis de 0,5 mg, el promedio de la vida media de eliminación vítrea de ranibizumab es de 9 días aproximadamente. Tras la administración intravítrea mensual de Lucentis 0,5 mg/ojo, se prevé que la Cmax de ranibizumab sérica alcanzada aproximadamente 1 día después de la administración, varíe en general en un rango de entre 0,79 y 2,90 ng/ml, y que la Cmin varíe en general en un rango de entre 0,07 y 0,49 ng/ml. Se prevé que las concentraciones séricas de ranibizumab sean aproximadamente 90.000 veces inferiores a las concentraciones vítreas de ranibizumab.
Pacientes con insuficiencia renal: No se han realizado estudios formales para investigar la farmacocinética de Lucentis en pacientes con insuficiencia renal. En un análisis farmacocinético poblacional de pacientes con DMAE neovascular, el 68% (136 de 200) de los pacientes tenían insuficiencia renal (leve en un 46,5% [50-80 ml/min], moderada en un 20% [30-50 ml/min] y grave en un 1,5% [<30 ml/min]). En el caso de pacientes con OVR, el 48,2% (253 de 525) tenían insuficiencia renal (leve en un 36,4%, moderada en un 9,5% y grave en un 2,3%). El aclaramiento sistémico fue ligeramente inferior, pero esto no fue clínicamente significativo.
Insuficiencia hepática: No se han realizado estudios formales para investigar la farmacocinética de Lucentis en pacientes con insuficiencia hepática.
5.3 Datos preclínicos sobre seguridad
La administración intravítrea bilateral de ranibizumab a macacos, a dosis de entre 0,25 mg/ojo y
2,0 mg/ojo una vez cada 2 semanas durante 26 semanas, ocasionó efectos oculares dosis-dependientes.
Intraocularmente, se observaron incrementos dosis-dependientes de exudados y de células en la cámara anterior, con un máximo a los 2 días después de la inyección. Por lo general, la gravedad de la respuesta inflamatoria disminuyó con las inyecciones posteriores o durante el periodo de recuperación. En el segmento posterior, hubo infiltración de células en la cámara vítrea y partículas flotantes, que tendieron también a ser dosis-dependientes y que, en general, persistieron hasta el final del periodo de tratamiento. En el ensayo a 26 semanas, la gravedad de la inflamación vítrea aumentó con el número de inyecciones. Sin embargo, se observó evidencia de reversibilidad tras el periodo de recuperación. La naturaleza y cronología de la inflamación en el segmento posterior sugiere una respuesta inmunitaria mediada por anticuerpos, que puede ser clínicamente irrelevante. En algunos animales se observó la formación de cataratas tras un periodo relativamente largo de inflamación intensa, lo cual sugiere que las alteraciones en el cristalino fueron secundarias a la inflamación grave. Tras las inyecciones intravítreas se observó un aumentó transitorio de la presión intraocular independiente de la dosis.
Los cambios oculares microscópicos fueron relacionados con la inflamación y no eran indicativos de procesos degenerativos. Se observaron cambios inflamatorios granulomatosos en el disco óptico de algunos ojos. Estas alteraciones en el segmento posterior disminuyeron, y en algunos casos se resolvieron, durante el periodo de recuperación.
Tras la administración intravítrea, no se detectaron signos de toxicidad sistémica. En un subgrupo de animales tratados se detectaron anticuerpos séricos y vítreos contra ranibizumab.
No se dispone de datos de carcinogenicidad ni de mutagenicidad.
En hembras de mono preñadas, el tratamiento con ranibizumab intravítreo resultando en exposiciones sistémicas máximas 0,9-7 veces la peor exposición clínica, no provocó toxicidad en el desarrollo ni teratogenicidad y no tuvo ningún efecto sobre el peso o la estructura de la placenta, aunque en base a su efecto farmacológico, ranibizumab debe considerarse potencialmente teratogénico y embrio/fetotóxico.
La ausencia de efectos mediados por ranibizumab sobre el desarrollo embrio-fetal está plausiblemente relacionado principalmente con la incapacidad del fragmento Fab de atravesar la placenta. Sin embargo, se describió un caso de niveles séricos de ranibizumab maternos elevados y presencia de ranibizumab en el suero fetal lo que sugiere que el anticuerpo contra ranibizumab actuó como proteína transportadora (conteniendo la región Fc) para ranibizumab, disminuyendo de ese modo su aclaramiento sérico materno y permitiendo su paso a la placenta. Dado que las investigaciones en el desarrollo embrio-fetal se llevaron a cabo en animales preñados sanos y las enfermedades (tales como la diabetes) pueden modificar la permeabilidad de la placenta para el fragmento Fab, el estudio debe interpretarse con cautela.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
a,a-trehalosa dihidrato
Hidrocloruro de histidina monohidrato
Histidina
Polisorbato 20
Agua para inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
3 años
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Antes de usar, el vial sin abrir se puede conservar a temperatura ambiente (25°C) durante un máximo de 24 horas.
6.5 Naturaleza y contenido del envase
0. 23.ml de solución estéril en un vial (vidrio tipo I) con un tapón (goma de clorobutilo) y 1 aguja roma con filtro (18G x 1V", 1,2 mm x 40 mm, 5 pm). El envase contiene 1 vial.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El vial y la aguja con filtro son para un solo uso. La reutilización puede dar lugar a una infección u otra enfermedad/lesión. Todos los componentes son estériles. No debe utilizarse ningún componente cuyo envase muestre signos de deterioro o manipulación. La esterilidad sólo se puede garantizar si el sellado del envase de los componentes se mantiene intacto.
Para la preparación y la inyección intravítrea se necesitan los siguientes productos sanitarios de un solo uso:
- una aguja con filtro de 5 pm (18G x 1V", 1,2 mm x 40 mm, suministrada)
- una jeringa estéril de 1 ml (no incluida en el envase de Lucentis)
- una aguja para inyección (30G x V"; no incluida en el envase de Lucentis)
Para la preparación de Lucentis para administración intravítrea, siga las siguientes instrucciones:
1. Antes de extraer la solución, se debe desinfectar la parte exterior del tapón de goma del vial.
2. Incorporar la aguja con filtro de 5 pm (18G x 1V", 1,2 mm x 40 mm, suministrada) a una
jeringa de 1 ml usando técnicas asépticas. Insertar la aguja roma con filtro en el centro del tapón del vial hasta que la aguja toque el extremo inferior del vial.
3. Extraer todo el líquido del vial, manteniendo el vial en posición vertical, ligeramente inclinado para facilitar la extracción completa.
4. Al vaciar el vial, asegurar que el émbolo se retira hacia atrás lo suficiente de forma que se vacíe por completo la aguja con filtro.
5. Dejar la aguja roma con filtro en el vial y desconectarla de la jeringa. La aguja con filtro se debe desechar tras extraer el contenido del vial, y no se debe utilizar para la inyección intravítrea.
6. Incorporar una aguja para inyección (30G x V") a la jeringa con firmeza y de forma aséptica.
7. Quitar la cápsula de cierre de la aguja para inyección cuidadosamente sin desconectar la aguja para inyección de la jeringa.
Nota: Sujetar la aguja para inyección por el cono mientras se retira la cápsula de cierre.
8. Expulsar el aire de la jeringa y ajustar la dosis hasta la marca de 0,05 ml en la jeringa cuidadosamente. La jeringa está lista para la inyección.
Nota: No secar la aguja para inyección. No tirar del émbolo hacia atrás.
Tras la inyección, no tapar la aguja con la cápsula de cierre ni separarla de la jeringa. Eliminar la jeringa usada junto con la aguja en un contenedor para objetos punzantes o eliminar de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/374/004
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 22 Enero 2007 Fecha de la última renovación: 24 Enero 2012
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de los fabricantes del principio activo biológico
Genentech, Inc.
1 DNA Way
South San Francisco, CA 94080-4990 EEUU
Roche Singapore Technical Operations Pte. Ltd.
10 Tuas Bay Link Singapore 637394 Singapur
Nombre y dirección del fabricante responsable de la liberación de los lotes
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nuremberg Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2)
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos;
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
• Medidas adicionales de minimización de riesgos
El TAC debe acordar el material informativo final con la Autoridad Nacional Competente en cada Estado Miembro antes del lanzamiento.
El TAC debe asegurar que, tras las conversaciones y acuerdos con las Autoridades Nacionales Competentes en cada Estado Miembro donde se comercializa Lucentis, se entregue durante y después del lanzamiento a todas las unidades oftalmológicas donde se espera que se use Lucentis, documentación informativa actualizada para el médico que contenga los siguientes elementos:
• Información para el médico
• Vídeo sobre el procedimiento de inyección intravítrea
• Pictograma sobre el procedimiento de inyección intravítrea
• Paquetes de información para el paciente
La información para el médico debe contener los siguientes elementos básicos:
• Ficha Técnica o Resumen de las Características del Producto
• Técnicas de esterilidad, incluyendo la desinfección ocular y periocular, para minimizar el riesgo de infección
• Utilización de povidona iodada o equivalente
• La necesidad de expulsar el exceso de volumen de la jeringa precargada antes de inyectar Lucentis para evitar una sobredosis
• Técnicas de inyección intravítrea
• Seguimiento del paciente tras la inyección IVT
• Signos y síntomas clave de los efectos adversos relacionados con la inyección IVT incluyendo aumento de la presión intraocular, catarata traumática y endoftalmitis
• Manejo de los efectos adversos relacionados con la inyección IVT
El paquete de información para el paciente se debe proporcionar tanto en la forma de folletos con información para el paciente como en un audio-CD, que contendrán los siguientes elementos básicos:
• Prospecto o información para el usuario
• Cómo prepararse para el tratamiento con Lucentis
• Pasos a seguir después del tratamiento con Lucentis
• Signos y síntomas clave de los efectos adversos graves incluyendo aumento de la presión intraocular, catarata traumática y endoftalmitis
• Cuándo deben requerir atención médica urgente
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
VIAL + AGUJA CON FILTRO + AGUJA PARA INYECCIÓN + JERINGA
1. NOMBRE DEL MEDICAMENTO
Lucentis 10 mg/ml solución inyectable Ranibizumab
2. PRINCIPIO(S) ACTIVO(S)
Un ml contiene 10 mg de ranibizumab. El vial contiene 2,3 mg de ranibizumab.
3. LISTA DE EXCIPIENTES
También contiene: a,a-trehalosa dihidrato; hidrocloruro de histidina monohidrato; histidina; polisorbato 20; agua para inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 vial con 0,23 ml de solución inyectable, 1 aguja para inyección (30G x A", 0,3 mm x 13 mm), 1 aguja con filtro (18G x ÍA", 1,2 mm x 40 mm, 5 ^m), 1 jeringa (1 ml).
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía intravítrea.
Vial, agujas y jeringa para un solo uso.
Leer el prospecto antes de utilizar este medicamento. La aguja con filtro no es para inyección.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Desechar las agujas usadas en un contenedor para objetos punzantes.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/374/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
ETIQUETA
VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Lucentis 10 mg/ml solución inyectable
Ranibizumab
Vía intravítrea
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
1 vial = 2,3 mg de ranibizumab.
6. OTROS
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
CAJA
VIAL
1. NOMBRE DEL MEDICAMENTO
Lucentis 10 mg/ml solución inyectable Ranibizumab
2. PRINCIPIO(S) ACTIVO(S)
Un ml contiene 10 mg de ranibizumab. El vial contiene 2,3 mg de ranibizumab.
3. LISTA DE EXCIPIENTES
También contiene: a,a-trehalosa dihidrato; hidrocloruro de histidina monohidrato; histidina; polisorbato 20; agua para inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 vial con 0,23 ml de solución inyectable.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía intravítrea.
Vial para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/374/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
ETIQUETA
VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Lucentis 10 mg/ml solución inyectable
Ranibizumab
Vía intravítrea
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
1 vial = 2,3 mg de ranibizumab.
6. OTROS
JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO
Lucentis 10 mg/ml solución inyectable en jeringa precargada Ranibizumab
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa precargada de 0,165 ml de solución contiene 1,65 mg de ranibizumab (10 mg/ml).
3. LISTA DE EXCIPIENTES
También contiene: a,a-trehalosa dihidrato; hidrocloruro de histidina monohidrato; histidina; polisorbato 20; agua para inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable 1 jeringa precargada de 0,165 ml.
Dosis única de 0,5 mg/0,05 ml.
El exceso de volumen se debe expulsar antes de la inyección.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Para un solo uso. Al abrir la bandeja sellada, se debe proceder bajo condiciones asépticas. Ajustar la dosis hasta la marca de dosis de 0,05 ml.
Leer el prospecto antes de utilizar este medicamento.
Vía intravítrea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar la jeringa precargada en su bandeja sellada en la caja para protegerla de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/374/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
LÁMINA DEL BLÍSTER
JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Lucentis 10 mg/ml solución inyectable en jeringa precargada
Ranibizumab
Vía intravítrea
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
0,165 ml
ETIQUETA
JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Lucentis 10 mg/ml solución inyectable
Ranibizumab
Vía intravítrea
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
0,165 ml
6. OTROS
VIAL + AGUJA CON FILTRO
1. NOMBRE DEL MEDICAMENTO
Lucentis 10 mg/ml solución inyectable Ranibizumab
2. PRINCIPIO(S) ACTIVO(S)
Un ml contiene 10 mg de ranibizumab. El vial contiene 2,3 mg de ranibizumab.
3. LISTA DE EXCIPIENTES
También contiene: a,a-trehalosa dihidrato; hidrocloruro de histidina monohidrato; histidina; polisorbato 20; agua para inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 vial con 0,23 ml de solución inyectable, 1 aguja con filtro (18G x IV2", 1,2 mm x 40 mm, 5 ^m).
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía intravítrea.
Vial y aguja con filtro para un solo uso.
Leer el prospecto antes de utilizar este medicamento. La aguja con filtro no es para inyección.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Desechar la aguja usada en un contenedor para objetos punzantes.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/374/004
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
ETIQUETA
VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Lucentis 10 mg/ml solución inyectable
Ranibizumab
Vía intravítrea
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
1 vial = 2,3 mg de ranibizumab.
6. OTROS
B. PROSPECTO
Lucentis 10 mg/ml solución inyectable
Ranibizumab
Lea todo el prospecto detenidamente antes de que le administren este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto.
Contenido del prospecto
1. Qué es Lucentis y para qué se utiliza
2. Qué necesita saber antes de que le administren Lucentis
3. Cómo se administra Lucentis
4. Posibles efectos adversos
5. Conservación de Lucentis
6. Contenido del envase e información adicional
1. Qué es Lucentis y para qué se utiliza Qué es Lucentis
Lucentis es una solución que se inyecta en el ojo. Lucentis pertenece a un grupo de medicamentos denominados agentes antineovascularización. Contiene el principio activo denominado ranibizumab.
Para qué se utiliza Lucentis
Lucentis se utiliza en adultos para tratar varias enfermedades oculares que causan alteración de la visión.
Estas enfermedades son el resultado de una lesión en la retina (capa sensible a la luz en la parte posterior del ojo) provocada por:
- El crecimiento de vasos sanguíneos anómalos, que pierden líquido (neovascularización coroidea, NVC). Esto se observa en enfermedades como la degeneración macular asociada a la edad (DMAE) o la miopía patológica (MP).
- Edema macular (hinchazón del centro de la retina). La causa de este hinchazón puede ser la diabetes (una enfermedad conocida como edema macular diabético (EMD)) o un bloqueo de las venas retinianas de la retina (una enfermedad conocida como oclusión de la vena de la retina (OVR)).
Cómo actúa Lucentis
Lucentis reconoce y se une de forma específica a una proteína denominada factor de crecimiento endotelial vascular A (VEGF-A) humano presente en los ojos. En exceso, el VEGF-A causa el crecimiento de vasos sanguíneos anómalos e hinchazón en el ojo que puede ocasionar una alteración de la visión en enfermedades como DMAE, MP, EMD u OVR. Mediante la unión al VEGF-A, Lucentis puede impedir que actúe y prevenir dicho crecimiento e hinchazón anómalos.
En estas enfermedades, Lucentis puede ayudar a estabilizar y, en muchos casos, mejorar su visión.
2. Qué necesita saber antes de que le administren Lucentis No le deben administrar Lucentis
- Si es alérgico al ranibizumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- Si tiene una infección en el ojo o alrededor del mismo.
- Si tiene dolor o enrojecimiento (inflamación intraocular grave) en el ojo.
Advertencias y precauciones
Consulte a su médico antes de que le administren Lucentis
- Lucentis se administra mediante una inyección en el ojo. Ocasionalmente, tras el tratamiento con Lucentis puede aparecer una infección en la parte interna del ojo, dolor o enrojecimiento (inflamación), desprendimiento o desgarro de una de las capas situadas en el fondo del ojo (desprendimiento o desgarro de la retina y desprendimiento o desgarro del epitelio pigmentario de la retina), o enturbiamiento del cristalino (catarata). Es importante identificar y tratar tal infección o desprendimiento de retina lo antes posible. Informe inmediatamente a su médico si nota signos como dolor en el ojo o aumento de las molestias en el ojo, si empeora el enrojecimiento en el ojo, visión borrosa o disminución de la visión, un aumento del número de pequeñas manchas en la visión o aumento de la sensibilidad a la luz.
- En algunos pacientes, después de la inyección la presión en el ojo puede aumentar durante un corto periodo de tiempo. Es posible que usted no se de cuenta de ello, por lo que puede que su médico le realice un seguimiento de la presión ocular después de cada inyección.
- Informe a su médico si ha tenido enfermedades en los ojos o ha recibido algún tratamiento en los ojos anteriormente, o si ha sufrido un accidente cerebrovascular o ha tenido signos pasajeros de accidente cerebrovascular (debilidad o parálisis de un miembro o cara, dificultad en el habla o en la comprensión). Esta información se tendrá en consideración para evaluar si Lucentis es el tratamiento apropiado para usted.
Niños y adolescentes (menos de 18 años)
No se recomienda el uso de Lucentis en niños y adolescentes ya que no se ha estudiado en estos grupos de edad.
Uso de Lucentis con otros medicamentos
Informe a su médico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
- Se aconseja a las mujeres que pueden quedarse embarazadas que utilicen un método anticonceptivo eficaz durante el tratamiento.
- No hay experiencia del uso de Lucentis en mujeres embarazadas; por tanto no se conocen los riesgos potenciales. Si está embarazada, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de iniciar el tratamiento con Lucentis.
- No se recomienda el uso de Lucentis durante la lactancia, porque se desconoce si Lucentis pasa a la leche materna. Consulte a su médico o farmacéutico antes del tratamiento con Lucentis.
Conducción y uso de máquinas
Después del tratamiento con Lucentis usted puede experimentar visión borrosa temporalmente. Si esto le ocurre, no conduzca ni use máquinas hasta que este síntoma desaparezca.
3. Cómo se administra Lucentis
Lucentis se administra por el oftalmólogo en forma de inyección única en el ojo bajo anestesia local. La dosis habitual de una inyección es 0,05 ml (que contiene 0,5 mg de principio activo). El intervalo entre dos dosis aplicadas en el mismo ojo debe ser como mínimo de cuatro semanas. Todas las inyecciones serán administradas por un oftalmólogo.
Para prevenir una infección, antes de la inyección su médico le lavará el ojo cuidadosamente. Su médico también le administrará un anestésico local para reducir o prevenir cualquier dolor que pudiera sentir con la inyección.
El tratamiento se inicia con una inyección de Lucentis cada mes. Su médico controlará la enfermedad de su ojo y dependiendo de cómo responda al tratamiento, decidirá si necesita o no recibir más tratamiento y cuándo necesita ser tratado.
Al final del prospecto en el apartado “Cómo preparar y administrar Lucentis” se dan instrucciones detalladas de uso.
Pacientes de edad avanzada (65 años y mayores)
Lucentis puede utilizarse en personas de 65 años de edad o más, y no es necesario un ajuste de la dosis.
Si no se le administra una dosis de Lucentis
Contacte con su médico o su hospital tan pronto como le sea posible para pedir una nueva cita.
Antes de interrumpir el tratamiento con Lucentis
Si usted se está planteando interrumpir el tratamiento con Lucentis, acuda a la siguiente consulta y coméntelo antes con su médico. Su médico le aconsejará y decidirá durante cuánto tiempo deberá ser tratado con Lucentis.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, Lucentis puede producir efectos adversos, aunque no todas las personas los sufran.
Los efectos adversos asociados con la administración de Lucentis se deben o al propio medicamento o al procedimiento de inyección y la mayoría afectan al ojo.
A continuación se describen los efectos adversos más graves:
Efectos adversos graves frecuentes (pueden afectar hasta 1 de cada 10 pacientes): Desprendimiento o desgarro de una capa en la parte interna del ojo (desprendimiento o desgarro de la retina), que da como resultado destellos de luz con partículas flotantes que progresan a una pérdida de visión transitoria o a un enturbiamiento del cristalino (catarata).
Efectos adversos graves poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes): Ceguera, infección del globo ocular (endoftalmitis) con inflamación de la parte interna del ojo.
Los síntomas que podría experimentar se describen en el apartado 2 de este prospecto (por favor lea el apartado 2 “Qué necesita saber antes de que le administren Lucentis”). Informe a su médico inmediatamente si presenta alguno de estos efectos adversos.
A continuación se describen los efectos adversos comunicados más frecuentemente:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes)
Los efectos adversos oculares incluyen: Inflamación del ojo, sangrado en la parte posterior del ojo (hemorragia en la retina), alteraciones visuales, dolor en el ojo, pequeñas partículas o manchas en la visión (partículas flotantes), sangre en el ojo, irritación del ojo, sensación de tener algo dentro del ojo, aumento de la producción de lágrima, inflamación o infección en el borde de los párpados, ojo seco, enrojecimiento o picor en el ojo y aumento de la presión en el ojo.
Los efectos adversos no oculares incluyen: Dolor de garganta, congestión nasal, goteo nasal, dolor de cabeza y dolor en las articulaciones.
A continuación se describen otros efectos adversos que pueden ocurrir tras el tratamiento con Lucentis:
Efectos adversos frecuentes
Los efectos adversos oculares incluyen: Disminución de la nitidez de la visión, hinchazón de una sección del ojo (úvea, córnea), inflamación de la córnea (parte delantera del ojo), pequeñas marcas en la superficie del ojo, visión borrosa, sangrado en el lugar de inyección, sangrado en el ojo, secreción del ojo con picor, enrojecimiento e hinchazón (conjuntivitis), sensibilidad a la luz, molestias en el ojo, hinchazón del párpado, dolor en el párpado.
Los efectos adversos no oculares incluyen: Infección de las vías urinarias, recuento de glóbulos rojos bajo (con síntomas tales como cansancio, dificultad al respirar, mareo, palidez), ansiedad, tos, náuseas, reacciones alérgicas tales como erupción, urticaria, picor y enrojecimiento de la piel.
Efectos adversos poco frecuentes
Los efectos adversos oculares incluyen: Inflamación y sangrado en la parte anterior del ojo, acúmulo de pus en el ojo, cambios en la parte central de la superficie ocular, dolor o irritación en el lugar de inyección, sensación anormal en el ojo, irritación del párpado.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Lucentis
- Mantener este medicamento fuera de la vista y del alcance de los niños.
- No utilice este medicamento después de la fecha de caducidad que aparece en la caja después
de CAD y en la etiqueta del vial después de EXP. La fecha de caducidad es el último día del mes que se indica.
- Conservar en nevera (entre 2°C y 8°C). No congelar.
- Antes de usar, el vial sin abrir se puede conservar a temperatura ambiente (25°C) durante un máximo de 24 horas.
- Conservar el vial en el embalaje exterior para protegerlo de la luz.
- No utilice ningún envase que esté dañado.
6. Contenido del envase e información adicional
Composición de Lucentis
- El principio activo es ranibizumab (10 mg/ml). Cada ml contiene 10 mg de ranibizumab.
- Los demás componentes son a,a-trehalosa dihidrato; hidrocloruro de histidina monohidrato; histidina; polisorbato 20; agua para inyectables.
Aspecto del producto y contenido del envase
Lucentis es una solución inyectable contenida en un vial (0,23 ml). La solución es transparente, de incolora a amarillo pálido y acuosa.
Lucentis se suministra en un envase que contiene un vial de vidrio con ranibizumab, con tapón de goma de clorobutilo, una aguja roma con filtro (18G x 1'A", 1,2 mm x 40 mm, 5 micrómetros) para extraer el contenido del vial, una aguja para inyección (30G x 'A'", 0,3 mm x 13 mm) y una jeringa (1 ml) para extraer el contenido del vial y para la inyección intravítrea. Todos los componentes son para un solo uso.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien Lietuva
Novartis Pharma N.V. Novartis Pharma Services Inc.
Tél/Tel: +32 2 246 16 11 Tel: +370 5 269 16 50
Etarapna
Novartis Pharma Services Inc. Tea.: +359 2 489 98 28
Ceská republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
ELXáóa
Novartis (Hellas) A.E.B.E. Tr(k: +30 210 281 17 12
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Osterreich
Novartis Pharma GmbH Tel: +43 1 86 6570
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kúnpoq Novartis Pharma Services Inc. Tni: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
|
Fecha de la última revisión de este prospecto: |
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
Ver también la sección 3 “Cómo se administra Lucentis”.
Cómo preparar y administrar Lucentis
Vial para un solo uso. Unicamente para vía intravítrea.
Lucentis debe ser administrado por un oftalmólogo que tenga experiencia en la administración de inyecciones intravítreas.
En la DMAE exudativa y en la alteración visual debida a EMD, a edema macular secundario a OVR o a NVC secundaria a MP, la dosis recomendada de Lucentis es 0,5 mg administrada en forma de inyección intravítrea única. Esto corresponde a un volumen de inyección de 0,05 ml. El intervalo entre dos dosis inyectadas en el mismo ojo debe ser como mínimo de cuatro semanas.
El tratamiento se inicia con una inyección al mes hasta alcanzar la agudeza visual máxima y/o no haya signos de actividad de la enfermedad, es decir ningún cambio en la agudeza visual ni en otros signos y síntomas de la enfermedad bajo tratamiento continuado. En pacientes con DMAE exudativa, EMD y OVR inicialmente pueden ser necesarias tres o más inyecciones consecutivas administradas mensualmente.
A partir de ese momento, los intervalos de monitorización y tratamiento se deben determinar según criterio médico y en base a la actividad de la enfermedad, valorada mediante la agudeza visual y/o parámetros anatómicos.
Se debe interrumpir el tratamiento con Lucentis si bajo criterio del médico, los parámetros visuales y anatómicos indican que el paciente no se está beneficiando del tratamiento continuado.
La monitorización para determinar la actividad de la enfermedad puede incluir examen clínico, control funcional o técnicas de imagen (p. ej. tomografía de coherencia óptica o angiografía con fluoresceína).
Si se está tratando a los pacientes de acuerdo a un régimen de tratar y extender, una vez se ha alcanzado la agudeza visual máxima y/o no hay signos de actividad de la enfermedad, los intervalos de tratamiento se pueden espaciar de forma gradual hasta que vuelvan a aparecer signos de actividad de la enfermedad o alteración visual. En el caso de la DMAE exudativa el intervalo de tratamiento no debe espaciarse en más de dos semanas cada vez y en el caso del EMD se puede espaciar hasta un mes cada vez. Para la OVR, los intervalos de tratamiento también pueden espaciarse de forma gradual, sin embargo los datos que hay no son suficientes para determinar la duración de estos intervalos. Si vuelve a aparecer actividad de la enfermedad, se debe acortar el intervalo de tratamiento de manera consecuente.
En el tratamiento de la alteración visual debida a la NVC secundaria a MP, muchos pacientes pueden necesitar únicamente una o dos inyecciones durante el primer año, mientras que algunos pacientes pueden necesitar tratamiento con mayor frecuencia.
Lucentis y fotocoagulación con láser en EMD y edema macular secundario a oclusión de la rama venosa retiniana (ORVR)
Existe alguna experiencia con Lucentis administrado concomitantemente con fotocoagulación con láser. Cuando se administren en el mismo día, Lucentis debe ser administrado como mínimo 30 minutos después de la fotocoagulación con láser. Lucentis puede administrarse en pacientes que han recibido fotocoagulación con láser previamente.
Lucentis y la terapia fotodinámica con Visudyne en la NVC secundaria a MP No hay experiencia en la administración concomitante de Lucentis y Visudyne.
Antes de la administración de Lucentis se debe comprobar visualmente la ausencia de partículas y decoloración.
El procedimiento de inyección deberá llevarse a cabo bajo condiciones asépticas, que incluyen el lavado quirúrgico de las manos, el uso de guantes estériles, un campo estéril, un blefarostato estéril para los párpados (o equivalente) y la disponibilidad de una paracentesis estéril (en caso necesario). Antes de realizar el procedimiento de inyección intravítrea, se deberá evaluar detalladamente la historia clínica del paciente en cuanto a reacciones de hipersensibilidad. Antes de la inyección se debe administrar una anestesia adecuada y un microbicida tópico de amplio espectro para desinfectar la piel de la zona periocular, párpado y superficie ocular, de acuerdo con la práctica local.
Para la preparación de Lucentis para administración intravítrea, siga las siguientes instrucciones:
Todos los componentes son estériles y para un solo uso. No debe utilizarse ningún componente cuyo envase muestre signos de deterioro o manipulación. La esterilidad sólo se puede garantizar si el sellado del envase de los componentes se mantiene intacto. La reutilización puede dar lugar a una infección u otra enfermedad/lesión.

1. Antes de extraer la solución, se debe desinfectar la parte exterior del tapón de goma del vial.
2. Incorporar la aguja con filtro de 5 pm (18G x 1'A",
1,2 mm x 40 mm, 5 pm, suministrada) a la jeringa de 1 ml (suministrada) usando técnicas asépticas. Insertar la aguja roma con filtro en el centro del tapón del vial hasta que la aguja toque el extremo inferior del vial.
3. Extraer todo el líquido del vial, manteniendo el vial en posición vertical, ligeramente inclinado para facilitar la extracción completa.

4. Al vaciar el vial, asegurar que el émbolo se retira hacia atrás lo suficiente de forma que se vacíe por completo la aguja con filtro.
5. Dejar la aguja roma con filtro en el vial y desconectarla de la jeringa. La aguja con filtro se debe desechar tras extraer el contenido del vial, y no se debe utilizar para la inyección intravítrea.
6. Incorporar la aguja para inyección (30G x V", 0,3 mm x 13 mm, suministrada) a la jeringa con firmeza y de forma aséptica.

7. Quitar la cápsula de cierre de la aguja para inyección cuidadosamente sin desconectar la aguja para inyección de la jeringa.
Nota: Sujetar la aguja para inyección por el cono amarillo mientras se retira la cápsula de cierre.
8. Expulsar el aire de la jeringa y ajustar la dosis hasta la marca de 0,05 ml en la jeringa cuidadosamente. La jeringa está lista para la inyección.
Nota: No secar la aguja para inyección. No tirar del émbolo hacia atrás.
La aguja para inyección se deberá introducir 3,5-4,0 mm por detrás del limbo en la cavidad vitrea, evitando el meridiano horizontal y en dirección al centro del globo. Seguidamente debe liberarse el volumen de inyección de 0,05 ml; las inyecciones siguientes deberán aplicarse cada vez en un punto escleral distinto.
Tras la inyección, no tapar la aguja con la cápsula de cierre ni separarla de la jeringa. Eliminar la jeringa usada junto con la aguja en un contenedor para objetos punzantes o eliminar de acuerdo con la normativa local.
Lucentis 10 mg/ml solución inyectable
Ranibizumab
Lea todo el prospecto detenidamente antes de que le administren este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto.
Contenido del prospecto
1. Qué es Lucentis y para qué se utiliza
2. Qué necesita saber antes de que le administren Lucentis
3. Cómo se administra Lucentis
4. Posibles efectos adversos
5. Conservación de Lucentis
6. Contenido del envase e información adicional
1. Qué es Lucentis y para qué se utiliza Qué es Lucentis
Lucentis es una solución que se inyecta en el ojo. Lucentis pertenece a un grupo de medicamentos denominados agentes antineovascularización. Contiene el principio activo denominado ranibizumab.
Para qué se utiliza Lucentis
Lucentis se utiliza en adultos para tratar varias enfermedades oculares que causan alteración de la visión.
Estas enfermedades son el resultado de una lesión en la retina (capa sensible a la luz en la parte posterior del ojo) provocada por:
- El crecimiento de vasos sanguíneos anómalos, que pierden líquido (neovascularización coroidea, NVC). Esto se observa en enfermedades como la degeneración macular asociada a la edad (DMAE) o la miopía patológica (MP).
- Edema macular (hinchazón del centro de la retina). La causa de este hinchazón puede ser la diabetes (una enfermedad conocida como edema macular diabético (EMD)) o un bloqueo de las venas retinianas de la retina (una enfermedad conocida como oclusión de la vena de la retina (OVR)).
Cómo actúa Lucentis
Lucentis reconoce y se une de forma específica a una proteína denominada factor de crecimiento endotelial vascular A (VEGF-A) humano presente en los ojos. En exceso, el VEGF-A causa el crecimiento de vasos sanguíneos anómalos e hinchazón en el ojo que puede ocasionar una alteración de la visión en enfermedades como DMAE, MP, EMD u OVR. Mediante la unión al VEGF-A, Lucentis puede impedir que actúe y prevenir dicho crecimiento e hinchazón anómalos.
En estas enfermedades, Lucentis puede ayudar a estabilizar y, en muchos casos, mejorar su visión.
2. Qué necesita saber antes de que le administren Lucentis No le deben administrar Lucentis
- Si es alérgico al ranibizumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- Si tiene una infección en el ojo o alrededor del mismo.
- Si tiene dolor o enrojecimiento (inflamación intraocular grave) en el ojo.
Advertencias y precauciones
Consulte a su médico antes de que le administren Lucentis
- Lucentis se administra mediante una inyección en el ojo. Ocasionalmente, tras el tratamiento con Lucentis puede aparecer una infección en la parte interna del ojo, dolor o enrojecimiento (inflamación), desprendimiento o desgarro de una de las capas situadas en el fondo del ojo (desprendimiento o desgarro de la retina y desprendimiento o desgarro del epitelio pigmentario de la retina), o enturbiamiento del cristalino (catarata). Es importante identificar y tratar tal infección o desprendimiento de retina lo antes posible. Informe inmediatamente a su médico si nota signos como dolor en el ojo o aumento de las molestias en el ojo, si empeora el enrojecimiento en el ojo, visión borrosa o disminución de la visión, un aumento del número de pequeñas manchas en la visión o aumento de la sensibilidad a la luz.
- En algunos pacientes, después de la inyección la presión en el ojo puede aumentar durante un corto periodo de tiempo. Es posible que usted no se de cuenta de ello, por lo que puede que su médico le realice un seguimiento de la presión ocular después de cada inyección.
- Informe a su médico si ha tenido enfermedades en los ojos o ha recibido algún tratamiento en los ojos anteriormente, o si ha sufrido un accidente cerebrovascular o ha tenido signos pasajeros de accidente cerebrovascular (debilidad o parálisis de un miembro o cara, dificultad en el habla o en la comprensión). Esta información se tendrá en consideración para evaluar si Lucentis es el tratamiento apropiado para usted.
Niños y adolescentes (menos de 18 años)
No se recomienda el uso de Lucentis en niños y adolescentes ya que no se ha estudiado en estos grupos de edad.
Uso de Lucentis con otros medicamentos
Informe a su médico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
- Se aconseja a las mujeres que pueden quedarse embarazadas que utilicen un método anticonceptivo eficaz durante el tratamiento.
- No hay experiencia del uso de Lucentis en mujeres embarazadas; por tanto no se conocen los riesgos potenciales. Si está embarazada, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de iniciar el tratamiento con Lucentis.
- No se recomienda el uso de Lucentis durante la lactancia, porque se desconoce si Lucentis pasa a la leche materna. Consulte a su médico o farmacéutico antes del tratamiento con Lucentis.
Conducción y uso de máquinas
Después del tratamiento con Lucentis usted puede experimentar visión borrosa temporalmente. Si esto le ocurre, no conduzca ni use máquinas hasta que este síntoma desaparezca.
3. Cómo se administra Lucentis
Lucentis se administra por el oftalmólogo en forma de inyección única en el ojo bajo anestesia local. La dosis habitual de una inyección es 0,05 ml (que contiene 0,5 mg de principio activo). El intervalo entre dos dosis aplicadas en el mismo ojo debe ser como mínimo de cuatro semanas. Todas las inyecciones serán administradas por un oftalmólogo.
Para prevenir una infección, antes de la inyección su médico le lavará el ojo cuidadosamente. Su médico también le administrará un anestésico local para reducir o prevenir cualquier dolor que pudiera sentir con la inyección.
El tratamiento se inicia con una inyección de Lucentis cada mes. Su médico controlará la enfermedad de su ojo y dependiendo de cómo responda al tratamiento, decidirá si necesita o no recibir más tratamiento y cuándo necesita ser tratado.
Al final del prospecto en el apartado “Cómo preparar y administrar Lucentis” se dan instrucciones detalladas de uso.
Pacientes de edad avanzada (65 años y mayores)
Lucentis puede utilizarse en personas de 65 años de edad o más, y no es necesario un ajuste de la dosis.
Si no se le administra una dosis de Lucentis
Contacte con su médico o su hospital tan pronto como le sea posible para pedir una nueva cita.
Antes de interrumpir el tratamiento con Lucentis
Si usted se está planteando interrumpir el tratamiento con Lucentis, acuda a la siguiente consulta y coméntelo antes con su médico. Su médico le aconsejará y decidirá durante cuánto tiempo deberá ser tratado con Lucentis.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, Lucentis puede producir efectos adversos, aunque no todas las personas los sufran.
Los efectos adversos asociados con la administración de Lucentis se deben o al propio medicamento o al procedimiento de inyección y la mayoría afectan al ojo.
A continuación se describen los efectos adversos más graves:
Efectos adversos graves frecuentes (pueden afectar hasta 1 de cada 10 pacientes): Desprendimiento o desgarro de una capa en la parte interna del ojo (desprendimiento o desgarro de la retina), que da como resultado destellos de luz con partículas flotantes que progresan a una pérdida de visión transitoria o a un enturbiamiento del cristalino (catarata).
Efectos adversos graves poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes): Ceguera, infección del globo ocular (endoftalmitis) con inflamación de la parte interna del ojo.
Los síntomas que podría experimentar se describen en el apartado 2 de este prospecto (por favor lea el apartado 2 “Qué necesita saber antes de que le administren Lucentis”). Informe a su médico inmediatamente si presenta alguno de estos efectos adversos.
A continuación se describen los efectos adversos comunicados más frecuentemente:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes)
Los efectos adversos oculares incluyen: Inflamación del ojo, sangrado en la parte posterior del ojo (hemorragia en la retina), alteraciones visuales, dolor en el ojo, pequeñas partículas o manchas en la visión (partículas flotantes), sangre en el ojo, irritación del ojo, sensación de tener algo dentro del ojo, aumento de la producción de lágrima, inflamación o infección en el borde de los párpados, ojo seco, enrojecimiento o picor en el ojo y aumento de la presión en el ojo.
Los efectos adversos no oculares incluyen: Dolor de garganta, congestión nasal, goteo nasal, dolor de cabeza y dolor en las articulaciones.
A continuación se describen otros efectos adversos que pueden ocurrir tras el tratamiento con Lucentis:
Efectos adversos frecuentes
Los efectos adversos oculares incluyen: Disminución de la nitidez de la visión, hinchazón de una sección del ojo (úvea, córnea), inflamación de la córnea (parte delantera del ojo), pequeñas marcas en la superficie del ojo, visión borrosa, sangrado en el lugar de inyección, sangrado en el ojo, secreción del ojo con picor, enrojecimiento e hinchazón (conjuntivitis), sensibilidad a la luz, molestias en el ojo, hinchazón del párpado, dolor en el párpado.
Los efectos adversos no oculares incluyen: Infección de las vías urinarias, recuento de glóbulos rojos bajo (con síntomas tales como cansancio, dificultad al respirar, mareo, palidez), ansiedad, tos, náuseas, reacciones alérgicas tales como erupción, urticaria, picor y enrojecimiento de la piel.
Efectos adversos poco frecuentes
Los efectos adversos oculares incluyen: Inflamación y sangrado en la parte anterior del ojo, acúmulo de pus en el ojo, cambios en la parte central de la superficie ocular, dolor o irritación en el lugar de inyección, sensación anormal en el ojo, irritación del párpado.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Lucentis
- Mantener este medicamento fuera de la vista y del alcance de los niños.
- No utilice este medicamento después de la fecha de caducidad que aparece en la caja después de CAD y en la etiqueta del vial después de EXP. La fecha de caducidad es el último día del mes que se indica.
- Conservar en nevera (entre 2°C y 8°C). No congelar.
- Antes de usar, el vial sin abrir se puede conservar a temperatura ambiente (25°C) durante un máximo de 24 horas.
- Conservar el vial en el embalaje exterior para protegerlo de la luz.
- No utilice ningún envase que esté dañado.
Composición de Lucentis
- El principio activo es ranibizumab (10 mg/ml). Cada ml contiene 10 mg de ranibizumab.
- Los demás componentes son a,a-trehalosa dihidrato; hidrocloruro de histidina monohidrato; histidina; polisorbato 20; agua para inyectables.
Aspecto del producto y contenido del envase
Lucentis es una solución inyectable contenida en un vial (0,23 ml). La solución es transparente, de incolora a amarillo pálido y acuosa.
Lucentis se suministra en un envase que contiene un vial de vidrio con ranibizumab, con tapón de goma de clorobutilo.El vial es para un solo uso.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien Lietuva
Novartis Pharma N.V. Novartis Pharma Services Inc.
Tél/Tel: +32 2 246 16 11 Tel: +370 5 269 16 50
Etarapna
Novartis Pharma Services Inc. Tea.: +359 2 489 98 28
Ceská republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kórcpog Novartis Pharma Services Inc. T^: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
|
Fecha de la última revisión de este prospecto: |
EXXáóa
Novartis (Helias) A.E.B.E. Tr(k: +30 210 281 17 12
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
Ver también la sección 3 “Cómo se administra Lucentis”.
Cómo preparar y administrar Lucentis
Vial para un solo uso. Únicamente para vía intravítrea.
Lucentis debe ser administrado por un oftalmólogo que tenga experiencia en la administración de inyecciones intravítreas.
En la DMAE exudativa y en la alteración visual debida a EMD, a edema macular secundario a OVR o a NVC secundaria a MP, la dosis recomendada de Lucentis es 0,5 mg administrada en forma de inyección intravítrea única. Esto corresponde a un volumen de inyección de 0,05 ml. El intervalo entre dos dosis inyectadas en el mismo ojo debe ser como mínimo de cuatro semanas.
El tratamiento se inicia con una inyección al mes hasta alcanzar la agudeza visual máxima y/o no haya signos de actividad de la enfermedad, es decir ningún cambio en la agudeza visual ni en otros signos y síntomas de la enfermedad bajo tratamiento continuado. En pacientes con DMAE exudativa, EMD y OVR inicialmente pueden ser necesarias tres o más inyecciones consecutivas administradas mensualmente.
A partir de ese momento, los intervalos de monitorización y tratamiento se deben determinar según criterio médico y en base a la actividad de la enfermedad, valorada mediante la agudeza visual y/o parámetros anatómicos.
Se debe interrumpir el tratamiento con Lucentis si bajo criterio del médico, los parámetros visuales y anatómicos indican que el paciente no se está beneficiando del tratamiento continuado.
La monitorización para determinar la actividad de la enfermedad puede incluir examen clínico, control funcional o técnicas de imagen (p. ej. tomografía de coherencia óptica o angiografía con fluoresceína).
Si se está tratando a los pacientes de acuerdo a un régimen de tratar y extender, una vez se ha alcanzado la agudeza visual máxima y/o no hay signos de actividad de la enfermedad, los intervalos de tratamiento se pueden espaciar de forma gradual hasta que vuelvan a aparecer signos de actividad de la enfermedad o alteración visual. En el caso de la DMAE exudativa el intervalo de tratamiento no debe espaciarse en más de dos semanas cada vez y en el caso del EMD se puede espaciar hasta un mes cada vez. Para la OVR, los intervalos de tratamiento también pueden espaciarse de forma gradual, sin embargo los datos que hay no son suficientes para determinar la duración de estos intervalos. Si vuelve a aparecer actividad de la enfermedad, se debe acortar el intervalo de tratamiento de manera consecuente.
En el tratamiento de la alteración visual debida a la NVC secundaria a MP, muchos pacientes pueden necesitar únicamente una o dos inyecciones durante el primer año, mientras que algunos pacientes pueden necesitar tratamiento con mayor frecuencia.
Lucentis y fotocoagulación con láser en EMD y edema macular secundario a oclusión de la rama venosa retiniana (ORVR)
Existe alguna experiencia con Lucentis administrado concomitantemente con fotocoagulación con láser. Cuando se administren en el mismo día, Lucentis debe ser administrado como mínimo 30 minutos después de la fotocoagulación con láser. Lucentis puede administrarse en pacientes que han recibido fotocoagulación con láser previamente.
Lucentis y la terapia fotodinámica con Visudyne en la NVC secundaria a MP No hay experiencia en la administración concomitante de Lucentis y Visudyne.
Antes de la administración de Lucentis se debe comprobar visualmente la ausencia de partículas y decoloración.
El procedimiento de inyección deberá llevarse a cabo bajo condiciones asépticas, que incluyen el lavado quirúrgico de las manos, el uso de guantes estériles, un campo estéril, un blefarostato estéril para los párpados (o equivalente) y la disponibilidad de una paracentesis estéril (en caso necesario). Antes de realizar el procedimiento de inyección intravítrea, se deberá evaluar detalladamente la historia clínica del paciente en cuanto a reacciones de hipersensibilidad. Antes de la inyección se debe administrar una anestesia adecuada y un microbicida tópico de amplio espectro para desinfectar la piel de la zona periocular, párpado y superficie ocular, de acuerdo con la práctica local.
Para la preparación de Lucentis para administración intravítrea, siga las siguientes instrucciones:
El vial es para un solo uso. Tras la inyección se debe desechar cualquier sobrante de producto no utilizado. No debe utilizarse ningún vial que muestre signos de deterioro o manipulación. La esterilidad sólo se puede garantizar si el sellado del envase se mantiene intacto.
Para la preparación y la inyección intravítrea se necesitan los siguientes productos sanitarios de un solo uso:
- una aguja con filtro de 5 pm (18G)
- una jeringa estéril de 1 ml
- una aguja para inyección (30G x 'A").
Estos productos sanitarios no se incluyen en el envase de Lucentis.

1. Antes de extraer la solución, se debe desinfectar la parte exterior del tapón de goma del vial.
2. Incorporar una aguja con filtro de 5 pm (18G) a una jeringa de 1 ml usando técnicas asépticas. Insertar la aguja con filtro en el centro del tapón del vial hasta que la aguja toque el extremo inferior del vial.
3. Extraer todo el líquido del vial, manteniendo el vial en posición vertical, ligeramente inclinado para facilitar la extracción completa.
4. Al vaciar el vial, asegurar que el émbolo se retira hacia atrás lo suficiente de forma que se vacíe por completo la aguja con filtro.
5. Dejar la aguja con filtro en el vial y desconectarla de la jeringa. La aguja con filtro se debe desechar tras extraer el contenido del vial, y no se debe utilizar para la inyección intravítrea.
6. Incorporar una aguja para inyección (30G x V") a la jeringa con firmeza y de forma aséptica.

7. Quitar la cápsula de cierre de la aguja para inyección cuidadosamente sin desconectar la aguja para inyección de la jeringa.
Nota: Sujetar la aguja para inyección por el cono mientras se retira la cápsula de cierre.
8. Expulsar el aire de la jeringa y ajustar la dosis hasta la marca de 0,05 ml en la jeringa cuidadosamente. La jeringa está lista para la inyección.
Nota: No secar la aguja para inyección. No tirar del émbolo hacia atrás.
La aguja para inyección se deberá introducir 3,5-4,0 mm por detrás del limbo en la cavidad vítrea, evitando el meridiano horizontal y en dirección al centro del globo. Seguidamente debe liberarse el volumen de inyección de 0,05 ml; las inyecciones siguientes deberán aplicarse cada vez en un punto escleral distinto.
Tras la inyección, no tapar la aguja con la cápsula de cierre ni separarla de la jeringa. Eliminar la jeringa usada junto con la aguja en un contenedor para objetos punzantes o eliminar de acuerdo con la normativa local.
Lucentis 10 mg/ml solución inyectable en jeringa precargada
Ranibizumab
Lea todo el prospecto detenidamente antes de que le administren este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto.
Contenido del prospecto
1. Qué es Lucentis y para qué se utiliza
2. Qué necesita saber antes de que le administren Lucentis
3. Cómo se administra Lucentis
4. Posibles efectos adversos
5. Conservación de Lucentis
6. Contenido del envase e información adicional
1. Qué es Lucentis y para qué se utiliza Qué es Lucentis
Lucentis es una solución que se inyecta en el ojo. Lucentis pertenece a un grupo de medicamentos denominados agentes antineovascularización. Contiene el principio activo denominado ranibizumab.
Para qué se utiliza Lucentis
Lucentis se utiliza en adultos para tratar varias enfermedades oculares que causan alteración de la visión.
Estas enfermedades son el resultado de una lesión en la retina (capa sensible a la luz en la parte posterior del ojo) provocada por:
- El crecimiento de vasos sanguíneos anómalos, que pierden líquido (neovascularización coroidea, NVC). Esto se observa en enfermedades como la degeneración macular asociada a la edad (DMAE) o la miopía patológica (MP).
- Edema macular (hinchazón del centro de la retina). La causa de este hinchazón puede ser la diabetes (una enfermedad conocida como edema macular diabético (EMD)) o un bloqueo de las venas retinianas de la retina (una enfermedad conocida como oclusión de la vena de la retina (OVR)).
Cómo actúa Lucentis
Lucentis reconoce y se une de forma específica a una proteína denominada factor de crecimiento endotelial vascular A (VEGF-A) humano presente en los ojos. En exceso, el VEGF-A causa el crecimiento de vasos sanguíneos anómalos e hinchazón en el ojo que puede ocasionar una alteración de la visión en enfermedades como DMAE, MP, EMD u OVR. Mediante la unión al VEGF-A, Lucentis puede impedir que actúe y prevenir dicho crecimiento e hinchazón anómalos.
En estas enfermedades, Lucentis puede ayudar a estabilizar y, en muchos casos, mejorar su visión.
2. Qué necesita saber antes de que le administren Lucentis No le deben administrar Lucentis
- Si es alérgico al ranibizumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- Si tiene una infección en el ojo o alrededor del mismo.
- Si tiene dolor o enrojecimiento (inflamación intraocular grave) en el ojo.
Advertencias y precauciones
Consulte a su médico antes de que le administren Lucentis
- Lucentis se administra mediante una inyección en el ojo. Ocasionalmente, tras el tratamiento con Lucentis puede aparecer una infección en la parte interna del ojo, dolor o enrojecimiento (inflamación), desprendimiento o desgarro de una de las capas situadas en el fondo del ojo (desprendimiento o desgarro de la retina y desprendimiento o desgarro del epitelio pigmentario de la retina), o enturbiamiento del cristalino (catarata). Es importante identificar y tratar tal infección o desprendimiento de retina lo antes posible. Informe inmediatamente a su médico si nota signos como dolor en el ojo o aumento de las molestias en el ojo, si empeora el enrojecimiento en el ojo, visión borrosa o disminución de la visión, un aumento del número de pequeñas manchas en la visión o aumento de la sensibilidad a la luz.
- En algunos pacientes, después de la inyección la presión en el ojo puede aumentar durante un corto periodo de tiempo. Es posible que usted no se de cuenta de ello, por lo que puede que su médico le realice un seguimiento de la presión ocular después de cada inyección.
- Informe a su médico si ha tenido enfermedades en los ojos o ha recibido algún tratamiento en los ojos anteriormente, o si ha sufrido un accidente cerebrovascular o ha tenido signos pasajeros de accidente cerebrovascular (debilidad o parálisis de un miembro o cara, dificultad en el habla o en la comprensión). Esta información se tendrá en consideración para evaluar si Lucentis es el tratamiento apropiado para usted.
Niños y adolescentes (menos de 18 años)
No se recomienda el uso de Lucentis en niños y adolescentes ya que no se ha estudiado en estos grupos de edad.
Uso de Lucentis con otros medicamentos
Informe a su médico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
- Se aconseja a las mujeres que pueden quedarse embarazadas que utilicen un método anticonceptivo eficaz durante el tratamiento.
- No hay experiencia del uso de Lucentis en mujeres embarazadas; por tanto no se conocen los riesgos potenciales. Si está embarazada, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de iniciar el tratamiento con Lucentis.
- No se recomienda el uso de Lucentis durante la lactancia, porque se desconoce si Lucentis pasa a la leche materna. Consulte a su médico o farmacéutico antes del tratamiento con Lucentis.
Conducción y uso de máquinas
Después del tratamiento con Lucentis usted puede experimentar visión borrosa temporalmente. Si esto le ocurre, no conduzca ni use máquinas hasta que este síntoma desaparezca.
3. Cómo se administra Lucentis
Lucentis se administra por el oftalmólogo en forma de inyección única en el ojo bajo anestesia local. La dosis habitual de una inyección es 0,05 ml (que contiene 0,5 mg de principio activo). La jeringa precargada contiene más cantidad que la dosis recomendada de 0,5 mg. El volumen extraíble no se administrará en su totalidad. El exceso de volumen se debe expulsar antes de la inyección. Si se inyecta todo el volumen de la jeringa precargada puede dar lugar a una sobredosis.
El intervalo entre dos dosis aplicadas en el mismo ojo debe ser como mínimo de cuatro semanas. Todas las inyecciones serán administradas por un oftalmólogo.
Para prevenir una infección, antes de la inyección su médico le lavará el ojo cuidadosamente. Su médico también le administrará un anestésico local para reducir o prevenir cualquier dolor que pudiera sentir con la inyección.
El tratamiento se inicia con una inyección de Lucentis cada mes. Su médico controlará la enfermedad de su ojo y dependiendo de cómo responda al tratamiento, decidirá si necesita o no recibir más tratamiento y cuándo necesita ser tratado.
Al final del prospecto en el apartado “Cómo preparar y administrar Lucentis” se dan instrucciones detalladas de uso.
Pacientes de edad avanzada (65 años y mayores)
Lucentis puede utilizarse en personas de 65 años de edad o más, y no es necesario un ajuste de la dosis.
Si no se le administra una dosis de Lucentis
Contacte con su médico o su hospital tan pronto como le sea posible para pedir una nueva cita.
Antes de interrumpir el tratamiento con Lucentis
Si usted se está planteando interrumpir el tratamiento con Lucentis, acuda a la siguiente consulta y coméntelo antes con su médico. Su médico le aconsejará y decidirá durante cuánto tiempo deberá ser tratado con Lucentis.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, Lucentis puede producir efectos adversos, aunque no todas las personas los sufran.
Los efectos adversos asociados con la administración de Lucentis se deben o al propio medicamento o al procedimiento de inyección y la mayoría afectan al ojo.
A continuación se describen los efectos adversos más graves:
Efectos adversos graves frecuentes (pueden afectar hasta 1 de cada 10 pacientes): Desprendimiento o desgarro de una capa en la parte interna del ojo (desprendimiento o desgarro de la retina), que da como resultado destellos de luz con partículas flotantes que progresan a una pérdida de visión transitoria o a un enturbiamiento del cristalino (catarata).
Efectos adversos graves poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes): Ceguera, infección del globo ocular (endoftalmitis) con inflamación de la parte interna del ojo.
Los síntomas que podría experimentar se describen en el apartado 2 de este prospecto (por favor lea el apartado 2 “Qué necesita saber antes de que le administren Lucentis”). Informe a su médico inmediatamente si presenta alguno de estos efectos adversos.
A continuación se describen los efectos adversos comunicados más frecuentemente:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes)
Los efectos adversos oculares incluyen: Inflamación del ojo, sangrado en la parte posterior del ojo (hemorragia en la retina), alteraciones visuales, dolor en el ojo, pequeñas partículas o manchas en la visión (partículas flotantes), sangre en el ojo, irritación del ojo, sensación de tener algo dentro del ojo, aumento de la producción de lágrima, inflamación o infección en el borde de los párpados, ojo seco, enrojecimiento o picor en el ojo y aumento de la presión en el ojo.
Los efectos adversos no oculares incluyen: Dolor de garganta, congestión nasal, goteo nasal, dolor de cabeza y dolor en las articulaciones.
A continuación se describen otros efectos adversos que pueden ocurrir tras el tratamiento con Lucentis:
Efectos adversos frecuentes
Los efectos adversos oculares incluyen: Disminución de la nitidez de la visión, hinchazón de una sección del ojo (úvea, córnea), inflamación de la córnea (parte delantera del ojo), pequeñas marcas en la superficie del ojo, visión borrosa, sangrado en el lugar de inyección, sangrado en el ojo, secreción del ojo con picor, enrojecimiento e hinchazón (conjuntivitis), sensibilidad a la luz, molestias en el ojo, hinchazón del párpado, dolor en el párpado.
Los efectos adversos no oculares incluyen: Infección de las vías urinarias, recuento de glóbulos rojos bajo (con síntomas tales como cansancio, dificultad al respirar, mareo, palidez), ansiedad, tos, náuseas, reacciones alérgicas tales como erupción, urticaria, picor y enrojecimiento de la piel.
Efectos adversos poco frecuentes
Los efectos adversos oculares incluyen: Inflamación y sangrado en la parte anterior del ojo, acúmulo de pus en el ojo, cambios en la parte central de la superficie ocular, dolor o irritación en el lugar de inyección, sensación anormal en el ojo, irritación del párpado.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Lucentis
- Mantener este medicamento fuera de la vista y del alcance de los niños.
- No utilice este medicamento después de la fecha de caducidad que aparece en la caja después de CAD y en la etiqueta de la jeringa precargada después de EXP. La fecha de caducidad es el último día del mes que se indica.
- Conservar en nevera (entre 2°C y 8°C). No congelar.
- Antes de usar, la bandeja sellada se puede conservar a temperatura ambiente (25°C) durante un máximo de 24 horas.
- Conservar la jeringa precargada en su bandeja sin abrir en la caja para protegerla de la luz.
- No utilice ningún envase que esté dañado.
Composición de Lucentis
- El principio activo es ranibizumab (10 mg/ml). Cada ml contiene 10 mg de ranibizumab.
- Los demás componentes son a,a-trehalosa dihidrato; hidrocloruro de histidina monohidrato; histidina; polisorbato 20; agua para inyectables.
Aspecto del producto y contenido del envase
Lucentis es una solución inyectable contenida en una jeringa precargada. La jeringa precargada contiene 0,165 ml de una solución acuosa, estéril, transparente, de incolora a amarillo pálido. La jeringa precargada contiene más cantidad que la dosis recomendada de 0,5 mg. El volumen extraíble no se administrará en su totalidad. El exceso de volumen se debe expulsar antes de la inyección. Si se inyecta todo el volumen de la jeringa precargada puede dar lugar a una sobredosis.
El tamaño de envase es de una jeringa precargada, envasada en una bandeja contenedora sellada. La jeringa precargada es para un solo uso.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Etarapna
Novartis Pharma Services Inc. Tea.: +359 2 489 98 28
Ceská republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Lietuva
Novartis Pharma Services Inc. Tel: +370 5 269 16 50
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Eesti
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Novartis Pharma Services Inc. Tel: +372 66 30 810
|
EXXáóa Novartis (Hellas) A.E.B.E. Tr(k: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kórcpog Novartis Pharma Services Inc. T^: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
|
Fecha de la última revisión de este prospecto: |
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
Ver también la sección 3 “Cómo se administra Lucentis”.
Cómo preparar y administrar Lucentis
Jeringa precargada para un solo uso. Unicamente para vía intravítrea.
Lucentis debe ser administrado por un oftalmólogo que tenga experiencia en la administración de inyecciones intravítreas.
En la DMAE exudativa y en la alteración visual debida a EMD, a edema macular secundario a OVR o a NVC secundaria a MP, la dosis recomendada de Lucentis es 0,5 mg administrada en forma de inyección intravítrea única. Esto corresponde a un volumen de inyección de 0,05 ml. El intervalo entre dos dosis inyectadas en el mismo ojo debe ser como mínimo de cuatro semanas.
El tratamiento se inicia con una inyección al mes hasta alcanzar la agudeza visual máxima y/o no haya signos de actividad de la enfermedad, es decir ningún cambio en la agudeza visual ni en otros signos y síntomas de la enfermedad bajo tratamiento continuado. En pacientes con DMAE exudativa, EMD y OVR inicialmente pueden ser necesarias tres o más inyecciones consecutivas administradas mensualmente.
A partir de ese momento, los intervalos de monitorización y tratamiento se deben determinar según criterio médico y en base a la actividad de la enfermedad, valorada mediante la agudeza visual y/o parámetros anatómicos.
Se debe interrumpir el tratamiento con Lucentis si bajo criterio del médico, los parámetros visuales y anatómicos indican que el paciente no se está beneficiando del tratamiento continuado.
La monitorización para determinar la actividad de la enfermedad puede incluir examen clínico, control funcional o técnicas de imagen (p. ej. tomografía de coherencia óptica o angiografía con fluoresceína).
Si se está tratando a los pacientes de acuerdo a un régimen de tratar y extender, una vez se ha alcanzado la agudeza visual máxima y/o no hay signos de actividad de la enfermedad, los intervalos de tratamiento se pueden espaciar de forma gradual hasta que vuelvan a aparecer signos de actividad de la enfermedad o alteración visual. En el caso de la DMAE exudativa el intervalo de tratamiento no debe espaciarse en más de dos semanas cada vez y en el caso del EMD se puede espaciar hasta un mes cada vez. Para la OVR, los intervalos de tratamiento también pueden espaciarse de forma gradual, sin embargo los datos que hay no son suficientes para determinar la duración de estos intervalos. Si vuelve a aparecer actividad de la enfermedad, se debe acortar el intervalo de tratamiento de manera consecuente.
En el tratamiento de la alteración visual debida a la NVC secundaria a MP, muchos pacientes pueden necesitar únicamente una o dos inyecciones durante el primer año, mientras que algunos pacientes pueden necesitar tratamiento con mayor frecuencia.
Lucentis y fotocoagulación con láser en EMD y edema macular secundario a oclusión de la rama venosa retiniana (ORVR)
Existe alguna experiencia con Lucentis administrado concomitantemente con fotocoagulación con láser. Cuando se administren en el mismo día, Lucentis debe ser administrado como mínimo 30 minutos después de la fotocoagulación con láser. Lucentis puede administrarse en pacientes que han recibido fotocoagulación con láser previamente.
Lucentis y la terapia fotodinámica con Visudyne en la NVC secundaria a MP No hay experiencia en la administración concomitante de Lucentis y Visudyne.
Antes de la administración de Lucentis se debe comprobar visualmente la ausencia de partículas y decoloración.
El procedimiento de inyección deberá llevarse a cabo bajo condiciones asépticas, que incluyen el lavado quirúrgico de las manos, el uso de guantes estériles, un campo estéril, un blefarostato estéril para los párpados (o equivalente) y la disponibilidad de una paracentesis estéril (en caso necesario). Antes de realizar el procedimiento de inyección intravítrea, se deberá evaluar detalladamente la historia clínica del paciente en cuanto a reacciones de hipersensibilidad. Antes de la inyección se debe administrar una anestesia adecuada y un microbicida tópico de amplio espectro para desinfectar la piel de la zona periocular, párpado y superficie ocular, de acuerdo con la práctica local.
La jeringa precargada es para un solo uso. La jeringa precargada es estéril. No use el producto si el envase está deteriorado. La esterilidad de la jeringa precargada sólo se puede garantizar si la bandeja se mantiene sellada. No use la jeringa precargada si la solución ha cambiado de color, está turbia o contiene partículas.
La jeringa precargada contiene más cantidad que la dosis recomendada de 0,5 mg. El volumen extraíble de la jeringa precargada (0,1 ml) no se administrará en su totalidad. El exceso de volumen se debe expulsar antes de la inyección. Si se inyecta todo el volumen de la jeringa precargada puede dar lugar a una sobredosis. Para expulsar las burbujas de aire y el exceso de medicamento, presione lentamente el émbolo hasta que el borde inferior de la cúpula que forma el extremo del tapón de goma quede alineado con la línea negra de dosificación de la jeringa (equivalente a 0,05 ml, es decir, 0,5 mg de ranibizumab).
Para la inyección intravítrea, se debe usar una aguja para inyección estéril 30G x A".
Para la preparación de Lucentis para administración intravítrea, siga las instrucciones de uso:
Lea todas las instrucciones detenidamente antes de usar la jeringa precargada.
La jeringa precargada es para un solo uso. La jeringa precargada es estéril. No usar el producto si el envase está deteriorado. La apertura de la bandeja sellada y los siguientes pasos se deben realizar bajo condiciones asépticas.
Nota: La dosis a administrar se debe ajustar a 0,05 ml._
Cápsula de cierre de la jeringa
Marca de dosis Aleta de sujeción de 0,05 ml
|
i- |
i |
* \\ | |
|
b ] | |||
|
_i— ' | |||
Luer lock Tapón de goma
Émbolo
1.
2.
4.
Asegurarse que el envase contiene:
una jeringa precargada estéril en una bandeja sellada.
Quitar la cubierta de la bandeja contenedora de la jeringa y, usando una
técnica aséptica, extraer la jeringa cuidadosamente._
Comprobar que:
la cápsula de cierre de la jeringa no esté separado del Luer lock. la jeringa no esté deteriorada. la solución tenga un aspecto transparente, de incolora a amarillo pálido y no contenga ninguna partícula.
Si alguno de los puntos anteriores no es cierto, desechar la jeringa precargada y
usar una nueva._
Tirar y desprender la cápsula de cierre de la jeringa (no lo gire ni lo retuerza) (ver Figura 2).
Desechar la cápsula de cierre de la jeringa (ver Figura 3).

Figura 2

Figura 3
7. Conectar con firmeza a la j eringa una aguja para inyección estéril 30G x A", enroscando bien la aguja en el Luer lock (ver Figura 4).
8. Quitar la cápsula de cierre de la aguja cuidadosamente, tirando directamente de él hacia fuera (ver Figura 5).
Nota: No secar la aguja en ningún momento.

Figura 4 Figura 5
|
Extraer las |
9. |
|
burbujas de |
c |
|
aire |
10. S s |
Mantener la jeringa en posición vertical con la aguja dirigida hacia arriba.
Si hay alguna burbuja de aire, golpear suavemente la jeringa con el dedo hasta que las burbujas asciendan a su parte superior (ver Figura 6).

Figura 6
11. Mantener la jeringa a la altura de los ojos y presionar cuidadosamente el émbolo hasta que el borde inferior de la cúpula que forma el extremo del tapón de goma quede alineado con la marca de dosis (ver Figura 7). Con esto se expulsará el aire y el exceso de solución y se ajustará la dosis a 0,05 ml.
Nota: El émbolo no está unido al tapón de goma - esto es para evitar la entrada de aire en la jeringa.

Figura 7
El procedimiento de inyección debe llevarse a cabo bajo condiciones asépticas.
12. La aguja para inyección se debe introducir 3,5-4,0 mm por detrás del limbo en la cavidad vítrea, evitando el meridiano horizontal y en dirección al centro del globo.
13. Inyectar lentamente hasta que el tapón de goma llegue a la parte inferior de la jeringa, con el fin de administrar el volumen de 0,05 ml.
14. Las inyecciones siguientes se deben aplicar cada vez en un punto escleral distinto.
15. Tras la inyección, no tapar la aguja con la cápsula de cierre ni separarla de la
jeringa. Eliminar la jeringa usada junto con la aguja en un contenedor para objetos punzantes o eliminar de acuerdo con la normativa local._
Lucentis 10 mg/ml solución inyectable
Ranibizumab
Lea todo el prospecto detenidamente antes de que le administren este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto.
Contenido del prospecto
1. Qué es Lucentis y para qué se utiliza
2. Qué necesita saber antes de que le administren Lucentis
3. Cómo se administra Lucentis
4. Posibles efectos adversos
5. Conservación de Lucentis
6. Contenido del envase e información adicional
1. Qué es Lucentis y para qué se utiliza Qué es Lucentis
Lucentis es una solución que se inyecta en el ojo. Lucentis pertenece a un grupo de medicamentos denominados agentes antineovascularización. Contiene el principio activo denominado ranibizumab.
Para qué se utiliza Lucentis
Lucentis se utiliza en adultos para tratar varias enfermedades oculares que causan alteración de la visión.
Estas enfermedades son el resultado de una lesión en la retina (capa sensible a la luz en la parte posterior del ojo) provocada por:
- El crecimiento de vasos sanguíneos anómalos, que pierden líquido (neovascularización coroidea, NVC). Esto se observa en enfermedades como la degeneración macular asociada a la edad (DMAE) o la miopía patológica (MP).
- Edema macular (hinchazón del centro de la retina). La causa de este hinchazón puede ser la diabetes (una enfermedad conocida como edema macular diabético (EMD)) o un bloqueo de las venas retinianas de la retina (una enfermedad conocida como oclusión de la vena de la retina (OVR)).
Cómo actúa Lucentis
Lucentis reconoce y se une de forma específica a una proteína denominada factor de crecimiento endotelial vascular A (VEGF-A) humano presente en los ojos. En exceso, el VEGF-A causa el crecimiento de vasos sanguíneos anómalos e hinchazón en el ojo que puede ocasionar una alteración de la visión en enfermedades como DMAE, MP, EMD u OVR. Mediante la unión al VEGF-A, Lucentis puede impedir que actúe y prevenir dicho crecimiento e hinchazón anómalos.
En estas enfermedades, Lucentis puede ayudar a estabilizar y, en muchos casos, mejorar su visión.
2. Qué necesita saber antes de que le administren Lucentis No le deben administrar Lucentis
- Si es alérgico al ranibizumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- Si tiene una infección en el ojo o alrededor del mismo.
- Si tiene dolor o enrojecimiento (inflamación intraocular grave) en el ojo.
Advertencias y precauciones
Consulte a su médico antes de que le administren Lucentis
- Lucentis se administra mediante una inyección en el ojo. Ocasionalmente, tras el tratamiento con Lucentis puede aparecer una infección en la parte interna del ojo, dolor o enrojecimiento (inflamación), desprendimiento o desgarro de una de las capas situadas en el fondo del ojo (desprendimiento o desgarro de la retina y desprendimiento o desgarro del epitelio pigmentario de la retina), o enturbiamiento del cristalino (catarata). Es importante identificar y tratar tal infección o desprendimiento de retina lo antes posible. Informe inmediatamente a su médico si nota signos como dolor en el ojo o aumento de las molestias en el ojo, si empeora el enrojecimiento en el ojo, visión borrosa o disminución de la visión, un aumento del número de pequeñas manchas en la visión o aumento de la sensibilidad a la luz.
- En algunos pacientes, después de la inyección la presión en el ojo puede aumentar durante un corto periodo de tiempo. Es posible que usted no se de cuenta de ello, por lo que puede que su médico le realice un seguimiento de la presión ocular después de cada inyección.
- Informe a su médico si ha tenido enfermedades en los ojos o ha recibido algún tratamiento en los ojos anteriormente, o si ha sufrido un accidente cerebrovascular o ha tenido signos pasajeros de accidente cerebrovascular (debilidad o parálisis de un miembro o cara, dificultad en el habla o en la comprensión). Esta información se tendrá en consideración para evaluar si Lucentis es el tratamiento apropiado para usted.
Niños y adolescentes (menos de 18 años)
No se recomienda el uso de Lucentis en niños y adolescentes ya que no se ha estudiado en estos grupos de edad.
Uso de Lucentis con otros medicamentos
Informe a su médico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
- Se aconseja a las mujeres que pueden quedarse embarazadas que utilicen un método anticonceptivo eficaz durante el tratamiento.
- No hay experiencia del uso de Lucentis en mujeres embarazadas; por tanto no se conocen los riesgos potenciales. Si está embarazada, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de iniciar el tratamiento con Lucentis.
- No se recomienda el uso de Lucentis durante la lactancia, porque se desconoce si Lucentis pasa a la leche materna. Consulte a su médico o farmacéutico antes del tratamiento con Lucentis.
Conducción y uso de máquinas
Después del tratamiento con Lucentis usted puede experimentar visión borrosa temporalmente. Si esto le ocurre, no conduzca ni use máquinas hasta que este síntoma desaparezca.
3. Cómo se administra Lucentis
Lucentis se administra por el oftalmólogo en forma de inyección única en el ojo bajo anestesia local. La dosis habitual de una inyección es 0,05 ml (que contiene 0,5 mg de principio activo). El intervalo entre dos dosis aplicadas en el mismo ojo debe ser como mínimo de cuatro semanas. Todas las inyecciones serán administradas por un oftalmólogo.
Para prevenir una infección, antes de la inyección su médico le lavará el ojo cuidadosamente. Su médico también le administrará un anestésico local para reducir o prevenir cualquier dolor que pudiera sentir con la inyección.
El tratamiento se inicia con una inyección de Lucentis cada mes. Su médico controlará la enfermedad de su ojo y dependiendo de cómo responda al tratamiento, decidirá si necesita o no recibir más tratamiento y cuándo necesita ser tratado.
Al final del prospecto en el apartado “Cómo preparar y administrar Lucentis” se dan instrucciones detalladas de uso.
Pacientes de edad avanzada (65 años y mayores)
Lucentis puede utilizarse en personas de 65 años de edad o más, y no es necesario un ajuste de la dosis.
Si no se le administra una dosis de Lucentis
Contacte con su médico o su hospital tan pronto como le sea posible para pedir una nueva cita.
Antes de interrumpir el tratamiento con Lucentis
Si usted se está planteando interrumpir el tratamiento con Lucentis, acuda a la siguiente consulta y coméntelo antes con su médico. Su médico le aconsejará y decidirá durante cuánto tiempo deberá ser tratado con Lucentis.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, Lucentis puede producir efectos adversos, aunque no todas las personas los sufran.
Los efectos adversos asociados con la administración de Lucentis se deben o al propio medicamento o al procedimiento de inyección y la mayoría afectan al ojo.
A continuación se describen los efectos adversos más graves:
Efectos adversos graves frecuentes (pueden afectar hasta 1 de cada 10 pacientes): Desprendimiento o desgarro de una capa en la parte interna del ojo (desprendimiento o desgarro de la retina), que da como resultado destellos de luz con partículas flotantes que progresan a una pérdida de visión transitoria o a un enturbiamiento del cristalino (catarata).
Efectos adversos graves poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes): Ceguera, infección del globo ocular (endoftalmitis) con inflamación de la parte interna del ojo.
Los síntomas que podría experimentar se describen en el apartado 2 de este prospecto (por favor lea el apartado 2 “Qué necesita saber antes de que le administren Lucentis”). Informe a su médico inmediatamente si presenta alguno de estos efectos adversos.
A continuación se describen los efectos adversos comunicados más frecuentemente:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes)
Los efectos adversos oculares incluyen: Inflamación del ojo, sangrado en la parte posterior del ojo (hemorragia en la retina), alteraciones visuales, dolor en el ojo, pequeñas partículas o manchas en la visión (partículas flotantes), sangre en el ojo, irritación del ojo, sensación de tener algo dentro del ojo, aumento de la producción de lágrima, inflamación o infección en el borde de los párpados, ojo seco, enrojecimiento o picor en el ojo y aumento de la presión en el ojo.
Los efectos adversos no oculares incluyen: Dolor de garganta, congestión nasal, goteo nasal, dolor de cabeza y dolor en las articulaciones.
A continuación se describen otros efectos adversos que pueden ocurrir tras el tratamiento con Lucentis:
Efectos adversos frecuentes
Los efectos adversos oculares incluyen: Disminución de la nitidez de la visión, hinchazón de una sección del ojo (úvea, córnea), inflamación de la córnea (parte delantera del ojo), pequeñas marcas en la superficie del ojo, visión borrosa, sangrado en el lugar de inyección, sangrado en el ojo, secreción del ojo con picor, enrojecimiento e hinchazón (conjuntivitis), sensibilidad a la luz, molestias en el ojo, hinchazón del párpado, dolor en el párpado.
Los efectos adversos no oculares incluyen: Infección de las vías urinarias, recuento de glóbulos rojos bajo (con síntomas tales como cansancio, dificultad al respirar, mareo, palidez), ansiedad, tos, náuseas, reacciones alérgicas tales como erupción, urticaria, picor y enrojecimiento de la piel.
Efectos adversos poco frecuentes
Los efectos adversos oculares incluyen: Inflamación y sangrado en la parte anterior del ojo, acúmulo de pus en el ojo, cambios en la parte central de la superficie ocular, dolor o irritación en el lugar de inyección, sensación anormal en el ojo, irritación del párpado.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Lucentis
- Mantener este medicamento fuera de la vista y del alcance de los niños.
- No utilice este medicamento después de la fecha de caducidad que aparece en la caja después de CAD y en la etiqueta del vial después de EXP. La fecha de caducidad es el último día del mes que se indica.
- Conservar en nevera (entre 2°C y 8°C). No congelar.
- Antes de usar, el vial sin abrir se puede conservar a temperatura ambiente (25°C) durante un máximo de 24 horas.
- Conservar el vial en el embalaje exterior para protegerlo de la luz.
- No utilice ningún envase que esté dañado.
Composición de Lucentis
- El principio activo es ranibizumab (10 mg/ml). Cada ml contiene 10 mg de ranibizumab.
- Los demás componentes son a,a-trehalosa dihidrato; hidrocloruro de histidina monohidrato; histidina; polisorbato 20; agua para inyectables.
Aspecto del producto y contenido del envase
Lucentis es una solución inyectable contenida en un vial (0,23 ml). La solución es transparente, de incolora a amarillo pálido y acuosa.
Lucentis se suministra en un envase que contiene un vial de vidrio con ranibizumab, con tapón de goma de clorobutilo y una aguja roma con filtro (18G x IV2", 1,2 mm x 40 mm, 5 micrómetros) para extraer el contenido del vial. Todos los componentes son para un solo uso.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien Lietuva
Novartis Pharma N.V. Novartis Pharma Services Inc.
Tél/Tel: +32 2 246 16 11 Tel: +370 5 269 16 50
Etarapna
Novartis Pharma Services Inc. Tea.: +359 2 489 98 28
Ceská republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kórcpog Novartis Pharma Services Inc. T^: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
|
Fecha de la última revisión de este prospecto: |
EXXáóa
Novartis (Helias) A.E.B.E. Tr(k: +30 210 281 17 12
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
Ver también la sección 3 “Cómo se administra Lucentis”.
Cómo preparar y administrar Lucentis
Vial para un solo uso. Unicamente para vía intravítrea.
Lucentis debe ser administrado por un oftalmólogo que tenga experiencia en la administración de inyecciones intravítreas.
En la DMAE exudativa y en la alteración visual debida a EMD, a edema macular secundario a OVR o a NVC secundaria a MP, la dosis recomendada de Lucentis es 0,5 mg administrada en forma de inyección intravítrea única. Esto corresponde a un volumen de inyección de 0,05 ml. El intervalo entre dos dosis inyectadas en el mismo ojo debe ser como mínimo de cuatro semanas.
El tratamiento se inicia con una inyección al mes hasta alcanzar la agudeza visual máxima y/o no haya signos de actividad de la enfermedad, es decir ningún cambio en la agudeza visual ni en otros signos y síntomas de la enfermedad bajo tratamiento continuado. En pacientes con DMAE exudativa, EMD y OVR inicialmente pueden ser necesarias tres o más inyecciones consecutivas administradas mensualmente.
A partir de ese momento, los intervalos de monitorización y tratamiento se deben determinar según criterio médico y en base a la actividad de la enfermedad, valorada mediante la agudeza visual y/o parámetros anatómicos.
Se debe interrumpir el tratamiento con Lucentis si bajo criterio del médico, los parámetros visuales y anatómicos indican que el paciente no se está beneficiando del tratamiento continuado.
La monitorización para determinar la actividad de la enfermedad puede incluir examen clínico, control funcional o técnicas de imagen (p. ej. tomografía de coherencia óptica o angiografía con fluoresceína).
Si se está tratando a los pacientes de acuerdo a un régimen de tratar y extender, una vez se ha alcanzado la agudeza visual máxima y/o no hay signos de actividad de la enfermedad, los intervalos de tratamiento se pueden espaciar de forma gradual hasta que vuelvan a aparecer signos de actividad de la enfermedad o alteración visual. En el caso de la DMAE exudativa el intervalo de tratamiento no debe espaciarse en más de dos semanas cada vez y en el caso del EMD se puede espaciar hasta un mes cada vez. Para la OVR, los intervalos de tratamiento también pueden espaciarse de forma gradual, sin embargo los datos que hay no son suficientes para determinar la duración de estos intervalos. Si vuelve a aparecer actividad de la enfermedad, se debe acortar el intervalo de tratamiento de manera consecuente.
En el tratamiento de la alteración visual debida a la NVC secundaria a MP, muchos pacientes pueden necesitar únicamente una o dos inyecciones durante el primer año, mientras que algunos pacientes pueden necesitar tratamiento con mayor frecuencia.
Lucentis y fotocoagulación con láser en EMD y edema macular secundario a oclusión de la rama venosa retiniana (ORVR)
Existe alguna experiencia con Lucentis administrado concomitantemente con fotocoagulación con láser. Cuando se administren en el mismo día, Lucentis debe ser administrado como mínimo 30 minutos después de la fotocoagulación con láser. Lucentis puede administrarse en pacientes que han recibido fotocoagulación con láser previamente.
Lucentis y la terapia fotodinámica con Visudyne en la NVC secundaria a MP No hay experiencia en la administración concomitante de Lucentis y Visudyne.
Antes de la administración de Lucentis se debe comprobar visualmente la ausencia de partículas y decoloración.
El procedimiento de inyección deberá llevarse a cabo bajo condiciones asépticas, que incluyen el lavado quirúrgico de las manos, el uso de guantes estériles, un campo estéril, un blefarostato estéril para los párpados (o equivalente) y la disponibilidad de una paracentesis estéril (en caso necesario). Antes de realizar el procedimiento de inyección intravítrea, se deberá evaluar detalladamente la historia clínica del paciente en cuanto a reacciones de hipersensibilidad. Antes de la inyección se debe administrar una anestesia adecuada y un microbicida tópico de amplio espectro para desinfectar la piel de la zona periocular, párpado y superficie ocular, de acuerdo con la práctica local.
Para la preparación de Lucentis para administración intravítrea, siga las siguientes instrucciones:
Todos los componentes son estériles y para un solo uso. No debe utilizarse ningún componente cuyo envase muestre signos de deterioro o manipulación. La esterilidad sólo se puede garantizar si el sellado del envase de los componentes se mantiene intacto. La reutilización puede dar lugar a una infección u otra enfermedad/lesión.
Para la preparación y la inyección intravítrea se necesitan los siguientes productos sanitarios de un solo uso:
- una aguja con filtro de 5 pm (18G x IV2", 1,2 mm x 40 mm, suministrada)
- una jeringa estéril de 1 ml (no incluida en el envase de Lucentis)
- una aguja para inyección (30G x A"; no incluida en el envase de Lucentis)

1. Antes de extraer la solución, se debe desinfectar la parte exterior del tapón de goma del vial.
2. Incorporar la aguja con filtro de 5 pm (18G x 1A",
1,2 mm x 40 mm, 5 pm, suministrada) a una jeringa de 1 ml usando técnicas asépticas. Insertar la aguja roma con filtro en el centro del tapón del vial hasta que la aguja toque el extremo inferior del vial.
3. Extraer todo el líquido del vial, manteniendo el vial en posición vertical, ligeramente inclinado para facilitar la extracción completa.
4. Al vaciar el vial, asegurar que el émbolo se retira hacia atrás lo suficiente de forma que se vacíe por completo la aguja con filtro.
5. Dejar la aguja roma con filtro en el vial y desconectarla de la jeringa. La aguja con filtro se debe desechar tras extraer el contenido del vial, y no se debe utilizar para la inyección intravítrea.
6. Incorporar una aguja para inyección (30G x V") a la jeringa con firmeza y de forma aséptica.

7. Quitar la cápsula de cierre de la aguja para inyección cuidadosamente sin desconectar la aguja para inyección de la jeringa.
Nota: Sujetar la aguja para inyección por el cono mientras se retira la cápsula de cierre.
8. Expulsar el aire de la jeringa y ajustar la dosis hasta la marca de 0,05 ml en la jeringa cuidadosamente. La jeringa está lista para la inyección.
Nota: No secar la aguja para inyección. No tirar del émbolo hacia atrás.
La aguja para inyección se deberá introducir 3,5-4,0 mm por detrás del limbo en la cavidad vítrea, evitando el meridiano horizontal y en dirección al centro del globo. Seguidamente debe liberarse el volumen de inyección de 0,05 ml; las inyecciones siguientes deberán aplicarse cada vez en un punto escleral distinto.
Tras la inyección, no tapar la aguja con la cápsula de cierre ni separarla de la jeringa. Eliminar la jeringa usada junto con la aguja en un contenedor para objetos punzantes o eliminar de acuerdo con la normativa local.
158