Lofton 150 Mg Comprimidos Recubiertos

MINISTERIO
DE SANIDAD, POLÍTICA SOCIAL E IGUALDAD
|
íit |
k agencia española de 1 medicamentos y | productos sanitarios |
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
LOFTON 150 mg comprimidos recubiertos LOFTON 150 mg/ml gotas orales en solución
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Lofton 150 mg comprimidos recubiertos:
Cada comprimido recubierto contiene: buflomedil hidrocloruro 150 mg Excipiente: cada comprimido recubierto contiene 56 mg de lactosa.
Lofton 150 mg/ml gotas orales en solución:
Cada ml contiene: buflomedil hidrocloruro 150 mg.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Lofton 150 mg comprimidos recubiertos: comprimidos blancos y biconvexos
Lofton 150 mg/ml gotas orales en solución: solución libre de partículas de color amarillo pálido.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento sintomático de la claudicación intermitente en la enfermedad arterial periférica oclusiva (en estadío II).
4.2 Posología y forma de administración
Dado el estrecho margen terapéutico de buflomedil, es necesario determinar la función renal y respetar la posología recomendada.
Función renal normal (aclaramiento de creatinina > 80 ml/min)
La dosis diaria recomendada en pacientes con la función renal normal es de 300 mg a 600 mg al día dividida en al menos 2 dosis. La dosis de 300 mg al día se alcanza con 1 comprimido de 150 mg o 1 ml de solución cada 12 horas. La dosis de 600 mg al día se alcanza con 2 comprimidos de 150 mg o 2 ml de solución cada 12 horas. La dosis máxima diaria no debe sobrepasar los 600 mg.
Insuficiencia renal de leve a moderada (aclaramiento de creatinina entre 30 y 80 ml/min)
En pacientes con insuficiencia renal de leve a moderada la dosis máxima diaria debe ser reducida a la mitad, es decir, un comprimido de 150 mg o 1 ml de solución cada 12 horas. En estos pacientes, la dosis máxima diaria no debe sobrepasar los 300 mg.
Insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min)
LOFTON está contraindicado en insuficiencia renal grave (ver sección 4.3).
Correo electrónicoI
C/ CAMPEZO, 1 - EDIFICIO 8 28022 MADRID
Se atenderán exclusivamente incidencias informáticas sobre la aplicación CIMA (http://www.aemps.gob.es/cima)
Monitorización de la función renal
Antes del inicio del tratamiento debe determinarse sistemáticamente la función renal. El cálculo del aclaramiento de creatinina es particularmente necesario en pacientes mayores de 65 años y en pacientes que pesen menos de 50 kg (ver sección 4.4).
Insuficiencia hepática
En caso de insuficiencia hepática debe reducirse la dosis a la mitad, es decir, un comprimido de 150 mg o 1 ml de solución cada 12 horas.
Si se excede la dosis máxima diaria o no se ajusta la dosis diaria en pacientes con insuficiencia renal o hepática, pueden darse casos de sobredosis, con manifestaciones graves a nivel neurológico y cardiovascular (ver sección 4.8 y 4.9)
Uso en ancianos (ver sección 4.4):
La experiencia clínica no ha mostrado diferencias entre pacientes de edad avanzada y pacientes jóvenes en la respuesta terapéutica. En general, la dosis en pacientes ancianos debe ser establecida con precaución, comenzando el tratamiento con una dosis diaria baja y sin exceder la dosis diaria de 600 mg.
Uso en niños
Dado que no hay datos clínicos disponibles, la seguridad y la eficacia de buflomedil en este grupo de pacientes no han sido establecidas, por lo que el uso de buflomedil en niños menores de 18 años no está recomendado.
Duración del tratamiento
No deben esperarse beneficios terapéuticos adicionales con dosis mayores que las indicadas. Aunque los síntomas de mejora subjetiva de los pacientes pueden ser rápidos, los beneficios clínicos aparecen gradualmente a medida que transcurre el tratamiento. Se recomienda advertir al paciente que el uso prolongado de LOFTON puede ser necesario tanto para alcanzar, como para mantener la mejoría clínica.
Modo de administración
LOFTON 150 mg comprimidos recubiertos: los comprimidos deben ingerirse enteros, sin masticar, con una cantidad suficiente de líquido, preferentemente agua.
LOFTON 150 mg/ml gotas orales en solución: para una dosificación exacta, los envases de LOFTON 150 mg/ml gotas orales en solución contienen una jeringa dosificadora de 2 ml. Se deben seguir los siguientes pasos en cada administración utilizando la jeringa dosificadora:
1. Agitar el frasco. Desenroscar el tapón.
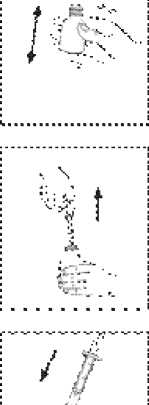


2. Introducir la jeringa en el frasco.
Tirar del émbolo de la jeringa hacia arriba hasta que el líquido alcance la cantidad indicada por su médico (generalmente la línea de 1 a 2 ml por toma).
3. Sacar la jeringa dosificadora del frasco.
Presionar el émbolo para vaciar el contenido de la jeringa en un vaso con una cantidad suficiente de líquido (preferiblemente agua) antes de la toma o directamente en la boca del paciente.
Es conveniente lavar la jeringa dosificadora con agua después de cada uso.
Una vez administrada la dosis, cerrar el tapón del frasco fuertemente y guardar el frasco y la jeringa dosificadora en un lugar seguro.
4.3 Contraindicaciones
- Hipersensibilidad al principio activo o a alguno de los excipientes.
- Postparto inmediato y presencia de hemorragia arterial severa.
- Insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min).
- Epilepsia.
4.4 Advertencias y precauciones especiales de empleo
En situaciones de desórdenes circulatorios de las extremidades inferiores, el tratamiento con Lofton debe ser acompañado de medidas dietéticas (evitar la ingesta excesiva de comida, reducir la cantidad de grasas y de colesterol en la dieta), dejar de fumar y un cuidado especial de los pies.
Dado el estrecho margen terapéutico de buflomedil, deben respetarse las recomendaciones de la dosis máxima.
Si no se respeta la posología recomendada o no se ajusta la dosis en pacientes con insuficiencia renal, puede producirse una sobredosis caracterizada por acontecimientos adversos neurológicos y cardiacos graves (ver secciones 4.8 y 4.9). Para el ajuste de la dosis diaria en pacientes con insuficiencia renal ver sección 4.2.
Dado que buflomedil se elimina por vía renal, debe medirse la creatinina sérica y calcularse el aclaramiento de creatinina antes de iniciar la terapia con buflomedil y a intervalos regulares durante el tratamiento:
• al menos una vez al año en pacientes con la función renal normal
• al menos dos veces al año en pacientes con la creatinina sérica en el límite superior normal, en pacientes mayores de 65 años y en pacientes con peso inferior a 50 kg (ver sección 4.2).
No existe experiencia clínica del uso de buflomedil en pacientes menores de 18 años.
Antes de prescribir buflomedil, el médico debe excluir la posibilidad de que los signos y síntomas del paciente surjan de un estado potencialmente reversible para el cual exista terapia específica.

Lofton debe utilizarse con precaución en pacientes con:
- Infarto agudo de miocardio
- Hipotensión severa (presión sistólica < 90 mm Hg)
- Insuficiencia hepática
- Insuficiencia renal
- Hipotonía severa
- Alteraciones de la conducción cardíaca
Los pacientes con antecedentes de depresión y/o antecedentes de sobredosis de fármacos deben ser controlados cuando estén en tratamiento con buflomedil.
Información sobre excipientes
Lofton 150 mg comprimidos recubiertos contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp (insuficiencia observada en ciertas poblaciones de Laponia) o malabsorción de glucosa o galactosa no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Inhibidores de CYP2D6 fluoxetina, paroxetina, quinidina)
Existe un mayor riesgo de efectos indeseables neurológicos (convulsiones) cuando buflomedil se administra de forma concomitante con inhibidores de CYP2D6 en pacientes con insuficiencia renal o hepática (ver sección 5.2).
Es aconsejable aumentar la monitorización clínica, particularmente en el anciano.
Buflomedil puede potenciar los efectos hipotensores de vasodilatadores, de antagonistas del calcio (ej: amlodipino, diltiazem y verapamilo) y de agentes antihipertensivos.
4.6 Embarazo y lactancia
Los estudios con buflomedil en animales no han mostrado efectos teratogénicos. No hay datos suficientes para evaluar un posible efecto malformativo o fetotóxico cuando se administra buflomedil en mujeres embarazadas. Por lo tanto, no se recomienda utilizar buflomedil durante el embarazo.
No hay datos suficientes para evaluar el uso de buflomedil durante la lactancia, por lo que buflomedil no está recomendado en mujeres en periodo de lactancia.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Dadas las diferencias interindividuales en la respuesta a buflomedil, se debe tener en cuenta que este medicamento puede producir vértigo o mareo por lo que puede disminuir la capacidad para conducir y utilizar máquinas, especialmente al inicio del tratamiento, tras ajuste de la dosis diaria o en combinación con alcohol
4.8 Reacciones adversas
Las reacciones adversas se enumeran a continuación, clasificadas por frecuencia, empezando por las más frecuentes, utilizando la siguiente convención: muy frecuentes (> 1/10); frecuentes (>
1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); raras (> 1/10.000 a < 1/1.000) muy raras

(< 1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas que se han dado con una frecuencia mayor al 1% en los pacientes tratados con buflomedil en ensayos clínicos comparativos con placebo se detallan en la tabla 1. En la mayoría de los casos, las reacciones adversas fueron leves.
|
Tabla 1: Reacciones adversas durante ensayos clínicos. | |||
|
Buflomedil |
Placebo | ||
|
Pacientes tratados, n (%) |
309 (100) |
264 (100) | |
|
Pacientes con reacciones adversas, n (%) |
92 (29,8) |
84 (31,8) | |
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia (%) | |
|
Trastornos del oído y del laberinto Frecuentes |
Vértigo |
6,5 |
3,4 |
|
Trastornos del sistema nervioso Frecuentes |
Cefalea Mareo |
4,2 1,9 |
7,2 3,0 |
|
Trastornos gastrointestinales Frecuentes |
Molestias gastrointestinales Náuseas |
4.2 3.2 |
5,3 3,0 |
|
Trastornos vasculares Frecuentes |
Vasodilatación |
2,3 |
0 |
Experiencia post-comercialización:
Trastornos de la piel y del tejido subcutáneo
Frecuencia no conocida: erupción cutánea, urticaria, rubor, psoriasis.
Trastornos del sistema inmune
Reacciones alérgicas como rash, taquicardia e hipotensión/shock
Trastornos renales y urinarios Poliuria
Trastornos respiratorios, torácicos y del mediastino Epistaxis
Trastornos del sistema reproductor y trastornos mamarios Menorragia
Trastornos del músculo esquelético y trastornos del tejido conectivo Aumento de creatinemia
Pueden aparecer efectos indeseables graves, fundamentalmente neurológicos y cardiovasculares, en el caso de una sobredosis (ver sección 4.9) o en pacientes con insuficiencia renal:
• Signos neurológicos: convulsiones, status epiléptico, mioclonias, somnolencia,
• Signos cardiovasculares: fibrilación atrial, taquicardia sinusal, hipotensión, hipertensión, trastornos graves del ritmo ventricular, trastornos de la conducción (particularmente intraventricular), susceptibles de evolucionar a paro circulatorio.

4.9 Sobredosis
En caso de sobredosis voluntaria o accidental, pueden aparecer con rapidez (15-90 minutos) síntomas neurológicos (convulsiones, status epiléptico) y pueden ir seguidos de trastornos cardiovasculares (taquicardia sinusal, hipotensión, arritmia ventricular severa, alteraciones de la conducción, especialmente alteraciones de la conducción intraventricular) y el paciente puede desarrollar rápidamente un coma y/o una parada cardiocirculatoria.
Las manifestaciones clínicas son muy similares a la de una sobredosis por antidepresivos tricíclicos, tales como la imipramina o la amitriptilina.
La ingestión intencionada o no intencionada de dosis excesivas de buflomedil puede ser fatal.
En el caso de una sobredosis, el paciente debe ser evacuado inmediatamente a un hospital mediante un servicio especializado de transporte de emergencias con el objetivo de instaurar sin demora una monitorización neurológica y electrocardiográfica continua, asistencia respiratoria y tratamiento inmediato de la intoxicación.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Otros vasodilatadores periféricos, C04AX.
Código ATC: C04AX20.
La actividad vascular de buflomedil está mediada por un efecto al y a2 adrenolítico y por la acción directa sobre las estructuras miocitarias microcirculatorias. Por su acción adrenolítica a inespecífica, se opone localmente a los efectos vasocontrictores de la adrenalina, del estrés y del frío. Esta acción se encuentra principalmente a nivel de las arterias ricas en receptores a: arterias periféricas musculares del circuito de distribución. Por su acción específica microcirculatoria, relacionada con el flujo de calcio a nivel de los miocitos perivasculares, el buflomedil dilata los esfínteres precapilares contraídos, mejorando la circulación músculo-cutánea.
Estudios farmacodinámicos en animales:
- Un estudio microcinematográfico cuantitativo directo de los abazones del hámster mostró un aumento en el diámetro de los microvasos entre el 16% y el 20% después de la administración local de buflomedil.
- El buflomedil administrado por vía intraarterial, intravenosa e intraduodenal, en perros despiertos o anestesiados, causó un aumento en el flujo sanguíneo femoral, cutáneo y muscular con disminución de las resistencias periféricas. Este aumento fue significativamente más alto que los obtenidos con los controles.
Estudios farmacodinámicos en humanos:
- Los exámenes capilaroscópicos realizados después de la administración oral o intravenosa de buflomedil mostraron un aumento en el número y el diámetro de los bucles capilares, así como relajación de los esfínteres precapilares contraídos y un aumento de la velocidad de circulación de los hematíes.
- El buflomedil no moviliza un volumen sanguíneo importante, por lo que no modifica de manera significativa la hemodinámica cardiaca (presión de eyección ventricular, gasto cardiaco total y sistólico, índice cardiaco y trabajo del ventrículo izquierdo). Esto ha sido comprobado en animales y en seres humanos mediante cateterismo cardiaco.

Ensayos clínicos:
En el estudio "Limbs International Medicinal Buflomedil" (LIMB) se incluyeron 2078 pacientes con enfermedad arterial periférica oclusiva, claudicación intermitente y un índice tobillo-brazo entre 0,30 y 0,80. Siguiendo un diseño doble-ciego, los pacientes fueron aleatorizados a buflomedil (300 mg dos veces al día en pacientes con aclaramiento de creatinina > 40 ml/min y 150 mg dos veces al día en pacientes con un aclaramiento de creatinina < 40 ml/min) o placebo, durante 2 a 4 años (media: 33 meses). La variable principal de eficacia fue la incidencia de eventos cardiovasculares críticos, definidos como la combinación de muerte cardiovascular, infarto de miocardio (IM) no fatal, accidente cerebrovascular (ACV) no fatal, deterioro sintomático de la enfermedad arterial periférica oclusiva, o amputación de la pierna. La incidencia de eventos cardiovasculares críticos fue significativamente más baja en el grupo de buflomedil con respecto al grupo placebo (9,1% versus 12,4%; Riesgo relativo, 0,74; intervalo de confianza al 95%, 0,60 a 0,92; P = 0,02). El factor fundamental para la diferencia obtenida a favor de buflomedil fue la reducción en el deterioro sintomático de la enfermedad arterial periférica, sin que hubiese diferencias significativas en cuanto a mortalidad total ni para cada uno de los componentes de la variable principal por separado. El índice tobillo-brazo aumentó en un 9,2% en los pacientes tratados con buflomedil y disminuyó en un 3,6% en los pacientes del grupo placebo (P < 0,001). La tolerabilidad de buflomedil fue comparable a la de placebo.
5.2 Propiedades farmacocinéticas
La farmacocinética de buflomedil es lineal dentro del intervalo de dosis estudiado (150, 300 y 450 mg por vía oral y 50, 100 y 200 mg por vía intravenosa).
Absorción
Buflomedil se absorbe rápidamente en el tracto gastrointestinal tras administración oral siendo su biodisponibilidad del 55%-80%). Cuando se administra por vía oral, la biodisponibilidad no se ve afectada por la ingesta de alimento. La biodisponibilidad tras administración intravenosa es equivalente a la obtenida tras administración intramuscular.
Distribución
La concentración sérica máxima tras administración oral se alcanza a las 1,5-3,0 horas. Buflomedil se distribuye ampliamente a través de los fluidos y tejidos corporales (volumen de distribución aparente de 80 a 100 litros). Los estudios in vitro de unión a proteínas de buflomedil en plasma humano han demostrado que un 81% del fármaco se une con 0,5 mcg/ml, un 61% con 5 mcg/ml y un 25% con 50 mcg/ml, por lo que el potencial para la interacción medicamentosa por desplazamiento de la unión a proteínas plasmáticas parece ser bajo.
Metabolismo
El buflomedil se metaboliza de forma extensa en el hígado, principalmente por la isoforma CYP2D6 del citocromo P450, tal y como lo demuestran estudios in vitro. Aproximadamente el 20% de la dosis absorbida sufre metabolismo hepático de primer paso. Un estudio farmacocinético realizado en sujetos sanos, metabolizadores lentos o rápidos para el CYP2D6, confirmó la preponderancia de esta vía metabólica para buflomedil. El uso de buflomedil en metabolizadotes lentos (7% de la población general), o de la administración concomitante con un inhibidor del CYP2D6 no debería tener consecuencias en términos de tolerabilidad del fármaco. No obstante, en pacientes con insuficiencia renal o hepática o en los ancianos, una inhibición del CYP2D6 puede causar un aumento en las concentraciones plasmáticas de buflomedil, con un aumento de riesgo de sus efectos indeseables, especialmente neurológicos (ver sección 4.5).

Eliminación
Buflomedil presenta una semivida plasmática relativamente corta (1,91 a 3,65 horas). En pacientes con insuficiencia renal grave, la semivida de eliminación plasmática es aproximadamente de 5 horas (± 3,4 h). La excreción es principalmente renal, en forma inalterada (12%-18%) y en forma de metabolitos glucuroconjugados.
5.3 Datos preclínicos sobre seguridad
En animales tratados con dosis altas de buflomedil se han referido convulsiones.
No se ha observado ninguna evidencia de teratogenicidad, potencial mutagénico o carcinogenicidad en los estudios llevados a cabo en animales.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
LOFTON 150 mg comprimidos recubiertos: lactosa, celulosa microcristalina, almidón de maíz, hipromelosa, estearato de magnesio, macrogol 8000, macrogol 400, dióxido de titanio (E-171), premezcla de recubrimiento blanca 13675 y agua purificada.
LOFTON 150 mg/ml gotas orales en solución: glicirrizinato de amonio, propilénglicol, sabor chocolate menta, solución de cloruro de benzalconio 500 mg/ml (50%) y agua purificada .
6.2 Incompatibilidades
No procede
6.3 Periodo de validez
LOFTON 150 mg comprimidos recubiertos: 4 años.
LOFTON 150 mg/ml gotas orales en solución: 2 años.
6.4 Precauciones especiales de conservación
LOFTON 150 mg comprimidos recubiertos y LOFTON 150 mg/ml gotas orales en solución: No conservar a temperatura superior a 30°C.
6.5. Naturaleza y contenido del envase
LOFTON 150 mg comprimidos recubiertos: blister con 50 comprimidos y envase clínico de 500 comprimidos.
LOFTON 150 mg/ml gotas orales en solución: frasco de 50 ml.
6.6 Precauciones especiales de eliminación
Ninguna especial.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN:
AMDIPHARM LIMITED Temple Chambers. 3 Burlington Road 4 Dublín - Irlanda

8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
LOFTON 150 mg comprimidos recubiertos: 55404 LOFTON 150 mg/ml gotas orales en solución: 56947
9. FECHA DE LA RENOVACIÓN DE LA AUTORIZACIÓN
LOFTON 150 mg comprimidos recubiertos: abril 2009 LOFTON 150 mg/ml gotas orales en solución: abril 2009
10. FECHA DE LA REVISIÓN DEL TEXTO
Marzo 2011.
Agencia española de
medicamentos y
productos sanitarios