Lisvy 0,06 Mg/0,013 Mg Cada 24 Horas Parche Transdermico
FICHA TÉCNICA
0. ADVERTENCIA TRIÁNGULO NEGRO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Lisvy 0,06 mg/0,013 mg cada 24 horas parche transdérmico
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada parche transdérmico de 11 cm2 contiene 2,10 mg de gestodeno y 0,55 mg de etinilestradiol.
Cada parche transdérmico libera 0,06 mg de gestodeno cada 24 horas y 0,013 mg de etinilestradiol (equivalente a dosis orales de 0,02 mg) cada 24 horas.
Para consultar la lista completa de excipientes ver sección 6.1 .
3. FORMA FARMACÉUTICA
Parche transdérmico.
Parche transdérmico de tipo matriz fina, constituido por cinco capas.
El parche es redondo, transparente y tiene un tamaño de 11 cm 2 . Por su cara adherente, está cubierto por un revestimiento protector claro y brillante de forma cuadrada dividido en dos partes.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Anticoncepción hormonal femenina. Lisvy está previsto para mujeres en edad fértil. Se ha establecido la seguridad y eficacia en mujeres de 18 a 45 años. La decisión de prescribir Lisvy debe tener en cuenta los factores de riesgo actuales de cada mujer en particular, concretamente los de tromboembolismo venoso (TEV), y cómo se compara el riesgo de TEV con Lisvy con el de otros anticonceptivos hormonales combinados (AHCs) (ver secciones 4.3 y 4.4).
4.2 Posología y forma de administración Posología
Lisvy se utiliza en un ciclo de 28 días (4 semanas):
Durante tres semanas consecutivas (21 días), se aplica un parche nuevo por semana y se retira el parche usado. En la semana cuatro no se usa ningún parche. Es de esperar que durante este tiempo aparezca una hemorragia por privación. Una semana después de la retirada del último parche, se inicia un nuevo ciclo de 4 semanas aplicando un nuevo parche (en el mismo día de la semana que antes, el “Día de recambio del parche"), independientemente de si la hemorragia por privación habitual aún persiste o ya se ha detenido. Ver “Reducción del control de los ciclos” en la sección 4.4 en caso de que no se produzca la hemorragia por privación habitual. Para consultar el calendario preciso de aplicación/retirada de los parches, ver "Día de recambio del parche" en la sección “Cómo usar Lisvy”.
Cuándo comenzar con Lisvy por primera vez
• Sin uso previo de anticonceptivos hormonales (en el mes anterior)
¡m
El parche debe aplicarse el primer día del ciclo natural de la mujer (es decir, el primer día de su sangrado menstrual). Está permitido comenzar los días 2-5, pero durante el primer ciclo es preciso utilizar un método de barrera durante los 7 días de uso del primer parche.
• Cambio a partir de un anticonceptivo hormonal combinado (anticonceptivo oral combinado [AOC], anillo vaginal o parche transdérmico diferente)
El parche debe aplicarse preferiblemente el día siguiente a la toma del último comprimido con contenido hormonal del AOC, pero, como muy tarde, el día siguiente al intervalo habitual sin comprimidos o sin hormonas del AOC.
Si el cambio se realiza a partir de un anillo vaginal o un parche transdérmico diferente, la mujer debe aplicar el parche preferiblemente el día de la retirada del último anillo o parche de un envase de ciclos, pero, como muy tarde, el día en el que le tocaría ponérselo de nuevo.
• Cambio a partir de un método exclusivamente con progestágenos (píldora, inyección, implante de solo progestágenos) o de un sistema de liberación intrauterino (SLI) de progestágeno.
La mujer puede cambiar cualquier día a partir de la píldora de progestágeno solo (en caso de un implante o un SLI, el día de su retirada; o en caso de un inyectable, el día en el que le tocaría la siguiente inyección). En todos estos casos, se debe aconsejar a la mujer que utilice adicionalmente un método de barrera durante los 7 días de uso del primer parche.
• Tras un aborto en el primer trimestre
La mujer puede comenzar inmediatamente. Cuando lo haga, no es necesario que tome medidas anticonceptivas adicionales.
• Tras un parto o un aborto en el segundo trimestre
Se debe aconsejar a las mujeres que inicien la aplicación durante los días 21 a 28 tras el parto o un aborto en el segundo trimestre. Si lo hace más tarde, se debe aconsejar a la mujer que utilice adicionalmente un método de barrera durante los 7 días de uso del primer parche. No obstante, si ya ha mantenido relaciones sexuales, es necesario descartar un embarazo antes de proceder al inicio de Lisvy o la mujer debe esperar a que aparezca su primer periodo menstrual.
P ara las muj eres en periodo de lactancia, ver sección 4.6 .
Cómo usar Lisvy
Lisvy se utiliza en un ciclo de 28 días (4 semanas) (1 parche por semana durante 3 semanas seguido de un intervalo de 7 días sin parche). Solo se usa un parche cada vez. Cada ciclo siguiente comienza inmediatamente después del intervalo sin parche del ciclo anterior independientemente de si la hemorragia por privación habitual aún persiste o ya se ha detenido.
• Día de recambio del parche
Cada parche nuevo debe aplicarse el mismo día de la semana. Este día se conoce como “Día de recambio del parche”. Por ej emplo, si el primer parche se aplica un domingo, todos los siguientes parches deberán aplicarse en domingo. Solo se usa un parche cada vez.
1 er parche
2° parche 3 er parche Sin parche
Día 1: aplicación del 1 er parche (para las mujeres que utilicen Lisvy por primera vez, consultar “Cuándo comenzar con Lisvy por primera vez”)
Día 8: retirada del 1 er parche y aplicación inmediata del 2° parche Día 15: retirada del 2° parche y aplicación inmediata del 3 er parche Día 22: retirada del 3 er parche (sin parche durante los días 22-28)
La retirada del parche se realiza el mismo día de la semana ("Día de recambio del parche"). Los cambios de parche pueden realizarse a cualquier hora del "Día de recambio del parche". Los ciclos siguientes se inician el mismo “Día de recambio del parche”, tras el intervalo de 7 días sin parche (días 22-28).
• Días sin parche
No se usa ningún parche desde el día 22 (tras la retirada del 3 er parche) hasta el día 28 (“semana 4").
Bajo ninguna circunstancia debe existir un intervalo sin parche mayor de 7 días entre los ciclos.
Si permanece sin parche más de siete días, ES POSIBLE QUE LA MUJER NO ESTÉ PROTEGIDA FRENTE A UN EMBARAZO. Se debe iniciar un nuevo ciclo aplicando un nuevo parche en cuanto la mujer se dé cuenta de que se ha olvidado del comienzo de un nuevo ciclo y utilizar un método
anticonceptivo de refuerzo, como preservativos, espermicida o diafragma, durante los siete días siguientes. Tal como ocurre con los AOCs, el riesgo de ovulación aumenta con cada día que pasa más allá del periodo sin parche recomendado.
Si se han mantenido relaciones sexuales durante un intervalo sin parche prolongado, debe considerarse la posibilidad de un embarazo.
Ver también “Cómo actuar en caso de desprendimiento, pérdida u omisión del recambio de los parches".
Forma de administración
Vía de administración: vía transdérmica
Dónde aplicar el parche
El parche siempre debe aplicarse en uno de los siguientes lugares de aplicación (ver la siguiente figura): abdomen, nalgas, parte superior y externa del brazo.
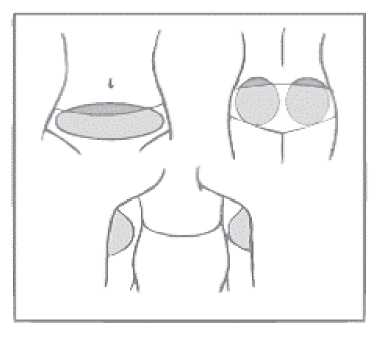
Deben evitarse las zonas en las que el parche puede desprenderse por el roce (p. ej., a la altura de la cintura de la ropa).
El parche debe aplicarse sobre piel limpia, seca, intacta, sana y preferiblemente sin vello.
Lisvy no debe colocarse en zonas de piel grasa, enrojecida, irritada o que hayan sufrido cortes u otros daños.
Los parches no deben aplicarse en las mamas.
Para prevenir interferencias con las propiedades adhesivas de Lisvy, no deben aplicarse maquillaje, cremas, lociones, polvos ni otros productos cutáneos en la zona de piel en la que se encuentra o va a aplicarse Lisvy.
Deben variarse los puntos de aplicación. Esto puede efectuarse utilizando diferentes zonas en el mismo lugar de aplicación. Por ejemplo, la mujer puede cambiar del lado izquierdo al derecho del abdomen o de la nalga o de la parte superior y externa del brazo.
La mujer también puede utilizar un lugar de aplicación distinto cada semana (p. ej., una semana la parte superior y externa del brazo, la siguiente el abdomen).
La mujer debe comprobar visualmente el parche a diario para asegurarse de que permanece bien adherido.
Importante
• Solo se usa un parche cada vez.
• Si el parche se aplica correctamente, la mujer puede bañarse o ducharse de la forma habitual.
• El parche transparente está protegido frente a la radiación UV/luz solar, por lo que puede exponerse al sol y no es necesario que permanezca cubierto por la ropa. En caso de irritación cutánea
Si el uso del parche da lugar a una incómoda irritación en el lugar de aplicación, se debe retirar y colocar un nuevo parche en un lugar diferente. Este parche se utilizará hasta el siguiente “Día de recambio del parche” programado.
¡m
Cómo preparar el parche para su aplicación
Lisvy se presenta en una caja que contiene: un folleto y 3, 9 o 18 sobres cerrados, que contienen cada uno un parche transdérmico Lisvy.

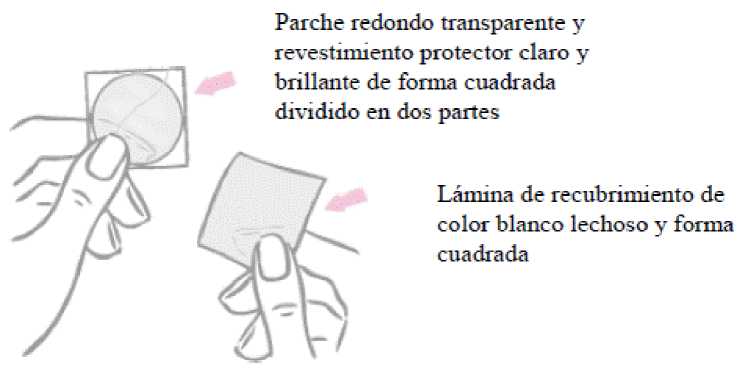
El parche es redondo y transparente:
Por su cara adherente, el parche está cubierto por un revestimiento protector claro y brillante de forma cuadrada dividido en dos partes. Este revestimiento protege la cara adherente que contiene los componentes activos del parche. También garantiza que la superficie adherente se mantenga hasta la aplicación.
Por la cara opuesta, el parche está cubierto por una lámina de recubrimiento de color blanco lechoso y forma cuadrada que impide que el parche quede atascado en el interior del sobre.
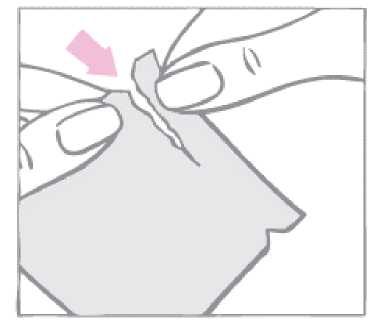
La mujer debe romper el sobre rasgándolo con los dedos a lo largo de su parte superior. Las hendiduras ayudarán a guiar el movimiento de rotura.
La mujer no debe utilizar tijeras ni cortar, dañar o alterar el parche en forma alguna, ya que podría reducir su efecto anticonceptivo.
El parche anticonceptivo redondo viene envuelto entre un revestimiento protector claro y brillante de forma cuadrada dividido en dos partes y una lámina de recubrimiento de color blanco lechoso y forma cuadrada. Es importante que extraiga el parche del sobre junto con su revestimiento protector claro y su lámina de recubrimiento de color blanco lechoso. El sobre no debe desecharse. Debe guardarse para eliminar el parche tras el uso.
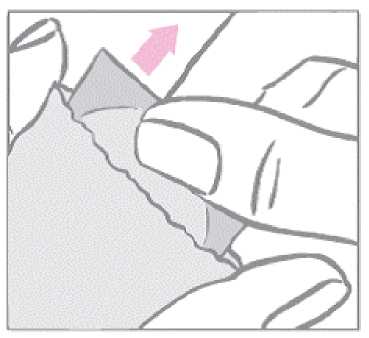
El parche debe aplicarse inmediatamente después de la apertura del sobre, del modo siguiente:
Primero, la mujer debe retirar de la cara superior del parche la lámina de recubrimiento de una sola pieza de color blanco lechoso y forma cuadrada.
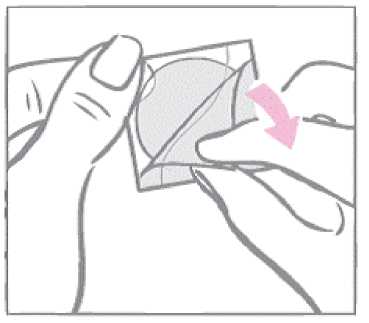
Una vez retirada del parche, esta lámina de recubrimiento de color blanco lechoso y forma cuadrada que impide que el parche quede atascado en el interior del sobre debe desecharse.
A continuación, la mujer debe retirar la mitad del revestimiento protector claro y brillante de forma cuadrada dividido en dos partes que recubre la cara inferior (adherente) del parche redondo transparente.

(La cara adherente contiene los fármacos activos.) Debe evitar tocar la superficie adherente del parche a fin de mantener su adhesividad.
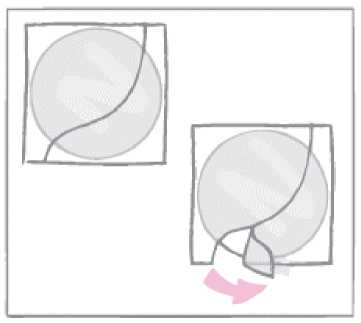
Mientras se sujeta por el borde que todavía está recubierto por la segunda mitad del revestimiento protector, el parche debe colocarse sobre la piel donde se va a llevar.
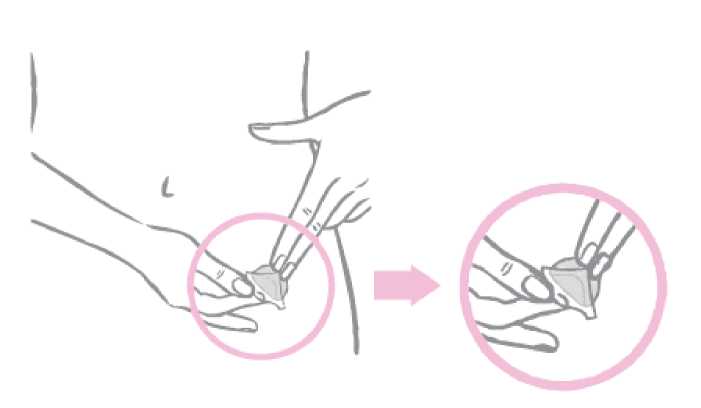
Con la mitad del parche suavemente adherido al lugar de aplicación, se debe retirar la segunda mitad del revestimiento protector.
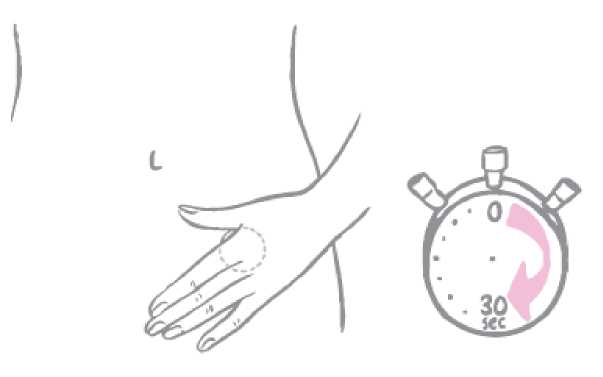
La mujer debe presionar firmemente sobre el parche con la palma de la mano durante 30 segundos y asegurarse de que sus bordes se adhieren bien.
Nota: el sobre no debe desecharse, ya que se necesitará para eliminar el parche después del uso. Eliminación de los parches
Ver sección 6.6.
siguiente
suprimir
Cómo actuar en caso de desprendimiento, pérdida u omisión del recambio de los parches Las normas de actuación en caso de desviaciones en la aplicación de los parches se basan en la regla:
Son necesarios al menos 7 días consecutivos con un parche correctamente aplicado para adecuadamente el eje hipotálamo-hipófisis-ovario con el fin de lograr eficacia anticonceptiva.
• En caso de desprendimiento parcial o total de un parche:
Durante menos de un día (hasta 24 horas):
La mujer debe intentar aplicarlo de nuevo en el mismo lugar o sustituirlo por un parche nuevo inmediatamente. No se necesitan métodos anticonceptivos de refuerzo. El “Día de recambio del parche” de la mujer seguirá siendo el mismo.
Durante más de un día (24 horas o más)o si la mujer no sabe con seguridad durante cuánto tiempo ha estado desprendido el parche:
ES POSIBLE QUE NO ESTÉ PROTEGIDA FRENTE A UN EMBARAZO. Debe interrumpir el ciclo anticonceptivo actual y comenzar un nuevo ciclo inmediatamente aplicando un nuevo parche. A partir de este momento, hay un nuevo “día 1” y un nuevo “Día de recambio del parche”. Deben utilizarse métodos anticonceptivos de refuerzo, como preservativos, espermicida o diafragma, durante la primera semana del nuevo ciclo.
No debe volverse a aplicar un parche si ya no es adherente, se ha pegado sobre sí mismo o a otra superficie, tiene otro material pegado a él o se ha aflojado o caído previamente. Si el parche no puede volverse a aplicar, debe aplicarse un parche nuevo inmediatamente. No deben emplearse adhesivos o envoltorios suplementarios para mantener Lisvy en su lugar.
• En caso de que la mujer olvide recambiar el parche:
Al inicio de cualquier ciclo de parches (semana uno/día 1):
ES POSIBLE QUE NO ESTÉ PROTEGIDA FRENTE A UN EMBARAZO. Debe aplicar el primer parche de su nuevo ciclo en cuanto se acuerde. A partir de este momento, hay un nuevo “Día de recambio del parche” y un nuevo “día 1”. La muj er debe utilizar métodos anticonceptivos de refuerzo, como preservativos, espermicida o diafragma, durante la primera semana del nuevo ciclo.
En el medio del ciclo de parches (semana dos/día 8 o semana 3/día 15):
*Durante uno o dos días (hasta 48 horas):
Debe aplicar un nuevo parche inmediatamente. El siguiente parche debe aplicarse el “Día de recambio del parche” habitual. No se necesitan métodos anticonceptivos de refuerzo.
*Durante más de dos días (48 horas o más):
ES POSIBLE QUE NO ESTÉ PROTEGIDA FRENTE A UN EMBARAZO. Debe interrumpir el ciclo anticonceptivo actual y comenzar un nuevo ciclo de 4 semanas inmediatamente aplicando un nuevo parche. A partir de este momento, hay un nuevo “Día de recambio del parche” y un nuevo “día 1”. La muj er debe utilizar métodos anticonceptivos de refuerzo durante una semana.
Al final del ciclo de parches (semana cuatro/día 22):
Si la mujer olvida retirar el parche el día 22, debe quitarlo en cuanto se acuerde (como muy tarde, el día 28). El ciclo siguiente debe iniciarse entonces con un nuevo parche (el día siguiente al DÍA 28: el "Día de recambio del parche" habitual), nunca más tarde. No se necesitan métodos anticonceptivos de refuerzo.
• Consecuencias del desprendimiento, pérdida u omisión del recambio de los parches y acciones necesarias:
Consecuencias del desprendimiento, pérdida u omisión del recambio de los parches y acciones necesarias.

|
Desprendimiento de los parches3 |
Plazo |
Consecuencias sobre la fiabilidad anticonceptiva,1 |
Acciones necesarias3 |
|
Desprendimiento del parche |
< 24 horas |
Eficacia anticonceptiva garantizada |
- Aplicar un nuevo parche inmediatamente - No se necesitan métodos anticonceptivos de refuerzo - "Día de recambio del parche" sin cambios |
|
> 24 horas |
Eficacia anticonceptiva en riesgo |
- Comenzar un nuevo ciclo de 4 semanas inmediatamente aplicando un nuevo parche - Utilizar métodos anticonceptivos de refuerzo durante los siguientes 7 días b - Anotar un nuevo "Día de recambio del parche" |

|
Omisión del recambio a tiempo de los parchesa |
Plazo |
Consecuencias sobre la fiabilidad anticonceptivaa |
Acciones necesarias*1 |
|
Omisión de la aplicación a tiempo del 1er parche (semana 1, día 1) |
Intervalo sin parche d >7 días |
Eficacia anticonceptiva en nesgo |
- Comenzar un nuevo ciclo de 4 semanas inmediatamente aplicando un nuevo parche - Utilizar métodos anticonceptivos de refuerzo durante los siguientes 7 díasb - Anotar un nuevo "Día de recambio del parche" |
|
Omisión de la aplicación a tiempo del 1er o 2° parche (semana 1/2 o 2/3) |
< 48 horas |
Eficacia anticonceptiva garantizada |
- Aplicar un nuevo parche inmediatamente - No se necesitan métodos anticonceptivos de refuerzo - "Día de recambio del parche" sin cambios |
|
> 48 horas |
Eficacia anticonceptiva en riesgo |
- Comenzar un nuevo ciclo de 4 semanas inmediatamente aplicando un nuevo parche - Utilizar métodos anticonceptivos de refuerzo durante los siguientes 7 díasb ’ - Anotar un nuevo "Día de recambio del parche" | |
|
Omisión de la aplicación a tiempo del 3 er parche (semana 3/4) |
Eficacia anticonceptiva garantizadac |
- Retirar el parche - Comenzar el siguiente ciclo de 4 semanas en el "Día de recambio del parche” habitual |
a Válido para cada ciclo.
bLos métodos anticonceptivos de refuerzo son cualquier método anticonceptivo no hormonal adicional, excepto el método del calendario y el método de la temperatura.
c Siempre que el 3a parche se haya sustituido por uno nuevo como muy tarde el día 1 habitual del nuevo ciclo de parches.
d Tiempo transcurrido desde la retirada del último parche del ciclo anterior.
La prescripción del siguiente envase debe realizarse a tiempo, es decir, antes del uso del último parche del envase para garantizar que la mujer no se quede sin parches.
Ajuste del "Día de recambio del parche"
Si la muj er desea cambiar su “Día de recambio del parche”, debe completar el ciclo actual retirando el tercer parche el día correspondiente. Durante la semana sin parche, puede seleccionar un “Día de recambio del parche” anterior al previo aplicando un nuevo parche el día deseado. En ningún caso se debe permanecer más de 7 días consecutivos sin parche.
Poblaciones especiales
¡m
Sexo
Lisvy solo está indicado en las mujeres.
Mujeres de edad avanzada
No aplicable. Lisvy no está indicado tras la menopausia.
Indice de masa corporal
Existen datos limitados sobre la eficacia anticonceptiva en las mujeres con índice de masa corporal > 30 kg/m2.
Insuficiencia renal
Lisvy no se ha estudiado en mujeres con insuficiencia renal. No se prevé un mayor riesgo en muj eres con insuficiencia renal (ver sección 5.2 ).
Insuficiencia hepática
Lisvy no se ha estudiado en mujeres con insuficiencia hepática. Lisvy está contraindicado en mujeres con hepatopatía grave o antecedentes de la misma, siempre y cuando los valores de función hepática no hayan retornado a la normalidad. Ver sección 4.3.
Diferencias étnicas
La farmacocinética del etinilestradiol se estudió en combinación con otro progestágeno en mujeres caucásicas, chinas y japonesas y no mostró diferencias clínicamente significativas. La farmacocinética de Lisvy no se ha estudiado específicamente en mujeres de distintas etnias. No se conocen enzimas polimórficas que contribuyan en un grado importante al metabolismo del gestodeno. Los datos disponibles en mujeres caucásicas, negras e hispanas no indican ninguna diferencia relevante en la farmacocinética de Lisvy entre las mujeres de distintas razas/etnias. Se disponen de datos muy limitados en mujeres asiáticas.
Población pediátrica
No se ha establecido la seguridad y eficacia en adolescentes menores de 18 años. No existe una recomendación de uso específica para Lisvy en niñas y adolescentes premenárquicas.
4.3 Contraindicaciones
No se deben utilizar AHCs en las siguientes condiciones. De producirse alguno de estos trastornos por primera vez durante el uso de Lisvy, el parche debe retirarse inmediatamente.
• Presencia o riesgo de tromboembolismo venoso (TEV).
o Tromboembolismo venoso: TEV (con anticoagulantes) o antecedentes del mismo (p. ej., trombosis venosa profunda (TVP) o embolia pulmonar (EP)). o Predisposición hereditaria o adquirida conocida al tromboembolismo venoso, como resistencia a la PCA (incluyendo el factor V Leiden), deficiencia de antitrombina III, deficiencia de proteína C, deficiencia de proteína S. o Cirugía mayor con inmovilización prolongada (ver sección 4.4 ).
o Riesgo elevado de tromboembolismo venoso debido a la presencia de varios factores de riesgo (ver sección 4.4 ).
• Presencia o riesgo de tromboembolismo arterial (TEA).
o Tromboembolismo arterial: tromboembolismo arterial, antecedentes del mismo (p. ej.
infarto de miocardio) o afección prodrómica (p. ej. angina de pecho). o Enfermedad cerebrovascular: ictus, antecedentes de ictus o afección prodrómica (p. ej. accidente isquémico transitorio, AIT).
o Predisposición hereditaria o adquirida conocida al tromboembolismo arterial, tal como hiperhomocisteinemia y anticuerpos antifosfolípidos (anticuerpos anticardiolipina, anticoagulante del lupus).
o Antecedentes de migraña con síntomas neurológicos focales.
o Riesgo elevado de tromboembolismo arterial debido a múltiples factores de riesgo (ver sección 4.4 ) o a la presencia de un factor de riesgo grave como:
■ diabetes mellitus con síntomas vasculares
¡m
■ hipertensión grave
■ dislipoproteinemia intensa
• Hepatopatía grave o o antecedentes de la misma, siempre y cuando los valores de función hepática no hayan retornado a la normalidad.
o Presencia o antecedentes de tumores hepáticos (benignos o malignos). o Presencia conocida o sospecha de neoplasias malignas influidas por esteroides sexuales (p.
ej., de los órganos genitales o las mamas). o Sangrado vaginal sin diagnosticar.
o Hipersensibilidad a los principios activos o a alguno de los excipientes incluidos en la sección 6.1 .
4.4 Advertencias y precauciones especiales de empleo Advertencias
Si alguno de los trastornos o factores de riesgo que se mencionan a continuación está presente, se debe comentar con la mujer la idoneidad de Lisvy. Si alguno de estos trastornos o de estos factores de riesgo se agrava o aparece por primera vez, se debe aconsejar a la mujer que consulte con su médico para determinar si se debe interrumpir el uso de Lisvy.
• Trastornos circulatorios Riesgo de tromboembolismo venoso (TEV)
El uso de cualquier AHC aumenta el riesgo de tromboembolismo venoso (TEV), comparado con la no utilización. Los medicamentos que contienen levonorgestrel, norgestimato o noretisterona se asocian con el riesgo más bajo de TEV.
No se sabe todavía cómo se compara el riesgo de Lisvy con el de estos medicamentos de menor riesgo. La decisión de utilizar cualquier medicamento diferente del que tiene el menor riesgo de TEV se debe tomar solamente después de comentarlo con la mujer para garantizar que comprende el riesgo de TEV con los AHCs, cómo afectan sus actuales factores de riesgo a este riesgo y que su riesgo de TEV es mayor durante el primer año de uso. También existen ciertas evidencias de que el riesgo aumenta cuando se reinicia el AHC después de una interrupción del uso de 4 semanas o más.
Entre las mujeres que no utilizan un AHC y que no están embarazadas, aproximadamente 2 de cada 10.000 presentarán un TEV en el plazo de un año. No obstante, el riesgo puede ser mucho mayor en cada mujer en particular, en función de sus factores de riesgo subyacentes (ver a continuación).
Se estima[1] que de cada 10.000 mujeres que utilizan un AOC que contiene gestodeno, entre 9 y 12 mujeres presentarán un TEV en un año; esto se compara con unas 6 [2] en mujeres que utilizan un AHC que contiene levonorgestrel.
En ambos casos, el número de TEVs por año es inferior al número esperado en mujeres durante el embarazo o en el período de posparto.
No se sabe todavía cómo se compara el riesgo de TEV con Lisvy, con el de los AHCs de dosis baja que contienen levonogestrel y AOCs que contienen gestodeno.
El TEV puede ser mortal en el 1-2% de los casos.
¡m

venas y arterias hepáticas, mesentéricas, renales o retinianas, en usuarias de AHC.
Factores de riesgo de TEV
El riesgo de complicaciones tromboembólicas venosas en usuarias de AHC puede aumentar sustancialmente en una mujer con factores de riesgo adicionales, en particular si existen varios factores de riesgo (ver tabla).
Lisvy está contraindicado si una mujer tiene varios factores de riesgo que la ponen en una situación de alto riesgo de trombosis venosa (ver sección 4.3 ). Si una mujer tiene más de un factor de riesgo, es posible que el aumento del riesgo sea mayor que la suma de los factores individuales; en este caso se debe tener en cuenta su riesgo total de TEV. Si se considera que la relación beneficio/riesgo es negativa, no se debe prescribir un AHC (ver sección 4.3 ).
Tabla: Factores de riesgo de TEV
|
Factor de riesgo |
Comentario |
|
Obesidad (índice de masa corporal (IMC) superior a 30 kg/m2) |
El riesgo aumenta de forma sustancial con el aumento del IMC. Especialmente importante en mujeres con factores de riesgo adicionales. |
¡m
|
Inmovilización prolongada, la cirugía mayor, cualquier intervención quirúrgica de las piernas o pelvis, neurocirugía o traumatismo importante. Nota: La inmovilización temporal, incluyendo los viajes en avión >4 horas, también puede ser un factor de riesgo de TEV, en especial en mujeres con otros factores de riesgo. |
En estas circunstancias es aconsejable interrumpir el uso del parche (en caso de intervención quirúrgica programada, al menos con cuatro semanas de antelación) y no reanudarlo hasta dos semanas después de que se recupere completamente la movilidad. Se debe utilizar otro método anticonceptivo para evitar un embarazo involuntario. Se debe considerar un tratamiento antitrombótico si no se ha interrumpido con antelación la toma de Lisvy. |
|
Antecedentes familiares positivos (algún caso de tromboembolismo venoso en un hermano o en un progenitor, especialmente a una edad relativamente temprana, p. ej. antes de los 50 años). |
Si se sospecha que existe una predisposición hereditaria, la mujer se debe derivar a un especialista antes de tomar la decisión de usar un AHC. |
|
Otras enfermedades asociadas al TEV. |
Cáncer, lupus eritematoso sistémico, síndrome urémico hemolítico, enfermedad inflamatoria intestinal crónica (enfermedad de Crohn o colitis ulcerosa) y anemia de células falciformes. |
|
Aumento de la edad. |
En especial por encima de los 35 años. |
No hay consenso sobre el posible papel de las venas varicosas y la tromboflebitis superficial en la aparición o progresión de la trombosis venosa.
Es preciso tener en cuenta el aumento del riesgo de tromboembolismo en el embarazo, y en particular en el período de 6 semanas del puerperio (para obtener información sobre “Embarazo y lactancia”, ver sección 4.6).
S
íntomas de TEV (trombosis venosa profunda y embolia pulmonar)
En el caso de que se produzcan síntomas, se debe aconsejar a la mujer que busque asistencia médica urgente y que informe al profesional sanitario de que está tomando un AHC.
Los síntomas de trombosis venosa profunda (TVP) pueden incluir:
• Hinchazón unilateral de la pierna y/o pie o a lo largo de una vena de la pierna.
• Dolor o sensibilidad en la pierna, que tal vez se advierta sólo al ponerse de pie o al caminar.
• Aumento de la temperatura en la pierna afectada; enrojecimiento o decoloración de la piel de la pierna.
Los síntomas de embolia pulmonar (EP) pueden incluir:
• Aparición repentina de disnea o respiración rápida injustificadas.
• Tos repentina que puede estar asociada a hemoptisis.
• Dolor torácico agudo.
• Aturdimiento intenso o mareo.
• Latidos cardíacos acelerados o irregulares.
Algunos de estos síntomas (p. ej ., “disnea”, “tos”) son inespecíficos y se pueden malinterpretar como trastornos más frecuentes o menos graves (p. ej. infecciones del tracto respiratorio).
Otros signos de oclusión vascular pueden incluir: dolor repentino, hinchazón y ligera coloración azul de una extremidad.
Si la oclusión se produce en el ojo, los síntomas pueden variar desde visión borrosa indolora, que puede evolucionar a pérdida de la visión. A veces la pérdida de la visión se puede producir casi de inmediato.
Riesgo de tromboembolismo arterial (TEA)
¡m
En ciertos estudios epidemiológicos se ha asociado el uso de los AHCs con un aumento del riesgo de tromboembolismo arterial (infarto de miocardio) o de accidente cerebrovascular (p. ej. accidente isquémico transitorio, ictus). Los episodios tromboembólicos arteriales pueden ser mortales.
Factores de riesgo de TEA
El riesgo de que se produzcan complicaciones tromboembólicas arteriales o un accidente cerebrovascular en las usuarias de AHC aumenta en mujeres con factores de riesgo (ver tabla). Lisvy está contraindicado si una mujer tiene un factor de riesgo grave o varios factores de riesgo de TEA que la ponen en una situación de alto riesgo de trombosis arterial (ver sección 4.3 ). Si una mujer tiene más de un factor de riesgo, es posible que el aumento del riesgo sea mayor que la suma de los factores individuales; en este caso se debe tener en cuenta su riesgo total. Si se considera que la relación beneficio/riesgo es negativa, no se debe prescribir un AE1C (ver sección 4.3).
Tabla: Factores de riesgo de TEA
|
Factor de riesgo |
Comentario |
|
Aumento de la edad |
En especial por encima de los 35 años. |
|
Tabaquismo |
Se debe aconsejar a las mujeres que no fumen si desean utilizar un AHC. Se debe aconsejar encarecidamente a las mujeres de más de 35 años que continúan fumando que utilicen un método anticonceptivo diferente. |
|
Hipertensión | |
|
Obesidad (índice de masa corporal superior a 30 kg/m2) |
El riesgo aumenta de forma sustancial con el aumento del IMC. Especialmente importante en mujeres con factores de riesgo adicionales. |
|
Antecedentes familiares positivos (algún caso de tromboembolismo arterial en un hermano o en un progenitor, especialmente a una edad relativamente temprana, p. ej. menos de 50 años) |
Si se sospecha que existe una predisposición hereditaria, la mujer debe ser derivada a un especialista antes de tomar la decisión de usar un AHC. |
|
Migraña |
Un aumento de la frecuencia o la intensidad de las migrañas durante el uso de AHC (que puede ser prodrómico de un acontecimiento cerebrovascular) puede motivar su interrupción inmediata. |
|
Otras enfermedades asociadas a acontecimientos vasculares adversos |
Diabetes mellitus, hiperhomocisteinemia, valvulopatía y fibrilación auricular, dislipoproteinemia y lupus eritematoso sistémico. |
Síntomas de TEA
En el caso de que se produzcan síntomas, se debe aconsejar a la mujer que busque asistencia médica urgente y que informe al profesional sanitario de que está tomando un AHC.
Los síntomas de un accidente cerebrovascular pueden incluir:
• Entumecimiento o debilidad repentinos de la cara, brazo o pierna, especialmente en un lado del cuerpo.
• Dificultad repentina para caminar, mareo, pérdida del equilibrio o de la coordinación.
• Confusión repentina, dificultad para hablar o para comprender.
• Dificultad repentina de visión en un ojo o en ambos.
• Cefalea repentina, intensa o prolongada sin causa conocida.
• Pérdida del conocimiento o desmayo, con o sin convulsiones.
Los síntomas temporales sugieren que el episodio es un accidente isquémico transitorio (AIT).
Los síntomas de infarto de miocardio (IM) pueden incluir:
¡m
• Dolor, molestias, presión, pesadez, sensación de opresión o plenitud en el tórax, brazo o debajo del esternón.
• Malestar que irradia a la espalda, la mandíbula, la garganta, el brazo o el estómago.
• Sensación de plenitud, indigestión o ahogo.
• Sudoración, náuseas, vómitos o mareo.
• Debilidad extrema, ansiedad o falta de aliento.
• Latidos cardíacos acelerados o irregulares.
Cuando considere la relación riesgo/beneficio, el médico debe tener en cuenta que el tratamiento adecuado de una enfermedad puede reducir el riesgo asociado de trombosis y que el riesgo asociado al embarazo es mayor que el asociado a los anticonceptivos hormonales combinados de dosis bajas (<0,05 mg de etinilestradiol).
■ Tumores
En algunos estudios epidemiológicos se ha notificado un riesgo aumentado de cáncer de cuello uterino en usuarias a largo plazo de AOCs (> 5 años), pero sigue existiendo controversia acerca del grado en que este hallazgo es atribuible a los efectos de confusión de la conducta sexual y otros factores como el virus del papiloma humano (VPH).
En un metaanálisis de 54 estudios epidemiológicos se notificó que las mujeres que están utilizando un AOC presentan un riesgo relativo ligeramente aumentado (RR = 1,24) de recibir un diagnóstico de cáncer de mama. El exceso de riesgo desaparece paulatinamente durante el transcurso de los 10 años siguientes a la suspensión del uso de AOC. Dado que el cáncer de mama es raro en mujeres menores de 40 años, el exceso numérico de diagnósticos de cáncer de mama en mujeres que están utilizando o han utilizado recientemente un AOC es pequeño en relación con el riesgo global de cáncer de mama. Estos estudios no aportan evidencias sobre las causas. El patrón observado de riesgo aumentado puede deberse a un diagnóstico más precoz del cáncer de mama en las usuarias de AOC, a los efectos biológicos de los AOCs o a una combinación de ambos. Los cánceres de mama diagnosticados en las mujeres que los utilizaron en alguna ocasión tienden a ser menos avanzados clínicamente que los diagnosticados en las mujeres que nunca los han utilizado.
Se han notificado casos raros de tumores hepáticos benignos y casos aún más raros de tumores hepáticos malignos en usuarias de AOC. En casos aislados, estos tumores han provocado hemorragias intraabdominales potencialmente mortales. Se debe considerar la existencia de un tumor hepático en el diagnóstico diferencial cuando una usuaria de anticonceptivos hormonales combinados presente dolor intenso en la región superior del abdomen, agrandamiento del hígado o signos de hemorragia intraabdominal.
Con el uso de AOCs que contienen más de 0,05 mg de etinilestradiol, el riesgo de cáncer de endometrio y ovario es reducido. Todavía está por confirmar si esto también es válido para los anticonceptivos hormonales combinados de dosis más bajas.
• Otros trastornos
Si se producen irritaciones cutáneas persistentes de forma repetida (p. ej., eritema o prurito persistentes en el lugar de aplicación) incluso cuando el lugar de aplicación se cambia de acuerdo con las instrucciones, se debe considerar la interrupción del tratamiento transdérmico.
Las mujeres con hipertrigliceridemia o antecedentes familiares de esta afección pueden presentar un riesgo aumentado de pancreatitis al usar anticonceptivos hormonales combinados.
Aunque se han notificado pequeños incrementos de la presión arterial en muchas mujeres usuarias de anticonceptivos hormonales combinados, los incrementos clínicamente relevantes son raros. No obstante, si aparece hipertensión sostenida clínicamente significativa durante el uso de Lisvy, es prudente que el médico retire la preparación y trate la hipertensión. Cuando se considere adecuado, el uso de Lisvy puede reanudarse si se logran valores tensionales normales con la terapia antihipertensiva.
¡m
Se ha notificado la aparición o empeoramiento de los siguientes trastornos tanto con el embarazo como con el uso de anticonceptivos hormonales combinados, pero los datos indicativos de una asociación con el uso de anticonceptivos hormonales combinados no son concluyentes: ictericia y/o prurito relacionados con colestasis, formación de cálculos biliares, porfiria, lupus eritematoso sistémico, síndrome urémico hemolítico, corea de Sydenham, herpes gestacional, pérdida de audición relacionada con otosclerosis.
En mujeres con angioedema hereditario, los estrógenos exógenos pueden inducir o exacerbar los síntomas de angioedema.
Las alteraciones agudas o crónicas de la función hepática pueden precisar la interrupción de Lisvy hasta que los marcadores de la función hepática retornen a la normalidad. La recurrencia de ictericia colestática aparecida por primera vez durante un embarazo o el uso previo de esteroides sexuales hace necesaria la interrupción de Lisvy.
Aunque los anticonceptivos hormonales combinados pueden ejercer un efecto sobre la resistencia periférica a la insulina y la tolerancia a la glucosa, no existen datos indicativos de una necesidad de modificar la pauta terapéutica en las diabéticas usuarias de anticonceptivos hormonales combinados de dosis bajas (que contengan < 0,05 mg de etinilestradiol). No obstante, se debe vigilar cuidadosamente a las mujeres diabéticas durante el uso de anticonceptivos hormonales combinados.
Se ha notificado empeoramiento de la depresión endógena, la epilepsia, la enfermedad de Crohn y la colitis ulcerosa durante el uso de anticonceptivos hormonales combinados.
En casos ocasionales puede aparecer cloasma, especialmente en las mujeres con antecedentes de cloasma gravídico. Las mujeres con tendencia a presentar cloasma deben evitar la exposición al sol o a la radiación ultravioleta mientras estén usando anticonceptivos hormonales combinados.
Se debe prestar especial atención a la interacción de los anticonceptivos hormonales combinados con lamotrigina (ver sección 4.5).
Exploración/consulta médica
Antes de iniciar o reanudar el tratamiento con Lisvy, se debe realizar una anamnesis completa (incluidos los antecedentes familiares) y descartar un posible embarazo. Se debe medir la tensión arterial y realizar una exploración física, guiada por las contraindicaciones (ver sección 4.3 ) y por las advertencias (ver sección 4.4 ). Es importante dirigir la atención de la mujer hacia la información sobre la trombosis venosa y arterial, incluido el riesgo de Lisvy en comparación con otros AHC, los síntomas de TEV y TEA, los factores de riesgo conocidos y qué debe hacer en caso de una sospecha de trombosis.
También se debe indicar a la mujer que lea cuidadosamente el prospecto y siga las instrucciones allí descritas. La frecuencia y la naturaleza de las exploraciones deben basarse en las directrices clínicas establecidas y se adaptarán a cada mujer en particular.
Debe advertirse a las mujeres que los anticonceptivos hormonales no protegen frente a la infección por VIH (SIDA) ni frente a otras enfermedades de transmisión sexual.
Reducción de la eficacia
La eficacia de Lisvy puede verse reducida, por ejemplo, en caso de:
• Omisión de la aplicación programada de un parche.
• Desprendimiento de un parche.
• Olvido del recambio de un parche (ver “Cómo actuar en caso de desprendimiento, pérdida u omisión del recambio de los parches” en la sección 4.2 ).
• Medicación concomitante (ver sección 4.5 ).
Reducción del control de los ciclos
Con todos los anticonceptivos hormonales combinados pueden producirse sangrados irregulares (manchado o hemorragias intermenstruales), especialmente durante los primeros meses de uso. En tales casos, debe continuarse la aplicación de Lisvy. La evaluación de cualquier sangrado irregular sólo es significativa tras
¡m
un intervalo de adaptación de aproximadamente tres ciclos de uso de Lisvy. El porcentaje de mujeres usuarias de Lisvy que presentaron sangrados intracíclicos tras este periodo de adaptación osciló entre el 7-12%.
Solo una minoría de mujeres, en el rango del 1% por ciclo, fueron amenorreicas.
Si las irregularidades hemorrágicas persisten o aparecen tras ciclos regulares con Lisvy, deben considerarse causas no hormonales y están indicadas medidas diagnósticas adecuadas para descartar un embarazo o una neoplasia maligna. Entre ellas puede encontrarse un legrado.
En algunas mujeres es posible que no se produzca una hemorragia por privación durante el intervalo sin parche. Si Lisvy se ha utilizado conforme a las instrucciones descritas en la sección 4.2, es improbable que la mujer esté embarazada. Sin embargo, si Lisvy no se ha utilizado conforme a estas instrucciones antes de la primera ausencia de hemorragia por privación o si no aparecen dos hemorragias por privación, se debe descartar un embarazo antes de continuar usando Lisvy.
1 [1] Estas incidencias se estimaron a partir de la totalidad de los datos de los estudios epidemiológicos, utilizando riesgos relativos para los diferentes productos en comparación con los AHCs que contienen levonorgestrel .
[2] Punto medio del rango de 5-7 por cada 10.000 mujeres-año ( MA), basado en un riesgo relativo para los AHCs que contienen levonorgestrel frente a la no utilización de aproximadamente 2,3 a 3,6 .
4.5 Interacción con otros medicamentos y otras formas de interacción
Nota: para identificar las posibles interacciones habrá que consultar la información sobre la prescripción de medicación concomitante.
Efectos de otros medicamentos sobre Lisvy
Pueden producirse interacciones con los fármacos que inducen las enzimas microsomales, lo que puede dar lugar a un aumento del aclaramiento de las hormonas sexuales y a hemorragias intermenstruales y/o fallos en la anticoncepción.
En la literatura se han publicado las siguientes interacciones.
Sustancias que aumentan el aclaramiento de los anticonceptivos hormonales combinados (disminución de la eficacia de los anticonceptivos hormonales combinados por inducción enzimática), p. ej.:
Barbitúricos, primidona, carbamazepina, fenitoína, primidona, rifampicina, rifabutina y medicación frente al VIH, ritonavir, nevirapina y efavirenz y posiblemente también felbamato, griseofulvina, oxcarbazepina, topiramato, eslicarbazepina, modafinilo y los productos que contienen la planta medicinal Hierba de San Juan (Hypericum perforatum).
Manejo
La inducción enzimática puede observarse ya después de algunos días de tratamiento. La inducción enzimática máxima se observa en unas semanas. Después de interrumpir el tratamiento, la inducción enzimática puede mantenerse durante unas 4 semanas.
Tratamiento a corto _plazo
Las mujeres en tratamiento con medicamentos inductores enzimáticos deben usar temporalmente un método de barrera o cualquier otro método anticonceptivo además del AHC. El método de barrera debe utilizarse durante todo el tiempo que dure el tratamiento concomitante y durante 28 días después de su interrupción.
¡m
Si la terapia se prolonga más allá del tercer parche de un ciclo de aplicación, el siguiente parche debe iniciarse justo después del anterior, omitiendo el intervalo habitual sin parche.
Tratamiento a largo _plazo
En mujeres en tratamiento a largo plazo con principios activos inductores enzimáticos, se recomienda otro método anticonceptivo no hormonal fiable.
Sustancias con efectos variables sobre el aclaramiento de los anticonceptivos hormonales combinados: Cuando se administran concomitantemente con anticonceptivos hormonales combinados, muchas combinaciones de inhibidores de la proteasa del VIH e inhibidores de la transcriptasa inversa no nucleosídica, que incluyen combinaciones con inhibidores del VHC pueden aumentar o reducir las concentraciones plasmáticas del estrógeno o el progestágeno. Los efectos netos de estos cambios pueden ser clínicamente relevantes en algunos casos.
Por lo tanto, se debe consultar la información de prescripción de los medicamentos concomitantes frente al VIH/VHC para identificar posibles interacciones y las recomendaciones relacionadas. En caso de duda, las mujeres en tratamiento con inhibidores de la proteasa o inhibidores de la transcriptasa inversa no nucleosídicos, deben utilizar un método anticonceptivo de barrera adicional.
Sustancias que aumentan las concentraciones farmacológicas de los anticonceptivos hormonales combinados (inhibidores enzimáticos)
Se ha constatado que el etoricoxib aumenta las concentraciones plasmáticas de etinilestradiol (50 a 60%) cuando se toma concomitantemente con un anticonceptivo hormonal trifásico oral. Se cree que el etoricoxib aumenta las concentraciones de etinilestradiol porque inhibe la actividad de las sulfotransferasas, con la consiguiente inhibición del metabolismo del etinilestradiol.
Efectos de los anticonceptivos hormonales combinados sobre otros medicamentos
Los anticonceptivos hormonales combinados pueden afectar al metabolismo de ciertos principios activos. En consecuencia, las concentraciones plasmática y tisular pueden aumentar (p. ej. ciclosporina) o disminuir (p. ej. lamotrigina).
Se debe prestar especial atención a la interacción de los anticonceptivos hormonales combinados con lamotrigina. No se recomienda el uso concomitante debido al riesgo de reducción de las concentraciones y de la eficacia de lamotrigina (ver sección 4.4). Se debe evitar iniciar un anticonceptivo hormonal combinado durante la determinación de la concentración de lamotrigina. En mujeres bajo tratamiento con lamotrigina, es preciso realizar un seguimiento clínico y ajustar la dosis de lamotrigina durante el inicio de los anticonceptivos hormonales combinados y tras suspenderlos.
Otras formas de interacciones
Análisis de laboratorio
La utilización de esteroides anticonceptivos puede influir sobre los resultados de algunos análisis de laboratorio, entre los que se encuentran parámetros bioquímicos del hígado, el tiroides, la función adrenal y renal, las concentraciones plasmáticas de las proteínas (transportadoras) como, p. ej., la globulina transportadora de corticosteroides y las fracciones de lípidos/lipoproteínas, parámetros del metabolismo de los carbohidratos y parámetros de la coagulación y la fibrinolisis. Los cambios generalmente se mantienen dentro del intervalo normal del laboratorio.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Lisvy no está indicado durante el embarazo. Si se produce un embarazo durante el uso de Lisvy, se debe retirar y suspender su uso ulterior. No obstante, amplios estudios epidemiológicos no han mostrado ni un riesgo aumentado de anomalías congénitas en los hijos de mujeres que usaron anticonceptivos hormonales
¡m
combinados antes del embarazo ni la existencia de efectos teratógenos en los casos en los que se utilizaron anticonceptivos hormonales combinados inadvertidamente durante el inicio del embarazo.
Se debe tener en cuenta el aumento de riesgo de TEV durante el periodo de posparto cuando se reinicia la administración con Lisvy (ver sección 4.2 y 4.4 ).
Lactancia
Los anticonceptivos hormonales combinados pueden influir sobre la lactancia, ya que pueden reducir la cantidad de leche materna y modificar su composición. Por lo tanto, no se debe recomendar en general el uso de anticonceptivos hormonales combinados hasta que la madre lactante haya destetado por completo al bebé. Pequeñas cantidades de esteroides anticonceptivos y/o sus metabolitos pueden excretarse con la leche.
Fertilidad
El uso de Lisvy no altera el curso de la fertilidad en el futuro. Una vez retirado Lisvy, las mujeres retornan a su fertilidad normal.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas. No se han observado efectos sobre la capacidad para conducir y utilizar máquinas en las usuarias de anticonceptivos hormonales combinados.
4.8 Reacciones adversas Resumen del perfil de seguridad
Las reacciones adversas más frecuentemente notificadas con Lisvy son reacciones en el lugar de aplicación (exantema, prurito, irritaciones, eritema e hipersensibilidad). Ocurren en el 20,9% de las usuarias. Las reacciones adversas raras y graves son los tromboembolismos arteriales y venosos.
Lista tabulada de reacciones adversas
En la siguiente tabla se resumen las frecuencias de las reacciones adversas notificadas en los ensayos clínicos de fases 2 y 3 con Lisvy (N = 3.5731). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Las frecuencias se definen como muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100) y raras (> 1/10.000 a < 1/1.000).
|
Clasificación de órganos del sistema MedDRA |
Muy frecuentes |
F recuentes |
Poco frecuentes |
Raras |
|
Trastornos psiquiátricos |
Labilidad emocional |
Depresión/estado de ánimo depresivo, disminución y pérdida de la libido | ||
|
Trastornos del sistema nervioso |
Migraña | |||
|
Trastornos vasculares |
Episodios tromboembólicos venosos y arteriales* | |||
|
Trastornos gastrointestinales |
Náuseas | |||
|
Trastornos de la piel y del tejido subcutáneo |
Reacción en el lugar de aplicación | |||
|
Trastornos del aparato reproductor y de la mama |
Hemorragia del tracto |
¡m
|
Clasificación de órganos del sistema MedDRA |
Muy frecuentes |
F recuentes |
Poco frecuentes |
Raras |
|
genital**, dolor mamario |
1 Las reacciones adversas de los estudios clínicos se codificaron utilizando el diccionario MedDRA (versión 14.1). Se emplea el término preferido del MedDRA para describir una cierta reacción y sus sinónimos y trastornos relacionados.Diferentes términos del MedDRA que representan el mismo fenómeno médico se han agrupado conjuntamente en una única reacción adversa para evitar diluir u oscurecer el verdadero efecto.
*- Frecuencia estimada a partir de estudios epidemiológicos que comprendían un grupo de anticonceptivos orales combinados. La frecuencia era muy cercana a muy rara. - “Episodios tromboembólicos venosos y arteriales” resume las siguientes entidades médicas: oclusión venosa profunda periférica, trombosis y embolia/oclusión vascular pulmonar, trombosis, embolia e infarto/infarto de miocardio/infarto cerebral e ictus no especificado como hemorrágico.
** Contiene las entidades médicas hemorragia del tracto genital femenino, hemorragia uterina no programada
Descripción de reacciones adversas seleccionadas
A continuación se enumeran las reacciones adversas de frecuencia muy baja o con retraso en el inicio de los síntomas que se consideran relacionadas con el grupo de los anticonceptivos hormonales combinados, incluidos los AOCs (ver secciones 4.3 y 4.4 ):
Trastornos circulatorios
• Se ha observado un aumento del riesgo de episodios trombóticos y tromboembólicos arteriales y venosos, entre ellos infarto de miocardio, ictus, accidentes isquémicos transitorios, trombosis venosa y embolia pulmonar, en muj eres que utilizan AHC, que se comentan con más detalle en la sección 4.4 .
Tumores
• La frecuencia del diagnóstico de cáncer de mama se encuentra muy ligeramente aumentada en las usuarias de anticonceptivos hormonales combinados. Dado que el cáncer de mama es raro en mujeres menores de 40 años, el exceso numérico es pequeño en relación con el riesgo global de cáncer de mama. Se desconoce la relación etiológica con el uso de anticonceptivos hormonales combinados.
• Tumores hepáticos (benignos y malignos).
Otros trastornos
• Eritema nudoso, eritema multiforme.
• Mujeres con hipertrigliceridemia (riesgo aumentado de pancreatitis al usar AOC).
• Hipertensión.
• Aparición o deterioro de trastornos cuya asociación con el uso de AOC no es concluyente: ictericia y/o prurito relacionados con colestasis, formación de cálculos biliares, porfiria, lupus eritematoso sistémico, síndrome urémico hemolítico, corea de Sydenham, herpes gestacional, pérdida de audición relacionada con otosclerosis.
• En mujeres con angioedema hereditario, los estrógenos exógenos pueden inducir o exacerbar los síntomas de angioedema.
• Alteraciones de la función hepática.
• Cambios en la tolerancia a la glucosa o efecto sobre la resistencia periférica a la insulina.
• Empeoramiento de la enfermedad de Crohn o de la colitis ulcerosa.
• Empeoramiento de la epilepsia.
• Cloasma.
• Hipersensibilidad (incluidos síntomas como exantema, urticaria).
¡m
Interacciones
Las interacciones de otros fármacos (inductores enzimáticos) con los anticonceptivos hormonales combinados pueden causar hemorragias intermenstruales y/o fallos en la anticoncepción (ver sección 4.5).
Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de medicamentos de Uso Humano: https://www.notificaram.es.
4.9 Sobredosis
De acuerdo con la experiencia general con los anticonceptivos orales combinados, los síntomas que podrían aparecer en este caso son: náuseas, vómitos y, en mujeres jóvenes, sangrado vaginal ligero. No hay antídotos y el tratamiento debe ser sintomático.
Los parches adicionales o inadecuadamente utilizados deben retirarse de la piel.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Hormonas sexuales y moduladores del sistema genital. Progestágenos y estrógenos, combinaciones fijas.
Código ATC: G03AA10 Mecanismo de acción
El efecto anticonceptivo de los anticonceptivos hormonales combinados se basa en la interacción de diversos factores, los más importantes de los cuales se consideran la inhibición de la ovulación y los cambios en la secreción cervical.
Eficacia clínica y seguridad
En el ensayo clínico realizado con Lisvy en la Unión Europea, Latinoamérica y Australia, se calcularon los siguientes índices de Pearl:
índice de Pearl (18-35 años, índice de masa corporal n 30 kg/m2)
Fallo del método: Índice de Pearl 0,82 (límite superior de confianza
del 95% 1,55)
Fallo del método y de la usuaria: Índice de Pearl 1,19 (límite superior de confianza del 95%
2,00)
Para la población europea se calcularon los siguientes índices de Pearl:
índice de Pearl (18-35 años, índice de masa corporal n 30 kg/m2)
Fallo del método: Índice de Pearl 0,40 (límite superior de confianza del 95%
1,18) ,
Fallo del método y de la usuaria: Índice de Pearl 0,76 (límite superior de confianza del 95%
1,66)
En el ensayo clínico realizado con Lisvy en EE.UU., se calcularon los siguientes índices de Pearl:
Índice de Pearl (18-35 años, sin restricciones con respecto al índice de masa corporal)
Fallo del método: Índice de Pearl 2,91 (límite superior de confianza del 95%
4,41)
Fallo del método y de la usuaria: Índice de Pearl 3,92 (límite superior de confianza del 95%
5,53)
La tasa de fallo puede aumentar cuando Lisvy se usa incorrectamente.
¡m
Se investigó la histología endometrial de 49 mujeres de un estudio clínico tras 13 ciclos de tratamiento. No hubo resultados anómalos.
5.2 Propiedades farmacocinéticas
Absorción
Tras la aplicación dérmica de Lisvy, el etinilestradiol y el gestodeno se absorben bien a través de la piel. El promedio de la liberación de etinilestradiol y gestodeno a lo largo del periodo de aplicación de 7 días de Lisvy genera la misma exposición sistémica (AUC) durante el estado estacionario que la observada tras la administración diaria de un anticonceptivo oral combinado con 0,02 mg de etinilestradiol y 0,06 mg de gestodeno.
Se determinaron las concentraciones séricas de etinilestradiol y gestodeno durante la tercera semana de diferentes ciclos de tratamiento (ciclo 1 a ciclo 7). Las concentraciones séricas máximas medias de etinilestradiol en el rango de 36-51 ng/l se alcanzaron aproximadamente 1 día después de la aplicación dérmica del parche. A partir de entonces, las concentraciones séricas de etinilestradiol disminuyeron a valores medios mínimos en el rango de 15-23 ng/l al final del intervalo de aplicación de 1 semana. El promedio de la concentración durante la semana 3 estuvo en el rango de 22-33 ng/l.
Las concentraciones séricas medias máximas de gestodeno total en el rango de 4,7-7,5 pg/l se alcanzaron aproximadamente 1-1,5 días después de la aplicación dérmica del parche. A partir de entonces, las concentraciones séricas disminuyeron a valores medios mínimos en el rango de 2,6-4,0 pg/l al final del intervalo de aplicación de 1 semana. El promedio de la concentración durante la semana 3 estuvo en el rango de 3,6-5,7 pg/l.
Influencia del peso corporal y del índice de masa corporal
Las concentraciones séricas de etinilestradiol y gestodeno durante el tratamiento con Lisvy dependen del peso corporal o el índice de masa corporal de la mujer. En mujeres obesas con un índice de masa corporal > 35 kg/m2, los promedios de las concentraciones séricas de etinilestradiol y gestodeno son un 24% y un 30% más bajos, respectivamente, que los observados en mujeres con un índice de masa corporal normal U 30 kg/m2. Existen datos limitados sobre la eficacia anticonceptiva en las mujeres con índice de masa corporal 30 kg/m2.
Influencia del calor, la humedad, el ejercicio y el lugar de aplicación
Se investigó la farmacocinética del etinilestradiol y el gestodeno tras la aplicación de Lisvy en condiciones específicas de calor, humedad y ejercicio, es decir, sauna, hidromasaje, natación y diferentes ejercicios físicos, en comparación con una actividad normal. En general, se demostró bioequivalencia para los parámetros Cmax y AUC del etinilestradiol y el gestodeno en estas condiciones específicas. Los resultados indican que no se observan diferencias clínicamente relevantes en la exposición al etinilestradiol y el gestodeno en las condiciones específicas propias de un gimnasio como sauna, hidromasaje, natación o diferentes ejercicios físicos en comparación con las actividades normales de la vida cotidiana.
En un estudio formal de farmacocinética (análisis no compartimental) que analizó 3 lugares de aplicación distintos, la exposición sistémica media al gestodeno y el etinilestradiol fue del 24% y el 31%, respectivamente, más elevada cuando el parche se aplicó en la parte superior y externa del brazo que en las nalgas o el abdomen. El rango de los datos de exposición para los tres lugares de aplicación presentó un amplio solapamiento. En un análisis de farmacocinética en poblaciones (meta-análisis), la exposición media geométrica a etinilestradiol (AUC (0-168) y Cmax) fue del 41% más elevada tras aplicar el parche en el brazo o nalgas comparado con la aplicación en el abdomen. Las correspondientes diferencias de los valores medios de AUC y Cmax para el gestodeno total fueron del 26% y 22%, respectivamente. No hay datos indicativos de que las diferencias en la exposición media afecten a la seguridad o eficacia de Lisvy.
Datos comparativos entre Lisvy transdérmico y anticonceptivos orales combinados
En un estudio de biodisponibilidad relativa, se compararon las concentraciones séricas en el estado estacionario y los parámetros farmacocinéticos del etinilestradiol y el gestodeno tras la aplicación de Lisvy con los de un anticonceptivo oral combinado con 0,020 mg de etinilestradiol y 0,075 mg de gestodeno. Los
¡m
valores medios de Cmax en el estado estacionario de etinilestradiol y gestodeno fueron en general un 30%-40% más bajos tras la aplicación de Lisvy que con el anticonceptivo oral combinado. La exposición (AUC y Cav) al etinilestradiol fue comparable tras ambas vías de administración, mientras que la exposición al gestodeno (concentración libre) fue un 18% más baja tras la aplicación de Lisvy. Estos datos generaron unos promedios de exposición/estimaciones posológicas para Lisvy equivalentes a la administración oral diaria de 0,020 mg de etinilestradiol y 0,060 mg de gestodeno. La variabilidad interindividual (%CV) de los principales parámetros farmacocinéticos como Cmax y AUC tras la aplicación de Lisvy fue más baja para el etinilestradiol, pero más alta para el gestodeno, en comparación con la determinada tras la administración oral.
Distribución
El etinilestradiol se une en gran medida, pero no de forma específica, a la albúmina sérica (en aproximadamente un 98%), pero no a la SHBG. El gestodeno se une en un alto grado a la albúmina sérica y a la SHBG. Solo alrededor del 1% de las concentraciones séricas farmacológicas totales se encuentran en forma de esteroide libre, el 40-80% se une a la SHBG. El etinilestradiol induce un gran aumento de las concentraciones séricas de SHBG, mientras que la administración de gestodeno genera una ligera disminución de la concentración de SHBG. Tras la aplicación dérmica repetida de Lisvy, las concentraciones séricas medias de SHBG en el estado estacionario se encuentran en el rango de 201237 nmol/l.
Tras la administración intravenosa de etinilestradiol, se determinó un volumen aparente de distribución de 3-9 l/kg. El respectivo volumen aparente de distribución del gestodeno es de aproximadamente 0,7 l/kg.
Biotransformación
El etinilestradiol (EE) se metaboliza principalmente mediante hidroxilación aromática, pero se forman una gran variedad de metabolitos hidroxilados y metilados que se encuentran presentes como metabolitos libres y como conjugados con glucurónidos y sulfato. La principal ruta metabólica del etinilestradiol es la 2-hidroxilación dependiente del CYP450 y la formación del catecol estrógeno 2-hidroxi-EE. La 2-hidroxilación del EE es catalizada por las familias genéticas CYP2C, CYP2E y CYP3A. La tasa de aclaramiento metabólica se encuentra en el rango de 2-7 ml/min/kg.
El gestodeno se metaboliza por completo a metabolitos generalmente más polares. El metabolismo del gestodeno se caracteriza por la hidroxilación en varias posiciones del núcleo esteroide y por la reducción de la función 3-ceto y el doble enlace delta-4. No se han descrito metabolitos activos. Además de CYP3A4, un grupo de diferentes enzimas del citocromo P450 puede contribuir en menor grado al metabolismo del gestodeno.
En dos estudios en los que se investigó el efecto de los inhibidores de CYP3A4 (ketoconazol, eritromicina), ninguno de los dos inhibidores influyó sobre las concentraciones séricas de etinilestradiol en el estado estacionario. En caso del gestodeno, la administración concomitante de los inhibidores generó un aumento del 11% y el 34% en la AUC (0-168) para el ketoconazol y la eritromicina, respectivamente. Este pequeño aumento, que da lugar a una exposición dentro del rango de los anticonceptivos orales combinados comercializados, no se considera clínicamente relevante.
En un estudio en el que se investigó el efecto de Lisvy sobre una administración única de midazolam, un sustrato modelo para las sustancias metabolizadas por CYP3A4, no se observó un aumento clínicamente relevante de las concentraciones plasmáticas de midazolam. La administración concomitante de midazolam generó un pequeño aumento del 7% y el 14% en la AUC (0-tlast) y la Cmax del midazolam, respectivamente.
Eliminación
El etinilestradiol no se excreta de forma inalterada en ningún grado significativo. Los metabolitos del etinilestradiol se excretan en una relación urinaria/biliar de 4:6. El descenso de las concentraciones séricas se caracteriza por al menos dos fases de eliminación con una semivida terminal de alrededor de 16 horas determinada tras la administración intravenosa, que genera concentraciones generalmente no cuantificables dos días después de la retirada del parche.
El gestodeno no se excreta de forma inalterada. Sus metabolitos se excretan en una relación urinaria/biliar de alrededor de 6:4. Tras la retirada del parche, las concentraciones séricas totales de gestodeno descienden más lentamente que las de etinilestradiol, con una semivida terminal media de aproximadamente 26 horas.
Linealidad / No linealidad
¡m
La farmacocinética del etinilestradiol es lineal con respecto a la dosis en el rango de 0,020 mg-0,100 mg. No se ha observado un cambio clínicamente relevante en la farmacocinética del etinilestradiol con el tiempo.
La farmacocinética del gestodeno depende de la concentración de SHBG, sobre a la que a su vez influyen los estrógenos, los andrógenos y también el gestodeno. Tras la aplicación dérmica repetida de Lisvy, se observan concentraciones de SHBG 3 a 4 veces superiores a los valores basales habituales. Concordantemente, las concentraciones séricas de gestodeno en el estado estacionario difieren de las observadas tras una única aplicación. Estos cambios dependientes de la SHBG provocan un cambio no lineal en la farmacocinética del gestodeno con el tiempo. Además, se determinó que la farmacocinética del gestodeno sin unir depende de la concentración de acuerdo con tres estudios en los que se investigó la farmacocinética de Lisvy a lo largo de un periodo de tres ciclos. Por lo tanto, la farmacocinética del gestodeno se considera no lineal con respecto al tiempo y la concentración.
Poblaciones especiales
Sexo
Lisvy solo está indicado en las mujeres.
Mujeres de edad avanzada
Lisvy no está indicado tras la menopausia.
Indice de masa corporal
Existen datos limitados sobre la eficacia anticonceptiva en mujeres con índice de masa corporal n 30 kg/m2.
Insuficiencia renal
Lisvy no se ha estudiado en mujeres con insuficiencia renal. Debido a la completa metabolización del etinilestradiol y el gestodeno a metabolitos inactivos antes de su eliminación y a la disponibilidad de una segunda ruta de excreción a través del hígado, no se espera un riesgo aumentado en las mujeres con insuficiencia renal.
Insuficiencia hepática
Lisvy no se ha estudiado en mujeres con insuficiencia hepática. Lisvy está contraindicado en mujeres con hepatopatía grave o antecedentes de la misma, siempre y cuando los valores de función hepática no hayan retornado a la normalidad. Ver también sección 4.3.
Diferencias étnicas
La farmacocinética del etinilestradiol se estudió en combinación con otro progestágeno en mujeres caucásicas, chinas y japonesas y no mostró diferencias clínicamente significativas. La farmacocinética de Lisvy no se ha estudiado específicamente en mujeres de distintas etnias. No se conocen enzimas polimórficas que contribuyan en un grado importante a la metabolización del gestodeno. Los datos disponibles en mujeres caucásicas, negras e hispanas no indican ninguna diferencia relevante en la farmacocinética de Lisvy entre las mujeres de distintas razas/etnias. Se disponen de datos muy limitados en mujeres asiáticas.
Tabaquismo
No existen datos indicativos de que el tabaquismo repercuta sobre la farmacocinética del etinilestradiol y el gestodeno.
Población pediátrica
No se ha establecido la seguridad y eficacia en adolescentes menores de 18 años. No existe una recomendación de uso específica para Lisvy en niñas y adolescentes premenárquicas.
¡m
5.3 Datos preclínicos sobre seguridad
Los datos preclínicos sobre los principios activos no muestran riesgos especiales para los seres humanos según los estudios convencionales de toxicidad a dosis repetidas, genotoxicidad, tolerabilidad local, potencial carcinogénico y toxicidad para la reproducción. No obstante, se debe tener presente que los esteroides sexuales pueden estimular el crecimiento de ciertos tejidos y tumores dependientes de hormonas. Los estudios de biocompatibilidad con el parche y sus materiales no mostraron riesgos especiales para los seres humanos con respecto a la seguridad local y sistémica del parche.
Evaluación del Riesgo Medioambiental (ERA)
Las sustancias activas gestodeno y etinilestradiol conllevan un riesgo para el medio ambiente, especialmente para los peces. Además el gestodeno y el etinilestradiol persisten en el medio ambiente. La eliminación de los parches utilizados o no utilizados se realizará de acuerdo con la normativa local . En caso de duda, se debe consultar al farmacéutico (ver sección 6.6 ).
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Capa de refuerzo:
Capa exterior de polietileno (PE) de baja densidad
Capa adherente:
Adhesivo compuesto por:
Éster de resina hidrogenada
Polibuteno
Poliisobutileno
Pentaeritritol tetrakis (3-(3,5-di-tert-butil-4-hidroxifenil) propionato) Bemotrizinol
Hoja de separación:
Película de tereftalato de polietileno (PET)
Matriz adherente:
Adhesivo compuesto por:
Éster de resina hidrogenada
Polibuteno
Poliisobutileno
Pentaeritritol tetrakis (3-(3,5-di-tert-butil-4-hidroxifenil) propionato) Revestimiento de liberación:
Película de tereftalato de polietileno (PET) siliconado
6.2 Incompatibilidades No procede.
6.3 Periodo de validez
3 años
6.4 Precauciones especiales de conservación
Conservar en el sobre original para protegerlo de la luz y la humedad.
6.5 Naturaleza y contenido del envase
Material de embalaje primario
Un sobre se compone de cuatro capas: una película de polietileno de baja densidad (capa más interna), una hoja de aluminio, una capa de papel y una película de tereftalato de polietileno.
Una hoja de separación de tereftalato de polietileno siliconado evita la adhesión del parche transdérmico en el interior del sobre.
Material de embalaje secundario
Los sobres se etiquetan y envasan junto a un prospecto (que incluye una tarjeta de recuerdo y unas pegatinas de recuerdo) en una caja de cartón.
Cada caja contiene 3, 9 o 18 parches transdérmicos Lisvy en sobres individuales.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El parche debe aplicarse inmediatamente después de extraerlo del sobre protector.
Para prevenir interferencias con las propiedades adhesivas de Lisvy, no deben aplicarse maquillaje, cremas, lociones, polvos ni otros productos cutáneos en la zona de piel en la que se encuentra o va a aplicarse Lisvy.
Las sustancias activas gestodeno y etinilestradiol conllevan un riesgo para el medio ambiente, especialmente para los peces. Además, el gestodeno y el etinilestradiol persisten en medio ambiente. Los parches usados no deben tirarse por el retrete ni introducirse en sistemas de eliminación de residuos líquidos. El parche usado debe eliminarse cuidadosamente de acuerdo con las siguientes instrucciones.
El sobre original debe guardarse para eliminar el parche tras el uso. El parche usado debe doblarse por la mitad con la cara adherente/pegajosa hacia dentro. Debe introducirse en el sobre original y cerrarse doblando el borde abierto.
Hay una etiqueta de dos páginas en el sobre. La primera página de la etiqueta debe levantarse y utilizarse para cerrar el borde doblado del sobre. Debajo de la primera página, pueden encontrarse las instrucciones de eliminación en la segunda página.
El parche debe eliminarse de forma segura fuera del alcance de los niños o mascotas.
La eliminación de los parches utilizados o no utilizados se realizará de acuerdo con la normativa local. En caso de duda, se debe consultar al farmacéutico.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
GEDEON RICHTER PLC.
Gyomroi ut 19-21 H-1103 Budapest, Hungria
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
79.138
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Octubre 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada y actualizada de este medicamento está disponible en la página Web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.gob.es/
27 de 27