Inovelon 100 Mg Comprimidos Recubiertos Con Pelicula
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Inovelon 100 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 100 mg de rufinamida.
Excipiente con efecto conocido: cada comprimido recubierto con película contiene 20 mg de lactosa monohidrato
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película.
Rosa, ‘ovalado’ ligeramente convexo, ranurado en ambos lados, con la inscripción ‘C261’ en un lado y nada en el otro.
El comprimido se puede dividir en dosis iguales.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Inovelon está indicado como terapia coadyuvante en el tratamiento de las crisis asociadas al síndrome de Lennox-Gastaut en pacientes de 4 años o mayores.
4.2 Posología y forma de administración
El tratamiento con rufinamida deberá iniciarlo un médico especializado en pediatría o neurología con experiencia en el tratamiento de la epilepsia.
Inovelon suspensión oral e Inovelon comprimidos recubiertos con película se pueden intercambiar en dosis iguales. Se debe vigilar a los pacientes durante el periodo de cambio.
Posología
Uso en niños de cuatro años o más y pesen menos de 30 kg Pacientes de <30 kg que no reciben valproato:
El tratamiento debe iniciarse con una dosis diaria de 200 mg. Según la respuesta clínica y la tolerabilidad, la dosis puede aumentarse a incrementos de 200 mg/día, cada dos días, hasta la dosis máxima recomendada de 1000 mg/día. Se han estudiado dosis de hasta 3600 mg/día en un número limitado de pacientes.
Pacientes de <30 kg que también reciban valproato:
Como el valproato disminuye significativamente el aclaramiento de la rufinamida, en los pacientes de <30 kg a los que se coadministre valproato se recomienda una dosis máxima más baja de Inovelon. El tratamiento debe iniciarse con una dosis diaria de 200 mg. Según la respuesta clínica y la tolerabilidad, después de al menos 2 días, la dosis podrá aumentarse en incrementos de 200 mg/día, hasta la dosis máxima recomendada de 600 mg/día.
Uso en adultos, adolescentes y niños de 4 años o mayores que pesen 30 kg o más El tratamiento debe iniciarse con una dosis diaria de 400 mg. Según la respuesta clínica y a la tolerabilidad, podrá aumentarse la dosis en incrementos de 400 mg/día, después de al menos 2 días, hasta la dosis máxima recomendada de la forma indicada en la siguiente tabla.
|
Rango de peso |
30,0-50,0 kg |
50,1-70,0 kg |
>70,1 kg |
|
Dosis máxima recomendada |
1800 mg/día |
2400 mg/día |
3200 mg/día |
Se han estudiado dosis de hasta 4000 mg/día (en el rango de 30-50 kg) o 4800 mg/día (en la categoría de más de 50 kg) en un número limitado de pacientes.
Interrupción del tratamiento
Cuando se vaya a interrumpir el tratamiento con rufinamida, debe hacerse gradualmente. En los ensayos clínicos, la interrupción del tratamiento con rufinamida se llevó a cabo reduciendo aproximadamente un 25 % de la dosis cada dos días.
En caso de olvidarse una o más dosis, será necesario aplicar un criterio clínico individualizado.
Los ensayos abiertos no controlados indican una eficacia a largo plazo sostenida, aunque no se ha realizado ningún ensayo controlado durante más de 3 meses.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de la rufinamida en niños de 4 años o menores.
No se dispone de datos.
Pacientes de edad avanzada
La información disponible sobre el uso de la rufinamida en pacientes de edad avanzada es limitada. Ya que la farmacocinética de la rufinamida no se altera en los pacientes de edad avanzada (ver sección 5.2), no se requieren ajustes de dosis en pacientes mayores de 65 años.
Insuficiencia renal
Un estudio realizado en pacientes con insuficiencia renal grave indicó que no se requieren ajustes de dosis en estos pacientes (ver sección 5.2).
Insuficiencia hepática
No se ha estudiado el uso en pacientes con insuficiencia hepática. Se recomienda precaución y un ajuste cuidadoso de la dosis en el tratamiento de pacientes con insuficiencia hepática de leve a moderada. No se recomienda el uso en pacientes con insuficiencia hepática severa.
Forma de administración
La rufinamida se administra por vía oral. Se debe tomar con agua dos veces al día, una por la mañana y otra por la noche, en dos dosis iguales. Como se ha observado un efecto con alimentos, Inovelon debe administrarse con alimentos (ver sección 5.2). Si el paciente tiene dificultad para tragar los comprimidos, los puede triturar y tomarlos disueltos en medio vaso de agua.
4.3 Contraindicaciones
Hipersensibilidad al principio activo, a los derivados triazólicos o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Estado epiléptico
Se han observado casos de estado epiléptico con rufinamida en los estudios de desarrollo clínico mientras que no se ha observado ningún caso con placebo. Estos efectos adversos ocasionaron la interrupción del tratamiento con rufinamida en el 20 % de los casos. Si los pacientes desarrollan nuevos tipos de convulsiones y/o experimentan un aumento de la frecuencia de estado epiléptico que sea diferente de la situación basal del paciente, debe reevaluarse el balance beneficio-riesgo de la terapia.
Retirada de la rufinamida
La rufinamida debe interrumpirse gradualmente para reducir la posibilidad de convulsiones durante la retirada. En los ensayos clínicos, la interrupción se llevó a cabo reduciendo aproximadamente un 25 % de la dosis cada dos días. No hay datos suficientes sobre la interrupción de tratamientos antiepilépticos concomitantes, una vez alcanzado el control de las convulsiones con la adición de rufinamida.
Reacciones en el sistema nervioso central
El tratamiento con rufinamida se ha asociado con mareos, somnolencia, ataxia y trastornos de la marcha, lo que puede incrementar la aparición de caídas accidentales en esta población (ver sección 4.8). Los pacientes y cuidadores deben tener precaución hasta que estén familiarizados con los posibles efectos de este medicamento.
Reacciones de hipersensibilidad
Se han producido el síndrome de hipersensibilidad a antiepilépticos grave incluyendo DRESS (Reacción al Fármaco con Eosinofilia y Síntomas Sistémicos) y síndrome de Stevens-Johnson asociado con la terapia con rufinamida. Los signos y los síntomas de este trastorno fueron diversos; sin embargo, los pacientes normalmente, aunque no de forma exclusiva, presentaron fiebre y erupción cutánea asociadas con afectación de otros órganos del sistema. Otras manifestaciones asociadas incluyeron linfadenopatía, anomalías en las pruebas de la función hepática y hematuria. Al tratarse de un trastorno que varía en su expresión pueden producirse otros signos y síntomas en los sistemas y órganos no citados aquí. Este síndrome de hipersensibilidad a antiepilépticos se asoció temporalmente al comienzo de la terapia con rufinamida y en la población pediátrica. Si se sospecha esta reacción, debe interrumpirse la administración de rufinamida y comenzar un tratamiento alternativo. Todos los pacientes que desarrollen erupción cutánea mientras tomen rufinamida deben monitorizarse cuidadosamente.
Acortamiento del intervalo QT
En un estudio minucioso del efecto sobre el intervalo QT, la rufinamida produjo un acortamiento del intervalo QTc proporcional a la concentración. Aunque se desconozcan el mecanismo subyacente y la relevencia para la seguridad de este hallazgo, los médicos deben seguir un criterio clínico cuando valoren la posible prescripción de rufinamida a pacientes que presenten un riesgo adicional de acortamiento del QTc (por ej.; síndrome de QT corto congénito o pacientes con una historia familiar de este tipo de síndrome).
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar anticonceptivos eficaces durante el tratamiento con Inovelon. Los médicos deben intentar asegurar que se utilizan métodos anticonceptivos apropiados, y deben seguir un criterio clínico a la hora de valorar si los anticonceptivos orales o las dosis de los componentes de los anticonceptivos orales son adecuados en función de la situación clínica de cada paciente (ver sección 4.5).
Inovelon contiene lactosa, por lo tanto los pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia de lactasa de Lapp o mala absorción de glucosa-galactosa no deben tomar este medicamento.
Pensamientos suicidas
Se han notificado pensamientos y conductas suicidas en pacientes tratados con antiepilépticos en varias indicaciones. Asimismo, un metanálisis de ensayos aleatorizados y controlados con placebo de antiepilépticos ha demostrado un pequeño aumento en el riesgo de pensamientos y conductas suicidas. Se desconoce el mecanismo de este riesgo y los datos disponibles no descartan la posibilidad de un aumento del riesgo con Inovelon.
Por lo tanto, se debe vigilar a los pacientes por si presentan signos de pensamientos y conductas suicidas y considerar el tratamiento adecuado. Se debe informar a los pacientes (y cuidadores de los pacientes) que acudan al médico si aparecen signos de pensamientos o conductas suicidas.
4.5 Interacción con otros medicamentos y otras formas de interacción
Posibilidad de que otros medicamentos afecten a la rufinamida
Otros antiepilépticos
Las concentraciones de rufinamida no están sujetas a cambios clínicamente relevantes al coadministrarse con antiepilépticos que se sabe que inducen enzimas.
En pacientes que estén en tratamiento con Inovelon y en los que se inicie la terapia con valproato, pueden producirse incrementos significativos en las concentraciones plasmáticas de rufinamida. Los incrementos más pronunciados se observaron en pacientes con bajo peso corporal (<30 kg). Por lo tanto, debe considerarse una reducción de la dosis de Inovelon en pacientes de <30 kg que inicien la terapia con valproato (ver sección 4.2).
La adición o interrupción de estos medicamentos o el ajuste de la dosis de estos medicamentos durante la terapia con rufinamida pueden requerir un ajuste de la dosis de la rufinamida.
No se observan cambios significativos en la concentración de rufinamida tras la coadministración de lamotrigina, topiramato o benzodiazepinas.
Posibilidad de que la rufinamida afecte a otros medicamentos
Otros antiepilépticos
Las interacciones farmacocinéticas entre la rufinamida y otros antiepilépticos se han evaluado en pacientes epilépticos utilizando modelos farmacocinéticos poblacionales. La rufinamida parece no tener ningún efecto clínicamente relevante sobre las concentraciones en estado estacionario de carbamazepina, lamotrigina, fenobarbital, topiramato, fenitoína o valproato.
Anticonceptivos orales
La coadministración de rufinamida 800 mg dos veces al día junto con un anticonceptivo oral combinado (etinilestradiol 35 microgramos y noretisterona 1 mg) durante 14 días dio lugar a una reducción media del AUC0-24 del etinilestradiol del 22 % y del AUC0-24 de la noretisterona del 14 %. No se han realizado estudios con otros anticonceptivos orales o implantables. A las mujeres en edad fértil que utilicen anticonceptivos hormonales, se les aconseja el uso de un método anticonceptivo seguro y eficaz adicional (ver secciones 4.4 y 4.6).
Enzimas del citocromo P450
La rufinamida se metaboliza mediante hidrólisis y no se metaboliza de forma notable por las enzimas del citocromo P450. Además, la rufinamida no inhibe la actividad de las enzimas del citocromo P450
5
(ver sección 5.2). Por lo tanto, es improbable que la rufinamida produzca interacciones clínicamente significativas por la inhibición del sistema del citocromo P450 Se ha demostrado que la rufinamida induce la enzima CYP3A4 del citocromo P450, y por lo tanto puede reducir las concentraciones plasmáticas de las sustancias metabolizadas por esta enzima. El efecto fue de pequeño a moderado. La actividad media de CYP3A4, evaluada como el aclaramiento de triazolam, aumentó en un 55 % tras 11 días de tratamiento con 400 mg de rufinamida dos veces al día. La exposición de triazolam se redujo en un 36 %. Dosis de rufinamida más altas pueden dar lugar a una inducción más pronunciada. No se puede descartar la posibilidad de que la rufinamida también pueda disminuir la exposición de sustancias metabolizadas por otras enzimas, o transportadas por proteínas transportadoras como la glucoproteína-P.
En los pacientes tratados con sustancias metabolizadas por el sistema enzimático CYP3A4, se recomienda una monitorización cuidadosa durante dos semanas al inicio del tratamiento con rufinamida o al finalizar el mismo, o después de cualquier cambio relevante en la dosis. Puede ser necesario considerar un ajuste de la dosis del medicamento administrado concomitantemente. Estas recomendaciones deben asimismo considerarse cuando se utilice la rufinamida concomitantemente con sustancias con un margen terapéutico estrecho como warfarina y digoxina.
Un estudio de interacción específico en sujetos sanos no reveló ninguna influencia de la rufinamida a una dosis de 400 mg dos veces al día sobre la farmacocinética de olanzapina, un sustrato de CYP1A2.
No hay datos disponibles sobre la interacción de la rufinamida con el alcohol.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Riesgo relacionado en general con la epilepsia y los antiepilépticos:
Se ha demostrado que la prevalencia de malformaciones en la descendencia de mujeres epilépticas, es dos o tres veces mayor que la tasa de aproximadamente el 3 % de población general. En la población tratada con politerapia, se ha observado un aumento de las malformaciones; sin embargo, no se ha elucidado hasta qué punto es responsabilidad del tratamiento y/o de la enfermedad.
Además, no debe interrumpirse una terapia antiepiléptica eficaz, ya que el agravamiento de la enfermedad va en detrimento tanto de la madre como del feto.
Riesgo relacionado a la rufinamida:
Los estudios en animales no han mostrado efectos teratogénicos aunque sí se observó fetotoxicidad en presencia de toxicidad materna (ver sección 5.3). Se desconoce el riesgo potencial en seres humanos.
No se dispone de datos clínicos sobre embarazos de riesgo para rufinamida.
Teniendo en cuenta estos datos, rufinamida no debe utilizarse durante el embarazo salvo que fuese claramente necesario ni en mujeres en edad fértil que no utilicen medidas anticonceptivas.
Las mujeres en edad fértil deben utilizar medidas anticonceptivas durante el tratamiento con rufinamida. Los médicos deben intentar asegurar que se utilicen anticonceptivos apropiados, y deberán seguir un criterio clínico a la hora de valorar si los anticonceptivos orales o las dosis de los componentes de los anticonceptivos orales son adecuados en función de la situación clínica de cada paciente (ver sección 4.5).
Si las mujeres que reciben tratamiento con rufinamida planean quedarse embarazadas, deberá sopesarse cuidadosamente la indicación de este producto. Durante el embarazo, no debe interrumpirse un tratamiento antiepiléptico eficaz con rufinamida, ya que el agravamiento de la enfermedad va en detrimento tanto de la madre como del feto.
Se desconoce si la rufinamida se excreta en la leche materna. Debido a los posibles efectos nocivos para los lactantes, debe evitarse la lactancia durante el tratamiento de la madre con rufinamida.
Fertilidad
No hay datos disponibles sobre los efectos en la fertilidad tras el tratamiento con rufinamida.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Inovelon puede producir mareos, somnolencia y visión borrosa. Dependiendo de la sensibilidad individual, la rufinamida puede tener una influencia de pequeña a importante en la capacidad para conducir y utilizar máquinas. Debe informarse a los pacientes que tengan cuidado en aquellas actividades que requieran mucha concentración, por ejemplo, conducir y utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
El programa de desarrollo clínico ha incluido a más de 1900 pacientes, con diferentes tipos de epilepsia, expuestos a rufinamida. Las reacciones adversas notificadas con mayor frecuencia en general fueron cefalea, mareos, fatiga y somnolencia. Las reacciones adversas más frecuentes y notificadas con una incidencia mayor que con el placebo en pacientes con síndrome de Lennox-Gastaut fueron somnolencia y vómitos. Las reacciones adversas generalmente presentaron una gravedad de leve a moderada. La tasa de interrupción del tratamiento en el síndrome de Lennox-Gastaut debido a las reacciones adversas fue del 8,2 % para los pacientes que recibían rufinamida y del 0 % para los pacientes que recibían el placebo. Las reacciones adversas más frecuentes que dieron lugar a la interrupción en el grupo tratado con rufinamida fueron erupción cutánea y vómitos.
Listado tabulado de reacciones adversas
Las reacciones adversas notificadas con una incidencia superior al placebo, durante los ensayos de doble ciego en el síndrome de Lennox-Gastaut o en la población global expuesta a rufinamida, se enumeran en la siguiente tabla siguiendo el sistema de clasificación por órganos MedDRA y en función de la frecuencia.
Las frecuencias se definen como: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000).
|
Clasificación de órganos del sistema |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
Infecciones e infestaciones |
Neumonía Gripe Nasofaringitis Infección de oído Sinusitis Rinitis | |||
|
Trastornos del sistema inmunológico |
Hipersensibilidad* | |||
|
Trastornos del metabolismo y de la nutrición |
Anorexia Trastorno del apetito Disminución del apetito | |||
|
Trastornos psiquiátricos |
Ansiedad Insomnio |
|
Clasificación de órganos del sistema |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
Trastornos del sistema nervioso |
Somnolencia* Cefalea Mareos* |
Estado epiléptico* Convulsión Coordinación anormal* Nistagmo Hiperactividad psicomotora | ||
|
Temblores | ||||
|
Trastornos oculares |
Diplopía Visión borrosa | |||
|
Trastornos del oído y del laberinto |
Vértigo | |||
|
Trastornos respiratorios, torácicos y mediastínicos |
Epistaxis | |||
|
Trastornos gastrointestinales |
Náuseas Vómitos |
Dolor abdomen superior Estreñimiento Dispepsia Diarrea | ||
|
Trastornos hepatobiliares |
Aumento de las enzimas hepáticas | |||
|
Trastornos de la piel y del tejido subcutáneo |
Erupción cutánea* Acné | |||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor de espalda | |||
|
Trastornos del aparato reproductor y de la mama |
Oligomenorrea | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Fatiga |
Trastorno de la marcha* | ||
|
Exploraciones complementarias |
Disminución de peso | |||
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Traumatismo craneal Contusión |
*consultar sección 4.4
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello
permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.4.9 Sobredosis
Tras una sobredosis aguda, se puede vaciar el estómago mediante lavado gástrico o mediante la inducción de vómitos. No hay ningún antídoto específico para la rufinamida. El tratamiento debe ser de apoyo y puede incluir la hemodiálisis (ver sección 5.2).
La administración de dosis múltiples de 7200 mg/día no se asoció con signos o síntomas importantes.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antiepilépticos, derivados de carboxamida; código ATC: N03AF03. Mecanismo de acción
La rufinamida modula la actividad de los canales de sodio, prolongando el estado inactivo. La rufinamida es activa en diversos modelos animales de epilepsia.
Experiencia clínica
En un ensayo de doble ciego y controlado con placebo, se administró Inovelon (comprimidos de rufinamida) en dosis de hasta 45 mg/kg/día durante 84 días, a 139 pacientes con convulsiones no controladas adecuadamente asociadas con el síndrome de Lennox-Gastaut (incluyendo crisis de ausencia atípica y episodios de caída). Se incluyeron hombres y mujeres (de entre 4 y 30 años) que recibían tratamiento concomitantemente con 1 a 3 antiepilépticos a dosis fijas. Cada paciente tenía que haber tenido al menos 90 convulsiones en el mes anterior a la entrada en el ensayo. Se observó una mejoría significativa en las tres variables primarias principales: el cambio porcentual en la frecuencia total de crisis cada 28 días durante la fase de mantenimiento respecto a la frecuencia basal (-35,8 % con Inovelon frente al -1,6 % con placebo, p=0,0006), el número de convulsiones tónicas-atónicas (42,9 % con Inovelon frente al 2,2 % con placebo, p = 0,0002), y la puntuación de la gravedad de las crisis a partir de la Evaluación Global realizada por los padres/representante legal al final de la fase de doble ciego (mejoría grande o muy grande en el 32,2 % con Inovelon frente al 14,5 % en el grupo de placebo, p=0,0041).
Los modelos farmacocinéticos/farmacodinámicos poblacionales demostraron que la reducción de las frecuencias de las convulsiones totales y las convulsiones tónicas-atónicas, la mejoría de la evaluación global de la gravedad de las convulsiones y el aumento de la probabilidad de reducción de la frecuencia de las convulsiones dependieron de las concentraciones de rufinamida.
5.2 Propiedades farmacocinéticas
Absorción
Los niveles plasmáticos máximos se alcanzan aproximadamente 6 horas después de la administración. La concentración máxima (Cmáx) y el AUC de plasma de rufinamida aumentan menos que proporcionalmente en relación con las dosis administradas a sujetos sanos tanto en ayunas como con alimentos y a pacientes, probablemente se deba a la absorción limitada de la dosis. Tras dosis únicas, la comida aumenta la biodisponibilidad (AUC) de la rufinamida en aproximadamente el 34 % y la concentración plasmática máxima en 56 %.
Inovelon suspensión oral e Inovelon comprimidos recubiertos con película han demostrado ser bioequivalentes.
Distribución
En los estudios in vitro, solo una pequeña fracción de rufinamida (34 %) se fijó a las proteínas séricas humanas de las que la albúmina supone aproximadamente el 80 % de esta fijación. Esto indica un riesgo mínimo de interacciones medicamentosas debidas al desplazamiento de los sitios de fijación durante la administración concomitante de otras sustancias. La rufinamida presentó una distribución uniforme entre los eritrocitos y el plasma.
Metabolismo
La rufinamida se elimina de forma casi exclusiva a través del metabolismo. La vía principal de metabolismo es la hidrólisis del grupo carboxilamida para formar el derivado ácido farmacológicamente inactivo, CGP 47292. El metabolismo mediado por el citocromo P450 es muy pequeño. No se puede excluir por completo la formación de pequeñas cantidades de conjugados de glutatión.
In vitro, la rufinamida ha demostrado tener una capacidad pequeña o no significativa para actuar como un inhibidor competitivo o basado en el mecanismo de las siguientes enzimas P450 humanas: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 o CYP4A9/11-2.
Eliminación
La semivida de eliminación plasmática es aproximadamente de 6-10 horas en sujetos sanos y en pacientes con epilepsia. Cuando se administra dos veces al día a intervalos de 12 horas, la rufinamida se acumula en el grado previsto a partir de su semivida terminal, lo que indica que la farmacocinética de la rufinamida es independiente del tiempo (es decir, no hay ninguna autoinducción del metabolismo).
En un ensayo con marcadores radiactivos en tres voluntarios sanos, el compuesto original (rufinamida) fue el principal componente radiactivo en el plasma, representando aproximadamente el 80 % de la radiactividad total, y el metabolito CGP 47292 solo supuso aproximadamente el 15 %. La excreción renal fue la vía predominante de eliminación para el material relacionado con el principio activo, representando el 84,7 % de la dosis.
Linealidad/no linealidad:
La biodisponibilidad de la rufinamida es dosis-dependiente. Al aumentar la dosis, disminuye la biodisponibilidad.
Farmacocinética en grupos especiales de pacientes
Sexo
Se han utilizado modelos farmacocinéticos poblacionales para evaluar la influencia del sexo en la farmacocinética de la rufinamida. Estas evaluaciones indican que el sexo no afecta a la farmacocinética de la rufinamida en un grado clínicamente relevante.
Insuficiencia renal
La farmacocinética de una sola dosis de 400 mg de rufinamida no se vio alterada en sujetos con insuficiencia renal crónica y severa en comparación con voluntarios sanos. Sin embargo, los niveles plasmáticos disminuyeron en aproximadamente un 30 % al utilizar la hemodiálisis tras la administración de rufinamida, lo que sugiere que puede tratarse de un procedimiento útil en caso de sobredosis (ver secciones 4.2 y 4.9).
Insuficiencia hepática
No se han realizado ensayos en pacientes con insuficiencia hepática y, por tanto, Inovelon no debe administrarse a pacientes con insuficiencia hepática grave (ver sección 4.2).
Niños (2-12 años)
En general, los niños presentan un menor aclaramiento de rufinamida que los adultos, y esta diferencia está relacionada con el tamaño corporal. No se han realizado ensayos en neonatos o en lactantes ni en niños menores de 2 años.
Pacientes de edad avanzada
Un estudio farmacocinético en voluntarios sanos de edad avanzada no mostró ninguna diferencia significativa en los parámetros farmacocinéticos comparados con los de adultos más jóvenes.
5.3 Datos preclínicos sobre seguridad
Los estudios convencionales de farmacología de seguridad no muestran riesgos especiales con las dosis clínicamente relevantes.
La toxicidad observada en perros con niveles similares a la exposición humana obtenida con la dosis máxima recomendada, fue cambios hepáticos incluyendo trombos biliares, colestasis y aumento de las enzimas hepáticas, que se cree que están relacionados con un aumento de la secreción biliar en esta especie. No hubo evidencia de ningún riesgo asociado en los estudios de toxicidad a dosis repetidas en ratas y monos.
En los estudios de toxicidad reproductiva y toxicidad durante el desarrollo, se redujo la supervivencia y crecimiento fetales y hubo algunos casos de mortinatalidad secundaria a la toxicidad materna. Sin embargo, no se observaron efectos en la morfología y la función de la descendencia, incluyendo el aprendizaje o la memoria. La rufinamida no fue teratogénica en ratones, ratas o conejos.
La rufinamida no resultó genotóxica y no presenta potencial carcinogénico. Las reacciones adversas no observadas en los ensayos clínicos aunque sí vistas en animales con niveles de exposición similares a los clínicos y con posible relevancia para el uso humano fue mielofibrosis de la médula ósea en el estudio de carcinogenicidad con ratón. Los neoplasmas óseos benignos (osteomas) y la hiperostosis observados en ratones se consideraron el resultado de la activación de un virus específico a ratón por los iones de fluoruro liberados durante el metabolismo oxidativo de la rufinamida.
Con respecto al potencial inmunotóxico, en un estudio en perros de 13 semanas de duración se observaron timo pequeño e involución del timo con respuesta significativa a la dosis alta en los machos. En el estudio de 13 semanas, se notificaron con incidencia baja cambios en la medula ósea y linfoides en hembras a las que se administro la dosis alta. Solo en el estudio de carcinogenicidad en ratas, observaron disminución celular de la médula ósea y atrofia del timo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo:
Lactosa monohidrato Celulosa, microcristalina Almidón de maíz Croscarmelosa sódica Hipromelosa Estearato de magnesio Laurilsulfato de sodio Sílice coloidal, anhidra
Recubrimiento:
Hipromelosa
Macrogol (8000)
Dióxido de titanio (E171)
Talco
Óxido férrico rojo (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30°C.
6.5 Naturaleza y contenido del envase
Blíster de aluminio/aluminio, envases de 10, 30, 50, 60 y 100 comprimidos recubiertos con película. Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
Ninguna especial.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Eisai Limited, European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/378/001-005
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16/enero/2007 Fecha de la última renovación: 16/enero/2012
10. FECHA DE LA REVISIÓN DEL TEXTO
{MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
NOMBRE DEL MEDICAMENTO
1.
Inovelon 200 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 200 mg de rufinamida.
Excipiente con efecto conocido: cada comprimido recubierto con película contiene 40 mg de lactosa monohidrato
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película.
Rosa, ‘ovalado’ ligeramente convexo, ranurado en ambos lados, con la inscripción ‘C262’ en un lado y nada en el otro.
El comprimido se puede dividir en dosis iguales.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Inovelon está indicado como terapia coadyuvante en el tratamiento de las crisis asociadas al síndrome de Lennox-Gastaut en pacientes de 4 años o mayores.
4.2 Posología y forma de administración
El tratamiento con rufinamida deberá iniciarlo un médico especializado en pediatría o neurología con experiencia en el tratamiento de la epilepsia.
Inovelon suspensión oral e Inovelon comprimidos recubiertos con película se pueden intercambiar en dosis iguales. Se debe vigilar a los pacientes durante el periodo de cambio.
Posología
Uso en niños de cuatro años o más y pesen menos de 30 kg Pacientes de <30 kg que no reciben valproato:
El tratamiento debe iniciarse con una dosis diaria de 200 mg. Según la respuesta clínica y la tolerabilidad, la dosis puede aumentarse a incrementos de 200 mg/día, cada dos días, hasta la dosis máxima recomendada de 1000 mg/día. Se han estudiado dosis de hasta 3600 mg/día en un número limitado de pacientes.
Pacientes de <30 kg que también reciban valproato:
Como el valproato disminuye significativamente el aclaramiento de la rufinamida, en los pacientes de <30 kg a los que se coadministre valproato se recomienda una dosis máxima más baja de Inovelon. El tratamiento debe iniciarse con una dosis diaria de 200 mg. Según la respuesta clínica y la tolerabilidad, después de al menos 2 días, la dosis podrá aumentarse en incrementos de 200 mg/día, hasta la dosis máxima recomendada de 600 mg/día.
Uso en adultos, adolescentes y niños de 4 años o mayores que pesen 30 kg o más El tratamiento debe iniciarse con una dosis diaria de 400 mg. Según la respuesta clínica y a la tolerabilidad, podrá aumentarse la dosis en incrementos de 400 mg/día, después de al menos 2 días, hasta la dosis máxima recomendada de la forma indicada en la siguiente tabla.
|
Rango de peso |
30,0-50,0 kg |
50,1-70,0 kg |
>70,1 kg |
|
Dosis máxima recomendada |
1800 mg/día |
2400 mg/día |
3200 mg/día |
Se han estudiado dosis de hasta 4000 mg/día (en el rango de 30-50 kg) o 4800 mg/día (en la categoría de más de 50 kg) en un número limitado de pacientes.
Interrupción del tratamiento
Cuando se vaya a interrumpir el tratamiento con rufinamida, debe hacerse gradualmente. En los ensayos clínicos, la interrupción del tratamiento con rufinamida se llevó a cabo reduciendo aproximadamente un 25 % de la dosis cada dos días.
En caso de olvidarse una o más dosis, será necesario aplicar un criterio clínico individualizado.
Los ensayos abiertos no controlados indican una eficacia a largo plazo sostenida, aunque no se ha realizado ningún ensayo controlado durante más de 3 meses.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de la rufinamida en niños de 4 años o menores.
No se dispone de datos.
Pacientes de edad avanzada
La información disponible sobre el uso de la rufinamida en pacientes de edad avanzada es limitada. Ya que la farmacocinética de la rufinamida no se altera en los pacientes de edad avanzada (ver sección 5.2), no se requieren ajustes de dosis en pacientes mayores de 65 años.
Insuficiencia renal
Un estudio realizado en pacientes con insuficiencia renal grave indicó que no se requieren ajustes de dosis en estos pacientes (ver sección 5.2).
Insuficiencia hepática
No se ha estudiado el uso en pacientes con insuficiencia hepática. Se recomienda precaución y un ajuste cuidadoso de la dosis en el tratamiento de pacientes con insuficiencia hepática de leve a moderada. No se recomienda el uso en pacientes con insuficiencia hepática severa.
Forma de administración
La rufinamida se administra por vía oral. Se debe tomar con agua dos veces al día, una por la mañana y otra por la noche, en dos dosis iguales. Como se ha observado un efecto con alimentos, Inovelon debe administrarse con alimentos (ver sección 5.2). Si el paciente tiene dificultad para tragar los comprimidos, los puede triturar y tomarlos disueltos en medio vaso de agua.
4.3 Contraindicaciones
Hipersensibilidad al principio activo, a los derivados triazólicos o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Estado epiléptico
Se han observado casos de estado epiléptico con rufinamida en los estudios de desarrollo clínico mientras que no se ha observado ningún caso con placebo. Estos efectos adversos ocasionaron la interrupción del tratamiento con rufinamida en el 20 % de los casos. Si los pacientes desarrollan nuevos tipos de convulsiones y/o experimentan un aumento de la frecuencia de estado epiléptico que sea diferente de la situación basal del paciente, debe reevaluarse el balance beneficio-riesgo de la terapia.
Retirada de la rufinamida
La rufinamida debe interrumpirse gradualmente para reducir la posibilidad de convulsiones durante la retirada. En los ensayos clínicos, la interrupción se llevó a cabo reduciendo aproximadamente un 25 % de la dosis cada dos días. No hay datos suficientes sobre la interrupción de tratamientos antiepilépticos concomitantes, una vez alcanzado el control de las convulsiones con la adición de rufinamida.
Reacciones en el sistema nervioso central
El tratamiento con rufinamida se ha asociado con mareos, somnolencia, ataxia y trastornos de la marcha, lo que puede incrementar la aparición de caídas accidentales en esta población (ver sección 4.8). Los pacientes y cuidadores deben tener precaución hasta que estén familiarizados con los posibles efectos de este medicamento.
Reacciones de hipersensibilidad
Se han producido el síndrome de hipersensibilidad a antiepilépticos grave incluyendo DRESS (Reacción al Fármaco con Eosinofilia y Síntomas Sistémicos) y síndrome de Stevens-Johnson asociado con la terapia con rufinamida. Los signos y los síntomas de este trastorno fueron diversos; sin embargo, los pacientes normalmente, aunque no de forma exclusiva, presentaron fiebre y erupción cutánea asociadas con afectación de otros órganos del sistema. Otras manifestaciones asociadas incluyeron linfadenopatía, anomalías en las pruebas de la función hepática y hematuria. Al tratarse de un trastorno que varía en su expresión pueden producirse otros signos y síntomas en los sistemas y órganos no citados aquí. Este síndrome de hipersensibilidad a antiepilépticos se asoció temporalmente al comienzo de la terapia con rufinamida y en la población pediátrica. Si se sospecha esta reacción, debe interrumpirse la administración de rufinamida y comenzar un tratamiento alternativo. Todos los pacientes que desarrollen erupción cutánea mientras tomen rufinamida deben monitorizarse cuidadosamente.
Acortamiento del intervalo QT
En un estudio minucioso del efecto sobre el intervalo QT, la rufinamida produjo un acortamiento del intervalo QTc proporcional a la concentración. Aunque se desconozcan el mecanismo subyacente y la relevencia para la seguridad de este hallazgo, los médicos deben seguir un criterio clínico cuando valoren la posible prescripción de rufinamida a pacientes que presenten un riesgo adicional de acortamiento del QTc (por ej.; síndrome de QT corto congénito o pacientes con una historia familiar de este tipo de síndrome).
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar anticonceptivos eficaces durante el tratamiento con Inovelon. Los médicos deben intentar asegurar que se utilizan métodos anticonceptivos apropiados, y deben seguir un criterio clínico a la hora de valorar si los anticonceptivos orales o las dosis de los componentes de los anticonceptivos orales son adecuados en función de la situación clínica de cada paciente (ver sección 4.5).
Inovelon contiene lactosa, por lo tanto los pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia de lactasa de Lapp o mala absorción de glucosa-galactosa no deben tomar este medicamento.
Pensamientos suicidas
Se han notificado pensamientos y conductas suicidas en pacientes tratados con antiepilépticos en varias indicaciones. Asimismo, un metanálisis de ensayos aleatorizados y controlados con placebo de antiepilépticos ha demostrado un pequeño aumento en el riesgo de pensamientos y conductas suicidas. Se desconoce el mecanismo de este riesgo y los datos disponibles no descartan la posibilidad de un aumento del riesgo con Inovelon.
Por lo tanto, se debe vigilar a los pacientes por si presentan signos de pensamientos y conductas suicidas y considerar el tratamiento adecuado. Se debe informar a los pacientes (y cuidadores de los pacientes) que acudan al médico si aparecen signos de pensamientos o conductas suicidas.
4.5 Interacción con otros medicamentos y otras formas de interacción
Posibilidad de que otros medicamentos afecten a la rufinamida
Otros antiepilépticos
Las concentraciones de rufinamida no están sujetas a cambios clínicamente relevantes al coadministrarse con antiepilépticos que se sabe que inducen enzimas.
En pacientes que estén en tratamiento con Inovelon y en los que se inicie la terapia con valproato, pueden producirse incrementos significativos en las concentraciones plasmáticas de rufinamida. Los incrementos más pronunciados se observaron en pacientes con bajo peso corporal (<30 kg). Por lo tanto, debe considerarse una reducción de la dosis de Inovelon en pacientes de <30 kg que inicien la terapia con valproato (ver sección 4.2).
La adición o interrupción de estos medicamentos o el ajuste de la dosis de estos medicamentos durante la terapia con rufinamida pueden requerir un ajuste de la dosis de la rufinamida.
No se observan cambios significativos en la concentración de rufinamida tras la coadministración de lamotrigina, topiramato o benzodiazepinas.
Posibilidad de que la rufinamida afecte a otros medicamentos
Otros antiepilépticos
Las interacciones farmacocinéticas entre la rufinamida y otros antiepilépticos se han evaluado en pacientes epilépticos utilizando modelos farmacocinéticos poblacionales. La rufinamida parece no tener ningún efecto clínicamente relevante sobre las concentraciones en estado estacionario de carbamazepina, lamotrigina, fenobarbital, topiramato, fenitoína o valproato.
Anticonceptivos orales
La coadministración de rufinamida 800 mg dos veces al día junto con un anticonceptivo oral combinado (etinilestradiol 35 microgramos y noretisterona 1 mg) durante 14 días dio lugar a una reducción media del AUC0-24 del etinilestradiol del 22 % y del AUC0-24 de la noretisterona del 14 %. No se han realizado estudios con otros anticonceptivos orales o implantables. A las mujeres en edad fértil que utilicen anticonceptivos hormonales, se les aconseja el uso de un método anticonceptivo seguro y eficaz adicional (ver secciones 4.4 y 4.6).
Enzimas del citocromo P450
La rufinamida se metaboliza mediante hidrólisis y no se metaboliza de forma notable por las enzimas del citocromo P450. Además, la rufinamida no inhibe la actividad de las enzimas del citocromo P450
16
(ver sección 5.2). Por lo tanto, es improbable que la rufinamida produzca interacciones clínicamente significativas por la inhibición del sistema del citocromo P450 Se ha demostrado que la rufinamida induce la enzima CYP3A4 del citocromo P450, y por lo tanto puede reducir las concentraciones plasmáticas de las sustancias metabolizadas por esta enzima. El efecto fue de pequeño a moderado. La actividad media de CYP3A4, evaluada como el aclaramiento de triazolam, aumentó en un 55 % tras 11 días de tratamiento con 400 mg de rufinamida dos veces al día. La exposición de triazolam se redujo en un 36 %. Dosis de rufinamida más altas pueden dar lugar a una inducción más pronunciada. No se puede descartar la posibilidad de que la rufinamida también pueda disminuir la exposición de sustancias metabolizadas por otras enzimas, o transportadas por proteínas transportadoras como la glucoproteína-P.
En los pacientes tratados con sustancias metabolizadas por el sistema enzimático CYP3A4, se recomienda una monitorización cuidadosa durante dos semanas al inicio del tratamiento con rufinamida o al finalizar el mismo, o después de cualquier cambio relevante en la dosis. Puede ser necesario considerar un ajuste de la dosis del medicamento administrado concomitantemente. Estas recomendaciones deben asimismo considerarse cuando se utilice la rufinamida concomitantemente con sustancias con un margen terapéutico estrecho como warfarina y digoxina.
Un estudio de interacción específico en sujetos sanos no reveló ninguna influencia de la rufinamida a una dosis de 400 mg dos veces al día sobre la farmacocinética de olanzapina, un sustrato de CYP1A2.
No hay datos disponibles sobre la interacción de la rufinamida con el alcohol.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Riesgo relacionado en general con la epilepsia y los antiepilépticos:
Se ha demostrado que la prevalencia de malformaciones en la descendencia de mujeres epilépticas, es dos o tres veces mayor que la tasa de aproximadamente el 3 % de población general. En la población tratada con politerapia, se ha observado un aumento de las malformaciones; sin embargo, no se ha elucidado hasta qué punto es responsabilidad del tratamiento y/o de la enfermedad.
Además, no debe interrumpirse una terapia antiepiléptica eficaz, ya que el agravamiento de la enfermedad va en detrimento tanto de la madre como del feto.
Riesgo relacionado a la rufinamida:
Los estudios en animales no han mostrado efectos teratogénicos aunque sí se observó fetotoxicidad en presencia de toxicidad materna (ver sección 5.3). Se desconoce el riesgo potencial en seres humanos.
No se dispone de datos clínicos sobre embarazos de riesgo para rufinamida.
Teniendo en cuenta estos datos, rufinamida no debe utilizarse durante el embarazo salvo que fuese claramente necesario ni en mujeres en edad fértil que no utilicen medidas anticonceptivas.
Las mujeres en edad fértil deben utilizar medidas anticonceptivas durante el tratamiento con rufinamida. Los médicos deben intentar asegurar que se utilicen anticonceptivos apropiados, y deberán seguir un criterio clínico a la hora de valorar si los anticonceptivos orales o las dosis de los componentes de los anticonceptivos orales son adecuados en función de la situación clínica de cada paciente (ver sección 4.5).
Si las mujeres que reciben tratamiento con rufinamida planean quedarse embarazadas, deberá sopesarse cuidadosamente la indicación de este producto. Durante el embarazo, no debe interrumpirse un tratamiento antiepiléptico eficaz con rufinamida, ya que el agravamiento de la enfermedad va en detrimento tanto de la madre como del feto.
Se desconoce si la rufinamida se excreta en la leche materna. Debido a los posibles efectos nocivos para los lactantes, debe evitarse la lactancia durante el tratamiento de la madre con rufinamida.
Fertilidad
No hay datos disponibles sobre los efectos en la fertilidad tras el tratamiento con rufinamida.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Inovelon puede producir mareos, somnolencia y visión borrosa. Dependiendo de la sensibilidad individual, la rufinamida puede tener una influencia de pequeña a importante en la capacidad para conducir y utilizar máquinas. Debe informarse a los pacientes que tengan cuidado en aquellas actividades que requieran mucha concentración, por ejemplo, conducir y utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
El programa de desarrollo clínico ha incluido a más de 1900 pacientes, con diferentes tipos de epilepsia, expuestos a rufinamida. Las reacciones adversas notificadas con mayor frecuencia en general fueron cefalea, mareos, fatiga y somnolencia. Las reacciones adversas más frecuentes y notificadas con una incidencia mayor que con el placebo en pacientes con síndrome de Lennox-Gastaut fueron somnolencia y vómitos. Las reacciones adversas generalmente presentaron una gravedad de leve a moderada. La tasa de interrupción del tratamiento en el síndrome de Lennox-Gastaut debido a las reacciones adversas fue del 8,2 % para los pacientes que recibían rufinamida y del 0 % para los pacientes que recibían el placebo. Las reacciones adversas más frecuentes que dieron lugar a la interrupción en el grupo tratado con rufinamida fueron erupción cutánea y vómitos.
Listado tabulado de reacciones adversas
Las reacciones adversas notificadas con una incidencia superior al placebo, durante los ensayos de doble ciego en el síndrome de Lennox-Gastaut o en la población global expuesta a rufinamida, se enumeran en la siguiente tabla siguiendo el sistema de clasificación por órganos MedDRA y en función de la frecuencia.
Las frecuencias se definen como: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000).
|
Clasificación de órganos del sistema |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
Infecciones e infestaciones |
Neumonía Gripe Nasofaringitis Infección de oído Sinusitis Rinitis | |||
|
Trastornos del sistema inmunológico |
Hipersensibilidad* | |||
|
Trastornos del metabolismo y de la nutrición |
Anorexia Trastorno del apetito Disminución del apetito | |||
|
Trastornos psiquiátricos |
Ansiedad Insomnio |
|
Clasificación de órganos del sistema |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
Trastornos del sistema nervioso |
Somnolencia* Cefalea Mareos* |
Estado epiléptico* Convulsión Coordinación anormal* | ||
|
Nistagmo Hiperactividad psicomotora Temblores | ||||
|
Trastornos oculares |
Diplopía Visión borrosa | |||
|
Trastornos del oído y del laberinto |
Vértigo | |||
|
Trastornos respiratorios, torácicos y mediastínicos |
Epistaxis | |||
|
Trastornos gastrointestinales |
Náuseas Vómitos |
Dolor abdomen superior Estreñimiento Dispepsia Diarrea | ||
|
Trastornos hepatobiliares |
Aumento de las enzimas hepáticas | |||
|
Trastornos de la piel y del tejido subcutáneo |
Erupción cutánea* Acné | |||
|
Trastornos musculoesquelético s y del tejido conjuntivo |
Dolor de espalda | |||
|
Trastornos del aparato reproductor y de la mama |
Oligomenorrea | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Fatiga |
Trastorno de la marcha* | ||
|
Exploraciones complementarias |
Disminución de peso | |||
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Traumatismo craneal Contusión |
*consultar sección 4.4
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación incluido en el Anexo V.
4.9 Sobredosis
Tras una sobredosis aguda, se puede vaciar el estómago mediante lavado gástrico o mediante la inducción de vómitos. No hay ningún antídoto específico para la rufinamida. El tratamiento debe ser de apoyo y puede incluir la hemodiálisis (ver sección 5.2).
La administración de dosis múltiples de 7200 mg/día no se asoció con signos o síntomas importantes.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antiepilépticos, derivados de carboxamida; código ATC: N03AF03. Mecanismo de acción
La rufinamida modula la actividad de los canales de sodio, prolongando el estado inactivo. La rufinamida es activa en diversos modelos animales de epilepsia.
Experiencia clínica
En un ensayo de doble ciego y controlado con placebo, se administró Inovelon (comprimidos de rufinamida) en dosis de hasta 45 mg/kg/día durante 84 días, a 139 pacientes con convulsiones no controladas adecuadamente asociadas con el síndrome de Lennox-Gastaut (incluyendo crisis de ausencia atípica y episodios de caída). Se incluyeron hombres y mujeres (de entre 4 y 30 años) que recibían tratamiento concomitantemente con 1 a 3 antiepilépticos a dosis fijas. Cada paciente tenía que haber tenido al menos 90 convulsiones en el mes anterior a la entrada en el ensayo. Se observó una mejoría significativa en las tres variables primarias principales: el cambio porcentual en la frecuencia total de crisis cada 28 días durante la fase de mantenimiento respecto a la frecuencia basal (-35,8 % con Inovelon frente al -1,6 % con placebo, p=0,0006), el número de convulsiones tónicas-atónicas (
42,9 % con Inovelon frente al 2,2 % con placebo, p = 0,0002), y la puntuación de la gravedad de las crisis a partir de la Evaluación Global realizada por los padres/representante legal al final de la fase de doble ciego (mejoría grande o muy grande en el 32,2 % con Inovelon frente al 14,5 % en el grupo de placebo, p=0,0041).
Los modelos farmacocinéticos/farmacodinámicos poblacionales demostraron que la reducción de las frecuencias de las convulsiones totales y las convulsiones tónicas-atónicas, la mejoría de la evaluación global de la gravedad de las convulsiones y el aumento de la probabilidad de reducción de la frecuencia de las convulsiones dependieron de las concentraciones de rufinamida.
5.2 Propiedades farmacocinéticas
Absorción
Los niveles plasmáticos máximos se alcanzan aproximadamente 6 horas después de la administración. La concentración máxima (Cmáx) y el AUC de plasma de rufinamida aumentan menos que proporcionalmente en relación con las dosis administradas a sujetos sanos tanto en ayunas como con alimentos y a pacientes, probablemente se deba a la absorción limitada de la dosis. Tras dosis únicas, la comida aumenta la biodisponibilidad (AUC) de la rufinamida en aproximadamente el 34 % y la concentración plasmática máxima en 56 %.
Inovelon suspensión oral e Inovelon comprimidos recubiertos con película han demostrado ser bioequivalentes.
Distribución
En los estudios in vitro, solo una pequeña fracción de rufinamida (34 %) se fijó a las proteínas séricas humanas de las que la albúmina supone aproximadamente el 80 % de esta fijación. Esto indica un riesgo mínimo de interacciones medicamentosas debidas al desplazamiento de los sitios de fijación durante la administración concomitante de otras sustancias. La rufinamida presentó una distribución uniforme entre los eritrocitos y el plasma.
Metabolismo
La rufinamida se elimina de forma casi exclusiva a través del metabolismo. La vía principal de metabolismo es la hidrólisis del grupo carboxilamida para formar el derivado ácido farmacológicamente inactivo, CGP 47292. El metabolismo mediado por el citocromo P450 es muy pequeño. No se puede excluir por completo la formación de pequeñas cantidades de conjugados de glutatión.
In vitro, la rufinamida ha demostrado tener una capacidad pequeña o no significativa para actuar como un inhibidor competitivo o basado en el mecanismo, de las siguientes enzimas P450 humanas: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 o CYP4A9/11-2.
Eliminación
La semivida de eliminación plasmática es aproximadamente de 6-10 horas en sujetos sanos y en pacientes con epilepsia. Cuando se administra dos veces al día a intervalos de 12 horas, la rufinamida se acumula en el grado previsto a partir de su semivida terminal, lo que indica que la farmacocinética de la rufinamida es independiente del tiempo (es decir, no hay ninguna autoinducción del metabolismo).
En un ensayo con marcadores radiactivos en tres voluntarios sanos, el compuesto original (rufinamida) fue el principal componente radiactivo en el plasma, representando aproximadamente el 80 % de la radiactividad total, y el metabolito CGP 47292 solo supuso aproximadamente el 15 %. La excreción renal fue la vía predominante de eliminación para el material relacionado con el principio activo, representando el 84,7 % de la dosis.
Linealidad/no linealidad:
La biodisponibilidad de la rufinamida es dosis-dependiente. Al aumentar la dosis, disminuye la biodisponibilidad.
Farmacocinética en grupos especiales de pacientes
Sexo
Se han utilizado modelos farmacocinéticos poblacionales para evaluar la influencia del sexo en la farmacocinética de la rufinamida. Estas evaluaciones indican que el sexo no afecta a la farmacocinética de la rufinamida en un grado clínicamente relevante.
Insuficiencia renal
La farmacocinética de una sola dosis de 400 mg de rufinamida no se vio alterada en sujetos con insuficiencia renal crónica y severa en comparación con voluntarios sanos. Sin embargo, los niveles plasmáticos disminuyeron en aproximadamente un 30 % al utilizar la hemodiálisis tras la administración de rufinamida, lo que sugiere que puede tratarse de un procedimiento útil en caso de sobredosis (ver secciones 4.2 y 4.9).
Insuficiencia hepática
No se han realizado ensayo en pacientes con insuficiencia hepática y, por tanto, Inovelon no debe administrarse a pacientes con insuficiencia hepática grave (ver sección 4.2).
21
Niños (2-12 años)
En general, los niños presentan un menor aclaramiento de rufinamida que los adultos, y esta diferencia está relacionada con el tamaño corporal. No se han realizado ensayo en neonatos o en lactantes ni en niños menores de 2 años.
Pacientes de edad avanzada
Un estudio farmacocinético en voluntarios sanos de edad avanzada no mostró ninguna diferencia significativa en los parámetros farmacocinéticos comparados con los de adultos más jóvenes.
5.3 Datos preclínicos sobre seguridad
Los estudios convencionales de farmacología de seguridad no muestran riesgos especiales con las dosis clínicamente relevantes.
La toxicidad observada en perros con niveles similares a la exposición humana obtenida con la dosis máxima recomendada, fue cambios hepáticos incluyendo trombos biliares, colestasis y aumento de las enzimas hepáticas, que se cree que están relacionados con un aumento de la secreción biliar en esta especie. No hubo evidencia de ningún riesgo asociado en los estudios de toxicidad a dosis repetidas en ratas y monos.
En los estudios de toxicidad reproductiva y toxicidad durante el desarrollo, se redujo la supervivencia y crecimiento fetales y hubo algunos casos de mortinatalidad secundaria a la toxicidad materna. Sin embargo, no se observaron efectos en la morfología y la función de la descendencia, incluyendo el aprendizaje o la memoria. La rufinamida no fue teratogénica en ratones, ratas o conejos.
La rufinamida no resultó genotóxica y no presenta potencial carcinogénico. Las reacciones adversas no observadas en los ensayos clínicos aunque sí vistas en animales con niveles de exposición similares a los clínicos y con posible relevancia para el uso humano fue mielofibrosis de la médula ósea en el estudio de carcinogenicidad con ratón. Los neoplasmas óseos benignos (osteomas) y la hiperostosis observados en ratones se consideraron el resultado de la activación de un virus específico a ratón por los iones de fluoruro liberados durante el metabolismo oxidativo de la rufinamida.
Con respecto al potencial inmunotóxico, en un estudio en perros de 13 semanas de duración se observaron timo pequeño e involución del timo con respuesta significativa a la dosis alta en los machos. En el estudio de 13 semanas, se notificaron con incidencia baja cambios en la medula ósea y linfoides en hembras a las que se administro la dosis alta. Solo en el estudio de carcinogenicidad en ratas, observaron disminución celular de la médula ósea y atrofia del timo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo:
Lactosa monohidrato Celulosa, microcristalina Almidón de maíz Croscarmelosa sódica Hipromelosa Estearato de magnesio Laurilsulfato de sodio Sílice coloidal, anhidra
Recubrimiento:
Hipromelosa Macrogol (8000)
Dióxido de titanio (E171)
Talco
Óxido férrico rojo (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30°C.
6.5 Naturaleza y contenido del envase
Blíster de aluminio/aluminio, envases de 10, 30, 50, 60 y 100 comprimidos recubiertos con película. Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
Ninguna especial.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Eisai Limited, European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/378/006-010
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16/enero/2007 Fecha de la última renovación: 16/enero/2012
10. FECHA DE LA REVISIÓN DEL TEXTO
{MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
NOMBRE DEL MEDICAMENTO
1.
Inovelon 400 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 400 mg de rufinamida.
Excipiente con efecto conocido: cada comprimido recubierto con película contiene 80 mg de lactosa monohidrato.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película.
Rosa, ‘ovalado’ ligeramente convexo, ranurado en ambos lados, con la inscripción ‘C263’ en un lado y nada en el otro.
El comprimido se puede dividir en dosis iguales.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Inovelon está indicado como terapia coadyuvante en el tratamiento de las crisis asociadas al síndrome de Lennox-Gastaut en pacientes de 4 años o mayores.
4.2 Posología y forma de administración
El tratamiento con rufinamida deberá iniciarlo un médico especializado en pediatría o neurología con experiencia en el tratamiento de la epilepsia.
Inovelon suspensión oral e Inovelon comprimidos recubiertos con película se pueden intercambiar en dosis iguales. Se debe vigilar a los pacientes durante el periodo de cambio.
Posología
Uso en niños de cuatro años o más y pesen menos de 30 kg Pacientes de <30 kg que no reciben valproato:
El tratamiento debe iniciarse con una dosis diaria de 200 mg. Según la respuesta clínica y la tolerabilidad, la dosis puede aumentarse a incrementos de 200 mg/día, cada dos días, hasta la dosis máxima recomendada de 1000 mg/día. Se han estudiado dosis de hasta 3600 mg/día en un número limitado de pacientes.
Pacientes de <30 kg que también reciban valproato:
Como el valproato disminuye significativamente el aclaramiento de la rufinamida, en los pacientes de <30 kg a los que se coadministre valproato se recomienda una dosis máxima más baja de Inovelon. El tratamiento debe iniciarse con una dosis diaria de 200 mg. Según la respuesta clínica y la tolerabilidad, después de al menos 2 días, la dosis podrá aumentarse en incrementos de 200 mg/día, hasta la dosis máxima recomendada de 600 mg/día.
Uso en adultos, adolescentes y niños de 4 años o mayores que pesen 30 kg o más El tratamiento debe iniciarse con una dosis diaria de 400 mg. Según la respuesta clínica y a la tolerabilidad, podrá aumentarse la dosis en incrementos de 400 mg/día, después de al menos 2 días, hasta la dosis máxima recomendada de la forma indicada en la siguiente tabla.
|
Rango de peso |
30,0-50,0 kg |
50,1-70,0 kg |
>70,1 kg |
|
Dosis máxima recomendada |
1800 mg/día |
2400 mg/día |
3200 mg/día |
Se han estudiado dosis de hasta 4000 mg/día (en el rango de 30-50 kg) o 4800 mg/día (en la categoría de más de 50 kg) en un número limitado de pacientes.
Interrupción del tratamiento
Cuando se vaya a interrumpir el tratamiento con rufinamida, debe hacerse gradualmente. En los ensayos clínicos, la interrupción del tratamiento con rufinamida se llevó a cabo reduciendo aproximadamente un 25 % de la dosis cada dos días.
En caso de olvidarse una o más dosis, será necesario aplicar un criterio clínico individualizado.
Los ensayos abiertos no controlados indican una eficacia a largo plazo sostenida, aunque no se ha realizado ningún ensayo controlado durante más de 3 meses.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de la rufinamida en niños de 4 años o menores.
No se dispone de datos.
Pacientes de edad avanzada
La información disponible sobre el uso de la rufinamida en pacientes de edad avanzada es limitada. Ya que la farmacocinética de la rufinamida no se altera en los pacientes de edad avanzada (ver sección 5.2), no se requieren ajustes de dosis en pacientes mayores de 65 años.
Insuficiencia renal
Un estudio realizado en pacientes con insuficiencia renal grave indicó que no se requieren ajustes de dosis en estos pacientes (ver sección 5.2).
Insuficiencia hepática
No se ha estudiado el uso en pacientes con insuficiencia hepática. Se recomienda precaución y un ajuste cuidadoso de la dosis en el tratamiento de pacientes con insuficiencia hepática de leve a moderada. No se recomienda el uso en pacientes con insuficiencia hepática severa.
Forma de administración
La rufinamida se administra por vía oral. Se debe tomar con agua dos veces al día, una por la mañana y otra por la noche, en dos dosis iguales. Como se ha observado un efecto con alimentos, Inovelon debe administrarse con alimentos (ver sección 5.2). Si el paciente tiene dificultad para tragar los comprimidos, los puede triturar y tomarlos disueltos en medio vaso de agua.
4.3 Contraindicaciones
Hipersensibilidad al principio activo, a los derivados triazólicos o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Estado epiléptico
Se han observado casos de estado epiléptico con rufinamida en los estudios de desarrollo clínico mientras que no se ha observado ningún caso con placebo. Estos efectos adversos ocasionaron la interrupción del tratamiento con rufinamida en el 20 % de los casos. Si los pacientes desarrollan nuevos tipos de convulsiones y/o experimentan un aumento de la frecuencia de estado epiléptico que sea diferente de la situación basal del paciente, debe reevaluarse el balance beneficio-riesgo de la terapia.
Retirada de la rufinamida
La rufinamida debe interrumpirse gradualmente para reducir la posibilidad de convulsiones durante la retirada. En los ensayos clínicos, la interrupción se llevó a cabo reduciendo aproximadamente un 25 % de la dosis cada dos días. No hay datos suficientes sobre la interrupción de tratamientos antiepilépticos concomitantes, una vez alcanzado el control de las convulsiones con la adición de rufinamida.
Reacciones en el sistema nervioso central
El tratamiento con rufinamida se ha asociado con mareos, somnolencia, ataxia y trastornos de la marcha, lo que puede incrementar la aparición de caídas accidentales en esta población (ver sección 4.8). Los pacientes y cuidadores deben tener precaución hasta que estén familiarizados con los posibles efectos de este medicamento.
Reacciones de hipersensibilidad
Se han producido el síndrome de hipersensibilidad a antiepilépticos grave incluyendo DRESS (Reacción al Fármaco con Eosinofilia y Síntomas Sistémicos) y síndrome de Stevens-Johnson asociado con la terapia con rufinamida. Los signos y los síntomas de este trastorno fueron diversos; sin embargo, los pacientes normalmente, aunque no de forma exclusiva, presentaron fiebre y erupción cutánea asociadas con afectación de otros órganos del sistema. Otras manifestaciones asociadas incluyeron linfadenopatía, anomalías en las pruebas de la función hepática y hematuria. Al tratarse de un trastorno que varía en su expresión pueden producirse otros signos y síntomas en los sistemas y órganos no citados aquí. Este síndrome de hipersensibilidad a antiepilépticos se asoció temporalmente al comienzo de la terapia con rufinamida y en la población pediátrica. Si se sospecha esta reacción, debe interrumpirse la administración de rufinamida y comenzar un tratamiento alternativo. Todos los pacientes que desarrollen erupción cutánea mientras tomen rufinamida deben monitorizarse cuidadosamente.
Acortamiento del intervalo QT
En un estudio minucioso del efecto sobre el intervalo QT, la rufinamida produjo un acortamiento del intervalo QTc proporcional a la concentración. Aunque se desconozcan el mecanismo subyacente y la relevencia para la seguridad de este hallazgo, los médicos deben seguir un criterio clínico cuando valoren la posible prescripción de rufinamida a pacientes que presenten un riesgo adicional de acortamiento del QTc (por ej.; síndrome de QT corto congénito o pacientes con una historia familiar de este tipo de síndrome).
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar anticonceptivos eficaces durante el tratamiento con Inovelon. Los médicos deben intentar asegurar que se utilizan métodos anticonceptivos apropiados, y deben seguir un criterio clínico a la hora de valorar si los anticonceptivos orales o las dosis de los componentes de los anticonceptivos orales son adecuados en función de la situación clínica de cada paciente (ver sección 4.5).
Lactosa
Inovelon contiene lactosa, por lo tanto los pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia de lactasa de Lapp o mala absorción de glucosa-galactosa no deben tomar este medicamento.
Pensamientos suicidas
Se han notificado pensamientos y conductas suicidas en pacientes tratados con antiepilépticos en varias indicaciones. Asimismo, un metanálisis de ensayos aleatorizados y controlados con placebo de antiepilépticos ha demostrado un pequeño aumento en el riesgo de pensamientos y conductas suicidas. Se desconoce el mecanismo de este riesgo y los datos disponibles no descartan la posibilidad de un aumento del riesgo con Inovelon.
Por lo tanto, se debe vigilar a los pacientes por si presentan signos de pensamientos y conductas suicidas y considerar el tratamiento adecuado. Se debe informar a los pacientes (y cuidadores de los pacientes) que acudan al médico si aparecen signos de pensamientos o conductas suicidas.
4.5 Interacción con otros medicamentos y otras formas de interacción
Posibilidad de que otros medicamentos afecten a la rufinamida
Otros antiepilépticos
Las concentraciones de rufinamida no están sujetas a cambios clínicamente relevantes al coadministrarse con antiepilépticos que se sabe que inducen enzimas.
En pacientes que estén en tratamiento con Inovelon y en los que se inicie la terapia con valproato, pueden producirse incrementos significativos en las concentraciones plasmáticas de rufinamida. Los incrementos más pronunciados se observaron en pacientes con bajo peso corporal (<30 kg). Por lo tanto, debe considerarse una reducción de la dosis de Inovelon en pacientes de <30 kg que inicien la terapia con valproato (ver sección 4.2).
La adición o interrupción de estos medicamentos o el ajuste de la dosis de estos medicamentos durante la terapia con rufinamida pueden requerir un ajuste de la dosis de rufinamida.
No se observan cambios significativos en la concentración de rufinamida tras la coadministración de lamotrigina, topiramato o benzodiazepinas.
Posibilidad de que la rufinamida afecte a otros medicamentos
Otros antiepilépticos
Las interacciones farmacocinéticas entre la rufinamida y otros antiepilépticos se han evaluado en pacientes epilépticos utilizando modelos farmacocinéticos poblacionales. La rufinamida parece no tener ningún efecto clínicamente relevante sobre las concentraciones en estado estacionario de carbamazepina, lamotrigina, fenobarbital, topiramato, fenitoína o valproato.
Anticonceptivos orales
La coadministración de rufinamida 800 mg dos veces al día junto con un anticonceptivo oral combinado (etinilestradiol 35 microgramos y noretisterona 1 mg) durante 14 días dio lugar a una reducción media del AUC0-24 del etinilestradiol del 22 % y del AUC0-24 de la noretisterona del 14 %. No se han realizado estudios con otros anticonceptivos orales o implantables. A las mujeres en edad fértil que utilicen anticonceptivos hormonales, se les aconseja el uso de un método anticonceptivo seguro y eficaz adicional (ver secciones 4.4 y 4.6).
Enzimas del citocromo P450
La rufinamida se metaboliza mediante hidrólisis y no se metaboliza de forma notable por las enzimas del citocromo P450. Además, la rufinamida no inhibe la actividad de las enzimas del citocromo P450 (ver sección 5.2). Por lo tanto, es improbable que la rufinamida produzca interacciones clínicamente significativas por la inhibición del sistema del citocromo P450 Se ha demostrado que la rufinamida induce la enzima CYP3A4 del citocromo P450, y por lo tanto puede reducir las concentraciones plasmáticas de las sustancias metabolizadas por esta enzima. El efecto fue de pequeño a moderado. La actividad media de CYP3A4, evaluada como el aclaramiento de triazolam, aumentó en un 55 % tras 11 días de tratamiento con 400 mg de rufinamida dos veces al día. La exposición de triazolam se redujo en un 36 %. Dosis de rufinamida más altas pueden dar lugar a una inducción más pronunciada. No se puede descartar la posibilidad de que la rufinamida también pueda disminuir la exposición de sustancias metabolizadas por otras enzimas, o transportadas por proteínas transportadoras como la glucoproteína-P.
En los pacientes tratados con sustancias metabolizadas por el sistema enzimático CYP3A4, se recomienda una monitorización cuidadosa durante dos semanas al inicio del tratamiento con rufinamida o al finalizar el mismo, o después de cualquier cambio relevante en la dosis. Puede ser necesario considerar un ajuste de la dosis del medicamento administrado concomitantemente. Estas recomendaciones deben asimismo considerarse cuando se utilice la rufinamida concomitantemente con sustancias con un margen terapéutico estrecho como warfarina y digoxina.
Un estudio de interacción específico en sujetos sanos no reveló ninguna influencia de la rufinamida a una dosis de 400 mg dos veces al día sobre la farmacocinética de olanzapina, un sustrato de CYP1A2.
No hay datos disponibles sobre la interacción de la rufinamida con el alcohol.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Riesgo relacionado en general con la epilepsia y los antiepilépticos:
Se ha demostrado que la prevalencia de malformaciones en la descendencia de mujeres epilépticas, es dos o tres veces mayor que la tasa de aproximadamente el 3 % de población general. En la población tratada con politerapia, se ha observado un aumento de las malformaciones; sin embargo, no se ha elucidado hasta qué punto es responsabilidad del tratamiento y/o de la enfermedad.
Además, no debe interrumpirse una terapia antiepiléptica eficaz, ya que el agravamiento de la enfermedad va en detrimento tanto de la madre como del feto.
Riesgo relacionado a la rufinamida:
Los estudios en animales no han mostrado efectos teratogénicos aunque sí se observó fetotoxicidad en presencia de toxicidad materna (ver sección 5.3). Se desconoce el riesgo potencial en seres humanos.
No se dispone de datos clínicos sobre embarazos de riesgo para rufinamida.
Teniendo en cuenta estos datos, rufinamida no debe utilizarse durante el embarazo salvo que fuese claramente necesario ni en mujeres en edad fértil que no utilicen medidas anticonceptivas.
Las mujeres en edad fértil deben utilizar medidas anticonceptivas durante el tratamiento con rufinamida. Los médicos deben intentar asegurar que se utilicen anticonceptivos apropiados, y deberán seguir un criterio clínico a la hora de valorar si los anticonceptivos orales o las dosis de los componentes de los anticonceptivos orales son adecuados en función de la situación clínica de cada paciente (ver sección 4.5).
Si las mujeres que reciben tratamiento con rufinamida planean quedarse embarazadas, deberá sopesarse cuidadosamente la indicación de este producto. Durante el embarazo, no debe interrumpirse
28
un tratamiento antiepiléptico eficaz con rufinamida, ya que el agravamiento de la enfermedad va en detrimento tanto de la madre como del feto.
Lactancia
Se desconoce si la rufinamida se excreta en la leche materna. Debido a los posibles efectos nocivos para los lactantes, debe evitarse la lactancia durante el tratamiento de la madre con rufinamida.
Fertilidad
No hay datos disponibles sobre los efectos en la fertilidad tras el tratamiento con rufinamida.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Inovelon puede producir mareos, somnolencia y visión borrosa. Dependiendo de la sensibilidad individual, la rufinamida puede tener una influencia de pequeña a importante en la capacidad para conducir y utilizar máquinas. Debe informarse a los pacientes que tengan cuidado en aquellas actividades que requieran mucha concentración, por ejemplo, conducir y utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
El programa de desarrollo clínico ha incluido a más de 1900 pacientes, con diferentes tipos de epilepsia, expuestos a rufinamida. Las reacciones adversas notificadas con mayor frecuencia en general fueron cefalea, mareos, fatiga y somnolencia. Las reacciones adversas más frecuentes y notificadas con una incidencia mayor que con el placebo en pacientes con síndrome de Lennox-Gastaut fueron somnolencia y vómitos. Las reacciones adversas generalmente presentaron una gravedad de leve a moderada. La tasa de interrupción del tratamiento en el síndrome de Lennox-Gastaut debido a las reacciones adversas fue del 8,2 % para los pacientes que recibían rufinamida y del 0 % para los pacientes que recibían el placebo. Las reacciones adversas más frecuentes que dieron lugar a la interrupción en el grupo tratado con rufinamida fueron erupción cutánea y vómitos.
Listado tabulado de reacciones adversas
Las reacciones adversas notificadas con una incidencia superior al placebo, durante los ensayo de doble ciego en el síndrome de Lennox-Gastaut o en la población global expuesta a rufinamida, se enumeran en la siguiente tabla siguiendo el sistema de clasificación por órganos MedDRA y en función de la frecuencia.
Las frecuencias se definen como: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000).
|
Clasificación de órganos del sistema |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
Infecciones e infestaciones |
Neumonía Gripe Nasofaringitis Infección de oído Sinusitis Rinitis | |||
|
Trastornos del sistema inmunológico |
Hipersensibilidad* | |||
|
Trastornos del |
Anorexia |
|
Clasificación de órganos del sistema |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
metabolismo y de la nutrición |
Trastorno del apetito Disminución del apetito | |||
|
Trastornos psiquiátricos |
Ansiedad Insomnio | |||
|
Trastornos del sistema nervioso |
Somnolencia* Cefalea Mareos* |
Estado epiléptico* Convulsión Coordinación anormal* | ||
|
Nistagmo Hiperactividad psicomotora Temblores | ||||
|
Trastornos oculares |
Diplopía Visión borrosa | |||
|
Trastornos del oído y del laberinto |
Vértigo | |||
|
Trastornos respiratorios, torácicos y mediastínicos |
Epistaxis | |||
|
Trastornos gastrointestinales |
Náuseas Vómitos |
Dolor abdomen superior Estreñimiento Dispepsia Diarrea | ||
|
Trastornos hepatobiliares |
Aumento de las enzimas hepáticas | |||
|
Trastornos de la piel y del tejido subcutáneo |
Erupción cutánea* Acné | |||
|
Trastornos musculoesquelético s y del tejido conjuntivo |
Dolor de espalda | |||
|
Trastornos del aparato reproductor y de la mama |
Oligomenorrea | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Fatiga |
Trastorno de la marcha* | ||
|
Exploraciones complementarias |
Disminución de peso | |||
|
Lesiones |
Traumatismo craneal |
|
Clasificación de órganos del sistema |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Contusión |
*consultar sección 4.4
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación incluido en el Anexo V.
4.9 Sobredosis
Tras una sobredosis aguda, se puede vaciar el estómago mediante lavado gástrico o mediante la inducción de vómitos. No hay ningún antídoto específico para la rufinamida. El tratamiento debe ser de apoyo y puede incluir la hemodiálisis (ver sección 5.2).
La administración de dosis múltiples de 7200 mg/día no se asoció con signos o síntomas importantes.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antiepilépticos, derivados de carboxamida; código ATC: N03AF03. Mecanismo de acción
La rufinamida modula la actividad de los canales de sodio, prolongando el estado inactivo. La rufinamida es activa en diversos modelos animales de epilepsia.
Experiencia clínica
En un ensayo de doble ciego y controlado con placebo, se administró Inovelon (comprimidos de rufinamida) en dosis de hasta 45 mg/kg/día durante 84 días, a 139 pacientes con convulsiones no controladas adecuadamente asociadas con el síndrome de Lennox-Gastaut (incluyendo crisis de ausencia atípica y episodios de caída). Se incluyeron hombres y mujeres (de entre 4 y 30 años) que recibían tratamiento concomitantemente con 1 a 3 antiepilépticos a dosis fijas. Cada paciente tenía que haber tenido al menos 90 convulsiones en el mes anterior a la entrada en el ensayo. Se observó una mejoría significativa en las tres variables primarias principales: el cambio porcentual en la frecuencia total de crisis cada 28 días durante la fase de mantenimiento respecto a la frecuencia basal (-35,8 % con Inovelon frente al -1,6 % con placebo, p=0,0006), el número de convulsiones tónicas-atónicas (-42,9 % con Inovelon frente al 2,2 % con placebo, p = 0,0002), y la puntuación de la gravedad de las crisis a partir de la Evaluación Global realizada por los padres/representante legal al final de la fase de doble ciego (mejoría grande o muy grande en el 32,2 % con Inovelon frente al 14,5 % en el grupo de placebo, p=0,0041).
Los modelos farmacocinéticos/farmacodinámicos poblacionales demostraron que la reducción de las frecuencias de las convulsiones totales y las convulsiones tónicas-atónicas, la mejoría de la evaluación global de la gravedad de las convulsiones y el aumento de la probabilidad de reducción de la frecuencia de las convulsiones dependieron de las concentraciones de rufinamida.
31
5.2 Propiedades farmacocinéticas
Absorción
Los niveles plasmáticos máximos se alcanzan aproximadamente 6 horas después de la administración. La concentración máxima (Cmáx) y el AUC de plasma de rufinamida aumentan menos que proporcionalmente en relación con las dosis administradas a sujetos sanos tanto en ayunas como con alimentos y a pacientes, probablemente se deba a la absorción limitada de la dosis. Tras dosis únicas, la comida aumenta la biodisponibilidad (AUC) de la rufinamida en aproximadamente el 34 % y la concentración plasmática máxima en 56 %.
Inovelon suspensión oral e Inovelon comprimidos recubiertos con película han demostrado ser bioequivalentes.
Distribución
En los estudios in vitro, solo una pequeña fracción de rufinamida (34 %) se fijó a las proteínas séricas humanas de las que la albúmina supone aproximadamente el 80 % de esta fijación. Esto indica un riesgo mínimo de interacciones medicamentosas debidas al desplazamiento de los sitios de fijación durante la administración concomitante de otras sustancias. La rufinamida presentó una distribución uniforme entre los eritrocitos y el plasma.
Metabolismo
La rufinamida se elimina de forma casi exclusiva a través del metabolismo. La vía principal de metabolismo es la hidrólisis del grupo carboxilamida para formar el derivado ácido farmacológicamente inactivo, CGP 47292. El metabolismo mediado por el citocromo P450 es muy pequeño. No se puede excluir por completo la formación de pequeñas cantidades de conjugados de glutatión.
In vitro, la rufinamida ha demostrado tener una capacidad pequeña o no significativa para actuar como un inhibidor competitivo o basado en el mecanismo, de las siguientes enzimas P450 humanas: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 o CYP4A9/11-2.
Eliminación
La semivida de eliminación plasmática es aproximadamente de 6-10 horas en sujetos sanos y en pacientes con epilepsia. Cuando se administra dos veces al día a intervalos de 12 horas, la rufinamida se acumula en el grado previsto a partir de su semivida terminal, lo que indica que la farmacocinética de la rufinamida es independiente del tiempo (es decir, no hay ninguna autoinducción del metabolismo).
En un ensayo con marcadores radiactivos en tres voluntarios sanos, el compuesto original (rufinamida) fue el principal componente radiactivo en el plasma, representando aproximadamente el 80 % de la radiactividad total, y el metabolito CGP 47292 solo supuso aproximadamente el 15 %. La excreción renal fue la vía predominante de eliminación para el material relacionado con el principio activo representando el 84,7 % de la dosis.
Linealidad/no linealidad:
La biodisponibilidad de la rufinamida es dosis-dependiente. Al aumentar la dosis, disminuye la biodisponibilidad.
Farmacocinética en grupos especiales de pacientes
Sexo
Se han utilizado modelos farmacocinéticos poblacionales para evaluar la influencia del sexo en la farmacocinética de la rufinamida. Estas evaluaciones indican que el sexo no afecta a la farmacocinética de la rufinamida en un grado clínicamente relevante.
Insuficiencia renal
La farmacocinética de una sola dosis de 400 mg de rufinamida no se vio alterada en sujetos con insuficiencia renal crónica y severa en comparación con voluntarios sanos. Sin embargo, los niveles plasmáticos disminuyeron en aproximadamente un 30 % al utilizar la hemodiálisis tras la administración de rufinamida, lo que sugiere que puede tratarse de un procedimiento útil en caso de sobredosis (ver secciones 4.2 y 4.9).
Insuficiencia hepática
No se han realizado ensayo en pacientes con insuficiencia hepática y, por tanto, Inovelon no debe administrarse a pacientes con insuficiencia hepática grave (ver sección 4.2).
Niños (2-12 años)
En general, los niños presentan un menor aclaramiento de rufinamida que los adultos, y esta diferencia está relacionada con el tamaño corporal. No se han realizado ensayo en neonatos o en lactantes ni en niños menores de 2 años.
Pacientes de edad avanzada
Un estudio farmacocinético en voluntarios sanos de edad avanzada no mostró ninguna diferencia significativa en los parámetros farmacocinéticos comparados con los de adultos más jóvenes.
5.3 Datos preclínicos sobre seguridad
Los estudios convencionales de farmacología de seguridad no muestran riesgos especiales con las dosis clínicamente relevantes.
La toxicidad observada en perros con niveles similares a la exposición humana obtenida con la dosis máxima recomendada, fue cambios hepáticos incluyendo trombos biliares, colestasis y aumento de las enzimas hepáticas, que se cree que están relacionados con un aumento de la secreción biliar en esta especie. No hubo evidencia de ningún riesgo asociado en los estudios de toxicidad a dosis repetidas en ratas y monos.
En los estudios de toxicidad reproductiva y toxicidad durante el desarrollo, se redujo la supervivencia y crecimiento fetales y hubo algunos casos de mortinatalidad secundaria a la toxicidad materna. Sin embargo, no se observaron efectos en la morfología y la función de la descendencia, incluyendo el aprendizaje o la memoria. La rufinamida no fue teratogénica en ratones, ratas o conejos.
La rufinamida no resultó genotóxica y no presenta potencial carcinogénico. Las reacciones adversas no observadas en los ensayos clínicos aunque sí vistas en animales con niveles de exposición similares a los clínicos y con posible relevancia para el uso humano fue mielofibrosis de la médula ósea en el estudio de carcinogenicidad con ratón. Los neoplasmas óseos benignos (osteomas) y la hiperostosis observados en ratones se consideraron el resultado de la activación de un virus específico a ratón por los iones de fluoruro liberados durante el metabolismo oxidativo de la rufinamida.
Con respecto al potencial inmunotóxico, en un estudio en perros de 13 semanas de duración se observaron timo pequeño e involución del timo con respuesta significativa a la dosis alta en los machos. En el estudio de 13 semanas, se notificaron con incidencia baja cambios en la medula ósea y linfoides en hembras a las que se administro la dosis alta. Solo en el estudio de carcinogenicidad en ratas, observaron disminución celular de la médula ósea y atrofia del timo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo:
Lactosa monohidrato Celulosa, microcristalina Almidón de maíz Croscarmelosa sódica Hipromelosa Estearato de magnesio Laurilsulfato de sodio Sílice coloidal, anhidra
Recubrimiento:
Hipromelosa Macrogol (8000)
Dióxido de titanio (E171)
Talco
Óxido férrico rojo (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30°C.
6.5 Naturaleza y contenido del envase
Blíster de aluminio/aluminio, envases de 10, 30, 50, 60, 100 y 200 comprimidos recubiertos con película.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
Ninguna especial.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Eisai Limited, European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/378/011-016
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16/enero/2007 Fecha de la última renovación: 16/enero/2012
10. FECHA DE LA REVISIÓN DEL TEXTO
{MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
1. NOMBRE DEL MEDICAMENTO
Inovelon 40 mg/ml suspensión oral
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada ml de suspensión oral contiene 40 mg de rufinamida.
1 frasco de 460 ml contiene 18 400 mg de rufinamida.
Excipientes con efecto conocido:
Cada ml de suspensión oral contiene 1,2 mg de parahidroxibenzoato de metilo (E218), 0,3 mg de parahidroxibenzoato de propilo (E216) y 250 mg de sorbitol (E420).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Suspensión oral.
Suspensión ligeramente viscosa de color blanco.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Inovelon está indicado como terapia adyuvante en el tratamiento de las crisis convulsivas asociadas al síndrome de Lennox-Gastaut en pacientes de 4 años o mayores.
4.2 Posología y forma de administración
El tratamiento con rufinamida deberá iniciarlo un médico especializado en pediatría o neurología con experiencia en el tratamiento de la epilepsia.
Inovelon suspensión oral e Inovelon comprimidos recubiertos con película se pueden intercambiar en dosis iguales. Se debe vigilar a los pacientes durante el periodo de cambio.
Posología
Uso en niños de cuatro años o mayores que pesan menos de 30 kg Pacientes de <30 kg que no reciben valproato:
El tratamiento se debe iniciar con una dosis diaria de 200 mg (5 ml de suspensión administrada en dos dosis de 2,5 ml, una por la mañana y una por la noche). Según la respuesta clínica y la tolerabilidad, se puede aumentar la dosis a incrementos de 200 mg/día, a intervalos de al menos dos días, hasta la dosis máxima recomendada de 1000 mg/día (25 ml/día). Se han estudiado dosis de hasta 3600 mg/día (90 ml/día) en un número limitado de pacientes.
Pacientes de <30 kg que también reciben valproato:
Como el valproato disminuye significativamente el aclaramiento de la rufinamida, en los pacientes de <30 kg a los que se coadministre valproato se recomienda una dosis máxima más baja de Inovelon. El tratamiento se debe iniciar con una dosis diaria de 200 mg. Según la respuesta clínica y la
tolerabilidad, después de al menos 2 días, la dosis se podrá aumentar en incrementos de 200 mg/día, hasta la dosis máxima recomendada de 600 mg/día (15 ml/día).
Uso en adultos, adolescentes y niños de 4 años o mayores que pesan 30 kg o más El tratamiento se debe iniciar con una dosis diaria de 400 mg (10 ml de suspensión administrada en dos dosis de 5 ml). Según la respuesta clínica y la tolerabilidad, se podrá aumentar la dosis en incrementos de 400 mg/día, a intervalos de al menos dos días, hasta la dosis máxima recomendada de la forma indicada en la siguiente tabla.
|
Rango de peso |
30,0-50,0 kg |
50,1-70,0 kg |
>70,1 kg |
|
Dosis máxima recomendada |
1800 mg/día o 45 ml/día |
2400 mg/día o 60 ml/día |
3200 mg/día o 80 ml/día |
Se han estudiado dosis de hasta 4000 mg/día (en el rango de 30-50 kg) o 4800 mg/día (120 ml/día) en la categoría de más de 50 kg en un número limitado de pacientes.
Interrupción del tratamiento
Cuando se vaya a interrumpir el tratamiento con rufinamida, debe hacerse gradualmente. En los ensayos clínicos, la interrupción del tratamiento con rufinamida se llevó a cabo reduciendo aproximadamente un 25 % de la dosis cada dos días.
En caso de olvidarse una o más dosis, será necesario aplicar un criterio clínico individualizado.
Los ensayos abiertos no controlados indican una eficacia a largo plazo sostenida, aunque no se ha realizado ningún ensayo controlado durante más de 3 meses.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de la rufinamida en niños de 4 años o menores.
No se dispone de datos.
Pacientes de edad avanzada
Hay información limitada sobre el uso de la rufinamida en pacientes de edad avanzada. No se requieren ajustes de la dosis en pacientes mayores de 65 años, ya que la farmacocinética de la rufinamida no se ve alterada en los pacientes de edad avanzada (ver sección 5.2).
Insuficiencia renal
Un ensayo realizado en pacientes con insuficiencia renal severa indicó que no se requieren ajustes de la dosis en estos pacientes (ver sección 5.2).
Insuficiencia hepática
No se ha estudiado el uso en pacientes con insuficiencia hepática. Se recomienda precaución y un ajuste cuidadoso de la dosis en el tratamiento de pacientes con insuficiencia hepática de leve a moderada. No se recomienda el uso en pacientes con insuficiencia hepática severa.
Forma de administración
La rufinamida se administra por vía oral. Se debe tomar dos veces al día, una por la mañana y otra por la noche, en dos dosis iguales. Como se ha observado un efecto con alimentos, se debe administrar con alimentos (ver sección 5.2).
Se debe agitar enérgicamente la suspensión oral antes de cada administración. Para más información, consultar la sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo, a los derivados triazólicos o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Estado epiléptico
Se han observado casos de estado epiléptico con rufinamida en los ensayos de desarrollo clínico mientras que no se ha observado ningún caso con placebo. Estos efectos adversos llevaron a la suspensión del tratamiento con rufinamida en el 20 % de los casos. Si los pacientes desarrollan nuevos tipos de crisis convulsivas y/o presentan un aumento de la frecuencia de estado epiléptico que sea diferente de la situación basal del paciente, se debe reevaluar la relación beneficio-riesgo de la terapia.
Retirada de la rufinamida
La rufinamida se debe interrumpir gradualmente para reducir la posibilidad de crisis convulsivas durante la retirada. En los ensayos clínicos, la suspensión se llevó a cabo reduciendo aproximadamente un 25 % de la dosis cada dos días. No hay datos suficientes sobre la interrupción de tratamientos antiepilépticos concomitantes una vez alcanzado el control de las crisis convulsivas con la adición de rufinamida.
Reacciones en el sistema nervioso central
El tratamiento con rufinamida se ha asociado con mareos, somnolencia, ataxia y trastornos de la marcha, lo que puede incrementar la aparición de caídas accidentales en esta población (ver sección 4.8). Los pacientes y cuidadores deben tener precaución hasta que estén familiarizados con los posibles efectos de este medicamento.
Reacciones de hipersensibilidad
Se han producido el síndrome de hipersensibilidad a antiepilépticos grave incluyendo DRESS (Reacción al Fármaco con Eosinofilia y Síntomas Sistémicos) y síndrome de Stevens-Johnson asociado con la terapia con rufinamida. Los signos y los síntomas de este trastorno fueron diversos; sin embargo, los pacientes normalmente, aunque no de forma exclusiva, presentaron fiebre y erupción cutánea asociadas con afectación de otros órganos del sistema. Otras manifestaciones asociadas incluyeron linfadenopatía, anomalías en las pruebas de la función hepática y hematuria. Ya que el trastorno se presenta de diversas formas, pueden producirse signos y síntomas en otros órganos del sistema no citados aquí. Este síndrome de hipersensibilidad a antiepilépticos se asoció temporalmente al comienzo de la terapia con rufinamida y se observó en la población pediátrica. Si se sospecha esta reacción, se debe suspender la administración de la rufinamida y comenzar un tratamiento alternativo. Se debe monitorizar de cerca a todos los pacientes que desarrollen erupción cutánea mientras toman rufinamida.
Acortamiento del intervalo QT
En un estudio minucioso del efecto sobre el intervalo QT, la rufinamida produjo un acortamiento del intervalo QTc proporcional a la concentración. Aunque se desconoce el mecanismo subyacente y la relevencia para la seguridad de este hallazgo, los médicos deben seguir un criterio clínico cuando valoren la posible prescripción de rufinamida a pacientes que presenten un riesgo de acortamiento adicional del intervalo QTc (por ej.: síndrome de QT corto congénito o pacientes con una historia familiar de este tipo de síndrome).
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar métodos anticonceptivos durante el tratamiento con Inovelon. Los médicos deben procurar que se utilicen métodos anticonceptivos apropiados, y deben seguir un criterio clínico a la hora de valorar si los anticonceptivos orales o las dosis de los componentes de los anticonceptivos orales son adecuados en función de la situación clínica de cada paciente (ver sección 4.5).
Parahidroxibenzoatos. sorbitol
Inovelon suspensión oral contiene parahidroxibenzoatos que pueden producir reacciones alérgicas (posiblemente tardías). Además contiene sorbitol y, por lo tanto, no debe administrarse a los pacientes con problemas hereditarios raros de intolerancia a la fructosa.
Pensamientos suicidas
Se han notificado pensamientos y conductas suicidas en pacientes tratados con antiepilépticos en varias indicaciones. Asimismo, un metanálisis de ensayos aleatorizados y controlados con placebo de antiepilépticos ha demostrado un pequeño aumento en el riesgo de pensamientos y conductas suicidas. Se desconoce el mecanismo de este riesgo y los datos disponibles no descartan la posibilidad de un aumento del riesgo con Inovelon.
Por lo tanto, se debe vigilar a los pacientes por si presentan signos de pensamientos y conductas suicidas y considerar el tratamiento adecuado. Se debe informar a los pacientes (y cuidadores de los pacientes) que acudan al médico si aparecen signos de pensamientos o conductas suicidas.
4.5 Interacción con otros medicamentos y otras formas de interacción
Posibilidad de que otros medicamentos afecten a la rufinamida
Otros antiepilépticos
Las concentraciones de rufinamida no están sujetas a cambios clínicamente relevantes al coadministrarse con antiepilépticos que se sabe que inducen enzimas.
En pacientes que estén en tratamiento con Inovelon y que empiecen la terapia con valproato, pueden producirse incrementos significativos en las concentraciones plasmáticas de la rufinamida. Los incrementos más pronunciados se observaron en pacientes con bajo peso corporal (<30 kg). Por lo tanto, se debe considerar reducir la dosis de Inovelon en pacientes de <30 kg que inicien la terapia con valproato (ver sección 4.2).
La adición o interrupción de estos medicamentos o el ajuste de la dosis de estos medicamentos durante la terapia con rufinamida pueden requerir un ajuste de la dosis de la rufinamida.
No se observan cambios significativos en la concentración de rufinamida tras la coadministración de lamotrigina, topiramato o benzodiazepinas.
Posibilidad de que rufinamida afecte a otros medicamentos
Otros antiepilépticos
Las interacciones farmacocinéticas entre la rufinamida y otros antiepilépticos se han evaluado en pacientes epilépticos utilizando modelos farmacocinéticos poblacionales. La rufinamida parece no tener ningún efecto clínicamente relevante en las concentraciones en estado estacionario de carbamazepina, lamotrigina, fenobarbital, topiramato, fenitoína o valproato.
Anticonceptivos orales
La coadministración de rufinamida 800 mg dos veces al día junto con un anticonceptivo oral combinado (etinilestradiol 35 microgramos y noretindrona 1 mg) durante 14 días dio lugar a una reducción media del 22 % en el AUC0-24 del etinilestradiol y del 14 % en el AUC0-24 de la noretindrona. No se han realizado estudios con otros anticonceptivos orales o implantables. A las mujeres en edad fértil que utilicen anticonceptivos hormonales se les aconseja el uso de un método anticonceptivo seguro y efectivo adicional (ver secciones 4.4 y 4.6).
Enzimas del citocromo P450
La rufinamida se metaboliza mediante hidrólisis y no se metaboliza de forma notable por las enzimas del citocromo P450. Además, la rufinamida no inhibe la actividad de las enzimas del citocromo P450
(ver sección 5.2). Por lo tanto, es improbable que la rufinamida produzca interacciones clínicamente significativas por la inhibición del sistema del citocromo P450. Se ha demostrado que la rufinamida induce la enzima CYP3A4 del citocromo P450, y por lo tanto puede reducir las concentraciones plasmáticas de los medicamentos metabolizados por esta enzima. El efecto fue de pequeño a moderado. La actividad media de CYP3A4, evaluada como el aclaramiento del triazolam, aumentó en un 55 % tras 11 días de tratamiento con 400 mg de rufinamida dos veces al día. La exposición al triazolam se redujo en un 36 %. Dosis de rufinamida más altas pueden dar lugar a una inducción más pronunciada. No se puede descartar la posibilidad de que la rufinamida también pueda disminuir la exposición de sustancias metabolizadas por otras enzimas, o transportadas por proteínas de transporte como la glucoproteína-P.
En los pacientes tratados con sustancias metabolizadas por el sistema enzimático CYP3A4, se recomienda una monitorización cuidadosa durante dos semanas al inicio del tratamiento con rufinamida o al finalizar el mismo, o después de cualquier cambio relevante en la dosis. Puede ser necesario considerar un ajuste de la dosis del medicamento administrado concomitantemente. Estas recomendaciones deben asimismo considerarse cuando se utilice la rufinamida concomitantemente con sustancias con un margen terapéutico estrecho como la warfarina y la digoxina.
Un estudio de interacciones específico en sujetos sanos no reveló ninguna influencia de la rufinamida a una dosis de 400 mg dos veces al día en la farmacocinética de la olanzapina, un sustrato de CYP1A2.
No hay datos disponibles sobre la interacción de la rufinamida con el alcohol.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Riesgo relacionado con la epilepsia y con los antiepilépticos en general:
Se ha demostrado que la prevalencia de malformaciones en los hijos de mujeres epilépticas es dos o tres veces mayor que la tasa de aproximadamente el 3 % en la población general. En la población tratada, se ha observado un aumento de las malformaciones con la politerapia; sin embargo, no se ha elucidado hasta qué punto se debe al tratamiento y/o a la enfermedad.
Además, no se debe interrumpir un tratamiento antiepiléptico eficaz, ya que el agravamiento de la enfermedad va en detrimento tanto de la madre como del feto.
Riesgo relacionado con la rufinamida:
Los estudios realizados en animales no han mostrado efectos teratogénicos aunque sí se observó fetotoxicidad en presencia de toxicidad materna (ver sección 5.3). Se desconoce el riesgo potencial en seres humanos.
No hay datos clínicos disponibles sobre embarazos de riesgo para rufinamida.
Teniendo en cuenta estos datos, no debe utilizarse la rufinamida durante el embarazo excepto si fuese claramente necesario, ni tampoco en mujeres en edad fértil que no utilicen métodos anticonceptivos.
Las mujeres en edad fértil deben utilizar métodos anticonceptivos durante el tratamiento con rufinamida. Los médicos deben procurar que se utilicen métodos anticonceptivos apropiados, y deben seguir un criterio clínico a la hora de valorar si los anticonceptivos orales o las dosis de los componentes de los anticonceptivos orales son adecuados en función de la situación clínica de cada paciente (ver sección 4.5).
Si las mujeres que reciben tratamiento con rufinamida tienen previsto quedarse embarazadas, deberá sopesarse cuidadosamente la indicación de este medicamento. Durante el embarazo, no se debe interrumpir un tratamiento antiepiléptico eficaz con rufinamida, ya que el agravamiento de la enfermedad va en detrimento tanto de la madre como del feto.
Lactancia
Se desconoce si la rufinamida se excreta en la leche materna. Debido a los posibles efectos nocivos para los lactantes, debe evitarse la lactancia durante el tratamiento de la madre con rufinamida.
Fertilidad
No hay datos disponibles sobre los efectos en la fertilidad tras el tratamiento con rufinamida.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La rufinamida puede producir mareos, somnolencia y visión borrosa. Dependiendo de la sensibilidad individual, la rufinamida puede tener una influencia de pequeña a importante en la capacidad para conducir y utilizar máquinas. Se debe informar a los pacientes que tengan cuidado en aquellas actividades que requieran mucha concentración, por ejemplo, conducir y utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
El programa de desarrollo clínico ha incluido a más de 1900 pacientes, con diferentes tipos de epilepsia, expuestos a rufinamida. Las reacciones adversas notificadas con mayor frecuencia en general fueron cefalea, mareos, fatiga y somnolencia. Las reacciones adversas más frecuentes y notificadas con una incidencia mayor que con el placebo en pacientes con síndrome de Lennox-Gastaut fueron somnolencia y vómitos. Las reacciones adversas generalmente presentaron una severidad de leve a moderada. La tasa de suspensión del tratamiento en el síndrome de Lennox-Gastaut debido a las reacciones adversas fue del 8,2 % en los pacientes que recibieron rufinamida y del 0 % en los pacientes que recibieron el placebo. Las reacciones adversas más frecuentes que dieron lugar a la suspensión en el grupo tratado con rufinamida fueron erupción cutánea y vómitos.
Listado tabulado de reacciones adversas
Las reacciones adversas notificadas con una incidencia superior al placebo, durante los ensayos de doble ciego en el síndrome de Lennox-Gastaut o en la población global expuesta a rufinamida, se enumeran en la siguiente tabla por término preferente, clasificación por órganos y sistemas y por frecuencia de MedDRA.
Las frecuencias se definen como: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco
frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000).
|
Clasificación de órganos del sistema |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
Infecciones e infestaciones |
Neumonía Gripe Nasofaringitis Infección de oído Sinusitis Rinitis | |||
|
Trastornos del sistema inmunológico |
Hipersensibilidad* | |||
|
Trastornos del metabolismo y de la nutrición |
Anorexia Trastorno alimenticio Disminución del apetito | |||
|
Trastornos psiquiátricos |
Ansiedad | |||
|
Insomnio | ||||
|
Trastornos del sistema nervioso |
Somnolencia* Cefalea Mareos* |
Estado epiléptico* Convulsión Coordinación anormal* Nistagmo Hiperactividad psicomotora Temblores | ||
|
Trastornos oculares |
Diplopía Visión borrosa | |||
|
Trastornos del oído y del laberinto |
Vértigo | |||
|
Trastornos respiratorios, torácicos y mediastínicos |
Epistaxis | |||
|
Trastornos gastrointestinales |
Náuseas Vómitos |
Dolor abdominal superior Estreñimiento Dispepsia Diarrea | ||
|
Trastornos hepatobiliares |
Aumento de las enzimas hepáticas | |||
|
Trastornos de la piel y del tejido subcutáneo |
Erupción cutánea* | |||
|
Acné | ||||
|
Trastornos musculoesquelé-ticos y del tejido |
Dolor de espalda |
|
Clasificación de órganos del sistema |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
conjuntivo | ||||
|
Trastornos del aparato reproductor y de la mama |
Oligomenorrea | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Fatiga |
Trastorno de la marcha* | ||
|
Exploraciones complementarias |
Disminución de peso | |||
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Traumatismo craneoencefálico | |||
|
Contusión |
*Referencia cruzada a sección 4.4.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Tras una sobredosis aguda, se puede vaciar el estómago mediante lavado gástrico o mediante la inducción de vómitos. No hay ningún antídoto específico para la rufinamida. El tratamiento debe ser de apoyo y puede incluir hemodiálisis (ver sección 5.2).
La administración de dosis múltiples de 7200 mg/día no se asoció a signos o síntomas importantes.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antiepilépticos, derivados de la carboxamida; código ATC: N03AF03. Mecanismo de acción
La rufinamida modula la actividad de los canales de sodio, prolongando el estado inactivo. La rufinamida es activa en diversos modelos animales de epilepsia.
Experiencia clínica
En un ensayo de doble ciego y controlado con placebo, se administró Inovelon (comprimidos de rufinamida) en dosis de hasta 45 mg/kg/día durante 84 días, a 139 pacientes con crisis convulsivas no controladas adecuadamente asociadas al síndrome de Lennox-Gastaut (incluidas crisis de ausencia atípica y episodios de caída). Se incluyó a hombres y mujeres (de entre 4 y 30 años) que recibieron tratamiento concomitantemente con 1 a 3 antiepilépticos a dosis fijas. Cada paciente tenía que haber tenido al menos 90 crisis convulsivas en el mes anterior a la entrada en el ensayo. Se observó una mejoría significativa en los tres criterios de valoración principales: el cambio porcentual en la frecuencia total de crisis convulsivas cada 28 días durante la fase de mantenimiento respecto a la frecuencia basal (-35,8 % con Inovelon frente al -1,6 % con placebo, p = 0,0006), el número de crisis convulsivas tónicas-atónicas (-42,9 % con Inovelon frente al 2,2 % con placebo, p = 0,0002), y la puntuación de la severidad de las crisis convulsivas a partir de la Evaluación Global realizada por los padres/representante legal al final de la fase de doble ciego (mejoría grande o muy grande en el 32,2 % con Inovelon frente al 14,5 % en el grupo de placebo, p = 0,0041).
Los modelos farmacocinéticos/farmacodinámicos poblacionales demostraron que la reducción de las frecuencias de las crisis convulsivas totales y las crisis convulsivas tónicas-atónicas, la mejoría de la evaluación global de la severidad de las crisis convulsivas y el aumento de la probabilidad de reducción de la frecuencia de las crisis convulsivas dependieron de las concentraciones de rufinamida.
5.2 Propiedades farmacocinéticas
Absorción
Los niveles plasmáticos máximos se alcanzan aproximadamente 6 horas después de la administración. La concentración máxima (Cmáx) y el AUC de la rufinamida en plasma aumentan menos que proporcionalmente con las dosis administradas a sujetos sanos tanto en ayunas como con alimentos y a pacientes, probablemente debido a la absorción limitada de la dosis. Tras dosis únicas, los alimentos aumentan la biodisponibilidad (AUC) de la rufinamida en aproximadamente el 34 % y la concentración plasmática máxima en el 56 %.
Inovelon suspensión oral e Inovelon comprimidos recubiertos con película han demostrado ser bioequivalentes.
Distribución
En los estudios in vitro, solo una pequeña fracción de rufinamida (34 %) se unió a las proteínas séricas humanas de las que la albúmina representa aproximadamente el 80 % de esta unión. Esto indica un riesgo mínimo de interacciones medicamentosas debidas al desplazamiento de los sitios de unión durante la administración concomitante de otras sustancias. La rufinamida presentó una distribución uniforme entre los eritrocitos y el plasma.
Biotransformación
La rufinamida se elimina de forma casi exclusiva a través del metabolismo. La vía principal de metabolismo es la hidrólisis del grupo carboxilamida para formar el derivado ácido farmacológicamente inactivo, CGP 47292. El metabolismo mediado por el citocromo P450 es muy pequeño. No se puede excluir por completo la formación de pequeñas cantidades de conjugados con glutatión.
In vitro, la rufinamida ha demostrado tener una capacidad pequeña o insignificante para actuar como un inhibidor competitivo o un inhibidor basado en el mecanismo de las siguientes enzimas P450 humanas: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 o CYP4A9/11-2.
Eliminación
La semivida de eliminación plasmática es aproximadamente de 6-10 horas en sujetos sanos y en pacientes con epilepsia. Cuando se administra dos veces al día a intervalos de 12 horas, la rufinamida se acumula en el grado previsto a partir de su semivida terminal, lo que indica que la farmacocinética de la rufinamida es independiente del tiempo (es decir, no hay ninguna autoinducción del metabolismo).
En un ensayo con marcadores radiactivos en tres voluntarios sanos, el compuesto original (rufinamida) fue el principal componente radiactivo en el plasma, representando aproximadamente el 80 % de la radiactividad total, y el metabolito CGP 47292 solo supuso el 15 % aproximadamente. La excreción renal fue la vía predominante de eliminación para el material relacionado con el principio activo, representando el 84,7 % de la dosis.
Linealidad/no linealidad
La biodisponibilidad de la rufinamida es dependiente de la dosis. Al aumentar la dosis, disminuye la biodisponibilidad.
Farmacocinética en grupos especiales de pacientes
Sexo
Se han utilizado modelos farmacocinéticos poblacionales para evaluar la influencia del sexo en la farmacocinética de la rufinamida. Estas evaluaciones indican que el sexo no afecta a la farmacocinética de la rufinamida en un grado clínicamente relevante.
Insuficiencia renal
La farmacocinética de una sola dosis de 400 mg de rufinamida no se vio alterada en sujetos con insuficiencia renal crónica y severa en comparación con voluntarios sanos. Sin embargo, los niveles plasmáticos disminuyeron en aproximadamente un 30 % al utilizar la hemodiálisis tras la administración de rufinamida, lo que sugiere que puede tratarse de un procedimiento útil en caso de sobredosis (ver secciones 4.2 y 4.9).
Insuficiencia hepática
No se han realizado ensayos en pacientes con insuficiencia hepática y, por tanto, Inovelon no debe administrarse a pacientes con insuficiencia hepática severa (ver sección 4.2).
Niños (2-12 años)
En general, los niños presentan un menor aclaramiento de la rufinamida que los adultos, y esta diferencia está relacionada con el tamaño corporal. No se han realizado ensayos en neonatos o en lactantes ni en niños menores de 2 años.
Pacientes de edad avanzada
Un estudio farmacocinético en voluntarios sanos de edad avanzada no mostró ninguna diferencia significativa en los parámetros farmacocinéticos en comparación con los de adultos más jóvenes.
5.3 Datos preclínicos sobre seguridad
Los estudios convencionales de farmacología de seguridad no muestran riesgos especiales a las dosis clínicamente relevantes.
Las toxicidades observadas en perros con niveles similares a la exposición humana con la dosis máxima recomendada fueron cambios hepáticos que incluyeron trombos biliares, colestasis y aumento de las enzimas hepáticas, que se cree que están relacionados con un aumento de la secreción biliar en esta especie. No hubo evidencia de ningún riesgo asociado en los estudios de toxicidad a dosis repetidas en ratas y monos.
En los estudios de toxicidad para la reproducción y de toxicidad durante el desarrollo, se redujo la supervivencia y crecimiento fetales y hubo algunos casos de mortinatalidad secundaria a la toxicidad materna. Sin embargo, no se observaron efectos en la morfología y función, incluidos el aprendizaje o la memoria, en la descendencia. La rufinamida no fue teratogénica en ratones, ratas o conejos.
La rufinamida no resultó genotóxica y no presenta potencial carcinogénico. Las reacciones adversas no observadas en los ensayos clínicos aunque sí observadas en animales con niveles de exposición similares a los clínicos y con posible relevancia para el uso humano fueron mielofibrosis de la médula ósea en el estudio de carcinogenicidad con ratón. Las neoplasias óseas benignas (osteomas) y las hiperostosis observadas en ratones se consideraron el resultado de la activación de un virus específico de los ratones por los iones de fluoruro liberados durante el metabolismo oxidativo de la rufinamida.
Con respecto al potencial inmunotóxico, en un estudio en perros de 13 semanas de duración se observaron timo pequeño e involución del timo con respuesta significativa a la dosis alta en los machos. En el estudio de 13 semanas, se notificaron con incidencia baja cambios en la medula ósea y linfoides en hembras que recibieron la dosis alta. En ratas, se observaron disminución de la celularidad de la médula ósea y atrofia del timo solo en el estudio de carcinogenicidad.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Celulosa microcristalina (E460)
Carmelosa sódica (E466)
Hidroxietilcelulosa Ácido cítrico, anhidro (E330)
Simeticona emulsión, 30 % que contiene ácido benzoico, ciclotetrasiloxano, dimeticona, estearato de glicol y distearato de glicerilo, metilcelulosa, estearato de PEG-40 (estearato de polietilenglicol), polisorbato 65, silica gel, ácido sórbico, ácido sulfúrico y agua.
Poloxámero 188
Parahidroxibenzoato de metilo (E218)
Parahidroxibenzoato de propilo (E216)
Propilenglicol (E1520)
Sorbato de potasio (E202)
Sorbitol (E420), líquido (no cristalizante)
Aroma de naranja Agua
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años.
Después de la primera apertura: 90 días.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación. Para las condiciones de conservación tras la primera apertura del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Frasco de tereftalato de polietileno orientado (o-PET) con un cierre de polipropileno (PP) de seguridad; cada frasco contiene 460 ml de suspensión en una caja exterior de cartón.
Cada caja contiene un frasco, dos jeringas idénticas de administración oral calibradas y un adaptador para el frasco a presión (PIBA). Las jeringas de administración oral están graduadas en incrementos de 0,5 ml.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Preparación: el adaptador para el frasco a presión (PIBA) que se suministra en la caja del producto se debe introducir firmemente en el cuello del frasco antes de usar, y debe permanecer ahí durante el uso del frasco. La jeringa de administración se debe introducir en el PIBA y extraer la dosis con el frasco invertido. Después de cada uso, se debe volver a poner el tapón. El tapón encaja perfectamente con el PIBA colocado.
Ninguna especial para su eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Eisai Limited, European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/378/017
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16/enero/2007 Fecha de la última renovación: 16/enero/ 2012
10. FECHA DE LA REVISIÓN DEL TEXTO
{MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES RELATIVAS AL USO SEGURO Y EFICAZ DEL MEDICAMENTO
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POSTAUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN EN CIRCUNSTANCIAS EXCEPCIONALES
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes
Eisai Manufacturing Limited
Mosquito Way
Hatfield
Hertfordshire
AL10 9SN
Reino Unido
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POSTAUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN EN CIRCUNSTANCIAS EXCEPCIONALES
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Inovelon 100 mg comprimidos recubiertos con película Rufinamida
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido contiene 100 mg de rufinamida.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
10
10 comprimidos recubiertos con película 30
30 comprimidos recubiertos con película 50
50 comprimidos recubiertos con película 60
60 comprimidos recubiertos con película 100
100 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30°C.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE
COMERCIALIZACIÓN
Eisai Ltd., European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Reino Unido.
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/378/001-005
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Inovelon 100 mg comprimidos
1. NOMBRE DEL MEDICAMENTO
Inovelon 100 mg comprimidos recubiertos con película Rufinamida
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Eisai Ltd.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
1. NOMBRE DEL MEDICAMENTO
Inovelon 200 mg comprimidos recubiertos con película Rufinamida
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido contiene 200 mg de rufinamida.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
10
10 comprimidos recubiertos con película 30
30 comprimidos recubiertos con película 50
50 comprimidos recubiertos con película 60
60 comprimidos recubiertos con película 100
100 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30°C.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE
COMERCIALIZACIÓN
Eisai Ltd., European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Reino Unido.
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/378/006-010
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Inovelon 200 mg comprimidos
1. NOMBRE DEL MEDICAMENTO
Inovelon 200 mg comprimidos recubiertos con película Rufinamida
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Eisai Ltd.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
1. NOMBRE DEL MEDICAMENTO
Inovelon 400 mg comprimidos recubiertos con película Rufinamida
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido contiene 400 mg de rufinamida.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
10
10 comprimidos recubiertos con película 30
30 comprimidos recubiertos con película 50
50 comprimidos recubiertos con película 60
60 comprimidos recubiertos con película 100
100 comprimidos recubiertos con película 200
200 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
CAD (MM/AAAA)
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30°C.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE
COMERCIALIZACIÓN
Eisai Ltd., European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Reino Unido.
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/378/011-016
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Inovelon 400 mg comprimidos
1. NOMBRE DEL MEDICAMENTO
Inovelon 400 mg comprimidos recubiertos con película Rufinamida
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Eisai Ltd.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR Y EL ACONDICIONAMIENTO PRIMARIO
1. NOMBRE DEL MEDICAMENTO
Inovelon 40 mg/ml suspensión oral Rufinamida
2. PRINCIPIO(S) ACTIVO(S)
1 ml de Inovelon suspensión oral contiene 40 mg de rufinamida. 1 frasco contiene 18 400 mg de rufinamida.
3. LISTA DE EXCIPIENTES
Además contiene parahidroxibenzoato de metilo (E218) parahidroxibenzoato de propilo (E216) sorbitol (E420)
Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Suspensión oral 460 ml.
Cada caja contiene 1 frasco, 2 jeringas y un adaptador para el frasco a presión (PIBA).
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Agitar bien antes de usar.
Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
CAD:
Después de la primera apertura: usar en 90 días.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Eisai Ltd., European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Reino Unido.
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/378/017
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Inovelon 40 mg/ml
B. PROSPECTO
Prospecto: información para el usuario
Inovelon 100 mg comprimidos recubiertos con película Inovelon 200 mg comprimidos recubiertos con película Inovelon 400 mg comprimidos recubiertos con película
Rufinamida
Lea todo el prospecto detenidamente antes de empezar a tomar el medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Inovelon y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Inovelon
3. Cómo usar Inovelon
4. Posibles efectos adversos
5. Conservación de Inovelon
6. Contenido del envase e información adicional
1. Qué es Inovelon y para qué se utiliza
Inovelon contiene un medicamento llamado rufinamida. Pertenece a un grupo de medicamentos llamados antiepilépticos, que se utilizan para tratar la epilepsia (una enfermedad que causas crisis convulsivas o ataques epilépticos).
Inovelon se utiliza con otros medicamentos para tratar las crisis convulsivas asociadas al síndrome de Lennox-Gastaut en adultos, adolescentes y niños mayores de 4 años. El síndrome de Lennox-Gastaut es el nombre que recibe un grupo de epilepsias severas en las que se pueden presentar crisis repetidas de varios tipos.
Su médico le ha recetado Inovelon para reducir el número de crisis o ataques.
2. Qué necesita saber antes de empezar a tomar Inovelon No tome Inovelon:
- si es alérgico a la rufinamida, a los derivados triazólicos o a cualquiera de los demás componentes de Inovelon (incluidos en la sección 6).
Advertencias y precauciones
Informe a su médico o farmacéutico:
- si tiene síndrome de QT corto congénito o historia familiar de este tipo de síndrome (alteración eléctrica del corazón), ya que el uso de la rufinamida puede empeorarlo.
- si padece problemas hepáticos. La información sobre el uso de la rufinamida en este grupo es limitada, por lo tanto puede ser necesario aumentar con más lentitud la dosis del medicamento. Si su enfermedad hepática es severa, el médico podrá decidir que Inovelon no es recomendable para usted.
- si desarrolla erupción cutánea o fiebre. Podrían ser signos de una reacción alérgica. Acuda al médico inmediatamente ya que muy ocasionalmente puede llegar a ser grave.
64
- si sufre un aumento en el número o severidad o duración de las crisis convulsivas, debe ponerse en contacto con su médico inmediatamente.
- si presenta dificultad para andar, movimientos anómalos, mareos o somnolencia, informe a su médico.
Por favor, consulte con su médico, incluso si estos casos le afectaron en algún momento en el pasado. Niños
Inovelon no se debe utilizar en niños menores de 4 años porque no hay información suficiente sobre el uso en este grupo de edad.
Uso de Inovelon con otros medicamentos
Informe a su médico o farmacéutico si está utilizando o ha utilizado recientemente otros medicamentos, incluso los adquiridos sin receta. Si está utilizando otros medicamentos que se eliminan del cuerpo por medio del sistema enzimático CYP3A4, puede necesitar que lo vigilen cuidadosamente durante dos semanas al comienzo o al final del tratamiento con rufinamida, o después de cualquier cambio importante en la dosis. Puede ser necesario cambiar la dosis de los otros medicamentos ya que pueden ser menos eficaces cuando se administran con rufinamida.
Informe a su médico si utiliza anticonceptivos orales/hormonales. Inovalon puede hacer que los anticonceptivos orales/hormonales, como la píldora anticonceptiva, sean menos eficaces. Por lo tanto, se recomienda que utilice además otro método anticonceptivo seguro y eficaz mientras utilice Inovelon.
Informe a su médico si utiliza adelgazantes de la sangre, como warfarina. Puede ser que el médico tenga que ajustarle la dosis.
Informe a su médico si utiliza digoxina (un medicamento que se utiliza para tratar enfermedades cardiacas). Puede ser que el médico tenga que ajustarle la dosis.
Si el médico le receta o le recomienda un tratamiento adicional para la epilepsia (por ej.: valproato), debe informarle que toma Inovelon ya que puede ser necesario ajustarle la dosis.
Toma de Inovelon con alimentos y bebidas
Ver sección 3 - ‘Cómo usar Inovelon’ para las recomendaciones sobre la toma de Inovelon con alimentos y bebidas.
Embarazo y lactancia
Si usted es mujer en edad fértil, debe utilizar métodos anticonceptivos mientras tome Inovelon.
Si está embarazada o cree que podría estarlo o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento. Solo debe tomar Inovelon durante el embarazo si el médico así lo indica.
Se le aconseja no dar el pecho mientras tome Inovelon ya que se desconoce si la rufinamida pasa a la leche materna.
Consulte a su médico o farmacéutico antes de utilizar cualquier medicamento.
Conducción y uso de máquinas
Inovelon puede hacer que se sienta mareado, somnoliento y afectarle a la visión, especialmente al comienzo del tratamiento o después de un aumento de la dosis. Si le sucede esto, no conduzca ni utilice máquinas.
Inovelon contiene lactosa
Si el médico le ha dicho que tiene intolerancia a algunos azúcares, póngase en contacto con el médico antes de tomar este medicamento.
3. Cómo usar Inovelon
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico.
En caso de duda, consulte de nuevo a su médico o farmacéutico.
Niños de 4 años o más que pesan menos de 30 kg (y no toman valproato)
La dosis de inicio recomendada es de 200 mg al día en dos dosis. El médico le ajustará la dosis y la podrá aumentar en 200 mg a intervalos de dos días, hasta alcanzar una dosis diaria que no supere los 1000 mg.
Niños de 4 años o más que pesan menos de 30 kg (y toman valproato)
Para los niños que pesan menos de 30 kg que toman valproato (un tratamiento para la epilepsia), la dosis diaria máxima recomendada de Inovelon es 600 mg al día.
La dosis de inicio recomendada es 200 mg al día en dos dosis. El médico le ajustará la dosis y la podrá aumentar en 200 mg a intervalos de dos días hasta alcanzar la dosis máxima recomendada de 600 mg al día.
Adultos y niños que pesan 30 kg o más
La dosis de inicio normal es de 400 mg al día en dos dosis. El médico le ajustará la dosis y la podrá aumentar en 400 mg a intervalos de dos días al día hasta alcanzar una dosis diaria que no supere los 3200 mg, dependiendo de su peso.
Algunos pacientes pueden responder a dosis menores y el médico podrá ajustarle la dosis en función de su respuesta al tratamiento.
Si presenta efectos adversos, el médico podrá aumentarle la dosis de forma más lenta.
Inovelon comprimidos se debe tomar dos veces al día con agua, por la mañana y por la noche.
Inovelon se debe tomar con alimentos. Si tiene dificultades para tragar, puede machacar el comprimido, a continuación, mezcle el polvo en aproximadamente medio vaso de agua (100 ml) y bébalo inmediatamente.
Si toma más Inovelon del que debe
Si puede haber tomado más Inovelon del que debe, informe al médico o al farmacéutico inmediatamente, o póngase en contacto con el servicio de urgencias del hospital más cercano, llevando el medicamento con usted.
Si olvidó tomar Inovelon
Si olvidó tomar una dosis, continúe tomando el medicamento de la forma habitual. No tome una dosis doble para compensar la dosis olvidada. Si olvida tomar más de una dosis, consulte al médico.
Si interrumpe el tratamiento con Inovelon
Si el médico le indica que deje el tratamiento, siga sus instrucciones respecto a la reducción paulatina de Inovelon para reducir el riesgo de un aumento de las crisis convulsivas.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, Inovelon puede producir efectos adversos, aunque no todas las personas los sufran.
Los siguientes efectos adversos pueden ser muy graves:
Erupción cutánea y/o fiebre. Pueden ser signos de una reacción alérgica. Si le ocurre, informe a su médico o acuda al hospital inmediatamente.
Cambio en los tipos de crisis que presenta/estado epiléptico más frecuente (crisis que duran un tiempo largo, crisis repetidas). Informe a su médico inmediatamente.
Un pequeño número de personas en tratamiento con antiepilépticos como Inovelon han tenido pensamientos autolesivos o suicidas. Si en algún momento tiene estos pensamientos, póngase en contacto con su médico inmediatamente.
Puede presentar los siguientes efectos adversos con este medicamento. Informe al médico si padece cualquiera de los siguientes efectos:
Los efectos adversos muy frecuentes (más de 1 de cada 10 pacientes) de Inovelon son:
Mareos, dolor de cabeza, náuseas, vómitos, somnolencia, fatiga.
Los efectos adversos frecuentes (más de 1 de cada 100 pacientes) de Inovelon son:
Problemas asociados con el sistema nervioso que incluyen: dificultad para caminar, movimientos anormales, convulsiones/crisis convulsivas, movimientos inusuales del ojo, visión borrosa, temblores.
Problemas asociados con el estómago que incluyen: dolor de estómago, estreñimiento, indigestión, heces blandas (diarrea), pérdida o cambios en el apetito, pérdida de peso.
Infecciones: infección de oído, gripe, congestión nasal, infección pulmonar
Además los pacientes han presentado: ansiedad, insomnio, s hemorragia nasal, acné, erupción cutánea, dolor de espalda, menstruaciones infrecuentes, moratones, lesiones craneoencefálicas (como consecuencia de una lesión accidental durante una crisis epiléptica).
Los efectos adversos poco frecuentes (entre 1 de cada 100 y 1 de cada 1.000 pacientes) de Inovelon son:
Reacciones alérgicas y un aumento de los marcadores de la función hepática (aumento de las enzimas hepáticas).
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Inovelon
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el blíster y en la caja. La fecha de caducidad es el último día del mes que se indica.
No conservar a temperatura superior a 30°C.
No utilice este medicamento si observa que ha cambiado el aspecto del medicamento.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de Inovelon
- El principio activo es rufinamida.
Cada comprimido recubierto con película de 100 mg contiene 100 mg de rufinamida.
Cada comprimido recubierto con película de 200 mg contiene 200 mg de rufinamida.
Cada comprimido recubierto con película de 400 mg contiene 400 mg de rufinamida.
- Los demás componentes son lactosa monohidrato, celulosa microcristalina, almidón de maíz, croscarmelosa sódica, hipromelosa, estearato de magnesio, laurilsulfato de sodio y sílice coloidal anhidra. El recubrimiento consiste en hipromelosa, macrogol (8000), dióxido de titanio (E171), talco y óxido férrico rojo (E172).
Aspecto de Inovelon y contenido del envase
- Los comprimidos de Inovelon 100 mg son comprimidos recubiertos con película, de color rosa, ovalados y ligeramente convexos, ranurados en ambos lados, con la inscripción ‘C261’ en un lado y sin inscripciones en el otro lado.
Está disponible en envases de 10, 30, 50, 60 y 100 comprimidos recubiertos con película.
- Los comprimidos de Inovelon 200 mg son comprimidos recubiertos con película, de color rosa, ovalados y ligeramente convexos, ranurados en ambos lados, con la inscripción ‘C262’ en un lado y sin inscripciones en el otro lado.
Está disponible en envases de 10, 30, 50, 60 y 100 comprimidos recubiertos con película.
- Los comprimidos de Inovelon 400 mg son comprimidos recubiertos con película, de color rosa, ovalados y ligeramente convexos, ranurados en ambos lados, con la inscripción ‘C263’ en un lado y sin inscripciones en el otro lado.
Está disponible en envases de 10, 30, 50, 60, 100 y 200 comprimidos recubiertos con película. Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización:
Eisai Ltd., European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Reino Unido.
Responsable de la fabricación:
Eisai Manufacturing Ltd, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Reino Unido
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Eisai Europe Ltd. Tél/Tel: + 32 (0) 2 502 58 04 |
Lietuva Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Jungtiné Karalysté) |
|
Bt^rapna Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (06egHHeHOTO KpaacTBo) |
Luxembourg/Luxemburg Eisai Europe Ltd. Tél/Tel: + 32 (0) 2 502 58 04 (Belgique/Belgien) |
|
Ceská republika Eisai GesmbH organizacní slozka Tel: + 420 242 485 839 |
Magyarország Eisai Ltd. Tel.: + 44 (0) 20 8600 1400 Egyesült Királyság (Nagy-Britannia) |
|
Danmark Eisai AB Tlf: +46 (0) 8 501 01 600 (Sverige) |
Malta Associated Drug Company Ltd Tel: + 356 (0) 2277 8000 |
|
Deutschland Eisai GmbH Tel: + 49 (0) 696 65 85-0 |
Nederland Eisai BV. Tel: + 31 (0) 900 575 3340 |
|
Eesti (Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Ühendkuningriik) |
Norge Eisai AB Tlf: + 46 (0) 8 501 01 600 (Sverige) |
|
EXláda Arriani Pharmaceuticals S.A. Tnh + 30 210 668 3000 |
Osterreich Eisai GesmbH Tel: + 43 (0) 1 535 1980-0 |
|
España Eisai Farmacéutica, S.A. Tel: +(34) 91 455 94 55 |
Polska Eisai Ltd. Tel.: + 44 (0) 20 8600 1400 (Wielka Brytania) |
|
France Eisai SAS Tél: + (33) 1 47 67 00 05 |
Portugal Eisai Farmacéutica, Unipessoal Lda Tel: + 351 21 487 55 40 |
|
Hrvatska Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Velika Britanija) |
Romania Eisai Ltd. Tel: + 44 (0) 208 600 1400 (Marea Britanie) |
|
Ireland Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (United Kingdom) |
Slovenija Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Velika Britanija) |
|
Ísland Eisai AB Sími: + 46 (0) 8 501 01 600 (Svífejóó) |
Slovenská republika Eisai GesmbH organizacní slozka Tel: + 420 242 485 839 (Ceská republika) |
|
Italia Eisai S.r.l. Tel: + 39 02 5181401 |
Suomi/Finland Eisai AB Puh/Tel: + 46 (0) 8 501 01 600 (Ruotsi/Sverige) |
|
Kúnpoq Arriani Pharmaceuticals S.A. Tnk: +30 210 668 3000 (EkkáSa) |
Sverige Eisai AB Tel: + 46 (0) 8 501 01 600 |
|
Latvija Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Liebritanija) |
United Kingdom Eisai Ltd. Tel: + 44 (0) 20 8600 1400 |
Fecha de la última revisión de este prospecto: {MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
Prospecto: información para el usuario
Inovelon 40 mg/ml suspensión oral
Rufinamida
Lea todo el prospecto detenidamente antes de empezar a tomar el medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted y no debe dárselo a otras personas, aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. (Ver sección 4).
Contenido del prospecto:
1. Qué es Inovelon y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Inovelon
3. Cómo usar Inovelon
4. Posibles efectos adversos
5. Conservación de Inovelon
6. Contenido del envase e información adicional
1. Qué es Inovelon y para qué se utiliza
Inovelon contiene un medicamento llamado rufinamida. Pertenece a un grupo de medicamentos llamados antiepilépticos, que se utilizan para tratar la epilepsia (una enfermedad que causas crisis convulsivas o ataques epilépticos).
Inovelon se utiliza con otros medicamentos para tratar las crisis convulsivas asociadas al síndrome de Lennox-Gastaut en adultos, adolescentes y niños mayores de 4 años. El síndrome de Lennox-Gastaut es el nombre que recibe un grupo de epilepsias severas en las que se pueden presentar crisis repetidas de varios tipos.
Su médico le ha recetado Inovelon para reducir el número de crisis o ataques.
2. Qué necesita saber antes de empezar a tomar INOVELON
No tome Inovelon
- si es alérgico a la rufinamida o a los derivados triazólicos o a cualquiera de los demás componentes de Inovelon (incluídos en la sección 6).
Advertencias y precauciones
Informe a su médico o farmacéutico:
- si tiene síndrome de QT corto congénito o historia familiar de este tipo de síndrome (alteración eléctrica del corazón), ya que el uso de la rufinamida puede empeorarlo.
- si padece problemas hepáticos. La información sobre el uso de la rufinamida en este grupo es limitada, por lo tanto puede ser necesario aumentar con más lentitud la dosis del medicamento. Si su enfermedad hepática es severa, el médico podrá decidir que Inovelon no es recomendable para usted.
- si desarrolla erupción cutánea o fiebre. Podrían ser signos de una reacción alérgica. Acuda al médico inmediatamente ya que muy ocasionalmente puede llegar a ser grave.
- si sufre un aumento en el número o severidad o duración de las crisis convulsivas, debe ponerse en contacto con su médico inmediatamente.
- si presenta dificultad para andar, movimientos anómalos, mareos o somnolencia, informe a su médico.
Por favor, consulte con su médico, incluso si estos casos le afectaron en algún momento en el pasado. Niños
Inovelon no se debe utilizar en niños menores de 4 años porque no hay información suficiente sobre el uso en este grupo de edad.
Uso de Inovelon con otros medicamentos
Informe a su médico o farmacéutico si está utilizando o ha utilizado recientemente otros medicamentos, incluso los adquiridos sin receta. Si está utilizando otros medicamentos que se eliminan del cuerpo por medio del sistema enzimático CYP3A4, puede necesitar que lo vigilen cuidadosamente durante dos semanas al comienzo o al final del tratamiento con rufinamida, o después de cualquier cambio importante en la dosis. Puede ser necesario cambiar la dosis de los otros medicamentos ya que pueden ser menos eficaces cuando se administran con rufinamida.
Informe a su médico si utiliza anticonceptivos orales/hormonales. Inovalon puede hacer que los anticonceptivos orales/hormonales, como la píldora anticonceptiva, sean menos eficaces. Por lo tanto, se recomienda que utilice además otro método anticonceptivo seguro y eficaz mientras utilice Inovelon.
Informe a su médico si utiliza adelgazantes de la sangre, como warfarina. Puede ser que el médico tenga que ajustarle la dosis.
Informe a su médico si utiliza digoxina (un medicamento que se utiliza para tratar enfermedades cardiacas). Puede ser que el médico tenga que ajustarle la dosis.
Si el médico le receta o le recomienda un tratamiento adicional para la epilepsia (por ej.: valproato) debe informarle que toma Inovelon ya que puede ser necesario ajustarle la dosis.
Toma de Inovelon con alimentos y bebidas
Ver sección 3 - ‘Cómo usar Inovelon’ para las recomendaciones sobre la toma de Inovelon con alimentos y bebidas.
Embarazo y lactancia
Si usted es mujer en edad fértil, debe utilizar métodos anticonceptivos mientras tome Inovelon.
Si está embarazada o cree que podría estarlo, o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento. Solo debe tomar Inovelon durante el embarazo si el médico así lo indica.
Se le aconseja no dar el pecho mientras tome Inovelon ya que se desconoce si la rufinamida pasa a la leche materna.
Consulte a su médico o farmacéutico antes de utilizar cualquier medicamento.
Conducción y uso de máquinas
Inovelon puede hacer que se sienta mareado, somnoliento y afectarle a la visión, especialmente al comienzo del tratamiento o después de un aumento de la dosis. Si le sucede esto, no conduzca ni utilice máquinas.
Inovelon contiene parahidroxibenzoato de metilo (E218) y parahidroxibenzoato de propilo (E216)
Estos componentes pueden causar reacciones alérgicas (posiblemente tardías).
3. Cómo usar Inovelon
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Niños de 4 años o más que pesan menos de 30 kg (y no toman valproato)
La dosis de inicio recomendada es de 200 mg al día en dos dosis. Esto es 5 ml de suspensión administrados en una dosis de 2,5 ml por la mañana y otra de 2,5 ml por la noche.
El médico le ajustará la dosis y la podrá aumentar en 200 mg a intervalos de dos días, hasta alcanzar una dosis diaria que no supere los 1000 mg (25 ml).
Niños de 4 años o más que pesan menos de 30 kg (y toman valproato)
Para los niños que pesan menos de 30 kg que toman valproato (un tratamiento para la epilepsia), la dosis diaria máxima recomendada de Inovelon es 600 mg al día.
La dosis de inicio recomendada es 200 mg al día en dos dosis. Esto es 5 ml de suspensión administrados en una dosis de 2,5 ml por la mañana y otra de 2,5 ml por la noche.
El médico le ajustará la dosis y la podrá aumentar en 200 mg a intervalos de dos días hasta alcanzar la dosis máxima recomendada de 600 mg al día (15 ml).
Adultos y niños que pesan 30 kg o más
La dosis de inicio normal es de 400 mg al día en dos dosis. Esto es 10 ml de suspensión administrados en una dosis de 5 ml por la mañana y otra de 5 ml por la noche.
El médico le ajustará la dosis y la podrá aumentar en 400 mg a intervalos de dos días hasta alcanzar una dosis diaria que no supere los 3200 mg, dependiendo de su peso.
Algunos pacientes pueden responder a dosis menores y el médico podrá ajustarle la dosis en función de su respuesta al tratamiento.
Si presenta efectos adversos, el médico podrá aumentarle la dosis de forma más lenta.
Inovelon se debe tomar dos veces al día, una por la mañana y otra por la noche. Se debe tomar con alimentos.
Forma de administración
Utilice la jeringa y el adaptador suministrados para la administración.
A continuación se facilitan las instrucciones de uso de la jeringa y del adaptador:
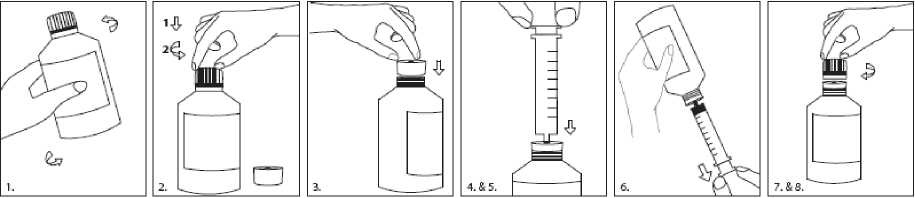
1. Agite bien antes del uso.
2. Presione y gire el tapón para abrir el frasco.
3. Introduzca el adaptador en el cuello del frasco hasta que quede herméticamente cerrado.
4. Meta totalmente el émbolo de la jeringa.
5. Introduzca la jeringa en la apertura del adaptador el máximo posible.
6. Ponga el frasco boca abajo y extraiga la cantidad prescrita de Inovelon.
7. Ponga el frasco boca arriba y saque la jeringa.
8. Deje el adaptador colocado y vuelva a poner el tapón al frasco. Lave la jeringa con agua limpia y séquela bien.
No reduzca la dosis ni deje de tomar el medicamento a menos que se lo indique el médico.
Si toma más Inovelon del que debe
Si puede haber tomado más Inovelon del que debe, informe al médico o farmacéutico inmediatamente, o póngase en contacto con el servicio de urgencias del hospital más cercano, y lleve el medicamento.
Si olvidó tomar Inovelon
Si olvidó tomar una dosis, continúe tomando el medicamento de la forma habitual. No tome una dosis doble para compensar la dosis olvidada. Si olvida tomar más de una dosis, consulte al médico.
Si interrumpe el tratamiento con Inovelon
Si el médico le indica que deje el tratamiento, siga sus instrucciones respecto a la reducción paulatina de Inovelon para reducir el riesgo de un aumento de las crisis convulsivas.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, Inovelon puede producir efectos adversos, aunque no todas las personas los sufran.
Los siguientes efectos adversos pueden ser muy graves:
Erupción cutánea y/o fiebre. Pueden ser signos de una reacción alérgica. Si le ocurre, informe a su médico o acuda al hospital inmediatamente.
Cambio en los tipos de crisis que presenta/ estado epiléptico más frecuente (crisis que duran un tiempo largo, crisis repetidas). Informe a su médico inmediatamente.
Un pequeño número de personas en tratamiento con antiepilépticos como Inovelon han tenido pensamientos autolesivos o suicidas. Si en algún momento tiene estos pensamientos, póngase en contacto con su médico inmediatamente.
Puede presentar los siguientes efectos adversos con este medicamento. Informe al médico si presenta cualquiera de los siguientes efectos:
Los efectos adversos muy frecuentes (más de 1 de cada 10 pacientes) de Inovelon son:
Mareos, dolor de cabeza, náuseas, vómitos, somnolencia, fatiga.
Los efectos adversos frecuentes (más de 1 de cada 100 pacientes) de Inovelon son:
Problemas asociados con el sistema nervioso que incluyen: dificultad para caminar, movimientos anómalos, convulsiones/crisis convulsivas, movimientos inusuales del ojo, visión borrosa, temblores.
Problemas asociados con el estómago que incluyen: dolor de estómago, estreñimiento, indigestión, heces blandas (diarrea), pérdida o cambios en el apetito, pérdida de peso.
Infecciones: infección de oído, gripe, congestión nasal, infección pulmonar.
Además, los pacientes han presentado: ansiedad, insomnio, hemorragia nasal, acné, erupción cutánea, dolor de espalda, menstruaciones infrecuentes, moratones, lesiones craneoencefálicas (como consecuencia de una lesión accidental durante una crisis epiléptica).
Los efectos adversos poco frecuentes (entre 1 de cada 100 y 1 de cada 1.000 pacientes) de Inovelon son:
Reacciones alérgicas y un aumento de los marcadores de la función hepática (aumento de las enzimas hepáticas).
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Inovelon
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta del frasco y en la caja. La fecha de caducidad es el último día del mes que se indica.
Si transcurren más de 90 días después de la primera apertura, la suspensión que quede en el frasco no se debe utilizar.
No utilice la suspensión si observa que el aspecto o el olor del medicamento han cambiado. Lleve el medicamento a su farmacéutico.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
Contenido del envase e información adicional
6.
Composición de Inovelon
- El principio activo es rufinamida. Cada mililitro contiene 40 mg de rufinamida. 5 ml contienen 200 mg de rufinamida.
- Los demás componentes son celulosa microcristalina y carmelosa sódica, ácido cítrico anhidro, simeticona emulsión 30 % (que contiene ácido benzoico, ciclotetrasiloxano, dimeticona, estearato de glicol y distearato de glicerilo, metilcelulosa, estearato de PEG-40 [estearato de polietilenglicol], polisorbato 65, silica gel, ácido sórbico, ácido sulfúrico y agua), poloxámero 188, hidroxietilcelulosa, parahidroxibenzoato de metilo (E218), parahidroxibenzoato de propilo (E216), sorbato potásico, propilenglicol (E1520), sorbitol, líquido (no cristalizante), aroma de naranja y agua.
Aspecto del producto y contenido del envase
- Inovelon es una suspensión ligeramente viscosa de color blanco. Viene en un frasco de 460 ml con dos jeringas idénticas y un adaptador para el frasco a presión (PIBA). Las jeringas están graduadas en incrementos de 0,5 ml.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización:
Eisai Ltd, European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire AL10 9SN, Reino Unido.
Responsable de la fabricación:
Eisai Manufacturing Ltd, Mosquito Way, Hatfield, Hertfordshire AL10 9SN, Reino Unido.
Pueden solicitar más información respecto a este titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Eisai Europe Ltd.
Tél/Tel: + 32 (0) 2 502 58 04
medicamento dirigiéndose al representante local del
Bt^rapnn
Eisai Ltd.
Tel: + 44 (0) 20 8600 1400 (06egHHeHOTO KpaacTBo)
Ceská republika
Eisai GesmbH organizacní slozka Tel: + 420 242 485 839
Danmark
Eisai AB
Tlf: +46 (0) 8 501 01 600 (Sverige)
Lietuva
Eisai Ltd.
Tel: + 44 (0) 20 8600 1400 (Jungtiné Karalysté)
Luxembourg/Luxemburg
Eisai Europe Ltd.
Tél/Tel: + 32 (0) 2 502 58 04 (Belgique/Belgien)
Magyarország Eisai Ltd.
Tel.: + 44 (0) 20 8600 1400 Egyesült Királyság (Nagy-Britannia)
Malta
Associated Drug Company Ltd Tel: + 356 (0) 2277 8000
|
Deutschland Eisai GmbH Tel: + 49 (0) 696 65 85-0 |
Nederland Eisai BV. Tel: + 31 (0) 900 575 3340 |
|
Eesti (Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Ühendkuningriik) |
Norge Eisai AB Tlf: + 46 (0) 8 501 01 600 (Sverige) |
|
EXlába Arriani Pharmaceuticals S.A. Tnk: + 30 210 668 3000 |
Osterreich Eisai GesmbH Tel: + 43 (0) 1 535 1980-0 |
|
España Eisai Farmacéutica, S.A. Tel: +(34) 91 455 94 55 |
Polska Eisai Ltd. Tel.: + 44 (0) 20 8600 1400 (Wielka Brytania) |
|
France Eisai SAS Tél: + (33) 1 47 67 00 05 |
Portugal Eisai Farmacéutica, Unipessoal Lda Tel: + 351 21 487 55 40 |
|
Hrvatska Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Velika Britanija) |
Romania Eisai Ltd. Tel: + 44 (0) 208 600 1400 (Marea Britanie) |
|
Ireland Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (United Kingdom) |
Slovenija Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Velika Britanija) |
|
Ísland Eisai AB Sími: + 46 (0) 8 501 01 600 (Sví^jóó) |
Slovenská republika Eisai GesmbH organizacní slozka Tel: + 420 242 485 839 (Ceská republika) |
|
Italia Eisai S.r.l. Tel: + 39 02 5181401 |
Suomi/Finland Eisai AB Puh/Tel: + 46 (0) 8 501 01 600 (Ruotsi/Sverige) |
|
Kúnpoq Arriani Pharmaceuticals S.A. Tnk: +30 210 668 3000 (EkkáSa) |
Sverige Eisai AB Tel: + 46 (0) 8 501 01 600 |
Fecha de la última revisión de este prospecto: {MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
78