Immunine 200 Ui Polvo Y Disolvente Para Solucion Inyectable O Para Perfusion
Información obsoleta, busque otroDE SANIDAD, POLÍTICA SOCIAL E IGUALDAD
|
1 | |
|
Oí |
k agencia española de 1 medicamentos y I productos sanitarios |
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
IMMUNINE 200 UI polvo y disolvente para solución inyectable o para perfusión
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
1 vial con polvo para solución inyectable contiene 200 UI de factor IX de coagulación humano.
1 ml de solución contiene aproximadamente 40 UI/ml de factor IX de coagulación humano cuando se reconstituye con 5 ml de agua para preparaciones inyectables.
La potencia de factor IX (UI) se determina por el método de coagulación de una etapa de la Farmacopea Europea.
La actividad específica de IMMUNINE es igual o superior a 50 UI de factor IX/mg de proteína.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable o para perfusión Polvo liofilizado o sólido friable de color blanco o amarillo claro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia B (deficiencia congénita de factor IX).
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Posología
La dosis y duración de la terapia de sustitución depende de la gravedad de la deficiencia del factor IX, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor IX administradas se expresa en Unidades Internacionales (UI), que se relacionan con el estándar actual de la OMS para productos de factor IX. La actividad del factor IX en el plasma se expresa bien como porcentaje (referido a plasma humano normal) o en Unidades Internacionales (referidas a un estándar internacional de concentrados de factor IX en plasma).
Una Unidad Internacional (UI) de actividad de factor IX es equivalente a la cantidad de factor IX existente en un ml de plasma humano normal.
El cálculo de la dosis de factor IX requerida se basa en el hallazgo empírico de que 1 UI de factor IX por kg de peso corporal incrementa la actividad del factor IX en el plasma en un 0,9 % de la actividad normal.
Correo electrónicoI
C / CAMPEZO, 1 - EDIFICIO 8 28022 MADRID
La dosis requerida se determina utilizando la siguiente fórmula:
UI requeridas = peso corporal (kg) x aumento de factor IX deseado ( %) (Ul/dl) x 1,1
La cantidad a administrar y la frecuencia de administración siempre deben orientarse a la eficacia clínica en cada caso individual. Los productos de factor IX raramente precisan administrarse más de una vez al día.
En caso de los episodios hemorrágicos siguientes, la actividad del factor IX no debe permitirse que descienda por debajo del nivel de actividad plasmática dada (en % del normal o en UI/dl) en el período de tiempo correspondiente.
En cirugía y en los episodios hemorrágicos, puede utilizarse la siguiente tabla como guía de dosificación:
|
Grado de hemorragia/Tipo de intervención quirúrgica |
Nivel de factor IX requerido (% del normal o UI/dl) |
Frecuencia de dosis (horas)/Duración del tratamiento (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral |
20-40 |
Repetir cada 24 horas al menos 1 día, hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma |
30-60 |
Repetir la perfusión cada 24 horas durante 3-4 días o más, hasta que cese el dolor y se resuelva la incapacidad aguda. |
|
Hemorragias con riesgo vital |
60-100 |
Repetir la perfusión cada 8-24 horas hasta superar el peligro. |
|
Cirugía | ||
|
Cirugía menor incluyendo las extracciones dentales |
30-60 |
Cada 24 horas, al menos durante 1 día, hasta la curación. |
|
Cirugía mayor |
80-100 (pre y postoperatorio) |
Repetir la perfusión cada 8-24 horas hasta la adecuada curación de la herida y continuar la terapia al menos durante otros 7 días, para mantener una actividad del factor IX del 30% al 60 % . |
Durante el curso del tratamiento, se aconseja determinar de forma adecuada los niveles de factor IX como guía de la dosis a administrar y la frecuencia de las infusiones repetidas. Particularmente, en el caso de intervenciones de cirugía mayor es indispensable una exacta monitorización de la terapia de sustitución por medio de análisis de la coagulación (actividad del factor IX plasmático). La respuesta al factor IX puede variar en cada paciente individualmente, alcanzando diferentes niveles de recuperación in vivo y mostrando diferentes vidas medias.
Para la profilaxis de larga duración frente a las hemorragias en pacientes con hemofilia B grave, las dosis normales son de 20 a 40 UI de factor IX por kg de peso corporal a intervalos de 3 a 4 días.
En algunos casos, especialmente en los pacientes más jóvenes, pueden requerirse intervalos de dosificación más cortos o dosis mayores.
No hay datos suficientes para recomendar la utilización de Immunine en niños menores de 6 años.
Los pacientes deben monitorizarse frente al desarrollo de inhibidores del factor IX. Si no se logran los niveles plasmáticos de actividad del factor IX esperados, o si no se controla la hemorragia con una dosis adecuada, deberá realizarse un ensayo para determinar si hay presencia de inhibidores del factor IX. En pacientes con altos niveles de inhibidor, la terapia de factor IX puede no ser efectiva y deberían considerarse otras opciones terapéuticas. El manejo de estos pacientes debe estar dirigido por médicos con experiencia en el tratamiento de pacientes hemofílicos.
Ver también la sección 4.4.
Forma de administración
Disolver el preparado según se indica en la sección 6.6. El producto debe administrarse por vía intravenosa. Se recomienda no administrar más de 2 ml por minuto.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes.
Coagulación intravascular diseminada (CID) y/o hiperfibrinolisis.
Después de un tratamiendo adecuado de estas situaciones, INMMUNINE debe ser administrado sólo en caso de episodios hemorrágicos con riesgo vital.
4.4 Advertencias y precauciones especiales de empleo
Como con cualquier otro producto de origen proteico para administración intravenosa, es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico.
El producto contiene trazas de proteínas humanas además del factor IX. Se debe informar a los pacientes de los signos iniciales de las reacciones de hipersensibilidad incluyendo urticaria local, urticaria generalizada, opresión del pecho, sibilancias, hipotensión y anafilaxis y se les debe recomendar que dejen inmediatamente de utilizar el producto y que se pongan en contacto con su médico si estos síntomas tienen lugar.
En caso de shock, para su tratamiento se deben seguir las pautas médicas actuales.
Los pacientes con riesgo de trombosis (por ejemplo, en pacientes con antecedentes de enfermedad hepática, trombofilia, estados de hipercoagulabilidad, angina de pecho, enfermedad coronaria o infarto agudo de miocardio, o en recién nacidos prematuros) deben someterse a una estrecha vigilancia para detectar la aparición de CID y/o trombosis.
En los pacientes con sospecha de CID, la terapia de sustitución debe suspenderse inmediatamente.
Para prevenir la transmisión de enfermedades infecciosas cuando se administran medicamentos derivados de sangre o plasma humanos se toman medidas estandar como la selección de donantes, análisis de marcadores específicos de infeccines en las donaciones individuales y en las mezclas de plasma, así como la inclusión de etapas en el proceso de fabricación para eliminar/inactivar virus. A pesar de esto, cuando se administran medicamentos derivados de sangre o plasma humanos, la posibilidad de transmisión de agentes infecciosos no puede excluirse totalmente. Esto también se refiere a virus y agentes infecciosos emergentes o de naturaleza desconocida.
Las medidas adoptadas se consideran eficaces para los virus envueltos como el VIH, VHB, VHC y para los no envueltos VHA. Los procedimientos de inactivación/eliminación pueden tener un valor limitado para virus no envueltos tales como el parvovirus B19.
La infección por parvovirus B19 puede ser grave para una mujer embarazada (infección fetal) y para sujetos con immunodeficiencias o con una producción aumentada de hematíes (p. ej., con anemia hemolítica).
En pacientes que reciben concentrados de factor IX obtenidos de plasma humano de forma regular o repetida, debe considerarse su posible vacunación frente a la hepatitis A y B.
Se recomienda encarecidamente que cada vez que se administre IMMUNINE a un paciente, se deje constancia del nombre y el número de lote del producto, para poder mantener la trazabilidad entre el paciente y el lote del producto administrado.
Puesto que la cantidad de sodio en la dosis diaria máxima puede exceder los 200 mg, puede ser perjudicial para personas que sigan una dieta baja en sodio.
Tras el tratamiento repetido con productos del factor IX humano de coagulación, debe monitorizarse a los pacientes frente al desarrollo de anticuerpos neutralizantes (inhibidores) que deben cuantificarse en Unidades Bethesda (UB) utilizando los ensayos biológicos adecuados.
En la bibliografía existen comunicaciones que muestran una correlación entre la existencia de inhibidores del factor IX y reacciones alérgicas. Por tanto, en los pacientes que hallan padecido reacciones alérgicas debe evaluarse la presencia de un inhibidor. Debe tenerse en cuenta que los pacientes con inhibidores del factor IX pueden presentar un mayor riesgo de reacción anafilactica si se les vuelve a administrar factor IX en el futuro.
Dado el riesgo de reacciones alérgicas con concentrados de factor IX, la administración inicial de factor IX debe realizarse, a juicio del médico encargado del tratamiento, bajo observación médica en la que se proporcione los cuidados médicos adecuados frente a las reacciones alérgicas.
Dado que el uso de concentrados de complejo de factor IX se ha asociado históricamente con el desarrollo de complicaciones tromboembólicas, el riesgo es mayor con preparados de poca pureza, el uso de productos que contienen factor IX, puede ser potencialmente peligroso en pacientes con signos de fibrinolisis y en pacientes con coagulación intravascular diseminada (CID). Dado el riesgo potencial de complicaciones trombóticas, al administrar este producto a pacientes con enfermedad hepática, postcirugía, recién nacidos o a pacientes con riesgo de fenómenos trombóticos o CID, se debe iniciar una vigilancia clínica frente a signos trombóticos iniciales y de coagulopatía de consumo, mediante ensayos biológicos adecuados. En cada una de estas situaciones, el beneficio del tratamiento con IMMUNINE debe sopesarse frente al riesgo de estas complicaciones.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se conocen interacciones del factor IX humano de coagulación con otros medicamentos.
4.6 Embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor IX. Puesto que los casos de hemofilia B son raros en mujeres, no se dispone de experiencia sobre el uso del factor IX durante el embarazo y la lactancia. Por tanto, el factor IX debe usarse únicamente si está claramente indicado.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Immunine no afecta a la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas
Las reacciones adversas que se enumeran a continuación se basan en notificaciones de ensayos clínicos y en la vigilancia posterior a la comercialización.
Su frecuencia se ha evaluado según los siguiente criterios: muy frecuentes (>1/10), frecuentes (>1/100; <
1/10), poco frecuentes (>1/1.000; <1/100), raros (>1/10.000; < 1/1.000), muy raros (<1/10.000).
Notificaciones de ensayos clínicos
Las siguientes reacciones adversas se consideran poco frecuentes (tasa de incidencia > 1/1.000; < 1/100).
Trastornos de la piel y del tejido subcutáneo
Prurito
Exantema
Trastornos respiratorios, torácicos y mediastínicos Irritación y dolor de garganta Tos seca
Notificaciones durante la vigilancia posterior a la comercialización
Las siguientes reacciones adversas se consideran muy raras (tasa de incidencia < 1/10.000).
Trastornos de la sangre y del sistema linfático
■ Anticuerpos neutralizantes (inhibidores) del factor IX,
■ Coagulación intravascular diseminada
Trastornos del sistema inmunológico
■ Reacciones alérgicas
■ Anafilaxis grave
■ Angioedema
■ Rubor
■ Urticaria generalizada
■ Urticaria localizada
Trastornos del sistema nervioso
■ Cefalea
■ Inquietud
■ Hormigueo
Trastornos cardiacos
• Taquicardia
• Infarto de miocardio
Trastornos vasculares
■ Hipotensión
■ Episodios tromboembólicos

Embolismo pulmonar Trombosis venosa
Trastornos respiratorios, torácicos y mediastínicos
■ Sibilancias
Trastornos gastrointestinales
■ Náuseas
■ Vómitos
Trastornos de la piel y del tejido subcutáneo
■ Exantema
Trastornos renales y urinarios
■ Síndrome nefrótico
Trastornos generales y alteraciones en el lugar de administración
■ Escalofríos
■ Quemazón y ardor en el lugar de la injección
■ Fiebre
■ Reacciones de hipersensibilidad
■ Letargo
■ Opresión del pecho
Posibles efectos adversos con concentrados del factor IX humano de la coagulación:
De manera poco frecuente en pacientes tratados con productos que contenían factor IX, se han observado reacciones de hipersensibilidad o alérgicas (que pueden incluir angioedema, quemazón y escozor en el lugar de la injección, escalofríos, rubor, urticaria generalizada, dolor de cabeza, urticaria local, hipotensión, letargo, náuseas, inquietud, taquicardia, opresión del pecho, hormigueo, vómitos, sibilancias). En algunos casos, estas reacciones pueden progresar hasta una anafilaxia grave y han ocurrido en estrecha relación temporal con el desarrollo de inhibidores del factor IX (ver también 4.4).
En pacientes con hemofilia B con inhibidores del factor IX e historia de reacciones alérgicas, se han notificado casos de síndrome nefrótico tras el intento de inducir tolerancia inmune.
En raras ocasiones se ha observado fiebre.
Los pacientes con hemofilia B pueden desarrollar anticuerpos neutralizantes (inhibidores) del factor IX. Si se producen estos inhibidores, se manifestarán como una respuesta clínica insuficiente. En tales casos, se recomienda contactar con un centro especializado en hemofilia.
Tras la administración de productos de factor IX, sobre todo si son de baja pureza, existe el riesgo potencial de episodios tromboembólicos. La utilización de productos de factor IX de baja pureza se ha relacionado con casos de infarto de miocardio, coagulación intravascular diseminada, trombosis venosa y embolismo pulmonar. En cambio, la utilización de factor IX de alta pureza rara vez se ha relacionado con esas reacciones adversas.
Para información adicional sobre la seguridad viral, ver 4.4.
4.9 Sobredosis

No se han notificado casos de sobredosis con el factor IX humano de coagulación.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor IX de la coagulación sanguínea Código ATC: B02BD04
El factor IX es una glucoproteína de cadena simple con una masa molecular de alrededor de 68.000 Daltons. . Es un factor de la coagulación dependiente de la vitamina K y se sintetiza en el hígado. El factor IX se activa por el factor XIa en la vía intrínseca de la coagulación y por el complejo factor VII/factor tisular en la vía extrínseca. El factor IX activado, en combinación con el factor VIII activado, activan al factor X. El factor X activado convierte a la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina y se forma el coágulo. La hemofilia B es una alteración hereditaria de la coagulación sanguínea ligada al sexo y que es debida a la disminución de los niveles de factor IX que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontanea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor IX aumentan, consiguiendo una corrección temporal de la deficiencia de factor y una corrección de las tendencias hemorrágicas.
5.2 Propiedades farmacocinéticas
La recuperación in vivo del factor IX es de 0,92 ± 0,06 UI/dl por UI/kg administrado (aproximadamente un 40 %) y la semivida biológica es de unas 17 horas. Tras la administración intravenosa, la concentración pico se alcanza después de 10-30 minutos.
Un estudio farmacocinético realizado en 26 pacientes arrojó los siguientes resultados:
|
Parámetro | ||||
|
Número |
Valor medio |
DE |
IC 95 % | |
|
Aclaramiento (ml/h/kg) |
26 |
8,89 |
2,91 |
7,72-10,06 |
|
Tiempo residual medio (h) |
26 |
23,86 |
5,09 |
1,85-25,88 |
5.3 Datos preclínicos sobre seguridad
IMMUNINE es un concentrado de factor IX altamente purificado que contiene sólo trazas de los factores II, VII y X. La administración de una dosis única de Immunine a animales de laboratorio no mostró signos de potencial toxicológico o trombogénico.
No tiene sentido realizar estudios no clínicos con la administración de dosis repetidas, debido al carácter heterólogo de las proteínas humanas en animales de laboratorio.
Dado que el factor IX es una proteína de origen humano, que en condiciones fisiológicas circula en el plasma, no es de esperar que se produzcan efectos sobre la reproducción, mutagénicos ni carcinogénicos.
6. DATOS FARMACÉUTICOS 6.1 Lista de excipientes
Polvo: Cloruro de sodio
Citrato de sodio
Disolvente: Agua para preparaciones inyectables
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros.
Sólo deben utilizarse los equipos de inyección/perfusión que se suministran dado que el tratamiento puede fallar como consecuencia de la adsorción del factor IX humano de coagulación a las superficies internas de algunos equipos de inyección/perfusión.
6.3 Periodo de validez
dos años
Se ha demostrado la estabilidad química y física de IMMUNINE reconstituido durante 3 horas por debajo de 25°C. Desde el punto de vista microbiológico, el producto debe utilizarse inmediatamente, a menos que el método de reconstitución elimine el riesgo de contaminación microbiológica (condiciones asépticas validadas). Si no se utiliza inmediatamente, la conservación y las condiciones de uso son responsabilidad del usuario. El producto reconstituido no debe volverse a refrigerar.
6.4 Precauciones especiales de conservación
Conservar en nevera (2°C-8°C). No congelar.
Conservar en el envase original para protegerlo de la luz.
Dentro del período de validez indicado, IMMUNINE puede conservarse por debajo de 25°C durante un período de 3 meses. Debe anotarse en el envase del producto el período de conservación por debajo de 25°C. Después de haberse conservado por debajo de 25°C, IMMUNINE no debe volverse a refrigerar, sino que debe utilizarse inmediatamente o desecharse.
6.5 Naturaleza y contenido del envase
El polvo de IMMUNINE se presenta en viales monodosis de vidrio neutro de clase hidrolítica II. El disolvente se presenta en viales monodosis de vidrio neutro de clase hidrolítica I. Los viales de producto se cierran con tapones de goma de clorobutilo. Los viales del disolvente se cierran con tapones de goma de bromobutilo.
Contenido del envase:
1 vial de IMMUNINE 200 UI 1 vial de 5 ml de agua para preparaciones inyectables 1 aguja de trasvasación 1 aguja de aireación 1 aguja filtro 1 aguja desechable 1 jeringa desechable (5 ml)
1 equipo de perfusión
Tamaño del envase: 1 x 200 UI

6.6 Precauciones especiales de eliminación y otras manipulaciones
Deben utilizarse únicamente los equipos de inyección/perfusión suministrados.
IMMUNINE debe reconstituirse sólo inmediatamente antes de su administración. La solución reconstituida debe administrarse lo antes posible (el producto no contiene conservantes). La solución debe ser transparente o ligeramente opalescente. No administrar si la solución está turbia o contiene depósitos. Los productos reconstituidos deben ser inspeccionados visualmente antes de su administración por si contienen alguna partícula o han perdido color.
Se aconseja lavar el acceso venoso común con solución salina isotónica antes y después de la perfusión de IMMUNINE.
Reconstitución del liofilizado para preparar una solución inyectable:
¡Usar una técnica aséptica!
1. Calentar el vial cerrado con tapón de goma que contiene el disolvente (agua para preparaciones inyectables) a temperatura ambiente (máx. 37 °C).
2. Quitar los protectores de los viales de polvo y de disolvente (fig. A) y desinfectar los tapones de goma de ambos viales.
3. Quitar el precinto que cubre uno de los extremos de la aguja de trasvasación suministrada girando y tirando de él. Introducir la aguja a través del tapón de goma del vial de disolvente (fig. B y C).
4. Quitar el precinto que cubre el otro extremo de la aguja de trasvasación, teniendo cuidado de no tocar el extremo expuesto.
5. Inviertir el vial de disolvente sobre el vial de polvo e introducir el extremo libre de la agua de trasvasación en el vial de polvo, perforando el tapón obturador (fig. D). El vacío existente en el vial de polvo aspirará el disolvente.
6. Después de que todo el disolvente haya pasado al vial de polvo, separar los dos viales retirando la aguja de trasvasación del vial de liofilizado (fig. E). Agitar suavemente o rotar el vial del polvo para acelerar la disolución.
7. Una vez que se ha disuelto completamente el polvo, introducir la aguja de aireación incluida (Fig. F) y desaparecerá la espuma que pudiera haberse formado. Retirar la aguja de aireación.
Inyección / Perfusión:
¡Usar una técnica aséptica!
1. Quitar el protector de la aguja filtro suministrada girando y tirando y colocarla en una jeringa desechable estéril. Aspirar la solución con la jeringa (fig. G).
2. Separar la jeringa de la aguja filtro e inyectar lentamente (velocidad máxima de inyección: 2 ml por minuto) la solución por vía intravenosa, utilizando la aguja mariposa suministrada (o la aguja desechable suministrada).
Si se administra por perfusión, utilizar un equipo de perfusión desechable con un filtro adecuado.


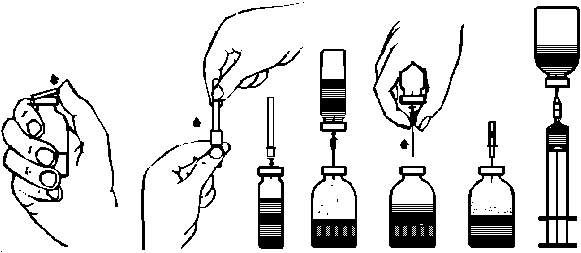
fig. A fig. B fig. C fig. D fig. Efig. F fig. G
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter, S.L.
Pouet de Camilo 2
46394 - Ribarroja del Turia (Valencia)
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
69601
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Febrero 2008
10. FECHA DE LA REVISIÓN DEL TEXTO
Agencia española de
medicamentos y
productos sanitarios