Feiba 500 Uf Polvo Y Disolvente Para Solucion Inyectable
FICHA TÉCNICA
1. Nombre del medicamento
Feiba 500 UF polvo y disolvente para solución inyectable
Feiba 1000 UF polvo y disolvente para solución inyectable
2. Composición cualitativa y cuantitativa
Feiba
Vial con polvo liofilizado: Principio activo:
Complejo coagulante antiinhibidor Proteína total (máx)
Excipientes con efecto conocido:
Citrato de sodio Cloruro de sodio
Vial de disolvente:
|
500 UF |
1000 |
UF |
|
500 UF* |
1000 |
UF* |
|
600 mg |
1200 |
mg |
|
80 mg |
80 |
mg |
|
160 mg |
160 |
mg |
Agua para preparaciones inyectables 20 ml 20 ml
Feiba contiene los factores II, IX y X principalmente no activados, así como el factor VII activado. El antígeno del Factor VIII coagulante (F VIII C: Ag) está presente en la concentración máxima de 0,1 UI/1 UF. El producto está libre, o contiene sólo trazas del sistema calicreína-cinina.
*1 UF acorta el tiempo de tromboplastina parcial activada (TTPA) de un plasma con inhibidor del factor VIII en un 50% del valor obtenido con solución tampón.
Para consultar la lista completa de excipientes ver sección 6.1.
3. Forma farmacéutica
Polvo y disolvente para solución inyectable.
4. Datos clínicos
4.1. Indicaciones terapéuticas
Tratamiento y profilaxis de hemorragias en pacientes con hemofilia A con inhibidor del factor VIII. Debe considerarse la gravedad del episodio hemorrágico, el título del inhibidor y la respuesta anamnésica del paciente si es conocida.
4.2. Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
4.2.1 Posología
Como guía general se recomiendan dosis de 50 a 100 UF/kg, sin exceder una dosis diaria de 200 UF/kg. No debe sobrepasarse 100 UF/kg/dosis, ni dosis diarias de 200 UF/kg, a menos que la gravedad de la hemorragia requiera y justifique el uso de dosis superiores. Ver sección 4.4.
La dosis es independiente del título de inhibidor del paciente. La respuesta al tratamiento puede variar de paciente a paciente.
La dosis y la duración de la terapia dependen de la gravedad de la alteración de la función hemostática, de la localización y gravedad de la hemorragia y del estado clínico del paciente.
IMPORTANTE
La dosis y la frecuencia de la administración se establecerán siempre en función de la efectividad clínica en cada caso.
Población pediátrica
La experiencia en niños menores de 6 años es limitada; se debe adaptar el mismo régimen posológico que en adultos a la situación clínica del niño.
Hemorragias en articulaciones, músculos o tejido blando:
Para hemorragias leves y moderadas se recomienda una dosis de 50 a 75 UF/kg, cada 12 horas.
El tratamiento debe continuar hasta que aparezcan claros signos de mejoría clínica, tales como disminución del dolor, reducción de la tumefacción o movilización de la articulación.
Hemorragias en membranas mucosas:
Se recomienda una dosis de 50 UF/kg cada 6 horas, bajo estricta vigilancia del paciente (lugar del sangrado, repetición del hematocrito).
Profilaxis de hemorragias perioperatorias:
Se recomienda administrar Feiba cada 6 a 12 horas, oscilando la dosis diaria entre 100-200 UF/kg de peso corporal.
4.2.2 Monitorización
En caso de una respuesta inadecuada al tratamiento con el producto, se recomienda realizar un recuento plaquetario, dado que se considera necesario un número suficiente de plaquetas funcionalmente intactas para que el tratamiento con el producto sea eficaz.
Debido al complejo mecanismo de acción, no se dispone de una monitorización directa de los principios activos. Las pruebas de coagulación como el tiempo de coagulación de sangre total (TCT), el tromboelastograma (TEG), y el tiempo parcial de tromboplastina activado (TTPa), generalmente muestran sólo pequeños acortamientos, que no se correlacionan necesariamente con la mejoría clínica. Por estas razones la utilidad de estos ensayos para monitorizar el tratamiento con Feiba, es muy limitada. Ver sección 4.4.
4.2.3 Forma de administración
Feiba debe ser administrado como una inyección o perfusión intravenosa. La velocidad de administración debe asegurar la comodidad del paciente y no debe exceder un máximo de 2 UF/kg/min.
4.3. Contraindicaciones
En las situaciones siguientes, Feiba no se debe utilizar si se dispone de alternativas terapéuticas:
- Hipersensibilidad a los principios activos o a alguno de los excipientes incluidos en la sección 6.1.
- Coagulación Intravascular Diseminada (CID).
- Trombosis aguda o embolia (incluyendo infarto de miocardio).
Ver sección 4.4.
4.4. Advertencias y precauciones especiales de empleo ADVERTENCIAS
Acontecimientos tromboembólicos
Durante el tratamiento con Feiba han ocurrido acontecimientos tromboembólicos, incluyendo Coagulación Intravascular Diseminada (CID), trombosis venosa, embolia pulmonar, infarto de miocardio e ictus.
Algunos de estos acontecimientos aparecieron con dosis superiores a 200 UF/kg/día o en pacientes con otros factores de riesgo (incluyendo CID, enfermedad aterosclerótica avanzada, síndrome de aplastamiento o septicemia) para acontecimientos tromboembólicos. El tratamiento concomitante con Factor VIIa recombinante puede incrementar el riesgo de desarrollar acontecimientos tromboembólicos. La posible presencia de esos factores de riesgo siempre debe tenerse en cuenta en pacientes con hemofilia congénita y adquirida.
Se debe utilizar Feiba con una precaución especial en pacientes con riesgo de CID y trombosis arterial o venosa. Ver sección 4.3.
A la aparición del primer signo o síntoma de acontecimiento tromboembólico, la perfusión se debe interrumpir inmediatamente y se deben adoptar las medidas de diagnóstico y terapéuticas necesarias.
No debe sobrepasarse una dosis única de 100 UF/kg, ni dosis diarias de 200 UF/kg a menos que la gravedad de la hemorragia justifique y requiera el uso de dosis elevadas.
Cuando se utiliza para parar una hemorragia, el producto se debe administrar únicamente el tiempo que sea absolutamente necesario para conseguir el efecto terapéutico deseado.
Reacciones de hipersensibilidad de tipo alérgico
Feiba puede producir la aparición de reacciones de hipersensibilidad que pueden incluir, urticaria, angioedema, alteraciones gastrointestinales, broncoespasmo e hipotensión; estas reacciones pueden ser graves y pueden ser sistémicas (p.e. anafilaxis con urticaria y angioedema, broncoespasmo, shock circulatorio). También pueden aparecer otras reacciones relacionadas con la perfusión, como escalofríos, pirexia e hipertensión.
La administración de Feiba se debe interrumpir al primer signo o síntoma de reacción de hipersensibilidad y se deben iniciar las medidas terapéuticas apropiadas.
Cuando se considere una reexposición a Feiba en pacientes con sospecha de hipersensibilidad o hipersensibilidad conocida al producto, se debe valorar cuidadosamente el beneficio esperado frente al riesgo de la reexposición, teniendo en cuenta el tipo de hipersensibilidad del paciente, conocida o sospechada (alérgica o no alérgica), incluyendo remedios potenciales y/o terapia preventiva o medicamentos alternativos.
En todos estos casos debe sopesarse la relación beneficio/riesgo a la hora de administrar Feiba. Monitorización de la terapia
No debe sobrepasarse dosis individuales de100 UF/kg, ni dosis diarias de 200 UF/kg. Los pacientes que reciben más de 100 UF/kg deben ser monitorizados para el desarrollo de CID, y/o isquemia coronaria aguda, signos u otros síntomas de acontecimientos tromboembólicos .
Pruebas de laboratorio y eficacia clínica
Los ensayos in vitro para controlar la eficacia, tales como TTPa, tiempo de coagulación total (TCT) y tromboelastrograma (TEG) no tienen necesariamente una correlación con la mejora clínica. Por esta razón, no se debe buscar la normalización de estos valores mediante un incremento de dosis de Feiba.
Importancia del recuento plaquetario
En caso de respuesta inadecuada al tratamiento con Feiba, se recomienda realizar un recuento plaquetario, dado que se considera necesario un número suficiente de plaquetas funcionalmente intactas para que el tratamiento con Feiba sea eficaz.
Medidas para prevenir la transmisión de agentes transmisibles
Para prevenir la transmisión de enfermedades infecciosas cuando se administran medicamentos derivados de sangre o plasma humanos se toman medidas estándar como la selección de donantes, análisis de marcadores específicos de infecciones en las donaciones individuales y en las mezclas de plasma, así como la inclusión de etapas en el proceso de fabricación para eliminar/inactivar virus. A pesar de esto, cuando se administran medicamentos derivados de sangre o plasma humanos, la posibilidad de transmisión de agentes infecciosos no puede excluirse totalmente. Esto también se refiere a virus y agentes infecciosos emergentes o de naturaleza desconocida.
Las medidas adoptadas se consideran eficaces para virus envueltos como el VIH, VHB y VHC y para los no envueltos como el VHA. Los procedimientos de inactivación/eliminación pueden tener un valor limitado para virus no envueltos tales como el parvovirus B19. La infección por parvovirus B19 puede ser grave para una mujer embarazada (infección fetal) y para sujetos con inmunodeficiencia o con una producción aumentada de hematíes (ej. con anemia hemolítica).
Cada vez que se administra Feiba a un paciente, se recomienda indicar el nombre y el número de lote del producto para mantener un vínculo entre el paciente y el lote del producto.
Deberá considerarse la conveniencia de una vacunación adecuada (hepatitis A y B) en pacientes que reciban derivados de plasma humano de forma regular/repetida.
PRECAUCIONES
Respuesta discorde a los agentes de by-pass
Debido a factores específicos de los pacientes, la respuesta a un agente de by-pass puede variar y en determinadas situaciones hemorrágicas, los pacientes pueden tener una respuesta insuficiente a un agente y pueden responder a otro. En caso de que se produzca una respuesta insuficiente a un determinado agente de by-pass, se debe considerar la utilización de otro.
Respuestas anamnésicas
La administración de Feiba en pacientes con inhibidores puede producir un incremento inicial “anamnéstico” de los niveles del inhibidor. Durante la administración continuada de Feiba, los niveles del inhibidor pueden descender a lo largo del tiempo.
Tanto los datos clínicos como los datos publicados sugieren que la eficacia de Feiba no se reduce. Anticuerpos de superficie de la hepatitis B e interpretación del test
Después de la administración de dosis elevadas de Feiba, el aumento transitorio de los anticuerpos de superficie de la hepatitis B transferidos pasivamente puede provocar una interpretación errónea de los resultados positivos del test serológico.
Población pediátrica
Los datos limitados de los ensayos clínicos y de las notificaciones sugieren que Feiba se puede utilizar en niños menores de 6 años de edad.
Uso en profilaxis
Solamente se encuentran disponibles datos clínicos limitados de la aplicación de Feiba para la profilaxis de hemorragias en pacientes hemofílicos.
Advertencias sobre excipientes
Este medicamento contiene aproximadamente 80 mg (3,48 mmol) de sodio por vial, lo que deberá tenerse en cuenta en el tratamiento de pacientes con dietas pobres en sodio.
4.5. Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios adecuados y bien controlados del uso combinado o secuencial de Feiba y Factor VIIa recombinante o antifibrinolíticos.
Cuando se utilizan antifibrinolíticos sistémicos, como ácido tranexámico o ácido aminocaproico, durante el tratamiento con Feiba se debe considerar la posibilidad de aparición de acontecimientos tromboembólicos.
Por lo tanto, no se deben utilizar antifibrinolíticos hasta aproximadamente 6 a 12 horas después de la administración de Feiba.
De acuerdo a los datos in vitro disponibles y a las observaciones clínicas, puede ocurrir una interacción potencial medicamentosa con el uso concomitante con Factor VIIa recombinante (que, potencialmente, produzca acontecimientos adversos como un acontecimiento tromboembólico).
4.6. Fertilidad, embarazo y lactancia
No hay datos adecuados relativos al uso de Feiba en mujeres embarazadas o en periodo de lactancia. Los profesionales sanitarios deben considerar detenidamente los riesgos potenciales y solo prescribir Feiba si es claramente necesario, teniendo en cuenta que el periodo de embarazo y de postparto se caracteriza por un aumento del riesgo de acontecimientos tromboembólicos y que algunas complicaciones del embarazo están asociadas con aumento del riesgo de CID.
No se han realizado estudios de reproducción en animales con Feiba y no se han establecido los efectos de Feiba sobre la fertilidad en ensayos clínicos controlados.
Para información sobre el riesgo de infección por parvovirus B19 en mujeres embarazadas, ver sección 4.4.
4.7. Efectos sobre la capacidad para conducir y utilizar máquinas
No hay información sobre los efectos de Feiba sobre la capacidad para conducir y utilizar máquinas.
4.8. Reacciones adversas
Las reacciones adversas descritas en esta sección se han notificado durante la experiencia postcomercialización y en dos estudios realizados con Feiba para el tratamiento de episodios hemorrágicos en pacientes pediátricos y adultos con hemofilia A o B e inhibidores de los factores VIII o IX. Uno de los estudios reclutó a pacientes con hemofilia adquirida con inhibidores del factor VIII (2 de 49 pacientes). Se han incluido las reacciones adversas notificadas en un tercer estudio que compara el tratamiento profiláctico con el tratamiento a demanda.
Las categorías de frecuencias se definen según la siguiente convención:
Muy frecuentes (>1/10)
Frecuentes (>1/100 a <1/10)
Poco frecuentes (>1/1.000 a <1/100)
Raras (>1/10.000 a <1/1.000)
Muy raras (<1/10.000)
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
Las reacciones adversas notificadas en los ensayos clínicos y durante la fase de postcomercialización se describen a continuación. La frecuencia no se puede estimar a partir de los datos disponibles por lo tanto se clasifican como de frecuencia no conocida.
Reacciones adversas
Clasificación de órganos-sistema (SOC)
Término preferente de MedDRA (version 17.0)
Frecuencia*
Trastornos de la sangre y del sistema linfático
Coagulación intravascular diseminada (CID)
Incremento del título de inhibidor (respuesta anamnéstica)*a
No conocida
No conocida
Trastornos del sistema inmunológico
Hipersensibilidad
Urticaria
Reacción anafiláctica
Frecuentes No conocida No conocida
Trastornos del sistema nervioso
|
Parestesia |
No conocida |
|
Hipoestesia |
No conocida |
|
Accidente trombótico |
No conocida |
|
Accidente embólico |
No conocida |
|
Cefaleac |
Frecuentes |
|
Somnolencia* |
No conocida |
|
Mareob |
Frecuentes |
|
Disgeusia* |
No conocida |
Trastornos cardiacos
Infarto de miocardio Taquicardia
No conocida No conocida
Trastornos vasculares
Trombosis Trombosis venosa Trombosis arterial Embolia (complicaciones tromboembólicas)
Hipotensión0
Hipertensión
Rubor
No conocida No conocida No conocida No conocida
Frecuentes No conocida No conocida
Trastornos respiratorios, torácicos y mediastínicos
Embolia pulmonar Broncoespasmo Sibilancias Tos
Disnea*
No conocida No conocida No conocida No conocida No conocida
Trastornos gastrointestinales
Vómitos
Diarrea
Malestar abdominal Náuseas*
No conocida No conocida No conocida No conocida
Trastornos de la piel y del tejido subcutáneo
Sensación de entumecimiento en la cara
Angioedema
Urticaria
Prurito
Erupción cutáneac
No conocida
No conocida No conocida No conocida Frecuentes
|
Trastornos generales y alteraciones en el lugar de administración |
Dolor en el lugar de la inyección Malestar general Sensación de calor Escalofríos* Pirexia* Dolor torácico* Malestar en el pecho* |
No conocida No conocida No conocida No conocida No conocida No conocida No conocida |
|
Exploraciones complementarias |
Disminución de la presión arterial Anticuerpo de superficie de la hepatitis B positivoc |
No conocida Frecuentes |
* No es posible realizar un estimado preciso de la frecuencia de estas reacciones adversas a partir de los datos disponibles.
a) Incremento del título de inhibidor (respuesta anamnésica) (no es término preferente de MedDRA) se corresponde con el incremento de los títulos de inhibidores que existían previamente y que ocurre después de la administración de Feiba. Ver sección 4.4.
b) Reacciones adversas notificadas en el ensayo original y en el de profilaxis. La frecuencia descrita se corresponde solo a la del ensayo de profilaxis.
c) Reacciones adversas notificadas en el ensayo de profilaxis. La frecuencia descrita se corresponde a la del ensayo de profilaxis.
Reacciones de clase
Otros síntomas de reacciones de hipersensibilidad a productos derivados de plasma incluyen letargía y cansancio.
Notificación de sospechas de reacciones adversas
Es importante notificar las sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaram.es
4.9. Sobredosis
Algunos de los acontecimientos tromboembólicos notificados ocurrieron con dosis superiores a 200 UF/kg. Si se observan signos o síntomas de acontecimientos tromboembólicos se debe interrumpir la perfusión inmediatamente y se deben adoptar las medidas de diagnóstico y terapéuticas necesarias. Ver sección 4.4.
5. Propiedades farmacológicas
5.1. Propiedades farmacodinámicas
Grupo farmacoterapéutico: inhibidor de la vía alternativa del factor VIII. Código ATC: B02BD03.
Feiba contiene un complejo coagulante antiinhibidor con actividad Feiba estandarizada (Actividad de corrección del inhibidor del Factor VIII).
La actividad de Feiba se basa en la llamada "Actividad de corrección del inhibidor de factor VIII" o la teoría por la cual la coagulación se induce en el punto donde el factor VIII o los factores IX, XI y XII; así como, teóricamente también otros factores excepto el factor V no es necesario. Sin embargo, no ha sido posible aún determinar exactamente cómo actúa el principio activo.
5.2. Propiedades farmacocinéticas
La farmacocinética de Feiba no se puede estudiar de forma convencional ya que los posibles agentes activos involucrados son proteínas plasmáticas o enzimas o compuestos moleculares cuyas estructuras y metabolismo son todavía desconocidos.
En base a la amplia experiencia clínica del producto pueden recomendarse intervalos posológicos de 6 a 12 horas.
5.3. Datos preclínicos sobre seguridad
Basado en estudios de toxicidad aguda en ratones con un gen objetivo de deficiencia de factor VIII y en ratones normales y ratas con dosis superiores a la dosis máxima diaria en humanos (p.e. mayor de 200 UF/kg de peso corporal), se puede concluir que los efectos adversos relacionados con Feiba son principalmente el resultado de la hipercoagulación inducida por las propiedades farmacológicas del producto.
Los estudios de toxicidad a dosis repetidas en animales son prácticamente inviables debido al desarrollo de anticuerpos de las proteínas heterólogas.Ya que la experiencia clínica no ha dado señales de efectos carcinogénicos o mutagénicos de los concentrados de complejo coagulante antiinhibidor de plasma humano, se consideran innecesarios los estudios experimentales, especialmente en especies heterólogas.
6 . Datos farmacéuticos
6.1. Lista de excipientes
Polvo: citrato de sodio y cloruro de sodio.
Disolvente: agua para preparaciones inyectables.
6.2. Incompatibilidades
No se han realizado estudios de compatibilidad con este producto. Por tanto, Feiba no debe mezclarse con otros medicamentos o disolventes. Es conveniente lavar la vía venosa con solución salina isotónica antes y después de la perfusión de Feiba.
Los factores de coagulación derivados del plasma humano pueden ser adsorbidos por las superficies internas de algunos equipos para inyección/perfusión. Si esto ocurre, puede producirse un fallo del tratamiento. Por lo tanto, sólo se deben utilizar los equipos de plástico para perfusión autorizados.
6.3. Periodo de validez 2 años.
tP.
n
Desde un punto de vista microbiológico, Feiba se debe utilizar inmediatamente después de su reconstitución. Si la solución reconstituida no se administra inmediatamente, las condiciones y los tiempos de conservación serán responsabilidad del usuario.
6.4. Precauciones especiales de conservación No conservar a temperatura superior a 25°C.
No administrar Feiba una vez pasada la fecha de caducidad que figura en el envase.
No congelar.
Conservar el medicamento en el embalaje exterior para protegerlo de la luz.
6.5. Naturaleza y contenido del envase
Feiba se presenta en viales de vidrio de clase hidrolítica I/II. Los tapones de ambos viales son de clorobutilo.
Cada envase contiene o:
- un vial con polvo liofilizado para administración intravenosa, un vial con disolvente, una jeringa desechable, una aguja filtro, una aguja de transferencia, una aguja de aireación, una aguja mariposa con pinza (equipo tipo mariposa para infusión) y una aguja desechable.
o
- un vial con polvo liofilizado para administración intravenosa, un vial con disolvente, un equipo BAXJECT II Hi-Flow (equipo para reconstitución sin aguja para transferir y mezclar los productos de los dos viales dentro de la jeringa), una jeringa desechable, una aguja desechable y una aguja mariposa con pinza (equipo tipo mariposa para infusión).
6.6. Precauciones especiales de eliminación y otras manipulaciones
Feiba debe conservarse liofilizado y reconstituirse inmediatamente antes de su administración. Usar una técnica aséptica durante todo el procedimiento.
La solución reconstituida debe administrarse lo antes posible por vía intravenosa (la solución no contiene conservantes).
Agitar suavemente hasta que todo el producto esté disuelto. Asegurar que todo el producto se encuentre disuelto ya que, de lo contrario, pasarán menos unidades de Feiba a través del filtro del equipo.
Después de la reconstitución, se debe inspeccionar la solución por si contiene partículas o está decolorada antes de su administración. No administrar si la solución está turbia o contiene depósitos. Desechar debidamente toda solución que no se hubiese administrado. No utilizar si el equipo para reconstitución sin aguja o el equipo para reconstitución con aguja, su sistema de barrera de esterilidad o su envase están dañados o muestran cualquier signo de deterioro.
an
Si se administra por perfusión, utilizar un equipo de perfusión desechable con un filtro adecuado de, al menos, 149 pm de tamaño de poro.
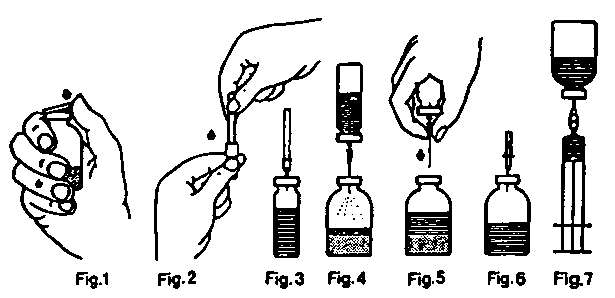
Reconstitución del liofilizado con el equipo de reconstitución con agujas:
1° Calentar el vial de disolvente, a temperatura ambiente (máximo 37°C), si es necesario.
2° Quitar los protectores de ambos viales (liofilizado y disolvente) y desinfectar los tapones obturadores.
(Fig. 1).
3° Romper el precinto de la aguja de transferencia (Fig. 2), quitar un protector e introducir el extremo libre de la aguja en el vial de disolvente (Fig. 3).
4° Sacar el otro protector de la aguja detransferencia, teniendo cuidado de no tocarla.
5° Invertir el vial de disolvente e introducir el extremo libre de la aguja de transferencia en el vial del liofilizado, perforando el tapón obturador. El vacío existente en este vial aspirará el disolvente (Fig. 4).
6° Separar los viales retirando la aguja del vial de concentrado (Fig. 5). Agitar suavemente o rotar el vial de concentrado para acelerar la disolución.
7° Disuelto completamente el liofilizado, introducir la aguja de aireación (Fig 6); desaparecerá la espuma que pudiera haberse formado. Quitar la aguja de aireación.
Inyección/Perfusión:
1° Quitar el protector de la aguja filtro y colocarla en una jeringa estéril desechable. Aspirar la solución con la jeringa a través de la aguja filtro (Fig. 7).
2° Separar la jeringa de la aguja filtro, colocar el equipo de inyección (o la aguja desechable) e inyectar la solución lentamente (velocidad máxima de inyección: 2 UF/kg por minuto) por vía intravenosa.
No exceder una velocidad de inyección/perfusión de 2 unidades de Feiba por kg de peso corporal por
minuto.
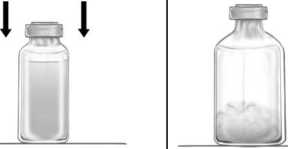
Reconstitución del liofilizado con el equipoBAXJECT II Hi-Flow:
1° Calentar el vial de disolvente (agua esterilizada para preparaciones inyectables) a temperatura ambiente (15°C-25°C), por ejemplo, utilizando un baño de agua durante varios minutos (máximo 37°C), si es necesario.
2° Quitar los protectores de los viales de Feiba y de disolvente y desinfectar los tapones de goma de ambos viales. Colocar los viales en una superficie lisa.
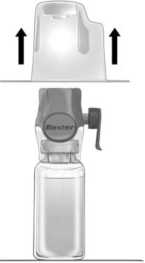
3° Abrir el envoltorio del accesorio BAXJECT II Hi-Flow quitando la tapa de papel sin tocar el interior (figura a). No sacar el equipo del envoltorio.
4° Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente (Figura b). Coger el envoltorio por su extremo y sacar el equipo BAXJECT II Hi-Flow de su envoltorio (Figura c). No quitar el protector azul del equipo BAXJECT II Hi-Flow.
5° Con el equipo BAXJECT II Hi-Flow unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico de color púrpura dentro del tapón de Feiba. El vacío hará que el disolvente penetre en el vial de Feiba (Figura d).
6° Agitar con suavidad, sin sacudir, todo el sistema hasta que todo el material se haya disuelto. Asegúrese de que Feiba esté completamente disuelto, de otra manera el material activo no pasará a través del filtro del equipo.
Figura a

Figura b

Figura c

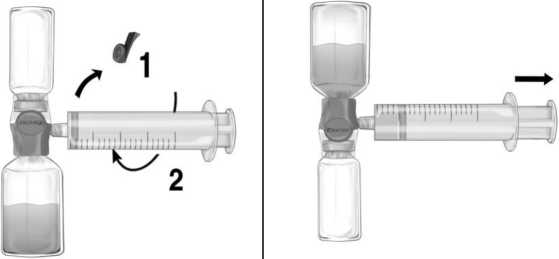
Instrucciones para la Inyección/Perfusión:
1° Quitar el protector azul del equipo BAXJECT II Hi-Flow. Conectar firmemente la jeringa al equipo BAXJECT II Hi-Flow (NO INTRODUCIR AIRE EN LA JERINGA) (Figura e). Se recomienda encarecidamente utilizar una jeringa luer lock con objeto de asegurar una conexión firme entre la jeringa y el equipo BAXJECT II Hi-Flow (girar la jeringa en el sentido de las agujas del reloj hasta que se pare cuando llegue al tope).
2° Invertir el sistema hasta que el producto disuelto se encuentre en la parte de arriba. Introducir la solución de Feiba en la jeringa, tirando del émbolo hacia atrás LENTAMENTE y asegurar que la conexión firme entre el BAXJECT II Hi-Flow y la jeringa se mantiene durante todo el proceso mientras se tira del émbolo de la jeringa (Figura f).
3° Desconectar la jeringa.
4° Si se produce espuma dentro de la jeringa, esperar a que la espuma se compacte. Administrar lentamente por vía intravenosa la solución con el equipo de perfusión suministrado (o una aguja desechable).
Figura d
I

Figura e
Figura f
No exceder una velocidad de inyección/perfusión de 2 unidades de Feiba por kg de peso corporal por minuto.
7. Titular de la autorización de comercialización
Baxalta Spain S.L.
Parque Empresarial San Fernando, Edificio Londres 28830- San Fernando de Henares, Madrid - España
8. Número(s) de autorización de comercialización
Feiba 500 UF 55.953 Feiba 1000 UF 55.954
9. Fecha de la primera autorización/ renovación de la autorización
Fecha de la renovación: agosto 2008
10. Fecha de la revisión del texto
Octubre 2016
13 de 13