Extavia 250 Microgramos/Ml Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Extavia 250 microgramos/ml, polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Extavia contiene 300 microgramos (9,6 millones de UI) de interferón beta-1b recombinante por vial*.
Después de la reconstitución, cada ml contiene 250 microgramos (8,0 millones de UI) de interferón beta-1b recombinante.
* obtenido por ingeniería genética a partir de una cepa de Escherichia coli.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo - de color blanco o casi blanco. Disolvente - solución clara/incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Extavia está indicado para el tratamiento de:
• Pacientes que presentan un único episodio desmielinizante, con un proceso inflamatorio activo, si es lo suficientemente grave como para justificar un tratamiento con corticosteroides intravenosos, si se han excluido otros diagnósticos, y si se determina que hay un riesgo elevado de desarrollar esclerosis múltiple clínicamente definida (ver sección 5.1).
• Pacientes con esclerosis múltiple remitente recidivante y dos o más recaídas en los dos últimos años.
• Pacientes con esclerosis múltiple secundaria progresiva que presentan enfermedad activa, demostrada por la aparición de recaídas.
4.2 Posología y forma de administración
El tratamiento con Extavia deberá iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de esta enfermedad.
Posología
Adultos y adolescentes de 12-17 años de edad
La dosis recomendada de Extavia es de 250 microgramos (8,0 millones de UI), correspondiente a 1 ml de solución reconstituida (ver sección 6.6), inyectada por vía subcutánea cada dos días.
En general, se recomienda ajustar la dosis al iniciar el tratamiento.
Se debe comenzar con 62,5 microgramos (0,25 ml) por vía subcutánea en días alternos e ir aumentando paulatinamente hasta una dosis de 250 microgramos (1,0 ml) en días alternos (ver Tabla A). El periodo de ajuste de la dosis puede modificarse si se presentan reacciones adversas significativas. Para obtener la eficacia adecuada deben alcanzarse dosis de 250 microgramos (1,0 ml) en días alternos.
Tabla A: Pauta de ajuste de dosis*
|
Día de tratamiento |
Dosis |
Volumen |
|
1, 3, 5 |
62,5 microgramos |
0,25 ml |
|
7, 9, 11 |
125 microgramos |
0,5 ml |
|
13, 15, 17 |
187,5 microgramos |
0,75 ml |
|
>19 |
250 microgramos |
1,0 ml |
* El periodo de ajuste de dosis puede modificarse si se presentan reacciones adversas significativas.
No está completamente establecida la dosis óptima.
A día de hoy, no se conoce durante cuánto tiempo debe ser tratado el paciente. Se dispone de datos de seguimiento de ensayos clínicos controlados de pacientes con esclerosis múltiple remitente-recidivante durante un máximo de 5 años y de pacientes con esclerosis múltiple secundaria progresiva durante un máximo de 3 años. Para la esclerosis múltiple remitente-recidivante se ha demostrado eficacia del tratamiento durante los primeros dos años. Los datos disponibles para los otros tres años son consistentes con una eficacia del tratamiento con Extavia mantenida durante todo el periodo.
Se ha demostrado eficacia durante un periodo de tres años en pacientes con un único acontecimiento clínico sugestivo de esclerosis múltiple.
No se recomienda el tratamiento en pacientes con esclerosis múltiple remitente recidivante que hayan sufrido menos de dos recaídas en los dos años anteriores, ni en pacientes con esclerosis múltiple secundaria progresiva que no hayan tenido enfermedad activa en los 2 años anteriores.
El tratamiento con Extavia se debe suspender si el paciente no responde a éste y, por ejemplo, tiene lugar una progresión continua según la Escala Ampliada del Estado de Discapacidad (EAED) durante 6 meses o requiere tratamiento adicional con hormona adrenocorticotropa o corticotropina (ACTH) o corticoides en tres ocasiones, como mínimo, durante un período de un año a pesar del tratamiento con Extavia.
Población pediátrica
No se han realizado ensayos clínicos ni estudios farmacocinéticos formales en niños o adolescentes. Sin embargo, los limitados datos publicados sugieren que el perfil de seguridad en los adolescentes de 12 a 17 años tratados con 8,0 millones de UI de Extavia por vía subcutánea en días alternos es similar al observado en los adultos. No se dispone de datos de Extavia en niños menores de 12 años y, por lo tanto, Extavia no debe emplearse en esta población.
Forma de administración
La solución reconstituida se debe inyectar por vía subcutánea en días alternos.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
- Hipersensibilidad al interferón beta natural o recombinante, albúmina humana o a alguno de los excipientes incluidos en la sección 6.1.
- Inicio del tratamiento en el embarazo (ver sección 4.6).
- Pacientes con depresión grave y/o ideación suicida (ver secciones 4.4 y 4.8).
- Pacientes con hepatopatía descompensada (ver secciones 4.4, 4.5, y 4.8).
4.4 Advertencias y precauciones especiales de empleo
Trastornos del sistema inmunológico
La administración de citocinas a pacientes que presentan gammapatía monoclonal preexistente se ha asociado al desarrollo del síndrome de extravasación capilar sistémica con síntomas parecidos al shock y desenlace fatal.
Trastornos gastrointestinales
Con el uso de Extavia se han observado casos raros de pancreatitis, asociada a menudo a hipertrigliceridemia.
Trastornos del sistema nervioso
Extavia debería administrarse con precaución a los pacientes con trastornos depresivos previos o actuales, particularmente a aquellos con antecedentes de ideación suicida (ver sección 4.3). Se sabe que la depresión y la ideación suicida ocurren con mayor frecuencia en la población con esclerosis múltiple y en asociación con el tratamiento con interferón. Se debería aconsejar a los pacientes tratados con Extavia que notifiquen inmediatamente a su médico cualquier síntoma de depresión y/o ideación suicida. Los pacientes que presenten depresión deben ser estrechamente vigilados durante el tratamiento con Extavia y tratados apropiadamente. Debería considerarse la interrupción del tratamiento con Extavia (ver también secciones 4.3 y 4.8).
Extavia se debe administrar con precaución a los pacientes que presentan antecedentes de trastornos convulsivos y a los pacientes que reciben tratamiento con anticonvulsivantes, y en particular a los pacientes con epilepsia que no están adecuadamente controlados con el tratamiento anticonvulsivante (ver secciones 4.5 y 4.8).
Este medicamento contiene albúmina humana y, por ello, conlleva un riesgo potencial de transmisión de enfermedades víricas. No puede excluirse un riesgo de transmisión de la enfermedad de Creutzfeld-Jacob (ECJ).
Pruebas de laboratorio
Se recomienda realizar pruebas de la función tiroidea con regularidad en pacientes que presentan antecedentes de disfunción tiroidea o cuando esté clínicamente indicado.
Además de aquellas pruebas de laboratorio normalmente requeridas para el seguimiento de pacientes con esclerosis múltiple, antes del inicio, a intervalos regulares tras comenzar el tratamiento con Extavia, y después periódicamente en ausencia de síntomas clínicos, se recomienda realizar recuentos hemáticos completos con fórmula leucocitaria, recuentos plaquetarios, y parámetros bioquímicos en sangre, incluyendo pruebas de función hepática (entre ellas, aspartato aminotransferasa o transaminasa glutámico-oxalacética sérica (SGOT), alanino aminotransferasa o transaminasa glutámico-pirúvica sérica (SGPT) y gama glutamil transferasa.
Los pacientes con anemia, trombocitopenia o leucopenia (aislada o en cualquier combinación) pueden requerir una vigilancia más frecuente de los recuentos hemáticos completos, con fórmula leucocitaria y recuento plaquetario. Los pacientes que desarrollen neutropenia deberán someterse a un cuidadoso seguimiento por la aparición de fiebre o infección. Se han notificado casos de trombocitopenia con reducciones importantes del número de plaquetas.
Trastornos hepatobiliares
Con mucha frecuencia, durante los ensayos clínicos se detectaron aumentos asintomáticos de las transaminasas séricas, en la mayoría de los casos moderados y pasajeros, en pacientes tratados con Extavia. Al igual que con otros interferones beta, se notificaron casos de daño hepático grave, incluyendo fallo hepático, en pacientes tratados con Extavia. Los acontecimientos más graves ocurrieron a menudo en pacientes expuestos a otros medicamentos o sustancias conocidas por estar asociadas a hepatotoxicidad, o en presencia de patologías concomitantes (p. ej. enfermedad maligna con metástasis, infección grave y sepsis, alcoholismo).
Se debe hacer un seguimiento de los pacientes para detectar signos de daño hepático. Si se detecta un aumento de las transaminasas en suero, debe llevarse a cabo un seguimiento cuidadoso y una investigación. Se debe considerar la retirada del tratamiento con Extavia si los niveles se incrementan de una manera significativa, o si van acompañados de síntomas como ictericia. En ausencia de evidencia clínica de que exista daño hepático, y después de la normalización de las enzimas hepáticas, puede considerarse una reanudación del tratamiento con un seguimiento apropiado de las funciones hepáticas.
Microangiopatía trombótica (MAT)
Se han notificado casos de microangiopatía trombótica, que se manifestaron como púrpura trombocitopénica trombótica (TTP) o síndrome hemolítico urémico (SHU), incluyendo casos mortales, con medicamentos que contienen interferón beta. También se notificaron casos en diferentes momentos del tratamiento y podrían ocurrir de varias semanas a varios años desde el inicio del tratamiento con interferón beta. Los síntomas y signos clínicos tempranos incluyen trombocitopenia, aparición de hipertensión, fiebre, síntomas del sistema nervioso central (p. ej.: confusión, paresis) y deterioro de la función renal. Los hallazgos de laboratorio sugieren que MAT incluye un descenso del contaje de plaquetas, incrementos séricos de lactato deshidrogenasa (LDH) debidos a la hemólisis, y esquistocitos (fragmentación de eritrocitos) en una frotis sanguíneo. Por lo tanto, si se observan características clínicas de MAT, se recomiendan pruebas adicionales de niveles de plaquetas en sangre, de LDH en suero, frotis sanguíneo y de función renal. Si se diagnostica MAT, se requiere tratamiento temprano (considerando el recambio plasmático) y se recomienda la inmediata suspensión de Extavia.
Trastornos renales y urinarios
Se debe extremar la precaución y considerar una estrecha vigilancia de los pacientes a los que se administre interferón beta y presentan fallo renal grave.
Síndrome nefrótico
Durante el tratamiento con interferones beta se han notificado casos de síndrome nefrótico con diferentes nefropatías subyacentes incluyendo glomeruloesclerosis focal y segmentaria (GEFS), enfermedad de cambios mínimos (ECM), glomerulonefritis membranoproliferativa (GNMP) y glomerulonefritis membranosa (GNM). Se notificaron casos en varios momentos del tratamiento y podrían ocurrir tras varios años de tratamiento con interferón beta. Se recomienda una monitorización periódica de signos o síntomas tempranos, p. ej.: edema, proteinuria y alteraciones de la función renal, especialmente en pacientes de alto riesgo de enfermedad renal. Se requiere un tratamiento temprano del síndrome nefrótico y se debe tener en consideración la interrupción del tratamiento con Extavia.
Trastornos cardiacos
Extavia también debe ser usado con precaución en pacientes que presentan antecedentes de trastornos cardiacos. Se debe vigilar un posible empeoramiento de la patología cardiaca de los pacientes con enfermedades cardiacas preexistentes significativas tales como insuficiencia cardiaca congestiva, enfermedad coronaria o arritmia, especialmente durante el inicio del tratamiento con Extavia.
Aunque no hay constancia de que Extavia tenga una toxicidad cardiaca directa, los síntomas del síndrome de tipo gripal asociado a los interferones beta puede suponer una sobrecarga para los pacientes que presentan enfermedades cardiacas preexistentes significativas. Durante el periodo postcomercialización, en raras ocasiones se han recibido casos de empeoramiento temporal del estado cardiológico al inicio del tratamiento con Extavia, en los pacientes que presentan enfermedad cardiaca preexistente significativa.
Se han notificado casos de miocardiopatía.. Si esto ocurriera, y se sospechara de alguna relación causal con Extavia, debe interrumpirse el tratamiento.
Trastornos generales y alteraciones en el lugar de administración
Pueden presentarse reacciones graves de hipersensibilidad (reacciones agudas graves, tales como broncoespasmo, anafilaxia y urticaria). Ante la aparición de reacciones graves, se debe suspender la administración de Extavia e instaurarse el tratamiento médico adecuado.
Se ha notificado necrosis en el lugar de inyección en pacientes que utilizan Extavia (ver sección 4.8). Puede ser extensa y podría incluir la fascia muscular así como el tejido adiposo, pudiendo por lo tanto dar como resultado la formación de cicatrices. Se ha requerido ocasionalmente un desbridamiento y, con menor frecuencia, un injerto de piel, pudiendo tardar la curación hasta 6 meses.
Si el paciente experimenta alguna rotura en la piel, que puede estar asociada a hinchazón o salida de fluido por el lugar de la inyección, se debe aconsejar al paciente que consulte con su médico antes de continuar con las inyecciones de Extavia.
Si el paciente presenta múltiples lesiones debe interrumpirse el tratamiento con Extavia hasta su curación. Los pacientes que presentan lesiones únicas pueden continuar con Extavia siempre que la necrosis no sea demasiado extensa, ya que en algunos pacientes se ha producido la curación de la necrosis en el lugar de inyección mientras continuaban con el tratamiento de Extavia.
Con objeto de minimizar el riesgo de necrosis en el lugar de inyección, debe aconsejarse a los pacientes:
- utilizar una técnica de inyección aséptica.
- alternar los lugares de inyección con cada dosis.
La incidencia de reacciones en el lugar de la inyección puede disminuir si se utiliza un autoinyector.
En el estudio fundamental en pacientes que presentan un único episodio clínico sugestivo de esclerosis múltiple se utilizó autoinyector en la mayoría de los pacientes. Las reacciones y la necrosis en el lugar de la inyección fueron observadas con menos frecuencia en este estudio que en los demás estudios fundamentales.
El procedimiento de auto-inyección por el paciente debe ser revisado de manera periódica, especialmente si han aparecido reacciones en el lugar de inyección.
Inmunogenicidad
Como con todas las proteínas terapéuticas, existe una inmunogenicidad potencial. En los ensayos clínicos controlados se recogieron muestras de sangre cada 3 meses para vigilar la aparición de anticuerpos frente a Extavia.
En los diferentes ensayos clínicos controlados, entre el 23% y el 41% de los pacientes desarrollaron actividad neutralizante en suero contra el interferón beta-1b, confirmada por títulos positivos en, al menos, dos ocasiones consecutivas. Entre el 43% y el 55% de estos pacientes evolucionaron hacia una estabilización, negativizando en suero los títulos de los anticuerpos (aparición de dos títulos negativos consecutivos) durante el período de observación posterior del estudio en cuestión.
La aparición de actividad neutralizante se asocia con una disminución de la eficacia clínica solamente en relación con la actividad de las recaídas. Algunos análisis sugieren que este efecto puede ser más pronunciado en los pacientes que presentan los títulos más elevados de actividad neutralizante.
En el estudio en pacientes que presentan un único acontecimiento clínico sugestivo de esclerosis múltiple se observó actividad neutralizante medida cada 6 meses al menos una vez en el 32%
(89 casos) de los pacientes tratados inmediatamente con Extavia. El 60% (53 casos) de estos pacientes volvieron a una situación negativa en cuanto a actividad neutralizante en base a la última evaluación disponible durante un periodo de 5 años. El desarrollo de actividad neutralizantese asoció a un incremento en nuevas lesiones activas y volumen de las lesiones en T2 en las imágenes de resonancia magnética. Sin embargo, esto no parece que esté asociado con una disminución de la eficacia clínica (con respecto al tiempo hasta la aparición de esclerosis múltiple clínicamente definida (EMCD), al tiempo transcurrido hasta la progresión confirmada de la EDSS y la tasa de recaídas).
No se han asociado nuevas reacciones adversas con la aparición de actividad neutralizante.
Se ha demostrado in vitro que Extavia presenta reacción cruzada con el interferón beta natural. Sin embargo, esto no se ha investigado in vivo y su significación clínica es incierta.
Son escasos y no concluyentes los datos de pacientes que, habiendo desarrollado actividad neutralizante, hayan completado el tratamiento con Extavia.
La decisión de continuar o suspender el tratamiento se debe basar en la evolución clínica de la enfermedad y no en el estado de la actividad neutralizante.
Excipientes
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis; esto es, esencialmente «exento de sodio».
Personas sensibles al látex
La cápsula de cierre extraíble de la jeringa precargada de Extavia contiene un derivado de látex de caucho natural. A pesar de que no se ha detectado látex de caucho natural en la cápsula de cierre, no se ha estudiado el uso seguro de la jeringa precargada de Extavia en personas sensibles al látex y por lo tanto existe un riesgo potencial de reacciones de hipersensibilidad, que no puede descartarse por completo.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones.
No se conoce el efecto de la administración de 250 microgramos (8,0 millones de UI) de Extavia, en días alternos, sobre el metabolismo de fármacos en pacientes de esclerosis múltiple. El tratamiento de las recidivas con corticosteroides o ACTH durante hasta 28 días ha sido bien tolerado en pacientes que están recibiendo Extavia.
No se recomienda el empleo concomitante de Extavia con otros inmunomoduladores, con excepción de corticosteroides o ACTH, por la falta de experiencia clínica en pacientes de esclerosis múltiple.
Se ha notificado que los interferones originan una reducción de la actividad de enzimas dependientes del citocromo hepático P450, tanto en animales como en seres humanos. Por ello se debe tener precaución al administrar Extavia en combinación con fármacos que tengan un estrecho índice terapéutico y dependan notablemente para su aclaramiento del sistema citocromo hepático P450, como por ejemplo los antiepilépticos. Deberá tenerse precaución adicional con cualquier medicación concomitante que afecte al sistema hematopoyético.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Las mujeres en edad fértil deben tomar las medidas anticonceptivas adecuadas.
Embarazo
La información sobre el empleo de Extavia durante el embarazo es limitada. Los datos disponibles indican que puede existir un aumento del riesgo de aborto espontáneo. El inicio del tratamiento está contraindicado durante el embarazo (ver sección 4.3). Si una paciente se queda embarazada o planea hacerlo mientras está en tratamiento con Extavia, debe ser informada de los peligros potenciales y se debe considerar la suspensión del tratamiento (ver sección 5.3). En pacientes que presentan una tasa de recaídas elevada antes del inicio del tratamiento, se debe sopesar el riesgo de una recaída grave tras la suspensión del tratamiento con Extavia en caso de embarazo frente al posible aumento del riesgo de aborto espontáneo.
Lactancia
Se desconoce si el interferón beta-1b se excreta en la leche materna. A causa de la posible inducción de reacciones adversas graves en los lactantes, se debe decidir si interrumpir la lactancia o el tratamiento con Extavia.
Fertilidad
No se han realizado investigaciones sobre la fertilidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas.
Los efectos adversos relacionados con el sistema nervioso central asociados al empleo de Extavia podrían afectar la capacidad de conducir vehículos y utilizar máquinas en pacientes susceptibles.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Al iniciarse el tratamiento son frecuentes las reacciones adversas, pero en general remiten al seguir con él. Las reacciones adversas que se presentan con más frecuencia son un complejo sintomático de tipo gripal (fiebre, escalofríos, artralgia, malestar, sudores, dolor de cabeza o mialgia), que se debe principalmente a los efectos farmacológicos del medicamento, y reacciones en el lugar de inyección. Las reacciones en el lugar de inyección se presentaron con frecuencia después de la administración de Extavia. Enrojecimiento, hinchazón, decoloración, inflamación, dolor, hipersensibilidad, necrosis y reacciones inespecíficas están asociadas significativamente al tratamiento con 250 microgramos (8,0 millones de UI) de Extavia.
En general, se recomienda ajustar la dosis al inicio del tratamiento para aumentar la tolerabilidad a Extavia (ver sección 4.2). La administración de medicamentos antiinflamatorios no esteroídicos también puede reducir la incidencia de los síntomas de tipo gripal. La incidencia de reacciones en el lugar de inyección puede reducirse mediante el uso de autoinyector.
Tabla de reacciones adversas
En las siguientes tablas se utiliza el término MedDRA más adecuado para describir una determinada reacción y sus sinónimos y trastornos relacionados.
Las siguientes listas de reacciones adversas están basadas en los informes de los ensayos clínicos (Tabla 1, acontecimientos adversos y alteraciones en las pruebas de laboratorio) y en la farmacovigilancia posterior a la comercialización (Tabla 2, frecuencias - cuando se conocen - basadas en el conjunto de ensayos clínicos (muy frecuentes >1/10, frecuentes >1/100 a <1/10, poco frecuentes >1/1.000 a <1/100, raras >1/10.000 a <1/1.000, muy raras <1/10.000)) del uso de Extavia. La experiencia con Extavia en pacientes con esclerosis múltiple (EM) es limitada, por consiguiente, puede que algunas reacciones adversas que se presenten en muy raras ocasiones no hayan sido observadas todavía.
Tabla 1 Acontecimientos adversos y alteraciones en las pruebas de laboratorio con índices de incidencia >10% y sus porcentajes correspondientes con placebo; reacciones adversas asociadas significativamente <10% basadas en los informes de los ensayos clínicos
|
Sistema de |
Acontecimiento |
Esclerosis |
Esclerosis |
Esclerosis |
|
clasificación de |
único sugestivo |
múltiple |
múltiple |
múltiple |
|
órganos |
de Esclerosis |
secundaria |
secundaria |
recidivante- |
|
Múltiple |
progresiva |
progresiva |
remitente | |
|
Acontecimientos |
(BENEFIT) |
(Estudio |
(Estudio | |
|
adversos y |
europeo) |
norteamericano) | ||
|
alteraciones en las |
Extavia |
Extavia |
Extavia |
Extavia |
|
pruebas de |
250 |
250 |
250 microgramos |
250 |
|
laboratorio |
microgramos |
microgramos |
(placebo) |
microgramos |
|
(placebo) |
(placebo) |
n=317 (n=308) |
(placebo) | |
|
n=292 (n=176) |
n=360 (n=358) |
n=124 (n=123) | ||
|
Infecciones e infestaciones | ||||
|
Infección |
6% (3%) |
13% (11%) |
11% (10%) |
14% (13%) |
|
Absceso |
0% (1%) |
4% (2%) |
4% (5%) |
1% (6%) |
|
Trastornos de la sangre y del sistema linfático | ||||
|
Descenso del recuento de linfocitos (<1500/mm3) x A ° |
79% (45%) |
53% (28%) |
88% (68%) |
82% (67%) |
|
Descenso del recuento absoluto de neutrófilos (<1500/mm3) x A * ° |
11% (2%) |
18% (5%) |
4% (10%) |
18% (5%) |
|
Descenso del recuento de leucocitos (<3000/mm3) x A * ° |
11% (2%) |
13% (4%) |
13% (4%) |
16% (4)% |
|
Linfoadenopatía |
1% (1%) |
3% (1%) |
11% (5%) |
14% (11%) |
|
Trastornos del metabo |
ismo y de la nutrición | |||
|
Descenso de la glucemia (<55 mg/dl) x |
3% (5%) |
27% (27%) |
5% (3%) |
15% (13%) |
|
Trastornos psiquiátricos | ||||
|
Depresión |
10% (11%) |
24% (31%) |
44% (41%) |
25% (24%) |
|
Ansiedad |
3% (5%) |
6% (5%) |
10% (11%) |
15% (13%) |
|
Trastornos del sistema nervioso | ||||
|
Cefalea A |
27% (17%) |
47% (41%) |
55% (46%) |
84% (77%) |
|
Mareo |
3% (4%) |
14% (14%) |
28% (26%) |
35% (28%) |
|
Insomnio |
8% (4%) |
12% (8%) |
26% (25%) |
31% (33%) |
|
Migraña |
2% (2%) |
4% (3%) |
5% (4%) |
12% (7%) |
|
Parestesias |
16% (17%) |
35% (39%) |
40% (43%) |
19% (21%) |
|
Trastornos oculares | ||||
|
Conjuntivitis |
1% (1%) |
2% (3%) |
6% (6%) |
12% (10%) |
|
Visión anormal A |
3% (1%) |
11% (15%) |
11% (11%) |
7% (4%) |
|
Trastornos del oído y t |
el laberinto | |||
|
Dolor de oído |
0% (1%) |
<1% (1%) |
6% (8%) |
16% (15%) |
|
Trastornos cardiacos | ||||
|
Palpitación * |
1% (1%) |
2% (3%) |
5% (2%) |
8% (2%) |
|
Trastornos vasculares | ||||
|
Vasodilatación |
0% (0%) |
6% (4%) |
13% (8%) |
18% (17%) |
|
Hipertensión ° |
2% (0%) |
4% (2%) |
9% (8%) |
7% (2%) |
|
Trastornos respiratorios, torácicos y met |
iastínicos | |||
|
Infección del tracto respiratorio superior |
18% (19%) |
3% (2%) | ||
|
Sinusitis |
4% (6%) |
6% (6%) |
16% (18%) |
36% (26%) |
|
Aumento de la tos |
2% (2%) |
5% (10%) |
11% (15%) |
31% (23%) |
|
Disnea * |
0% (0%) |
3% (2%) |
8% (6%) |
8% (2%) |
|
Trastornos gastrointestinales | ||||
|
Diarrea |
4% (2%) |
7% (10%) |
21% (19%) |
35% (29%) |
|
Estreñimiento |
1% (1%) |
12% (12%) |
22% (24%) |
24% (18%) |
|
Náuseas |
3% (4%) |
13% (13%) |
32% (30%) |
48% (49%) |
|
Vómitos A |
5% (1%) |
4% (6%) |
10% (12%) |
21% (19%) |
|
Dolor abdominal ° |
5% (3%) |
11%(6%) |
18% (16%) |
32% (24%) |
|
Trastornos hepatobiliares | ||||
|
Aumento de alanina aminotransferasa (SGPT >5 veces el valor basal) x A * ° |
18% (5%) |
14% (5%) |
4% (2%) |
19% (6%) |
|
Aumento de aspartato aminotransferasa (SGOT >5 veces el valor basal) x A * ° |
6% (1%) |
4% (1%) |
2% (1%) |
4% (0%) |
|
Trastornos de la piel y del tejido subcutáneo | ||||
|
Trastorno de la piel |
1% (0%) |
4% (4%) |
19% (17%) |
6% (8%) |
|
Erupción cutánea A ° |
11% (3%) |
20% (12%) |
26% (20%) |
27% (32%) |
|
Trastornos musculoesq |
ueléticos y del tejido conjuntivo | |||
|
Hipertonía° |
2% (1%) |
41% (31%) |
57% (57%) |
26% (24%) |
|
Mialgia * ° |
8% (8%) |
23% (9%) |
19% (29%) |
44% (28%) |
|
Miastenia |
2% (2%) |
39% (40%) |
57% (60%) |
13% (10%) |
|
Dolor de espalda |
10% (7%) |
26% (24%) |
31% (32%) |
36% (37%) |
|
Dolor en las extremidades |
6% (3%) |
14% (12%) |
0% (0%) | |
|
Trastornos renales y urinarios | ||||
|
Retención urinaria |
1% (1%) |
4% (6%) |
15% (13%) | |
|
Proteinuria positiva (>1+)x |
25% (26%) |
14% (11%) |
5% (5%) |
5% (3%) |
|
Frecuencia urinaria |
1% (1%) |
6% (5%) |
12% (11%) |
3% (5%) |
|
Incontinencia urinaria |
1% (1%) |
8% (15%) |
20% (19%) |
2% (1%) |
|
Urgencia urinaria |
1% (1%) |
8% (7%) |
21% (17%) |
4% (2%) |
|
Trastornos del aparato reproductor y de la mama | ||||
|
Dismenorrea |
2% (0%) |
<1% (<1%) |
6% (5%) |
18% (11%) |
|
Trastornos menstruales * |
1% (2%) |
9% (13%) |
10% (8%) |
17% (8%) |
|
Metrorragia |
2% (0%) |
12% (6%) |
10% (10%) |
15% (8%) |
|
Impotencia |
1% (0%) |
7% (4%) |
10% (11%) |
2% (1%) |
|
Trastornos generales y alteraciones en el lugar de administración | ||||
|
Reacción en el lugar de inyección (varios tipos) A * ° § |
52% (11%) |
78% (20%) |
89% (37%) |
85% (37%) |
|
Necrosis en el lugar de inyección * ° |
1% (0%) |
5% (0%) |
6% (0%) |
5% (0%) |
|
Complejo sintomático de tipo gripal & A *° |
44% (18%) |
61% (40%) |
43% (33%) |
52% (48%) |
|
Fiebre A * ° |
13% (5%) |
40% (13%) |
29% (24%) |
59% (41%) |
|
Dolor |
4% (4%) |
31% (25%) |
59% (59%) |
52% (48%) |
|
Dolor torácico ° |
1% (0%) |
5% (4%) |
15% (8%) |
15% (15%) |
|
Edema periférico |
0% (0%) |
7% (7%) |
21% (18%) |
7% (8%) |
|
Astenia * |
22% (17%) |
63% (58%) |
64% (58%) |
49% (35%) |
|
Escalofríos A * ° |
5% (1%) |
23% (7%) |
22% (12%) |
46% (19%) |
|
Sudoración * |
2% (1%) |
6% (6%) |
10% (10%) |
23% (11%) |
|
Malestar * |
0% (1%) |
8% (5%) |
6% (2%) |
15% (3%) |
|
El término MedDRA más adecuado se utiliza para describir una determinada reacción y sus sinónimos y trastornos relacionados. x Alteración en una prueba de laboratorio A Asociado significativamente al tratamiento con Extavia en pacientes con un primer episodio sugestivo de EM, p<0,05 * Asociado significativamente al tratamiento con Extavia en la EMRR, p<0,05 ° Asociado significativamente al tratamiento con Extavia en la EMSP, p<0,05 § Reacción en el lugar de inyección (varios tipos): Comprende todas los acontecimientos adversos que se producen en el lugar de inyección, es decir, los siguientes términos: hemorragia en el lugar de inyección, hipersensibilidad en el lugar de inyección, inflamación en el lugar de inyección, nódulo en el lugar de inyección, necrosis en el lugar de inyección, dolor en el lugar de inyección, reacción en el lugar de inyección, edema en el lugar de inyección y atrofia en el lugar de inyección. & «Complejo sintomático de tipo gripal»: Se refiere a un síndrome y/o combinación donde se producen, al menos, dos acontecimientos adversos de entre los siguientes: fiebre, escalofríos, mialgia, malestar y sudoración. | ||||
Tabla 2 Reacciones adversas a medicamentos (RA) identificadas durante la vigilancia
postcomercialización (las frecuencias - cuando se conocen - se han calculado en base al conjunto de ensayos clínicos N=1.093)
|
Sistema de clasificación de órganos |
Muy frecuentes (>1/10) |
Frecuentes (>1/100 a <1/10) |
Poco frecuentes (>1/1.000 a <1/100) |
Raras (>1/10.000 a <1/1.000) |
Frecuencia no conocida |
|
Trastornos de la sangre y del sistema linfático |
Anemia |
Trombocitopenia |
Microangioptaía trombótica incluyendo púrpura trombocitopénica trombótica/síndro me hemolítico , . # urémico | ||
|
Trastornos del sistema inmunológico |
Reacciones anafilácticas |
Síndrome de extravasación capilar en gammapatía monoclonal preexistente* | |||
|
Trastornos endocrinos |
Hipotiroidismo |
Hipertiroidismo, Trastorno del tiroides |
|
Trastornos del metabolismo y de la nutrición |
Peso aumentado, Peso disminuido |
Aumento de los triglicéridos en sangre |
Anorexia* | ||
|
Trastornos psiquiátricos |
Estado de confusión |
Intento de suicidio (ver también sección 4.4), Labilidad emocional | |||
|
Trastornos del sistema nervioso |
Convulsión | ||||
|
Trastornos cardiacos |
Taquicardia |
Miocardiopatía* | |||
|
Trastornos respiratorios, torácicos y mediastínicos |
Broncoespasmo* |
Hipertensión arterial pulmonar** | |||
|
Trastornos gastrointestinales |
Pancreatitis | ||||
|
Trastornos hepatobiliares |
Aumento de la bilirrubina sanguínea |
Aumento de la gamma-glutamil-transferasa, Hepatitis |
Daño hepático (incluyendo hepatitis), Fallo hepático* | ||
|
Trastornos de la piel y del tejido subcutáneo |
Urticaria, Prurito, Alopecia |
Decoloración de la piel | |||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia |
Lupus eritematoso inducido por el fármaco | |||
|
Trastornos renales y urinarios |
Síndrome nefrótico, glomeruloesclerosis (ver sección 4.4)* , # | ||||
|
Trastornos del aparato reproductor y de la mama |
Menorragia |
* RA’s derivadas solo durante la post-comercialización.
# Efecto de clase para medicamentos con interferon beta (ver sección 4.4).
** Ficha técnica de los productos que contienen interferón, ver debajo de “Hipertensión arterial pulmonar”.
Hipertensión arterial pulmonar
Se han notificado casos de hipertensión arterial pulmonar (HAP) con medicamentos que contienen interferón beta. Los episodios se notificaron en distintos momentos, incluso varios años después de comenzar el tratamiento con interferón beta.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Interferón beta-1b ha sido administrado a pacientes adultos con cáncer, en dosis de hasta
5.500 microgramos (176 millones de UI) por vía intravenosa, tres veces/semana, sin efectos adversos
graves que comprometieran funciones vitales.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inmunoestimulantes, interferones, código ATC: L03AB08
Los Interferones pertenecen a la familia de las citoquinas, que son proteínas naturales. Los Interferones tienen pesos moleculares comprendidos entre 15.000 y 21.000 Dalton. Se han identificado tres clases principales de Interferones denominados alfa, beta y gamma. Interferón alfa, beta y gamma tienen actividades biológicas diferentes, aunque se solapan parcialmente. Las actividades del Interferón beta-1b están restringidas a la especie y, por tanto, la información farmacológica de mayor interés es la que se deriva de los estudios realizados sobre cultivos de células humanas o los estudios in vivo en humanos.
Mecanismo de acción
Interferón beta-1b ha demostrado poseer actividad antivírica e inmunorreguladora. Los mecanismos mediante los cuales ejerce sus acciones en la esclerosis múltiple aún no están totalmente aclarados. Sin embargo, se sabe que las propiedades modificadoras de respuesta biológica de Interferón beta-1b están mediadas por sus interacciones con receptores celulares específicos que se localizan en la superficie de las células humanas. La unión del Interferón beta-1b a estos receptores induce la expresión de un número de productos genéticos que se supone que son los mediadores de las acciones biológicas del Interferón beta-1b. Algunos de estos productos han sido determinados en el suero y en fracciones celulares de sangre recogida de pacientes tratados con Interferón beta-1b. Interferón beta-1b reduce la afinidad de unión y aumenta la internalización y degradación del receptor de Interferón gamma. Interferón beta-1b también aumenta la actividad supresora de las células mononucleares de sangre periférica.
Eficacia clínica y seguridad
No se han realizado ensayos específicos acerca de la influencia de Extavia sobre el sistema cardiovascular, aparato respiratorio ni sobre la función de órganos endocrinos.
Esclerosis múltiple remitente recidivante (EM RR)
Se realizó un ensayo clínico controlado de Extavia en pacientes con esclerosis múltiple remitente recidivante incapaces de caminar sin ayuda (EDSS basal de 0 a 5,5). En los pacientes tratados con Extavia hubo una reducción de la frecuencia (30%) y la gravedad de las recidivas clínicas y en el número de hospitalizaciones causadas por la enfermedad. Se observó además una prolongación del intervalo sin enfermedad. No hubo indicios de un efecto de Extavia sobre la duración de las recaídas ni sobre los síntomas entre las recaídas, ni se observó un efecto importante sobre la progresión de la enfermedad en la esclerosis múltiple remitente recidivante.
Esclerosis múltiple secundaria progresiva (EM SP)
Se efectuaron dos ensayos clínicos controlados con Extavia en los que participaron un total de 1.657 pacientes con esclerosis múltiples secundaria progresiva (EDSS basal de 3 a 6,5, es decir, los pacientes eran capaces de andar). No se estudió a pacientes con enfermedad leve ni a los que no podían andar. Los dos estudios no mostraron resultados uniformes para el criterio de valoración primario, el tiempo hasta la progresión confirmada, indicativo del retraso de la progresión de la incapacidad:
Uno de los dos estudios puso de manifiesto un retraso estadísticamente significativo del tiempo hasta la progresión de la incapacidad (cociente de riesgo = 0,69, intervalo de confianza del 95% (0,55, 0,86), p=0,0010, equivalente a una reducción del riesgo del 31% por Extavia) y del tiempo hasta la dependencia de una silla de ruedas (cociente de riesgo = 0,61, intervalo de confianza del 95% (0,44, 0,85), p=0,0036, equivalente a una reducción del riesgo del 39% por Extavia) en los pacientes que recibieron Extavia. Este efecto continuó durante el período de observación, hasta 33 meses. El efecto del tratamiento se producía en pacientes con todos los niveles de incapacidad investigados y con independencia de la actividad de recaída.
En el segundo ensayo de Extavia en la esclerosis múltiple secundaria progresiva, no se observó un retraso del tiempo hasta la progresión de la incapacidad. Existen indicios de que los pacientes incluidos en este estudio presentaban una enfermedad global menos activa que en el otro estudio sobre la esclerosis múltiple secundaria progresiva.
En los metaanálisis retrospectivos en los que se incluyeron los datos de los dos estudios, se halló un efecto global del tratamiento estadísticamente significativo (p=0,0076; 8,0 millones de UI de Extavia frente a todos los pacientes con placebo).
Los análisis retrospectivos de subgrupos mostraron que la probabilidad máxima de un efecto del tratamiento en la progresión de la incapacidad se da en los pacientes con enfermedad activa antes del inicio del tratamiento [cociente de riesgo 0,72, intervalo de confianza del 95% (0,59, 0,88), p=0.0011, equivalente a una reducción del riesgo del 28% debida a Extavia en los pacientes con recaídas o progresión acusada de la EDSS, 8,0 millones de UI de Extavia frente a todos los pacientes con placebo]. Estos análisis retrospectivos de subgrupos aportaron datos indicativos de que tanto las recaídas como la progresión acusada de la EDSS (EDSS > 1 punto o > 0,5 puntos para una EDSS > 6 en los dos años anteriores) pueden ayudar a identificar a los pacientes con enfermedad activa.
En ambos ensayos hubo una reducción (30%) de la frecuencia de las recaídas clínicas en los pacientes con esclerosis múltiple secundaria progresiva tratados con Extavia. No existen pruebas de que Extavia tenga un efecto sobre la duración de las recaídas.
Episodio desmielinizante único sugestivo de esclerosis múltiple
Se realizó un ensayo clínico controlado con Extavia en pacientes con un único acontecimiento clínico y hallazgos en la Resonancia magnética (RM) sugestivos de esclerosis múltiple (al menos dos lesiones clínicamente silentes en la RM ponderada en T2). Se incluyeron pacientes con inicio monofocal o multifocal de la enfermedad (es decir, pacientes que presentaban signos clínicos de una única lesión o de, al menos, dos lesiones respectivamente, del sistema nervioso central). Se tenía que excluir cualquier otra enfermedad que no fuera la esclerosis múltiple y que pudiera explicar mejor los signos y síntomas del paciente. Este ensayo constaba de dos fases: una fase controlada con placebo seguida de una fase de seguimiento planificada previamiente. La fase controlada con placebo duró 2 años o hasta que el paciente desarrollara una esclerosis múltiple clínicamente definida (EMCD), según cuál de estas dos circunstancias ocurriese primero. Tras la fase controlada con placebo, el paciente entraba en una fase de seguimiento planificada previamiente con Extavia cuyo fin era evaluar los efectos del inicio inmediato frente al inicio diferido del tratamiento con Extavia, comparando a los pacientes inicialmente aleatorizados a Extavia («grupo de tratamiento inmediato») o a placebo («grupo de tratamiento diferido»). Los pacientes y los investigadores siguieron sin conocer las asignaciones iniciales del tratamiento.
En la fase controlada con placebo, Extavia retrasó la progresión desde el primer acontecimiento clínico hasta la esclerosis múltiple clínicamente definida (EMCD) de forma significativa, tanto estadística como clínicamente, correspondiente a una reducción del riesgo del 47% (Cociente de Riesgos = 0,53; intervalo de confianza 95% (0,39; 0,73), p<0,0001). En el periodo del estudio de dos años, se desarrolló la EMCD en el 45% del grupo tratado con placebo comparado con el 28% en el grupo tratado con Extavia (estimaciones de Kaplan-Meier). Extavia prolongó el tiempo hasta la EMCD en 363 días, desde 255 días en el grupo tratado con placebo hasta 618 días en el grupo tratado con Extavia (según los percentiles 25). Este efecto terapéutico seguía siendo evidente tras el año adicional de seguimiento, momento en el que la reducción del riesgo era del 41% (cociente de riesgos = 0,59, intervalo de confianza del 95% (0,42, 0,83), p=0,0011).Dentro del periodo de estudio de tres años, se produjo una EMCD en el 51% de los pacientes del grupo de tratamiento diferido, frente a un 37% de los del grupo de tratamiento inmediato (estimaciones de Kaplan-Meier). Se observó persistencia del efecto terapéutico aunque la mayoría de los pacientes del grupo placebo recibieron tratamiento con Extavia en el tercer año del ensayo.
La solidez del efecto del tratamiento también se demostró por el retraso de la progresión a esclerosis múltiple según los criterios de McDonald. En dos años, el riesgo fue 85% en el grupo placebo y 69% en el grupo de Extavia (cociente de riesgos = 0,57; intervalo de confianza 95% (0,46; 0,71),
p<0,00001).
Después de tres años y en un análisis intermedio previamente planificado se halló que la progresión de EDSS (incremento confirmado en la escala EDSS de un punto (1.0) o superior en comparación con los valores basales), ocurrió en un 24% de los pacientes que habían iniciado el tratamiento de forma diferida en comparación con el grupo de pacientes que recibió el tratamiento de forma inmediata que mostró un 16% (cociente de riesgos = 0.6, intervalo de confianza 95% (0.39, 0.92, p=0.022). No hay evidencia de mejora en lo que se refiere a la evolución de la discapacidad confirmada en la mayor parte de los pacientes que recibieron el tratamiento de forma «inmediata». El seguimiento de pacientes continúa para obtener más datos. No se vió mejora en la calidad de vida (medida mediante la escala FAMS-Functional Assesment of MS: Treatment Outcomes Index) que pueda ser atribuible a Extavia.
Los análisis de los subgrupos según los parámetros basales demostraron evidencia de eficacia en todos los subgrupos evaluados. También se obtuvieron efectos significativos en pacientes con una enfermedad menos diseminada y menos activa en el momento del primer acontecimiento. El riesgo de progresión a EMCD en un plazo de dos años en pacientes con inicio monofocal fue 47% para placebo y 24% para Extavia, sin contraste con gadolinio (Gd) 41% y 20%, con menos de 9 lesiones en T2 39% y 18%. Los análisis adicionales de los subgrupos indicaron un alto riesgo de progresión a EMCD en 2 años en pacientes monofocales con, al menos, 9 lesiones en T2 (55% de riesgo para placebo, 26% para Extavia) o captación de Gd (63% frente a 33%). En pacientes multifocales, el riesgo de EMCD fue independiente de los hallazgos de la RM basales, indicando un alto riesgo de EMCD debido a la diseminación de la enfermedad basada en los hallazgos clínicos. Sin embargo, se desconoce el impacto a largo plazo del tratamiento precoz con Extavia, incluso en estos subgrupos de alto riesgo, ya que el estudio fue diseñado principalmente para evaluar el tiempo hasta la EMCD y no la evolución a largo plazo de la enfermedad. Además, en estos momentos no existe una definición bien establecida de paciente de alto riesgo, aunque una aproximación conservadora es aceptar, al menos, nueve lesiones hiperintensas en T2 en la resonancia inicial y, al menos, una nueva lesión en T2 o una nueva lesión captante de Gd en una resonancia de seguimiento realizada, al menos, 1 mes después de la resonancia inicial. En cualquier caso, el tratamiento sólo se debería plantear en pacientes clasificados como de alto riesgo.
El tratamiento con Extavia fue bien aceptado en el estudio en pacientes con un único acontecimiento clínico, como se constató por la elevada tasa de finalización del estudio (92,8% en el grupo de Extavia). Para aumentar la tolerabilidad de Extavia en el estudio en pacientes con un primer acontecimiento clínico, se procedió a un ajuste de la dosis y se administraron medicamentos antiinflamatorios no esteroídicos al inicio del tratamiento. Además, se utilizó un autoinyector en la mayoría de los pacientes a lo largo del estudio.
EM RR, EM SP y acontecimiento único desmielinizante sugestivo de EM En todos los estudios de esclerosis múltiple Extavia fue eficaz reduciendo la actividad de la enfermedad (inflamación aguda en el sistema nervioso central y alteraciones permanentes de los tejidos), según las determinaciones realizadas mediante imágenes de resonancia magnética (RM). Actualmente no se conoce de modo completo la relación entre la actividad patológica de la esclerosis múltiple determinada por RM y el resultado clínico.
5.2 Propiedades farmacocinéticas
Los niveles séricos de Extavia se determinaron en pacientes y voluntarios sanos mediante un bioensayo que no era completamente específico, detectándose valores máximos en suero de aproximadamente 40 UI/ml en el período de 1-8 horas tras la inyección subcutánea de 500 microgramos (16,0 millones de UI) de Interferón beta-1b. Las tasas medias de aclaramiento sérico y los valores de las semividas de las fases de eliminación en suero se han estimado, a partir de varios estudios, en no más de 30 mlmin'1kg'1 y de 5 horas, respectivamente.
La administración de las inyecciones de Extavia en días altemos no da lugar a elevación de la concentración sérica del fármaco y la farmacocinética no parece modificarse durante el tratamiento.
La biodisponibilidad absoluta de Interferón beta-1b en administración subcutánea fue aproximadamente del 50%.
5.3 Datos preclínicos sobre seguridad
No se han realizado estudios de toxicidad aguda. Puesto que el Interferón beta humano no es activo en los roedores, los estudios de administración repetida se efectuaron en monos rhesus. Se observó hipertermia transitoria, así como un aumento significativo de los linfocitos y un descenso significativo de plaquetas y neutrófilos segmentados.
No se han realizado estudios a largo plazo. Los estudios sobre reproducción en monos rhesus revelaron toxicidad materna y fetal, originando mortalidad prenatal. No se observaron malformaciones en los animales supervivientes.
No se han realizado investigaciones sobre la fertilidad. No se ha observado influencia alguna sobre el ciclo estral en monos. La experiencia con otros interferones sugiere un deterioro potencial de la fertilidad de machos y hembras.
En un único estudio de genotoxicidad (test de Ames) no se observó efecto mutagénico. No se han realizado estudios de carcinogénesis. Un ensayo de transformación celular in vitro no mostró indicios de potencial tumorigénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo
Albúmina humana Manitol (E421)
Disolvente Cloruro de sodio
Agua para preparaciones inyectables
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros, excepto con el disolvente que lo acompaña mencionado en sección 6.6.
6.3 Periodo de validez
2 años.
Se recomienda el uso inmediato del producto tras su reconstitución. No obstante, se ha demostrado su estabilidad durante 3 horas, a 2°C - 8°C.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 25°C.
No congelar.
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Polvo
Vial (vidrio transparente tipo I) de 3 ml con un tapón de caucho butílico (tipo I) y cierre de cápsula de aluminio conteniendo 300 microgramos (9,6 millones de IU) de polvo (interferon beta-1b recombinante).
Disolvente
Jeringa precargada graduada (con marcas de dosis de: 0,25 ml, 0,5 ml, 0,75 ml, 1,0 ml) de 2,25 ml (vidrio tipo I) con 1,2 ml de disolvente.
Tamaños de envase
- Envase conteniendo 5 viales con polvo y 5 jeringas precargadas con disolvente.
- Envase conteniendo 14 viales con polvo y 14 jeringas precargadas con disolvente.
- Envase conteniendo 15 viales con polvo y 15 jeringas precargadas con disolvente.
- Envase conteniendo 14 viales con polvo y 15 jeringas precargadas con disolvente.
- Multienvase para 3 meses conteniendo 42 (3x14) viales con polvo y 42 (3x14) jeringas
precargadas con disolvente.
- Multienvase para 3 meses conteniendo 45 (3x15) viales con polvo y 45 (3x15) jeringas precargadas con disolvente.
- Multienvase para 3 meses conteniendo 42 (3x14) viales con polvo y 45 (3x15) jeringas precargadas con disolvente.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
La cápsula de cierre de la jeringa precargada contiene un derivado de látex de caucho natural. Por lo tanto, la cápsula de cierre puede contener látex de caucho natural y no debe ser manipulada por personas sensibles a esta sustancia.
Reconstitución
Para reconstituir el polvo, utilizar la jeringa precargada con disolvente que se suministra con una aguja o un adaptador para el vial para inyectar los 1,2 ml de disolvente (solución de cloruro de sodio con 5,4 mg/ml (0,54%) solución inyectable) en el vial con Extavia. Disolver completamente el polvo sin agitar. Después de la reconstitución, extraiga 1,0 ml del vial con la jeringa para administrar 250 microgramos de Extavia.
Inspección antes de su empleo
Inspeccionar la solución reconstituida, antes de su empleo. El producto reconstituido oscila entre incoloro y amarillo claro y entre ligeramente opalescente y opalescente.
Desechar el medicamento si contiene partículas o está coloreado.
Eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/454/008-014
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 20 Mayo 2008 Fecha de la última renovación: 20 Mayo 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINSTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante del principio activo biológico
Boehringer Ingelheim RCV GmbH & Co KG Dr.-Boehringer-Gasse 5-11 A-1121 Viena Austria
Nombre y dirección del fabricante responsable de la liberación de los lotes
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nuremberg Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos. El ciclo de IPS para Extavia está en línea con el medicament de referencia, Betaferon, hasta que el CHMP acuerde algo distinto.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Extavia 250 microgramos/ml, Polvo y disolvente para solución inyectable interferón beta-1b
2. PRINCIPIO(S) ACTIVO(S)
1 vial contiene 300 microgramos (9,6 millones de IU) de interferon beta-1b.
1 ml contiene 250 microgramos (8,0 millones de UI) de interferón beta-1b cuando se reconstituye.
3. LISTA DE EXCIPIENTES
Excipientes:
Polvo: Albúmina humana, manitol.
Disolvente: Cloruro de sodio, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable.
5 viales con polvo y 5 jeringas precargadas con 1,2 ml de disolvente.
14 viales con polvo y 14 jeringas precargadas con 1,2 ml de disolvente.
15 viales con polvo y 15 jeringas precargadas con 1,2 ml de disolvente.
14 viales con polvo y 15 jeringas precargadas con 1,2 ml de disolvente.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Para vía subcutánea tras reconstitución con 1,2 ml de disolvente. Un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD
Se recomienda utilizar el producto inmediatamente después de su reconstitución. Estabilidad demostrada durante 3 horas, a 2°C - 8°C.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 25°C. No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
|
EU/1/08/454/008 |
15 viales con polvo y 15 jeringas precargadas con disolvente |
|
EU/1/08/454/010 |
5 viales con polvo y 5 jeringas precargadas con disolvente |
|
EU/1/08/454/011 EU/1/08/454/013 |
14 viales con polvo y 14 jeringas precargadas con disolvente 14 viales con polvo y 15 jeringas precargadas con disolvente |
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
1. NOMBRE DEL MEDICAMENTO
Extavia 250 microgramos/ml, Polvo y disolvente para solución inyectable interferón beta-1b
2. PRINCIPIO(S) ACTIVO(S)
1 vial contiene 300 microgramos (9,6 millones de IU) de interferón beta-1b.
1 ml contiene 250 microgramos (8,0 millones de UI) de interferón beta-1b cuando se reconstituye.
3. LISTA DE EXCIPIENTES
Excipientes:
Polvo: Albúmina humana, manitol.
Disolvente: Cloruro de sodio, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
Multienvase para 3 meses: 42 (3 envases de 14) viales con polvo y 42 (3 envases de 14) jeringas precargadas con 1,2 ml de disolvente.
|
precargadas con 1,2 ml de disolvente. | |
|
Multienvase para 3 meses: 42 (3 envases de 14) viales con polvo y 45 (3 envases de 15) jeringas | |
|
precargadas con 1,2 ml de disolvente. | |
Multienvase para 3 meses 45 (3 envases de 15) viales con polvo y 45 (3 envases de 15) jeringas
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Para vía subcutánea tras reconstitución con 1,2 ml de disolvente. Un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD
Se recomienda utilizar el producto inmediatamente después de su reconstitución. Estabilidad demostrada durante 3 horas, a 2°C - 8°C.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 25°C. No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
|
EU/1/08/454/009 |
Multienvase para 3 meses compuesto de 45 viales con polvo y 45 jeringas | |
|
precargadas con disolvente | ||
|
EU/1/08/454/012 |
Multienvase para 3 meses compuesto de 42 viales con polvo y 42 jeringas | |
|
precargadas con disolvente | ||
|
EU/1/08/454/014 |
Multienvase para 3 meses compuesto de 42 viales con polvo y 45 jeringas | |
precargadas con disolvente
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
1. NOMBRE DEL MEDICAMENTO
Extavia 250 microgramos/ml, Polvo y disolvente para solución inyectable interferón beta-1b
2. PRINCIPIO(S) ACTIVO(S)
1 vial contiene 300 microgramos (9,6 millones de IU) de interferón beta-1b.
1 ml contiene 250 microgramos (8,0 millones de UI) de interferón beta-1b cuando se reconstituye.
3. LISTA DE EXCIPIENTES
Excipientes:
Polvo: Albúmina humana, manitol.
Disolvente: Cloruro de sodio, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
14 viales con polvo y 14 jeringas precargadas con 1,2 ml de disolvente. Componente de un multienvase para 3 meses. No se venden separadamente.
15 viales con polvo y 15 jeringas precargadas con 1,2 ml de disolvente. Componente de un multienvase para 3 meses. No se venden separadamente.
14 viales con polvo y 15 jeringas precargadas con 1,2 ml de disolvente. Componente de un multienvase para 3 meses. No se venden separadamente.
5. FORMA Y VÍA DE ADMINISTRACIÓN
Para vía subcutánea tras reconstitución con 1,2 ml de disolvente. Un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD
Se recomienda utilizar el producto inmediatamente después de su reconstitución. Estabilidad demostrada durante 3 horas, a 2°C - 8°C.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 25°C. No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
|
EU/1/08/454/009 |
Multienvase para 3 meses compuesto de 45 viales con polvo y 45 jeringas | |
|
precargadas con disolvente | ||
|
EU/1/08/454/012 |
Multienvase para 3 meses compuesto de 42 viales con polvo y 42 jeringas | |
|
precargadas con disolvente | ||
|
EU/1/08/454/014 |
Multienvase para 3 meses compuesto de 42 viales con polvo y 45 jeringas | |
precargadas con disolvente
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
ETIQUETA DEL VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Extavia 250 microgramos/ml, Polvo para solución inyectable interferón beta-1b Vía subcutánea.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
Se recomienda utilizar el producto inmediatamente después de su reconstitución. Estabilidad demostrada durante 3 horas, a 2°C - 8°C.
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
250 microgramos (8,0 millones de UI) por ml tras la reconstitución.
6. OTROS
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS
BLISTER DE JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO
Disolvente para la reconstitución de Extavia
1,2 ml de solución de cloruro de sodio con 5,4 mg/ml
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
Leer el prospecto antes de utilizar este medicamento.
ETIQUETA DE JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Disolvente para Extavia
Para vía subcutánea tras reconstitución.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
1,2 ml de solución de cloruro de sodio con 5,4 mg/ml.
6. OTROS
0,25 / 0,5 / 0,75 / 1,0
B. PROSPECTO
Prospecto: información para el usuario
Extavia 250 microgramos/ml, polvo y disolvente para solución inyectable
Interferón beta-1b
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas auque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Extavia y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Extavia
3. Cómo usar Extavia
4. Posibles efectos adversos
5. Conservación de Extavia
6. Contenido del envase e información adicional Anexo - Procedimiento para la auto-inyección
1. Qué és Extavia y para qué se utiliza Qué es Extavia
Extavia es un tipo de medicamento conocido como interferón, utilizado en el tratamiento de la esclerosis múltiple. Los interferones son proteínas producidas por el organismo que le ayudan a combatir contra los ataques al sistema inmunitario, tales como las infecciones víricas.
Cómo actúa Extavia
La esclerosis múltiple (EM) es un trastorno a largo plazo que afecta al sistema nervioso central (SNC), en particular al funcionamiento del cerebro y de la médula espinal. En la EM, la inflamación destruye la cubierta protectora (llamada mielina) que envuelve los nervios del SNC e impide que los nervios funcionen correctamente. A esto se le llama desmielinización.
La causa exacta de la EM se desconoce. Se piensa que en el proceso que daña el SNC juega un importante papel la aparición de una respuesta anormal por parte del sistema inmunitario.
El daño al SNC puede ocurrir durante un ataque de EM (recaída). Puede causar una incapacidad temporal, como dificultad para caminar. Los síntomas pueden desaparecer completa o parcialmente.
Se ha demostrado que el interferón beta-1b cambia la respuesta del sistema inmunitario y contribuye a reducir la actividad de la enfermedad.
Cómo ayuda Extavia a combatir su enfermedad
Episodio clinico único, sugestivo de un alto riesgo de desarrollo de esclerosis múltiple: Se ha
mostrado que Extavia retrasa la progresión hacia una esclerosis multiple definida.
Esclerosis múltiple remitente-recidivante: Las personas con EM remitente-recidivante tienen ataques ocasionales o recaídas, durante las cuales los síntomas empeoran considerablemente. Extavia ha demostrado reducir el número de ataques y hacerlos menos graves, reduce el número de estancias hospitalarias debidas a la enfermedad y prolonga el tiempo sin recaídas.
Esclerosis múltiple secundaria y progresiva: En algunos casos, las personas con EM remitente y recidivante notan que sus síntomas aumentan y progresan hacia otra forma de EM llamada EM secundaria progresiva. En esta forma, las personas se sienten cada vez más discapacitadas, tengan o no tengan recaídas. Extavia puede reducir el número y la severidad de los ataques, y retrasar la progresión de la discapacidad.
Para qué se utiliza Extavia Extavia es para uso en pacientes
► Que han sufrido por primera vez síntomas que indiquen un alto riesgo de desarrollar esclerosis múltiple. Su médico descartará otras causas que pudieran explicar estos síntomas antes de administrarle el tratamiento.
► Que sufren esclerosis múltiple remitente y recidivante, con la aparición de al menos dos recaídas en los dos años anteriores.
► Que sufren esclerosis múltiple secundaria y progresiva, con enfermedad activa demostrada por la aparición de recaídas.
2. Qué necesita saber antes de empezar a usar Extavia No use Extavia
- Si es alérgico al interferón beta natural o recombinante, a la albúmina humana o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- Si está embarazada. No debe iniciar el tratamiento con Extavia (ver la sección inferior «Embarazo»).
- Si se queda o planea quedarse embarazada. Debe interrumpir su tratamiento con Extavia e informar a su médico (ver la sección inferior «Embarazo»).
- Si padece actualmente depresión grave y/o ideas suicidas (ver «Advertencias y precauciones» y sección 4 «Posibles efectos adversos»).
- Si tiene una enfermedad hepática grave.(ver «Advertencias y precauciones », «Uso de Extavia con otros medicamentos» y sección 4 «Posibles efectos adversos»).
► Informe a su médico, si algo de lo anterior es de aplicación en su caso.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar Extavia:
- Si padece gammapatía monoclonal, una enfermedad del sistema inmunitario en la que se encuentra una proteína anómala en la sangre. Pueden aparecer problemas (síndrome de extravasación capilar sistémica) en los vasos sanguíneos de menor tamaño (capilares) al utilizar medicamentos como Extavia. Esto puede llegar a originar un shock (colapso) e incluso ser mortal.
- Si ha tenido o tiene depresión o anteriormente ha tenido ideas de suicidio. Su médico le vigilará de cerca durante el tratamiento. Si su depresión y/o ideas suicidas son graves, no se le recetará Extavia (ver también «No use Extavia»).
- Si ha sufrido convulsiones en alguna ocasión, o si está usando medicamentos para tratar la epilepsia fanti-epilépticosj, su médico vigilará atentamente su tratamiento (ver también «Uso de Extavia con otros medicamentos» y sección 4 «Posibles efectos adversos»).
- Si tiene problemas graves de riñón, su médico puede vigilar su función renal durante el tratamiento.
- Si ha tenido una reacción alérgica al látex en alguna ocasión. La cápsula de cierre de la jeringa precargada contiene un derivado de látex de caucho natural. Por lo tanto, la cápsula de cierre puede contener látex de caucho natural.
Su médico también debe conocer las siguientes circunstancias mientras está usando Extavia:
- Si experimenta síntomas tales como picor por todo el cuerpo, hinchazón de la cara y/o la lengua, o dificultad respiratoria repentina. Pueden ser síntomas de una reacción alérgica grave, que podría convertirse en mortal.
- Si se siente considerablemente más triste o desesperanzado que antes de iniciar el tratamiento con Extavia, o si presenta ideas de suicidio. Si se deprime mientras está en tratamiento con Extavia, puede que necesite tratamiento especial, y su médico le vigilará de cerca y puede considerar también interrumpir su tratamiento. Si padece depresión grave y/o ideas suicidas, no será tratado con Extavia (ver también «No use Extavia»).
- Si nota que le salen cardenales con facilidad, que sangra demasiado cuando tiene heridas, o que contrae muchas infecciones. Pueden ser síntomas de un descenso del recuento de células sanguíneas o del número de plaquetas de la sangre (células que contribuyen a la coagulación de la sangre). Puede necesitar un seguimiento más estrecho por parte de su médico.
- Si experimenta pérdida de apetito, cansancio, mareos (náuseas), vómitos repetidos, y especialmente si nota picor difuso, la aparición de un color amarillento en la piel o en la parte blanca de los ojos o que le salen cardenales fácilmente. Estos síntomas pueden indicar problemas con su hígado. En algunos estudios clínicos se han observado cambios en los valores de la función del hígado en pacientes tratados con Extavia. Al igual que con otros interferones beta, en los pacientes que toman Extavia se ha comunicado, en raras ocasiones, un daño hepático grave, que incluye casos de insuficiencia hepática. Los más graves se comunicaron en los pacientes que tomaban otros medicamentos o que padecían enfermedades que pueden afectar al hígado (p.ej., abuso del alcohol, infección grave).
- Si experimenta síntomas tales como irregularidades en los latidos de su corazón o hinchazón en los tobillos o en las piernas, o dificultad respiratoria. Esto puede indicar una enfermedad del músculo del corazón (miocardiopatía), que se ha comunicado en pacientes que usaban Extavia.
- Si nota dolor en el vientre que irradia hacia la espalda, o si se marea o tiene fiebre. Esto puede indicar una inflamación del páncreas (pancreatitis), que se ha comunicado con el uso de Extavia. Esto se asocia con frecuencia a un aumento de ciertas grasas en sangre (triglicéridos).
► Interrumpa el uso de Extavia e informe a su médico inmediatamente si le ocurre cualquiera de ellos.
Otras cosas a tener en cuenta al usar Extavia:
- Será necesario un análisis de sangre para determinar el número de sus células sanguíneas, la bioquímica de la sangre y las enzimas hepáticas. Esto se hará antes de que comience a usar Extavia, regularmente después de haber iniciado el tratamiento con Extavia y después, durante el tratamiento, periódicamente, incluso si no tiene síntomas específicos. Estos análisis de sangre se harán además de los análisis que se realizan normalmente para vigilar su EM.
- Si tiene una enfermedad cardiaca, los síntomas de tipo gripal que con frecuencia ocurren al inicio del tratamiento pueden suponer una sobrecarga para usted. Extavia debe usarse con precaución, y su médico vigilará un posible empeoramiento de su patología cardiaca, sobre todo al inicio del tratamiento. Extavia por sí mismo no afecta al corazón directamente.
- Se le realizará una comprobación de la función de la glándula tiroides, regularmente o siempre que su médico lo considere necesario por otros motivos.
- Extavia contiene albúmina humana y, por lo tanto, conlleva un riesgo potencial de transmisión de enfermedades víricas. No se puede descartar un riesgo de transmisión de la enfermedad de Creutzfeld-Jacob (ECJ).
- Durante el tratamiento con Extavia, su organismo puede producir sustancias llamadas anticuerpos neutralizantes, los cuales pueden reaccionar con Extavia. No está claro si estos anticuerpos neutralizantes reducen la eficacia del tratamiento. Los anticuerpos neutralizantes no se producen en todos los pacientes. Actualmente no es posible predecir qué pacientes pertenecen a este grupo.
- Durante el tratamiento con Extavia, puede tener problemas de riñón que reduzcan la función renal, incluyendo esclerosis (glomeruloesclerosis). Su médico puede hacerle pruebas para comprobar su función renal.
- Durante el tratamiento puede tener coágulos de sangre en los vasos sanguíneos pequeños.
Estos coágulos de sangre pueden afectar a sus riñones. Esto puede ocurrir de varias semanas a varios años después de empezar a usar Extavia. Su médico puede hacerle pruebas para comprobar su presión sanguínea, sangre (recuento de plaquetas) y la función de los riñones.
Reacciones en el lugar de la inyección
Durante el tratamiento con Extavia, es probable que experimente reacciones en el lugar de la inyección. Los síntomas consisten en enrojecimiento, hinchazón, cambio en el color de la piel, inflamación, dolor e hipersensibilidad. Con menos frecuencia, se observan piel estropeada y daño de tejido (necrosis) alrededor del lugar de la inyección. Las reacciones en el lugar de la inyección suelen volverse menos frecuentes con el tiempo.
La rotura de la piel y su destrucción pueden dar como resultado la formación de cicatrices. Si éstas son graves, un médico puede tener que realizar una eliminación de materia extraña y tejido muerto (desbridamientoj y, con menor frecuencia, un injerto de piel, pudiendo tardar la curación hasta 6 meses.
Para reducir el riesgo de tener una reacción en el lugar de la inyección debe:
- Usar una técnica de inyección estéril (aséptica/
- Cambiar el lugar de la inyección después de cada inyección (ver el anexo Procedimiento para la auto-inyección).
Las reacciones en el lugar de la inyección pueden ocurrir con menos frecuencia si utiliza un dispositivo auto-inyector. Su médico o enfermero puede informarle acerca de esto.
Si sufre una rotura de la piel asociada a hinchazón o pérdida de líquidos por el lugar de inyección:
► Interrumpa la inyección de Extavia e informe a su médico
► Si presenta un único lugar de la inyección ulcerado (lesión) y la destrucción del tejido (necrosis) no es demasiado extensa, puede continuar usando Extavia.
► Si presenta más de un lugar de la inyección ulcerado (lesiones múltiples) debe dejar de usar Extavia hasta que su piel se haya recuperado.
Su médico comprobará regularmente la manera en que usted se autoinyecta, sobre todo si ha sufrido reacciones en el lugar de inyección.
Niños y adolescentes
No se han realizado ensayos clínicos formales en niños o adolescentes.
Sin embargo, se dispone de algunos datos en adolescentes de 12 a 17 años de edad que sugieren que el perfil de seguridad de Estavia en este grupo es el mismo que el de los adultos. Extavia no debe utilizarse en niños menores de 12 años, ya que no hay información disponible para este grupo de edad.
Uso de Extavia con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
No se han llevado a cabo estudios de interacción formales para averiguar si Extavia afecta a otros medicamentos o es afectado por ellos.
No se recomienda el uso de Extavia con otros medicamentos que modifican la respuesta del sistema inmunitario, excepto los medicamentos antiinflamatorios llamados corticosteroides o la hormona adrenocorticotrópica (ACTH).
Extavia debe utilizarse con precaución con:
- Los medicamentos que necesitan un determinado sistema enzimático hepático para su eliminación del organismo (conocido como sistema citocromo P450), por ejemplo, los medicamentos usados para tratar la epilepsia (como la fenitoina).
- Los medicamentos que afectan a la producción de las células sanguíneas.
Uso de Extavia con alimentos y bebidas
Extavia se inyecta bajo la piel, por lo que no se considera que ninguna comida o bebida que usted consuma tendrá algún efecto sobre Extavia.
Embarazo
Si se puede quedar embarazada, use medidas anticonceptivas adecuadas durante el tratamiento con Extavia.
► Si está embarazada o piensa que puede estar embarazada, informe a su médico. El tratamiento con Extavia no se debe iniciar si está embarazada (ver también «No use Extavia»).
► Si desea quedarse embarazada, antes de iniciar el tratamiento con Extavia lo debe discutir primero con el médico.
► Si se queda embarazada mientras utiliza Extavia, debe suspender el tratamiento y contactar con su médico inmediatamente. Usted y su médico decidirán si continuar su tratamiento con Extavia.
Consulte a su médico o farmacéutico antes de utilizar este medicamento.
Lactancia
No se sabe si el interferón beta-1b pasa a la leche materna humana. Sin embargo, teóricamente sería posible que Extavia cause efectos adversos en un lactante.
► Antes de iniciar el tratamiento con Extavia coméntelo primero con su médico, para decidir si interrumpe la lactancia natural para poder utilizar Extavia.
Consulte a su médico o farmacéutico antes de utilizar este medicamento.
Conducción y uso de máquinas
Extavia puede causar efectos adversos en el sistema nervioso central (ver sección 4. «Posibles efectos adversos»). Si es especialmente sensible, esto puede influir sobre su capacidad para conducir o utilizar máquinas.
Extavia contiene sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por ml; esto es, esencialmente «exento de sodio».
3. Cómo usar Extavia
El tratamiento con Extavia debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la esclerosis múltiple.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
La dosis recomendada es en días alternos (una vez cada dos días) 1,0 ml de la solución preparada se inyecta bajo la piel (subcutáneamente) (ver el Anexo «Procedimiento para la auto-inyección» en la segunda parte del prospecto). Esto equivale a 250 microgramos (8,0 millones de UI) de interferón beta-1b.
En general, el tratamiento debe iniciarse con una dosis baja de 0,25 ml (62,5 microgramos). Sus dosis irán aumentando gradualmente hasta la dosis completa de 1,0 ml (250 microgramos).
La dosis debe ir aumentando cada cuatro inyecciones en cuatro pasos (0,25 ml; 0,5 ml; 0,75 ml;
1,0 ml). Su médico puede decidir junto con usted cambiar los intervalos de tiempo para el aumento de la dosis dependiendo de los efectos adversos que pueda sufrir al inicio del tratamiento.
Preparación de la inyección
Antes de la inyección, se prepara la solución para inyección de Extavia a partir de un vial de Extavia polvo y 1,2 ml de líquido de una de las jeringas precargadas de disolvente. Esto lo hará su médico ó enfermera/o, incluso usted mismo, una vez que se le haya instruido de manera cuidadosa en la técnica.
Se incluyen instrucciones detalladas para la auto-inyección de Extavia debajo de la piel en el
Anexo al dorso de este prospecto en las que también se explica cómo preparar la solución de Extavia para inyección.
Hay que cambiar el lugar de inyección regularmente. Ver sección 2 «Advertencias y precauciones» y seguir las instrucciones que se indican en el apartado «Rotación de los lugares de inyección» en el Anexo al dorso de este prospecto.
Duración del tratamiento
A día de hoy, no se conoce durante cuánto tiempo debe ser tratado el paciente. La duración del tratamiento debe decidirla el médico junto con usted.
Si usa más Extavia del que debe
La administración de dosis de Extavia muy superiores a las recomendadas para la esclerosis múltiple no ha dado lugar a situaciones que ponen en peligro la vida.
► Informe a su médico si se ha inyectado demasiado Extavia o lo ha inyectado con demasiada frecuencia.
Si olvidó usar Extavia
Si ha olvidado administrarse una inyección en el horario previsto, hágalo tan pronto como sea posible y continúe con la siguiente 48 horas más tarde.
No inyecte una dosis doble para compensar las dosis olvidadas.
Si interrupe el tratamiento con Extavia
Hable con su médico si interrumpe o desea interrumpir el tratamiento. Se desconoce si la interrupción de Extavia causa síntomas agudos de abstinencia.
► Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Extavia puede causar efectos adversos graves. Si considera que alguno de los efectos adversos que sufre es grave o si aprecia cualquier efecto adverso no mencionado en este prospecto, informe a su médico, farmacéutico o enfermero.
► Informe a su médico inmediatamente e interrumpa el uso de Extavia:
- Si experimenta síntomas tales como picor por todo el cuerpo, hinchazón de la cara y/o la lengua, o dificultad respiratoria repentina.
- Si se siente considerablemente más triste o desesperanzado que antes de iniciar el tratamiento con Extavia, o si tiene ideas de suicidio.
- Si nota que le salen cardenales con facilidad, que sangra demasiado cuando tiene heridas, o que contrae muchas infecciones.
- Si tiene pérdida de apetito, cansancio, mareos (náuseas), vómitos repetidos, especialmente si nota picor difuso, la aparición de un color amarillento en la piel ,o en la parte blanca de los ojos, o que salgan cardenales fácilmente.
- Si presenta síntomas como irregularidades en los latidos de su corazón o hinchazón en los tobillos o las piernas, o dificultad respiratoria.
- Si nota dolor en el vientre que irradia hacia la espalda, o si se marea o tiene fiebre.
► Informe a su médico inmediatamente:
- Si tiene alguno de estos síntomas o todos: orina espumosa, cansancio, hinchazón, especialmente en los tobillos y párpados, aumento de peso, ya quie podrían ser signos de un posible problema renal.
Al iniciar el tratamiento son habituales los efectos adversos, pero en general disminuyen al seguir con él.
Los efectos adversos más frecuentes son:
► Un complejo de síntomas de tipo gripal, como fiebre, escalofríos, dolor en las articulaciones, malestar, sudores, dolor de cabeza o dolor muscular.Estos síntomas se pueden reducir tomando paracetamol o medicamentos antiinflamatorios no esterodeos, como el ibuprofeno
► Reacciones en el lugar de inyección. Los síntomas pueden ser enrojecimiento, hinchazón, decoloración, inflamación, dolor, hipersensibilidad, daño de tejido (necrosis). Ver «Advertencias y precauciones» en la sección 2 para más información y qué hacer si experimenta una reacción en el lugar de la inyección. Éstas se pueden reducir mediante el uso de un dispositivo auto-inyector. Hable con su médico, farmacéutico o enfermero para información adicional.
Para reducir el riesgo de efectos adversos al inicio del tratamiento, su médico debe comenzar con una dosis baja de Extavia y aumentarla gradualmente (ver sección 3 «Cómo usar Extavia»).
La lista de efectos adversos a continuación está basada en las comunicaciones de los ensayos clínicos con Extavia (Lista 1) y en los efectos adversos comunicados del producto comercializado (Lista 2).
Lista 1: Efectos adversos muy frecuentes que han ocurrido en los ensayos clínicos con Extavia
(pueden afectar a más de 1 de cada 10 personas) y en un porcentaje mayor a los que se observaron con placebo. La lista también incluye los efectos adversos que ocurrieron frecuentemente (pueden afectar hasta 1 de cada 10 personas) pero que estuvieron significativamente asociados con el tratamiento:
- infección, absceso
- reducción de glóbulos blancos, hinchazón de los ganglios linfáticos (linfoadenopatía)
- disminución de la cantidad de glucosa en la sangre (hipoglicemia)
- depresión, ansiedad
- dolor de cabeza, mareo, insomnio, migraña, sensación de sueño u hormigueos (parestesia)
- inflamación del ojo (conjuntivitis), visión anormal
- dolor de oído
- latidos más rápidos e irregulares, o palpitaciones del corazón (palpitación)
- enrojecimiento y/o eritema facial debido a la dilatación de los vasos sanguíneos (vasodilatación), aumento de la presión arterial (hipertensión)
- goteo nasal, tos, ronquera debido a infección de las vías respiratorias altas, sinusitis, aumento de la tos, dificultad respiratoria (disnea)
- diarrea, estreñimiento, náuseas, vómitos, dolor abdominal
- aumentos en los niveles en sangre de las enzimas hepáticas que se revelan en análisis de sangre)
- trastorno de la piel, erupción cutánea
- rigidez muscular (hipertonía), dolor muscular (mialgia), debilidad muscular (miastenia),dolor de espalda, dolor en las extremidades como en los dedos de las manos y los pies
- retención urinaria, aparición de proteínas en la orina (se revelará en el análisis de orina) frecuencia urinaria, incapacidad para contener la micción (incontinencia urinaria), urgencia urinaria
- reglas (menstruación) dolorosas (dismenorrea), trastorno menstrual,
hemorragia uterina abundante (metrorragia), especialmente entre periodos menstruales, impotencia
- reacciones en el lugar de inyección (entre las que están enrojecimiento, hinchazón, decoloración, inflamación, dolor, reacción alérgica; ver sección 2 «Advertencias y precauciones»), rotura de la piel y daño del tejido (necrosis) en el lugar de inyección (ver sección 2 «Advertencias y precauciones»)
- síntomas de tipo gripal, fiebre, dolor, dolor en el pecho, acumulación de líquidos en el brazo, la pierna o la cara (edema periférico), falta/pérdida de fuerza (astenia), escalofríos, sudoración, sensación de malestar general
Además, las siguientes reacciones adversas han sido identificadas durante la experiencia postcomercialización.
Lista 2: Reacciones adversas notificadas del producto comercializado (las frecuencias - cuando
son conocidas - se basan en los ensayos clínicos).
► Muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes):
- dolor en articulaciones (artralgia).
► Frecuentes (pueden afectar hasta 1 de cada 10 pacientes):
- puede descender el número de glóbulos rojos en la sangre (anemia),
- la glándula tiroides no funciona adecuadamente (se produce demasiado poca hormona) (hipotoriodismo),
- aumento o disminución de peso,
- confusión,
- latido del corazón anormalmente rápido (taquicardia),
- puede aparecer un pigmento amarillo rojizo (bilirrubina) producido por el hígado (esto se revelará en el análisis de sangre),
- manchas cutáneas o membranas mucosas elevadas, edematosas y que suelen picar (urticaria),
- picor (prurito),
- caída del cabello (alopecia),
- trastornos menstruales (menorragia).
► Poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes):
- puede descender el número de plaquetas (que contribuyen a que la sangre coagule) (trombocitopenia),
- puede aumentar una fracción de las grasas de la sangre (triglicéridos), (se revelará en el análisis de sangre) ver sección 2 «Advertencias y precauciones»,
- intento de suicidio,
- inestabilidad emocional,
- convulsiones,
- puede aumentar una enzima específica del hígado (gamma GT) producida por el hígado (esto se revelará en el análisis de sangre),
- inflamación del hígado (hepatitis),
- decoloración de la piel.
► Raras (pueden afectar hasta 1 de cada 1.000 pacientes):
- reacciones alérgicas (anafilácticas) graves,
- la glándula tiroides no funciona correctamente (alteración tiroidea) se produce demasiada hormona (hipertiroidismo),
- inflamación del páncreas (pancreatitis), ver sección 2 «Advertencias y precauciones»,
- coágulos de sangre en los vasos sanguíneos pequeños que pueden afectar a sus riñones (púrpura trombocitopénica trombótica o síndrome hemolítico urémico). Los síntomas pueden incluir hematoma, sangrado, fiebre, debilidad extrema, dolor de cabeza, mareo o vahídos. Su médico puede encontrar cambios en su sangre o en la función de sus riñones.
Reacciones adversas derivadas solo durante la postcomercialización:
- problemas renales incluyendo esclerosis (glomeruloesclerosis) que puede reducir su función renal (poco frecuentes).
- pérdida de apetito grave que produce pérdida de peso (anorexia), (raras).
- enfermedad del músculo del corazón (miocardiopatía), (raras).
- dificultad respiratoria repentina (broncoespasmo), (raras).
- el hígado no funciona correctamente (daño hepático [incluyendo hepatitis], fallo hepático), (raras).
- se pueden desarrollar problemas en sus vasos sanguíneos pequeños (capilares) cuando se usan medicamentos como Extavia (síndrome de extravasación capilar sistémica), (frecuencia no conocida).
- erupción cutánea, enrojecimiento de la piel de la cara, dolor en las articulaciones, fiebre, debilidad y otros síntomas causados por el medicamento (lupus eritematoso inducido por el fármaco), frecuencia no conocida.
- gran estrechamiento de los vasos sanguíneos de los pulmones que provoca un aumento de la presión en los vasos sanguíneos que transportan la sangre del corazón a los pulmones (hipertensión arterial pulmonar), (frecuencia no conocida). La hipertensión arterial pulmonar se notificó en distintos momentos, incluso varios años después del inicio del tratamiento con Extavia.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Extavia
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase. La fecha de caducidad es el último día del mes que se indica.
No conservar a temperatura superior a 25°C. No congelar.
Debe usar inmediatamente la solución tras su preparación. No obstante, si no le es posible hacerlo, la solución se puede utilizar durante 3 horas, si se conserva en una nevera (2°C - 8°C).
No utilice este medicamento si observa que contiene partículas o si está decolorado.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Extavia
- El principio activo es interferón beta-1b. Cada vial contiene 300 microgramos (9,6 millones de UI) de interferón beta-1b por vial. Después de la reconstitución, cada mililitro contiene
250 microgramos (8,0 millones de UI) de interferón beta-1b.
- Los demás componentes son
- en el polvo: manitol y albúmina humana
- en el disolvente: cloruro de sodio, agua para preparaciones inyectables.
La cápsula de cierre de la jeringa precargada contiene un derivado de látex de caucho natural. Por lo tanto, la cápsula de cierre puede contener látex de caucho natural.
Aspecto del producto y contenido del envase
Extavia es un polvo y un disolvente para solución para inyección.
El polvo es de color blanco o casi blanco.
El polvo de Extavia se suministra en un vial de 3 mililitros.
El disolvente es una solución clara/incolora.
El disolvente para Extavia se suministra en una jeringa precargada de 2,25 ml y contiene 1,2 ml de cloruro de sodio 5,4 mg/ml (0,54% p/v solución para inyección.
Extavia se suministra en envases de:
- 5 viales de interferón beta-1b y 5 jeringas precargadas que contienen el disolvente.
- 14 viales de interferón beta-1b y 14 jeringas precargadas que contienen el disolvente.
- 15 viales de interferón beta-1b y 15 jeringas precargadas que contienen el disolvente.
- 14 viales de interferón beta-1b y 15 jeringas precargadas que contienen el disolvente.
beta-1b y 42 (3x14) beta-1b y 45 (3x15) beta-1b y 45 (3x15)
- Multienvase para 3 meses conteniendo 42 (3x14) viales con interferón jeringas precargadas que contienen el disolvente.
- Multienvase para 3 meses conteniendo 45 (3x15) viales con interferón jeringas precargadas que contienen el disolvente.
- Multienvase para 3 meses conteniendo 42 (3x14) viales con interferón jeringas precargadas que contienen el disolvente.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización.
Belgie/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Btnrapun
Novartis Pharma Services Inc. Ten.: +359 2 489 98 28
Ceská republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Lietuva
Novartis Pharma Services Inc. Tel: +370 5 269 16 50
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Eesti
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Novartis Pharma Services Inc. Tel: +372 66 30 810
|
EXXáóa Novartis (Hellas) A.E.B.E. Tr(k: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kórcpog Novartis Pharma Services Inc. Tr(k: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
|
Fecha de la última revisión de este prospecto: |
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
Las siguientes instrucciones e imágenes explican cómo debe preparar la solución de Extavia para inyección y cómo debe proceder para inyectársela usted mismo. Por favor, lea cuidadosamente las instrucciones y sígalas paso a paso. Su médico o enfermero le instruirá y adiestrará en el procedimiento y la técnica de autoadministración. No intente la autoadministración hasta estar seguro de haber comprendido cómo ha de preparar la solución para inyección y cómo ha de inyectársela.
PARTE I: INSTRUCCIONES PASO A PASO
Las instrucciones incluyen los siguientes pasos principales:
A) Consejos generales
B) Preparándose para inyectar
C) Reconstitución y preparación de la solución para inyección, paso a paso
D) Preparación para inyección manual (para realizar la inyección con el autoinyector
ExtaviPro 30G, ver las instrucciones de uso suministradas con el autoinyector)
A) Consejos generales
• ¡Empiece bien!
Comprobará que, en unas pocas semanas, el tratamiento formará parte de su vida. Para empezar, lo siguiente le puede ayudar:
- Tenga preparada permanentemente una zona apropiada en su casa, fuera de la vista y del alcance de los niños donde le sea fácil encontrar Extavia y los demás utensilios.
Consulte las condiciones de conservación en la sección 5 del prospecto, «Conservación de Extavia».
- Intente inyectarse cada día a la misma hora, ya que así le será más fácil recordarlo y reservarse un tiempo en el que no le interrumpan.
Consulte otros detalles sobre el uso de Extavia en la sección 3 del prospecto, «Cómo usar Extavia».
- Prepare cada dosis sólo cuando esté listo para inyectarse, ya que debe inyectarse inmediatamente después de reconstituir Extavia (si no usa inmediatamente este medicamento, consulte las instrucciones para guardarlo en la sección 5 del prospecto, «Conservación de Extavia»).
• Consejos importantes que debe tener en cuenta
- Sea constante. Use este medicamento tal como se explica en la sección 3 del prospecto, «Cómo usar Extavia». Compruebe siempre dos veces la dosis preparada.
- Mantenga el contenedor en el que elimina las jeringas y las propias jeringas fuera de la vista y del alcance de los niños. Cierre el material bajo llave, si es posible.
- No reutilice nunca ni las jeringas ni las agujas.
- Use siempre una técnica estéril (aséptica), como se explica a continuación.
- Deseche siempre las jeringas usadas sólo en el contenedor apropiado.
B) Preparándose para inyectar
• ¿Cómo elegir el lugar para la inyección?
Antes de preparar la inyección de Extavia, decida dónde se va a inyectar. Debe inyectarse este medicamento en la capa grasa, entre la piel y el músculo (es decir, en el tejido subcutáneo, entre 8 mm y 12 mm por debajo de la piel). Los mejores lugares para la inyección son aquellos en los que la piel es blanda y suave, y lejos de las articulaciones, los nervios y los huesos, por ejemplo, el abdomen, el brazo, el muslo o las nalgas.
Importante:
La cápsula de cierre de la jeringa precargada contiene un derivado de látex de caucho natural. Por lo tanto, la cápsula de cierre puede contener látex de caucho natural. Si es alérgico al látex, consulte a su médico antes de empezar a usar Extavia.
No se inyecte en las zonas en las que usted perciba bultos, hematomas, nódulos firmes, dolor o una zona en la cual la piel esté decolorada, deprimida, con costra o con una rotura abierta. Hable con su médico o enfermero acerca de éstas u otras condiciones poco habituales que pueda hallar.
Debe alternar el lugar de inyección cada vez que se inyecte siguiendo ciclos de rotación. Si alguna zona es demasiado difícil de alcanzar, es posible que algún familiar o un amigo tenga que ayudarle a administrarse esas inyecciones. Siga la secuencia que se describe en el esquema que se incluye al final del Anexo (ver Parte II «Rotación de los lugares de inyección») y volverá al primer lugar en que se inyectó después de 8 inyecciones (16 días). En ese tiempo, cada lugar de la inyección se recuperará completamente antes de recibir la siguiente inyección.
Consulte el esquema de rotaciones que se encuentra al final de este Anexo para saber cómo elegir el lugar de inyección. También se incluye un ejemplo de «Calendario para la administración» (ver Anexo Parte III). Con esto, tendrá una idea de cómo controlar los lugares y las fechas de sus inyecciones.
• Medicación
Necesitará la medicación:
• 1 vial de Extavia (con polvo para solución inyectable)
• 1 jeringa precargada con disolvente para Extavia (solución de cloruro de sodio)
Para reconstituir e inyectar su medicación necesitará utilizar el kit de aplicación ExtaviPro 30G (se proporciona por separado de su medicación), el cual contiene los siguientes componentes y las instrucciones de cómo utilizarlos:
• Adaptadores para el vial para utilizar cuando se reconstituya su medicación
• Agujas de calibre 30 para inyecctar su medicación
• Toallitas con alcohol
También necesitará un contenedor de desechos para eliminar las jeringas y las agujas usadas.
Las agujas de calibre 30 suministradas con el kit de aplicación para la administración de este medicamento pueden utilizarse para la inyección manual O con un autoinyector ExtaviPro 30G.
Utilice un desinfectante adecuado para desinfectar la piel recomendado por su farmacéutico.
1 - Lávese las manos cuidadosamente con agua y jabón antes de comenzar este proceso.
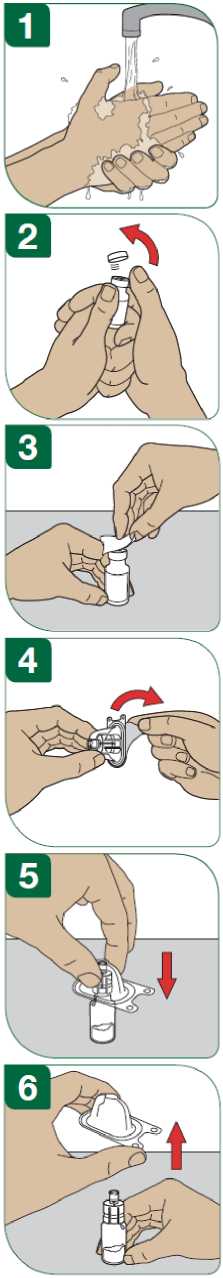
2 - Quite el tapón del vial de Extavia. Es mejor usar el pulgar que las uñas, porque sus uñas se podrían partir. Ponga el vial en la mesa.
3 - Limpie la parte superior del vial con una toallita con alcohol, moviéndola sólo en una dirección, Déjela apoyada encima del vial.
4 - Despegue y quite la cubierta de la envoltura del adaptador del vial. No quite el adaptador del vial de su envoltura.
5 - Quite el hisopo de algodón de la parte superior del vial. Utilize el envoltorio para manejar el adaptador del vial. Conéctelo al vial empujando hacia abajo hasta que el adaptador del vial penetre y se encaje en la parte superior del vial.
6 - Manteniendo los bordes de manera segura, quite y
deseche el envoltorio asegurándose de que el adaptador del vial permanece en el vial.
7 - Saque de su envase la jeringa precargada de disolvente. Desenganche y deseche la punta de la jeringa precargada. Nota: Tenga cuidado de no tocar el extremo expuesto del émbolo de la jeringa. No empuje el émbolo.
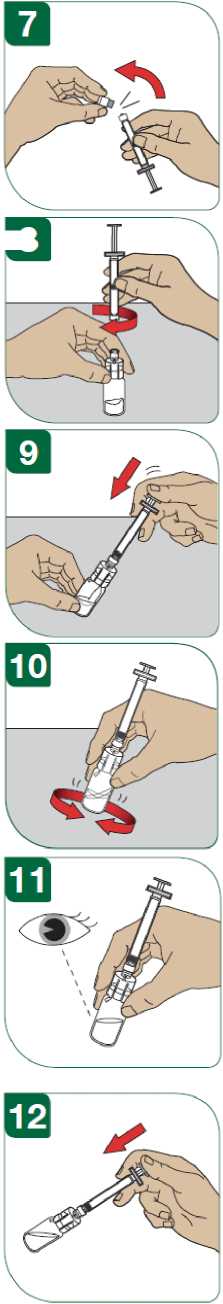
• •
8 - Mientras sostiene el vial de y el adaptador de manera segura, enrosque la jeringa completamente en el vial.
Así se obtiene el sistema jeringa-vial.
9 - Mantenga el sistema jeringa-vial ligeramente inclinado. Empuje lentamente el émbolo hacia abajo, para así permitir que el líquido penetre en el interior del vial.
Transfiera todo el disolvente hacia el vial.
Nota: No agite el vial ya que podría producir un exceso de espuma.
10 - Sostenga el vial entre sus dedos pulgar y corazón. Gire suavemente el sistema jeringa-vial hasta que el polvo esté completamente disuelto.
Nota: No agite el vial.
11 - Examine la solución cuidadosamente. Debe ser incolora (transparente) y no debe presentar partículas.
Nota: Si la solución está coloreada o tiene partículas, deséchela y comience de nuevo, utilizando un jeringa nueva y un vial nuevo del envase.
Si aparece excesiva espuma, lo que puede suceder cuando el vial se agita o se mueve en círculos demasiado vigorosamente, déjelo reposar hasta que desaparezca la espuma.
12 - Compruebe que el émbolo se mantiente completamente presionado antes de proceder con el siguiente paso, ya que podría haberse movido.
13 - Gire el sistema jeringa-vial, de modo que el vial se encuentre arriba. Tire lentamente del émbolo hacia atrás, para extraer toda la solución del vial a la jeringa.
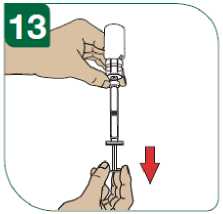
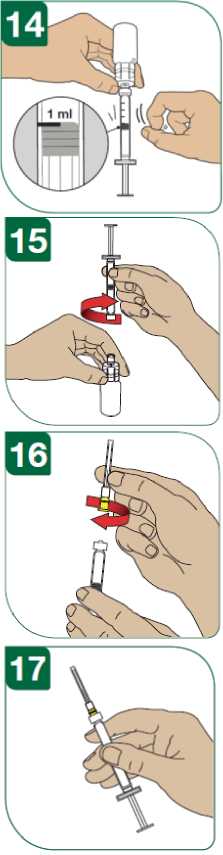
14 - Elimine cualquier exceso de burbujas de aire, dando unos suaves golpecitos a la jeringa. Empuje el émbolo hasta la marca de 1 ml, (o hasta el volumen recetado por su médico).
Nota: Puede ser necesario ajustar la posición del émbolo hacia atrás y hacerlo sucesivamente para comprobar que las burbujas de aire han desaparecido y que hay 1 ml de solución en la jeringa.
15 - Desenganche la jeringa, dejando el adaptador del vial en el vial.
Deseche el vial y la porción restante no utilizada de la solución en la unidad de desecho.
16 - Saque la aguja de su envoltura y enrósquela firmemente sobre la punta de la jeringa.
17 - Deje el tapón de la aguja puesto. Ya está listo para la inyección manual por usted mismo o para usar el auto-inyector ExtaviPro 30G para la administración de Extavia.
Conservación tras la reconstitución
Si por algún motivo, no puede inyectarse Extavia inmediatamente, puede mantener refrigerada la solución reconsituída hasta un máximo de 3 horas antes de utilizarla. No congele la solución, y no espere más de 3 horas para inyectarla. Si pasan más de 3 horas, deseche el medicamento y prepare una nueva inyección. Cuando utilice la solución, caliéntela manteniéndo la jeringa o vial en sus manos antes de inyectarla para evitar el dolor.
D) Preparación para inyección manual (para realizar la inyección con el autoinyector ExtaviPro 30G, ver las instrucciones de uso suministradas con el autoinyector)
1 - Elija un lugar para la inyección (ver sección ¿Cómo elegir el lugar para la inyección? y los diagramas al final de este prospecto) y anótela en su «Calendario para la administración».
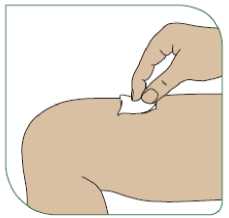

2 - Emplee una toallita con alcohol para limpiar la piel en el lugar de la inyección: Deje secar la piel al aire. Tire la toallita.
3 - Quite el capuchón de la aguja, tirando del mismo y sin girarlo.
4 - Donde sea posible pellizque suavemente la piel que rodea el punto de la inyección previamente desinfectado (para levantarla un poco).
5 - Sujete la jeringa como un lápiz o un dardo; empuje la aguja en la piel, manteniéndola recta en un ángulo de 90°, con un movimiento rápido y sin titubeos.
6 - Inyecte el medicamento, (presionando el émbolo lenta y contantemente hasta el final hasta que la jeringa quede vacía).
7 - Deseche la jeringa y la aguja, depositándolas en el contenedor de desechos.
Debe elegir un nuevo lugar para cada inyección, para permitir a la zona tiempo de recuperación, ayudando a prevenir una infección. Se proporciona consejo sobre qué áreas elegir en la primera parte de este Anexo. Es una buena idea conocer dónde se va a aplicar la inyección antes de preparar la jeringa. El esquema mostrado en el diagrama más adelante le ayudará a variar los lugares de forma adecuada. Por ejemplo, administre la primera inyección en el lado derecho del abdomen, elija el lado izquierdo para la segunda inyección; después, desplácese hacia el muslo derecho para la tercera, y así, a través del diagrama, hasta que se hayan utilizado todas las zonas apropiadas posibles del cuerpo. Conserve una anotación de dónde y cuándo se inyectó la última vez. Una forma de hacerlo es anotar esta información en el «Calendario para la administración», que se acompaña.
Siguiendo este esquema, volverá usted al lugar inicial (es decir, al lado derecho del abdomen), después de ocho inyecciones (16 días). Esto es lo que se conoce como «ciclo de rotación». En nuestro calendario de ejemplo, cada una de las ocho áreas corporales se ha dividido a su vez en seis lugares de inyección (sumándolos todos, hay 48 lugares en los que se administran las inyecciones): izquierdo y derecho: partes superior, media e inferior de cada área corporal. Cuando vuelva a un área de inyección después de terminar un Ciclo de Rotación, elija la zona más alejada dentro de esa área. Si aparece alguna úlcera, consulte con su médico o enfermera antes de elegir otros lugares de inyección.
Calendario de rotación
Para ayudarle a rotar de forma adecuada los lugares de inyección, le recomendamos que rellene un registro con la fecha y el lugar de inyección. Puede usar el siguiente esquema de rotación.
Trabaje cada ciclo de rotación sucesivamente. Cada ciclo será de ocho inyecciones (16 días), administradas desde la primera área hasta la octava área, por turno. Siguiendo esta secuencia, dará a cada área la oportunidad de recuperarse antes de recibir otra inyección.
Primer ciclo de rotación: Segundo ciclo de rotación: Tercer ciclo de rotación: Cuarto ciclo de rotación: Quinto ciclo de rotación: Sexto ciclo de rotación:
Sección superior izquierda de cada área Sección inferior derecha de cada área Sección central izquierda de cada área Sección superior derecha de cada área Sección inferior izquierda de cada área Sección central derecha de cada área
Instrucciones para controlar los lugares y las fechas de inyección
- -Comience con su primera inyección (o con la última inyección, si no es un usuario nuevo de Extavia).
- Seleccione un sitio de inyección. Si usted ya ha estado utilizando Extavia, comience con un área que no haya sido utilizada durante el último ciclo de rotación (es decir, en las últimos 16 días).
- Después de la inyección, rellene el lugar de inyección y la fecha en el «Calendario para la administración» (ver el ejemplo en: «Instrucciones para controlar los lugares y las fechas de inyección»).
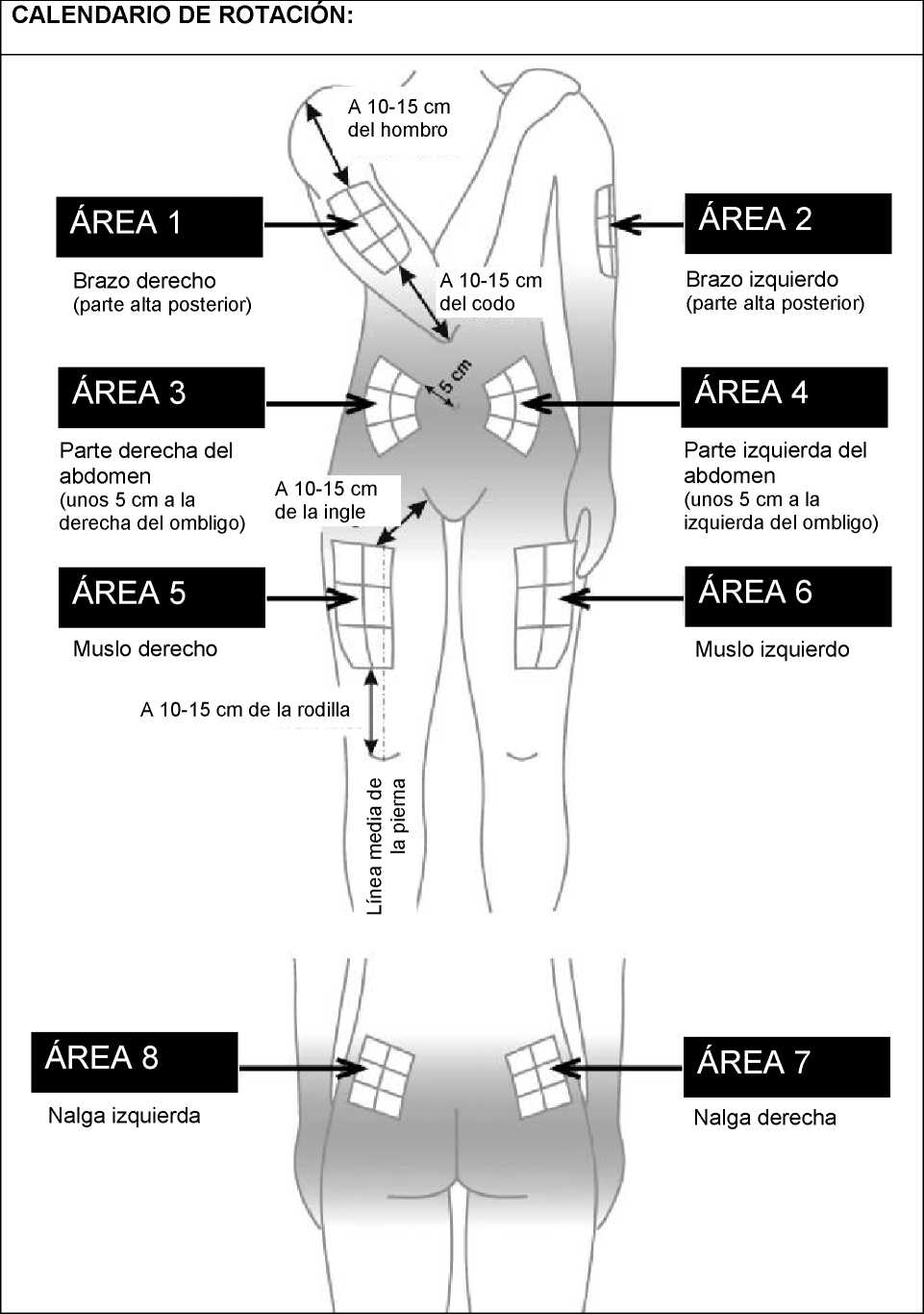
EJEMPLO DE CALENDARIO PARA LA ADMINISTRACION:
Mantenga un registro de los lugares y fechas de las inyecciones
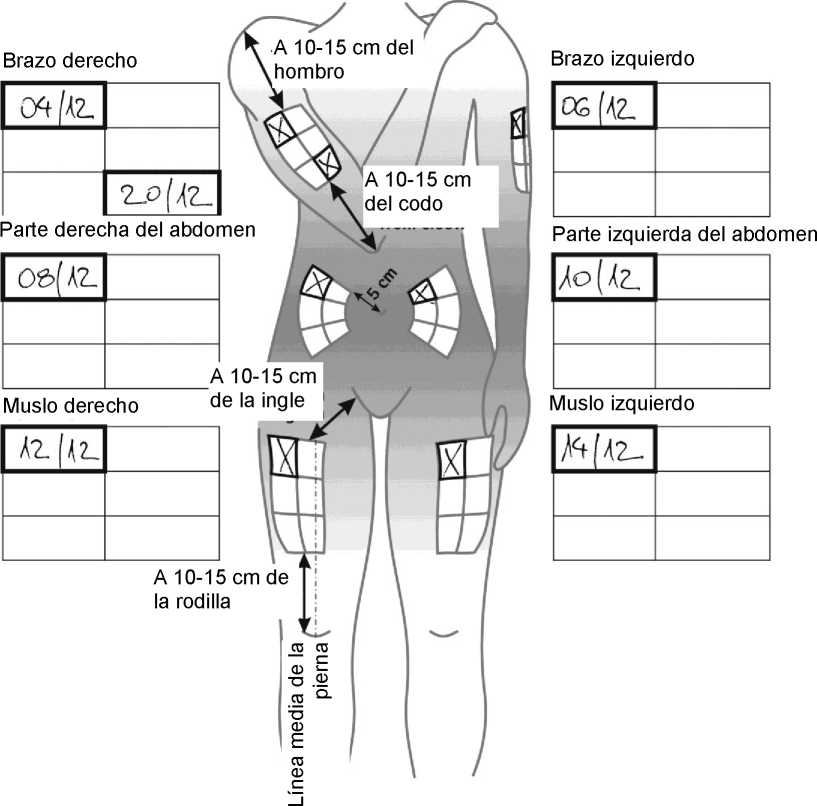
Nalga izquierda
|
xs | |
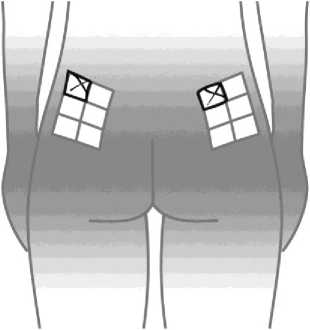
Nalga derecha
|
K&[KL | |
54