Etinilestradiol/Drospirenona Diario Quasset 0.03 Mg/3 Mg Comprimidos Recubiertos Con Pelicula Efg
Información obsoleta, busque otroRESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Etinilestradiol/Drospirenona Diario Quasset 0,03 mg/3 mg comprimidos recubiertos con película EFG
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA Comprimidos amarillos (comprimidos activos):
Cada comprimido recubierto con película contiene 0,03 mg de etinilestradiol y 3 mg de Drospirenona Excipiente con efecto conocido:
62 mg de lactosa monohidrato
Comprimidos blancos (comprimidos placebo):
El comprimido no contiene principios activos Excipiente con efecto conocido:
89,5 mg de lactosa anhidra
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película.
Comprimidos activos: Comprimidos recubiertos con película, amarillos, redondos Comprimidos placebo: Comprimidos recubiertos con película, blancos, redondos.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Anticoncepción oral
La decisión de prescribir Etinilestradiol/Drospirenona Diario Quasset debe tener en cuenta los factores de riesgo actuales de cada mujer en particular, concretamente los de tromboembolismo venoso (TEV), y cómo se compara el riesgo de TEV con Etinilestradiol/Drospirenona Diario Quasset con el de otros anticonceptivos hormonales combinados (AHCs) (ver secciones 4.3 y 4.4).
4.2 Posología y forma de administración Vía de administración: uso oral.
Cómo tomar Etinilestradiol/Drospirenona Diario Quasset
Los comprimidos deben tomarse todos los días aproximadamente a la misma hora, con un poco de líquido si es necesario, en el orden que se indica en el blíster. La toma de comprimidos de Etinilestradiol/Drospirenona Diario Quasset es continua. Se debe tomar un comprimido diariamente durante 28 días consecutivos. Cada nuevo envase se empieza al día siguiente de terminar el último comprimido del envase anterior. La hemorragia por privación suele dar comienzo 2-3 días después de empezar a tomar los comprimidos placebo (última fila) y es posible que no haya cesado antes de empezar el siguiente envase.
Cómo comenzar a tomar Etinilestradiol/Drospirenona Diario Quasset
• Si no se ha usado ningún anticonceptivo hormonal (en el mes anterior)
Los comprimidos se empezarán a tomar el día 1 del ciclo natural de la mujer (es decir, el primer día de la hemorragia menstrual).
• Para sustituir otro método anticonceptivo hormonal combinado (anticonceptivo oral combinado (AOC), anillo vaginal o parche ransdérmico).
La mujer debe empezar a tomar Etinilestradiol/Drospirenona Diario Quasset al día siguiente de finalizar el intervalo habitual sin toma de hormonas (intervalo sin comprimidos o con comprimidos placebo) de su método anticonceptivo oral combinado previo. En caso de uso de un anillo vaginal o parche transdérmico, debe empezar a tomar Etinilestradiol/Drospirenona Diario Quasset preferentemente el día de su extracción, o a más tardar cuando se hubiera tenido que volver a aplicar el parche o anillo.
• Para sustituir un método basado exclusivamente en progestágenos (píldora, inyección o implante basado sólo en progestágenos) o un dispositivo intrauterino de liberación de progestágenos (DIU).
La mujer puede sustituir cualquier día la píldora basada sólo en progestágenos (si se trata de un implante o de un DIU, se sustituirá el mismo día de su extracción y si se trata de un inyectable, el día que corresponda la siguiente inyección) pero, en todos estos casos, se debe recomendar el uso adicional de un método de barrera durante los 7 primeros días de la toma de comprimidos.
• Tras un aborto en el primer trimestre
La mujer puede empezar inmediatamente. En tal caso, no es necesario que tome medidas anticonceptivas adicionales.
• Tras el parto o un aborto en el segundo trimestre
Se debe recomendar a la mujer que empiece entre 21 y 28 días después del parto o del aborto en el segundo trimestre. Si lo hace más tarde, se aconsejará a la mujer que utilice adicionalmente un método de barrera durante los 7 primeros días. No obstante, si ya ha mantenido relaciones sexuales, hay que descartar que se haya producido un embarazo antes de iniciar el uso de AOC, o bien esperar hasta su primer período menstrual.
Para mujeres en período de lactancia, ver sección 4.6.
Procedimiento a seguir en caso de no tomar algún comprimido
El olvido de la toma de los comprimidos de última fila del blíster (comprimidos placebo) no tiene consecuencias. No obstante, éstos deben ser desechados para evitar prolongar de forma no intencionada la fase de comprimidos placebo.
La siguiente advertencia se refiere al olvido de de la toma de los comprimidos activos (filas 1 a 3 del blister):
Si se retrasa menos de 12 horas en la toma de algún comprimido activo, la protección anticonceptiva no se ve reducida. La mujer debe tomar el comprimido tan pronto como se acuerde e ingerir los comprimidos siguientes a la hora habitual.
Si se retrasa más de 12 horas en la toma de algún comprimido activo, la protección anticonceptiva puede verse reducida. La pauta a seguir en caso de olvido de comprimidos se rige por estas dos normas básicas:
1. Nunca se debe suspender la toma de comprimidos durante más de 7 días consecutivos.
2. Se requiere tomar los comprimidos de forma ininterrumpida durante 7 días para conseguir una supresión adecuada del eje hipotálamo-hipófisis-ovario.
En consecuencia, en la práctica diaria se puede recomendar lo siguiente:
• Semana 1
La mujer debe tomar el último comprimido olvidado tan pronto como se acuerde, incluso si esto significa tomar dos comprimidos a la vez. Posteriormente seguirá tomando los comprimidos restantes a su hora habitual. Además, durante los 7 días siguientes debe utilizar un método de barrera, como un preservativo. Si la mujer ha mantenido relaciones sexuales en los 7 días previos, se debe considerar la posibilidad de un embarazo. Cuantos más comprimidos hayan sido olvidados y cuanto más cerca estén de la fase de comprimidos placebo, mayor es el riesgo de embarazo.
• Semana 2
La mujer debe tomar el último comprimido olvidado tan pronto como se acuerde, incluso si esto significa tomar dos comprimidos a la vez. Posteriormente seguirá tomando los comprimidos restantes a su hora habitual. Siempre que en los 7 días anteriores al primer comprimido olvidado la mujer haya tomado los comprimidos correctamente, no es necesario utilizar medidas anticonceptivas adicionales. Sin embargo, si ha olvidado tomar más de un comprimido, se le aconsejará que tome precauciones anticonceptivas adicionales durante los 7 días siguientes.
• Semana 3
El riesgo de reducción de la fiabilidad anticonceptiva es inminente debido a la cercanía de la fase de 7 días de comprimidos placebo.
No obstante, ajustando el calendario de toma de comprimidos, aún se puede prevenir la reducción de la protección anticonceptiva. Por consiguiente, si sigue una de las dos opciones siguientes, no necesitará adoptar precauciones anticonceptivas adicionales, siempre que en los 7 días anteriores al primer comprimido olvidado la mujer haya tomado todos los comprimidos correctamente. Si este no es el caso, se le aconsejará que siga la primera de estas dos opciones, y que además tome precauciones anticonceptivas adicionales durante los 7 días siguientes.
1. La mujer debe tomar el último comprimido olvidado tan pronto como se acuerde, incluso si esto significa tomar dos comprimidos a la vez.
Posteriormente, seguirá tomando los comprimidos a su hora habitual hasta acabar los comprimidos activos. Los 7 comprimidos de la última fila (comprimidos placebo) deben ser desechados. El siguiente envase se debe empezar inmediatamente. Probablemente no haya una hemorragia por privación hasta el final de la toma de los comprimidos activos del segundo envase, pero puede presentarse manchado o hemorragia por privación en los días de toma de comprimidos.
2. También se debe recomendar a la mujer que deje de tomar los comprimidos activos del envase actual. A continuación, debe tomar los comprimidos de la última fila (comprimidos placebo) durante 7 días como máximo, incluidos los días en que olvidó los comprimidos, y continuar con el siguiente envase.
Si la mujer olvida tomar varios comprimidos y posteriormente no presenta hemorragia por privación en la fase de comprimidos placebo, se debe considerar la posibilidad de embarazo.
Recomendación en caso de trastornos gastrointestinales
En caso de alteraciones gastrointestinales graves (por ejemplo, vómitos o diarrea grave), la absorción puede no ser completa, y se deben tomar medidas anticonceptivas adicionales. Si se producen vómitos en las 3-4 horas siguientes a la toma del comprimido activo, se debe tomar un nuevo comprimido lo antes posible. El nuevo comprimido se debe tomar, si es posible, no más de 12 horas después de la hora habitual a la que se toman los comprimidos.
Si han transcurrido más de 12 horas, se deberán seguir las recomendaciones referentes al olvido de la toma de comprimidos, tal como se expone en la sección 4.2. “Procedimiento a seguir en caso de no tomar algún comprimido”. Si la mujer no desea cambiar su pauta normal de toma de comprimidos, deberá tomar el/los comprimido/s adicional/es necesario/s de otro envase.
Cómo retrasar una hemorragia por privación
Para retrasar un período, la mujer debe continuar con otro envase de Etinilestradiol/Drospirenona Diario Quasset sin tomar los comprimidos placebo del envase actual. Puede mantener esta extensión tanto como desee hasta que se terminen los comprimidos activos del segundo envase. Durante la extensión, la mujer puede experimentar manchado o hemorragia intermenstrual. Posteriormente, la toma regular de Etinilestradiol/Drospirenona Diario Quasset se reanuda tras la fase de comprimidos placebo.
Para cambiar sus períodos a otro día de la semana al que la mujer está acostumbrada, se le puede aconsejar que acorte la siguiente fase de comprimidos placebo tantos días como desee. Cuanto más corto sea, mayor es el riesgo de que no tenga hemorragia por privación, y de que experimente manchado y hemorragia intermenstrual durante la toma del envase siguiente (igual que cuando se retrasa un período).
4.3 Contraindicaciones
No se deben utilizar anticonceptivos hormonales combinados (AHCs) en las siguientes condiciones. Si cualquiera de estos cuadros aparece por primera vez durante el uso de AHC, se debe suspender inmediatamente el tratamiento.
• Presencia o riesgo de tromboembolismo venoso (TEV)
o Tromboembolismo venoso: TEV actual (con anticoagulantes) o antecedentes del mismo (p. ej., trombosis venosa profunda (TVP) o embolia pulmonar (EP)). o Predisposición hereditaria o adquirida conocida al tromboembolismo venoso, como resistencia a la PCA (incluyendo el factor V Leiden), deficiencia de antitrombina III, deficiencia de proteína C, deficiencia de proteína S. o Cirugía mayor con inmovilización prolongada (ver sección 4.4). o Riesgo elevado de tromboembolismo venoso debido a la presencia de varios factores de riesgo (ver sección 4.4).
• Presencia o riesgo de tromboembolismo arterial (TEA).
o Tromboembolismo arterial: tromboembolismo arterial actual, antecedentes de tromboembolismo (p. ej. infarto de miocardio) o afección prodrómica (p. ej. angina de pecho).
o Enfermedad cerebrovascular: ictus actual, antecedentes de ictus o afección prodrómica (p. ej. accidente isquémico transitorio, AIT).
o Predisposición hereditaria o adquirida conocida al tromboembolismo arterial, tal como hiperhomocisteinemia y anticuerpos antifosfolípidos (anticuerpos anticardiolipina, anticoagulante del lupus).
o Antecedentes de migraña con síntomas neurológicos focales.
o Riesgo elevado de tromboembolismo arterial debido a múltiples factores de riesgo (ver sección 4.4) o a la presencia de un factor de riesgo grave como
• diabetes mellitus con síntomas vasculares
• hipertensión grave
• dislipoproteinemia intensa
• Presencia o antecedentes de pancreatitis, si se asocia a hipertrigliceridemia grave.
• Presencia o antecedentes de enfermedad hepática grave, siempre que los valores de las pruebas de
función hepática no se hayan normalizado.
• Insuficiencia renal grave o fallo renal agudo.
• Presencia o antecedentes de tumores hepáticos (benignos o malignos).
• Procesos malignos, conocidos o sospechados dependientes de los esteroides sexuales (por ejemplo,
de los órganos genitales o de las mamas).
• Hemorragia vaginal no diagnosticada.
• Hipersensibilidad a los principios activos o a alguno de los excipientes de
Etinilestradiol/Drospirenona Diario Quasset.
4.4 Advertencias y precauciones especiales de empleo Advertencias
Si alguna de las afecciones o factores de riesgo que se mencionan a continuación está presente, se debe comentar con la mujer la idoneidad de Etinilestradiol/Drospirenona Diario Quasset.
Si alguna de estas afecciones o de estos factores de riesgo se agrava o aparece por primera vez, se debe aconsejar a la mujer que consulte a su médico para determinar si se debe interrumpir el uso de Etinilestradiol/Drospirenona Diario Quasset.
En caso de sospecha o confirmación de TEV o TEA, se debe suspender el uso de AHC. En caso de iniciar un tratamiento anticoagulante, se deberá empezar un método de anticoncepción alternativo adecuado debido a la teratogenicidad del tratamiento anticoagulante (cumarinas).
Riesgo de tromboembolismo venoso (TEV)
El uso de cualquier anticonceptivo hormonal combinado (AHC) aumenta el riesgo de tromboembolismo venoso (TEV), comparado con la no utilización. Los medicamentos que contienen levonorgestrel, norgestimato o noretisterona se asocian con el riesgo más bajo de TEV. Otros medicamentos como Etinilestradiol/Drospirenona Diario Quasset pueden tener hasta el doble de este nivel de riesgo. La decisión de utilizar cualquier medicamento diferente del que tiene el menor riesgo de TEV se debe tomar solamente después de comentarlo con la mujer para garantizar que comprende el riesgo de TEV con Etinilestradiol/Drospirenona Diario Quasset, cómo afectan sus actuales factores de riesgo a este riesgo y que su riesgo de TEV es mayor durante el primer año de uso. También existen ciertas
evidencias de que el riesgo aumenta cuando se reinicia el AHC después de una interrupción del uso de 4 semanas o más.
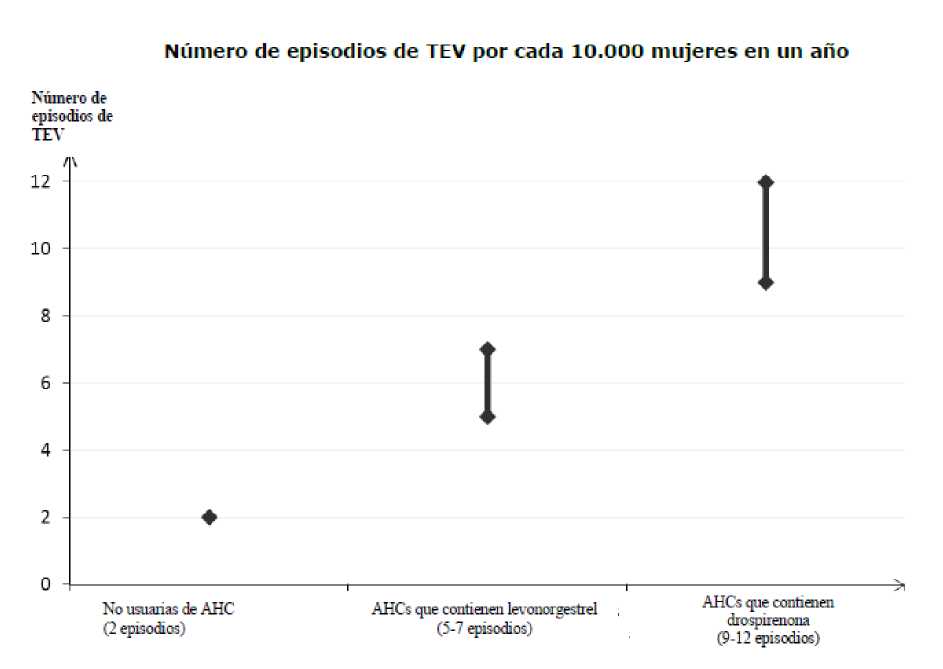
Entre las mujeres que no utilizan un AHC y que no están embarazadas, aproximadamente 2 de cada 10.000 presentarán un TEV en el plazo de un año. No obstante, el riesgo puede ser mucho mayor en cada mujer en particular, en función de sus factores de riesgo subyacentes (ver a continuación).
Se estima1 que de cada 10.000 mujeres que utilizan un AHC que contiene drospirenona, entre 9 y 12 mujeres presentarán un TEV en un año; esto se compara con unas 62 en mujeres que utilizan un AHC que contiene levonorgestrel.
En ambos casos, el número de TEVs por año es inferior al número esperado en mujeres durante el embarazo o en el período de posparto.
El TEV puede ser mortal en el 1-2 % de los casos.
'Estas incidencias se estimaron a partir de la totalidad de los datos de estudios epidemiológicos, utilizando riesgos relativos para los diferentes medicamentos comparados con los AHCs que contienen levonorgestrel
2Punto medio del intervalo 5-7 por cada 10.000 mujeres-año (MA), basado en un riesgo relativo para los AHCs que contienen levonorgestrel frente a la no utilización de aproximadamente 2,3 a 3,6.
De forma extremadamente rara, se han notificado casos de trombosis en otros vasos sanguíneos, por ejemplo en venas y arterias hepáticas, mesentéricas, renales, o retinianas, en usuarias de AHC.
Factores de riesgo de TEV
El riesgo de complicaciones tromboembólicas venosas en usuarias de AHC puede aumentar sustancialmente en una mujer con factores de riesgo adicionales, en particular si existen varios factores de riesgo (ver tabla).
Etinilestradiol/Drospirenona Diario Quasset está contraindicado si una mujer tiene varios factores de riesgo que la ponen en una situación de alto riesgo de trombosis venosa (ver sección 4.3). Si una mujer tiene más de un factor de riesgo, es posible que el aumento del riesgo sea mayor que la suma de los factores individuales; en este caso se debe tener en cuenta su riesgo total de TEV. Si se considera que la relación beneficio/riesgo es negativa, no se debe prescribir un AHC (ver sección 4.3).
|
Tabla: Factores de riesgo de TEV | |
|
Factor de riesgo |
Comentario |
|
Obesidad (índice de masa corporal (IMC) superior a 30 kg/m2). |
El riesgo aumenta de forma sustancial con el aumento del IMC. Especialmente importante en mujeres con factores de riesgo adicionales. |
|
Inmovilización prolongada, la cirugía mayor, cualquier intervención quirúrgica de las piernas o pelvis, neurocirugía o traumatismo importante. Nota: La inmovilización temporal, incluyendo los viajes en avión >4 horas, también puede ser un factor de riesgo de TEV, en especial en mujeres con otros factores de riesgo. |
En estas circunstancias es aconsejable interrumpir el uso del parche/comprimido/anillo (en caso de intervención quirúrgica programada, al menos con cuatro semanas de antelación) y no reanudarlo hasta dos semanas después de que se recupere completamente la movilidad. Se debe utilizar otro método anticonceptivo para evitar un embarazo involuntario. Se debe considerar un tratamiento antitrombótico si no se ha interrumpido con antelación la toma de Etinilestradiol/Drospirenona Diario Quasset. |
|
Antecedentes familiares positivos (algún caso de tromboembolismo venoso en un hermano o en un progenitor, especialmente a una edad relativamente temprana, p. ej. antes de los 50 años). |
Si se sospecha que existe una predisposición hereditaria, la mujer se debe derivar a un especialista antes de tomar la decisión de usar un AHC. |
|
Otras enfermedades asociadas al TEV. |
Cáncer, lupus eritematoso sistémico, síndrome urémico hemolítico, enfermedad intestinal inflamatoria crónica (enfermedad de Crohn o colitis ulcerosa) y anemia de células falciformes. |
|
Aumento de la edad. |
En especial por encima de los 35 años. |
No hay consenso sobre el posible papel de las venas varicosas y la tromboflebitis superficial en la aparición o progresión de la trombosis venosa.
Es preciso tener en cuenta el aumento del riesgo de tromboembolismo en el embarazo, y en particular en el período de 6 semanas del puerperio (para obtener información sobre “Embarazo y lactancia”, ver sección 4.6).
Síntomas de TEV (trombosis venosa profunda y embolia pulmonar)
En el caso de que se produzcan síntomas, se debe aconsejar a la mujer que busque asistencia médica urgente y que informe al profesional sanitario de que está tomando un AHC.
Los síntomas de trombosis venosa profunda (TVP) pueden incluir:
- Hinchazón unilateral de la pierna y/o pie o a lo largo de una vena de la pierna.
- Dolor o sensibilidad en la pierna, que tal vez se advierta sólo al ponerse de pie o al caminar.
- Aumento de la temperatura en la pierna afectada; enrojecimiento o decoloración de la piel de
la pierna.
Los síntomas de embolia pulmonar (EP) pueden incluir:
- Aparición repentina de falta de aliento o respiración rápida injustificadas.
- Tos repentina que puede estar asociada a hemoptisis.
- Dolor torácico agudo.
- Aturdimiento intenso o mareo.
- Latidos cardíacos acelerados o irregulares.
Algunos de estos síntomas (p. ej. “falta de aliento”, “tos”) son inespecíficos y se pueden confundir con acontecimientos más frecuentes o menos graves (p. ej. infecciones del tracto respiratorio).
Otros signos de oclusión vascular pueden incluir: dolor repentino, hinchazón y ligera coloración azul de una extremidad.
Si la oclusión se produce en el ojo, los síntomas pueden variar desde visión borrosa indolora, que puede evolucionar hasta pérdida de la visión. A veces la pérdida de la visión se puede producir casi de inmediato.
Riesgo de tromboembolismo arterial (TEA)
Estudios epidemiológicos han asociado el uso de los AHCs con un aumento del riesgo de tromboembolismo arterial (infarto de miocardio) o de accidente cerebrovascular (p. ej. accidente isquémico transitorio, ictus). Los episodios tromboembólicos arteriales pueden ser mortales.
Factores de riesgo de TEA
El riesgo de que se produzcan complicaciones tromboembólicas arteriales o un accidente cerebrovascular en usuarias de AHC aumenta en mujeres con factores de riesgo (ver tabla). Etinilestradiol/Drospirenona Diario Quasset está contraindicado si una mujer presenta varios factores de riesgo de TEA o uno grave que la ponen en una situación de alto riesgo de trombosis arterial (ver sección 4.3). Si una mujer tiene más de un factor de riesgo, es posible que el aumento del riesgo sea mayor que la suma de los factores individuales; en este caso se debe tener en cuenta su riesgo total. Si se considera que la relación beneficio/riesgo es negativa, no se debe prescribir un AHC (ver sección 4.3).
Tabla: Factores de riesgo de TEA
|
Factor de riesgo |
Comentario |
|
Aumento de la edad. |
En especial por encima de los 35 años. |
|
Tabaquismo. |
Se debe aconsejar a las mujeres que no fumen si desean utilizar un AHC. Se debe aconsejar encarecidamente a |
|
las mujeres de más de 35 años que continúan fumando que utilicen un método anticonceptivo diferente. | |
|
Hipertensión arterial | |
|
Obesidad (índice de masa corporal superior a 30 kg/m2). |
El riesgo aumenta de forma sustancial con el aumento del IMC. Especialmente importante en mujeres con factores de riesgo adicionales. |
|
Antecedentes familiares positivos (algún caso de tromboembolismo arterial en un hermano o en un progenitor, especialmente a una edad relativamente temprana, p. ej. menos de 50 años). |
Si se sospecha que existe una predisposición hereditaria, la mujer debe ser derivada a un especialista antes de tomar la decisión de usar un AHC. |
|
Migraña. |
Un aumento de la frecuencia o la intensidad de las migrañas durante el uso de AHC (que puede ser prodrómico de un acontecimiento cerebrovascular) puede motivar su interrupción inmediata. |
|
Otras enfermedades asociadas a acontecimientos vasculares adversos. |
Diabetes mellitus, hiperhomocisteinemia, valvulopatía y fibrilación auricular, dislipoproteinemia y lupus eritematoso sistémico. |
Síntomas de TEA
En el caso de que se produzcan síntomas, se debe aconsejar a la mujer que busque asistencia médica urgente y que informe al profesional sanitario de que está tomando un AHC.
Los síntomas de un accidente cerebrovascular pueden incluir:
- Entumecimiento o debilidad repentinos de la cara, brazo o pierna, especialmente en un lado del cuerpo.
- Dificultad repentina para caminar, mareo, pérdida del equilibrio o de la coordinación.
- Confusión repentina, dificultad para hablar o para comprender.
- Dificultad repentina de visión en un ojo o en ambos.
- Cefalea repentina, intensa o prolongada sin causa conocida.
- Pérdida del conocimiento o desmayo, con o sin convulsiones.
Los síntomas temporales sugieren que el episodio es un accidente isquémico transitorio (AIT).
Los síntomas de infarto de miocardio (IM) pueden incluir:
- Dolor, molestias, presión, pesadez, sensación de opresión o plenitud en el tórax, brazo o debajo del esternón.
- Malestar que irradia a la espalda, la mandíbula, la garganta, el brazo o el estómago.
- Sensación de plenitud, indigestión o ahogo.
- Sudoración, náuseas, vómitos o mareo.
- Debilidad extrema, ansiedad o falta de aliento.
- Latidos cardíacos acelerados o irregulares.
• Tumores
En algunos estudios epidemiológicos se ha asociado un aumento del riesgo de cáncer cervicouterino al uso de AOC durante largos períodos de tiempo;(> 5 años) ,sin embargo, sigue existiendo controversia acerca de hasta qué punto este hallazgo puede ser atribuido a factores relacionados con la conducta sexual u otros factores como el virus del papiloma humano (VPH).
En un metaanálisis de 54 estudios epidemiológicos se observó que existe un ligero aumento del riesgo relativo (RR = 1,24) de que se diagnostique cáncer de mama en mujeres que están tomando AOC. Este aumento del riesgo desaparece gradualmente en los 10 años siguientes a la discontinuación de los AOC. Dado que el cáncer de mama es raro en las mujeres de menos de 40 años, el aumento de casos diagnosticados de cáncer de mama en las mujeres que toman AHC o que los han tomado recientemente es pequeño en relación con el riesgo total de cáncer de mama. Estos estudios no aportan datos sobre las causas. El patrón observado de aumento del riesgo puede deberse a que el diagnóstico de cáncer de mama es más precoz en usuarias de AOC, a los efectos biológicos de los AOC o a una combinación de ambos factores. Los cánceres de mama diagnosticados en mujeres que han usado un AOC en alguna ocasión suelen estar menos avanzados, desde el punto de vista clínico, que los diagnosticados en quienes nunca los han tomado.
En raros casos se han notificado tumores hepáticos benignos, y aún más raramente malignos, en usuarias de AOC. En casos aislados, estos tumores han dado lugar a hemorragias intraabdominales potencialmente mortales. Se debe considerar la posibilidad de que exista un tumor hepático en el diagnóstico diferencial en mujeres que toman AOC y que presentan dolor intenso en la parte superior del abdomen, hepatomegalia o signos de hemorragia intraabdominal.
Con el uso de dosis altas de AOC (50 p de etinilestradiol) el riesgo de cáncer de ovario y endometrio se reduce, aunque aún no se ha confirmado que esto se pueda extrapolar a dosis menores de AOC.
• Otras situaciones
El componente progestágeno de Etinilestradiol/Drospirenona Diario Quasset es un antagonista de la aldosterona con propiedades ahorradoras de potasio. En la mayoría de los casos no es de esperar un aumento de los niveles de potasio. No obstante, en un estudio clínico realizado en pacientes con insuficiencia renal leve o moderado y uso concomitante de medicamentos ahorradores de potasio, los niveles séricos de potasio aumentaron de forma leve, aunque no significativa, durante la toma de drospirenona. Por lo tanto, se recomienda monitorizar el potasio sérico durante el primer ciclo de tratamiento en pacientes que presenten insuficiencia renal y niveles séricos de potasio previos al tratamiento en el límite superior de los valores de referencia, principalmente durante el uso concomitante de medicamentos ahorradores de potasio. Ver sección 4.5.
Las mujeres con hipertrigliceridemia, o con antecedentes familiares de dicho trastorno, pueden tener mayor riesgo de pancreatitis cuando usan AOC.
Aunque se han notificado pequeños aumentos de la presión arterial en muchas mujeres que toman AOC, son raros los casos con relevancia clínica. La discontinuación inmediata del uso de AOC sólo está justificada en estos casos raros. Si durante el uso de un AHC en pacientes con hipertensión preexistente se observan valores constantemente elevados de la presión arterial o un aumento significativo de ésta que no responden adecuadamente al tratamiento antihipertensivo, debe retirarse el AOC. Si con el tratamiento antihipertensivo se alcanzan valores normales de presión arterial, puede reanudarse la toma de AOC si se considera apropiado.
Se ha notificado que las siguientes afecciones pueden aparecer o empeorar tanto durante el embarazo como durante el uso de AOC, pero los datos relativos a su asociación con los AOC no son concluyentes: ictericia y/o prurito relacionado con la colestasis, cálculos biliares, porfiria, lupus eritematoso sistémico, síndrome hemolítico urémico, corea de Sydenham, herpes gestacional, perdida de la audición relacionada con la otosclerosis.
En las mujeres con angioedema hereditario, los estrógenos exógenos pueden inducir o exacerbar los síntomas de angioedema.
Las alteraciones agudas o crónicas de la función hepática pueden requerir la discontinuación del uso de AOC hasta que los marcadores de la función hepática retornen a valores normales. La recurrencia de una ictericia colestásica y/o un prurito asociado a colestasis que se hayan manifestado previamente durante un embarazo, o durante el uso previo de esteroides sexuales requiere la discontinuación de los AOC.
Aunque los AOC pueden afectar a la resistencia periférica a la insulina y la tolerancia a la glucosa, no existen datos que indiquen que sea necesario alterar la pauta terapéutica en diabéticas que usan AOC a dosis bajas (con < 0,05 mg de etinilestradiol). En cualquier caso, las mujeres diabéticas deben ser vigiladas cuidadosamente, especialmente durante la etapa inicial del uso de AOC.
Durante el uso de AOC se ha notificado empeoramiento de la depresión endógena, de la epilepsia, de la enfermedad de Crohn y de la colitis ulcerosa.
Ocasionalmente puede aparecer cloasma, especialmente en las mujeres con antecedentes de cloasma gravídico. Las mujeres con tendencia al cloasma deben evitar la exposición al sol o a la radiación ultravioleta mientras están tomando AOC.
Cada comprimido amarillo contiene 44 mg de lactosa por comprimido, cada comprimido blanco contiene 89,5 mg de lactosa por comprimido. Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp (insuficiencia observada en ciertas poblaciones de Laponia) o malabsorción de glucosa o galactosa no deben tomar este medicamento.
Exploración/consulta médica
Antes de iniciar o reanudar el tratamiento con Etinilestradiol/Drospirenona Diario Quasset, se debe realizar una anamnesis completa (incluidos los antecedentes familiares) y descartarse un posible embarazo. Se debe medir la tensión arterial y realizar una exploración física guiada por las contraindicaciones (ver sección 4.3) y por las advertencias (ver sección 4.4). Es importante dirigir a la mujer hacia la información sobre la trombosis venosa y arterial, inlcuido el riesgo de Etinilestradiol/Drospirenona Diario Quasset en comparación con otros AHCs, los síntomas de TEV y TEA, los factores de riesgo conocidos y qué debe hacer en caso de una sospecha de trombosis.
También se debe indicar a la mujer que lea cuidadosamente el prospecto y siga las instrucciones allí descritas. La frecuencia y la naturaleza de las exploraciones deben basarse en las directrices clínicas establecidas y se adaptarán a cada mujer en particular.
Se debe advertir a las mujeres que los anticonceptivos hormonales no protegen frente a la infección por VIH (SIDA) ni frente a otras enfermedades de transmisión sexual.
Disminución de la eficacia
La eficacia de los AOC puede disminuir, por ejemplo, en caso de olvido de la toma de los comprimidos activos (ver sección 4.2), trastornos gastrointestinales, como vómitos o diarrea grave (ver sección 4.2) o uso de medicación concomitante (ver sección 4.5).
Reducción del control de los ciclos
Todos los AOC pueden dar lugar a sangrados irregulares (manchado o hemorragia intermenstrual), especialmente durante los primeros meses de uso. Por lo tanto, la evaluación de cualquier sangrado irregular sólo es significativa tras un intervalo de adaptación de unos tres ciclos.
Si las irregularidades en el sangrado persisten o se producen tras ciclos previos regulares, deberán tenerse en cuenta posibles causas no hormonales, y están indicadas medidas diagnósticas adecuadas para excluir un tumor maligno o un embarazo. Éstas pueden incluir el legrado.
En algunas mujeres puede no producirse la hemorragia por privación durante la fase de comprimidos placebo. Si el AOC se ha tomado siguiendo las instrucciones descritas en la sección 4.2, es poco probable que la mujer esté embarazada. Sin embargo, si el AOC no se ha tomado siguiendo estas indicaciones antes de producirse la primera falta de hemorragia por privación, o si faltan dos hemorragias por privación, se debe descartar un embarazo antes de continuar tomando el AOC.
4.5 Interacción con otros medicamentos y otras formas de interacción
Nota: deben consultarse las fichas técnicas de los medicamentos concomitantes con el fin de identificar interacciones potenciales.
• Influencia de otros medicamentos sobre Etinilestradiol/Drospirenona Diario Quasset
Las interacciones entre anticonceptivos orales y otros medicamentos pueden producir hemorragia por privación y/o fallo de la anticoncepción. Las siguientes interacciones han sido comunicadas a través de la literatura médica.
Metabolismo hepático
Se pueden dar interacciones con otros medicamentos que inducen las encimas hepáticas lo cual puede dar lugar a un aumento del aclaramiento de las hormonas sexuales (ej. Fenitoina, barbitúricos, primidona, cabamazepina, rifampicina, bosentano y medicamentos para el VIH (ej.ritonavir, nevirapina) y posiblemente también oxcarbazepina, topiramato, felbamato, griseofulvina y la planta de San Juan (Hypericum perforatum). Habitualmente la inducción enzimática máxima se observa a los 10 días, pero puede mantenerse al menos hasta 4 semanas despúes de la discontinuación del tratamiento.
Interferencia con la circulación enterohepártica
También se han notificado fallos de los anticonceptivos con los antibióticos como ampicilinas y tetraciclinas. No se ha dilucidado el mecanismo de este efecto.
Procedimiento a seguir
Las mujeres tratadas durante períodos cortos (hasta de una semana) con cualquiera de los grupos de medicamentos mencionados anteriormente, o con los principios activos individuales, deben usar temporalmente un método de barrera además del AOC, es decir, durante el tiempo de administración concomitante de los medicamentos y en los 7 días siguientes a la discontinuación.
Las mujeres tratadas con rifampicina deben utilizar un método de barrera además del AOC mientras dure la administración de rifampicina y durante los 28 días siguientes a su discontinuación.
En mujeres sometidas a tratamiento a largo plazo con principios activos inductores de las enzimas hepáticas, los expertos recomiendan incrementar las dosis de esteroides anticonceptivos.
Las mujeres en tratamiento con antibióticos (además de rifampicina, ver más arriba) deben utilizar el método de barrera hasta 7 días después de la interrupción.
Si la administración concomitante de estos medicamentos continúa más allá del final de los comprimidos en el blister de AOC, se deben descartar los comprimidos de placebo e iniciar el siguiente envase de AOC.
Los principales metabolitos de drospirenona en plasma humano se generan sin la participación del sistema del citocromo P450. Por lo tanto, es poco probable que los inhibidores de este sistema enzimático influyan sobre el metabolismo de drospirenona.
• Influencia de Etinilestradiol/Drospirenona Diario Quasset sobre otros medicamentos
Los anticonceptivos orales pueden influir en el metabolismo de otros principios activos. En consecuencia, los niveles plasmáticos y tisulares pueden aumentar (p. ej., ciclosporina) o disminuir (p. ej., lamotrigina).
Basándose en estudios de inhibición in vitro y en estudios de interacción in vivo, realizados en voluntarias que empleaban omeprazol, simvastatina y midazolam como sustrato marcador, se establece que es poco probable que se produzca una interacción de drospirenona a dosis de 3 mg con el metabolismo de otros principios activos.
• Otras interacciones
En pacientes sin insuficiencia renal, el uso concomitante de drospirenona e inhibidores de la ECA o AINE no mostró un efecto significativo sobre los niveles séricos de potasio. No obstante, no ha sido estudiado el uso concomitante de Etinilestradiol/Drospirenona Diario Quasset con antagonistas de la aldosterona o diuréticos ahorradores de potasio. En este caso, debe monitorizarse el potasio sérico durante el primer ciclo de tratamiento. (Ver sección 4.4).
• Pruebas de laboratorio
El uso de esteroides anticonceptivos puede afectar a los resultados de ciertas pruebas de laboratorio, como son los parámetros bioquímicos de la función hepática, tiroidea, suprarrenal y renal, los niveles plasmáticos de proteínas (transportadoras), por ejemplo la globulina transportadora de corticoesteroides y las fracciones lípido/lipoproteínas, los parámetros del metabolismo de los hidratos de carbono y los parámetros de la coagulación y la fibrinólisis. Por lo general, los cambios permanecen dentro de los límites normales. Drospirenona produce un aumento de la actividad de la renina plasmática y de la aldosterona plasmática, inducido por su leve actividad antimineralcorticoide.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Etinilestradiol/Drospirenona Diario Quasset no está indicado durante el embarazo.
Si se produjera un embarazo durante el tratamiento con Etinilestradiol/Drospirenona Diario Quasset, debe suspenderse su administración inmediatamente. Algunos estudios epidemiológicos extensos no han revelado ni un aumento del riesgo de defectos congénitos en niños nacidos de madres usuarias de AOC antes del embarazo, ni un efecto teratogénico cuando los AOC fueron tomados de forma inadvertida durante el embarazo.
En estudios con animales se han observado efectos adversos durante el embarazo y la lactancia (ver sección 5.3). En función de estos datos en animales, no se pueden descartar las reacciones adversas
debidas a la acción hormonal de los principios activos. Sin embargo, la experiencia general con AOC durante el embarazo no proporciona datos indicativos de una reacción adversa en humanos.
Los datos disponibles acerca del uso de Etinilestradiol/Drospirenona Diario Quasset durante el embarazo son demasiado limitados para extraer conclusiones relativas a las reacciones adversas de Etinilestradiol/Drospirenona Diario Quasset sobre el embarazo y la salud del feto o del recién nacido. Hasta la fecha de hoy, no se dispone de datos epidemiológicos relevantes.
Se debe tener en cuenta el aumento de riesgo de TEV durante el periodo de posparto cuando se reinicia la administración con Etinilestradiol/Drospirenona Diario Quasset (ver sección 4.2 y 4.4)
Lactancia
La lactancia puede resultar afectada por los AOC, dado que éstos pueden reducir la cantidad de leche materna y alterar su composición. Por consiguiente, en general no debe recomendarse el uso de AOC hasta que la madre haya cesado completamente la lactancia de su hijo. Durante el uso de AOC pueden eliminarse a través de la leche pequeñas cantidades de esteroides anticonceptivos y/o de sus metabolitos. Estas cantidades pueden afectar al lactante.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas. No se han observado efectos sobre la capacidad para conducir y utilizar máquinas en usuarias de AOC.
4.8 Reacciones adversas
Los efectos adversos graves de los AOC se indican en la sección 4.4
Se han descrito las siguientes reacciones adversas durante el uso de Etinilestradiol/Drospirenona Diario Quasset:
La siguiente tabla enumera las reacciones adversas según la clasificación por órganos y sistemas de MedDRA (MedDRA SOCs).
|
Clasificación por órganos y sistemas |
Frecuencia de las reacciones adversas | ||
|
Frecuentes |
Poco frecuentes |
Raras | |
|
(> 1/100 a < 1/10)> |
(> 1/1000 a < 1/100)> |
>10.000 a <1/1.000 | |
|
Infecciones e infestaciones |
Candidiasis Herpes simple | ||
|
Trastornos del sistema inmunológico |
Reacción alérgica |
Asma | |
|
Trastornos del metabolismo y de la nutrición |
Aumento del apetito | ||
|
Trastornos psiquiátricos |
Inestabilidad emocional |
Depresión Nerviosismo Trastornos del sueño Disminución de la libido | |
|
Trastornos del sistema nervioso |
Cefalea |
Parestesia Mareo | |
|
Trastornos del oído y del laberinto |
Hipacusia | ||
|
Trastornos oculares |
Molestias visuales | ||
|
Trastornos cardíacos |
Extrasístoles Taquicardia | ||
|
Trastornos vasculares |
Embolia pulmonar Hipertensión Hipotensión Migrañas Venas varicosas |
Tromboembolismo venoso (TEV), Tromboembolismo arterial (TEA) | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Faringitis | ||
|
Trastornos gastrointestinales |
Dolor abdominal |
Náuseas Vómitos Gastroenteritis Diarrea Estreñimiento Trastornos gastrointestinales | |
|
Trastornos de la piel y del tejido subcutáneo |
Acné |
Angioedema Alopecia Eccema Prurito Exantema Sequedad cutánea Seborrea Trastornos de la piel |
Eritema nudoso Eritema multiforme |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor de cuello Dolor en las extremidades Calambres musculares | ||
|
Trastornos renales y urinarios |
Cistitis | ||
|
Trastornos del aparato reproductor y de la mama |
Dolor de mama Aumento del tamaño de las mamas Dismenorrea Metrorragia |
Neoplasia de mama Mama fibroquística Galactorrea Quiste ovárico Sofocos Trastornos menstruales Amenorrea Menorragia Candidosis vaginal Vaginitis Flujo vaginal Trastorno vulvovaginal Sequedad vaginal Dolor pélvico |
|
Frotis de Papanicolau anormal | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Edema Astenia Dolor Sed excesiva Aumento de la sudoración | ||
|
Exploraciones complementarias |
Aumento de peso |
Pérdida de peso |
El término MedDRA más adecuado se utiliza para describir una determinada reacción y sus sinónimos y trastornos relacionados.
Descripción de reacciones adversas seleccionadas
Se ha observado un aumento del riesgo de episodios trombóticos y tromboembólicos arteriales y venosos, entre ellos infarto de miocardio, accidente cerebrovascular, accidentes isquémicos transitorios, trombosis venosa y embolia pulmonar, en mujeres que utilizan AHCs, que se comentan con más detalle en la sección 4.4.
Se han notificado los siguientes acontecimientos adversos graves en las mujeres usuarias de AOC, que se detallan en la sección 4.4:
• Trastornos tromboembólicos venosos;
• Trastornos tromboembólicos arteriales;
• Hipertensión;
• Tumores hepáticos;
• Aparición o deterioro de cuadros en los que la asociación con un AOC no resulta concluyente: enfermedad de Crohn, colitis ulcerosa, epilepsia, migraña, endometriosis, mioma uterino, porfiria, lupus eritematoso sistémico, herpes gestacional, corea de Sydenham, síndrome hemolítico urémico, ictericia colestásica;
• Cloasma;
• Los trastornos agudos o crónicos de la función hepática pueden requerir la discontinuación del uso de AOC hasta que los marcadores de la función hepática retornen a la normalidad.
• En mujeres con angioedema hereditario, los estrógenos exógenos pueden inducir o exacerbar los síntomas de angioedema.
La frecuencia del diagnóstico de cáncer de mama entre usuarias de AOC es ligeramente mayor. Dado que el cáncer de mama es raro en mujeres menores de 40 años, este aumento es pequeño con relación al riesgo global de cáncer de mama. Se desconoce la causalidad relacionada con el uso de AOC. Para más información, ver las secciones 4.3 y 4.4.
Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de medicamentos de Uso Humano: https://www.notificaram.es.
4.9 Sobredosis
No se han notificado casos de sobredosis con Etinilestradiol/Drospirenona Diario Quasset. Según la experiencia general con los anticonceptivos orales combinados, los síntomas que posiblemente pueden producirse en caso de sobredosis de comprimidos activos son: náuseas, vómitos y, en las chicas jóvenes, hemorragia vaginal leve. No existe antídoto y el tratamiento debe ser sintomático.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico (ATC): Progestágenos y estrógenos, preparados de dosis fijas. Código ATC: G03AA12
Índice de Pearl por promedio de falla: 0,11 (límite superior, intervalo de confianza del 95%, 0,60%).
Índice de Pearl: 0,31 (límite superior, intervalo de confianza del 97,5%: 0,91)
El efecto anticonceptivo de Etinilestradiol/Drospirenona Diario Quasset se basa en la interacción de diversos factores; los más importantes son la inhibición de la ovulación y los cambios en el endometrio.
Etinilestradiol/Drospirenona Diario Quasset es un anticonceptivo oral combinado compuesto por etinilestradiol y el progestágeno drospirenona. A dosis terapéuticas, la drospirenona posee también propiedades antiandrogénicas y antimineralocorticoides leves. Carece de cualquier tipo de actividad estrogénica, glucocorticoide o antiglucocorticoide. Esto otorga a la drospirenona un perfil farmacológico muy parecido al de la progesterona natural.
Datos procedentes de estudios clínicos indican que las leves propiedades antimineralocorticoides de Etinilestradiol/Drospirenona Diario Quasset producen un efecto antimineralocorticoide ligero.
5.2 Propiedades farmacocinéticas Drospirenona
Absorción
Drospirenona se absorbe de forma rápida y casi completa por vía oral. Tras la administración de una dosis única se alcanzan concentraciones séricas máximas del principio activo de aproximadamente 38 ng/ml entre 1-2 horas después de la ingestión. La biodisponibilidad oscila entre el 76% y el 85%. La ingestión concomitante de alimentos no influye en la biodisponibilidad de drospirenona.
Distribución
Tras su administración por vía oral, los niveles de drospirenona en suero disminuyen con una semivida terminal de 31 horas.
Drospirenona se une a la albúmina sérica pero no a la globulina fijadora de hormonas sexuales (SHBG) ni a la globulina fijadora de corticoides (CBG). Sólo entre el 3% y el 5% de la concentración total del principio activo en suero está presente en forma de esteroide libre. El aumento de la SHBG inducido por etinilestradiol no afecta a la unión de drospirenona a las proteínas séricas. El volumen de distribución medio aparente de drospirenona es de 3,7 l/kg ± 1,2 l/kg.
Metabolismo
Drospirenona se metaboliza intensamente tras su administración por vía oral. En plasma, los principales metabolitos son la forma ácida de drospirenona, que se genera por la apertura del anillo lactona, y el 4,5-dihidro-Drospirenona-3-sulfato; los dos se forman sin la intervención del sistema P450. Una pequeña parte de drospirenona es metabolizada por el citocromo P450 3A4, y ha demostrado tener capacidad de inhibir esta enzima y también los citocromos P450 1A1, P450 2C9 y P450 2C19, in vitro.
Eliminación
La tasa de aclaramiento metabólico de drospirenona en suero es de 1,5 ml/min/kg ± 0,2 ml/min/kg. Drospirenona se elimina sólo en cantidades mínimas en forma inalterada. Los metabolitos de Drospirenona se eliminan por heces y orina, con una tasa de eliminación de aproximadamente 1,2 a 1,4. La semivida de eliminación de los metabolitos por orina y heces es de unas 40 horas.
Condiciones en estado estacionario
Durante un ciclo de tratamiento se alcanzan concentraciones séricas máximas de drospirenona en estado estacionario de unos 70 ng/ml después de unos 8 días de tratamiento. Los niveles séricos de drospirenona se acumularon en un factor de aproximadamente 3, como consecuencia de la tasa entre la semivida terminal y el intervalo de administración.
Poblaciones especiales de pacientes
Efecto de la insuficiencia renal
Los niveles séricos de Drospirenona en estado estacionario en mujeres con insuficiencia renal leve (aclaramiento de creatinina CLCr, 50 ml/min - 80 ml/min) fueron comparables a los de mujeres con función renal normal. Los niveles séricos de Drospirenona en mujeres con insuficiencia renal moderada (CLCr, 30 ml/min - 50 ml/min) fueron un 37% superiores, como media, comparados con los de mujeres con función renal normal. El tratamiento con Drospirenona fue bien tolerado también por las mujeres con insuficiencia renal leve y moderada. El tratamiento con drospirenona no mostró ningún efecto clínicamente significativo sobre las concentraciones séricas de potasio.
Efecto de la insuficiencia hepática
En un estudio de dosis única, el aclaramiento oral (CL/F) disminuyó aproximadamente un 50% en voluntarias con insuficiencia hepática moderada, comparado con las voluntarias con función hepática normal. La disminución en el aclaramiento de Drospirenona observada en voluntarias con insuficiencia hepática moderada no se tradujo en ninguna diferencia aparente en términos de niveles séricos de potasio. Incluso en presencia de diabetes y tratamiento concomitante con espironolactona (dos factores que pueden predisponer al paciente a hiperpotasiemia), no se observó un incremento de las concentraciones séricas de potasio por encima del límite superior del rango normal. Puede concluirse que drospirenona es bien tolerada en pacientes con insuficiencia hepática leve o moderada (Child-Pugh B).
Grupos étnicos
No se han observado diferencias clínicamente relevantes en la farmacocinética de drospirenona, ni de etinilestradiol, entre mujeres japonesas y caucásicas.
Etinilestradiol
Absorción
Etinilestradiol se absorbe de forma rápida y completa después de administración oral.
Tras la administración de una única dosis, se alcanzan concentraciones plasmáticas máximas de 33 pg/ml al cabo de 1-2 horas después de la ingestión. Etinilestradiol está sujeto a un intenso metabolismo de primer paso, que muestra grandes variaciones de unos individuos a otros. La biodisponibilidad absoluta es del 60% aproximadamente. La ingesta concomitante de alimentos redujo la biodisponibilidad de etinilestradiol aproximadamente en el 25% de las pacientes investigadas, mientras que no se observó ningún cambio en el resto.
Distribución
Los niveles séricos de etinilestradiol disminuyen en dos fases, la fase de disposición terminal está caracterizada por una semivida de aproximadamente 24 horas. Etinilestradiol se une, mayoritariamente pero no de forma específica, a la albúmina sérica (aproximadamente en un 98,5%), e induce un aumento de las concentraciones séricas de SHBG y de la globulina fijadora de corticoides (CBG). Se ha determinado un volumen de distribución aparente de aproximadamente 5 l/kg.
Metabolismo
La metabolización del etinilestradiol está sujeta a la conjugación presistémica tanto en la mucosa del intestino delgado como en el hígado. Etinilestradiol se metaboliza, principalmente, por hidroxilación aromática aunque se forman una amplia variedad de metabolitos hidroxilados y metilados, que se presentan como metabolitos libres y como conjugados con glucuronides y sulfato Etinilestradiol se metaboliza completamente (aclaramiento plasmático metabólico: 5 ml/min/kg).
Eliminación
Etinilestradiol no se elimina en forma intacta en grado significativo. La proporción de eliminación urinaria: biliar de los metabolitos de etinilestradiol es de 4:6. La semivida de eliminación de los metabolitos es de 1 día aproximadamente.
Condiciones en estado estacionario
Las condiciones en estado estacionario se alcanzan durante la segunda mitad de un ciclo de tratamiento, y los niveles séricos de etinilestradiol se acumulan en un factor de aproximadamente 2,0 a 2,3.
5.3 Datos preclínicos sobre seguridad
En animales de laboratorio los efectos de drospirenona y etinilestradiol se limitaron a los asociados a la acción farmacológica conocida. En particular, los estudios de toxicidad para la reproducción revelaron efectos embriotóxicos y fetotóxicos en animales, considerados como específicos de especie. Tras la exposición a dosis de drospirenona superiores a las de las usuarias de Etinilestradiol/Drospirenona Diario Quasset, se observaron efectos sobre la diferenciación sexual en fetos de rata, pero no en monos.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Comprimidos activos (comprimidos amarillos):
Núcleo del comprimido:
Lactosa monohidrato
Almidón de maíz
Almidón de maíz pregelatinizado
Crospovidona
Povidona
Polisorbato 80
Estearato de magnesio Cubierta:
Alcohol polivinílico parcialmente hidrolizado Dióxido de titanio (E171)
Macrogol 3350 Talco
Óxido de hierro amarillo (E172)
Comprimidos placebo (comprimidos blancos):
Núcleo del comprimido:
Lactosa anhidra Povidona
Estearato de magnesio
Cubierta:
Alcohol polivinílico parcialmente hidrolizado Dióxido de titanio (E171)
Macrogol 3350 Talco
6.2 Incompatibilidades
No procede.
6.3 Período de validez
3 años.
6.4 Precauciones especiales de conservación
Este medicamento no requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Blísteres con lámina de aluminio de apertura mediante presión y película de PVC/PVDC.
Tamaños de envase:
1 x 28 comprimidos recubiertos con película (21 activos y 7 placebo)
2 x 28 comprimidos recubiertos con película (21 activos y 7 placebo)
3 x 28 comprimidos recubiertos con película (21 activos y 7 placebo)
6 x 28 comprimidos recubiertos con película (21 activos y 7 placebo)
13 x 28 comprimidos recubiertos con película (21 activos y 7 placebo)
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Qualitec Europa, S.L.
Paseo Pintor Rosales 42 28008 Madrid España
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
77273
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: Marzo 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
Mayo 2015
21/21