Emend 80 Mg Capsulas Duras
Información obsoleta, busque otroANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
EMEND 40 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula contiene 40 mg de aprepitant.
Excipiente con efecto conocido:
Cada cápsula contiene 40 mg de sacarosa.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura.
Las cápsulas son opacas con cuerpo blanco y tapa amarillo mostaza, con “464” y “40 mg” impreso en forma radial en tinta negra en el cuerpo.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
EMEND 40 mg está indicado para la prevención de náuseas y vómitos posquirúrgicos (NVPQ) en adultos.
4.2 Posología y forma de administración
Posología
Se deben considerar las directrices de tratamiento clínico en cuanto a la necesidad de tratamiento preventivo frente a las náuseas y vómitos posquirúrgicos (NVPQ).
La dosis oral recomendada de EMEND es una dosis única de 40 mg en el transcurso de las 3 horas anteriores a la inducción de la anestesia.
Poblaciones especiales
Pacientes de edad avanzada (> 65 años)
No es necesario ajustar la dosis en los pacientes de edad avanzada (ver sección 5.2).
Sexo
No es necesario ajustar la dosis según el sexo (ver sección 5.2).
Insuficiencia renal
No es necesario ajustar la dosis en los pacientes con insuficiencia renal ni en los pacientes con nefropatía terminal sometidos a hemodiálisis (ver sección 5.2).
Insuficiencia hepática
No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. Aprepitant se debe usar con precaución en estos pacientes (ver secciones 4.4 y 5.2).
Población pediátrica
No s e ha establecido la seguridad y eficacia de EMEND en niños y adolescentes menores de 18 años de edad. No se dispone de datos.
Forma de administración
Las cápsulas duras se deben tragar enteras.
EMEND puede tomarse con o sin alimentos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Pacientes con insuficiencia hepática moderada a grave
Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. EMEND se debe usar con precaución en estos pacientes (ver sección 5.2).
Interacciones con el CYP3A4
EMEND (40 mg) se debe usar con precaución en pacientes que estén recibiendo de forma concomitante la administración de pimozida, terfenadina, astemizol, cisaprida o derivados de los alcaloides del cornezuelo. La inhibición de la isoenzima 3A4 del citocromo P450 (CYP3A4) por aprepitant podría dar lugar a elevaciones de las concentraciones plasmáticas de estos principios activos, lo que podría provocar reacciones adversas graves (ver sección 4.5).
Se debe evitar la administración concomitante de EMEND con principios activos que sean potentes inductores de la actividad del CYP3A4 (por ejemplo, rifampicina, fenitoína, carbamazepina, fenobarbital), puesto que la combinación puede provocar un descenso en las concentraciones plasmáticas de aprepitant (ver sección 4.5). No se recomienda la administración concomitante de EMEND con preparados a base de plantas que contienen hipérico (Hypericum perforatum; también conocido como Hierba de San Juan).
Se debe tener especial precaución cuando se administre EMEND de forma concomitante con principios activos que sean inhibidores de la actividad del CYP3A4 (por ejemplo, ketoconazol, itraconazol, voriconazol, posaconazol, claritromicina, telitromicina, nefazodona e inhibidores de la proteasa), puesto que la combinación se espera que provoque un aumento de las concentraciones plasmáticas de aprepitant (ver sección 4.5).
Administración conjunta con anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver sección 4.5).
Para más información sobre la posible interacción de aprepitant a dosis elevadas y múltiples, consulte la Ficha Técnica o Resumen de las Características del Producto de EMEND 80 mg cápsulas duras y EMEND 125 mg cápsulas duras.
Excipientes
EMEND contiene sacarosa. Los pacientes con intolerancia hereditaria a la fructosa, problemas de absorción de glucosa o galactosa, o insuficiencia de sacarasa-isomaltasa, no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Aprepitant es un sustrato, y un inhibidor dependiente de la dosis, y un inductor de CYP3A4.
Aprepitant es también un inductor de CYP2C9. Durante el tratamiento, la dosis única de 40 mg de aprepitant recomendada para NVPQ da lugar a una inhibición débil de CYP3A4. Después del tratamiento, EMEND causa una inducción transitoria suave de CYP2C9, CYP3A4 y glucuronidación. Aprepitant se ha estudiado a dosis más elevadas. Durante el tratamiento de las náuseas y los vómitos inducidos por la quimioterapia (NVIQ), el tratamiento de 3 días con un régimen de 125 mg/80 mg de aprepitant da lugar a una inhibición moderada de CYP3A4. Aprepitant no parece que interaccione con el transportador de la P-glucoproteína, como sugiere la falta de interacción de aprepitant con digoxina.
Efecto de aprepitant sobre la farmacocinética de otros principios activos
Inhibición de CYP3A4
Como inhibidor débil de CYP3A4, aprepitant (40 mg) puede aumentar las concentraciones plasmáticas de los principios activos coadministrados por vía oral, que se metabolizan a través de CYP3A4. La exposición total de los sustratos de CYP3A4 que se administran por vía oral puede aumentar hasta aproximadamente 1,5 veces después de una dosis única de 40 mg de aprepitant; se estima que el efecto de aprepitant sobre las concentraciones plasmáticas de los sustratos de CYP3A4 que se administran por vía intravenosa sea menor.
EMEND 40 mg se debe usar con precaución en pacientes que estén recibiendo pimozida, terfenadina, astemizol, cisaprida o derivados de los alcaloides del cornezuelo. La inhibición de CYP3A4 por aprepitant podría dar lugar a elevaciones de las concentraciones plasmáticas de estos principios activos, lo que podría provocar reacciones graves.
Corticoesteroides
Dexametasona: Una dosis única de 40 mg de aprepitant, cuando se administra conjuntamente con una dosis única oral de 20 mg de dexametasona, aumenta el AUC de dexametasona en 1,45 veces. No se recomienda el ajuste de dosis.
Metilprednisolona: Aunque no se ha estudiado la administración concomitante de metilprednisolona con una dosis única de 40 mg de aprepitant, una dosis única de 40 mg de aprepitant produce una inhibición débil de CYP3A4 y no se espera que altere las concentraciones plasmáticas de metilprednisolona a un nivel clínicamente significativo. Por tanto, no se recomienda el ajuste de dosis.
Midazolam
El AUC de midazolam aumentó en 1,2 veces cuando una dosis única de 40 mg de aprepitant se administró conjuntamente con una dosis oral única de 2 mg de midazolam; este efecto no se consideró clínicamente importante.
Inducción
Como inductor suave de CYP2C9, CYP3A4 y glucuronidación, aprepitant puede disminuir las concentraciones plasmáticas de sustratos eliminados por estas vías durante las dos semanas posteriores al inicio del tratamiento. Para los sustratos CYP2C9 y CYP3A4 la inducción es transitoria con un efecto máximo alcanzado después de 3-5 días. El efecto se puede mantener durante unos pocos días y se espera que sea clínicamente insignificante a las 2 semanas después de terminar el tratamiento con EMEND. No existen datos relativos a los efectos sobre CYP2C8 y CYP2C19. La administración conjunta de EMEND con principios activos que se sabe que son metabolizados por CYP2C9 (por ej. fenitoína, warfarina) puede dar lugar a concentraciones plasmásticas más bajas de estos principios activos. En base a los ensayos de interacciones con tolbutamida y anticonceptivos orales, la exposición total de principios activos metabolizados por CYP2C9 o CYP3A4 administrados conjuntamente puede reducirse hasta el 15-30%.
Anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses
siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo.
Antagonistas 5-HT3
En ensayos clínicos de interacción, aprepitant no tuvo efectos clínicamente importantes sobre la farmacocinética de ondansetrón, granisetrón ni hidrodolasetrón (el metabolito activo de dolasetrón).
Efecto de otros medicamentos sobre la farmacocinética de aprepitant
La administración concomitante de EMEND con principios activos que inhiben la actividad de CYP3A4 (por ejemplo, ketoconazol, itraconazol, voriconazol, posaconazol, claritromicina, telitromicina, nefazodona e inhibidores de la proteasa) se debe abordar con precaución, ya que la combinación se espera que provoque una elevación de varias veces las concentraciones plasmáticas de aprepitant (ver sección 4.4).
La administración concomitante de EMEND con principios activos que inducen fuertemente la actividad de CYP3A4 (por ejemplo, rifampicina, fenitoína, carbamazepina, fenobarbital) se debe evitar ya que la combinación provoca reducciones en las concentraciones plasmáticas de aprepitant lo que podría provocar una disminución de la eficacia. No se recomienda la administración concomitante de EMEND con plantas medicinales que contienen hipérico (Hypericum perforatum).
Ketoconazol
Al administrarse una dosis única de 125 mg de aprepitant el día 5 de un régimen de 10 días de 400 mg/día de ketoconazol, un potente inhibidor de CYP3A4, el AUC de aprepitant aumentó aproximadamente 5 veces y la semivida terminal media de aprepitant aumentó aproximadamente 3 veces.
Rifampicina
Al administrarse una dosis única de 375 mg de aprepitant el día 9 de un régimen de 14 días de 600 mg/día de rifampicina, un potente inductor de CYP3A4, el AUC de aprepitant disminuyó un 91% y la semivida terminal media disminuyó un 68%.
4.6 Fertilidad, embarazo y lactancia
Anticoncepción en varones y mujeres
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de su administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver secciones 4.4 y 4.5).
Embarazo
No hay datos clínicos disponibles sobre la exposición a aprepitant durante el embarazo. En estudios en animales, no hubo indicios de efectos perjudiciales directos ni indirectos en términos del embarazo, el desarrollo embrionario/fetal, el parto o el desarrollo posnatal (ver sección 5.3). Se desconocen los posibles efectos sobre la reproducción de alteraciones en la regulación de la neurocinina. EMEND no se debe utilizar durante el embarazo excepto si fuese claramente necesario.
Lactancia
Aprepitant se excreta en la leche de ratas lactantes. Se desconoce si aprepitant se excreta en la leche materna; por consiguiente, no se recomienda la lactancia durante el tratamiento con EMEND.
Fertilidad
Los estudios de fertilidad no sugirieron efectos perjudiciales directos ni indirectos en términos del estado de apareamiento, fertilidad, desarrollo embrionario/fetal, o recuento de esperma y movilidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de EMEND sobre la capacidad para conducir y utilizar máquinas es pequeña. Se pueden producir mareos y fatiga después de la administración de EMEND (ver sección 4.8).
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad de aprepitant se evaluó en aproximadamente 6.500 sujetos.
Las reacciones adversas se notificaron en aproximadamente un 4% de los pacientes tratados con 40 mg de aprepitant en comparación con aproximadamente un 6% de los pacientes que recibieron 4 mg de ondansetrón vía intravenosa. En ensayos clínicos controlados en pacientes recibiendo anestesia general, se administraron 40 mg de aprepitant por vía oral a 564 pacientes y se administraron 4 mg de ondansetrón vía intravenosa a 538 pacientes. La mayoría de las reacciones adversas en estos ensayos clínicos fueron descritas con intensidad de leve a moderada.
La reacción adversa más frecuente, notificada con mayor incidencia en pacientes tratados con 40 mg de aprepitant (1,1 %) que con ondansetrón (1,0 %) fue ALT elevada.
Lista tabulada de reacciones adversas
Las reacciones adversas siguientes se observaron en ensayos en NVPQ en pacientes tratados con aprepitant con una incidencia mayor que con ondansetrón o en el uso después de la comercialización:
Las frecuencias se definen como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000) y muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Clasificación de órganos del Sistema |
Reacción adversa |
Frecuencia |
|
Trastornos del sistema inmunológico |
reacciones de hipersensibilidad incluyendo reacciones anafilácticas |
no conocida |
|
Trastornos psiquiátricos |
insomnio |
poco frecuentes |
|
Trastornos del sistema nervioso |
disartria, hipoestesia, alteración sensitiva |
poco frecuentes |
|
Trastornos oculares |
miosis, agudeza visual disminuida |
poco frecuentes |
|
Trastornos cardiacos |
bradicardia |
poco frecuentes |
|
Trastornos respiratorios, torácicos y mediastínicos |
disnea, sibilancia |
poco frecuentes |
|
Trastornos gastrointestinales |
dolor abdominal alto, ruidos intestinales anormales, boca seca, náuseas, molestias en el estómago, estreñimiento*, subíleo* |
poco frecuentes |
|
Trastornos de la piel y del tejido subcutáneo |
prurito, erupción, urticaria, síndrome de Stevens-Johnson/necrolisis epidérmica tóxica |
no conocida |
|
Exploraciones complementarias |
ALT elevada |
frecuentes |
*Notificadas en pacientes que tomaban una dosis más alta de aprepitant.
Descripción de reacciones adversas seleccionadas
Se observaron otras reacciones adversas en pacientes tratados con el régimen de aprepitant (125 mg/80 mg) para náuseas y vómitos inducidos por quimioterapia (NVIQ), con una incidencia mayor que con el tratamiento estándar: distensión abdominal, dolor abdominal, acné, anemia, ansiedad, AST elevada, astenia, fosfatasa alcalina aumentada, sodio disminuido en sangre, candidiasis, trastorno cardiovascular, malestar torácico, trastorno cognoscitivo, conjuntivitis, tos, apetito disminuido, desorientación, mareo, perforación de úlcera duodenal, disgeusia, dispepsia, disuria, eructos, estado de ánimo eufórico, heces duras, fatiga, neutropenia febril, flatulencia, alteración de la marcha, enfermedad por reflujo gastroesofágico, presencia de glucosuria, hipo, acaloramiento, hiperhidrosis, letargía, malestar general, espasmos musculares, debilidad muscular, náuseas*, colitis neutropénica, recuento disminuido de neutrófilos, edema, dolor orofaríngeo, palpitaciones, reacción de fotosensibilidad, polaquiuria, polidipsia, goteo postnasal, erupción prurítica, hematíes en orina positivos, seborrea, lesión de la piel, estornudos, somnolencia, infección estafilocócica, estomatitis, irritación de garganta, acúfenos, excreción urinaria aumentada, vómitos*, peso disminuido.
*Náuseas y vómitos fueron parámetros de eficacia en los 5 primeros días de tratamiento post-quimioterapia y sólo después se notificaron como reacciones adversas.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
En caso de sobredosis, se debe suspender la administración de EMEND y proporcionar tratamiento de apoyo general y vigilancia. Debido a la actividad antiemética de aprepitant, es posible que la emesis inducida por un medicamento no resulte eficaz.
Aprepitant no puede eliminarse mediante hemodiálisis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antieméticos y antinauseosos, código ATC: A04AD12
Aprepitant es un antagonista selectivo de alta afinidad por los receptores de la sustancia P neurocinina 1 (NKi) humana.
En 2 ensayos clínicos en fase III de grupos paralelos, multicéntricos, aleatorizados, doble ciego, controlados con un comparador activo, se comparó aprepitant con ondansetrón en la prevención de NVPQ en 1.658 pacientes sometidos a cirugía abdominal abierta. La mayoría de los pacientes eran mujeres (>90%), principalmente sometidas a cirugía ginecológica. Los pacientes fueron aleatorizados a recibir 40 mg de aprepitant, 125 mg de aprepitant o 4 mg de ondansetrón. Se administró aprepitant por vía oral con 50 ml de agua de 1 a 3 horas antes de la anestesia. Ondansetrón se administró por vía intravenosa inmediatamente antes de la inducción de la anestesia. Se evaluó la actividad entiemética de aprepitant durante el periodo de 0 a 48 horas después de finalizar la operación.
Los resultados muestran que un porcentaje mayor de pacientes posquirúrgicos experimentaron una respuesta completa (sin emesis y sin utilizar medicación de rescate) con 40 mg de aprepitant que con 4 mg de ondansetrón (el límite inferior del IC es 0,0, lo que indica significación limítrofe), como se describe en la Tabla 1:
Tabla 1
Porcentaje de pacientes posquirúrgicos que respondieron por grupo de tratamiento Resultados combinados de 2 ensayos en fase III
|
Aprepitant 40 mg, vía oral (N=541) |
Ondansetrón 4 mg, vía intravenosa (N=526) |
Diferencia de puntos de porcentaje (%) § e IC del 95% # | ||||
|
n/m |
(%) |
n/m |
(%) |
% |
IC del 95% | |
|
Respuesta completa (0-24 horas) t |
298/541 |
(55,1) |
258/526 |
(49,0) |
5,9 |
(0,0, 11,8) |
f Respuesta completa: sin emesis y sin medicación de rescate § Diferencia (%) calculada como aprepitant 40 mg menos ondansetrón 4 mg # Diferencia (%) e IC del 95% calculado usando el método estratificado de Miettinen-Nurminen, usando los pesos de Cochran-Mantel-Haenszel
La reducción del riesgo de un episodio de vómitos en el periodo de 0 a 24 horas, con 40 mg de aprepitant en relación con 4 mg de ondansetrón fue del 53,3 % (IC del 95%: 35,3 a 66,3), en un análisis que no tuvo en cuenta a los pacientes en el momento de usar la medicación de rescate.
Población pediátrica
Los ensayos clínicos para evaluar el uso de aprepitant en pacientes pediátricos están en curso (ver sección 4.2 para la información sobre el uso pediátrico).
5.2 Propiedades farmacocinéticas
Aprepitant muestra una farmacocinética no lineal. Tanto el aclaramiento como la biodisponibilidad absoluta disminuyen al aumentar la dosis.
Absorción
La biodisponibilidad oral absoluta media de aprepitant es de 67% para la cápsula de 80 mg y de 59% para la cápsula de 125 mg. La concentración plasmática máxima media (Cmax) de aprepitant se alcanzó aproximadamente a las 4 horas (tnax).
Tras la administración oral de una dosis única de 40 mg de EMEND en ayunas, el AUC0-OT (media ± DE) fue 8,0 ±2,1 ^g • h/ml y la Cmax fue de 0,7 ± 0,24 ^g/ml. La tnax media fue de 3,0 h.
La ingesta concomitante de una dosis de 40 mg con un desayuno estándar, sólo disminuyó la Cmax de aprepitant en un 18 % pero no afectó al AUC. Esto no se consideró clínicamente importante.
Distribución
Aprepitant se une fuertemente a proteínas, con una media del 97%. La media geométrica del volumen aparente de distribución en el estado equilibrio (Vdee) es aproximadamente de 66 litros en el ser humano.
Biotransformación
Aprepitant se metaboliza extensamente. En adultos jóvenes sanos, aprepitant representa aproximadamente el 19% de la radiactividad plasmática durante 72 horas después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant, lo que indica una importante presencia de metabolitos en el plasma. En el plasma humano se han identificado doce metabolitos de aprepitant. El metabolismo de aprepitant se produce en gran medida por oxidación en el anillo de morfolina y sus cadenas laterales y los metabolitos resultantes sólo fueron débilmente activos. Estudios in vitro en los que se usaron microsomas hepáticos humanos indicaron que aprepitant se metaboliza principalmente a través de CYP3A4 y posiblemente con una contribución menor a través de CYP1A2 y CYP2C19.
Eliminación
Aprepitant no se elimina inalterado en la orina. Los metabolitos se eliminan en la orina y a través de excreción biliar en las heces. Después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant a sujetos sanos, el 57% de la radiactividad se recuperó en la orina y el 45% en las heces.
El aclaramiento plasmático de aprepitant es dependiente de la dosis, disminuyendo al aumentar la dosis y oscilando aproximadamente entre 60 a 72 ml/min en el intervalo de la dosis terapéutica. La semivida terminal es de aproximadamente 9 horas después de la administración de una dosis única de 40 mg.
Farmacocinética en poblaciones especiales
Pacientes de edad avanzada: Tras la administración oral de una dosis única de 125 mg de aprepitant el día 1 y 80 mg una vez al día los días 2 a 5, el AUC0-24h de aprepitant fue un 21% superior el día 1 y un 36% superior el día 5 en pacientes de edad avanzada (>65 años) respecto de los adultos más jóvenes. La Cmax fue un 10% superior el día 1 y un 24% superior el día 5 en pacientes de edad avanzada respecto de los adultos más jóvenes. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en los pacientes de edad avanzada.
Sexo: Tras la administración oral de una dosis única de 125 mg de aprepitant, la Cmax de aprepitant es un 16% superior en las mujeres en comparación con los varones. La semivida de aprepitant es un 25% inferior en las mujeres en comparación con los varones y su tmax se produce en aproximadamente el mismo tiempo. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en función del sexo.
Insuficiencia hepática: La insuficiencia hepática leve (Child-Pugh clase A) no afecta a la farmacocinética de aprepitant en un grado clínicamente relevante. No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. De los datos disponibles no pueden extraerse conclusiones relativas a la influencia de la insuficiencia hepática moderada (Child-Pugh clase B) sobre la farmacocinética de aprepitant. No existen datos clínicos ni farmacocinéticos de pacientes con insuficiencia hepática grave (Child-Pugh clase C).
Insuficiencia renal: Se administró una dosis única de 240 mg de aprepitant a pacientes con insuficiencia renal grave (CrCl<30 ml/min) y a pacientes con nefropatía terminal que requería hemodiálisis.
En los pacientes con insuficiencia renal grave, el AUC0-<X) de aprepitant total (no unido y unido a proteínas) disminuyó en un 21 % y la Cmax disminuyó en un 32 %, respecto de los sujetos sanos. En los pacientes con nefropatía terminal sometidos a hemodiálisis, el AUC0-<X) de aprepitant total disminuyó en un 42 % y la Cmax disminuyó en un 32 %. Debido a los modestos descensos en la unión a proteínas de aprepitant en los pacientes con enfermedad renal, el AUC de aprepitant no unido farmacológicamente activo no se vio significativamente afectado en los pacientes con insuficiencia renal en comparación con los sujetos sanos. La hemodiálisis realizada 4 ó 48 horas después de la administración no tuvo efectos significativos sobre la farmacocinética de aprepitant; en el dializado se recuperó menos de 0,2 % de la dosis.
En pacientes con insuficiencia renal o en pacientes con nefropatía terminal sometidos a hemodiálisis no es necesario ajustar la dosis de EMEND.
Relación entre concentración y efecto
Usando un trazador altamente específico del receptor NKb los estudios de tomografía por emisión de positrones (PET) en varones jóvenes sanos han demostrado que aprepitant penetra en el cerebro y ocupa los receptores NK de forma dependiente de la dosis y de la concentración plasmática. Se predice que las concentraciones plasmáticas de aprepitant alcanzadas con el régimen de 3 días de EMEND proporcionarán una ocupación superior al 95 % de los receptores NK cerebrales.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de toxicidad, genotoxicidad, potencial carcinogénico y toxicidad para la reproducción a dosis únicas y repetidas. Se deberá tener en cuenta que la exposición sistémica en ratas macho fue inferior a la exposición terapéutica en seres humanos a la dosis de 40 mg. Consecuentemente, no se puede hacer una evaluación adecuada de los posibles efectos sobre la fertilidad en ratas macho. Sin embargo, en un estudio de 9 meses en perros, no se observaron cambios en el peso de los órganos, ni hallazgos macroscópicos ni histomorfológicos en órganos reproductores masculinos a niveles de exposición sistémica 35 veces superior a la exposición terapéutica en seres humanos a la dosis de 40 mg. Aunque no se observaron efectos adversos en los estudios sobre la
reproducción cuando animales hembra se expusieron a 3,5 a 4 veces por encima de la exposición terapéutica en seres humanos con 40 mg, se desconocen los posibles efectos sobre la reproducción de alteraciones en la regulación de la neurocinina.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula Sacarosa
Celulosa microcristalina (E 460)
Hidroxipropilcelulosa (E 463)
Lauril sulfato de sodio
Cubierta de la cápsula Gelatina
Dióxido de titanio (E 171)
Óxido férrico amarillo (E 172)
Tinta para impresión Laca
Hidróxido de potasio Óxido férrico negro (E 172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años
6.4 Precauciones especiales de conservación
Conservar en el embalaje original para protegerlo de la humedad.
6.5 Naturaleza y contenido del envase
Están disponibles diferentes tamaños de envase:
Blister de aluminio conteniendo una cápsula de 40 mg.
5 blisters de aluminio conteniendo cada uno una cápsula de 40 mg.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
Ninguna especial para su eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/007
EU/1/03/262/008
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 11/noviembre/2003 Fecha de la última renovación: 11/noviembre/2008
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
1. NOMBRE DEL MEDICAMENTO
EMEND 80 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula contiene 80 mg de aprepitant.
Excipiente con efecto conocido:
Cada cápsula contiene 80 mg de sacarosa.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura.
Las cápsulas son opacas con cuerpo y tapa blancos y con “461” y “80 mg” impreso en forma radial en tinta negra en el cuerpo.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Prevención de las náuseas y los vómitos agudos y diferidos que se asocian con la quimioterapia antineoplásica altamente emetógena basada en el cisplatino en adultos.
Prevención de las náuseas y los vómitos que se asocian con la quimioterapia antineoplásica moderadamente emetógena en adultos.
EMEND 80 mg se administra como parte de un tratamiento de combinación (ver sección 4.2).
4.2 Posología y forma de administración
Posología
EMEND se administra durante 3 días como parte de un régimen que incluye un corticoesteroide y un antagonista 5-HT3. La dosis recomendada es de 125 mg por vía oral, una vez al día, una hora antes de empezar la quimioterapia el día 1 y 80 mg por vía oral, una vez al día, los días 2 y 3.
Se recomiendan los siguientes regímenes de administración para la prevención de las náuseas y los vómitos asociados con la quimioterapia antineoplásica emetógena:
|
Día 1 |
Día 2 |
Día 3 |
Día 4 | |
|
EMEND |
125 mg vía oral |
80 mg vía oral |
80 mg vía oral |
nada |
|
Dexametasona |
12 mg vía oral |
8 mg vía oral |
8 mg vía oral |
8 mg vía oral |
|
Antagonistas 5-HT3 |
Dosis habituales de los antagonistas 5-HT3. Verla información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada |
nada |
nada |
nada |
Dexametasona se debe administrar 30 minutos antes de la quimioterapia el día 1 y por la mañana los días 2 a 4. La dosis de dexametasona es responsable de las interacciones del principio activo.
Régimen de quimioterapia moderadamente emetógena
|
Día 1 |
Día 2 |
Día 3 | |
|
EMEND |
125 mg vía oral |
80 mg vía oral |
80 mg vía oral |
|
Dexametasona |
12 mg vía oral |
nada |
nada |
|
Antagonistas 5-HT3 |
Dosis habituales de los antagonistas 5-HT3. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada |
nada |
nada |
Dexametasona se debe administrar 30 minutos antes de la quimioterapia el día 1. La dosis de dexametasona es responsable de las interacciones del principio activo.
Los datos de eficacia en combinación con otros corticoesteroides y antagonistas 5-HT3 son limitados. Para información adicional sobre la co-administración con corticoesteroides, ver sección 4.5.Consultar la Ficha Técnica o Resumen de las Características del Producto de los medicamentos antagonistas 5-HT3 coadministrados.
Poblaciones especiales
Pacientes de edad avanzada (> 65 años)
No es necesario ajustar la dosis en los pacientes de edad avanzada (ver sección 5.2).
Sexo
No es necesario ajustar la dosis según el sexo (ver sección 5.2).
Insuficiencia renal
No es necesario ajustar la dosis en los pacientes con insuficiencia renal ni en los pacientes con nefropatía terminal sometidos a hemodiálisis (ver sección 5.2).
Insuficiencia hepática
No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. Aprepitant se debe usar con precaución en estos pacientes (ver secciones 4.4 y 5.2).
Población pediátrica
No s e ha establecido la seguridad y eficacia de EMEND en niños y adolescentes menores de 18 años de edad. No se dispone de datos.
Forma de administración
Las cápsulas duras se deben tragar enteras.
EMEND puede tomarse con o sin alimentos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Co-administración con pimozida, terfenadina, astemizol o cisaprida (ver sección 4.5).
4.4 Advertencias y precauciones especiales de empleo Pacientes con insuficiencia hepática moderada a grave
Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. EMEND se debe usar con precaución en estos pacientes (ver sección 5.2).
Interacciones con el CYP3A4
EMEND se debe usar con precaución en pacientes que estén recibiendo de forma concomitante principios activos, administrados por vía oral, metabolizados principalmente a través de CYP3A4 y con un rango terapéutico estrecho, tales como ciclosporina, tacrolimus, sirolimus, everolimus, alfentanilo, diergotamina, ergotamina, fentanilo y quinidina (ver sección 4.5). Adicionalmente, la administración concomitante con irinotecano se debe abordar con especial prudencia ya que esta combinación puede provocar un aumento de la toxicidad.
La administración conjunta de EMEND con derivados de los alcaloides del cornezuelo, que son sustratos de CYP3A4, puede dar lugar a elevaciones de las concentraciones plasmáticas de estos principios activos. Por lo tanto, se aconseja precaución debido al riesgo potencial de toxicidad relacionada con el cornezuelo del centeno.
Se debe evitar la administración concomitante de EMEND con principios activos que sean potentes inductores de la actividad del CYP3A4 (por ejemplo, rifampicina, fenitoína, carbamazepina, fenobarbital), puesto que la combinación puede provocar un descenso en las concentraciones plasmáticas de aprepitant (ver sección 4.5). No se recomienda la administración concomitante de EMEND con preparados a base de plantas que contienen hipérico (Hypericum perforatum; también conocido como Hierba de San Juan).
Se debe tener especial precaución cuando se administre EMEND de forma concomitante con principios activos que sean inhibidores de la actividad del CYP3A4 (por ejemplo, ketoconazol, itraconazol, voriconazol, posaconazol, claritromicina, telitromicina, nefazodona e inhibidores de la proteasa), puesto que la combinación se espera que provoque un aumento de las concentraciones plasmáticas de aprepitant (ver sección 4.5).
Administración conjunta con warfarina (un sustrato de CYP2C9)
La administración conjunta de EMEND con warfarina da lugar a una disminución del tiempo de protrombina, comunicado como coeficiente internacional normalizado (INR, International Normalised Ratio). En pacientes en tratamiento crónico con warfarina, el INR se debe vigilar estrechamente durante el tratamiento con EMEND y durante 14 días después de cada ciclo de 3 días de EMEND (ver sección 4.5).
Administración conjunta con anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver sección 4.5).
Excipientes
EMEND contiene sacarosa. Los pacientes con intolerancia hereditaria a la fructosa, problemas de absorción de glucosa o galactosa, o insuficiencia de sacarasa-isomaltasa, no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Aprepitant (125 mg/80 mg) es un sustrato, un inhibidor moderado, y un inductor de CYP3A4. Aprepitant es también un inductor de CYP2C9. Durante el tratamiento con EMEND, CYP3A4 se inhibe. Después de terminar el tratamiento, EMEND produce una inducción transitoria suave de CYP2C9, de CYP3A4 y glucuronidación. Aprepitant no parece que interaccione con el transportador de la P-glucoproteína, como sugiere la falta de interacción de aprepitant con digoxina.
Efecto de aprepitant sobre la farmacocinética de otros principios activos
Inhibición de CYP3A4
Como inhibidor moderado de CYP3A4, aprepitant (125 mg/80 mg) puede aumentar las concentraciones plasmáticas de los principios activos que se metabolizan a través de CYP3A4 cuando se administran conjuntamente. La exposición total de los sustratos de CYP3A4 que se administran por vía oral puede aumentar hasta aproximadamente tres veces durante el tratamiento de 3 días con EMEND; se estima que el efecto de aprepitant sobre las concentraciones plasmáticas de los sustratos de CYP3A4 que se administran por vía intravenosa sea menor. EMEND no se debe usar simultáneamente con pimozida, terfenadina, astemizol o cisaprida (ver sección 4.3). La inhibición de CYP3A4 por aprepitant podría dar lugar a elevaciones de las concentraciones plasmáticas de estos principios activos, lo que podría provocar reacciones graves o potencialmente mortales. Se aconseja precaución durante la administración concomitante de EMEND y principios activos, administrados por vía oral, metabolizados principalmente a través de CYP3A4 y con un rango terapéutico estrecho, tales como ciclosporina, tacrolimus, sirolimus, everolimus, alfentanilo, diergotamina, ergotamina, fentanilo y quinidina (ver sección 4.4).
Corticoesteroides
Dexametasona: La dosis habitual de dexametasona oral se debe reducir aproximadamente en un 50 % cuando se administra conjuntamente con un tratamiento de 125 mg/80 mg de EMEND. La dosis de dexametasona utilizada en los ensayos clínicos de náuseas y vómitos inducidos por quimioterapia, se eligió en función de las interacciones del principio activo (ver sección 4.2). EMEND, administrado en un régimen de 125 mg conjuntamente con 20 mg de dexametasona oral el día 1, y EMEND, administrado en un régimen de 80 mg/día conjuntamente con 8 mg de dexametasona oral los días 2 a 5, aumentó el AUC de dexametasona, un sustrato de CYP3A4, 2,2 veces los días 1 y 5.
Metilprednisolona: La dosis habitual de metilprednisolona intravenosa se debe reducir aproximadamente un 25 %, y la dosis habitual de metilprednisolona oral se debe reducir aproximadamente un 50 % al administrarse conjuntamente con un tratamiento de 125 mg/80 mg de EMEND. EMEND, administrado en un régimen de 125 mg el día 1 y 80 mg/día los días 2 y 3, aumentó el AUC de metilprednisolona, un sustrato de CYP3A4, 1,3 veces el día 1 y 2,5 veces el día 3, al administrarse conjuntamente metilprednisolona por vía intravenosa, 125 mg el día 1, y por vía oral, 40 mg los días 2 y 3.
Durante el tratamiento continuo con metilprednisolona, el AUC de metilprednisolona puede disminuir en puntos de tiempo posteriores en el transcurso de las 2 semanas siguientes al inicio de la administración de EMEND, debido al efecto inductor de aprepitant sobre CYP3A4. Puede ser que este efecto sea más pronunciado para metilprednisolona administrada oralmente.
Antineoplásicos
En ensayos farmacocinéticos, EMEND, administrado en un régimen de 125 mg el día 1 y 80 mg/día los días 2 y 3, no influyó en la farmacocinética de docetaxel administrado por vía intravenosa el día 1 ni en la de vinorelbina administrada por vía intravenosa el día 1 o el día 8. Debido a que el efecto de EMEND sobre la farmacocinética de sustratos de CYP3A4 administrados por vía oral es mayor que el efecto de EMEND sobre la farmacocinética de sustratos de CYP3A4 administrados por vía intravenosa, no puede excluirse una interacción con medicamentos antineoplásicos administrados por vía oral que se metabolizan principal o parcialmente a través de CYP3A4 (p. ej. etopósido, vinorelbina). En pacientes que reciben medicamentos que se metabolizan principal o parcialmente a través de CYP3A4, se aconseja precaución y puede ser conveniente una vigilancia adicional (ver sección 4.4). Se han notificado acontecimientos adversos de neurotoxicidad postcomercialización, una reacción adversa potencial de ifosfamida, tras la administración simultánea de aprepitant e ifosfamida.
Inmunosupresores
Durante el régimen de tratamiento de 3 días de las náuseas y vómitos inducidos por quimioterapia, se espera un incremento moderado transitorio seguido de una leve disminución en la exposición de los inmunosupresores metabolizados por CYP3A4 (por ej. ciclosporina, tacrolimus, everolimus y sirolimus). Dada la corta duración del régimen de tratamiento de 3 días y los cambios limitados en la exposición dependientes del tiempo, no se recomienda una reducción de dosis de los inmunosupresores durante los 3 días de la administración conjunta con EMEND.
Midazolam
Los posibles efectos de aumentos en las concentraciones plasmáticas de midazolam u otras benzodiazepinas metabolizadas a través de CYP3A4 (alprazolam, triazolam) se deben tener en cuenta al administrar estos medicamentos conjuntamente con EMEND (125 mg/80 mg).
EMEND aumentó el AUC de midazolam, un sustrato sensible de CYP3A4, 2,3 veces el día 1 y 3,3 veces el día 5, al administrarse conjuntamente una dosis oral única de 2 mg de midazolam los días 1 y 5 de un régimen de EMEND 125 mg el día 1 y 80 mg/día los días 2 a 5.
En otro ensayo con administración intravenosa de midazolam, EMEND se administró como 125 mg el día 1 y 80 mg/día los días 2 y 3, y 2 mg de midazolam se administró por vía intravenosa antes de la administración del régimen de 3 días de EMEND y en los días 4, 8 y 15. EMEND aumentó el AUC de midazolam un 25 % el día 4 y disminuyó el AUC de midazolam un 19% el día 8 y un 4% el día 15. Estos efectos no se consideraron clínicamente importantes.
En un tercer ensayo con administración intravenosa y oral de midazolam, se administraron 125 mg de EMEND en el día 1 y 80 mg/día en los días 2 y 3, junto con 32 mg de ondansetrón el día 1, 12 mg de dexametasona el día 1 y 8 mg los días 2-4. Esta combinación (esto es, EMEND, ondansetrón y dexametasona) disminuyó el AUC de midazolam oral un 16 % el día 6, un 9 % el día 8, un 7 % el día 15 y un 17 % el día 22. Estos efectos no se consideraron clínicamente importantes.
Se finalizó un ensayo adicional con administración intravenosa de midazolam y EMEND. Una hora después de la administración oral de una dosis única de EMEND 125 mg, se administraron por vía intravenosa 2 mg de midazolam. El AUC plasmático de midazolam aumentó en 1,5 veces. Este efecto no se consideró clínicamente importante.
Inducción
Como inductor suave de CYP2C9, de CYP3A4 y de la glucuronidación, aprepitant puede disminuir las concentraciones plasmáticas de los sustratos eliminados por estas vías durante las dos semanas posteriores al inicio del tratamiento. Este efecto puede hacerse evidente únicamente después de finalizar el tratamiento de 3 días con EMEND. Para los sustratos de CYP2C9 y CYP3A4, la inducción es transitoria con un efecto máximo alcanzado a los 3-5 días después de finalizar el tratamiento de 3 días con EMEND. El efecto se mantiene durante unos pocos días, después desciende lentamente y es clínicamente insignificante a las 2 semanas después de finalizar el tratamiento con EMEND. La inducción suave de la glucuronidación también se observa con 80 mg de aprepitant oral administrado durante 7 días. Se carece de datos relativos a los efectos sobre CYP2C8 y CYP2C19. Se aconseja precaución al administrar durante este periodo de tiempo warfarina, acenocumarol, tolbutamida, fenitoína u otros principios activos que se sabe que son metabolizados por CYP2C9.
Warfarina
En pacientes en tratamiento crónico con warfarina, el tiempo de protrombina (INR) se debe vigilar estrechamente durante el tratamiento con EMEND y durante 2 semanas después de cada ciclo de 3 días de EMEND en el caso de náuseas y vómitos inducidos por quimioterapia (ver sección 4.4). Al administrarse una dosis única de 125 mg de EMEND el día 1 y 80 mg/día los días 2 y 3 a sujetos sanos estabilizados en un tratamiento crónico con warfarina, no se observó ningún efecto de EMEND sobre el AUC plasmático de R(+) o S(-) warfarina determinado el día 3; sin embargo, se observó un descenso del 34% en la concentración mínima de S(-) warfarina (un sustrato de CYP2C9) acompañado de un descenso del 14% en el INR 5 días después de finalizar el tratamiento con EMEND.
Tolbutamida
EMEND, administrado como 125 mg el día 1 y 80 mg/día los días 2 y 3, disminuyó el AUC de tolbutamida (un sustrato de CYP2C9) en un 23% el día 4, un 28% el día 8 y un 15% el día 15, al administrarse una dosis única de tolbutamida 500 mg por vía oral antes de la administración de un régimen de 3 días de EMEND y en los días 4, 8 y 15.
Anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo.
En un ensayo clínico, dosis únicas de un anticonceptivo oral que contenía etinil estradiol y noretindrona se administraron en los días 1 hasta el 21 con EMEND, administrado con una pauta posológica de 125 mg en el día 8 y 80 mg/día en los días 9 y 10 con ondansetrón 32 mg vía intravenosa en el día 8 y dexametasona oral administrada como 12 mg en el día 8 y 8 mg/día los Días 9, 10 y 11. Durante los días 9 hasta el 21 en este ensayo, hubo un descenso hasta del 64 % en las concentraciones mínimas de etinil estradiol y hasta del 60% en las concentraciones mínimas de noretindrona.
Antagonistas 5-HT3
En ensayos clínicos de interacción, aprepitant no tuvo efectos clínicamente importantes sobre la farmacocinética de ondansetrón, granisetrón ni hidrodolasetrón (el metabolito activo de dolasetrón).
Efecto de otros medicamentos sobre la farmacocinética de aprepitant
La administración concomitante de EMEND con principios activos que inhiben la actividad de CYP3A4 (por ejemplo, ketoconazol, itraconazol, voriconazol, posaconazol, claritromicina, telitromicina, nefazodona e inhibidores de la proteasa) se debe abordar con precaución, ya que la combinación se espera que provoque una elevación de varias veces las concentraciones plasmáticas de aprepitant (ver sección 4.4).
La administración concomitante de EMEND con principios activos que inducen fuertemente la actividad de CYP3A4 (por ejemplo, rifampicina, fenitoína, carbamazepina, fenobarbital) se debe evitar ya que la combinación provoca reducciones en las concentraciones plasmáticas de aprepitant lo que podría provocar una disminución de la eficacia de EMEND. No se recomienda la administración concomitante de EMEND con plantas medicinales que contienen hipérico (Hypericum perforatum).
Ketoconazol
Al administrarse una dosis única de 125 mg de aprepitant el día 5 de un régimen de 10 días de 400 mg/día de ketoconazol, un potente inhibidor de CYP3A4, el AUC de aprepitant aumentó aproximadamente 5 veces y la semivida terminal media de aprepitant aumentó aproximadamente 3 veces.
Rifampicina
Al administrarse una dosis única de 375 mg de aprepitant el día 9 de un régimen de 14 días de
600 mg/día de rifampicina, un potente inductor de CYP3A4, el AUC de aprepitant disminuyó un 91 %
y la semivida terminal media disminuyó un 68 %.
4.6 Fertilidad, embarazo y lactancia
Anticoncepción en varones y mujeres
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de su administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver secciones 4.4 y 4.5).
Embarazo
No hay datos clínicos disponibles sobre la exposición a aprepitant durante el embarazo. La capacidad de aprepitant para provocar toxicidad sobre la reproducción no se ha caracterizado completamente, ya que en los estudios animales no se pudieron alcanzar niveles de exposición por encima de la exposición terapéutica en seres humanos a la dosis de 125 mg/80 mg. Estos estudios no sugirieron efectos perjudiciales directos ni indirectos en términos del embarazo, el desarrollo embrionario/fetal, el parto o el desarrollo posnatal (ver sección 5.3). Se desconocen los posibles efectos sobre la reproducción de alteraciones en la regulación de la neurocinina. EMEND no se debe utilizar durante el embarazo excepto si fuese claramente necesario.
Lactancia
Aprepitant se excreta en la leche de ratas lactantes. Se desconoce si aprepitant se excreta en la leche materna; por consiguiente, no se recomienda la lactancia durante el tratamiento con EMEND.
Fertilidad
El potencial efecto de aprepitant sobre la fertilidad no se ha caracterizado completamente, ya que en los estudios en animales no se pudieron alcanzar niveles de exposición por encima de la exposición terapéutica en humanos. Estos estudios de fertilidad no sugirieron efectos perjudiciales directos ni indirectos en términos del estado de apareamiento, fertilidad, desarrollo embrionario/fetal, o recuento de esperma y movilidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de EMEND sobre la capacidad para conducir y utilizar máquinas es pequeña. Se pueden producir mareos y fatiga después de la administración de EMEND (ver sección 4.8).
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad de aprepitant se evaluó en aproximadamente 6.500 sujetos.
Las reacciones adversas más frecuentes que se notificaron con una mayor incidencia en pacientes tratados con el régimen de aprepitant que con el tratamiento estándar en pacientes que estaban recibiendo quimioterapia altamente emetógena (HEC) fueron: hipo (4,6 % versus 2,9 %), alanina aminotransferasa (ALT) elevada (2,8 % versus 1,1 %), dispepsia (2,6 % versus 2,0 %), estreñimiento (2,4 % versus 2,0 %), cefalea (2,0 % versus 1,8 %) y apetito disminuido (2,0 % versus 0,5 %). La reacción adversa más frecuente notificada con una mayor incidencia en pacientes tratados con el régimen de aprepitant que con el tratamiento estándar en pacientes que estaban recibiendo quimioterapia moderadamente emetógena (MEC) fue fatiga (1,4 % versus 0,9 %).
Lista tabulada de reacciones adversas
Las reacciones adversas siguientes se observaron en un análisis combinado de los ensayos en HEC y en MEC con una incidencia mayor con aprepitant que con el tratamiento estándar o en el uso después de la comercialización:
Las frecuencias se definen como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000) y muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Clasificación de órganos del Sistema |
Reacción adversa |
Frecuencia |
|
Infecciones e infestaciones |
candidiasis, infección estafilocócica |
raras |
|
Trastornos de la sangre y del sistema linfático |
neutropenia febril, anemia |
poco frecuentes |
|
Trastornos del sistema inmunológico |
reacciones de hipersensibilidad incluyendo reacciones anafilácticas |
no conocida |
|
Trastornos del metabolismo y de la nutrición |
apetito disminuido |
frecuentes |
|
polidipsia |
raras | |
|
Trastornos psiquiátricos |
ansiedad |
poco frecuentes |
|
desorientación, estado de ánimo eufórico |
raras | |
|
Trastornos del sistema nervioso |
cefalea |
frecuentes |
|
mareo, somnolencia |
poco frecuentes | |
|
trastorno cognoscitivo, letargia, disgeusia |
raras | |
|
Trastornos oculares |
conjuntivitis |
raras |
|
Trastornos del oído y del laberinto |
acúfenos |
raras |
|
Trastornos cardiacos |
palpitaciones |
poco frecuentes |
|
bradicardia, trastorno cardiovascular |
raras | |
|
Trastornos respiratorios, torácicos y mediastínicos |
hipo |
frecuentes |
|
dolor orofaríngeo, estornudos, tos, goteo postnasal, irritación de garganta |
raras | |
|
Trastornos gastrointestinales |
estreñimiento, dispepsia |
frecuentes |
|
eructos, náuseas*, vómitos*, enfermedad por reflujo gastroesofágico, dolor abdominal, boca seca, flatulencia |
poco frecuentes | |
|
perforación de úlcera de duodeno, estomatitis, distensión abdominal, heces duras, colitis neutropénica |
raras | |
|
Trastornos de la piel y del tejido subcutáneo |
erupción, acné |
poco frecuentes |
|
reacciones de fotosensibilidad, hiperhidrosis, piel grasa, lesión en la piel, exantema pruriginoso, síndrome de Stevens-Johnson/necrolisis epidérmica tóxica |
raras | |
|
prurito, urticaria |
frecuencia no conocida | |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
debilidad muscular, calambres musculares |
raras |
|
Trastornos renales y urinarios |
disuria |
poco frecuentes |
|
polaquiuria |
raras | |
|
Trastornos generales y alteraciones en el lugar de administración |
fatiga |
frecuentes |
|
astenia, malestar general |
poco frecuentes | |
|
edema, malestar torácico, alteración de la marcha |
raras | |
|
Exploraciones complementarias |
ALT elevada |
frecuentes |
|
AST elevada, fosfatasa alcalina en sangre aumentada |
poco frecuentes |
|
Clasificación de órganos del Sistema |
Reacción adversa |
Frecuencia |
|
hematíes en orina positivos, sodio disminuido en sangre, peso disminuido, recuento disminuido de neutrófilos, presencia de glucosuria, excreción urinaria aumentada |
raras |
* Náuseas y vómitos fueron parámetros de eficacia en los 5 primeros días de tratamiento post-quimioterapia y sólo después se notificaron como experiencias adversas.
Descripción de reacciones adversas seleccionadas
Los perfiles de reacciones adversas en la extensión de Ciclos Múltiples de ensayos en HEC y MEC que se prolongó durante 6 ciclos adicionales de quimioterapia fueron por lo general similares a los observados en el Ciclo 1.
En un ensayo clínico adicional con controlador activo en 1.169 pacientes que estaban recibiendo aprepitant y HEC, el perfil de reacciones adversas fue generalmente similar al observado en los otros ensayos HEC con aprepitant.
Se observaron otras reacciones adversas en pacientes tratados con aprepitant para las náuseas y los vómitos posquirúrgicos (NVPQ), con una incidencia mayor que con ondansetrón: dolor en la zona superior del abdomen, ruidos intestinales anormales, estreñimiento*, disartria, disnea, hipoestesia, insomnio, miosis, náuseas, alteración sensitiva, molestias en el estómago, subíleo*, agudeza visual disminuida, sibilancia.
*Notificado en pacientes que tomaron una dosis más alta de aprepitant.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
En caso de sobredosis, se debe suspender la administración de EMEND y proporcionar tratamiento de apoyo general y vigilancia. Debido a la actividad antiemética de aprepitant, es posible que la emesis inducida por un medicamento no resulte eficaz.
Aprepitant no puede eliminarse mediante hemodiálisis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antieméticos y antinauseosos, código ATC: A04AD12
Aprepitant es un antagonista selectivo de alta afinidad por los receptores de la sustancia P neurocinina 1 (NKi) humana.
Régimen de tratamiento de 3 días de aprepitant
En 2 ensayos aleatorizados, doble ciego, en los que se incluyó un total de 1.094 pacientes que recibían quimioterapia que incluía cisplatino >70 mg/m2, aprepitant en combinación con un régimen de ondansetrón/dexametasona (ver sección 4.2) se comparó con un régimen estándar (placebo más ondansetrón 32 mg administrados por vía intravenosa el día 1 más dexametasona 20 mg por vía oral el día 1 y 8 mg por vía oral dos veces al día los días 2 a 4). Aunque en ensayos clínicos se usó una dosis intravenosa de 32 mg de ondansetrón, ésta ya no es la dosis recomendada. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada.
La eficacia se basó en la evaluación de las siguientes medidas compuestas: respuesta completa (definida como ausencia de episodios eméticos sin uso de tratamiento de rescate) principalmente durante el Ciclo 1. Los resultados se evaluaron para cada ensayo individual y para los 2 ensayos combinados.
En la Tabla 1 se muestra un resumen de los resultados clave del ensayo obtenidos del análisis combinado.
Tabla 1
Porcentaje de pacientes que estaban recibiendo quimioterapia altamente emetógena que respondieron por
grupo de tratamiento y fase - Ciclo 1
Régimen con Tratamiento estándar Diferencias*
|
MEDIDAS COMPUESTAS |
aprepitant (N= 521) + % |
ll Z |
% |
(IC del 95%) |
|
Respuesta completa (sin emesis |
y sin tratamiento de rescate) | |||
|
Global (0-120 horas) |
67,7 |
47,8 |
19,9 |
(14,0, 25,8) |
|
0-24 horas |
86,0 |
73,2 |
12,7 |
(7,9, 17,6) |
|
25-120 horas |
71,5 |
51,2 |
20,3 |
(14,5, 26,1) |
MEDIDAS INDIVIDUALES_
Sin emesis (sin episodios eméticos independientemente del uso de tratamiento de rescate)
|
Global (0-120 horas) |
71,9 |
49,7 |
22,2 |
(16,4, 28,0) |
|
0-24 horas |
86,8 |
74,0 |
12,7 |
(8,0, 17,5) |
|
25-120 horas |
76,2 |
53,5 |
22,6 |
(17,0, 28,2) |
Sin náuseas significativas (EAV máxima <25 mm en una escala de 0-100 mm)_
Global (0-120 horas) 72,1 64,9 7,2 (1,6, 12,8)
25-120 horas_74,0_66,9_7,1 (1,5, 12,6)
* Los intervalos de confianza se calcularon sin ajuste por sexo ni quimioterapia concomitante, los cuales fueron incluidos en el análisis primario de cociente de posibilidades y modelos logísticos. t Sólo un paciente en el régimen de aprepitant tuvo datos en la fase aguda y se excluyó de los análisis de fase retardada y general; sólo un paciente en el régimen estándar tuvo datos en la fase retardada y se excluyó del análisis de fase aguda y general.
El tiempo estimado hasta la primera emesis en el análisis combinado se representa en el gráfico Kapl an-Meier de la Figura 1.
Figura 1
Porcentaje de pacientes que estaban recibiendo quimioterapia altamente emetógena que siguieron sin padecer emesis con el tiempo - Ciclo 1
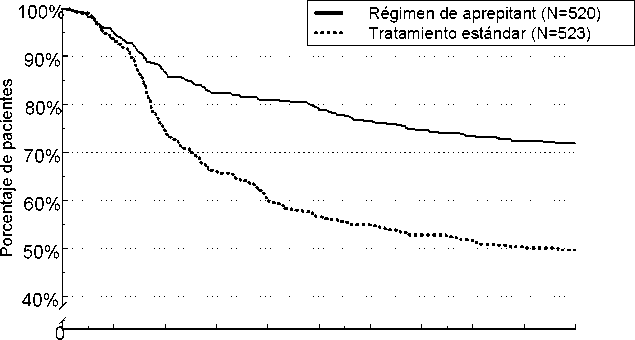
0 12 24 36 48 60 72 84 96 108 120
Tiempo (horas)
También se observaron diferencias estadísticamente significativas en eficacia en cada uno de los 2 ensayos individuales.
En los mismos 2 ensayos clínicos, 851 pacientes continuaron en la extensión de Ciclos Múltiples durante 5 ciclos adicionales de quimioterapia. La eficacia del régimen de aprepitant se mantuvo aparentemente durante todos los ciclos.
En un ensayo aleatorizado, doble ciego con un total de 866 pacientes (864 mujeres, 2 varones) que estaban recibiendo quimioterapia, que incluía ciclofosfamida 750-1500 mg/m2; o ciclofosfamida 500-1500 mg/m2 y doxorrubicina (<60 mg/m2) o epirubicina (<100 mg/m2), se comparó aprepitant en combinación con un régimen de ondansetrón/dexametasona (ver sección 4.2) con el tratamiento estándar (placebo más ondansetrón 8 mg por vía oral (dos veces en el día 1 y cada 12 horas en los días 2 y 3) más dexametasona 20 mg por vía oral en el día 1).
La eficacia se basó en la evaluación de las medidas compuestas: respuesta completa (definida como ausencia de episodios eméticos sin uso de tratamiento de rescate) principalmente durante el Ciclo 1.
Tabla 2
Porcentaje de pacientes que respondieron por grupo de tratamiento y fase - Ciclo 1 Quimioterapia moderadamente emetógena
|
Régimen |
Tratamiento | |
|
con |
estándar | |
|
MEDIDAS COMPUESTAS aprepitant |
(N= 424) | |
|
(N= 433) f |
% |
% (IC del 95%) |
|
% |
Respuesta completa (sin emesis y sin tratamiento de rescate)_
Global (0-120 horas) 50,8 42,5 8,3 (1,6, 15,0)
0-24 horas 75,7 69,0 6,7 (0,7, 12,7)
25-120 horas_55,4_49,1_6,3 (-0,4, 13,0)
MEDIDAS INDIVIDUALES_
Sin emesis (sin episodios eméticos independientemente del uso de tratamiento de rescate)
Global (0-120 horas) 75,7 58,7 17,0 (10,8, 23,2)
0-24 horas 87,5 77,3 10,2 (5,1, 15,3)
25-120 horas_80,8_69,1_11,7 (5,9, 17,5)
Sin náuseas significativas (EAV máxima <25 mm en una escala de 0-100 mm)_
Global (0-120 horas) 60,9 55,7 5,3 (-1,3, 11,9)
0-24 horas 79,5 78,3 1,3 (-4,2, 6,8)
Tabla 3
Porcentaje de pacientes que respondieron por grupo de tratamiento y fase en el ensayo 2 - Ciclo 1
Quimioterapia moderadamente emetógena
|
Régimen |
Tratamiento |
Diferencias* |
|
con |
estándar | |
|
aprepitant |
(N= 406) | |
|
(N= 425) |
% |
% (IC del 95 %) |
|
% |
|
Respuesta completa (sin emesis y |
sin tratamiento de |
rescate) | |||
|
Global (0-120 horas) |
68,7 |
56,3 |
12,4 |
(5,9, |
18,9) |
|
0-24 horas |
89,2 |
80,3 |
8,9 |
(4,0, |
13,8) |
|
25-120 horas |
70,8 |
60,9 |
9,9 |
(3,5, |
16,3) |
|
Sin emesis (sin episodios eméticos |
independientemente del uso de |
tratamiento | |||
|
de rescate) | |||||
|
Global (0-120 horas) |
76,2 |
62,1 |
14,1 |
(7,9, |
20,3) |
|
0-24 horas |
92,0 |
83,7 |
8,3 |
(3,9, |
12,7) |
|
25-120 horas |
77,9 |
66,8 |
11,1 |
(5,1, |
17,1) |
|
Sin nauseas significativas (EAV máxima <25 mm en |
una escala de 0-100 mm) | ||||
|
Global (0-120 horas) |
73,6 |
66,4 |
7,2 |
(1,0, |
13,4) |
|
0-24 horas |
90,9 |
86,3 |
4,6 |
(0,2, |
9,0) |
|
25-120 horas |
74,9 |
69,5 |
5,4 |
(-0,7 |
, 11,5) |
*Los intervalos de confianza se calcularon sin ajuste por sexo ni región, los cuales fueron incluidos en el análisis primario utilizando modelos logísticos.
El beneficio del tratamiento en combinación de aprepitant en la población del ensayo total fue dirigido principalmente por los resultados observados en pacientes con bajo control con el régimen estándar como en mujeres, aunque los resultados fueron numéricamente mejores independientemente de la edad, tipo de tumor o sexo. La respuesta completa al régimen de aprepitant y al tratamiento estándar, respectivamente, se alcanzó en 209/324 (65 %) y 161/320 (50 %) en mujeres y 83/101 (82 %) y 68/87 (78 %) de varones.
Población pediátrica
Los ensayos clínicos para evaluar el uso de aprepitant en pacientes pediátricos están en curso (ver sección 4.2 para la información sobre el uso pediátrico).
5.2 Propiedades farmacocinéticas
Aprepitant muestra una farmacocinética no lineal. Tanto el aclaramiento como la biodisponibilidad absoluta disminuyen al aumentar la dosis.
Absorción
La biodisponibilidad oral absoluta media de aprepitant es de 67 % para la cápsula de 80 mg y de 59 % para la cápsula de 125 mg. La concentración plasmática máxima media (Cmax) de aprepitant se alcanzó aproximadamente a las 4 horas (tmax). La administración oral de la cápsula con un desayuno estándar de aproximadamente 800 Kcal ocasionó un aumento de hasta el 40 % en el AUC de aprepitant. Este aumento no se consideró clínicamente de interés.
La farmacocinética de aprepitant es no lineal en el intervalo de la dosis clínica. En adultos jóvenes sanos, el aumento en el AUC0-a, fue un 26 % mayor que proporcional a la dosis entre las dosis únicas de 80 mg y 125 mg administradas en estado posprandial.
Tras la administración oral de una dosis única de 125 mg de EMEND el día 1 y 80 mg una vez al día los días 2 y 3, el AUC0-24h (media ± DE) fue 19,6 ± 2,5 ^g • h/ml y 21,2 ± 6,3 ^g • h/ml los días 1 y 3, respectivamente. La Cmax fUe 1,6±0,36 ^g/ml y 1,4 ± 0,22 microgramos/ml los días 1 y 3, respectivamente.
Distribución
Aprepitant se une fuertemente a proteínas, con una media del 97 %. La media geométrica del volumen aparente de distribución en el estado equilibrio (Vdee) es aproximadamente de 66 litros en el ser humano.
Biotransformación
Aprepitant se metaboliza extensamente. En adultos jóvenes sanos, aprepitant representa aproximadamente el 19% de la radiactividad plasmática durante 72 horas después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant, lo que indica una importante presencia de metabolitos en el plasma. En el plasma humano se han identificado doce metabolitos de aprepitant. El metabolismo de aprepitant se produce en gran medida por oxidación en el anillo de morfolina y sus cadenas laterales y los metabolitos resultantes sólo fueron débilmente activos. Estudios in vitro en los que se usaron microsomas hepáticos humanos indicaron que aprepitant se metaboliza principalmente a través de CYP3A4 y posiblemente con una contribución menor a través de CYP1A2 y CYP2C19.
Eliminación
Aprepitant no se elimina inalterado en la orina. Los metabolitos se eliminan en la orina y a través de excreción biliar en las heces. Después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant a sujetos sanos, el 57% de la radiactividad se recuperó en la orina y el 45% en las heces.
El aclaramiento plasmático de aprepitant es dependiente de la dosis, disminuyendo al aumentar la dosis y oscilando aproximadamente entre 60 a 72 ml/min en el intervalo de la dosis terapéutica. La semivida terminal osciló entre aproximadamente 9 a 13 horas.
Farmacocinética en poblaciones especiales
Pacientes de edad avanzada: Tras la administración oral de una dosis única de 125 mg de aprepitant el día 1 y 80 mg una vez al día los días 2 a 5, el AUC0-24h de aprepitant fue un 21% superior el día 1 y un 36% superior el día 5 en pacientes de edad avanzada (>65 años) respecto de los adultos más jóvenes. La Cmax fue un 10% superior el día 1 y un 24% superior el día 5 en pacientes de edad avanzada respecto de los adultos más jóvenes. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en los pacientes de edad avanzada.
Sexo: Tras la administración oral de una dosis única de 125 mg de aprepitant, la Cmax de aprepitant es un 16% superior en las mujeres en comparación con los varones. La semivida de aprepitant es un 25% inferior en las mujeres en comparación con los varones y su tmax se produce en aproximadamente el mismo tiempo. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en función del sexo.
Insuficiencia hepática: La insuficiencia hepática leve (Child-Pugh clase A) no afecta a la farmacocinética de aprepitant en un grado clínicamente relevante. No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. De los datos disponibles no pueden extraerse conclusiones relativas a la influencia de la insuficiencia hepática moderada (Child-Pugh clase B) sobre la farmacocinética de aprepitant. No existen datos clínicos ni farmacocinéticos de pacientes con insuficiencia hepática grave (Child-Pugh clase C).
Insuficiencia renal: Se administró una dosis única de 240 mg de aprepitant a pacientes con insuficiencia renal grave (CrCl<30 ml/min) y a pacientes con nefropatía terminal que requería hemodiálisis.
En los pacientes con insuficiencia renal grave, el AUC0-<X) del aprepitant total (no unido y unido a proteínas) disminuyó en un 21% y la Cmax disminuyó en un 32%, respecto de los sujetos sanos. En los pacientes con nefropatía terminal sometidos a hemodiálisis, el AUC0-<X) del aprepitant total disminuyó en un 42% y la Cmax disminuyó en un 32%. Debido a los modestos descensos en la unión a proteínas de aprepitant en los pacientes con enfermedad renal, el AUC de aprepitant no unido farmacológicamente activo no se vio significativamente afectado en los pacientes con insuficiencia renal en comparación con los sujetos sanos. La hemodiálisis realizada 4 ó 48 horas después de la administración no tuvo efectos significativos sobre la farmacocinética de aprepitant; en el dializado se recuperó menos de 0,2% de la dosis.
En pacientes con insuficiencia renal o en pacientes con nefropatía terminal sometidos a hemodiálisis no es necesario ajustar la dosis de EMEND.
Relación entre concentración y efecto
Usando un trazador altamente específico del receptor NKi, los estudios de tomografía por emisión de positrones (PET) en varones jóvenes sanos han demostrado que aprepitant penetra en el cerebro y ocupa los receptores NK1 de forma dependiente de la dosis y de la concentración plasmática. Se predice que las concentraciones plasmáticas de aprepitant alcanzadas con el régimen de 3 días de EMEND proporcionarán una ocupación superior al 95% de los receptores NK1 cerebrales.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de toxicidad, genotoxicidad, potencial carcinogénico y toxicidad para la reproducción a dosis únicas y repetidas. Sin embargo, se deberá tener en cuenta que la exposición sistémica en roedores fue similar o incluso inferior a la exposición terapéutica en seres humanos a la dosis de 125 mg/80 mg. En especial, aunque no se observaron efectos adversos en los estudios sobre la reproducción a los niveles de exposición en seres humanos, las exposiciones en animales no son suficientes para hacer una valoración de riesgo adecuada en el hombre.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula Sacarosa
Celulosa microcristalina (E 460) Hidroxipropilcelulosa (E 463)
Lauril sulfato de sodio
Cubierta de la cápsula Gelatina
Dióxido de titanio (E 171)
Tinta para impresión Laca
Hidróxido de potasio Óxido férrico negro (E 172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
6.4 Precauciones especiales de conservación
Conservar en el embalaje original para protegerlo de la humedad.
6.5 Naturaleza y contenido del envase
Están disponibles diferentes tamaños de envase con concentraciones diferentes.
Blister de aluminio conteniendo una cápsula de 80 mg.
Blister de aluminio conteniendo dos cápsulas de 80 mg.
5 blisters de aluminio conteniendo cada uno una cápsula de 80 mg.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación Ninguna especial para su eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/001
EU/1/03/262/002
EU/1/03/262/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 11/noviembre/2003 Fecha de la última renovación: 11/noviembre/2008
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
1. NOMBRE DEL MEDICAMENTO
EMEND 125 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula contiene 125 mg de aprepitant.
Excipiente con efecto conocido:
Cada cápsula contiene 125 mg de sacarosa.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura.
Las cápsulas son opacas con cuerpo blanco y tapa rosa con “462” y “125 mg” impreso en forma radial en tinta negra en el cuerpo.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Prevención de las náuseas y los vómitos agudos y diferidos que se asocian con la quimioterapia antineoplásica altamente emetógena basada en el cisplatino en adultos.
Prevención de las náuseas y los vómitos que se asocian con la quimioterapia antineoplásica moderadamente emetógena en adultos.
EMEND 125 mg se administra como parte de un tratamiento de combinación (ver sección 4.2).
4.2 Posología y forma de administración Posología
EMEND se administra durante 3 días como parte de un régimen que incluye un corticoesteroide y un antagonista 5-HT3. La dosis recomendada es de 125 mg por vía oral una vez al día una hora antes de empezar la quimioterapia el día 1 y 80 mg por vía oral, una vez al día, los días 2 y 3.
Se recomiendan los siguientes regímenes de administración para la prevención de las náuseas y los vómitos asociados con la quimioterapia antineoplásica emetógena:
|
Día 1 |
Día 2 |
Día 3 |
Día 4 | |
|
EMEND |
125 mg vía oral |
80 mg vía oral |
80 mg vía oral |
nada |
|
Dexametasona |
12 mg vía oral |
8 mg vía oral |
8 mg vía oral |
8 mg vía oral |
|
Antagonistas 5-HT3 |
Dosis habituales de los antagonistas 5-HT3. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada |
nada |
nada |
nada |
|
Dexametasona se d |
ebe administrar 30 minutos antes de la quimioterapia el día 1 y por la mañana los | |||
días 2 a 4. La dosis de dexametasona es responsable de las interacciones del principio activo.
Régimen de quimioterapia moderadamente emetógena
|
Día 1 |
Día 2 |
Día 3 | |
|
EMEND |
125 mg vía oral |
80 mg vía oral |
80 mg vía oral |
|
Dexametasona |
12 mg vía oral |
nada |
nada |
|
Antagonistas 5-HT3 |
Dosis habituales de los antagonistas 5-HT3. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada |
nada |
nada |
Dexametasona se debe administrar 30 minutos antes de la quimioterapia el día 1. La dosis de dexametasona es responsable de las interacciones del principio activo.
Los datos de eficacia en combinación con otros corticoesteroides y antagonistas 5HT3 son limitados. Para información adicional sobre la co-administración con corticoesteroides, ver sección 4.5. Consultar la Ficha Técnica o Resumen de las Características del Producto de los medicamentos antagonistas 5-HT3 coadministrados.
Poblaciones especiales
Pacientes de edad avanzada (>65 años)
No es necesario ajustar la dosis en los pacientes de edad avanzada (ver sección 5.2).
Sexo
No es necesario ajustar la dosis según el sexo (ver sección 5.2).
Insuficiencia renal
No es necesario ajustar la dosis en los pacientes con insuficiencia renal ni en los pacientes con nefropatía terminal sometidos a hemodiálisis (ver sección 5.2).
Insuficiencia hepática
No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. Aprepitant se debe usar con precaución en estos pacientes (ver secciones 4.4 y 5.2).
Población pediátrica
No s e ha establecido la seguridad y eficacia de EMEND en niños y adolescentes menores de 18 años de edad. No se dispone de datos.
Forma de administración
Las cápsulas duras se deben tragar enteras.
EMEND puede tomarse con o sin alimentos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Co-administración con pimozida, terfenadina, astemizol o cisaprida (ver sección 4.5).
4.4 Advertencias y precauciones especiales de empleo Pacientes con insuficiencia hepática moderada a grave
Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. EMEND se debe usar con precaución en estos pacientes (ver sección 5.2).
Interacciones con el CYP3A4
EMEND se debe usar con precaución en pacientes que estén recibiendo de forma concomitante principios activos, administrados por vía oral, metabolizados principalmente a través de CYP3A4 y con un rango terapéutico estrecho, tales como ciclosporina, tacrolimus, sirolimus, everolimus, alfentanilo, diergotamina, ergotamina, fentanilo y quinidina (ver sección 4.5). Adicionalmente, la administración concomitante con irinotecano se debe abordar con especial prudencia ya que esta combinación puede provocar un aumento de la toxicidad.
La administración conjunta de EMEND con derivados de los alcaloides del cornezuelo, que son sustratos de CYP3A4, puede dar lugar a elevaciones de las concentraciones plasmáticas de estos principios activos. Por lo tanto, se aconseja precaución debido al riesgo potencial de toxicidad relacionada con el cornezuelo del centeno.
Se debe evitar la administración concomitante de EMEND con principios activos que sean potentes inductores de la actividad del CYP3A4 (por ejemplo, rifampicina, fenitoína, carbamazepina, fenobarbital), puesto que la combinación puede provocar un descenso en las concentraciones plasmáticas de aprepitant (ver sección 4.5). No se recomienda la administración concomitante de fosaprepitant con preparados a base de plantas que contienen hipérico (Hypericum perforatum; también conocido como Hierba de San Juan).
Se debe tener especial precaución cuando se administre EMEND de forma concomitante con principios activos que sean inhibidores de la actividad del CYP3A4 (por ejemplo, ketoconazol, itraconazol, voriconazol, posaconazol, claritromicina, telitromicina, nefazodona e inhibidores de la proteasa), puesto que la combinación se espera que provoque un aumento de las concentraciones plasmáticas de aprepitant (ver sección 4.5).
Administración conjunta con warfarina (un sustrato CYP2C9)
La administración conjunta de EMEND con warfarina da lugar a una disminución del tiempo de protrombina, comunicado como coeficiente internacional normalizado (INR, International Normalised Ratio). En pacientes en tratamiento crónico con warfarina, el INR se debe vigilar estrechamente durante el tratamiento con EMEND y durante 14 días después de cada ciclo de 3 días de EMEND (ver sección 4.5).
Administración conjunta con anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver sección 4.5).
Excipientes
EMEND contiene sacarosa. Los pacientes con intolerancia hereditaria a la fructosa, problemas de absorción de glucosa o galactosa, o insuficiencia de sacarasa-isomaltasa, no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Aprepitant (125 mg/80 mg) es un sustrato, un inhibidor moderado, y un inductor de CYP3A4. Aprepitant es también un inductor de CYP2C9. Durante el tratamiento con EMEND, CYP3A4 se inhibe. Después de terminar el tratamiento, EMEND produce una inducción transitoria suave de CYP2C9, de CYP3A4 y glucuronidación. Aprepitant no parece que interaccione con el transportador de la P-glucoproteína, como sugiere la falta de interacción de aprepitant con digoxina.
Efecto de aprepitant sobre la farmacocinética de otros principios activos
Inhibición de CYP3A4
Como inhibidor moderado de CYP3A4, aprepitant (125 mg/80 mg) puede aumentar las concentraciones plasmáticas de los principios activos que se metabolizan a través de CYP3A4 cuando se administran conjuntamente. La exposición total de los sustratos de CYP3A4 que se administran por vía oral puede aumentar hasta aproximadamente tres veces durante el tratamiento de 3 días con EMEND; se estima que el efecto de aprepitant sobre las concentraciones plasmáticas de los sustratos de CYP3A4 que se administran por vía intravenosa sea menor. EMEND no se debe usar simultáneamente con pimozida, terfenadina, astemizol o cisaprida (ver sección 4.3). La inhibición de CYP3A4 por aprepitant podría dar lugar a elevaciones de las concentraciones plasmáticas de estos principios activos, lo que podría provocar reacciones graves o potencialmente mortales. Se aconseja precaución durante la administración concomitante de EMEND y principios activos, administrados por vía oral, metabolizados principalmente a través de CYP3A4 y con un rango terapéutico estrecho, tales como ciclosporina, tacrolimus, sirolimus, everolimus, alfentanilo, diergotamina, ergotamina, fentanilo y quinidina (ver sección 4.4).
Corticoesteroides
Dexametasona: La dosis habitual de dexametasona oral se debe reducir aproximadamente en un 50% cuando se administra conjuntamente con un tratamiento de 125 mg/80 mg de EMEND. La dosis de dexametasona utilizada en los ensayos clínicos de náuseas y vómitos inducidos por quimioterapia, se eligió en función de las interacciones del principio activo (ver sección 4.2). EMEND, administrado en un régimen de 125 mg conjuntamente con 20 mg de dexametasona oral el día 1, y EMEND, administrado en un régimen de 80 mg/día conjuntamente con 8 mg de dexametasona oral los días 2 a 5, aumentó el AUC de dexametasona, un sustrato de CYP3A4, 2,2 veces los días 1 y 5.
Metilprednisolona: La dosis habitual de metilprednisolona intravenosa se debe reducir aproximadamente un 25%, y la dosis habitual de metilprednisolona oral se debe reducir aproximadamente un 50% al administrarse conjuntamente con un tratamiento de 125 mg/80 mg de EMEND. EMEND, administrado en un régimen de 125 mg el día 1 y 80 mg/día los días 2 y 3, aumentó el AUC de metilprednisolona, un sustrato de CYP3A4, 1,3 veces el día 1 y 2,5 veces el día 3, al administrarse conjuntamente metilprednisolona por vía intravenosa, 125 mg el día 1, y por vía oral, 40 mg los días 2 y 3.
Durante el tratamiento continuo con metilprednisolona, el AUC de metilprednisolona puede disminuir en puntos de tiempo posteriores en el transcurso de las 2 semanas siguientes al inicio de la administración de EMEND, debido al efecto inductor de aprepitant sobre CYP3A4. Puede ser que este efecto sea más pronunciado para metilprednisolona administrada oralmente.
Antineoplásicos
En ensayos farmacocinéticos, EMEND, administrado en un régimen de 125 mg el día 1 y 80 mg/día los días 2 y 3, no influyó en la farmacocinética de docetaxel administrado por vía intravenosa el día 1 ni en la de vinorelbina administrada por vía intravenosa el día 1 o el día 8. Debido a que el efecto de EMEND sobre la farmacocinética de sustratos de CYP3A4 administrados por vía oral es mayor que el efecto de EMEND sobre la farmacocinética de sustratos de CYP3A4 administrados por vía intravenosa, no puede excluirse una interacción con medicamentos antineoplásicos administrados por vía oral que se metabolizan principal o parcialmente a través de CYP3A4 (p. ej. etopósido, vinorelbina). En pacientes que reciben medicamentos que se metabolizan principal o parcialmente a través de CYP3A4, se aconseja precaución y puede ser conveniente una vigilancia adicional (ver sección 4.4). Se han notificado acontecimientos adversos de neurotoxicidad postcomercialización, una reacción adversa potencial de ifosfamida, tras la administración simultánea de aprepitant e ifosfamida.
Inmunosupresores
Durante el régimen de tratamiento de 3 días de las náuseas y vómitos inducidos por quimioterapia, se espera un incremento moderado transitorio seguido de una leve disminución en la exposición de los inmunosupresores metabolizados por CYP3A4 (por ej. ciclosporina, tacrolimus, everolimus y sirolimus). Dada la corta duración del régimen de tratamiento de 3 días y los cambios limitados en la exposición dependientes del tiempo, no se recomienda una reducción de dosis de los inmunosupresores durante los 3 días de la administración conjunta con EMEND.
Midazolam
Los posibles efectos de aumentos en las concentraciones plasmáticas de midazolam u otras benzodiazepinas metabolizadas a través de CYP3A4 (alprazolam, triazolam) se deben tener en cuenta al administrar estos medicamentos conjuntamente con EMEND (125 mg/80 mg).
EMEND aumentó el AUC de midazolam, un sustrato sensible de CYP3A4, 2,3 veces el día 1 y
3,3 veces el día 5, al administrarse conjuntamente una dosis oral única de 2 mg de midazolam los días 1 y 5 de un régimen de EMEND 125 mg el día 1 y 80 mg/día los días 2 a 5.
En otro ensayo con administración intravenosa de midazolam, EMEND se administró como 125 mg el día 1 y 80 mg/día los días 2 y 3, y 2 mg de midazolam se administró por vía intravenosa antes de la administración del régimen de 3 días de EMEND y en los días 4, 8 y 15. EMEND aumentó el AUC de midazolam un 25% el día 4 y disminuyó el AUC de midazolam un 19% el día 8 y un 4% el día 15. Estos efectos no se consideraron clínicamente importantes.
En un tercer ensayo con administración intravenosa y oral de midazolam, se administraron 125 mg de EMEND en el día 1 y 80 mg/día en los días 2 y 3, junto con 32 mg de ondansetrón el día 1, 12 mg de dexametasona el día 1 y 8 mg los días 2-4. Esta combinación (esto es, EMEND, ondansetrón y dexametasona) disminuyó el AUC de midazolam oral un 16 % el día 6, un 9 % el día 8, un 7 % el día 15 y un 17 % el día 22. Estos efectos no se consideraron clínicamente importantes.
Se finalizó un ensayo adicional con administración intravenosa de midazolam y EMEND. Una hora después de la administración oral de una dosis única de EMEND 125 mg, se administraron por vía intravenosa 2 mg de midazolam. El AUC plasmático de midazolam aumentó en 1,5 veces. Este efecto no se consideró clínicamente importante.
Inducción
Como inductor suave de CYP2C9, de CYP3A4 y de la glucuronidación, aprepitant puede disminuir las concentraciones plasmáticas de los sustratos eliminados por estas vías durante las dos semanas posteriores al inicio del tratamiento. Este efecto puede hacerse evidente únicamente después de finalizar el tratamiento de 3 días con EMEND. Para los sustratos de CYP2C9 y CYP3A4, la inducción es transitoria con un efecto máximo alcanzado a los 3-5 días después de finalizar el tratamiento de 3 días con EMEND. El efecto se mantiene durante unos pocos días, después desciende lentamente y es clínicamente insignificante a las 2 semanas después de finalizar el tratamiento con EMEND. La inducción suave de la glucuronidación también se observa con 80 mg de aprepitant oral administrado durante 7 días. Se carece de datos relativos a los efectos sobre CYP2C8 y CYP2C19. Se aconseja precaución al administrar durante este periodo de tiempo warfarina, acenocumarol, tolbutamida, fenitoína u otros principios activos que se sabe que son metabolizados por CYP2C9.
Warfarina
En pacientes en tratamiento crónico con warfarina, el tiempo de protrombina (INR) se debe vigilar estrechamente durante el tratamiento con EMEND y durante 2 semanas después de cada ciclo de 3 días de EMEND en el caso de náuseas y vómitos inducidos por quimioterapia (ver sección 4.4). Al administrarse una dosis única de 125 mg de EMEND el día 1 y 80 mg/día los días 2 y 3 a sujetos sanos estabilizados en un tratamiento crónico con warfarina, no se observó ningún efecto de EMEND sobre el AUC plasmático de R(+) o S(-) warfarina determinado el día 3; sin embargo, se observó un descenso del 34% en la concentración mínima de S(-) warfarina (un sustrato de CYP2C9) acompañado de un descenso del 14% en el INR 5 días después de finalizar el tratamiento con EMEND.
Tolbutamida
EMEND, administrado como 125 mg el día 1 y 80 mg/día los días 2 y 3, disminuyó el AUC de tolbutamida (un sustrato de CYP2C9) en un 23% el día 4, un 28% el día 8 y un 15% el día 15, al administrarse una dosis única de tolbutamida 500 mg por vía oral antes de la administración de un régimen de 3 días de EMEND y en los días 4, 8 y 15.
Anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo.
En un ensayo clínico, dosis únicas de un anticonceptivo oral que contenía etinil estradiol y noretindrona se administraron en los días 1 hasta el 21 con EMEND, administrado con una pauta posológica de 125 mg en el día 8 y 80 mg/día en los días 9 y 10 con ondansetrón 32 mg vía intravenosa en el día 8 y dexametasona oral administrada como 12 mg en el día 8 y 8 mg/día los días 9, 10 y 11. Durante los días 9 hasta el 21 en este ensayo, hubo un descenso hasta del 64% en las concentraciones mínimas de etinil estradiol y hasta del 60% en las concentraciones mínimas de noretindrona.
Antagonistas 5-HT3
En ensayos clínicos de interacción, aprepitant no tuvo efectos clínicamente importantes sobre la farmacocinética de ondansetrón, granisetrón ni hidrodolasetrón (el metabolito activo de dolasetrón).
Efecto de otros medicamentos sobre la farmacocinética de aprepitant
La administración concomitante de EMEND con principios activos que inhiben la actividad de CYP3A4 (por ejemplo, ketoconazol, itraconazol, voriconazol, posaconazol, claritromicina, telitromicina, nefazodona e inhibidores de la proteasa) se debe abordar con precaución, ya que la combinación se espera que provoque una elevación de varias veces las concentraciones plasmáticas de aprepitant (ver sección 4.4).
La administración concomitante de EMEND con principios activos que inducen fuertemente la actividad de CYP3A4 (por ejemplo, rifampicina, fenitoína, carbamazepina, fenobarbital) se debe evitar ya que la combinación provoca reducciones en las concentraciones plasmáticas de aprepitant lo que podría provocar una disminución de la eficacia de EMEND. No se recomienda la administración concomitante de EMEND con plantas medicinales que contienen hipérico (Hypericum perforatum).
Ketoconazol
Al administrarse una dosis única de 125 mg de aprepitant el día 5 de un régimen de 10 días de 400 mg/día de ketoconazol, un potente inhibidor de CYP3A4, el AUC de aprepitant aumentó aproximadamente 5 veces y la semivida terminal media de aprepitant aumentó aproximadamente 3 veces.
Rifampicina
Al administrarse una dosis única de 375 mg de aprepitant el día 9 de un régimen de 14 días de 600 mg/día de rifampicina, un potente inductor de CYP3A4, el AUC de aprepitant disminuyó un 91% y la semivida terminal media disminuyó un 68%.
4.6 Fertilidad, embarazo y lactancia
Anticoncepción en varones y mujeres
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de su administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver secciones 4.4 y 4.5).
Embarazo
No hay datos clínicos disponibles sobre la exposición a aprepitant durante el embarazo. La capacidad de aprepitant para provocar toxicidad sobre la reproducción no se ha caracterizado completamente, ya que en los estudios animales no se pudieron alcanzar niveles de exposición por encima de la exposición terapéutica en seres humanos a la dosis de 125 mg/80 mg. Estos estudios no sugirieron efectos perjudiciales directos ni indirectos en términos del embarazo, el desarrollo embrionario/fetal, el parto o el desarrollo posnatal (ver sección 5.3). Se desconocen los posibles efectos sobre la reproducción de alteraciones en la regulación de la neurocinina. EMEND no se debe utilizar durante el embarazo excepto si fuese claramente necesario.
Lactancia
Aprepitant se excreta en la leche de ratas lactantes. Se desconoce si aprepitant se excreta en la leche materna; por consiguiente, no se recomienda la lactancia durante el tratamiento con EMEND.
Fertilidad
El potencial efecto de aprepitant sobre la fertilidad no se ha caracterizado completamente, ya que en los estudios en animales no se pudieron alcanzar niveles de exposición por encima de la exposición terapéutica en humanos. Estos estudios de fertilidad no sugirieron efectos perjudiciales directos ni indirectos en términos del estado de apareamiento, fertilidad, desarrollo embrionario/fetal, o recuento de esperma y movilidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de EMEND sobre la capacidad para conducir y utilizar máquinas es pequeña. Se pueden producir mareos y fatiga después de la administración de EMEND (ver sección 4.8).
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad de aprepitant se evaluó en aproximadamente 6.500 sujetos.
Las reacciones adversas más frecuentes que se notificaron con una mayor incidencia en pacientes tratados con el régimen de aprepitant que con el tratamiento estándar en pacientes que estaban recibiendo quimioterapia altamente emetógena (HEC) fueron: hipo (4,6 % versus 2,9 %), alanina aminotransferasa (ALT) elevada (2,8 % versus 1,1 %), dispepsia (2,6 % versus 2,0 %), estreñimiento (2,4 % versus 2,0 %), cefalea (2,0 % versus 1,8 %) y apetito disminuido (2,0 % versus 0,5 %). La reacción adversa más frecuente notificada con una mayor incidencia en pacientes tratados con el régimen de aprepitant que con el tratamiento estándar en pacientes que estaban recibiendo quimioterapia moderadamente emetógena (MEC) fue fatiga (1,4 % versus 0,9%).
Lista tabulada de reacciones adversas
Las reacciones adversas siguientes se observaron en un análisis combinado de los ensayos en HEC y en MEC con una incidencia mayor con aprepitant que con el tratamiento estándar o en el uso después de la comercialización:
Las frecuencias se definen como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000) y muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Clasificación de órganos del Sistema |
Reacción adversa |
Frecuencia |
|
Infecciones e infestaciones |
candidiasis, infección estafilocócica |
raras |
|
Trastornos de la sangre y del sistema linfático |
neutropenia febril, anemia |
poco frecuentes |
|
Trastornos del sistema inmunológico |
reacciones de hipersensibilidad incluyendo reacciones anafilácticas |
no conocida |
|
Trastornos del metabolismo y de la nutrición |
apetito disminuido |
frecuentes |
|
polidipsia |
raras | |
|
Trastornos psiquiátricos |
ansiedad |
poco frecuentes |
|
desorientación, estado de ánimo eufórico |
raras | |
|
Trastornos del sistema nervioso |
cefalea |
frecuentes |
|
mareo, somnolencia |
poco frecuentes | |
|
trastorno cognoscitivo, letargia, disgeusia |
raras | |
|
Trastornos oculares |
conjuntivitis |
raras |
|
Trastornos del oído y del laberinto |
acúfenos |
raras |
|
Trastornos cardiacos |
palpitaciones |
poco frecuentes |
|
bradicardia, trastorno cardiovascular |
raras | |
|
Trastornos vasculares |
acaloramiento |
poco frecuentes |
|
Trastornos respiratorios, torácicos y mediastínicos |
hipo |
frecuentes |
|
dolor orofaríngeo, estornudos, tos, goteo postnasal, irritación de garganta |
raras | |
|
Trastornos gastrointestinales |
estreñimiento, dispepsia |
frecuentes |
|
eructos, náuseas*, vómitos*, enfermedad por reflujo gastroesofágico, dolor abdominal, boca seca, flatulencia |
poco frecuentes | |
|
perforación de úlcera de duodeno, estomatitis, distensión abdominal, heces duras, colitis neutropénica |
raras | |
|
Trastornos de la piel y del tejido subcutáneo |
erupción, acné |
poco frecuentes |
|
reacción de fotosensibilidad, hiperhidrosis, seborrea, lesión de la piel, erupción prurítica, síndrome de Stevens-Johnson/necrolisis epidérmica tóxica |
raras | |
|
prurito, urticaria |
no conocida | |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
debilidad muscular, calambres musculares |
raras |
|
Trastornos renales y urinarios |
disuria |
poco frecuentes |
|
polaquiuria |
raras | |
|
Trastornos generales y alteraciones en el lugar de administración |
fatiga |
frecuentes |
|
astenia, malestar general |
poco frecuentes | |
|
edema, malestar torácico, alteración de la marcha |
raras | |
|
Exploraciones complementarias |
ALT elevada |
frecuentes |
|
AST elevada, fosfatasa alcalina en sangre aumentada |
poco frecuentes |
|
Clasificación de órganos del Sistema |
Reacción adversa |
Frecuencia |
|
hematíes en orina positivos, sodio disminuido en sangre, peso disminuido, recuento disminuido de neutrófilos, presencia de glucosuria, excreción urinaria aumentada |
raras |
* Náuseas y vómitos fueron parámetros de eficacia en los 5 primeros días de tratamiento post-quimioterapia y sólo después se notificaron como experiencias adversas.
Descripción de reacciones adversas seleccionadas
Los perfiles de reacciones adversas en la extensión de Ciclos Múltiples de ensayos en HEC y MEC que se prolongó durante 6 ciclos adicionales de quimioterapia fueron por lo general similares a los observados en el Ciclo 1.
En un ensayo clínico adicional con controlador activo en 1.169 pacientes que estaban recibiendo aprepitant y HEC, el perfil de reacciones adversas fue generalmente similar al observado en los otros ensayos HEC con aprepitant.
Se observaron otras reacciones adversas en pacientes tratados con aprepitant para las náuseas y los vómitos posquirúrgicos (NVPQ), con una incidencia mayor que con ondansetrón: dolor en la zona superior del abdomen, ruidos intestinales anormales, estreñimiento*, disartria, disnea, hipoestesia, insomnio, miosis, náuseas, alteración sensitiva, molestias en el estómago, subíleo*, agudeza visual disminuida, sibilancia.
*Notificado en pacientes que tomaron una dosis más alta de aprepitant.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
En caso de sobredosis, se debe suspender la administración de EMEND y proporcionar tratamiento de apoyo general y vigilancia. Debido a la actividad antiemética de aprepitant, es posible que la emesis inducida por un medicamento no resulte eficaz.
Aprepitant no puede eliminarse mediante hemodiálisis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antieméticos y antinauseosos, código ATC: A04A D12
Aprepitant es un antagonista selectivo de alta afinidad por los receptores de la sustancia P neurocinina 1 (NKi) humana.
Régimen de tratamiento de 3 días de aprepitant
En 2 ensayos aleatorizados, doble ciego, en los que se incluyó un total de 1.094 pacientes que recibían quimioterapia que incluía cisplatino >70 mg/m2, aprepitant en combinación con un régimen de ondansetrón/dexametasona (ver sección 4.2) se comparó con un régimen estándar (placebo más ondansetrón 32 mg administrados por vía intravenosa el día 1 más dexametasona 20 mg por vía oral el día 1 y 8 mg por vía oral dos veces al día los días 2 a 4). Aunque en ensayos clínicos se usó una dosis intravenosa de 32 mg de ondansetrón, ésta ya no es la dosis recomendada. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada.
La eficacia se basó en la evaluación de las siguientes medidas compuestas: respuesta completa (definida como ausencia de episodios eméticos sin uso de tratamiento de rescate) principalmente durante el Ciclo 1. Los resultados se evaluaron para cada ensayo individual y para los 2 ensayos combinados.
En la Tabla 1 se muestra un resumen de los resultados clave del ensayo obtenidos del análisis combinado.
Tabla 1
Porcentaje de pacientes que estaban recibiendo quimioterapia altamente emetógena que respondieron
por grupo de tratamiento y fase - Ciclo 1
Régimen con Tratamiento estándar Diferencias*
aprepitant (N= 524)f
MEDIDAS COMPUESTAS (N= 521)f %
_%_% (IC del 95%)
Respuesta completa (sin emesis y sin tratamiento de rescate)_
|
Global (0-120 horas) |
67,7 |
47,8 |
19,9 |
(14,0, 25,8) |
|
0-24 horas |
86,0 |
73,2 |
12,7 |
(7,9, 17,6) |
|
25-120 horas |
71,5 |
51,2 |
20,3 |
(14,5, 26,1) |
MEDIDAS INDIVIDUALES
Sin emesis (sin episodios eméticos independientemente del uso de tratamiento de rescate)
|
Global (0-120 horas) |
71,9 |
49,7 |
22,2 |
(16,4, 28,0) |
|
0-24 horas |
86,8 |
74,0 |
12,7 |
(8,0, 17,5) |
|
25-120 horas |
76,2 |
53,5 |
22,6 |
(17,0, 28,2) |
|
Sin náuseas significativas (EAV |
máxima <25 mm en |
una escala de 0-100 mm) | ||
Global (0-120 horas) 72,1 64,9 7,2 (1,6, 12,8)
25-120 horas_74,0_66,9_7,1 (1,5, 12,6)
*Los intervalos de confianza se calcularon sin ajuste por sexo ni quimioterapia concomitante, los cuales fueron incluidos en el análisis primario de cociente de posibilidades y modelos logísticos. t Sólo un paciente en el régimen de aprepitant tuvo datos en la fase aguda y se excluyó de los análisis de fase retardada y general; sólo un paciente en el régimen estándar tuvo datos en la fase retardada y se excluyó del análisis de fase aguda y general.
El tiempo estimado hasta la primera emesis en el análisis combinado se representa en el gráfico Kapl an-Meier de la Figura 1.
Figura 1
Porcentaje de pacientes que estaban recibiendo quimioterapia altamente emetógena que siguieron sin padecer emesis con el tiempo - Ciclo 1
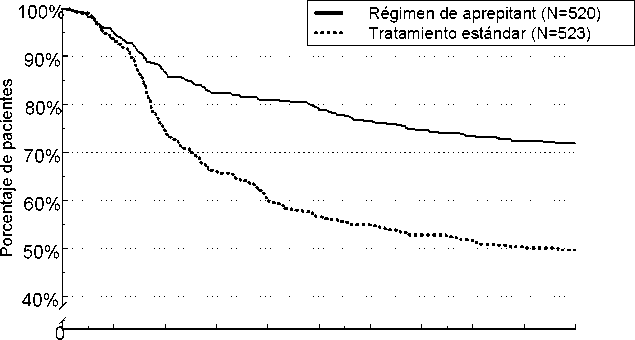
0 12 24 36 48 60 72 84 96 108 120
Tiempo (horas)
También se observaron diferencias estadísticamente significativas en eficacia en cada uno de los 2 ensayos individuales.
En los mismos 2 ensayos clínicos, 851 pacientes continuaron en la extensión de Ciclos Múltiples durante 5 ciclos adicionales de quimioterapia. La eficacia del régimen de aprepitant se mantuvo aparentemente durante todos los ciclos.
En un ensayo aleatorizado, doble ciego con un total de 866 pacientes (864 mujeres, 2 varones) que estaban recibiendo quimioterapia, que incluía ciclofosfamida 750-1.500 mg/m2; o ciclofosfamida 500-1500 mg/m2 y doxorrubicina (<60 mg/m2) o epirubicina (<100 mg/m2), se comparó aprepitant en combinación con un régimen de ondansetrón/dexametasona (ver sección 4.2) con el tratamiento estándar (placebo más ondansetrón 8 mg por vía oral (dos veces en el día 1 y cada 12 horas en los días 2 y 3) más dexametasona 20 mg por vía oral en el día 1).
La eficacia se basó en la evaluación de las medidas compuestas: respuesta completa (definida como ausencia de episodios eméticos sin uso de tratamiento de rescate) principalmente durante el Ciclo 1.
Tabla 2
Porcentaje de pacientes que respondieron por grupo de tratamiento y fase - Ciclo 1 Quimioterapia moderadamente emetógena
|
Régimen |
Tratamiento | |
|
con |
estándar | |
|
MEDIDAS COMPUESTAS aprepitant |
(N= 424) | |
|
(N= 433) f |
% |
% (IC del 95%) |
|
% |
Respuesta completa (sin emesis y sin tratamiento de rescate)_
Global (0-120 horas) 50,8 42,5 8,3 (1,6, 15,0)
0-24 horas 75,7 69,0 6,7 (0,7, 12,7)
25-120 horas_55,4_49,1_6,3 (-0,4, 13,0)
MEDIDAS INDIVIDUALES_
Sin emesis (sin episodios eméticos independientemente del uso de tratamiento de rescate)
Global (0-120 horas) 75,7 58,7 17,0 (10,8, 23,2)
0-24 horas 87,5 77,3 10,2 (5,1, 15,3)
25-120 horas_80,8_69,1_11,7 (5,9, 17,5)
Sin náuseas significativas (EAV máxima <25 mm en una escala de 0-100 mm)_
Global (0-120 horas) 60,9 55,7 5,3 (-1,3, 11,9)
0-24 horas 79,5 78,3 1,3 (-4,2, 6,8)
Tabla 3
Porcentaje de pacientes que respondieron por grupo de tratamiento y fase en el ensayo 2 - Ciclo 1
Quimioterapia moderadamente emetógena
|
Régimen |
Tratamiento |
Diferencias* |
|
con |
estándar | |
|
aprepitant |
(N= 406) | |
|
(N= 425) |
% |
% (IC del 95 %) |
|
% |
Respuesta completa (sin emesis y sin tratamiento de rescate)
|
Global (0-120 horas) |
68,7 |
56,3 |
12,4 |
(5,9, 18,9) |
|
0-24 horas |
89,2 |
80,3 |
8,9 |
(4,0, 13,8) |
|
25-120 horas |
70,8 |
60,9 |
9,9 |
(3,5, 16,3) |
Sin emesis (sin episodios eméticos independientemente del uso de tratamiento de rescate)_
|
Global (0-120 horas) |
76,2 |
62,1 |
14,1 |
(7,9, 20,3) |
|
0-24 horas |
92,0 |
83,7 |
8,3 |
(3,9, 12,7) |
|
25-120 horas |
77,9 |
66,8 |
11,1 |
(5,1, 17,1) |
|
Sin nauseas significativas (EAV |
máxima <25 mm en |
una escala |
de 0-100 mm) | |
|
Global (0-120 horas) |
73,6 |
66,4 |
7,2 |
(1,0, 13,4) |
|
0-24 horas |
90,9 |
86,3 |
4,6 |
(0,2, 9,0) |
|
25-120 horas |
74,9 |
69,5 |
5,4 |
(-0,7, 11,5) |
*Los intervalos de confianza se calcularon sin ajuste por sexo ni región, los cuales fueron incluidos en el análisis primario utilizando modelos logísticos.
El beneficio del tratamiento en combinación de aprepitant en la población del ensayo total fue dirigido principalmente por los resultados observados en pacientes con bajo control con el régimen estándar como en mujeres, aunque los resultados fueron numéricamente mejores independientemente de la edad, tipo de tumor o sexo. La respuesta completa al régimen de aprepitant y al tratamiento estándar, respectivamente, se alcanzó en 209/324 (65 %) y 161/320 (50 %) en mujeres y 83/101 (82 %) y 68/87 (78 %) de varones.
Población pediátrica
Los ensayos clínicos para evaluar el uso de aprepitant en pacientes pediátricos están en curso (ver sección 4.2 para la información sobre el uso pediátrico).
5.2 Propiedades farmacocinéticas
Aprepitant muestra una farmacocinética no lineal. Tanto el aclaramiento como la biodisponibilidad absoluta disminuyen al aumentar la dosis.
Absorción
La biodisponibilidad oral absoluta media de aprepitant es de 67% para la cápsula de 80 mg y de 59% para la cápsula de 125 mg. La concentración plasmática máxima media (Cmax) de aprepitant se alcanzó aproximadamente a las 4 horas (tmax). La administración oral de la cápsula con un desayuno estándar de aproximadamente 800 Kcal ocasionó un aumento de hasta el 40% en el AUC de aprepitant. Este aumento no se consideró clínicamente de interés.
La farmacocinética de aprepitant es no lineal en el intervalo de la dosis clínica. En adultos jóvenes sanos, el aumento en el AUC0-<X) fue un 26% mayor que proporcional a la dosis entre las dosis únicas de 80 mg y 125 mg administradas en estado posprandial.
Tras la administración oral de una dosis única de 125 mg de EMEND el día 1 y 80 mg una vez al día los días 2 y 3, el AUC0-24h (media ± DE) fue 19,6 ± 2,5 ^,g»h/ml y 21,2 ± 6,3 ^,g»h/ml los días 1 y 3, respectivamente. La Cmax fue 1,6 ± 0,36 ^g/ml y 1,4 ± 0,22 ^g/ml los días 1 y 3, respectivamente.
Distribución
Aprepitant se une fuertemente a proteínas, con una media del 97%. La media geométrica del volumen aparente de distribución en el estado equilibrio (Vdee) es aproximadamente de 66 litros en el ser humano.
Biotransformación
Aprepitant se metaboliza extensamente. En adultos jóvenes sanos, aprepitant representa aproximadamente el 19% de la radiactividad plasmática durante 72 horas después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant, lo que indica una importante presencia de metabolitos en el plasma. En el plasma humano se han identificado doce metabolitos de aprepitant. El metabolismo de aprepitant se produce en gran medida por oxidación en el anillo de morfolina y sus cadenas laterales y los metabolitos resultantes sólo fueron débilmente activos. Estudios in vitro en los que se usaron microsomas hepáticos humanos indicaron que aprepitant se metaboliza principalmente a través de CYP3A4 y posiblemente con una contribución menor a través de CYP1A2 y CYP2C19.
Eliminación
Aprepitant no se elimina inalterado en la orina. Los metabolitos se eliminan en la orina y a través de excreción biliar en las heces. Después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant a sujetos sanos, el 57% de la radiactividad se recuperó en la orina y el 45% en las heces.
El aclaramiento plasmático de aprepitant es dependiente de la dosis, disminuyendo al aumentar la dosis y oscilando aproximadamente entre 60 a 72 ml/min en el intervalo de la dosis terapéutica. La semivida terminal osciló entre aproximadamente 9 a 13 horas.
Farmacocinética en poblaciones especiales
Pacientes de edad avanzada: Tras la administración oral de una dosis única de 125 mg de aprepitant el día 1 y 80 mg una vez al día los días 2 a 5, el AUC0-24h de aprepitant fue un 21% superior el día 1 y un 36% superior el día 5 en pacientes de edad avanzada (>65 años) respecto de los adultos más jóvenes. La Cmax fue un 10% superior el día 1 y un 24% superior el día 5 en pacientes de edad avanzada respecto de los adultos más jóvenes. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en los pacientes de edad avanzada.
Sexo: Tras la administración oral de una dosis única de 125 mg de aprepitant, la Cmax de aprepitant es un 16% superior en las mujeres en comparación con los varones. La semivida de aprepitant es un 25% inferior en las mujeres en comparación con los varones y su tmax se produce en aproximadamente el mismo tiempo. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en función del sexo.
Insuficiencia hepática: La insuficiencia hepática leve (Child-Pugh clase A) no afecta a la farmacocinética de aprepitant en un grado clínicamente relevante. No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. De los datos disponibles no pueden extraerse conclusiones relativas a la influencia de la insuficiencia hepática moderada (Child-Pugh clase B) sobre la farmacocinética de aprepitant. No existen datos clínicos ni farmacocinéticos de pacientes con insuficiencia hepática grave (Child-Pugh clase C).
Insuficiencia renal: Se administró una dosis única de 240 mg de aprepitant a pacientes con insuficiencia renal grave (CrCl<30 ml/min) y a pacientes con nefropatía terminal que requería hemodiálisis.
En los pacientes con insuficiencia renal grave, el AUC0-a, del aprepitant total (no unido y unido a proteínas) disminuyó en un 21% y la Cmax disminuyó en un 32%, respecto de los sujetos sanos. En los pacientes con nefropatía terminal sometidos a hemodiálisis, el AUC0-<X) del aprepitant total disminuyó en un 42% y la Cmax disminuyó en un 32%. Debido a los modestos descensos en la unión a proteínas de aprepitant en los pacientes con enfermedad renal, el AUC de aprepitant no unido farmacológicamente activo no se vio significativamente afectado en los pacientes con insuficiencia renal en comparación con los sujetos sanos. La hemodiálisis realizada 4 ó 48 horas después de la administración no tuvo efectos significativos sobre la farmacocinética de aprepitant; en el dializado se recuperó menos de 0,2% de la dosis.
En pacientes con insuficiencia renal o en pacientes con nefropatía terminal sometidos a hemodiálisis no es necesario ajustar la dosis de EMEND.
Relación entre concentración y efecto
Usando un trazador altamente específico del receptor NK1, los estudios de tomografía por emisión de positrones (PET) en varones jóvenes sanos han demostrado que aprepitant penetra en el cerebro y ocupa los receptores NK1 de forma dependiente de la dosis y de la concentración plasmática. Se predice que las concentraciones plasmáticas de aprepitant alcanzadas con el régimen de 3 días de EMEND proporcionarán una ocupación superior al 95% de los receptores NK1 cerebrales.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de toxicidad, genotoxicidad, potencial carcinogénico y toxicidad para la reproducción a dosis únicas y repetidas. Sin embargo, se deberá tener en cuenta que la exposición sistémica en roedores fue similar o incluso inferior a la exposición terapéutica en seres humanos a la dosis de 125 mg/80 mg. En especial, aunque no se observaron efectos adversos en los estudios sobre la reproducción a los niveles de exposición en seres humanos, las exposiciones en animales no son suficientes para hacer una valoración de riesgo adecuada en el hombre.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula Sacarosa
Celulosa microcristalina (E 460) Hidroxipropilcelulosa (E 463)
Lauril sulfato de sodio
Cubierta de la cápsula Gelatina
Dióxido de titanio (E 171)
Óxido férrico rojo (