Cosentyx 150Mg Solucion Inyectable En Pluma Precargada
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Cosentyx 150 mg polvo para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial de polvo contiene 150 mg de secukinumab*. Tras la reconstitución, 1 ml de solución contiene 150 mg de secukinumab.
*Secukinumab es un anticuerpo monoclonal recombinante, íntegramente humano, selectivo a la interleuquina17A. Secukinumab es un anticuerpo de tipo IgG1/K producido en células ováricas de hámster chino.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para solución inyectable El polvo es un polvo liofilizado blanco.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Psoriasis en placas
Cosentyx está indicado para el tratamiento de la psoriasis en placas de moderada a grave en adultos candidatos a tratamientos sistémicos.
Artritis psoriásica
Cosentyx, solo o en combinación con metotrexato (MTX), está indicado para el tratamiento de la artritis psoriásica activa en pacientes adultos que han mostrado una respuesta inadecuada a tratamientos previos con fármacos antirreumáticos modificadores de la enfermedad (FAME) (ver sección 5.1).
Espondilitis anquilosante
Cosentyx está indicado para el tratamiento de la espondilitis anquilosante activa en adultos que no han respondido adecuadamente al tratamiento convencional.
4.2 Posología y forma de administración
Cosentyx se ha de utilizar bajo la dirección y la supervisión de un médico con experiencia en el diagnóstico y tratamiento de las enfermedades para las que Cosentyx está indicado.
Posología
Psoriasis en _placas
La dosis recomendada es de 300 mg de secukinumab por inyección subcutánea, que se administra inicialmente en la semana 0, 1, 2 y 3 y, luego mensualmente, comenzando en la semana 4, durante la fase de mantenimiento. Cada dosis de 300 mg se administra de forma repartida en dos inyecciones subcutáneas de 150 mg.
Artritis _ psoriásica
Para pacientes que padecen psoriasis en placas de moderada a grave de forma concomitante o que son respondedores inadecuados (RI) a anti-TNFa, la dosis recomendada es de 300 mg por inyección subcutánea, que se administra inicialmente en la semana 0, 1, 2 y 3 y, luego durante la fase de mantenimiento, mensualmente comenzando en la semana 4. Cada dosis de 300 mg se administra en dos inyecciones subcutáneas de 150 mg.
Para el resto de pacientes, la dosis recomendada es de 150 mg por inyección subcutánea, que se administra inicialmente en la semana 0, 1, 2 y 3 y, luego durante la fase de mantenimiento, mensualmente comenzando en la semana 4.
Espondilitis anquilosante
La dosis recomendada es de 150 mg por inyección subcutánea, que se administra inicialmente en la semana 0, 1, 2 y 3 y, luego durante la fase de mantenimiento, mensualmente comenzando en la semana 4.
Para todas las indicaciones anteriores, los datos disponibles sugieren que una respuesta clínica se alcanza normalmente en las 16 semanas de tratamiento. Se debe considerar interrumpir el tratamiento en los pacientes que no han mostrado respuesta a las 16 semanas de tratamiento. Algunos pacientes con una respuesta parcial al inicio, pueden mejorar posteriormente con un tratamiento continuado de más de 16 semanas.
Poblaciones especiales
Pacientes de edad avanzada (mayores de 65 años)
No es necesario un ajuste de la dosis (ver sección 5.2).
Insuficiencia renal / insuficiencia hepática
No se ha estudiado Cosentyx en estas poblaciones de pacientes. No se puede hacer ninguna recomendación posológica.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de Cosentyx en niños menores de 18 años. No se dispone de datos.
Forma de administración
Cosentyx se ha de administrar por inyección subcutánea. En la medida de lo posible, se deben evitar como lugares de inyección las zonas de la piel que presenten signos de psoriasis. El polvo para solución inyectable se debe reconstituir antes de utilizarlo. Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6 y también, las Instrucciones de Uso del prospecto.
Reacciones de hipersensibilidad graves al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Infecciones activas clínicamente importantes (p.ej. tuberculosis activa; ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
Infecciones
Cosentyx puede aumentar el riesgo de infecciones. En los ensayos clínicos se han observado infecciones en los pacientes que recibieron Cosentyx (ver sección 4.8). La mayoría fueron infecciones leves o moderadas de las vías respiratorias altas como rinofaringitis que no requirieron interrumpir el tratamiento.
Relacionado con el mecanismo de acción de Cosentyx, en los ensayos clínicos de psoriasis se han notificado infecciones mucocutáneas no graves por cándida más frecuentemente con secukinumab que con placebo (3,55 por 100 pacientes-año con secukinumab 300 mg frente a 1,00 por 100 paciente-año con placebo) (ver sección 4.8).
Se debe tener precaución cuando se valore la administración de Cosentyx en pacientes con infecciones crónicas o con antecedentes de infecciones recurrentes.
Se debe indicar al paciente que consulte al médico cuando padezca signos o síntomas indicativos de una infección. El paciente que contraiga una infección grave debe ser objeto de una estrecha observación y no debe recibir Cosentyx hasta que la infección se haya resuelto.
No se han notificado en los ensayos clínicos una mayor sensibilidad a la tuberculosis. Aun así, Cosentyx no se debe administrar a pacientes con tuberculosis activa. Se debe considerar la posibilidad de administrar un tratamiento antituberculoso antes de comenzar el tratamiento con Cosentyx en los pacientes con tuberculosis latente.
Enfermedad de Crohn
Se debe tener precaución cuando se prescriba Cosentyx a pacientes con enfermedad de Crohn. En los ensayos clínicos se ha observado exacerbaciones, en algunos casos graves, tanto en el grupo de pacientes de Cosentyx como en el de placebo. Se deben vigilar estrechamente los pacientes tratados con Cosentyx con enfermedad de Crohn.
Reacciones de hipersensibilidad
En los ensayos clínicos se han observado, en raras ocasiones, reacciones anafilácticas en pacientes que estaban recibiendo Cosentyx. Si aparecen reacciones anafilácticas u otras reacciones alérgicas graves, se debe interrumpir inmediatamente el tratamiento con Cosentyx e iniciar otro tratamiento alternativo.
Vacunas
No se deben administrar simultáneamente las vacunas elaboradas con microorganismos vivos con Cosentyx.
Los pacientes tratados con Cosentyx pueden recibir de forma simultánea vacunas inactivadas o no elaboradas con microorganismos vivos. En un estudio, después de la administración de vacunas antigripales inactivadas y antimeningocócicas, los voluntarios sanos tanto del grupo de 150 mg de secukinumab como de placebo, fueron capaces de generar una respuesta inmunitaria satisfactoria que por lo menos cuadruplicó los títulos de anticuerpos contra esas vacunas. Los datos indican que Cosentyx no inhibe la respuesta inmunitaria humoral a las vacunas antimeningocócicas o antigripales.
Tratamiento inmunosupresor concomitante
En los estudios de psoriasis, no se ha evaluado la seguridad y eficacia de Cosentyx en combinación con inmunosupresores, incluidos biológicos, o fototerapia (ver también sección 4.5).
4.5 Interacción con otros medicamentos y otras formas de interacción
No se deben administrar las vacunas elaboradas con microorganismos vivos simultáneamente con Cosentyx (ver también sección 4.4).
No se han realizado estudios de interacciones en humanos. No existe una evidencia directa del papel de IL-17A en la expresión de las enzimas CYP450. La formación de algunas enzimas P450 se suprime debido al aumento de citoquinas durante la inflamación crónica. Por tanto los tratamientos antiinflamatorios, tales como secukinumab, un inhibidor de IL17A, podría normalizar los niveles de CYP450 y en consecuencia, disminuir la exposición de las medicaciones concomitantes metabolizadas por el CYP450. Por lo que no se puede excluir que exista un efecto clínico relevante en los medicamentos de estrecho margen terapéutico, donde la dosis se ajusta de forma individual (p. ej. warfarina), que sean sustratos de CYP450. Cuando se inicie el tratamiento con secukinumab en pacientes tratados con este tipo de medicamentos, se debe considerar el realizar monitorización terapéutica.
No se observó interacción cuando Cosentyx se administró de forma concomitante con metotrexato (MTX) y/o corticosteroides en ensayos en artritis (incluyendo pacientes con artritis psoriásica y espondilitis anquilosante).
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar un método anticonceptivo durante el tratamiento y durante al menos 20 semanas después del tratamiento.
Embarazo
No se dispone de datos suficientes sobre el uso de secukinumab en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para el embarazo, el desarrollo embrionario o fetal, el parto o el desarrollo postnatal (ver sección 5.3). Como medida de precaución, es preferible evitar el uso de Cosentyx en el embarazo.
Lactancia
Se desconoce si secukinumab se excreta en la leche materna. Las inmunoglobulinas pasan a la leche materna y se desconoce si secukinumab se absorbe sistémicamente tras su ingestión. Debido a las posibles reacciones adversas de secukinumab en los lactantes, se debe decidir si interrumpir la lactancia durante el tratamiento y hasta 20 semanas después del tratamiento o interrumpir el tratamiento con Cosentyx, teniendo en cuenta el beneficio de la lactancia para el niño o el beneficio del tratamiento con Cosentyx para la mujer.
Fertilidad
No se ha evaluado el efecto de secukinumab sobre la fertilidad humana. Los estudios en animales no indican que Cosentyx tenga efectos nocivos directos o indirectos sobre la fertilidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Cosentyx sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Un total de 6.804 pacientes han recibido Cosentyx en los ensayos clínicos con y sin enmascaramiento en diversas indicaciones (psoriasis en placas, artritis psoriásica, espondilitis anquilosante y otras enfermedades autoinmunes). De éstos, 3.671 pacientes se han expuesto a Cosentyx durante al menos un año, lo que representa una exposición de 6.450 paciente-año.
Reacciones adversas en psoriasis en placas
Se agruparon los datos de los cuatro estudios de fase III controlados con placebo en psoriasis en placas para evaluar la seguridad de Cosentyx hasta 12 semanas después del inicio del tratamiento. Se evaluaron 2.076 pacientes en total (de los cuales, 692 pacientes recibieron la dosis de 150 mg, 690, la de 300 mg y 694, el placebo).
Las reacciones adversas notificadas con mayor frecuencia fueron las infecciones de las vías respiratorias altas (con mayor frecuencia rinofaringitis y rinitis). La mayoría de las reacciones fueron de naturaleza leve o moderada.
Reacciones adversas en artritis psoriásica
Cosentyx se estudió en dos ensayos controlados con placebo en artritis psoriásica con 1.003 pacientes (703 pacientes en Cosentyx y 300 pacientes en placebo) para una exposición total de 1.061 pacientes-años de exposición del ensayo (duración media de exposición para los pacientes tratados con secukinumab: 456 días en el Ensayo 1 en artritis psoriásica y 245 días en el Ensayo 2 en artritis psoriásica). El perfil de seguridad observado en pacientes con artritis psoriásica tratados con Cosentyx es consistente con el perfil de seguridad en psoriasis.
Reacciones adversas en espondilitis anquilosante
Cosentyx se estudió en dos ensayos controlados con placebo en espondilitis anquilosante con 590 pacientes (394 pacientes en Cosentyx y 196 pacientes en placebo) para un total de 755 pacientes-años de exposición del ensayo (duración media de exposición para los pacientes tratados con secukinumab: 469 días en el Ensayo 1 en espondilitis anquilosante y 460 días en el Ensayo 2 en espondilitis anquilosante). El perfil de seguridad observado en pacientes con espondilitis anquilosante tratados con Cosentyx es consistente con el perfil de seguridad en psoriasis.
Las reacciones adversas de los ensayos clínicos en psoriasis, artritis psoriásica y espondilitis anquilosante (Tabla 1) se presentan según la clasificación de órganos del MedDRA. Dentro de cada órgano y sistema, las reacciones adversas se ordenan por frecuencia, las más frecuentes primero. Las reacciones adversas se incluyen en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Además, las categorías de frecuencia se definen utilizando los siguientes criterios: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000).
Tabla 1 Lista de las reacciones adversas en los ensayos clínicos1-1
|
Sistema de Clasificación de Órganos |
Frecuencia |
Reacción adversa |
|
Infecciones e infestaciones |
Muy frecuentes |
Infecciones de las vías respiratorias altas |
|
Frecuentes |
Herpes oral | |
|
Poco frecuentes |
Candidiasis oral | |
|
Pie de atleta | ||
|
Otitis externa | ||
|
Trastornos de la sangre y del sistema linfático |
Poco frecuentes |
Neutropenia |
|
Trastornos del sistema inmunológico |
Raras |
Reacciones anafilácticas |
|
Trastornos oculares |
Poco frecuentes |
Conjuntivitis |
|
Trastornos respiratorios, torácicos y mediastínicos |
Frecuentes |
Rinorrea |
|
Trastornos gastrointestinales |
Frecuentes |
Diarrea |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
Urticaria |
|
]) En los ensayos clínicos controlados con placebo (fase III) en psoriasis en placas, artritis psoriásica y espondilitis anquilosante, los pacientes recibieron 300 mg, 150 mg, 75 mg o placebo durante 12 semanas (psoriasis) o 16 semanas (artritis psoriásica y espondilitis anquilosante) de duración del tratamiento | ||
Descripción de las reacciones adversas seleccionadas Infecciones
Durante la fase comparativa con placebo de los ensayos clínicos en psoriasis en placas (en los que un total de 1.382 pacientes recibieron Cosentyx y 694, el placebo, durante un período de hasta 12 semanas), se notificaron infecciones en el 28,7% de los pacientes del grupo de Cosentyx y en el 18,9% de los pacientes que recibieron el placebo. La mayoría de las infecciones se consideraron no graves, infecciones leves o moderadas de las vías respiratorias altas, como rinofaringitis, que no requirieron interrupción del tratamiento. Hubo un aumento de candidiasis en mucosa y piel, consistente con el mecanismo de acción, no graves, de leves a moderadas y que respondieron al tratamiento estándar sin tener que interrumpir el tratamiento. Las infecciones graves aparecieron en un 0,14% de los pacientes tratados con Cosentyx y en un 0,3% de los pacientes tratados con placebo (ver sección 4.4).
Durante todo el periodo de tratamiento (un total de 3.430 pacientes tratados con Cosentyx durante 52 semanas, en la mayoría de los pacientes), se notificaron infecciones en el 47,5% de los pacientes tratados con Cosentyx (0,9 por paciente-año de seguimiento). El 1,2% de los pacientes tratados con Cosentyx notificaron infecciones graves (0,015 por paciente-año de seguimiento).
La tasa de infecciones observada en los ensayos clínicos en artritis psoriásica y espondilitis anquilosante fue similar a la observada en los ensayos en psoriasis.
Neutropenia
En los ensayos clínicos de fase 3 en psoriasis, se ha observado con mayor frecuencia neutropenia con secukinumab que con placebo, pero en la mayoría de los casos fue leve, transitoria y reversible. Se notificó neutropenia <1,0-0,5x109/l (CTCAE Grado 3) en 18 de los 3.430 (0,5%) pacientes con secukinumab, independientemente de la dosis y sin una relación temporal con la infección en 15 de los 18 casos. No se notificaron casos de neutropenia más grave. Los otros 3 casos restantes fueron infecciones no graves que respondieron al tratamiento estándar y no requirieron interrumpir el tratamiento con Cosentyx.
La frecuencia de neutropenia en artritis psoriásica y espondilitis anquilosante es similar a psoriasis.
Se notificaron raros casos de neutropenia <0,5x109/l (CTCAE Grado 4).
Reacciones de hipersensibilidad
En los ensayos clínicos se ha observado urticaria y raros casos de reacción anafiláctica a Cosentyx (ver también sección 4.4).
Inmunogenicidad
En ensayos clínicos en psoriasis, artritis psoriásica y espondilitis anquilosante, menos del 1% de los pacientes tratados con Cosentyx desarrollaron anticuerpos a secukinumab a lo largo de 52 semanas de tratamiento. Aproximadamente la mitad de los anticuerpos antifármaco producidos durante el tratamiento fueron neutralizantes pero esto no se asoció a una pérdida de eficacia o a trastornos farmacocinéticos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han notificado casos de sobredosis en los ensayos clínicos.
Durante los estudios clínicos se han administrado por vía intravenosa dosis de hasta 30 mg/kg (aproximadamente de 2.000 a 3.000 mg) sin que haya aparecido toxicidad limitante de la dosis. En caso de sobredosis se recomienda vigilar al paciente en busca de signos o síntomas de reacciones adversas e instaurar inmediatamente el tratamiento sintomático más adecuado.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inmunosupresores, inhibidores de la interleucina, código ATC: L04AC10 Mecanismo de acción
Secukinumab es un anticuerpo de tipo IgG1/K monoclonal, íntegramente humano, que se une selectivamente y neutraliza una citoquina proinflamatoria, la interleuquina 17A (IL-17A). Secukinumab actúa dirigiéndose a IL-17A e inhibe su interacción con el receptor de IL-17, que se encuentra en varios tipos de células, incluidos los queratinocitos. Como resultado, secukinumab inhibe la liberación de citoquinas proinflamatorias, de quimioquinas y de mediadores del daño tisular, y reduce los efectos mediados por la IL-17A, que participan en la enfermedad autoinmunitaria e inflamatoria. A la piel llegan concentraciones clínicamente importantes de secukinumab y reducen los marcadores inflamatorios locales. Como consecuencia directa, el tratamiento con secukinumab reduce el eritema, la induración y la descamación presentes en las lesiones de la psoriasis en placas.
IL-17A es una citoquina natural que participa en reacciones inmunitarias e inflamatorias normales. IL-17A desempeña una función clave en la patogenia de la psoriasis en placas, artritis psoriásica y espondilitis anquilosante y se encuentra concentrada en la piel lesionada, a diferencia de la piel no lesionada de los pacientes con psoriasis en placas y en el tejido sinovial de los pacientes con artritis psoriásica. La frecuencia de células productoras de IL-17, también fue significativamente superior en la médula ósea subcondral de las articulaciones facetarías de pacientes con espondilitis anquilosante.
Efectos farmacodinámicos
Las concentraciones séricas de IL-17A total (libre y unida a secukinumab) aumentan inicialmente en los pacientes que reciben secukinumab. Después disminuye lentamente debido a un aclaramiento reducido del complejo secukinumab-IL-17A, lo que indica que secukinumab es capaz de fijarse selectivamente a la IL-17A libre, la cual desempeña un papel fundamental en la patogenia de la psoriasis en placas.
En un estudio con secukinumab, los neutrófilos epidérmicos infiltrantes y los distintos marcadores asociados a neutrófilos, presentes en gran número en la piel lesionada de los pacientes con psoriasis en placas, disminuyeron significativamente al cabo de una o dos semanas de tratamiento.
Secukinumab ha demostrado que reduce (entre 1 y 2 semanas de tratamiento) los niveles de proteína C reactiva, que es un marcador de la inflamación.
Eficacia clínica y seguridad
Psoriasis en placas
La seguridad y la eficacia de Cosentyx se evaluaron en cuatro estudios de fase III, aleatorizados, con doble enmascaramiento y comparativos con placebo, llevados a cabo en pacientes con psoriasis en placas moderada o grave que eran candidatos de fototerapia o de tratamientos sistémicos [ERASURE, FIXTURE, FEATURE y JUNCTURE]. La eficacia y la seguridad de Cosentyx 150 mg y 300 mg se evaluaron frente a placebo y etanercept. Además, en otro estudio [SCULPTURE] se evaluó un régimen terapéutico crónico en comparación con la pauta de “repetición del tratamiento en caso de necesidad”.
De los 2.403 pacientes que participaron en los estudios comparativos con placebo, el 79% carecía de antecedentes de tratamiento biológico, el 45% procedían de fracasos con tratamientos no biológicos, un 8% procedía de fracasos de tratamientos biológicos (el 6% de fracasos con anti-TNF y el 2% de tratamientos anti-p40). Entre el 15 y el 25% de los pacientes de los estudios de fase III tenían artritis psoriásica al inicio.
En el estudio 1 sobre psoriasis (ERASURE) se evaluaron 738 pacientes. Los pacientes asignados aleatoriamente al grupo de Cosentyx recibieron dosis de 150 ó 300 mg en las semanas 0, 1, 2 y 3 y, luego una vez al mes, comenzando en la semana 4, la misma dosis. En el estudio 2 sobre psoriasis (FIXTURE) se evaluaron 1.306 pacientes. Los pacientes asignados aleatoriamente al grupo de Cosentyx recibieron dosis de 150 ó 300 mg en las semanas 0, 1, 2 y 3 y, luego una vez al mes, comenzando en la semana 4, la misma dosis. Los pacientes asignados aleatoriamente al grupo del etanercept recibieron dosis de 50 mg dos veces por semana durante 12 semanas y, luego, 50 mg una vez por semana. En ambos estudios, estudio 1 y 2, los pacientes asignados aleatoriamente al grupo del placebo que no respondían al cabo de 12 semanas pasaron a recibir Cosentyx (150 ó 300 mg) a las semanas 12, 13, 14 y 15 y, luego una vez al mes, comenzando en la semana 16, la misma dosis. Desde la primera administración del tratamiento del estudio, todos los pacientes fueron sometidos a un seguimiento de hasta 52 semanas de duración.
En el estudio 3 sobre psoriasis (FEATURE) se evaluaron 177 pacientes usando una jeringa precargada en comparación con el placebo después de 12 semanas de tratamiento para evaluar la seguridad, la tolerabilidad y la autoadministración de Cosentyx con la jeringa precargada. En el estudio 4 sobre psoriasis (JUNCTURE) se evaluaron 182 pacientes usando una pluma precargada en comparación con el placebo después de 12 semanas de tratamiento para evaluar la seguridad, la tolerabilidad y la autoadministración de Cosentyx con la pluma precargada. En ambos estudios, estudio 3 y 4, los pacientes asignados aleatoriamente al grupo de Cosentyx, recibieron dosis de 150 ó 300 mg a las semanas 0, 1, 2 y 3 y, luego una vez al mes, comenzando en la semana 4, la misma dosis. También se aleatorizaron pacientes para que recibiesen el placebo las semanas 0, 1, 2 y 3 y, luego una vez al mes, comenzando en la semana 4, la misma dosis.
En el estudio 5 sobre psoriasis (SCULPTURE) se evaluaron 966 pacientes. Todos los pacientes recibieron Cosentyx en dosis de 150 ó 300 mg a las semanas 0, 1, 2, 3, 4, 8 y 12 y, luego, se les asignó de forma aleatoria un régimen de mantenimiento mensual con la misma dosis a partir de la semana 12, o bien una pauta de “repetición del tratamiento en caso de necesidad” con la misma dosis. Los pacientes asignados aleatoriamente al grupo de “repetición del tratamiento en caso de necesidad” que no consiguieron un mantenimiento satisfactorio de la respuesta, se recomendó un régimen de mantenimiento con dosis mensuales fijas.
Las co-variables principales en los estudios con placebo y con comparador fueron la proporción de pacientes que consiguieron una respuesta de PASI 75 y una respuesta IGA mod 2011 de “blanqueamiento completo” o “prácticamente completo”, en comparación con placebo al cabo de 12 semanas (ver Tabla 2 y 3). Con la dosis de 300 mg se obtuvo una mejoría en la piel, en particular, un “blanqueamiento completo” o “prácticamente completo” con criterios de eficacia de PASI 90,
PASI 100, e IGA mod 2011 0 ó 1 en todos los estudios, con efectos máximos a la semana 16, de modo que se recomienda esta dosis.
Tabla 2 Resumen de las respuestas PASI 50/75/90/100 “blanqueamiento completo” o
“prácticamente completo” en IGA* mod 2011 en los estudios 1, 3 y 4 en psoriasis (ERASURE, FEATURE y JUNCTURE)
|
Semana 12 |
Semana 16 |
Semana 52 | |||||
|
Placebo |
150 mg |
300 mg |
150 mg |
300 mg |
150 mg |
300 mg | |
|
Estudio 1 | |||||||
|
Número de pacientes |
246 |
244 |
245 |
244 |
245 |
244 |
245 |
|
n (%) de respuesta PASI 50 |
22 (8,9%) |
203 (83,5%) |
222 (90,6%) |
212 (87,2%) |
224 (91,4%) |
187 (77%) |
207 (84,5%) |
|
n (%) de respuesta PASI 75 |
11 (4,5%) |
174 (71,6%)** |
200 (81,6%)** |
188 (77,4%) |
211 (86,1%) |
146 (60,1%) |
182 (74,3%) |
|
n (%) de respuesta PASI 90 |
3 (1,2%) |
95 (39,1%)** |
145 (59,2%)** |
130 (53,5%) |
171 (69,8%) |
88 (36,2%) |
147 (60,0%) |
|
n (%) de respuesta PASI 100 |
2 (0,8%) |
31 (12,8%) |
70 (28,6%) |
51 (21,0%) |
102 (41,6%) |
49 (20,2%) |
96 (39,2%) |
|
n (%) de “blanqueamiento |
6 (2,40%) |
125 |
160 |
142 |
180 |
101 |
148 |
|
completo” o “prácticamente completo” según IGA mod 2011 Estudio 3 |
(51,2%)** |
(65,3%)** |
(58,2%) |
(73,5%) |
(41,4%) |
(60,4%) | |
|
Número de pacientes |
59 |
59 |
58 |
- |
- |
- |
- |
|
n (%) de respuesta PASI 50 |
3 (5,1%) |
51 (86,4%) |
51 (87,9%) |
- |
- |
- |
- |
|
n (%) de respuesta PASI 75 |
0 (0,0%) |
41 (69,5%)** |
44 (75,9%)** |
- |
- |
- |
- |
|
n (%) de respuesta PASI 90 |
0 (0,0%) |
27 (45,8%) |
35 (60,3%) |
- |
- |
- |
- |
|
n (%) de respuesta PASI 100 |
0 (0,0%) |
5 (8,5%) |
25 (43,1%) |
- |
- |
- |
- |
|
n (%) de “blanqueamiento completo” o “prácticamente completo” según IGA mod 2011 Estudio 4 |
0 (0,0%) |
31 (52,5%)** |
40 (69,0%)** | ||||
|
Número de pacientes |
61 |
60 |
60 |
- |
- |
- |
- |
|
n (%) de respuesta PASI 50 |
5 (8,2%) |
48 (80,0%) |
58 (96,7%) |
- |
- |
- |
- |
|
n (%) de respuesta PASI 75 |
2 (3,3%) |
43 (71,7%)** |
52 (86,7%)** |
- |
- |
- |
- |
|
n (%) de respuesta PASI 90 |
0 (0,0%) |
24 (40,0%) |
33 (55,0%) |
- |
- |
- |
- |
|
n (%) de respuesta PASI 100 |
0 (0,0%) |
10 (16,7%) |
16 (26,7%) |
- |
- |
- |
- |
|
n (%) de “blanqueamiento completo” o “prácticamente |
0 (0,0%) |
32 (53,3%)** |
44 (73,3%)** |
- |
- |
- |
- |
completo” según IGA mod 2011
* La IGA mod 2011 es una escala de 5 categorías: “0 = blanqueamiento completo total”, “1 = blanqueamiento prácticamente completo”, “2 = (psoriasis) leve”, “3 = moderada” o “4 = grave” que indica la evaluación global del médico sobre la intensidad de la psoriasis en función de la induración, el eritema y la descamación. Se definió como éxito terapéutico, “remisión total” o “remisión casi total”, la ausencia de signos de psoriasis o bien una coloración normal o rosada de las lesiones cutáneas, ausencia de induración de la placa y ninguna o una mínima descamación focal.
** valores de p con respecto al placebo, ajustados en función de la multiplicidad: p<0,0001._
Tabla 3 Resumen de la respuesta clínica del estudio 2 de psoriasis (FIXTURE)
Semana 12 Semana 16 Semana 52
|
Placebo |
150 mg |
300 mg |
Etanercept |
150 mg |
300 mg |
Etanercept |
150 mg |
300 mg |
Etanercept | |
|
Número de |
324 |
327 |
323 |
323 |
327 |
323 |
323 |
327 |
323 |
323 |
|
pacientes n (%) de respuesta |
49 |
266 |
296 |
226 |
290 |
302 |
257 (79,6%) |
249 |
274 |
234 (72,4%) |
|
PASI 50 |
(15,1%) |
(81,3%) |
(91,6%) |
(70,0%) |
(88,7%) |
(93,5%) |
(76,1%) |
(84,8%) | ||
|
n (%) de respuesta |
16 |
219 |
249 |
142 |
247 |
280 |
189 (58,5%) |
215 |
254 |
179 (55,4%) |
|
PASI 75 |
(4,9%) |
(67,0%) ** |
(77,1%) ** |
(44,0%) |
(75,5%) |
(86,7%) |
(65,7%) |
(78,6%) | ||
|
n (%) de respuesta |
5 (1,5%) |
137 |
175 |
67 (20,7%) |
176 |
234 |
101 (31,3%) |
147 |
210 |
108 (33,4%) |
|
PASI 90 |
(41,9%) |
(54,2%) |
(53,8%) |
(72,4%) |
(45,0%) |
(65,0%) | ||||
|
n (%) de respuesta |
0 (0%) |
47 |
78 |
14 (4,3%) |
84 |
119 |
24 (7,4%) |
65 |
117 |
32 (9,9%) |
|
PASI 100 |
(14,4%) |
(24,1%) |
(25,7%) |
(36,8%) |
(19,9%) |
(36,2%) | ||||
|
n (%) de de |
9 (2,8%) |
167 |
202 |
88 (27,2%) |
200 |
244 |
127 (39,3%) |
168 |
219 |
120 (37,2%) |
|
“blanqueamiento |
(51,1%) |
(62,5%) |
(61,2%) |
(75,5%) |
(51,4%) |
(67,8%) | ||||
|
completo” o |
** |
** |
“prácticamente completo” en la
IGA mod 2011_
** valores p con respecto al etanercept: p=0,0250
En un ensayo adicional en psoriasis (CLEAR) se evaluaron 676 pacientes. Secukinumab 300 mg alcanzó las variables principal y secundaria mostrando superioridad a ustekinumab de acuerdo a la respuesta de PASI 90 a la semana 16 y a la velocidad de inicio de respuesta de PASI 75 a la semana 4. Se observó una mayor eficacia para secukinumab comparado con ustekinumab para los criterios de respuesta de PASI 75/90/100 e IGA mod 2011 0 ó 1 (“blanqueamiento completo” o “prácticamente completo”) de forma temprana y continua hasta la semana 16.
Tabla 4 Resumen de la respuesta clínica en el ensayo CLEAR
|
Semana 4 |
Semana 16 | |||
|
Secukinumab 300 mg |
Ustekinumab* |
Secukinumab 300 mg |
Ustekinumab* | |
|
Número de pacientes |
334 |
335 |
334 |
335 |
|
n (%) de respuesta PASI 75 |
167 (50,0%)** |
69 (20,6%) |
311 (93,1%) |
277 (82,7%) |
|
n (%) de respuesta PASI 90 |
70 (21,0%) |
18 (5,4%) |
264 (79,0%)** |
193 (57,6%) |
|
n (%) de respuesta PASI 100 |
14 (4,2%) |
3 (0,9%) |
148 (44,3%) |
95 (28,4%) |
|
n (%) de respuesta IGA mod 2011 “blanqueamiento completo” o “prácticamente completo” |
126 (37,7%) |
41 (12,2%) |
277 (82,9%) |
226 (67,5%) |
* Los pacientes tratados con secukinumab recibieron dosis de 300 mg a las semanas 0, 1, 2 y 3, seguidas por la misma dosis a las semanas 4, 8 y 12. Los pacientes tratados con ustekinumab recibieron 45 mg o 90 mg a las semanas 0 y 4 (dosificadas por peso según la posología aprobada)
** valores p con respecto a ustekinumab: p<0,0001
Cosentyx fue eficaz en pacientes sin antecedentes de tratamiento sistémico, sin antecedentes de tratamiento biológico, en pacientes previamente expuestos a un tratamiento biológico anti-TNF y en los pacientes que habían fracasado con un tratamiento biológico anti-TNF. Al inicio del estudio, las mejoras en PASI 75 en pacientes con artritis psoriásica concomitante fueron similares a las de la población general con psoriasis en placas.
Cosentyx se asociaba a un efecto de inicio rápido, con un 50% de reducción en la puntuación media del PASI en la semana 3 con la dosis de 300 mg.
Figura 1 Variación porcentual de la puntuación media del PASI con respecto a la inicial durante el estudio 1 (ERASURE)
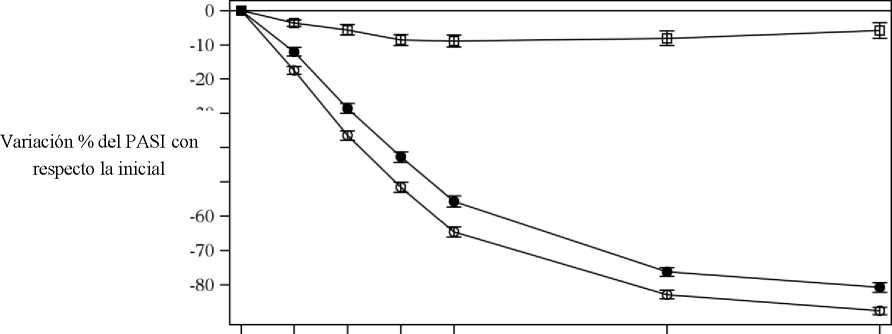
|
0 1 2 |
Semanas de tratamiento n=número de pacientes evaluables |
12 |
|
• Cosentyx 150 mg (n=243) o Cosentyx 300 mg (n=245) □ Placebo (n=245) | ||
Localizaciones/formas específicas de psoriasis en placas
En dos ensayos adicionales controlados con placebo, se observó mejoría en psoriasis ungueal (TRANSFIGURE, 198 pacientes) y en psoriasis en placa palmoplantar (GESTURE, 205 pacientes).
En el ensayo TRANSFIGURE, secukinumab fue superior a placebo a la semana 16 (46,1% para 300 mg, 38,4% para 150 mg y 11,7% para placebo) según lo evaluado por la mejoría significativa desde el periodo basal en el Índice de Gravedad de Psoriasis Ungueal (NAPSI %) para pacientes con psoriasis en placas de moderada a grave con afectación ungueal. En el ensayo GESTURE, secukinumab fue superior a placebo a la semana 16 (33,3% para 300 mg, 22,1% para 150 mg, y 1,5% para placebo) según lo evaluado por la mejoría significativa de respuesta de ppIGA 0 ó 1 (“blanqueamiento completo” o “ prácticamente completo”) para pacientes con psoriasis en placas palmoplantar de moderada a grave.
Calidad de vida/resultados percibidos por los pacientes
En la semana 12, el índice de calidad de vida en dermatología (DLQI) había mejorado estadísticamente de manera significativa en comparación con placebo respecto al inicio (estudios 1-4). La disminución media (mejoras) en DLQI respecto al inicio se puntuó desde -10,4 a -11,6 con secukinumab 300 mg, desde -7,7 a -10,1 con secukinumab 150 mg, frente al -1,1 y -1,9 de placebo en la semana 12. Estas mejoras se mantuvieron durante las 52 semanas (estudios 1 y 2).
El 40% de los participantes de los estudios 1 y 2 completaron el diario de síntomas de psoriasis (Psoriasis Symptom Diary®). De los participantes de cada uno de estos estudios que completaron el diario, mostraron mejoras estadísticamente significativas en los signos y síntomas de picor, dolor y descamación percibidos por los pacientes, a la semana 12 con respecto al inicio en comparación con el placebo.
Artritis _ psoriásica
Se evaluaron la seguridad y eficacia de Cosentyx en 1.003 pacientes en dos ensayos de fase III controlados con placebo, doble ciegos, aleatorizados, en pacientes con artritis psoriásica activa (>3 articulaciones inflamadas y >3 articulaciones dolorosas) a pesar del tratamiento con medicamentos antiinflamatorios no esteroideos (NSAID), corticosteroides o fármacos antirreumáticos modificadores de la enfermedad (FAME). En estos ensayos se reclutaron pacientes con cada subtipo de PsA, incluidas la artritis poliarticular sin evidencia de nódulos reumatoides, espondilitis con artritis periférica, artritis periférica asimétrica, participación interfalángica distal y artritis mutilante. Los pacientes que participaron en estos ensayos tenían un diagnóstico de PsA durante una media de 3,9 a
5,3 años. La mayoría de los pacientes también presentaban lesiones cutáneas de psoriasis activa o un historial documentado de psoriasis. Más del 62% y del 47% de los pacientes con PsA presentaban entesitis y dactilitis basales, respectivamente. Para ambos ensayos, la variable principal fue la respuesta 20 de la American College of Rheumatology (ACR) a la semana 24.
En el ensayo 1 en artritis psoriásica (Ensayo 1 en PsA) y en el ensayo 2 en artritis psoriásica (Ensayo 2 en PsA), el 29% y el 35% de los pacientes, respectivamente, fueron previamente tratados con un medicamento anti-TNFa e interrumpieron dicho tratamiento por falta de eficacia o intolerancia (pacientes con respuesta inadecuada a anti-TNFa).
En el Ensayo 1 en PsA (FUTURE 1) se evaluaron 606 pacientes, de los cuales el 60,7% recibían MTX de forma concomitante. Los pacientes aleatorizados a Cosentyx recibieron 10 mg/kg vía intravenosa a las semanas 0, 2, y 4, seguidas por 75 mg o 150 mg vía subcutánea mensualmente empezando en la semana 8. Los pacientes aleatorizados a placebo que eran no respondedores a la semana 16 (rescate temprano) y el resto de pacientes de placebo a la semana 24 se cruzaron para recibir Cosentyx (75 mg o 150 mg vía subcutánea) seguido por la misma dosis mensualmente.
En el Ensayo 2 en PsA (FUTURE 2) se evaluaron 397 pacientes, de los cuales el 46,6% recibían MTX de forma concomitante. Los pacientes aleatorizados a Cosentyx recibieron 75 mg, 150 mg o 300 mg vía subcutánea a las semanas 0, 1, 2, y 3, seguidas por la misma dosis mensualmente empezando en la semana 4. Los pacientes aleatorizados a recibir placebo que no eran respondedores a la semana 16 (rescate temprano) se cruzaron para recibir Cosentyx (150 mg o 300 mg vía subcutánea) a la semana 16 seguido por la misma dosis mensualmente. Los pacientes aleatorizados a recibir placebo que eran respondedores a la semana 16 se cruzaron para recibir Cosentyx (150 mg o 300 mg vía subcutánea) a la semana 24 seguido por la misma dosis mensualmente.
Signos y síntomas
El tratamiento con Cosentyx dio como resultado una mejora significativa en cuanto a las medidas de la actividad de la enfermedad en comparación con placebo a la semana 24 (ver Tabla 5).
Tabla 5 Respuesta clínica en el Ensayo 2 en PsA a la semana 24
|
Semana 24 | ||||
|
Placebo |
75 mg |
150 mg |
300 mg | |
|
Número de pacientes aleatorizados |
98 |
99 |
100 |
100 |
|
n (%) de respuesta ACR20 |
15 (15,3%) |
29 (29,3%*) |
51 (51,0%***) |
54 (54,0%***) |
|
n (%) de respuesta ACR50 |
7 (7,1%) |
18 (18,2%) |
35 (35,0%) |
35 (35,0%**) |
|
n (%) de respuesta ACR70 |
1 (1,0%) |
6 (6,1%) |
21 (21,0%**) |
20 (20,0%**) |
|
DAS28-CRP |
-0,96 |
-1,12 |
-1,58** |
-1,61** |
|
Número de pacientes con >3% BSA afectada de psoriasis al inicio del estudio |
43 (43,9%) |
50 (50,5%) |
58 (58,0%) |
41 (41,0%) |
|
n (%) de respuesta PASI 75 |
7 (16,3%) |
14 (28,0%) |
28 (48,3%**) |
26 (63,4%***) |
|
n (%) de respuesta PASI 90 |
4 (9,3%) |
6 (12,0%) |
19 (32,8%**) |
20 (48,8%***) |
|
n (%) de resolución de Dactilitis f |
4 (14,8%) |
10 (30,3%) |
16 (50,0%**) |
26 (56,5%**) |
|
n (%) de resolución de Entesitis $ |
14 (21,5%) |
22 (32,4%) |
27 (42,2%*) |
27 (48,2%**) |
* p<0,05, ** p<0,01, *** p<0,001; frente a placebo
Todos los valores-p se ajustan en función de la multiplicidad de ensayos de acuerdo a la jerarquía predefinida, excepto para ACR70, Dactilitis y Entesitis, que eran variables exploratorias.
Imputación de no respondedor utilizada para la variable binaria que falta.
ACR: American College of Rheumatology; PASI: Índice de Severidad y Área de Psoriasis; DAS: Puntuación de Actividad de la Enfermedad; BSA: Área de Superficie Corporal fEn pacientes con dactilitis al inicio del estudio (n=27, 33, 32, 46, respectivamente)
{En pacientes con entesitis al inicio del estudio (n=65, 68, 64, 56, respectivamente)_
El inicio de acción de Cosentyx se produjo tan pronto como en la semana 2. En la semana 3 se alcanzó una diferencia estadísticamente significativa en el ACR 20 frente a placebo. En la semana 16, los pacientes tratados con Cosentyx demostraron mejoras significativas en los signos y síntomas, entre los cuales se encuentran, respuestas significativamente mayores en ACR 20 (33,3%, 60,0% y 57,0% para 75 mg, 150 mg y 300 mg, respectivamente) comparado con placebo (18,4%).
En la Figura 2 se muestra el porcentaje de pacientes por visita que alcanzaron la respuesta ACR 20. Figure 2 Respuesta ACR20 en el Ensayo 2 de PsA a lo largo del tiempo hasta la semana 24
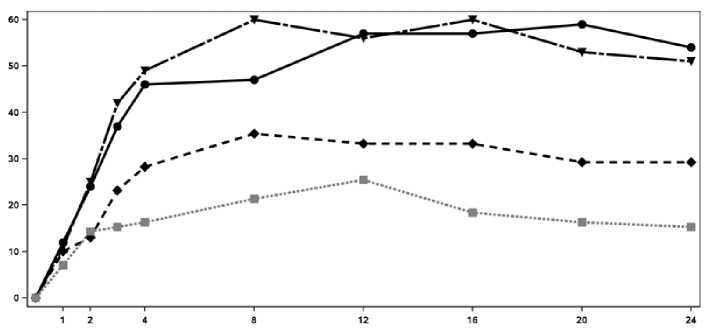
Tiempo (Semanas)
■ Cosentyx 150 mg Cosentyx 300 mg ■ ■ ■ • Placebo
Porcentaje de respondedores
Se observaron respuestas similares para las variables primaria y secundaria principal en pacientes con PsA a pesar de que estuviesen o no con tratamiento concomitante con MTX. En la semana 24, los pacientes tratados con Cosentyx y tratamiento concomitante con MTX solían tener una respuesta ACR 20 (44,7%, 47,7% y 54,4% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 20,0%) y ACR 50 (27,7%, 31,8% y 38,6% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 8,0%) más elevada. Los pacientes tratados con Cosentyx sin el uso concomitante de MTX solían tener una respuesta ACR 20 (15,4%, 53,6% y 53,6% para 75 mg,
150 mg y 300 mg, respectivamente, comparado con placebo 10,4%) y ACR 50 (9,6%, 37,5% y 32,1% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 6,3%) más elevada.
Los pacientes tratados con Cosentyx que eran “naíve” para anti-TNFa o con RI a anti-TNFa, presentaron una respuesta ACR 20 significativamente mayor comparado con placebo a la semana 24, con una respuesta ligeramente superior en el grupo “naíve” para anti-TNFa (“naíve” para anti-TNFa: 37%, 64% y 58% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 15,9%; RI a anti-TNFa: 15%, 30% y 46% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 14,3%). En el subgrupo de pacientes con RI a anti-TNFa, solo la dosis de 300 mg mostró una tasa de respuesta significativamente mayor para ACR 20 comparado con placebo (p<0,05) y demostró un beneficio clínico significativo sobre 150 mg en las variables secundarias múltiples. Se observaron mejorías en la respuesta PASI 75 en ambos subgrupos y la dosis de 300 mg mostró un beneficio estadísticamente significativo en los pacientes con RI a anti-TNFa.
El número de pacientes con PsA con afectación axial era demasiado pequeño como para permitir una evaluación significativa.
Se mostraron mejorías en todos los componentes de las puntuaciones ACR, incluyendo la evaluación del dolor del paciente. La proporción de pacientes que alcanzaron una respuesta Criterio de Respuesta a PsA (PsARC) modificada fue superior en los pacientes tratados con Cosentyx (38,4%, 62,0% y 63,0% para 75 mg, 150 mg y 300 mg, respectivamente) comparado con placebo (29,6%) a la semana 24.
En el Ensayo 1 en PsA y en el Ensayo 2 en PsA, la eficacia se mantuvo hasta la semana 52. En el Ensayo 2 en PsA, entre los 200 pacientes inicialmente aleatorizados a Cosentyx 150 mg y 300 mg, 178 (89%) pacientes aún estaban en tratamiento en la semana 52. De los 100 pacientes aleatorizados a Cosentyx 150 mg, 64, 39 y 20 presentaron una respuesta ACR 20/50/70, respectivamente. De los 100 pacientes aleatorizados a Cosentyx 300 mg, 64, 44 y 24 presentaron una respuesta ACR 20/50/70, respectivamente.
Respuesta radiográfica
No se ha demostrado la inhibición de la progresión del daño estructural en PsA, utilizando el régimen de carga vía subcutánea aprobado para el uso clínico.
En el Ensayo 1 en PsA, se evaluó radiográficamente la inhibición de la progresión del daño estructural y se expresó como el cambio en la puntuación total de Sharp modificada (mTSS) y sus componentes, en la puntuación de la erosión (ES) y en la puntuación del estrechamiento del espacio articular (JSN) a la semana 24 y 52, comparado con el periodo basal. En la Tabla 6 se presentan los datos a la semana 24.
Tabla 6 Cambio en la puntuación total de Sharp modificada en artritis psoriásica
|
Placebo N=179 |
Cosentyx 75 mg1 N=181 |
Cosentyx 150 mg1 N=185 | |
|
Puntuación total | |||
|
Periodo basal |
28,4 |
20,4 |
22,3 |
|
(DE) |
(63,5) |
(39,4) |
(48,0) |
|
Cambio medio a la semana 24 |
0,57 |
0,02* |
0,13* |
|
*p<0,05 de acuerdo al valor-p nominal pero no ajustado '10 mg/kg a las semanas 0, 2 y 4 seguido por dosis subcutáneas de 75 mg o |
150 mg | ||
La inhibición del daño estructural se mantuvo con el tratamiento con Cosentyx hasta la semana 52.
El porcentaje de pacientes sin progresión de la enfermedad (definido como un cambio desde el periodo basal en mTSS de <0,5) desde la aleatorización hasta la semana 24 fue del 92,3% en secukinumab 10 mg/kg como carga vía intravenosa - 75 mg como mantenimiento vía subcutánea, 82,3% en secukinumab 10 mg/kg como carga vía intravenosa - 150 mg como mantenimiento vía subcutánea y 75,7% en placebo. El porcentaje de pacientes sin progresión de la enfermedad desde la semana 24 hasta la semana 52 para secukinumab 10 mg/kg como carga vía intravenosa - seguido por 75 mg o 150 mg como mantenimiento vía subcutánea y para los pacientes con placebo que cambiaron a 75 mg o 150 mg vía subcutánea cada 4 semanas a la semana 16 o semana 24, fue del 85,8%, 85,7% y 86,8%, respectivamente.
Función física y calidad de vida relacionada con la salud
En el Ensayo 2 en PsA, los pacientes tratados con Cosentyx 150 mg (p=0,0555) y 300 mg (p=0,0040) mostraron mejoría en la función física comparado con los pacientes tratados con placebo evaluado por el índice de discapacidad del cuestionario de evaluación de la salud (HAQ-DI) a la semana 24. Se observaron mejorías en las puntuaciones HAQ-DI independientemente de la exposición previa a anti-TNFa. Se observaron respuestas similares en el Ensayo 1 en PsA.
Los pacientes tratados con Cosentyx notificaron mejorías significativas en la calidad de vida relacionada con la salud medido según la puntuación del resumen del componente físico del cuestionario de salud Short Form-36 (SF-36 PCS) (p<0,001). Asimismo, se observaron mejorías estadísticamente significativas demostradas en las variables exploratorias valoradas según la evaluación funcional para el tratamiento de enfermedades crónicas - Fatiga (FACIT-F) para 150 mg y 300 mg comparado con placebo (7,97, 5,97 comparado con 1,63, respectivamente). Se observaron respuestas similares en el ensayo 1 en PsA y la eficacia se mantuvo hasta la semana 52.
Espondilitis anquilosante
Se evaluó la seguridad y eficacia de Cosentyx en 590 pacientes en dos ensayos fase III controlados con placebo, doble ciego, aleatorizados, en pacientes con espondilitis anquilosante (AS) activa con un índice de actividad de la enfermedad espondilitis anquilosante de Bath (BASDAI) >4 a pesar del tratamiento con medicamentos antiinflamatorios no esteroideos (NSAID), corticosteroides o fármacos antirreumáticos modificadores de la enfermedad (FAME). Los pacientes que participaron en estos ensayos tenían un diagnóstico de AS durante una media de 2,7 a 5,8 años. Para ambos ensayos, la variable principal fue como mínimo una mejoría del 20% según los criterios de la Assessment of Spondyloarthritis International Society (ASAS 20) a la semana 16.
En el Ensayo 1 en espondilitis anquilosante (Ensayo 1 en AS) y en el Ensayo 2 en espondilitis anquilosante (Ensayo 2 en AS) el 27,0% y el 38,8% de los pacientes, respectivamente, se trataron previamente con un medicamento anti-TNFa y discontinuaron el tratamiento por falta de eficacia o intolerancia (pacientes con respuesta inadecuada a anti-TNFa).
En el Ensayo 1 en AS (MEASURE 1) se evaluaron 371 pacientes, de los cuales el 14,8% y el 33,4% utilizaron MTX o sulfasalacina de forma concomitante, respectivamente. Los pacientes aleatorizados a Cosentyx recibieron 10 mg/kg vía intravenosa a las semanas 0, 2, y 4, seguido por 75 mg o 150 mg vía subcutánea mensualmente empezando en la semana 8. Los pacientes aleatorizados a placebo que eran no respondedores a la semana 16 (rescate temprano) y todo el resto de pacientes de placebo a la semana 24 se cruzaron para recibir Cosentyx (75 mg o 150 mg vía subcutánea) seguido por la misma dosis mensualmente.
En el Ensayo 2 en AS (MEASURE 2) se evaluaron 219 pacientes, de los cuales el 11,9% y el 14,2% utilizaron MTX o sulfasalacina de forma concomitante, respectivamente. Los pacientes aleatorizados a Cosentyx recibieron 75 mg o 150 mg vía subcutánea a las semanas 0, 1, 2, y 3, seguida por la misma dosis mensualmente empezando en la semana 4. En la semana 16, los pacientes que fueron aleatorizados a recibir placebo al inicio del tratamiento, se volvieron a aleatorizar a recibir Cosentyx (75 mg o 150 mg vía subcutánea) mensualmente.
Signos y síntomas
En el Ensayo 2 en AS, el tratamiento con Cosentyx 150 mg dio como resultado una mejora superior en cuanto a las medidas de la actividad de la enfermedad en comparación con placebo a la semana 16 (ver Tabla 7).
Tabla 7 Respuesta clínica en el Ensayo 2 en AS a la semana 16
|
Resultado (valor-p frente a placebo) |
Placebo (n = 74) |
75 mg (n = 73) |
150 mg (n = 72) |
|
% de respuesta ASAS 20 |
28,4 |
41,1 |
61,1*** |
|
% de respuesta ASAS 40 |
10,8 |
26,0 |
36,1*** |
|
hsCRP, (tasa post-BSL/BSL) |
1,13 |
0,61 |
0,55*** |
|
% de ASAS 5/6 |
8,1 |
34,2 |
43 1*** |
|
% de remisión parcial de ASAS |
4,1 |
15,1 |
13,9 |
|
% de BASDAI 50 |
10,8 |
24,7* |
30,6** |
|
Mejora importante de ASDAS-CRP |
4,1 |
15,1* |
25,0*** |
|
* p<0,05, ** p<0,01, *** p<0,001; frente a placebo Todos los valores-p se ajustan en función de la multiplicidad de ensayos de acuerdo a la jerarquía predefinida, excepto para BASDAI 50 y ASDAS-CRP Imputación de no respondedor utilizada para la variable binaria que falta. | |||
|
ASAS: criterio de la Sociedad Internacional de evaluación de la espondiloartritis; BASDAI: índice de actividad de la enfermedad espondilitis anquilosante de Bath; hsCRP: proteína-C reactiva de alta sensibilidad; ASDAS: puntuación de la actividad de la enfermedad espondilitis anquilosante; BSL: periodo basal | |||
El inicio de acción de Cosentyx 150 mg se produjo tan pronto como en la semana 1 para ASAS 20 y semana 2 para ASAS 40 (superior a placebo) en el Ensayo 2 en AS.
Las respuestas ASAS 20 mejoraron en la semana 16 en los pacientes naíve para anti-TNFa (68,2% frente a 31,1%; p<0,05) y en los pacientes con RI para anti-TNFa (50,0% frente a 24,1%; p<0,05) para Cosentyx 150 mg comparado con placebo, respectivamente.
En ambos ensayos en AS, los pacientes tratados con Cosentyx (150 mg en el Ensayo 2 en AS y ambos regímenes en el Ensayo 1 en AS) demostraron signos y síntomas de mejoría significativa a la semana 16, con una magnitud de respuesta y eficacia comparable, mantenida hasta la semana 52 en pacientes naíve para anti-TNFa y en pacientes con RI para anti-TNFa-IR. En el Ensayo 2 en AS, de los 72 pacientes aleatorizados inicialmente a Cosentyx 150 mg, 61 (84,7%) pacientes estaban aún en tratamiento a la semana 52. De los 72 pacientes aleatorizados a Cosentyx 150 mg, 45 y 35 presentaron una respuesta ASAS 20/40, respectivamente.
Movilidad espinal
Los pacientes tratados con Cosentyx 150 mg mostraron mejorías en la movilidad espinal medido por el cambio desde el periodo basal según BASMI hasta la semana 16 para ambos ensayos, Ensayo 1 en AS (-0,40 vs -0,12 para placebo; p=0,0114) y Ensayo 2 en AS (-0,51 vs -0,22 para placebo; p=0,0533). Estas mejorías se mantuvieron hasta la semana 52.
Función física y calidad de vida relacionada con la salud
En el Ensayo 1 y Ensayo 2 en AS, los pacientes tratados con Cosentyx 150 mg mostraron mejoría en la calidad de vida relacionada con la salud medida por el cuestionario de calidad de vida en AS (ASQoL) (p=0,001) y en el resumen del componente físico del SF-36 (SF-36 PCS) (p<0,001). Los pacientes tratados con Cosentyx 150 mg también mostraron mejorías estadísticamente significativas en las variables exploratorias en la función física evaluado por el índice funcional de la espondilitis anquilosante de Bath (BASFI) comparado con placebo (-2,15 vs -0,68) y en la fatiga evaluado según la escala para la evaluación funcional para el tratamiento de enfermedades crónicas - Fatiga (FACIT-Fatiga) comparado con placebo (8,10 vs 3,30). Estas mejorías se mantuvieron hasta la semana 52.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Cosentyx en pacientes pediátricos, desde recién nacidos a menores de 6 años, con psoriasis en placas y desde recién nacidos a menores de 2 años, con artritis idiopática crónica (ver sección 4.2. para consultar la información sobre el uso en la población pediátrica).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Cosentyx en pacientes pediátricos, de 6 años a menores de 18 años, con psoriasis en placas y de 2 años a menores de 18 años, con artritis idiopática crónica (ver sección 4.2. para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Psoriasis en placas
Absorción
Tras una sola dosis subcutánea de 300 mg como formulación líquida en voluntarios sanos, secukinumab alcanzó las concentraciones máximas séricas de 43,2 ±10,4 pg/ml entre 2 y 14 días de la dosis.
Basado en el análisis farmacocinético de la población, con una sola administración subcutánea de 150 ó 300 mg de secukinumab a pacientes con psoriasis en placas se logra una concentración sérica máxima de secukinumab de 13,7 ± 4,8 pg/ml ó 27,3 ± 9,5 pg/ml, respectivamente, 5 ó 6 días después de la administración.
El tiempo transcurrido hasta alcanzar la concentración máxima después del primer mes de tratamiento con dosis semanales fue de 31 a 34 días, basado en el análisis farmacocinético de la población.
Sobre la base de datos simulados, las concentraciones máximas del estado estacionario (Cmáx,ss), tras la administración subcutánea de 150 ó 300 mg, son de 27,6 pg/ml o 55,2 pg/ml, respectivamente. El análisis farmacocinético de la población sugiere que el estado estacionario se alcanza después de 20 semanas con regímenes de administración mensuales.
El análisis farmacocinético de la población mostró que durante la fase de mantenimiento de administración mensual repetida, los pacientes presentan concentraciones séricas máximas y un área bajo la curva (AUC) dos veces mayor que las obtenidas con una sola administración.
El análisis farmacocinético de la población mostró que secukinumab se absorbió con una biodisponibilidad media absoluta del 73% en pacientes con psoriasis en placas. En todos los estudios, la biodisponibilidad absoluta se calculó que se encontraba entre el 60 y el 77%.
Distribución
El volumen medio de distribución durante la fase terminal (Vz) tras una sola administración intravenosa varía entre 7,10 y 8,60 litros en los pacientes con psoriasis en placas, lo que sugiere que la distribución del secukinumab hacia los compartimientos periféricos es limitada.
Biotransformación
La mayor parte de la eliminación de IgG ocurre mediante el catabolismo intracelular, tras endocitosis de la fase líquida o mediada por receptor.
Eliminación
El aclaramiento medio sistémico (CL) tras la administración única intravenosa en pacientes con psoriasis en placas fue de 0,13 a 0,36 l/día. En el análisis farmacocinético de la población, el aclaramiento medio sistémico (CL) fue de 0,19 l/día en los pacientes con psoriasis en placas. El CL no se ve afectado por el sexo. El aclaramiento no es dosis ni tiempo dependiente.
El análisis farmacocinético de la población, la vida de eliminación media se estimó en 27 días en los pacientes con psoriasis en placas, con un rango de 18 a 46 días en todos los estudios de psoriasis con administración intravenosa.
Linealidad/No linealidad
La farmacocinética tras dosis únicas y repetidas del secukinumab en pacientes con psoriasis en placas se determinó en varios estudios en los que se usaron tanto dosis intravenosas de entre 1 x 0,3 mg/kg a 3 x 10 mg/kg, como dosis subcutáneas de entre 1 x 25 mg a múltiples dosis de 300 mg. En todos los casos, la exposición resultó proporcional a la dosis.
Artritis psoriásica
Las propiedades farmacocinéticas de secukinumab observadas en pacientes con artritis psoriásica fueron similares a las que se muestran en pacientes con psoriasis en placas. La biodisponibilidad de secukinumab en pacientes con PsA fue del 85% de acuerdo al modelo farmacocinético poblacional.
Espondilitis anquilosante
Las propiedades farmacocinéticas de secukinumab observadas en pacientes con espondilitis anquilosante fueron similares a las que se muestran en pacientes con psoriasis en placas.
Poblaciones especiales
Pacientes de edad avanzada
De los 3.430 pacientes con psoriasis en placas que recibieron Cosentyx en los ensayos clínicos,
230 pacientes tenían más de 65 años de edad y 32, eran mayores de 75.
De los 974 pacientes con artritis psoriásica que recibieron Cosentyx en los ensayos clínicos, un total de 85 pacientes tenían a partir de 65 años de edad y 4 pacientes tenían a partir de 75 años.
De los 571 pacientes con espondilitis anquilosante que recibieron Cosentyx en los ensayos clínicos, un total de 24 pacientes tenían a partir de 65 años de edad y 3 pacientes tenían a partir de 75 años.
Según el análisis farmacocinético de la población con un número limitado de pacientes de edad avanzada (n=71 de más de 65 años y n=7 de más de 75 años), el aclaramiento fue similar en los pacientes de edad avanzada que en los menores de 65 años.
Pacientes con insuficiencia renal o hepática
No se disponen de datos farmacocinéticos en pacientes con insuficiencia renal o hepática. Se estima que la cantidad de Cosentyx sin metabolizar, un anticuerpo monoclonal de IgG, que se elimina por vía renal, es baja y de menor importancia. Las IgGs se eliminan principalmente por catabolismo, por lo que la insuficiencia hepática no se espera que afecte al aclaramiento de Cosentyx.
Efecto del _peso en la _farmacocinética
El aclaramiento de secukinumab y el volumen de distribución incrementan con el aumento del peso corporal.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios de reactividad cruzada en tejidos, farmacología de seguridad, estudios de toxicidad de la función reproductora y de dosis repetidas realizados con secukinumab o con un anticuerpo murino anti- IL-17A murina equivalente.
Como secukinumab se une a la IL-17A del ser humano y del mono cinomolgus, la seguridad se estudió en el mono cinomolgus. No se han observado efectos indeseados con secukinumab tras su administración a mono cinomolgus tanto por vía subcutánea, durante un período de hasta 13 semanas, como por vía intravenosa, durante un período de hasta 26 semanas (incluyendo evaluaciones farmacocinéticas, farmacodinamias, de inmunogenicidad y de inmunotoxicidad, p.ej., respuesta inmunitaria dependiente de linfocitos T y actividad de linfocitos citolíticos naturales). La concentración sérica media determinada en monos después de la administración de 13 dosis subcutáneas de 150 mg/kg una vez por semana es mayor que la concentración sérica media prevista para los pacientes con psoriasis que vayan a recibir la dosis clínica mayor. Se detectaron anticuerpos anti-secukinumáb en solo uno de los animales expuestos. No se observaron signos de reactividad cruzada tisular inespecífica tras la aplicación del secukinumab sobre tejidos humanos normales.
No se han realizado estudios en animales para evaluar el potencial carcinogénico de secukinumab.
Un estudio de desarrollo embriofetal efectuado en mono cinomolgus, no mostró toxicidad materna, embriotoxicidad ni teratogenicidad cuando secukinumab se administró durante la organogénesis o hacia el final de la gestación.
No se han observado efectos indeseados con un anticuerpo murino anti-IL-17A murina equivalente en los estudios de fecundidad y desarrollo embrionario temprano o de desarrollo pre- y postnatal del ratón. La dosis más alta que se usó en tales estudios era superior a la dosis máxima eficaz en cuanto a inhibición y actividad de la IL-17A (ver sección 4.6).
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Sacarosa
L-histidina
Hidrocloruro de L-histidina monohidratado Polisorbato 80
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez
3 años
Tras la reconstitución
Una vez reconstituido, se ha demostrado una estabilidad química y física de 24 horas de 2°C a 8°C. Desde un punto de vista microbiológico, salvo que el método de reconstitución excluya el riesgo de contaminación microbiana, el medicamento se debe utilizar inmediatamente.
En el caso que no se utilice de forma inmediata, el tiempo y las condiciones de almacenamiento una vez reconstituido, son de responsabilidad del usuario.
6.4 Precauciones especiales de conservación
Conservar en nevera (2°C - 8°C).
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Cosentyx se comercializa en un vial de vidrio con tapón de goma gris y cápsula de aluminio con tapa blanca que contiene 150 mg de secukinumab.
Cosentyx está disponible en un envase que contiene un vial.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El vial de un único uso contiene 150 mg de secukinumab que se debe reconstituir con agua estéril para preparaciones inyectables. La solución resultante debe ser transparente e incolora tirando a ligeramente amarillenta. No utilizar si el polvo liofilizado no se ha disuelto por completo o si el líquido contiene partículas visibles, está turbio o tiene un color claramente marrón. En el prospecto se incluye información detallada sobre las instrucciones de uso.
La eliminación del medicamento no utilizado y de los materiales sobrantes que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/980/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
15.01.2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Cosentyx 150 mg solución inyectable en jeringa precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada jeringa precargada contiene 150 mg de secukinumab* en 1 ml.
*Secukinumab es un anticuerpo monoclonal recombinante, íntegramente humano, selectivo a la interleuquina17A. Secukinumab es un anticuerpo de tipo IgG1/K producido en células ováricas de hámster chino.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable en jeringa precargada (inyectable) La solución es clara e incolora, ligeramente amarillenta.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Psoriasis en placas
Cosentyx está indicado para el tratamiento de la psoriasis en placas de moderada a grave en adultos candidatos a tratamientos sistémicos.
Artritis psoriásica
Cosentyx, solo o en combinación con metotrexato (MTX), está indicado para el tratamiento de la artritis psoriásica activa en pacientes adultos que han mostrado una respuesta inadecuada a tratamientos previos con fármacos antirreumáticos modificadores de la enfermedad (FAME) (ver sección 5.1).
Espondilitis anquilosante
Cosentyx está indicado para el tratamiento de la espondilitis anquilosante activa en adultos que no han respondido adecuadamente al tratamiento convencional.
4.2 Posología y forma de administración
Cosentyx se ha de utilizar bajo la dirección y la supervisión de un médico con experiencia en el diagnóstico y tratamiento de las enfermedades para las que Cosentyx está indicado.
Posología
Psoriasis en placas
La dosis recomendada es de 300 mg de secukinumab por inyección subcutánea, que se administra inicialmente en la semana 0, 1, 2 y 3 y, luego mensualmente, comenzando en la semana 4, durante la fase de mantenimiento. Cada dosis de 300 mg se administra de forma repartida en dos inyecciones subcutáneas de 150 mg.
Artritis psoriásica
Para pacientes que padecen psoriasis en placas de moderada a grave de forma concomitante o que son respondedores inadecuados (RI) a anti-TNFa, la dosis recomendada es de 300 mg por inyección subcutánea, que se administra inicialmente en la semana 0, 1, 2 y 3 y, luego durante la fase de mantenimiento, mensualmente comenzando en la semana 4. Cada dosis de 300 mg se administra en dos inyecciones subcutáneas de 150 mg.
Para el resto de pacientes, la dosis recomendada es de 150 mg por inyección subcutánea, que se administra inicialmente en la semana 0, 1, 2 y 3 y, luego durante la fase de mantenimiento, mensualmente comenzando en la semana 4.
Espondilitis anquilosante
La dosis recomendada es de 150 mg por inyección subcutánea, que se administra inicialmente en la semana 0, 1, 2 y 3 y, luego durante la fase de mantenimiento, mensualmente comenzando en la semana 4.
Para todas las indicaciones anteriores, los datos disponibles sugieren que una respuesta clínica se alcanza normalmente en las 16 semanas de tratamiento. Se debe considerar interrumpir el tratamiento en los pacientes que no han mostrado respuesta a las 16 semanas de tratamiento. Algunos pacientes con una respuesta parcial al inicio, pueden mejorar posteriormente con un tratamiento continuado de más de 16 semanas.
Poblaciones especiales
Pacientes de edad avanzada (mayores de 65 años)
No es necesario un ajuste de la dosis (ver sección 5.2).
Insuficiencia renal / insuficiencia hepática
No se ha estudiado Cosentyx en estas poblaciones de pacientes. No se puede hacer ninguna recomendación posológica.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de Cosentyx en niños menores de 18 años. No se dispone de datos.
Forma de administración
Cosentyx se ha de administrar por inyección subcutánea. En la medida de lo posible, se deben evitar como lugares de inyección las zonas de la piel que presenten signos de psoriasis.
Si el médico lo considera oportuno, los pacientes se pueden autoinyectar Cosentyx después de haber aprendido correctamente la técnica de inyección subcutánea. No obstante, el médico se debe asegurar, haciendo un adecuado seguimiento de los pacientes. Se debe indicar a los pacientes que se inyecten toda la cantidad de Cosentyx conforme a las instrucciones del prospecto. Las instrucciones completas de administración se pueden consultar en el prospecto.
4.3 Contraindicaciones
Reacciones de hipersensibilidad graves al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Infecciones activas clínicamente importantes (p.ej. tuberculosis activa; ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
Infecciones
Cosentyx puede aumentar el riesgo de infecciones. En los ensayos clínicos se han observado infecciones en los pacientes que recibieron Cosentyx (ver sección 4.8). La mayoría fueron infecciones leves o moderadas de las vías respiratorias altas como rinofaringitis que no requirieron interrumpir el tratamiento.
Relacionado con el mecanismo de acción de Cosentyx, en los ensayos clínicos de psoriasis se han notificado infecciones mucocutáneas no graves por cándida más frecuentemente con secukinumab que con placebo (3,55 por 100 pacientes-año con secukinumab 300 mg frente a 1,00 por 100 paciente-año con placebo) (ver sección 4.8).
Se debe tener precaución cuando se valore la administración de Cosentyx en pacientes con infecciones crónicas o con antecedentes de infecciones recurrentes.
Se debe indicar al paciente que consulte al médico cuando padezca signos o síntomas indicativos de una infección. El paciente que contraiga una infección grave debe ser objeto de una estrecha observación y no debe recibir Cosentyx hasta que la infección se haya resuelto.
No se han notificado en los ensayos clínicos una mayor sensibilidad a la tuberculosis. Aun así, Cosentyx no se debe administrar a pacientes con tuberculosis activa. Se debe considerar la posibilidad de administrar un tratamiento antituberculoso antes de comenzar el tratamiento con Cosentyx en los pacientes con tuberculosis latente.
Enfermedad de Crohn
Se debe tener precaución cuando se prescriba Cosentyx a pacientes con enfermedad de Crohn. En los ensayos clínicos se ha observado exacerbaciones, en algunos casos graves, tanto en el grupo de pacientes de Cosentyx como en el de placebo. Se deben vigilar estrechamente los pacientes tratados con Cosentyx con enfermedad de Crohn.
Reacciones de hipersensibilidad
En los ensayos clínicos se han observado, en raras ocasiones, reacciones anafilácticas en pacientes que estaban recibiendo Cosentyx. Si aparecen reacciones anafilácticas u otras reacciones alérgicas graves, se debe interrumpir inmediatamente el tratamiento con Cosentyx e iniciar otro tratamiento alternativo.
Individuos sensibles al látex
El capuchón extraíble de la aguja de la jeringa precargada de Cosentyx contiene un derivado de la goma látex natural. Hasta la fecha no se ha detectado goma látex natural en el capuchón extraíble de la aguja. Sin embargo tampoco se ha estudiado el uso de las jeringas precargadas de Cosentyx en individuos sensibles al látex y por tanto, hay un riesgo potencial de reacciones de hipersensibilidad que no puede ser completamente descartado.
Vacunas
No se deben administrar simultáneamente las vacunas elaboradas con microorganismos vivos con Cosentyx.
Los pacientes tratados con Cosentyx pueden recibir de forma simultánea vacunas inactivadas o no elaboradas con microorganismos vivos. En un estudio, después de la administración de vacunas antigripales inactivadas y antimeningocócicas, los voluntarios sanos tanto del grupo de 150 mg de secukinumab como de placebo, fueron capaces de generar una respuesta inmunitaria satisfactoria que por lo menos cuadruplicó los títulos de anticuerpos contra esas vacunas. Los datos indican que Cosentyx no inhibe la respuesta inmunitaria humoral a las vacunas antimeningocócicas o antigripales.
Tratamiento inmunosupresor concomitante
En los estudios de psoriasis, no se ha evaluado la seguridad y eficacia de Cosentyx en combinación con inmunosupresores, incluidos biológicos, o fototerapia (ver también sección 4.5).
4.5 Interacción con otros medicamentos y otras formas de interacción
No se deben administrar las vacunas elaboradas con microorganismos vivos simultáneamente con Cosentyx (ver también sección 4.4).
No se han realizado estudios de interacciones en humanos. No existe una evidencia directa del papel de IL-17A en la expresión de las enzimas CYP450. La formación de algunas enzimas P450 se suprime debido al aumento de citoquinas durante la inflamación crónica. Por tanto los tratamientos antiinflamatorios, tales como secukinumab, un inhibidor de IL17A, podría normalizar los niveles de CYP450 y en consecuencia, disminuir la exposición de las medicaciones concomitantes metabolizadas por el CYP450. Por lo que no se puede excluir que exista un efecto clínico relevante en los medicamentos de estrecho margen terapéutico, donde la dosis se ajusta de forma individual (p. ej. warfarina), que sean sustratos de CYP450. Cuando se inicie el tratamiento con secukinumab en pacientes tratados con este tipo de medicamentos, se debe considerar el realizar monitorización terapéutica.
No se observó interacción cuando Cosentyx se administró de forma concomitante con metotrexato (MTX) y/o corticosteroides en ensayos en artritis (incluyendo pacientes con artritis psoriásica y espondilitis anquilosante).
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar un método anticonceptivo durante el tratamiento y durante al menos 20 semanas después del tratamiento.
Embarazo
No se dispone de datos suficientes sobre el uso de secukinumab en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para el embarazo, el desarrollo embrionario o fetal, el parto o el desarrollo postnatal (ver sección 5.3). Como medida de precaución, es preferible evitar el uso de Cosentyx en el embarazo.
Lactancia
Se desconoce si secukinumab se excreta en la leche materna. Las inmunoglobulinas pasan a la leche materna y se desconoce si secukinumab se absorbe sistémicamente tras su ingestión. Debido a las posibles reacciones adversas de secukinumab en los lactantes, se debe decidir si interrumpir la lactancia durante el tratamiento y hasta 20 semanas después del tratamiento o interrumpir el tratamiento con Cosentyx, teniendo en cuenta el beneficio de la lactancia para el niño o el beneficio del tratamiento con Cosentyx para la mujer.
Fertilidad
No se ha evaluado el efecto de secukinumab sobre la fertilidad humana. Los estudios en animales no indican que Cosentyx tenga efectos nocivos directos o indirectos sobre la fertilidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Cosentyx sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Un total de 6.804 pacientes han recibido Cosentyx en los ensayos clínicos con y sin enmascaramiento en diversas indicaciones (psoriasis en placas, artritis psoriásica, espondilitis anquilosante y otras enfermedades autoinmunes). De éstos, 3.671 pacientes se han expuesto a Cosentyx durante al menos un año, lo que representa una exposición de 6.450 paciente-año.
Reacciones adversas en psoriasis en placas
Se agruparon los datos de los cuatro estudios de fase III controlados con placebo en psoriasis en placas para evaluar la seguridad de Cosentyx hasta 12 semanas después del inicio del tratamiento. Se evaluaron 2.076 pacientes en total (de los cuales, 692 pacientes recibieron la dosis de 150 mg, 690, la de 300 mg y 694, el placebo).
Las reacciones adversas notificadas con mayor frecuencia fueron las infecciones de las vías respiratorias altas (con mayor frecuencia rinofaringitis y rinitis). La mayoría de las reacciones fueron de naturaleza leve o moderada.
Reacciones adversas en artritis psoriásica
Cosentyx se estudió en dos ensayos controlados con placebo en artritis psoriásica con 1.003 pacientes (703 pacientes en Cosentyx y 300 pacientes en placebo) para una exposición total de 1.061 pacientes-años de exposición del ensayo (duración media de exposición para los pacientes tratados con secukinumab: 456 días en el Ensayo 1 en artritis psoriásica y 245 días en el Ensayo 2 en artritis psoriásica). El perfil de seguridad observado en pacientes con artritis psoriásica tratados con Cosentyx es consistente con el perfil de seguridad en psoriasis.
Reacciones adversas en espondilitis anquilosante
Cosentyx se estudió en dos ensayos controlados con placebo en espondilitis anquilosante con 590 pacientes (394 pacientes en Cosentyx y 196 pacientes en placebo) para un total de 755 pacientes-años de exposición del ensayo (duración media de exposición para los pacientes tratados con secukinumab: 469 días en el Ensayo 1 en espondilitis anquilosante y 460 días en el Ensayo 2 en espondilitis anquilosante). El perfil de seguridad observado en pacientes con espondilitis anquilosante tratados con Cosentyx es consistente con el perfil de seguridad en psoriasis.
Las reacciones adversas de los ensayos clínicos en psoriasis, artritis psoriásica y espondilitis anquilosante (Tabla 1) se presentan según la clasificación de órganos del MedDRA. Dentro de cada órgano y sistema, las reacciones adversas se ordenan por frecuencia, las más frecuentes primero. Las reacciones adversas se incluyen en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Además, las categorías de frecuencia se definen utilizando los siguientes criterios: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000).
Tabla 1 Lista de las reacciones adversas en los ensayos clínicos1-1
|
Sistema de Clasificación de Órganos |
Frecuencia |
Reacción adversa |
|
Infecciones e infestaciones |
Muy frecuentes |
Infecciones de las vías respiratorias altas |
|
Frecuentes |
Herpes oral | |
|
Poco frecuentes |
Candidiasis oral | |
|
Pie de atleta | ||
|
Otitis externa | ||
|
Trastornos de la sangre y del sistema linfático |
Poco frecuentes |
Neutropenia |
|
Trastornos del sistema inmunológico |
Raras |
Reacciones anafilácticas |
|
Trastornos oculares |
Poco frecuentes |
Conjuntivitis |
|
Trastornos respiratorios, torácicos y mediastínicos |
Frecuentes |
Rinorrea |
|
Trastornos gastrointestinales |
Frecuentes |
Diarrea |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
Urticaria |
|
En los ensayos clínicos controlados con placebo (fase III) en psoriasis en placas, artritis psoriásica y espondilitis anquilosante, los pacientes recibieron 300 mg, 150 mg, 75 mg o placebo durante 12 semanas (psoriasis) o 16 semanas (artritis psoriásica y espondilitis anquilosante) de duración del tratamiento | ||
Descripción de las reacciones adversas seleccionadas Infecciones
Durante la fase comparativa con placebo de los ensayos clínicos en psoriasis en placas (en los que un total de 1.382 pacientes recibieron Cosentyx y 694, el placebo, durante un período de hasta 12 semanas), se notificaron infecciones en el 28,7% de los pacientes del grupo de Cosentyx y en el 18,9% de los pacientes que recibieron el placebo. La mayoría de las infecciones se consideraron no graves, infecciones leves o moderadas de las vías respiratorias altas, como rinofaringitis, que no requirieron interrupción del tratamiento. Hubo un aumento de candidiasis en mucosa y piel, consistente con el mecanismo de acción, no graves, de leves a moderadas y que respondieron al tratamiento estándar sin tener que interrumpir el tratamiento. Las infecciones graves aparecieron en un 0,14% de los pacientes tratados con Cosentyx y en un 0,3% de los pacientes tratados con placebo (ver sección 4.4).
Durante todo el periodo de tratamiento (un total de 3.430 pacientes tratados con Cosentyx durante 52 semanas, en la mayoría de los pacientes), se notificaron infecciones en el 47,5% de los pacientes tratados con Cosentyx (0,9 por paciente-año de seguimiento). El 1,2% de los pacientes tratados con Cosentyx notificaron infecciones graves (0,015 por paciente-año de seguimiento).
La tasa de infecciones observada en los ensayos clínicos en artritis psoriásica y espondilitis anquilosante fue similar a la observada en los ensayos en psoriasis.
Neutropenia
En los ensayos clínicos de fase 3 en psoriasis, se ha observado con mayor frecuencia neutropenia con secukinumab que con placebo, pero en la mayoría de los casos fue leve, transitoria y reversible. Se notificó neutropenia <1,0-0,5x109/l (CTCAE Grado 3) en 18 de los 3.430 (0,5%) pacientes con secukinumab, independientemente de la dosis y sin una relación temporal con la infección en 15 de los 18 casos. No se notificaron casos de neutropenia más grave. Los otros 3 casos restantes fueron infecciones no graves que respondieron al tratamiento estándar y no requirieron interrumpir el tratamiento con Cosentyx.
La frecuencia de neutropenia en artritis psoriásica y espondilitis anquilosante es similar a psoriasis.
Se notificaron raros casos de neutropenia <0,5x109/l (CTCAE Grado 4).
Reacciones de hipersensibilidad
En los ensayos clínicos se ha observado urticaria y raros casos de reacción anafiláctica a Cosentyx (ver también sección 4.4).
Inmunogenicidad
En ensayos clínicos en psoriasis, artritis psoriásica y espondilitis anquilosante, menos del 1% de los pacientes tratados con Cosentyx desarrollaron anticuerpos a secukinumab a lo largo de 52 semanas de tratamiento. Aproximadamente la mitad de los anticuerpos antifármaco producidos durante el tratamiento fueron neutralizantes pero esto no se asoció a una pérdida de eficacia o a trastornos farmacocinéticos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han notificado casos de sobredosis en los ensayos clínicos.
Durante los estudios clínicos se han administrado por vía intravenosa dosis de hasta 30 mg/kg (aproximadamente de 2.000 a 3.000 mg) sin que haya aparecido toxicidad limitante de la dosis. En caso de sobredosis se recomienda vigilar al paciente en busca de signos o síntomas de reacciones adversas e instaurar inmediatamente el tratamiento sintomático más adecuado.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inmunosupresores, inhibidores de la interleucina, código ATC: L04AC10 Mecanismo de acción
Secukinumab es un anticuerpo de tipo IgG1/K monoclonal, íntegramente humano, que se une selectivamente y neutraliza una citoquina proinflamatoria, la interleuquina 17A (IL-17A). Secukinumab actúa dirigiéndose a IL-17A e inhibe su interacción con el receptor de IL-17, que se encuentra en varios tipos de células, incluidos los queratinocitos. Como resultado, secukinumab inhibe la liberación de citoquinas proinflamatorias, de quimioquinas y de mediadores del daño tisular, y reduce los efectos mediados por la IL-17A, que participan en la enfermedad autoinmunitaria e inflamatoria. A la piel llegan concentraciones clínicamente importantes de secukinumab y reducen los marcadores inflamatorios locales. Como consecuencia directa, el tratamiento con secukinumab reduce el eritema, la induración y la descamación presentes en las lesiones de la psoriasis en placas.
IL-17A es una citoquina natural que participa en reacciones inmunitarias e inflamatorias normales. IL-17A desempeña una función clave en la patogenia de la psoriasis en placas, artritis psoriásica y espondilitis anquilosante y se encuentra concentrada en la piel lesionada, a diferencia de la piel no lesionada de los pacientes con psoriasis en placas y en el tejido sinovial de los pacientes con artritis psoriásica. La frecuencia de células productoras de IL-17, también fue significativamente superior en la médula ósea subcondral de las articulaciones facetarías de pacientes con espondilitis anquilosante.
Efectos farmacodinámicos
Las concentraciones séricas de IL-17A total (libre y unida a secukinumab) aumentan inicialmente en los pacientes que reciben secukinumab. Después disminuye lentamente debido a un aclaramiento reducido del complejo secukinumab-IL-17A, lo que indica que secukinumab es capaz de fijarse selectivamente a la IL-17A libre, la cual desempeña un papel fundamental en la patogenia de la psoriasis en placas.
En un estudio con secukinumab, los neutrófilos epidérmicos infiltrantes y los distintos marcadores asociados a neutrófilos, presentes en gran número en la piel lesionada de los pacientes con psoriasis en placas, disminuyeron significativamente al cabo de una o dos semanas de tratamiento.
Secukinumab ha demostrado que reduce (entre 1 y 2 semanas de tratamiento) los niveles de proteína C reactiva, que es un marcador de la inflamación.
Eficacia clínica y seguridad
Psoriasis en _placas
La seguridad y la eficacia de Cosentyx se evaluaron en cuatro estudios de fase III, aleatorizados, con doble enmascaramiento y comparativos con placebo, llevados a cabo en pacientes con psoriasis en placas moderada o grave que eran candidatos de fototerapia o de tratamientos sistémicos [ERASURE, FIXTURE, FEATURE y JUNCTURE]. La eficacia y la seguridad de Cosentyx 150 mg y 300 mg se evaluaron frente a placebo y etanercept. Además, en otro estudio [SCULPTURE] se evaluó un régimen terapéutico crónico en comparación con la pauta de “repetición del tratamiento en caso de necesidad”.
De los 2.403 pacientes que participaron en los estudios comparativos con placebo, el 79% carecía de antecedentes de tratamiento biológico, el 45% procedían de fracasos con tratamientos no biológicos, un 8% procedía de fracasos de tratamientos biológicos (el 6% de fracasos con anti-TNF y el 2% de tratamientos anti-p40). Entre el 15 y el 25% de los pacientes de los estudios de fase III tenían artritis psoriásica al inicio.
En el estudio 1 sobre psoriasis (ERASURE) se evaluaron 738 pacientes. Los pacientes asignados aleatoriamente al grupo de Cosentyx recibieron dosis de 150 ó 300 mg en las semanas 0, 1, 2 y 3 y, luego una vez al mes, comenzando en la semana 4, la misma dosis. En el estudio 2 sobre psoriasis (FIXTURE) se evaluaron 1.306 pacientes. Los pacientes asignados aleatoriamente al grupo de Cosentyx recibieron dosis de 150 ó 300 mg en las semanas 0, 1, 2 y 3 y, luego una vez al mes, comenzando en la semana 4, la misma dosis. Los pacientes asignados aleatoriamente al grupo del etanercept recibieron dosis de 50 mg dos veces por semana durante 12 semanas y, luego, 50 mg una vez por semana. En ambos estudios, estudio 1 y 2, los pacientes asignados aleatoriamente al grupo del placebo que no respondían al cabo de 12 semanas pasaron a recibir Cosentyx (150 ó 300 mg) a las semanas 12, 13, 14 y 15 y, luego una vez al mes, comenzando en la semana 16, la misma dosis. Desde la primera administración del tratamiento del estudio, todos los pacientes fueron sometidos a un seguimiento de hasta 52 semanas de duración.
En el estudio 3 sobre psoriasis (FEATURE) se evaluaron 177 pacientes usando una jeringa precargada en comparación con el placebo después de 12 semanas de tratamiento para evaluar la seguridad, la tolerabilidad y la autoadministración de Cosentyx con la jeringa precargada. En el estudio 4 sobre psoriasis (JUNCTURE) se evaluaron 182 pacientes usando una pluma precargada en comparación con el placebo después de 12 semanas de tratamiento para evaluar la seguridad, la tolerabilidad y la autoadministración de Cosentyx con la pluma precargada. En ambos estudios, estudio 3 y 4, los pacientes asignados aleatoriamente al grupo de Cosentyx, recibieron dosis de 150 ó 300 mg a las semanas 0, 1, 2 y 3 y, luego una vez al mes, comenzando en la semana 4, la misma dosis. También se aleatorizaron pacientes para que recibiesen el placebo las semanas 0, 1, 2 y 3 y, luego una vez al mes, comenzando en la semana 4, la misma dosis.
En el estudio 5 sobre psoriasis (SCULPTURE) se evaluaron 966 pacientes. Todos los pacientes recibieron Cosentyx en dosis de 150 ó 300 mg a las semanas 0, 1, 2, 3, 4, 8 y 12 y, luego, se les asignó de forma aleatoria un régimen de mantenimiento mensual con la misma dosis a partir de la semana 12, o bien una pauta de “repetición del tratamiento en caso de necesidad” con la misma dosis. Los pacientes asignados aleatoriamente al grupo de “repetición del tratamiento en caso de necesidad” que no consiguieron un mantenimiento satisfactorio de la respuesta, se recomendó un régimen de mantenimiento con dosis mensuales fijas.
Las co-variables principales en los estudios con placebo y con comparador fueron la proporción de pacientes que consiguieron una respuesta de PASI 75 y una respuesta IGA mod 2011 de “blanqueamiento completo” o “prácticamente completo”, en comparación con placebo al cabo de 12 semanas (ver Tabla 2 y 3). Con la dosis de 300 mg se obtuvo una mejoría en la piel, en particular, un “blanqueamiento completo” o “prácticamente completo” con criterios de eficacia de PASI 90,
PASI 100, e IGA mod 2011 0 ó 1 en todos los estudios, con efectos máximos a la semana 16, de modo que se recomienda esta dosis.
Tabla 2 Resumen de las respuestas PASI 50/75/90/100 “blanqueamiento completo” o
“prácticamente completo” en IGA* mod 2011 en los estudios 1, 3 y 4 en psoriasis (ERASURE, FEATURE y JUNCTURE)
|
Número de pacientes |
61 |
60 |
60 |
- |
- |
- |
- |
|
n (%) de respuesta PASI 50 |
5 (8,2%) |
48 (80,0%) |
58 (96,7%) |
- |
- |
- |
- |
|
n (%) de respuesta PASI 75 |
2 (3,3%) |
43 (71,7%)** |
52 (86,7%)** |
- |
- |
- |
- |
|
n (%) de respuesta PASI 90 |
0 (0,0%) |
24 (40,0%) |
33 (55,0%) |
- |
- |
- |
- |
|
n (%) de respuesta PASI 100 |
0 (0,0%) |
10 (16,7%) |
16 (26,7%) |
- |
- |
- |
- |
|
n (%) de “blanqueamiento completo” o “prácticamente completo” según IGA |
0 (0,0%) |
32 (53,3%)** |
44 (73,3%)** |
mod 2011
* La IGA mod 2011 es una escala de 5 categorías: “0 = blanqueamiento completo total”, “1 = blanqueamiento prácticamente completo”, “2 = (psoriasis) leve”, “3 = moderada” o “4 = grave” que indica la evaluación global del médico sobre la intensidad de la psoriasis en función de la induración, el eritema y la descamación. Se definió como éxito terapéutico, “remisión total” o “remisión casi total”, la ausencia de signos de psoriasis o bien una coloración normal o rosada de las lesiones cutáneas, ausencia de induración de la placa y ninguna o una mínima descamación focal.
** valores de p con respecto al placebo, ajustados en función de la multiplicidad: p<0,0001._
|
Semana 12 |
Semana 16 |
Semana 52 | |||||
|
Placebo |
150 mg |
300 mg |
150 mg |
300 mg |
150 mg |
300 mg | |
|
Estudio 1 | |||||||
|
Número de pacientes |
246 |
244 |
245 |
244 |
245 |
244 |
245 |
|
n (%) de respuesta PASI 50 |
22 (8,9%) |
203 |
222 (90,6%) |
212 |
224 |
187 |
207 |
|
(83,5%) |
(87,2%) |
(91,4%) |
(77%) |
(84,5%) | |||
|
n (%) de respuesta PASI 75 |
11 (4,5%) |
174 |
200 |
188 |
211 |
146 |
182 |
|
(71,6%)** |
(81,6%)** |
(77,4%) |
(86,1%) |
(60,1%) |
(74,3%) | ||
|
n (%) de respuesta PASI 90 |
3 (1,2%) |
95 |
145 |
130 |
171 |
88 |
147 |
|
(39,1%)** |
(59,2%)** |
(53,5%) |
(69,8%) |
(36,2%) |
(60,0%) | ||
|
n (%) de respuesta PASI 100 |
2 (0,8%) |
31 (12,8%) |
70 (28,6%) |
51 |
102 |
49 |
96 (39,2%) |
|
(21,0%) |
(41,6%) |
(20,2%) | |||||
|
n (%) de “blanqueamiento |
6 (2,40%) |
125 |
160 |
142 |
180 |
101 |
148 |
|
completo” o “prácticamente |
(51,2%)** |
(65,3%)** |
(58,2%) |
(73,5%) |
(41,4%) |
(60,4%) | |
|
completo” según IGA | |||||||
|
mod 2011 | |||||||
|
Estudio 3 | |||||||
|
Número de pacientes |
59 |
59 |
58 |
- |
- |
- |
- |
|
n (%) de respuesta PASI 50 |
3 (5,1%) |
51 (86,4%) |
51 (87,9%) |
- |
- |
- |
- |
|
n (%) de respuesta PASI 75 |
0 (0,0%) |
41 |
44 (75,9%)** |
- |
- |
- |
- |
|
(69,5%)** | |||||||
|
n (%) de respuesta PASI 90 |
0 (0,0%) |
27 (45,8%) |
35 (60,3%) |
- |
- |
- |
- |
|
n (%) de respuesta PASI 100 |
0 (0,0%) |
5 (8,5%) |
25 (43,1%) |
- |
- |
- |
- |
|
n (%) de “blanqueamiento |
0 (0,0%) |
31 |
40 (69,0%)** |
- |
- |
- |
- |
|
completo” o “prácticamente |
(52,5%)** | ||||||
completo” según IGA mod 2011 Estudio 4
Tabla 3 Resumen de la respuesta clínica del estudio 2 de psoriasis (FIXTURE)
Semana 12 Semana 16 Semana 52
|
Placebo |
150 mg |
300 mg |
Etanercept |
150 mg |
300 mg |
Etanercept |
150 mg |
300 mg |
Etanercept | |
|
Número de |
324 |
327 |
323 |
323 |
327 |
323 |
323 |
327 |
323 |
323 |
|
pacientes n (%) de respuesta |
49 |
266 |
296 |
226 |
290 |
302 |
257 (79,6%) |
249 |
274 |
234 (72,4%) |
|
PASI 50 |
(15,1%) |
(81,3%) |
(91,6%) |
(70,0%) |
(88,7%) |
(93,5%) |
(76,1%) |
(84,8%) | ||
|
n (%) de respuesta |
16 |
219 |
249 |
142 |
247 |
280 |
189 (58,5%) |
215 |
254 |
179 (55,4%) |
|
PASI 75 |
(4,9%) |
(67,0%) ** |
(77,1%) ** |
(44,0%) |
(75,5%) |
(86,7%) |
(65,7%) |
(78,6%) | ||
|
n (%) de respuesta |
5 (1,5%) |
137 |
175 |
67 (20,7%) |
176 |
234 |
101 (31,3%) |
147 |
210 |
108 (33,4%) |
|
PASI 90 |
(41,9%) |
(54,2%) |
(53,8%) |
(72,4%) |
(45,0%) |
(65,0%) | ||||
|
n (%) de respuesta |
0 (0%) |
47 |
78 |
14 (4,3%) |
84 |
119 |
24 (7,4%) |
65 |
117 |
32 (9,9%) |
|
PASI 100 |
(14,4%) |
(24,1%) |
(25,7%) |
(36,8%) |
(19,9%) |
(36,2%) | ||||
|
n (%) de de |
9 (2,8%) |
167 |
202 |
88 (27,2%) |
200 |
244 |
127 (39,3%) |
168 |
219 |
120 (37,2%) |
|
“blanqueamiento |
(51,1%) |
(62,5%) |
(61,2%) |
(75,5%) |
(51,4%) |
(67,8%) | ||||
|
completo” o |
** |
** |
“prácticamente completo” en la
IGA mod 2011_
** valores p con respecto al etanercept: p=0,0250
En un ensayo adicional en psoriasis (CLEAR) se evaluaron 676 pacientes. Secukinumab 300 mg alcanzó las variables principal y secundaria mostrando superioridad a ustekinumab de acuerdo a la respuesta de PASI 90 a la semana 16 y a la velocidad de inicio de respuesta de PASI 75 a la semana 4. Se observó una mayor eficacia para secukinumab comparado con ustekinumab para los criterios de respuesta de PASI 75/90/100 e IGA mod 2011 0 ó 1 (“blanqueamiento completo” o “prácticamente completo”) de forma temprana y continua hasta la semana 16.
Tabla 4 Resumen de la respuesta clínica en el ensayo CLEAR
|
Semana 4 |
Semana 16 | |||
|
Secukinumab 300 mg |
Ustekinumab* |
Secukinumab 300 mg |
Ustekinumab* | |
|
Número de pacientes |
334 |
335 |
334 |
335 |
|
n (%) de respuesta PASI 75 |
167 (50,0%)** |
69 (20,6%) |
311 (93,1%) |
277 (82,7%) |
|
n (%) de respuesta PASI 90 |
70 (21,0%) |
18 (5,4%) |
264 (79,0%)** |
193 (57,6%) |
|
n (%) de respuesta PASI 100 |
14 (4,2%) |
3 (0,9%) |
148 (44,3%) |
95 (28,4%) |
|
n (%) de respuesta IGA mod 2011 “blanqueamiento completo” o “prácticamente completo” |
126 (37,7%) |
41 (12,2%) |
277 (82,9%) |
226 (67,5%) |
* Los pacientes tratados con secukinumab recibieron dosis de 300 mg a las semanas 0, 1, 2 y 3, seguidas por la misma dosis a las semanas 4, 8 y 12. Los pacientes tratados con ustekinumab recibieron 45 mg o 90 mg a las semanas 0 y 4 (dosificadas por peso según la posología aprobada)
** valores p con respecto a ustekinumab: p<0,0001
Cosentyx fue eficaz en pacientes sin antecedentes de tratamiento sistémico, sin antecedentes de tratamiento biológico, en pacientes previamente expuestos a un tratamiento biológico anti-TNF y en los pacientes que habían fracasado con un tratamiento biológico anti-TNF. Al inicio del estudio, las mejoras en PASI 75 en pacientes con artritis psoriásica concomitante fueron similares a las de la población general con psoriasis en placas.
Cosentyx se asociaba a un efecto de inicio rápido, con un 50% de reducción en la puntuación media del PASI en la semana 3 con la dosis de 300 mg.
Figura 1 Variación porcentual de la puntuación media del PASI con respecto a la inicial durante el estudio 1 (ERASURE)
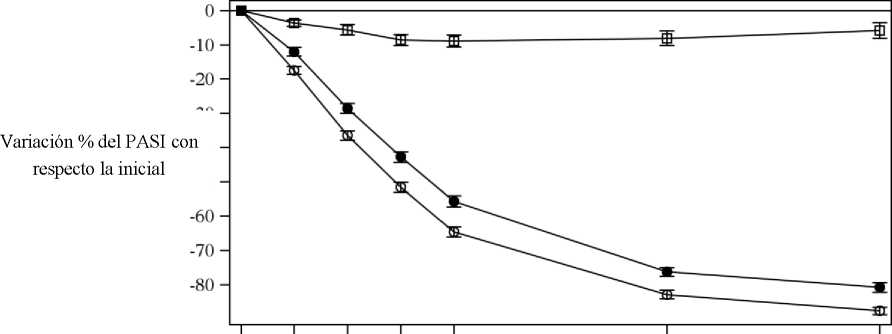
|
0 1 2 |
Semanas de tratamiento n=número de pacientes evaluables |
12 |
|
• Cosentyx 150 mg (n=243) o Cosentyx 300 mg (n=245) □ Placebo (n=245) | ||
Localizaciones/formas específicas de psoriasis en placas
En dos ensayos adicionales controlados con placebo, se observó mejoría en psoriasis ungueal (TRANSFIGURE, 198 pacientes) y en psoriasis en placa palmoplantar (GESTURE, 205 pacientes).
En el ensayo TRANSFIGURE, secukinumab fue superior a placebo a la semana 16 (46,1% para 300 mg, 38,4% para 150 mg y 11,7% para placebo) según lo evaluado por la mejoría significativa desde el periodo basal en el Índice de Gravedad de Psoriasis Ungueal (NAPSI %) para pacientes con psoriasis en placas de moderada a grave con afectación ungueal. En el ensayo GESTURE, secukinumab fue superior a placebo a la semana 16 (33,3% para 300 mg, 22,1% para 150 mg, y 1,5% para placebo) según lo evaluado por la mejoría significativa de respuesta de ppIGA 0 ó 1 (“blanqueamiento completo” o “ prácticamente completo”) para pacientes con psoriasis en placas palmoplantar de moderada a grave.
Calidad de vida/resultados percibidos por los pacientes
En la semana 12, el índice de calidad de vida en dermatología (DLQI) había mejorado estadísticamente de manera significativa en comparación con placebo respecto al inicio (estudios 1-4). La disminución media (mejoras) en DLQI respecto al inicio se puntuó desde -10,4 a -11,6 con secukinumab 300 mg, desde -7,7 a -10,1 con secukinumab 150 mg, frente al -1,1 y -1,9 de placebo en la semana 12. Estas mejoras se mantuvieron durante las 52 semanas (estudios 1 y 2).
El 40% de los participantes de los estudios 1 y 2 completaron el diario de síntomas de psoriasis (Psoriasis Symptom Diary®). De los participantes de cada uno de estos estudios que completaron el diario, mostraron mejoras estadísticamente significativas en los signos y síntomas de picor, dolor y descamación percibidos por los pacientes, a la semana 12 con respecto al inicio en comparación con el placebo.
Artritis _ psoriásica
Se evaluaron la seguridad y eficacia de Cosentyx en 1.003 pacientes en dos ensayos de fase III controlados con placebo, doble ciegos, aleatorizados, en pacientes con artritis psoriásica activa (>3 articulaciones inflamadas y >3 articulaciones dolorosas) a pesar del tratamiento con medicamentos antiinflamatorios no esteroideos (NSAID), corticosteroides o fármacos antirreumáticos modificadores de la enfermedad (FAME). En estos ensayos se reclutaron pacientes con cada subtipo de PsA, incluidas la artritis poliarticular sin evidencia de nódulos reumatoides, espondilitis con artritis periférica, artritis periférica asimétrica, participación interfalángica distal y artritis mutilante. Los pacientes que participaron en estos ensayos tenían un diagnóstico de PsA durante una media de 3,9 a 5,3 años. La mayoría de los pacientes también presentaban lesiones cutáneas de psoriasis activa o un historial documentado de psoriasis. Más del 62% y del 47% de los pacientes con PsA presentaban entesitis y dactilitis basales, respectivamente. Para ambos ensayos, la variable principal fue la respuesta 20 de la American College of Rheumatology (ACR) a la semana 24.
En el ensayo 1 en artritis psoriásica (Ensayo 1 en PsA) y en el ensayo 2 en artritis psoriásica (Ensayo 2 en PsA), el 29% y el 35% de los pacientes, respectivamente, fueron previamente tratados con un medicamento anti-TNFa e interrumpieron dicho tratamiento por falta de eficacia o intolerancia (pacientes con respuesta inadecuada a anti-TNFa).
En el Ensayo 1 en PsA (FUTURE 1) se evaluaron 606 pacientes, de los cuales el 60,7% recibían MTX de forma concomitante. Los pacientes aleatorizados a Cosentyx recibieron 10 mg/kg vía intravenosa a las semanas 0, 2, y 4, seguidas por 75 mg o 150 mg vía subcutánea mensualmente empezando en la semana 8. Los pacientes aleatorizados a placebo que eran no respondedores a la semana 16 (rescate temprano) y el resto de pacientes de placebo a la semana 24 se cruzaron para recibir Cosentyx (75 mg o 150 mg vía subcutánea) seguido por la misma dosis mensualmente.
En el Ensayo 2 en PsA (FUTURE 2) se evaluaron 397 pacientes, de los cuales el 46,6% recibían MTX de forma concomitante. Los pacientes aleatorizados a Cosentyx recibieron 75 mg, 150 mg o 300 mg vía subcutánea a las semanas 0, 1, 2, y 3, seguidas por la misma dosis mensualmente empezando en la semana 4. Los pacientes aleatorizados a recibir placebo que no eran respondedores a la semana 16 (rescate temprano) se cruzaron para recibir Cosentyx (150 mg o 300 mg vía subcutánea) a la semana 16 seguido por la misma dosis mensualmente. Los pacientes aleatorizados a recibir placebo que eran respondedores a la semana 16 se cruzaron para recibir Cosentyx (150 mg o 300 mg vía subcutánea) a la semana 24 seguido por la misma dosis mensualmente.
Signos y síntomas
El tratamiento con Cosentyx dio como resultado una mejora significativa en cuanto a las medidas de la actividad de la enfermedad en comparación con placebo a la semana 24 (ver Tabla 5).
Tabla 5 Respuesta clínica en el Ensayo 2 en PsA a la semana 24
|
Semana 24 | ||||
|
Placebo |
75 mg |
150 mg |
300 mg | |
|
Número de pacientes aleatorizados |
98 |
99 |
100 |
100 |
|
n (%) de respuesta ACR20 |
15 |
29 |
51 |
54 |
|
(15,3%) |
(29,3%*) |
(51,0%***) |
(54,0%***) | |
|
n (%) de respuesta ACR50 |
7 |
18 |
35 |
35 |
|
(7,1%) |
(18,2%) |
(35,0%) |
(35,0%**) | |
|
n (%) de respuesta ACR70 |
1 |
6 |
21 |
20 |
|
(1,0%) |
(6,1%) |
(21,0%**) |
(20,0%**) | |
|
DAS28-CRP |
-0,96 |
-1,12 |
-1,58** |
-1,61** |
|
Número de pacientes con >3% |
43 |
50 |
58 |
41 |
|
BSA afectada de psoriasis al inicio |
(43,9%) |
(50,5%) |
(58,0%) |
(41,0%) |
|
del estudio | ||||
|
n (%) de respuesta PASI 75 |
7 |
14 |
28 |
26 |
|
(16,3%) |
(28,0%) |
(48,3%**) |
(63,4%***) | |
|
n (%) de respuesta PASI 90 |
4 |
6 |
19 |
20 |
|
(9,3%) |
(12,0%) |
(32,8%**) |
(48,8%***) | |
|
n (%) de resolución de Dactilitis f |
4 |
10 |
16 |
26 |
|
(14,8%) |
(30,3%) |
(50,0%**) |
(56,5%**) | |
|
n (%) de resolución de Entesitis $ |
14 |
22 |
27 |
27 |
|
(21,5%) |
(32,4%) |
(42,2%*) |
(48,2%**) | |
* p<0,05, ** p<0,01, *** p<0,001; frente a placebo
Todos los valores-p se ajustan en función de la multiplicidad de ensayos de acuerdo a la jerarquía predefinida, excepto para ACR70, Dactilitis y Entesitis, que eran variables exploratorias.
Imputación de no respondedor utilizada para la variable binaria que falta.
ACR: American College of Rheumatology; PASI: Índice de Severidad y Área de Psoriasis; DAS: Puntuación de Actividad de la Enfermedad; BSA: Área de Superficie Corporal fEn pacientes con dactilitis al inicio del estudio (n=27, 33, 32, 46, respectivamente)
{En pacientes con entesitis al inicio del estudio (n=65, 68, 64, 56, respectivamente)_
El inicio de acción de Cosentyx se produjo tan pronto como en la semana 2. En la semana 3 se alcanzó una diferencia estadísticamente significativa en el ACR 20 frente a placebo. En la semana 16, los pacientes tratados con Cosentyx demostraron mejoras significativas en los signos y síntomas, entre los cuales se encuentran, respuestas significativamente mayores en ACR 20 (33,3%, 60,0% y 57,0% para 75 mg, 150 mg y 300 mg, respectivamente) comparado con placebo (18,4%).
En la Figura 2 se muestra el porcentaje de pacientes por visita que alcanzaron la respuesta ACR 20. Figure 2 Respuesta ACR20 en el Ensayo 2 de PsA a lo largo del tiempo hasta la semana 24
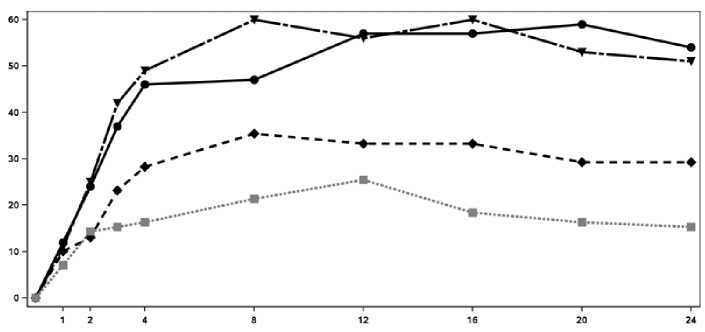
Tiempo (Semanas)
■ Cosentyx 150 mg Cosentyx 300 mg ■ ■ ■ • Placebo
Porcentaje de respondedores
Se observaron respuestas similares para las variables primaria y secundaria principal en pacientes con PsA a pesar de que estuviesen o no con tratamiento concomitante con MTX. En la semana 24, los pacientes tratados con Cosentyx y tratamiento concomitante con MTX solían tener una respuesta ACR 20 (44,7%, 47,7% y 54,4% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 20,0%) y ACR 50 (27,7%, 31,8% y 38,6% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 8,0%) más elevada. Los pacientes tratados con Cosentyx sin el uso concomitante de MTX solían tener una respuesta ACR 20 (15,4%, 53,6% y 53,6% para 75 mg,
150 mg y 300 mg, respectivamente, comparado con placebo 10,4%) y ACR 50 (9,6%, 37,5% y 32,1% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 6,3%) más elevada.
Los pacientes tratados con Cosentyx que eran “naíve” para anti-TNFa o con RI a anti-TNFa, presentaron una respuesta ACR 20 significativamente mayor comparado con placebo a la semana 24, con una respuesta ligeramente superior en el grupo “naíve” para anti-TNFa (“naíve” para anti-TNFa: 37%, 64% y 58% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 15,9%; RI a anti-TNFa: 15%, 30% y 46% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 14,3%). En el subgrupo de pacientes con RI a anti-TNFa, solo la dosis de 300 mg mostró una tasa de respuesta significativamente mayor para ACR 20 comparado con placebo (p<0,05) y demostró un beneficio clínico significativo sobre 150 mg en las variables secundarias múltiples. Se observaron mejorías en la respuesta PASI 75 en ambos subgrupos y la dosis de 300 mg mostró un beneficio estadísticamente significativo en los pacientes con RI a anti-TNFa.
El número de pacientes con PsA con afectación axial era demasiado pequeño como para permitir una evaluación significativa.
Se mostraron mejorías en todos los componentes de las puntuaciones ACR, incluyendo la evaluación del dolor del paciente. La proporción de pacientes que alcanzaron una respuesta Criterio de Respuesta a PsA (PsARC) modificada fue superior en los pacientes tratados con Cosentyx (38,4%, 62,0% y 63,0% para 75 mg, 150 mg y 300 mg, respectivamente) comparado con placebo (29,6%) a la semana 24.
En el Ensayo 1 en PsA y en el Ensayo 2 en PsA, la eficacia se mantuvo hasta la semana 52. En el Ensayo 2 en PsA, entre los 200 pacientes inicialmente aleatorizados a Cosentyx 150 mg y 300 mg, 178 (89%) pacientes aún estaban en tratamiento en la semana 52. De los 100 pacientes aleatorizados a Cosentyx 150 mg, 64, 39 y 20 presentaron una respuesta ACR 20/50/70, respectivamente. De los 100 pacientes aleatorizados a Cosentyx 300 mg, 64, 44 y 24 presentaron una respuesta ACR 20/50/70, respectivamente.
Respuesta radiográfica
No se ha demostrado la inhibición de la progresión del daño estructural en PsA, utilizando el régimen de carga vía subcutánea aprobado para el uso clínico.
En el Ensayo 1 en PsA, se evaluó radiográficamente la inhibición de la progresión del daño estructural y se expresó como el cambio en la puntuación total de Sharp modificada (mTSS) y sus componentes, en la puntuación de la erosión (ES) y en la puntuación del estrechamiento del espacio articular (JSN) a la semana 24 y 52, comparado con el periodo basal. En la Tabla 6 se presentan los datos a la semana 24.
Tabla 6 Cambio en la puntuación total de Sharp modificada en artritis psoriásica
|
Placebo N=179 |
Cosentyx 75 mg1 N=181 |
Cosentyx 150 mg1 N=185 | |
|
Puntuación total | |||
|
Periodo basal |
28,4 |
20,4 |
22,3 |
|
(DE) |
(63,5) |
(39,4) |
(48,0) |
|
Cambio medio a la semana 24 |
0,57 |
0,02* |
0,13* |
|
*p<0,05 de acuerdo al valor-p nominal pero no ajustado '10 mg/kg a las semanas 0, 2 y 4 seguido por dosis subcutáneas de 75 mg o |
150 mg | ||
La inhibición del daño estructural se mantuvo con el tratamiento con Cosentyx hasta la semana 52.
El porcentaje de pacientes sin progresión de la enfermedad (definido como un cambio desde el periodo basal en mTSS de <0,5) desde la aleatorización hasta la semana 24 fue del 92,3% en secukinumab 10 mg/kg como carga vía intravenosa - 75 mg como mantenimiento vía subcutánea, 82,3% en secukinumab 10 mg/kg como carga vía intravenosa - 150 mg como mantenimiento vía subcutánea y 75,7% en placebo. El porcentaje de pacientes sin progresión de la enfermedad desde la semana 24 hasta la semana 52 para secukinumab 10 mg/kg como carga vía intravenosa - seguido por 75 mg o 150 mg como mantenimiento vía subcutánea y para los pacientes con placebo que cambiaron a 75 mg o 150 mg vía subcutánea cada 4 semanas a la semana 16 o semana 24, fue del 85,8%, 85,7% y 86,8%, respectivamente.
Función física y calidad de vida relacionada con la salud
En el Ensayo 2 en PsA, los pacientes tratados con Cosentyx 150 mg (p=0,0555) y 300 mg (p=0,0040) mostraron mejoría en la función física comparado con los pacientes tratados con placebo evaluado por el índice de discapacidad del cuestionario de evaluación de la salud (HAQ-DI) a la semana 24. Se observaron mejorías en las puntuaciones HAQ-DI independientemente de la exposición previa a anti-TNFa. Se observaron respuestas similares en el Ensayo 1 en PsA.
Los pacientes tratados con Cosentyx notificaron mejorías significativas en la calidad de vida relacionada con la salud medido según la puntuación del resumen del componente físico del cuestionario de salud Short Form-36 (SF-36 PCS) (p<0,001). Asimismo, se observaron mejorías estadísticamente significativas demostradas en las variables exploratorias valoradas según la evaluación funcional para el tratamiento de enfermedades crónicas - Fatiga (FACIT-F) para 150 mg y 300 mg comparado con placebo (7,97, 5,97 comparado con 1,63, respectivamente). Se observaron respuestas similares en el ensayo 1 en PsA y la eficacia se mantuvo hasta la semana 52.
Espondilitis anquilosante
Se evaluó la seguridad y eficacia de Cosentyx en 590 pacientes en dos ensayos fase III controlados con placebo, doble ciego, aleatorizados, en pacientes con espondilitis anquilosante (AS) activa con un índice de actividad de la enfermedad espondilitis anquilosante de Bath (BASDAI) >4 a pesar del tratamiento con medicamentos antiinflamatorios no esteroideos (NSAID), corticosteroides o fármacos antirreumáticos modificadores de la enfermedad (FAME). Los pacientes que participaron en estos ensayos tenían un diagnóstico de AS durante una media de 2,7 a 5,8 años. Para ambos ensayos, la variable principal fue como mínimo una mejoría del 20% según los criterios de la Assessment of Spondyloarthritis International Society (ASAS 20) a la semana 16.
En el Ensayo 1 en espondilitis anquilosante (Ensayo 1 en AS) y en el Ensayo 2 en espondilitis anquilosante (Ensayo 2 en AS) el 27,0% y el 38,8% de los pacientes, respectivamente, se trataron previamente con un medicamento anti-TNFa y discontinuaron el tratamiento por falta de eficacia o intolerancia (pacientes con respuesta inadecuada a anti-TNFa).
En el Ensayo 1 en AS (MEASURE 1) se evaluaron 371 pacientes, de los cuales el 14,8% y el 33,4% utilizaron MTX o sulfasalacina de forma concomitante, respectivamente. Los pacientes aleatorizados a Cosentyx recibieron 10 mg/kg vía intravenosa a las semanas 0, 2, y 4, seguido por 75 mg o 150 mg vía subcutánea mensualmente empezando en la semana 8. Los pacientes aleatorizados a placebo que eran no respondedores a la semana 16 (rescate temprano) y todo el resto de pacientes de placebo a la semana 24 se cruzaron para recibir Cosentyx (75 mg o 150 mg vía subcutánea) seguido por la misma dosis mensualmente.
En el Ensayo 2 en AS (MEASURE 2) se evaluaron 219 pacientes, de los cuales el 11,9% y el 14,2% utilizaron MTX o sulfasalacina de forma concomitante, respectivamente. Los pacientes aleatorizados a Cosentyx recibieron 75 mg o 150 mg vía subcutánea a las semanas 0, 1, 2, y 3, seguida por la misma dosis mensualmente empezando en la semana 4. En la semana 16, los pacientes que fueron aleatorizados a recibir placebo al inicio del tratamiento, se volvieron a aleatorizar a recibir Cosentyx (75 mg o 150 mg vía subcutánea) mensualmente.
Signos y síntomas
En el Ensayo 2 en AS, el tratamiento con Cosentyx 150 mg dio como resultado una mejora superior en cuanto a las medidas de la actividad de la enfermedad en comparación con placebo a la semana 16 (ver Tabla 7).
Tabla 7 Respuesta clínica en el Ensayo 2 en AS a la semana 16
|
Resultado (valor-p frente a placebo) |
Placebo (n = 74) |
75 mg (n = 73) |
150 mg (n = 72) |
|
% de respuesta ASAS 20 |
28,4 |
41,1 |
61,1*** |
|
% de respuesta ASAS 40 |
10,8 |
26,0 |
36,1*** |
|
hsCRP, (tasa post-BSL/BSL) |
1,13 |
0,61 |
0,55*** |
|
% de ASAS 5/6 |
8,1 |
34,2 |
43 1*** |
|
% de remisión parcial de ASAS |
4,1 |
15,1 |
13,9 |
|
% de BASDAI 50 |
10,8 |
24,7* |
30,6** |
|
Mejora importante de ASDAS-CRP |
4,1 |
15,1* |
25,0*** |
|
* p<0,05, ** p<0,01, *** p<0,001; frente a placebo Todos los valores-p se ajustan en función de la multiplicidad de ensayos de acuerdo a la jerarquía predefinida, excepto para BASDAI 50 y ASDAS-CRP Imputación de no respondedor utilizada para la variable binaria que falta. | |||
|
ASAS: criterio de la Sociedad Internacional de evaluación de la espondiloartritis; BASDAI: índice de actividad de la enfermedad espondilitis anquilosante de Bath; hsCRP: proteína-C reactiva de alta sensibilidad; ASDAS: puntuación de la actividad de la enfermedad espondilitis anquilosante; BSL: periodo basal | |||
El inicio de acción de Cosentyx 150 mg se produjo tan pronto como en la semana 1 para ASAS 20 y semana 2 para ASAS 40 (superior a placebo) en el Ensayo 2 en AS.
Las respuestas ASAS 20 mejoraron en la semana 16 en los pacientes naíve para anti-TNFa (68,2% frente a 31,1%; p<0,05) y en los pacientes con RI para anti-TNFa (50,0% frente a 24,1%; p<0,05) para Cosentyx 150 mg comparado con placebo, respectivamente.
En ambos ensayos en AS, los pacientes tratados con Cosentyx (150 mg en el Ensayo 2 en AS y ambos regímenes en el Ensayo 1 en AS) demostraron signos y síntomas de mejoría significativa a la semana 16, con una magnitud de respuesta y eficacia comparable, mantenida hasta la semana 52 en pacientes naíve para anti-TNFa y en pacientes con RI para anti-TNFa-IR. En el Ensayo 2 en AS, de los 72 pacientes aleatorizados inicialmente a Cosentyx 150 mg, 61 (84,7%) pacientes estaban aún en tratamiento a la semana 52. De los 72 pacientes aleatorizados a Cosentyx 150 mg, 45 y 35 presentaron una respuesta ASAS 20/40, respectivamente.
Movilidad espinal
Los pacientes tratados con Cosentyx 150 mg mostraron mejorías en la movilidad espinal medido por el cambio desde el periodo basal según BASMI hasta la semana 16 para ambos ensayos, Ensayo 1 en AS (-0,40 vs -0,12 para placebo; p=0,0114) y Ensayo 2 en AS (-0,51 vs -0,22 para placebo; p=0,0533). Estas mejorías se mantuvieron hasta la semana 52.
Función física y calidad de vida relacionada con la salud
En el Ensayo 1 y Ensayo 2 en AS, los pacientes tratados con Cosentyx 150 mg mostraron mejoría en la calidad de vida relacionada con la salud medida por el cuestionario de calidad de vida en AS (ASQoL) (p=0,001) y en el resumen del componente físico del SF-36 (SF-36 PCS) (p<0,001). Los pacientes tratados con Cosentyx 150 mg también mostraron mejorías estadísticamente significativas en las variables exploratorias en la función física evaluado por el índice funcional de la espondilitis anquilosante de Bath (BASFI) comparado con placebo (-2,15 vs -0,68) y en la fatiga evaluado según la escala para la evaluación funcional para el tratamiento de enfermedades crónicas - Fatiga (FACIT-Fatiga) comparado con placebo (8,10 vs 3,30). Estas mejorías se mantuvieron hasta la semana 52.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Cosentyx en pacientes pediátricos, desde recién nacidos a menores de 6 años, con psoriasis en placas y desde recién nacidos a menores de 2 años, con artritis idiopática crónica (ver sección 4.2. para consultar la información sobre el uso en la población pediátrica).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Cosentyx en pacientes pediátricos, de 6 años a menores de 18 años, con psoriasis en placas y de 2 años a menores de 18 años, con artritis idiopática crónica (ver sección 4.2. para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Psoriasis en placas
Absorción
Tras una sola dosis subcutánea de 300 mg como formulación líquida en voluntarios sanos, secukinumab alcanzó las concentraciones máximas séricas de 43,2 ±10,4 pg/ml entre 2 y 14 días de la dosis.
Basado en el análisis farmacocinético de la población, con una sola administración subcutánea de 150 ó 300 mg de secukinumab a pacientes con psoriasis en placas se logra una concentración sérica máxima de secukinumab de 13,7 ± 4,8 pg/ml ó 27,3 ± 9,5 pg/ml, respectivamente, 5 ó 6 días después de la administración.
El tiempo transcurrido hasta alcanzar la concentración máxima después del primer mes de tratamiento con dosis semanales fue de 31 a 34 días, basado en el análisis farmacocinético de la población.
Sobre la base de datos simulados, las concentraciones máximas del estado estacionario (Cmáx,ss), tras la administración subcutánea de 150 ó 300 mg, son de 27,6 pg/ml o 55,2 pg/ml, respectivamente. El análisis farmacocinético de la población sugiere que el estado estacionario se alcanza después de 20 semanas con regímenes de administración mensuales.
El análisis farmacocinético de la población mostró que durante la fase de mantenimiento de administración mensual repetida, los pacientes presentan concentraciones séricas máximas y un área bajo la curva (AUC) dos veces mayor que las obtenidas con una sola administración.
El análisis farmacocinético de la población mostró que secukinumab se absorbió con una biodisponibilidad media absoluta del 73% en pacientes con psoriasis en placas. En todos los estudios, la biodisponibilidad absoluta se calculó que se encontraba entre el 60 y el 77%.
Distribución
El volumen medio de distribución durante la fase terminal (Vz) tras una sola administración intravenosa varía entre 7,10 y 8,60 litros en los pacientes con psoriasis en placas, lo que sugiere que la distribución del secukinumab hacia los compartimientos periféricos es limitada.
Biotransformación
La mayor parte de la eliminación de IgG ocurre mediante el catabolismo intracelular, tras endocitosis de la fase líquida o mediada por receptor.
Eliminación
El aclaramiento medio sistémico (CL) tras la administración única intravenosa en pacientes con psoriasis en placas fue de 0,13 a 0,36 l/día. En el análisis farmacocinético de la población, el aclaramiento medio sistémico (CL) fue de 0,19 l/día en los pacientes con psoriasis en placas. El CL no se ve afectado por el sexo. El aclaramiento no es dosis ni tiempo dependiente.
El análisis farmacocinético de la población, la vida de eliminación media se estimó en 27 días en los pacientes con psoriasis en placas, con un rango de 18 a 46 días en todos los estudios de psoriasis con administración intravenosa.
Linealidad/No linealidad
La farmacocinética tras dosis únicas y repetidas del secukinumab en pacientes con psoriasis en placas se determinó en varios estudios en los que se usaron tanto dosis intravenosas de entre 1 x 0,3 mg/kg a 3 x 10 mg/kg, como dosis subcutáneas de entre 1 x 25 mg a múltiples dosis de 300 mg. En todos los casos, la exposición resultó proporcional a la dosis.
Artritis psoriásica
Las propiedades farmacocinéticas de secukinumab observadas en pacientes con artritis psoriásica fueron similares a las que se muestran en pacientes con psoriasis en placas. La biodisponibilidad de secukinumab en pacientes con PsA fue del 85% de acuerdo al modelo farmacocinético poblacional.
Espondilitis anquilosante
Las propiedades farmacocinéticas de secukinumab observadas en pacientes con espondilitis anquilosante fueron similares a las que se muestran en pacientes con psoriasis en placas.
Poblaciones especiales
Pacientes de edad avanzada
De los 3.430 pacientes con psoriasis en placas que recibieron Cosentyx en los ensayos clínicos,
230 pacientes tenían más de 65 años de edad y 32, eran mayores de 75.
De los 974 pacientes con artritis psoriásica que recibieron Cosentyx en los ensayos clínicos, un total de 85 pacientes tenían a partir de 65 años de edad y 4 pacientes tenían a partir de 75 años.
De los 571 pacientes con espondilitis anquilosante que recibieron Cosentyx en los ensayos clínicos, un total de 24 pacientes tenían a partir de 65 años de edad y 3 pacientes tenían a partir de 75 años.
Según el análisis farmacocinético de la población con un número limitado de pacientes de edad avanzada (n=71 de más de 65 años y n=7 de más de 75 años), el aclaramiento fue similar en los pacientes de edad avanzada que en los menores de 65 años.
Pacientes con insuficiencia renal o hepática
No se disponen de datos farmacocinéticos en pacientes con insuficiencia renal o hepática. Se estima que la cantidad de Cosentyx sin metabolizar, un anticuerpo monoclonal de IgG, que se elimina por vía renal, es baja y de menor importancia. Las IgGs se eliminan principalmente por catabolismo, por lo que la insuficiencia hepática no se espera que afecte al aclaramiento de Cosentyx.
Efecto del _peso en la _farmacocinética
El aclaramiento de secukinumab y el volumen de distribución incrementan con el aumento del peso corporal.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios de reactividad cruzada en tejidos, farmacología de seguridad, estudios de toxicidad de la función reproductora y de dosis repetidas realizados con secukinumab o con un anticuerpo murino anti- IL-17A murina equivalente.
Como secukinumab se une a la IL-17A del ser humano y del mono cinomolgus, la seguridad se estudió en el mono cinomolgus. No se han observado efectos indeseados con secukinumab tras su administración a mono cinomolgus tanto por vía subcutánea, durante un período de hasta 13 semanas, como por vía intravenosa, durante un período de hasta 26 semanas (incluyendo evaluaciones farmacocinéticas, farmacodinamias, de inmunogenicidad y de inmunotoxicidad, p.ej., respuesta inmunitaria dependiente de linfocitos T y actividad de linfocitos citolíticos naturales). La concentración sérica media determinada en monos después de la administración de 13 dosis subcutáneas de 150 mg/kg una vez por semana es mayor que la concentración sérica media prevista para los pacientes con psoriasis que vayan a recibir la dosis clínica mayor. Se detectaron anticuerpos anti-secukinumáb en solo uno de los animales expuestos. No se observaron signos de reactividad cruzada tisular inespecífica tras la aplicación del secukinumab sobre tejidos humanos normales.
No se han realizado estudios en animales para evaluar el potencial carcinogénico de secukinumab.
Un estudio de desarrollo embriofetal efectuado en mono cinomolgus, no mostró toxicidad materna, embriotoxicidad ni teratogenicidad cuando secukinumab se administró durante la organogénesis o hacia el final de la gestación.
No se han observado efectos indeseados con un anticuerpo murino anti-IL-17A murina equivalente en los estudios de fecundidad y desarrollo embrionario temprano o de desarrollo pre- y postnatal del ratón. La dosis más alta que se usó en tales estudios era superior a la dosis máxima eficaz en cuanto a inhibición y actividad de la IL-17A (ver sección 4.6). 1
18 meses
6.4 Precauciones especiales de conservación
Conservar en nevera (2°C - 8°C). No congelar.
Conservar las jeringas en el embalaje exterior para protegerlo de la luz.
6.5 Naturaleza y contenido del envase
Cosentyx se comercializa en una jeringa precargada de 1 ml de cristal con un émbolo recubierto de Fluor Tec, con una aguja de 27G x A" protegida en un tapón de caucho de estireno butadieno montado en un dispositivo de seguridad de policarbonato.
Cosentyx está disponible en envases unitarios que contienen 1 ó 2 jeringas precargadas y en envases múltiples que contienen 6 (3 envases de 2) jeringas precargadas.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Cosentyx 150 mg solución inyectable se comercializa en una jeringa precargada de único uso para uso individual. No debe agitar ni congelar la jeringa. La jeringa se debe sacar de la nevera 20 minutos antes de la administración para que se atempere.
Antes de utilizar la jeringa precargada se recomienda hacer una revisión visual. El líquido debe ser transparente. Su color puede variar de incoloro a ligeramente amarillento. Puede ver alguna burbuja de aire pequeña, que es normal. No utilizar si el líquido contiene partículas, está turbio o tiene un color claramente marrón. En el prospecto se incluye información detallada sobre las instrucciones de uso.
La eliminación del medicamento no utilizado y de los materiales sobrantes que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/980/002
EU/1/14/980/003
EU/1/14/980/006
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
15.01.2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Cosentyx 150 mg solución inyectable en pluma precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada pluma precargada contiene 150 mg de secukinumab* en 1 ml.
*Secukinumab es un anticuerpo monoclonal recombinante, íntegramente humano, selectivo a la interleuquina17A. Secukinumab es un anticuerpo de tipo IgG1/K producido en células ováricas de hámster chino.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable en pluma precargada (pluma SensoReady) La solución es clara e incolora, ligeramente amarillenta.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Psoriasis en placas
Cosentyx está indicado para el tratamiento de la psoriasis en placas de moderada a grave en adultos candidatos a tratamientos sistémicos.
Artritis psoriásica
Cosentyx, solo o en combinación con metotrexato (MTX), está indicado para el tratamiento de la artritis psoriásica activa en pacientes adultos que han mostrado una respuesta inadecuada a tratamientos previos con fármacos antirreumáticos modificadores de la enfermedad (FAME) (ver sección 5.1).
Espondilitis anquilosante
Cosentyx está indicado para el tratamiento de la espondilitis anquilosante activa en adultos que no han respondido adecuadamente al tratamiento convencional.
4.2 Posología y forma de administración
Cosentyx se ha de utilizar bajo la dirección y la supervisión de un médico con experiencia en el diagnóstico y tratamiento de las enfermedades para las que Cosentyx está indicado.
Posología
Psoriasis en placas
La dosis recomendada es de 300 mg de secukinumab por inyección subcutánea, que se administra inicialmente en la semana 0, 1, 2 y 3 y, luego mensualmente, comenzando en la semana 4, durante la fase de mantenimiento. Cada dosis de 300 mg se administra de forma repartida en dos inyecciones subcutáneas de 150 mg.
Artritis psoriásica
Para pacientes que padecen psoriasis en placas de moderada a grave de forma concomitante o que son respondedores inadecuados (RI) a anti-TNFa, la dosis recomendada es de 300 mg por inyección subcutánea, que se administra inicialmente en la semana 0, 1, 2 y 3 y, luego durante la fase de mantenimiento, mensualmente comenzando en la semana 4. Cada dosis de 300 mg se administra en dos inyecciones subcutáneas de 150 mg.
Para el resto de pacientes, la dosis recomendada es de 150 mg por inyección subcutánea, que se administra inicialmente en la semana 0, 1, 2 y 3 y, luego durante la fase de mantenimiento, mensualmente comenzando en la semana 4.
Espondilitis anquilosante
La dosis recomendada es de 150 mg por inyección subcutánea, que se administra inicialmente en la semana 0, 1, 2 y 3 y, luego durante la fase de mantenimiento, mensualmente comenzando en la semana 4.
Para todas las indicaciones anteriores, los datos disponibles sugieren que una respuesta clínica se alcanza normalmente en las 16 semanas de tratamiento. Se debe considerar interrumpir el tratamiento en los pacientes que no han mostrado respuesta a las 16 semanas de tratamiento. Algunos pacientes con una respuesta parcial al inicio, pueden mejorar posteriormente con un tratamiento continuado de más de 16 semanas.
Poblaciones especiales
Pacientes de edad avanzada (mayores de 65 años)
No es necesario un ajuste de la dosis (ver sección 5.2).
Insuficiencia renal / insuficiencia hepática
No se ha estudiado Cosentyx en estas poblaciones de pacientes. No se puede hacer ninguna recomendación posológica.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de Cosentyx en niños menores de 18 años. No se dispone de datos.
Forma de administración
Cosentyx se ha de administrar por inyección subcutánea. En la medida de lo posible, se deben evitar como lugares de inyección las zonas de la piel que presenten signos de psoriasis.
Si el médico lo considera oportuno, los pacientes se pueden autoinyectar Cosentyx después de haber aprendido correctamente la técnica de inyección subcutánea. No obstante, el médico se debe asegurar, haciendo un adecuado seguimiento de los pacientes. Se indicará a los pacientes que se inyecten toda la cantidad de Cosentyx conforme a las instrucciones del prospecto. Las instrucciones completas de administración pueden consultarse en el prospecto.
Reacciones de hipersensibilidad graves al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Infecciones activas clínicamente importantes (p.ej. tuberculosis activa; ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
Infecciones
Cosentyx puede aumentar el riesgo de infecciones. En los ensayos clínicos se han observado infecciones en los pacientes que recibieron Cosentyx (ver sección 4.8). La mayoría fueron infecciones leves o moderadas de las vías respiratorias altas como rinofaringitis que no requirieron interrumpir el tratamiento.
Relacionado con el mecanismo de acción de Cosentyx, en los ensayos clínicos de psoriasis se han notificado infecciones mucocutáneas no graves por cándida más frecuentemente con secukinumab que con placebo (3,55 por 100 pacientes-año con secukinumab 300 mg frente a 1,00 por 100 paciente-año con placebo) (ver sección 4.8).
Se debe tener precaución cuando se valore la administración de Cosentyx en pacientes con infecciones crónicas o con antecedentes de infecciones recurrentes.
Se debe indicar al paciente que consulte al médico cuando padezca signos o síntomas indicativos de una infección. El paciente que contraiga una infección grave debe ser objeto de una estrecha observación y no debe recibir Cosentyx hasta que la infección se haya resuelto.
No se han notificado en los ensayos clínicos una mayor sensibilidad a la tuberculosis. Aun así, Cosentyx no se debe administrar a pacientes con tuberculosis activa. Se debe considerar la posibilidad de administrar un tratamiento antituberculoso antes de comenzar el tratamiento con Cosentyx en los pacientes con tuberculosis latente.
Enfermedad de Crohn
Se debe tener precaución cuando se prescriba Cosentyx a pacientes con enfermedad de Crohn. En los ensayos clínicos se ha observado exacerbaciones, en algunos casos graves, tanto en el grupo de pacientes de Cosentyx como en el de placebo. Se deben vigilar estrechamente los pacientes tratados con Cosentyx con enfermedad de Crohn.
Reacciones de hipersensibilidad
En los ensayos clínicos se han observado, en raras ocasiones, reacciones anafilácticas en pacientes que estaban recibiendo Cosentyx. Si aparecen reacciones anafilácticas u otras reacciones alérgicas graves, se debe interrumpir inmediatamente el tratamiento con Cosentyx e iniciar otro tratamiento alternativo.
Individuos sensibles al látex
El capuchón extraíble de la aguja de la pluma precargada de Cosentyx contiene un derivado de la goma látex natural. Hasta la fecha no se ha detectado goma látex natural en el capuchón extraíble de la aguja. Sin embargo tampoco se ha estudiado el uso de las plumas precargadas de Cosentyx en individuos sensibles al látex y por tanto, hay un riesgo potencial de reacciones de hipersensibilidad que no puede ser completamente descartado.
Vacunas
No se deben administrar simultáneamente las vacunas elaboradas con microorganismos vivos con Cosentyx.
Los pacientes tratados con Cosentyx pueden recibir de forma simultánea vacunas inactivadas o no elaboradas con microorganismos vivos. En un estudio, después de la administración de vacunas antigripales inactivadas y antimeningocócicas, los voluntarios sanos tanto del grupo de 150 mg de secukinumab como de placebo, fueron capaces de generar una respuesta inmunitaria satisfactoria que por lo menos cuadruplicó los títulos de anticuerpos contra esas vacunas. Los datos indican que Cosentyx no inhibe la respuesta inmunitaria humoral a las vacunas antimeningocócicas o antigripales.
Tratamiento inmunosupresor concomitante
En los estudios de psoriasis, no se ha evaluado la seguridad y eficacia de Cosentyx en combinación con inmunosupresores, incluidos biológicos, o fototerapia (ver también sección 4.5).
4.5 Interacción con otros medicamentos y otras formas de interacción
No se deben administrar las vacunas elaboradas con microorganismos vivos simultáneamente con Cosentyx (ver también sección 4.4).
No se han realizado estudios de interacciones en humanos. No existe una evidencia directa del papel de IL-17A en la expresión de las enzimas CYP450. La formación de algunas enzimas P450 se suprime debido al aumento de citoquinas durante la inflamación crónica. Por tanto los tratamientos antiinflamatorios, tales como secukinumab, un inhibidor de IL17A, podría normalizar los niveles de CYP450 y en consecuencia, disminuir la exposición de las medicaciones concomitantes metabolizadas por el CYP450. Por lo que no se puede excluir que exista un efecto clínico relevante en los medicamentos de estrecho margen terapéutico, donde la dosis se ajusta de forma individual (p. ej. warfarina), que sean sustratos de CYP450. Cuando se inicie el tratamiento con secukinumab en pacientes tratados con este tipo de medicamentos, se debe considerar el realizar monitorización terapéutica.
No se observó interacción cuando Cosentyx se administró de forma concomitante con metotrexato (MTX) y/o corticosteroides en ensayos en artritis (incluyendo pacientes con artritis psoriásica y espondilitis anquilosante).
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar un método anticonceptivo durante el tratamiento y durante al menos 20 semanas después del tratamiento.
Embarazo
No se dispone de datos suficientes sobre el uso de secukinumab en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para el embarazo, el desarrollo embrionario o fetal, el parto o el desarrollo postnatal (ver sección 5.3). Como medida de precaución, es preferible evitar el uso de Cosentyx en el embarazo.
Lactancia
Se desconoce si secukinumab se excreta en la leche materna. Las inmunoglobulinas pasan a la leche materna y se desconoce si secukinumab se absorbe sistémicamente tras su ingestión. Debido a las posibles reacciones adversas de secukinumab en los lactantes, se debe decidir si interrumpir la lactancia durante el tratamiento y hasta 20 semanas después del tratamiento o interrumpir el tratamiento con Cosentyx, teniendo en cuenta el beneficio de la lactancia para el niño o el beneficio del tratamiento con Cosentyx para la mujer.
Fertilidad
No se ha evaluado el efecto de secukinumab sobre la fertilidad humana. Los estudios en animales no indican que Cosentyx tenga efectos nocivos directos o indirectos sobre la fertilidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Cosentyx sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Un total de 6.804 pacientes han recibido Cosentyx en los ensayos clínicos con y sin enmascaramiento en diversas indicaciones (psoriasis en placas, artritis psoriásica, espondilitis anquilosante y otras enfermedades autoinmunes). De éstos, 3.671 pacientes se han expuesto a Cosentyx durante al menos un año, lo que representa una exposición de 6.450 paciente-año.
Reacciones adversas en psoriasis en placas
Se agruparon los datos de los cuatro estudios de fase III controlados con placebo en psoriasis en placas para evaluar la seguridad de Cosentyx hasta 12 semanas después del inicio del tratamiento. Se evaluaron 2.076 pacientes en total (de los cuales, 692 pacientes recibieron la dosis de 150 mg, 690, la de 300 mg y 694, el placebo).
Las reacciones adversas notificadas con mayor frecuencia fueron las infecciones de las vías respiratorias altas (con mayor frecuencia rinofaringitis y rinitis). La mayoría de las reacciones fueron de naturaleza leve o moderada.
Reacciones adversas en artritis psoriásica
Cosentyx se estudió en dos ensayos controlados con placebo en artritis psoriásica con 1.003 pacientes (703 pacientes en Cosentyx y 300 pacientes en placebo) para una exposición total de 1.061 pacientes-años de exposición del ensayo (duración media de exposición para los pacientes tratados con secukinumab: 456 días en el Ensayo 1 en artritis psoriásica y 245 días en el Ensayo 2 en artritis psoriásica). El perfil de seguridad observado en pacientes con artritis psoriásica tratados con Cosentyx es consistente con el perfil de seguridad en psoriasis.
Reacciones adversas en espondilitis anquilosante
Cosentyx se estudió en dos ensayos controlados con placebo en espondilitis anquilosante con 590 pacientes (394 pacientes en Cosentyx y 196 pacientes en placebo) para un total de 755 pacientes-años de exposición del ensayo (duración media de exposición para los pacientes tratados con secukinumab: 469 días en el Ensayo 1 en espondilitis anquilosante y 460 días en el Ensayo 2 en espondilitis anquilosante). El perfil de seguridad observado en pacientes con espondilitis anquilosante tratados con Cosentyx es consistente con el perfil de seguridad en psoriasis.
Las reacciones adversas de los ensayos clínicos en psoriasis, artritis psoriásica y espondilitis anquilosante (Tabla 1) se presentan según la clasificación de órganos del MedDRA. Dentro de cada órgano y sistema, las reacciones adversas se ordenan por frecuencia, las más frecuentes primero. Las reacciones adversas se incluyen en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Además, las categorías de frecuencia se definen utilizando los siguientes criterios: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000).
Tabla 1 Lista de las reacciones adversas en los ensayos clínicos1-1
|
Sistema de Clasificación de Órganos |
Frecuencia |
Reacción adversa |
|
Infecciones e infestaciones |
Muy frecuentes |
Infecciones de las vías respiratorias altas |
|
Frecuentes |
Herpes oral | |
|
Poco frecuentes |
Candidiasis oral | |
|
Pie de atleta | ||
|
Otitis externa | ||
|
Trastornos de la sangre y del sistema linfático |
Poco frecuentes |
Neutropenia |
|
Trastornos del sistema inmunológico |
Raras |
Reacciones anafilácticas |
|
Trastornos oculares |
Poco frecuentes |
Conjuntivitis |
|
Trastornos respiratorios, torácicos y mediastínicos |
Frecuentes |
Rinorrea |
|
Trastornos gastrointestinales |
Frecuentes |
Diarrea |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
Urticaria |
|
En los ensayos clínicos controlados con placebo (fase III) en psoriasis en placas, artritis psoriásica y espondilitis anquilosante, los pacientes recibieron 300 mg, 150 mg, 75 mg o placebo durante 12 semanas (psoriasis) o 16 semanas (artritis psoriásica y espondilitis anquilosante) de duración del tratamiento | ||
Descripción de las reacciones adversas seleccionadas Infecciones
Durante la fase comparativa con placebo de los ensayos clínicos en psoriasis en placas (en los que un total de 1.382 pacientes recibieron Cosentyx y 694, el placebo, durante un período de hasta 12 semanas), se notificaron infecciones en el 28,7% de los pacientes del grupo de Cosentyx y en el 18,9% de los pacientes que recibieron el placebo. La mayoría de las infecciones se consideraron no graves, infecciones leves o moderadas de las vías respiratorias altas, como rinofaringitis, que no requirieron interrupción del tratamiento. Hubo un aumento de candidiasis en mucosa y piel, consistente con el mecanismo de acción, no graves, de leves a moderadas y que respondieron al tratamiento estándar sin tener que interrumpir el tratamiento. Las infecciones graves aparecieron en un 0,14% de los pacientes tratados con Cosentyx y en un 0,3% de los pacientes tratados con placebo (ver sección 4.4).
Durante todo el periodo de tratamiento (un total de 3.430 pacientes tratados con Cosentyx durante 52 semanas, en la mayoría de los pacientes), se notificaron infecciones en el 47,5% de los pacientes tratados con Cosentyx (0,9 por paciente-año de seguimiento). El 1,2% de los pacientes tratados con Cosentyx notificaron infecciones graves (0,015 por paciente-año de seguimiento).
La tasa de infecciones observada en los ensayos clínicos en artritis psoriásica y espondilitis anquilosante fue similar a la observada en los ensayos en psoriasis.
Neutropenia
En los ensayos clínicos de fase 3 en psoriasis, se ha observado con mayor frecuencia neutropenia con secukinumab que con placebo, pero en la mayoría de los casos fue leve, transitoria y reversible. Se notificó neutropenia <1,0-0,5x102 3 4 5 6/l (CTCAE Grado 3) en 18 de los 3.430 (0,5%) pacientes con secukinumab, independientemente de la dosis y sin una relación temporal con la infección en 15 de los 18 casos. No se notificaron casos de neutropenia más grave. Los otros 3 casos restantes fueron infecciones no graves que respondieron al tratamiento estándar y no requirieron interrumpir el tratamiento con Cosentyx.
La frecuencia de neutropenia en artritis psoriásica y espondilitis anquilosante es similar a psoriasis.
Se notificaron raros casos de neutropenia <0,5x106/l (CTCAE Grado 4).
Reacciones de hipersensibilidad
En los ensayos clínicos se ha observado urticaria y raros casos de reacción anafiláctica a Cosentyx (ver también sección 4.4).
Inmunogenicidad
En ensayos clínicos en psoriasis, artritis psoriásica y espondilitis anquilosante, menos del 1% de los pacientes tratados con Cosentyx desarrollaron anticuerpos a secukinumab a lo largo de 52 semanas de tratamiento. Aproximadamente la mitad de los anticuerpos antifármaco producidos durante el tratamiento fueron neutralizantes pero esto no se asoció a una pérdida de eficacia o a trastornos farmacocinéticos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han notificado casos de sobredosis en los ensayos clínicos.
Durante los estudios clínicos se han administrado por vía intravenosa dosis de hasta 30 mg/kg (aproximadamente de 2.000 a 3.000 mg) sin que haya aparecido toxicidad limitante de la dosis. En caso de sobredosis se recomienda vigilar al paciente en busca de signos o síntomas de reacciones adversas e instaurar inmediatamente el tratamiento sintomático más adecuado.
el eritema, la induración y la descamación presentes en las lesiones de la psoriasis en placas.
IL-17A es una citoquina natural que participa en reacciones inmunitarias e inflamatorias normales. IL-17A desempeña una función clave en la patogenia de la psoriasis en placas, artritis psoriásica y espondilitis anquilosante y se encuentra concentrada en la piel lesionada, a diferencia de la piel no lesionada de los pacientes con psoriasis en placas y en el tejido sinovial de los pacientes con artritis psoriásica. La frecuencia de células productoras de IL-17, también fue significativamente superior en la médula ósea subcondral de las articulaciones facetarías de pacientes con espondilitis anquilosante.
Efectos farmacodinámicos
Las concentraciones séricas de IL-17A total (libre y unida a secukinumab) aumentan inicialmente en los pacientes que reciben secukinumab. Después disminuye lentamente debido a un aclaramiento reducido del complejo secukinumab-IL-17A, lo que indica que secukinumab es capaz de fijarse selectivamente a la IL-17A libre, la cual desempeña un papel fundamental en la patogenia de la psoriasis en placas.
En un estudio con secukinumab, los neutrófilos epidérmicos infiltrantes y los distintos marcadores asociados a neutrófilos, presentes en gran número en la piel lesionada de los pacientes con psoriasis en placas, disminuyeron significativamente al cabo de una o dos semanas de tratamiento.
Secukinumab ha demostrado que reduce (entre 1 y 2 semanas de tratamiento) los niveles de proteína C reactiva, que es un marcador de la inflamación.
Eficacia clínica y seguridad
Psoriasis en placas
La seguridad y la eficacia de Cosentyx se evaluaron en cuatro estudios de fase III, aleatorizados, con doble enmascaramiento y comparativos con placebo, llevados a cabo en pacientes con psoriasis en placas moderada o grave que eran candidatos de fototerapia o de tratamientos sistémicos [ERASURE, FIXTURE, FEATURE y JUNCTURE]. La eficacia y la seguridad de Cosentyx 150 mg y 300 mg se evaluaron frente a placebo y etanercept. Además, en otro estudio [SCULPTURE] se evaluó un régimen terapéutico crónico en comparación con la pauta de “repetición del tratamiento en caso de necesidad”.
De los 2.403 pacientes que participaron en los estudios comparativos con placebo, el 79% carecía de antecedentes de tratamiento biológico, el 45% procedían de fracasos con tratamientos no biológicos, un 8% procedía de fracasos de tratamientos biológicos (el 6% de fracasos con anti-TNF y el 2% de tratamientos anti-p40). Entre el 15 y el 25% de los pacientes de los estudios de fase III tenían artritis psoriásica al inicio.
En el estudio 1 sobre psoriasis (ERASURE) se evaluaron 738 pacientes. Los pacientes asignados aleatoriamente al grupo de Cosentyx recibieron dosis de 150 ó 300 mg en las semanas 0, 1, 2 y 3 y, luego una vez al mes, comenzando en la semana 4, la misma dosis. En el estudio 2 sobre psoriasis (FIXTURE) se evaluaron 1.306 pacientes. Los pacientes asignados aleatoriamente al grupo de Cosentyx recibieron dosis de 150 ó 300 mg en las semanas 0, 1, 2 y 3 y, luego una vez al mes, comenzando en la semana 4, la misma dosis. Los pacientes asignados aleatoriamente al grupo del etanercept recibieron dosis de 50 mg dos veces por semana durante 12 semanas y, luego, 50 mg una vez por semana. En ambos estudios, estudio 1 y 2, los pacientes asignados aleatoriamente al grupo del placebo que no respondían al cabo de 12 semanas pasaron a recibir Cosentyx (150 ó 300 mg) a las semanas 12, 13, 14 y 15 y, luego una vez al mes, comenzando en la semana 16, la misma dosis. Desde la primera administración del tratamiento del estudio, todos los pacientes fueron sometidos a un seguimiento de hasta 52 semanas de duración.
En el estudio 3 sobre psoriasis (FEATURE) se evaluaron 177 pacientes usando una jeringa precargada en comparación con el placebo después de 12 semanas de tratamiento para evaluar la seguridad, la tolerabilidad y la autoadministración de Cosentyx con la jeringa precargada. En el estudio 4 sobre psoriasis (JUNCTURE) se evaluaron 182 pacientes usando una pluma precargada en comparación con el placebo después de 12 semanas de tratamiento para evaluar la seguridad, la tolerabilidad y la autoadministración de Cosentyx con la pluma precargada. En ambos estudios, estudio 3 y 4, los pacientes asignados aleatoriamente al grupo de Cosentyx, recibieron dosis de 150 ó 300 mg a las semanas 0, 1, 2 y 3 y, luego una vez al mes, comenzando en la semana 4, la misma dosis. También se aleatorizaron pacientes para que recibiesen el placebo las semanas 0, 1, 2 y 3 y, luego una vez al mes, comenzando en la semana 4, la misma dosis.
En el estudio 5 sobre psoriasis (SCULPTURE) se evaluaron 966 pacientes. Todos los pacientes recibieron Cosentyx en dosis de 150 ó 300 mg a las semanas 0, 1, 2, 3, 4, 8 y 12 y, luego, se les asignó de forma aleatoria un régimen de mantenimiento mensual con la misma dosis a partir de la semana 12, o bien una pauta de “repetición del tratamiento en caso de necesidad” con la misma dosis. Los pacientes asignados aleatoriamente al grupo de “repetición del tratamiento en caso de necesidad” que no consiguieron un mantenimiento satisfactorio de la respuesta, se recomendó un régimen de mantenimiento con dosis mensuales fijas.
Las co-variables principales en los estudios con placebo y con comparador fueron la proporción de pacientes que consiguieron una respuesta de PASI 75 y una respuesta IGA mod 2011 de “blanqueamiento completo” o “prácticamente completo”, en comparación con placebo al cabo de 12 semanas (ver Tabla 2 y 3). Con la dosis de 300 mg se obtuvo una mejoría en la piel, en particular, un “blanqueamiento completo” o “prácticamente completo” con criterios de eficacia de PASI 90,
PASI 100, e IGA mod 2011 0 ó 1 en todos los estudios, con efectos máximos a la semana 16, de modo que se recomienda esta dosis.
Tabla 2 Resumen de las respuestas PASI 50/75/90/100 “blanqueamiento completo” o
“prácticamente completo” en IGA* mod 2011 en los estudios 1, 3 y 4 en psoriasis (ERASURE, FEATURE y JUNCTURE)
Semana 12 Semana 16 Semana 52
|
Placebo |
150 mg |
300 mg |
150 mg |
300 mg |
150 mg |
300 mg | |
|
Estudio 1 | |||||||
|
Número de pacientes |
246 |
244 |
245 |
244 |
245 |
244 |
245 |
|
n (%) de respuesta PASI 50 |
22 (8,9%) |
203 (83,5%) |
222 (90,6%) |
212 (87,2%) |
224 (91,4%) |
187 (77%) |
207 (84,5%) |
|
n (%) de respuesta PASI 75 |
11 (4,5%) |
174 (71,6%)** |
200 (81,6%)** |
188 (77,4%) |
211 (86,1%) |
146 (60,1%) |
182 (74,3%) |
|
n (%) de respuesta PASI 90 |
3 (1,2%) |
95 (39,1%)** |
145 (59,2%)** |
130 (53,5%) |
171 (69,8%) |
88 (36,2%) |
147 (60,0%) |
|
n (%) de respuesta PASI 100 |
2 (0,8%) |
31 (12,8%) |
70 (28,6%) |
51 (21,0%) |
102 (41,6%) |
49 (20,2%) |
96 (39,2%) |
|
n (%) de “blanqueamiento |
6 (2,40%) |
125 |
160 |
142 |
180 |
101 |
148 |
|
completo” o “prácticamente completo” según IGA mod 2011 Estudio 3 |
(51,2%)** |
(65,3%)** |
(58,2%) |
(73,5%) |
(41,4%) |
(60,4%) | |
|
Número de pacientes |
59 |
59 |
58 |
- |
- |
- |
- |
|
n (%) de respuesta PASI 50 |
3 (5,1%) |
51 (86,4%) |
51 (87,9%) |
- |
- |
- |
- |
|
n (%) de respuesta PASI 75 |
0 (0,0%) |
41 (69,5%)** |
44 (75,9%)** |
- |
- |
- |
- |
|
n (%) de respuesta PASI 90 |
0 (0,0%) |
27 (45,8%) |
35 (60,3%) |
- |
- |
- |
- |
|
n (%) de respuesta PASI 100 |
0 (0,0%) |
5 (8,5%) |
25 (43,1%) |
- |
- |
- |
- |
|
n (%) de “blanqueamiento completo” o “prácticamente completo” según IGA mod 2011 Estudio 4 |
0 (0,0%) |
31 (52,5%)** |
40 (69,0%)** | ||||
|
Número de pacientes |
61 |
60 |
60 |
- |
- |
- |
- |
|
n (%) de respuesta PASI 50 |
5 (8,2%) |
48 (80,0%) |
58 (96,7%) |
- |
- |
- |
- |
|
n (%) de respuesta PASI 75 |
2 (3,3%) |
43 (71,7%)** |
52 (86,7%)** |
- |
- |
- |
- |
|
n (%) de respuesta PASI 90 |
0 (0,0%) |
24 (40,0%) |
33 (55,0%) |
- |
- |
- |
- |
|
n (%) de respuesta PASI 100 |
0 (0,0%) |
10 (16,7%) |
16 (26,7%) |
- |
- |
- |
- |
|
n (%) de “blanqueamiento completo” o “prácticamente |
0 (0,0%) |
32 (53,3%)** |
44 (73,3%)** |
- |
- |
- |
- |
completo” según IGA mod 2011
* La IGA mod 2011 es una escala de 5 categorías: “0 = blanqueamiento completo total”, “1 = blanqueamiento prácticamente completo”, “2 = (psoriasis) leve”, “3 = moderada” o “4 = grave” que indica la evaluación global del médico sobre la intensidad de la psoriasis en función de la induración, el eritema y la descamación. Se definió como éxito terapéutico, “remisión total” o “remisión casi total”, la ausencia de signos de psoriasis o bien una coloración normal o rosada de las lesiones cutáneas, ausencia de induración de la placa y ninguna o una mínima descamación focal.
** valores de p con respecto al placebo, ajustados en función de la multiplicidad: p<0,0001._
Tabla 3 Resumen de la respuesta clínica del estudio 2 de psoriasis (FIXTURE)
Semana 12 Semana 16 Semana 52
|
Placebo |
150 mg |
300 mg |
Etanercept |
150 mg |
300 mg |
Etanercept |
150 mg |
300 mg |
Etanercept | |
|
Número de |
324 |
327 |
323 |
323 |
327 |
323 |
323 |
327 |
323 |
323 |
|
pacientes n (%) de respuesta |
49 |
266 |
296 |
226 |
290 |
302 |
257 (79,6%) |
249 |
274 |
234 (72,4%) |
|
PASI 50 |
(15,1%) |
(81,3%) |
(91,6%) |
(70,0%) |
(88,7%) |
(93,5%) |
(76,1%) |
(84,8%) | ||
|
n (%) de respuesta |
16 |
219 |
249 |
142 |
247 |
280 |
189 (58,5%) |
215 |
254 |
179 (55,4%) |
|
PASI 75 |
(4,9%) |
(67,0%) ** |
(77,1%) ** |
(44,0%) |
(75,5%) |
(86,7%) |
(65,7%) |
(78,6%) | ||
|
n (%) de respuesta |
5 (1,5%) |
137 |
175 |
67 (20,7%) |
176 |
234 |
101 (31,3%) |
147 |
210 |
108 (33,4%) |
|
PASI 90 |
(41,9%) |
(54,2%) |
(53,8%) |
(72,4%) |
(45,0%) |
(65,0%) | ||||
|
n (%) de respuesta |
0 (0%) |
47 |
78 |
14 (4,3%) |
84 |
119 |
24 (7,4%) |
65 |
117 |
32 (9,9%) |
|
PASI 100 |
(14,4%) |
(24,1%) |
(25,7%) |
(36,8%) |
(19,9%) |
(36,2%) | ||||
|
n (%) de de |
9 (2,8%) |
167 |
202 |
88 (27,2%) |
200 |
244 |
127 (39,3%) |
168 |
219 |
120 (37,2%) |
|
“blanqueamiento |
(51,1%) |
(62,5%) |
(61,2%) |
(75,5%) |
(51,4%) |
(67,8%) | ||||
|
completo” o |
** |
** |
“prácticamente completo” en la
IGA mod 2011_
** valores p con respecto al etanercept: p=0,0250
En un ensayo adicional en psoriasis (CLEAR) se evaluaron 676 pacientes. Secukinumab 300 mg alcanzó las variables principal y secundaria mostrando superioridad a ustekinumab de acuerdo a la respuesta de PASI 90 a la semana 16 y a la velocidad de inicio de respuesta de PASI 75 a la semana 4. Se observó una mayor eficacia para secukinumab comparado con ustekinumab para los criterios de respuesta de PASI 75/90/100 e IGA mod 2011 0 ó 1 (“blanqueamiento completo” o “prácticamente completo”) de forma temprana y continua hasta la semana 16.
Tabla 4 Resumen de la respuesta clínica en el ensayo CLEAR
|
Semana 4 |
Semana 16 | |||
|
Secukinumab 300 mg |
Ustekinumab* |
Secukinumab 300 mg |
Ustekinumab* | |
|
Número de pacientes |
334 |
335 |
334 |
335 |
|
n (%) de respuesta PASI 75 |
167 (50,0%)** |
69 (20,6%) |
311 (93,1%) |
277 (82,7%) |
|
n (%) de respuesta PASI 90 |
70 (21,0%) |
18 (5,4%) |
264 (79,0%)** |
193 (57,6%) |
|
n (%) de respuesta PASI 100 |
14 (4,2%) |
3 (0,9%) |
148 (44,3%) |
95 (28,4%) |
|
n (%) de respuesta IGA mod 2011 “blanqueamiento completo” o “prácticamente completo” |
126 (37,7%) |
41 (12,2%) |
277 (82,9%) |
226 (67,5%) |
* Los pacientes tratados con secukinumab recibieron dosis de 300 mg a las semanas 0, 1, 2 y 3, seguidas por la misma dosis a las semanas 4, 8 y 12. Los pacientes tratados con ustekinumab recibieron 45 mg o 90 mg a las semanas 0 y 4 (dosificadas por peso según la posología aprobada) ** valores p con respecto a ustekinumab: p<0,0001
Cosentyx fue eficaz en pacientes sin antecedentes de tratamiento sistémico, sin antecedentes de tratamiento biológico, en pacientes previamente expuestos a un tratamiento biológico anti-TNF y en los pacientes que habían fracasado con un tratamiento biológico anti-TNF. Al inicio del estudio, las mejoras en PASI 75 en pacientes con artritis psoriásica concomitante fueron similares a las de la población general con psoriasis en placas.
Cosentyx se asociaba a un efecto de inicio rápido, con un 50% de reducción en la puntuación media del PASI en la semana 3 con la dosis de 300 mg.
Figura 1 Variación porcentual de la puntuación media del PASI con respecto a la inicial durante el estudio 1 (ERASURE)
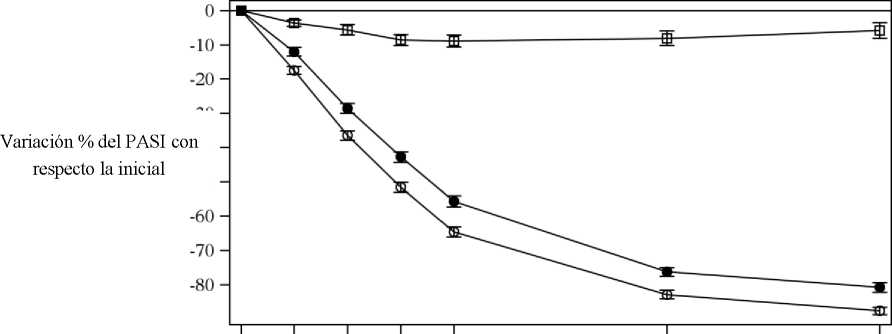
|
0 1 2 |
Semanas de tratamiento n=número de pacientes evaluables |
12 |
|
• Cosentyx 150 mg (n=243) o Cosentyx 300 mg (n=245) □ Placebo (n=245) | ||
Localizaciones/formas específicas de psoriasis en placas
En dos ensayos adicionales controlados con placebo, se observó mejoría en psoriasis ungueal (TRANSFIGURE, 198 pacientes) y en psoriasis en placa palmoplantar (GESTURE, 205 pacientes).
En el ensayo TRANSFIGURE, secukinumab fue superior a placebo a la semana 16 (46,1% para 300 mg, 38,4% para 150 mg y 11,7% para placebo) según lo evaluado por la mejoría significativa desde el periodo basal en el Índice de Gravedad de Psoriasis Ungueal (NAPSI %) para pacientes con psoriasis en placas de moderada a grave con afectación ungueal. En el ensayo GESTURE, secukinumab fue superior a placebo a la semana 16 (33,3% para 300 mg, 22,1% para 150 mg, y 1,5% para placebo) según lo evaluado por la mejoría significativa de respuesta de ppIGA 0 ó 1 (“blanqueamiento completo” o “ prácticamente completo”) para pacientes con psoriasis en placas palmoplantar de moderada a grave.
Calidad de vida/resultados percibidos por los pacientes
En la semana 12, el índice de calidad de vida en dermatología (DLQI) había mejorado estadísticamente de manera significativa en comparación con placebo respecto al inicio (estudios 1-4). La disminución media (mejoras) en DLQI respecto al inicio se puntuó desde -10,4 a -11,6 con secukinumab 300 mg, desde -7,7 a -10,1 con secukinumab 150 mg, frente al -1,1 y -1,9 de placebo en la semana 12. Estas mejoras se mantuvieron durante las 52 semanas (estudios 1 y 2).
El 40% de los participantes de los estudios 1 y 2 completaron el diario de síntomas de psoriasis (Psoriasis Symptom Diary®). De los participantes de cada uno de estos estudios que completaron el diario, mostraron mejoras estadísticamente significativas en los signos y síntomas de picor, dolor y descamación percibidos por los pacientes, a la semana 12 con respecto al inicio en comparación con el placebo.
Artritis _ psoriásica
Se evaluaron la seguridad y eficacia de Cosentyx en 1.003 pacientes en dos ensayos de fase III controlados con placebo, doble ciegos, aleatorizados, en pacientes con artritis psoriásica activa (>3 articulaciones inflamadas y >3 articulaciones dolorosas) a pesar del tratamiento con medicamentos antiinflamatorios no esteroideos (NSAID), corticosteroides o fármacos antirreumáticos modificadores de la enfermedad (FAME). En estos ensayos se reclutaron pacientes con cada subtipo de PsA, incluidas la artritis poliarticular sin evidencia de nódulos reumatoides, espondilitis con artritis periférica, artritis periférica asimétrica, participación interfalángica distal y artritis mutilante. Los pacientes que participaron en estos ensayos tenían un diagnóstico de PsA durante una media de 3,9 a
5,3 años. La mayoría de los pacientes también presentaban lesiones cutáneas de psoriasis activa o un historial documentado de psoriasis. Más del 62% y del 47% de los pacientes con PsA presentaban entesitis y dactilitis basales, respectivamente. Para ambos ensayos, la variable principal fue la respuesta 20 de la American College of Rheumatology (ACR) a la semana 24.
En el ensayo 1 en artritis psoriásica (Ensayo 1 en PsA) y en el ensayo 2 en artritis psoriásica (Ensayo 2 en PsA), el 29% y el 35% de los pacientes, respectivamente, fueron previamente tratados con un medicamento anti-TNFa e interrumpieron dicho tratamiento por falta de eficacia o intolerancia (pacientes con respuesta inadecuada a anti-TNFa).
En el Ensayo 1 en PsA (FUTURE 1) se evaluaron 606 pacientes, de los cuales el 60,7% recibían MTX de forma concomitante. Los pacientes aleatorizados a Cosentyx recibieron 10 mg/kg vía intravenosa a las semanas 0, 2, y 4, seguidas por 75 mg o 150 mg vía subcutánea mensualmente empezando en la semana 8. Los pacientes aleatorizados a placebo que eran no respondedores a la semana 16 (rescate temprano) y el resto de pacientes de placebo a la semana 24 se cruzaron para recibir Cosentyx (75 mg o 150 mg vía subcutánea) seguido por la misma dosis mensualmente.
En el Ensayo 2 en PsA (FUTURE 2) se evaluaron 397 pacientes, de los cuales el 46,6% recibían MTX de forma concomitante. Los pacientes aleatorizados a Cosentyx recibieron 75 mg, 150 mg o 300 mg vía subcutánea a las semanas 0, 1, 2, y 3, seguidas por la misma dosis mensualmente empezando en la semana 4. Los pacientes aleatorizados a recibir placebo que no eran respondedores a la semana 16 (rescate temprano) se cruzaron para recibir Cosentyx (150 mg o 300 mg vía subcutánea) a la semana 16 seguido por la misma dosis mensualmente. Los pacientes aleatorizados a recibir placebo que eran respondedores a la semana 16 se cruzaron para recibir Cosentyx (150 mg o 300 mg vía subcutánea) a la semana 24 seguido por la misma dosis mensualmente.
Signos y síntomas
El tratamiento con Cosentyx dio como resultado una mejora significativa en cuanto a las medidas de la actividad de la enfermedad en comparación con placebo a la semana 24 (ver Tabla 5).
Tabla 5 Respuesta clínica en el Ensayo 2 en PsA a la semana 24
|
Semana 24 | ||||
|
Placebo |
75 mg |
150 mg |
300 mg | |
|
Número de pacientes aleatorizados |
98 |
99 |
100 |
100 |
|
n (%) de respuesta ACR20 |
15 |
29 |
51 |
54 |
|
(15,3%) |
(29,3%*) |
(51,0%***) |
(54,0%***) | |
|
n (%) de respuesta ACR50 |
7 |
18 |
35 |
35 |
|
(7,1%) |
(18,2%) |
(35,0%) |
(35,0%**) | |
|
n (%) de respuesta ACR70 |
1 |
6 |
21 |
20 |
|
(1,0%) |
(6,1%) |
(21,0%**) |
(20,0%**) | |
|
DAS28-CRP |
-0,96 |
-1,12 |
-1,58** |
-1,61** |
|
Número de pacientes con >3% |
43 |
50 |
58 |
41 |
|
BSA afectada de psoriasis al inicio |
(43,9%) |
(50,5%) |
(58,0%) |
(41,0%) |
|
del estudio | ||||
|
n (%) de respuesta PASI 75 |
7 |
14 |
28 |
26 |
|
(16,3%) |
(28,0%) |
(48,3%**) |
(63,4%***) | |
|
n (%) de respuesta PASI 90 |
4 |
6 |
19 |
20 |
|
(9,3%) |
(12,0%) |
(32,8%**) |
(48,8%***) | |
|
n (%) de resolución de Dactilitis f |
4 |
10 |
16 |
26 |
|
(14,8%) |
(30,3%) |
(50,0%**) |
(56,5%**) | |
|
n (%) de resolución de Entesitis $ |
14 |
22 |
27 |
27 |
|
(21,5%) |
(32,4%) |
(42,2%*) |
(48,2%**) | |
* p<0,05, ** p<0,01, *** p<0,001; frente a placebo
Todos los valores-p se ajustan en función de la multiplicidad de ensayos de acuerdo a la jerarquía predefinida, excepto para ACR70, Dactilitis y Entesitis, que eran variables exploratorias.
Imputación de no respondedor utilizada para la variable binaria que falta.
ACR: American College of Rheumatology; PASI: Índice de Severidad y Área de Psoriasis; DAS: Puntuación de Actividad de la Enfermedad; BSA: Área de Superficie Corporal fEn pacientes con dactilitis al inicio del estudio (n=27, 33, 32, 46, respectivamente)
{En pacientes con entesitis al inicio del estudio (n=65, 68, 64, 56, respectivamente)_
El inicio de acción de Cosentyx se produjo tan pronto como en la semana 2. En la semana 3 se alcanzó una diferencia estadísticamente significativa en el ACR 20 frente a placebo. En la semana 16, los pacientes tratados con Cosentyx demostraron mejoras significativas en los signos y síntomas, entre los cuales se encuentran, respuestas significativamente mayores en ACR 20 (33,3%, 60,0% y 57,0% para 75 mg, 150 mg y 300 mg, respectivamente) comparado con placebo (18,4%).
En la Figura 2 se muestra el porcentaje de pacientes por visita que alcanzaron la respuesta ACR 20. Figure 2 Respuesta ACR20 en el Ensayo 2 de PsA a lo largo del tiempo hasta la semana 24
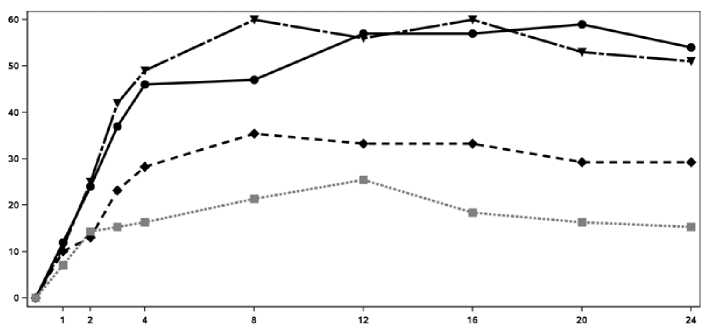
Tiempo (Semanas)
■ Cosentyx 150 mg Cosentyx 300 mg ■ ■ ■ • Placebo
Porcentaje de respondedores
Se observaron respuestas similares para las variables primaria y secundaria principal en pacientes con PsA a pesar de que estuviesen o no con tratamiento concomitante con MTX. En la semana 24, los pacientes tratados con Cosentyx y tratamiento concomitante con MTX solían tener una respuesta ACR 20 (44,7%, 47,7% y 54,4% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 20,0%) y ACR 50 (27,7%, 31,8% y 38,6% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 8,0%) más elevada. Los pacientes tratados con Cosentyx sin el uso concomitante de MTX solían tener una respuesta ACR 20 (15,4%, 53,6% y 53,6% para 75 mg,
150 mg y 300 mg, respectivamente, comparado con placebo 10,4%) y ACR 50 (9,6%, 37,5% y 32,1% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 6,3%) más elevada.
Los pacientes tratados con Cosentyx que eran “naíve” para anti-TNFa o con RI a anti-TNFa, presentaron una respuesta ACR 20 significativamente mayor comparado con placebo a la semana 24, con una respuesta ligeramente superior en el grupo “naíve” para anti-TNFa (“naíve” para anti-TNFa: 37%, 64% y 58% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 15,9%; RI a anti-TNFa: 15%, 30% y 46% para 75 mg, 150 mg y 300 mg, respectivamente, comparado con placebo 14,3%). En el subgrupo de pacientes con RI a anti-TNFa, solo la dosis de 300 mg mostró una tasa de respuesta significativamente mayor para ACR 20 comparado con placebo (p<0,05) y demostró un beneficio clínico significativo sobre 150 mg en las variables secundarias múltiples. Se observaron mejorías en la respuesta PASI 75 en ambos subgrupos y la dosis de 300 mg mostró un beneficio estadísticamente significativo en los pacientes con RI a anti-TNFa.
El número de pacientes con PsA con afectación axial era demasiado pequeño como para permitir una evaluación significativa.
Se mostraron mejorías en todos los componentes de las puntuaciones ACR, incluyendo la evaluación del dolor del paciente. La proporción de pacientes que alcanzaron una respuesta Criterio de Respuesta a PsA (PsARC) modificada fue superior en los pacientes tratados con Cosentyx (38,4%, 62,0% y 63,0% para 75 mg, 150 mg y 300 mg, respectivamente) comparado con placebo (29,6%) a la semana 24.
En el Ensayo 1 en PsA y en el Ensayo 2 en PsA, la eficacia se mantuvo hasta la semana 52. En el Ensayo 2 en PsA, entre los 200 pacientes inicialmente aleatorizados a Cosentyx 150 mg y 300 mg, 178 (89%) pacientes aún estaban en tratamiento en la semana 52. De los 100 pacientes aleatorizados a Cosentyx 150 mg, 64, 39 y 20 presentaron una respuesta ACR 20/50/70, respectivamente. De los 100 pacientes aleatorizados a Cosentyx 300 mg, 64, 44 y 24 presentaron una respuesta ACR 20/50/70, respectivamente.
Respuesta radiográfica
No se ha demostrado la inhibición de la progresión del daño estructural en PsA, utilizando el régimen de carga vía subcutánea aprobado para el uso clínico.
En el Ensayo 1 en PsA, se evaluó radiográficamente la inhibición de la progresión del daño estructural y se expresó como el cambio en la puntuación total de Sharp modificada (mTSS) y sus componentes, en la puntuación de la erosión (ES) y en la puntuación del estrechamiento del espacio articular (JSN) a la semana 24 y 52, comparado con el periodo basal. En la Tabla 6 se presentan los datos a la semana 24.
Tabla 6 Cambio en la puntuación total de Sharp modificada en artritis psoriásica
|
Placebo N=179 |
Cosentyx 75 mg1 N=181 |
Cosentyx 150 mg1 N=185 | |
|
Puntuación total | |||
|
Periodo basal |
28,4 |
20,4 |
22,3 |
|
(DE) |
(63,5) |
(39,4) |
(48,0) |
|
Cambio medio a la semana 24 |
0,57 |
0,02* |
0,13* |
|
*p<0,05 de acuerdo al valor-p nominal pero no ajustado '10 mg/kg a las semanas 0, 2 y 4 seguido por dosis subcutáneas de 75 mg o |
150 mg | ||
La inhibición del daño estructural se mantuvo con el tratamiento con Cosentyx hasta la semana 52.
El porcentaje de pacientes sin progresión de la enfermedad (definido como un cambio desde el periodo basal en mTSS de <0,5) desde la aleatorización hasta la semana 24 fue del 92,3% en secukinumab 10 mg/kg como carga vía intravenosa - 75 mg como mantenimiento vía subcutánea, 82,3% en secukinumab 10 mg/kg como carga vía intravenosa - 150 mg como mantenimiento vía subcutánea y 75,7% en placebo. El porcentaje de pacientes sin progresión de la enfermedad desde la semana 24 hasta la semana 52 para secukinumab 10 mg/kg como carga vía intravenosa - seguido por 75 mg o 150 mg como mantenimiento vía subcutánea y para los pacientes con placebo que cambiaron a 75 mg o 150 mg vía subcutánea cada 4 semanas a la semana 16 o semana 24, fue del 85,8%, 85,7% y 86,8%, respectivamente.
Función física y calidad de vida relacionada con la salud
En el Ensayo 2 en PsA, los pacientes tratados con Cosentyx 150 mg (p=0,0555) y 300 mg (p=0,0040) mostraron mejoría en la función física comparado con los pacientes tratados con placebo evaluado por el índice de discapacidad del cuestionario de evaluación de la salud (HAQ-DI) a la semana 24. Se observaron mejorías en las puntuaciones HAQ-DI independientemente de la exposición previa a anti-TNFa. Se observaron respuestas similares en el Ensayo 1 en PsA.
Los pacientes tratados con Cosentyx notificaron mejorías significativas en la calidad de vida relacionada con la salud medido según la puntuación del resumen del componente físico del cuestionario de salud Short Form-36 (SF-36 PCS) (p<0,001). Asimismo, se observaron mejorías estadísticamente significativas demostradas en las variables exploratorias valoradas según la evaluación funcional para el tratamiento de enfermedades crónicas - Fatiga (FACIT-F) para 150 mg y 300 mg comparado con placebo (7,97, 5,97 comparado con 1,63, respectivamente). Se observaron respuestas similares en el ensayo 1 en PsA y la eficacia se mantuvo hasta la semana 52.
Espondilitis anquilosante
Se evaluó la seguridad y eficacia de Cosentyx en 590 pacientes en dos ensayos fase III controlados con placebo, doble ciego, aleatorizados, en pacientes con espondilitis anquilosante (AS) activa con un índice de actividad de la enfermedad espondilitis anquilosante de Bath (BASDAI) >4 a pesar del tratamiento con medicamentos antiinflamatorios no esteroideos (NSAID), corticosteroides o fármacos antirreumáticos modificadores de la enfermedad (FAME). Los pacientes que participaron en estos ensayos tenían un diagnóstico de AS durante una media de 2,7 a 5,8 años. Para ambos ensayos, la variable principal fue como mínimo una mejoría del 20% según los criterios de la Assessment of Spondyloarthritis International Society (ASAS 20) a la semana 16.
En el Ensayo 1 en espondilitis anquilosante (Ensayo 1 en AS) y en el Ensayo 2 en espondilitis anquilosante (Ensayo 2 en AS) el 27,0% y el 38,8% de los pacientes, respectivamente, se trataron previamente con un medicamento anti-TNFa y discontinuaron el tratamiento por falta de eficacia o intolerancia (pacientes con respuesta inadecuada a anti-TNFa).
En el Ensayo 1 en AS (MEASURE 1) se evaluaron 371 pacientes, de los cuales el 14,8% y el 33,4% utilizaron MTX o sulfasalacina de forma concomitante, respectivamente. Los pacientes aleatorizados a Cosentyx recibieron 10 mg/kg vía intravenosa a las semanas 0, 2, y 4, seguido por 75 mg o 150 mg vía subcutánea mensualmente empezando en la semana 8. Los pacientes aleatorizados a placebo que eran no respondedores a la semana 16 (rescate temprano) y todo el resto de pacientes de placebo a la semana 24 se cruzaron para recibir Cosentyx (75 mg o 150 mg vía subcutánea) seguido por la misma dosis mensualmente.
En el Ensayo 2 en AS (MEASURE 2) se evaluaron 219 pacientes, de los cuales el 11,9% y el 14,2% utilizaron MTX o sulfasalacina de forma concomitante, respectivamente. Los pacientes aleatorizados a Cosentyx recibieron 75 mg o 150 mg vía subcutánea a las semanas 0, 1, 2, y 3, seguida por la misma dosis mensualmente empezando en la semana 4. En la semana 16, los pacientes que fueron aleatorizados a recibir placebo al inicio del tratamiento, se volvieron a aleatorizar a recibir Cosentyx (75 mg o 150 mg vía subcutánea) mensualmente.
Signos y síntomas
En el Ensayo 2 en AS, el tratamiento con Cosentyx 150 mg dio como resultado una mejora superior en cuanto a las medidas de la actividad de la enfermedad en comparación con placebo a la semana 16 (ver Tabla 7).
Tabla 7 Respuesta clínica en el Ensayo 2 en AS a la semana 16
|
Resultado (valor-p frente a placebo) |
Placebo (n = 74) |
75 mg (n = 73) |
150 mg (n = 72) |
|
% de respuesta ASAS 20 |
28,4 |
41,1 |
61,1*** |
|
% de respuesta ASAS 40 |
10,8 |
26,0 |
36,1*** |
|
hsCRP, (tasa post-BSL/BSL) |
1,13 |
0,61 |
0,55*** |
|
% de ASAS 5/6 |
8,1 |
34,2 |
43 1*** |
|
% de remisión parcial de ASAS |
4,1 |
15,1 |
13,9 |
|
% de BASDAI 50 |
10,8 |
24,7* |
30,6** |
|
Mejora importante de ASDAS-CRP |
4,1 |
15,1* |
25,0*** |
|
* p<0,05, ** p<0,01, *** p<0,001; frente a placebo Todos los valores-p se ajustan en función de la multiplicidad de ensayos de acuerdo a la jerarquía predefinida, excepto para BASDAI 50 y ASDAS-CRP Imputación de no respondedor utilizada para la variable binaria que falta. | |||
|
ASAS: criterio de la Sociedad Internacional de evaluación de la espondiloartritis; BASDAI: índice de actividad de la enfermedad espondilitis anquilosante de Bath; hsCRP: proteína-C reactiva de alta sensibilidad; ASDAS: puntuación de la actividad de la enfermedad espondilitis anquilosante; BSL: periodo basal | |||
El inicio de acción de Cosentyx 150 mg se produjo tan pronto como en la semana 1 para ASAS 20 y semana 2 para ASAS 40 (superior a placebo) en el Ensayo 2 en AS.
Las respuestas ASAS 20 mejoraron en la semana 16 en los pacientes naíve para anti-TNFa (68,2% frente a 31,1%; p<0,05) y en los pacientes con RI para anti-TNFa (50,0% frente a 24,1%; p<0,05) para Cosentyx 150 mg comparado con placebo, respectivamente.
En ambos ensayos en AS, los pacientes tratados con Cosentyx (150 mg en el Ensayo 2 en AS y ambos regímenes en el Ensayo 1 en AS) demostraron signos y síntomas de mejoría significativa a la semana 16, con una magnitud de respuesta y eficacia comparable, mantenida hasta la semana 52 en pacientes naíve para anti-TNFa y en pacientes con RI para anti-TNFa-IR. En el Ensayo 2 en AS, de los 72 pacientes aleatorizados inicialmente a Cosentyx 150 mg, 61 (84,7%) pacientes estaban aún en tratamiento a la semana 52. De los 72 pacientes aleatorizados a Cosentyx 150 mg, 45 y 35 presentaron una respuesta ASAS 20/40, respectivamente.
Movilidad espinal
Los pacientes tratados con Cosentyx 150 mg mostraron mejorías en la movilidad espinal medido por el cambio desde el periodo basal según BASMI hasta la semana 16 para ambos ensayos, Ensayo 1 en AS (-0,40 vs -0,12 para placebo; p=0,0114) y Ensayo 2 en AS (-0,51 vs -0,22 para placebo; p=0,0533). Estas mejorías se mantuvieron hasta la semana 52.
Función física y calidad de vida relacionada con la salud
En el Ensayo 1 y Ensayo 2 en AS, los pacientes tratados con Cosentyx 150 mg mostraron mejoría en la calidad de vida relacionada con la salud medida por el cuestionario de calidad de vida en AS (ASQoL) (p=0,001) y en el resumen del componente físico del SF-36 (SF-36 PCS) (p<0,001). Los pacientes tratados con Cosentyx 150 mg también mostraron mejorías estadísticamente significativas en las variables exploratorias en la función física evaluado por el índice funcional de la espondilitis
anquilosante de Bath (BASFI) comparado con placebo (-2,15 vs -0,68) y en la fatiga evaluado según la escala para la evaluación funcional para el tratamiento de enfermedades crónicas - Fatiga (FACIT-Fatiga) comparado con placebo (8,10 vs 3,30). Estas mejorías se mantuvieron hasta la semana 52.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Cosentyx en pacientes pediátricos, desde recién nacidos a menores de 6 años, con psoriasis en placas y desde recién nacidos a menores de 2 años, con artritis idiopática crónica (ver sección 4.2. para consultar la información sobre el uso en la población pediátrica).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Cosentyx en pacientes pediátricos, de 6 años a menores de 18 años, con psoriasis en placas y de 2 años a menores de 18 años, con artritis idiopática crónica (ver sección 4.2. para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Psoriasis en placas
Absorción
Tras una sola dosis subcutánea de 300 mg como formulación líquida en voluntarios sanos, secukinumab alcanzó las concentraciones máximas séricas de 43,2 ±10,4 pg/ml entre 2 y 14 días de la dosis.
Basado en el análisis farmacocinético de la población, con una sola administración subcutánea de 150 ó 300 mg de secukinumab a pacientes con psoriasis en placas se logra una concentración sérica máxima de secukinumab de 13,7 ± 4,8 pg/ml ó 27,3 ± 9,5 pg/ml, respectivamente, 5 ó 6 días después de la administración.
El tiempo transcurrido hasta alcanzar la concentración máxima después del primer mes de tratamiento con dosis semanales fue de 31 a 34 días, basado en el análisis farmacocinético de la población.
Sobre la base de datos simulados, las concentraciones máximas del estado estacionario (Cmáx,ss), tras la administración subcutánea de 150 ó 300 mg, son de 27,6 pg/ml o 55,2 pg/ml, respectivamente. El análisis farmacocinético de la población sugiere que el estado estacionario se alcanza después de 20 semanas con regímenes de administración mensuales.
El análisis farmacocinético de la población mostró que durante la fase de mantenimiento de administración mensual repetida, los pacientes presentan concentraciones séricas máximas y un área bajo la curva (AUC) dos veces mayor que las obtenidas con una sola administración.
El análisis farmacocinético de la población mostró que secukinumab se absorbió con una biodisponibilidad media absoluta del 73% en pacientes con psoriasis en placas. En todos los estudios, la biodisponibilidad absoluta se calculó que se encontraba entre el 60 y el 77%.
Distribución
El volumen medio de distribución durante la fase terminal (Vz) tras una sola administración intravenosa varía entre 7,10 y 8,60 litros en los pacientes con psoriasis en placas, lo que sugiere que la distribución del secukinumab hacia los compartimientos periféricos es limitada.
Biotransformación
La mayor parte de la eliminación de IgG ocurre mediante el catabolismo intracelular, tras endocitosis de la fase líquida o mediada por receptor.
El aclaramiento medio sistémico (CL) tras la administración única intravenosa en pacientes con psoriasis en placas fue de 0,13 a 0,36 l/día. En el análisis farmacocinético de la población, el aclaramiento medio sistémico (CL) fue de 0,19 l/día en los pacientes con psoriasis en placas. El CL no se ve afectado por el sexo. El aclaramiento no es dosis ni tiempo dependiente.
El análisis farmacocinético de la población, la vida de eliminación media se estimó en 27 días en los pacientes con psoriasis en placas, con un rango de 18 a 46 días en todos los estudios de psoriasis con administración intravenosa.
Linealidad/No linealidad
La farmacocinética tras dosis únicas y repetidas del secukinumab en pacientes con psoriasis en placas se determinó en varios estudios en los que se usaron tanto dosis intravenosas de entre 1 x 0,3 mg/kg a 3 x 10 mg/kg, como dosis subcutáneas de entre 1 x 25 mg a múltiples dosis de 300 mg. En todos los casos, la exposición resultó proporcional a la dosis.
Artritis psoriásica
Las propiedades farmacocinéticas de secukinumab observadas en pacientes con artritis psoriásica fueron similares a las que se muestran en pacientes con psoriasis en placas. La biodisponibilidad de secukinumab en pacientes con PsA fue del 85% de acuerdo al modelo farmacocinético poblacional.
Espondilitis anquilosante
Las propiedades farmacocinéticas de secukinumab observadas en pacientes con espondilitis anquilosante fueron similares a las que se muestran en pacientes con psoriasis en placas.
Poblaciones especiales
Pacientes de edad avanzada
De los 3.430 pacientes con psoriasis en placas que recibieron Cosentyx en los ensayos clínicos,
230 pacientes tenían más de 65 años de edad y 32, eran mayores de 75.
De los 974 pacientes con artritis psoriásica que recibieron Cosentyx en los ensayos clínicos, un total de 85 pacientes tenían a partir de 65 años de edad y 4 pacientes tenían a partir de 75 años.
De los 571 pacientes con espondilitis anquilosante que recibieron Cosentyx en los ensayos clínicos, un total de 24 pacientes tenían a partir de 65 años de edad y 3 pacientes tenían a partir de 75 años.
Según el análisis farmacocinético de la población con un número limitado de pacientes de edad avanzada (n=71 de más de 65 años y n=7 de más de 75 años), el aclaramiento fue similar en los pacientes de edad avanzada que en los menores de 65 años.
Pacientes con insuficiencia renal o hepática
No se disponen de datos farmacocinéticos en pacientes con insuficiencia renal o hepática. Se estima que la cantidad de Cosentyx sin metabolizar, un anticuerpo monoclonal de IgG, que se elimina por vía renal, es baja y de menor importancia. Las IgGs se eliminan principalmente por catabolismo, por lo que la insuficiencia hepática no se espera que afecte al aclaramiento de Cosentyx.
Efecto del peso en la farmacocinética
El aclaramiento de secukinumab y el volumen de distribución incrementan con el aumento del peso corporal.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios de reactividad cruzada en tejidos, farmacología de seguridad, estudios de toxicidad de la función reproductora y de dosis repetidas realizados con secukinumab o con un anticuerpo murino anti- IL-17A murina equivalente.
Como secukinumab se une a la IL-17A del ser humano y del mono cinomolgus, la seguridad se estudió en el mono cinomolgus. No se han observado efectos indeseados con secukinumab tras su administración a mono cinomolgus tanto por vía subcutánea, durante un período de hasta 13 semanas, como por vía intravenosa, durante un período de hasta 26 semanas (incluyendo evaluaciones farmacocinéticas, farmacodinamias, de inmunogenicidad y de inmunotoxicidad, p.ej., respuesta inmunitaria dependiente de linfocitos T y actividad de linfocitos citolíticos naturales). La concentración sérica media determinada en monos después de la administración de 13 dosis subcutáneas de 150 mg/kg una vez por semana es mayor que la concentración sérica media prevista para los pacientes con psoriasis que vayan a recibir la dosis clínica mayor. Se detectaron anticuerpos anti-secukinumáb en solo uno de los animales expuestos. No se observaron signos de reactividad cruzada tisular inespecífica tras la aplicación del secukinumab sobre tejidos humanos normales.
No se han realizado estudios en animales para evaluar el potencial carcinogénico de secukinumab.
Un estudio de desarrollo embriofetal efectuado en mono cinomolgus, no mostró toxicidad materna, embriotoxicidad ni teratogenicidad cuando secukinumab se administró durante la organogénesis o hacia el final de la gestación.
No se han observado efectos indeseados con un anticuerpo murino anti-IL-17A murina equivalente en los estudios de fecundidad y desarrollo embrionario temprano o de desarrollo pre- y postnatal del ratón. La dosis más alta que se usó en tales estudios era superior a la dosis máxima eficaz en cuanto a inhibición y actividad de la IL-17A (ver sección 4.6).
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Trehalosa dihidrato L-histidina
Hidrocloruro de L-histidina monohidratado
L-metionina
Polisorbato 80
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
18 meses
6.4 Precauciones especiales de conservación
Conservar en nevera (2°C - 8°C). No congelar.
Conservar las plumas en el embalaje exterior para protegerlo de la luz.
6.5 Naturaleza y contenido del envase
Cosentyx se comercializa en una jeringa precargada de un único uso montado en una pluma de forma triangular con visor transparente y etiqueta (pluma SensoReady). La jeringa precargada dentro de la pluma es una jeringa de 1 ml de cristal con un émbolo recubierto de Fluor Tec, con una aguja de 27G x A" protegida en un tapón de caucho de estireno butadieno.
Cosentyx está disponible en envases unitarios que contienen 1 ó 2 plumas precargadas y en envases múltiples que contienen 6 (3 envases de 2) plumas precargadas.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Cosentyx 150 mg solución inyectable se comercializa en una pluma precargada de único uso para uso individual. No debe agitar ni congelar la pluma. La pluma se debe sacar de la nevera 20 minutos antes de la administración para que se atempere.
Antes de utilizar la pluma precargada se recomienda hacer una revisión visual. El líquido debe ser transparente. Su color puede variar de incoloro a ligeramente amarillento. Puede ver alguna burbuja de aire pequeña, que es normal. No utilizar si el líquido contiene partículas, está turbio o tiene un color claramente marrón. En el prospecto se incluye información detallada sobre las instrucciones de uso.
La eliminación del medicamento no utilizado y de los materiales sobrantes que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/980/004
EU/1/14/980/005
EU/1/14/980/007
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
15.01.2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSIBLE DE LA LIBERACIÓN DE LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSIBLE DE LA LIBERACIÓN DE LOTES
Nombre y dirección del fabricante del principio active biológico
Novartis Pharma S.A.S.
Centre de Biotechnologie 8, rae de l’Industrie F-68330 Huningue Francia
Nombre y fabricante del fabricante responsable de la liberación de lotes
Novartis Pharma GmbH RoonstraBe 25 D-90429 Nuremberg Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará el primer informe periódico de seguridad para este medicamento en un plazo de 6 meses después de la autorización. Posteriormente, el titular de la autorización de comercialización presentará informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, (párrafo 7), de la Directiva 2001/83/CE y publicados en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo
acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de
Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden
presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Cosentyx 150 mg polvo para solución inyectable secukinumab
2. PRINCIPIO(S) ACTIVO(S)
Un vial contiene 150 mg de secukinumab. Tras la reconstitución, 1 ml de solución contiene 150 mg de secukinumab.
3. LISTA DE EXCIPIENTES
También contiene: Sacarosa, L-histidina, hidrocloruro de L-histidina monohidratado, polisorbato 80.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para solución inyectable 1 vial
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/980/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Cosentyx 150 mg
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETA DEL VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Cosentyx 150 mg polvo para solución inyectable
secukinumab
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
1. NOMBRE DEL MEDICAMENTO
Cosentyx 150 mg solución inyectable en jeringa precargada secukinumab
2. PRINCIPIO(S) ACTIVO(S)
Una jeringa precargada contiene 150 mg de secukinumab en 1 ml de solución.
3. LISTA DE EXCIPIENTES
También contiene: Trehalosa dihidrato, L-histidina, monohidrocloruro de L-histidina monohidrato, L-metionina, polisorbato 80, agua para preparaciones inyectables.
4. PHARMACEUTICAL FORM AND CONTENTS
Solución inyectable
1 jeringa precargada
2 jeringas precargadas
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea
Leer el prospecto antes de utilizar este medicamento. Para un único uso.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
Conservar en nevera. No congelar.
Conservar la jeringa precargada en el embalaje exterior para protegerla de la luz. Conservar las jeringas precargadas en el embalaje exterior para protegerlas de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/980/002 Envase conteniendo 1 jeringa precargada
EU/1/14/980/003 Envase conteniendo 2 jeringas precargadas
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Cosentyx 150 mg
CAJA DEL ENVASE MÚLTIPLE (INCLUYENDO BLUE BOX) - jeringa precargada
1. NOMBRE DEL MEDICAMENTO
Cosentyx 150 mg solución inyectable en jeringa precargada secukinumab
2. PRINCIPIO(S) ACTIVO(S)
Una jeringa precargada contiene 150 mg de secukinumab en 1 ml de solución.
3. LISTA DE EXCIPIENTES
También contiene: Trehalosa dihidrato, L-histidina, monohidrocloruro de L-histidina monohidrato, L-metionina, polisorbato 80, agua para preparaciones inyectables.
4. PHARMACEUTICAL FORM AND CONTENTS
Solución inyectable
Envase múltiple: 6 (3 envases de 2 jeringas precargadas)
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea
Leer el prospecto antes de utilizar este medicamento. Para un único uso.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera. No congelar.
Conservar las jeringas precargadas en el embalaje exterior para protegerlas de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/980/006 Envase multiple conteniendo 6 (3x2) jeringas precargadas
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Cosentyx 150 mg
CARTONAJE DEL ENVASE INTERMEDIO DEL ENVASE MÚLTIPLE (SIN BLUE BOX) -jeringa precargada
1. NOMBRE DEL MEDICAMENTO
Cosentyx 150 mg solución inyectable en jeringa precargada secukinumab
2. PRINCIPIO(S) ACTIVO(S)
Una jeringa precargada contiene 150 mg de secukinumab en 1 ml de solución.
3. LISTA DE EXCIPIENTES
También contiene: Trehalosa dihidrato, L-histidina, monohidrocloruro de L-histidina monohidrato, L-metionina, polisorbato 80, agua para preparaciones inyectables.
4. PHARMACEUTICAL FORM AND CONTENTS
Solución inyectable
2 jeringas precargadas. Componente de un envase múltiple. No vender individualmente.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea
Leer el prospecto antes de utilizar este medicamento. Para un único uso.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
Conservar en nevera. No congelar.
Conservar las jeringas precargadas en el embalaje exterior para protegerlas de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/980/006 Envase multiple conteniendo 6 (3 x2) jeringas precargadas
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Cosentyx 150 mg
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLÍSTER DE LA JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO
Cosentyx 150 mg solución inyectable en jeringa precargada secukinumab
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETA DE LA JERINGA
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Cosentyx 150 mg inyectable
secukinumab
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
1. NOMBRE DEL MEDICAMENTO
Cosentyx 150 mg solución inyectable en pluma precargada secukinumab
2. PRINCIPIO(S) ACTIVO(S)
Una pluma precargada contiene 150 mg de secukinumab en 1 ml de solución.
3. LISTA DE EXCIPIENTES
También contiene: Trehalosa dihidrato, L-histidina, monohidrocloruro de L-histidina monohidrato, L-metionina, polisorbato 80, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable.
1 pluma SensoReady precargada
2 plumas SensoReady precargadas
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea
Leer el prospecto antes de utilizar este medicamento. Para un único uso.
6. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
Conservar en nevera. No congelar.
Conservar la pluma SensoReady precargada en el embalaje exterior para protegerla de la luz. Conservar las plumas SensoReady precargadas en el embalaje exterior para protegerlas de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/980/004 El envase contiene 1 pluma SensoReady precargada
EU/1/14/980/005 El envase contiene 2 plumas SensoReady precargadas
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Cosentyx 150 mg
1. NOMBRE DEL MEDICAMENTO
Cosentyx 150 mg solución inyectable en pluma precargada secukinumab
2. PRINCIPIO(S) ACTIVO(S)
Una pluma precargada contiene 150 mg de secukinumab en 1 ml de solución.
3. LISTA DE EXCIPIENTES
También contiene: Trehalosa dihidrato, L-histidina, monohidrocloruro de L-histidina monohidrato, L-metionina, polisorbato 80, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable.
Envase múltiple: 6 (3 envases de 2 plumas SensoReady precargadas)
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea
Leer el prospecto antes de utilizar este medicamento. Para un único uso.
6. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
Conservar en nevera. No congelar.
Conservar las plumas SensoReady precargadas en el embalaje exterior para protegerlas de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/980/007 El envase múltiple contiene 6 (3 x2) plumas SensoReady precargadas
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Cosentyx 150 mg
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
CARTONAJE DEL ENVASE INTERMEDIO DEL ENVASE MÚLTIPLE (SIN BLUE BOX) -pluma precargada
1. NOMBRE DEL MEDICAMENTO
Cosentyx 150 mg solución inyectable en pluma precargada secukinumab
2. PRINCIPIO(S) ACTIVO(S)
Una pluma precargada contiene 150 mg de secukinumab en 1 ml de solución.
3. LISTA DE EXCIPIENTES
También contiene: Trehalosa dihidrato, L-histidina, monohidrocloruro de L-histidina monohidrato, L-metionina, polisorbato 80, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable.
2 plumas SensoReady precargadas. Componente de un envase múltiple. No vender individualmente.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea
Leer el prospecto antes de utilizar este medicamento. Para un único uso.
6. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
Conservar en nevera. No congelar.
Conservar las plumas SensoReady precargadas en el embalaje exterior para protegerlas de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/980/007 El envase multiple contiene 6 (3 x2) plumas SensoReady precargadas
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Cosentyx 150 mg
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETA DE LA PLUMA
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Cosentyx 150 mg solución inyectable en pluma precargada
secukinumab
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
Pluma SensoReady
B. PROSPECTO
Prospecto: información para el paciente Cosentyx 150 mg polvo para solución inyectable
Secukinumab
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Cosentyx y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Cosentyx
3. Cómo usar Cosentyx
4. Posibles efectos adversos
5. Conservación de Cosentyx
6. Contenido del envase e información adicional
1. Qué es Cosentyx y para qué se utiliza
Cosentyx contiene el principio activo secukinumab. Secukinumab es un anticuerpo monoclonal. Los anticuerpos monoclonales son unas proteínas que reconocen y se unen específicamente a ciertas proteínas del cuerpo.
Cosentyx pertenece a un grupo de medicamentos conocidos como “inhibidores de interieuquinas”. Actúa neutralizando la actividad de una proteína denominada IL-17A, que está presente en cantidades elevadas en enfermedades como la psoriasis, la artritis psoriásica y la espondilitis anquilosante.
Cosentyx se utiliza para el tratamiento de las siguientes enfermedades inflamatorias:
• Psoriasis en placas
• Artritis psoriásica
• Espondilitis anquilosante
Psoriasis en placas
Cosentyx se usa para el tratamiento de un trastorno de la piel conocido como “psoriasis en placas” que provoca inflamación en la piel. Cosentyx reduce la inflamación y otros síntomas de la enfermedad. Cosentyx se usa en personas adultas que padecen psoriaris en placas de naturaleza moderada a grave.
Utilizar Cosentyx para psoriasis en placas le beneficiará ya que produce mejorías en el aspecto de la piel y la disminución de síntomas tales como la descamación, el prurito y el dolor.
Artritis psoriásica
Cosentyx se usa para el tratamiento de un trastorno conocido como “artritis psoriásica”. Se trata de una enfermedad inflamatoria de las articulaciones, a menudo acompañada de psoriasis. Si padece artritis psoriásica activa, recibirá primero otros medicamentos. Si no responde suficientemente bien a estos medicamentos, recibirá Cosentyx para reducir los signos y síntomas de la artritis psoriásica activa, mejorar la función física y ralentizar el daño en el cartílago y huesos de las articulaciones involucradas en la enfermedad.
Cosentyx se usa en adultos con artritis psoriásica activa y se puede utilizar solo o con otro medicamento denominado metotrexato.
Utilizar Cosentyx para artritis psoriásica le beneficiará ya que reduce los signos y síntomas de la enfermedad, ralentizando el daño en los cartílagos y huesos de las articulaciones y mejorando su habilidad para realizar las actividades diarias normales.
Espondilitis anquilosante
Cosentyx se usa para el tratamiento de un trastorno conocido como “espondilitis anquilosante”. Se trata de una enfermedad inflamatoria que afecta principalmente a la columna causando inflamación de las articulaciones de la columna. Si padece espondilitis anquilosante, recibirá primero otros medicamentos. Si no responde suficientemente bien a estos medicamentos, recibirá Cosentyx para reducir los signos y síntomas de la enfermedad, reducir la inflamación y mejorar su función física.
Cosentyx se usa en adultos con espondilitis anquilosante activa.
Utilizar Cosentyx para espondilitis anquilosante le beneficiará ya que reduce los signos y síntomas de la enfermedad y mejora la función física.
2. Qué necesita saber antes de empezar a usar Cosentyx
No use Cosentyx:
• si es alérgico al secukinumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Si usted sospecha que es alérgico, consulte con su médico antes de usar Cosentyx.
• si tiene alguna infección que su médico considere importante.
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de usar Cosentyx:
• si ha contraído una infección
• si padece infecciones repetidas o prolongadas.
• si tiene tuberculosis.
• si padece la enfermedad de Crohn.
• si ha sido vacunado recientemente o va a ser vacunado durante el tratamiento con Cosentyx.
• si sigue algún otro tratamiento para la psoriasis, como por ejemplo, si usa otros inmunosupresores o fototerapia con luz ultravioleta (UV).
Vigile la aparición de infecciones y reacciones alérgicas
Cosentyx puede ocasionar potencialmente efectos adversos graves, incluidas infecciones y reacciones alérgicas. Debe vigilar la aparición de signos de estas enfermedades mientras use Cosentyx.
Interrumpa el tratamiento con Cosentyx y avise a su médico o busque asistencia médica inmediatamente si nota alguno de los signos que indican una posible infección o reacción alérgica graves. Estos signos se incluyen en la sección 4 “Posibles efectos adversos”.
Niños y adolescentes
No se recomienda el uso de Cosentyx en niños y adolescentes (menores de 18 años) pues no se ha
estudiado el medicamento en personas de esta edad.
Uso de Cosentyx con otros medicamentos
Informe a su médico o farmacéutico:
• Si está tomando, ha tomado o podría tener que tomar cualquier otro medicamento.
• Si ha sido vacunado recientemente o va a ser vacunado próximamente. No le deben administrar ciertos tipos de vacunas (vacunas vivas) mientras use Cosentyx.
Embarazo, lactancia y fertilidad
• Es preferible que evite el uso de Cosentyx durante el embarazo. Se desconoce el efecto de este medicamento en mujeres embarazadas. Si es una mujer en edad fértil, se aconseja que evite quedarse embarazada y debe utilizar un anticonceptivo adecuado mientras use Cosentyx y durante al menos 20 semanas después de la última dosis de Cosentyx. Consulte a su médico si está embarazada, cree que podría estar embarazada o tiene la intención de quedarse embarazada.
• Consulte a su médico si están dando el pecho o tiene previsto dar el pecho. Usted y su médico deben decidir si va a dar el pecho o va a utilizar Cosentyx. No puede hacer las dos cosas. Después de utilizar Cosentyx no debe dar el pecho durante al menos 20 semanas después de la última dosis.
Conducción y uso de máquinas
La influencia de Cosentyx sobre la capacidad para conducir o utilizar máquinas es nula o
insignificante.
3. Cómo usar Cosentyx
Cosentyx se administra por medio de una inyección debajo de la piel (es decir, por vía subcutánea) con la ayuda de un profesional sanitario.
Asegúrese de acordar con su médico cuándo recibirá las inyecciones y cuándo tendrá las visitas de seguimiento.
En el apartado “Instrucciones de Uso de Cosentyx polvo para solución inyectable” que se encuentra al final de este prospecto, se dan instrucciones detalladas de cómo reconstituir e inyectar Cosentyx.
Cuánto Cosentyx debe administrarse y durante cuánto tiempo
Su medico decidirá cuánto Consentyx usted necesita y durante cuánto tiempo.
Psoriasis en placas
• La dosis recomendada es 300 mg por inyección subcutánea.
• Una dosis de 300 mg se administra mediante dos inyecciones de 150 mg.
Después de la primera dosis, usted recibirá inyecciones semanales en las semanas 1, 2 y 3. A partir de la semana 4, las inyecciones serán mensuales. En cada cita, usted recibirá una dosis de 300 mg repartida en dos inyecciones de 150 mg.
Artritis psoriásica
Para pacientes con artritis psoriásica que además padecen psoriasis en placas de moderada a grave o pacientes que no respondieron bien a medicamentos conocidos como bloqueadores del factor de necrosis tumoral (TNF):
• La dosis recomendada es 300 mg por inyección subcutánea.
• Cada dosis de 300 mg se administra mediante dos inyecciones de 150 mg.
Después de la primera dosis, usted recibirá inyecciones semanales en las semanas 1, 2 y 3. A partir de la semana 4, las inyecciones serán mensuales. En cada cita, usted recibirá una dosis de 300 mg repartida en dos inyecciones de 150 mg.
Para el resto de pacientes con artritis psoriásica:
• La dosis recomendada es 150 mg por inyección subcutánea.
Después de la primera dosis, usted recibirá inyecciones semanales en las semanas 1, 2 y 3. A partir de la semana 4, las inyecciones serán mensuales.
Espondilitis anquilosante
• La dosis recomendada es 150 mg por inyección subcutánea.
Después de la primera dosis, usted recibirá invecciones semanales en las semanas 1, 2 y 3. A partir de la semana 4, las invecciones serán mensuales.
Cosentyx es un tratamiento de larga duración. Su médico controlará periódicamente el estado de su enfermedad para comprobar si el tratamiento surte el efecto deseado.
Si usa más Cosentyx del que debe
Si recibe más Cosentyx del que debe o la dosis ha sido administrada antes del tiempo indicado por su médico, informe a su médico.
Si olvidó usar Cosentyx
Si olvidó una inyección de Cosentyx, hable con su médico.
Si interrumpe el tratamiento con Cosentyx
No es peligroso dejar de usar Cosentyx. No obstante, si lo hace, es posible que reaparezcan los síntomas de psoriasis, artritis psoriásica o espondilitis anquilosante.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos graves
Interrumpa el tratamiento con Cosentyx e informe a su médico o busque inmediatamente asistencia médica si nota alguno de los siguientes efectos adversos:
Posible infección grave - los signos pueden incluir:
• fiebre, síntomas gripales, sudores nocturnos
• sensación de cansancio o dificultad para respirar, tos persistente
• piel caliente, enrojecida y dolorosa a la palpación, o erupción dolorosa con ampollas
• ardor al orinar.
Reacción alérgica grave - los signos pueden incluir:
• dificultad para respirar o tragar
• tensión arterial baja, que puede causar mareo o un ligero aturdimiento
• hinchazón del rostro, labios o garganta
• picor (prurito) intenso de la piel acompañado de erupción o ampollas.
Su médico decidirá si debe y cuándo reiniciar el tratamiento.
Otros efectos adversos
La mayoría de los siguientes efectos adversos son leves o moderados. Si en algún caso se convierten en graves, informe a su médico, farmacéutico o enfermero.
Algunos efectos adversos son muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
• infecciones de las vías respiratorias altas con síntomas tales como dolor de garganta y congestión nasal (rinofaringitis, rinitis).
Agunos efectos adversos son frecuentes (pueden afectar hasta 1 de cada 10 personas):
• úlceras bucales (herpes oral)
• diarrea
• secreción nasal (rinorrea)
Algunos efectos adversos son poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
• aftas bucales (candidiasis oral)
• signos de escasez de glóbulos blancos, como fiebre, dolor de garganta o úlceras bucales debido a infecciones (neutropenia)
• pié de atleta (tinea pedis)
• infección del oído externo (otitis externa)
• supuración del ojo con picor, enrojecimiento e hinchazón (conjuntivitis)
• erupción con picor (urticaria)
Algunos efectos adversos son raros (pueden afectar hasta 1 de cada 1.000 personas):
• reacción alérgica grave con shock (reacción anafiláctica)
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Cosentyx
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la caja o en el vial después de “CADT’EXP”.
Antes de la reconstitución: Conserve el vial en la nevera entre 2°C y 8°C.
Después de la reconstitución: La solución se puede usar inmediatamente o conservarse entre 2 y 8°C durante 24 horas. No congelar. La solución se debe utilizar en el plazo de 1 hora después de haberla sacado de la nevera a 2-8°C.
Este medicamento es de un único uso. Pregunte a su farmacéutico cómo deshacerse de los medicamentos que ya no necesita.
Contenido del envase e información adicional
6.
Composición de Cosentyx
- El principio activo es secukinumab. Cada vial de polvo para solución inyectable contiene 150 mg de secukinumab. Tras la reconstitución, 1 ml de solución contiene 150 mg de secukinumab.
- Los demás componentes son sacarosa, L-histidina, hidrocloruro de L-histidina monohidrato, y polisorbato 80.
Aspecto de Cosentyx y contenido del envase
Cosentyx polvo para solución inyectable es un polvo sólido y blanco en un vial de vidrio. No utilizar si el polvo liofilizado no se ha disuelto completamente o si el líquido contiene partículas fácilmente visibles, está turbio o es claramente marrón. Cosentyx se presenta en un envase que contiene 1 vial.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH RoonstraBe 25 D-90429 Nuremberg Alemania
medicamento dirigiéndose al representante local del
Lietuva
Novartis Pharma Services Inc. Tel: +370 5 269 16 50
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Osterreich
Novartis Pharma GmbH Tel: +43 1 86 6570
Pueden solicitar más información respecto a este titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Etarapna
Novartis Pharma Services Inc.
Tea: +359 2 489 98 28
Ceská republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc.
Tel: +372 66 30 810
EkXáóa
Novartis (Hellas) A.E.B.E.
Tqk: +30 210 281 17 12
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A. Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kórcpog Novartis Pharma Services Inc. TpL +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
|
Fecha de la última revisión de este prospecto: |
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Instrucciones de uso de Cosentyx polvo para solución inyectable Esta información está destinada únicamente a profesionales médicos o del sector sanitario.
Conserve el vial de polvo en nevera entre 2°C y 8°C.
El vial es de un único uso, contiene 150 mg de secukinumab que ha de reconstituirse con agua estéril para preparaciones inyectables. No utilice el vial después de la fecha de caducidad que aparece en la caja o en el vial. Si hubiera caducado, devuelva el envase completo a la farmacia.
La preparación de la solución inyectable para la vía subcutánea se debe hacer sin interrupción, teniendo cuidado de usar una técnica aséptica. El tiempo de preparación desde la perforación del tabique de goma hasta el final de la reconstitución de la solución es de unos 20 minutos de media y no debe exceder los 90 minutos.
Para preparar Cosentyx 150 mg polvo para solución inyectable por favor siga estas instrucciones.
Instrucciones para la reconstitución de Cosentyx 150 mg polvo para solución inyectable:
1. Deje que el vial de polvo se atempere y asegúrese de que el agua estéril para preparaciones inyectables esté a temperatura ambiente.
2. Extraiga un volumen ligeramente superior a 1,0 ml de agua estéril para preparaciones inyectables con una jeringa graduada desechable de 1 ml y ajústelo hasta 1,0 ml.
3. Quite la tapa de plástico del vial.
4. Inserte la aguja de la jeringa por el centro del tapón de goma del vial que contiene el polvo e inyecte lentamente 1,0 ml de agua estéril para inyectables dentro del vial para reconstituirlo. El flujo de agua para preparaciones inyectables debe ir dirigido hacia el polvo.


5. Incline el vial a 45° aproximadamente y hágalo girar con suavidad con la punta de los dedos durante alrededor de 1 minuto. No lo sacuda ni invierta.
6. Déjelo reposar a temperatura ambiente durante 10 minutos como mínimo para favorecer la disolución. Puede que note la formación de espuma en la solución.
7. Incline el vial a 45° aproximadamente y hágalo girar con suavidad con la punta de los dedos durante alrededor de 1 minuto. No lo sacuda ni invierta.

8. Deje el vial en reposo a temperatura ambiente unos 5 minutos. La solución resultante debe ser transparente. Su color puede variar de incoloro a ligeramente amarillenta. No lo utilice si el polvo liofilizado no se ha disuelto por completo o si el líquido contiene partículas visibles, está turbio o tiene un color claramente marrón.
9. Prepare el número necesario de viales (2 viales para la dosis de 300 mg).
Después de la preparación, la solución para inyección subcutánea se puede usar inmediatamente o conservarse a 2-8°C durante 24 horas. No congelar. Tras conservar a 2-8°C, es necesario atemperar la solución durante 20 minutos antes de la administración. La solución se debe utilizar en el plazo de 1 hora después de haberla sacado de la nevera a 2-8°C.
Instrucciones para la administración de la solución de Cosentyx
1. Incline el vial a 45° aproximadamente y coloque la punta de la aguja en el fondo de la solución
del vial mientras extraiga la solución a la jeringa. NO invierta el vial.

2. Extraiga cuidadosamente un poco más de 1,0 ml de la solución para inyección subcutánea del vial en una jeringa graduada desechable de 1 ml utilizando una aguja adecuada (21G x 2"). Esta aguja solo se utilizará para trasvasar Cosentyx a la jeringa desechable. Prepare el número de jeringas necesarias (2 jeringas para la dosis de 300 mg).
3. Con la aguja apuntando hacia arriba, golpee la jeringa para que asciendan las burbujas de aire presentes.

4.
Remplace la aguja de la jeringa por otra de 27G x Vi".

5. Expulse las burbujas de aire y desplace el émbolo hasta la marca de 1,0 ml.
6. Desinfecte el lugar de inyección con una toallita humedecida con alcohol.
7. Inyecte por vía subcutánea la solución de Cosentyx en la parte superior de los muslos, o en la parte inferior del abdomen (pero no en un áreade 5 cm alrededor del ombligo) o en la parte superior de los brazos. Elija cada vez un nuevo lugar para la inyección. No inyecte en ninguna zona donde la piel esté sensible, dañada, enrojecida, descamada o dura. Evite las zonas con cicatrices o estrías.

8. Los restos de solución que queden en el vial no se deben utilizar y se eliminarán de acuerdo a la normativa local. Los viales son para un solo uso. Deseche la jeringa utilizada en el cubo de eliminación de objetos punzantes (recipiente cerrado y resistente a pinchazos). Por motivos de seguridad y de salud de usted y de otras personas, las agujas y las jeringas nunca se deben reutilizar.
Prospecto: información para el paciente Cosentyx 150 mg solución inyectable en jeringa precargada
Secukinumab
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Cosentyx y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Cosentyx
3. Cómo usar Cosentyx
4. Posibles efectos adversos
5. Conservación de Cosentyx
6. Contenido del envase e información adicional
1. Qué es Cosentyx y para qué se utiliza
Cosentyx contiene el principio activo secukinumab. Secukinumab es un anticuerpo monoclonal. Los anticuerpos monoclonales son unas proteínas que reconocen y se unen específicamente a ciertas proteínas del cuerpo.
Cosentyx pertenece a un grupo de medicamentos conocidos como “inhibidores de interleuquinas”. Actúa neutralizando la actividad de una proteína denominada IL-17A, que está presente en cantidades elevadas en enfermedades como la psoriasis, la artritis psoriásica y la espondilitis anquilosante.
Cosentyx se utiliza para el tratamiento de las siguientes enfermedades inflamatorias:
• Psoriasis en placas
• Artritis psoriásica
• Espondilitis anquilosante
Psoriasis en placas
Cosentyx se usa para el tratamiento de un trastorno de la piel conocido como “psoriasis en placas” que provoca inflamación en la piel. Cosentyx reduce la inflamación y otros síntomas de la enfermedad. Cosentyx se usa en personas adultas que padecen psoriaris en placas de naturaleza moderada a grave.
Utilizar Cosentyx para psoriasis en placas le beneficiará ya que produce mejorías en el aspecto de la piel y la disminución de síntomas tales como la descamación, el prurito y el dolor.
Artritis psoriásica
Cosentyx se usa para el tratamiento de un trastorno conocido como “artritis psoriásica”. Se trata de una enfermedad inflamatoria de las articulaciones, a menudo acompañada de psoriasis. Si padece artritis psoriásica activa, recibirá primero otros medicamentos. Si no responde suficientemente bien a estos medicamentos, recibirá Cosentyx para reducir los signos y síntomas de la artritis psoriásica activa, mejorar la función física y ralentizar el daño en el cartílago y huesos de las articulaciones involucradas en la enfermedad.
Cosentyx se usa en adultos con artritis psoriásica activa y se puede utilizar solo o con otro medicamento denominado metotrexato.
Utilizar Cosentyx para artritis psoriásica le beneficiará ya que reduce los signos y síntomas de la enfermedad, ralentizando el daño en los cartílagos y huesos de las articulaciones y mejorando su habilidad para realizar las actividades diarias normales.
Espondilitis anquilosante
Cosentyx se usa para el tratamiento de un trastorno conocido como “espondilitis anquilosante”. Se trata de una enfermedad inflamatoria que afecta principalmente a la columna causando inflamación de las articulaciones de la columna. Si padece espondilitis anquilosante, recibirá primero otros medicamentos. Si no responde suficientemente bien a estos medicamentos, recibirá Cosentyx para reducir los signos y síntomas de la enfermedad, reducir la inflamación y mejorar su función física.
Cosentyx se usa en adultos con espondilitis anquilosante activa.
Utilizar Cosentyx para espondilitis anquilosante le beneficiará ya que reduce los signos y síntomas de la enfermedad y mejora la función física.
2. Qué necesita saber antes de empezar a usar Cosentyx No use Cosentyx:
• si es alérgico al secukinumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Si usted sospecha que es alérgico, consulte con su médico antes de usar Cosentyx.
• si tiene alguna infección que su médico considere importante.
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de usar Cosentyx:
• si ha contraído una infección
• si padece infecciones repetidas o prolongadas.
• si tiene tuberculosis.
• si padece la enfermedad de Crohn.
• si ha sido vacunado recientemente o va a ser vacunado durante el tratamiento con Cosentyx.
• si sigue algún otro tratamiento para la psoriasis, como por ejemplo, si usa otros inmunosupresores o fototerapia con luz ultravioleta (UV).
Vigile la aparición de infecciones y reacciones alérgicas
Cosentyx puede ocasionar potencialmente efectos adversos graves, incluidas infecciones y reacciones alérgicas. Debe vigilar la aparición de signos de estas enfermedades mientras use Cosentyx.
Interrumpa el tratamiento con Cosentyx y avise a su médico o busque asistencia médica inmediatamente si nota alguno de los signos que indican una posible infección o reacción alérgica graves. Estos signos se incluyen en la sección 4 “Posibles efectos adversos”.
Niños y adolescentes
No se recomienda el uso de Cosentyx en niños y adolescentes (menores de 18 años) pues no se ha
estudiado el medicamento en personas de esta edad.
Uso de Cosentyx con otros medicamentos
Informe a su médico o farmacéutico:
• Si está tomando, ha tomado o podría tener que tomar cualquier otro medicamento.
• Si ha sido vacunado recientemente o va a ser vacunado próximamente. No le deben administrar ciertos tipos de vacunas (vacunas vivas) mientras use Cosentyx.
Embarazo, lactancia y fertilidad
• Es preferible que evite el uso de Cosentyx durante el embarazo. Se desconoce el efecto de este medicamento en mujeres embarazadas. Si es una mujer en edad fértil, se aconseja que evite quedarse embarazada y debe utilizar un anticonceptivo adecuado mientras use Cosentyx y durante al menos 20 semanas después de la última dosis de Cosentyx. Consulte a su médico si está embarazada, cree que podría estar embarazada o tiene la intención de quedarse embarazada.
• Consulte a su médico si están dando el pecho o tiene previsto dar el pecho. Usted y su médico deben decidir si va a dar el pecho o va a utilizar Cosentyx. No puede hacer las dos cosas. Después de utilizar Cosentyx no debe dar el pecho durante al menos 20 semanas después de la última dosis.
Conducción y uso de máquinas
La influencia de Cosentyx sobre la capacidad para conducir o utilizar máquinas es nula o
insignificante.
3. Cómo usar Cosentyx
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico.
En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero.
Cosentyx se administra por medio de una inyección debajo de la piel (es decir, por vía subcutánea). Usted y su médico deben decidir si ha de ser usted mismo quien se va a inyectar Cosentyx.
Es importante que no intente inyectarse el medicamento hasta que el médico, enfermero o farmacéutico le hayan enseñado a hacerlo. La persona que lo cuida también puede ponerle la inyección de Cosentyx después de haber recibido la formación oportuna.
En el apartado “Instructiones de uso de Cosentyx en jeringa precargada” que se encuentra al final de este prospecto, se dan instrucciones detalladas de cómo administrar Cosentyx.
Cuánto Cosentyx debe administrarse y durante cuánto tiempo
Su medico decidirá cuánto Consentyx usted necesita y durante cuánto tiempo.
Psoriasis en placas
• La dosis recomendada es 300 mg por inyección subcutánea.
• Una dosis de 300 mg se administra mediante dos inyecciones de 150 mg.
Después de la primera dosis, usted recibirá inyecciones semanales en las semanas 1, 2 y 3. A partir de la semana 4, las inyecciones serán mensuales. En cada cita, usted recibirá una dosis de 300 mg repartida en dos inyecciones de 150 mg.
Artritis psoriásica
Para pacientes con artritis psoriásica que además padecen psoriasis en placas de moderada a grave o pacientes que no respondieron bien a medicamentos conocidos como bloqueadores del factor de necrosis tumoral (TNF):
• La dosis recomendada es 300 mg por inyección subcutánea.
• Cada dosis de 300 mg se administra mediante dos inyecciones de 150 mg.
Después de la primera dosis, usted recibirá inyecciones semanales en las semanas 1, 2 y 3. A partir de la semana 4, las inyecciones serán mensuales. En cada cita, usted recibirá una dosis de 300 mg repartida en dos inyecciones de 150 mg.
Para el resto de pacientes con artritis psoriásica:
• La dosis recomendada es 150 mg por inyección subcutánea.
Después de la primera dosis, usted recibirá inyecciones semanales en las semanas 1, 2 y 3. A partir de la semana 4, las inyecciones serán mensuales.
Espondilitis anquilosante
• La dosis recomendada es 150 mg por inyección subcutánea.
Después de la primera dosis, usted recibirá invecciones semanales en las semanas 1, 2 y 3. A partir de la semana 4, las invecciones serán mensuales.
Cosentyx es un tratamiento de larga duración. Su médico controlará periódicamente el estado de su enfermedad para comprobar si el tratamiento surte el efecto deseado.
Si usa más Cosentyx del que debe
Si recibe más Cosentyx del que debe o la dosis ha sido administrada antes del tiempo indicado por su médico, informe a su médico.
Si olvidó usar Cosentyx
Si olvidó una inyección de Cosentyx, hable con su médico.
Si interrumpe el tratamiento con Cosentyx
No es peligroso dejar de usar Cosentyx. No obstante, si lo hace, es posible que reaparezcan los síntomas de psoriasis, artritis psoriásica o espondilitis anquilosante.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos graves
Interrumpa el tratamiento con Cosentyx e informe a su médico o busque inmediatamente asistencia médica si nota alguno de los siguientes efectos adversos:
Posible infección grave - los signos pueden incluir:
• fiebre, síntomas gripales, sudores nocturnos
• sensación de cansancio o dificultad para respirar, tos persistente
• piel caliente, enrojecida y dolorosa a la palpación, o erupción dolorosa con ampollas
• ardor al orinar.
Reacción alérgica grave - los signos pueden incluir:
• dificultad para respirar o tragar
• tensión arterial baja, que puede causar mareo o un ligero aturdimiento
• hinchazón del rostro, labios o garganta
• picor (prurito) intenso de la piel acompañado de erupción o ampollas.
Su médico decidirá si debe y cuándo reiniciar el tratamiento.
Otros efectos adversos
La mayoría de los siguientes efectos adversos son leves o moderados. Si en algún caso se convierten en graves, informe a su médico, farmacéutico o enfermero.
Algunos efectos adversos son muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
• infecciones de las vías respiratorias altas con síntomas tales como dolor de garganta y congestión nasal (rinofaringitis, rinitis).
Agunos efectos adversos son frecuentes (pueden afectar hasta 1 de cada 10 personas):
• úlceras bucales (herpes oral)
• diarrea
• secreción nasal (rinorrea)
Algunos efectos adversos son poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
• aftas bucales (candidiasis oral)
• signos de escasez de glóbulos blancos, como fiebre, dolor de garganta o úlceras bucales debido a infecciones (neutropenia)
• pié de atleta (tinea pedis)
• infección del oído externo (otitis externa)
• supuración del ojo con picor, enrojecimiento e hinchazón (conjuntivitis)
• erupción con picor (urticaria)
Algunos efectos adversos son raros (pueden afectar hasta 1 de cada 1.000 personas):
• reacción alérgica grave con shock (reacción anafiláctica)
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento:
• después de la fecha de caducidad que aparece en la caja o en la etiqueta de la jeringa después de “CAD”
• si el líquido contiene partículas fácilmente visibles, está turbio o tiene un color claramente marrón.
Conservar la jeringa precintada en su caja para protegerla de la luz. Conservar en la nevera entre 2°C y 8°C. No congelar. No agitar.
Este medicamento es de un único uso. Pregunte a su farmacéutico cómo deshacerse de los medicamentos que ya no necesita.
6. Contenido del envase e información adicional Composición de Cosentyx
- El principio activo es secukinumab. Cada jeringa precargada contiene 150 mg de secukinumab.
- Los demás componentes son trehalosa dihidrato, L-histidina, hidrocloruro de L-histidina monohidrato, L-metionina, polisorbato 80 y agua para preparaciones inyectables.
Aspecto de Cosentyx y contenido del envase
Cosentyx solución inyectable es un líquido transparente. Su color varía de incoloro a ligeramente amarillo. No utilizar si el líquido contiene partículas fácilmente visibles, está turbio o es claramente marrón. Cosentyx se presenta en envases unitarios de 1 o 2 jeringa(s) precargada(s) y en envases múltiples que contienen 6 (3 envases de 2) jeringas precargadas. Puede que no estén comercializados todos los tamaños de envase.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH RoonstraBe 25 D-90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Etarapun Novartis Pharma Services Inc. Tea: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Ceská republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXXáda Novartis (Hellas) A.E.B.E. T^k: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Suomi/Finland
Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Lea TODAS las instrucciones antes de inyectar el medicamento. Es importante que no intente inyectarse el medicamento hasta que el médico, enfermero o farmacéutico le hayan enseñado a hacerlo. La caja contiene la(s) jeringa(s) precargada(s) de Cosentyx dentro de un blíster de plástico.
Su Cosentyx en jeringa precargada
Protector de la jeringa Sujeción para colocar los dedos Émbolo

Una vez que el medicamento se haya inyectado, el protector para cubrir la aguja se activará. Éste está concebido para proteger de lesiones por pinchazos accidentales a los profesionales sanitarios, a los pacientes que se autoinyectan el medicamento prescrito por el médico y a los individuos que ayudan a los pacientes a autoinyectarse el medicamento.

Qué más necesita para la inyección:
• Toallita humedecida en alcohol.
• Algodón o gasa.
• Cubo de eliminación de objetos punzantes.
Información importante de seguridad
Advertencia: Mantenga la jeringa fuera de la vista y del alcance de los niños.
1. El capuchón de la aguja de la jeringa puede contener goma seca (látex) que no deben tocarlas las personas sensibles a ella.
2. No abra la caja hasta que esté listo para ponerse este medicamento.
3. No use este medicamento si el precinto de la caja o el blíster está roto, ya que puede que no sea seguro utilizarlo.
4. Nunca deje la jeringa en lugares donde otras personas puedan tocarlo.
5. No agite la j eringa.
6. Tenga mucho cuidado de no tocar los clips de activación antes de su utilización. Si lo hace, se disparará el protector de la aguja antes de tiempo.
7. No quite el capuchón de la aguja hasta justo antes de ponerse la inyección.
8. No se puede reutilizar la jeringa. Una vez utilizada, deseche la jeringa al cubo de eliminación de objetos punzantes.
Conservación de Cosentyx en jeringa precargada
1. Conservar este medicamento precintado dentro de su caja para protegerla de la luz. Conservar en nevera entre 2°C y 8°C. NO CONGELAR.
2. Recuerde sacar la jeringa de la nevera para que se atempere antes de preparar la inyección (15-30 minutos).
3. No utilice la jeringa después de la fecha de caducidad que aparece en la caja o en la etiqueta de la jeringa después de “CAD”/”EXP”. Si ha caducado, devuelva el envase completo a la farmacia.
Lugares de inyección


El lugar de la inyección es el sitio donde usted se pondrá la jeringa.
• Se recomienda utilizar la parte superior de los muslos. También puede utilizar la parte inferior de abdomen, pero no en un área de 5 cm alrededor del ombligo.
• Elija un lugar diferente cada vez que se ponga la inyección.
• No se inyecte en zonas donde la piel sea sensible, esté dañada, enrojecida, descamada o endurecida. Evite las zonas con cicatrices o estrías.
Si quien pone la inyección es la persona que cuida al paciente, entonces también se puede utilizar la parte superior de los brazos.
Preparación de Cosentyx en jeringa precargada lista para utilizar
Nota: Para la dosis de 300 mg, prepare 2 jeringas precargadas e inyecte el contenido de ambas.
1. Saque de la nevera la caja con la jeringa y déjela sin abrir unos 15-30 minutos para que se atempere.
2. Cuando esté listo para utilizar la jeringa, lávese bien las manos con agua y jabón.
3. Desinfecte bien la zona de inyección con una toallita humedecida con alcohol.
4. Saque la jeringa de la caja y abra el blíster.
5. Inspeccione la jeringa. El líquido debe ser transparente. Su color puede variar de incoloro a ligeramente amarillento. Puede haber alguna burbuja de aire pequeña, que es normal. NO UTILIZAR si el líquido contiene partículas, está turbio o tiene un color claramente marrón. NO UTILIZAR si la jeringa está rota. En todos estos casos, devuelva el envase completo a la farmacia.
Cómo utilizar la jeringa precargada de Cosentyx

Retire con cuidado el capuchón de la aguja de la jeringa. Deséchelo. Puede que observe una gota en la punta de la aguja. Esto es normal.
Pellizque suavemente la piel del lugar de la inyección e inserte la aguja como muestra la figura. Introduzca la aguja por completo para garantizar que se administre todo el medicamento.


Instrucciones de eliminación

Sujete la jeringa como se muestra. Presione lentamente el émbolo hasta el final de tal manera que la cabeza del émbolo quede encajada en los clips de activación del protector.
Mantenga presionado el émbolo mientras mantiene la jeringa en esa posición durante 5 segundos.
Sin soltar el émbolo, retire cuidadosamente la aguja del lugar de inyección.
Suelte el émbolo lentamente y deje que el protector tape automáticamente la aguja.
Puede que haya un poquito de sangre en el lugar de inyección. Puede presionar durante 10 segundos con un algodón o una gasa la zona de inyección. No se frote el lugar de inyección. Se puede poner una tirita si lo necesita.
Deseche la jeringa usada en un cubo de eliminación de objetos punzantes (recipiente cerrado y resistente a pinchazos). Por motivos de seguridad y de salud (de usted y de otras personas), las agujas y las jeringas usadas nunca se deben reutilizar.
Prospecto: información para el paciente Cosentyx 150 mg solución inyectable en pluma precargada
Secukinumab
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Cosentyx y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Cosentyx
3. Cómo usar Cosentyx
4. Posibles efectos adversos
5. Conservación de Cosentyx
6. Contenido del envase e información adicional
1. Qué es Cosentyx y para qué se utiliza
Cosentyx contiene el principio activo secukinumab. Secukinumab es un anticuerpo monoclonal. Los anticuerpos monoclonales son unas proteínas que reconocen y se unen específicamente a ciertas proteínas del cuerpo.
Cosentyx pertenece a un grupo de medicamentos conocidos como “inhibidores de interleuquinas”. Actúa neutralizando la actividad de una proteína denominada IL-17A, que está presente en cantidades elevadas en enfermedades como la psoriasis, la artritis psoriásica y la espondilitis anquilosante.
Cosentyx se utiliza para el tratamiento de las siguientes enfermedades inflamatorias:
• Psoriasis en placas
• Artritis psoriásica
• Espondilitis anquilosante
Psoriasis en placas
Cosentyx se usa para el tratamiento de un trastorno de la piel conocido como “psoriasis en placas” que provoca inflamación en la piel. Cosentyx reduce la inflamación y otros síntomas de la enfermedad. Cosentyx se usa en personas adultas que padecen psoriaris en placas de naturaleza moderada a grave.
Utilizar Cosentyx para psoriasis en placas le beneficiará ya que produce mejorías en el aspecto de la piel y la disminución de síntomas tales como la descamación, el prurito y el dolor.
Artritis psoriásica
Cosentyx se usa para el tratamiento de un trastorno conocido como “artritis psoriásica”. Se trata de una enfermedad inflamatoria de las articulaciones, a menudo acompañada de psoriasis. Si padece artritis psoriásica activa, recibirá primero otros medicamentos. Si no responde suficientemente bien a estos medicamentos, recibirá Cosentyx para reducir los signos y síntomas de la artritis psoriásica activa, mejorar la función física y ralentizar el daño en el cartílago y huesos de las articulaciones involucradas en la enfermedad.
Cosentyx se usa en adultos con artritis psoriásica activa y se puede utilizar solo o con otro medicamento denominado metotrexato.
Utilizar Cosentyx para artritis psoriásica le beneficiará ya que reduce los signos y síntomas de la enfermedad, ralentizando el daño en los cartílagos y huesos de las articulaciones y mejorando su habilidad para realizar las actividades diarias normales.
Espondilitis anquilosante
Cosentyx se usa para el tratamiento de un trastorno conocido como “espondilitis anquilosante”. Se trata de una enfermedad inflamatoria que afecta principalmente a la columna causando inflamación de las articulaciones de la columna. Si padece espondilitis anquilosante, recibirá primero otros medicamentos. Si no responde suficientemente bien a estos medicamentos, recibirá Cosentyx para reducir los signos y síntomas de la enfermedad, reducir la inflamación y mejorar su función física.
Cosentyx se usa en adultos con espondilitis anquilosante activa.
Utilizar Cosentyx para espondilitis anquilosante le beneficiará ya que reduce los signos y síntomas de la enfermedad y mejora la función física.
2. Qué necesita saber antes de empezar a usar Cosentyx No use Cosentyx:
• si es alérgico al secukinumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Si usted sospecha que es alérgico, consulte con su médico antes de usar Cosentyx.
• si tiene alguna infección que su médico considere importante.
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de usar Cosentyx:
• si ha contraído una infección
• si padece infecciones repetidas o prolongadas.
• si tiene tuberculosis.
• si padece la enfermedad de Crohn.
• si ha sido vacunado recientemente o va a ser vacunado durante el tratamiento con Cosentyx.
• si sigue algún otro tratamiento para la psoriasis, como por ejemplo, si usa otros inmunosupresores o fototerapia con luz ultravioleta (UV).
Vigile la aparición de infecciones y reacciones alérgicas
Cosentyx puede ocasionar potencialmente efectos adversos graves, incluidas infecciones y reacciones alérgicas. Debe vigilar la aparición de signos de estas enfermedades mientras use Cosentyx.
Interrumpa el tratamiento con Cosentyx y avise a su médico o busque asistencia médica inmediatamente si nota alguno de los signos que indican una posible infección o reacción alérgica graves. Estos signos se incluyen en la sección 4 “Posibles efectos adversos”.
Niños y adolescentes
No se recomienda el uso de Cosentyx en niños y adolescentes (menores de 18 años) pues no se ha
estudiado el medicamento en personas de esta edad.
Uso de Cosentyx con otros medicamentos
Informe a su médico o farmacéutico:
• Si está tomando, ha tomado o podría tener que tomar cualquier otro medicamento.
• Si ha sido vacunado recientemente o va a ser vacunado próximamente. No le deben administrar ciertos tipos de vacunas (vacunas vivas) mientras use Cosentyx.
Embarazo, lactancia y fertilidad
• Es preferible que evite el uso de Cosentyx durante el embarazo. Se desconoce el efecto de este medicamento en mujeres embarazadas. Si es una mujer en edad fértil, se aconseja que evite quedarse embarazada y debe utilizar un anticonceptivo adecuado mientras use Cosentyx y durante al menos 20 semanas después de la última dosis de Cosentyx. Consulte a su médico si está embarazada, cree que podría estar embarazada o tiene la intención de quedarse embarazada.
• Consulte a su médico si están dando el pecho o tiene previsto dar el pecho. Usted y su médico deben decidir si va a dar el pecho o va a utilizar Cosentyx. No puede hacer las dos cosas. Después de utilizar Cosentyx no debe dar el pecho durante al menos 20 semanas después de la última dosis.
Conducción y uso de máquinas
La influencia de Cosentyx sobre la capacidad para conducir o utilizar máquinas es nula o
insignificante.
3. Cómo usar Cosentyx
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero.
Cosentyx se administra por medio de una inyección debajo de la piel (es decir, por vía subcutánea). Usted y su médico deben decidir si ha de ser usted mismo quien se va a inyectar Cosentyx.
Lo importante es que usted no intente inyectarse el medicamento hasta que su médico, enfermero o farmacéutico le hayan enseñado cómo hacerlo. La persona que lo cuida también puede ponerle la inyección de Cosentyx después de haber recibido la formación oportuna.
En el apartado “Instrucciones de uso de Cosentyx en pluma SensoReady” que se encuentra al final de este prospecto, se dan instrucciones sobre cómo inyectar Cosentyx.
Cuánto Cosentyx debe administrarse y durante cuánto tiempo
Su medico decidirá cuánto Consentyx usted necesita y durante cuánto tiempo.
Psoriasis en placas
• La dosis recomendada es 300 mg por inyección subcutánea.
• Una dosis de 300 mg se administra mediante dos inyecciones de 150 mg.
Después de la primera dosis, usted recibirá inyecciones semanales en las semanas 1, 2 y 3. A partir de la semana 4, las inyecciones serán mensuales. En cada cita, usted recibirá una dosis de 300 mg repartida en dos inyecciones de 150 mg.
Artritis psoriásica
Para pacientes con artritis psoriásica que además padecen psoriasis en placas de moderada a grave o pacientes que no respondieron bien a medicamentos conocidos como bloqueadores del factor de necrosis tumoral (TNF):
• La dosis recomendada es 300 mg por inyección subcutánea.
• Cada dosis de 300 mg se administra mediante dos inyecciones de 150 mg.
Después de la primera dosis, usted recibirá inyecciones semanales en las semanas 1, 2 y 3. A partir de la semana 4, las inyecciones serán mensuales. En cada cita, usted recibirá una dosis de 300 mg repartida en dos inyecciones de 150 mg.
Para el resto de pacientes con artritis psoriásica:
• La dosis recomendada es 150 mg por inyección subcutánea.
Después de la primera dosis, usted recibirá inyecciones semanales en las semanas 1, 2 y 3. A partir de la semana 4, las inyecciones serán mensuales.
Espondilitis anquilosante
• La dosis recomendada es 150 mg por inyección subcutánea.
Después de la primera dosis, usted recibirá invecciones semanales en las semanas 1, 2 y 3. A partir de la semana 4, las invecciones serán mensuales.
Cosentyx es un tratamiento de larga duración. Su médico controlará periódicamente el estado de su enfermedad para comprobar si el tratamiento surte el efecto deseado.
Si usa más Cosentyx del que debe
Si recibe más Cosentyx del que debe o la dosis ha sido administrada antes del tiempo indicado por su médico, informe a su médico.
Si olvidó usar Cosentyx
Si olvidó una inyección de Cosentyx, hable con su médico.
Si interrumpe el tratamiento con Cosentyx
No es peligroso dejar de usar Cosentyx. No obstante, si lo hace, es posible que reaparezcan los síntomas de psoriasis, artritis psoriásica o espondilitis anquilosante.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos graves
Interrumpa el tratamiento con Cosentyx e informe a su médico o busque inmediatamente asistencia médica si nota alguno de los siguientes efectos adversos:
Posible infección grave - los signos pueden incluir:
• fiebre, síntomas gripales, sudores nocturnos
• sensación de cansancio o dificultad para respirar, tos persistente
• piel caliente, enrojecida y dolorosa a la palpación, o erupción dolorosa con ampollas
• ardor al orinar.
Reacción alérgica grave - los signos pueden incluir:
• dificultad para respirar o tragar
• tensión arterial baja, que puede causar mareo o un ligero aturdimiento
• hinchazón del rostro, labios o garganta
• picor (prurito) intenso de la piel acompañado de erupción o ampollas.
Su médico decidirá si debe y cuándo reiniciar el tratamiento.
Otros efectos adversos
La mayoría de los siguientes efectos adversos son leves o moderados. Si en algún caso se convierten en graves, informe a su médico, farmacéutico o enfermero.
Algunos efectos adversos son muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
• infecciones de las vías respiratorias altas con síntomas tales como dolor de garganta y congestión nasal (rinofaringitis, rinitis).
Agunos efectos adversos son frecuentes (pueden afectar hasta 1 de cada 10 personas):
• úlceras bucales (herpes oral)
• diarrea
• secreción nasal (rinorrea)
Algunos efectos adversos son poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
• aftas bucales (candidiasis oral)
• signos de escasez de glóbulos blancos, como fiebre, dolor de garganta o úlceras bucales debido a infecciones (neutropenia)
• pié de atleta (tinea pedis)
• infección del oído externo (otitis externa)
• supuración del ojo con picor, enrojecimiento e hinchazón (conjuntivitis)
• erupción con picor (urticaria)
Algunos efectos adversos son raros (pueden afectar hasta 1 de cada 1.000 personas):
• reacción alérgica grave con shock (reacción anafiláctica)
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento:
• después de la fecha de caducidad que aparece en la caja o en la etiqueta de la pluma después de “CAD”
• si el líquido contiene partículas fácilmente visibles, está turbio o tiene un color claramente marrón.
Conservar la pluma precintada dentro de su caja para protegerla de la luz. Conservar en la nevera entre 2°C y 8°C. No congelar. No agitar.
Este medicamento es de un único uso. Pregunte a su farmacéutico cómo deshacerse de los medicamentos que ya no necesita.
6. Contenido del envase e información adicional Composición de Cosentyx
- El principio activo es secukinumab. Cada pluma precargada contiene 150 mg de secukinumab.
- Los demás componentes son trehalosa dihidrato, L-histidina, hidrocloruro de L-histidina monohidrato, L-metionina, polisorbato 80 y agua para preparaciones inyectables.
Aspecto de Cosentyx y contenido del envase
Cosentyx solución inyectable es un líquido transparente. Su color varía de incoloro a ligeramente amarillo. No utilizar si el líquido contiene partículas fácilmente visibles, está turbio o es claramente marrón. Cosentyx se presenta en envases unitarios de 1 o 2 pluma(s) precargada(s) y en envases múltiples que contienen 6 (3 envases de 2) plumas precargadas. Puede que no estén comercializados todos los tamaños de envase.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH RoonstraBe 25 D-90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Etarapun Novartis Pharma Services Inc. Tea: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Ceská republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXXáda Novartis (Hellas) A.E.B.E. T^k: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Suomi/Finland
Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Cosentyx 150 mg en pluma SensoReady

Solución para inyección en pluma precargada Secukinumab
Instrucciones de uso para el paciente

Lea TODAS las instrucciones antes de inyectarse el medicamento.
Estas instrucciones le van a ayudar a ponerse correctamente Cosentyx en pluma SensoReady.
Es importante que usted no intente inyectarse el medicamento hasta que el médico, la enfermera o el farmacéutico le hayan enseñado cómo hacerlo.

Cosentyx en pluma SensoReady sin tapa. No quite la tapa hasta que esté listo para ponerse la inyección.
Qué más necesita para la inyección:
Incluido en la caja:
Conserve la caja con la pluma en la nevera entre 2°C y 8°C y fuera del alcance de los niños.
• No congelar la pluma.
• No agitar la pluma.
• No utilizar la pluma si se cae sin la tapa.
Para una inyección más agradable, saque de la nevera la pluma 15-30 minutos antes para que se atempere.
No incluido en la caja:
Una pluma SensoReady de Cosentyx nueva y sin usar (se necesitan 2 plumas para la dosis de
300 mg).

• Toallita humedecida en alcohol.
• Algodón y gasa.
• Cubo de eliminación de objetos punzantes.

1. Antes de inyectarse compruebe los aspectos importantes de seguridad:



El líquido debe ser transparente. Su color puede variar de incoloro a ligeramente amarillento.
No utilizar si el líquido contiene partículas, está turbio o tiene un color claramente marrón. Puede ver pequeñas burbujas de aire, que es normal.
No utilice la pluma si ha pasado la fecha de caducidad.
No utilizar si el precinto está roto.
Contacte con su farmacéutico si la pluma no cumple con alguno de estos requisitos.
2a. Elija un lugar de inyección:
• Se recomienda utilizar la parte superior de los muslos. También puede utilizar la parte inferior de abdomen, pero no en un área de 5 cm alrededor del ombligo.
• Elija cada vez un nuevo lugar para la inyección.
• No inyecte en ninguna zona donde la piel esté sensible, dañada, enrojecida, descamada o dura. Evite las zonas con cicatrices o estrías.
2b. Solo cuidadores o profesionales sanitarios:
• Si quien pone la inyección es la persona que cuida al paciente o profesional sanitario, entonces también se puede utilizar la parte superior de los brazos.
3. Desinfecte el lugar de inyección:
• Lávese antes las manos con agua caliente y jabón.
• Desinfecte el lugar de inyección con una toallita humedecida con alcohol, haciendo un movimiento circular. Déjelo secar antes de ponerse la inyección.
• No vuelva a tocar la zona limpia antes de la inyección.
4. Quite la tapa:

• Quite la tapa solo cuando esté listo para utilizar la pluma.
• Desenrosque la tapa en la dirección de la flecha.
• Una vez retirado, deséchelo. No intente enroscarlo de nuevo.
• Utilice la pluma en los 5 minutos posteriores a haberle quitado la tapa.
5. Sujete su pluma:
• Sujete la pluma a 90° del lugar de inyección desinfectado.

DEBE LEER LO SIGUIENTE ANTES DE LA INYECCIÓN.
Durante la inyección, usted oirá 2 clics intensos.
El 1er clic indica el inicio de la inyección. Después de unos segundos, el 2° clic indicará que la inyección está a punto de finalizar.
Mantenga la pluma firmemente presionada contra la piel hasta que el indicador verde llene la ventana y haya dejado de moverse.

6. Inicio de la inyección:
• Presione con firmeza la pluma contra la piel para iniciar la inyección.
• El 1er clic indica el inicio de la inyección.
• Mantenga la pluma firmemente presionada contra la piel.
• El indicador verde le indicará el progreso de la inyección.
7. Final de la inyección:
• Escucha el 2° clic. Esto indica que la inyección está a punto de finalizar.
• Verifique que el indicador verde llene la ventana y haya dejado de moverse.
• Ahora puede retirar la pluma.

8. Verifique que el indicador verde llene la ventana:
• Esto significa que el medicamento ha sido administrado. En el caso que no se viera el indicador verde, póngase en contacto con su médico.
• Puede presionar durante 10 segundos con un algodón o una gasa la zona de inyección. No se frote el lugar de inyección. Se puede poner una tirita si lo necesita.
9. Eliminación de Cosentyx en pluma SensoReady:
• Deseche la pluma usada en un cubo de eliminación de objetos punzantes (recipiente cerrado y resistente a pinchazos o similar).
• Nunca trate de reutilizar la pluma.
124
DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Trehalosa dihidrato L-histidina
Hidrocloruro de L-histidina monohidratado
L-metionina
Polisorbato 80
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inmunosupresores, inhibidores de la interleucina, código ATC: L04AC10
Mecanismo de acción
Secukinumab es un anticuerpo de tipo IgG1/K monoclonal, íntegramente humano, que se une selectivamente y neutraliza una citoquina proinflamatoria, la interleuquina 17A (IL-17A). Secukinumab actúa dirigiéndose a IL-17A e inhibe su interacción con el receptor de IL-17, que se encuentra en varios tipos de células, incluidos los queratinocitos. Como resultado, secukinumab inhibe la liberación de citoquinas proinflamatorias, de quimioquinas y de mediadores del daño tisular, y reduce los efectos mediados por la IL-17A, que participan en la enfermedad autoinmunitaria e inflamatoria. A la piel llegan concentraciones clínicamente importantes de secukinumab y reducen los marcadores inflamatorios locales. Como consecuencia directa, el tratamiento con secukinumab reduce