Corifina 1 Mg/Ml Solución Para Pulverización Nasal
FICHA TÉCNICA O RESUMEN DE CARACTERÍSTICAS DE PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Corifina 1 mg/ml solución para pulverización nasal
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA:
Cada ml de solución contiene 1 mg de azelastina hidrocloruro.
Una aplicación (0,14 ml) contiene 0,14 mg de clorhidrato de azelastina.
Para consultar la lista completa de excipientes ver sección 6.1.
FORMA FARMACÉUTICA
Solución para pulverización nasal, transparente e incolora. DATOS CLÍNICOS
3.
4.
4.1 Indicaciones terapéuticas
Para el tratamiento sintomático de la rinitis alérgica estacional y exacerbaciones agudas de la rinitis alérgica perenne.
4.2 Posología y forma de administración
Vía nasal.
Posología:
Adultos: una aplicación (0,14 ml) en cada fosa nasal, dos veces al día (0,56 mg de azelastina hidrocloruro).
Personas de edad avanzada: no se han realizado estudios específicos en personas de edad avanzada.
Población pediátrica:
Niños mayores de 6 años: una aplicación (0,14 ml) en cada fosa nasal, dos veces al día (0,56 mg de azelastina hidrocloruro).
Forma de administración:
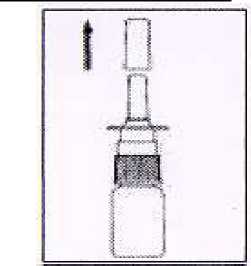
1. Quitar tapa de protección
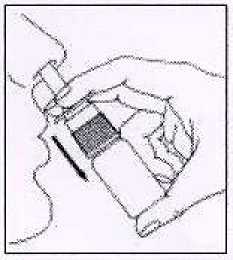
3. Atomizar una vez en cada fosa nasal manteniendo la cabeza vertical.
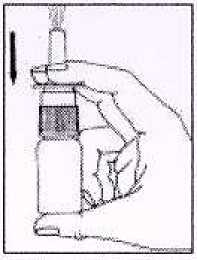
2. Antes de usar, pulsar la bomba unas veces (3-4 veces) hasta pulverización constante
4. Limpiar y colocar la tapa protectora.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes..
4.4 Advertencias y precauciones especiales de empleo
Si bien la experiencia clínica con el spray nasal de Azelastina en la población pediátrica de edad comprendida entre los 6 y 12 años es inferior a la obtenida en pacientes mayores de 12 años, los ensayos clínicos realizados hasta la fecha no muestran diferencias respecto de la eficacia y seguridad del producto en esta población.
4.5 Interacciones con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones.
4.6 Fertilidad, embarazo y lactancia
En los estudios de toxicidad durante la reproducción en animales, con altas dosis, 500 veces la dosis oral diaria propuesta para humanos, se ha producido muerte fetal, retraso del crecimiento y un incremento de la incidencia de anormalidades esqueléticas.
Debido a la vía de administración nasal y a la baja dosis administrada se puede esperar una exposición sistémica mínima. Sin embargo, así como ocurre con todos los medicamentos, se deben tomar precauciones durante el uso en embarazo y lactancia.
4.7 Efecto sobre la capacidad para conducir vehículos y utilizar máquinas
No se han descrito
4.8 Reacciones adversas
Las frecuencias de las reacciones adversas se definen como: frecuentes (>1/100, <1/10), poco frecuentes (>1/1.000, <1/100), raras (>1/10.000, <1/1.000) o desconocidas (no se pueden estimar a partir de los datos disponibles).
Trastornos generales y reacciones en el lugar de administración:
Frecuentes: Tras la administración puede aparecer un sabor amargo debido a un método de aplicación incorrecto, por ejemplo, con la cabeza demasiado inclinada hacia atrás. En ocasiones, este sabor amargo puede originar náuseas.
Poco frecuentes: irritación de la mucosa nasal que se puede manifestar con escozor, picor, estornudos.
Frecuencia desconocida: En casos aislados puede aparecer epistaxis.
4.9 Sobredosis
Los resultados de los estudios en animales muestran que las dosis tóxicas pueden producir síntomas sobre el SNC, por ejemplo, excitación, temblor, convulsiones. Si esto ocurriese en humanos, se iniciará un tratamiento sintomático y de apoyo puesto que no existe un antídoto específico. Si la sobredosis es reciente se recomienda un lavado gástrico.
Con la vía de administración nasal no se prevén reacciones de sobredosis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: otros antihistamínicos para uso sistémico.
Código ATC: R06A X 19
Azelastina, derivado de la ftalazinona de nueva estructura, está clasificado como un potente antialérgico de acción prolongada con propiedades antagonistas H1 especialmente fuertes.
Datos de estudios en animales muestran que cuando se alcanzan niveles altos de azelastina se produce tanto la inhibición como la liberación de los mediadores químicos (ej., leucotrienos, histamina, serotonina) involucrados en la reacción alérgica.
5.2 Propiedades farmacocinéticas
Después de aplicaciones nasales repetidas (0,14 mg en cada fosa nasal dos veces al día), los niveles plasmáticos de azelastina fueron alrededor de 0,26 ng/ml. Los niveles del metabolito activo demetilazelastina fueron detectados en el límite inferior de cuantificación (0.12 ng/ml) o por debajo de él.
Después de administraciones orales repetidas, se determinó la Cmax media de los niveles plasmáticos en equilibrio estacionario, siendo 3.9 ng/ml para azelastina y 1,86 ng/ml para demetilazelastina después de administrar 2.2 mg de Azelastina dos veces al día, que es la dosis oral terapéutica para el tratamiento de la rinitis alérgica. Tras la administración oral, azelastina se absorbe rápidamente mostrando una biodisponibilidad absoluta del 81%. Los alimentos no tienen influencia sobre la absorción. El volumen de distribución es alto indicando una distribución predominantemente en los tejidos periféricos.
El grado de unión a proteínas es bajo (80-95% un nivel demasiado bajo como para afectar a las reacciones de desplazamiento del fármaco).
La vida media de eliminación plasmática tras la dosis única de azelastina es de aproximadamente 20 horas para Azelastina y de unas 45 horas para N-demetil Azelastina (metabolito terapéuticamente activo). La excreción se produce principalmente por vía fecal. La eliminación prolongada de pequeñas cantidades de la dosis en heces sugiere que puede tener lugar una circulación enterohepática.
6. DATOS FARMACÉUTICOS
6.1 Relación de excipientes:
Hidroxipropilmetilcelulosa, edetato de disodio, ácido cítrico anhidro, fosfato de sodio, cloruro de sodio, agua purificada.
6.2 Incompatibilidades:
No procede.
6.3 Período de validez:
3 años.
6.4 Precauciones especiales de conservación
No almacenarse por debajo de 8°C. No refrigerar.
6.5 Naturaleza y contenido del recipiente
Frascos de cristal ámbar de 10 ml y 20 ml con válvula dosificadora incorporada.
6.6 Precauciones especiales reeliminación y otras manipulaciones
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN.
MEDA PHARMA S.A.U
Avda. de Castilla, 2. 28830- San Femando de Henares (Madrid)
8. NÚMERO DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN.
61.056
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 27 de Diciembre 1996 Fecha de la última revalidación: 18 Agosto 2007
10. FECHA DE LA REVISIÓN DEL TEXTO
Julio 2000