Colfinair 1 Millon De Ui Polvo Para Solucion Para Inhalacion Por Nebulizador
Información obsoleta, busque otro
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Colfinair 1 millón de UI polvo para solución para inhalación por nebulizador.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial de 10 ml contiene 1 millón de UI equivalente a 80 mg de colistimetato sódico.
3. FORMA FARMACÉUTICA
Polvo para solución para inhalación por nebulizador.
Polvo blanco.
4. DATOS CLÍNICOS
4.1. Indicaciones terapéuticas
Colfinair está indicado para el tratamiento por vía inhalatoria de la infección pulmonar por Pseudomonas aeruginosa en pacientes con fibrosis quística (FQ).
Deben tenerse en cuenta las recomendaciones oficiales sobre el uso adecuado de agentes antibacterianos.
4.1 Indicaciones terapéuticas
Colfinair está indicado en adultos y pacientes pediátricos para el tratamiento de infecciones pulmonares crónicas causadas Pseudomonas aeruginosa, en pacientes con fibrosis quística (ver sección 5.1).
Se deben tener en cuenta las recomendaciones oficiales sobre el uso adecuado de agentes antibacterianos.
4.2 Posología y forma de administración
Se recomienda que el colistimetato de sodio (CMS) se administre bajo la supervisión de médicos con la experiencia adecuada en su uso.
Posología
La dosificación se puede ajustar en función de la gravedad de la enfermedad y de la respuesta clínica. Intervalo de dosis recomendado:
Administración por vía inhalatoria
Adultos, adolescentes y niños > 2 años
1-2 MUI dos o tres veces al día (máx. 6 MUI/día)
Niños < 2 años
0,5-1 MUI dos veces al día (máx. 2 MUI/día)
SAN IDAD, POLITICA LITIGA
SOCIAL E IGUALDAD IALDAD
Agencia esparto» de cSacse-
medicamentos y ¡y
proouctcs san-íanos taws
Deben observarse las guías clínicas pertinentes sobre pautas posológicas, incluyendo la duración del tratamiento, la periodicidad del tratamiento y la administración combinada de otros agentes antibacterianos.
Pacientes de edad avanzada
No se considera necesario el ajuste de la dosis
Insuficiencia renal
No se considera necesario el ajuste de la dosis, sin embargo, se recomienda precaución en pacientes con insuficiencia renal (ver las secciones 4.4 y 5.2).
Insuficiencia hepática
No se considera necesario el ajuste de la dosis Forma de administración Vía inhalatoria
En prematuros y neonatos se debe tener un cuidado especial ya que la función renal está insuficientemente desarrollada en esta población.
Para su uso en niños menores de 2 años de edad se recomienda PARI LC Sprint Baby (adaptador de tobera roja) con mascarilla.
El contenido de un vial de Colfinair de 1 millón de UI debe disolverse en 3 ml de solución de cloruro sódico isotónica.
Consulte en la sección 6.6 las instrucciones de dilución del producto antes de la administración.
Características de dispensación del fármaco previstas según estudios in vitro (in vivo) con distintos dispositivos nebulizadores para Colfinair 1,0 millón de UI disuelto en 3 ml de cloruro de sodio isotónico. (mín - máx)
|
Sistema nebulizador |
PARI LC SPRINT con compresor PARI Boy S |
Nebulizador eFlowrapid |
|
Total de fármaco dispensado |
25 mg CMS (22,1 - 27,2) |
27 mg CMS (19,9 - 30,5 |
|
Masa de partículas finas < 5 pm |
15 mg CMS (12,7 - 16,8) |
18mg CMS (13,0 - 20,8) |
|
Tasa de dispensación del fármaco |
4,6 mg CMS/ min (4,3 - 5,0) |
7,0 mg CMS/ min (5,2 - |
|
Diámetro aerodinámico mediana de la masa |
3,8 pm (3,3 - 4,3) |
4,1 pm (4,0 - 4,4) |
|
Desviación estándar geométrica |
2,2 |
1,6 |
80 mg CMS corresponde aproximadamente a 1 millón de UI
• El tiempo de nebulización puede incrementarse durante 60 ciclos de nebulización desde ~ 3 minutos a ~ 4,5 minutos con el dispositivo nebulizador de mano eFlowrapid.
• El nebulizador debe mantenerse en posición horizontal durante su utilización.
• El paciente debe estar sentado en posición erguida durante la inhalación. La inhalación debe realizarse aplicando un patrón de respiración normal sin interrupción.
SAN IDAD, POLITICA LITIGA
SOCIAL E IGUALDAD IALDAD
Agencia esparto» de cSacse-
medicamentos y ¡y
Sfoouctos san-tanos taws
• El nebulizador debe limpiarse y desinfectarse tras el uso tal como se describe en las instrucciones de uso del nebulizador.
El colistimetato de sodio en solución acuosa se hidroliza al principio activo colistina.
Consulte las precauciones especiales de eliminación y de manipulación de soluciones reconstituidas en la sección 6.6.
Si está tomando otros tratamientos, debe tomarlos en el orden recomendado por su médico. Tabla de conversión de dosis:
En la UE, la dosis de colistimetato de sodio (CMS) se debe prescribir y administrar únicamente en forma de Unidades Internacionales (UI). La etiqueta del producto indica el número de UI por vial.
Se han producido confusiones y errores de medicación debido a las diferentes formas de expresar la dosis en términos de potencia. En EE.UU. y en otras partes del mundo, la dosis se expresa como miligramos de actividad de colistina base (mg CBA).
La siguiente tabla de conversión ha sido preparada a título informativo y los valores recogidos se deben considerar solamente nominales y aproximados.
Tabla de conversión de CMS
|
Potencia |
~ masa de CMS (mg)* | |
|
U.I. |
~ mg CBA | |
|
12.500 |
0,4 |
1 |
|
150.000 |
5 |
12 |
|
1.000.000 |
34 |
80 |
|
4.500.000 |
150 |
360 |
|
9.000.000 |
300 |
720 |
principio activo = 12.500 UI/mg
*Potencia nominal de
4.3 Contraindicaciones
Hipersensibilidad al colistimetato sódico o a la polimixina B.
Miastenia gravis: Se sabe que el colistimetato sódico reduce la cantidad de acetilcolina liberada de la unión neuromuscular presináptica y por tanto no debe usarse en pacientes con miastenia gravis.
4.4 Advertencias y precauciones especiales de empleo
Con la inhalación de antibióticos puede producirse tos y broncoespasmo.
Se recomienda administrar la primera dosis bajo supervisión médica. Se recomienda usar un broncodilatador antes de cada dosis. Esto debería ser rutinario, especialmente si el broncodilatador es parte del régimen terapéutico actual del paciente. Se debería evaluar el VEFj antes y después de la dosis. Si hay evidencia de hiperreactividad bronquial inducida por el colistimetato sódico en un paciente que no recibe pre-tratamiento con broncodilatadores se debe repetir la prueba en otra ocasión usando un broncodilatador. La evidencia de hiperreactividad bronquial en presencia de un broncodilatador puede indicar una respuesta alérgica y deberá interrumpirse la administración de Colfinair. El broncoespasmo se debe tratar como esté indicado clínicamente.
La hiperreactividad bronquial como respuesta al colistimetato sódico puede desarrollarse con el uso continuado a lo largo del tiempo y se recomienda que el VEF1 antes y después del tratamiento se evalúe en las visitas periódicas a la consulta.
En caso de hipersensibilidad con respecto a las dosis y volúmenes recomendados se deben usar soluciones más diluidas, añadiendo alrededor de 1 - 3 ml de solución salina isotónica a los volúmenes y concentraciones de dosis recomendados.
Usar con extrema precaución en pacientes con porfiria, ya que el colistimetato sódico puede causar una exacerbación de la misma.
Pueden darse nefrotoxicidad o neurotoxicidad si se excede la dosis parenteral recomendada. El riesgo se reduce debido a la baja biodisponibilidad durante la inhalación, pero Colfinair debe usarse con precaución en pacientes con insuficiencia renal (véase la sección 4.2). Debe monitorizarse la aparición de reacciones neurotóxicas así como la función renal.
Insuficiencia renal
El colistimetato sódico se excreta por vía renal y es nefrotóxico si se alcanzan altas concentraciones séricas. Aunque esto es poco probable durante el tratamiento por inhalación, se recomienda que se hagan estimaciones de la concentración sérica especialmente en pacientes con insuficiencia renal.
Resistencia microbiana
Se ha notificado resistencia adquirida al colistimetato sódico de Pseudomonas aeruginosa mucoide durante el uso clínico. Deberían hacerse pruebas de sensibilidad a los pacientes que se tratan a largo plazo, en las visitas periódicas a la consulta, y siempre que un paciente experimente una exacerbación (véase la sección 5.1).
4.5 Interacción con otros medicamentos y otras formas de interacción
Debe evitarse el uso concomitante de colistimetato sódico con otros medicamentos de potencial neurotóxico o nefrotóxico (p. ej. cefalosporinas, aminoglucósidos, ciclosporina), incluidos aquellos que se administran por vía intravenosa o intramuscular.
Durante el uso concomitante de narcóticos por vía inhalatoria (p. ej. éter, halotano), relajantes musculares y aminoglucósidos con Colfinair se deberá monitorizar exhaustivamente la aparición de reacciones neurotóxicas debido al efecto de prolongación de la inhalación de narcóticos.
Debido a los efectos del colistimetato sódico en la liberación de acetilcolina, los relajantes musculares no despolarizantes deben utilizarse con suma precaución en pacientes que reciban Colfinair ya que sus efectos podrían prolongarse.
4.6 Fertilidad, embarazo y lactancia
No existen datos adecuados del uso de colistimetato sódico en mujeres embarazadas. Estudios de dosis única en el embarazo mostraron que el colistimetato sódico atraviesa la barrera placentaria y puede haber riesgo de toxicidad fetal si se administran dosis repetidas a pacientes embarazadas. Los estudios en animales no son suficientes con relación al efecto del colistimetato sódico en la reproducción y el desarrollo (véase la sección 5.3). El colistimetato sódico no debe usarse en el embarazo a menos que el beneficio para la madre supere el riesgo potencial para el feto.
tp.
n
El colistimetato sódico se excreta en la leche materna. El colistimetato sódico debe administrarse a pacientes en periodo de lactancia solo cuando está claramente indicado y el beneficio para la madre supera el riesgo potencial para el lactante.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Colfinair sobre la capacidad de conducir y utilizar máquinas es moderada.Durante el tratamiento con colistimetato sódico puede darse neurotoxicidad con posibilidad de mareos, confusión o perturbación visual. Se debe advertir a los pacientes que no conduzcan ni operen maquinaria si ocurren estos efectos.
4.8 Reacciones adversas
Las reacciones adversas más frecuentes tras la nebulización de colistimetato sódico son tos y broncoespasmo, en aproximadamente el 10 % de los pacientes. En pacientes con fibrosis quística tratados por inyección por vía intravenosa o intramuscular, se han notificado acontecimientos neurológicos en hasta un 27 % de pacientes.
Resumen tabulado de reacciones adversas
Las reacciones adversas se enumeran en la Tabla 1 de acuerdo con la clasificación de órganos del sistema MedDRA. Dentro de cada clase de órganos del sistema, las reacciones adversas se enumeran por frecuencia, siendo las primeras las reacciones más frecuentes. Además, para cada reacción adversa se indica la categoría de frecuencia correspondiente según la convención siguiente (CIOMS III): Muy frecuentes (> 1/10); frecuentes (> 1/100 a <1/10); poco frecuentes (> 1/1.000 a <1/100); raras (> 1/10.000 a <1/1.000); muy raras (<1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
La probabilidad de acontecimientos adversos puede estar relacionada con la edad, la función renal y el estado del paciente.
Tabla 1 Reacciones adversas
|
Reacciones adversas |
Categoría de frecuencia |
|
Trastornos respiratorios, torácicos y mediastínicos | |
|
Dolor faringolaríngeo |
Muy frecuente |
|
Molestias faringolaríngeas |
Muy frecuente |
|
Tos |
Muy frecuente |
|
Disnea |
Muy frecuente |
|
Sibilancias |
Muy frecuente |
|
Dificultad respiratoria |
Muy frecuente |
|
Volumen espiratorio forzado disminuido |
Muy frecuente |
|
Apnea |
Muy frecuente |
|
Trastornos psiquiátricos | |
|
Estado de confusión |
No conocida |
|
Trastorno psicótico |
No conocida |
|
Trastornos del sistema nervioso | |
|
Mareo |
No conocida |
|
Parestesia |
No conocida |
|
Disartria |
No conocida |
|
Desequilibrio del sistema nervioso autónomo |
No conocida |
|
Trastornos oculares Perturbación visual |
No conocida |
|
Trastornos del oído y del laberinto Vértigo |
No conocida |
|
Trastornos renales y urinarios Insuficiencia renal |
No conocida |
Los pacientes con una insuficiencia renal grave y a las dosis más altas pueden experimentar efectos adversos conocidos para la administración intravenosa.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaram.es
4.9 Sobredosis
Síntomas:
La sobredosis puede causar debilidad muscular, apnea y posible parada respiratoria así como insuficiencia renal aguda caracterizada por una disminución de la producción urinaria y concentraciones séricas incrementadas de nitrogeno ureico en sangre (NUS) y creatinina.
No hay ningún antídoto específico.
Tratamiento:
La gestión de la sobredosis se hará mediante un tratamiento de apoyo y medidas para incrementar la tasa de eliminación de colistina como la diuresis con manitol, hemodiálisis prolongada o diálisis peritoneal.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: otros antibacterianos, polimixinas, código ATC: J01XB01 Mecanismo de acción
El colistimetato sódico es un antibiótico polipéptido cíclico derivado de Bacillus polymyxa var. colistinus. Los antibióticos polimixinas son agentes catiónicos que actúan dañando la pared celular. Los efectos fisiológicos resultantes son letales para las bacterias. Las polimixinas son selectivas para las bacterias Gramnegativas que tienen una membrana externa hidrofóbica.
Puntos de corte
Sensible (S) < 4 mg/l Resistente (R) > 8 mg/l Resistencia
Las bacterias resistentes se caracterizan por la modificación de los grupos fosfato en los lipopolisacáridos que se sustituyen por etanolamina o aminoarabinosa. Las bacterias Gram negativas intrínsecamente
resistentes, como Proteus mirabilis y Burkholderia cepacia, muestran una sustitución completa de sus fosfolípidos con etanolamina o aminoarabinosa.
Se ha notificado la resitencia adquirida al colistimetato sódico de la Pseudomonas aeruginosa en aproximadamente un 3 %. Deben realizarse pruebas de sensibilidad a los pacientes que se estén tratando a largo plazo.
Resistencia cruzada
Es de esperar una resistencia cruzada entre el colistimetato sódico y la polimixina B. Puesto que el mecanismo de acción de las polimixinas es distinto al de otros antibióticos, la resistencia al colistimetato sódico y la polimixina por el mecanismo arriba indicado únicamente no es de esperar que provoque una resistencia a otras clases de antibióticos.
Sensibilidad
La prevalencia de resistencia adquirida puede variar geográficamente y con el tiempo para las especies seleccionadas, por lo que sería deseable disponer de información local sobre resistencias, especialmente cuando se tratan infecciones graves. Cuando sea necesario, se debe obtener asesoramiento de expertos si la prevalencia local de la resistencia es tal que la utilidad del agente en al menos algunos tipos de infecciones sea cuestionable.
Especies comúnmente sensibles
Pseudomonas aeruginosa
Especies para las que la resistencia adquirida podría ser un problema
Enterobacter spp Klebsiella spp
Organismos resistentes inherentemente
Brucella spp
Burkholderia cepacia y especies relacionadas.
Neisseria spp Proteus spp Providencia spp Serratia spp
Anaerobios
Todos los organismos Gram positivos
*Los resultados in vitro pueden no correlacionarse con las respuestas clínicas en el caso de la Acinetobacter spp.
PARI LC PLUS y PARI LC STAR son nebulizadores que se han utilizado en el pasado para la nebulización del colistimetato sódico. Estos nebulizadores tipo "jet" se compararon con el eFlowrapid para la nebulización de colistimetato sódico de 1 millón de UI disuelto en 3 ml de solución salina isotónica mediante pruebas invitro:
Nebulizador: eFlowrapid PARI LC PLUS
PARI LC STAR
Total de fármaco dispensado [mg ± IC 95 %] 26,6 ± 1,62 26,0 ± 0,33
27,5 ± 2,06
Tasa de dispensación del fármaco [mg/min ± IC 95 %] 7,0 ± 0,39 4,3 ± 0,14 3,2
± 0,27
5.2 Propiedades farmacocinéticas
Absorción
La absorción en el tracto gastrointestinal es insignificante.
Cuando se administra por nebulización, se ha notificado absorción variable que puede depender del tamaño de la partícula del aerosol, del sistema nebulizador y del estado pulmonar. Los estudios en voluntarios sanos y pacientes con infecciones varias han informado de niveles séricos que van desde nulos a concentraciones potencialmente terapéuticas de 4 mg/l o superiores. Por tanto, debe tenerse siempre en cuenta la posibilidad de absorción sistémica cuando se trate a los pacientes por vía inhalatoria.
Distribución
Tras la administración a pacientes con fibrosis quística de 7,5 mg/kg/día en dosis divididas administradas como infusiones intravenosas de 30 minutos hasta el estado estacionario, la Cmáx se determinó en 23 ± 6 mg/l y la Cmín a las 8 h fue de 4,5 ± 4 mg/l. En otro estudio en pacientes similares a quienes se administró
2 millones de unidades cada 8 horas durante 12 días la C máx fue de 12,9 mg/l (5,7 - 29,6 mg/l) y la Cmín fue de 2,76 mg/l (1,0 - 6,2 mg/l). En voluntarios sanos a quienes se administró una inyección en bolo de 150 mg (2 MIU aprox.), los valores séricos máximos de 18 mg/l se observaron 10 minutos tras la inyección.
La unión a las proteínas es baja. Las polimixinas persisten en el hígado, riñón, cerebro, corazón y músculos. Un estudio en pacientes con fibrosis quística da un volumen de distribución en estado estacionario de 0,09 l/kg.
Metabolismo
El colistimetato sódico se convierte en su base in vivo. Puesto que el 80 % de la dosis puede recuperarse sin alterar en la orina, y no hay excreción biliar, se puede asumir que el resto del principio activo está activo en los tejidos. El mecanismo se desconoce.
Eliminación
La principal vía de eliminación tras la administración parenteral es por excreción renal, con el 40 % de una dosis parenteral recuperada en la orina en el plazo de 8 horas y alrededor del 80% en 24 horas. Puesto que el colistimetato sódico se excreta mayoritariamente en la orina, es necesaria una reducción de dosis en la insuficiencia renal para evitar la acumulación. Consulte la tabla de la sección 4.2.
Tras la administración intravenosa a adultos sanos, la semivida de eliminación es de unas 1,5 horas. En un estudio en pacientes con fibrosis quística a quienes se les administró una única infusión intravenosa de 30 minutos, la semivida de eliminación fue de 3,4 ± 1,4 horas.
La eliminación del colistimetato sódico tras la inhalación no se ha estudiado. Un estudio en pacientes de fibrosis quística no detectó colistimetato sódico en orina tras inhalar 1 millón de UI dos veces al día durante
3 meses.
La cinética del colistimetato sódico parece ser similar en niños y adultos, incluidos los pacientes de edad avanzada, siempre que la función renal sea normal. Los datos disponibles son limitados sobre el uso en neonatos sugieren que la cinética es similar a la de los niños y adultos, pero se debe considerar la posibilidad de unos valores séricos máximos más altos y una semivida prolongada en estos pacientes y se deben monitorizar los valores séricos.
Concentraciones séricas y farmacocinética en 5 pacientes que recibieron colistimetato sódico por inhalación
|
Parámetro |
160 mg (aproximadamente 2 millones de UI) de colistimetato sódico nebulizado |
|
AUCo_4 (h/mg/l) |
165,9 ± 76,5 |
|
Cmáx (mg/l) |
0,051 ± 0,0244 |
|
Tmáx (h) |
1,9 ± 1,2 |
|
Ka (h-1 ) |
3,0 ± 1,8 |
|
U (h) |
10,4 ± 3,6 |
|
Cl/F |
0,27 ± 0,15 |
5.3 Datos preclínicos sobre seguridad
Los datos sobre la genotoxicidad potencial son limitados y no existen datos sobre carcinogenicidad para el colistimetato sódico. Se ha demostrado que el colistimetato sódico induce aberraciones cromosómicas en los linfocitos humanos in vitro. Este efecto puede estar relacionado con una reducción en el índice mitótico, que también se observó.
Los estudios de toxicidad en la reproducción en ratas y ratones no indican propiedades teratogénicas. Sin embargo, el colistimetato sódico administrado por vía intramuscular durante la organogénesis a conejos a 4,15 y 9,3 mg/kg provocó pie varo en 2,6 y 2,9 % de los fetos, respectivamente. Estas dosis son 0,5 y 1,2 veces la dosis humana diaria máxima. Además, se dio un aumento de la resorción a 9,3 mg/kg.
No hay otros datos de seguridad preclinica relevantes para el médico que prescribe que puedan sumarse a los datos de seguridad derivados de la exposición del paciente y ya incluidos en otras secciones de la Ficha Técnica.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Ninguno
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
3 años
Soluciones reconstituidas:
Colfinair se puede conservar tras su reconstitución con una solución salina al 0,9% durante 24 horas por debajo de los 25 °C. No se recomienda la conservación durante más de 24 horas debido al riesgo de contaminación microbiana de la solución reconstituida.
]£
Por favor, siga las instrucciones del fabricante sobre el uso correcto del nebulizador seleccionado para su uso con la solución de Colfinair.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 25°C. Mantener el vial dentro del envase externo de cartón para protegerlo de la luz.
Consulte en la sección 6.3 las condiciones de conservación del medicamento reconstituido.
6.5 Naturaleza y contenido del envase
Colfinair 1 millón de UI viales de vidrio incoloro de 10 ml con tapones rojos con perforación ("levantar y rasgar").
Presentación:
2 cajas de cartón
3 ml cada una (60 viales) y

Caja de cartón que contiene 8 cajas de cartón de 7 viales cada una (56 viales),
INQUA Cloruro
de sodio 9 mg/ml (0.9%) solución para inhalación que contienen 30 viales de un
dispositivo nebulizador de mano eFlowrapid.
6.6 Precauciones especiales de eliminación Precauciones especiales de eliminación y otras manipulaciones
La dosis necesaria de Colfinair debe disolverse agitándola con suavidad en el volumen respectivo de solución de cloruro de sodio isotónica estéril. Debido a las propiedades espumosas de Colfinair, se debe evitar agitar vigorosamente. La solución resultante para la nebulización debe ser transparente y se debe transferir con cuidado al depósito de medicación del nebulizador. Si desea más instrucciones para el manejo y uso, consulte las instrucciones de uso del nebulizador.
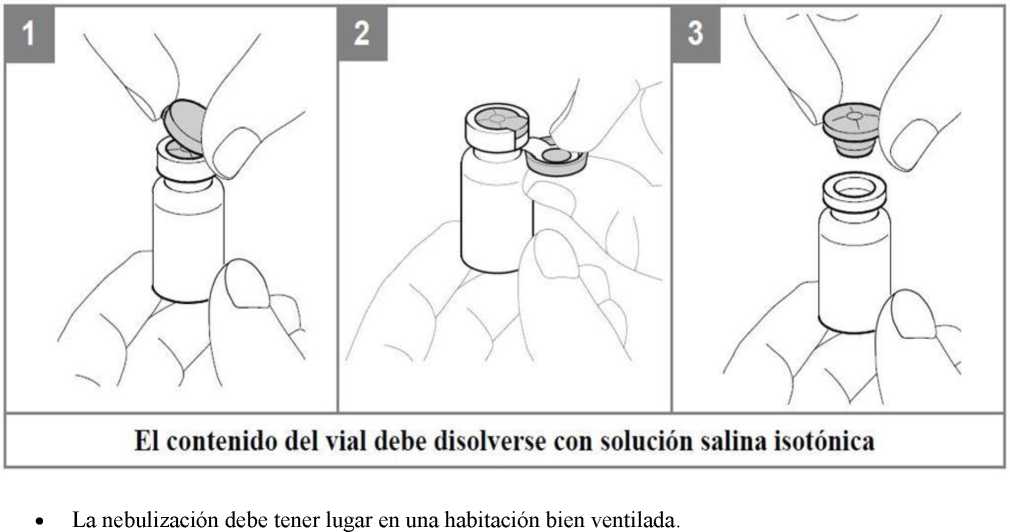

La solución es de un solo uso, y toda la solución restante debe desecharse.
Para obtener información más detallada sobre el dispositivo, consulte el manual de instrucciones del nebulizador.
Consulte en la sección 4.2 las instrucciones de dilución del producto antes de la administración. El aspecto de la solución tras su reconstitución debe ser transparente.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
PARI Pharma GmbH Moosstrasse 3 82319 Starnberg Alemania
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
75706
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
26/02/2014
10. FECHA DE LA REVISIÓN DEL TEXTO
03/2015
MINISTER10DE )E
SAN IDAD, POLITICA LITIGA
SOCIAL E IGUALDAD IALDAD
Agencia esparto» de cSacse-
medicamentos y ¡y
proouctcs san-íanos taws