Beriplex 500 Ui Polvo Y Disolvente Para Solucion Para Inyeccion
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Beriplex 500 UI, polvo y disolvente para solución inyectable. Beriplex 1000 UI, polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Composición cualitativa y cuantitativa
Beriplex se presenta como polvo y disolvente para solución inyectable que contiene el complejo de protrombina humano. El producto contiene nominalmente las siguientes UI de los factores de coagulación humano que a continuación se citan en la siguiente tabla.
|
Nombre del componente |
Contenido después de la reconstitución (UI/ml) |
Beriplex 500 UI contenido por vial (UI) |
Beriplex 1000 UI contenido por vial (Uü |
|
Principios activos | |||
|
Factor II de la coagulación, humano |
20 - 48 |
400 - 960 |
800 - 1920 |
|
Factor VII de la coagulación, humano |
10 - 25 |
200 - 500 |
400 - 1000 |
|
Factor IX de la coagulación, humano |
20 - 31 |
400 - 620 |
800 - 1240 |
|
Factor X de la coagulación, humano |
22 - 60 |
440 - 1200 |
880 - 2400 |
|
Otros principios activos | |||
|
Proteína C |
15 - 45 |
300 - 900 |
600 - 1800 |
|
Proteína S |
12-38 |
240-760 |
480 - 1520 |
El contenido total de proteínas es de 6-14 mg/ml de solución reconstituida.
La actividad específica del factor IX es de 2,5 UI por mg de proteína total.
Las actividades de todos los factores de la coagulación, así como de las proteínas C y S (antígenos) se analizan de acuerdo con los actuales estándares internacionales válidos de la OMS.
Excipiente(s) con efecto conocido
Hasta 343 mg de sodio (aproximadamente 15 mmol) por 100 ml de solución.
La lista completa de excipientes se encuentra en la Sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo blanco o ligeramente coloreado o sólido friable.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
- Tratamiento y profilaxis perioperativa de sangrados en deficiencia adquirida de los factores de la coagulación del complejo de protrombina, como la deficiencia causada por el tratamiento con antagonistas de la vitamina K, o en caso de sobredosis de antagonistas de la vitamina K, cuando es necesaria una rápida corrección de la deficiencia de los mismos.
- Tratamiento y profilaxis perioperativa en casos de sangrados en la deficiencia congénita de alguno de los factores de la coagulación dependientes de la vitamina K, cuando no se dispone de productos purificados del factor específico.
4.2 Posología y forma de administración
Posología
Las pautas posológicas que a continuación se dan, sólo tienen un carácter general. El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de los trastornos de la coagulación. La posología y la duración de la terapia de sustitución dependen de la gravedad de la deficiencia, de la localización e importancia de la hemorragia y del cuadro clínico del paciente.
La posología y la frecuencia de administración se establecerán de forma individual para cada paciente. Los intervalos de dosificación deben adaptarse a las vidas medias circulantes de los respectivos factores de la coagulación del complejo de protrombina (ver Sección 5.2). Los requerimientos posológicos individuales sólo pueden ser identificados sobre la base de una determinación periódica de los niveles plasmáticos de los factores de la coagulación en cuestión, o del análisis global de los niveles del complejo de protrombina (INR, Prueba de Quick) y la monitorización continua de la situación clínica del paciente.
En el caso de intervenciones de cirugía mayor es imprescindible una monitorización precisa de la terapia de sustitución, por medio de análisis de la coagulación (ensayos específicos de los factores de la coagulación y/o análisis globales para medir los niveles del complejo de protrombina).
- Sangrado y profilaxis perioperativa de sangrados durante el tratamiento con medicamentos antagonistas de la vitamina K:
La posología dependerá del valor del INR antes del tratamiento y del INR que se desea conseguir. En la siguiente tabla se dan dosis aproximadas (ml de solución reconstituida del producto/kg de peso corporal y UI de FIX/kg de peso corporal) que se requieren para normalizar el INR (p.ej. < 1.3) a diferentes niveles iniciales de INR.
|
INR inicial |
2,0 - 3,9 |
4,0 - 6,0 |
> 6,0 |
|
Dosis aproximada (ml/kg de peso corporal) |
1 |
1,4 |
2 |
|
Dosis aproximada de Factor IX |
25 |
35 |
50 |
|
(UI)/kg de peso corporal |
La dosis máxima recomendada no debe exceder un máximo de 5000 UI de FIX.
La corrección del trastorno de la hemostasia inducido por los antagonistas de la vitamina K se produce como muy tarde 30 minutos después de la inyección y persistirá durante 6 - 8 horas aproximadamente. Sin embargo, el efecto de la vitamina K, si se administra de manera concomitante, se alcanza normalmente transcurridas de 4 a 6 horas. Por eso, no se requiere, usualmente, un tratamiento repetido con el complejo humano de protrombina cuando se ha administrado vitamina K.
Estas recomendaciones están basadas en datos de ensayos clínicos con un número limitado de individuos. La recuperación y la duración del efecto pueden variar, por lo que la monitorización del INR, durante el tratamiento es obligatoria.
- Los sangrados y la profilaxis perioperativa en deficiencia congénita de algunos de los factores de la coagulación dependientes de la vitamina K, cuando no se dispone de productos del factor de la coagulación específico:
El cálculo de la dosis requerida del concentrado del complejo de protrombina se basa en datos de ensayos clínicos:
• 1 UI de factor IX por kilo de peso corporal puede incrementar la actividad plasmática del factor IX en un 1,3 % (0,013 UI/ml) de la actividad normal.
• 1 UI de factor VII por kg de peso corporal incrementa la actividad plasmática del factor VII en un 1,7 % (0,017 UI/ml) de la actividad normal.
• 1 UI de factor II por kg de peso corporal incrementa la actividad plasmática del factor II en un 1,9 % (0,019 UI/ml) de la actividad normal.
• 1 UI de factor X por kg de peso corporal incrementa la actividad plasmática del factor X en un 1,8 % (0,018 UI/ml) de la actividad normal.
La posología de un factor específico administrado se expresa en Unidades Internacionales (UI), que están relacionadas con el estándar vigente de la OMS para cada factor. La actividad plasmática de un factor de la coagulación específico se expresa, bien como un porcentaje (relativo al plasma normal) o en Unidades Internacionales (relativas al estándar internacional para el factor de la coagulación específico).
Una Unidad Internacional (UI) de actividad de un factor de la coagulación es equivalente a la cantidad contenida en un ml de plasma humano normal.
Por ejemplo, el cálculo de la dosis requerida de factor X se basa en el hallazgo de que 1 Unidad Internacional (UI) de factor X por kg de peso corporal incrementa la actividad plasmática del factor X en 0,018 UI/ml.
La dosis requerida se determina usando la siguiente fórmula:
Unidades requeridas = peso corporal [kg] x incremento deseado de factor X [UI/ml] x 56 Donde 56 (ml/kg) es el valor recíproco de la recuperación estimada.
Si se conoce la recuperación individual, este valor debe usarse en el cálculo.
Población pediátrica
Todavía no se ha establecido la seguridad y eficacia de Beriplex en niños y adolescentes mediante ensayos clínicos controlados (ver sección 4.4).
Población geriátrica
La posología y la forma de administración en pacientes ancianos (mayores de 65 años) se corresponden con las recomendaciones generales.
Forma de administración
Ver la Sección 6.6 para las instrucciones de reconstitución del medicamento antes de su administración. La solución reconstituida debe administrarse por vía intravenosa (no más de 8 ml/min*).
* en ensayos clínicos con Beriplex, se administraron las dosis con una velocidad máxima de infusión de 0,12 ml/kg/min (menos de 8 ml/min) a pacientes con un peso <70 kg.
4.3 Contraindicaciones
Hipersensibilidad conocida al principio activo o a alguno de los excipientes listados en la sección 6.1.
En el caso de una coagulación intravascular diseminada, los medicamentos que contienen el complejo de protrombina sólo pueden administrarse tras finalizar la situación de consumo.
Historia conocida de trombocitopenia inducida por heparina.
4.4 Advertencias y precauciones especiales de empleo
Se debe consultar a un especialista con experiencia en el manejo de los trastornos de la coagulación.
En pacientes con deficiencia adquirida de factores de la coagulación dependientes de la vitamina K (p.ej. la inducida por el tratamiento con antagonistas de la vitamina K), Beriplex únicamente debe utilizarse cuando sea necesaria una rápida corrección de los niveles del complejo de protrombina, como en las hemorragias graves o la cirugía de urgencia. En los demás casos, por lo general será suficiente la reducción de la dosis del antagonista de la vitamina K y/o la administración de vitamina K.
Los pacientes tratados con un antagonista de la vitamina K pueden presentar un estado subyacente de hipercoagulación y la perfusión de complejo de protrombina podría exacerbar dicha condición.
En la deficiencia congénita de cualquiera de los factores dependientes de la vitamina K se deben utilizar productos del factor de coagulación específico, siempre que estén disponibles.
En caso de producirse reacciones de tipo alérgico o anafiláctico, se interrumpirá inmediatamente la administración de Beriplex (p.ej. interrumpir la inyección) y se instaurará el tratamiento apropiado. Las medidas terapéuticas dependerán del tipo y gravedad del efecto adverso. Se observarán los estándares clínicos actuales para el tratamiento del shock.
Cuando se tratan pacientes con deficiencia congénita o adquirida con complejo de protrombina humano, en especial en administraciones repetidas existe riesgo de trombosis o de coagulación intravascular diseminada. Dicho riesgo puede ser más elevado en el tratamiento de la deficiencia aislada de factor VII, puesto que los demás factores de la coagulación dependientes de la vitamina
K, con vidas medias más prolongadas, se pueden acumular hasta niveles notablemente superiores a los normales. Los pacientes a los que se administra complejo de protrombina humano deben ser sometidos a estrecha observación para detectar la presencia de signos o síntomas de coagulación intravascular diseminada o trombosis.
Debido al riesgo potencial de complicaciones tromboembólicas se debe tener precaución al administrar Beriplex a pacientes con antecedentes de enfermedad cardiaca coronaria o de infarto de miocardio, a pacientes con enfermedad hepática, a pacientes en el pre o postoperatorio, a neonatos o a pacientes con riesgo de fenómenos tromboembólicos o de coagulación intravascular diseminada o deficiencia simultánea de inhibidor de la coagulación. En cada una de estas situaciones, el beneficio potencial del tratamiento con Beriplex se deberá sopesar frente al riesgo potencial de tales complicaciones. En pacientes con coagulación intravascular diseminada y sepsis el tratamiento sustitutivo de antitrombina III deberá considerase antes de la administración con Beriplex.
En los pacientes con coagulación intravascular diseminada puede ser necesario, en determinadas circunstancias, sustituir los factores de la coagulación del complejo de protrombina. No obstante, dicha sustitución sólo se puede realizar una vez finalizada la situación de consumo (p.ej. por tratamiento de la causa subyacente con normalización persistente del nivel de antitrombina III).
Cuando se utiliza Beriplex para normalizar la coagulación alterada, deberá considerarse la administración profiláctica de heparina.
No se dispone de datos relativos al uso de Beriplex en el caso de hemorragia perinatal en neonatos debida a la deficiencia de vitamina K.
Beriplex contiene hasta 343 mg de sodio (aproximadamente 15 mmol) por 100 ml. Esto deberá ser tenido en cuenta por los pacientes bajo dieta controlada en sodio.
Seguridad vírica
Las medidas estándares para la prevención de infecciones resultantes del uso de medicamentos derivados de la sangre o el plasma humanos incluyen la selección de donantes, el análisis de detección en las donaciones individuales y en las mezclas de plasmas de marcadores específicos de infección así como la inclusión de etapas de fabricación eficaces en la inactivación/eliminación de virus. A pesar de ello, cuando se administran medicamentos derivados de sangre o plasma humanos, no puede excluirse totalmente la posibilidad de transmisión de agentes infecciosos. Ello también es cierto en el caso de virus y demás patógenos desconocidos o emergentes.
Las medidas aplicadas se consideran eficaces para los virus envueltos tales como el virus de la inmunodeficiencia humana (VIH), virus de la hepatitis B (VHB), virus de la hepatitis C (VHC) y para los virus no envueltos como el virus de la hepatitis A (VHA) y parvovirus B19.
Deberá considerarse la conveniencia de una vacunación adecuada (hepatitis A y B) en pacientes bajo administración periódica/repetida de medicamentos del complejo protrombínico derivados del plasma humano.
Se recomienda encarecidamente que cada vez que se administre Beriplex a un paciente, se registre el nombre y número de lote del producto con el fin de mantener la trazabilidad entre el paciente y el lote de producto.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los productos conteniendo complejo de protrombina humano neutralizan el efecto del tratamiento con antagonistas de la vitamina K, pero no se conocen interacciones con otros medicamentos.
Cuando se realizan pruebas de coagulación sensibles a la heparina, en pacientes tratados con altas dosis de complejo de protrombina humano, se debe considerar la heparina contenida en el producto administrado.
4.6 Fertilidad, embarazo y lactancia
No se ha establecido la seguridad del complejo de protrombina humano para su uso durante el embarazo o lactancia humanas con estudios clínicos controlados. No se dispone de estudios de experimentación animal con Beriplex para evaluar la seguridad en el embarazo, desarrollo fetal / embrionario, parto o desarrollo postnatal.
Por lo tanto, el complejo de protrombina humano sólo deberá utilizarse durante el embarazo y la lactancia si está claramente indicado.
Fertilidad
No se dispone de datos sobre fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se ha realizado estudios sobre la capacidad de conducir o usar maquinaria.
4.8 Reacciones adversas
Las siguientes reacciones adversas se basan en la experiencia postcomercialización así como en la literatura científica.
Trastornos del sistema inmunológico:
Muy raramente se ha observado hipersensibilidad o reacciones alérgicas (que pueden incluir angioedema, quemazón y punzadas en el lugar de la inyección, escalofríos, sofoco, urticaria generalizada, cefalea, ronchas, hipotensión, letargia, náuseas, agitación, taquicardia, angina de pecho, hormigueo, vómitos o dificultad respiratoria) en pacientes tratados con productos conteniendo factor IX. En algunos casos, dichas reacciones han progresado hasta anafilaxia grave y se han producido en estrecha asociación temporal con el desarrollo de inhibidores del factor IX (ver Sección 4.4).
Si se producen reacciones alérgico-anafilácticas, se debe interrumpir inmediatamente la administración de Beriplex (p.ej. interrumpir la inyección) e instaurar el tratamiento apropiado (ver Sección 4.4).
En muy raros casos se puede producir el desarrollo de anticuerpos frente a uno o varios factores del complejo de protrombina. Si se presentan dichos inhibidores, dicha condición se pone de manifiesto a través de una escasa respuesta clínica. En tales casos se recomienda contactar con un centro especializado en hemofilia.
Las reacciones adversas pueden incluir el desarrollo de una trombocitopenia de tipo II inducida por heparina (TIH, tipo II). Un signo característico de la TIH es un descenso del recuento
plaquetario superior al 50 por ciento y/o la aparición de nuevas complicaciones tromboembólicas inesperadas durante el tratamiento con heparina. La aparición de la TIH tiene lugar de manera típica entre los 4 a 14 días después del inicio del tratamiento con heparina, pero puede presentarse dentro de las 10 primeras horas en pacientes expuestos recientemente a la heparina (en los 100 días previos).
Trastornos generales y alteraciones en el lugar de administración:
En casos muy raros se ha observado aumento de la temperatura corporal.
Trastornos vasculares:
Existe un riesgo de complicaciones tromboembólicas tras la administración del complejo de protrombina humano (ver sección 4.4).
Trastornos renales y urinarios:
Se han notificado casos aislados de síndrome nefrótico tras intentar la inducción de tolerancia inmune en pacientes con hemofilia B con inhibidores del factor IX e historial de reacciones alérgicas.
Para mayor información sobre seguridad en relación con agentes transmisibles, ver sección 4.4. Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de medicamentos de Uso Humano: https://www.notificaram.es.
4.9 Sobredosis
Para evitar la sobredosis, está indicada la realización de controles periódicos del estado de coagulación durante el tratamiento ya que el uso de dosis elevadas de concentrado de complejo de protrombina (sobredosis) se ha asociado a episodios de infarto de miocardio, coagulación intravascular diseminada, trombosis venosa y embolia pulmonar. En caso de sobredosis aumenta el riesgo de complicaciones tromboembólicas o coagulación intravascular diseminada en los pacientes con riesgo de padecer dichas complicaciones.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: hemostáticos, combinación de factores II, VII, IX y X de la coagulación sanguínea.
Código ATC: B02B D01
Los factores II, VII, IX y X de la coagulación, que se sintetizan en el hígado con ayuda de la vitamina K, reciben comúnmente la denominación de complejo de protrombina. Además de los factores de la coagulación, Beriplex contiene los inhibidores de la coagulación dependientes de la vitamina K, Proteína C y Proteína S.
El factor VII es el zimógeno del factor proteasa de serina activo VIIa, por medio del cual se inicia la vía extrínseca de la coagulación sanguínea. El complejo de factor tromboplastina tisular-factor VIIa activa los factores de la coagulación IX y X, dando lugar a la formación de los factores IXa y
Xa. Con la posterior activación de la cascada de la coagulación, la protrombina (factor II) se activa y se transforma en trombina. Por acción de la trombina, el fibrinógeno se convierte en fibrina, lo que resulta en la formación del coágulo. La normal generación de trombina también es de vital importancia para la función plaquetaria, como parte de la hemostasia primaria.
La deficiencia grave de factor VII aislada conduce a una reducida formación de trombina y a una tendencia al sangrado debido a la disminución en la formación de fibrina y al deterioro de la hemostasia primaria. La deficiencia aislada de factor IX constituye una de las hemofilias clásicas (hemofilia B). La deficiencia aislada de factor II o factor X es muy rara pero en su forma grave causa una tendencia al sangrado similar a la observada en la hemofilia clásica.
Los demás componentes, los inhibidores de la coagulación Proteína C y Proteína S, también se sintetizan en el hígado. La actividad biológica de la Proteína C se potencia por el cofactor Proteína S.
La Proteína C activada inhibe la coagulación por inactivación de los factores de la coagulación Va y VIIIa. La Proteína S como cofactor de la Proteína C favorece la inactivación de la coagulación. La deficiencia de Proteína C se asocia a un riesgo aumentado de trombosis.
La deficiencia adquirida de factores de la coagulación dependientes de la vitamina K aparece durante el tratamiento con antagonistas de la vitamina K. Cuando la deficiencia es grave da lugar a una grave tendencia al sangrado, caracterizada por hemorragias retroperitoneales o cerebrales más bien que por las que afectan a músculos y articulaciones. La insuficiencia hepática grave también origina niveles notablemente reducidos de los factores de la coagulación dependientes de la vitamina K y una tendencia al sangrado clínicamente relevante que, sin embargo, con frecuencia es compleja debido a la coagulación intravascular de grado menor simultánea, niveles reducidos de plaquetas, deficiencia de los inhibidores de la coagulación y fibrinólisis alterada.
La administración de complejo de protrombina humano genera un aumento en los niveles plasmáticos de los factores de la coagulación dependientes de la vitamina K y puede corregir temporalmente el defecto de la coagulación de los pacientes con deficiencia de uno o varios de estos factores.
5.2 Propiedades farmacocinéticas
Los valores de vida media plasmática son los que se indican a continuación (datos obtenidos de un estudio clínico que incluyó 15 voluntarios sanos, valores medios, rango):
|
Factor II: |
60 |
(25 - 135) |
horas |
|
Factor VII: |
4 |
(2 - 9) |
horas |
|
Factor IX: |
17 |
(10 - 127) |
horas* |
|
Factor X: |
31 |
(17 - 44) |
horas |
|
Proteína C: |
47 |
(9 - 122) |
horas* |
|
Proteína S: |
49 |
(33 - 83) |
horas* |
*vida media terminal, modelo bicompartimental
Beriplex se distribuye y se metaboliza en el organismo del mismo modo que los factores de la coagulación II, VII, IX y X endógenos.
La administración intravenosa implica que el preparado está disponible de forma inmediata y la biodisponibilidad es proporcional a la dosis administrada.
5.3 Datos preclínicos sobre seguridad
Beriplex contiene como principios activos los factores del complejo de protrombina (factores II, VII, IX y X). Se obtienen a partir de plasma humano y actúan de igual modo que los componentes endógenos del plasma.
Los estudios con dosis únicas de 200 UI/kg (máxima dosis experimentada) del producto antecesor (pasteurizado, pero no nanofiltrado) a ratones reveló una toxicidad moderada. Los estudios no clínicos con administración de dosis repetidas (toxicidad crónica, cancerogénesis y toxicidad reproductiva) son impracticables en modelos animales convencionales debido al desarrollo de anticuerpos tras la administración de proteínas humanas heterólogas.
En conejo se demostró tolerancia local tras administración intravenosa de Beriplex. Un estudio de neoantigenicidad realizado en conejos no evidenció indicación alguna de generación de neoepitopos debidos al proceso de pasteurización.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo: heparina, albúmina humana, antitrombina III humana, cloruro sódico, citrato sódico y ácido clorhídrico o hidróxido sódico (en cantidades reducidas para el ajuste de pH).
Disolvente:
Agua para preparaciones inyectables.
6.2 Incompatibilidades
Beriplex no debe mezclarse con otros medicamentos, diluyentes o disolventes.
6.3 Periodo de validez 3 años.
Tras la reconstitución, desde un punto de vista microbiológico y dado que Beriplex no contiene conservantes, el producto reconstituido se debe usar inmediatamente. La estabilidad fisicoquímica ha sido demostrada para un periodo de 24 horas a temperatura ambiente (max. 25° C). En caso de no utilizarse inmediatamente, no debe conservarse durante más de 8 horas a temperatura ambiente.
6.4 Precauciones especiales de conservación
No conservar Beriplex a temperatura superior a 25°C. No congelar.
Mantener el envase en el cartonaje, para protegerlo de la luz.
Ver sección 6.3 sobre las condiciones de almacenamiento tras reconstitución del medicamento.
6.5 Naturaleza y contenido del envase Envase primario
.<ítp.
Beriplex 500 UI:
Viales con el polvo: Vial de inyección de vidrio incoloro (Tipo II), sellado con tapón sin látex para infusión, cápsula de aluminio y precinto desgarrable de plástico.
Vial de disolvente: 20 ml de Agua para preparaciones inyectables en vial de inyección de vidrio incoloro (Tipo I), sellado con tapón sin látex para infusión (tapón de clorobutilo), cápsula de aluminio y precinto desgarrable de plástico.
Accesorio para la inyección: 1 trasvasador con filtro 20/20.
Beriplex 1000 UI:
Viales con el polvo: Vial de inyección de vidrio incoloro (Tipo II), sellado con tapón sin látex para infusión, cápsula de aluminio y precinto desgarrable de plástico.
Vial de disolvente: 40 ml de Agua para preparaciones inyectables en vial de inyección de vidrio incoloro (Tipo I), sellado con tapón sin látex para infusión (tapón de clorobutilo), cápsula de aluminio y precinto desgarrable de plástico.
Accesorio para la inyección: 1 trasvasador con filtro 20/20.
No tienen porqué estar comercializadas todas las presentaciones.
6.6 Precauciones especiales de eliminación y otras manipulaciones
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
Método de administración
Instrucciones generales
- La solución debe ser transparente o ligeramente opalescente. Tras la filtración/extracción (ver más abajo), y antes de su administración el producto reconstituido se debe inspeccionar visualmente para detectar presencia de partículas extrañas y decoloración. No utilizar soluciones turbias o que contengan residuos.
- La reconstitución y la extracción se deben realizar en condiciones asépticas. Reconstitución
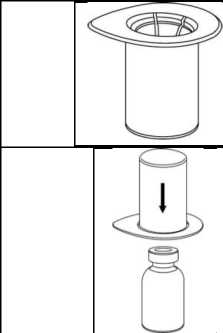
Llevar el disolvente a temperatura ambiente. Comprobar que se han retirado los precintos de los viales de producto y disolvente y que se han humedecido los tapones con solución aséptica dejando que se seque antes de abrir el envase del dispositivo Mix2Vial.
1
2
1. Abra el blister del Mix2Vial desprendiendo el precinto. No retire el Mix2Vial del blister
2. Coloque el vial del disolvente sobre una superficie limpia y plana y sujételo con firmeza. Sujete el Mix2Vial junto con el blister y empuje el terminal azul hacia abajo encajándolo en el tapón del vial del disolvente.
3
4
5
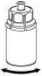
6
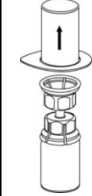
3. Retire, con cuidado, el blister del Mix2Vial sujetando el borde y tirando verticalmente hacia arriba. Asegúrese de que sólo retira el blister y no el Mix2Vial.
4. Coloque el vial del polvo liofilizado sobre una superficie plana y firme. Invierta el vial del disolvente con el Mix2Vial acoplado y empuje el terminal del adaptador transparente hacia abajo encajándolo en el tapón del vial con el polvo. El disolvente se transferirá automáticamente al vial del polvo liofilizado.
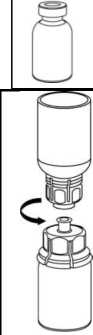
5. Con una mano, sujete el vial con el producto con el Mix2Vial y, con la otra mano, sujete el vial del disolvente y desenrosque con cuidado el sistema separándolo en dos piezas. Deseche el vial del disolvente con el adaptador azul del Mix2Vial acoplado.
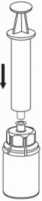
7
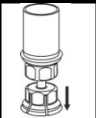
6. Someta el vial de la solución reconstituida
con el adaptador transparente acoplado a movimientos de rotación suaves hasta que la sustancia se haya disuelto por completo. No lo agite._
7. Llene de aire una jeringa vacía y estéril. Manteniendo el vial con la solución en posición vertical, conecte la jeringa al adaptador Luer Lock del Mix2Vial. Inyecte el aire en el vial de la solución.
Extracción y administración
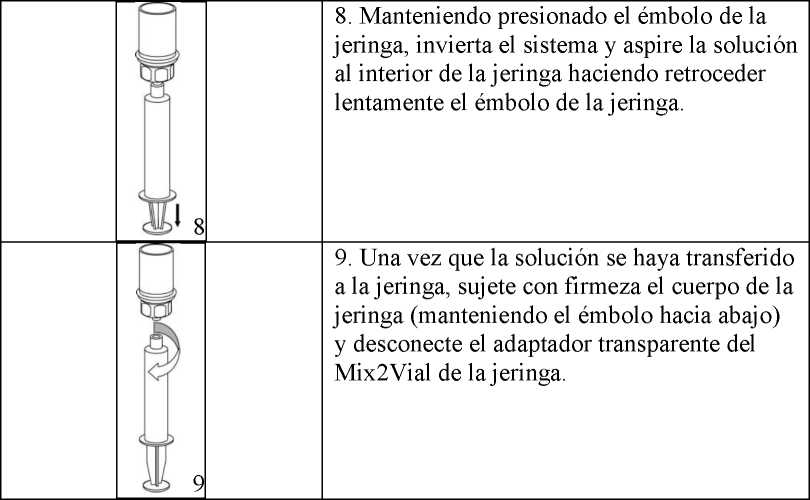
Se debe tener la precaución de que no penetre sangre en la jeringa cargada con el producto puesto que existe el riesgo de que la sangre coagule en la jeringa y que con ello se administren coágulos de fibrina al paciente.
Si se necesita más de un vial de Beriplex, es posible agrupar varios viales de Beriplex en una única perfusión mediante un accesorio para perfusión disponible en el mercado.
La solución Beriplex no se debe diluir.
La solución reconstituida se debe administrar por vía intravenosa (no más de 8 ml/min*).
La eliminación de los productos no utilizados o material de desecho se realizará de acuerdo con las exigencias locales.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
CSL Behring GmbH Emil-von-Behring-Str. 76 35041 Marburg Alemania
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
Beriplex 500 UI, polvo y disolvente para solución inyectable: 69890.
Beriplex 1000 UI, polvo y disolvente para solución inyectable: 76961.
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Beriplex 500 UI, polvo y disolvente para solución inyectable: 13 junio 2008 / 9 noviembre 2012. Beriplex 1000 UI, polvo y disolvente para solución inyectable: Febrero 2014.
10. FECHA DE LA REVISIÓN DEL TEXTO
Noviembre 2012.
La información detallada y actualizada de este medicamento está disponible en la página Web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.gob.es
* en ensayos clínicos con Beriplex, se administraron las dosis con una velocidad máxima de infusión de 0,12 ml/kg/min (menos de 8 ml/min) a pacientes con un peso <70 kg.
13 de 13