Anbinex Polvo Y Disolvente Para Solucion Para Perfusion
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Anbinex 50 Ul/ml Polvo y disolvente para solución para perfusión.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Antitrombina humana.
Anbinex se presenta como polvo liofilizado conteniendo nominalmente 500 UI ó 1000 UI de antitrombina derivada de plasma humano por vial.
El producto contiene aproximadamente 500 UI/10 ml ó 1000 UI/20 ml de antitrombina derivada de plasma humano cuando se reconstituye con 10 ml ó 20 ml de agua para inyectables.
La potencia (UI) se determina por el método cromogénico de la Farmacopea Europea. La actividad específica de Anbinex es como mínimo de 5 UI/mg de proteína.
Anbinex contiene 1,45 mmol (33,35 mg) de sodio por 10 ml (jeringa) para la presentación de 500 UI, y 2,90 mmol (66,7 mg) de sodio por 20 ml (jeringa) para la presentación de 1000 UI.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución para perfusión.
Vial conteniendo polvo o sólido friable blanco, higroscópico y jeringa precargada con agua para inyectables (disolvente).
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
- Pacientes con déficit congénito de antitrombina:
a) Profilaxis de la trombosis venosa profunda y el tromboembolismo en situaciones de riesgo clínico (especialmente durante la cirugía o durante el período peri-parto), en asociación con heparina si está indicado.
b) Prevención de la progresión de la trombosis venosa profunda y el tromboembolismo en asociación con heparina si está indicado.
- Déficit adquirido de antitrombina
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de pacientes con déficit de antitrombina.
Posología
En el déficit congénito, la dosis debe individualizarse para cada paciente, teniendo en cuenta la historia familiar por lo que respecta a episodios tromboembólicos, los factores de riesgo clínico del paciente y las pruebas de laboratorio.
La dosis y duración de la terapia de sustitución en el déficit adquirido depende del nivel de antitrombina plasmática, la presencia de signos de movilización aumentada, el trastorno subyacente y la gravedad del cuadro clínico del paciente. La cantidad de producto a administrar, así como la frecuencia de administración debe estar siempre basada en la eficacia clínica y las pruebas de laboratorio en cada caso en particular.
El número de unidades de antitrombina administradas se expresa en Unidades Internacionales (UI), en relación con el estándar de la Organización Mundial de la Salud (OMS) vigente para la antitrombina. La actividad plasmática de antitrombina se expresa como un porcentaje (en relación con el plasma humano normal) o en Unidades Internacionales (en relación con un estándar internacional para antitrombina en plasma).
Una unidad internacional (UI) de actividad de antitrombina es equivalente a la cantidad de antitrombina presente en 1 ml de plasma humano normal. La estimación de la dosis necesaria de Anbinex se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de antitrombina por kg de peso corporal, da lugar a un aumento de la actividad de antitrombina plasmática de entre 1,1% y 1,6%.
La dosis inicial se determina de acuerdo con la siguiente fórmula:
Unidades a administrar = peso corporal (kg) x (100 - actividad de antitrombina basal (%)) x 0,8
Inicialmente, la actividad de antitrombina a alcanzar depende del estado clínico. Cuando se establece que la reposición con antitrombina está indicada, la dosis debería ser la suficiente para alcanzar la actividad de antitrombina deseada y para mantener un nivel efectivo. La dosis se establecerá y se monitorizará de acuerdo con los resultados analíticos de actividad de antitrombina. Esta analítica se realizará al menos dos veces al día hasta que el paciente se estabilice, después una vez al día y siempre inmediatamente antes de la siguiente infusión. El ajuste de la dosis debe tener en cuenta signos de movilización aumentada de antitrombina de acuerdo con las pruebas de laboratorio y el curso clínico. La actividad de antitrombina debe mantenerse por encima del 80% durante el tratamiento, a no ser que el estado clínico indicase un nivel efectivo distinto.
La dosis inicial habitual en el déficit congénito sería de 30 - 50 UI/kg.
Por lo tanto, la dosis y la frecuencia de administración, así como la duración del tratamiento deben ajustarse a los datos biológicos y al estado clínico del paciente.
Población pediátrica
No hay datos disponibles.
Forma de administración
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6. El producto debe administrarse por vía intravenosa. La velocidad de administración no debe superar 0,08 ml/kg/min.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes.
4.4 Advertencias y precauciones especiales de empleo
Al igual que con cualquier producto proteico para administración intravenosa, es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico. Se requiere una estrecha monitorización y cuidadosa observación de los pacientes para detectar cualquier síntoma durante el periodo de perfusión. Se debe informar a los pacientes acerca de los signos iniciales de las reacciones de hipersensibilidad, que incluyen erupciones cutáneas que pueden llegar a urticaria generalizada, opresión torácica, dificultad al respirar, hipotensión y anafilaxia. Si se producen estos síntomas después de la administración, contactar con el médico.
En caso de shock, se seguirán las recomendaciones vigentes para tratamiento del shock.
Para prevenir la transmisión de enfermedades infecciosas cuando se administran medicamentos derivados de la sangre o plasma humanos, se toman medidas estándar como la selección de donantes, análisis de marcadores específicos de infecciones en las donaciones individuales y en las mezclas de plasma, así como la inclusión de etapas en el proceso de fabricación para eliminar / inactivar virus. A pesar de esto, cuando se administran medicamentos derivados de la sangre o plasma humanos, la posibilidad de transmisión de agentes infecciosos no se puede excluir totalmente. Esto también se refiere a virus y agentes infecciosos emergentes o de naturaleza desconocida.
Las medidas tomadas se consideran efectivas para virus envueltos tales como el VIH, el VHB y el VHC y para el virus no envuelto de la hepatitis A. Las medidas tomadas pueden tener un valor limitado para virus no envueltos tales como el parvovirus B19.
La infección por parvovirus B19 puede ser grave para una mujer embarazada (infección fetal) y para sujetos con inmunodeficiencia o con una producción aumentada de hematíes (ej. con anemia hemolítica).
Es altamente recomendable que cada vez que se administre Anbinex a un paciente se deje constancia del nombre del medicamento y n° de lote administrado a fin de mantener una relación entre el paciente y el lote de producto.
Se recomienda la vacunación apropiada (hepatitis A y B) para los pacientes que reciban regularmente concentrados de antitrombina derivada de plasma humano.
Monitorización Clínica y Biológica en caso de administración conjunta de antitrombina y heparina:
- A fin de ajustar la dosis de heparina y evitar una excesiva hipocoagulabilidad, se deben realizar regularmente controles del alcance de la anticoagulación (APPT, y cuando proceda actividad anti-FXa), a intervalos cortos y en especial en los primeros minutos/horas posteriores al inicio de la administración de la antitrombina.
- Determinación diaria de los niveles de antitrombina, a fin de ajustar la dosis individual, debido al riesgo de disminución de los niveles de antitrombina como consecuencia de un tratamiento prolongado con heparina no fraccionada.
Precauciones especiales respecto a los excipientes: Anbinex contiene 1,45 mmol (33,35 mg) de sodio por 10 ml (jeringa) para la presentación de 500 UI, y 2,90 mmol (66,7 mg) de sodio por 20 ml (jeringa) para la presentación de 1000 UI. Esto deberá tenerse en cuenta en el tratamiento de pacientes con dietas pobres en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción
Heparina: la reposición de antitrombina durante la administración de heparina en dosis terapéuticas aumenta el riesgo de sangrado. El efecto de la antitrombina se ve potenciado en gran medida por la heparina. La semivida de la antitrombina puede disminuir considerablemente por el tratamiento concomitante con heparina, debido a la movilización acelerada de la antitrombina. Por consiguiente, la
administración simultánea de heparina y antitrombina a un paciente con riesgo elevado de sangrado se debe monitorizar clínica y biológicamente.
4.6 Fertilidad, embarazo y lactancia
La experiencia en relación con la seguridad de los productos de antitrombina humana para su uso en embarazo humano es limitada.
Anbinex debe administrarse a mujeres embarazadas o lactantes con déficit de antitrombina solamente si está claramente indicado, teniendo en cuenta que el embarazo confiere un aumento del riesgo de episodios tromboembólicos en estas pacientes.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Anbinex sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Rara vez se han observado reacciones alérgicas o de hipersensibilidad (que puede incluir angioedema, sensación de ardor y picor en el lugar de infusión, escalofríos, enrojecimiento, erupciones cutáneas que pueden llegar a urticaria generalizada, cefalea, hipotensión, somnolencia, náuseas, inquietud, taquicardia, opresión torácica, hormigueo, vómitos, dificultad al respirar) en pacientes tratados con productos que contienen antitrombina. En ciertos casos, estas reacciones han progresado hasta anafilaxia grave (incluyendo shock).
En raras ocasiones se ha observado fiebre.
No existen datos robustos de la frecuencia de reacciones adversas en ensayos clínicos y en la experiencia post-comercialización.
Para la seguridad con respecto a agentes transmisibles, ver sección 4.4.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es
4.9 Sobredosis
No se han notificado casos de sobredosis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes antitrombóticos: grupo de la heparina, código ATC: B01AB02.
La antitrombina, una glicoproteína de 432 aminoácidos y 58 kD, pertenece a la superfamilia de las serpinas (inhibidores de la serinproteasa). Es uno de los inhibidores naturales de la coagulación sanguínea más importantes. Los factores más intensamente inhibidos son la trombina y el factor Xa; pero también factores de activación por contacto, el sistema intrínseco y el complejo factor VIIa/factor tisular. La actividad de la antitrombina se ve altamente aumentada por la heparina, y los efectos anticoagulantes de la heparina
dependen de la presencia de antitrombina.
La antitrombina contiene dos dominios funcionalmente importantes. El primero contiene el centro reactivo y proporciona un lugar de segmentación para las proteinasas como la trombina, un pre-requisito para la formación de un complejo inhibidor-proteinasa estable. El segundo es un dominio de unión a glicosaminoglicanos, responsable de la interacción con la heparina y las sustancias relacionadas, que acelera la inhibición de la trombina. Los complejos inhibidor-enzima de la coagulación son eliminados por el sistema retículoendotelial.
La actividad de la antitrombina es de un 80 - 120% en adultos. En recién nacidos , los niveles están alrededor del 40 - 60 %.
5.2 Propiedades farmacocinéticas
Los estudios farmacocinéticos realizados con antitrombina muestran un valor medio de semivida biológica de aproximadamente 3 días. La vida media puede disminuir a 1,5 días debido al tratamiento conjunto con heparina. La vida media puede disminuir a horas en condiciones de alto consumo.
Los resultados obtenidos en el ensayo clínico con Anbinex en pacientes con deficiencia congénita de antitrombina se estimaron por modelo independiente y son los siguientes:
- Incremento de la recuperación 1,3 ± 0,2 (DS) con un rango de 1,1 a 1,6% basado en análisis funcionales.
- El área bajo la curva (AUC) = 66.461 ± 15.445 IU h/l
- La vida media terminal es de 98,1 ± 45,0 h basada en análisis funcionales.
- El tiempo de residencia (MRT) = 121,7 ± 52,1 h.
- Aclaramiento 0,931 ± 0,214 ml/h/kg.
5.3 Datos preclínicos sobre seguridad
La antitrombina es un componente normal del plasma humano.
Las pruebas de toxicidad a dosis única son poco relevantes y no permiten la estimación de la dosis tóxica o letal o de la relación dosis-efecto.
Las pruebas de toxicidad a dosis repetidas en animales son impracticables debido a la formación de anticuerpos.
No se han descrito signos de toxicidad aguda en animales de experimentación.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
D-Manitol Cloruro sódico Citrato sódico
Agua para inyectables (disolvente)
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
6.3 Periodo de validez
El tiempo de validez del producto sin reconstituir es de 3 años.
Tras la reconstitución el producto es estable química y físicamente durante 12 horas a 25° C. Desde un punto de vista microbiológico, el producto debe utilizarse inmediatamente. Si no se utiliza inmediatamente, el tiempo y las condiciones de conservación antes de su uso son responsabilidad del usuario y, normalmente no serán más de 24 horas a 2° C - 8° C a menos que la reconstitución se haya realizado en condiciones asépticas controladas y validadas.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30 °C.
No congelar.
Para las condiciones de conservación del medicamento reconstituido, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Anbinex se presenta en un vial de vidrio tipo II que contiene 500 ó 1000 UI de antitrombina humana (polvo para perfusión) tapado con un tapón de caucho de bromobutilo, y una jeringa precargada de vidrio tipo I con tapón de émbolo (caucho de bromobutilo) con 10 ml (500 UI) o 20 ml (1000 UI) de agua para inyectables (disolvente).
Cada vial de Anbinex va acompañado de los accesorios necesarios para su reconstitución: adaptador de vial y microfiltro.
Contenido de la caja: 1 vial de producto liofilizado, 1 jeringa precargada de disolvente y accesorios.
Puede que solamente estén comercializados algunos tamaños de envases.
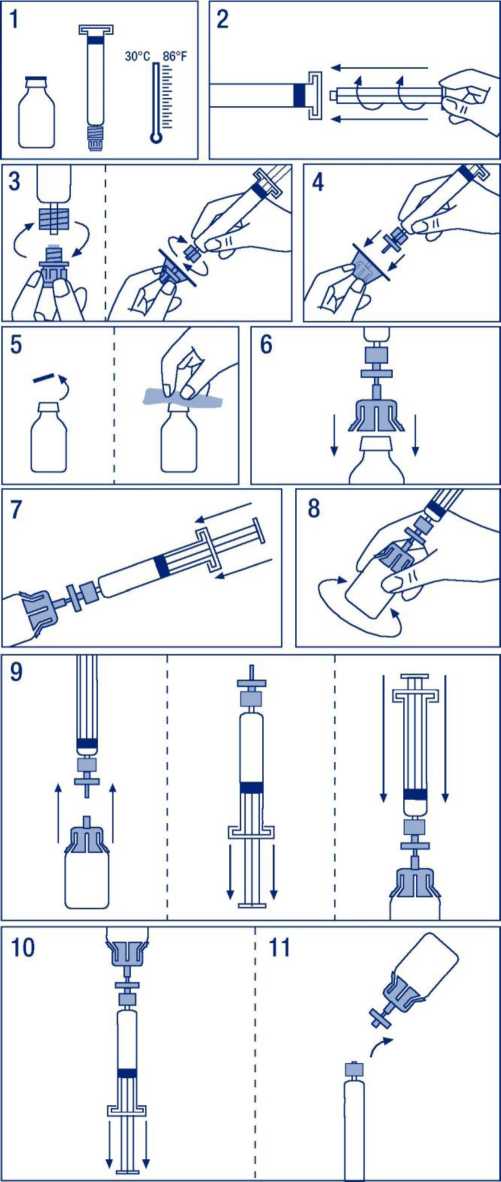
6.6 Precauciones especiales de eliminación y otras manipulaciones Preparación de la solución:
1. Atemperar el vial y la jeringa del disolvente sin sobrepasar los 30 °C.
2. Acoplar el émbolo a la j eringa del disolvente.
3. Desprecintar el filtro. Separar el tapón del cono de la jeringa del disolvente y acoplarla al filtro.
4. Desprecintar el adaptador de vial y acoplarlo al conjunto filtro-jeringa.
5. Desprecintar el vial, desinfectando el tapón con una toallita antiséptica.
6. Introducir la espina del adaptador en el vial.
7. Trasvasar todo el disolvente de la jeringa al vial.
8. Agitar el vial mediante giros suaves hasta la total disolución.
9. Separar el conjunto filtro - jeringa del resto. Aspirar aire suficiente para el volumen total de la solución. Acoplar de nuevo el conjunto filtro - jeringa al vial.
10. Invertir el vial y aspirar el contenido en la jeringa.
11. Separar la jeringa y administrar lentamente por vía intravenosa. La velocidad de administración no debe superar los 0,08 ml/kg/min.
Generalmente la solución es clara o ligeramente opalescente. No utilizar las soluciones que presenten turbidez o sedimento.
Una vez reconstituida, la solución debe desecharse si se observan partículas en su interior o algún tipo de decoloración.
En ningún caso se aprovechará la fracción que no se haya utilizado.
No deben reutilizarse los equipos de administración.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Instituto Grifols, S.A.
Can Guasch, 2 - Parets del Valles 08150 Barcelona - ESPAÑA
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
Anbinex, N° de Reg.: 59259
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Autorización: 14/10/1992. Última renovación: 14/10/2007
10. FECHA DE LA REVISIÓN DEL TEXTO
Julio 2016
La información detallada y actualizada de este medicamento está disponible en la página Web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.es/

8 de 8