Accofil 48 Mu/0,5 Ml Solucion Inyectable Y Para Perfusion En Jeringa Precargada
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Accofil 30 MU/0,5 ml solución inyectable y para perfusión en jeringa precargada.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada ml de solución contiene 60 millones de unidades (MU; equivalentes a 600 microgramos [^g]) de filgrastim.
Cada jeringa precargada contiene 30 MU (equivalentes a 300 microgramos) de filgrastim en 0,5 ml de solución inyectable y para perfusión.
Filgrastim es un factor metionil recombinante estimulador de las colonias de granulocitos humanos por tecnología de ADN recombinante en Escherichia coli (BL21).
Excipientes con efecto conocido:
Cada ml de solución contiene 50 mg de sorbitol (E420)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable y para perfusión. Solución transparente, incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Accofil está indicado para la reducción de la duración de la neutropenia y de la incidencia de neutropenia febril en pacientes tratados con quimioterapia citotóxica establecida para neoplasias malignas (con la excepción de la leucemia mielógena crónica y los síndromes mielodisplásicos) y para la reducción de la duración de la neutropenia en pacientes que reciben tratamiento mieloablativo seguido de un trasplante de médula ósea que se considere que presentan un aumento del riesgo de neutropenia grave prolongada. La seguridad y la eficacia de Accofil son similares en adultos y en niños que reciben quimioterapia citotóxica.
Accofil está indicado para la movilización de células progenitoras de sangre periférica (CPSP).
En pacientes (niños o adultos) con neutropenia congénita, cíclica o idiopática grave que presentan un recuento absoluto de neutrófilos (RAN) < 0,5 x 109/l y antecedentes de infecciones intensas o recurrentes, la administración de Accofil a largo plazo está indicada para elevar los recuentos de neutrófilos y reducir la incidencia y la duración de los acontecimientos relacionados con las infecciones.
Accofil está indicado para el tratamiento de la neutropenia persistente (RAN igual o inferior a 1,0 x 109/l) en pacientes con infección avanzada por el VIH, con objeto de reducir el riesgo de infecciones bacterianas cuando otras opciones para el control de la neutropenia no son adecuadas.
4.2 Posología y forma de administración
El tratamiento con Accofil debe administrarse únicamente en colaboración con un centro de oncología que tenga experiencia en el uso del factor estimulante de las colonias de granulocitos (G-CSF) y en hematología y que disponga de las instalaciones necesarias para el diagnóstico. Los procedimientos de movilización y aféresis deben realizarse en colaboración con un centro de oncología-hematología con experiencia suficiente en este campo y en el que el seguimiento de las células progenitoras hematopoyéticas pueda realizarse de una forma correcta.
Posología
Quimioterapia citotóxica establecida
La dosis recomendada de filgrastim es de 0,5 MU/kg/día (5 microgramos/kg/día). La primera dosis de Accofil no debe administrarse menos de 24 horas después de la quimioterapia citotóxica. En ensayos clínicos aleatorizados, se utilizó una dosis subcutánea de 230 microgramos/m2/día (4,0 a 8,4 microgramos/kg/día).
La administración diaria de filgrastim debe continuar hasta que se haya sobrepasado el nadir teórico de neutrófilos y su recuento retorne a su intervalo normal. Después de la quimioterapia establecida en tumores sólidos, linfomas y leucemias linfoides, se prevé que la duración del tratamiento necesario para alcanzar estos criterios sea de hasta 14 días. Tras el tratamiento de inducción y consolidación en la leucemia mieloide aguda, la duración del tratamiento puede ser bastante mayor (hasta 38 días) en función del tipo, la posología y las pautas de administración de la quimioterapia citotóxica.
Los pacientes sometidos a quimioterapia citotóxica experimentan un aumento transitorio del recuento de neutrófilos que ocurre habitualmente 1 a 2 días después de iniciar la administración de filgrastim.
Sin embargo, para conseguir una respuesta terapéutica sostenida, no se debe suspender el tratamiento con filgrastim hasta que no se haya sobrepasado el nadir teórico de neutrófilos y su recuento retorne a su intervalo normal. No se recomienda la interrupción prematura del tratamiento con filgrastim antes de alcanzar el nadir teórico de neutrófilos.
Pacientes sometidos a tratamiento mieloablativo seguido de trasplante de médula ósea
La dosis inicial recomendada de filgrastim es de 1,0 MU/kg/día (10 microgramos/kg/día). La primera dosis de filgrastim debe administrarse por lo menos 24 horas después de la quimioterapia citotóxica y por lo menos 24 horas después de la infusión de médula ósea.
Una vez sobrepasado el nadir de neutrófilos, la dosis diaria de filgrastim se ajustará en función de la respuesta de los neutrófilos del siguiente modo:
|
Recuento absoluto de neutrófilos (RAN) |
Ajuste de la dosis de filgrastim |
|
RAN > 1,0 x 109/L durante 3 días consecutivos |
Reducir a 0,5 MU/kg/día (5 microgramos/kg/día) |
|
Después, si el RAN permanece > 1,0 x 109/L durante 3 días consecutivos más |
Suspender filgrastim |
|
Si el RAN desciende a < 1,0 x 109/L durante el período de tratamiento, se debe reajustar de nuevo la dosis de filgrastim siguiendo los pasos previamente indicados | |
Movilización de las células progenitoras sanguíneas periféricas (CPSP)
Pacientes sometidos a tratamiento mielodepresor o mieloablativo seguido de trasplante autólogo de CPSP
La dosis recomendada de filgrastim para la movilización de PBPC cuando se usa solo es de
1,0 MU/kg/día (10 microgramos/kg/día) durante 5-7 días consecutivos. El tiempo de leucocitaféresis: 1
o 2 leucocitaféresis en los días 5 y 6, que es a menudo suficiente. En otras circunstancias,
leucocitaféresis adicionales pueden ser necesarias. La administración de filgrastim debe mantenerse hasta la última leucocitaféresis.
La dosis recomendada de filgrastim para la movilización de PBPC tras una quimioterapia mielosupresora es de 0,5 MU/kg/día (5 microgramos/kg/día) administrados diariamente desde el primer día tras concluir la quimioterapia hasta sobrepasar el nadir teórico de neutrófilos y el recuento de neutrófilos retorne al rango normal. La leucocitaféresis se debe realizar durante el período en que el ANC se incrementa de < 0,5 x 109/L a > 5,0 x 109/L. Para los pacientes que no han sido sometidos a quimioterapia, una leucocitaféresis suele ser suficiente. En otras circunstancias, se recomienda la realización de leucocitaféresis adicionales.
Para la movilización de CPSP en donantes sanos antes del alotrasplante de CPSP
Para la movilización de PBPC en donantes sanos, filgrastim debe administrarse a 1,0 MU/kg/day (10 microgramos/kg/día) durante 4-5 días consecutivos. La leucocitaféresis debe iniciarse en el día 5 y se prolongarse hasta el día 6 si es necesario con el fin de recoger 4 x 106 células CD34+/kg del peso del cuerpo receptor.
En pacientes con neutropenia crónica grave (NCG)
Neutropenia congénita
La dosis inicial recomendada es de 1,2 MU/kg/día (12 microgramos/kg/día) en dosis única o dividida en varias dosis.
Neutropenia idiopática o cíclica
La dosis inicial recomendada es de 0,5 MU/kg/día (5 microgramos/kg/día) en dosis única o dividida en varias dosis.
Ajustes de la dosis: filgrastim se debe administrar diariamente por inyección subcutánea hasta alcanzar y poder mantener el recuento de neutrófilos por encima de 1,5 x 109/L. Una vez alcanzada la respuesta se establecerá la dosis mínima efectiva para mantener este nivel. Si se desea mantener un nivel de neutrófilos adecuado, es necesaria la administración diaria de filgrastim a largo plazo. La dosis inicial se puede duplicar o reducir a la mitad al cabo de 1 a 2 semanas de tratamiento, en función de la respuesta del paciente. Posteriormente, la dosis se puede ajustar individualmente en intervalos de 1-2 semanas para mantener un recuento medio de neutrófilos entre 1,5 x 109/L y 10 x 109/L. En los pacientes con infecciones graves se puede contemplar un aumento más rápido de la dosis. En los ensayos clínicos, el 97% de los pacientes que respondieron al tratamiento presentaron una respuesta completa a dosis < 2,4 MU/kg/día (24 microgramos/kg/día). En pacientes con NCG, no se ha establecido la seguridad a largo plazo de la administración de filgrastim por encima de 2,4 MU/kg/día (24 microgramos/kg/día).
Pacientes con infección por VIH
Para la recuperación de la neutropenia
La dosis inicial recomendada de filgrastim es de 0,1 MU/kg/día (1 microgramos/kg/día) se administra diariamente, ajustando la dosis hasta un máximo de 0,4 MU/kg/día (4 microgramos/kg/día) hasta que se alcanza un recuento de neutrófilos normal y se puede mantener (RAN > 2,0 x 109/L). En los estudios clínicos, más del 90% de los pacientes respondieron a estas dosis, logrando una recuperación de la neutropenia en una mediana de 2 días. En un pequeño número de pacientes (<10%), dosis de hasta 1,0 MU/kg/día (10 microgramos/kg/día) fueron obligados a revertir la neutropenia.
Para mantener el recuento normal de neutrófilos
Una vez alcanzada la recuperación de la neutropenia, se establecerá la dosis mínima efectiva para mantener un recuento normal de neutrófilos. Se recomienda comenzar el ajuste de dosis administrando 30 MU/kg/día (300 microgramos/kg/día) cada dos días. En función del RAN del paciente podrán ser necesarios nuevos ajustes de la dosis para mantener un recuento de neutrófilos > 2,0 x 109/L. En los ensayos clínicos, se requirió la administración de 30 MU/kg/día (300 microgramos/kg/día) de 1 a 7 días a la semana para mantener un RAN > 2,0 x 109/L, siendo la mediana de la frecuencia de dosis de 3 días a la semana. Puede ser necesaria una administración prolongada para mantener el RAN > 2,0 x 109/L.
Poblaciones especiales Pacientes de edad avanzada
Los ensayos clínicos con filgrastim han incluido un pequeño número de pacientes de edad avanzada, pero los estudios especiales no se han llevado a cabo en este grupo y por lo tanto, las recomendaciones posológicas específicas no se pueden hacer.
Pacientes con insuficiencia renal o hepática
Los estudios del filgrastim en pacientes con alteración grave de la función hepática o renal demuestran que presenta un perfil farmacodinámico y farmacocinético similar al observado en individuos sanos.
En estos casos no se requiere ajuste de dosis.
Pacientes pediátricos en el marco de la NCG y el cáncer
Sesenta y cinco por ciento de los pacientes estudiados en el programa de ensayo sobre NCG con la administración de filgrastim, eran menores de 18 años de edad. La eficacia del tratamiento era clara para este grupo de edad, que incluía a la mayoría de los pacientes con neutropenia congénita. No existían diferencias en los perfiles de seguridad para pacientes pediátricos tratados por NCG.
Los datos de estudios clínicos en pacientes pediátricos indican que la seguridad y la eficacia del filgrastim son similares en adultos y en niños que reciben quimioterapia citotóxica.
Las recomendaciones posológicas para pacientes pediátricos son iguales que las correspondientes a los adultos que reciben quimioterapia citotóxica mielosupresora.
Forma de administración
Quimioterapia citotóxica establecida
Filgrastim puede administrarse como una inyección subcutánea diaria o, alternativamente, como una perfusión intravenosa diaria diluido en glucosa 50 mg / ml (5%) solución durante 30 minutos. Para obtener más instrucciones sobre la dilución antes de la perfusión, ver sección 6.6. Se prefiere la vía subcutánea en la mayoría de los casos. Existe cierta evidencia de un estudio de administración de dosis única que la dosificación intravenosa puede acortar la duración del efecto. La relevancia clínica de este hallazgo en la administración de dosis múltiples no está clara. La elección de la vía de administración depende de la situación clínica individual. En los estudios clínicos aleatorizados, se utilizó una dosis subcutánea de 23 MU/m2/día (230 microgramos/m2/día) o bien 4-8,4 microgramos/kg/día.
Pacientes sometidos a tratamiento mieloablativo seguido de trasplante de médula ósea
Filgrastim se administra en perfusión intravenosa breve a lo largo de 30 minutos o en perfusión subcutánea o intravenosa continua a lo largo de 24 horas, en ambos casos tras su dilución en 20 ml de solución de glucosa de 50 mg/ml (5%). Para obtener instrucciones adicionales acerca de su dilución con solución de glucosa de 50 mg/ml (5%) antes de la perfusión, ver sección 6.6.
En pacientes con la movilización de PBPC
Filgrastim para la movilización de PBPC cuando se usa solo:
Filgrastim se puede administrar como una sola inyección subcutánea diaria o una perfusión continua subcutánea 24 horas. Para las perfusiones filgrastim debe diluirse en 20 ml de solución de glucosa (ver sección 6.6).
Filgrastim para la movilización de PBPC después de la quimioterapia mielsupresiva:
Filgrastim se debe administrar por inyección subcutánea.
Para la movilización de PBPC en donantes normales antes de un alotransplante PBPC Filgrastim se debe administrar por inyección subcutánea.
En pacientes con NCG
Neutropenia congénita, idiopática o cíclica: Filgrastim se debe administrar por vía inyección subcutánea.
En pacientes con infección por el VIH
Para la recuperación de la neutropenia y mantenimiento del recuento de neutrófilos en pacientes con infección por VIH, filgrastim se administra por vía subcutánea
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Advertencias especiales
Filgrastim no debe utilizarse para incrementar la dosis de quimioterapia citotóxica más allá de las pautas posológicas establecidas.
No se debe administrar filgrastim a pacientes con neutropenia congénita grave que presenten leucemia o muestren evidencia de evolución leucémica.
Se ha notificado hipersensibilidad, incluidas reacciones anafilácticas, que se producen al inicio del tratamiento o posteriormente en pacientes tratados con filgrastim. Suspenda de manera permanente el filgrastim en pacientes con hipersensibilidad clínicamente significativa. No administre filgrastim a pacientes con antecedentes de hipersensibilidad a filgrastim o pegfilgrastim.
Como ocurre con todas las proteínas terapéuticas, existe un potencial de inmunogenicidad. La velocidad de generación de anticuerpos contra filgrastim generalmente es baja. Los anticuerpos de fijación se producen como se espera con todos los productos biológicos; sin embargo, no se han asociado con actividad neutralizante en la actualidad.
Precauciones especiales en pacientes con leucemia mieloide aguda (LMA)
Crecimiento de las células malignas
El G-CSF puede estimular el crecimiento de células granulocíticas in vitro y se pueden observar efectos similares en algunas células no granulocíticas in vitro.
No se ha establecido la seguridad y la eficacia de la administración de filgrastim en los pacientes con síndrome mielodisplásico o leucemia mieloide crónica. Por lo tanto, filgrastim no está indicado en estas enfermedades. Se debe prestar especial atención para distinguir el diagnóstico de leucemia mieloide crónica en transformación blástica del de leucemia mieloide aguda.
Considerando que los datos disponibles sobre la seguridad y la eficacia en pacientes con LMA secundaria son limitados, filgrastim debe administrarse con precaución. No se ha establecido la seguridad y la eficacia de la administración de filgrastim en pacientes menores de 55 años y con LMA de novo con buena citogenética [t (8; 21), t (15; 17) e inv (16)].
Otras precauciones especiales
La monitorización de la densidad ósea puede estar indicada en pacientes tratados con filgrastim durante más de 6 meses que presenten enfermedad osteoporótica subyacente.
Se han notificado reacciones adversas pulmonares, en particular neumonía intersticial, tras la administración de G-CSF. Los pacientes con antecedentes recientes de infiltrados pulmonares o neumonía pueden presentar mayor riesgo. La aparición de síntomas respiratorios tales como tos, fiebre y disnea, en asociación con signos radiológicos de infiltrados pulmonares y deterioro de la función pulmonar, pueden ser los síntomas preliminares del síndrome de dificultad respiratoria aguda (SDRA). Se debe suspender la administración de filgrastim e instaurar el tratamiento adecuado en estos casos.
Se han notificado casos de síndrome de extravasación capilar después de la administración de factor estimulante de colonias de granulocitos, que se caracterizan por hipotensión, hipoalbuminemia, edema y hemoconcentración. Los pacientes que desarrollan síntomas de síndrome de extravasación capilar se deben controlar de cerca y deben recibir tratamiento sintomático estándar, que puede incluir la necesidad de cuidados intensivos (ver sección 4.8).
Precauciones especiales en los pacientes con cáncer
Se han notificado casos de esplenomegalia y ruptura esplénica de forma poco frecuente tras la administración de filgrastim. Algunos casos de ruptura esplénica fueron mortales. Los pacientes que recibieron filgrastim que notificaron dolor abdominal superior y/o extremo del hombro se deben evaluar para agrandamiento del bazo o ruptura esplénica.
Leucocitosis
En menos del 5% de los pacientes tratados con filgrastim a dosis superiores a 0,3 MU/kg/día (3 ^g/kg/día) se ha observado un recuento leucocitario de 100 x 109/L o superior. No se ha notificado ninguna reacción adversa directamente atribuible a este grado de leucocitosis. No obstante, ante los posibles riesgos asociados a la leucocitosis intensa, se debe realizar periódicamente un recuento de leucocitos durante el tratamiento con filgrastim. Si el recuento leucocitario supera 50 x 109/L después del nadir teórico, se debe interrumpir inmediatamente el tratamiento con filgrastim. Sin embargo, durante el período de administración de filgrastim para la movilización de CPSP, se debe interrumpir la administración de filgrastim o disminuir la dosis si el recuento de leucocitos aumenta por encima de 70 x 109/L.
Riesgos asociados con el aumento de la dosis de la quimioterapia
Se debe tener especial precaución en los pacientes tratados con quimioterapia en dosis altas, ya que no se ha demostrado una mejora de los resultados obtenidos sobre el tumor y la intensificación de las dosis de quimioterapia puede ocasionar mayor toxicidad cardíaca, pulmonar, neurológica y dermatológica (consulte la ficha técnica de los agentes quimioterápicos concretos utilizados).
El tratamiento con filgrastim en monoterapia no impide la trombocitopenia y anemia secundaria a la quimioterapia mielodepresora. Ante la posibilidad de recibir dosis más altas de quimioterapia (p. ej., dosis completas según el calendario prescrito), el paciente puede correr mayor riesgo de trombocitopenia y anemia. Por eso, se recomienda vigilar periódicamente el recuento plaquetario y el hematocrito. Se deben tomar medidas de precaución especiales cuando se administren agentes quimioterápicos, tanto solos como combinados, susceptibles de provocar trombocitopenia grave.
Se ha demostrado que el uso de CPSP movilizadas por filgrastim reduce la intensidad y duración de la trombocitopenia tras la quimioterapia mieloablativa o mielodepresora.
Otras precauciones especiales
Aún no se conoce el efecto de filgrastim en los pacientes con una reducción considerable de los progenitores mieloides. Filgrastim actúa fundamentalmente sobre los precursores de los neutrófilos, aumentando el recuento de estas células. Por tanto, la respuesta de neutrófilos podría disminuir en los pacientes con disminución de las células precursoras (como aquellos tratados con radioterapia o quimioterapia intensiva, o aquellos con infiltración tumoral de médula ósea).
De forma esporádica se han notificado trastornos vasculares, entre ellos flebopatía oclusiva y alteraciones de la volemia, en pacientes tratados con dosis altas de quimioterapia seguidas de trasplante.
Se han notificado casos de enfermedad de injerto contra huésped (EICH) y muertes en pacientes que recibieron G-CSF tras un alotrasplante de médula ósea (ver secciones 4.8 y 5.1).
El aumento de la actividad hematopoyética de la médula ósea en respuesta al tratamiento con factor de crecimiento se ha relacionado con anomalías transitorias en las imágenes óseas. Esto se debe tener en cuenta cuando se interpreten resultados de imágenes óseas.
Precauciones especiales en pacientes sometidos a movilización de PBPC
Movilización de PBPC
No existen comparaciones aleatorizadas de forma prospectiva de los dos métodos de movilización recomendados (filgrastim en monoterapia o en combinación con quimioterapia mielodepresora) en la misma población de pacientes. El grado de variación entre pacientes individuales y entre pruebas analíticas de células CD34+ significa que la comparación directa entre estudios diferentes es difícil. Por consiguiente, es difícil recomendar un método óptimo. La elección de método de movilización debe contemplarse en relación con los objetivos generales del tratamiento para un paciente individual.
Exposición previa a agentes citotóxicos
Los pacientes que se han sometido a tratamiento mielodepresor previo muy intenso pueden no manifestar una movilización suficiente de CPSP para alcanzar el rendimiento mínimo recomendado (2,0 x 106 células CD34+kg) o una aceleración en la recuperación plaquetaria, en el mismo grado.
Algunos agentes citotóxicos muestran toxicidad especial en el reservorio progenitor hematopoyético y pueden afectar negativamente a la movilización de las células progenitoras. Agentes tales como melfalán, carmustina (BCNU) y carboplatino cuando se administran durante períodos prolongados previos al intento de movilización de células progenitoras, pueden reducir el rendimiento de las células progenitoras. Sin embargo, la administración de melfalán, carboplatino o carmustina (BCNU), junto con filgrastim ha demostrado ser eficaz para la movilización de las células progenitoras. Cuando se prevé un trasplante de células progenitoras de sangre periférica, se recomienda planificar el procedimiento de movilización de células madre al comienzo del periodo de tratamiento del paciente. Particular atención debe prestarse a la cantidad de células progenitoras movilizadas en estos pacientes antes de la administración de altas dosis de quimioterapia. Si los rendimientos no son adecuados, según lo medido por los criterios anteriores, se deben considerar formas alternativas de tratamiento que no requieran el soporte de células progenitoras.
Evaluación del rendimiento de células progenitoras
Se recomienda prestar especial atención al método de cuantificación para evaluar el número de células progenitoras recolectadas en los pacientes tratados con filgrastim. Los resultados de los análisis por citometría de flujo del número de células CD34+ varían en función de precisión de la metodología utilizada; por tanto, deben interpretarse con precaución las recomendaciones numéricas basadas en estudios realizados en otros laboratorios.
Los análisis estadísticos de la relación entre el número de células CD34+ nuevamente perfundidas y la velocidad de recuperación plaquetaria tras altas dosis de quimioterapia indican que dicha relación es compleja pero continua.
La recomendación de un rendimiento mínimo de > 2,0 x 106 células CD34+/kg se basa en las experiencias publicadas resultantes de una reconstitución hematológica adecuada. Los rendimientos superiores parecen estar en correlación con una recuperación más rápida, y los inferiores con una recuperación más lenta.
Precauciones especiales en donantes sanos sometidos a movilización de células progenitoras sanguíneas periféricas
La movilización de CPSP no ofrece ningún beneficio clínico directo a los donantes sanos y solamente debe considerarse en el marco de un trasplante alogénico de células progenitoras hematopoyéticas.
Solo debe contemplarse la movilización de CPSP en donantes que cumplan los criterios normales de idoneidad clínica y analítica para la donación de células madre, prestando especial atención a los valores hematológicos y a las enfermedades infecciosas. No se ha evaluado la seguridad y la eficacia del filgrastim en donantes sanos menores de 16 años o mayores de 60 años.
Se ha notificado trombocitopenia de forma muy frecuente en pacientes a los que se administra filgrastim. Por tanto, se debe controlar estrechamente el recuento plaquetario.
Después de la administración de filgrastim y de la leucocitaféresis se ha observado trombocitopenia transitoria (plaquetas < 100 x 109/L) en el 35% de los sujetos estudiados. Entre ellos, se notificaron dos casos con plaquetas < 50 x 109/L, que se atribuyeron al procedimiento de leucocitaféresis. Si hace falta más de una leucocitaféresis, se debe prestar especial atención a los donantes cuyas plaquetas sean <
100 x 109/L antes de la leucocitaféresis; en general no se debe realizar aféresis si las plaquetas son < 75 x 109/L.
No deben realizarse leucocitaféresis a donantes tratados con anticoagulantes o con defectos conocidos en la homeostasis. Se debe suspender la administración de filgrastim o reducir la dosis si el recuento de leucocitos se eleva a > 70 x 109/L. Los donantes tratados con G-CSF para la movilización de CPSP deben controlarse hasta que los índices hematológicos vuelvan a los valores normales.
Anomalías citogenéticas transitorias se han observado en los donantes sanos tratados con G-CSF. Se desconoce la importancia de estos cambios. Sin embargo, no se puede excluir el riesgo de estimulación de un clon mieloide maligno. Se recomienda que el centro de aféresis lleve un control y seguimiento sistemático de los donantes de células progenitoras hematopoyéticas durante al menos 10 años para garantizar la seguridad a largo plazo.
Se han notificado casos frecuentes pero generalmente asintomáticos de esplenomegalia y poco frecuentes los casos de ruptura esplénica en donantes sanos y en pacientes tras la administración de G-CSF. Algunos casos de ruptura esplénica fueron mortales. Por lo tanto, el tamaño del bazo debe controlarse estrechamente (p. ej., examen clínico, ultrasonido). Se debe considerar un diagnóstico de ruptura esplénica en los donantes y/o pacientes que refieran dolor abdominal superior izquierda o el extremo del hombro.
En donantes sanos, la disnea se ha notificado con frecuencia y otros eventos adversos pulmonares (hemoptisis, hemorragia pulmonar, infiltrados pulmonares, e hipoxia) se han notificado de forma poco frecuente. En caso de eventos adversos pulmonares sospechosos o confirmados, se debe considerar la interrupción del tratamiento con filgrastim y administrar la atención médica adecuada.
Precauciones especiales para los receptores de CPSP movilizadas con filgrastim
Los datos disponibles indican que, las interacciones inmunitarias entre el aloinjerto de CPSP y el receptor pueden estar asociadas a un incremento del riesgo de EICH aguda o crónica en comparación con el trasplante de médula ósea.
Precauciones especiales en los pacientes con NCG
Se ha notificado de forma frecuente trombocitopenia en pacientes a los que se administra filgrastim. El recuento plaquetario se debe controlar estrechamente, sobre todo durante las primeras semanas de tratamiento con filgrastim. En los pacientes que presenten trombocitopenia, es decir, en aquellos con un recuento de plaquetas persistentemente < 100,000/mm3, debe valorarse la posibilidad de suspender el tratamiento con filgrastim de forma intermitente o, al menos, reducir la dosis.
Existen también otros cambios del hemograma, como anemia y aumento transitorio de los progenitores mieloides, que obligan a vigilar estrechamente el recuento celular.
Transformación hacia leucemia o síndrome mielodisplásico
Conviene establecer cuidadosamente el diagnóstico de las NCG y diferenciarlas de otros trastornos hematopoyéticos como anemia aplásica, mielodisplasia y leucemia mieloide. Antes del tratamiento debe realizarse un hemograma completo con fórmula leucocitaria y recuento de plaquetas, así como un estudio de la morfología y del cariotipo de la médula ósea.
Hubo una baja frecuencia (aproximadamente 3%) de síndrome mielodisplásico (SMD) o leucemia en pacientes incluidos en ensayos clínicos con NCG tratados con filgrastim. Esta observación sólo se ha hecho en pacientes con neutropenia congénita. SMD y las leucemias son complicaciones naturales de la enfermedad y su relación con el tratamiento con filgrastim es incierta. Un subconjunto de aproximadamente el 12% de pacientes con evaluaciones citogenéticas normales al inicio del estudio posteriormente se encontró que tenían anormalidades, incluyendo monosomía 7, en la evaluación repetida habitual. Si los pacientes con NCG desarrollan una citogenética anormal, los riesgos y beneficios de continuar filgrastim debe sopesarse cuidadosamente; filgrastim debe interrumpir si se desarrolla síndrome mielodisplásico o leucemia. Actualmente no está claro si el tratamiento a largo plazo de los pacientes con NCG predispone hacia anormalidades citogenéticas, SMD o transformación leucémica. Se recomienda realizar exámenes de médula ósea morfológicos y citogenéticos en pacientes a intervalos regulares (aproximadamente cada 12 meses).
Otras precauciones especiales
Se deben excluir las causas que provoquen neutropenia transitoria, como es el caso de las infecciones víricas.
Se han notificado de forma muy frecuente casos de esplenomegalia y casos de ruptura esplénica se han notificado de forma frecuente tras la administración de filgrastim. Los pacientes que recibieron filgrastim que notificaron dolor abdominal superior y/o dolor de extremo del hombro se deben evaluar para agrandamiento del bazo o ruptura esplénica.
La esplenomegalia es una consecuencia directa del tratamiento con filgrastim. En los ensayos, el treinta y uno por ciento (31%) de los pacientes presentaron esplenomegalia detectable por palpación.
El aumento del volumen, medido radiográficamente, se presentó al comienzo del tratamiento con filgrastim y tendió a estabilizarse más tarde durante el tratamiento. La progresión del aumento del tamaño del bazo disminuyó o se detuvo al reducir la dosis y solo un 3% de los pacientes requirieron esplenectomía. El tamaño del bazo debe evaluarse de forma regular. Para detectar un aumento anómalo del volumen esplénico basta con realizar una palpación abdominal.
La hematuria era frecuente y la proteinuria ocurre en un pequeño número de pacientes. Se deben realizar análisis de orina normal para controlar este evento.
No se ha establecido la seguridad y la eficacia en recién nacidos ni en pacientes con neutropenia autoinmunitaria.
Precauciones especiales en pacientes con infección por VIH
Se han notificado de forma frecuente casos de esplenomegalia tras la administración de filgrastim. Los pacientes que recibieron filgrastim que notificaron dolor abdominal superior y/o dolor de extremo del hombro se deben evaluar debido al agrandamiento del bazo o ruptura esplénica.
Hay que controlar estrechamente el RAN, sobre todo durante las primeras semanas de tratamiento con filgrastim. Algunos pacientes pueden responder muy rápidamente a la dosis inicial de filgrastim y con un aumento considerable del recuento de neutrófilos. Se recomienda la medición diaria del RAN durante los 2 a 3 primeros días de la administración de filgrastim. Después, se recomienda medir el RAN al menos dos veces por semana durante las dos primeras semanas y posteriormente una vez a la semana o una vez cada dos semanas durante la terapia de mantenimiento. Durante la administración intermitente de 30 MU (300 microgramos)/día de filgrastim pueden producirse grandes fluctuaciones del RAN del paciente a lo largo del tiempo. Con objeto de determinar el RAN valle o nadir del paciente, se recomienda tomar muestras de sangre para medir el RAN inmediatamente antes de la administración de cualquier dosis prevista de filgrastim.
Riesgos asociados con dosis más altas de medicamentos mielodepresores
El tratamiento con filgrastim en monoterapia no impide la trombocitopenia y anemia secundaria a los medicamentos mielodepresores. Como consecuencia de la posibilidad de recibir dosis más altas o un mayor número de estos medicamentos con el tratamiento con filgrastim, el paciente puede presentar mayor riesgo de adquirir trombocitopenia y anemia. Se recomienda vigilar el recuento sanguíneo de forma regular (ver más arriba).
Infecciones y neoplasias que causan mielodepresión
La neutropenia puede deberse a infecciones oportunistas por infiltración de la médula ósea tales como el complejo Mycobacterium avium o a tumores como los linfomas. En los pacientes con tumores o infecciones conocidos que han infiltrado la médula ósea, se debe considerar la administración de un tratamiento adecuado para la enfermedad subyacente, además de la administración de filgrastim tratar la neutropenia. No se ha establecido adecuadamente el efecto del filgrastim sobre la neutropenia causada por tumores o por infecciones con infiltración de la médula ósea.
Precauciones especiales en la anemia drepanocítica
Se han notificado crisis drepanocíticas, en algunos casos ocasiones mortales, en pacientes con anemia drepanocítica a quienes se les administró filgrastim. Los médicos deberán tener precaución al considerar la administración de filgrastim a pacientes con anemia drepanocítica y evaluar minuciosamente los posible beneficios y riesgos.
Todos los _pacientes
Accofil contiene sorbitol (E420) como excipiente a una concentración de 50 mg/ml. Los pacientes con intolerancia hereditaria a la fructosa no deben utilizar este medicamento.
El protector de la aguja de la jeringa precargada contiene caucho natural seco (un derivado del látex), que puede provocar reacciones alérgicas.
Para mejorar la trazabilidad de los factores estimulantes de las colonias de granulocitos (G-CSF), se debe registrar claramente el nombre comercial del producto administrado en el historial del paciente.
4.5 Interacción con otros medicamentos y otras formas de interacción
La seguridad y eficacia de filgrastim administrado el mismo día que la quimioterapia citotóxica mielosupresora no ha sido establecido definitivamente. Teniendo en cuenta la sensibilidad de las células mieloides en rápida división a la quimioterapia citotóxica mielosupresora, no se recomienda el empleo de filgrastim desde 24 horas antes y 24 horas después de la quimioterapia. La evidencia preliminar de un pequeño número de pacientes tratados de forma concomitante con filgrastim y 5-Fluorouracilo indican que la gravedad de la neutropenia puede ser exacerbada.
Todavía no se ha investigado en ensayos clínicos la posible interacción con otros factores de crecimiento hematopoyético y citocinas.
Como el litio estimula la liberación de neutrófilos, es probable que potencie el efecto del filgrastim. Aunque esta interacción no se ha investigado formalmente, no hay evidencia de que tal interacción pueda ser nociva.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos o estos son limitados relativos al uso de filgrastim en mujeres embarazadas. Los estudios en animales han mostrado toxicidad para la reproducción. Se ha observado una mayor incidencia de pérdida embrionaria en conejos con múltiples altas dosis de exposición clínica y en presencia de toxicidad materna (ver sección 5.3). Existen informes en la literatura en los que se demuestra el paso transplacentario de filgrastim en mujeres embarazadas. No se recomienda filgrastim durante el embarazo.
Lactancia
Se desconoce si filgrastim// metabolitos se excretan en la leche materna. No se puede excluir un riesgo para el recién nacido/lactante. Se debe decidir si es necesario interrumpir la lactancia o interrumpir el tratamiento con filgrastim tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la mujer.
Fertilidad
Filgrastim no afectó la capacidad reproductiva o la fertilidad en ratas machos o hembras (ver sección
5.3) .
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios sobre los efectos en la capacidad de conducir y utilizar máquinas.
4.8 Reacciones adversas Resumen del perfil de seguridad
En los ensayos clínicos en pacientes con cáncer tratados con filgrastim, el efecto adverso más frecuente fue dolor musculoesquelético que fue leve o moderada en el 10% y en el 3% de los pacientes respectivamente.
También se ha notificado enfermedad injerto contra huésped (EICH).
En la movilización de PBPC en donantes sanos la reacción adversa más frecuente fue el dolor musculoesquelético. Se ha observado leucocitosis en los donantes y también se observó trombocitopenia después de filgrastim y leucocitaféresis de donantes. También se notificó esplenomegalia y ruptura esplénica. Algunos casos de ruptura esplénica fueron mortales.
En pacientes con NCG las reacciones adversas más frecuentes atribuibles a filgrastim son el dolor óseo, dolor musculoesquelético general y esplenomegalia. Los síndromes mielodisplásicos (SMD) o leucemia han desarrollado en pacientes con neutropenia congénita tratados con filgrastim (ver sección
4.4) .
El síndrome de extravasación capilar, que puede ser potencialmente mortal si se demora su tratamiento, fue raramente notificado (>1/1.000 a <1/100) en pacientes con cáncer sometidos a quimioterapia y en donantes sanos sometidos a movilización de células progenitoras de sangre periférica tras la administración de factores estimulantes de colonias de granulocitos (ver sección 4.4 e inciso C de la sección 4.8).
En los estudios clínicos con la administración de filgrastim a pacientes con VIH, los únicos efectos
adversos considerados de forma consistente en relación con la administración de filgrastim fueron dolor musculoesquelético, dolor óseo y mialgia.
Tabla resumen de reacciones adversas
Los datos de las tablas siguientes describen las reacciones adversas notificadas en los ensayos clínicos y en las notificaciones espontáneas. Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Los datos se presentan por separado para pacientes con cáncer, movilización de CPSP en donantes sanos, pacientes con NCG y pacientes con VIH, lo que refleja los diferentes perfiles de reacciones adversas de estas poblaciones.
La evaluación de las reacciones adversas se basa en los datos de frecuencia siguientes:
Muy frecuentes: > 1/10 Frecuentes: > 1/100 a < 1/10 Poco frecuentes: > 1/1,000 a < 1/100 Raras: > 1/10,000, < 1/1,000 Muy raras: < 1/10,000
Frecuencia no conocida: no puede estimarse a partir de los datos disponibles.
Pacientes con cáncer
|
Clasificación de órganos del sistema MedDRA |
Reacciones adversas | ||||
|
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras | |
|
Trastornos de la sangre y del sistema linfático |
Crisis drepanocíticaa Esplenomegaliaa Ruptura esplénicaa | ||||
|
Trastornos del sistema inmunológico |
Hipersensibilidad al fármacoa |
Enfermedad de injerto contra huésped b | |||
|
Trastornos del metabolismo y de la nutrición |
Aumento del ácido úrico en la sangre Aumento de la lactato- deshidrogenasa en la sangre Disminución del apetito3 |
Condrocalcinosisb | |||
|
Trastornos del sistema nervioso |
Cefalea3 | ||||
|
Trastornos vasculares |
Hipotensión |
Flebopatía oclusivad Alteraciones de la volemia Síndrome de extravasación capilar1 | |||
|
Trastornos respiratorios, torácicos y |
Dolor orofaríngeoa Tos3 |
Hemoptisise |
Síndrome de dificultad respiratoria | ||
|
Clasificación de órganos del sistema MedDRA |
Reacciones adversas | ||||
|
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras | |
|
mediastínicos |
Disnea |
agudaa Insuficiencia respiratoriaa Edema pulmonara Neumopatía intersticiala Infiltración pulmonara Hemorragia pulmonar | |||
|
Trastornos gastrointestinales |
Diarreaa Vómitos3 Estreñimiento3 Náuseasa | ||||
|
Trastornos hepatobiliares |
Aumento de la y-glutamiltransferasa Aumento de la fosfatasa alcalina en la sangre | ||||
|
Trastornos de la piel y del tejido subcutáneo |
Exantemaa Alopeciaa |
Síndrome de Sweet Vasculitis cutáneaa | |||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor musculoesquelético3 |
Exacerbación de la artritis reumatoide | |||
|
Trastornos renales y urinarios |
Disuria |
Anomalías urinarias | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Asteniaa Fatigaa Inflamación mucosaa |
Dolor torácicoa |
Dolor a | ||
aVer sección 4.8, Descripción de reacciones adversas seleccionadas.
bSe han notificado casos de EICH y muertes en pacientes tras un alotrasplante de médula ósea (ver sección 4.8, Descripción de reacciones adversas seleccionadas).
cIncluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en las extremidades, dolor musculoesquelético, dolor torácico musculoesquelético, dolor de cuello.
dSe observaron casos con filgrastim en situación posterior a la comercialización en pacientes sometidos a
trasplante de médula ósea o a movilización de CPSP.
eSe observaron casos en el entorno de ensayos clínicos con filgrastim.
|
Clasificación de órganos del sistema MedDRA |
Reacciones adversas | ||||
|
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras | |
|
Trastornos de la sangre y del sistema linfático |
T rombocitopenia Leucocitosis |
Esplenomega liaa |
Ruptura esplénica Crisis drepanocíticaa | ||
|
Trastornos del sistema inmunológico |
Reacción anafiláctica | ||||
|
Trastornos del metabolismo y de la nutrición |
Aumento de la lactato-deshidrogena sa en la sangre |
Hiperuricemia (aumento del ácido úrico en la sangre) | |||
|
Trastornos del sistema nervioso |
Cefalea | ||||
|
Trastornos vasculares |
Síndrome de extravasación capilara | ||||
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Hemorragia pulmonar Hemoptisis Infiltración pulmonar Hipoxia | |||
|
Trastornos hepatobiliares |
Aumento de la fosfatasa alcalina en la sangre |
Aumento de la aspartato-aminotransfer asa | |||
|
Trastornos musculoesquelétic os y del tejido conjuntivo |
Dolor musculoesquelético * |
Agravamiento de la artritis reumatoide | |||
aVer sección 4.8, Descripción de reacciones adversas seleccionadas
*Incluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en las extremidades, dolor
musculoesquelético, dolor torácico musculoesquelético, dolor de cuello.
Pacientes con NCG
|
Clasificación de |
Reacciones adversas | ||||
|
órganos del sistema MedDRA |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras |
|
Trastornos de la sangre y del sistema linfático |
Esplenomegalia Anaemia |
Trombocitopenia Ruptura esplénica |
Crisis drepanocíticaa | ||
|
Trastornos del metabolismo y de la nutrición |
Hiperuricemia Disminución de | ||||
|
Clasificación de |
Reacciones adversas | ||||
|
la glucosa en la sangre Aumento de la lactato- deshidrogenasa en la sangre | |||||
|
Trastornos del sistema nervioso |
Cefalea | ||||
|
Trastornos respiratorios, torácicos y mediastínicos |
Epistaxis | ||||
|
Trastornos gastrointestinales |
Diarrea | ||||
|
Trastornos hepatobiliares |
Hepatomegalia Aumento de la fosfatasa alcalina en la sangre | ||||
|
Trastornos de la piel y del tejido subcutáneo |
Exantema |
Vasculitis cutánea Alopecia | |||
|
Trastornos musculoesquelético s y del tejido conjuntivo |
Dolor musculoesqueléti co* Artralgia |
Osteoporosis | |||
|
Trastornos renales y urinarios |
Hematuria |
Proteinuria | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección | ||||
aVer sección 4.8, Descripción de reacciones adversas seleccionadas
*Incluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en las extremidades, dolor
musculoesquelético, dolor torácico musculoesquelético, dolor de cuello.
Pacientes con VIH
|
Clasificación de órganos del sistema MedDRA |
Reacciones adversas | ||||
|
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras | |
|
Trastornos de la sangre y del sistema linfático |
Esplenomegalia |
Crisis drepanocítica3 | |||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor musculoesquelét ico * | ||||
aVer sección 4.8, Descripción de reacciones adversas seleccionadas
*Incluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en las extremidades, dolor
musculoesquelético, dolor torácico musculoesquelético, dolor de cuello.
Descripción de reacciones adversas seleccionadas
Se han notificado casos de EICH y muertes en pacientes que recibieron G-CSF tras un trasplante alogénico de médula ósea (ver secciones 4.4 y 5.1).
Se han notificado casos de síndrome de extravasación capilar en la fase de pos-comercialización con el uso de factor estimulante de colonias de granulocitos. Por lo general, estos casos se han presentado en pacientes con neoplasias malignas avanzadas, sepsis, pacientes que toman múltiples medicamentos antineoplásicos o sometidos a aféresis (ver sección 4.4).
Pacientes con cáncer
En los ensayos clínicos aleatorizados controlados con placebo, filgrastim no incrementó la incidencia de reacciones adversas asociadas a la quimioterapia citotóxica. En dichos ensayos clínicos se notificaron ciertas reacciones adversas con la misma frecuencia en pacientes con cáncer tratados con filgrastim/quimioterapia y placebo/quimioterapia, entre ellas náuseas y vómitos, alopecia, diarrea, fatiga, anorexia, mucositis, cefalea, tos, exantema, dolor torácico, debilidad generalizada, dolor faríngeo, estreñimiento y dolor.
En situación posterior a la comercialización se ha notificado vasculitis cutánea en pacientes tratados con filgrastim. Se desconoce el mecanismo de la vasculitis en los pacientes que reciben filgrastim. A partir de los datos de los ensayos clínicos se estima que es poco frecuente.
Se han notificado casos de síndrome de Sweet (dermatosis febril aguda) posteriores a la comercialización. A partir de los datos de los ensayos clínicos se estima que son poco frecuentes.
En los estudios clínicos y en situación posterior a la comercialización se han notificado efectos adversos pulmonares, entre ellos neumopatía intersticial, edema pulmonar e infiltración pulmonar, que en algunos casos han ocasionado insuficiencia respiratoria o síndrome de dificultad respiratoria aguda (SDRA), que pueden ser mortales (ver sección 4.4).
Se han notificado de manera poco frecuente de casos de esplenomegalia y ruptura esplénica tras la administración de filgrastim. Algunos casos de ruptura esplénica fueron mortales (ver sección 4.4).
Reacciones de tipo hipersensibilidad incluyendo anafilaxia, erupción cutánea, urticaria, angioedema, disnea e hipotensión ocurrieron el tratamiento inicial o posterior en estudios clínicos y en la experiencia post-comercialización. En general, las notificaciones fueron más frecuentes después de la administración intravenosa. En algunos casos, los síntomas han reaparecido tras reexposición al fármaco, lo que sugiere una relación causal. Filgrastim se debe interrumpir de forma permanente en pacientes que desarrollen alguna reacción alérgica grave.
En la post-comercialización, se han notificado casos aislados de crisis de células falciformes en pacientes con enfermedad de células falciformes (ver sección 4.4). La frecuencia se estimó como poco frecuente a partir de los datos de ensayos clínicos.
Se ha notificado pseudogota en pacientes con cáncer tratados con filgrastim, y la frecuencia se estimó como poco frecuente a partir de los datos de ensayos clínicos.
Movilización de CPSP en donantes sanos
Se han notificado casos frecuentes pero generalmente asintomáticos de esplenomegalia y casos poco frecuentes de ruptura esplénica en pacientes y en donantes sanos después de la administración de filgrastim (ver sección 4.4).
Se han notificado acontecimientos adversos pulmonares tales como hemoptisis, hemorragia pulmonar, infiltración pulmonar, disnea e hipoxia (ver sección 4.4).
Se ha notificado con poca frecuencia exacerbación de los síntomas de artritis.
Se ha observado leucocitosis (leucocitos > 50 x 109/L) en el 41% de los donantes y se ha observado trombocitopenia transitoria (plaquetas < 100 x 109/L) después del tratamiento con filgrastim y la leucocitaféresis se observó en el 35% de los donantes.
Pacientes con NCG
Entre las reacciones adversas figuran la esplenomegalia, que puede ser progresiva en una minoría de los casos, y la trombocitopenia (ver sección 4.4).
Otras reacciones adversas posiblemente relacionadas con el tratamiento con filgrastim y observadas habitualmente en < 2% de los pacientes con NCG fueron reacción en el lugar de la inyección, cefalea, hepatomegalia, artralgia, alopecia, osteoporosis y exantema.
Durante el empleo prolongado se ha notificado vasculitis cutánea en el 2% de los pacientes con NCG. Pacientes con VIH
La esplenomegalia se notificó relacionada con el tratamiento con filgrastim en <3% de los pacientes.
En todos los casos de aumento del tamaño del bazo en pacientes con VIH, este se consideró leve o moderado durante la exploración física y la evolución clínica fue benigna; a ningún paciente se le diagnosticó hiperesplenismo y ninguno se sometió a una esplenectomía. Como el aumento del tamaño del bazo es frecuente en los pacientes con infección por VIH y la mayoría de los pacientes con SIDA lo presentan en mayor o menor grado, la relación con el tratamiento con filgrastim no está clara (ver sección 4.4).
Población pediátrica
Los datos de estudios clínicos con filgrastim en pacientes pediátricos indican que la seguridad y la eficacia del filgrastim son similares en adultos y en niños que reciben quimioterapia citotóxica, lo cual sugiere que no existen diferencias basadas en la edad en la farmacocinética del filgrastim. La única reacción adversa notificada de forma regular fue dolor musculoesquelético, lo cual no es diferente de la experiencia en la población adulta. No hay suficientes datos para evaluar más el uso de filgrastim en los pacientes pediátricos.
Otras poblaciones especiales
Pacientes de edad avanzada
No se observaron diferencias generales en la seguridad o eficacia entre los sujetos mayores de 65 años de edad en comparación con adultos más jóvenes (> 18 años de edad) de los sujetos que reciben quimioterapia citotóxica y la experiencia clínica no ha identificado diferencias en las respuestas entre los pacientes de edad avanzada y jóvenes. No hay datos suficientes para evaluar el uso de Accofil en pacientes de edad avanzada para otras indicaciones Accofil aprobadas.
Pacientes con NCG pediátricos
Los casos de disminución de la densidad ósea y la osteoporosis se han notificado en pacientes pediátricos con neutropenia crónica grave en tratamiento crónico con filgrastim. La frecuencia se estimó como "frecuente" según los datos de los ensayos clínicos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han establecido los efectos de la sobredosis de Accofil. La interrupción del tratamiento con filgrastim suele acompañarse de una disminución del 50% de los neutrófilos circulantes al cabo de 1 a 2 días, con una normalización al cabo de 1 a 7 días.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: citocinas, código ATC: L03AA02.
Accofil es un medicamento biosimilar. La información detallada sobre este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Efectos farmacodinámicos
El G-CSF humano es una glucoproteína que regula la producción y liberación de neutrófilos funcionales de la médula ósea. Accofil, que contiene r-metHuG-CSF (filgrastim), aumenta considerablemente el recuento de neutrófilos en la sangre periférica a las 24 horas, y mínimamente el de monocitos. En algunos pacientes con NCG, filgrastim puede inducir también un leve aumento del número de eosinófilos y basófilos circulantes con relación a los valores iniciales; algunos de estos pacientes pueden presentar eosinofilia o basofilia ya antes del tratamiento. El incremento de los recuentos de neutrófilos depende de la dosis, con la posología recomendada. Los neutrófilos producidos en respuesta al filgrastim muestran una función normal o superior a la habitual, de acuerdo con las pruebas de la función quimiotáctica y fagocitaria. Después de interrumpir el tratamiento con filgrastim, el recuento de neutrófilos circulantes se reduce un 50% al cabo de 1 a 2 días y se normaliza en el plazo de 1 a 7 días.
El empleo de filgrastim en pacientes sometidos a quimioterapia citotóxica reduce de forma significativa la incidencia, gravedad y duración de la neutropenia y de la neutropenia febril. El tratamiento con filgrastim reduce significativamente la duración de la neutropenia febril, el uso de antibióticos y la hospitalización después de la quimioterapia de inducción en la leucemia mieloide aguda o tras un tratamiento mieloablativo seguido de trasplante de médula ósea. La incidencia de fiebre y de infecciones documentadas no se redujo en ninguno de los dos casos. Tampoco se redujo la duración de la fiebre en los pacientes sometidos a tratamiento mieloablativo seguido de trasplante de médula ósea.
El empleo de filgrastim, bien en monoterapia o tras la quimioterapia, moviliza las células progenitoras hematopoyéticas hacia la sangre periférica. Estas CPSP autólogas se pueden recolectar y perfundir después de altas dosis de tratamiento citotóxico, bien junto con un trasplante de médula ósea o en lugar de éste. La perfusión de las CPSP acelera la recuperación hematopoyética, reduciendo el período de riesgo de complicaciones hemorrágicas y la necesidad de transfusiones plaquetarias. Los receptores de trasplantes alogénicos de CPSP movilizadas con filgrastim experimentaron una recuperación hematopoyética significativamente más rápida que los tratados con trasplante alogénico de médula ósea, ocasionando una disminución significativa del tiempo hasta la recuperación de las plaquetas sin soporte externo.
Un estudio retrospectivo europeo que evaluó el uso de G-CSF tras un trasplante alogénico de médula ósea en pacientes con leucemias agudas sugirió un aumento del riesgo de EICH, mortalidad relacionada con el tratamiento (MRT) y mortalidad cuando se administraba G-CSF. En un estudio retrospectivo internacional independiente en pacientes con leucemias mieloides agudas y crónicas, no se observó efecto alguno sobre el riesgo de EICH, MRT ni mortalidad. Un metanálisis de estudios de trasplantes alogénicos, que incluyó los resultados de nueve ensayos prospectivos aleatorizados, 8 estudios retrospectivos y 1 estudio de caso-control, no detectó efecto alguno sobre los riesgos de EICH aguda, EICH crónica ni mortalidad temprana relacionada con el tratamiento.
Riesgo relativo (IC del 95%) de EICH y MRT
|
tras el tratamiento con G-CSF después de un trasplante de médula ósea (MO) | |||||
|
Publicación |
Período de estudio |
N |
EICH aguda de grado II-IV |
EICH crónica |
MRT |
|
Metanálisis (2003) |
a 1986-2001 |
1.198 |
1,08 (0,87, 1,33) |
1,02 (0,82, 1,26) |
0,70 (0,38, 1,31) |
|
Estudio retrospectivo europeo (2004) |
b 1992-2002 |
1.789 |
1,33 (1,08, 1,64) |
1,29 (1,02, 1,61) |
1,73 (1,30, 2,32) |
|
Estudio retrospectivo internacional (2006) |
b 1995-2000 |
2.110 |
1,11 (0,86, 1,42) |
1,10 (0,86, 1,39) |
1,26 (0,95, 1,67) |
a
GM-CSF.
El análisis incluye estudios con trasplante de MO durante este período; algunos estudios utilizaron
b
El análisis incluye a pacientes que recibieron trasplantes de MO durante este período.
Empleo de filgrastim para la movilización de CPSP en donantes sanos antes de un trasplante alogénico de CPSP.
En donantes sanos, a 10 microgramos/kg/día de dosis administradas por vía subcutánea durante 4-5 días consecutivos permite obtener > 4 x 106 células CD34+/kg de peso del receptor en la mayoría de los donantes después de dos leucocitaféresis.
El empleo de filgrastim en los adultos, con NCG (neutropenia congénita grave, cíclica y neutropenia idiopática) induce un aumento sostenido de la ANC en la sangre periférica y una reducción de las infecciones y procesos relacionados.
El empleo de filgrastim en pacientes con infección por VIH mantiene el recuento de neutrófilos en los niveles normales permitiendo la administración programada de tratamiento antiviral y otros tratamientos mielodepresores. No hay pruebas de que los pacientes con infección por VIH tratados con filgrastim presenten un aumento de la replicación del VIH.
Al igual que otros factores de crecimiento hematopoyético, el factor estimulante de colonias de granulocitos ha demostrado tener propiedades estimuladoras de las células endoteliales humanas in
vitro.
5.2 Propiedades farmacocinéticas
Absorción
Las concentraciones séricas se mantuvieron por encima de 10 ng/ml durante 8-16 horas después de la administración subcutánea de las dosis recomendadas.
Distribución
El volumen de distribución en la sangre es de unos 150 ml/kg. Eliminación
Se ha demostrado que el aclaramiento del filgrastim sigue una farmacocinética de primer orden, tras su administración subcutánea o intravenosa. La semivida de eliminación del filgrastim es de unas 3,5 horas con una tasa de aclaramiento de unos 0,6 ml/min/kg. La perfusión continua de Accofil a lo largo de un período de hasta de 28 días en pacientes que se recuperan de un trasplante de médula ósea autóloga no dio muestras de acumulación farmacológica y las semividas de eliminación fueron comparables.
Linealidad
Existe una correlación lineal positiva entre la dosis y la concentración sérica de filgrastim, tanto en administración intravenosa como subcutánea. Tras la administración subcutánea de las dosis recomendadas, las concentraciones séricas se mantuvieron por encima de 10 ng/ml durante 8 a 16 horas. El volumen de distribución en sangre es de aproximadamente 150 ml/kg.
5.3 Datos preclínicos sobre seguridad
Se investigó filgrastim en estudios repetidos de toxicidad de dosis de hasta 1 año de duración que revelaron cambios atribuibles a las acciones farmacológicas esperadas que incluían aumentos de leucocitos, hiperplasia mieloide en médula ósea, granulopoyesis extramedular y aumento de tamaño del bazo. Estos cambios revertían después de suspender el tratamiento.
Se han estudiado los efectos de filgrastim en el desarrollo prenatal en ratas y conejos. La administración intravenosa (80 pg/kg/día) de filgrastim a conejos durante el periodo de organogénesis fue tóxica para la madre y aumentaba el aborto espontáneo, se observó la pérdida postimplantación y menor tamaño medio de cría viva y peso fetal.
Basándose en los datos notificados para otro producto filgrastim similar al de Accofil, se observaron hallazgos comparables más un aumento de las malformaciones fetales con 100 pg/kg/día, una dosis tóxica materna que correspondía a una exposición sistémica de aproximadamente 50 a 90 veces las exposiciones observadas en pacientes tratados con la dosis clínica de 5 pg/kg/día. El nivel de efecto adverso no observado para la toxicidad embriofetal en este estudio era de 10 pg/kg/día, que correspondía a una exposición sistemática de aproximadamente 3 a 5 veces las exposiciones observadas en pacientes tratados con la dosis clínica.
En ratas preñadas, no se observó toxicidad materna o fetal en dosis de hasta 575 pg/kg/día. Las crías de rata a las que se les administró filgrastim durante los periodos perinatales y de lactancia, mostraron un retraso en la diferenciación externa y retardo de crecimiento (>20 pg/kg/día) y una tasa de supervivencia ligeramente reducida (100 pg/kg/día).
Filgrastim no tenía un efecto observado sobre la fertilidad de las ratas machos o hembras.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Ácido acético glacial.
Hidróxido de sodio.
Sorbitol (E420).
Polisorbato 80.
Agua para preparaciones inyectables.
6.2 Incompatibilidades
Accofil no debe diluirse con soluciones salinas.
Filgrastim diluido puede adsorberse al vidrio y materiales plásticos.
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
6.3 Período de validez
36 meses.
Se ha demostrado la estabilidad química y física de la solución diluida para perfusión durante el uso hasta 24 horas entre 2 ° C y 8 °C. Desde el punto de vista microbiológico, el producto debe utilizarse inmediatamente. Si no se utiliza inmediatamente, el tiempo y las condiciones de conservación durante el uso y las condiciones antes de su uso son responsabilidad del usuario, y no se deberían superar normalmente las 24 horas entre 2 °C y 8 °C, salvo que la dilución haya tenido lugar en condiciones asépticas controladas y validadas.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C-8 °C). No congelar.
La exposición accidental por una sola vez a temperaturas de congelación no afecta de forma adversa a la estabilidad de Accofil. Si la exposición ha superado las 24 horas o se ha congelado más de una vez, entonces NO se debe utilizar Accofil.
Conservar la jeringa en el embalaje exterior para protegerlo de la luz.
Dentro de su periodo de validez y para su uso ambulatorio, el paciente puede sacar el medicamento de la nevera y almacenarlo a temperatura ambiente (no a temperatura superior a 25°C) durante un único periodo máximo de 15 días. Después de este periodo, el medicamento no se debe volver a refrigerar y se debe desechar.
Para las condiciones de conservación del medicamento diluido, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Jeringa precargada con aguja para inyección, con o sin protector de seguridad para la aguja.
Envases con una, tres, cinco, siete o diez jeringas precargadas, con o sin blister y gasas impregnadas en alcohol. Los envases sin blister no tienen protector de seguridad. Los envases blister son para jeringas individuales con protector de seguridad para la aguja prefijada. Las jeringas precargadas son de vidrio tipo I con tienen una aguja de acero inoxidable permanentemente incorporada en su extremo y tienen marcas impresas 1/40 para graduaciones de 0.1 ml a 1 ml en el cilindro. El protector de la aguja de la jeringa precargada contiene caucho natural seco (ver sección 4.4). Cada jeringa precargada contiene 0,5 ml de solución.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Si es necesario, Accofil se puede diluir en glucosa al 5%. No se recomienda en ningún caso diluir a una concentración final inferior a 0,2 MU (2 pg) por ml.
La solución debe inspeccionarse visualmente antes de usarla. Solamente deben utilizarse soluciones transparentes sin partículas. No agite.
Para los pacientes tratados con filgrastim diluido a concentraciones inferiores a 1,5 MU (15 pg) por ml, debe añadirse seroalbúmina humana (SAH) a una concentración final de 2 mg/ml. Ejemplo: en un volumen de inyección final de 20 ml, las dosis totales de filgrastim inferiores a 30 MU (300 pg) deben administrarse con adición de 0,2 ml de solución de albúmina humana 200 mg/ml (20%).
Accofil no contiene conservantes. En vista del posible riesgo de contaminación microbiana, las jeringas precargadas de Accofil se deben utilizar una sola vez.
Cuando se diluye en glucosa al 5%, Accofil es compatible con el vidrio y con diversos plásticos como PVC, poliolefina (copolímero de polipropileno y polietileno) y polipropileno.
Uso de la jeringa precargada con protector de seguridad para la aguja
El protector de seguridad para la aguja cubre la aguja tras haber realizado la inyección con el fin de prevenir pinchazos accidentales, lo cual no afecta a la forma de utilizar la jeringa. Empujar el émbolo lenta y uniformemente hasta que se haya administrado la totalidad de la dosis y el émbolo no pueda seguir avanzando. Retirar la jeringa manteniendo la presión sobre el émbolo. El protector de seguridad para la aguja cubrirá la aguja una vez que se suelte el émbolo.
Uso de la jeringa precargada sin protector de seguridad para la aguja
Administrar la dosis según el protocolo estándar.
Eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Accord Healthcare Limited Sage House, 319 Pinner Road,
North Harrow, Middlesex HA1 4HF,
Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/946/001
EU/1/14/946/002
EU/1/14/946/005
EU/1/14/946/006
EU/1/14/946/007
EU/1/14/946/008
EU/1/14/946/009
EU/1/14/946/010
EU/1/14/946/017
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 18.09.2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Accofil 48 MU/0,5 ml solución inyectable y para perfusión en jeringa precargada.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada ml de solución contiene 96 millones de unidades (MU; equivalentes a 960 microgramos [^g]) de filgrastim.
Cada jeringa precargada contiene 48 MU (equivalentes a 480 microgramos) de filgrastim en 0,5 ml de solución inyectable y para perfusión.
Filgrastim es un factor metionil recombinante estimulador de las colonias de granulocitos humanos por tecnología de ADN recombinante en Escherichia coli (BL21).
Excipientes con efecto conocido:
Cada mi de solución contiene 50 mg de sorbitol (E420)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable y para perfusión. Solución transparente, incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Accofil está indicado para la reducción de la duración de la neutropenia y de la incidencia de neutropenia febril en pacientes tratados con quimioterapia citotóxica establecida para neoplasias malignas (con la excepción de la leucemia mielógena crónica y los síndromes mielodisplásicos) y para la reducción de la duración de la neutropenia en pacientes que reciben tratamiento mieloablativo seguido de un trasplante de médula ósea que se considere que presentan un aumento del riesgo de neutropenia grave prolongada. La seguridad y la eficacia de Accofil son similares en adultos y en niños que reciben quimioterapia citotóxica.
Accofil está indicado para la movilización de células progenitoras de sangre periférica (CPSP).
En pacientes (niños o adultos) con neutropenia congénita, cíclica o idiopática grave que presentan un recuento absoluto de neutrófilos (RAN) < 0,5 x 109/l y antecedentes de infecciones intensas o recurrentes, la administración de Accofil a largo plazo está indicada para elevar los recuentos de neutrófilos y reducir la incidencia y la duración de los acontecimientos relacionados con las infecciones.
Accofil está indicado para el tratamiento de la neutropenia persistente (RAN igual o inferior a 1,0 x 109/l) en pacientes con infección avanzada por el VIH, con objeto de reducir el riesgo de infecciones bacterianas cuando otras opciones para el control de la neutropenia no son adecuadas.
4.2 Posología y forma de administración
El tratamiento con Accofil debe administrarse únicamente en colaboración con un centro de oncología que tenga experiencia en el uso del factor estimulante de las colonias de granulocitos (G-CSF) y en hematología y que disponga de las instalaciones necesarias para el diagnóstico. Los procedimientos de movilización y aféresis deben realizarse en colaboración con un centro de oncología-hematología con experiencia suficiente en este campo y en el que el seguimiento de las células progenitoras hematopoyéticas pueda realizarse de una forma correcta.
Posología
Quimioterapia citotóxica establecida
La dosis recomendada de filgrastim es de 0,5 MU/kg/día (5 microgramos/kg/día). La primera dosis de Accofil no debe administrarse menos de 24 horas después de la quimioterapia citotóxica. En estudios clínicos aleatorizados, se utilizó una dosis subcutánea de 230 microgramos/m2/día (4,0 a 8,4 microgramos/kg/día).
La administración diaria de filgrastim debe continuar hasta que se haya sobrepasado el nadir teórico de neutrófilos y su recuento retorne a su intervalo normal. Después de la quimioterapia establecida en tumores sólidos, linfomas y leucemias linfoides, se prevé que la duración del tratamiento necesario para alcanzar estos criterios sea de hasta 14 días. Tras el tratamiento de inducción y consolidación en la leucemia mieloide aguda, la duración del tratamiento puede ser bastante mayor (hasta 38 días) en función del tipo, la posología y las pautas de administración de la quimioterapia citotóxica.
Los pacientes sometidos a quimioterapia citotóxica experimentan un aumento transitorio del recuento de neutrófilos que ocurre habitualmente 1 a 2 días después de iniciar la administración de filgrastim.
Sin embargo, para conseguir una respuesta terapéutica sostenida, no se debe suspender el tratamiento con filgrastim hasta que no se haya sobrepasado el nadir teórico de neutrófilos y su recuento retorne a su intervalo normal. No se recomienda la interrupción prematura del tratamiento con filgrastim antes de alcanzar el nadir teórico de neutrófilos.
Pacientes sometidos a tratamiento mieloablativo seguido de trasplante de médula ósea
La dosis inicial recomendada de filgrastim es de 1,0 MU/kg/día (10 microgramos/kg/día). La primera dosis de filgrastim debe administrarse por lo menos 24 horas después de la quimioterapia citotóxica y por lo menos 24 horas después de la infusión de médula ósea.
Una vez sobrepasado el nadir de neutrófilos, la dosis diaria de filgrastim se ajustará en función de la respuesta de los neutrófilos del siguiente modo:
|
Recuento absoluto de neutrófilos (RAN) |
Ajuste de la dosis de filgrastim |
|
RAN > 1,0 x 109/L durante 3 días consecutivos |
Reducir a 0,5 MU/kg/día (5 microgramos/kg/día) |
|
Después, si el RAN permanece > 1,0 x 109/L durante 3 días consecutivos más |
Suspender filgrastim |
|
Si el RAN desciende a < 1,0 x 109/L durante el período de tratamiento, se debe reajustar de nuevo la dosis de filgrastim siguiendo los pasos previamente indicados | |
Movilización de las células progenitoras sanguíneas periféricas (CPSP)
Pacientes sometidos a tratamiento mielodepresor o mieloablativo seguido de trasplante autólogo de CPSP
La dosis recomendada de filgrastim para la movilización de PBPC cuando se usa solo es de 1,0 MU/kg/día (10 microgramos/kg/día) durante 5-7 días consecutivos. El tiempo de leucocitaféresis: 1 o 2 leucocitaféresis en los días 5 y 6, que es a menudo suficiente. En otras circunstancias, leucocitaféresis adicionales pueden ser necesarias. La administración de filgrastim debe mantenerse hasta la última leucocitaféresis.
La dosis recomendada de filgrastim para la movilización de PBPC tras una quimioterapia mielosupresora es de 0,5 MU/kg/día (5 microgramos/kg/día) administrados diariamente desde el primer día tras concluir la quimioterapia hasta sobrepasar el nadir teórico de neutrófilos y el recuento de neutrófilos retorne al rango normal. La leucocitaféresis se debe realizar durante el período en que el ANC se levanta de < 0,5 x 109/L de > 5,0 x 109/L. Para los pacientes que no han sido sometidos a quimioterapia, una leucocitaféresis suele ser suficiente. En otras circunstancias, se recomienda la realización de leucocitaféresis adicionales.
Para la movilización de CPSP en donantes sanos antes del alotrasplante de CPSP
Para la movilización de PBPC en donantes sanos, filgrastim debe administrarse a los 10 microgramos/kg/día durante 4-5 días consecutivos. Las leucocitaféresis deben iniciarse en el día 5 y se prolongó hasta el día 6 si es necesario con el fin de recoger 4 x 106 células CD34+/kg del peso del cuerpo receptor.
En pacientes con neutropenia crónica grave (NCG)
Neutropenia congénita
La dosis inicial recomendada es de 1,2 MU/kg/día (12 microgramos/kg/día) en dosis única o dividida en varias dosis.
Neutropenia idiopática o cíclica
La dosis inicial recomendada es de 0,5 MU/kg/día (5 microgramos/kg/día) en dosis única o dividida en varias dosis.
Ajustes de la dosis: filgrastim se debe administrar diariamente por inyección subcutánea hasta alcanzar y poder mantener el recuento de neutrófilos por encima de 1,5 x 109/L. Una vez alcanzada la respuesta se establecerá la dosis mínima efectiva para mantener este nivel. Si se desea mantener un nivel de neutrófilos adecuado, es necesaria la administración diaria de filgrastim a largo plazo. La dosis inicial se puede duplicar o reducir a la mitad al cabo de 1 a 2 semanas de tratamiento, en función de la respuesta del paciente. Posteriormente, la dosis se puede ajustar individualmente en intervalos de 1-2 semanas para mantener un recuento medio de neutrófilos entre 1,5 x 109/L y 10 x 109/L. En los pacientes con infecciones graves se puede contemplar un aumento más rápido de la dosis. En los ensayos clínicos, el 97% de los pacientes que respondieron al tratamiento presentaron una respuesta completa a dosis < 2,4 MU/kg/día (24 microgramos/kg/día). En pacientes con NCG, no se ha establecido la seguridad a largo plazo de la administración de filgrastim por encima de 2,4 MU/kg/día (24 microgramos/kg/día).
Pacientes con infección por VIH
Para la recuperación de la neutropenia
La dosis inicial recomendada de filgrastim es de 0,1 MU/kg/día (1 microgramos/kg/día) se administra diariamente, ajustando la dosis hasta un máximo de 0,4 MU/kg/día (4 microgramos/kg/día) hasta que se alcanza un recuento de neutrófilos normal y se puede mantener (RAN > 2,0 x 109/L). En los estudios clínicos, más del 90% de los pacientes respondieron a estas dosis, logrando una recuperación de la neutropenia en una mediana de 2 días. En un pequeño número de pacientes (<10%), dosis de hasta 1,0 MU/kg/día (10 microgramos/kg/día) fueron obligados a revertir la neutropenia.
Para mantener el recuento normal de neutrófilos
Una vez alcanzada la recuperación de la neutropenia, se establecerá la dosis mínima efectiva para mantener un recuento normal de neutrófilos. Se recomienda comenzar el ajuste de dosis administrando 30 MU/kg/día (300 microgramos/kg/día) cada dos días. En función del RAN del paciente podrán ser necesarios nuevos ajustes de la dosis para mantener un recuento de neutrófilos > 2,0 x 109/L. En los ensayos clínicos, se requirió la administración de 30 MU/kg/día (300 microgramos/kg/día) de 1 a 7 días a la semana para mantener un RAN > 2,0 x 109/L, siendo la mediana de la frecuencia de dosis de 3 días a la semana. Puede ser necesaria una administración prolongada para mantener el RAN > 2,0 x 109/L.
Poblaciones especiales Pacientes de edad avanzada
Los ensayos clínicos con filgrastim han incluido un pequeño número de pacientes de edad avanzada, pero los estudios especiales no se han llevado a cabo en este grupo y por lo tanto, las recomendaciones posológicas específicas no se pueden hacer.
Pacientes con insuficiencia renal o hepática
Los estudios del filgrastim en pacientes con alteración grave de la función hepática o renal demuestran que presenta un perfil farmacodinámico y farmacocinético similar al observado en individuos sanos.
En estos casos no se requiere ajuste de dosis.
Pacientes pediátricos en el marco de la NCG y el cáncer
Sesenta y cinco por ciento de los pacientes estudiados en el programa de ensayo sobre NCG con la administración de filgrastim, eran menores de 18 años de edad. La eficacia del tratamiento era clara para este grupo de edad, que incluía a la mayoría de los pacientes con neutropenia congénita. No existían diferencias en los perfiles de seguridad para pacientes pediátricos tratados por NCG.
Los datos de estudios clínicos en pacientes pediátricos indican que la seguridad y la eficacia del filgrastim son similares en adultos y en niños que reciben quimioterapia citotóxica.
Las recomendaciones posológicas para pacientes pediátricos son iguales que las correspondientes a los adultos que reciben quimioterapia citotóxica mielosupresora.
Forma de administración
Quimioterapia citotóxica establecida
Filgrastim puede administrarse como una inyección subcutánea diaria o, alternativamente, como una perfusión intravenosa diaria diluido en glucosa 50 mg / ml (5%) solución durante 30 minutos. Para obtener más instrucciones sobre la dilución antes de la perfusión, ver sección 6.6. Se prefiere la vía subcutánea en la mayoría de los casos. Existe cierta evidencia de un estudio de administración de dosis única que la dosificación intravenosa puede acortar la duración del efecto. La relevancia clínica de este hallazgo en la administración de dosis múltiples no está clara. La elección de la vía de administración depende de la situación clínica individual. En los estudios clínicos aleatorizados, se utilizó una dosis subcutánea de 23 MU/m2/día (230 microgramos/m2/día) o bien 4-8,4 microgramos/kg/día.
Pacientes sometidos a tratamiento mieloablativo seguido de trasplante de médula ósea
Filgrastim se administra en perfusión intravenosa breve a lo largo de 30 minutos o en perfusión subcutánea o intravenosa continua a lo largo de 24 horas, en ambos casos tras su dilución en 20 ml de solución de glucosa de 50 mg/ml (5%). Para obtener instrucciones adicionales acerca de su dilución con solución de glucosa de 50 mg/ml (5%) antes de la perfusión, ver sección 6.6.
En pacientes con la movilización de PBPC
Filgrastim para la movilización de PBPC cuando se usa solo:
Filgrastim se puede administrar como pefusión continua subcutánea de 24 horas o por inyección sucutánea
Filgrastim para la movilización de PBPC después de la quimioterapia mielosupresora:
Filgrastim se debe administrar por inyección subcutánea.
Para la movilización de PBPC en donante normal antes del alotransplante PBPC Figrastim se debe administrar por inyección subcutánea.
En pacientes con NCG
Neutropenia congénita, idiopática o cíclica: Filgrastim se debe administrar por inyección subcutánea.
En pacientes con infección por el VIH
Para la recuperación de la neutropenia y mantenimiento del recuento de neutrófilos en pacientes con infección por VIH, filgrastim se administra por vía subcutánea
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Advertencias especiales
Filgrastim no debe utilizarse para incrementar la dosis de quimioterapia citotóxica más allá de las pautas posológicas establecidas.
No se debe administrar filgrastim a pacientes con neutropenia congénita grave que presenten leucemia o muestren evidencia de evolución leucémica.
Se ha notificado hipersensibilidad, incluidas reacciones anafilácticas, que se producen al inicio del tratamiento o posteriormente en pacientes tratados con filgrastim. Suspenda de manera permanente el filgrastim en pacientes con hipersensibilidad clínicamente significativa. No administre filgrastim a pacientes con antecedentes de hipersensibilidad a filgrastim o pegfilgrastim.
Como ocurre con todas las proteínas terapéuticas, existe un potencial de inmunogenicidad. La velocidad de generación de anticuerpos contra filgrastim generalmente es baja. Los anticuerpos de fijación se producen como se espera con todos los productos biológicos; sin embargo, no se han asociado con actividad neutralizante en la actualidad.
Precauciones especiales en pacientes con leucemia mieloide aguda (LMA)
Crecimiento de las células malignas
El G-CSF puede estimular el crecimiento de células granulocíticas in vitro y se pueden observar efectos similares en algunas células no granulocíticas in vitro.
No se ha establecido la seguridad y la eficacia de la administración de filgrastim en los pacientes con síndrome mielodisplásico o leucemia mieloide crónica. Por lo tanto, filgrastim no está indicado en estas enfermedades. Se debe prestar especial atención para distinguir el diagnóstico de leucemia mieloide crónica en transformación blástica del de leucemia mieloide aguda.
Considerando que los datos disponibles sobre la seguridad y la eficacia en pacientes con LMA secundaria son limitados, filgrastim debe administrarse con precaución. No se ha establecido la seguridad y la eficacia de la administración de filgrastim en pacientes menores de 55 años y con LMA de novo con buena citogenética [t (8; 21), t (15; 17) e inv (16)].
La monitorización de la densidad ósea puede estar indicada en pacientes tratados con filgrastim durante más de 6 meses que presenten enfermedad osteoporótica subyacente.
Se han notificado reacciones adversas pulmonares, en particular neumonía intersticial, tras la administración de G-CSF. Los pacientes con antecedentes recientes de infiltrados pulmonares o neumonía pueden presentar mayor riesgo. La aparición de síntomas respiratorios tales como tos, fiebre y disnea, en asociación con signos radiológicos de infiltrados pulmonares y deterioro de la función pulmonar, pueden ser los síntomas preliminares del síndrome de dificultad respiratoria aguda (SDRA). Se debe suspender la administración de filgrastim e instaurar el tratamiento adecuado en estos casos.
Se han notificado casos de síndrome de extravasación capilar después de la administración de factor estimulante de colonias de granulocitos, que se caracterizan por hipotensión, hipoalbuminemia, edema y hemoconcentración. Los pacientes que desarrollan síntomas de síndrome de extravasación capilar se deben controlar de cerca y deben recibir tratamiento sintomático estándar, que puede incluir la necesidad de cuidados intensivos (ver sección 4.8).
Precauciones especiales en los pacientes con cáncer
Se han notificado casos de esplenomegalia y ruptura esplénica de forma poco frecuente tras la administración de filgrastim. Algunos casos de ruptura esplénica fueron mortales. Los pacientes que recibieron filgrastim que notificaron dolor abdominal superior y/o extremo del hombro se deben evaluar para agrandamiento del bazo o ruptura esplénica.
Leucocitosis
En menos del 5% de los pacientes tratados con filgrastim a dosis superiores a 0,3 MU/kg/día (3 ^g/kg/día) se ha observado un recuento leucocitario de 100 x 109/L o superior. No se ha notificado ninguna reacción adversa directamente atribuible a este grado de leucocitosis. No obstante, ante los posibles riesgos asociados a la leucocitosis intensa, se debe realizar periódicamente un recuento de leucocitos durante el tratamiento con filgrastim. Si el recuento leucocitario supera 50 x 109/L después del nadir teórico, se debe interrumpir inmediatamente el tratamiento con filgrastim. Sin embargo, durante el período de administración de filgrastim para la movilización de CPSP, se debe interrumpir la administración de filgrastim o disminuir la dosis si el recuento de leucocitos aumenta por encima de 70 x 109/L.
Riesgos asociados con el aumento de la dosis de la quimioterapia
Se debe tener especial precaución en los pacientes tratados con quimioterapia en dosis altas, ya que no se ha demostrado una mejora de los resultados obtenidos sobre el tumor y la intensificación de las dosis de quimioterapia puede ocasionar mayor toxicidad cardíaca, pulmonar, neurológica y dermatológica (consulte la ficha técnica de los agentes quimioterápicos concretos utilizados).
El tratamiento con filgrastim en monoterapia no impide la trombocitopenia y anemia secundaria a la quimioterapia mielodepresora. Ante la posibilidad de recibir dosis más altas de quimioterapia (p. ej., dosis completas según el calendario prescrito), el paciente puede correr mayor riesgo de trombocitopenia y anemia. Por eso, se recomienda vigilar periódicamente el recuento plaquetario y el hematocrito. Se deben tomar medidas de precaución especiales cuando se administren agentes quimioterápicos, tanto solos como combinados, susceptibles de provocar trombocitopenia grave.
Se ha demostrado que el uso de CPSP movilizadas por filgrastim reduce la intensidad y duración de la trombocitopenia tras la quimioterapia mieloablativa o mielodepresora.
Otras precauciones especiales
Aún no se conoce el efecto de filgrastim en los pacientes con una reducción considerable de los progenitores mieloides. Filgrastim actúa fundamentalmente sobre los precursores de los neutrófilos,
aumentando el recuento de estas células. Por tanto, la respuesta de neutrófilos podría disminuir en los pacientes con disminución de las células precursoras (como aquellos tratados con radioterapia o quimioterapia intensiva, o aquellos con infiltración tumoral de médula ósea).
De forma esporádica se han notificado trastornos vasculares, entre ellos flebopatía oclusiva y alteraciones de la volemia, en pacientes tratados con dosis altas de quimioterapia seguidas de trasplante.
Se han notificado casos de enfermedad de injerto contra huésped (EICH) y muertes en pacientes que recibieron G-CSF tras un alotrasplante de médula ósea (ver secciones 4.8 y 5.1).
El aumento de la actividad hematopoyética de la médula ósea en respuesta al tratamiento con factor de crecimiento se ha relacionado con anomalías transitorias en las imágenes óseas. Esto se debe tener en cuenta cuando se interpreten resultados de imágenes óseas.
Precauciones especiales en pacientes sometidos a movilización de PBPC
Movilización de PBPC
No existen comparaciones aleatorizadas de forma prospectiva de los dos métodos de movilización recomendados (filgrastim en monoterapia o en combinación con quimioterapia mielodepresora) en la misma población de pacientes. El grado de variación entre pacientes individuales y entre pruebas analíticas de células CD34+ significa que la comparación directa entre estudios diferentes es difícil. Por consiguiente, es difícil recomendar un método óptimo. La elección de método de movilización debe contemplarse en relación con los objetivos generales del tratamiento para un paciente individual.
Exposición previa a agentes citotóxicos
Los pacientes que se han sometido a tratamiento mielodepresor previo muy intenso pueden no manifestar una movilización suficiente de CPSP para alcanzar el rendimiento mínimo recomendado (2,0 x 106 células CD34+kg) o una aceleración en la recuperación plaquetaria, en el mismo grado.
Algunos agentes citotóxicos muestran toxicidad especial en el reservorio progenitor hematopoyético y pueden afectar negativamente a la movilización de las células progenitoras. Agentes tales como melfalán, carmustina (BCNU) y carboplatino cuando se administran durante períodos prolongados previos al intento de movilización de células progenitoras, pueden reducir el rendimiento de las células progenitoras. Sin embargo, la administración de melfalán, carboplatino o carmustina (BCNU), junto con filgrastim ha demostrado ser eficaz para la movilización de las células progenitoras. Cuando se prevé un trasplante de células progenitoras de sangre periférica, se recomienda planificar el procedimiento de movilización de células madre al comienzo del periodo de tratamiento del paciente. Particular atención debe prestarse a la cantidad de células progenitoras movilizadas en estos pacientes antes de la administración de altas dosis de quimioterapia. Si los rendimientos no son adecuados, según lo medido por los criterios anteriores, se deben considerar formas alternativas de tratamiento que no requieran el soporte de células progenitoras.
Evaluación del rendimiento de células progenitoras
Se recomienda prestar especial atención al método de cuantificación para evaluar el número de células progenitoras recolectadas en los pacientes tratados con filgrastim. Los resultados de los análisis por citometría de flujo del número de células CD34+ varían en función de precisión de la metodología utilizada; por tanto, deben interpretarse con precaución las recomendaciones numéricas basadas en estudios realizados en otros laboratorios.
Los análisis estadísticos de la relación entre el número de células CD34+ nuevamente perfundidas y la velocidad de recuperación plaquetaria tras altas dosis de quimioterapia indican que dicha relación es compleja pero continua.
La recomendación de un rendimiento mínimo de > 2,0 x 106 células CD34+/kg se basa en las experiencias publicadas resultantes de una reconstitución hematológica adecuada. Los rendimientos superiores parecen estar en correlación con una recuperación más rápida, y los inferiores con una recuperación más lenta.
Precauciones especiales en donantes sanos sometidos a movilización de células progenitoras sanguíneas periféricas
La movilización de CPSP no ofrece ningún beneficio clínico directo a los donantes sanos y solamente debe considerarse en el marco de un trasplante alogénico de células progenitoras hematopoyéticas.
Solo debe contemplarse la movilización de CPSP en donantes que cumplan los criterios normales de idoneidad clínica y analítica para la donación de células madre, prestando especial atención a los valores hematológicos y a las enfermedades infecciosas. No se ha evaluado la seguridad y la eficacia del filgrastim en donantes sanos menores de 16 años o mayores de 60 años.
Se ha notificado trombocitopenia de forma muy frecuente en pacientes a los que se administra filgrastim. Por tanto, se debe controlar estrechamente el recuento plaquetario.
Después de la administración de filgrastim y de la leucocitaféresis se ha observado trombocitopenia transitoria (plaquetas < 100 x 109/L) en el 35% de los sujetos estudiados. Entre ellos, se notificaron dos casos con plaquetas < 50 x 109/L, que se atribuyeron al procedimiento de leucocitaféresis. Si hace falta más de una leucocitaféresis, se debe prestar especial atención a los donantes cuyas plaquetas sean <
100 x 109/L antes de la leucocitaféresis; en general no se debe realizar aféresis si las plaquetas son < 75 x 109/L.
No deben realizarse leucocitaféresis a donantes tratados con anticoagulantes o con defectos conocidos en la homeostasis. Se debe suspender la administración de filgrastim o reducir la dosis si el recuento de leucocitos se eleva a > 70 x 109/L. Los donantes tratados con G-CSF para la movilización de CPSP deben controlarse hasta que los índices hematológicos vuelvan a los valores normales.
Anomalías citogenéticas transitorias se han observado en los donantes sanos tratados con G-CSF. Se desconoce la importancia de estos cambios. Sin embargo, no se puede excluir el riesgo de estimulación de un clon mieloide maligno. Se recomienda que el centro de aféresis lleve un control y seguimiento sistemático de los donantes de células progenitoras hematopoyéticas durante al menos 10 años para garantizar la seguridad a largo plazo.
Se han notificado casos frecuentes pero generalmente asintomáticos de esplenomegalia y poco frecuentes los casos de ruptura esplénica en donantes sanos y en pacientes tras la administración de G-CSF. Algunos casos de ruptura esplénica fueron mortales. Por lo tanto, el tamaño del bazo debe controlarse estrechamente (p. ej., examen clínico, ultrasonido). Se debe considerar un diagnóstico de ruptura esplénica en los donantes y/o pacientes que refieran dolor abdominal superior izquierda o el extremo del hombro.
En donantes sanos, la disnea se ha notificado con frecuencia y otros eventos adversos pulmonares (hemoptisis, hemorragia pulmonar, infiltrados pulmonares, e hipoxia) se han notificado de forma poco frecuente. En caso de eventos adversos pulmonares sospechosos o confirmados, se debe considerar la interrupción del tratamiento con filgrastim y administrar la atención médica adecuada.
Precauciones especiales para los receptores de CPSP movilizadas con filgrastim
Los datos disponibles indican que, las interacciones inmunitarias entre el aloinjerto de CPSP y el receptor pueden estar asociadas a un incremento del riesgo de EICH aguda o crónica en comparación con el trasplante de médula ósea.
Precauciones especiales en los pacientes con NCG
Se ha notificado de forma frecuente trombocitopenia en pacientes a los que se administra filgrastim. El recuento plaquetario se debe controlar estrechamente, sobre todo durante las primeras semanas de tratamiento con filgrastim. En los pacientes que presenten trombocitopenia, es decir, en aquellos con un recuento de plaquetas persistentemente < 100,000/mm3, debe valorarse la posibilidad de suspender el tratamiento con filgrastim de forma intermitente o, al menos, reducir la dosis.
Existen también otros cambios del hemograma, como anemia y aumento transitorio de los progenitores mieloides, que obligan a vigilar estrechamente el recuento celular.
Transformación hacia leucemia o síndrome mielodisplásico
Conviene establecer cuidadosamente el diagnóstico de las NCG y diferenciarlas de otros trastornos hematopoyéticos como anemia aplásica, mielodisplasia y leucemia mieloide. Antes del tratamiento debe realizarse un hemograma completo con fórmula leucocitaria y recuento de plaquetas, así como un estudio de la morfología y del cariotipo de la médula ósea.
Hubo una baja frecuencia (aproximadamente 3%) de síndrome mielodisplásico (SMD) o leucemia en pacientes incluidos en ensayos clínicos con NCG tratados con filgrastim. Esta observación sólo se ha hecho en pacientes con neutropenia congénita. SMD y las leucemias son complicaciones naturales de la enfermedad y su relación con el tratamiento con filgrastim es incierta. Un subconjunto de aproximadamente el 12% de pacientes con evaluaciones citogenéticas normales al inicio del estudio posteriormente se encontró que tenían anormalidades, incluyendo monosomía 7, en la evaluación repetida habitual. Si los pacientes con NCG desarrollan una citogenética anormal, los riesgos y beneficios de continuar filgrastim debe sopesarse cuidadosamente; filgrastim debe interrumpir si se desarrolla síndrome mielodisplásico o leucemia. Actualmente no está claro si el tratamiento a largo plazo de los pacientes con NCG predispone hacia anormalidades citogenéticas, SMD o transformación leucémica. Se recomienda realizar exámenes de médula ósea morfológicos y citogenéticos en pacientes a intervalos regulares (aproximadamente cada 12 meses).
Otras precauciones especiales
Se deben excluir las causas que provoquen neutropenia transitoria, como es el caso de las infecciones víricas.
Se han notificado de forma muy frecuente casos de esplenomegalia y casos de ruptura esplénica se han notificado de forma frecuente tras la administración de filgrastim. Los pacientes que recibieron filgrastim que notificaron dolor abdominal superior y/o dolor de extremo del hombro se deben evaluar para agrandamiento del bazo o ruptura esplénica
La esplenomegalia es una consecuencia directa del tratamiento con filgrastim. En los ensayos, el treinta y uno por ciento (31%) de los pacientes presentaron esplenomegalia detectable por palpación.
El aumento del volumen, medido radiográficamente, se presentó al comienzo del tratamiento con filgrastim y tendió a estabilizarse más tarde durante el tratamiento. La progresión del aumento del tamaño del bazo disminuyó o se detuvo al reducir la dosis y solo un 3% de los pacientes requirieron esplenectomía. El tamaño del bazo debe evaluarse de forma regular. Para detectar un aumento anómalo del volumen esplénico basta con realizar una palpación abdominal.
La hematuria era frecuente y la proteinuria ocurre en un pequeño número de pacientes. Se deben realizar análisis de orina normal para controlar este evento.
No se ha establecido la seguridad y la eficacia en recién nacidos ni en pacientes con neutropenia autoinmunitaria.
Precauciones especiales en pacientes con infección por VIH
Se han notificado de forma frecuente casos de esplenomegalia tras la administración de filgrastim. Los pacientes que recibieron filgrastim que notificaron dolor abdominal superior y/o dolor de extremo del hombro se deben evaluar debido al agrandamiento del bazo o ruptura esplénica.
Hay que controlar estrechamente el RAN, sobre todo durante las primeras semanas de tratamiento con filgrastim. Algunos pacientes pueden responder muy rápidamente a la dosis inicial de filgrastim y con un aumento considerable del recuento de neutrófilos. Se recomienda la medición diaria del RAN durante los 2 a 3 primeros días de la administración de filgrastim. Después, se recomienda medir el RAN al menos dos veces por semana durante las dos primeras semanas y posteriormente una vez a la semana o una vez cada dos semanas durante la terapia de mantenimiento. Durante la administración intermitente de 30 MU (300 microgramos)/día de filgrastim pueden producirse grandes fluctuaciones del RAN del paciente a lo largo del tiempo. Con objeto de determinar el RAN valle o nadir del paciente, se recomienda tomar muestras de sangre para medir el RAN inmediatamente antes de la administración de cualquier dosis prevista de filgrastim.
Riesgos asociados con dosis más altas de medicamentos mielodepresores
El tratamiento con filgrastim en monoterapia no impide la trombocitopenia y anemia secundaria a los medicamentos mielodepresores. Como consecuencia de la posibilidad de recibir dosis más altas o un mayor número de estos medicamentos con el tratamiento con filgrastim, el paciente puede presentar mayor riesgo de adquirir trombocitopenia y anemia. Se recomienda vigilar el recuento sanguíneo de forma regular (ver más arriba).
Infecciones y neoplasias que causan mielodepresión
La neutropenia puede deberse a infecciones oportunistas por infiltración de la médula ósea tales como el complejo Mycobacterium avium o a tumores como los linfomas. En los pacientes con tumores o infecciones conocidos que han infiltrado la médula ósea, se debe considerar la administración de un tratamiento adecuado para la enfermedad subyacente, además de la administración de filgrastim tratar la neutropenia. No se ha establecido adecuadamente el efecto del filgrastim sobre la neutropenia causada por tumores o por infecciones con infiltración de la médula ósea.
Precauciones especiales en la anemia drepanocítica
Se han notificado crisis drepanocíticas, en algunos casos ocasiones mortales, en pacientes con anemia drepanocítica a quienes se les administró filgrastim. Los médicos deberán tener precaución al considerar la administración de filgrastim a pacientes con anemia drepanocítica y evaluar minuciosamente los posible beneficios y riesgos.
Todos los _pacientes
Accofil contiene sorbitol (E420) como excipiente a una concentración de 50 mg/ml. Los pacientes con intolerancia hereditaria a la fructosa no deben utilizar este medicamento.
El protector de la aguja de la jeringa precargada contiene caucho natural seco (un derivado del látex), que puede provocar reacciones alérgicas.
Para mejorar la trazabilidad de los factores estimulantes de las colonias de granulocitos (G-CSF), se debe registrar claramente el nombre comercial del producto administrado en el historial del paciente.
4.5 Interacción con otros medicamentos y otras formas de interacción
La seguridad y eficacia de filgrastim administrado el mismo día que la quimioterapia citotóxica mielosupresora no ha sido establecido definitivamente. Teniendo en cuenta la sensibilidad de las células mieloides en rápida división a la quimioterapia citotóxica mielosupresora, no se recomienda el empleo de filgrastim desde 24 horas antes y 24 horas después de la quimioterapia. La evidencia preliminar de un pequeño número de pacientes tratados de forma concomitante con filgrastim y 5-Fluorouracilo indican que la gravedad de la neutropenia puede ser exacerbada.
Todavía no se ha investigado en ensayos clínicos la posible interacción con otros factores de crecimiento hematopoyético y citocinas.
Como el litio estimula la liberación de neutrófilos, es probable que potencie el efecto del filgrastim. Aunque esta interacción no se ha investigado formalmente, no hay evidencia de que tal interacción pueda ser nociva.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos o estos son limitados relativos al uso de filgrastim en mujeres embarazadas. Los estudios en animales han mostrado toxicidad para la reproducción. Se ha observado una mayor incidencia de pérdida embrionaria en conejos con múltiples altas dosis de exposición clínica y en presencia de toxicidad materna (ver sección 5.3). Existen informes en la literatura en los que se demuestra el paso transplacentario de filgrastim en mujeres embarazadas.
No se recomienda filgrastim durante el embarazo.
Lactancia
Se desconoce si filgrastim / metabolitos se excretan en la leche materna. No se puede excluir un riesgo para el recién nacido/lactante. Se debe decidir si es necesario interrumpir la lactancia o interrumpir el tratamiento con filgrastim tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la mujer.
Fertilidad
Filgrastim no afectó la capacidad reproductiva o la fertilidad en ratas machos o hembras (ver sección
5.3) .
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios sobre los efectos en la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas Resumen del perfil de seguridad
En los ensayos clínicos en pacientes con cáncer tratados con filgrastim, el efecto adverso más frecuente fue dolor musculoesquelético que fue leve o moderada en el 10% y en el 3% de los pacientes respectivamente.
También se ha notificado enfermedad injerto contra huésped (EICH).
En la movilización de PBPC en donantes sanos la reacción adversa más frecuente fue el dolor musculoesquelético. Se ha observado leucocitosis en los donantes y también se observó trombocitopenia después de filgrastim y leucocitaféresis de donantes. También se notificó esplenomegalia y ruptura esplénica. Algunos casos de ruptura esplénica fueron mortales.
En pacientes con NCG las reacciones adversas más frecuentes atribuibles a filgrastim son el dolor óseo, dolor musculoesquelético general y esplenomegalia. Los síndromes mielodisplásicos (SMD) o leucemia han desarrollado en pacientes con neutropenia congénita tratados con filgrastim (ver sección
4.4) .
El síndrome de extravasación capilar, que puede ser potencialmente mortal si se demora su tratamiento, fue raramente notificado (>1/1,000 a <1/100) en pacientes con cáncer sometidos a quimioterapia y en donantes sanos sometidos a movilización de células progenitoras de sangre periférica tras la administración de factores estimulantes de colonias de granulocitos (ver sección 4.4 e inciso C de la sección 4.8).
En los estudios clínicos con la administración de filgrastim a pacientes con VIH, los únicos efectos
adversos considerados de forma consistente en relación con la administración de filgrastim fueron dolor musculoesquelético, dolor óseo y mialgia.
Tabla resumen de reacciones adversas
Los datos de las tablas siguientes describen las reacciones adversas notificadas en los ensayos clínicos y en las notificaciones espontáneas. Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Los datos se presentan por separado para pacientes con cáncer, movilización de CPSP en donantes sanos, pacientes con NCG y pacientes con VIH, lo que refleja los diferentes perfiles de reacciones adversas de estas poblaciones.
La evaluación de las reacciones adversas se basa en los datos de frecuencia siguientes:
Muy frecuentes: > 1/10 Frecuentes: > 1/100 a < 1/10 Poco frecuentes: > 1/1,000 a < 1/100 Raras: > 1/10,000, < 1/1,000 Muy raras: < 1/10,000
Frecuencia no conocida: no puede estimarse a partir de los datos disponibles.
Pacientes con cáncer
|
Clasificación de órganos del sistema MedDRA |
Reacciones adversas | ||||
|
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras | |
|
Trastornos de la sangre y del sistema linfático |
Crisis drepanocíticaa Esplenomegalia Ruptura esplénica | ||||
|
Trastornos del sistema inmunológico |
Hipersensibilidad al fármacoa |
Enfermedad de injerto contra huésped b | |||
|
Trastornos del metabolismo y de la nutrición |
Aumento del ácido úrico en la sangre Aumento de la lactato- deshidrogenasa en la sangre Disminución del apetito3 |
Condrocalcinosisb | |||
|
Trastornos del sistema nervioso |
Cefalea3 | ||||
|
Trastornos vasculares |
Hipotensión |
Flebopatía oclusivad Alteraciones de la volemia Síndrome de extravasación capilar1 | |||
|
Trastornos respiratorios, torácicos y mediastínicos |
Dolor orofaríngeoa Tos3 |
Hemoptisise |
Síndrome de dificultad respiratoria agudaa | ||
|
Clasificación de órganos del sistema MedDRA |
Reacciones adversas | ||||
|
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras | |
|
Disnea |
Insuficiencia respiratoriaa Edema pulmonar1 Neumopatía intersticiala Infiltración pulmonara Hemorragia pulmonar | ||||
|
Trastornos gastrointestinales |
Diarrea3 Vómitos3 Estreñimientoa Náuseasa | ||||
|
Trastornos hepatobiliares |
Aumento de la y-glutamiltransferasa Aumento de la fosfatasa alcalina en la sangre | ||||
|
Trastornos de la piel y del tejido subcutáneo |
Exantemaa Alopeciaa |
Síndrome de Sweet Vasculitis cutáneaa | |||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor musculoesquelético3 |
Exacerbación de la artritis reumatoide | |||
|
Trastornos renales y urinarios |
Disuria |
Anomalías urinarias | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Asteniaa Fatigaa Inflamación mucosaa |
Dolor torácicoa |
Dolor a | ||
|
aVer sección 4.8, Descripción de reacciones ac |
versas seleccionadas. | ||||
bSe han notificado casos de EICH y muertes en pacientes tras un alotrasplante de médula ósea (Ver sección 4.8, Descripción de reacciones adversas seleccionadas).
cIncluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en las extremidades, dolor musculoesquelético, dolor torácico musculoesquelético, dolor de cuello.
dSe observaron casos con filgrastim en situación posterior a la comercialización en pacientes sometidos a
trasplante de médula ósea o a movilización de CPSP.
eSe observaron casos en el entorno de ensayos clínicos con filgrastim.
|
Clasificación de órganos del sistema MedDRA |
Reacciones adversas | ||||
|
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras | |
|
Trastornos de la sangre y del sistema linfático |
T rombocitopenia Leucocitosis |
Esplenomegalia a |
Ruptura esplénica Crisis drepanocítica3 | ||
|
Trastornos del sistema inmunológico |
Reacción anafiláctica | ||||
|
Trastornos del metabolismo y de la nutrición |
Aumento de la lactato- deshidrogenasa en la sangre |
Hiperuricemia (aumento del ácido úrico en la sangre) | |||
|
Trastornos del sistema nervioso |
Cefalea | ||||
|
Trastornos vasculares |
Síndrome de extravasación capilar3 | ||||
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Hemorragia pulmonar Hemoptisis Infiltración pulmonar Hipoxia | |||
|
Trastornos hepatobiliares |
Aumento de la fosfatasa alcalina en la sangre |
Aumento de la aspartato-aminotransfera sa | |||
|
Trastornos musculoesquelétic os y del tejido conjuntivo |
Dolor musculoesquelétic o* |
Agravamiento de la artritis reumatoide | |||
aver sección 4.8, Descripción de reacciones adversas seleccionadas * Incluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en las extremidades, dolor musculoesquelético, dolor torácico musculoesquelético, dolor de cuello.
Pacientes con NCG
|
Clasificación de |
Reacciones adversas | ||||
|
órganos del sistema MedDRA |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras |
|
Trastornos de la sangre y del sistema linfático |
Esplenomegalia Anaemia |
Trombocitopeni a Ruptura esplénica |
Crisis drepanocíticaa | ||
|
Trastornos del metabolismo y de la nutrición |
Hiperuricemia Disminución de | ||||
|
Clasificación de órganos del sistema MedDRA |
Reacciones adversas | ||||
|
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras | |
|
la glucosa en la sangre Aumento de la lactato- deshidrogenasa en la sangre | |||||
|
Trastornos del sistema nervioso |
Cefalea | ||||
|
Trastornos respiratorios, torácicos y mediastínicos |
Epistaxis | ||||
|
Trastornos gastrointestinales |
Diarrea | ||||
|
Trastornos hepatobiliares |
Hepatomegalia Aumento de la fosfatasa alcalina en la sangre | ||||
|
Trastornos de la piel y del tejido subcutáneo |
Exantema |
Vasculitis cutánea Alopecia | |||
|
Trastornos musculoesquelético s y del tejido conjuntivo |
Dolor musculoesqueléti co* Artralgia |
Osteoporosis | |||
|
Trastornos renales y urinarios |
Hematuria |
Proteinuria | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Reacción en el lugar de la inyección | ||||
aVer sección 4.8, Descripción de reacciones adversas seleccionadas
*Incluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en las extremidades, dolor
musculoesquelético, dolor torácico musculoesquelético, dolor de cuello.
Pacientes con VIH
|
Clasificación de órganos del sistema MedDRA |
Reacciones adversas | ||||
|
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras | |
|
Trastornos de la sangre y del sistema linfático |
Esplenomegalia |
Crisis drepanocítica3 | |||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor musculoesquelét ico * | ||||
aVer sección 4.8, Descripción de reacciones adversas seleccionadas
*Incluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en las extremidades, dolor musculoesquelético, dolor torácico musculoesquelético, dolor de cuello.
Descripción de reacciones adversas seleccionadas
Se han notificado casos de EICH y muertes en pacientes que recibieron G-CSF tras un trasplante alogénico de médula ósea (ver secciones 4.4 y 5.1).
Se han notificado casos de síndrome de extravasación capilar en la fase de pos-comercialización con el uso de factor estimulante de colonias de granulocitos. Por lo general, estos casos se han presentado en pacientes con neoplasias malignas avanzadas, sepsis, pacientes que toman múltiples medicamentos antineoplásicos o sometidos a aféresis (ver sección 4.4).
Pacientes con cáncer
En los ensayos clínicos aleatorizados controlados con placebo, filgrastim no incrementó la incidencia de reacciones adversas asociadas a la quimioterapia citotóxica. En dichos ensayos clínicos se notificaron ciertas reacciones adversas con la misma frecuencia en pacientes con cáncer tratados con filgrastim/quimioterapia y placebo/quimioterapia, entre ellas náuseas y vómitos, alopecia, diarrea, fatiga, anorexia, mucositis, cefalea, tos, exantema, dolor torácico, debilidad generalizada, dolor faríngeo, estreñimiento y dolor.
En situación posterior a la comercialización se ha notificado vasculitis cutánea en pacientes tratados con filgrastim. Se desconoce el mecanismo de la vasculitis en los pacientes que reciben filgrastim. A partir de los datos de los ensayos clínicos se estima que es poco frecuente.
Se han notificado casos de síndrome de Sweet (dermatosis febril aguda) posteriores a la comercialización. A partir de los datos de los ensayos clínicos se estima que son poco frecuentes.
En los estudios clínicos y en situación posterior a la comercialización se han notificado efectos adversos pulmonares, entre ellos neumopatía intersticial, edema pulmonar e infiltración pulmonar, que en algunos casos han ocasionado insuficiencia respiratoria o síndrome de dificultad respiratoria aguda (SDRA), que pueden ser mortales (ver sección 4.4).
Se han notificado de manera poco frecuente de casos de esplenomegalia y ruptura esplénica tras la administración de filgrastim. Algunos casos de ruptura esplénica fueron mortales (ver sección 4.4).
Reacciones de tipo hipersensibilidad incluyendo anafilaxia, erupción cutánea, urticaria, angioedema, disnea e hipotensión ocurrieron el tratamiento inicial o posterior en estudios clínicos y en la experiencia post-comercialización. En general, las notificaciones fueron más frecuentes después de la administración intravenosa. En algunos casos, los síntomas han reaparecido tras reexposición al fármaco, lo que sugiere una relación causal. Filgrastim se debe interrumpir de forma permanente en pacientes que desarrollen alguna reacción alérgica grave.
En la post-comercialización, se han notificado casos aislados de crisis de células falciformes en pacientes con enfermedad de células falciformes (ver sección 4.4). La frecuencia se estimó como poco frecuente a partir de los datos de ensayos clínicos.
Se ha notificado pseudogota en pacientes con cáncer tratados con filgrastim, y la frecuencia se estimó como poco frecuente a partir de los datos de ensayos clínicos.
Movilización de CPSP en donantes sanos
Se han notificado casos frecuentes pero generalmente asintomáticos de esplenomegalia y casos poco frecuentes de ruptura esplénica en pacientes y en donantes sanos después de la administración de filgrastim (ver sección 4.4).
Se han notificado acontecimientos adversos pulmonares tales como hemoptisis, hemorragia pulmonar, infiltración pulmonar, disnea e hipoxia (ver sección 4.4).
Se ha notificado con poca frecuencia exacerbación de los síntomas de artritis.
Se ha observado leucocitosis (leucocitos > 50 x 109/L) en el 41% de los donantes y se ha observado trombocitopenia transitoria (plaquetas < 100 x 109/L) después del tratamiento con filgrastim y la leucocitaféresis se observó en el 35% de los donantes.
Pacientes con NCG
Entre las reacciones adversas figuran la esplenomegalia, que puede ser progresiva en una minoría de los casos, y la trombocitopenia (ver sección 4.4).
Otras reacciones adversas posiblemente relacionadas con el tratamiento con filgrastim y observadas habitualmente en < 2% de los pacientes con NCG fueron reacción en el lugar de la inyección, cefalea, hepatomegalia, artralgia, alopecia, osteoporosis y exantema.
Durante el empleo prolongado se ha notificado vasculitis cutánea en el 2% de los pacientes con NCG. Pacientes con VIH
La esplenomegalia se notificó relacionada con el tratamiento con filgrastim en <3% de los pacientes.
En todos los casos de aumento del tamaño del bazo en pacientes con VIH, este se consideró leve o moderado durante la exploración física y la evolución clínica fue benigna; a ningún paciente se le diagnosticó hiperesplenismo y ninguno se sometió a una esplenectomía. Como el aumento del tamaño del bazo es frecuente en los pacientes con infección por VIH y la mayoría de los pacientes con SIDA lo presentan en mayor o menor grado, la relación con el tratamiento con filgrastim no está clara (ver sección 4.4).
Población pediátrica
Los datos de estudios clínicos con filgrastim en pacientes pediátricos indican que la seguridad y la eficacia del filgrastim son similares en adultos y en niños que reciben quimioterapia citotóxica, lo cual sugiere que no existen diferencias basadas en la edad en la farmacocinética del filgrastim. La única reacción adversa notificada de forma regular fue dolor musculoesquelético, lo cual no es diferente de la experiencia en la población adulta. No hay suficientes datos para evaluar aún más el uso de filgrastim en pacientes pediátricos.
Otras poblaciones especiales
Pacientes de edad avanzada
No se observaron diferencias generales en la seguridad o eficacia entre los sujetos mayores de 65 años de edad en comparación con adultos más jóvenes (> 18 años de edad) de los sujetos que reciben quimioterapia citotóxica y la experiencia clínica no ha identificado diferencias en las respuestas entre los pacientes de edad avanzada y jóvenes. No hay datos suficientes para evaluar el uso de Accofil en pacientes de edad avanzada para otras indicaciones Accofil aprobadas.
Pacientes con NCG pediátricos
Los casos de disminución de la densidad ósea y la osteoporosis se han notificado en pacientes pediátricos con neutropenia crónica grave en tratamiento crónico con filgrastim. La frecuencia se estimó como "frecuente" según los datos de los ensayos clínicos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han establecido los efectos de la sobredosis de Accofil. La interrupción del tratamiento con filgrastim suele acompañarse de una disminución del 50% de los neutrófilos circulantes al cabo de 1 a 2 días, con una normalización al cabo de 1 a 7 días.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: citocinas, código ATC: L03AA02.
Accofil es un medicamento biosimilar. La información detallada sobre este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Efectos farmacodinámicos
El G-CSF humano es una glucoproteína que regula la producción y liberación de neutrófilos funcionales de la médula ósea. Accofil, que contiene r-metHuG-CSF (filgrastim), aumenta considerablemente el recuento de neutrófilos en la sangre periférica a las 24 horas, y mínimamente el de monocitos. En algunos pacientes con NCG, filgrastim puede inducir también un leve aumento del número de eosinófilos y basófilos circulantes con relación a los valores iniciales; algunos de estos pacientes pueden presentar eosinofilia o basofilia ya antes del tratamiento. El incremento de los recuentos de neutrófilos depende de la dosis, con la posología recomendada. Los neutrófilos producidos en respuesta al filgrastim muestran una función normal o superior a la habitual, de acuerdo con las pruebas de la función quimiotáctica y fagocitaria. Después de interrumpir el tratamiento con filgrastim, el recuento de neutrófilos circulantes se reduce un 50% al cabo de 1 a 2 días y se normaliza en el plazo de 1 a 7 días.
El empleo de filgrastim en pacientes sometidos a quimioterapia citotóxica reduce de forma significativa la incidencia, gravedad y duración de la neutropenia y de la neutropenia febril. El tratamiento con filgrastim reduce significativamente la duración de la neutropenia febril, el uso de antibióticos y la hospitalización después de la quimioterapia de inducción en la leucemia mieloide aguda o tras un tratamiento mieloablativo seguido de trasplante de médula ósea. La incidencia de fiebre y de infecciones documentadas no se redujo en ninguno de los dos casos. Tampoco se redujo la duración de la fiebre en los pacientes sometidos a tratamiento mieloablativo seguido de trasplante de médula ósea.
El empleo de filgrastim, bien en monoterapia o tras la quimioterapia, moviliza las células progenitoras hematopoyéticas hacia la sangre periférica. Estas CPSP autólogas se pueden recolectar y perfundir después de altas dosis de tratamiento citotóxico, bien junto con un trasplante de médula ósea o en lugar de éste. La perfusión de las CPSP acelera la recuperación hematopoyética, reduciendo el período de riesgo de complicaciones hemorrágicas y la necesidad de transfusiones plaquetarias.
Los receptores de trasplantes alogénicos de CPSP movilizadas con filgrastim experimentaron una recuperación hematopoyética significativamente más rápida que los tratados con trasplante alogénico de médula ósea, ocasionando una disminución significativa del tiempo hasta la recuperación de las plaquetas sin soporte externo.
Un estudio retrospectivo europeo que evaluó el uso de G-CSF tras un trasplante alogénico de médula ósea en pacientes con leucemias agudas sugirió un aumento del riesgo de EICH, mortalidad relacionada con el tratamiento (MRT) y mortalidad cuando se administraba G-CSF. En un estudio retrospectivo internacional independiente en pacientes con leucemias mieloides agudas y crónicas, no se observó efecto alguno sobre el riesgo de EICH, MRT ni mortalidad. Un metanálisis de estudios de trasplantes alogénicos, que incluyó los resultados de nueve ensayos prospectivos aleatorizados, 8 estudios retrospectivos y 1 estudio de caso-control, no detectó efecto alguno sobre los riesgos de EICH aguda, EICH crónica ni mortalidad temprana relacionada con el tratamiento.
|
Riesgo relativo (IC del 95%) de EICH y MRT tras el tratamiento con G-CSF después de un trasplante de médula ósea (MO) | |||||
|
Publicación |
Período de estudio |
N |
EICH aguda de grado II-IV |
EICH crónica |
MRT |
|
Metanálisis (2003) |
a 1986-2001 |
1.198 |
1,08 (0,87, 1,33) |
1,02 (0,82, 1,26) |
0,70 (0,38, 1,31) |
|
Estudio retrospectivo europeo (2004) |
b 1992-2002 |
1.789 |
1,33 (1,08, 1,64) |
1,29 (1,02, 1,61) |
1,73 (1,30, 2,32) |
|
Estudio retrospectivo internacional (2006) |
b 1995-2000 |
2.110 |
1,11 (0,86, 1,42) |
1,10 (0,86, 1,39) |
1,26 (0,95, 1,67) |
a
El análisis incluye estudios con trasplante de MO durante este período; algunos estudios utilizaron
GM-CSF.
b
El análisis incluye a pacientes que recibieron trasplantes de MO durante este período.
Empleo de filgrastim para la movilización de CPSP en donantes sanos antes de un trasplante d alogénico de CPSP.
En donantes sanos, a 10 microgramos/kg/día de dosis administradas por vía subcutánea durante 4-5 días consecutivos permite obtener > 4 x 101 células CD34+/kg de peso del receptor en la mayoría de los donantes después de dos leucocitaféresis.
El empleo de filgrastim en los adultos, con NCG (neutropenia congénita grave, cíclica y neutropenia idiopática) induce un aumento sostenido de la ANC en la sangre periférica y una reducción de las infecciones y procesos relacionados.
El empleo de filgrastim en pacientes con infección por VIH mantiene el recuento de neutrófilos en los niveles normales permitiendo la administración programada de tratamiento antiviral y otros tratamientos mielodepresores. No hay pruebas de que los pacientes con infección por VIH tratados con filgrastim presenten un aumento de la replicación del VIH.
Al igual que otros factores de crecimiento hematopoyético, el factor estimulante de colonias de granulocitos ha demostrado tener propiedades estimuladoras de las células endoteliales humanas in
vitro.
5.2 Propiedades farmacocinéticas
Absorción
Las concentraciones séricas se mantuvieron por encima de 10 ng/ml durante 8-16 horas después de la administración subcutánea de las dosis recomendadas.
Distribución
El volumen de distribución en la sangre es de unos 150 ml/kg. Eliminación
Se ha demostrado que el aclaramiento del filgrastim sigue una farmacocinética de primer orden, tras su administración subcutánea o intravenosa. La semivida de eliminación del filgrastim es de unas 3,5 horas con una tasa de aclaramiento de unos 0,6 ml/min/kg. La perfusión continua de Accofil a lo largo de un período de hasta de 28 días en pacientes que se recuperan de un trasplante de médula ósea autóloga no dio muestras de acumulación farmacológica y las semividas de eliminación fueron comparables.
Linealidad
Existe una correlación lineal positiva entre la dosis y la concentración sérica de filgrastim, tanto en administración intravenosa como subcutánea. Tras la administración subcutánea de las dosis recomendadas, las concentraciones séricas se mantuvieron por encima de 10 ng/ml durante 8 a 16 horas. El volumen de distribución en sangre es de aproximadamente 150 ml/kg.
5.3 Datos preclínicos sobre seguridad
Se investigó filgrastim en estudios repetidos de toxicidad de dosis de hasta 1 año de duración que revelaron cambios atribuibles a las acciones farmacológicas esperadas que incluían aumentos de leucocitos, hiperplasia mieloide en médula ósea, granulopoyesis extramedular y aumento de tamaño del bazo. Estos cambios revertían después de suspender el tratamiento.
Se han estudiado los efectos de filgrastim en el desarrollo prenatal en ratas y conejos. La administración intravenosa (80 pg/kg/día) de filgrastim a conejos durante el periodo de organogénesis fue tóxica para la madre y aumentaba el aborto espontáneo, se observó la pérdida postimplantación y menor tamaño medio de cría viva y peso fetal.
Basándose en los datos notificados para otro producto filgrastim similar al de Accofil, se observaron hallazgos comparables más un aumento de las malformaciones fetales con 100 pg/kg/día, una dosis tóxica materna que correspondía a una exposición sistémica de aproximadamente 50 a 90 veces las exposiciones observadas en pacientes tratados con la dosis clínica de 5 pg/kg/día. El nivel de efecto adverso no observado para la toxicidad embriofetal en este estudio era de 10 pg/kg/día, que correspondía a una exposición sistemática de aproximadamente 3 a 5 veces las exposiciones observadas en pacientes tratados con la dosis clínica.
En ratas preñadas, no se observó toxicidad materna o fetal en dosis de hasta 575 pg/kg/día. Las crías de rata a las que se les administró filgrastim durante los periodos perinatales y de lactancia, mostraron un retraso en la diferenciación externa y retardo de crecimiento (>20 pg/kg/día) y una tasa de supervivencia ligeramente reducida (100 pg/kg/día).
Filgrastim no tenía un efecto observado sobre la fertilidad de las ratas machos o hembras. 1
Se ha demostrado la estabilidad química y física de la solución diluida para perfusión durante el uso hasta 24 horas entre 2 °C y 8 °C. Desde el punto de vista microbiológico, el producto debe utilizarse inmediatamente. Si no se utiliza inmediatamente, el tiempo y las condiciones de conservación durante el uso y las condiciones antes de su uso son responsabilidad del usuario, y no se deberían superar normalmente las 24 horas entre 2 °C y 8 °C, salvo que la dilución haya tenido lugar en condiciones asépticas controladas y validadas.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C-8 °C). No congelar.
La exposición accidental por una sola vez a temperaturas de congelación no afecta de forma adversa a la estabilidad de Accofil. Si la exposición ha superado las 24 horas o se ha congelado más de una vez, entonces NO se debe utilizar Accofil.
Dentro de su periodo de validez y para su uso ambulatorio, el paciente puede sacar el medicamento de la nevera y almacenarlo a temperatura ambiente (no a temperatura superior a 25°C) durante un único periodo máximo de 15 días. Después de este periodo, el medicamento no se debe volver a refrigerar y se debe desechar.
Conservar la jeringa en el embalaje exterior para protegerlo de la luz.
Para las condiciones de conservación del medicamento diluido, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Jeringa precargada con aguja para inyección, con o sin protector de seguridad para la aguja. Envases con una, tres, cinco, siete o diez jeringas precargadas, con o sin blíster y gasas impregnadas en alcohol. Los envases sin blister no tienen protector de seguridad. Los envases blister son para jeringas individuales con protector de seguridad para la aguja prefijada. Las jeringas precargadas son de vidrio tipo I contienen una aguja de acero inoxidable permanentemente incorporada en su extremo y tienen marcas impresas 1/40 para graduaciones de 0.1 ml a 1 ml en el cilindro. El protector de la aguja de la jeringa precargada contiene caucho natural seco (ver sección 4.4). Cada jeringa precargada contiene 0,5 ml de solución.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Si es necesario, Accofil se puede diluir en glucosa al 5%. No se recomienda en ningún caso diluir a una concentración final inferior a 0,2 MU (2 pg) por ml.
La solución debe inspeccionarse visualmente antes de usarla. Solamente deben utilizarse soluciones transparentes sin partículas. No agite.
Para los pacientes tratados con filgrastim diluido a concentraciones inferiores a 1,5 MU (15 pg) por ml, debe añadirse seroalbúmina humana (SAH) a una concentración final de 2 mg/ml. Ejemplo: en un volumen de inyección final de 20 ml, las dosis totales de filgrastim inferiores a 30 MU (300 pg) deben administrarse con adición de 0,2 ml de solución de albúmina humana 200 mg/ml (20%).
Accofil no contiene conservantes. En vista del posible riesgo de contaminación microbiana, las jeringas precargadas de Accofil se deben utilizar una sola vez.
Cuando se diluye en glucosa al 5%, Accofil es compatible con el vidrio y con diversos plásticos como PVC, poliolefina (copolímero de polipropileno y polietileno) y polipropileno.
Uso de la jeringa precargada con protector de seguridad para la aguja
El protector de seguridad para la aguja cubre la aguja tras haber realizado la inyección con el fin de prevenir pinchazos accidentales, lo cual no afecta a la forma de utilizar la jeringa. Empujar el émbolo lenta y uniformemente hasta que se haya administrado la totalidad de la dosis y el émbolo no pueda seguir avanzando. Retirar la jeringa manteniendo la presión sobre el émbolo. El protector de seguridad para la aguja cubrirá la aguja una vez que se suelte el émbolo.
Uso de la jeringa precargada sin protector de seguridad para la aguja
Administrar la dosis según el protocolo estándar.
Eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Accord Healthcare Limited Sage House, 319 Pinner Road,
North Harrow, Middlesex HA1 4HF,
Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/946/003
EU/1/14/946/004
EU/1/14/946/011
EU/1/14/946/012
EU/1/14/946/013
EU/1/14/946/014
EU/1/14/946/015
EU/1/14/946/016
EU/1/14/946/018
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 18.09.2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
Nombre y dirección del (de los) fabricante(s) del (de los) principio(s) activo(s) biológico(s)
Intas Pharmaceuticals Ltd.
Plot no: 423 P/A
Sarkhej Bavla Highway
Village Moraiya; Taluka: Sanand,
Ahmedabad - 382213 Gujarat, India
Nombre y dirección del (de los) fabricante(s) responsable(s) de la liberación de los lotes
Accord Healthcare Limited Sage House, 319 Pinner Road,
North Harrow,
Middlesex HA1 4HF,
Reino Unido
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de gestión de riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado
en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en
cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR Caja exterior
1. NOMBRE DEL MEDICAMENTO
Accofil 30 MU/0,5 ml solución inyectable y para perfusión. filgrastim
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa de 0,5 ml contiene 30 MU de filgrastim (0,6 mg/ml).
3. LISTA DE EXCIPIENTES
Ácido acético, hidróxido de sodio, sorbitol (E420), polisorbato 80 y agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 jeringa precargada (0,5 ml) + 1 gasa impregnada en alcohol “5 jeringas precargadas (0,5 ml) + 5 gasas impregnadas en alcohol”
“3 jeringas precargadas (0,5 ml) + 3 gasas impregnadas en alcohol” “10 jeringas precargadas (0,5 ml) + 10 gasas impregnadas en alcohol”
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
Vía subcutánea o vía intravenosa.
No agitar.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD:
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Accord Healthcare Limited Sage House, 319 Pinner Road,
North Harrow, Middlesex HA1 4HF, Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/946/001 1 jeringa precargada EU/1/14/946/002 5 jeringas precargadas EU/1/14/946/006 - 3 jeringas precargadas EU/1/14/946/009 - 10 jeringas precargadas
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
Caja exterior - Jeringa precargada con protector de seguridad para la aguja en envase blíster
1. NOMBRE DEL MEDICAMENTO
Accofil 30 MU/0,5 ml solución inyectable y para perfusión. filgrastim
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa de 0,5 ml contiene 30 MU de filgrastim (0,6 mg/ml).
3. LISTA DE EXCIPIENTES
Ácido acético, hidróxido de sodio, sorbitol (E420), polisorbato 80 y agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 jeringa precargada (0,5 ml) + 1 gasa impregnada en alcohol “3 jeringas precargadas (0,5 ml) + 3 gasas impregnadas en alcohol”
“5 jeringas precargadas (0,5 ml) + 5 gasas impregnadas en alcohol” “10 jeringas precargadas (0,5 ml) + 10 gasas impregnadas en alcohol” “7 jeringas precargadas (0,5 ml) + 7 gasas impregnadas en alcohol”
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
Vía subcutánea o vía intravenosa.
No agitar.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD:
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Accord Healthcare Limited Sage House, 319 Pinner Road,
North Harrow, Middlesex HA1 4HF, Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/946/005 - 1 jeringa precargada con protector de seguridad para la aguja EU/1/14/946/008 - 5 jeringas precargadas con protector de seguridad para la aguja EU/1/14/946/007 - 3 jeringas precargadas con protector de seguridad para la aguja EU/1/14/946/010 - 10 jeringas precargadas con protector de seguridad para la aguja EU/1/14/946/017 - 7 jeringas precargadas con protector de seguridad para la aguja
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Accofil 30 MU/0,5 ml solución inyectable y para perfusión.
filgrastim
SC/IV
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
0,5 ml
6. OTROS
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR Caja exterior
1. NOMBRE DEL MEDICAMENTO
Accofil 48 MU/0,5 ml solución inyectable y para perfusión. filgrastim
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa de 0,5 ml contiene 48 MU de filgrastim (0,96 mg/ml).
3. LISTA DE EXCIPIENTES
Ácido acético, hidróxido de sodio, sorbitol (E420), polisorbato 80 y agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 jeringa precargada (0,5 ml) + 1 gasa impregnada en alcohol “5 jeringas precargadas (0,5 ml) + 5 gasas impregnadas en alcohol”
“3 jeringas precargadas (0,5 ml) + 3 gasas impregnadas en alcohol” “10 jeringas precargadas (0,5 ml) + 10 gasas impregnadas en alcohol”
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
Vía subcutánea o vía intravenosa.
No agitar.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD:
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Accord Healthcare Limited Sage House, 319 Pinner Road,
North Harrow, Middlesex HA1 4HF, Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/946/003 - 1 jeringa precargada EU/1/14/946/004 - 5 jeringas precargadas EU/1/14/946/012 - 3 jeringas precargadas EU/1/14/946/015 - 10 jeringas precargadas
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
Caja exterior - Jeringa precargada con protector de seguridad para la aguja en envase blíster
1. NOMBRE DEL MEDICAMENTO
Accofil 48 MU/0,5 ml solución inyectable y para perfusión. filgrastim
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa de 0,5 ml contiene 48 MU de filgrastim (0,96 mg/ml).
3. LISTA DE EXCIPIENTES
Ácido acético, hidróxido de sodio, sorbitol (E420), polisorbato 80 y agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 jeringa precargada (0,5 ml) + 1 gasa impregnada en alcohol “3 jeringas precargadas (0,5 ml) + 3 gasas impregnadas en alcohol”
“5 jeringas precargadas (0,5 ml) + 5 gasas impregnadas en alcohol” “10 jeringas precargadas (0,5 ml) + 10 gasas impregnadas en alcohol” “7 jeringas precargadas (0,5 ml) + 7 gasas impregnadas en alcohol”
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
Vía subcutánea o vía intravenosa.
No agitar.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD:
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera. No congelar.
Conservar la jeringa en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Accord Healthcare Limited Sage House, 319 Pinner Road,
North Harrow, Middlesex HA1 4HF, Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/946/011 1 jeringa precargada con protector de seguridad para la aguja EU/1/14/946/014 5 jeringas precargadas con protector de seguridad para la aguja EU/1/14/946/013 3 jeringas precargadas con protector de seguridad para la aguja EU/1/14/946/016 10 jeringas precargadas con protector de seguridad para la aguja EU/1/14/946/018 7 jeringas precargadas con protector de seguridad para la aguja
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Accofil 48 MU/0,5 ml
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Accofil 48 MU/0,5 ml solución inyectable y para perfusión.
filgrastim
SC/IV
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
0,5 ml
6. OTROS
B. PROSPECTO
PROSPECTO: INFORMACIÓN PARA EL USUARIO
Accofil 30 MU/0,5 ml (0,6 mg/ml) solución inyectable y para perfusión en jeringa precargada
fílgrastim
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
• Conserve este prospecto, ya que puede tener que volver a leerlo.
• Si tiene alguna duda, consulte a su médico, enfermero o farmacéutico.
• Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
• Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Accofil y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Accofil
3. Cómo usar Accofil
4. Posibles efectos adversos
5. Conservación de Accofil
6. Contenido del envase e información adicional
1. Qué es Accofil y para qué se utiliza Qué es Accofil
Accofil contiene el principio activo filgrastim. Filgrastim es una proteína producida en la bacteria Escherichia coli mediante la tecnología del ADN recombinante. Pertenece a un grupo de proteínas llamadas citoquinas y es muy similar a una proteína natural (granulocitos y factor estimulante de colonias [G-CSF]) producido por su propio cuerpo. Filgrastim estimula la médula ósea (el tejido donde se producen nuevas células sanguíneas) para producir más glóbulos blancos que ayudan a combatir las infecciones.
Para qué se utiliza Accofil
Su médico le ha recetado Accofil para ayudar a su cuerpo a producir más glóbulos blancos. Su médico le dirá por qué usted está siendo tratado con Accofil. Accofil es útil en varias situaciones diferentes que son:
• la quimioterapia
• trasplante de médula ósea
• Neutropenia crónica grave (bajo número de un tipo de células blancas de la sangre)
• La neutropenia (bajo número de un tipo de células blancas de la sangre) en pacientes con infección
por VIH
• Movilización de células madre de sangre periférica (para estimular las células madre para entrar en el torrente sanguíneo para ser recogidos y utilizados en el trasplante de médula ósea).
2. Qué necesita saber antes de empezar a usar Accofil No use Accofil
- Si es alérgico a filgrastim o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a usar Accofil:
Antes de iniciar el tratamiento dígale a su médico si tiene:
- anemia drepanocítica, accofil puede provocar crisis drepanocitaria.
- osteoporosis (enfermedad de los huesos)
Informe a su médico inmediatamente durante el tratamiento con Accofil si:
- Tiene dolor en el abdomen (barriga) superior izquierdo, dolor debajo de las costillas izquierdas o
en el extremo del hombro izquierdo (estos pueden ser síntomas de agrandamiento del bazo o posiblemente ruptura del bazo).
- Nota hemorragia poco habitual o hematomas que pueden ser síntomas de menor número de
plaquetas en sangre (trombocitopenia) con una capacidad reducida de coagular la sangre.
- Tiene signos repentinos de alergia tales como erupciones, picores o urticaria en la piel, hinchazón
de la cara, labios, lengua u otras partes del cuerpo, falta de aliento, sibilancias (sonido silbante que se produce al respirar) o problemas al respirar ya que podrían ser signos de una reacción alérgica grave.
Es posible que tenga que hacerse análisis de sangre periódicos mientras está siendo tratado con Accofil para contar el número de neutrófilos y otros glóbulos blancos en la sangre. Esto le dirá a su médico cómo está funcionando el tratamiento y también indicará si necesita tratamiento debe continuarse.
La cubierta de la aguja de la jeringa precargada contiene caucho natural (un derivado del látex) que puede causar una reacción alérgica.
Otros medicamentos y Accofil
Usted no debe recibir Accofil en las 24 horas antes y 24 horas después de recibir la quimioterapia.
Por favor, informe a su médico o farmacéutico si está tomando o ha tomado recientemente otros medicamentos, incluso los adquiridos sin receta.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que puede estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de tomar este medicamento.
Accofil no ha sido estudiado en mujeres embarazadas. Su médico puede decidir que usted no debería usar este medicamento.
Se desconoce si filgrastim pasa a la leche materna. Por lo tanto, su médico puede decidir que usted no debe usar este medicamento si está en periodo de lactancia.
Conducción y uso de máquinas
Si usted se encuentra fatigado, no conduzca automóviles ni maneje máquinas.
Información importante sobre algunos de los componentes de Accofil
Este medicamento contiene sorbitol. Si su médico le ha indicado que padece una intolerancia a ciertos azúcares (fructosa), consulte con él antes de tomar este medicamento. Este medicamento también contiene sodio menos de 1 mmol de sodio (0.035 mg) por dosis, es decir, esencialmente "exento de sodio".
3. Cómo usar Accofil
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
La cantidad de Accofil que usted necesita dependerá de la enfermedad para la que está tomando Accofil y de su peso corporal.
Dosis
Accofil y neutropenia (bajo número de un tipo de células blancas de la sangre) asociados con la quimioterapia
La dosis habitual es de 0,5 millones de unidades (5 microgramos) por kilogramo de peso corporal al día. Por ejemplo, si usted pesa 60 kilogramos, su dosis diaria será de 30 millones de unidades (300 microgramos). El tratamiento con Accofil generalmente durará unos 14 días. No obstante, en algunos tipos de enfermedad puede ser necesario un tratamiento más largo con una duración de hasta aproximadamente un mes.
Accofil y trasplante de médula ósea
La dosis inicial habitual es de 1 millón de unidades (10 microgramos) por kilogramo de peso corporal al día administrada en perfusión. Por ejemplo, si usted pesa 60 kg su dosis diaria será de 60 millones de unidades (600 microgramos). Normalmente recibirá su primera dosis de Accofil al menos 24 horas después de la quimioterapia y al menos 24 horas después de recibir su trasplante de médula ósea. Luego, el médico le examinará la sangre para ver cómo está funcionando el tratamiento y cuánto tiempo durará.
Accofil neutropenia crónica y grave (bajo número de un tipo de células blancas de la sangre)
La dosis inicial habitual es de 0,5 millones de unidades (5 microgramos) y 1,2 millones de unidades (12 microgramos) por kilogramo de peso corporal al día en una dosis única o dividida. Luego, el médico le examinará la sangre para ver qué tan bien su tratamiento con Accofil está trabajando y para encontrar la dosis más adecuada para usted. Se requiere un tratamiento a largo plazo con Accofil para la reducción de la neutropenia.
Accofil y neutropenia (bajo número de un tipo de células blancas de la sangre) en pacientes con infección por el VIH
La dosis inicial habitual es de 0,1 millones de unidades (1 microgramos) y 0,4 millones de unidades (4 microgramos) por kilogramo de peso corporal por día. El médico puede examinar su sangre con regularidad para ver si el tratamiento con Accofil está funcionando. Una vez que el número de glóbulos blancos en su sangre ha vuelto a la normalidad, puede ser posible reducir la frecuencia de la dosis a menos de una vez al día. El tratamiento a largo plazo con Accofil puede ser necesario para mantener un número normal de células blancas en la sangre.
Accofil y trasplante de células madre de sangre periférica (células madre recogidas desde la sangre a usar en el trasplante de médula ósea)
Si va a donar células madre para usted, la dosis habitual es de 0,5 millones de unidades (5 microgramos) a 1 millón de unidades (10 microgramos) por kilogramo de peso corporal por día. El tratamiento con Accofil tendrá una duración de hasta 2 semanas. Su médico controlará su sangre para determinar el mejor momento para recoger las células madre.
Si actúa como donante de células madre para otra persona, la dosis habitual es de 1 millón de unidades (10 microgramos) por kilogramo de peso corporal al día. El tratamiento con Accofil tendrá una duración de 4 a 5 días. Su médico le realizará análisis de sangre para determinar el mejor momento para recoger las células madre.
Forma de administración
Este medicamento se administra mediante una inyección, ya sea a través de una perfusión (goteo) intravenosa (IV) o mediante una inyección subcutánea (SC) en el tejido debajo de la piel.
Si usted está recibiendo este medicamento en inyección subcutánea, su médico puede sugerir que usted aprenda cómo ponerse las inyecciones. Su médico o enfermera le dará instrucciones sobre cómo hacer esto (ver información a continuación las instrucciones para inyectar Accofil). No intente auto-inyectarse sin estas instrucciones. Parte de la información que usted requiere se da al final de este prospecto, pero el tratamiento adecuado de su enfermedad requiere una estrecha y constante colaboración con su médico.
Información para autoinyectarse
Esta sección contiene información sobre cómo administrarse a uno mismo una inyección de Accofil. Es importante que no trate de administrarse una inyección sin haber recibido la formación necesaria por parte de su médico o enfermero. Si no está seguro de poder inyectarse o si tiene cualquier duda, consulte a su médico o enfermero.
¿Cómo he de inyectarme Accofil?
Deberá administrarse la inyección en el tejido justo bajo la piel. Esto se conoce como inyección subcutánea. La inyección deberá administrarse todos los días aproximadamente a la misma hora.
Equipo necesario
Para ponerse una inyección subcutánea, necesitará:
• una j eringa precargada de Accofil y
• gasa impregnada con alcohol o similar.
¿Qué debo hacer antes de administrarme una inyección subcutánea de Accofil?
Asegurarse de que la cubierta de la aguja permanece en la jeringa hasta justo antes de estar preparado para inyectarse.
a. Sacar la jeringa precargada de Accofil del frigorífico.
b. Comprobar la fecha de caducidad que indica la jeringa precargada (CAD). No usarla si la fecha
es superior al último día del mes que aparece o si se ha mantenido fuera del frigorífico durante más de 15 días o ha caducado de otro modo.
c. Comprobar la apariencia de Accofil. Debe ser un líquido transparente e incoloro. Si hay
partículas en el interior, no debe utilizarlo.
d. Para una inyección más cómoda, deje reposar la jeringa precargada durante 30 minutos a
temperatura ambiente o sostenga la jeringa precargada con suavidad en sus manos durante unos minutos. No caliente Accofil de otra manera (por ejemplo, no lo caliente en un microondas ni en agua caliente)
e. Lávese las manos cuidadosamente.
f. Buscar un lugar cómodo y bien iluminado y colocar todo lo necesario al alcance (la jeringa
precargada de Accofil, y la gasa impregnada con alcohol).
¿Cómo debo preparar la inyección de Accofil?
Antes de inyectar Accofil hay que:
1. Coger la jeringa y quitar la cubierta protectora de la aguj a suavemente sin inclinarla. Separar
tal como se indica en las figuras 1 y 2. No toque la aguja ni empuje el embolo.
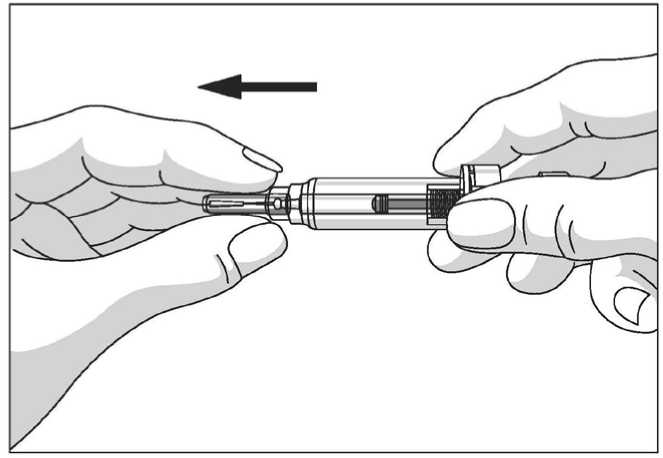
2
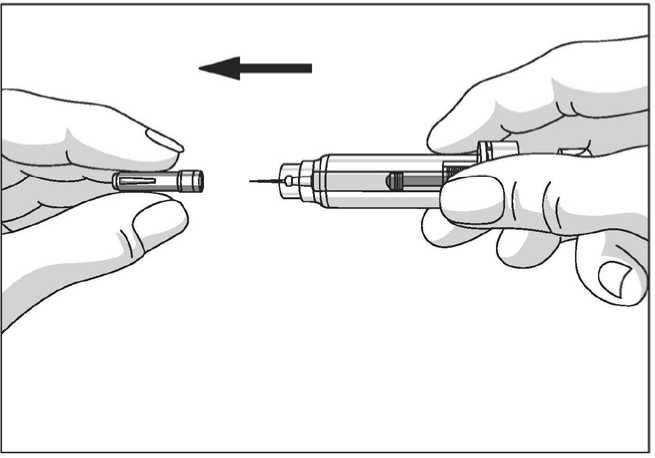
2. Puede aparecer una pequeña burbuja de aire en la jeringa precargada. Si hay burbujas, de golpecitos con sus dedos en la jeringa hasta que las burbujas vayan al final de la jeringa. Con la jeringa apuntando hacia arriba extraiga todo el aire de la jeringa empujando el embolo.
3. La jeringa puede contener más líquido del que necesita. Use la escala de la jeringa como se indica a continuación para ajustar la dosis correcta de Accofil que su médico le ha recetado. Expulse el líquido innecesario empujando el émbolo hasta el número (ml) de la jeringa que corresponde con la dosis recetada.
4. Compruebe de nuevo que la dosis de Accofil es la correcta.
5. Ahora puede utilizar la jeringa precargada.
¿En qué lugar debo poner la inyección?
Los sitios más adecuados para la inyección son:
• la parte superior de los muslos, y
• el abdomen, excepto la zona alrededor del ombligo (ver imagen 3).
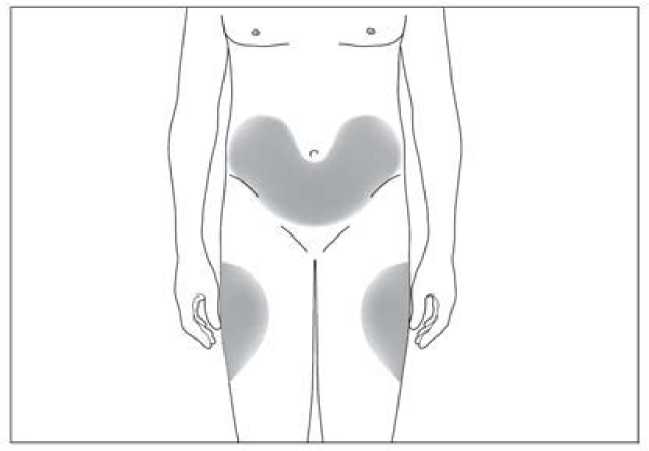
Si alguien le administra la inyección también podrá utilizar la parte posterior de sus antebrazos (ver imagen 4).
4
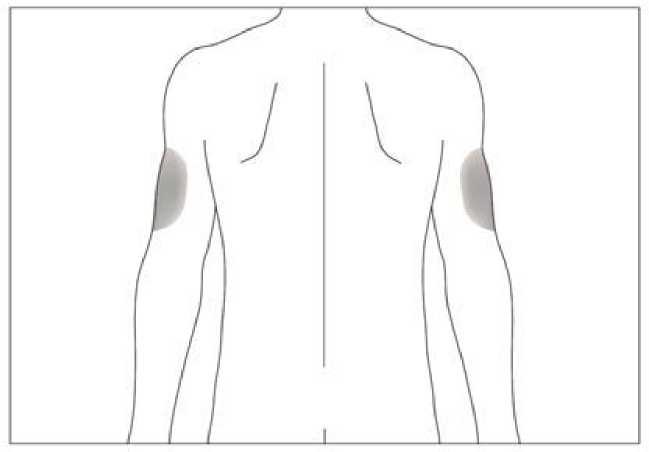
Para evitar el riesgo de dolor en un punto concreto es mejor cambiar cada día el lugar de la inyección. ¿Cómo debo inyectarme?
a. Desinfecte el lugar de la inyección usando una gasa con alcohol y pellizcar la piel entre el pulgar y el índice, sin apretar (ver imagen 5).
Jeringa precargada sin protector de seguridad para la aguja
b. Ponga la aguja totalmente en la piel tal como le indicó su enfermero o médico (véase la imagen 6).
c. Tire ligeramente del émbolo para comprobar que no se ha pinchado un vaso sanguíneo. Si ve sangre en la jeringa, retire la aguja y vuelva a insertarla en otro lugar.
d. Manteniendo la piel pellizcada, empuje el émbolo lentamente hasta haber administrado toda la dosis, hasta que el émbolo ya no pueda seguir avanzando. ¡No deje de presionar el émbolo!
e. Sólo inyecte la dosis recetada por su médico.
f. Tras inyectar el líquido, retire la aguja manteniendo la presión en el émbolo, y luego suelte la piel.
g. Coloque la jeringa en el recipiente especial para su desecho. Cada jeringa es para una sola inyección.
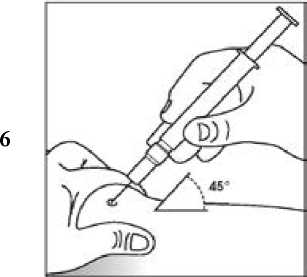
Jeringa precargada con protector de seguridad para la aguja
h. Insertar completamente la aguja en la piel tal como le indicó su enfermero o médico (ver imagen
7).
i. Tirar ligeramente del émbolo para comprobar que no se ha pinchado un vaso sanguíneo. Si se
ve sangre en la jeringa, retirar la aguja y vuelva a insertarla en otro lugar.
j. Inyecte únicamente la dosis que le ha indicado su médico siguiendo las instrucciones de abajo.
k. Manteniendo la piel pellizcada, empuje el émbolo lentamente y de manera uniforme mientras agarra el borde del dedo hasta haber administrado toda la dosis, hasta que el émbolo ya no pueda seguir avanzando. ¡No deje de presionar el émbolo!
l. Tras inyectar el líquido, retire la aguja manteniendo la jeringa en el mismo ángulo y manteniendo
la presión en el émbolo, y luego suelte la piel. La funda protectora cubrirá automáticamente la aguja y se oirá un "clic" audible para confirmar que se ha activado la protección (ver imagen 8). La protección de la aguja no se activará a menos que se haya administrado toda la dosis.
7
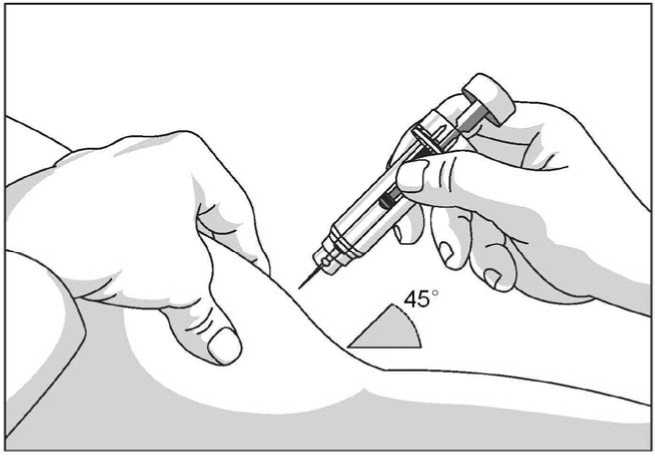
8
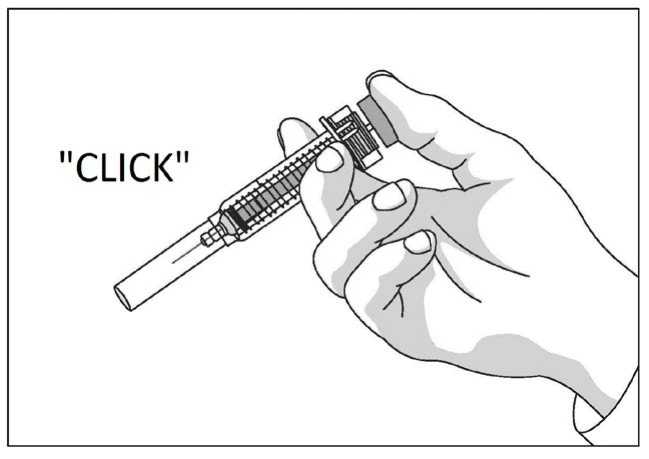
Recuerde
Si tiene cualquier duda, pida ayuda y consejos a su médico o enfermero.
Cómo desechar las jeringas usadas
• El protector de seguridad de la aguja evita pinchazos con la aguja tras su uso, y por lo tanto, no se necesitan precauciones especiales para su eliminación. Elimine la jeringa de la forma indicada por su médico, enfermero o farmacéutico.
Si usa más Accofil del que debe
Si usa más Accofil del que debe, consulte a su médico o farmacéutico lo antes posible.
Si olvidó usar Accofil
No se administre una dosis doble para compensar las dosis olvidadas. Póngase en contacto con su médico para discutir cuándo debe inyectarse la siguiente dosis.
Si interrumpe el tratamiento con Accofil
Su médico le indicará cuándo debe interrumpir el tratamiento con Accofil. Es muy normal tener un número de ciclos de tratamiento con Accofil.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Informe a su médico de inmediato durante el tratamiento:
• Si experimenta una reacción alérgica con debilidad, descenso de la presión sanguínea, dificultad para respirar, hinchazón en la cara (anafilaxia), exantema cutáneo, erupción cutánea (urticaria), hinchazón en la cara, labios, boca, lengua o garganta (angioedema) y falta de aliento (disnea). La hipersensibilidad es frecuente en pacientes con cáncer.
• Si experimenta tos, fiebre y dificultad para respirar (disnea), ya que esto puede ser un signo de síndrome de dificultad respiratoria aguda (SDRA). El SDRA es poco frecuente en pacientes con cáncer.
• Si nota dolor en la parte superior izquierda del abdomen, dolor debajo de la cavidad torácica izquierda o dolor en el extremo del hombro, ya que esto puede estar relacionado con un problema del bazo (esplenomegalia). Esto es muy frecuente en pacientes con neutropenia crónica grave, frecuente en pacientes con VIH y poco frecuente en donantes sanos de células madre.
• Si está en tratamiento por neutropenia crónica grave y tiene sangre en la orina (hematuria). Su médico le puede analizar la orina periódicamente si experimenta este efecto adverso o si se encuentran proteínas en su orina (proteinuria).
• Si usted experimenta alguno de los siguientes efectos secundarios o una combinación de los mismos: inflamación o hinchazón, que puede estar asociada con reducción de la frecuencia de la micción, dificultad para respirar, hinchazón abdominal y sensación de saciedad, y una sensación general de cansancio Estos síntomas por lo general se desarrollan rápidamente. Éstos podrían ser síntomas de una afección poco común (que puede afectar hasta 1 de cada 1.00 personas) llamada síndrome de extravasación capilar, que hace que la sangre se escape de los pequeños vasos sanguíneos del cuerpo y que precisa atención médica urgente.
Un efecto adverso muy frecuente con el uso de Accofil es el dolor en los músculos o huesos (dolor musculoesquelético), que se puede aliviar tomando analgésicos convencionales. En pacientes sometidos a trasplante de células madre o de médula ósea se puede producir enfermedad de injerto contra huésped (EICH): se trata de una reacción de las células del donante contra el paciente que recibe el trasplante; entre los signos y síntomas figuran erupción cutánea en las palmas de las manos y en las plantas de los pies y úlceras y llagas en la boca, intestino, hígado, piel o en los ojos, pulmones, vagina y articulaciones. En los donantes sanos de células madre se observa con mucha frecuencia un aumento de los glóbulos blancos (leucocitosis) y una disminución de las plaquetas que reduce la capacidad de la sangre para coagularse (trombocitopenia); su médico controlará estas reacciones.
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos muy frecuentes (observados en más de 1 de cada 10 personas que usan Accofil): en pacientes con cáncer
• cambios en la química sanguínea.
• aumento de ciertas enzimas de la sangre.
• disminución del apetito.
• dolor de cabeza.
• dolor en la boca y en la garganta (dolor orofaríngeo).
• tos.
• diarrea.
• vómitos.
• estreñimiento.
• náuseas.
• erupción cutánea.
• pérdida o debilitamiento del pelo (alopecia).
• dolor en los músculos o huesos (dolor musculoesquelético).
• debilidad generalizada (astenia).
• cansancio (fatiga).
• dolor e hinchazón del revestimiento del tubo digestivo que va desde la boca hasta el ano (inflamación mucosa).
• falta de aliento (disnea).
donantes sanos de células madre
• disminución de las plaquetas, que reduce la capacidad de la sangre para coagularse (trombocitopenia).
• aumento de los glóbulos blancos (leucocitosis).
• dolor de cabeza.
• dolor en los músculos o huesos (dolor musculoesquelético).
pacientes con neutropenia crónica grave
• dilatación del bazo (esplenomegalia).
• bajo recuento de glóbulos rojos (anemia).
• cambios en la química de la sangre.
• aumento de ciertas enzimas de la sangre.
• dolor de cabeza.
• hemorragias nasales (epistaxis).
• diarrea.
• dilatación del hígado (hepatomegalia).
• erupción cutánea.
• dolor en los músculos o huesos (dolor musculoesquelético).
• dolor en las articulaciones (artralgia).
pacientes con VIH
• dolor en los músculos o huesos (dolor musculoesquelético).
Efectos adversos frecuentes (observados en más de 1 de cada 100 personas que usan Accofil): pacientes con cáncer
• reacción alérgica (hipersensibilidad al medicamento).
• baj a tensión arterial (hipotensión).
• dolor al orinar (disuria).
• dolor en el pecho.
• Toser con sangre (hemoptisis).
donantes sanos de células madre
• aumento de ciertas enzimas de la sangre.
• falta de aliento (disnea).
pacientes con neutropenia crónica grave
• disminución de las plaquetas, que reduce la capacidad de la sangre para coagularse (trombocitopenia).
• cambios en la química de la sangre.
• inflamación de los vasos sanguíneos de la piel (vasculitis cutánea).
• pérdida o debilitamiento del pelo (alopecia).
• enfermedad que vuelve los huesos menos densos, haciéndolos más débiles, más frágiles y propensos a romperse (osteoporosis).
• sangre en la orina (hematuria).
• dolor en el lugar de la inyección.
pacientes con VIH
• dilatación del bazo (esplenomegalia).
Efectos adversos poco frecuentes (observados en más de 1 de cada 1000 personas que usan Accofil): pacientes con cáncer
• dolor intenso en los huesos, el tórax, el intestino o las articulariones (crisis drepanocítica).
• rechazo de la médula ósea trasplantada (enfermedad de injerto contra huésped).
• dolor e hinchazón de las articulaciones, similar a la gota (condrocalcinosis).
• inflamación pulmonar grave que provoca dificultad respiratoria (síndrome de dificultad respiratoria aguda).
• funcionamiento anómalo de los pulmones, que provoca falta de aliento (insuficiencia respiratoria).
• hinchazón o líquido en los pulmones (edema pulmonar).
• inflamación de los pulmones (neumopatía intersticial).
• anomalía radiográfica de los pulmones (infiltración pulmonar).
• lesiones dolorosas de color ciruela y con relieve en las extremidades, y a veces en la cara y cuello, junto con fiebre (Síndrome de Sweet).
• inflamación de los vasos sanguíneos de la piel (vasculitis cutánea).
• empeoramiento de la artritis reumatoide.
• cambios raros de la orina.
• dolor.
• lesión del hígado causada por el bloqueo de las venas pequeñas dentro del hígado (flebopatía oclusiva).
• sangrado del pulmón (hemorragia pulmonar).
• cambio del modo en que el organismo regula los líquidos corporales, que puede ocasionar hinchazón.
donantes sanos de células madre
• rotura del bazo.
• reacción alérgica repentina con riesgo de muerte (reacción anafiláctica).
• cambios en la química de la sangre.
• sangrado del pulmón (hemorragia pulmonar).
• Toser con sangre (hemoptisis).
• anomalía radiográfica de los pulmones (infiltración pulmonar).
• falta de absorción del oxígeno en el pulmón (hipoxia).
• aumento de ciertas enzimas de la sangre.
• empeoramiento de la artritis reumatoide.
pacientes con neutropenia crónica grave
• exceso de proteínas en la orina (proteinuria).
• rotura del bazo.
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto.
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Accofil
Mantener fuera de la vista y del alcance de los niños.
No use este medicamento después de la fecha de caducidad que aparece en el cartón y en la jeringa precargada después de CAD. La fecha de caducidad es el último día del mes.
Conservar en nevera (entre 2 °C-8 °C). No congelar.
La jeringa se puede sacar de la nevera y dejarse a temperatura ambiente (pero a una temperatura no superior a 25°C) durante un periodo único de hasta un máximo de 15 días y nunca superior a la fecha de caducidad de la etiqueta, Después de este periodo, el medicamento no se debe volver a refrigerar y se debe desechar.
Conservar la jeringa precargada en el embalaje exterior para de protegerlo de la luz.
No utilice Accofil si observa turbidez o hay decoloración o si hay partículas en ella.
No coloque la tapa en las agujas usadas, ya que puede pincharse accidentalmente. Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los medicamentos que no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Accofil
• El principio activo es filgrastim. Cada jeringa precargada contiene 30 MU (300 microgramos) de filgrastim en 0,5 ml lo que corresponde a 0,6 mg/ml.
• Los demás componentes son ácido acético, hidróxido de sodio, sorbitol (E420), polisorbato 80, agua para preparaciones inyectables.
Aspecto del producto y contenido del envase
Accofil es una solución transparente e incolora para inyección o infusión en una jeringa precargada marcado con 1/40 marcas impresas de 0,1 ml a 1 ml en el cilindro de la jeringa con una aguja para inyección. Cada jeringa precargada contiene 0,5 ml de solución.
Accofil está disponible en envases de 1, 3, 5, 7 y 10 jeringas precargadas, con protector de seguridad para la aguja prefijada, en envase blíster individual, o sin protector de seguridad para la aguja, en envase blíster y gasas impregnadas en alcohol.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y Responsable de la fabricación
Accord Healthcare Limited Sage House, 319 Pinner Road,
North Harrow, Middlesex HA1 4HF, Reino Unido
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
FR
Arrow Génériques
Tél: +33 4 72 72 60 72
IT
Accord Healthcare Limited Tel: +39 02 94323700
AT/BE/BG/CY/CZ/DE/DK/ EE / EL / FI / HR / HU / IS / LT / LV / LX/ MT / NL / NO / PT /
PL / RO / SE / SI / SK / UK
Accord Healthcare Limited Tel : +44 (0)208 863 1427
ES
Accord Healthcare S.L.U.
Tel: +34 93 301 00 64
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
La siguiente información está destinada únicamente a médicos o profesionales del sector sanitario:
Accofil no contiene conservantes. En vista de un posible riesgo de contaminación microbiana, las jeringas precargadas de Accofil son para un solo uso.
La exposición accidental a temperaturas de congelación hasta un máximo de 24 horas no afecta a la estabilidad de Accofil. Si la exposición supera las 24 horas o si se congela más de una vez, NO se debe usar Accofil.
Con el fin de mejorar la trazabilidad de los factores estimulantes de colonias de granulocitos, el nombre del producto (Accofil) y el número de lote del producto administrado deben estar claramente registrados en el historial del paciente
Accofil no se debe diluir con cloruro de sodio. Este medicamento no debe mezclarse con otros medicamentos excepto con los mencionados a continuación. La dilución de filgrastim puede ser adsorbida por el vidrio y materiales plásticos excepto si se diluye como se menciona a continuación.
Si es necesario, Accofil puede ser diluido en 5% de glucosa. La dilución a una concentración final de menos de 0,2 MU (2 g) por ml, no se recomienda en cualquier momento.
La solución debe inspeccionarse visualmente antes de su uso. Sólo deben utilizarse soluciones transparentes sin partículas.
Para los pacientes tratados con filgrastim diluido a concentraciones inferiores a 1,5 MU (15 g) por ml, se debe ser añadir albúmina sérica humana (HSA) a una concentración final de 2 mg/ml. Ejemplo: en un volumen de inyección final de 20 ml, las dosis totales de filgrastim inferiores a 30 MU (300 ^g) deben administrarse con adición de 0,2 ml de solución de albúmina humana 200 mg/ml (20%).
Cuando se diluye en 5% de glucosa, Accofil es compatible con el vidrio y una variedad de plásticos como PVC, poliolefina (un copolímero de polipropileno y polietileno) y polipropileno.
Tras la dilución:
La estabilidad química y física de la solución diluida para perfusión ha sido demostrada durante 24 horas almacenada entre 2 y 8 °C. Desde el punto de vista microbiológico, el medicamento se debe usar inmediatamente. Si no se usa inmediatamente, el tiempo y las condiciones de almacenamiento de la solución diluida son responsabilidad del usuario y normalmente no deberían sobrepasar las 24 horas entre 2 y 8 °C, a no ser que la dilución se haya realizado en condiciones de asepsia validadas y controladas.
Uso de la jeringa recargada con protector de seguridad de la aguja
El protector de seguridad cubre la aguja tras la inyección para evitar que se produzcan pinchazos con la aguja, Esto no afecta el funcionamiento normal de la jeringa. Presione el émbolo de la jeringa lenta y uniformemente hasta haber administrado toda la dosis, hasta que el émbolo ya no pueda avanzar más. Manteniendo la presión en el émbolo, retire la jeringa del paciente. El protector de seguridad de la aguja cubrirá la aguja al soltar el émbolo.
Uso de la jeringa precargada sin protector de seguridad para la aguja Administre la dosis siguiendo el protocolo estándar.
Eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
PROSPECTO: INFORMACIÓN PARA EL USUARIO
Accofil 48 MU/0,5 ml (0,96 mg/ml) solución inyectable y para perfusión en jeringa precargada
fílgrastim
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
• Conserve este prospecto, ya que puede tener que volver a leerlo.
• Si tiene alguna duda, consulte a su médico, enfermero o farmacéutico.
• Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
• Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Accofil y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Accofil
3. Cómo usar Accofil
4. Posibles efectos adversos
5. Conservación de Accofil
6. Contenido del envase e información adicional
1. Qué es Accofil y para qué se utiliza Qué es Accofil
Accofil contiene el principio activo filgrastim. Filgrastim es una proteína producida en la bacteria Escherichia coli mediante tecnología de ADN recombinante. Pertenece a un grupo de proteínas llamadas citoquinas y es muy similar a una proteína natural (granulocitos y factor estimulante de colonias [G-CSF]) producido por su propio cuerpo. Filgrastim estimula la médula ósea (el tejido donde se producen nuevas células sanguíneas) para producir más glóbulos blancos que ayudan a combatir las infecciones.
Para que se utiliza Accofil
Su médico le ha recetado Accofil para ayudar a su cuerpo a producir más glóbulos blancos. Su médico le dirá por qué usted está siendo tratado con Accofil. Accofil es útil para varias enfermedades diferentes que son:
• la quimioterapia
• trasplante de médula ósea
• Neutropenia crónica grave (bajo número de un tipo de células blancas de la sangre)
• La neutropenia (bajo número de un tipo de células blancas de la sangre) en pacientes con infección
por VIH
• Movilización de células madre de sangre periférica (para estimular las células madre para entrar en el torrente sanguíneo para ser recogidos y utilizados en el trasplante de médula ósea).
2. Qué necesita saber antes de empezar a usar Accofil No use Accofil
- Si es alérgico afilgrastim o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Advertencias y precauciones
Consulte con su médico antes de usar Accofil:
Consulte con su médico, farmacéutico o enfermero antes de empezar a usar Accofil:
Antes de iniciar el tratamiento dígale a su médico si tiene:
- anemia drepanocítica, accofil puede provocar crisis drepanocitaria.
- osteoporosis (enfermedad de los huesos)
Informe a su médico inmediatamente durante el tratamiento con Accofil si:
- tiene dolor en el abdomen (barriga) superior izquierdo, dolor debajo de las costillas izquierdas o en el extremo del hombro izquierdo (estos pueden ser síntomas de agrandamiento del bazo o posiblemente ruptura del bazo).
- nota hemorragia poco habitual o hematomas que pueden ser síntomas de menor número de plaquetas en sangre (trombocitopenia) con una capacidad reducida de coagular la sangre.
- tiene signos repentinos de alergia tales como erupciones, picores o urticaria en la piel, hinchazón de la cara, labios, lengua u otras partes del cuerpo, falta de aliento, sibilancias (sonido silbante que se produce al respirar) o problemas al respirar ya que podrían ser signos de una reacción alérgica grave.
Es posible que tenga que hacerse análisis de sangre periódicos mientras está siendo tratado con Accofil para contar el número de neutrófilos y otros glóbulos blancos en la sangre. Esto le dirá a su médico cómo está funcionando el tratamiento y también indicará si necesita tratamiento debe continuarse.
La cubierta de la aguja de la jeringa precargada contiene caucho natural (un derivado del látex) que puede causar una reacción alérgica.
Otros medicamentos y Accofil
Usted no debe recibir Accofil en las 24 horas antes y 24 horas después de recibir la quimioterapia.
Por favor, informe a su médico o farmacéutico si está tomando o ha tomado recientemente otros medicamentos, incluso los adquiridos sin receta.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que puede estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de tomar este medicamento.
Accofil no ha sido estudiado en mujeres embarazadas. Su médico puede decidir que usted no debería usar este medicamento.
Se desconoce si filgrastim pasa a la leche materna. Por lo tanto, su médico puede decidir que usted no debe usar este medicamento si está en periodo de lactancia.
Conducción y uso de máquinas
Si usted está experimentando la fatiga, no conduzca automóviles ni maneje máquinas.
Información importante sobre algunos de los componentes de Accofil
Este medicamento contiene sorbitol. Si usted ha si su médico le ha indicado que padece una intolerancia a ciertos azúcares (fructosa), consulte con él antes de tomar este medicamento. Este medicamento también contiene sodio a menos de 1 mmol de sodio (0.035 mg) por dosis, es decir, esencialmente "exento de sodio".
3. Cómo usar Accofil
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
La cantidad de Accofil usted necesita dependerá de la enfermedad por la que está tomando Accofil por y de su peso corporal.
Dosis
Accofil y neutropenia (bajo número de un tipo de células blancas de la sangre) asociados con la quimioterapia
La dosis habitual es de 0,5 millones de unidades (5 microgramos) por kilogramo de peso corporal al día. Por ejemplo, si usted pesa 60 kilogramos, su dosis diaria será de 30 millones de unidades (300 microgramos). El tratamiento con Accofil generalmente durará unos 14 días. No obstante, en algunos tipos de enfermedad puede ser necesario un tratamiento más largo con una duración de hasta aproximadamente un mes.
Accofil y trasplante de médula ósea
La dosis inicial habitual es de 1 millón de unidades (10 microgramos) por kilogramo de peso corporal al día administrada en perfusión. Por ejemplo, si usted pesa 60 kg su dosis diaria será de 60 millones de unidades (600 microgramos). Normalmente recibirá su primera dosis de Accofil al menos 24 horas después de la quimioterapia y al menos 24 horas después de recibir su trasplante de médula ósea. Luego, el médico le examinará la sangre para ver cómo está funcionando el tratamiento y cuánto tiempo durará.
Accofil neutropenia crónica y grave (bajo número de un tipo de células blancas de la sangre)
La dosis inicial habitual es de 0,5 millones de unidades (5 microgramos) y 1,2 millones de unidades (12 microgramos) por kilogramo de peso corporal al día en una dosis única o dividida. Luego, el médico le examinará la sangre para ver qué tan bien su tratamiento con Accofil está trabajando y para encontrar la dosis más adecuada para usted. Se requiere un tratamiento a largo plazo con Accofil para la reducción de la neutropenia.
Accofil y neutropenia (bajo número de un tipo de células blancas de la sangre) en pacientes con infección por el VIH
La dosis inicial habitual es de 0,1 millones de unidades (1 microgramos) y 0,4 millones de unidades (4 microgramos) por kilogramo de peso corporal por día. El médico puede examinar su sangre con regularidad para ver si el tratamiento con Accofil está funcionando. Una vez que el número de glóbulos blancos en su sangre ha vuelto a la normalidad, puede ser posible reducir la frecuencia de la dosis a menos de una vez al día. El tratamiento a largo plazo con Accofil puede ser necesario para mantener un número normal de células blancas en la sangre.
Accofil y trasplante de células madre de sangre periférica (células madre recogidas desde la sangre a usar en el trasplante de médula ósea)
Si va a donar células madre para usted, la dosis habitual es de 0,5 millones de unidades (5 microgramos) a 1 millón de unidades (10 microgramos) por kilogramo de peso corporal por día. El tratamiento con
Accofil tendrá una duración de hasta 2 semanas. Su médico controlará su sangre para determinar el mejor momento para recoger las células madre.
Si actúa como donante de células madre para otra persona, la dosis habitual es de 1 millón de unidades (10 microgramos) por kilogramo de peso corporal al día. El tratamiento con Accofil tendrá una duración de 4 a 5 días. Su médico le realizará análisis de sangre para determinar el mejor momento para recoger las células madre.
Forma de administración
Este medicamento se administra mediante una inyección, ya sea a través de una perfusión (goteo) intravenosa (IV) o mediante una inyección subcutánea (SC) en el tejido debajo de la piel.
Si usted está recibiendo este medicamento en inyección subcutánea, su médico puede sugerir que usted aprenda cómo ponerse las inyecciones. Su médico o enfermera le dará instrucciones sobre cómo hacer esto (ver información a continuación las instrucciones para inyectar Accofil). No intente auto-inyectarse sin estas instrucciones. Parte de la información que usted requiere se da al final de este prospecto, pero el tratamiento adecuado de su enfermedad requiere una estrecha y constante colaboración con su médico.
Información para autoinyectarse
Esta sección contiene información sobre cómo administrarse a uno mismo una inyección de Accofil. Es importante que no trate de administrarse una inyección sin haber recibido la formación necesaria por parte de su médico o enfermero. Si no está seguro de poder inyectarse o si tiene cualquier duda, consulte a su médico o enfermero.
¿Cómo he de inyectarme Accofil?
Deberá administrarse la inyección en el tejido justo bajo la piel. Esto se conoce como inyección subcutánea. La inyección deberá administrarse todos los días aproximadamente a la misma hora.
Equipo necesario
Para ponerse una inyección subcutánea, necesitará:
• una j eringa precargada de Accofil y
• gasa impregnada con alcohol o similar.
¿Qué debo hacer antes de administrarme una inyección subcutánea de Accofil?
Asegurarse de que la cubierta de la aguja permanece en la jeringa hasta justo antes de estar preparado para inyectarse
a. Sacar la jeringa precargada de Accofil del frigorífico.
b. Comprobar la fecha de caducidad que indica la jeringa precargada (CAD). No usarla si la fecha
es superior al último día del mes que aparece o si se ha dejado fuera del frigorífico durante más de 15 días o ha caducado de otro modo.
c. Comprobar la apariencia de Accofil. Debe ser un líquido transparente e incoloro. Si hay
partículas en el interior, no debe utilizarlo.
d. Para una inyección más cómoda, deje reposar la jeringa precargada durante 30 minutos a
temperatura ambiente o sostenga la jeringa precargada con suavidad en sus manos durante unos minutos. No caliente Accofil de otra manera (por ejemplo, no lo caliente en un microondas ni en agua caliente)
e. Lávese las manos cuidadosamente.
f. Buscar un lugar cómodo y bien iluminado y colocar todo lo necesario al alcance (la jeringa
precargada de Accofil, y la gasa impregnada con alcohol).
¿Cómo debo preparar la inyección de Accofil?
Antes de inyectar Accofil hay que:
1. Coger la jeringa y quitar la cubierta protectora de la aguj a suavemente sin inclinarla.
Separar tal como se indica en las figuras 1 y 2. No toque la aguja ni empuje el embolo.

2

2. Puede aparecer una pequeña burbuja de aire en la jeringa precargada. Si hay burbujas, de golpecitos con sus dedos en la jeringa hasta que las burbujas vayan al final de la jeringa. Con la jeringa apuntando hacia arriba extraiga todo el aire de la jeringa empujando el embolo.
3. La jeringa puede contener más líquido del que necesita. Use la escala de la jeringa como se indica a continuación para ajustar la dosis correcta de Accofil que su médico le ha recetado. Expulse el líquido innecesario empujando el émbolo hasta el número (ml) de la jeringa que corresponde con la dosis recetada.
4. Compruebe de nuevo que la dosis de Accofil es la correcta.
5. Ahora puede utilizar la jeringa precargada.
¿En qué lugar debo poner la inyección?
Los sitios más adecuados para la inyección son:
• la parte superior de los muslos, y
• el abdomen, excepto la zona alrededor del ombligo (ver imagen 3).

Si alguien le administra la inyección también podrá utilizar la parte posterior de sus antebrazos (ver imagen 4).
4

Para evitar el riesgo de dolor en un punto concreto es mejor cambiar cada día el lugar de la inyección.
¿Cómo debo inyectarme?
a. Desinfecte el lugar de la inyección usando una gasa con alcohol y pellizcar la piel entre el pulgar y el índice, sin apretar (ver imagen 5).

Jeringa precargada sin protector de seguridad para la aguja
b. Ponga la aguja totalmente en la piel tal como le indicó su enfermero o su médico (véase la imagen 6)
c. Tire ligeramente del émbolo para comprobar que no se haya pinchado un vaso sanguíneo. Si ve
sangre en la jeringa, retire la aguja y vuelva a insertarla en otro lugar.
d. Manteniendo la piel pellizcada, empuje el émbolo lentamente hasta haber administrado toda la dosis, hasta que el émbolo ya no pueda seguir avanzando. ¡No deje de presionar el émbolo!
e. Sólo inyecte la dosis recetada por su médico.
f. Tras inyectar el líquido, retire la aguja manteniendo la presión en el émbolo, y luego suelte la
piel.
g. Coloque la jeringa en el recipiente especial para su desecho. Cada jeringa es para una sola
inyección.

Jeringa precargada con protector de seguridad para la aguja
h. Insertar completamente la aguja en la piel tal como le indicó su enfermero o médico (ver imagen
7).
i. Tirar ligeramente del émbolo para comprobar que no se ha pinchado un vaso sanguíneo. Si se ve sangre en la jeringa, retirar la aguja y vuelva a insertarla en otro lugar.
j. Inyecte solo la dosis que su médico le ha dicho siguiendo las instrucciones de abajo.
k. Manteniendo la piel pellizcada, empuje el émbolo lentamente y de manera constante mientras
agarra el borde del dedo hasta haber administrado toda la dosis, hasta que el émbolo ya no pueda seguir avanzando. ¡No deje de presionar el émbolo!
l. Después de inyectar el líquido, retire la aguja mientras mantiene la jeringa en el mismo ángulo y mantiene la presión en el émbolo, a continuación suelte la piel. La funda protectora cubrirá automáticamente la aguja y se oirá un "clic" audible para confirmar la activación de la protección (ver imagen 8). La protección de la aguja no se activará a menos que se haya administrado toda la dosis.
7

8

Recuerde
Si tiene cualquier duda, pida ayuda y consejos a su médico o enfermero.
Cómo desechar las jeringas usadas
• El protector de seguridad de la aguja evita pinchazos con la aguja tras su uso, y por lo tanto, no se necesitan precauciones especiales para su eliminación. Elimine la jeringa de la forma indicada por su médico, enfermera o farmacéutico.
Si usa más Accofil del que debe
Si usa más Accofil del que debe, consulte a su médico o farmacéutico lo antes posible.
Si olvidó usar Accofil
No se administre una dosis doble para compensar las dosis olvidadas. Póngase en contacto con su médico para discutir cuándo debe inyectarse la siguiente dosis.
Si interrumpe el tratamiento con Accofil
Su médico le indicará cuándo debe interrumpe el tratamiento con Accofil. Es muy normal tener un número de ciclos de tratamiento con Accofil.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Informe a su médico de inmediato durante el tratamiento:
• Si experimenta una reacción alérgica con debilidad, descenso de la presión sanguínea, dificultad para respirar, hinchazón en la cara (anafilaxia), exantema cutáneo, erupción cutánea (urticaria), hinchazón en la cara, labios, boca, lengua o garganta (angioedema) y falta de aliento (disnea).
La hipersensibilidad es frecuente en pacientes con cáncer.
• Si experimenta tos, fiebre y dificultad para respirar (disnea), ya que esto puede ser un signo de síndrome de dificultad respiratoria aguda (SDRA). El SDRA es poco frecuente en pacientes con cáncer.
• Si nota dolor en la parte superior izquierda del abdomen, dolor debajo de la cavidad torácica izquierda o dolor en el extremo del hombro, ya que esto puede estar relacionado con un problema del bazo (esplenomegalia). Esto es muy frecuente en pacientes con neutropenia crónica grave, frecuente en pacientes con VIH y poco frecuente en donantes sanos de células madre.
• Si está en tratamiento por neutropenia crónica grave y tiene sangre en la orina (hematuria). Su médico le puede analizar la orina periódicamente si experimenta este efecto adverso o si se encuentran proteínas en su orina (proteinuria).
• Si usted experimenta alguno de los siguientes efectos secundarios o una combinación de los mismos: inflamación o hinchazón, que puede estar asociada con reducción de la frecuencia de la micción, dificultad para respirar, hinchazón abdominal y sensación de saciedad, y una sensación general de cansancio Estos síntomas por lo general se desarrollan rápidamente. Éstos podrían ser síntomas de una afección poco común (que puede afectar hasta 1 de cada 1.00 personas) llamada síndrome de extravasación capilar, que hace que la sangre se escape de los pequeños vasos sanguíneos del cuerpo y que precisa atención médica urgente.
Un efecto adverso muy frecuente con el uso de Accofil es el dolor en los músculos o huesos (dolor musculoesquelético), que se puede aliviar tomando analgésicos convencionales. En pacientes sometidos a trasplante de células madre o de médula ósea se puede producir enfermedad de injerto contra huésped (EICH): se trata de una reacción de las células del donante contra el paciente que recibe el trasplante; entre los signos y síntomas figuran erupción cutánea en las palmas de las manos y en las plantas de los pies y úlceras y llagas en la boca, intestino, hígado, piel o en los ojos, pulmones, vagina y articulaciones. En los donantes sanos de células madre se observa con mucha frecuencia un aumento de los glóbulos blancos (leucocitosis) y una disminución de las plaquetas que reduce la capacidad de la sangre para coagularse (trombocitopenia); su médico controlará estas reacciones.
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos muy frecuentes (observados en más de 1 de cada 10 personas que usan Accofil): en pacientes con cáncer
• cambios en la química sanguínea.
• aumento de ciertas enzimas de la sangre.
• disminución del apetito.
• dolor de cabeza.
• dolor en la boca y en la garganta (dolor orofaríngeo).
• tos.
• diarrea.
• vómitos.
• estreñimiento.
• náuseas.
• erupción cutánea.
• pérdida o debilitamientodel pelo (alopecia).
• dolor en los músculos o huesos (dolor musculoesquelético).
• debilidad generalizada (astenia).
• cansancio (fatiga).
• dolor e hinchazón del revestimiento del tubo digestivo que va desde la boca hasta el ano (inflamación mucosa).
• falta de aliento (disnea).
donantes sanos de células madre
• disminución de las plaquetas, que reduce la capacidad de la sangre para coagularse (trombocitopenia).
• aumento de los glóbulos blancos (leucocitosis).
• dolor de cabeza.
• dolor en los músculos o huesos (dolor musculoesquelético).
pacientes con neutropenia crónica grave
• dilatación del bazo (esplenomegalia).
• bajo recuento de glóbulos rojos (anemia).
• cambios en la química de la sangre.
• aumento de ciertas enzimas de la sangre.
• dolor de cabeza.
• hemorragias nasales (epistaxis).
• diarrea.
• dilatación del hígado (hepatomegalia).
• erupción cutánea.
• dolor en los músculos o huesos (dolor musculoesquelético).
• dolor en las articulaciones (artralgia).
pacientes con VIH
• dolor en los músculos o huesos (dolor musculoesquelético).
Efectos adversos frecuentes (observados en más de 1 de cada 100 personas que usan Accofil): pacientes con cáncer
• reacción alérgica (hipersensibilidad al medicamento).
• baja tensión arterial (hipotensión).
• dolor al orinar (disuria).
• dolor en el pecho.
• Toser con sangre (hemoptisis).
donantes sanos de células madre
• aumento de ciertas enzimas de la sangre.
• falta de aliento (disnea).
dilatación del bazo (esplenomegalia).
pacientes con neutropenia crónica grave
• disminución de las plaquetas, que reduce la capacidad de la sangre para coagularse (trombocitopenia).
• cambios en la química de la sangre.
• inflamación de los vasos sanguíneos de la piel (vasculitis cutánea).
• pérdida o debilitamientodel pelo (alopecia).
• enfermedad que vuelve los huesos menos densos, haciéndolos más débiles, más frágiles y propensos a romperse (osteoporosis).
• sangre en la orina (hematuria).
• dolor en el lugar de la inyección.
pacientes con VIH
• dilatación del bazo (esplenomegalia).
Efectos adversos poco frecuentes (observados en más de 1 de cada 1000 personas que usan Accofil): pacientes con cáncer
• dolor intenso en los huesos, el tórax, el intestino o las articulariones (crisis drepanocítica).
• rechazo de la médula ósea trasplantada (enfermedad de injerto contra huésped).
• dolor e hinchazón de las articulaciones, similar a la gota (condrocalcinosis).
• inflamación pulmonar grave que provoca dificultad respiratoria (síndrome de dificultad respiratoria aguda).
• funcionamiento anómalo de los pulmones, que provoca falta de aliento (insuficiencia respiratoria).
• hinchazón o líquido en los pulmones (edema pulmonar).
• inflamación de los pulmones (neumopatía intersticial).
• anomalía radiográfica de los pulmones (infiltración pulmonar).
• lesiones dolorosas de color ciruela y con relieve en las extremidades, y a veces en la cara y cuello, junto con fiebre (Síndrome de Sweet).
• inflamación de los vasos sanguíneos de la piel (vasculitis cutánea).
• empeoramiento de la artritis reumatoide.
• cambios raros de la orina.
• dolor.
• lesión del hígado causada por el bloqueo de las venas pequeñas dentro del hígado (flebopatía oclusiva).
• sangrado del pulmón (hemorragia pulmonar).
• cambio del modo en que el organismo regula los líquidos corporales, que puede ocasionar hinchazón.
donantes sanos de células madre
• rotura del bazo.
• reacción alérgica repentina con riesgo de muerte (reacción anafiláctica).
• cambios en la química de la sangre.
• sangrado del pulmón (hemorragia pulmonar).
• toser con sangre (hemoptisis).
• anomalía radiográfica de los pulmones (infiltración pulmonar).
• falta de absorción del oxígeno en el pulmón (hipoxia).
• aumento de ciertas enzimas de la sangre.
• empeoramiento de la artritis reumatoide.
pacientes con neutropenia crónica grave
• exceso de proteínas en la orina (proteinuria).
• rotura del bazo.
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Accofil
Mantener fuera de la vista y del alcance de los niños.
No use este medicamento después de la fecha de caducidad que aparece en el cartón y en la jeringa precargada después de CAD. La fecha de caducidad es el último día del mes.
Conservar en nevera (entre 2 °C-8 °C) No congelar.
La jeringa se puede sacar de la nevera y dejarse a temperatura ambiente (pero a una temperatura no superior a 25°C), durante un periodo único de hasta un máximo de 15 días y nunca superior a la fecha de caducidad de la etiqueta, Después de este periodo, el medicamento no se debe volver a refrigerar y se debe desechar.
Conservar la jeringa precargada en el embalaje con el fin de protegerlo de la luz.
No utilice Accofil si observa turbidez o hay decoloración o si hay partículas en ella.
No coloque la tapa en las agujas usadas, ya que puede pincharse accidentalmente. Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los medicamentos que no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Accofil
• El principio activo es filgrastim. Cada jeringa precargada contiene 48 MU (480 microgramos) de filgrastim en 0,5 ml lo que corresponde a 0,96 mg/ml.
• Los demás componentes son ácido acético, hidróxido de sodio, sorbitol (E420), polisorbato 80, agua para preparaciones inyectables.
Aspecto del producto y contenido del envase
Accofil es una solución transparente e incolora para inyección o infusión en una jeringa precargada marcado con 1/40 marcas impresas de 0,1 ml a 1 ml en el cilindro de la jeringa con una aguja para inyección. Cada jeringa precargada contiene 0,5 ml de solución.
Accofil está disponible en envases de 1, 3, 5, 7 y 10 jeringas precargadas, con protector de seguridad para la aguja prefijada, en envase blister individual, o sin protector de seguridad para la aguja, en envase blister y gasas impregnadas en alcohol.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y Responsable de la fabricación
Accord Healthcare Limited Sage House, 319 Pinner Road,
North Harrow,
Middlesex HA1 4HF,
Reino Unido
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
FR
Arrow Génériques
Tél: +33 4 72 72 60 72
IT
Accord Healthcare Limited Tel: +39 02 94323700
AT/BE/BG/CY/CZ/DE/DK/ EE / EL / FI / HR / HU / IS / LT / LV/LX MT/NL/NO/PT/
PL / RO / SE / SI / SK / UK
Accord Healthcare Limited Tel : +44 (0)208 863 1427
ES
Accord Healthcare S.L.U.
Tel: +34 93 301 00 64
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
La siguiente información está destinada únicamente a médicos o profesionales del sector sanitario:
Accofil no contiene conservantes. En vista de un posible riesgo de contaminación microbiana, las jeringas precargadas de Accofil son para un solo uso.
La exposición accidental a temperaturas de congelación hasta un máximo de 24 horas no afecta a la estabilidad de Accofil. Si la exposición supera las 24 horas o si se congela más de una vez, NO se debe usar Accofil.
Con el fin de mejorar la trazabilidad de los factores estimulantes de colonias de granulocitos, el nombre del producto (Accofil) y el número de lote del producto administrado deben estar claramente registrados en el historial del paciente
Accofil no se debe diluir con cloruro de sodio. Este medicamento no debe mezclarse con otros medicamentos excepto con los mencionados a continuación. La dilución de filgrastim puede ser adsorbida por el vidrio y materiales plásticos excepto si se diluye como se menciona a continuación.
Si es necesario, Accofil puede ser diluido en 5% de glucosa. La dilución a una concentración final de menos de 0,2 MU (2 g) por ml, no se recomienda en cualquier momento.
La solución debe inspeccionarse visualmente antes de su uso. Sólo deben utilizarse soluciones transparentes sin partículas.
Para los pacientes tratados con filgrastim diluido a concentraciones inferiores a 1,5 MU (15 g) por ml, se debe ser añadir albúmina sérica humana (HSA) a una concentración final de 2 mg/ml. Ejemplo: en un volumen de inyección final de 20 ml, las dosis totales de filgrastim inferiores a 30 MU (300 ^g) deben administrarse con adición de 0,2 ml de solución de albúmina humana 200 mg/ml (20%).
Cuando se diluye en 5% de glucosa, Accofil es compatible con el vidrio y una variedad de plásticos como PVC, poliolefina (un copolímero de polipropileno y polietileno) y polipropileno.
Tras la dilución:
La estabilidad química y física de la solución diluida para perfusión ha sido demostrada durante 24 horas almacenada entre 2 y 8 °C. Desde el punto de vista microbiológico, el medicamento se debe usar inmediatamente. Si no se usa inmediatamente, el tiempo y las condiciones de almacenamiento de la solución diluida son responsabilidad del usuario y normalmente no deberían sobrepasar las 24 horas entre 2 y 8 °C, a no ser que la dilución se haya realizado en condiciones de asepsia validadas y controladas.
Uso de la jeringa recargada con protector de seguridad de la aguja
El protector de seguridad cubre la aguja tras la inyección para evitar que se produzcan pinchazos con la aguja, Esto no afecta el funcionamiento normal de la jeringa. Presione el émbolo de la jeringa lenta y uniformemente hasta haber administrado toda la dosis, hasta que el émbolo ya no pueda avanzar más. Manteniendo la presión en el émbolo, retire la jeringa del paciente. El protector de seguridad de la aguja cubrirá la aguja al soltar el émbolo.
Uso de la jeringa precargada sin protector de seguridad para la aguja Administre la dosis siguiendo el protocolo estándar.
Eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
88
DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Ácido acético glacial.
Hidróxido de sodio.
Sorbitol (E420).
Polisorbato 80.
Agua para preparaciones inyectables.
6.2 Incompatibilidades
Accofil no debe diluirse con soluciones salinas.
Filgrastim diluido puede adsorberse al vidrio y materiales plásticos.
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
6.3 Período de validez 36 meses.