Zydelig 150 Mg Comprimidos Recubiertos Con Pelicula
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Zydelig 100 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 100 mg de idelalisib.
Excipiente(s) con efecto conocido: Cada comprimido contiene 0,1 mg de amarillo anaranjado FCF (E110) (ver sección 4.4).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película.
Comprimido recubierto con película de color naranja y forma ovalada, con unas dimensiones de 9,7 mm por 6,0 mm y con “GSI” grabado por un lado y “100” por el otro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Zydelig está indicado en combinación con rituximab para el tratamiento de los pacientes adultos con leucemia linfocítica crónica (LLC):
• que han recibido al menos un tratamiento anterior (ver sección 4.4), o bien
• para continuar el tratamiento en los pacientes con deleción en 17p o mutación de TP53 que no eran adecuados para quimioinmunoterapia y en los que ya se había iniciado Zydelig como tratamiento de primera línea (ver sección 4.4).
Zydelig está indicado en monoterapia para el tratamiento de los pacientes adultos con linfoma folicular (LF) refractario a dos líneas de tratamiento anteriores (ver sección 4.4).
4.2 Posología y forma de administración
El tratamiento con Zydelig debe ser realizado por un médico con experiencia en el uso de terapias anticancerosas.
Posología
La dosis recomendada de Zydelig es de 150 mg, administrados por vía oral dos veces al día. Se debe continuar el tratamiento hasta la progresión de la enfermedad o la aparición de efectos tóxicos inaceptables.
Si un paciente omite una dosis de Zydelig y han transcurrido un máximo de 6 horas con respecto a la hora a la que lo toma normalmente, debe tomar la dosis omitida lo antes posible y proseguir con su horario posológico habitual. Si un paciente omite una dosis y han transcurrido más de 6 horas, no debe tomar la dosis omitida, sino simplemente proseguir con su horario posológico habitual.
Modificación de la dosis
Elevación de las transaminasas hepáticas
Se debe interrumpir el tratamiento con Zydelig en caso de elevación de grado 3 ó 4 de las aminotransferasas (alanina aminotransferasa [ALT]/aspartato aminotransferasa [AST] >5 veces el límite superior de la normalidad [LSN]). Una vez que los valores hayan retornado al grado 1 ó inferior (ALT/AST <3 veces el LSN), se puede reanudar el tratamiento en dosis de 100 mg dos veces al día.
Si el problema no recurre, la dosis se puede incrementar de nuevo a 150 mg dos veces al día según el criterio del médico responsable del tratamiento.
Si el problema recurre, se debe interrumpir el tratamiento con Zydelig hasta que los valores retornen al grado 1 ó inferior, después de lo cual se puede considerar el reinicio en dosis de 100 mg dos veces al día según el criterio del médico (ver secciones 4.4 y 4.8).
Diarrea/colitis
Se debe interrumpir el tratamiento con Zydelig en caso de diarrea/colitis de grado 3 ó 4. Una vez que la diarrea/colitis haya retornado al grado 1 ó inferior, se puede reanudar el tratamiento en dosis de 100 mg dos veces al día. Si la diarrea/colitis no recurre, la dosis se puede incrementar de nuevo a 150 mg dos veces al día según el criterio del médico responsable del tratamiento (ver sección 4.8).
Neumonitis
Se debe interrumpir el tratamiento con Zydelig en caso de sospecha de neumonitis. Una vez resuelta la neumonitis y si resulta adecuado un nuevo tratamiento, se puede considerar la reanudación del tratamiento con dosis de 100 mg dos veces al día (ver secciones 4.4 y 4.8).
Exantema
Se debe interrumpir el tratamiento con Zydelig en caso de exantema de grado 3 ó 4. Una vez que el exantema haya retornado al grado 1 ó inferior, se puede reanudar el tratamiento en dosis de 100 mg dos veces al día. Si el exantema no recurre, la dosis se puede incrementar de nuevo a 150 mg dos veces al día según el criterio del médico responsable del tratamiento (ver sección 4.8).
Neutropenia
Se debe interrumpir el tratamiento con Zydelig en los pacientes mientras su recuento absoluto de neutrófilos (RAN) sea inferior a 500 por mm3. Se debe realizar RAN al menos semanalmente hasta que el RAN sea >500 por mm3 cuando se puede reanudar el tratamiento en dosis de 100 mg dos veces al día (ver sección 4.4).
|
RAN 1.000 a <1.500/mm3 |
RAN 500 to <1.000/mm3 |
RAN <500/mm3 |
|
Mantener el tratamiento con |
Mantener el tratamiento con |
Interrumpir el tratamiento con |
|
Zydelig. |
Zydelig. |
Zydelig. |
|
Realizar el RAN al menos |
Realizar el RAN al menos | |
|
semanalmente. |
semanalmente hasta que sea >500/mm3 y, a continuación, se puede reanudar el tratamiento con Zydelig en dosis de 100 mg dos veces al día. |
Poblaciones especiales de pacientes Pacientes de edad avanzada
No es necesario ajustar de forma específica la dosis en los pacientes de edad avanzada (edad >65 años) (ver sección 5.2).
Insuficiencia hepática
No es necesario ajustar la dosis al iniciar el tratamiento con Zydelig en los pacientes con insuficiencia hepática leve o moderada, pero se recomienda intensificar la vigilancia de las reacciones adversas (ver secciones 4.4 y 5.2).
No hay datos suficientes para realizar recomendaciones posológicas para pacientes con insuficiencia hepática grave. Por tanto, se recomienda precaución cuando se administre Zydelig en esta población y se recomienda intensificar la vigilancia de las reacciones adversas (ver secciones 4.4 y 5.2).
Población pediátrica
No se ha establecido la seguridad y eficacia de Zydelig en niños menores de 18 años. No se dispone de datos.
Forma de administración
Zydelig se administra por vía oral. Se debe indicar a los pacientes que traguen el comprimido entero. El comprimido recubierto con película no se debe masticar ni machacar. El comprimido recubierto con película se puede tomar acompañado o no de alimentos (ver sección 5.2).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Infecciones graves
No se debe iniciar el tratamiento con Zydelig en los pacientes en presencia de infección sistémica en curso bacteriana, fúngica o vírica.
Se han producido infecciones graves y mortales con idelalisib, incluidas infecciones oportunistas como neumonía por Pneumocystisjirovecii (NPJ) y por citomegalovirus (CMV). Por lo tanto, se debe administrar profilaxis contra la NPJ a todos los pacientes durante todo el tratamiento con idelalisib.
Se debe vigilar la aparición de signos y síntomas respiratorios en los pacientes durante todo el tratamiento e indicarles que notifiquen con rapidez nuevos síntomas respiratorios.
Se debe realizar una valoración clínica y analítica periódica para la detección de infecciones por CMV. El tratamiento con Zydelig se debe interrumpir en los pacientes que presenten infección o viremia.
Neutropenia
Durante el tratamiento han surgido casos de neutropenia de grado 3 o 4, incluida neutropenia febril, en pacientes tratados con idelalisib. Se deben realizar recuentos sanguíneos en todos los pacientes al menos cada 2 semanas durante los primeros 6 meses de tratamiento con idelalisib, y al menos semanalmente en los pacientes mientras el RAN sea inferior a 1.000 por mm3 (ver sección 4.2).
Elevaciones de las transaminasas
En los ensayos clínicos con idelalisib se han observado elevaciones de la ALT y la AST de grados 3 y 4 (>5 veces el LSN). Estos hallazgos analíticos se observaron fundamentalmente durante las 12 primeras semanas de tratamiento, fueron generalmente asintomáticos y revirtieron con la interrupción de la dosis del medicamento. La mayoría de los pacientes reanudó el tratamiento a una dosis más baja sin recurrencias (ver sección 4.2). Se debe controlar la ALT, la AST y la bilirrubina total en todos los pacientes cada 2 semanas durante los 3 primeros meses de tratamiento y cuando esté clínicamente indicado a partir de entonces. Si se observan elevaciones de la ALT y/o la AST de
grado 2 ó superior, se debe controlar semanalmente a los pacientes hasta que los valores retomen al grado 1 ó inferior.
Diarrea/c olitis
Se produjeron casos de colitis grave relacionada con el medicamento, relativamente tarde (meses) con respecto al inicio de la terapia, a veces con agravación rápida, pero se resolvieron en pocas semanas con la interrupción de la dosis del medicamento y tratamiento sintomático adicional (p. ej., medicamentos antinflamatorios, como budesonida entérica).
La experiencia del tratamiento de pacientes con antecedentes de enfermedad inflamatoria intestinal es muy limitada.
Neumonitis
Se han notificado casos de neumonitis en ensayos clínicos con idelalisib. Se debe evaluar una posible neumonitis medicamentosa en los pacientes que presentan acontecimientos pulmonares graves que no responden al tratamiento antimicrobiano convencional. Si se sospecha neumonitis, se debe interrumpir la administración de idelalisib y tratar al paciente en consecuencia. Se debe interrumpir el tratamiento en caso de neumonitis sintomática moderada o grave.
Síndrome de Stevens-Johnson y necrólisis epidérmica tóxica
Se han notificado casos de síndrome de Stevens-Johnson (SSJ) y necrólisis epidérmica tóxica (NET) con desenlace mortal cuando idelalisib se administró de forma concomitante con otros medicamentos asociados a estos síndromes. Si se sospecha SSJ o NET, se debe interrumpir inmediatamente la administración de idelalisib y tratar al paciente en consecuencia.
Inductores de CYP3A
La exposición a idelalisib se puede ver reducida cuando se administra de forma concomitante con inductores de CYP3A como la rifampicina, la fenitoína, la hierba de San Juan (Hypericum perforatum) o la carbamazepina. Como una reducción de la concentración plasmática de idelalisib puede ocasionar una disminución de la eficacia, se debe evitar la administración concomitante de Zydelig con inductores moderados o potentes de CYP3A (ver sección 4.5).
Sustratos de CYP3A
El principal metabolito de idelalisib, GS-563117, es un potente inhibidor de CYP3A4. Por tanto, idelalisib tiene el potencial de interactuar con medicamentos metabolizados por CYP3A, lo que puede dar lugar a un aumento de las concentraciones séricas del otro medicamento (ver sección 4.5). Cuando se administra idelalisib de forma concomitante con otros medicamentos, se debe consultar la ficha técnica o resumen de las características del producto del otro medicamento para conocer las recomendaciones acerca de la administración concomitante con inhibidores de CYP3A4. Se debe evitar el tratamiento concomitante de idelalisib con sustratos de CYP3A con reacciones adversas graves y/o potencialmente mortales (p. ej. afuzosina, amiodarona, cisaprida, pimozida, quinidina, ergotamina, dihidroergotamina, quetiapina, lovastatina, simvastatina, sildenafilo, midazolam, triazolam) y utilizar si es posible medicamentos alternativos menos sensibles a la inhibición por CYP3A4.
Insuficiencia hepática
Se recomienda intensificar la vigilancia de las reacciones adversas en pacientes con insuficiencia hepática, ya que se espera que la exposición aumente en esta población, en particular en pacientes con insuficiencia hepática grave. No se incluyeron pacientes con insuficiencia hepática grave en los ensayos clínicos de idelalisib. Se recomienda precaución cuando se administre Zydelig en esta población.
Hepatitis crónica
No se ha estudiado idelalisib en pacientes con hepatitis crónica activa incluyendo hepatitis vírica. Se debe actuar con precaución cuando se administre Zydelig en pacientes con hepatitis activa.
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar métodos anticonceptivos muy efectivos durante el tratamiento con idelalisib y durante el mes siguiente a su interrupción (ver sección 4.6). Las mujeres que usan anticonceptivos hormonales deben añadir un método de barrera como segundo método anticonceptivo, ya que actualmente se desconoce si idelalisib puede reducir la efectividad de los anticonceptivos hormonales.
Excipientes
Zydelig contiene el colorante azo amarillo anaranjado FCF (E110), que puede causar reacciones alérgicas.
4.5 Interacción con otros medicamentos y otras formas de interacción
Idelalisib se metaboliza principalmente a través de una aldehído oxidasa y, en menor grado, de CYP3A y una glucuronidación (UGT1A4). Su principal metabolito es GS-563117, que carece de actividad farmacológica. Idelalisib y GS-563117 son sustratos de P-gp y BCRP.
Efecto de otros medicamentos sobre la farmacocinética de idelalisib
Inductores de CYP3A
En un ensayo clínico de interacciones medicamentosas se constató que la administración concomitante de una dosis única de 150 mg de idelalisib con rifampicina (un potente inductor de CYP3A) generaba una reducción de aproximadamente el 75 % en la AUCinf de idelalisib. Se debe evitar la administración concomitante de Zydelig con inductores moderados o potentes de CYP3A como la rifampicina, la fenitoína, la hierba de San Juan o la carbamazepina, ya que esto puede ocasionar una disminución de la eficacia (ver sección 4.4).
Inhibidores de CYP3A/P-gp
En un ensayo clínico de interacciones medicamentosas se constató que la administración concomitante de una dosis única de 400 mg de idelalisib con 400 mg una vez al día de ketoconazol (un potente inhibidor de CYP3A, P-gp y BCRP) generaba un aumento del 26 % en la Cmax y un aumento del 79 % en la AUCinf de idelalisib. No se considera necesario efectuar un ajuste inicial de la dosis de idelalisib cuando se administra con inhibidores de CYP3A/P-gp, pero se recomienda intensificar la vigilancia de las reacciones adversas.
Efecto de idelalisib sobre la farmacocinética de otros medicamentos
Sustratos de CYP3A
El principal metabolito de idelalisib, GS-563117, es un potente inhibidor de CYP3A En un ensayo clínico de interacciones medicamentosas se constató que la administración concomitante de idelalisib con midazolam (un sustrato sensible de CYP3A) generaba un aumento de aproximadamente el 140 % en la y de alrededor del 440 % en la AUCinf del midazolam debido a la inhibición de CYP3A por GS-563117. La administración concomitante de idelalisib con sustratos de CYP3A puede aumentar sus exposiciones sistémicas y aumentar o prolongar su actividad terapéutica y sus reacciones adversas. In vitro, la inhibición de CYP3A4 fue irreversible y se espera por tanto que se tarde varios días en regresar a una actividad enzimática normal después de interrumpir la administración de idelalisib.
En la Tabla 1 se enumeran las posibles interacciones entre idelalisib y los medicamentos administrados de forma concomitante que son sustratos de CYP3A (el aumento se indica como “|”). Esta lista no es exhaustiva y solo pretende servir de orientación. En general, se debe consultar la ficha técnica del otro medicamento para conocer las recomendaciones acerca de la administración concomitante con inhibidores de CYP3A4 (ver sección 4.4).
Tabla 1: Interacciones entre idelalisib y otros medicamentos que son sustratos de CYP3A
|
Medicamento |
Efecto esperado de idelalisib sobre los niveles del medicamento |
Recomendación clínica para su administración concomitante con idelalisib |
|
ANTAGONISTAS DE LOS RECEPTORES ADRENÉRGICOS ALFA 1 | ||
|
Alfuzosina |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con alfuzosina. |
|
ANALGÉSICOS | ||
|
Fentanilo, alfentanilo, metadona, buprenorfma/naloxona |
t concentraciones séricas |
Se recomienda vigilar con atención las reacciones adversas (p. ej., depresión respiratoria, sedación). |
|
ANTIARRÍTMICOS | ||
|
Amiodarona, quinidina |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con amiodarona o quinidina. |
|
Bepridil, disopiramida, lidocaína |
t concentraciones séricas |
Se recomienda vigilancia clínica. |
|
ANTINEOPLÁSICOS | ||
|
Inhibidores de la tirosina quinasa como dasatinib y nilotinib, también vincristina y vinblastina |
t concentraciones séricas |
Se recomienda vigilar con atención la tolerancia a estos antineoplásicos. |
|
ANTICOAGULANTES | ||
|
Warfarina |
t concentraciones séricas |
Se recomienda vigilar el cociente internacional normalizado (INR) durante la administración concomitante y después de interrumpir el tratamiento con idelalisib. |
|
ANTICONVULSIVANTES | ||
|
Carbamazepina |
t concentraciones séricas |
Se deben vigilar los niveles de anticonvulsivantes. |
|
ANTIDEPRESIVOS | ||
|
Trazodona |
t concentraciones séricas |
Se recomienda un cuidadoso ajuste de la dosis del antidepresivo y vigilar la respuesta al mismo. |
|
ANTIGOTOSOS | ||
|
Colchicina |
t concentraciones séricas |
Se pueden requerir reducciones de la dosis de colchicina. No se debe administrar idelalisib de forma concomitante con colchicina a pacientes con insuficiencia renal o hepática. |
|
ANTIHIPERTENSIVOS | ||
|
Amlodipino, diltiazem, felodipino, nifedipino, nicardipino |
t concentraciones séricas |
Se recomienda vigilancia clínica del efecto terapéutico y de las reacciones adversas. |
|
ANTINFECCIOSOS | ||
|
Antifúngicos | ||
|
Ketoconazol, itraconazol, posaconazol, voriconazol |
t concentraciones séricas |
Se recomienda vigilancia clínica. |
|
Medicamento |
Efecto esperado de idelalisib sobre los niveles del medicamento |
Recomendación clínica para su administración concomitante con idelalisib |
|
Antimicobacterianos | ||
|
Rifabutina |
t concentraciones séricas |
Se recomienda aumentar la vigilancia de las reacciones adversas asociadas a rifabutina, entre ellas neutropenia y uveítis. |
|
Inhibidores de la proteasa del VHC | ||
|
Boceprevir, telaprevir |
t concentraciones séricas Se recomienda vigilancia clínica. | |
|
Antibióticos macrólidos | ||
|
Claritromicina, telitromicina |
t concentraciones séricas |
No se requiere ajuste de la dosis de claritromicina en los pacientes con función renal normal o insuficiencia renal leve (aclaramiento de creatinina [CrCl] 60-90 ml/min). Se recomienda vigilancia clínica en los pacientes con CrCl <90 ml/min. En los pacientes con CrCl <60 ml/min, se deben considerar antibacterianos alternativos. Se recomienda vigilancia clínica para la telitromicina. |
|
ANTIPSICÓTICOS/NEUROLÉPTICOS | ||
|
Quetiapina, pimozida |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con quetiapina o pimozida. Pueden considerarse medicamentos alternativos, como la olanzapina. |
|
ANTAGONISTAS DE LOS RECEPTORES DE LA ENDO |
TELINA | |
|
Bosentano |
t concentraciones séricas |
Hay que actuar con precaución y observar estrechamente a los pacientes en busca de toxicidad relacionada con bosentano. |
|
ALCALOIDES ERGÓTICOS | ||
|
Ergotamina, dihidroergotamina |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con ergotamina o dihidroergotamina. |
|
FÁRMACOS ESTIMULANTES DE LA MOTILIDAD GASTROINTESTINAL | ||
|
Cisaprida |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con cisaprida. |
|
GLUCOCORTICOIDES | ||
|
Corticosteroides inhalados/nasales: Budesonida, fluticasona Budesonida oral |
t concentraciones séricas t concentraciones séricas |
Se recomienda vigilancia clínica. Se recomienda vigilancia clínica en busca de un aumento de los signos/síntomas de los efectos de los corticosteroides. |
|
Medicamento |
Efecto esperado de idelalisib sobre los niveles del medicamento |
Recomendación clínica para su administración concomitante con idelalisib |
|
INHIBIDORES DE LA HMG-CoA REDUCTASA | ||
|
Lovastatina, simvastatina |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con lovastatina o simvastatina. |
|
Atorvastatina |
t concentraciones séricas |
Se recomienda vigilancia clínica y se puede considerar una dosis inicial más baja de atorvastatina. Alternativamente, se puede considerar el cambio a pravastatina, rosuvastatina o pitavastatina. |
|
IMMUNOSUPRESORES | ||
|
Ciclosporina, sirolimus, tacrolimus |
t concentraciones séricas |
Se recomienda vigilancia terapéutica. |
|
AGONISTA BETA INHALADO |
3 | |
|
Salmeterol |
t concentraciones séricas |
No se recomienda la administración concomitante de salmeterol e idelalis ib. La c ombinac ión puede ocasionar un mayor riesgo de acontecimientos adversos cardiovasculares asociados al salmeterol, entre ellos prolongación del QT, palpitaciones y taquicardia sinusal. |
|
INHIBIDORES DE LA FOSFO |
DIESTERASA | |
|
Para la hipertensión arterial pulmonar: | ||
|
Sildenafilo |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con sildenafilo. |
|
Tadalafilo |
t concentraciones séricas |
Hay que actuar con precaución, considerando incluso una reducción de la dosis, cuando se administre tadalafilo de forma concomitante con idelalisib. |
|
Para la disfunción eréctil: | ||
|
Sildenafilo, tadalafilo |
t concentraciones séricas |
Hay que actuar con especial precaución y se puede considerar una reducción de la dosis cuando se prescriba sildenafilo o tadalafilo con idelalisib, con mayor vigilancia de los acontecimientos adversos. |
|
SEDANTES/HIPNÓ TICOS | ||
|
Midazolam (oral), triazolam |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con midazolam (oral) o triazolam. |
|
Buspirona, clorazepato, diazepam, estazolam, flurazepam, zolpidem |
t concentraciones séricas |
Se recomienda vigilar la concentración de los sedantes/hipnóticos y puede considerarse una reducción de la dosis. |
Sustratos de CYP2C8
In vitro, idelalisib inhibió e indujo CYP2C8, pero se desconoce si esto se traduce en algún efecto in vivo sobre los sustratos de CYP2C8. Se recomienda precaución si se utiliza Zydelig junto con medicamentos con índices terapéuticos estrechos que son sustratos de CYP2C8 (paclitaxel).
Sustratos de enzimas inducibles (p. ej., CYP2C9, CYP2C19, CYP2B6y UGT)
In vitro, idelalisib fue un inductor de varias enzimas y no se puede excluir un riesgo de menor exposición y por tanto disminución de la eficacia de los sustratos de enzimas inducibles como CYP2C9, CYP2C19, CYP2B6 y UGT. Se recomienda precaución si se utiliza Zydelig junto con medicamentos con índices terapéuticos estrechos que son sustratos de estas enzimas (warfarina, fenitoína, S-mefenitoína).
Sustratos de BCRP, OATP1B1, OATP1B3y P-gp
La administración concomitante de varias dosis de idelalisib 150 mg dos veces al día a sujetos sanos ocasionó exposiciones similares a rosuvastatina (AUC IC del 90 %: 87, 121) y digoxina (AUC IC del 90 %: 98, 111), lo que sugiere que idelalisib no provoca una inhibición clínicamente relevante de BCRP, OATP1B1/1B3 o P-gp sistémica. No se puede excluir un riesgo de inhibición de la P-gp en el tubo digestivo, que podría ocasionar una mayor exposición a los sustratos sensibles a la P-gp intestinal como dabigatrán etexilato.
Población pediátrica
Los ensayos de interacciones se han realizado solo en adultos.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
De acuerdo con los hallazgos en animales, idelalisib puede causar daños fetales. Las mujeres deben evitar quedarse embarazadas durante el tratamiento con Zydelig y hasta un mes después de su finalización. Por tanto, las mujeres en edad fértil deben utilizar métodos anticonceptivos muy efectivos durante el tratamiento con Zydelig y durante el mes siguiente a su interrupción. Actualmente se desconoce si idelalisib puede reducir la efectividad de los anticonceptivos hormonales y por tanto las mujeres que usan anticonceptivos hormonales deben añadir un método de barrera como segundo método anticonceptivo.
Embarazo
No hay datos o éstos son limitados relativos al uso de idelalisib en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3).
No se recomienda utilizar Zydelig durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Lactancia
Se desconoce si idelalisib y sus metabolitos se excretan en la leche materna.
No se puede excluir el riesgo en recién nacidos/niños.
Se debe interrumpir la lactancia durante el tratamiento con Zydelig.
Fertilidad
No se dispone de datos en humanos acerca del efecto de idelalisib sobre la fertilidad. Los estudios en animales sugieren la posibilidad de efectos perjudiciales de idelalisib sobre la fertilidad y el desarrollo fetal (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Zydelig sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
La evaluación de las reacciones adversas se basa en un ensayo de fase 3 y siete ensayos de fases 1 y 2. El ensayo 312-0116 de fase 3 fue un ensayo aleatorizado, a doble ciego y controlado con placebo en el que se asignó aleatoriamente en una proporción 1:1 a 220 pacientes con LLC tratada anteriormente a recibir idelalisib + rituximab o placebo + rituximab. En los ensayos de fases 1 y 2 se evaluó la seguridad de idelalisib en 490 pacientes con neoplasias hematológicas malignas, que incluían a 354 sujetos que recibieron idelalisib (cualquier dosis) como medicamento único y a 136 sujetos que recibieron idelalisib en combinación con anticuerpos monoclonales contra CD20.
Durante el tratamiento con idelalisib, las reacciones adversas al medicamento notificadas con más frecuencia se enumeran en la T abla 2.
Tabla de reacciones adversas
En la Tabla 2 se presentan las reacciones adversas al medicamento notificadas con idelalisib solo o en combinación con anticuerpos monoclonales contra CD20. Las reacciones adversas se enumeran por sistema de clasificación de órganos y frecuencia. Las frecuencias se definen del siguiente modo: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000), muy raras (<1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 2: Reacciones adversas al medicamento notificadas en los ensayos clínicos en pacientes con neoplasias hematológicas malignas que recibieron idelalisib
|
Reacción |
Cualquier grado |
Grado >3 |
|
Infecciones e infestaciones | ||
|
Infecciones |
Muy frecuentes |
Muy frecuentes |
|
Trastornos de la sangre y del sistema linfático | ||
|
Neutropenia |
Muy frecuente |
Muy frecuente |
|
Trastornos respiratorios, torácicos y mediastínicos | ||
|
Neumonitis |
Frecuente |
Frecuente |
|
Trastornos gastrointestinales | ||
|
Diarrea/c olitis |
Muy frecuente |
Muy frecuente |
|
Trastornos hepatobiliares | ||
|
Aumento de las transaminasas |
Muy frecuente |
Muy frecuente |
|
Trastornos de la piel y del tejido subcutáneo | ||
|
Exantema* |
Muy frecuente |
Frecuente |
|
Síndrome de Stevens-Johnson / necrólisis epidérmica tóxica |
Rara |
Rara |
|
Trastornos generales y alteraciones en el lugar de administración | ||
|
Pirexia |
Muy frecuente |
Frecuente |
|
Exploraciones complementarias | ||
|
Aumento de los triglicéridos Muy frecuente |
Frecuente | |
* Incluye los términos preferidos dermatitis exfoliativa, erupción medicamentosa, exantema, exantema eritematoso, exantema generalizado, exantema macular, exantema maculopapular, exantema papular, exantema pruriginoso y exantema exfoliativo.
Descripción de reacciones adversas seleccionadas
Exantema
El exantema fue por lo general leve o moderado y ocasionó la interrupción del tratamiento en el 2 % de los pacientes aproximadamente. En el ensayo 312-0116, se produjo exantema (notificado como dermatitis exfoliativa, erupción medicamentosa, exantema, exantema eritematoso, exantema generalizado, exantema macular, exantema maculopapular, exantema papular y exantema pruriginoso) en el 24,5 % de los sujetos que recibieron idelalisib + rituximab y en el 6,5 % de los sujetos que recibieron placebo + rituximab. De estos, el 3,6 % de los que recibieron idelalisib + rituximab y el 0,9 % de los que recibieron placebo + rituximab presentaron exantema de grado 3, y ningún sujeto sufrió un acontecimiento adverso de grado 4. El exantema se resolvió habitualmente con tratamiento (p. ej., esteroides tópicos y/u orales, difenhidramina) e interrupción de la dosis en los casos graves (ver sección 5.3, fototoxicidad).
Síndrome de Stevens-Johnson y necrólisis epidérmica tóxica (ver sección 4.4)
En raras ocasiones, se han producido casos de SSJ y NET cuando idelalisib se administró de forma concomitante con otros medicamentos asociados a estos síndromes (bendamustina, rituximab, alopurinol y amoxicilina). El SSJ o la NET aparecieron dentro del mes siguiente tras la combinación de medicamentos y se han producido casos con desenlace mortal.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
En caso de sobredosis, se deberá vigilar al paciente por si hay evidencia de toxicidad (ver sección 4.8). El tratamiento de la sobredosis de Zydelig consiste en medidas generales de apoyo, incluida la vigilancia de las constantes vitales, así como la observación del estado clínico del paciente.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: agentes antineoplásicos, otros agentes antineoplásicos, código ATC: L01XX47
Mecanismo de acción
Idelalisib inhibe la fosfatidilinositol 3-quinasa p1105 (PI3K5), que es hiperactiva en las neoplasias malignas de linfocitos B y fundamental para numerosas vías de señalización que impulsan la proliferación, supervivencia, migración y retención de células malignas en los tejidos linfoides y en la médula ósea. Idelalisib es un inhibidor selectivo de la unión del adenosina-5’-trifosfato (ATP) al dominio catalítico de PI3K5, lo que genera una inhibición de la fosforilación del fosfatidilinositol, un segundo mensajero lipídico clave, y la prevención de la fosforilación de Akt (proteína quinasa B).
Idelalisib induce la apoptosis e inhibe la proliferación de las líneas celulares derivadas de los linfocitos B malignos y de las células tumorales primarias. Mediante la inhibición de la señalización a través de los receptores de quimiocinas CXCR4 y CXCR5 inducida por las quimiocinas CXCL12 y CXCL13, respectivamente, idelalisib inhibe la migración y retención de los linfocitos B malignos en el microambiente tumoral, incluidos los tejidos linfoides y la médula ósea.
Efectos farmacodinámicos
Se evaluó el efecto de idelalisib (150 mg y 400 mg) sobre el intervalo QT/QTc en un ensayo cruzado controlado con placebo y con control positivo (moxifloxacino 400 mg) en 40 sujetos sanos. A una dosis de 2,7 veces la dosis máxima recomendada, idelalisib no prolongó el intervalo QT/QTc (es decir, <10 ms).
Eficacia clínica en la leucemia linfocítica crónica
Idelalisib en combinación con inmunoterapia
El ensayo 312-0116 fue un ensayo aleatorizado, a doble ciego y controlado con placebo de fase 3 en 220 sujetos con LLC tratada anteriormente que precisaban tratamiento pero no se consideraban candidatos adecuados para quimioterapia citotóxica. Los sujetos se asignaron aleatoriamente en proporción 1:1 a recibir 8 ciclos de rituximab (primer ciclo en dosis de 375 mg/m2 de superficie corporal [SC], ciclos subsiguientes en dosis de 500 mg/m2 de SC) en combinación con un placebo por vía oral dos veces al día o con idelalisib en dosis de 150 mg administrados dos veces al día hasta la progresión de la enfermedad o la aparición de efectos tóxicos inaceptables.
La mediana de la edad era de 71 años (intervalo: 47 a 92) con un 78,2 % de los pacientes mayores de 65 años; el 65,5 % eran varones y el 90,0 % blancos; el 64,1 % se encontraban en estadio III o IV de Rai y el 55,9 % en estadio C de Binet. La mayoría de los sujetos presentaban factores citogenéticos con pronóstico adverso: el 43,2 % tenían una deleción cromosómica en 17p y/o una mutación de la proteína tumoral 53 (TP53) y el 83,6 % genes no mutados de la región variable de las cadenas pesadas de las inmunoglobulinas (IGHV). La mediana del tiempo transcurrido desde el diagnóstico de LLC hasta la aleatorización era de 8,5 años. Los sujetos presentaban una mediana de 8 en la puntuación de calificación de enfermedad acumulada (Cumulative Illness Rating Score, CIRS). La mediana del número de terapias previas era de 3,0. Casi todos (95,9 %) los sujetos habían recibido anticuerpos monoclonales contra CD20 con anterioridad. El criterio principal de valoración fue la supervivencia sin progresión (SSP). Los resultados de eficacia se resumen en las Tablas 3 y 4. En la Figura 1 se presenta la curva de Kaplan-Meier para la SSP.
En comparación con rituximab + placebo, el tratamiento con idelalisib + rituximab ocasionó mejorías estadísticamente significativas y clínicamente relevantes en el bienestar físico, el bienestar social, el bienestar funcional, así como en las subescalas específicas para la leucemia de los instrumentos de evaluación funcional del tratamiento del cáncer: leucemia (Functional Assessment of Cancer Therapy: Leukaemia, FACT-LEU), y mejorías estadísticamente significativas y clínicamente relevantes en la ansiedad, la depresión y las actividades habituales, medidas con el instrumento EuroCdV de cinco parámetros (EuroQoL Five-Dimensions, EQ-5D).
Tabla 3: Resultados de eficacia del ensayo 312-0116
|
Idelalisib + R N = 110 |
Placebo + R N = 110 | |
|
SSP Mediana (meses) (IC del 95 %) |
19,4 (12,3; NA) |
6,5 (4,0; 7,3) |
|
Razón de riesgo (IC del 95 %) |
0,15 (0,09; 0,25) | |
|
Valor p |
<0,0001 | |
|
TRG* n (%) (IC del 95 %) |
92 (83,6 %) (75,4; 90,0) |
17 (15,5 %) (9,3; 23,6) |
|
Razón de probabilidades (IC del 95 %) |
27,76 (13,40; 57,49) | |
|
Valor p |
<0,0001 | |
|
RGL** n/N (%) (IC del 95 %) |
102/106 (96,2 %) (90,6; 99,0) |
7/104 (6,7 %) (2,7; 13,4) |
|
Razón de probabilidades (IC del 95 %) |
225,83 (65,56; 777,94) | |
|
Valor p |
<0,0001 | |
|
SGA Mediana (meses) (IC del 95 %) |
NA (NA; NA) |
20,8 (14,8; NA) |
|
Razón de riesgo (IC del 95 %) |
0,34 (0,19; 0,60) | |
|
Valor p |
0,0001 | |
IC: intervalo de confianza; R: rituximab; n: número de sujetos con respuesta; N: número de sujetos por grupo; NA: no alcanzada. Los análisis de la SSP, tasa de respuesta global (TRG) y tasa de respuesta de los ganglios linfáticos (RGL) se basaron en la evaluación de un comité de revisión independiente (CRI).
*TRG definida como el porcentaje de sujetos que alcanzaron una respuesta completa (RC) o una respuesta parcial (RP) de acuerdo con los criterios de respuesta de la National Comprehensive CáncerNetwork (NCCN) de 2013 y de Cheson (2012).
**RGL definida como el porcentaje de sujetos que alcanzaron una reducción >50 % en la suma de los productos de los diámetros perpendiculares máximos de las lesiones índice. En este análisis solo se incluyó a los sujetos que tenían evaluaciones tanto en la situación basal como en >1 ocasión valorable posterior a la situación basal.
A El análisis de la supervivencia global (SG) incluye datos de sujetos que recibieron placebo + R en el ensayo 312-0116 y posteriormente recibieron idelalisib en un ensayo de extensión, basado en un análisis por intención de tratar.
Tabla 4: Resumen de la SSP y de las tasas de respuesta en los subgrupos predefinidos del ensayo 312-0116
|
Idelalisib + R |
Placebo + R | |
|
Deleción en 17p/mutación de TP53 |
N = 46 |
N = 49 |
|
Mediana de SSP (meses) (IC del 95 %) |
NA (12,3; NA) |
4,0 (3,7; 5,7) |
|
Razón de riesgo (IC del 95 %) |
0,13 (0,07; 0,27) | |
|
TRG (IC del 95 %) |
84,8 % (71,1; 93,7) |
12,2 % (4,6; 24,8) |
|
IGHV no mutados |
N = 91 |
N = 93 |
|
Mediana de SSP (meses) (IC del 95 %) |
19,4 (13,9; NA) |
5,6 (4,0; 7,2) |
|
Razón de riesgo (IC del 95 %) |
0,14 (0,08; 0,23) | |
|
TRG (IC del 95 %) |
82,4 % (73,0; 89,6) |
15,1 % (8,5; 24,0) |
|
Edad >65 años |
N = 89 |
N = 83 |
|
Mediana de SSP (meses) (IC del 95 %) |
19,4 (12,3; NA) |
5,7 (4,0; 7,3) |
|
Razón de riesgo (IC del 95 %) |
0,14 (0,08; 0,25) | |
|
TRG (IC del 95 %) |
84,3 % (75,0; 91,1) |
16,9 % (9,5; 26,7) |
IC: intervalo de confianza; R: rituximab; N: número de sujetos por grupo; NA: no alcanzada
Figura 1: Curva de Kaplan-Meier de la SSP del ensayo 312-0116 (población por intención de tratar)
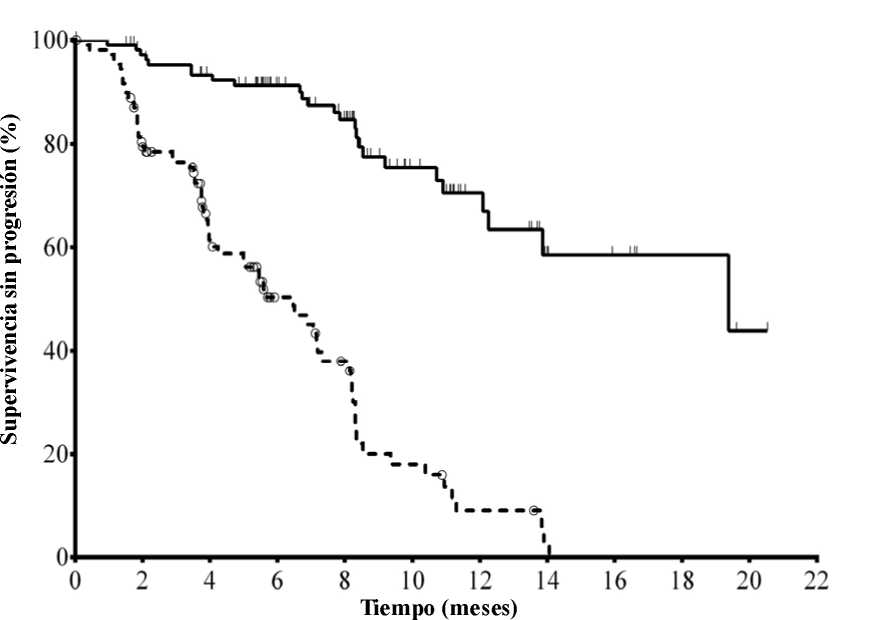
N° en riesgo (acontecimientos)
Idelalisib + R110 (0) 101 (3) 93 (7) 73 (9) 59 (14) 31 (19) 20 (21) 9 (24) 7 (24) 4 (24) 1 (25) 0 (25) Placebo + R 110 (0) 84 (21) 48 (38) 29 (46) 20 (53) 9 (63) 4 (67) 1 (69) 0 (70) 0 (70) 0 (70) 0 (70)
Línea continua: idelalisib + R(N = 110), línea discontinua: placebo + R(N = 110)
R: rituximab; N: número de sujetos por grupo
El análisis de la SSP se basó en la evaluación de un CRI. Para los sujetos del grupo tratado con placebo + R, el resumen incluye datos hasta la primera dosificación de idelalisib en el ensayo de extensión. Mediana de la SSP: idelalisib + R = 19,4 meses, placebo + R = 6,5 meses. Razón de riesgo = 0,15; IC del 95 % (0,09; 0,24);
p <0,0001.
En el ensayo 101-08/99 se reclutó a 64 sujetos con LLC sin tratamiento previo, incluidos 5 sujetos con linfoma linfocítico de células pequeñas (LLCP). Los sujetos recibieron idelalisib en dosis de 150 mg dos veces al día y rituximab en dosis de 375 mg/m2 de SC semanalmente. La TRG fue del 96,9 %, con 12 RC (18,8 %) y 50 RP (78,1 %), entre ellos 3 RC y 6 RP en sujetos con deleción en 17p y/o mutación de TP53 y 2 RC y 34 RP en sujetos con IGHV no mutados. No se ha alcanzado la mediana de la duración de la respuesta (DR).
Eficacia clínica en el linfoma folicular
La seguridad y eficacia de idelalisib se evaluó en un ensayo clínico multicéntrico de un solo grupo (ensayo 101-09) realizado en 125 sujetos con linfoma no-Hodgkin de linfocitos B indolente (LNHi, que incluye: LF, n = 72; LLCP, n = 28; linfoma linfoplasmocítico/macroglobulinemia de Waldenstrom [LLP/MW], n = 10; y linfoma de zona marginal [LZM], n = 15). Todos los sujetos eran resistentes al tratamiento con rituximab y 124 de 125 sujetos eran resistentes al tratamiento con al menos un agente alquilante. Ciento doce (89,6 %) sujetos habían sido resistentes a su última pauta terapéutica antes de la entrada en el ensayo.
De los 125 sujetos reclutados, 80 (64 %) eran varones, la mediana de la edad era de 64 años (intervalo: 33 a 87) y 110 (89 %) eran blancos. Los sujetos recibieron 150 mg de idelalisib por vía oral dos veces al día hasta que presentaron datos indicativos de progresión de la enfermedad o de la aparición de efectos tóxicos inaceptables.
El criterio principal de valoración fue la TRG, definida como el porcentaje de sujetos que alcanzaron una RC o una RP (de acuerdo con los criterios de respuesta revisados para el linfoma maligno [Cheson]) y, para los sujetos con macroglobulinemia de Waldenstrom, una respuesta menor (RM) (de acuerdo con la evaluación de la respuesta para la macroglobulinemia de Waldenstrom [Owen]). La DR fue un criterio de valoración secundario y se definió como el tiempo transcurrido desde la primera respuesta documentada (RC, RP o RM) hasta la primera documentación de progresión de la enfermedad o muerte por cualquier causa. Los resultados de eficacia se resumen en la T abla 5.
Tabla 5: Resumen de la respuesta en los sujetos con LF tratados con idelalisib (evaluación por parte de un CRI)
|
Característica |
Sujetos del ensayo n (%) |
|
TRG (linfoma folicular)* |
39 (54,2) |
|
IC del 95 % |
42,0 - 66,0 |
|
TRG (todos los sujetos)* |
71 (56,8) |
|
IC del 95 % |
47,6 - 65,6 |
|
Categoría de la respuesta (linfoma folicular)* RC |
6 (8,3) |
|
RP |
33 (45,8) |
IC: intervalo de confianza; n: número de sujetos con respuesta
* Respuesta según la valoración de un comité de revisión independiente (CRI), donde TRG= respuesta completa (RC) + respuesta parcial (RP).
La mediana de la DR para todos los sujetos fue de 12,5 meses (12,5 meses para los sujetos con LLCP, y no alcanzada para los sujetos con LF, LLP/MW y LZM). Entre los 122 sujetos con ganglios linfáticos medibles tanto en la situación basal como posteriormente, 67 (54,9 %) alcanzaron una reducción >50 % con respecto a la situación basal en la suma de los productos de los diámetros (SPD) de las lesiones índice. De los sujetos que no respondieron, 10 (8,0 %) presentaban progresión de la enfermedad como mejor respuesta y 2 (1,6 %) no eran evaluables. La mediana de la SG, incluyendo el seguimiento a largo plazo de los 125 sujetos, fue de 20,3 meses.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con idelalisib en uno o más grupos de la población pediátrica en el tratamiento de las neoplasias de linfocitos B maduros (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Tras la administración oral de una dosis única de idelalisib, se observaron concentraciones plasmáticas máximas 2 a 4 horas después de la administración con alimentos y tras 0,5 a 1,5 horas después en condiciones de ayuno.
Tras la administración de 150 mg de idelalisib dos veces al día, la media (intervalo) de la Cmax y del AUC en estado estacionario fue de 1.953 (272; 3.905) ng/ml y 10.439 (2.349; 29.315) ng»h/ml para idelalisib y de 4.039 (669; 10.897) ng/ml y 39.744 (6.002; 119.770) ng*h/ml para GS-563117, respectivamente. Las exposiciones (Cmax y AUC) a idelalisib son aproximadamente proporcionales a la dosis entre 50 mg y 100 mg y menores que la proporcional a la dosis por encima de 100 mg.
Efectos de los alimentos
En comparación con las condiciones de ayuno, la administración de una formulación inicial de idelalisib en cápsulas con una comida de alto contenido graso no produjo cambios en la Cmax, pero generó un aumento del 36 % en la AUC¡nf media. Idelalisib se puede administrar sin tener en cuenta la ingesta de alimentos.
Distribución
Idelalisib se une en un 93 % a 94 % a las proteínas plasmáticas humanas en las concentraciones observadas clínicamente. La relación media de las concentraciones sanguíneas/plasmáticas fue de aproximadamente 0,5. El volumen de distribución aparente de idelalisib (media) fue de unos 96 l.
Biotransformación
Idelalisib se metaboliza principalmente a través de una aldehido oxidasa y, en menor grado, de CYP3A y UGT1A4. Su principal y único metabolito circulante, GS-563117, es inactivo frente a PI3K5.
Eliminación
La semivida de eliminación terminal de idelalisib fue de 8,2 (intervalo: 1,9; 37,2) horas y el aclaramiento aparente de idelalisib fue de 14,9 (intervalo: 5,1; 63,8) l/h tras la administración oral de 150 mg de idelalisib dos veces al día. Tras una dosis única por vía oral de 150 mg de idelalisib marcado con [14C], se excretó aproximadamente un 78 % y un 15 % con las heces y la orina, respectivamente. Idelalisib sin modificar constituyó el 23 % de la radiactividad total recuperada en la orina durante 48 horas y el 12 % de la radiactividad total recuperada en las heces durante 144 horas.
Datos de interacción in vitro
Los datos in vitro indicaron que idelalisib no es un inhibidor de las enzimas metabolizadoras CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A o UGT1A1 ni de los transportadores OAT1, OAT3 u OCT2.
GS-563117 no es un inhibidor de las enzimas metabolizadoras CYP1A2, CYP2B6, CYP2C8,
CYP2C9, CYP2C19, CYP2D6 o UGT1A1 ni de los transportadores P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3 u OCT2.
Poblaciones especiales
Sexo y raza
Los análisis farmacocinéticos poblacionales indicaron que el sexo y la raza no ejercen un efecto clínicamente relevante sobre las exposiciones a idelalisib o a GS-563117.
Pacientes de edad avanzada
Los análisis farmacocinéticos poblacionales indicaron que la edad no ejerce un efecto clínicamente relevante sobre las exposiciones a idelalisib o a GS-563117, incluidos los sujetos de edad avanzada (65 años o más) en comparación con los sujetos más jóvenes.
Insuficiencia renal
Se realizó un ensayo de la farmacocinética y la seguridad de idelalisib en sujetos sanos y sujetos con insuficiencia renal grave (CrCl estimado 15 a 29 ml/min). Tras una dosis única de 150 mg, no se observaron cambios clínicamente relevantes en las exposiciones a idelalisib o a GS-563117 en los sujetos con insuficiencia renal grave en comparación con los sujetos sanos.
Insuficiencia hepática
Se realizó un ensayo de la farmacocinética y la seguridad de idelalisib en sujetos sanos y sujetos con insuficiencia hepática moderada (clase B de Child-Pugh) o grave (clase C de Child-Pugh). Tras una dosis única de 150 mg, el AUC de idelalisib (total, es decir, unido más no unido) fue aproximadamente un 60 % mayor en la insuficiencia hepática moderada o grave en comparación con controles emparejados. El AUC de idelalisib (no unido), tras considerar las diferencias en la unión a proteínas, fue aproximadamente un 80 % (1,8 veces) mayor en la insuficiencia moderada y aproximadamente un 152 % (2,5 veces) mayor en la insuficiencia grave en comparación con controles emparejados.
Población pediátrica
No se ha establecido la farmacocinética de idelalisib en los pacientes pediátricos (ver sección 4.2).
5.3 Datos preclínicos sobre seguridad
Toxicidad a dosis repetidas
Idelalisib indujo una disminución del número de linfocitos en el bazo, el timo, los ganglios linfáticos y el tejido linfoide asociado al tubo digestivo. En general, las áreas dependientes de los linfocitos B se vieron más afectadas que las dependientes de los linfocitos T. En ratas, idelalisib tiene el potencial de inhibir las respuestas de anticuerpos dependientes de los linfocitos T. No obstante, idelalisib no inhibió la respuesta normal del huésped a Staphylococcus aureus y no exacerbó el efecto mielosupresor de la ciclofosfamida. No se considera que idelalisib tenga una actividad inmunosupresora amplia.
Idelalisib indujo alteraciones inflamatorias tanto en ratas como en perros. En estudios de hasta 4 semanas en ratas y perros, se observó necrosis hepática a 7 y 5 veces, respectivamente, la exposición en humanos basada en el AUC. Las elevaciones de las transaminasas séricas se correlacionaban con necrosis hepáticas en perros, pero no se observaron en las ratas. No se observó insuficiencia hepática ni elevaciones crónicas de las transaminasas en ratas ni en perros en estudios de 13 semanas de duración o más.
Genotoxicidad
Idelalisib no indujo mutaciones en el ensayo de mutagénesis microbiana (Ames), no fue clastogénico en el ensayo de aberraciones cromosómicas in vitro con linfocitos de sangre periférica humana y no fue genotóxico en el estudio de micronúcleos de rata in vivo.
Carcinogenicidad
No se han realizado estudios de carcinogenicidad con idelalisib.
T oxicidad para la reproducción y el desarrollo
En un estudio de desarrollo embriofetal en ratas, se observó un aumento de la pérdida post-implantación, malformaciones (ausencia de vértebras caudales y, en algunos casos, también de vértebras sacras), cambios esqueléticos y menores pesos corporales fetales. Se observaron malformaciones a exposiciones a partir de 12 veces la exposición en humanos basadas en el AUC. No se investigaron los efectos sobre el desarrollo embriofetal en una segunda especie.
Se observó degeneración de los túbulos seminíferos testiculares en estudios a dosis repetidas de 2 a 13 semanas en perros y ratas, pero no en estudios de 26 semanas de duración o más. En un estudio de fertilidad masculina en ratas, se observaron reducciones en el peso de los epidí dimos y de los testículos, pero no se observaron efectos adversos sobre los parámetros de apareamiento y fertilidad ni degeneración o pérdida de la espermatogénesis. La fertilidad femenina no se vio afectada en las ratas.
Fototoxicidad
La evaluación del potencial de fototoxicidad en la línea celular de fibroblastos embrionarios murinos BALB/c 3T3 no fue concluyente para idelalisib debido a citotoxicidad en el ensayo in vitro. El principal metabolito, GS-563117, puede potenciar la fototoxicidad cuando las células se exponen simultáneamente a luz UVA. Existe un riesgo potencial de que idelalisib, a través de su principal metabolito, GS-563117, pueda provocar fotosensibilidad en los pacientes tratados.
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Núcleo del comprimido Celulosa microcristalina Hidroxipropilcelulosa (E463)
Croscarmelosa sódica Glic olato de almidón sódico Estearato de magnesio
Recubrimiento pelicular Alcohol polivinílico (E1203)
Macrogol 3350 (E1521)
Dióxido de titanio (E171)
Talco (E553B)
Amarillo anaranjado FCF (E110)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Frasco de polietileno de alta densidad (HDPE), tapado con un cierre de seguridad de polipropileno a prueba de niños, que contiene 60 comprimidos recubiertos con película y un relleno de poliéster.
Cada caja contiene 1 frasco.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/938/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 18/septiembre/2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Zydelig 150 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 150 mg de idelalisib. Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película.
Comprimido recubierto con película de color rosa y forma ovalada, con unas dimensiones de 10,0 mm por 6,8 mm y con “GSI” grabado por un lado y “150” por el otro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Zydelig está indicado en combinación con rituximab para el tratamiento de los pacientes adultos con leucemia linfocítica crónica (LLC):
• que han recibido al menos un tratamiento anterior (ver sección 4.4), o bien
• para continuar el tratamiento en los pacientes con deleción en 17p o mutación de TP53 que no eran adecuados para quimioinmunoterapia y en los que ya se había iniciado Zydelig como tratamiento de primera línea (ver sección 4.4).
Zydelig está indicado en monoterapia para el tratamiento de los pacientes adultos con linfoma folicular (LF) refractario a dos líneas de tratamiento anteriores (ver sección 4.4).
4.2 Posología y forma de administración
El tratamiento con Zydelig debe ser realizado por un médico con experiencia en el uso de terapias anticancerosas.
Posología
La dosis recomendada de Zydelig es de 150 mg, administrados por vía oral dos veces al día. Se debe continuar el tratamiento hasta la progresión de la enfermedad o la aparición de efectos tóxicos inaceptables.
Si un paciente omite una dosis de Zydelig y han transcurrido un máximo de 6 horas con respecto a la hora a la que lo toma normalmente, debe tomar la dosis omitida lo antes posible y proseguir con su horario posológico habitual. Si un paciente omite una dosis y han transcurrido más de 6 horas, no debe tomar la dosis omitida, sino simplemente proseguir con su horario posológico habitual.
Modificación de la dosis
Elevación de las transaminasas hepáticas
Se debe interrumpir el tratamiento con Zydelig en caso de elevación de grado 3 ó 4 de las aminotransferasas (alanina aminotransferasa [ALT]/aspartato aminotransferasa [AST] >5 veces el límite superior de la normalidad [LSN]). Una vez que los valores hayan retomado al grado 1 ó inferior (ALT/AST <3 veces el LSN), se puede reanudar el tratamiento en dosis de 100 mg dos veces al día.
Si el problema no recurre, la dosis se puede incrementar de nuevo a 150 mg dos veces al día según el criterio del médico responsable del tratamiento.
Si el problema recurre, se debe interrumpir el tratamiento con Zydelig hasta que los valores retornen al grado 1 ó inferior, después de lo cual se puede considerar el reinicio en dosis de 100 mg dos veces al día según el criterio del médico (ver secciones 4.4 y 4.8).
Diarrea/colitis
Se debe interrumpir el tratamiento con Zydelig en caso de diarrea/colitis de grado 3 ó 4. Una vez que la diarrea/colitis haya retornado al grado 1 ó inferior, se puede reanudar el tratamiento en dosis de 100 mg dos veces al día. Si la diarrea/colitis no recurre, la dosis se puede incrementar de nuevo a 150 mg dos veces al día según el criterio del médico responsable del tratamiento (ver sección 4.8).
Neumonitis
Se debe interrumpir el tratamiento con Zydelig en caso de sospecha de neumonitis. Una vez resuelta la neumonitis y si resulta adecuado un nuevo tratamiento, se puede considerar la reanudación del tratamiento con dosis de 100 mg dos veces al día (ver secciones 4.4 y 4.8).
Exantema
Se debe interrumpir el tratamiento con Zydelig en caso de exantema de grado 3 ó 4. Una vez que el exantema haya retornado al grado 1 ó inferior, se puede reanudar el tratamiento en dosis de 100 mg dos veces al día. Si el exantema no recurre, la dosis se puede incrementar de nuevo a 150 mg dos veces al día según el criterio del médico responsable del tratamiento (ver sección 4.8).
Neutropenia
Se debe interrumpir el tratamiento con Zydelig en los pacientes mientras su recuento absoluto de neutrófilos (RAN) sea inferior a 500 por mm3. Se debe realizar RAN al menos semanalmente hasta que el RAN sea >500 por mm3 cuando se puede reanudar el tratamiento en dosis de 100 mg dos veces al día (ver sección 4.4).
|
RAN 1.000 a <1.500/mm3 |
RAN 500 to <1.000/mm3 |
RAN<500/mm3 |
|
Mantener el tratamiento con |
Mantener el tratamiento con |
Interrumpir el tratamiento con |
|
Zydelig. |
Zydelig. |
Zydelig. |
|
Realizar el RAN al menos |
Realizar el RAN al menos | |
|
semanalmente. |
semanalmente hasta que sea >500/mm3 y, a continuación, se puede reanudar el tratamiento con Zydelig en dosis de 100 mg dos veces al día. |
Poblaciones especiales de pacientes Pacientes de edad avanzada
No es necesario ajustar de forma específica la dosis en los pacientes de edad avanzada (edad >65 años) (ver sección 5.2).
Insuficiencia renal
No es necesario ajustar la dosis en los pacientes con insuficiencia renal leve, moderada o grave (ver sección 5.2).
Insuficiencia hepática
No es necesario ajustar la dosis al iniciar el tratamiento con Zydelig en los pacientes con insuficiencia hepática leve o moderada, pero se recomienda intensificar la vigilancia de las reacciones adversas (ver secciones 4.4 y 5.2).
No hay datos suficientes para realizar recomendaciones posológicas para pacientes con insuficiencia hepática grave. Por tanto, se recomienda precaución cuando se administre Zydelig en esta población y se recomienda intensificar la vigilancia de las reacciones adversas (ver secciones 4.4 y 5.2).
Población pediátrica
No se ha establecido la seguridad y eficacia de Zydelig en niños menores de 18 años. No se dispone de datos.
Forma de administración
Zydelig se administra por vía oral. Se debe indicar a los pacientes que traguen el comprimido entero. El comprimido recubierto con película no se debe masticar ni machacar. El comprimido recubierto con película se puede tomar acompañado o no de alimentos (ver sección 5.2).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Infecciones graves
No se debe iniciar el tratamiento con Zydelig en los pacientes en presencia de infección sistémica en curso bacteriana, fúngica o vírica.
Se han producido infecciones graves y mortales con idelalisib, incluidas infecciones oportunistas como neumonía por Pneumocystisjirovecii (NPJ) y por citomegalovirus (CMV). Por lo tanto, se debe administrar profilaxis contra la NPJ a todos los pacientes durante todo el tratamiento con idelalisib.
Se debe vigilar la aparición de signos y síntomas respiratorios en los pacientes durante todo el tratamiento e indicarles que notifiquen con rapidez nuevos síntomas respiratorios.
Se debe realizar una valoración clínica y analítica periódica para la detección de infecciones por CMV. El tratamiento con Zydelig se debe interrumpir en los pacientes que presenten infección o viremia.
Neutropenia
Durante el tratamiento han surgido casos de neutropenia de grado 3 o 4, incluida neutropenia febril, en pacientes tratados con idelalisib. Se deben realizar recuentos sanguíneos en todos los pacientes al menos cada 2 semanas durante los primeros 6 meses de tratamiento con idelalisib, y al menos semanalmente en los pacientes mientras el RAN sea inferior a 1.000 por mm3 (ver sección 4.2).
Elevaciones de las transaminasas
En los ensayos clínicos con idelalisib se han observado elevaciones de la ALT y la AST de grados 3 y 4 (>5 veces el LSN). Estos hallazgos analíticos se observaron fundamentalmente durante las 12 primeras semanas de tratamiento, fueron generalmente asintomáticos y revirtieron con la interrupción de la dosis del medicamento. La mayoría de los pacientes reanudó el tratamiento a una dosis más baja sin recurrencias (ver sección 4.2). Se debe controlar la ALT, la AST y la bilirrubina total en todos los pacientes cada 2 semanas durante los 3 primeros meses de tratamiento y cuando esté clínicamente indicado a partir de entonces. Si se observan elevaciones de la ALT y/o la AST de grado 2 ó superior, se debe controlar semanalmente a los pacientes hasta que los valores retornen al grado 1 ó inferior.
Diarrea/c olitis
Se produjeron casos de colitis grave relacionada con el medicamento, relativamente tarde (meses) con respecto al inicio de la terapia, a veces con agravación rápida, pero se resolvieron en pocas semanas con la interrupción de la dosis del medicamento y tratamiento sintomático adicional (p. ej., medicamentos antinflamatorios, como budesonida entérica).
La experiencia del tratamiento de pacientes con antecedentes de enfermedad inflamatoria intestinal es muy limitada.
Neumonitis
Se han notificado casos de neumonitis en ensayos clínicos con idelalisib. Se debe evaluar una posible neumonitis medicamentosa en los pacientes que presentan acontecimientos pulmonares graves que no responden al tratamiento antimicrobiano convencional. Si se sospecha neumonitis, se debe interrumpir la administración de idelalisib y tratar al paciente en consecuencia. Se debe interrumpir el tratamiento en caso de neumonitis sintomática moderada o grave.
Síndrome de Stevens-Johnson y necrólisis epidérmica tóxica
Se han notificado casos de síndrome de Stevens-Johnson (SSJ) y necrólisis epidérmica tóxica (NET) con desenlace mortal cuando idelalisib se administró de forma concomitante con otros medicamentos asociados a estos síndromes. Si se sospecha SSJ o NET, se debe interrumpir inmediatamente la administración de idelalisib y tratar al paciente en consecuencia.
Inductores de CYP3A
La exposición a idelalisib se puede ver reducida cuando se administra de forma concomitante con inductores de CYP3A como la rifampicina, la fenitoína, la hierba de San Juan (Hypericum perforatum) o la carbamazepina. Como una reducción de la concentración plasmática de idelalisib puede ocasionar una disminución de la eficacia, se debe evitar la administración concomitante de Zydelig con inductores moderados o potentes de CYP3A (ver sección 4.5).
Sustratos de CYP3A
El principal metabolito de idelalisib, GS-563117, es un potente inhibidor de CYP3A4. Por tanto, idelalisib tiene el potencial de interactuar con medicamentos metabolizados por CYP3A, lo que puede dar lugar a un aumento de las concentraciones séricas del otro medicamento (ver sección 4.5). Cuando se administra idelalisib de forma concomitante con otros medicamentos, se debe consultar la ficha técnica o resumen de las características del producto del otro medicamento para conocer las recomendaciones acerca de la administración concomitante con inhibidores de CYP3A4. Se debe evitar el tratamiento concomitante de idelalisib con sustratos de CYP3A con reacciones adversas graves y/o potencialmente mortales (p. ej. alfuzosina, amiodarona, cisaprida, pimozida, quinidina, ergotamina, dihidroergotamina, quetiapina, lovastatina, simvastatina, sildenafilo, midazolam, triazolam) y utilizar si es posible medicamentos alternativos menos sensibles a la inhibición por CYP3A4.
Insuficiencia hepática
Se recomienda intensificar la vigilancia de las reacciones adversas en pacientes con insuficiencia hepática, ya que se espera que la exposición aumente en esta población, en particular en pacientes con insuficiencia hepática grave. No se incluyeron pacientes con insuficiencia hepática grave en los ensayos clínicos de idelalisib. Se recomienda precaución cuando se administre Zydelig en esta población.
Hepatitis crónica
No se ha estudiado idelalisib en pacientes con hepatitis crónica activa incluyendo hepatitis vírica. Se debe actuar con precaución cuando se administre Zydelig en pacientes con hepatitis activa.
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar métodos anticonceptivos muy efectivos durante el tratamiento con idelalisib y durante el mes siguiente a su interrupción (ver sección 4.6). Las mujeres que usan anticonceptivos hormonales deben añadir un método de barrera como segundo método anticonceptivo, ya que actualmente se desconoce si idelalisib puede reducir la efectividad de los anticonceptivos hormonales.
4.5 Interacción con otros medicamentos y otras formas de interacción
Idelalisib se metaboliza principalmente a través de una aldehído oxidasa y, en menor grado, de CYP3A y una glucuronidación (UGT1A4). Su principal metabolito es GS-563117, que carece de actividad farmacológica. Idelalisib y GS-563117 son sustratos de P-gp y BCRP.
Efecto de otros medicamentos sobre la farmacocinética de idelalisib
Inductores de CYP3A
En un ensayo clínico de interacciones medicamentosas se constató que la administración concomitante de una dosis única de 150 mg de idelalisib con rifampicina (un potente inductor de CYP3A) generaba una reducción de aproximadamente el 75 % en la AUCinf de idelalisib. Se debe evitar la administración concomitante de Zydelig con inductores moderados o potentes de CYP3A como la rifampicina, la fenitoína, la hierba de San Juan o la carbamazepina, ya que esto puede ocasionar una disminución de la eficacia (ver sección 4.4).
Inhibidores de CYP3A/P-gp
En un ensayo clínico de interacciones medicamentosas se constató que la administración concomitante de una dosis única de 400 mg de idelalisib con 400 mg una vez al día de ketoconazol (un potente inhibidor de CYP3A, P-gp y BCRP) generaba un aumento del 26 % en la Cmax y un aumento del 79 % en la AUCinf de idelalisib. No se considera necesario efectuar un ajuste inicial de la dosis de idelalisib cuando se administra con inhibidores de CYP3A/P-gp, pero se recomienda intensificar la vigilancia de las reacciones adversas.
Efecto de idelalisib sobre la farmacocinética de otros medicamentos
Sustratos de CYP3A
El principal metabolito de idelalisib, GS-563117, es un potente inhibidor de CYP3A En un ensayo clínico de interacciones medicamentosas se constató que la administración concomitante de idelalisib con midazolam (un sustrato sensible de CYP3A) generaba un aumento de aproximadamente el 140 % en la y de alrededor del 440 % en la AUCinf del midazolam debido a la inhibición de CYP3A por GS-563117. La administración concomitante de idelalisib con sustratos de CYP3A puede aumentar sus exposiciones sistémicas y aumentar o prolongar su actividad terapéutica y sus reacciones adversas. In vitro, la inhibición de CYP3A4 fue irreversible y se espera por tanto que se tarde varios días en regresar a una actividad enzimática normal después de interrumpir la administración de idelalisib.
En la Tabla 1 se enumeran las posibles interacciones entre idelalisib y los medicamentos administrados de forma concomitante que son sustratos de CYP3A (el aumento se indica como “|”). Esta lista no es exhaustiva y solo pretende servir de orientación. En general, se debe consultar la ficha técnica del otro medicamento para conocer las recomendaciones acerca de la administración concomitante con inhibidores de CYP3A4 (ver sección 4.4).
Tabla 1: Interacciones entre idelalisib y otros medicamentos que son sustratos de CYP3A
|
Medicamento |
Efecto esperado de idelalisib sobre los niveles del medicamento |
Recomendación clínica para su administración concomitante con idelalisib |
|
ANTAGONISTAS DE LOS RECEPTORES ADRENÉRGICOS ALFA 1 | ||
|
Alfuzosina |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con alfuzosina. |
|
ANALGESICOS | ||
|
Fentanilo, alfentanilo, metadona, buprenorfina/naloxona |
t concentraciones séricas |
Se recomienda vigilar con atención las reacciones adversas (p. ej., depresión respiratoria, sedación). |
|
Medicamento |
Efecto esperado de idelalisib sobre los niveles del medicamento |
Recomendación clínica para su administración concomitante con idelalisib |
|
antiarrítmicos | ||
|
Amiodarona, quinidina |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con amiodarona o quinidina. |
|
Bepridil, disopiramida, lidocaína |
t concentraciones séricas |
Se recomienda vigilancia clínica. |
|
ANTINEOPLASICOS | ||
|
Inhibidores de la tirosina quinasa como dasatinib y nilotinib, también vincristina y vinblastina |
t concentraciones séricas |
Se recomienda vigilar con atención la tolerancia a estos antineoplásicos. |
|
ANTI COAGULANTES | ||
|
Warfarina |
t concentraciones séricas |
Se recomienda vigilar el cociente internacional normalizado (INR) durante la administración concomitante y después de interrumpir el tratamiento con idelalisib. |
|
ANTICONVULSIVANTES | ||
|
Carbamazepina |
t concentraciones séricas |
Se deben vigilar los niveles de anticonvulsivantes. |
|
ANTIDEPRESIVOS | ||
|
Trazodona |
t concentraciones séricas |
Se recomienda un cuidadoso ajuste de la dosis del antidepresivo y vigilar la respuesta al mismo. |
|
ANTIGOTOSOS | ||
|
Colchicina |
t concentraciones séricas |
Se pueden requerir reducciones de la dosis de colchicina. No se debe administrar idelalisib de forma concomitante con colchicina a pacientes con insuficiencia renal o hepática. |
|
ANTIHIPERTENSIVOS | ||
|
Amlodipino, diltiazem, felodipino, nifedipino, nicardipino |
t concentraciones séricas |
Se recomienda vigilancia clínica del efecto terapéutico y de las reacciones adversas. |
|
ANTINFECCIOSOS | ||
|
Antifúngicos | ||
|
Ketoconazol, itraconazol, posaconazol, voriconazol |
t concentraciones séricas |
Se recomienda vigilancia clínica. |
|
Antimicobacterianos | ||
|
Rifabutina |
t concentraciones séricas |
Se recomienda aumentar la vigilancia de las reacciones adversas asociadas a rifabutina, entre ellas neutropenia y uveítis. |
|
Inhibidores de la proteasa del VHC | ||
|
Boceprevir, telaprevir |
t concentraciones séricas |
Se recomienda vigilancia clínica. |
|
Medicamento |
Efecto esperado de idelalisib sobre los niveles del medicamento |
Recomendación clínica para su administración concomitante con idelalisib |
|
Antibióticos macrólidos | ||
|
Claritromicina, telitromicina |
t concentraciones séricas |
No se requiere ajuste de la dosis de claritromicina en los pacientes con función renal normal o insuficiencia renal leve (aclaramiento de creatinina [CrCl] 60-90 ml/min). Se recomienda vigilancia clínica en los pacientes con CrCl <90 ml/min. En los pacientes con CrCl <60 ml/min, se deben considerar antibacterianos alternativos. Se recomienda vigilancia clínica para la telitromicina. |
|
ANTIPSICOIICOS/NEUROLEPIICOS | ||
|
Quetiapina, pimozida |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con quetiapina o pimozida. Pueden considerarse medicamentos alternativos, como la olanzapina. |
|
ANTAGONISTAS DE LOS RECEPTORES DE LA ENDO |
TELINA | |
|
Bosentano |
t concentraciones séricas |
Hay que actuar con precaución y observar estrechamente a los pacientes en busca de toxicidad relacionada con bosentano. |
|
ALCALOIDES ERGOIICOS | ||
|
Ergotamina, dihidroergotamina |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con ergotamina o dihidroergotamina. |
|
FÁRMACOS ESTIMULANTES DE LA MOTILIDAD GASTROINTESTINAL | ||
|
Cisaprida |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con cisaprida. |
|
GLUCOCORTICOIDES | ||
|
Corticosteroides inhalados/nasales: Budesonida, fluticasona Budesonida oral |
t concentraciones séricas t concentraciones séricas |
Se recomienda vigilancia clínica. Se recomienda vigilancia clínica en busca de un aumento de los signos/síntomas de los efectos de los corticosteroides. |
|
INHIBIDORES DE LA HMG-CoA REDUCTASA | ||
|
Lovastatina, simvastatina Atorvastatina |
t concentraciones séricas t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con lovastatina o simvastatina. Se recomienda vigilancia clínica y se puede considerar una dosis inicial más baja de atorvastatina. Alternativamente, se puede considerar el cambio a pravastatina, rosuvastatina o pitavastatina. |
|
Medicamento |
Efecto esperado de idelalisib sobre los niveles del medicamento |
Recomendación clínica para su administración concomitante con idelalisib |
|
IMMUNOSUPRESORES | ||
|
Ciclosporina, sirolimus, tacrolimus |
t concentraciones séricas |
Se recomienda vigilancia terapéutica. |
|
AGONISTA BETA INHALADO |
3 | |
|
Salmeterol |
t concentraciones séricas |
No se recomienda la administración concomitante de salmeterol e idelalisib. La combinación puede ocasionar un mayor riesgo de acontecimientos adversos cardiovasculares asociados al salmeterol, entre ellos prolongación del QT, palpitaciones y taquicardia sinusal. |
|
INHIBIDORES DE LA FOSFO |
DIESTERASA | |
|
Para la hipertensión arterial pulmonar: | ||
|
Sildenafilo |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con sildenafilo. |
|
Tadalafilo |
t concentraciones séricas |
Hay que actuar con precaución, considerando incluso una reducción de la dosis, cuando se administre tadalafilo de forma concomitante con idelalisib. |
|
Para la disfunción eréctil: | ||
|
Sildenafilo, tadalafilo |
t concentraciones séricas |
Hay que actuar con especial precaución y se puede considerar una reducción de la dosis cuando se prescriba sildenafilo o tadalafilo con idelalisib, con mayor vigilancia de los acontecimientos adversos. |
|
SEDANTES/HIPNÓ TICOS | ||
|
Midazolam (oral), triazolam |
t concentraciones séricas |
No se debe administrar idelalisib de forma concomitante con midazolam (oral) o triazolam. |
|
Buspirona, clorazepato, diazepam, estazolam, flurazepam, zolpidem |
t concentraciones séricas |
Se recomienda vigilar la concentración de los sedantes/hipnóticos y puede considerarse una reducción de la dosis. |
Sustratos de CYP2C8
In vitro, idelalisib inhibió e indujo CYP2C8, pero se desconoce si esto se traduce en algún efecto in vivo sobre los sustratos de CYP2C8. Se recomienda precaución si se utiliza Zydelig junto con medicamentos con índices terapéuticos estrechos que son sustratos de CYP2C8 (paclitaxel).
Sustratos de enzimas inducibles (p. ej., CYP2C9, CYP2C19, CYP2B6y UGT)
In vitro, idelalisib fue un inductor de varias enzimas y no se puede excluir un riesgo de menor exposición y por tanto disminución de la eficacia de los sustratos de enzimas inducibles como CYP2C9, CYP2C19, CYP2B6 y UGT. Se recomienda precaución si se utiliza Zydelig junto con medicamentos con índices terapéuticos estrechos que son sustratos de estas enzimas (warfarina, fenitoína, S-mefenitoína).
Sustratos de BCRP, OATP1B1, OATP1B3y P-gp
La administración concomitante de varias dosis de idelalisib 150 mg dos veces al día a sujetos sanos ocasionó exposiciones similares a rosuvastatina (AUC IC del 90 %: 87, 121) y digoxina (AUC IC del 90 %: 98, 111), lo que sugiere que idelalisib no provoca una inhibición clínicamente relevante de BCRP, OATP1B1/1B3 o P-gp sistémica. No se puede excluir un riesgo de inhibición de la P-gp en el tubo digestivo, que podría ocasionar una mayor exposición a los sustratos sensibles a la P-gp intestinal como dabigatrán etexilato.
Población pediátrica
Los ensayos de interacciones se han realizado solo en adultos.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
De acuerdo con los hallazgos en animales, idelalisib puede causar daños fetales. Las mujeres deben evitar quedarse embarazadas durante el tratamiento con Zydelig y hasta un mes después de su finalización. Por tanto, las mujeres en edad fértil deben utilizar métodos anticonceptivos muy efectivos durante el tratamiento con Zydelig y durante el mes siguiente a su interrupción. Actualmente se desconoce si idelalisib puede reducir la efectividad de los anticonceptivos hormonales y por tanto las mujeres que usan anticonceptivos hormonales deben añadir un método de barrera como segundo método anticonceptivo.
Embarazo
No hay datos o éstos son limitados relativos al uso de idelalisib en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3).
No se recomienda utilizar Zydelig durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Lactancia
Se desconoce si idelalisib y sus metabolitos se excretan en la leche materna.
No se puede excluir el riesgo en recién nacidos/niños.
Se debe interrumpir la lactancia durante el tratamiento con Zydelig.
Fertilidad
No se dispone de datos en humanos acerca del efecto de idelalisib sobre la fertilidad. Los estudios en animales sugieren la posibilidad de efectos perjudiciales de idelalisib sobre la fertilidad y el desarrollo fetal (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Zydelig sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Resumen del perfil de seguridad
La evaluación de las reacciones adversas se basa en un ensayo de fase 3 y siete ensayos de fases 1 y 2. El ensayo 312-0116 de fase 3 fue un ensayo aleatorizado, a doble ciego y controlado con placebo en el que se asignó aleatoriamente en una proporción 1:1 a 220 pacientes con LLC tratada anteriormente a recibir idelalisib + rituximab o placebo + rituximab. En los ensayos de fases 1 y 2 se evaluó la seguridad de idelalisib en 490 pacientes con neoplasias hematológicas malignas, que incluían a 354 sujetos que recibieron idelalisib (cualquier dosis) como medicamento único y a 136 sujetos que recibieron idelalisib en combinación con anticuerpos monoclonales contra CD20.
Durante el tratamiento con idelalisib, las reacciones adversas al medicamento notificadas con más frecuencia se enumeran en la T abla 2.
Tabla de reacciones adversas
En la Tabla 2 se presentan las reacciones adversas al medicamento notificadas con idelalisib solo o en combinación con anticuerpos monoclonales contra CD20. Las reacciones adversas se enumeran por sistema de clasificación de órganos y frecuencia. Las frecuencias se definen del siguiente modo: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000), muy raras (<1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 2: Reacciones adversas al medicamento notificadas en los ensayos clínicos en pacientes con neoplasias hematológicas malignas que recibieron idelalisib
|
Reacción |
Cualquier grado |
Grado >3 |
|
Infecciones e infestaciones | ||
|
Infecciones |
Muy frecuentes |
Muy frecuentes |
|
Trastornos de la sangre y del sistema linfático | ||
|
Neutropenia |
Muy frecuente |
Muy frecuente |
|
Trastornos respiratorios, torácicos y mediastínicos | ||
|
Neumonitis |
Frecuente |
Frecuente |
|
Trastornos gastrointestinales | ||
|
Diarrea/c olitis |
Muy frecuente |
Muy frecuente |
|
Trastornos hepatobiliares | ||
|
Aumento de las transaminasas |
Muy frecuente |
Muy frecuente |
|
Trastornos de la piel y del tejido subcutáneo | ||
|
Exantema* |
Muy frecuente |
Frecuente |
|
Síndrome de Stevens-Johnson / necrólisis epidérmica tóxica |
Rara |
Rara |
|
Trastornos generales y alteraciones en el lugar de administración | ||
|
Pirexia |
Muy frecuente |
Frecuente |
|
Exploraciones complementarias | ||
|
Aumento de los triglicéridos |
Muy frecuente |
Frecuente |
* Incluye los términos preferidos dermatitis exfoliativa, erupción medicamentosa, exantema, exantema eritematoso, exantema generalizado, exantema macular, exantema maculopapular, exantema papular, exantema pruriginoso y exantema exfoliativo.
Descripción de reacciones adversas seleccionadas
Exantema
El exantema fue por lo general leve o moderado y ocasionó la interrupción del tratamiento en el 2 % de los pacientes aproximadamente. En el ensayo 312-0116, se produjo exantema (notificado como dermatitis exfoliativa, erupción medicamentosa, exantema, exantema eritematoso, exantema generalizado, exantema macular, exantema maculopapular, exantema papular y exantema pruriginoso) en el 24,5 % de los sujetos que recibieron idelalisib + rituximab y en el 6,5 % de los sujetos que recibieron placebo + rituximab. De estos, el 3,6 % de los que recibieron idelalisib + rituximab y el 0,9 % de los que recibieron placebo + rituximab presentaron exantema de grado 3, y ningún sujeto sufrió un acontecimiento adverso de grado 4. El exantema se resolvió habitualmente con tratamiento (p. ej., esteroides tópicos y/u orales, difenhidramina) e interrupción de la dosis en los casos graves (ver sección 5.3, fototoxicidad).
Síndrome de Stevens-Johnson y necrólisis epidérmica tóxica (ver sección 4.4)
En raras ocasiones, se han producido casos de SSJ y NET cuando idelalisib se administró de forma concomitante con otros medicamentos asociados a estos síndromes (bendamustina, rituximab, alopurinol y amoxicilina). El SSJ o la NET aparecieron dentro del mes siguiente tras la combinación de medicamentos y se han producido casos con desenlace mortal.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
En caso de sobredosis, se deberá vigilar al paciente por si hay evidencia de toxicidad (ver sección 4.8). El tratamiento de la sobredosis de Zydelig consiste en medidas generales de apoyo, incluida la vigilancia de las constantes vitales, así como la observación del estado clínico del paciente.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: agentes antineoplásicos, otros agentes antineoplásicos, código ATC: L01XX47
Mecanismo de acción
Idelalisib inhibe la fosfatidilinositol 3-quinasa p1105 (PI3K5), que es hiperactiva en las neoplasias malignas de linfocitos B y fundamental para numerosas vías de señalización que impulsan la proliferación, supervivencia, migración y retención de células malignas en los tejidos linfoides y en la médula ósea. Idelalisib es un inhibidor selectivo de la unión del adenosina-5’-trifosfato (ATP) al dominio catalítico de PI3K5, lo que genera una inhibición de la fosforilación del fosfatidilinositol, un segundo mensajero lipídico clave, y la prevención de la fosforilación de Akt (proteína quinasa B).
Idelalisib induce la apoptosis e inhibe la proliferación de las líneas celulares derivadas de los linfocitos B malignos y de las células tumorales primarias. Mediante la inhibición de la señalización a través de los receptores de quimiocinas CXCR4 y CXCR5 inducida por las quimiocinas CXCL12 y CXCL13, respectivamente, idelalisib inhibe la migración y retención de los linfocitos B malignos en el microambiente tumoral, incluidos los tejidos linfoides y la médula ósea.
Efectos farmacodinámicos
Se evaluó el efecto de idelalisib (150 mg y 400 mg) sobre el intervalo QT/QTc en un ensayo cruzado controlado con placebo y con control positivo (moxifloxacino 400 mg) en 40 sujetos sanos. A una dosis de 2,7 veces la dosis máxima recomendada, idelalisib no prolongó el intervalo QT/QTc (es decir, <10 ms).
Eficacia clínica en la leucemia linfocítica crónica
Idelalisib en combinación con inmunoterapia
El ensayo 312-0116 fue un ensayo aleatorizado, a doble ciego y controlado con placebo de fase 3 en 220 sujetos con LLC tratada anteriormente que precisaban tratamiento pero no se consideraban candidatos adecuados para quimioterapia citotóxica. Los sujetos se asignaron aleatoriamente en proporción 1:1 a recibir 8 ciclos de rituximab (primer ciclo en dosis de 375 mg/m2 de superficie corporal [SC], ciclos subsiguientes en dosis de 500 mg/m2 de SC) en combinación con un placebo por vía oral dos veces al día o con idelalisib en dosis de 150 mg administrados dos veces al día hasta la progresión de la enfermedad o la aparición de efectos tóxicos inaceptables.
La mediana de la edad era de 71 años (intervalo: 47 a 92) con un 78,2 % de los pacientes mayores de 65 años; el 65,5 % eran varones y el 90,0 % blancos; el 64,1 % se encontraban en estadio III o IV de Rai y el 55,9 % en estadio C de Binet. La mayoría de los sujetos presentaban factores citogenéticos con pronóstico adverso: el 43,2 % tenían una deleción cromosómica en 17p y/o una mutación de la proteína tumoral 53 (TP53) y el 83,6 % genes no mutados de la región variable de las cadenas pesadas de las inmunoglobulinas (IGHV). La mediana del tiempo transcurrido desde el diagnóstico de LLC hasta la aleatorización era de 8,5 años. Los sujetos presentaban una mediana de 8 en la puntuación de calificación de enfermedad acumulada (Cumulative Illness Rating Score, CIRS). La mediana del número de terapias previas era de 3,0. Casi todos (95,9 %) los sujetos habían recibido anticuerpos monoclonales contra CD20 con anterioridad. El criterio principal de valoración fue la supervivencia sin progresión (SSP). Los resultados de eficacia se resumen en las Tablas 3 y 4. En la Figura 1 se presenta la curva de Kaplan-Meier para la SSP.
En comparación con rituximab + placebo, el tratamiento con idelalisib + rituximab ocasionó mejorías estadísticamente significativas y clínicamente relevantes en el bienestar físico, el bienestar social, el bienestar funcional, así como en las subescalas específicas para la leucemia de los instrumentos de evaluación funcional del tratamiento del cáncer: leucemia (Functional Assessment of Cancer Therapy: Leukaemia, FACT-LEU), y mejorías estadísticamente significativas y clínicamente relevantes en la ansiedad, la depresión y las actividades habituales, medidas con el instrumento EuroCdV de cinco parámetros (EuroQoL Five-Dimensions, EQ-5D).
Tabla 3: Resultados de eficacia del ensayo 312-0116
|
Idelalisib + R N = 110 |
Placebo + R N = 110 | |
|
SSP Mediana (meses) (IC del 95 %) |
19,4 (12,3; NA) |
6,5 (4,0; 7,3) |
|
Razón de riesgo (IC del 95 %) |
0,15 (0,09; 0,25) | |
|
Valor p |
<0,0001 | |
|
TRG* n (%) (IC del 95 %) |
92 (83,6 %) (75,4; 90,0) |
17 (15,5 %) (9,3; 23,6) |
|
Razón de probabilidades (IC del 95 %) |
27,76 (13,40; 57,49) | |
|
Valor p |
<0,0001 | |
|
RGL** n/N (%) (IC del 95 %) |
102/106 (96,2 %) (90,6; 99,0) 7/104 (6,7 %) (2,7; 13,4) | |
|
Razón de probabilidades (IC del 95 %) |
225,83 (65,56; 777,94) | |
|
Valor p |
<0,0001 | |
|
SGA Mediana (meses) (IC del 95 %) |
NA (NA; NA) |
20,8 (14,8; NA) |
|
Razón de riesgo (IC del 95 %) |
0,34 (0,19; 0,60) | |
|
Valor p |
0,0001 | |
IC: intervalo de confianza; R: rituximab; n: número de sujetos con respuesta; N: número de sujetos por grupo; NA: no alcanzada. Los análisis de la SSP, tasa de respuesta global (TRG) y tasa de respuesta de los ganglios linfáticos (RGL) se basaron en la evaluación de un comité de revisión independiente (CRI).
*TRGdefinida como el porcentaje de sujetos que alcanzaron una respuesta completa (RC) o una respuesta parcial (RP) de acuerdo con los criterios de respuesta de la National Comprehensive Cancer Network (NCCN) de 2013 y de Cheson (2012).
* *RGL definida como el porcentaje de sujetos que alcanzaron una reducción >50 % en la suma de los productos de los diámetros perpendiculares máximos de las lesiones índice. En este análisis solo se incluyó a los sujetos que tenían evaluaciones tanto en la situación basal como en >1 ocasión valorable posterior a la situación basal.
A El análisis de la supervivencia global (SG) incluye datos de sujetos que recibieron placebo + R en el ensayo 312-0116 y posteriormente recibieron idelalisib en un ensayo de extensión, basado en un análisis por intención de tratar.
Tabla 4: Resumen de la SSP y de las tasas de respuesta en los subgrupos predefinidos del ensayo 312-0116
|
Idelalisib + R |
Placebo + R | |
|
Deleción en 17p/mutación de TP53 |
N = 46 |
N = 49 |
|
Mediana de SSP (meses) (IC del 95 %) |
NA (12,3; NA) |
4,0 (3,7; 5,7) |
|
Razón de riesgo (IC del 95 %) |
0,13 (0,07; 0,27) | |
|
TRG (IC del 95 %) |
84,8 % (71,1; 93,7) |
12,2 % (4,6; 24,8) |
|
IGHV no mutados |
N = 91 |
N = 93 |
|
Mediana de SSP (meses) (IC del 95 %) |
19,4 (13,9; NA) |
5,6 (4,0; 7,2) |
|
Razón de riesgo (IC del 95 %) |
0,14 (0,08; 0,23) | |
|
TRG (IC del 95 %) |
82,4 % (73,0; 89,6) |
15,1 % (8,5; 24,0) |
|
Edad >65 años |
N = 89 |
N = 83 |
|
Mediana de SSP (meses) (IC del 95 %) |
19,4 (12,3; NA) |
5,7 (4,0; 7,3) |
|
Razón de riesgo (IC del 95 %) |
0,14 (0,08; 0,25) | |
|
TRG (IC del 95 %) |
84,3 % (75,0; 91,1) |
16,9 % (9,5; 26,7) |
IC: intervalo de confianza; R: rituximab; N: número de sujetos por grupo; NA: no alcanzada
Figura 1: Curva de Kaplan-Meier de la SSP del ensayo 312-0116 (población por intención de tratar)
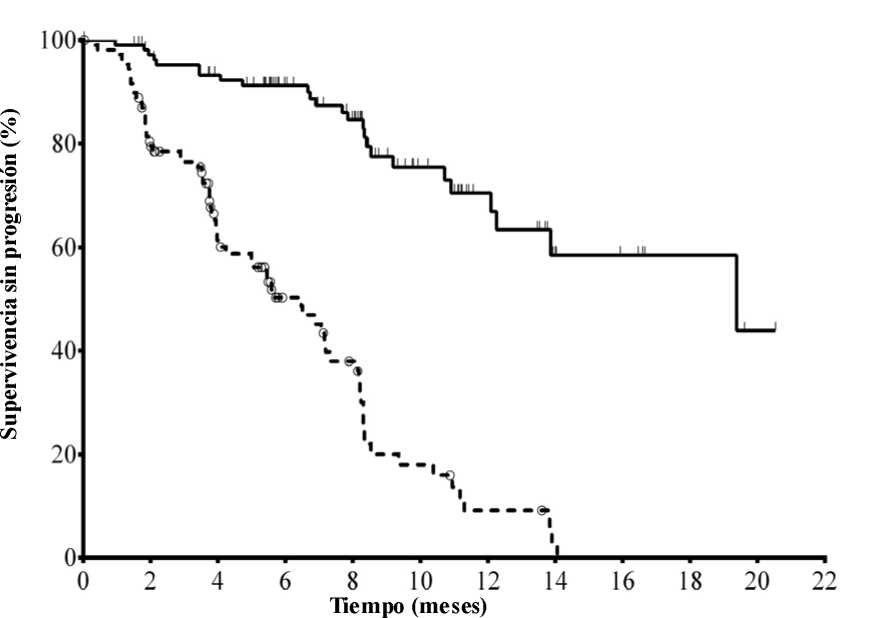
N° en riesgo (acontecimientos)
Idelalisib + R 110 (0) 101 (3) 93 (7) 73 (9) 59 (14) 31 (19) 20 (21) 9 (24) 7 (24) 4 (24) 1 (25) 0 (25) Placebo + R 110 (0) 84 (21) 48 (38) 29 (46) 20 (53) 9 (63) 4 (67) 1 (69) 0 (70) 0 (70) 0 (70) 0 (70)
Línea continua: idelalisib + R (N = 110), línea discontinua: placebo + R(N = 110)
R: rituximab; N: número de sujetos por grupo
El análisis de la SSP se basó en la evaluación de un CRI. Para los sujetos del grupo tratado con placebo + R, el resumen incluye datos hasta la primera dosificación de idelalisib en el ensayo de extensión. Mediana de la SSP: idelalisib + R = 19,4 meses, placebo + R = 6,5 meses. Razón de riesgo = 0,15; IC del 95 % (0,09; 0,24);
p <0,0001.
En el ensayo 101-08/99 se reclutó a 64 sujetos con LLC sin tratamiento previo, incluidos 5 sujetos con linfoma linfocítico de células pequeñas (LLCP). Los sujetos recibieron idelalisib en dosis de 150 mg dos veces al día y rituximab en dosis de 375 mg/m2 de SC semanalmente. La TRG fue del 96,9 %, con 12 RC (18,8 %) y 50 RP (78,1 %), entre ellos 3 RC y 6 RP en sujetos con deleción en 17p y/o mutación de TP53 y 2 RC y 34 RP en sujetos con IGHV no mutados. No se ha alcanzado la mediana de la duración de la respuesta (DR).
Eficacia clínica en el linfoma folicular
La seguridad y eficacia de idelalisib se evaluó en un ensayo clínico multicéntrico de un solo grupo (ensayo 101-09) realizado en 125 sujetos con linfoma no-Hodgkin de linfocitos B indolente (LNHi, que incluye: LF, n = 72; LLCP, n = 28; linfoma linfoplasmocítico/macroglobulinemia de Waldenstrom [LLP/MW], n = 10; y linfoma de zona marginal [LZM], n = 15). Todos los sujetos eran resistentes al tratamiento con rituximab y 124 de 125 sujetos eran resistentes al tratamiento con al menos un agente alquilante. Ciento doce (89,6 %) sujetos habían sido resistentes a su última pauta terapéutica antes de la entrada en el ensayo.
De los 125 sujetos reclutados, 80 (64 %) eran varones, la mediana de la edad era de 64 años (intervalo: 33 a 87) y 110 (89 %) eran blancos. Los sujetos recibieron 150 mg de idelalisib por vía oral dos veces al día hasta que presentaron datos indicativos de progresión de la enfermedad o de la aparición de efectos tóxicos inaceptables.
El criterio principal de valoración fue la TRG, definida como el porcentaje de sujetos que alcanzaron una RC o una RP (de acuerdo con los criterios de respuesta revisados para el linfoma maligno [Cheson]) y, para los sujetos con macroglobulinemia de Waldenstrom, una respuesta menor (RM) (de acuerdo con la evaluación de la respuesta para la macroglobulinemia de Waldenstrom [Owen]). La DR fue un criterio de valoración secundario y se definió como el tiempo transcurrido desde la primera respuesta documentada (RC, RP o RM) hasta la primera documentación de progresión de la enfermedad o muerte por cualquier causa. Los resultados de eficacia se resumen en la T abla 5.
Tabla 5: Resumen de la respuesta en los sujetos con LF tratados con idelalisib (evaluación por parte de un CRI)
|
Característica |
Sujetos del ensayo n (%) |
|
TRG (linfoma folicular)* |
39 (54,2) |
|
IC del 95 % |
42,0 - 66,0 |
|
TRG (todos los sujetos)* |
71 (56,8) |
|
IC del 95 % |
47,6 - 65,6 |
|
Categoría de la respuesta (linfoma folicular)* RC |
6 (8,3) |
|
RP |
33 (45,8) |
IC: intervalo de confianza; n: número de sujetos con respuesta
* Respuestasegún la valoración de un comité de revisión independiente (CRI), donde TRG= respuesta completa (RC) + respuesta parcial (RP).
La mediana de la DR para todos los sujetos fue de 12,5 meses (12,5 meses para los sujetos con LLCP, y no alcanzada para los sujetos con LF, LLP/MW y LZM). Entre los 122 sujetos con ganglios linfáticos medibles tanto en la situación basal como posteriormente, 67 (54,9 %) alcanzaron una reducción >50 % con respecto a la situación basal en la suma de los productos de los diámetros (SPD) de las lesiones índice. De los sujetos que no respondieron, 10 (8,0 %) presentaban progresión de la enfermedad como mejor respuesta y 2 (1,6 %) no eran evaluables. La mediana de la SG, incluyendo el seguimiento a largo plazo de los 125 sujetos, fue de 20,3 meses.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con idelalisib en uno o más grupos de la población pediátrica en el tratamiento de las neoplasias de linfocitos B maduros (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Tras la administración oral de una dosis única de idelalisib, se observaron concentraciones plasmáticas máximas 2 a 4 horas después de la administración con alimentos y tras 0,5 a 1,5 horas después en condiciones de ayuno.
Tras la administración de 150 mg de idelalisib dos veces al día, la media (intervalo) de la Cmax y del AUC en estado estacionario fue de 1.953 (272; 3.905) ng/ml y 10.439 (2.349; 29.315) ng»h/ml para idelalisib y de 4.039 (669; 10.897) ng/ml y 39.744 (6.002; 119.770) ng*h/ml para GS-563117, respectivamente. Las exposiciones (Cmax y AUC) a idelalisib son aproximadamente proporcionales a la dosis entre 50 mg y 100 mg y menores que la proporcional a la dosis por encima de 100 mg.
Efectos de los alimentos
En comparación con las condiciones de ayuno, la administración de una formulación inicial de idelalisib en cápsulas con una comida de alto contenido graso no produjo cambios en la Cmax, pero generó un aumento del 36 % en la AUCW media. Idelalisib se puede administrar sin tener en cuenta la ingesta de alimentos.
Distribución
Idelalisib se une en un 93 % a 94 % a las proteínas plasmáticas humanas en las concentraciones observadas clínicamente. La relación media de las concentraciones sanguíneas/plasmáticas fue de aproximadamente 0,5. El volumen de distribución aparente de idelalisib (media) fue de unos 96 l.
Biotransformación
Idelalisib se metaboliza principalmente a través de una aldehído oxidasa y, en menor grado, de CYP3A y UGT1A4. Su principal y único metabolito circulante, GS-563117, es inactivo frente a PI3K5.
Eliminación
La semivida de eliminación terminal de idelalisib fue de 8,2 (intervalo: 1,9; 37,2) horas y el aclaramiento aparente de idelalisib fue de 14,9 (intervalo: 5,1; 63,8) l/h tras la administración oral de 150 mg de idelalisib dos veces al día. Tras una dosis única por vía oral de 150 mg de idelalisib marcado con [14C], se excretó aproximadamente un 78 % y un 15 % con las heces y la orina, respectivamente. Idelalisib sin modificar constituyó el 23 % de la radiactividad total recuperada en la orina durante 48 horas y el 12 % de la radiactividad total recuperada en las heces durante 144 horas.
Datos de interacción in vitro
Los datos in vitro indicaron que idelalisib no es un inhibidor de las enzimas metabolizadoras CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A o UGT1A1 ni de los transportadores OAT1, OAT3 u OCT2.
GS-563117 no es un inhibidor de las enzimas metabolizadoras CYP1A2, CYP2B6, CYP2C8,
CYP2C9, CYP2C19, CYP2D6 o UGT1A1 ni de los transportadores P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3 u OCT2.
Poblaciones especiales
Sexo y raza
Los análisis farmacocinéticos poblacionales indicaron que el sexo y la raza no ejercen un efecto clínicamente relevante sobre las exposiciones a idelalisib o a GS-563117.
Pacientes de edad avanzada
Los análisis farmacocinéticos poblacionales indicaron que la edad no ejerce un efecto clínicamente relevante sobre las exposiciones a idelalisib o a GS-563117, incluidos los sujetos de edad avanzada (65 años o más) en comparación con los sujetos más jóvenes.
Insuficiencia renal
Se realizó un ensayo de la farmacocinética y la seguridad de idelalisib en sujetos sanos y sujetos con insuficiencia renal grave (CrCl estimado 15 a 29 ml/min). Tras una dosis única de 150 mg, no se observaron cambios clínicamente relevantes en las exposiciones a idelalisib o a GS-563117 en los sujetos con insuficiencia renal grave en comparación con los sujetos sanos.
Insuficiencia hepática
Se realizó un ensayo de la farmacocinética y la seguridad de idelalisib en sujetos sanos y sujetos con insuficiencia hepática moderada (clase B de Child-Pugh) o grave (clase C de Child-Pugh). Tras una dosis única de 150 mg, el AUC de idelalisib (total, es decir, unido más no unido) fue aproximadamente un 60 % mayor en la insuficiencia hepática moderada o grave en comparación con controles emparejados. El AUC de idelalisib (no unido), tras considerar las diferencias en la unión a proteínas, fue aproximadamente un 80 % (1,8 veces) mayor en la insuficiencia moderada y aproximadamente un 152 % (2,5 veces) mayor en la insuficiencia grave en comparación con controles emparejados.
Población pediátrica
No se ha establecido la farmacocinética de idelalisib en los pacientes pediátricos (ver sección 4.2).
5.3 Datos preclínicos sobre seguridad
Toxicidad a dosis repetidas
Idelalisib indujo una disminución del número de linfocitos en el bazo, el timo, los ganglios linfáticos y el tejido linfoide asociado al tubo digestivo. En general, las áreas dependientes de los linfocitos B se vieron más afectadas que las dependientes de los linfocitos T. En ratas, idelalisib tiene el potencial de inhibir las respuestas de anticuerpos dependientes de los linfocitos T. No obstante, idelalisib no inhibió la respuesta normal del huésped a Staphylococcus aureus y no exacerbó el efecto mielosupresor de la ciclofosfamida. No se considera que idelalisib tenga una actividad inmunosupresora amplia.
Idelalisib indujo alteraciones inflamatorias tanto en ratas como en perros. En estudios de hasta 4 semanas en ratas y perros, se observó necrosis hepática a 7 y 5 veces, respectivamente, la exposición en humanos basada en el AUC. Las elevaciones de las transaminasas séricas se correlacionaban con necrosis hepáticas en perros, pero no se observaron en las ratas. No se observó insuficiencia hepática ni elevaciones crónicas de las transaminasas en ratas ni en perros en estudios de 13 semanas de duración o más.
Genotoxicidad
Idelalisib no indujo mutaciones en el ensayo de mutagénesis microbiana (Ames), no fue clastogénico en el ensayo de aberraciones cromosómicas in vitro con linfocitos de sangre periférica humana y no fue genotóxico en el estudio de micronúcleos de rata in vivo.
Carcinogenicidad
No se han realizado estudios de carcinogenicidad con idelalisib.
T oxicidad para la reproducción y el desarrollo
En un estudio de desarrollo embriofetal en ratas, se observó un aumento de la pérdida post-implantación, malformaciones (ausencia de vértebras caudales y, en algunos casos, también de vértebras sacras), cambios esqueléticos y menores pesos corporales fetales. Se observaron malformaciones a exposiciones a partir de 12 veces la exposición en humanos basadas en el AUC. No se investigaron los efectos sobre el desarrollo embriofetal en una segunda especie.
Se observó degeneración de los túbulos seminíferos testiculares en estudios a dosis repetidas de 2 a 13 semanas en perros y ratas, pero no en estudios de 26 semanas de duración o más. En un estudio de fertilidad masculina en ratas, se observaron reducciones en el peso de los epidí dimos y de los testículos, pero no se observaron efectos adversos sobre los parámetros de apareamiento y fertilidad ni degeneración o pérdida de la espermatogénesis. La fertilidad femenina no se vio afectada en las ratas.
Fototoxicidad
La evaluación del potencial de fototoxicidad en la línea celular de fibroblastos embrionarios murinos BALB/c 3T3 no fue concluyente para idelalisib debido a citotoxicidad en el ensayo in vitro. El principal metabolito, GS-563117, puede potenciar la fototoxicidad cuando las células se exponen simultáneamente a luz UVA. Existe un riesgo potencial de que idelalisib, a través de su principal metabolito, GS-563117, pueda provocar fotosensibilidad en los pacientes tratados.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido Celulosa microcristalina Hidroxipropilcelulosa (E463)
Croscarmelosa sódica Glic olato de almidón sódico Estearato de magnesio
Recubrimiento pelicular Alcohol polivinílico (E1203)
Macrogol 3350 (E1521)
Dióxido de titanio (E171)
Talco (E553B)
Óxido de hierro rojo (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Frasco de polietileno de alta densidad (HDPE), tapado con un cierre de seguridad de polipropileno a prueba de niños, que contiene 60 comprimidos recubiertos con película y un relleno de poliéster.
Cada caja contiene 1 frasco.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/938/002
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 18/septiembre/2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y
USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
Nombre y dirección del (de los) fabricante(s) responsable(s) de la liberación de los lotes
Gilead Sciences Ireland UC
IDA Business & Technology Park
Carrigtohill
County Cork
Irlanda
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha T écnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará el primer informe periódico de seguridad para este medicamento en un plazo de 8 meses después de la autorización. Posteriormente, el titular de la autorización de comercialización presentará informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, párrafo 7, de la Directiva 2001/83/CE y publicados en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo
acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de
Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden
presentar conjuntamente.
Obligación de llevar a cabo medidas post-autorización
El TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas
|
Descripción |
Fecha límite |
|
El solicitante debe presentar el informe final del ensayo de extensión de fase 3 GS-US-312-0117, para evaluar la eficacia y seguridad de idelalisib (GS-1101) en combinación con rituximab para la LLC tratada anteriormente. Se deben presentar actualizaciones de SSP, SG y duración de la respuesta para los pacientes con o sin deleción en 17p/mutación de TP53 y para la población completa. |
31 de diciembre de 2017 |
|
El solicitante debe presentar el informe final del ensayo de fase 2 101-09 para evaluar la eficacia y seguridad de idelalisib en sujetos con LNH de linfocitos B indolente refractario al rituximab y a los agentes alquilantes. Se deben presentar actualizaciones de los resultados de seguridad y eficacia, entre ellos la supervivencia global, y actualizaciones de los análisis de sujetos con linfopenia en la situación basal. |
31 de diciembre de 2016 |
|
El solicitante debe presentar el informe final del ensayo de extensión 101-99. |
30 de septiembre de 2017 |
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Zydelig 100 mg comprimidos recubiertos con película Idelalisib
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 100 mg de idelalisib.
3. LISTA DE EXCIPIENTES
Contiene amarillo anaranjado FCF (E110); ver el prospecto para obtener más información.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Comprimidos recubiertos con película 60 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/938/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Zydelig 100 mg
1. NOMBRE DEL MEDICAMENTO
Zydelig 100 mg comprimidos recubiertos con película Idelalisib
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 100 mg de idelalisib.
3. LISTA DE EXCIPIENTES
Contiene amarillo anaranjado FCF (E110); ver el prospecto para obtener más información.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Comprimidos recubiertos con película 60 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/938/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
1. NOMBRE DEL MEDICAMENTO
Zydelig 150 mg comprimidos recubiertos con película Idelalisib
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 150 mg de idelalisib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Comprimidos recubiertos con película 60 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/938/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Zydelig 150 mg
1. NOMBRE DEL MEDICAMENTO
Zydelig 150 mg comprimidos recubiertos con película Idelalisib
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 150 mg de idelalisib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Comprimidos recubiertos con película 60 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/938/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
B. PROSPECTO
Prospecto: información para el paciente
Zydelig 100 mg comprimidos recubiertos con película
Idelalisib
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Zydelig y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Zydelig
3. Cómo tomar Zydelig
4. Posibles efectos adversos
5. Cons ervación de Zydelig
6. Contenido del envase e información adicional
1. Qué es Zydelig y para qué se utiliza
Zydelig es un medicamento contra el cáncer que contiene el principio activo idelalisib. Actúa bloqueando los efectos de una enzima que participa en la multiplicación y supervivencia de ciertos glóbulos blancos llamados linfocitos. Como esta enzima está excesivamente activada en ciertos glóbulos blancos cancerosos, al bloquearla, Zydelig eliminará células cancerosas y reducirá su número.
Zydelig se puede utilizar para el tratamiento de dos cánceres diferentes:
La leucemia linfocítica crónica
La leucemia linfocítica crónica (LLC) es un cáncer de un tipo de glóbulos blancos llamados linfocitos B. En esta enfermedad, los linfocitos se multiplican con demasiada rapidez y viven demasiado tiempo, así que hay demasiados circulando en la sangre.
En la LLC, se puede continuar el tratamiento con Zydelig en combinación con otro medicamento llamado rituximab en pacientes que tienen ciertos factores de alto riesgo y ya están tomando Zydelig. En la LLC, también se puede utilizar en combinación con rituximab en pacientes cuyo cáncer ha reaparecido después de al menos un tratamiento anterior.
El linforma folicular
El linfoma folicular (LF) es un cáncer de un tipo de glóbulos blancos llamados linfocitos B. En el linforma folicular, los linfocitos B se multiplican con demasiada rapidez y viven demasiado tiempo, así que hay demasiados en los ganglios linfáticos. En el LF, Zydelig se utiliza por sí solo en los pacientes cuyo cáncer no ha respondido a dos tratamientos previos contra el cáncer.
2. Qué necesita saber antes de empezar a tomar Zydelig
No tome Zydelig
• si es alérgico a idelalisib o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
^ Consulte a su médico si este es su caso.
Advertencias y precauciones
Consulte a su médico antes de empezar a tomar Zydelig. Informe a su médico:
• si padece problemas hepáticos
• si padece cualquier otra enfermedad o dolencia (especialmente una infección o fiebre)
Se han producido infecciones graves y mortales en pacientes tratados con Zydelig. Debe tomar un medicamento adicional que le proporcionará su médico mientras esté tomando Zydelig para prevenir un tipo de infección. Su médico le vigilará en busca de signos de infección. Informe a su médico inmediatamente si se pone enfermo (especialmente si tiene fiebre, tos o dificultades para respirar) mientras está tomando Zydelig.
Necesitará hacerse análisis de sangre periódicos antes y durante el transcurso del tratamiento con Zydelig. Esto es para comprobar que no tiene una infección, que el hígado funciona correctamente y que sus recuentos sanguíneos son normales. Si es necesario, su médico puede tomar la decisión de suspender el tratamiento durante algún tiempo, antes de iniciarlo de nuevo a la misma dosis o a una dosis más baja. Su médico también puede tomar la decisión de suspender definitivamente el tratamiento con Zydelig.
Zydelig puede provocar diarrea grave. Informe a su médico de inmediato al primer síntoma de diarrea.
Zydelig puede provocar inflamación de los pulmones. Informe a su médico de inmediato:
• si padece una tos nueva o un empeoramiento de la tos
• si padece falta de aliento o dificultad para respirar
Se han comunicado enfermedades de la piel ampollosas graves en algunas personas tratadas con Zydelig mientras también estaban recibiendo otros medicamentos que se sabe que causan estas enfermedades potencialmente mortales. La formación de ampollas también puede afectar al revestimiento de la boca, los genitales y/o los ojos. El desprendimiento de la piel puede causar una infección grave. Informe a su médico de inmediato:
• si presenta enrojecimiento y formación de ampollas en la piel
• si presenta hinchazón y formación de ampollas en el revestimiento de la boca, los genitales y/o los ojos
Niños y adolescentes
No le dé este medicamento a niños y adolescentes menores de 18 años porque no se ha estudiado en este grupo de edad.
Toma de Zydelig con otros medicamentos
Zydelig no se debe usar con otros medicamentos, a menos que su médico le haya indicado que es seguro hacerlo.
Informe a su médico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento. Esto es extremadamente importante, ya que utilizar más de un medicamento al mismo tiempo puede potenciar o debilitar sus efectos.
Tomar Zydelig con ciertos medicamentos puede hacer que no funcionen adecuadamente o que sus
efectos adversos empeoren. En particular, informe a su médico si está tomando alguno de los
siguientes:
• alfuzosina, un medicamento utilizado para tratar el aumento de tamaño de la próstata
• dabigatrán, warfarina, medicamentos utilizados para hacer menos espesa la sangre
• amiodarona, bepridil, disopiramida, lidocaína, quinidina, medicamentos utilizados para tratar problemas cardiacos
• dihidroergotamina, ergometrina, medicamentos utilizados para tratar las migrañas
• cisaprida, un medicamento utilizado para aliviar ciertos problemas del estómago
• pimozida, un medicamento utilizado para tratar pensamientos o sentimientos anómalos
• midazolam, triazolam, cuando se toman por la boca para ayudar a dormir y/o aliviar la ansiedad
• quetiapina, un medicamento utilizado para tratar la esquizofrenia, el trastorno bipolar y la depresión mayor
• amlodipino, diltiazem, felodipino, nicardipino, nifedipino, medicamentos utilizados para tratar la hipertensión arterial y los problemas cardiacos
• bosentano, un medicamento utilizado para tratar la hipertensión arterial pulmonar
• sildenafilo, tadalafilo, medicamentos utilizados para tratar la impotencia y la hipertensión pulmonar, una enfermedad del pulmón que dificulta la respiración
• budesonida, fluticasona, medicamentos utilizados para tratar la alergia al polen y el asma, y salmeterol, utilizado para tratar el asma
• rifabutina, un medicamento utilizado para tratar infecciones bacterianas, entre ellas la tuberculosis
• itraconazol, ketoconazol, pos aconazol, voriconazol, medic amentos utilizados para tratar infecciones fúngicas (hongos)
• boceprevir, telaprevir, medicamentos utilizados para tratar la hepatitis C
• carbamazepina, S-mefenitoína, fenitoína, medicamentos utilizados para prevenir las crisis convulsivas
• rifampicina, un medicamento utilizado para prevenir y tratar la tuberculosis y otras infecciones
• hierba de San Juan (Hypericumperforatum), un remedio a base de plantas utilizado contra la depresión y la ansiedad
• alfentanilo, fentanilo, metadona, buprenorfina/naloxona, medicamentos utilizados para aliviar el dolor
• ciclosporina, sirolimus, tacrolimus, medicamentos utilizados para controlar la respuesta inmunitaria del cuerpo después de un trasplante
• colchicina, un medicamento utilizado para tratar la gota
• trazodona, un medicamento utilizado para tratar la depresión
• buspirona, clorazepato, diazepam, estazolam, flurazepam, zolpidem, medicamentos utilizados para tratar los trastornos del sistema nervioso
• dasatinib, nilotinib, paclitaxel, vinblastina, vincristina, medicamentos utilizados para tratar el cáncer
• anticonceptivos hormonales orales o implantados, utilizados para prevenir el embarazo
• claritromicina, telitromicina, medicamentos utilizados para tratar infecciones bacterianas
• atorvastatina, lovastatina, simvastatina, medicamentos utilizados para reducir el colesterol
Zydelig se puede prescribir en combinación con otros medicamentos para el tratamiento de la LLC.
Es muy importante que también lea los prospectos suministrados con estos medicamentos.
Pregunte a su médico si tiene alguna duda sobre alguno de sus medicamentos.
Embarazo y lactancia
• Zydelig no se debe utilizar durante el embarazo. No se dispone de información sobre la seguridad de este medicamento en las mujeres embarazadas.
• Utilice un método anticonceptivo fiable para evitar quedarse embarazada mientras esté en tratamiento con Zydelig y durante el mes siguiente a su último tratamiento.
• Zydelig puede hacer que la “píldora” anticonceptiva y los anticonceptivos hormonales implantados funcionen peor. Debe utilizar también un método anticonceptivo de barrera como condones o un DIU mientras toma Zydelig y durante el mes siguiente a su último tratamiento.
• Informe a su médico inmediatamente si se queda embarazada.
No debe dar el pecho mientras toma Zydelig. Si está dando el pecho actualmente, consulte con su
médico antes de iniciar el tratamiento. Se desconoce si el principio activo de Zydelig pasa a la leche
materna.
Conducción y uso de máquinas
Es improbable que Zydelig afecte a su capacidad para conducir o usar máquinas.
Zydelig contiene amarillo anaranjado FCF (E110)
Informe a su médico si es alérgico al amarillo anaranjado FCF (E110). Zydelig contiene amarillo
anaranjado FCF, que puede causar reacciones alérgicas.
3. Cómo tomar Zydelig
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico.
La dosis recomendada es de150 mg por la boca dos veces al día. No obstante, su médico puede reducir esta dosis a 100 mg dos veces al día si experimenta ciertos efectos adversos.
Zydelig puede tomarse acompañado o no de alimentos.
Trague el comprimido entero. No mastique ni machaque el comprimido. Informe a su médico si tiene problemas para tragar los comprimidos.
Si toma más Zydelig del que debe
Si toma accidentalmente una cantidad mayor de Zydelig que la dosis recomendada, puede presentar un mayor riesgo de efectos adversos con este medicamento (ver sección 4, Posibles efectos adversos).
Póngase en contacto con su médico o con el servicio de urgencias más cercano inmediatamente para que le aconsejen. Lleve el frasco y este prospecto consigo para que pueda describir fácilmente lo que ha tomado.
Si olvidó tomar Zydelig
Procure no omitir ninguna dosis de Zydelig. Si omite una dosis y han transcurrido menos de 6 horas, tome la dosis omitida inmediatamente. Después, tome la siguiente dosis de la forma habitual. Si omite una dosis y han transcurrido más de 6 horas, espere y tome la siguiente dosis a la hora habitual.
No deje de tomar Zydelig
No deje de tomar este medicamento a menos que su médico se lo indique.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Algunos efectos adversos podrían ser graves.
DEJE de tomar Zydelig y busque ayuda médica de inmediato si presenta alguno de los siguientes síntomas:
• enrojecimiento y formación de ampollas en la piel
• hinchazón y formación de ampollas en el revestimiento de la boca, los genitales y/o los ojos Otros efectos adversos
Efectos adversos muy frecuentes
(pueden afectar a más de 1 de cada 10 personas)
• diarrea/inflamación del intestino grueso
• erupción
• disminución del número de glóbulos blancos
• infecciones
• fiebre
Efectos adversos frecuentes
(pueden afectar hasta a 1 de cada 10 personas)
• inflamación de los pulmones
Los análisis de sangre también pueden mostrar:
• aumento de la concentración sanguínea de enzimas hepáticas o grasas Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Zydelig
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el frasco y en la caja después de {CAD}. La fecha de caducidad es el último día del mes que se indica.
No requiere condiciones especiales de conservación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Zydelig
• El principio activo es idelalisib. Cada comprimido recubierto con película contiene 100 mg de idelalisib.
• Los demás componentes son:
Núcleo del comprimido:
Celulosa microcristalina, hidroxipropilcelulosa (E463), croscarmelosa sódica, glicolato de almidón sódico, estearato de magnesio.
Recubrimiento pelicular:
Alcohol polivinílico (E1203), macrogol 3350 (E1521), dióxido de titanio (E171), talco (E553B), amarillo anaranjado FCF (E110).
Aspecto del producto y contenido del envase
Los comprimidos recubiertos con película son comprimidos de color naranja y forma ovalada, con “GSI” grabado por un lado y “100” por el otro.
Está disponible el siguiente tamaño de envase: caja exterior con 1 frasco de plástico de 60 comprimidos recubiertos con película.
Titular de la autorización de comercialización
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Reino Unido
Responsable de la fabricación
Gilead Sciences Ireland UC
IDA Business & Technology Park
Carrigtohill
County Cork
Irlanda
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Gilead Sciences Belgium SPRL-BVBA Tél/Tel: + 32 (0) 24 01 35 50
Etarapna
Gilead Sciences International Ltd Tea.: + 44 (0) 20 7136 8820
Ceská republika
Gilead Sciences s.r.o.
Tel: + 420 910 871 986
Danmark
Gilead Sciences Sweden AB Tlf: + 46 (0) 8 5057 1849
Deutschland
Gilead Sciences GmbH Tel: + 49 (0) 89 899890-0
Eesti
Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849
Lietuva
Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849
Luxembourg/Luxemburg
Gilead Sciences Belgium SPRL-BVBA Tél/Tel: + 32 (0) 24 01 35 50
Magyarország
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Malta
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Nederland
Gilead Sciences Netherlands B.V.
Tel: + 31 (0) 20 718 36 98
Norge
Gilead Sciences Sweden AB Tlf: + 46 (0) 8 5057 1849
|
ELLá8a Gilead Sciences EXkác, M. El IE Tr|L: + 30 210 8930 100 |
Osterreich Gilead Sciences GesmbH Tel: + 43 1 260 830 |
|
España Gilead Sciences, S.L. Tel: + 34 91 378 98 30 |
Polska Gilead Sciences Poland Sp. z o.o. Tel: + 48 22 262 8702 |
|
France Gilead Sciences Tél: + 33 (0) 1 46 09 41 00 |
Portugal Gilead Sciences, Lda. Tel: + 351 21 7928790 |
|
Hrvatska Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820 |
Románia Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820 |
|
Ireland Gilead Sciences Ltd Tel: + 44 (0) 1223 897555 |
Slovenija Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820 |
|
Ísland Gilead Sciences Sweden AB Sími: + 46 (0) 8 5057 1849 |
Slovenská republika Gilead Sciences Slovakia s.r.o. Tel: + 421 232 121 210 |
|
Italia Gilead Sciences S.r.l. Tel: + 39 02 439201 |
Suomi/Finland Gilead Sciences Sweden AB Puh/Tel: + 46 (0) 8 5057 1849 |
|
Kúnpo^ Gilead Sciences ELLác M. ENE. Tr|L: + 30 210 8930 100 |
Sverige Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849 |
|
Latvija Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849 |
United Kingdom Gilead Sciences Ltd Tel: + 44 (0) 1223 897555 |
|
Fecha de la última revisión de este prospecto |
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www. ema. europa. eu
Prospecto: información para el paciente
Zydelig 150 mg comprimidos recubiertos con película
Idelalisib
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Zydelig y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Zydelig
3. Cómo tomar Zy delig
4. Posibles efectos adversos
5. Cons ervación de Zydelig
6. Contenido del envase e información adicional
1. Qué es Zydelig y para qué se utiliza
Zydelig es un medicamento contra el cáncer que contiene el principio activo idelalisib. Actúa bloqueando los efectos de una enzima que participa en la multiplicación y supervivencia de ciertos glóbulos blancos llamados linfocitos. Como esta enzima está excesivamente activada en ciertos glóbulos blancos cancerosos, al bloquearla, Zydelig eliminará células cancerosas y reducirá su número.
Zydelig se puede utilizar para el tratamiento de dos cánceres diferentes:
La leucemia linfocítica crónica
La leucemia linfocítica crónica (LLC) es un cáncer de un tipo de glóbulos blancos llamados linfocitos B. En esta enfermedad, los linfocitos se multiplican con demasiada rapidez y viven demasiado tiempo, así que hay demasiados circulando en la sangre.
En la LLC, se puede continuar el tratamiento con Zydelig en combinación con otro medicamento llamado rituximab en pacientes que tienen ciertos factores de alto riesgo y ya están tomando Zydelig. En la LLC, también se puede utilizar en combinación con rituximab en pacientes cuyo cáncer ha reaparecido después de al menos un tratamiento anterior.
El linforma folicular
El linfoma folicular (LF) es un cáncer de un tipo de glóbulos blancos llamados linfocitos B. En el linforma folicular, los linfocitos B se multiplican con demasiada rapidez y viven demasiado tiempo, así que hay demasiados en los ganglios linfáticos. En el LF, Zydelig se utiliza por sí solo en los pacientes cuyo cáncer no ha respondido a dos tratamientos previos contra el cáncer.
2. Qué necesita saber antes de empezar a tomar Zydelig
No tome Zydelig
• si es alérgico a idelalisib o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
^ Consulte a su médico si este es su caso.
Advertencias y precauciones
Consulte a su médico antes de empezar a tomar Zydelig. Informe a su médico:
• si padece problemas hepáticos
• si padece cualquier otra enfermedad o dolencia (especialmente una infección o fiebre)
Se han producido infecciones graves y mortales en pacientes tratados con Zydelig. Debe tomar un medicamento adicional que le proporcionará su médico mientras esté tomando Zydelig para prevenir un tipo de infección. Su médico le vigilará en busca de signos de infección. Informe a su médico inmediatamente si se pone enfermo (especialmente si tiene fiebre, tos o dificultades para respirar) mientras está tomando Zydelig.
Necesitará hacerse análisis de sangre periódicos antes y durante el transcurso del tratamiento con Zydelig. Esto es para comprobar que no tiene una infección, que el hígado funciona correctamente y que sus recuentos sanguíneos son normales. Si es necesario, su médico puede tomar la decisión de suspender el tratamiento durante algún tiempo, antes de iniciarlo de nuevo a la misma dosis o a una dosis más baja. Su médico también puede tomar la decisión de suspender definitivamente el tratamiento con Zydelig.
Zydelig puede provocar diarrea grave. Informe a su médico de inmediato al primer síntoma de diarrea.
Zydelig puede provocar inflamación de los pulmones. Informe a su médico de inmediato:
• si padece una tos nueva o un empeoramiento de la tos
• si padece falta de aliento o dificultad para respirar
Se han comunicado enfermedades de la piel ampollosas graves en algunas personas tratadas con Zydelig mientras también estaban recibiendo otros medicamentos que se sabe que causan estas enfermedades potencialmente mortales. La formación de ampollas también puede afectar al revestimiento de la boca, los genitales y/o los ojos. El desprendimiento de la piel puede causar una infección grave. Informe a su médico de inmediato:
• si presenta enrojecimiento y formación de ampollas en la piel
• si presenta hinchazón y formación de ampollas en el revestimiento de la boca, los genitales y/o los ojos
Niños y adolescentes
No le dé este medicamento a niños y adolescentes menores de 18 años porque no se ha estudiado en este grupo de edad.
Toma de Zydelig con otros medicamentos
Zydelig no se debe usar con otros medicamentos, a menos que su médico le haya indicado que es seguro hacerlo.
Informe a su médico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento. Esto es extremadamente importante, ya que utilizar más de un medicamento al mismo tiempo puede potenciar o debilitar sus efectos.
Tomar Zydelig con ciertos medicamentos puede hacer que no funcionen adecuadamente o que sus
efectos adversos empeoren. En particular, informe a su médico si está tomando alguno de los
siguientes:
• alfuzosina, un medicamento utilizado para tratar el aumento de tamaño de la próstata
• dabigatrán, warfarina, medicamentos utilizados para hacer menos espesa la sangre
• amiodarona, bepridil, disopiramida, lidocaína, quinidina, medicamentos utilizados para tratar problemas cardiacos
• dihidroergotamina, ergometrina, medicamentos utilizados para tratar las migrañas
• cisaprida, un medicamento utilizado para aliviar ciertos problemas del estómago
• pimozida, un medicamento utilizado para tratar pensamientos o sentimientos anómalos
• midazolam, triazolam, cuando se toman por la boca para ayudar a dormir y/o aliviar la ansiedad
• quetiapina, un medicamento utilizado para tratar la esquizofrenia, el trastorno bipolar y la depresión mayor
• amlodipino, diltiazem, felodipino, nicardipino, nifedipino, medicamentos utilizados para tratar la hipertensión arterial y los problemas cardiacos
• bosentano, un medicamento utilizado para tratar la hipertensión arterial pulmonar
• sildenafilo, tadalafilo, medicamentos utilizados para tratar la impotencia y la hipertensión pulmonar, una enfermedad del pulmón que dificulta la respiración
• budesonida, fluticasona, medicamentos utilizados para tratar la alergia al polen y el asma, y salmeterol, utilizado para tratar el asma
• rifabutina, un medicamento utilizado para tratar infecciones bacterianas, entre ellas la tuberculosis
• itraconazol, ketoconazol, pos aconazol, voriconazol, medic amentos utilizados para tratar infecciones fúngicas (hongos)
• boceprevir, telaprevir, medicamentos utilizados para tratar la hepatitis C
• carbamazepina, S-mefenitoína, fenitoína, medicamentos utilizados para prevenir las crisis convulsivas
• rifampicina, un medicamento utilizado para prevenir y tratar la tuberculosis y otras infecciones
• hierba de San Juan (Hypericumperforatum), un remedio a base de plantas utilizado contra la depresión y la ansiedad
• alfentanilo, fentanilo, metadona, buprenorfina/naloxona, medicamentos utilizados para aliviar el dolor
• ciclosporina, sirolimus, tacrolimus, medicamentos utilizados para controlar la respuesta inmunitaria del cuerpo después de un trasplante
• colchicina, un medicamento utilizado para tratar la gota
• trazodona, un medicamento utilizado para tratar la depresión
• buspirona, clorazepato, diazepam, estazolam, flurazepam, zolpidem, medicamentos utilizados para tratar los trastornos del sistema nervioso
• dasatinib, nilotinib, paclitaxel, vinblastina, vincristina, medicamentos utilizados para tratar el cáncer
• anticonceptivos hormonales orales o implantados, utilizados para prevenir el embarazo
• claritromicina, telitromicina, medicamentos utilizados para tratar infecciones bacterianas
• atorvastatina, lovastatina, simvastatina, medicamentos utilizados para reducir el colesterol
Zydelig se puede prescribir en combinación con otros medicamentos para el tratamiento de la LLC.
Es muy importante que también lea los prospectos suministrados con estos medicamentos.
Pregunte a su médico si tiene alguna duda sobre alguno de sus medicamentos.
Embarazo y lactancia
• Zydelig no se debe utilizar durante el embarazo. No se dispone de información sobre la seguridad de este medicamento en las mujeres embarazadas.
• Utilice un método anticonceptivo fiable para evitar quedarse embarazada mientras esté en tratamiento con Zydelig y durante el mes siguiente a su último tratamiento.
• Zydelig puede hacer que la “píldora” anticonceptiva y los anticonceptivos hormonales implantados funcionen peor. Debe utilizar también un método anticonceptivo de barrera como condones o un DIU mientras toma Zydelig y durante el mes siguiente a su último tratamiento.
• Informe a su médico inmediatamente si se queda embarazada.
No debe dar el pecho mientras toma Zydelig. Si está dando el pecho actualmente, consulte con su
médico antes de iniciar el tratamiento. Se desconoce si el principio activo de Zydelig pasa a la leche
materna.
Conducción y uso de máquinas
Es improbable que Zydelig afecte a su capacidad para conducir o usar máquinas.
3. Cómo tomar Zydelig
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico.
La dosis recomendada es de150 mg por la boca dos veces al día. No obstante, su médico puede reducir esta dosis a 100 mg dos veces al día si experimenta ciertos efectos adversos.
Zydelig puede tomarse acompañado o no de alimentos.
Trague el comprimido entero. No mastique ni machaque el comprimido. Informe a su médico si tiene problemas para tragar los comprimidos.
Si toma más Zydelig del que debe
Si toma accidentalmente una cantidad mayor de Zydelig que la dosis recomendada, puede presentar un mayor riesgo de efectos adversos con este medicamento (ver sección 4, Posibles efectos adversos).
Póngase en contacto con su médico o con el servicio de urgencias más cercano inmediatamente para que le aconsejen. Lleve el frasco y este prospecto consigo para que pueda describir fácilmente lo que ha tomado.
Si olvidó tomar Zydelig
Procure no omitir ninguna dosis de Zydelig. Si omite una dosis y han transcurrido menos de 6 horas, tome la dosis omitida inmediatamente. Después, tome la siguiente dosis de la forma habitual. Si omite una dosis y han transcurrido más de 6 horas, espere y tome la siguiente dosis a la hora habitual.
No deje de tomar Zydelig
No deje de tomar este medicamento a menos que su médico se lo indique.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Algunos efectos adversos podrían ser graves.
DEJE de tomar Zydelig y busque ayuda médica de inmediato si presenta alguno de los siguientes síntomas:
• enrojecimiento y formación de ampollas en la piel
• hinchazón y formación de ampollas en el revestimiento de la boca, los genitales y/o los ojos Otros efectos adversos
Efectos adversos muy frecuentes
(pueden afectar a más de 1 de cada 10 personas)
• diarrea/inflamación del intestino grueso
• erupción
• disminución del número de glóbulos blancos
• infecciones
• fiebre
Efectos adversos frecuentes
(pueden afectar hasta a 1 de cada 10 personas)
• inflamación de los pulmones
Los análisis de sangre también pueden mostrar:
• aumento de la concentración sanguínea de enzimas hepáticas o grasas Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Zydelig
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el frasco y en la caja después de {CAD}. La fecha de caducidad es el último día del mes que se indica.
No requiere condiciones especiales de conservación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Zydelig
• El principio activo es idelalisib. Cada comprimido recubierto con película contiene 150 mg de idelalisib.
• Los demás componentes son:
Núcleo del comprimido:
Celulosa microcristalina, hidroxipropilcelulosa (E463), croscarmelosa sódica, glicolato de almidón sódico, estearato de magnesio.
Recubrimiento pelicular:
Alcohol polivinílico (E1203), macrogol 3350 (E1521), dióxido de titanio (E171), talco (E553B), óxido de hierro rojo (E172).
Aspecto del producto y contenido del envase
Los comprimidos recubiertos con película son comprimidos de color rosa y forma ovalada, con “GSI” grabado por un lado y “150” por el otro.
Está disponible el siguiente tamaño de envase: caja exterior con 1 frasco de plástico de 60 comprimidos recubiertos con película.
Titular de la autorización de comercialización
Gilead Sciences International Ltd
Cambridge
CB21 6GT
Reino Unido
Responsable de la fabricación
Gilead Sciences Ireland UC
IDA Business & Technology Park
Carrigtohill
County Cork
Irlanda
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Gilead Sciences Belgium SPRL-BVBA Tél/Tel: + 32 (0) 24 01 35 50
Etarapna
Gilead Sciences International Ltd Tea.: + 44 (0) 20 7136 8820
Ceská republika
Gilead Sciences s.r.o.
Tel: + 420 910 871 986
Danmark
Gilead Sciences Sweden AB Tlf: + 46 (0) 8 5057 1849
Deutschland
Gilead Sciences GmbH Tel: + 49 (0) 89 899890-0
Eesti
Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849
Lietuva
Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849
Luxembourg/Luxemburg
Gilead Sciences Belgium SPRL-BVBA Tél/Tel: + 32 (0) 24 01 35 50
Magyarország
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Malta
Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820
Nederland
Gilead Sciences Netherlands B.V.
Tel: + 31 (0) 20 718 36 98
Norge
Gilead Sciences Sweden AB Tlf: + 46 (0) 8 5057 1849
|
ELLá8a Gilead Sciences EXkác, M. El IE Tr|L: + 30 210 8930 100 |
Osterreich Gilead Sciences GesmbH Tel: + 43 1 260 830 |
|
España Gilead Sciences, S.L. Tel: + 34 91 378 98 30 |
Polska Gilead Sciences Poland Sp. z o.o. Tel: + 48 22 262 8702 |
|
France Gilead Sciences Tél: + 33 (0) 1 46 09 41 00 |
Portugal Gilead Sciences, Lda. Tel: + 351 21 7928790 |
|
Hrvatska Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820 |
Románia Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820 |
|
Ireland Gilead Sciences Ltd Tel: + 44 (0) 1223 897555 |
Slovenija Gilead Sciences International Ltd Tel: + 44 (0) 20 7136 8820 |
|
Ísland Gilead Sciences Sweden AB Sími: + 46 (0) 8 5057 1849 |
Slovenská republika Gilead Sciences Slovakia s.r.o. Tel: + 421 232 121 210 |
|
Italia Gilead Sciences S.r.l. Tel: + 39 02 439201 |
Suomi/Finland Gilead Sciences Sweden AB Puh/Tel: + 46 (0) 8 5057 1849 |
|
Kúnpo^ Gilead Sciences ELLác M. ENE. Tr|L: + 30 210 8930 100 |
Sverige Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849 |
|
Latvija Gilead Sciences Sweden AB Tel: + 46 (0) 8 5057 1849 |
United Kingdom Gilead Sciences Ltd Tel: + 44 (0) 1223 897555 |
|
Fecha de la última revisión de este prospecto |
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www. ema. europa. eu
66