Zyclara 3,75% Crema
ANEXO I
RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Zyclara 3,75% crema
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada sobre contiene 9,375 mg de imiquimod en 250 mg de crema (3,75 %). Cada gramo de crema contiene 37,5 mg de imiquimod.
Excipientes con efecto conocido:
Parahidroxibenzoato de metilo (E 218) 2,0 mg/g crema Parahidroxibenzoato de propilo (E 216) 0,2 mg/g crema Alcohol cetílico 22,0 mg/g crema Alcohol estearílico 31,0 mg/g crema
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Crema.
Crema de color blanco a ligeramente amarillento, de aspecto uniforme.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Zyclara está indicado en el tratamiento tópico de las queratosis actínicas (QA) clínicamente típicas, no hiperqueratósicas, no hipertróficas, visibles o palpables, de todo el rostro o del cuero cabelludo alopécico en adultos inmunocompetentes, cuando otros tratamientos tópicos están contraindicados o son menos apropiados.
4.2 Posología y forma de administración
El tratamiento debe ser iniciado y monitorizado por un médico.
Posología
Zyclara (por aplicación: hasta 2 sobres, 250 mg de crema de imiquimod por sobre) se debe aplicar una vez al día, antes de acostarse, sobre la piel de la zona (área) de tratamiento afectada, durante dos ciclos de tratamiento de dos semanas cada uno, separados por un ciclo de 2 semanas sin tratamiento, o según las instrucciones del médico.
La zona de tratamiento es todo el rostro o el cuero cabelludo alopécico.
Debido a su modo de acción, son previsibles, y en parte frecuentes, las reacciones cutáneas locales en la zona de tratamiento (ver sección 4.4). Se puede establecer un periodo de descanso de varios días si así lo requiere el paciente a causa de las molestias o la gravedad de las reacciones cutáneas locales.
Sin embargo, no se deberá prolongar ninguno de los ciclos de tratamiento de 2 semanas con motivo de dosis omitidas o periodos de descanso.
Durante el tratamiento se puede producir un incremento transitorio del recuento de lesiones de queratosis actínica debido al efecto probable de imiquimod para revelar y tratar lesiones subclínicas. La respuesta al tratamiento no se puede evaluar de forma adecuada hasta la resolución de las reacciones cutáneas locales. El paciente deberá continuar el tratamiento según se le ha prescrito. El tratamiento se deberá continuar durante la totalidad del ciclo terapéutico, aun cuando parezcan haber desaparecido todas las lesiones de queratosis actínica.
El resultado clínico de la terapia tiene que ser determinado tras la regeneración de la piel tratada, aproximadamente 8 semanas después de haber finalizado el tratamiento y en intervalos apropiados posteriores en base a un juicio clínico. Las lesiones que no hayan respondido por completo al tratamiento a las 8 semanas después del segundo ciclo terapéutico deberán ser cuidadosamente reevaluadas y se deberá reconsiderar el tratamiento.
Población pediátrica
No se ha establecido la seguridad y eficacia de imiquimod en queratosis actínicas en niños y adolescentes menores de 18 años. No existen datos disponibles.
Forma de administración
Zyclara es de uso exclusivamente externo. Se debe evitar el contacto con ojos, labios y fosas nasales. La zona de tratamiento no se debe vendar ni ocluir de ninguna forma.
El responsable de la prescripción debe enseñar al paciente la técnica de aplicación correcta para optimizar el beneficio de la terapia de Zyclara.
Zyclara se debe aplicar una vez al día, antes de acostarse, sobre la piel de la zona afectada de tratamiento, y permanecer sobre la piel durante aproximadamente 8 horas. Durante este periodo, se debe evitar tomar duchas o baños. Antes de aplicarse la crema, el paciente debe lavar la zona de tratamiento con un jabón neutro y agua, dejando que la piel se seque totalmente. Zyclara se debe aplicar en forma de película delgada sobre toda la zona de tratamiento, realizando un suave masaje hasta que la crema desaparezca. Se pueden aplicar hasta 2 sobres de Zyclara en la zona de tratamiento (todo el rostro o cuero cabelludo, pero no ambos) en cada aplicación diaria. Los sobres parcialmente usados deben ser desechados y no se reutilizarán. Zyclara debe permanecer sobre la piel durante aproximadamente 8 horas; una vez transcurrido este plazo, es fundamental retirar la crema lavando la zona y las manos con jabón neutro y agua.
Las manos se deben lavar detenidamente tanto antes como después de aplicar la crema.
Dosis olvidada
En caso de olvidar una dosis, el paciente debe esperar hasta la noche siguiente para aplicar Zyclara, continuando seguidamente con el programa regular de tratamiento. La crema no debe aplicarse más de una vez al día. Cada ciclo de tratamiento no debe prolongarse más allá de 2 semanas a causa de dosis olvidadas o periodos de descanso.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Instrucciones generales para el tratamiento
Las lesiones atípicas desde el punto de vista clínico para QA o sospechosas de ser malignas deberán someterse a una biopsia para determinar el tratamiento apropiado.
Se debe evitar el contacto con ojos, labios y fosas nasales, puesto que no se ha evaluado la acción de imiquimod en el tratamiento de queratosis actínicas en los párpados, el interior de las fosas nasales u orejas, ni en la zona de los labios dentro de la línea sonrosada.
No se recomienda el uso de la terapia con imiquimod crema hasta que la piel se haya recuperado por completo del tratamiento anterior con cualquier medicamento o cirugía. La aplicación sobre piel no intacta podría dar lugar a una absorción sistémica de imiquimod dando lugar a un mayor riesgo de reacciones adversas (ver secciones 4.8 y 4.9).
En relación con un aumento de la susceptibilidad a sufrir quemaduras solares, se recomienda el uso de pantallas solares; los pacientes deberán reducir o evitar la exposición a la luz solar natural o artificial (sistemas de bronceado o tratamiento UVA/B) mientras utilicen Zyclara. La zona de piel tratada deberá estar protegida de la exposición al sol.
No hay datos disponibles sobre el uso de imiquimod al 3,75% para el tratamiento de queratosis actínicas en regiones anatómicas distintas del rostro y cuero cabelludo.
No se recomienda el uso de imiquimod para el tratamiento de lesiones de QA con una hiperqueratosis o hipertrofia marcadas, como se aprecia en los cuernos cutáneos.
Reacciones cutáneas locales
Durante la terapia y hasta su curación, es probable que la piel tratada muestre un aspecto marcadamente diferente del de la piel normal. Las reacciones cutáneas locales son frecuentes, pero normalmente su intensidad disminuye durante la terapia o se resuelven después de interrumpir el tratamiento con imiquimod crema. En ocasiones aisladas, pueden producirse intensas reacciones inflamatorias, que incluyen heridas supurantes o erosiones de la piel al cabo de tan sólo unas pocas aplicaciones de imiquimod crema.
Existe una asociación entre el índice de aclaramiento completo y la intensidad de las reacciones cutáneas locales (ej., eritema). Estas reacciones cutáneas locales pueden estar relacionadas con la estimulación de la respuesta inmunitaria local. Adicionalmente, imiquimod tiene el potencial de exacerbar las afecciones inflamatorias de la piel. Si lo requieren las molestias del paciente o la intensidad de la reacción cutánea local, se puede instaurar un periodo de descanso de varios días. El tratamiento con imiquimod crema se puede reanudar después de que la reacción cutánea se haya moderado. La intensidad de las reacciones cutáneas locales tiende a ser menor en el segundo ciclo de tratamiento con Zyclara comparado con el primero.
Reacciones sistémicas
Las reacciones cutáneas locales de carácter intenso pueden estar acompañadas o, incluso, precedidas, por signos y síntomas de tipo gripal y pueden incluir fatiga, náuseas, fiebre, mialgias, artralgias y escalofríos. Se debe considerar una interrupción de la administración o un ajuste de la dosis (ver sección 4.8).
Imiquimod debe utilizarse con precaución en pacientes con una reserva hematológica reducida (ver sección 4.8).
Poblaciones especiales
En los ensayos clínicos no se han incluido pacientes con trastornos cardíacos, hepáticos o renales. Deben tomarse precauciones en estos pacientes.
Uso en pacientes inmunocomprometidos y/o en pacientes con enfermedades autoinmunes No se ha establecido la seguridad y eficacia de Zyclara en pacientes inmuno-comprometidos (ej., pacientes con trasplantes de órganos) y/o pacientes con enfermedades autoinmunes. Por lo tanto, imiquimod crema deberá usarse con precaución en este tipo de pacientes (ver sección 4.5). En estos pacientes, se debe considerar la relación entre el beneficio del tratamiento con imiquimod frente al riesgo asociado tanto de un posible rechazo del órgano o una enfermedad injerto contra huésped, como un posible empeoramiento del trastorno autoinmune.
Re-tratamiento
No hay datos disponibles para el re-tratamiento de queratosis actínicas que hayan remitido después de dos ciclos de tratamiento y, posteriormente, vuelvan a aparecer.
Excipientes
El alcohol estearílico y el alcohol cetílico pueden producir reacciones locales en la piel (como dermatitis de contacto).
El parahidroxibenzoato de metilo (E 218) y el parahidroxibenzoato de propilo (E 216) pueden producir reacciones alérgicas (posiblemente retardadas).
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han llevado a cabo estudios de interacciones, incluidos estudios con medicamentos inmunosupresores. Las interacciones con medicamentos sistémicos estarían limitadas por la mínima absorción percutánea de imiquimod crema.
Debido a sus propiedades inmunoestimulantes, imiquimod crema se deberá utilizar con precaución en pacientes que estén bajo tratamiento con medicamentos inmunosupresores (ver sección 4.4).
Se debe evitar el uso simultáneo de Zyclara y cualquier otra crema de imiquimod en la misma zona de tratamiento, dado que contienen el mismo ingrediente activo (imiquimod) y pueden elevar el riesgo y la intensidad de las reacciones cutáneas locales.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos clínicos disponibles sobre embarazos expuestos a imiquimod. Los estudios en animales no evidencian efectos perjudiciales directos o indirectos en relación con el embarazo, el desarrollo embrionario/fetal, el parto o el desarrollo postnatal (ver sección 5.3).
Cuando se prescriba Zyclara en mujeres embarazadas, se debe proceder con precaución. Zyclara se debe usar durante el embarazo sólo si el beneficio potencial justifica el potencial riesgo para el feto.
Lactancia
No se pueden dar recomendaciones específicas sobre si el producto se debe utilizar o no en madres durante el periodo de lactancia.
Fertilidad
No se dispone de datos clínicos, por lo que el riesgo potencial para el ser humano es desconocido.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Zyclara sobre la capacidad para conducir o usar máquinas es nula o despreciable.
4.8 Reacciones adversas
Resumen del perfil de seguridad:
Los datos que se describen a continuación reflejan la exposición a Zyclara o al vehículo en 319 sujetos que participaron en dos estudios doble-ciego. Los sujetos aplicaron al día hasta dos sobres de Zyclara 3,75% crema o el vehículo sobre la piel de la zona afectada (todo el rostro o el cuero cabelludo alopécico, pero no ambas) durante dos ciclos de tratamiento de 2 semanas cada uno, separados por un ciclo de 2 semanas sin tratamiento.
La mayor parte de los pacientes que usaron Zyclara para el tratamiento de la QA experimentaron reacciones cutáneas locales (con mayor frecuencia, eritema, costras y exfoliación/sequedad en la zona de aplicación) en el sitio de administración. Sin embargo, solamente un 11% (17/160) de los pacientes incluidos en ensayos clínicos con Zyclara requirió un periodo de descanso (interrupción del tratamiento) a causa de las reacciones adversas locales. Algunos pacientes tratados con imiquimod informaron sobre reacciones adversas sistémicas, que incluyeron cefaleas y fatiga.
Lista tabulada de reacciones adversas
Los datos recogidos en la tabla siguiente reflejan:
- Exposición a Zyclara o al vehículo en los estudios mencionados anteriormente (muy frecuentes a poco frecuentes, y con mayor frecuencia tras el vehículo).
- Experiencia con imiquimod 5% crema
Definición de frecuencias:
Muy frecuente (> 1/10);
Frecuente (> 1/100 a < 1/10);
Poco frecuente (> 1/1.000 a < 1/100);
Rara (> 1/10.000 a < 1/1.000);
Muy rara (<1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
|
Clasificación de órganos y sistemas |
Frecuencia |
Reacciones adversas |
|
Infecciones e Infestaciones |
Frecuente |
Herpes simple |
|
Poco frecuente |
Infección | |
|
Pústulas |
|
Frecuencia no conocida |
Infección cutánea | |
|
Trastornos de la sangre y del sistema linfático |
Frecuente |
Linfadenopatía |
|
Frecuencia no conocida |
Hemoglobina reducida | |
|
Recuento de leucocitos reducido | ||
|
Recuento de neutrófilos reducido | ||
|
Recuento de plaquetas reducido | ||
|
Trastornos del sistema inmunológico |
Rara |
Exacerbación de las enfermedades autoinmunes |
|
Trastornos del metabolismo y de la nutrición |
Frecuente |
Anorexia |
|
Aumento de la glucosa en sangre | ||
|
Trastornos psiquiátricos |
Frecuente |
Insomnio |
|
Poco frecuente |
Depresión | |
|
Irritabilidad | ||
|
Trastornos del sistema nervioso |
Frecuente |
Cefaleas |
|
Vértigo | ||
|
Trastornos oculares |
Poco frecuente |
Irritación de la conjuntiva |
|
Edema palpebral | ||
|
Trastornos respiratorios, torácicos y mediastínicos |
Poco frecuente |
Congestión nasal |
|
Dolor de faringe y laringe | ||
|
Trastornos hepatobiliares |
Frecuencia no conocida |
Elevación de las enzimas hepáticas |
|
Trastornos gastrointestinales |
Frecuente |
Náuseas |
|
Diarrea | ||
|
Vómitos | ||
|
Poco frecuente |
Sequedad de boca | |
|
Trastornos de la piel y tejidos subcutáneos |
Muy frecuente |
Eritema |
|
Costras | ||
|
Exfoliación de la piel | ||
|
Edema cutáneo | ||
|
Úlceras cutáneas | ||
|
Hipopigmentación de la piel | ||
|
Frecuente |
Dermatitis | |
|
Poco frecuente |
Edema facial | |
|
Rara |
Reacción dermatológica en sitio alejado | |
|
Frecuencia no conocida |
Alopecia | |
|
Eritema multiforme | ||
|
Síndrome de Stevens Johnson | ||
|
Lupus eritematoso cutáneo | ||
|
Hiperpigmentación cutánea | ||
|
Trastornos musculo-esqueléticos y del tejido conjuntivo |
Frecuente |
Mialgia |
|
Artralgia | ||
|
Poco frecuente |
Lumbalgia | |
|
Dolor en extremidades | ||
|
Trastornos generales y alteraciones en el lugar de administración |
Muy frecuente |
Eritema en el sitio de aplicación |
|
Costras en el sitio de aplicación | ||
|
Exfoliación en el sitio de aplicación | ||
|
Sequedad del sitio de aplicación | ||
|
Edema en el sitio de aplicación | ||
|
Úlcera en el sitio de aplicación | ||
|
Supuración en el sitio de aplicación | ||
|
Frecuente |
Reacción en el sitio de aplicación | |
|
Prurito en el sitio de aplicación | ||
|
Dolor en el sitio de aplicación |
|
Tumefacción en el sitio de aplicación | |
|
Sensación de quemadura en el sitio de aplicación | |
|
Irritación en el sitio de aplicación | |
|
Exantema en el sitio de aplicación | |
|
Fatiga | |
|
Pirexia | |
|
Síntomas gripales | |
|
Dolor | |
|
Dolor torácico | |
|
Poco frecuente |
Dermatitis en el sitio de aplicación |
|
Hemorragia en el sitio de aplicación | |
|
Pápulas en el sitio de aplicación | |
|
Parestesias en el sitio de aplicación | |
|
Hiperestesia en el sitio de aplicación | |
|
Inflamación del sitio de aplicación | |
|
Cicatriz en el sitio de aplicación | |
|
Degradación de la piel del sitio de aplicación | |
|
Vesículas en el sitio de aplicación | |
|
Calor en el sitio de aplicación | |
|
Astenia | |
|
Escalofríos | |
|
Letargo | |
|
Molestias | |
|
Inflamación |
Descripción de reacciones adversas seleccionadas
Trastornos del sistema sanguíneo:
En ensayos clínicos realizados sobre el uso de imiquimod 5% crema, se han observado reducciones de hemoglobina, recuento de leucocitos, recuento absoluto de neutrófilos y de plaquetas. Estas reducciones no se consideran clínicamente significativas en pacientes con una reserva hematológica normal. En ensayos clínicos, no se han estudiado pacientes con reservas hematológicas reducidas. En estudios post-comercialización se ha informado de parámetros hematológicos reducidos, que han requerido intervención clínica.
Infecciones cutáneas:
Durante el tratamiento con imiquimod se han observado infecciones de la piel. Aunque no se han registrado secuelas graves, siempre se debe considerar la posibilidad de infección en la piel con lesiones.
Hipopigmentación e hiperpigmentación:
Se han recibido informes de casos localizados de hipopigmentación e hiperpigmentación tras el uso de imiquimod 5% crema. Los análisis de seguimiento indican que estos cambios de color de la piel pueden ser permanentes en algunos pacientes.
Reacción dermatológica en sitio alejado:
En ensayos clínicos de terapia con imiquimod 5% crema, se han comunicado casos aislados de reacciones dermatológicas en sitios alejados, que incluyen eritema multiforme.
Alopecia:
Estudios clínicos sobre el uso de imiquimod 5% crema en el tratamiento de la queratosis actínica han detectado una frecuencia de 0,4% (5/1214) de alopecia en el sitio de tratamiento o zona adyacente.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
Cuando se administra por vía tópica, es improbable que se produzca una sobredosis sistémica con la crema de imiquimod debido a su mínima absorción a través de la piel. Estudios en conejos han revelado una dosis dérmica letal de imiquimod superior a 5 g/kg. La sobredosis tópica persistente de crema de imiquimod podría dar como resultado graves reacciones cutáneas locales, pudiendo incrementar el riesgo de reacciones sistémicas.
Tras la ingestión accidental, podrían producirse náuseas, vómitos, cefaleas, mialgia y fiebre después de una dosis única de 200 mg de imiquimod, que corresponde al contenido de más de 21 sobres de Zyclara. El acontecimiento adverso clínicamente más grave comunicado tras múltiples dosis orales de > 200 mg fue hipotensión, que se resolvió después de la administración oral o intravenosa de líquidos.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antibióticos y quimioterapéuticos de uso dermatológico, antivirales, Código ATC: D06BB10
Efectos farmacodinámicos
Imiquimod es un modificador de la respuesta inmunitaria. Es el compuesto principal de la familia de las imidazolinas. Los estudios de unión saturable indican la existencia de receptores de membrana para imiquimod en las células respondedoras, denominados receptores de tipo Toll 7 y 8. Imiquimod induce la liberación de interferón alfa (IFN-a) y otras citoquinas a partir de diversas células humanas y animales (ej., a partir de monocitos/macrófagos y queratinocitos humanos). La aplicación tópica in vivo de imiquimod crema sobre la piel del ratón dio como resultado un incremento de las concentraciones de IFN y del factor de necrosis tumoral (TNF) en comparación con la piel de ratones no tratados. El grupo de citoquinas inducidas varía con el origen del tejido celular. Adicionalmente, en diversos estudios en animales de laboratorio y humanos se indujo la liberación de citoquinas tras la aplicación dérmica y la administración oral de imiquimod. En modelos animales, imiquimod es eficaz frente a infecciones víricas y actúa como agente antitumoral principalmente a través de la inducción de la liberación de interferón alfa y otras citoquinas.
En estudios humanos se han observado también incrementos de los niveles sistémicos de interferón alfa y otras citoquinas tras la aplicación tópica de imiquimod.
Eficacia clínica y seguridad
La eficacia de Zyclara se estudió en dos ensayos clínicos doble-ciego, aleatorizados y controlados con el vehículo. Los pacientes exhibieron 5-20 lesiones de QA típicas, visibles o palpables, en una zona que fue mayor de 25 cm2 en el rostro o en el cuero cabelludo alopécico. 319 sujetos con QA fueron tratados con hasta 2 sobres, una vez al día, de imiquimod 3,75% crema, o una crema de vehículo equivalente, durante dos ciclos de tratamiento de 2 semanas, separados por un ciclo sin tratamiento de 2 semanas. Para los ensayos combinados, el índice de aclaramiento completo de todo el rostro o del cuero cabelludo alopécico bajo la acción de imiquimod 3,75% crema fue de 35,6 % (57/160 pacientes, IC 28,2%, 43,6 %), y bajo la acción del vehículo fue de 6,3% (10/159 pacientes, IC 3,1%, 11,3%) en la visita post-tratamiento a las 8 semanas. No se registraron diferencias globales en cuanto a seguridad o eficacia entre pacientes de 65 o más años y pacientes más jóvenes. Se diagnosticó un carcinoma de células escamosas (SCC) en 1,3% (2/160) de los pacientes tratados con imiquimod, y en 0,6% (1/159) de los tratados con el vehículo. Esta diferencia no fue estadísticamente significativa.
En un estudio de seguimiento en el que se controló a los pacientes con un aclaramiento inicial causado por imiquimod durante al menos 14 meses sin ningún tratamiento adicional de QA, un 40,5% de los pacientes mostró un aclaramiento completo sostenido de toda la zona de tratamiento (rostro completo o cuero cabelludo). No existen datos sobre aclaramiento a largo plazo más allá de este periodo.
Población pediátrica
La queratosis actínica no es una afección habitual en la población pediátrica y no se ha estudiado.
5.2 Propiedades farmacocinéticas Absorción
Menos del 0,9% de una dosis única de imiquimod marcado radioactivamente, administrada por vía tópica, se absorbió a través de la piel de sujetos humanos.
La exposición sistémica (penetración percutánea) se calculó a partir del carbono 14 recuperado de [14C]-imiquimod en orina y heces fecales.
Durante un estudio farmacocinético realizado con imiquimod 3,75% crema se observó que, tras la aplicación de 2 sobres una vez al día (18,75 mg de imiquimod/día) durante hasta tres semanas sobre todo el rostro y/o el cuero cabelludo (aproximadamente 200 cm2), se registró una baja absorción sistémica en pacientes con QA. Los niveles en el estado de equilibrio se alcanzaron al cabo de 2 semanas y el tiempo hasta alcanzar las concentraciones máximas (Tmax) osciló dentro de un rango de 6 a 9 horas después de la última aplicación.
Distribución
La concentración máxima media en suero de imiquimod al final del estudio farmacocinético fue de 0,323 ng/ml.
Biotransformación
Imiquimod administrado de forma oral es rápida e intensamente metabolizado en sus dos metabolitos principales.
Eliminación
La pequeña cantidad del medicamento que se absorbió en la circulación sistémica se excretó rápidamente por vías urinaria y fecal en una proporción media de aproximadamente 3 a 1.
En el estudio farmacocinético, la semivida aparente tras la administración tópica de crema de imiquimod 3,75% se estableció en aproximadamente 29 horas.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanossegún los estudios convencionales de farmacología de seguridad, mutagenicidad y teratogenicidad.
En un estudio de toxicidad dérmica en ratas, de cuatro meses de duración, se observaron un descenso significativo del peso corporal y un aumento significativo del peso del bazo a las dosis de 0,5 y 2,5 mg/kg; no se registraron efectos similares en un estudio dérmico en ratones, de cuatro meses de duración. En ambas especies se observó irritación localizada de la dermis, especialmente con las dosis más altas.
En un estudio de 18 meses de carcinogenicidad en el ratón, por administración dérmica tres días a la semana, no se produjo aparición de tumores en la zona de aplicación. Solamente en los ratones hembras, la incidencia de adenomas hepatocelulares fue levemente superior a la de los controles. La incidencia es perfectamente compatible con el espectro de tumores espontáneos conocido para el ratón, en correspondencia con su edad. Por lo tanto, se considera que estos hallazgos son incidentales. Dado que imiquimod muestra una baja absorción sistémica desde la piel humana, y que no es mutagénico, cualquier riesgo para el ser humano derivado de la exposición sistémica es probablemente bajo. Además, en un estudio de carcinogenicidad oral de 2 años de duración en ratas, no se observaron tumores en ninguna zona.
Imiquimod crema fue evaluado en un bioensayo de fotocarcinogenicidad en ratones sin pelo, albinos, expuestos a radiación ultravioleta solar (UVR) simulada. Los animales recibieron imiquimod crema tres veces a la semana y fueron sometidos a irradiación 5 días a la semana durante 40 semanas. Los ratones se conservaron durante 12 semanas adicionales. Aparecieron tumores más precozmente y en mayor número en el grupo de ratones tratados con la crema de vehículo, en comparación con el grupo de control de UVR baja. Se desconoce la importancia para el ser humano. La administración tópica de imiquimod crema no produjo una potenciación de tumores a ninguna dosis, en comparación con el grupo que recibió el vehículo en crema.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Ácido isoesteárico Alcohol bencílico Alcohol cetílico Alcohol estearílico Parafina blanca ligera Polisorbato 60 Estearato de sorbitan Glicerol
Parahidroxibenzoato de metilo (E 218)
Parahidroxibenzoato de propilo (E 216)
Goma xantam Agua purificada
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
30 meses
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 25°C.
Los sobres no se deben reutilizar una vez abiertos.
6.5 Naturaleza y contenido del envase
Cajas con 14, 28 y 56 sobres de uso único, de poliéster/polietileno de baja densidad, blanco/lámina de aluminio, que contienen 250 mg de crema.
Puede que solamente estén comercializados algunos tamaños de envase.
6.6 Precauciones especiales de eliminación
Ninguna especial.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Meda AB Pipers vag 2A 170 73 Solna Suecia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/783/001-003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 23/08/2012
10. FECHA DE LA REVISIÓN DEL TEXTO
Información detallada sobre este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES RELATIVAS AL USO SEGURO Y EFICAZ DEL MEDICAMENTO
A. FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de losfabricantes responsables de la liberación de los lotes
3M Health Care Limited
Derby Road
Loughborough
Leicester
LE11 5SF
Reino Unido.
MEDA Pharma GmbH & Co. KG BenzstraBe 1 61352 Bad Homburg Alemania
El prospecto impreso del medicamento debe especificar el nombre y la dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica.
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Reisgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de una
nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Zyclara 3,75% crema imiquimod
2. PRINCIPIO(S) ACTIVO(S)
Cada sobre contiene 9,375 mg de imiquimod en 250 mg de crema (3,75 %). Cada gramo de crema contiene 37,5 mg de imiquimod
3. LISTA DE EXCIPIENTES
Excipientes: ácido isoesteárico, alcohol bencílico, alcohol cetílico, alcohol estearílico, parafina blanca ligera, polisorbato 60, estearato de sorbitan, glicerol, parahidroxibenzoato de metilo (E 218), parahidroxibenzoato de propilo (E 216), goma xantam, agua purificada.
Leer el prospecto antes de utilizar este medicamento.
4. FORMA FARMACÉUTICA Y CONTENIDO
Crema 14 sobres 28 sobres 56 sobres
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Uso cutáneo.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7._OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Exclusivamente para un único uso.
Deseche el resto de crema que quede en el sobre después del uso.
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar por debajo de 25 °C
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Meda AB Box 906 170 09 Solna Suecia
12. NÚMERO(S) DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/783/001 - 14 sobres EU/1/12/783/002 - 28 sobres EU/1/12/783/003 - 56 sobres
|
13. |
NÚMERO DE LOTE |
|
Lote | |
|
14. |
CONDICIONES GENERALES DE DISPENSACIÓN |
|
MEDICAMENTO SUJETO A PRESCRIPCIÓN MÉDICA. | |
|
15. |
INSTRUCCIONES DE USO |
16. INFORMACIÓN EN BRAILLE
Zyclara
TEXTO DEL SOBRE
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Zyclara 3,75% crema
imiquimod
Uso cutáneo
2. FORMA DE ADMINISTRACIÓN
|
3. |
FECHA DE CADUCIDAD |
|
CAD | |
|
4. |
NÚMERO DE LOTE |
|
Lote | |
|
5. |
CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES |
|
250 |
mg. |
|
6. |
OTROS |
B. PROSPECTO
Prospecto: Información para el paciente
Zyclara 3,75% crema Imiquimod
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Zyclara y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Zyclara
3. Cómo usar Zyclara
4. Posibles efectos adversos
5. Conservación de Zyclara
6. Contenido del envase e información adicional
1. Qué es Zyclara y para qué se utiliza
Zyclara 3,75% crema contiene la sustancia activa imiquimod.
Este medicamento se prescribe para el tratamiento de la queratosis actínica en adultos.
Este medicamento estimula su propio sistema inmunitario para producir sustancias naturales que contribuyen a combatir su queratosis actínica.
Las queratosis actínicas aparecen como zonas de piel rugosa presentes en personas que han estado expuestas a gran cantidad de luz solar en el transcurso de su vida. Estas zonas pueden ser del mismo color que su piel, o ser grisáceas, rosadas, rojas o marrones. Pueden ser planas y escamosas, o elevadas, rugosas, duras y verrugosas.
Este medicamento se debe usar exclusivamente en queratosis actínicas del rostro o del cuero cabelludo, si su médico ha decidido que es el tratamiento más adecuado para usted.
2. Qué necesita saber antes de empezar a usar Zyclara
No use Zyclara
- si es alérgico al imiquimod o a cualquiera de los demás componentes de este medicamento
(incluidos en la sección 6).
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar usar Zyclara:
• si ha utilizado anteriormente este medicamento u otro preparado similar, en una concentración diferente.
• si sufre problemas con su sistema inmunitario, o si toma medicamentos para suprimir el sistema inmunitario (ej., después de trasplante de un órgano).
• si tiene recuento sanguíneo anormal.
Instrucciones generales durante el tratamiento
• Si se ha sometido recientemente a tratamiento quirúrgico o farmacológico, espere hasta que la zona a tratar haya cicatrizado antes de usar este medicamento.
• Evite el contacto con ojos, labios y fosas nasales. En caso de contacto accidental, elimine la crema lavando la zona con agua.
• Utilice la crema sólo de forma externa (sobre la piel del rostro o cuero cabelludo).
• No use una cantidad de crema mayor que la recomendada por su médico.
• No cubra la zona tratada con vendajes u otros elementos después de haber aplicado Zyclara.
• Si desarrolla molestias excesivas en la zona tratada, retire la crema con un jabón neutro y agua. Una vez que hayan remitido las molestias, puede reanudar el tratamiento de la forma recomendada. La crema no se debe aplicar más de una vez al día.
• No utilice lámparas solares ni soláriums, y evite en la mayor medida posible la luz del sol durante el tratamiento con este medicamento. Si sale al aire libre durante el día, utilice pantalla solar y vista prendas protectoras, así como un sombrero de ala ancha.
Reacciones cutáneas locales
Al igual que la mayoría de los pacientes, mientras utilice Zyclara puede experimentar reacciones cutáneas locales debido a la forma en que el medicamento actúa sobre su piel. Estas reacciones pueden indicar que el medicamento está actuando del modo previsto.
Durante el uso de Zyclara y hasta la curación, la zona de tratamiento puede presentar un aspecto claramente diferente al de la piel normal. Existe también la posibilidad de que una inflamación preexistente empeore temporalmente.
Este medicamento también puede provocar síntomas similares a los de la gripe (incluidos cansancio, náuseas, fiebre, dolores musculares y articulares, y escalofríos) antes o durante la aparición de las reacciones cutáneas locales.
La respuesta al tratamiento no se puede evaluar adecuadamente hasta la resolución de las reacciones cutáneas locales. Deberá continuar el tratamiento de la forma prescrita.
Este medicamento puede poner de manifiesto y tratar queratosis actínicas que no hayan sido vistas o percibidas previamente, y que posteriormente pueden desaparecer. La aplicación deberá continuar durante la totalidad del ciclo de tratamiento aun cuando todas las queratosis actínicas parezcan haber desaparecido.
Niños y adolescentes
Este medicamento no se debe administrar a niños menores de 18 años, porque no se han establecido la seguridad y eficacia en pacientes menores de dicha edad. No se dispone de datos acerca del uso de imiquimod en niños y adolescentes.
Uso de Zyclara con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento, incluidos los de venta sin receta médica.
Embarazo, lactancia y fertilidad
Embarazo
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Lactancia
No se pueden dar consejos específicos sobre el uso o no de este medicamento en madres lactantes. Conducción y uso de máquinas
La influencia de este medicamento sobre la capacidad para conducir y usar máquinas es nula o despreciable.
Zyclara contiene parahidroxibenzoato de metilo, parahidroxibenzoato de propilo, alcohol cetílico y alcohol estearílico.
Puede producir reacciones alérgicas (posiblemente retardadas) porque contiene parahidroxibenzoato de metilo (E 218) y parahidroxibenzoato de propilo (E 216).
Este medicamento puede producir reacciones locales en la piel (como dermatitis de contacto) porque contiene alcohol cetílico y alcohol estearílico.
3. Cómo usar Zyclara
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico. No utilice este medicamento hasta que su médico le haya enseñado la manera correcta de hacerlo.
Este medicamento sólo se debe usar para las queratosis actínicas del rostro y cuero cabelludo. Dosificación
Aplique este medicamento sobre la zona afectada una vez al día, inmediatamente antes de acostarse. La dosis diaria máxima es de 2 sobres (500 mg = 2 sobres de 250 mg cada uno).
Este medicamento no se debe aplicar sobre zonas de tamaño mayor que todo el rostro o el cuero cabelludo alopécico.
Forma de administración
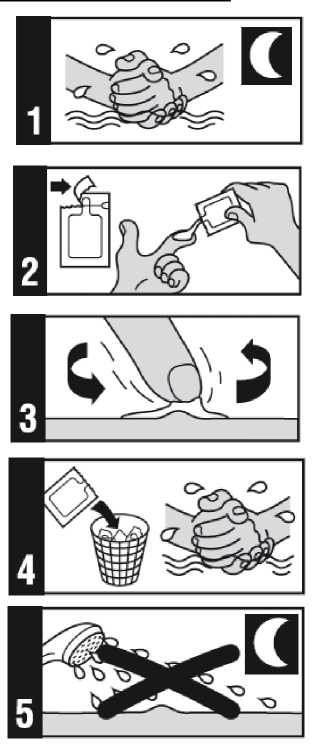
1. Antes de acostarse, lávese cuidadosamente las manos y la zona de tratamiento con un jabón neutro y agua. Séquese perfectamente las manos y deje secar la zona de tratamiento.
2. Abra un sobre nuevo de Zyclara inmediatamente antes de usarlo y deposite una porción de crema sobre la yema del dedo. No se deben usar más de 2 sobres por aplicación.
3. Aplique una capa fina de Zyclara sobre la zona afectada. Realice un suave masaje sobre la zona hasta que la crema se absorba. Evite el contacto con los ojos, los labios y las fosas nasales.
4. Después de aplicar la crema, deseche el sobre abierto. Lávese bien las manos con agua y jabón.
5. Zyclara debe permanecer sobre la piel durante aproximadamente 8 horas. No se duche ni moje la zona durante este periodo. No cubra la zona tratada con vendajes ni otros elementos.
6. Después de aproximadamente 8 horas, lave la zona sobre la que se aplicó Zyclara con agua y jabón neutro.
Duración del tratamiento
El tratamiento se inicia con una aplicación diaria durante dos semanas, seguido de una pausa sin aplicación durante dos semanas, y finaliza con una aplicación diaria durante dos semanas nuevamente.
Si usa más Zyclara del que debe
Si se ha aplicado demasiada crema, retire el exceso lavando la zona con agua y jabón neutro.
Cuando haya remitido cualquier reacción cutánea, puede reanudar el tratamiento según el programa regular recomendado. No se debe aplicar la crema más de una vez al día.
Si traga accidentalmente este medicamento, póngase de inmediato en contacto con su médico.
Si olvidó usar Zyclara
Si olvidó una dosis de Zyclara, espere hasta la noche siguiente para aplicarla y, a continuación, prosiga con el régimen habitual. La crema no se debe aplicar más de una vez al día. Cada ciclo de tratamiento no deberá durar más de dos semanas, incluso si ha omitido algunas dosis.
Si interrumpe el tratamiento con Zyclara
Consulte con su médico antes de suspender el tratamiento con Zyclara.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Solicite atención médica inmediata si aparece cualquiera de estos efectos adversos graves durante el uso de este medicamento:
Reacciones cutáneas graves, con lesiones cutáneas o manchas en la piel que se inician como pequeñas zonas de color rojo y evolucionan hasta alcanzar el aspecto de pequeñas dianas, posiblemente con síntomas tales como picor, fiebre, malestar general, dolores articulares, problemas de visión, sensación de quemadura, dolores o picores oculares y llagas bucales. Si sufre estos trastornos, interrumpa el uso de este medicamento e informe inmediatamente a su médico.
En algunas personas se ha observado un descenso de la fórmula sanguínea. Este hecho podría aumentar su susceptibilidad a sufrir infecciones, desarrollar hematomas o provocar cansancio. Si percibe cualquiera de estos síntomas, informe a su médico.
Si hay presencia de pus u otro signo de infección, consúltelo con su médico.
Muchos de los efectos secundarios de este medicamento se deben a su acción local sobre la piel. Las reacciones cutáneas locales pueden indicar que el medicamento está actuando de la forma prevista. Si su piel reacciona mal o sufre molestias excesivas durante el uso de este medicamento, suspenda la aplicación de la crema y lave la zona con agua y un jabón neutro. Seguidamente, póngase en contacto con su médico o farmacéutico, quien puede aconsejarle que interrumpa la aplicación de este medicamento durante algunos días (es decir, realizar un breve descanso durante el tratamiento).
Se han comunicado los siguientes efectos adversos con imiquimod:
Muy frecuentes (puede afectar a más de 1 de cada 10 personas)
- Enrojecimiento de la piel, costras, descamación cutánea, supuración, sequedad de la piel, hinchazón cutánea, úlceras cutáneas y reducción de la pigmentación cutánea en el sitio de aplicación.
Frecuentes (puede afectar hasta 1 de cada 10 personas)
- Otras reacciones adicionales en el sitio de aplicación, por ej., inflamación de la piel, prurito, dolor, sensación de quemadura, irritación y exantema cutáneo
- Glándulas inflamadas
- Cefaleas
- Vértigo
- Pérdida de apetito
- Náuseas
- Diarrea
- Vómitos
- Síntomas semejantes a la gripe
- Fiebre
- Dolor
- Dolores musculares y articulares
- Dolor torácico
- Insomnio
- Cansancio
- Infección vírica (herpes simple)
- Aumento de la glucosa en sangre
Poco frecuentes (puede afectar hasta 1 de cada 100 personas)
- Alteraciones en la zona de aplicación, por ej., sangrado, pequeñas zonas hinchadas en la piel, inflamación, hormigueo, aumento de la sensibilidad al contacto, formación de cicatrices, sensación de calor, lesiones de la piel, ampollas o pústulas
- Debilidad
- Escalofríos
- Falta de energía (letargo)
- Molestias
- Tumefacción del rostro
- Lumbalgia
- Dolor de extremidades
- Nariz taponada
- Dolor de garganta
- Irritación de los ojos
- Hinchazón de los párpados
- Depresión
- Irritabilidad
- Boca seca
Raros (puede afectar hasta 1 de cada 1.000 personas)
- Brotes de enfermedades autoinmunes (una enfermedad autoinmune es una enfermedad causada por una respuesta inmunitaria anormal)
- Reacciones cutáneas alejadas del sitio de aplicación
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
- Cambios de color de la piel
Algunos pacientes han experimentado cambios de color de la piel en la zona sobre la que se aplicó Zyclara. Aunque estos cambios tienden a mejorar con el tiempo, en algunos pacientes pueden ser permanentes
- Pérdida de pelo
Un número reducido de pacientes ha experimentado pérdida de pelo en la zona de tratamiento o inmediatamente adyacente
Elevación de las enzimas hepáticas
Ha habido informes de un incremento de las enzimas hepáticas.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Zyclara
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y en el envase exterior después de CAD.
La fecha de caducidad es el último día del mes que se indica.
No conservar a temperatura superior a 25°C.
Una vez abiertos, los sobres no se deben reutilizar.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de Zyclara
- El principio activo es imiquimod. Cada sobre contiene 9,375 mg de imiquimod en 250 mg de crema (100 mg de crema contienen 3,75 mg de imiquimod).
- Los demás componentes son: ácido isoesteárico, alcohol bencílico, alcohol cetílico, alcohol estearílico, parafina blanca ligera, polisorbato 60, estearato de sorbitan, glicerol, parahidroxibenzoato de metilo (E 218), parahidroxibenzoato de propilo (E 216), goma xantán, agua purificada.
Aspecto de Zyclara y contenido del envase
- Cada sobre de Zyclara 3,75% crema contiene 250 mg de una crema de color blanco a ligeramente amarillenta, de aspecto uniforme.
- Cada envase contiene 14, 28 ó 56 sobres de un solo uso de poliéster/ polietileno de baja densidad blanco/lámina de aluminio.
Puede que sólo estén comercializados algunos tamaños de envase.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización
Meda AB
Pipers vag 2A
170 73 Solna
Suecia
Responsable de la fabricación
3M Health Care Limited
Derby Road
Loughborough
Leicestershire
LE11 5SF, Reino Unido
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Meda Pharma S.A./N.V. Chaussée de la Hulpe 166 Terhulpsesteenweg 1170 Brussels Tél/Tel: +32 (0)2 5 04 08 11 |
Luxembourg/Luxemburg Meda Pharma S.A./N.V. Chaussée de la Hulpe 166 Terhulpsesteenweg B-1170 Brussels Belgique / Belgien Tél/Tel: +32 (0)2 5 04 08 11 |
|
Etarapun MEDA Pharmaceuticals Switzerland GmbH npegcTaBHTencTBO 3a Etarapna OgpHH 71-75 1303 Co^na Tea: +35924177977 |
Magyarország MEDA PHARMA Hungary Kereskedelmi Kft. 1139 Budapest Váci ut 91 Tel: +36 1 236 3410 |
|
Ceská republika MEDA Pharma s.r.o. Kodañská 1441 / 46 100 10 Praha 10 Tel: +420 234 064 203 |
Malta Alfred Gera and Sons Ltd. 10,Triq il -Masgar Qormi QRM3217 Tel: +356 21 446205 |
|
Danmark Meda AS Solvang 8 3450 Allerod Tlf: +45 44 52 88 88 |
Nederland MEDA Pharma B.V. Krijgsman 20 1186 DM Amstelveen Tel: +31 (0)20 751 65 00 |
|
Deutschland MEDA Pharma GmbH & Co. KG BenzstraEe 1 61352 Bad Homburg Tel: +49 (0) 6172 888 01 |
Norge Meda A/S Askerveien 61 1384 Asker Tlf: +47 66 75 33 00 |
|
Eesti Meda Pharma SIA Parda tn 4 10151 Tallinn Tel: +372 62 61 025 |
Osterreich MEDA Pharma GmbH Guglgasse 15 1110 Wien Tel: + 43 (0)1 86 390 0 |
|
EXXáda MEDA Pharmaceuticals A.E. Enpnxavíag, 3 GR-15231 Xa^ávSpi-Axxixq TqU +30 210 6 77 5690 |
Polska Meda Pharmaceuticals Sp.z.o.o. ul. Domaniewska 39A 02-672 Warszawa Tel: +48 22 697 7100 |
|
España Meda Pharma S.L. Avenida de Castilla, 2 Parque Empresarial San Fernando Edificio Berlín 28830 San Fernando de Henares (Madrid) Tel: +34 91 669 93 00 |
Portugal MEDA Pharma - Produtos Farmacéuticos, S.A Rua do Centro Cultural, 13 1749-066 Lisboa Tel: +351 21 842 0300 |
|
France MEDA Pharma 42 rue Washington 75008 Paris Tél: +33 (0)1 56 64 10 70 |
Romania MEDA Pharmaceuticals Switzerland GmbH Reprezentanta Romania Calea Floreasca 141-143, et4 014467 Bucuresti Tel.: +40 21 230 90 30 |
|
Hrvatska Medical Intertrade d.o.o. Dr. Franje Tudmana 3 10431 Sveta Nedelja Tel: +385 1 3374 010 |
Slovenija MEDA Pharmaceuticals Switzerland GmbH Podruznica Ljubljana Cesta 24. junija 23 Ljubljana Tel: +386 59 096 951 |
|
Ireland Meda Health Sales Ireland Ltd. Unit 34 / 35, Block A, Dunboyne Business Park, Dunboyne, Co Meath, Ireland Tel: +353 1 802 66 24 |
Slovenská republika MEDA Pharma spol. s. r.o. Trnavská cesta 50 821 02 Bratislava Tel: +421 2 4914 0172 |
|
Ísland Meda AB Box 906 170 09 Solna Sví^jóó Sími: +46 8 630 1900 |
Suomi/Finland Meda Oy Vaisalantie 4/Vaisalavagen 4 FIN-02130 Espoo/Esbo Puh/Tel: +358 20 720 9550 |
|
Italia Meda Pharma S.p.A. Via Felice Casati, 20 20124 Milano Tel: +39 039 73901 |
Sverige Meda AB Box 906 170 09 Solna Tel: +46 (0)8 630 1900 |
|
Kúnpoq Xp.r. nana^o'í'Zon AxS As©^. KrAxíg 35, 2234 Aaxorá TqV +357 22 49 03 05 Latvija SIA Meda Pharma Vienibas gatve 109 Riga LV-1058 Talr: +371 67616137 |
United Kingdom Meda Pharmaceuticals Ltd. Skyway House Parsonage Road Takeley Bishops Stortford CM22 6PU Tel.: + 44 845 460 0000 |
Lietuva
Meda Pharma SIA Ukmergés Street 369A LT-12142 Vilnius Tel. + 370 52059367
Fecha de la última revisión de este prospecto: (MM/AAAA)
La información detallada de este medicamento está disponible en la página Web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
28