Xolair 75 Mg Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Xolair 75 mg polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un vial contiene 75 mg de omalizumab*.
Después de la reconstitución un vial contiene 125 mg/ml de omalizumab (75 mg en 0,6 ml).
*Omalizumab es un anticuerpo monoclonal humanizado obtenido mediante la tecnología del ADN recombinante, a partir de una línea celular mamífera de ovario de hámster chino (OHC).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: liofilizado de color blanco a blanquecino. Disolvente: solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Xolair está indicado en adultos, adolescentes y niños (de 6 a <12 años).
El tratamiento con Xolair deberá ser considerado únicamente para pacientes con asma mediada de forma convincente por IgE (inmunoglobulina E) (ver sección 4.2).
Adultos y adolescentes (a partir de 12 años de edad)
Xolair está indicado para mejorar el control del asma cuando se administra como tratamiento adicional en pacientes con asma alérgica grave persistente que presentan test cutáneo positivo o reactividad in vitro a aeroalergenos perennes y con función pulmonar reducida (FEV1 <80%) así como, síntomas frecuentes durante el día o despertares por la noche y que han presentado múltiples exacerbaciones asmáticas graves documentadas, a pesar de utilizar corticosteroides diarios inhalados a dosis altas, más un agonista beta2 inhalado de larga duración.
Niños (6 a <12 años de edad)
Xolair está indicado para mejorar el control del asma cuando se administra como tratamiento adicional en pacientes con asma alérgica grave persistente que presentan test cutáneo positivo o reactividad in vitro a aeroalergenos perennes y síntomas frecuentes durante el día o despertares por la noche y que han presentado múltiples exacerbaciones asmáticas graves documentadas, a pesar de utilizar corticosteroides diarios inhalados a dosis altas, más un agonista beta2 inhalado de larga duración.
4.2 Posología y forma de administración
El tratamiento con Xolair debe iniciarlo un médico experimentado en el diagnóstico y tratamiento del asma grave persistente.
Posología
La dosis apropiada y la frecuencia de administración de Xolair se determina a partir de la concentración basal de IgE (UI/ml), medida antes de iniciar el tratamiento, y del peso corporal (kg). A efectos de la asignación de la dosis, antes de la administración de la dosis inicial se debe determinar la concentración de IgE en los pacientes mediante cualquier método comercial que analice la IgE plasmática total. En base a estas determinaciones, podrán ser necesarios en cada administración de 75 a 600 mg de Xolair en 1 a 4 inyecciones.
Era menos probable que experimentaran beneficio los pacientes con un valor de IgE inferior a 76 UI/ml (ver sección 5.1). Los médicos prescriptores deberán asegurar que los pacientes adultos y adolescentes con una IgE por debajo de 76 UI/ml y los niños (6 a < 12 años de edad) con una IgE por debajo de 200 UI/ml presenten una reactividad in vitro inequívoca (RAST) al alergeno perenne antes de iniciar el tratamiento.
Ver Tabla 1 de conversión y Tablas 2 y 3 para la determinación de dosis en adultos, adolescentes y niños (de 6 a <12 años).
No debe administrarse Xolair a pacientes cuya concentración basal de IgE o peso corporal en kilogramos, excedan los límites indicados en la tabla de dosis.
La dosis máxima recomendada es de 600 mg de omalizumab cada dos semanas.
Tabla 1: Conversión de la dosis al número de viales, número de inyecciones y volumen de inyección total para cada administración
|
Dosis (mg) Número de viales Número de inyecciones 75 mg a 150 mg b |
Volumen total de inyección (ml) | |||
|
75 |
1c |
0 |
1 |
0,6 |
|
150 |
0 |
1 |
1 |
1,2 |
|
225 |
1c |
1 |
2 |
1,8 |
|
300 |
0 |
2 |
2 |
2,4 |
|
375 |
1c |
2 |
3 |
3,0 |
|
450 |
0 |
3 |
3 |
3,6 |
|
525 |
1c |
3 |
4 |
4,2 |
|
600 |
0 |
4 |
4 |
4,8 |
a 0,6 ml = volumen máximo proporcionado por vial (Xolair 75 mg). b 1,2 ml = volumen máximo proporcionado por vial (Xolair 150 mg). c o utilizar 0,6 ml de un vial de 150 mg.
|
Peso corporal (kg) | ||||||||||
|
IgE basal |
>20- |
>25- |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>90- |
>125- |
|
(UI/ml) |
25 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
125 |
150 |
|
>30-100 |
75 |
75 |
75 |
150 |
150 |
150 |
150 |
150 |
300 |
300 |
|
>100-200 |
150 |
150 |
150 |
300 |
300 |
300 |
300 |
300 |
450 |
600 |
|
>200-300 |
150 |
150 |
225 |
300 |
300 |
450 |
450 |
450 |
600 | |
|
>300-400 |
225 |
225 |
300 |
450 |
450 |
450 |
600 |
600 | ||
|
>400-500 |
225 |
300 |
450 |
450 |
600 |
600 | ||||
|
>500-600 |
300 |
300 |
450 |
600 |
600 | |||||
|
>600-700 |
300 |
450 |
600 | |||||||
|
>700-800 | ||||||||||
|
>800-900 |
ADMINISTRACIÓN CADA | |||||||||
|
>900- 1000 |
2 SEMANAS VER TABLA 3 | |||||||||
|
>1000- 1100 | ||||||||||
|
Peso corporal (kg) | ||||||||||
|
IgE basal |
>20- |
>25- |
>30- |
>40- |
>50 |
>60- |
>70- |
>80- |
>90- |
>125- |
|
(UI/ml) |
25 |
30 |
40 |
50 |
-60 |
70 |
80 |
90 |
125 |
150 |
|
>30-100 |
ADMINISTRACIÓN CADA | |||||||||
|
>100-200 |
4 SEMANAS VER TABLA 2 | |||||||||
|
>200-300 |
375 | |||||||||
|
>300-400 |
450 |
525 | ||||||||
|
>400-500 |
375 |
375 |
525 |
600 | ||||||
|
>500-600 |
375 |
450 |
450 |
600 | ||||||
|
>600-700 |
225 |
375 |
450 |
450 |
525 | |||||
|
>700-800 |
225 |
225 |
300 |
375 |
450 |
450 |
525 |
600 | ||
|
>800-900 |
225 |
225 |
300 |
375 |
450 |
525 |
600 | |||
|
>900- |
225 |
300 |
375 |
450 |
525 |
600 | ||||
|
1000 | ||||||||||
|
>1000- 1100 |
225 |
300 |
375 |
450 |
600 | |||||
|
>1100- |
300 |
300 |
450 |
525 |
600 |
NO ADMINISTRAR - |
no se dispone de | |||
|
1200 |
datos para la recomendación de dosis | |||||||||
|
>1200- 1300 |
300 |
375 |
450 |
525 | ||||||
|
>1300- 1500 |
300 |
375 |
525 |
600 | ||||||
Duración del tratamiento, monitorización y ajuste de dosis
Xolair está indicado para tratamiento a largo plazo. Los ensayos clínicos han demostrado que son necesarias un mínimo de 12-16 semanas para que el tratamiento con Xolair demuestre efectividad. A las 16 semanas de iniciar el tratamiento con Xolair, los pacientes deberán ser evaluados por su médico con respecto a la efectividad del tratamiento antes de administrar inyecciones posteriores. La decisión de continuar con Xolair tras las 16 semanas, o en ocasiones posteriores, debe estar basada en si se observa una notable mejoría en el control global del asma (ver sección 5.1; Valoración global del médico con respecto a la efectividad del tratamiento).
La interrupción del tratamiento con Xolair generalmente da lugar a un retorno de las concentraciones elevadas de IgE libre y de los síntomas asociados. Los valores de IgE total son elevados durante el tratamiento y siguen siéndolo hasta un año después de la interrupción del mismo. Por lo tanto, no puede utilizarse la reevaluación de los valores de IgE durante el tratamiento con Xolair como guía para la determinación de la dosis. La determinación de la dosis tras interrupciones de tratamiento de menos de un año de duración debe basarse en las concentraciones plasmáticas de IgE obtenidas en la determinación de dosis inicial. Si el tratamiento con Xolair se ha interrumpido por más de un año deberán de volver a medirse las concentraciones plasmáticas de IgE total para la determinación de la dosis.
Las dosis deberán ajustarse en caso de variaciones significativas del peso corporal (ver Tablas 2 y 3).
Poblaciones especiales
Pacientes de edad avanzada (mayores de 65 años)
Aunque se dispone de datos limitados sobre el uso de Xolair en pacientes mayores de 65 años, no existe evidencia de que los pacientes de edad avanzada requieran una dosis diferente de la de pacientes adultos más jóvenes.
Insuficiencia renal o hepática
No se dispone de estudios sobre el efecto de la insuficiencia renal o hepática en la farmacocinética de Xolair. Debido a que el aclaramiento de omalizumab a dosis clínicas se lleva a cabo fundamentalmente por el sistema reticuloendotelial (SRE), es improbable que se vea alterado en caso de insuficiencia renal o hepática. Xolair deberá administrarse con precaución en estos pacientes, mientras no se recomiende un ajuste especial de la dosis (ver sección 4.4).
Población pediátrica
No se ha establecido la seguridad y eficacia de Xolair en niños menores de 6 años. No se dispone de datos.
Forma de administración
Para administración subcutánea únicamente. No administrar por vía intravenosa o intramuscular.
Las inyecciones se administran vía subcutánea en la región deltoidea del brazo. Si por alguna razón no pueden administrarse en esta zona, podrán administrase alternativamente en el muslo.
Existe experiencia limitada con respecto a la autoadministración de Xolair. Por lo tanto, está previsto que el tratamiento sea administrado únicamente por el profesional sanitario.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6 y también la sección “información para el profesional sanitario” del prospecto.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
General
Xolair no está indicado para el tratamiento de las exacerbaciones asmáticas, broncoespasmo o estados asmáticos de carácter agudo.
No se ha estudiado el efecto de Xolair en pacientes con síndrome de hiperinmunoglobulinemia E o aspergilosis broncopulmonar alérgica, ni en la prevención de reacciones anafilácticas, incluyendo las provocadas por alergias alimentarias, dermatitis atópica, o rinitis alérgica. Xolair no está indicado en el tratamiento de estas patologías.
El tratamiento con Xolair tampoco se ha estudiado en pacientes con enfermedades autoinmunes, procesos mediados por inmunocomplejos, o insuficiencia renal o hepática preexistente (ver sección 4.2). Se deberá tener precaución cuando se administre Xolair en esta población de pacientes.
No se recomienda la interrupción brusca de los corticosteroides sistémicos o inhalados tras la iniciación del tratamiento con Xolair. El descenso de los corticosteroides debe realizarse bajo la supervisión directa de un médico y puede ser necesario que se realice gradualmente.
Trastornos del sistema inmulógico
Reacciones alérgicas tipo I
Pueden producirse reacciones alérgicas tipo I locales o sistémicas, incluyendo anafilaxia y shock anafiláctico durante el tratamiento con omalizumab, incluso con inicio tras un tratamiento de larga duración. La mayoría de estas reacciones se produjeron durante las 2 horas posteriores a la primera y siguientes inyecciones de Xolair, pero algunas se iniciaron pasadas las 2 horas e incluso pasadas 24 horas tras la inyección. Por lo tanto, se deberán tener siempre disponibles medicamentos para el tratamiento inmediato de reacciones anafilácticas tras la administración de Xolair. Se deberá informar al paciente de que estas reacciones son posibles y que si se producen deberán solicitar atención médica de inmediato. Un antecedente de anafilaxia no asociado a omalizumab puede suponer un factor de riesgo de sufrir una reacción anafiláctica tras la administración de Xolair.
Se han detectado anticuerpos contra omalizumab en un pequeño número de pacientes en ensayos clínicos (ver sección 4.8). No se conoce bien la relevancia clínica de anticuerpos anti-Xolair.
Enfermedad del suero
Se ha observado enfermedad del suero y reacciones semejantes a la enfermedad del suero, que son reacciones alérgicas tipo III retardadas, en pacientes tratados con anticuerpos monoclonales humanizados incluido omalizumab. El supuesto mecanismo fisiopatológico incluye formación y deposición de inmunocomplejos debido al desarrollo de anticuerpos contra omalizumab. El inicio del cuadro se produce normalmente a los 1-5 días tras la administración de la primera o siguientes inyecciones e incluso tras un tratamiento de larga duración. Los síntomas que sugieren la enfermedad del suero incluyen artritis/artralgias, rash (urticaria u otras formas), fiebre y linfoadenopatía. Los antihistamínicos y corticosteroides pueden ser útiles para prevenir o tratar estas alteraciones y se deberá advertir a los pacientes que notifiquen cualquier síntoma sospechoso.
Síndrome de Churg-Strauss y síndrome hipereosinofílico
Los pacientes con asma grave pueden presentar raramente síndrome hipereosinofílico sistémico o vasculitis granulomatosa eosinofílica alérgica (Síndrome de Churg-Strauss), los cuales son normalmente tratados con corticosteroides sistémicos.
En raras ocasiones, los pacientes en tratamiento con medicamentos antiasmáticos, incluyendo omalizumab, pueden presentar o desarrollar eosinofilia sistémica y vasculitis. Estas reacciones están normalmente asociadas con la reducción del tratamiento con corticosteroides orales.
En estos pacientes, los médicos deberán estar alerta ante el desarrollo de eosinofilia importante, rash vasculítico, empeoramiento de los síntomas pulmonares, anormalidades en el seno paranasal, complicaciones cardíacas, y/o neuropatía.
Deberá considerarse la interrupción del tratamiento con omalizumab en todos aquellos casos graves que cursen con alteraciones del sistema inmune mencionadas anteriormente.
Infecciones parasitarias (helmínticos)
Las IgE pueden estar involucradas en la respuesta inmunológica a algunas infecciones helmínticas. En pacientes con un elevado riesgo crónico de infección helmíntica, un ensayo controlado con placebo demostró un ligero incremento en la proporción de infección con omalizumab, aunque no se modificó el curso, gravedad y respuesta al tratamiento de la infección. La proporción de infección helmíntica en el programa clínico global, el cual no fue diseñado para detectar este tipo de infecciones, fue inferior a 1 en 1.000 pacientes. Sin embargo, deberá garantizarse precaución en pacientes con elevado riesgo de infección helmíntica, en particular cuando viajen a zonas donde las infecciones helmínticas son endémicas. Si los pacientes no responden al tratamiento antihelmíntico recomendado, deberá considerarse la interrupción del tratamiento con Xolair.
4.5 Interacción con otros medicamentos y otras formas de interacción
Dado que las IgE pueden estar relacionadas con la respuesta immunológica a algunas infecciones por helmínticos, Xolair puede reducir indirectamente la eficacia de medicamentos utilizados para el tratamiento de infecciones helmínticas o por otros parásitos (ver sección 4.4).
Las enzimas del citocromo P450, las bombas de eflujo y los mecanismos de unión a proteínas no se hallan implicados en el aclaramiento de omalizumab; por ello, existe un bajo potencial de interacciones farmacológicas. No se han realizado estudios de interacción de otros medicamentos o vacunas con Xolair. No existe un motivo farmacológico para esperar que los medicamentos prescritos frecuentemente en el tratamiento del asma interaccionen con omalizumab.
En los ensayos clínicos Xolair se utilizó frecuentemente asociado a corticosteroides inhalados y orales, beta agonistas inhalados de corta y larga duración, antagonistas de los leucotrienos, teofilinas y antihistamínicos orales. No hubo indicios de que estos medicamentos utilizados habitualmente como antiasmáticos puedan afectar a la seguridad de Xolair. Se dispone de datos limitados sobre el uso de Xolair en combinación con inmunoterapia específica (terapia de hiposensibilización). En un ensayo clínico donde Xolair se administró conjuntamente con inmunoterapia, se observó que la seguridad y eficacia de Xolair en combinación con inmunoterapia específica, no fue diferente a la de Xolair solo.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Hay datos limitados relativos al uso de omalizumab en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción (ver sección 5.3). Omalizumab atraviesa la barrera placentaria y se desconoce el daño potencial sobre el feto. Omalizumab se ha asociado con descensos de las plaquetas sanguíneas, dependientes de la edad en primates no humanos, con una sensibilidad relativamente superior en animales jóvenes (ver sección 5.3). Xolair no debería utilizarse durante el embarazo excepto si fuese claramente necesario.
Lactancia
Se desconoce si omalizumab se excreta en la leche materna. Los datos disponibles en primates no humanos muestran que omalizumab se excreta en la leche (ver sección 5.3).. No se puede excluir el riesgo en recién nacidos/niños. Omalizumab no se debe administrar durante la lactancia.
Fertilidad
No hay datos de fertilidad en humanos para omalizumab. En estudios de fertilidad no clínicos diseñados específicamente en primates no humanos, incluidos los estudios de apareamiento, no se observó empeoramiento de la fertilidad en machos o hembras tras dosis repetidas con omalizumab de hasta 75 mg/kg. Además, no se observaron efectos genotóxicos en estudios separados no clínicos de genotoxicidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Xolair sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Resúmen del perfil de seguridad
Las reacciones adversas notificadas más frecuentemente durante los ensayos clínicos en pacientes adultos y adolescentes a partir de 12 años de edad fueron cefalea y reacciones en el lugar de la inyección, que incluían dolor, tumefacción, eritema y prurito. Las reacciones adversas notificadas más frecuentemente en los ensayos clínicos en niños de 6 a <12 años de edad fueron cefalea, pirexia y dolor abdominal superior. La mayoría de las reacciones fueron de gravedad leve a moderada.
Tabla de reacciones adversas
En la Tabla 4 se incluyen las reacciones adversas registradas en la población total de seguridad tratada con Xolair en los ensayos clínicos, por sistema de clasificación de órganos y frecuencia de MedDRA. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Las categorías de frecuencia se definen como: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000) y muy raras (<1/10.000). Las reacciones notificadas en la fase de postcomercialización se enumeran con frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 4: Reacciones adversas
|
Infecciones e infestaciones | |
|
Poco frecuentes Raras |
Faringitis Infección parasitaria |
|
Trastornos de la sangre y del sistema linfático | |
|
No conocida |
Trombocitopenia idiopática, incluyendo casos graves |
|
Trastornos del sistema inmunológico | |
|
Raras No conocida |
Reacción anafiláctica, otros procesos alérgicos graves, desarrollo de anticuerpos frente a omalizumab Enfermedad del suero que puede cursar con fiebre y linfoadenopatía |
|
Trastornos del sistema nervioso | |
|
Frecuentes Poco frecuentes |
Cefalea* Síncope, parestesia, somnolencia, mareo |
|
Trastornos vasculares | |
|
Poco frecuentes |
Hipotensión postural, rubor |
|
Trastornos respiratorios, torácicos y mediastínicos | |
|
Poco frecuentes Raras No conocida |
Broncoespasmo alérgico, tos Laringoedema Vasculitis granulomatosa alérgica (es decir, síndrome de Churg Strauss) |
|
Trastornos gastrointestinales | |
|
Frecuentes Poco frecuentes |
Dolor abdominal superior** Signos y síntomas dispépticos, diarrea, náuseas |
|
Trastornos de la piel y del tejido subcutáneo | |
|
Poco frecuentes Raras No conocida |
Fotosensibilidad, urticaria, rash, prurito Angioedema Alopecia |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |
|
Raras |
Lupus eritematoso sistémico (LES) |
|
No conocidas |
Artralgia, mialgia, tumefacción de las articulaciones |
|
Trastornos generales y alteraciones en el lugar de administración | |
|
Muy frecuentes Frecuentes Poco frecuentes |
Pirexia** Reacciones en el lugar de la inyección tales como tumefacción, eritema, dolor, prurito Enfermedad pseudo-gripal, brazos hinchados, incremento de peso, fatiga |
*: Muy frecuentes en niños de 6 a <12 años de edad **: En niños de 6 a <12 años de edad
Descripcción de reacciones adversas seleccionadas Trastornos del sistema inmunológico Para mayor información, ver sección 4.4.
Anafilaxia
Rara vez se observaron reacciones anafilácticas en los ensayos clínicos. Sin embargo, después de una búsqueda acumulada en la base de datos de seguridad, se recogieron un total de 898 casos de anafilaxia post comercialización. En base a una exposición estimada de 566.923 paciente tratado años, resultó en una frecuencia aproximada de 0,20%.
Efectos tromboembólicos arteriales (ETA)
En ensayos clínicos controlados y durante los análisis intermedios de un estudio observacional, se observó un desequilibrio numérico de ETA. La definición de la variable compuesta ETA incluye derrame cerebral, ataque isquémico transitorio, infarto de miocardio, angina inestable y muerte cardiovascular (incluyendo muerte por causa desconocida). En el análisis final del estudio observacional, el índice de ETA por 1.000 pacientes años fue de 7,52 (115/15.286 pacientes años) para los pacientes tratados con Xolair y de 5,12 (51/9.963 pacientes años) para los pacientes control. En un análisis multivariado para controlar los factores basales disponibles de riesgo cardiovascular, la proporción de riesgo fue de 1,32 (intervalo de confianza del 95% 0,91-1,91). En un análisis segregado de un conjunto de ensayos clínicos, el cual incluyó todos los ensayos clínicos aleatorizados, doble ciego, controlados con placebo de 8 semanas o más de duración, el índice de ETA por 1.000 pacientes años fue de 2,69 (5/1.856 pacientes años) para los pacientes tratados con Xolair y de 2,38 (4/1.680 pacientes años) para los pacientes tratados con placebo (tasa de incidencia 1,13, intervalo de confianza del 95% 0,24-5,71).
Plaquetas
En los ensayos clínicos, pocos pacientes presentaron recuentos de plaquetas por debajo del límite inferior del intervalo normal de laboratorio. Ninguno de estos cambios se asoció con episodios hemorrágicos o con una disminución de la hemoglobina. En los seres humanos (pacientes a partir de 6 años de edad), a diferencia de los primates no humanos, no se ha observado ningún patrón de disminución persistente en el recuento de plaquetas (ver sección 5.3), aunque se han notificado casos aislados de trombocitopenia idiopática, incluyendo casos graves, en la fase de postcomercialización.
Infecciones _ parasitarias
En pacientes con un elevado riesgo crónico de infección helmíntica, un ensayo controlado con placebo demostró un ligero incremento numérico en la proporción de infección con omalizumab que no fue estadísticamente significativo. No se modificaron el curso, gravedad y respuesta al tratamiento de la infección (ver sección 4.4).
Lupus eritematoso sistémico
En pacientes con asma moderada a grave y urticaria crónica espontánea (UCE) se han notificado casos de lupus eritematoso sistémico (LES) en ensayos clínicos y etapa post comercialización. La patogénesis del lupus eritematoso sistémico no se conoce bien.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
No se ha determinado la dosis máxima tolerada de Xolair. Se han administrado dosis únicas intravenosas de hasta 4.000 mg a pacientes sin evidencia de toxicidad dependiente de la dosis. La mayor dosis acumulada que se administró a los pacientes durante un periodo de 20-semanas fue de 44.000 mg y esta dosis no produjo ningún efecto adverso agudo.
En caso de sospecha de una sobredosis, se deberá monitorizar al paciente para cualquier signo o síntoma anormal. Se deberá buscar e instaurar tratamiento médico adecuado.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: fármacos para enfermedades obstructivas de las vías respiratorias, otros agentes sistémicos para enfermedades obstructivas de las vías respiratorias, código ATC: R03DX05
Omalizumab es un anticuerpo monoclonal humanizado derivado del ADN recombinante que se une selectivamente a la inmunoglobulina E humana (IgE). El anticuerpo es una IgG1 kappa que contiene regiones de estructura humana con regiones determinantes de complementariedad de un anticuerpo de origen murino que se une a la IgE.
Mecanismo de acción
Omalizumab se une a la IgE y previene la unión de ésta al FcsRI (receptor de IgE de alta afinidad), reduciendo así la cantidad de IgE libre disponible para desencadenar la cascada alérgica. El tratamiento de pacientes atópicos con omalizumab dio como resultado una disminución acentuada de los receptores FcsRI en los basófilos.
Efectos farmacodinámicos
La liberación in vitro de histamina de los basófilos aislados de pacientes que habían sido tratados con Xolair se redujo aproximadamente en un 90% tras la estimulación con un alergeno en comparación con los valores previos al tratamiento.
En los ensayos clínicos, las concentraciones plasmáticas de IgE libre disminuyeron de manera dosis dependiente en la hora posterior a la primera dosis y se mantuvieron reducidas entre dosis. Un año después de interrumpir el tratamiento con Xolair los niveles de IgE volvieron a los niveles previos al tratamiento, sin que se observase efecto de rebote en los niveles de IgE después del periodo de blanqueo del medicamento.
Eficacia clínica y seguridad
Adultos y adolescentes >12 años de edad
La eficacia y seguridad de Xolair se demostró en un ensayo doble ciego controlado con placebo de 28 semanas (estudio 1) en el que participaron 419 pacientes con asma alérgica grave, de edades comprendidas entre los 12 y 79 años, con función pulmonar reducida (FEVi 40-80% del valor teórico) y un pobre control de los síntomas del asma a pesar de recibir dosis elevadas de corticosteroides inhalados y un agonista beta2 de larga duración. Los pacientes escogidos habían sufrido exacerbaciones asmáticas múltiples habiendo necesitado tratamiento con corticosteroides sistémicos o habiendo sido hospitalizados o atendidos de urgencia debido a una exacerbación asmática grave en el pasado año a pesar del tratamiento continuo con dosis elevadas de corticosteroides inhalados y un agonista beta2 de larga duración. Se administró Xolair vía subcutánea o placebo como tratamiento adicional a >1.000 microgramos de dipropionato de beclometasona (o equivalente) más un agonista beta2 de larga duración. Como tratamiento de mantenimiento se permitieron los corticosteroides orales, la teofilina y los antagonistas de los leucotrienos (22%, 27%, y 35% de pacientes, respectivamente).
Se tomó como variable principal la proporción de exacerbaciones asmáticas que requirieron tratamiento con dosis altas de corticosteroides sistémicos. Omalizumab redujo la proporción de exacerbaciones asmáticas en un 19% (p = 0,153). Posteriores evaluaciones que mostraron significancia estadística (p<0,05) en favor de Xolair incluyeron reducciones de las exacerbaciones graves (donde la función pulmonar de los pacientes se redujo por debajo del 60% del mejor valor personal, necesitando corticosteroides sistémicos) y de las visitas de urgencia relacionadas con el asma (incluyendo hospitalizaciones, sala de urgencias, y visitas al médico no programadas), y mejoría en la valoración global del médico con respecto a la efectividad del tratamiento, calidad de vida relacionada con el asma (AQL), síntomas del asma y función pulmonar.
En un análisis de subgrupos, era más probable que los pacientes con un valor total de IgE >76 Ul/ml previo al tratamiento experimentaran un beneficio clínicamente significativo a Xolair. En estos pacientes en el estudio 1, Xolair redujo la proporción de exacerbaciones asmáticas en un 40% (p = 0,002). Además, un mayor número de pacientes presentaron respuestas clínicamente significativas en la población con un valor total de IgE >76 UI/ml a través de un programa con Xolair de asma grave. La Tabla 5 incluye resultados de la población del estudio 1.
Tabla 5: Resultados del estudio 1
Población total estudio 1 Xolair Placebo
_N=209_N=210
Exacerbaciones asmáticas
Proporción por periodo de 0,74 0,92
28 semanas
Reducción %, valor p de razón de 19,4%, p = 0,153
tasas
Exacerbaciones de asma grave
Proporción por periodo de 0,24 0,48
28 semanas
Reducción %, valor p de razón de 50,1%, p = 0,002
tasas
Visitas de urgencia
Proporción por periodo de 0,24 0,43
28 semanas
Reducción %, valor p de razón de 43,9%, p = 0,038
tasas
Valoración global del médico
42,8%
47,8%
respondedores1 % 60,5%
Valor p2 <0,001
Mejoría AQL
% de pacientes con una mejoría 60,8%
>0,5
Valor p 0,008
En los estudios 3, 4 y 5, los pacientes tratados con Xolair presentaron reducciones en la proporción de exacerbaciones asmáticas del 37,5% (p = 0,027), 40,3% (p<0,001) y 57,6% (p<0,001), respectivamente, comparado con placebo.
En el estudio 6, los pacientes con asma alérgica significativamente más grave tratados con Xolair pudieron reducir su dosis de fluticasona a <500 microgramos/día sin deterioro del control del asma (60,3%) comparado con el grupo placebo (45,8%, p<0,05).
La puntuación de calidad de vida se midió utilizando el cuestionario de calidad de vida relacionada con el asma de Juniper. En los 6 estudios hubo una mejoría estadísticamente significativa con respecto a la puntuación de calidad de vida basal para los pacientes tratados con Xolair con respecto al grupo placebo o control.
Valoración global del médico con respecto a la efectividad del tratamiento:
La valoración global del médico se realizó en cinco de los estudios anteriormente mencionados como una amplia medida del control del asma realizada por el médico que trata al paciente. El médico pudo tener en cuenta el PEF (flujo espiratorio máximo), los síntomas durante el día y la noche, el uso de medicación de rescate, la espirometría y las exacerbaciones. En los cinco estudios una proporción significativamente superior de los pacientes tratados con Xolair declararon haber alcanzado una notable mejoría o un control completo del asma comparado con los pacientes tratados con placebo.
Niños de 6 a <12 años de edad
El soporte principal para la eficacia y seguridad de Xolair en el grupo de edad de 6 a <12 años se obtiene de un ensayo multicéntrico, randomizado, doble ciego, controlado con placebo (estudio 7).
El estudio 7 fue un ensayo controlado con placebo que incluyó un subgrupo específico (n=235) de pacientes tal y como se define en la presente indicación, quienes fueron tratados con corticoides inhalados a dosis altas (>500 pg/día de fluticasona o equivalente) más agonista beta de larga duración.
Según la opinión clínica del investigador, una exacerbación clínicamente significativa se definió como un empeoramiento de los síntomas del asma, la cual requería una dosis doble de corticosteroide inhalado con respecto al valor basal, durante un mínimo de 3 días y/o tratamiento de rescate con corticosteroides sistémicos (oral o intravenoso) durante un mínimo de 3 días.
En el subgrupo específico de pacientes con corticoides inhalados a dosis alta, el grupo de omalizumab tuvo una tasa inferior estadísticamente significativa de exacerbaciones asmáticas clínicamente significativas que el grupo placebo. A las 24 semanas, la diferencia en las tasas entre los grupos de tratamiento representó un descenso del 34% (razón de tasas 0,662, p = 0,047) relativo a placebo para los pacientes con omalizumab. En el segundo periodo de tratamiento doble ciego de 28 semanas la diferencia en las tasas entre los grupos de tratamiento representó un descenso del 63% (razón de tasas 0,37, p<0,001) relativo a placebo para los pacientes con omalizumab.
Durante el periodo de tratamiento doble ciego de 52 semanas (incluyendo la fase de esteroides a dosis fija de 24 semanas y la fase de ajuste de esteroides de 28 semanas) la diferencia en las tasas entre los grupos de tratamiento representó un descenso relativo del 50% (razón de tasas 0,504, p<0,001) en exacerbaciones para los pacientes con omalizumab.
El grupo de omalizumab mostró descensos mayores en el uso de agonistas beta como medicación de rescate que el grupo placebo al final del periodo de tratamiento de 52 semanas, aunque la diferencia entre los grupos de tratamiento no fue estadísticamente significativa. De la evaluación global de la efectividad del tratamiento al final del periodo de tratamiento doble ciego de 52 semanas en el subgrupo de pacientes graves con corticoides inhalados a dosis alta más agonistas beta de larga duración, la proporción de pacientes calificados como que tuvieron una efectividad de tratamiento “excelente” fue superior, y la proporción que tuvo una efectividad de tratamiento “moderada” o “débil” fue inferior en el grupo de omalizumab comparado con el grupo placebo; la diferencia entre grupos fue estadísticamente significativa (p<0,001), mientras que no hubo diferencias entre los grupos de omalizumab y placebo en las valoraciones de calidad de vida subjetiva de los pacientes.
5.2 Propiedades farmacocinéticas
Se ha estudiado la farmacocinética de omalizumab en pacientes adultos y adolescentes con asma alérgica.
Absorción
Tras la administración subcutánea, omalizumab se absorbe con una biodisponibilidad absoluta media del 62%. Después de administrar una dosis subcutánea única a pacientes adultos y adolescentes con asma, omalizumab se absorbió lentamente alcanzando concentraciones plasmáticas máximas después de una media de 7-8 días. La farmacocinética de omalizumab es lineal a dosis mayores de 0,5 mg/kg. Tras dosis múltiples de omalizumab, las áreas bajo la curva de concentración plasmática-tiempo del Día 0 al Día 14 en estado estacionario fueron de hasta 6-veces las obtenidas tras la primera dosis.
La administración de Xolair fabricado como una formulación liofilizada o líquida dió como resultado perfiles similares de concentración sérica-tiempo de omalizumab.
Distribución
In vitro, omalizumab forma complejos de tamaño limitado con la IgE. No se han observado tanto in vitro como in vivo complejos precipitantes ni complejos con pesos moleculares superiores a un millón de Daltons. Tras la administración subcutánea el volumen de distribución aparente en los pacientes fue de 78 ± 32 ml/kg.
Eliminación
El aclaramiento de omalizumab comprende procesos de aclaramiento de las IgG, así como, aclaramiento a través de uniones específicas y formación de complejos con su ligando específico, la IgE. La eliminación hepática de IgG incluye una degradación en el sistema reticuloendotelial y células endoteliales. La IgG inalterada también se excreta en la bilis. La vida media de eliminación plasmática de omalizumab en pacientes asmáticos fue de un promedio de 26 días, con un aclaramiento aparente promedio de 2,4 ± 1,1 ml/kg/día. Además, la duplicación del peso corporal hace que el aclaramiento aparente sea de aproximadamente el doble.
Características demográficas
Edad, Raza/Grupo étnico, Sexo, Indice de Masa Corporal
Se analizó la farmacocinética poblacional de Xolair con el fin de evaluar los efectos de las características demográficas. El análisis de los escasos datos disponibles indica que no es necesario efectuar un ajuste de la dosis en función de la edad (6-76 años), raza/grupo étnico, sexo o Índice de Masa Corporal (ver sección 4.2).
Insuficiencia renal y hepática
No se dispone de datos farmacocinéticos o farmacodinámicos en pacientes con insuficiencia renal o hepática (ver secciones 4.2 y 4.4).
5.3 Datos preclínicos sobre seguridad
Se ha estudiado la seguridad de omalizumab en monos cinomolgos, ya que el omalizumab se une a las IgE del cinomolgos y humanas con una afinidad similar. Se detectaron anticuerpos a omalizumab en algunos monos tras la administración subcutánea o intravenosa repetida. Sin embargo, no se observó toxicidad aparente, tales como enfermedad mediada por inmunocomplejos o citotoxicidad dependiente de complemento. No hubo evidencia de una respuesta anafiláctica debido a la desgranulación de los mastocitos en monos cinomolgos.
La administración crónica de omalizumab a niveles de dosis de hasta 250 mg/kg (al menos 14 veces la dosis clínica más alta recomendada en mg/kg de acuerdo con la tabla de dosis recomendada) fue bien tolerada en primates no humanos (tanto en animales adultos como en jóvenes), con la excepción de un descenso en las plaquetas sanguíneas dependiente de la dosis y de la edad, con una mayor sensibilidad en animales jóvenes. La concentración plasmática necesaria para alcanzar un descenso del 50% en las plaquetas con respecto al valor basal en monos adultos cinomolgos fue aproximadamente de 4 a 20 veces más elevada que la concentración plasmática clínica máxima anticipada. Además, se observó inflamación y hemorragia aguda en el lugar de la inyección en monos cinomolgos.
No se han realizado estudios de carcinogenicidad formal con omalizumab.
En los estudios de reproducción en monos cinomolgos, dosis subcutáneas de hasta 75 mg/kg por semana (al menos 8 veces la dosis clínica más alta recomendada en mg/kg durante un periodo de 4-semanas) no provocaron toxicidad materna, embriotoxicidad o teratogenicidad cuando se administró durante toda la organogénesis y no provocó efectos adversos sobre el crecimiento fetal o neonatal cuando se administró durante la fase final de la gestación, parto y lactancia.
Omalizumab se excretó en la leche materna en monos cinomolgos. Los niveles de omalizumab en la leche fueron del 0,15% con respecto a la concentración sérica materna.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo
Sacarosa
L-histidina
Hidrocloruro de L-histidina monohidrato Polisorbato 20
Disolvente
Agua para preparaciones inyectables
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez 4 años.
Después de la reconstitución
Se ha demostrado la estabilidad física y química del medicamento reconstituido durante 8 horas entre 2°C y 8°C y durante 4 horas a 30°C.
Desde el punto de vista microbiológico, el medicamento deberá utilizarse inmediatamente después de su reconstitución. Si no se utiliza inmediatamente, el tiempo y las condiciones de conservación previas a su utilización son responsabilidad del manipulador y no deberían ser normalmente superiores a 8 horas entre 2°C y 8°C o 2 horas a 25°C.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Vial de polvo: Vial de vidrio incoloro tipo I, transparente, con tapón de caucho butilo y sello extraíble de color gris.
Ampolla de disolvente: Ampolla de vidrio incoloro tipo I, transparente, conteniendo 2 ml de agua para inyectables.
El envase contiene un vial de polvo para solución inyectable y una ampolla de agua para inyectables.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El medicamento liofilizado necesita entre 15 y 20 minutos para disolverse, aunque en algunos casos puede requerir más tiempo. El medicamento completamente reconstituido tiene un aspecto transparente a ligeramente opalescente, incoloro a amarillo parduzco claro y puede presentar unas cuantas burbujas pequeñas o espuma alrededor del borde del vial. Debido a la viscosidad del medicamento reconstituido deberá tenerse precaución de extraer todo el medicamento del vial antes de eliminar el aire o el exceso de solución de la jeringa con el fin de obtener los 0,6 ml.
Para preparar los viales de Xolair 75 mg para la administración subcutánea, siga por favor las siguientes instrucciones:
1. Retirar 0,9 ml de agua para inyectables de la ampolla con una jeringa equipada con una aguja gruesa de extracción (calibre 18).
2. Con el vial colocado en posición vertical sobre una superficie plana, insertar la aguja e inocular el agua para inyectables en el vial conteniendo el polvo liofilizado utilizando las técnicas asépticas estándar, dirigiendo el agua para inyectables directamente sobre el polvo.
3. Manteniendo el vial en posición vertical, removerlo vigorosamente (sin agitar) durante 1 minuto aproximadamente para humedecer el polvo uniformemente.
4. Para ayudar a la disolución tras completar el paso 3, remover suavemente el vial durante 5-10 segundos aproximadamente cada 5 minutos con el fin de disolver el polvo restante.
Observe que en ocasiones puede necesitar más de 20 minutos para disolver el polvo completamente. Si este es el caso, repita el paso 4 hasta que desaparezcan las partículas gelatinosas de la solución.
Una vez el medicamento se haya disuelto completamente, no deben quedar partículas gelatinosas visibles en la solución. Las pequeñas burbujas o espuma alrededor del borde del vial son completamente normales. El medicamento reconstituido tiene un aspecto transparente a ligeramente opalescente, incoloro a amarillo parduzco claro. No utilice el producto si observa partículas sólidas.
5. Invertir el vial durante un mínimo de 15 segundos con el fin de que la solución fluya hacia el tapón. Utilizando una jeringa nueva de 3-ml equipada con una aguja gruesa de extracción (calibre 18), insertar la aguja en el vial invertido. Manteniendo el vial en posición invertida colocar el extremo de la aguja al final de la solución en el vial cuando extraiga la solución con la jeringa. Antes de extraer la aguja del vial, tire del émbolo y llévelo hasta el fondo del cilindro de la jeringa con el fin de extraer toda la solución del vial invertido.
6. Reemplazar la aguja de calibre 18 por una de calibre 25 para inyección subcutánea.
7. Eliminar el aire, las burbujas grandes y cualquier exceso de solución con el fin de obtener la dosis requerida de 0,6 ml. Puede quedar una fina capa de pequeñas burbujas en la superficie de la solución contenida en la jeringa. Como la solución es ligeramente viscosa, la administración de la solución por inyección subcutánea puede durar entre 5 y 10 segundos.
El vial proporciona 0,6 ml (75 mg) de Xolair.
8. Las inyecciones se administran por vía subcutánea en la región deltoidea del brazo o en el muslo.
Xolair 75 mg polvo para solución inyectable se suministra en un vial de un solo uso.
Desde el punto de vista microbiológico, el medicamento debe utilizarse inmediatamente después de la
reconstitución (ver sección 6.3).
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto
con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/319/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 25 Octubre 2005 Fecha de la última renovación: 22 Junio 2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
1. NOMBRE DEL MEDICAMENTO
Xolair 150 mg polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un vial contiene 150 mg de omalizumab*.
Después de la reconstitución un vial contiene 125 mg/ml de omalizumab (150 mg en 1,2 ml).
*Omalizumab es un anticuerpo monoclonal humanizado obtenido mediante la tecnología del ADN recombinante, a partir de una línea celular mamífera de ovario de hámster chino (OHC).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: liofilizado de color blanco a blanquecino.
Disolvente: solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Asma alérgica
Xolair está indicado en adultos, adolescentes y niños (de 6 a <12 años).
El tratamiento con Xolair deberá ser considerado únicamente para pacientes con asma mediada de forma convincente por IgE (inmunoglobulina E) (ver sección 4.2).
Adultos y adolescentes (a _partir de 12 años de edad)
Xolair está indicado para mejorar el control del asma cuando se administra como tratamiento adicional en pacientes con asma alérgica grave persistente que presentan test cutáneo positivo o reactividad in vitro a aeroalergenos perennes y con función pulmonar reducida (FEVi <80%) así como, síntomas frecuentes durante el día o despertares por la noche y que han presentado múltiples exacerbaciones asmáticas graves documentadas, a pesar de utilizar corticosteroides diarios inhalados a dosis altas, más un agonista beta2 inhalado de larga duración.
Niños (6 a <12 años de edad)
Xolair está indicado para mejorar el control del asma cuando se administra como tratamiento adicional en pacientes con asma alérgica grave persistente que presentan test cutáneo positivo o reactividad in vitro a aeroalergenos perennes y síntomas frecuentes durante el día o despertares por la noche y que han presentado múltiples exacerbaciones asmáticas graves documentadas, a pesar de utilizar corticosteroides diarios inhalados a dosis altas, más un agonista beta2 inhalado de larga duración.
Urticaria crónica espontánea (UCE)
Xolair está indicado como tratamiento adicional, para el tratamiento de la urticaria crónica espontánea en pacientes adultos y adolescentes (a partir de 12 años) con respuesta inadecuada al tratamiento con antihistamínicos H1.
4.2 Posología y forma de administración
El tratamiento con Xolair debe iniciarlo un médico experimentado en el diagnóstico y tratamiento del asma grave persistente o urticaria crónica espontánea.
Asma alérgica
Posología
La dosis apropiada y la frecuencia de administración de Xolair se determina a partir de la concentración basal de IgE (UI/ml), medida antes de iniciar el tratamiento, y del peso corporal (kg). A efectos de la asignación de la dosis, antes de la administración de la dosis inicial se debe determinar la concentración de IgE en los pacientes mediante cualquier método comercial que analice la IgE plasmática total. En base a estas determinaciones, podrán ser necesarios en cada administración de 75 a 600 mg de Xolair en 1 a 4 inyecciones.
Era menos probable que experimentaran beneficio los pacientes con un valor de IgE inferior a 76 UI/ml (ver sección 5.1). Los médicos prescriptores deberán asegurar que los pacientes adultos y adolescentes con una IgE por debajo de 76 UI/ml y los niños (6 a < 12 años de edad) con una IgE por debajo de 200 UI/ml presenten una reactividad in vitro inequívoca (RAST) al alergeno perenne antes de iniciar el tratamiento.
Ver Tabla 1 de conversión y Tablas 2 y 3 para la determinación de dosis en adultos, adolescentes y niños (de 6 a <12 años).
No debe administrarse Xolair a pacientes cuya concentración basal de IgE o peso corporal en kilogramos, excedan los límites indicados en la tabla de dosis.
La dosis máxima recomendada es de 600 mg de omalizumab cada dos semanas.
Tabla 1: Conversión de la dosis al número de viales, número de inyecciones y volumen de inyección total para cada administración
|
Dosis (mg) Número de viales Número de inyecciones 75 mg a 150 mg b |
Volumen total de inyección (ml) | |||
|
75 |
1c |
0 |
1 |
0,6 |
|
150 |
0 |
1 |
1 |
1,2 |
|
225 |
1c |
1 |
2 |
1,8 |
|
300 |
0 |
2 |
2 |
2,4 |
|
375 |
1c |
2 |
3 |
3,0 |
|
450 |
0 |
3 |
3 |
3,6 |
|
525 |
1c |
3 |
4 |
4,2 |
|
600 |
0 |
4 |
4 |
4,8 |
a 0,6 ml = volumen máximo proporcionado por vial (Xolair 75 mg). b 1,2 ml = volumen máximo proporcionado por vial (Xolair 150 mg). c o utilizar 0,6 ml de un vial de 150 mg.
|
Peso corporal (kg) | ||||||||||
|
IgE basal |
>20 |
>25 |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>90- |
>125 |
|
(UI/ml) |
-25 |
-30 |
40 |
50 |
60 |
70 |
80 |
90 |
125 |
-150 |
|
>30-100 |
75 |
75 |
75 |
150 |
150 |
150 |
150 |
150 |
300 |
300 |
|
>100-200 |
150 |
150 |
150 |
300 |
300 |
300 |
300 |
300 |
450 |
600 |
|
>200-300 |
150 |
150 |
225 |
300 |
300 |
450 |
450 |
450 |
600 | |
|
>300-400 |
225 |
225 |
300 |
450 |
450 |
450 |
600 |
600 | ||
|
>400-500 |
225 |
300 |
450 |
450 |
600 |
600 | ||||
|
>500-600 |
300 |
300 |
450 |
600 |
600 | |||||
|
>600-700 |
300 |
450 |
600 | |||||||
|
>700-800 | ||||||||||
|
>800-900 |
ADMINISTRACIÓN CADA | |||||||||
|
>900- 1000 |
2 SEMANAS VER TABLA 3 | |||||||||
|
>1000- 1100 | ||||||||||
|
Peso corporal (kg) | ||||||||||
|
IgE basal |
>20- |
>25- |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>90- |
>125- |
|
(UI/ml) |
25 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
125 |
150 |
|
>30-100 |
ADMINISTRACIÓN CADA | |||||||||
|
>100-200 |
4 SEMANAS VER TABLA 2 | |||||||||
|
>200-300 |
375 | |||||||||
|
>300-400 |
450 |
525 | ||||||||
|
>400-500 |
375 |
375 |
525 |
600 | ||||||
|
>500-600 |
375 |
450 |
450 |
600 | ||||||
|
>600-700 |
225 |
375 |
450 |
450 |
525 | |||||
|
>700-800 |
225 |
225 |
300 |
375 |
450 |
450 |
525 |
600 | ||
|
>800-900 |
225 |
225 |
300 |
375 |
450 |
525 |
600 | |||
|
>900- |
225 |
300 |
375 |
450 |
525 |
600 | ||||
|
1000 | ||||||||||
|
>1000- 1100 |
225 |
300 |
375 |
450 |
600 | |||||
|
>1100- |
300 |
300 |
450 |
525 |
600 |
NO ADMINISTRAR - |
no se dispone de | |||
|
1200 |
datos para la recomendación de dosis | |||||||||
|
>1200- 1300 |
300 |
375 |
450 |
525 | ||||||
|
>1300- 1500 |
300 |
375 |
525 |
600 | ||||||
Duración del tratamiento, monitorización y ajuste de dosis
Xolair está indicado para tratamiento a largo plazo. Los ensayos clínicos han demostrado que son necesarias un mínimo de 12-16 semanas para que el tratamiento con Xolair demuestre efectividad. A las 16 semanas de iniciar el tratamiento con Xolair, los pacientes deberán ser evaluados por su médico con respecto a la efectividad del tratamiento antes de administrar inyecciones posteriores. La decisión de continuar con Xolair tras las 16 semanas, o en ocasiones posteriores, debe estar basada en si se observa una notable mejoría en el control global del asma (ver sección 5.1; Valoración global del médico con respecto a la efectividad del tratamiento).
La interrupción del tratamiento con Xolair generalmente da lugar a un retorno de las concentraciones elevadas de IgE libre y de los síntomas asociados. Los valores de IgE total son elevados durante el tratamiento y siguen siéndolo hasta un año después de la interrupción del mismo. Por lo tanto, no puede utilizarse la reevaluación de los valores de IgE durante el tratamiento con Xolair como guía para la determinación de la dosis. La determinación de la dosis tras interrupciones de tratamiento de menos de un año de duración debe basarse en las concentraciones plasmáticas de IgE obtenidas en la determinación de dosis inicial. Si el tratamiento con Xolair se ha interrumpido por más de un año deberán de volver a medirse las concentraciones plasmáticas de IgE total para la determinación de la dosis.
Las dosis deberán ajustarse en caso de variaciones significativas del peso corporal (ver Tablas 2 y 3).
Urticaria crónica espontánea (UCE)
Posología
La dosis recomendada es de 300 mg por inyección subcutánea cada cuatro semanas.
Se recomienda a los prescriptores que reevaluen periódicamente la necesidad de continuar el tratamiento.
La experiencia en ensayos clínicos del tratamiento a largo plazo durante más de 6 meses en esta indicación es limitada.
Poblaciones especiales
Pacientes de edad avanzada (a _partir de 65 años)
Aunque se dispone de datos limitados sobre el uso de Xolair en pacientes mayores de 65 años, no existe evidencia de que los pacientes de edad avanzada requieran una dosis diferente de la de pacientes adultos más jóvenes.
Insuficiencia renal o hepática
No se dispone de estudios sobre el efecto de la insuficiencia renal o hepática en la farmacocinética de omalizumab. Debido a que el aclaramiento de omalizumab a dosis clínicas se lleva a cabo fundamentalmente por el sistema reticuloendotelial (SRE), es improbable que se vea alterado en caso de insuficiencia renal o hepática. Xolair deberá administrarse con precaución en estos pacientes, mientras no se recomiende un ajuste especial de la dosis (ver sección 4.4).
Población pediátrica
En asma alérgica, no se ha establecido la seguridad y eficacia de Xolair en pacientes pediátricos menores de 6 años. No se dispone de datos.
En UCE, no se ha establecido la seguridad y eficacia de Xolair en pacientes pediátricos menores de 12 años.
Forma de administración
Para administración subcutánea únicamente. No administrar por vía intravenosa o intramuscular.
Las inyecciones se administran vía subcutánea en la región deltoidea del brazo. Si por alguna razón no pueden administrarse en esta zona, podrán administrase alternativamente en el muslo.
Existe experiencia limitada con respecto a la autoadministración de Xolair. Por lo tanto, está previsto que el tratamiento sea administrado únicamente por el profesional sanitario.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6 y también la sección “información para el profesional sanitario” del prospecto.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
General
Xolair no está indicado para el tratamiento de las exacerbaciones asmáticas, broncoespasmo o estados asmáticos de carácter agudo.
No se ha estudiado el efecto de Xolair en pacientes con síndrome de hiperinmunoglobulinemia E o aspergilosis broncopulmonar alérgica, ni en la prevención de reacciones anafilácticas, incluyendo las provocadas por alergias alimentarias, dermatitis atópica, o rinitis alérgica. Xolair no está indicado en el tratamiento de estas patologías.
El tratamiento con Xolair tampoco se ha estudiado en pacientes con enfermedades autoinmunes, procesos mediados por inmunocomplejos, o insuficiencia renal o hepática preexistente (ver sección 4.2). Se deberá tener precaución cuando se administre Xolair en esta población de pacientes.
No se recomienda la interrupción brusca de los corticosteroides sistémicos o inhalados tras la iniciación del tratamiento con Xolair. El descenso de los corticosteroides debe realizarse bajo la supervisión directa de un médico y puede ser necesario que se realice gradualmente.
Trastornos del sistema inmulógico
Reacciones alérgicas tipo I
Pueden producirse reacciones alérgicas tipo I locales o sistémicas, incluyendo anafilaxia y shock anafiláctico durante el tratamiento con omalizumab, incluso con inicio tras un tratamiento de larga duración. La mayoría de estas reacciones se produjeron durante las 2 horas posteriores a la primera y siguientes inyecciones de Xolair, pero algunas se iniciaron pasadas las 2 horas e incluso pasadas 24 horas tras la inyección. Por lo tanto, se deberán tener siempre disponibles medicamentos para el tratamiento inmediato de reacciones anafilácticas tras la administración de Xolair. Se deberá informar al paciente de que estas reacciones son posibles y que si se producen deberán solicitar atención médica de inmediato. Un antecedente de anafilaxia no asociado a omalizumab puede suponer un factor de riesgo de sufrir una reacción anafiláctica tras la administración de Xolair.
Se han detectado anticuerpos contra omalizumab en un pequeño número de pacientes en ensayos clínicos (ver sección 4.8). No se conoce bien la relevancia clínica de anticuerpos anti-Xolair.
Enfermedad del suero
Se ha observado enfermedad del suero y reacciones semejantes a la enfermedad del suero, que son reacciones alérgicas tipo III retardadas, en pacientes tratados con anticuerpos monoclonales humanizados incluido omalizumab. El supuesto mecanismo fisiopatológico incluye formación y deposición de inmunocomplejos debido al desarrollo de anticuerpos contra omalizumab. El inicio del cuadro se produce normalmente a los 1-5 días tras la administración de la primera o siguientes inyecciones e incluso tras un tratamiento de larga duración. Los síntomas que sugieren la enfermedad del suero incluyen artritis/artralgias, rash (urticaria u otras formas), fiebre y linfoadenopatía. Los antihistamínicos y corticosteroides pueden ser útiles para prevenir o tratar estas alteraciones y se deberá advertir a los pacientes que notifiquen cualquier síntoma sospechoso.
Síndrome de Churg-Strauss y síndrome hipereosinofílico
Los pacientes con asma grave pueden presentar raramente síndrome hipereosinofílico sistémico o vasculitis granulomatosa eosinofílica alérgica (Síndrome de Churg-Strauss), los cuales son normalmente tratados con corticosteroides sistémicos.
En raras ocasiones, los pacientes en tratamiento con medicamentos antiasmáticos, incluyendo omalizumab, pueden presentar o desarrollar eosinofilia sistémica y vasculitis. Estas reacciones están normalmente asociadas con la reducción del tratamiento con corticosteroides orales.
En estos pacientes, los médicos deberán estar alerta ante el desarrollo de eosinofilia importante, rash vasculítico, empeoramiento de los síntomas pulmonares, anormalidades en el seno paranasal, complicaciones cardíacas, y/o neuropatía.
Deberá considerarse la interrupción del tratamiento con omalizumab en todos aquellos casos graves que cursen con alteraciones del sistema inmune mencionadas anteriormente.
Infecciones parasitarias (helmínticos)
Las IgE pueden estar involucradas en la respuesta inmunológica a algunas infecciones helmínticas. En pacientes con un elevado riesgo crónico de infección helmíntica, un ensayo controlado con placebo en pacientes alérgicos demostró un ligero incremento en la proporción de infección con omalizumab, aunque no se modificó el curso, gravedad y respuesta al tratamiento de la infección. La proporción de infección helmíntica en el programa clínico global, el cual no fue diseñado para detectar este tipo de infecciones, fue inferior a 1 en 1.000 pacientes. Sin embargo, deberá garantizarse precaución en pacientes con elevado riesgo de infección helmíntica, en particular cuando viajen a zonas donde las infecciones helmínticas son endémicas. Si los pacientes no responden al tratamiento antihelmíntico recomendado, deberá considerarse la interrupción del tratamiento con Xolair.
4.5 Interacción con otros medicamentos y otras formas de interacción
Dado que las IgE pueden estar relacionadas con la respuesta immunológica a algunas infecciones por helmínticos, Xolair puede reducir indirectamente la eficacia de medicamentos utilizados para el tratamiento de infecciones helmínticas o por otros parásitos (ver sección 4.4).
Las enzimas del citocromo P450, las bombas de eflujo y los mecanismos de unión a proteínas no se hallan implicados en el aclaramiento de omalizumab; por ello, existe un bajo potencial de interacciones farmacológicas. No se han realizado estudios de interacción de otros medicamentos o vacunas con Xolair. No existe un motivo farmacológico para esperar que los medicamentos prescritos frecuentemente en el tratamiento del asma o UCE interaccionen con omalizumab.
Asma alérgica
En los ensayos clínicos Xolair se utilizó frecuentemente asociado a corticosteroides inhalados y orales, beta agonistas inhalados de corta y larga duración, antagonistas de los leucotrienos, teofilinas y antihistamínicos orales. No hubo indicios de que estos medicamentos utilizados habitualmente como antiasmáticos puedan afectar a la seguridad de Xolair. Se dispone de datos limitados sobre el uso de Xolair en combinación con inmunoterapia específica (terapia de hiposensibilización). En un ensayo clínico donde Xolair se administró conjuntamente con inmunoterapia, se observó que la seguridad y eficacia de Xolair en combinación con inmunoterapia específica, no fue diferente a la de Xolair solo.
Urticaria crónica espontánea (UCE)
En ensayos clínicos en UCE, Xolair se utilizó junto con antihistamínicos (anti-H1, anti-H2) y antagonistas del receptor de leucotrieno (ARLTs). No hubo evidencia de que la seguridad de omalizumab se alterase cuando se utilizaba con estos medicamentos en relación a su perfil de seguridad conocido en asma alérgica. Además, un análisis farmacocinético poblacional no mostró un efecto relevante de los antihistamínicos H2 y ARLTs sobre la farmacocinética de omalizumab (ver sección 5.2).
Población pediátrica
Los ensayos clínicos en UCE incluyeron algunos pacientes de 12 a 17 años de edad que usaron Xolair junto con antihistamínicos (anti-H1, anti-H2) y ARLTs. No se realizaron ensayos en niños menores de 12 años.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Hay datos limitados relativos al uso de omalizumab en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción (ver sección 5.3). Omalizumab atraviesa la barrera placentaria y se desconoce el daño potencial sobre el feto. Omalizumab se ha asociado con descensos de las plaquetas sanguíneas, dependientes de la edad en primates no humanos, con una sensibilidad relativamente superior en animales jóvenes (ver sección 5.3). Xolair no debería utilizarse durante el embarazo excepto si fuese claramente necesario.
Lactancia
Se desconoce si omalizumab se excreta en la leche materna. Los datos disponibles en primates no humanos muestran que omalizumab se excreta en la leche (ver sección 5.3). No se puede excluir el riesgo en recién nacidos/niños. Omalizumab no se debe administrar durante la lactancia.
Fertilidad
No hay datos de fertilidad en humanos para omalizumab. En estudios de fertilidad no clínicos diseñados específicamente en primates no humanos, incluidos los estudios de apareamiento, no se observó empeoramiento de la fertilidad en machos o hembras tras dosis repetidas con omalizumab de hasta 75 mg/kg. Además, no se observaron efectos genotóxicos en estudios separados no clínicos de genotoxicidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Xolair sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Asma alérgica
Resúmen del perfil de seguridad
Las reacciones adversas notificadas más frecuentemente durante los ensayos clínicos en pacientes adultos y adolescentes a partir de 12 años de edad fueron cefalea y reacciones en el lugar de la inyección, que incluían dolor, tumefacción, eritema y prurito. Las reacciones adversas notificadas más frecuentemente en los ensayos clínicos en niños de 6 a <12 años de edad fueron cefalea, pirexia y dolor abdominal superior. La mayoría de las reacciones fueron de gravedad leve a moderada.
Tabla de reacciones adversas
En la Tabla 4 se incluyen las reacciones adversas registradas en la población total de seguridad tratada con Xolair en los ensayos clínicos, por sistema de clasificación de órganos y frecuencia de MedDRA. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Las categorías de frecuencia se definen como: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000) y muy raras (<1/10.000). Las reacciones notificadas en la fase de postcomercialización se enumeran con frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 4: Reacciones adversas en asma alérgica
|
Infecciones e infestaciones | |
|
Poco frecuentes Raras |
Faringitis Infección parasitaria |
|
Trastornos de la sangre y del sistema linfático | |
|
No conocida |
Trombocitopenia idiopática, incluyendo casos graves |
|
Trastornos del sistema inmunológico | |
|
Raras No conocida |
Reacción anafiláctica, otros procesos alérgicos graves, desarrollo de anticuerpos frente a omalizumab Enfermedad del suero que puede cursar con fiebre y linfoadenopatía |
|
Trastornos del sistema nervioso | |
|
Frecuentes Poco frecuentes |
Cefalea* Síncope, parestesia, somnolencia, mareo |
|
Trastornos vasculares | |
|
Poco frecuentes |
Hipotensión postural, rubor |
|
Trastornos respiratorios, torácicos y mediastínicos | |
|
Poco frecuentes Raras No conocida |
Broncoespasmo alérgico, tos Laringoedema Vasculitis granulomatosa alérgica (es decir, síndrome de Churg Strauss) |
|
Trastornos gastrointestinales | |
|
Frecuentes Poco frecuentes |
Dolor abdominal superior** Signos y síntomas dispépticos, diarrea, náuseas |
|
Trastornos de la piel y del tejido subcutáneo | |
|
Poco frecuentes Raras No conocida |
Fotosensibilidad, urticaria, rash, prurito Angioedema Alopecia |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |
|
Raras |
Lupus eritematoso sistémico (LES) |
|
No conocidas |
Artralgia, mialgia, tumefacción de las articulaciones |
|
Trastornos generales y alteraciones en el lugar de administración | |
|
Muy frecuentes Frecuentes Poco frecuentes |
Pirexia** Reacciones en el lugar de la inyección tales como tumefacción, eritema, dolor, prurito Enfermedad pseudo-gripal, brazos hinchados, incremento de peso, fatiga |
*: Muy frecuentes en niños de 6 a <12 años de edad **: En niños de 6 a <12 años de edad
Urticaria crónica espontánea (UCE)
Resúmen del perfil de seguridad
Se investigó la seguridad y tolerancia de omalizumab con dosis de 75 mg, 150 mg y 300 mg cada cuatro semanas en 975 pacientes con UCE, 242 de los cuales recibieron placebo. En global,
733 pacientes se trataron con omalizumab durante 12 semanas y 490 pacientes durante 24 semanas. De estos, 412 pacientes se trataron durante 12 semanas y 333 pacientes se trataron durante 24 semanas a la dosis de 300 mg.
Tabla de reacciones adversas
En otra tabla (Tabla 5) se muestran las reacciones adversas para la indicación de UCE como resultado de las diferentes dosis y poblaciones de tratamiento (con factores de riesgo significativamente diferentes, comorbilidades, medicaciones concomitantes y edades [p.ej. ensayos clínicos en asma que incluyeron niños de 6-12 años de edad]).
En la Tabla 5 se incluyen las reacciones adversas (acontecimientos ocurridos en >1% de los pacientes en cualquiera de los grupos de tratamiento y >2% más frecuentemente en cualquiera de los grupos de tratamiento con omalizumanb que con placebo (después de la revisión médica)) notificadas con la dosis de 300 mg en los tres ensayos de fase III agrupados. Las reacciones adversas presentadas se dividen en dos grupos: las identificadas en los periodos de tratamiento de 12-semanas y de 24-semanas.
Las reacciones adversas se incluyen por sistema de clasificación de órganos de MedDRA. Las reacciones adversas se clasifican por frecuencia dentro de cada sistema de órganos, incluyendo primero las reacciones más frecuentes. Para cada reacción adversa, la correspondiente categoría de frecuencia se basa en la siguiente convención: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 5: Reacciones adversas de la base de datos de seguridad de UCE agrupada (día 1 a semana 24) a la dosis de 300 mg de omalizumab
Ensayos con omalizumab 1, 2 y 3 Categoría de frecuencia
12-Semanas _agrupados_
|
Placebo N=242 |
300 mg N=412 | ||
|
Infecciones e infestaciones | |||
|
Sinusitis |
5 (2,1%) |
20 (4,9%) |
Frecuente |
|
Trastornos del sistema nervioso | |||
|
Cefalea |
7 (2,9%) |
25 (6,1%) |
Frecuente |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |||
|
Artralgia |
1 (0,4%) |
12 (2,9%) |
Frecuente |
|
Trastornos generales y alteraciones en el lugar de administración | |||
|
Reacción en el lugar de la inyección* |
2 (0,8%) |
11 (2,7%) |
Frecuente |
|
Ensayos con omalizumab 1 y 3 agrupados |
Categoría de frecuencia | ||
|
24-Semanas |
Placebo N=163 |
300 mg N=333 | |
|
Infecciones e infestaciones Infección de las vías altas del tracto respiratorio |
5 (3,1%) |
19 (5,7%) |
Frecuente |
* A pesar de no mostrar una diferencia del 2% con respecto a placebo, se incluyeron las reacciones en el lugar de la inyección ya que todos los casos fueron evaluados como relacionados causalmente al tratamiento en estudio.
Descripción de reacciones adversas seleccionadas en relación a las indicaciones de asma alérgica y UCE
No se han obtenido datos relevantes en los ensayos clínicos en UCE que requiriesen una modificación de las secciones siguientes.
Trastornos del sistema inmunológico Para mayor información, ver sección 4.4.
Anafilaxia
Rara vez se observaron reacciones anafilácticas en los ensayos clínicos. Sin embargo, después de una búsqueda acumulada en la base de datos de seguridad, se recogieron un total de 898 casos de anafilaxia post comercialización. En base a una exposición estimada de 566.923 paciente tratado años, resultó en una frecuencia aproximada de 0,20%.
Efectos tromboembólicos arteriales (ETA)
En ensayos clínicos controlados y durante los análisis intermedios de un estudio observacional, se observó un desequilibrio numérico de ETA. La definición de la variable compuesta ETA incluye derrame cerebral, ataque isquémico transitorio, infarto de miocardio, angina inestable y muerte cardiovascular (incluyendo muerte por causa desconocida). En el análisis final del estudio observacional, el índice de ETA por 1.000 pacientes años fue de 7,52 (115/15.286 pacientes años) para los pacientes tratados con Xolair y de 5,12 (51/9.963 pacientes años) para los pacientes control. En un análisis multivariado para controlar los factores basales disponibles de riesgo cardiovascular, la proporción de riesgo fue de 1,32 (intervalo de confianza del 95% 0,91-1,91). En un análisis segregado de un conjunto de ensayos clínicos, el cual incluyó todos los ensayos clínicos aleatorizados, doble ciego, controlados con placebo de 8 semanas o más de duración, el índice de ETA por 1.000 pacientes años fue de 2,69 (5/1.856 pacientes años) para los pacientes tratados con Xolair y de 2,38 (4/1.680 pacientes años) para los pacientes tratados con placebo (tasa de incidencia 1,13, intervalo de confianza del 95% 0,24-5,71).
Plaquetas
En los ensayos clínicos, pocos pacientes presentaron recuentos de plaquetas por debajo del límite inferior del intervalo normal de laboratorio. Ninguno de estos cambios se asoció con episodios hemorrágicos o con una disminución de la hemoglobina. En los seres humanos (pacientes a partir de 6 años de edad), a diferencia de los primates no humanos, no se ha observado ningún patrón de disminución persistente en el recuento de plaquetas (ver sección 5.3), aunque se han notificado casos aislados de trombocitopenia idiopática, incluyendo casos graves, en la fase de postcomercialización.
Infecciones _ parasitarias
En pacientes alérgicos con un elevado riesgo crónico de infección helmíntica, un ensayo controlado con placebo demostró un ligero incremento numérico en la proporción de infección con omalizumab que no fue estadísticamente significativo. No se modificaron el curso, gravedad y respuesta al tratamiento de la infección (ver sección 4.4).
Lupus eritematoso sistémico
En pacientes con asma moderada a grave y urticaria crónica espontánea (UCE) se han notificado casos de lupus eritematoso sistémico (LES) en ensayos clínicos y etapa post comercialización. La patogénesis del lupus eritematoso sistémico no se conoce bien.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
No se ha determinado la dosis máxima tolerada de Xolair. Se han administrado dosis únicas intravenosas de hasta 4.000 mg a pacientes sin evidencia de toxicidad dependiente de la dosis. La mayor dosis acumulada que se administró a los pacientes durante un periodo de 20-semanas fue de 44.000 mg y esta dosis no produjo ningún efecto adverso agudo.
En caso de sospecha de una sobredosis, se deberá monitorizar al paciente para cualquier signo o síntoma anormal. Se deberá buscar e instaurar tratamiento médico adecuado.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: fármacos para enfermedades obstructivas de las vías respiratorias, otros agentes sistémicos para enfermedades obstructivas de las vías respiratorias, código ATC: R03DX05
Omalizumab es un anticuerpo monoclonal humanizado derivado del ADN recombinante que se une selectivamente a la inmunoglobulina E humana (IgE). El anticuerpo es una IgG1 kappa que contiene regiones de estructura humana con regiones determinantes de complementariedad de un anticuerpo de origen murino que se une a la IgE.
Asma alérgica
Mecanismo de acción
Omalizumab se une a la IgE y previene la unión de ésta al FcsRI (receptor de IgE de alta afinidad) en basófilos y mastocitos, reduciendo así la cantidad de IgE libre disponible para desencadenar la cascada alérgica. El tratamiento de pacientes atópicos con omalizumab dio como resultado una disminución acentuada de los receptores FcsRI en los basófilos.
Efectos _ farmacodinámicos
La liberación in vitro de histamina de los basófilos aislados de pacientes que habían sido tratados con Xolair se redujo aproximadamente en un 90% tras la estimulación con un alergeno en comparación con los valores previos al tratamiento.
En los ensayos clínicos en pacientes con asma alérgica, las concentraciones plasmáticas de IgE libre disminuyeron de manera dosis dependiente en la hora posterior a la primera dosis y se mantuvieron reducidas entre dosis. Un año después de interrumpir el tratamiento con Xolair los niveles de IgE volvieron a los niveles previos al tratamiento, sin que se observase efecto de rebote en los niveles de IgE después del periodo de blanqueo del medicamento.
Urticaria crónica espontánea (UCE)
Mecanismo de acción
Omalizumab se une a la IgE y a los niveles bajos de IgE libre. Como consecuencia, disminuyen los receptores de IgE (FCsRI) en las células. No se entiende completamente como esto se traduce en una mejoría de los síntomas de la UCE.
Efectos _ farmacodinámicos
En los ensayos clínicos en pacientes con UCE se observó la supresión máxima de IgE libre tres días después de la primera dosis subcutánea. Después de dosis repetidas una vez cada 4 semanas, los niveles predosis de IgE libre en suero permanecieron estables entre 12 y 24 semanas de tratamiento. Tras la interrupción de Xolair, los niveles de IgE libre incrementaron hacia los niveles pretratamiento tras un periodo de seguimiento superior a 16 semanas libre de tratamiento.
Eficacia clínica y seguridad en asma alérgica
Adultos y adolescentes >12 años de edad
La eficacia y seguridad de Xolair se demostró en un ensayo doble ciego controlado con placebo de 28 semanas (estudio 1) en el que participaron 419 pacientes con asma alérgica grave, de edades comprendidas entre los 12 y 79 años, con función pulmonar reducida (FEVi 40-80% del valor teórico) y un pobre control de los síntomas del asma a pesar de recibir dosis elevadas de corticosteroides inhalados y un agonista beta2 de larga duración. Los pacientes escogidos habían sufrido exacerbaciones asmáticas múltiples habiendo necesitado tratamiento con corticosteroides sistémicos o habiendo sido hospitalizados o atendidos de urgencia debido a una exacerbación asmática grave en el pasado año a pesar del tratamiento continuo con dosis elevadas de corticosteroides inhalados y un agonista beta2 de larga duración. Se administró Xolair vía subcutánea o placebo como tratamiento adicional a >1.000 microgramos de dipropionato de beclometasona (o equivalente) más un agonista beta2 de larga duración. Como tratamiento de mantenimiento se permitieron los corticosteroides orales, la teofilina y los antagonistas de los leucotrienos (22%, 27%, y 35% de pacientes, respectivamente).
Se tomó como variable principal la proporción de exacerbaciones asmáticas que requirieron tratamiento con dosis altas de corticosteroides sistémicos. Omalizumab redujo la proporción de exacerbaciones asmáticas en un 19% (p = 0,153). Posteriores evaluaciones que mostraron significancia estadística (p<0,05) en favor de Xolair incluyeron reducciones de las exacerbaciones graves (donde la función pulmonar de los pacientes se redujo por debajo del 60% del mejor valor personal, necesitando corticosteroides sistémicos) y de las visitas de urgencia relacionadas con el asma (incluyendo hospitalizaciones, sala de urgencias, y visitas al médico no programadas), y mejoría en la valoración global del médico con respecto a la efectividad del tratamiento, calidad de vida relacionada con el asma (AQL), síntomas del asma y función pulmonar.
En un análisis de subgrupos, era más probable que los pacientes con un valor total de IgE >76 Ul/ml previo al tratamiento experimentaran un beneficio clínicamente significativo a Xolair. En estos pacientes en el estudio 1, Xolair redujo la proporción de exacerbaciones asmáticas en un 40% (p = 0,002). Además, un mayor número de pacientes presentaron respuestas clínicamente significativas en la población con un valor total de IgE >76 Ul/ml a través de un programa con Xolair de asma grave. La Tabla 6 incluye resultados de la población del estudio 1.
Tabla 6: Resultados del estudio 1
Población total estudio 1 Xolair Placebo
_N=209_N=210
Exacerbaciones asmáticas
Proporción por periodo de 0,74 0,92
28 semanas
Reducción %, valor p de razón de 19,4%, p = 0,153
tasas
Exacerbaciones de asma grave
Proporción por periodo de 0,24 0,48
28 semanas
Reducción %, valor p de razón de 50,1%, p = 0,002
tasas
Visitas de urgencia
Proporción por periodo de 0,24 0,43
28 semanas
Reducción %, valor p de razón de 43,9%, p = 0,038
tasas
Valoración global del médico
42,8%
47,8%
respondedores3 % 60,5%
Valor p4 <0,001
Mejoría AQL
% de pacientes con una mejoría 60,8%
>0,5
Valor p 0,008
El estudio 2 valoró la eficacia y seguridad de Xolair en una población de 312 pacientes con asma alérgica grave que equiparaban la población del estudio 1. En este estudio abierto, el tratamiento con Xolair condujo a una reducción del 61% en la proporción de exacerbaciones asmáticas que fue clínicamente significativa comparado con el tratamiento actual del asma solo.
Cuatro amplios estudios adicionales de apoyo controlados con placebo de 28 a 52 semanas de duración en 1.722 adultos y adolescentes (estudios 3, 4, 5, 6) valoraron la eficacia y seguridad de Xolair en pacientes con asma persistente grave. La mayoría de los pacientes estuvo inadecuadamente controlado pero recibieron menos tratamiento concomitante del asma que los pacientes en los estudios 1 ó 2. Los estudios 3-5 utilizaron la exacerbación como variable principal, mientras que el estudio 6 evaluó principalmente el ahorro de corticosteroides inhalados.
En los estudios 3, 4 y 5, los pacientes tratados con Xolair presentaron reducciones en la proporción de exacerbaciones asmáticas del 37,5% (p = 0,027), 40,3% (p<0,001) y 57,6% (p<0,001), respectivamente, comparado con placebo.
En el estudio 6, los pacientes con asma alérgica significativamente más grave tratados con Xolair pudieron reducir su dosis de fluticasona a <500 microgramos/día sin deterioro del control del asma (60,3%) comparado con el grupo placebo (45,8%, p<0,05).
La puntuación de calidad de vida se midió utilizando el cuestionario de calidad de vida relacionada con el asma de Juniper. En los 6 estudios hubo una mejoría estadísticamente significativa con respecto a la puntuación de calidad de vida basal para los pacientes tratados con Xolair con respecto al grupo placebo o control.
Valoración global del médico con respecto a la efectividad del tratamiento:
La valoración global del médico se realizó en cinco de los estudios anteriormente mencionados como una amplia medida del control del asma realizada por el médico que trata al paciente. El médico pudo tener en cuenta el PEF (flujo espiratorio máximo), los síntomas durante el día y la noche, el uso de medicación de rescate, la espirometría y las exacerbaciones. En los cinco estudios una proporción significativamente superior de los pacientes tratados con Xolair declararon haber alcanzado una notable mejoría o un control completo del asma comparado con los pacientes tratados con placebo.
Niños de 6 a <12 años de edad
El soporte principal para la eficacia y seguridad de Xolair en el grupo de edad de 6 a <12 años se obtiene de un ensayo multicéntrico, randomizado, doble ciego, controlado con placebo (estudio 7).
El estudio 7 fue un ensayo controlado con placebo que incluyó un subgrupo específico (n=235) de pacientes tal y como se define en la presente indicación, quienes fueron tratados con corticoides inhalados a dosis altas (>500 pg/día de fluticasona o equivalente) más agonista beta de larga duración.
Según la opinión clínica del investigador, una exacerbación clínicamente significativa se definió como un empeoramiento de los síntomas del asma, la cual requería una dosis doble de corticosteroide inhalado con respecto al valor basal, durante un mínimo de 3 días y/o tratamiento de rescate con corticosteroides sistémicos (oral o intravenoso) durante un mínimo de 3 días.
En el subgrupo específico de pacientes con corticoides inhalados a dosis alta, el grupo de omalizumab tuvo una tasa inferior estadísticamente significativa de exacerbaciones asmáticas clínicamente significativas que el grupo placebo. A las 24 semanas, la diferencia en las tasas entre los grupos de tratamiento representó un descenso del 34% (razón de tasas 0,662, p = 0,047) relativo a placebo para los pacientes con omalizumab. En el segundo periodo de tratamiento doble ciego de 28 semanas la diferencia en las tasas entre los grupos de tratamiento representó un descenso del 63% (razón de tasas 0,37, p<0,001) relativo a placebo para los pacientes con omalizumab.
Durante el periodo de tratamiento doble ciego de 52 semanas (incluyendo la fase de esteroides a dosis fija de 24 semanas y la fase de ajuste de esteroides de 28 semanas) la diferencia en las tasas entre los grupos de tratamiento representó un descenso relativo del 50% (razón de tasas 0,504, p<0,001) en exacerbaciones para los pacientes con omalizumab.
El grupo de omalizumab mostró descensos mayores en el uso de agonistas beta como medicación de rescate que el grupo placebo al final del periodo de tratamiento de 52 semanas, aunque la diferencia entre los grupos de tratamiento no fue estadísticamente significativa. De la evaluación global de la efectividad del tratamiento al final del periodo de tratamiento doble ciego de 52 semanas en el subgrupo de pacientes graves con corticoides inhalados a dosis alta más agonistas beta de larga duración, la proporción de pacientes calificados como que tuvieron una efectividad de tratamiento “excelente” fue superior, y la proporción que tuvo una efectividad de tratamiento “moderada” o “débil” fue inferior en el grupo de omalizumab comparado con el grupo placebo; la diferencia entre grupos fue estadísticamente significativa (p<0,001), mientras que no hubo diferencias entre los grupos de omalizumab y placebo en las valoraciones de calidad de vida subjetiva de los pacientes.
Eficacia clínica y seguridad en urticaria crónica espontánea (UCE)
Se demostró la eficacia y seguridad de Xolair en dos ensayos de fase III aleatorizados, controlados con placebo (ensayos 1 y 2) en pacientes con UCE que permanecieron sintomáticos a pesar del tratamiento antihistamínico H1 a la dosis aprobada. Un tercer ensayo (ensayo 3) evaluó principalmente la seguridad de Xolair en pacientes con UCE que permanecieron sintomáticos a pesar del tratamiento con antihistamínicos H1 hasta cuatro veces la dosis aprobada y antihistamínicos H2 y/o tratamiento con ARLT. Los tres ensayos incluyeron 975 pacientes de edades comprendidas entre 12 y 75 años (edad media 42,3 años; 39 pacientes de 12-17 años, 54 pacientes >65 años; 259 hombres y 716 mujeres). Se requirió que todos los pacientes presentaran un control inadecuado de los síntomas, valorado a través de una escala semanal de la actividad de la urticaria (UAS7, intervalo 0-42) de >16, y una puntuación semanal de la gravedad del prurito (que es un componente de la UAS7; intervalo 0-21) de >8 para los 7 días anteriores a la aleatorización, a pesar de haber usado un antihistamínico durante al menos 2 semanas antes.
En los ensayos 1 y 2, los pacientes presentaron una puntuación semanal media de la gravedad del prurito de entre 13,7 y 14,5 en el periodo basal y una puntuación UAS7 media de 29,5 y 31,7, respectivamente. Los pacientes en el ensayo 3 de seguridad presentaron una puntuación semanal media de la gravedad del prurito de 13,8 y una puntuación UAS7 media de 31,2 en el periodo basal.
En los tres ensayos, los pacientes notificaron que recibían una media de 4 a 6 medicaciones (incluyendo antihistamínicos H1) para los síntomas de la UCE antes de ser incluidos en el ensayo.
Los pacientes recibieron Xolair a las dosis de 75 mg, 150 mg o 300 mg o placebo por inyección subcutánea cada 4 semanas durante 24 y 12 semanas en los ensayos 1 y 2, respectivamente, y 300 mg o placebo por inyección subcutánea cada 4 semanas durante 24 semanas en el ensayo 3. Todos los ensayos tuvieron un periodo de seguimiento libre de tratamiento de 16 semanas.
La variable primaria fue el cambio del periodo basal a la semana 12 en la puntuación semanal de la gravedad del prurito. Omalizumab a la dosis de 300 mg redujo la puntuación semanal de la gravedad del prurito de 8,55 a 9,77 (p <0,0001) comparado con una reducción de 3,63 a 5,14 para placebo (ver Tabla 7). Además se observaron resultados estadísticamente significativos en las tasas de respondedores para UAS7<6 (a la semana 12), las cuales fueron superiores para los grupos de tratamiento de 300 mg, que fueron del 52-66% (p<0,0001) comparado con el 11-19% para los grupos placebo, y se alcanzó la respuesta completa (UAS7=0) en el 34-44% (p<0,0001) de los pacientes tratados con la dosis de 300 mg comparado con el 5-9% de los pacientes en los grupos placebo. Los pacientes en los grupos de tratamiento de 300 mg alcanzaron la proporción media más elevada de días libres de angioedema desde la semana 4 a la semana 12, (91,0-96,1%; p<0,001) comparado con los grupos placebo (88,1-89,2%). El cambio medio del periodo basal a la semana 12 en el DLQI global para los grupos de tratamiento de 300 mg fue superior (p<0,001) que para placebo mostrando una mejora que osciló entre 9,7-10,3 puntos comparado con 5,1-6,1 puntos para los correspondientes grupos placebo.
Tabla 7: Cambio del periodo basal a la semana 12 en la puntuación semanal de la gravedad del prurito, ensayos 1, 2 y 3 (población ITTm5)
|
Placebo |
Omalizumab 300 mg | |
|
Ensayo 1 | ||
|
N |
80 |
81 |
|
Media (DE) |
-3,63 (5,22) |
-9,40 (5,73) |
|
Diferencia en LS media vs. placebo1 |
- |
-5,80 |
|
IC de 95% para diferencia |
- |
-7,49,-4,10 |
|
Valor-P vs. placebo2 |
- |
<0,0001 |
|
Ensayo 2 | ||
|
N |
79 |
79 |
|
Media (DE) |
-5,14 (5,58) |
-9,77 (5,95) |
|
Diferencia en LS media vs. placebo1 |
- |
-4,81 |
|
IC de 95% para diferencia |
- |
-6,49,-3,13 |
|
Valor-P vs. placebo2 |
- |
<0,0001 |
|
Ensayo 3 | ||
|
N |
83 |
252 |
|
Media (DE) |
-4,01 (5,87) |
-8,55 (6,01) |
|
Diferencia en LS media vs. placebo1 |
- |
-4,52 |
|
IC de 95% para diferencia |
- |
-5,97, -3,08 |
|
Valor-P vs. placebo2 |
- |
<0,0001 |
La Figura 1 muestra la puntuación semanal media de la gravedad del prurito a lo largo del tiempo en el ensayo 1. Las puntuaciones semanales medias de la gravedad del prurito descendieron con un efecto máximo alrededor de la semana 12, el cual se mantuvo a lo largo del periodo de tratamiento de 24 semanas. Los resultados fueron similares en el ensayo 3.
En los tres ensayos la puntuación semanal media de la gravedad del prurito se incrementó gradualmente durante el periodo de seguimiento libre de tratamiento de 16 semanas, consistente con la reaparición de los síntomas. Los valores medios al final del periodo de seguimiento fueron similares a los del grupo placebo, pero inferiores a los valores basales medios respectivos.
Figura 1: Puntuación semanal media de la gravedad del prurito a lo largo del tiempo, ensayo 1 (población ITTm)
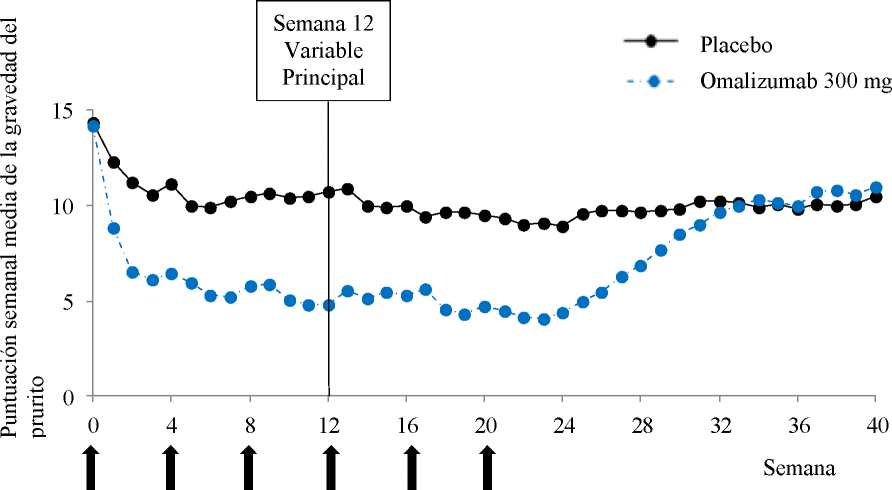
Omalizumab o placebo administrado
BOCF=observación basal llevada adelante; ITTm=población por intención de tratar modificada
Eficacia después de 24 semanas de tratamiento
La magnitud de los resultados de eficacia observados a la semana 24 de tratamiento fue comparable a la observada a la semana 12:
Para 300 mg, en los ensayos 1 y 3, el descenso medio en relación al periodo basal en la puntuación semanal de la gravedad del prurito fue de 9,8 y 8,6, la proporción de pacientes con UAS7<6 fue del 61,7% y 55,6%, y la proporción de pacientes con respuesta completa (UAS7=0) fue del 48,1% y 42,5%, respectivamente, (todos p<0,0001, cuando se comparó con placebo).
La experiencia clínica en el retratamiento de pacientes con omalizumab es limitada.
Los datos de los ensayos clínicos en adolescentes (12 a 17 años) incluyeron un total de 39 pacientes, de los cuales 11 recibieron la dosis de 300 mg. Se dispone de resultados para la dosis de 300 mg de 9 pacientes a la semana 12 y de 6 pacientes a la semana 24, y mostró una magnitud de respuesta similar al tratamiento con omalizumab comparado a la población adulta. El cambio medio con respecto al valor basal en relación a la puntuación semanal de la gravedad del prurito mostró una reducción de 8,25 a la semana 12 y de 8,95 a la semana 24. Las tasas de respondedores fueron: 33% a la semana 12 y 67% a la semana 24 para UAS7=0, y del 56% a la semana 12 y del 67% a la semana 24 para UAS7<6.
5.2 Propiedades farmacocinéticas
Se ha estudiado la farmacocinética de omalizumab en pacientes adultos y adolescentes con asma alérgica, así como en pacientes adultos y adolescentes con UCE. Las características farmacocinéticas generales de omalizumab son similares en estas poblaciones.
Absorción
Tras la administración subcutánea, omalizumab se absorbe con una biodisponibilidad absoluta media del 62%. Después de administrar una dosis subcutánea única a pacientes adultos y adolescentes con asma o UCE, omalizumab se absorbió lentamente alcanzando concentraciones plasmáticas máximas después de una media de 6-8 días. En pacientes con asma, tras dosis múltiples de omalizumab, las áreas bajo la curva de concentración plasmática-tiempo del Día 0 al Día 14 en estado estacionario fueron de hasta 6-veces las obtenidas tras la primera dosis.
La farmacocinética de omalizumab es lineal a dosis mayores de 0,5 mg/kg. Tras dosis de 75 mg,
150 mg o 300 mg cada 4 semanas en pacientes con UCE, las concentraciones séricas mínimas de omalizumab incrementaron proporcionalmente con el nivel de dosis.
La administración de Xolair fabricado como una formulación liofilizada o líquida dió como resultado perfiles similares de concentración sérica-tiempo de omalizumab.
Distribución
In vitro, omalizumab forma complejos de tamaño limitado con la IgE. No se han observado tanto in vitro como in vivo complejos precipitantes ni complejos con pesos moleculares superiores a un millón de Daltons. En base a la farmacocinética poblacional, la distribución de omalizumab fue similar en pacientes con asma alérgica y en pacientes con UCE. Tras la administración subcutánea el volumen de distribución aparente en los pacientes con asma fue de 78 ± 32 ml/kg.
Eliminación
El aclaramiento de omalizumab comprende procesos de aclaramiento de las IgG, así como, aclaramiento a través de uniones específicas y formación de complejos con su ligando específico, la IgE. La eliminación hepática de IgG incluye una degradación en el sistema reticuloendotelial y células endoteliales. La IgG inalterada también se excreta en la bilis. La vida media de eliminación plasmática de omalizumab en pacientes asmáticos fue de un promedio de 26 días, con un aclaramiento aparente promedio de 2,4 ± 1,1 ml/kg/día. La duplicación del peso corporal hace que el aclaramiento aparente sea de aproximadamente el doble. En pacientes con UCE, en base a las simulaciones de la farmacocinética poblacional, la semivida de eliminación sérica de omalizumab en estado estacionario fue de una media de 24 días y el aclaramiento aparente en estado estacionario para un paciente de 80 kg de peso fue de 3,0 ml/kg/día.
Características demográficas
Pacientes con asma: Se analizó la farmacocinética poblacional de omalizumab con el fin de evaluar los efectos de las características demográficas. El análisis de los escasos datos disponibles indica que no es necesario efectuar un ajuste de la dosis en pacientes con asma en función de la edad (6-76 años), raza/grupo étnico, sexo o índice de masa corporal (ver sección 4.2).
Pacientes con UCE: En base a la farmacocinética poblacional se evaluaron los efectos de las características demográficas y otros factores sobre la exposición de omalizumab. Además, se evaluaron los efectos de covarianza analizando la relación entre las concentraciones de omalizumab y la respuesta clínica. Estos análisis indicaron que no son necesarios ajustes de dosis en pacientes con UCE en función de la edad (12-75 años), raza/grupo étnico, sexo, peso corporal, índice de masa corporal, IgE basal, anticuerpos anti-FcsRI o uso concomitante de antihistamínicos H2 o ARLTs.
Insuficiencia renal y hepática
No se dispone de datos farmacocinéticos o farmacodinámicos en pacientes con asma alérgica o UCE, con insuficiencia renal o hepática (ver secciones 4.2 y 4.4).
5.3 Datos preclínicos sobre seguridad
Se ha estudiado la seguridad de omalizumab en monos cinomolgos, ya que el omalizumab se une a las IgE del cinomolgos y humanas con una afinidad similar. Se detectaron anticuerpos a omalizumab en algunos monos tras la administración subcutánea o intravenosa repetida. Sin embargo, no se observó toxicidad aparente, tales como enfermedad mediada por inmunocomplejos o citotoxicidad dependiente de complemento. No hubo evidencia de una respuesta anafiláctica debido a la desgranulación de los mastocitos en monos cinomolgos.
La administración crónica de omalizumab a niveles de dosis de hasta 250 mg/kg (al menos 14 veces la dosis clínica más alta recomendada en mg/kg de acuerdo con la tabla de dosis recomendada) fue bien tolerada en primates no humanos (tanto en animales adultos como en jóvenes), con la excepción de un descenso en las plaquetas sanguíneas dependiente de la dosis y de la edad, con una mayor sensibilidad en animales jóvenes. La concentración plasmática necesaria para alcanzar un descenso del 50% en las plaquetas con respecto al valor basal en monos adultos cinomolgos fue aproximadamente de 4 a 20 veces más elevada que la concentración plasmática clínica máxima anticipada. Además, se observó inflamación y hemorragia aguda en el lugar de la inyección en monos cinomolgos.
No se han realizado estudios de carcinogenicidad formal con omalizumab.
En los estudios de reproducción en monos cinomolgos, dosis subcutáneas de hasta 75 mg/kg por semana (al menos 8 veces la dosis clínica más alta recomendada en mg/kg durante un periodo de 4-semanas) no provocaron toxicidad materna, embriotoxicidad o teratogenicidad cuando se administró durante toda la organogénesis y no provocó efectos adversos sobre el crecimiento fetal o neonatal cuando se administró durante la fase final de la gestación, parto y lactancia.
Omalizumab se excretó en la leche materna en monos cinomolgos. Los niveles de omalizumab en la leche fueron del 0,15% con respecto a la concentración sérica materna.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo
Sacarosa
L-histidina
Hidrocloruro de L-histidina monohidrato Polisorbato 20
Disolvente
Agua para preparaciones inyectables
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez
4 años.
Después de la reconstitución
Se ha demostrado la estabilidad física y química del medicamento reconstituido durante 8 horas entre 2°C y 8°C y durante 4 horas a 30°C.
Desde el punto de vista microbiológico, el medicamento deberá utilizarse inmediatamente después de su reconstitución. Si no se utiliza inmediatamente, el tiempo y las condiciones de conservación previas a su utilización son responsabilidad del manipulador y no deberían ser normalmente superiores a 8 horas entre 2°C y 8°C o 2 horas a 25°C.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Vial de polvo: Vial de vidrio incoloro tipo I, transparente, con tapón de caucho butilo y sello extraíble de color azul.
Ampolla de disolvente: Ampolla de vidrio incoloro tipo I, transparente, conteniendo 2 ml de agua para inyectables.
Envases conteniendo 1 viale de polvo y 1 ampolla de agua para inyectables, y envases multiples que contienen 4 (4 envases de 1+1) o 10 (10 envases 1+1) con viales de polvo y ampollas de agua para inyectables.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El medicamento liofilizado necesita entre 15 y 20 minutos para disolverse, aunque en algunos casos puede requerir más tiempo. El medicamento completamente reconstituido tiene un aspecto transparente o a ligeramente opalescente, incoloro a amarillo parduzco claro y puede presentar unas cuantas burbujas pequeñas o espuma alrededor del borde del vial. Debido a la viscosidad del medicamento reconstituido deberá tenerse precaución de extraer todo el medicamento del vial antes de eliminar el aire o el exceso de solución de la jeringa con el fin de obtener los 1,2 ml.
Para preparar los viales de Xolair 150 mg para la administración subcutánea, siga por favor las siguientes instrucciones:
1. Retirar 1,4 ml de agua para inyectables de la ampolla con una jeringa equipada con una aguja gruesa de extracción (calibre 18).
2. Con el vial colocado en posición vertical sobre una superficie plana, insertar la aguja e inocular el agua para inyectables en el vial conteniendo el polvo liofilizado utilizando las técnicas asépticas estándar, dirigiendo el agua para inyectables directamente sobre el polvo.
3. Manteniendo el vial en posición vertical, removerlo vigorosamente (sin agitar) durante 1 minuto aproximadamente para humedecer el polvo uniformemente.
4. Para ayudar a la disolución tras completar el paso 3, remover suavemente el vial durante
5-10 segundos aproximadamente cada 5 minutos con el fin de disolver el polvo restante.
Observe que en ocasiones puede necesitar más de 20 minutos para disolver el polvo completamente. Si este es el caso, repita el paso 4 hasta que desaparezcan las partículas gelatinosas de la solución.
Una vez el medicamento se haya disuelto completamente, no deben quedar partículas gelatinosas visibles en la solución. Las pequeñas burbujas o espuma alrededor del borde del vial son completamente normales. El medicamento reconstituido tiene un aspecto transparente a ligeramente opalescente, incoloro a amarillo parduzco claro. No utilice el producto si observa partículas sólidas.
5. Invertir el vial durante un mínimo de 15 segundos con el fin de que la solución fluya hacia el tapón. Utilizando una jeringa nueva de 3-ml equipada con una aguja gruesa de extracción (calibre 18), insertar la aguja en el vial invertido. Manteniendo el vial en posición invertida colocar el extremo de la aguja al final de la solución en el vial cuando extraiga la solución con la jeringa. Antes de extraer la aguja del vial, tire del émbolo y llévelo hasta el fondo del cilindro de la jeringa con el fin de extraer toda la solución del vial invertido.
6. Reemplazar la aguja de calibre 18 por una de calibre 25 para inyección subcutánea.
7. Eliminar el aire, las burbujas grandes y cualquier exceso de solución con el fin de obtener la dosis requerida de 1,2 ml. Puede quedar una fina capa de pequeñas burbujas en la superficie de la solución contenida en la jeringa. Como la solución es ligeramente viscosa, la administración de la solución por inyección subcutánea puede durar entre 5 y 10 segundos.
El vial proporciona 1,2 ml (150 mg) de Xolair. Para obtener una dosis de 75 mg retirar 0,6 ml con la jeringa y desechar la solución restante.
8. Las inyecciones se administran por vía subcutánea en la región deltoidea del brazo o en el muslo.
Xolair 150 mg polvo para solución inyectable se suministra en un vial de un solo uso.
Desde el punto de vista microbiológico, el medicamento debe utilizarse inmediatamente después de la
reconstitución (ver sección 6.3).
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto
con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/319/002
EU/1/05/319/003
EU/1/05/319/004
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 25 Octubre 2005 Fecha de la última renovación: 22 Junio 2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
1. NOMBRE DEL MEDICAMENTO
Xolair 75 mg solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada jeringa precargada de 0,5 ml de solución contiene 75 mg de omalizumab*.
*Omalizumab es un anticuerpo monoclonal humanizado obtenido mediante la tecnología del ADN recombinante, a partir de una línea celular mamífera de ovario de hámster chino (OHC).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución de transparente a ligeramente opalescente, incolora a color amarillo parduzco claro..
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Xolair está indicado en adultos, adolescentes y niños (de 6 a <12 años).
El tratamiento con Xolair deberá ser considerado únicamente para pacientes con asma mediada de forma convincente por IgE (inmunoglobulina E) (ver sección 4.2).
Adultos y adolescentes (a partir de 12 años de edad)
Xolair está indicado para mejorar el control del asma cuando se administra como tratamiento adicional en pacientes con asma alérgica grave persistente que presentan test cutáneo positivo o reactividad in vitro a aeroalergenos perennes y con función pulmonar reducida (FEV1 <80%) así como, síntomas frecuentes durante el día o despertares por la noche y que han presentado múltiples exacerbaciones asmáticas graves documentadas, a pesar de utilizar corticosteroides diarios inhalados a dosis altas, más un agonista beta2 inhalado de larga duración.
Niños (6 a <12 años de edad)
Xolair está indicado para mejorar el control del asma cuando se administra como tratamiento adicional en pacientes con asma alérgica grave persistente que presentan test cutáneo positivo o reactividad in vitro a aeroalergenos perennes y síntomas frecuentes durante el día o despertares por la noche y que han presentado múltiples exacerbaciones asmáticas graves documentadas, a pesar de utilizar corticosteroides diarios inhalados a dosis altas, más un agonista beta2 inhalado de larga duración.
4.2 Posología y forma de administración
El tratamiento con Xolair debe iniciarlo un médico experimentado en el diagnóstico y tratamiento del asma grave persistente.
Posología
La dosis apropiada y la frecuencia de administración de Xolair se determina a partir de la concentración basal de IgE (UI/ml), medida antes de iniciar el tratamiento, y del peso corporal (kg). A efectos de la asignación de la dosis, antes de la administración de la dosis inicial se debe determinar la concentración de IgE en los pacientes mediante cualquier método comercial que analice la IgE plasmática total. En base a estas determinaciones, podrán ser necesarios en cada administración de 75 a 600 mg de Xolair en 1 a 4 inyecciones.
Era menos probable que experimentaran beneficio los pacientes con un valor de IgE inferior a 76 UI/ml (ver sección 5.1). Los médicos prescriptores deberán asegurar que los pacientes adultos y adolescentes con una IgE por debajo de 76 UI/ml y los niños (6 a < 12 años de edad) con una IgE por debajo de 200 UI/ml presenten una reactividad in vitro inequívoca (RAST) al alergeno perenne antes de iniciar el tratamiento.
Ver Tabla 1 de conversión y Tablas 2 y 3 para la determinación de dosis en adultos, adolescentes y niños (de 6 a <12 años).
No debe administrarse Xolair a pacientes cuya concentración basal de IgE o peso corporal en kilogramos, excedan los límites indicados en la tabla de dosis.
La dosis máxima recomendada es de 600 mg de omalizumab cada dos semanas.
Tabla 1: Conversión de la dosis al número de jeringas, número de inyecciones y volumen de inyección total para cada administración
|
Dosis (mg) Número de jeringas Número de inyecciones 75 mg 150 mg |
Volumen total de inyección (ml) | |||
|
75 |
1 |
0 |
1 |
0,5 |
|
150 |
0 |
1 |
1 |
1,0 |
|
225 |
1 |
1 |
2 |
1,5 |
|
300 |
0 |
2 |
2 |
2,0 |
|
375 |
1 |
2 |
3 |
2,5 |
|
450 |
0 |
3 |
3 |
3,0 |
|
525 |
1 |
3 |
4 |
3,5 |
|
600 |
0 |
4 |
4 |
4,0 |
|
Peso corporal (kg) | ||||||||||
|
IgE basal |
>20- |
>25- |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>90- |
>125- |
|
(UI/ml) |
25 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
125 |
150 |
|
>30-100 |
75 |
75 |
75 |
150 |
150 |
150 |
150 |
150 |
300 |
300 |
|
>100-200 |
150 |
150 |
150 |
300 |
300 |
300 |
300 |
300 |
450 |
600 |
|
>200-300 |
150 |
150 |
225 |
300 |
300 |
450 |
450 |
450 |
600 | |
|
>300-400 |
225 |
225 |
300 |
450 |
450 |
450 |
600 |
600 | ||
|
>400-500 |
225 |
300 |
450 |
450 |
600 |
600 | ||||
|
>500-600 |
300 |
300 |
450 |
600 |
600 | |||||
|
>600-700 |
300 |
450 |
600 | |||||||
|
>700-800 | ||||||||||
|
>800-900 |
ADMINISTRACIÓN CADA | |||||||||
|
>900- 1000 |
2 SEMANAS VER TABLA 3 | |||||||||
|
>1000- 1100 | ||||||||||
|
Peso corporal (kg) | ||||||||||
|
IgE basal |
>20- |
>25- |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>90- |
>125- |
|
(UI/ml) |
25 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
125 |
150 |
|
>30-100 |
ADMINISTRACIÓN CADA | |||||||||
|
>100-200 |
4 SEMANAS VER TABLA 2 | |||||||||
|
>200-300 |
375 | |||||||||
|
>300-400 |
450 |
525 | ||||||||
|
>400-500 |
375 |
375 |
525 |
600 | ||||||
|
>500-600 |
375 |
450 |
450 |
600 | ||||||
|
>600-700 |
225 |
375 |
450 |
450 |
525 | |||||
|
>700-800 |
225 |
225 |
300 |
375 |
450 |
450 |
525 |
600 | ||
|
>800-900 |
225 |
225 |
300 |
375 |
450 |
525 |
600 | |||
|
>900- |
225 |
300 |
375 |
450 |
525 |
600 | ||||
|
1000 | ||||||||||
|
>1000- 1100 |
225 |
300 |
375 |
450 |
600 | |||||
|
>1100- |
300 |
300 |
450 |
525 |
600 |
NO ADMINISTRAR - |
no se dispone de | |||
|
1200 |
datos para la recomendación de dosis | |||||||||
|
>1200- 1300 |
300 |
375 |
450 |
525 | ||||||
|
>1300- 1500 |
300 |
375 |
525 |
600 | ||||||
Duración del tratamiento, monitorización y ajuste de dosis
Xolair está indicado para tratamiento a largo plazo. Los ensayos clínicos han demostrado que son necesarias un mínimo de 12-16 semanas para que el tratamiento con Xolair demuestre efectividad. A las 16 semanas de iniciar el tratamiento con Xolair, los pacientes deberán ser evaluados por su médico con respecto a la efectividad del tratamiento antes de administrar inyecciones posteriores. La decisión de continuar con Xolair tras las 16 semanas, o en ocasiones posteriores, debe estar basada en si se observa una notable mejoría en el control global del asma (ver sección 5.1; Valoración global del médico con respecto a la efectividad del tratamiento).
La interrupción del tratamiento con Xolair generalmente da lugar a un retorno de las concentraciones elevadas de IgE libre y de los síntomas asociados. Los valores de IgE total son elevados durante el tratamiento y siguen siéndolo hasta un año después de la interrupción del mismo. Por lo tanto, no puede utilizarse la reevaluación de los valores de IgE durante el tratamiento con Xolair como guía para la determinación de la dosis. La determinación de la dosis tras interrupciones de tratamiento de menos de un año de duración debe basarse en las concentraciones plasmáticas de IgE obtenidas en la determinación de dosis inicial. Si el tratamiento con Xolair se ha interrumpido por más de un año deberán de volver a medirse las concentraciones plasmáticas de IgE total para la determinación de la dosis.
Las dosis deberán ajustarse en caso de variaciones significativas del peso corporal (ver Tablas 2 y 3).
Poblaciones especiales
Pacientes de edad avanzada (mayores de 65 años)
Aunque se dispone de datos limitados sobre el uso de Xolair en pacientes mayores de 65 años, no existe evidencia de que los pacientes de edad avanzada requieran una dosis diferente de la de pacientes adultos más jóvenes.
Insuficiencia renal o hepática
No se dispone de estudios sobre el efecto de la insuficiencia renal o hepática en la farmacocinética de Xolair. Debido a que el aclaramiento de omalizumab a dosis clínicas se lleva a cabo fundamentalmente por el sistema reticuloendotelial (SRE), es improbable que se vea alterado en caso de insuficiencia renal o hepática. Xolair deberá administrarse con precaución en estos pacientes, mientras no se recomiende un ajuste especial de la dosis (ver sección 4.4).
Población pediátrica
No se ha establecido la seguridad y eficacia de Xolair en niños menores de 6 años. No se dispone de datos.
Forma de administración
Para administración subcutánea únicamente. No administrar por vía intravenosa o intramuscular.
Las inyecciones se administran vía subcutánea en la región deltoidea del brazo. Si por alguna razón no pueden administrarse en esta zona, podrán administrase alternativamente en el muslo.
Existe experiencia limitada con respecto a la autoadministración de Xolair. Por lo tanto, está previsto que el tratamiento sea administrado únicamente por el profesional sanitario.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6 y también la sección “información para el profesional sanitario” del prospecto.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
General
Xolair no está indicado para el tratamiento de las exacerbaciones asmáticas, broncoespasmo o estados asmáticos de carácter agudo.
No se ha estudiado el efecto de Xolair en pacientes con síndrome de hiperinmunoglobulinemia E o aspergilosis broncopulmonar alérgica, ni en la prevención de reacciones anafilácticas, incluyendo las provocadas por alergias alimentarias, dermatitis atópica, o rinitis alérgica. Xolair no está indicado en el tratamiento de estas patologías.
El tratamiento con Xolair tampoco se ha estudiado en pacientes con enfermedades autoinmunes, procesos mediados por inmunocomplejos, o insuficiencia renal o hepática preexistente (ver sección 4.2). Se deberá tener precaución cuando se administre Xolair en esta población de pacientes.
No se recomienda la interrupción brusca de los corticosteroides sistémicos o inhalados tras la iniciación del tratamiento con Xolair. El descenso de los corticosteroides debe realizarse bajo la supervisión directa de un médico y puede ser necesario que se realice gradualmente.
Trastornos del sistema inmulógico
Reacciones alérgicas tipo I
Pueden producirse reacciones alérgicas tipo I locales o sistémicas, incluyendo anafilaxia y shock anafiláctico durante el tratamiento con omalizumab, incluso con inicio tras un tratamiento de larga duración. La mayoría de estas reacciones se produjeron durante las 2 horas posteriores a la primera y siguientes inyecciones de Xolair, pero algunas se iniciaron pasadas las 2 horas e incluso pasadas 24 horas tras la inyección. Por lo tanto, se deberán tener siempre disponibles medicamentos para el tratamiento inmediato de reacciones anafilácticas tras la administración de Xolair. Se deberá informar al paciente de que estas reacciones son posibles y que si se producen deberán solicitar atención médica de inmediato. Un antecedente de anafilaxia no asociado a omalizumab puede suponer un factor de riesgo de sufrir una reacción anafiláctica tras la administración de Xolair.
Se han detectado anticuerpos contra omalizumab en un pequeño número de pacientes en ensayos clínicos (ver sección 4.8). No se conoce bien la relevancia clínica de anticuerpos anti-Xolair.
Enfermedad del suero
Se ha observado enfermedad del suero y reacciones semejantes a la enfermedad del suero, que son reacciones alérgicas tipo III retardadas, en pacientes tratados con anticuerpos monoclonales humanizados incluido omalizumab. El supuesto mecanismo fisiopatológico incluye formación y deposición de inmunocomplejos debido al desarrollo de anticuerpos contra omalizumab. El inicio del cuadro se produce normalmente a los 1-5 días tras la administración de la primera o siguientes inyecciones e incluso tras un tratamiento de larga duración. Los síntomas que sugieren la enfermedad del suero incluyen artritis/artralgias, rash (urticaria u otras formas), fiebre y linfoadenopatía. Los antihistamínicos y corticosteroides pueden ser útiles para prevenir o tratar estas alteraciones y se deberá advertir a los pacientes que notifiquen cualquier síntoma sospechoso.
Síndrome de Churg-Strauss y síndrome hipereosinofílico
Los pacientes con asma grave pueden presentar raramente síndrome hipereosinofílico sistémico o vasculitis granulomatosa eosinofílica alérgica (Síndrome de Churg-Strauss), los cuales son normalmente tratados con corticosteroides sistémicos.
En raras ocasiones, los pacientes en tratamiento con medicamentos antiasmáticos, incluyendo omalizumab, pueden presentar o desarrollar eosinofilia sistémica y vasculitis. Estas reacciones están normalmente asociadas con la reducción del tratamiento con corticosteroides orales.
En estos pacientes, los médicos deberán estar alerta ante el desarrollo de eosinofilia importante, rash vasculítico, empeoramiento de los síntomas pulmonares, anormalidades en el seno paranasal, complicaciones cardíacas, y/o neuropatía.
Deberá considerarse la interrupción del tratamiento con omalizumab en todos aquellos casos graves que cursen con alteraciones del sistema inmune mencionadas anteriormente.
Infecciones parasitarias (helmínticos)
Las IgE pueden estar involucradas en la respuesta inmunológica a algunas infecciones helmínticas. En pacientes con un elevado riesgo crónico de infección helmíntica, un ensayo controlado con placebo demostró un ligero incremento en la proporción de infección con omalizumab, aunque no se modificó el curso, gravedad y respuesta al tratamiento de la infección. La proporción de infección helmíntica en el programa clínico global, el cual no fue diseñado para detectar este tipo de infecciones, fue inferior a 1 en 1.000 pacientes. Sin embargo, deberá garantizarse precaución en pacientes con elevado riesgo de infección helmíntica, en particular cuando viajen a zonas donde las infecciones helmínticas son endémicas. Si los pacientes no responden al tratamiento antihelmíntico recomendado, deberá considerarse la interrupción del tratamiento con Xolair.
Personas sensibles al látex
La cápsula protectora extraíble de la aguja de esta jeringa precargada contiene un derivado de látex de caucho natural. Hasta la fecha no se ha detectado látex de caucho natural en la cápsula protectora extraíble de la aguja. Sin embargo, no se ha estudiado el uso de Xolair solución inyectable en jeringa precargada en personas sensibles al látex y por lo tanto, existe un riesgo potencial de reacciones de hipersensibilidad, las cuales no se pueden descartar completamente.
4.5 Interacción con otros medicamentos y otras formas de interacción
Dado que las IgE pueden estar relacionadas con la respuesta immunológica a algunas infecciones por helmínticos, Xolair puede reducir indirectamente la eficacia de medicamentos utilizados para el tratamiento de infecciones helmínticas o por otros parásitos (ver sección 4.4).
Las enzimas del citocromo P450, las bombas de eflujo y los mecanismos de unión a proteínas no se hallan implicados en el aclaramiento de omalizumab; por ello, existe un bajo potencial de interacciones farmacológicas. No se han realizado estudios de interacción de otros medicamentos o vacunas con Xolair. No existe un motivo farmacológico para esperar que los medicamentos prescritos frecuentemente en el tratamiento del asma interaccionen con omalizumab.
En los ensayos clínicos Xolair se utilizó frecuentemente asociado a corticosteroides inhalados y orales, beta agonistas inhalados de corta y larga duración, antagonistas de los leucotrienos, teofilinas y antihistamínicos orales. No hubo indicios de que estos medicamentos utilizados habitualmente como antiasmáticos puedan afectar a la seguridad de Xolair. Se dispone de datos limitados sobre el uso de Xolair en combinación con inmunoterapia específica (terapia de hiposensibilización). En un ensayo clínico donde Xolair se administró conjuntamente con inmunoterapia, se observó que la seguridad y eficacia de Xolair en combinación con inmunoterapia específica, no fue diferente a la de Xolair solo.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Hay datos limitados relativos al uso de omalizumab en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción (ver sección 5.3). Omalizumab atraviesa la barrera placentaria y se desconoce el daño potencial sobre el feto. Omalizumab se ha asociado con descensos de las plaquetas sanguíneas, dependientes de la edad en primates no humanos, con una sensibilidad relativamente superior en animales jóvenes (ver sección 5.3). Xolair no debería utilizarse durante el embarazo excepto si fuese claramente necesario.
Lactancia
Se desconoce si omalizumab se excreta en la leche materna. Los datos disponibles en primates no humanos muestran que omalizumab se excreta en la leche (ver sección 5.3). No se puede excluir el riesgo en recién nacidos/niños. Omalizumab no se debe administrar durante la lactancia.
Fertilidad
No hay datos de fertilidad en humanos para omalizumab. En estudios de fertilidad no clínicos diseñados específicamente en primates no humanos, incluidos los estudios de apareamiento, no se observó empeoramiento de la fertilidad en machos o hembras tras dosis repetidas con omalizumab de hasta 75 mg/kg. Además, no se observaron efectos genotóxicos en estudios separados no clínicos de genotoxicidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Xolair sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Resúmen del perfil de seguridad
Las reacciones adversas notificadas más frecuentemente durante los ensayos clínicos en pacientes adultos y adolescentes a partir de 12 años de edad fueron cefalea y reacciones en el lugar de la inyección, que incluían dolor, tumefacción, eritema y prurito. Las reacciones adversas notificadas más frecuentemente en los ensayos clínicos en niños de 6 a <12 años de edad fueron cefalea, pirexia y dolor abdominal superior. La mayoría de las reacciones fueron de gravedad leve a moderada.
Tabla de reacciones adversas
En la Tabla 4 se incluyen las reacciones adversas registradas en la población total de seguridad tratada con Xolair en los ensayos clínicos, por sistema de clasificación de órganos y frecuencia de MedDRA. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Las categorías de frecuencia se definen como: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000) y muy raras (<1/10.000). Las reacciones notificadas en la fase de postcomercialización se enumeran con frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 4: Reacciones adversas
|
Infecciones e infestaciones | |
|
Poco frecuentes Raras |
Faringitis Infección parasitaria |
|
Trastornos de la sangre y del sistema linfático | |
|
No conocida |
Trombocitopenia idiopática, incluyendo casos graves |
|
Trastornos del sistema inmunológico | |
|
Raras No conocida |
Reacción anafiláctica, otros procesos alérgicos graves, desarrollo de anticuerpos frente a omalizumab Enfermedad del suero que puede cursar con fiebre y linfoadenopatía |
|
Trastornos del sistema nervioso | |
|
Frecuentes Poco frecuentes |
Cefalea* Síncope, parestesia, somnolencia, mareo |
|
Trastornos vasculares | |
|
Poco frecuentes |
Hipotensión postural, rubor |
|
Trastornos respiratorios, torácicos y mediastínicos | |
|
Poco frecuentes Raras No conocida |
Broncoespasmo alérgico, tos Laringoedema Vasculitis granulomatosa alérgica (es decir, síndrome de Churg Strauss) |
|
Trastornos gastrointestinales | |
|
Frecuentes Poco frecuentes |
Dolor abdominal superior** Signos y síntomas dispépticos, diarrea, náuseas |
|
Trastornos de la piel y del tejido subcutáneo | |
|
Poco frecuentes Raras No conocida |
Fotosensibilidad, urticaria, rash, prurito Angioedema Alopecia |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |
|
Raras |
Lupus eritematoso sistémico (LES) |
|
No conocidas |
Artralgia, mialgia, tumefacción de las articulaciones |
|
Trastornos generales y alteraciones en el lugar de administración | |
|
Muy frecuentes |
Pirexia** |
|
Frecuentes |
Reacciones en el lugar de la inyección tales como tumefacción, eritema, dolor, prurito |
|
Poco frecuentes |
Enfermedad pseudo-gripal, brazos hinchados, incremento de peso, fatiga |
*: Muy frecuentes en niños de 6 a <12 años de edad **: En niños de 6 a <12 años de edad
Descripción de reacciones adversas seleccionadas Trastornos del sistema inmunológico Para mayor información, ver sección 4.4.
Anafilaxia
Rara vez se observaron reacciones anafilácticas en los ensayos clínicos. Sin embargo, después de una búsqueda acumulada en la base de datos de seguridad, se recogieron un total de 898 casos de anafilaxia post comercialización. En base a una exposición estimada de 566.923 paciente tratado años, resultó en una frecuencia aproximada de 0,20%.
Efectos tromboembólicos arteriales (ETA)
En ensayos clínicos controlados y durante los análisis intermedios de un estudio observacional, se observó un desequilibrio numérico de ETA. La definición de la variable compuesta ETA incluye derrame cerebral, ataque isquémico transitorio, infarto de miocardio, angina inestable y muerte cardiovascular (incluyendo muerte por causa desconocida). En el análisis final del estudio observacional, el índice de ETA por 1.000 pacientes años fue de 7,52 (115/15.286 pacientes años) para los pacientes tratados con Xolair y de 5,12 (51/9.963 pacientes años) para los pacientes control. En un análisis multivariado para controlar los factores basales disponibles de riesgo cardiovascular, la proporción de riesgo fue de 1,32 (intervalo de confianza del 95% 0,91-1,91). En un análisis segregado de un conjunto de ensayos clínicos, el cual incluyó todos los ensayos clínicos aleatorizados, doble ciego, controlados con placebo de 8 semanas o más de duración, el índice de ETA por 1.000 pacientes años fue de 2,69 (5/1.856 pacientes años) para los pacientes tratados con Xolair y de 2,38 (4/1.680 pacientes años) para los pacientes tratados con placebo (tasa de incidencia 1,13, intervalo de confianza del 95% 0,24-5,71).
Plaquetas
En los ensayos clínicos, pocos pacientes presentaron recuentos de plaquetas por debajo del límite inferior del intervalo normal de laboratorio. Ninguno de estos cambios se asoció con episodios hemorrágicos o con una disminución de la hemoglobina. En los seres humanos (pacientes a partir de 6 años de edad), a diferencia de los primates no humanos, no se ha observado ningún patrón de disminución persistente en el recuento de plaquetas (ver sección 5.3), aunque se han notificado casos aislados de trombocitopenia idiopática, incluyendo casos graves, en la fase de postcomercialización.
Infecciones _ parasitarias
En pacientes con un elevado riesgo crónico de infección helmíntica, un ensayo controlado con placebo demostró un ligero incremento numérico en la proporción de infección con omalizumab que no fue estadísticamente significativo. No se modificaron el curso, gravedad y respuesta al tratamiento de la infección (ver sección 4.4).
Lupus eritematoso sistémico
En pacientes con asma moderada a grave y urticaria crónica espontánea (UCE) se han notificado casos de lupus eritematoso sistémico (LES) en ensayos clínicos y etapa post comercialización. La patogénesis del lupus eritematoso sistémico no se conoce bien.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
No se ha determinado la dosis máxima tolerada de Xolair. Se han administrado dosis únicas intravenosas de hasta 4.000 mg a pacientes sin evidencia de toxicidad dependiente de la dosis. La mayor dosis acumulada que se administró a los pacientes durante un periodo de 20-semanas fue de 44.000 mg y esta dosis no produjo ningún efecto adverso agudo.
En caso de sospecha de una sobredosis, se deberá monitorizar al paciente para cualquier signo o síntoma anormal. Se deberá buscar e instaurar tratamiento médico adecuado.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: fármacos para enfermedades obstructivas de las vías respiratorias, otros agentes sistémicos para enfermedades obstructivas de las vías respiratorias, código ATC: R03DX05
Omalizumab es un anticuerpo monoclonal humanizado derivado del ADN recombinante que se une selectivamente a la inmunoglobulina E humana (IgE). El anticuerpo es una IgG1 kappa que contiene regiones de estructura humana con regiones determinantes de complementariedad de un anticuerpo de origen murino que se une a la IgE.
Mecanismo de acción
Omalizumab se une a la IgE y previene la unión de ésta al FcsRI (receptor de IgE de alta afinidad), reduciendo así la cantidad de IgE libre disponible para desencadenar la cascada alérgica. El tratamiento de pacientes atópicos con omalizumab dio como resultado una disminución acentuada de los receptores FcsRI en los basófilos.
Efectos farmacodinámicos
La liberación in vitro de histamina de los basófilos aislados de pacientes que habían sido tratados con Xolair se redujo aproximadamente en un 90% tras la estimulación con un alergeno en comparación con los valores previos al tratamiento.
En los ensayos clínicos, las concentraciones plasmáticas de IgE libre disminuyeron de manera dosis dependiente en la hora posterior a la primera dosis y se mantuvieron reducidas entre dosis. Un año después de interrumpir el tratamiento con Xolair los niveles de IgE volvieron a los niveles previos al tratamiento, sin que se observase efecto de rebote en los niveles de IgE después del periodo de blanqueo del medicamento.
Eficacia clínica y seguridad
Adultos y adolescentes >12 años de edad
La eficacia y seguridad de Xolair se demostró en un ensayo doble ciego controlado con placebo de 28 semanas (estudio 1) en el que participaron 419 pacientes con asma alérgica grave, de edades comprendidas entre los 12 y 79 años, con función pulmonar reducida (FEVi 40-80% del valor teórico) y un pobre control de los síntomas del asma a pesar de recibir dosis elevadas de corticosteroides inhalados y un agonista beta2 de larga duración. Los pacientes escogidos habían sufrido exacerbaciones asmáticas múltiples habiendo necesitado tratamiento con corticosteroides sistémicos o habiendo sido hospitalizados o atendidos de urgencia debido a una exacerbación asmática grave en el pasado año a pesar del tratamiento continuo con dosis elevadas de corticosteroides inhalados y un agonista beta2 de larga duración. Se administró Xolair vía subcutánea o placebo como tratamiento adicional a >1.000 microgramos de dipropionato de beclometasona (o equivalente) más un agonista beta2 de larga duración. Como tratamiento de mantenimiento se permitieron los corticosteroides orales, la teofilina y los antagonistas de los leucotrienos (22%, 27%, y 35% de pacientes, respectivamente).
Se tomó como variable principal la proporción de exacerbaciones asmáticas que requirieron tratamiento con dosis altas de corticosteroides sistémicos. Omalizumab redujo la proporción de exacerbaciones asmáticas en un 19% (p = 0,153). Posteriores evaluaciones que mostraron significancia estadística (p<0,05) en favor de Xolair incluyeron reducciones de las exacerbaciones graves (donde la función pulmonar de los pacientes se redujo por debajo del 60% del mejor valor personal, necesitando corticosteroides sistémicos) y de las visitas de urgencia relacionadas con el asma (incluyendo hospitalizaciones, sala de urgencias, y visitas al médico no programadas), y mejoría en la valoración global del médico con respecto a la efectividad del tratamiento, calidad de vida relacionada con el asma (AQL), síntomas del asma y función pulmonar.
En un análisis de subgrupos, era más probable que los pacientes con un valor total de IgE >76 Ul/ml previo al tratamiento experimentaran un beneficio clínicamente significativo a Xolair. En estos pacientes en el estudio 1, Xolair redujo la proporción de exacerbaciones asmáticas en un 40% (p = 0,002). Además, un mayor número de pacientes presentaron respuestas clínicamente significativas en la población con un valor total de IgE >76 UI/ml a través de un programa con Xolair de asma grave. La Tabla 5 incluye resultados de la población del estudio 1.
Tabla 5: Resultados del estudio 1
Población total estudio 1 Xolair Placebo
_N=209_N=210
Exacerbaciones asmáticas
Proporción por periodo de 0,74 0,92
28 semanas
Reducción %, valor p de razón de 19,4%, p = 0,153
tasas
Exacerbaciones de asma grave
Proporción por periodo de 0,24 0,48
28 semanas
Reducción %, valor p de razón de 50,1%, p = 0,002
tasas
Visitas de urgencia
Proporción por periodo de 0,24 0,43
28 semanas
Reducción %, valor p de razón de 43,9%, p = 0,038
tasas
Valoración global del médico
42,8%
47,8%
respondedores6 % 60,5%
Valor p7 <0,001
Mejoría AQL
% de pacientes con una mejoría 60,8%
>0,5
Valor p 0,008
En los estudios 3, 4 y 5, los pacientes tratados con Xolair presentaron reducciones en la proporción de exacerbaciones asmáticas del 37,5% (p = 0,027), 40,3% (p<0,001) y 57,6% (p<0,001), respectivamente, comparado con placebo.
En el estudio 6, los pacientes con asma alérgica significativamente más grave tratados con Xolair pudieron reducir su dosis de fluticasona a <500 microgramos/día sin deterioro del control del asma (60,3%) comparado con el grupo placebo (45,8%, p<0,05).
La puntuación de calidad de vida se midió utilizando el cuestionario de calidad de vida relacionada con el asma de Juniper. En los 6 estudios hubo una mejoría estadísticamente significativa con respecto a la puntuación de calidad de vida basal para los pacientes tratados con Xolair con respecto al grupo placebo o control.
Valoración global del médico con respecto a la efectividad del tratamiento:
La valoración global del médico se realizó en cinco de los estudios anteriormente mencionados como una amplia medida del control del asma realizada por el médico que trata al paciente. El médico pudo tener en cuenta el PEF (flujo espiratorio máximo), los síntomas durante el día y la noche, el uso de medicación de rescate, la espirometría y las exacerbaciones. En los cinco estudios una proporción significativamente superior de los pacientes tratados con Xolair declararon haber alcanzado una notable mejoría o un control completo del asma comparado con los pacientes tratados con placebo.
Niños de 6 a <12 años de edad
El soporte principal para la eficacia y seguridad de Xolair en el grupo de edad de 6 a <12 años se obtiene de un ensayo multicéntrico, randomizado, doble ciego, controlado con placebo (estudio 7).
El estudio 7 fue un ensayo controlado con placebo que incluyó un subgrupo específico (n=235) de pacientes tal y como se define en la presente indicación, quienes fueron tratados con corticoides inhalados a dosis altas (>500 pg/día de fluticasona o equivalente) más agonista beta de larga duración.
Según la opinión clínica del investigador, una exacerbación clínicamente significativa se definió como un empeoramiento de los síntomas del asma, la cual requería una dosis doble de corticosteroide inhalado con respecto al valor basal, durante un mínimo de 3 días y/o tratamiento de rescate con corticosteroides sistémicos (oral o intravenoso) durante un mínimo de 3 días.
En el subgrupo específico de pacientes con corticoides inhalados a dosis alta, el grupo de omalizumab tuvo una tasa inferior estadísticamente significativa de exacerbaciones asmáticas clínicamente significativas que el grupo placebo. A las 24 semanas, la diferencia en las tasas entre los grupos de tratamiento representó un descenso del 34% (razón de tasas 0,662, p = 0,047) relativo a placebo para los pacientes con omalizumab. En el segundo periodo de tratamiento doble ciego de 28 semanas la diferencia en las tasas entre los grupos de tratamiento representó un descenso del 63% (razón de tasas 0,37, p<0,001) relativo a placebo para los pacientes con omalizumab.
Durante el periodo de tratamiento doble ciego de 52 semanas (incluyendo la fase de esteroides a dosis fija de 24 semanas y la fase de ajuste de esteroides de 28 semanas) la diferencia en las tasas entre los grupos de tratamiento representó un descenso relativo del 50% (razón de tasas 0,504, p<0,001) en exacerbaciones para los pacientes con omalizumab.
El grupo de omalizumab mostró descensos mayores en el uso de agonistas beta como medicación de rescate que el grupo placebo al final del periodo de tratamiento de 52 semanas, aunque la diferencia entre los grupos de tratamiento no fue estadísticamente significativa. De la evaluación global de la efectividad del tratamiento al final del periodo de tratamiento doble ciego de 52 semanas en el subgrupo de pacientes graves con corticoides inhalados a dosis alta más agonistas beta de larga duración, la proporción de pacientes calificados como que tuvieron una efectividad de tratamiento “excelente” fue superior, y la proporción que tuvo una efectividad de tratamiento “moderada” o “débil” fue inferior en el grupo de omalizumab comparado con el grupo placebo; la diferencia entre grupos fue estadísticamente significativa (p<0,001), mientras que no hubo diferencias entre los grupos de omalizumab y placebo en las valoraciones de calidad de vida subjetiva de los pacientes.
5.2 Propiedades farmacocinéticas
Se ha estudiado la farmacocinética de omalizumab en pacientes adultos y adolescentes con asma alérgica.
Absorción
Tras la administración subcutánea, omalizumab se absorbe con una biodisponibilidad absoluta media del 62%. Después de administrar una dosis subcutánea única a pacientes adultos y adolescentes con asma, omalizumab se absorbió lentamente alcanzando concentraciones plasmáticas máximas después de una media de 7-8 días. La farmacocinética de omalizumab es lineal a dosis mayores de 0,5 mg/kg. Tras dosis múltiples de omalizumab, las áreas bajo la curva de concentración plasmática-tiempo del Día 0 al Día 14 en estado estacionario fueron de hasta 6-veces las obtenidas tras la primera dosis.
La administración de Xolair fabricado como una formulación liofilizada o líquida dió como resultado perfiles similares de concentración sérica-tiempo de omalizumab.
Distribución
In vitro, omalizumab forma complejos de tamaño limitado con la IgE. No se han observado tanto in vitro como in vivo complejos precipitantes ni complejos con pesos moleculares superiores a un millón de Daltons. Tras la administración subcutánea el volumen de distribución aparente en los pacientes fue de 78 ± 32 ml/kg.
Eliminación
El aclaramiento de omalizumab comprende procesos de aclaramiento de las IgG, así como, aclaramiento a través de uniones específicas y formación de complejos con su ligando específico, la IgE. La eliminación hepática de IgG incluye una degradación en el sistema reticuloendotelial y células endoteliales. La IgG inalterada también se excreta en la bilis. La vida media de eliminación plasmática de omalizumab en pacientes asmáticos fue de un promedio de 26 días, con un aclaramiento aparente promedio de 2,4 ± 1,1 ml/kg/día. Además, la duplicación del peso corporal hace que el aclaramiento aparente sea de aproximadamente el doble.
Características demográficas
Edad, Raza/Grupo étnico, Sexo, Indice de Masa Corporal
Se analizó la farmacocinética poblacional de Xolair con el fin de evaluar los efectos de las características demográficas. El análisis de los escasos datos disponibles indica que no es necesario efectuar un ajuste de la dosis en función de la edad (6-76 años), raza/grupo étnico, sexo o Índice de Masa Corporal (ver sección 4.2).
Insuficiencia renal y hepática
No se dispone de datos farmacocinéticos o farmacodinámicos en pacientes con insuficiencia renal o hepática (ver secciones 4.2 y 4.4).
5.3 Datos preclínicos sobre seguridad
Se ha estudiado la seguridad de omalizumab en monos cinomolgos, ya que el omalizumab se une a las IgE del cinomolgos y humanas con una afinidad similar. Se detectaron anticuerpos a omalizumab en algunos monos tras la administración subcutánea o intravenosa repetida. Sin embargo, no se observó toxicidad aparente, tales como enfermedad mediada por inmunocomplejos o citotoxicidad dependiente de complemento. No hubo evidencia de una respuesta anafiláctica debido a la desgranulación de los mastocitos en monos cinomolgos.
La administración crónica de omalizumab a niveles de dosis de hasta 250 mg/kg (al menos 14 veces la dosis clínica más alta recomendada en mg/kg de acuerdo con la tabla de dosis recomendada) fue bien tolerada en primates no humanos (tanto en animales adultos como en jóvenes), con la excepción de un descenso en las plaquetas sanguíneas dependiente de la dosis y de la edad, con una mayor sensibilidad en animales jóvenes. La concentración plasmática necesaria para alcanzar un descenso del 50% en las plaquetas con respecto al valor basal en monos adultos cinomolgos fue aproximadamente de 4 a 20 veces más elevada que la concentración plasmática clínica máxima anticipada. Además, se observó inflamación y hemorragia aguda en el lugar de la inyección en monos cinomolgos.
No se han realizado estudios de carcinogenicidad formal con omalizumab.
En los estudios de reproducción en monos cinomolgos, dosis subcutáneas de hasta 75 mg/kg por semana (al menos 8 veces la dosis clínica más alta recomendada en mg/kg durante un periodo de 4-semanas) no provocaron toxicidad materna, embriotoxicidad o teratogenicidad cuando se administró durante toda la organogénesis y no provocó efectos adversos sobre el crecimiento fetal o neonatal cuando se administró durante la fase final de la gestación, parto y lactancia.
Omalizumab se excretó en la leche materna en monos cinomolgos. Los niveles de omalizumab en la leche fueron del 0,15% con respecto a la concentración sérica materna.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Hidrocloruro de L-arginina Hidrocloruro de L-histidina L-histidina Polisorbato 20
Agua para preparaciones inyectables
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros medicamentos.
6.3 Periodo de validez
15 meses.
El periodo de validez incluye posibles desviaciones de temperatura. El medicamento puede conservarse durante un total de 4 horas a 25°C. Si es necesario, el medicamento puede volver a guardarse en la nevera para utilizarlo más tarde, pero esto no debe hacerse más de una vez.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
6.5 Naturaleza y contenido del envase
0. 5.ml de solución en un cilindro de jeringa precargada (vidrio tipo I) con aguja fijada (acero inoxidable), tapón del émbolo (tipo I) y capuchón de la aguja.
Envase conteniendo 1 jeringa precargada, y envases multiples que contienen 4 (4 envases de 1) o 10 (10 envases de 1) jeringas precargadas.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Antes de completar la inyección, evite el contacto con las pinzas de activación del dispositivo para evitar que la aguja quede prematuramente cubierta por el protector.
Uso de la jeringa
1. Manteniendo la jeringa con la aguja mirando hacia arriba, retire con cuidado de la jeringa, la cápsula protectora de la aguja y deséchela. No toque la aguja expuesta. Posteriormente, golpee suavemente la jeringa con su dedo hasta que las burbujas de aire asciendan a la superficie de la jeringa. Presione lentamente el émbolo para forzar la expulsión de las burbujas de aire fuera de la jeringa sin expulsar solución inadvertidamente.
2. Pellizque suavemente la piel en el lugar de la inyección e inserte la aguja.
3. Sosteniendo por la aleta de sujeción, presione lentamente el émbolo hasta donde sea posible. Si gotea solución por el lugar de la inyección, inserte más la aguja.
4. Manteniendo el émbolo completamente presionado, extraiga la aguja cuidadosamente y por completo del lugar de la inyección.
5. Suelte lentamente el émbolo y deje que el protector de la aguja cubra automáticamente la aguja expuesta.
Instrucciones de eliminación
Deseche inmediatamente la jeringa usada en un contenedor para material cortante.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/319/005
EU/1/05/319/006
EU/1/05/319/007
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 25 Octubre 2005 Fecha de la última renovación: 22 Junio 2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
1. NOMBRE DEL MEDICAMENTO
Xolair 150 mg solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada jeringa precargada de 1 ml de solución contiene 150 mg de omalizumab*.
*Omalizumab es un anticuerpo monoclonal humanizado obtenido mediante la tecnología del ADN recombinante, a partir de una línea celular mamífera de ovario de hámster chino (OHC).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución de transparente a ligeramente opalescente, incolora a color amarillo parduzco claro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Asma alérgica
Xolair está indicado en adultos, adolescentes y niños (de 6 a <12 años).
El tratamiento con Xolair deberá ser considerado únicamente para pacientes con asma mediada de forma convincente por IgE (inmunoglobulina E) (ver sección 4.2).
Adultos y adolescentes (a _partir de 12 años de edad)
Xolair está indicado para mejorar el control del asma cuando se administra como tratamiento adicional en pacientes con asma alérgica grave persistente que presentan test cutáneo positivo o reactividad in vitro a aeroalergenos perennes y con función pulmonar reducida (FEVi <80%) así como, síntomas frecuentes durante el día o despertares por la noche y que han presentado múltiples exacerbaciones asmáticas graves documentadas, a pesar de utilizar corticosteroides diarios inhalados a dosis altas, más un agonista beta2 inhalado de larga duración.
Niños (6 a <12 años de edad)
Xolair está indicado para mejorar el control del asma cuando se administra como tratamiento adicional en pacientes con asma alérgica grave persistente que presentan test cutáneo positivo o reactividad in vitro a aeroalergenos perennes y síntomas frecuentes durante el día o despertares por la noche y que han presentado múltiples exacerbaciones asmáticas graves documentadas, a pesar de utilizar corticosteroides diarios inhalados a dosis altas, más un agonista beta2 inhalado de larga duración.
Urticaria crónica espontánea (UCE)
Xolair está indicado como tratamiento adicional, para el tratamiento de la urticaria crónica espontánea en pacientes adultos y adolescentes (a partir de 12 años) con respuesta inadecuada al tratamiento con antihistamínicos H1.
4.2 Posología y forma de administración
El tratamiento con Xolair debe iniciarlo un médico experimentado en el diagnóstico y tratamiento del asma grave persistente o urticaria crónica espontánea.
Asma alérgica
Posología
La dosis apropiada y la frecuencia de administración de Xolair se determina a partir de la concentración basal de IgE (UI/ml), medida antes de iniciar el tratamiento, y del peso corporal (kg). A efectos de la asignación de la dosis, antes de la administración de la dosis inicial se debe determinar la concentración de IgE en los pacientes mediante cualquier método comercial que analice la IgE plasmática total. En base a estas determinaciones, podrán ser necesarios en cada administración de 75 a 600 mg de Xolair en 1 a 4 inyecciones.
Era menos probable que experimentaran beneficio los pacientes con un valor de IgE inferior a 76 UI/ml (ver sección 5.1). Los médicos prescriptores deberán asegurar que los pacientes adultos y adolescentes con una IgE por debajo de 76 UI/ml y los niños (6 a < 12 años de edad) con una IgE por debajo de 200 UI/ml presenten una reactividad in vitro inequívoca (RAST) al alergeno perenne antes de iniciar el tratamiento.
Ver Tabla 1 de conversión y Tablas 2 y 3 para la determinación de dosis en adultos, adolescentes y niños (de 6 a <12 años).
No debe administrarse Xolair a pacientes cuya concentración basal de IgE o peso corporal en kilogramos, excedan los límites indicados en la tabla de dosis.
La dosis máxima recomendada es de 600 mg de omalizumab cada dos semanas.
Tabla 1: Conversión de la dosis al número de jeringas, número de inyecciones y volumen de inyección total para cada administración
|
Dosis (mg) Número de jeringas Número de inyecciones 75 mg 150 mg |
Volumen total de inyección (ml) | |||
|
75 |
1 |
0 |
1 |
0,5 |
|
150 |
0 |
1 |
1 |
1,0 |
|
225 |
1 |
1 |
2 |
1,5 |
|
300 |
0 |
2 |
2 |
2,0 |
|
375 |
1 |
2 |
3 |
2,5 |
|
450 |
0 |
3 |
3 |
3,0 |
|
525 |
1 |
3 |
4 |
3,5 |
|
600 |
0 |
4 |
4 |
4,0 |
|
Peso corporal (kg) | ||||||||||
|
IgE basal |
>20- |
>25- |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>90- |
>125- |
|
(UI/ml) |
25 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
125 |
150 |
|
>30-100 |
75 |
75 |
75 |
150 |
150 |
150 |
150 |
150 |
300 |
300 |
|
>100-200 |
150 |
150 |
150 |
300 |
300 |
300 |
300 |
300 |
450 |
600 |
|
>200-300 |
150 |
150 |
225 |
300 |
300 |
450 |
450 |
450 |
600 | |
|
>300-400 |
225 |
225 |
300 |
450 |
450 |
450 |
600 |
600 | ||
|
>400-500 |
225 |
300 |
450 |
450 |
600 |
600 | ||||
|
>500-600 |
300 |
300 |
450 |
600 |
600 | |||||
|
>600-700 |
300 |
450 |
600 | |||||||
|
>700-800 | ||||||||||
|
>800-900 |
ADMINISTRACIÓN CADA | |||||||||
|
>900- 1000 |
2 SEMANAS VER TABLA 3 | |||||||||
|
>1000- 1100 | ||||||||||
|
Peso corporal (kg) | ||||||||||
|
IgE basal |
>20- |
>25- |
>30- |
>40- |
>50- |
>60- |
>70- |
>80- |
>90- |
>125- |
|
(UI/ml) |
25 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
125 |
150 |
|
>30-100 |
ADMINISTRACIÓN CADA | |||||||||
|
>100-200 |
4 SEMANAS VER TABLA 2 | |||||||||
|
>200-300 |
375 | |||||||||
|
>300-400 |
450 |
525 | ||||||||
|
>400-500 |
375 |
375 |
525 |
600 | ||||||
|
>500-600 |
375 |
450 |
450 |
600 | ||||||
|
>600-700 |
225 |
375 |
450 |
450 |
525 | |||||
|
>700-800 |
225 |
225 |
300 |
375 |
450 |
450 |
525 |
600 | ||
|
>800-900 |
225 |
225 |
300 |
375 |
450 |
525 |
600 | |||
|
>900- |
225 |
300 |
375 |
450 |
525 |
600 | ||||
|
1000 | ||||||||||
|
>1000- 1100 |
225 |
300 |
375 |
450 |
600 | |||||
|
>1100- |
300 |
300 |
450 |
525 |
600 |
NO ADMINISTRAR - |
no se dispone | |||
|
1200 |
de datos para la recomendación de | |||||||||
|
dosis | ||||||||||
|
>1200- 1300 |
300 |
375 |
450 |
525 | ||||||
|
>1300- 1500 |
300 |
375 |
525 |
600 | ||||||
Duración del tratamiento, monitorización y ajuste de dosis
Xolair está indicado para tratamiento a largo plazo. Los ensayos clínicos han demostrado que son necesarias un mínimo de 12-16 semanas para que el tratamiento con Xolair demuestre efectividad. A las 16 semanas de iniciar el tratamiento con Xolair, los pacientes deberán ser evaluados por su médico con respecto a la efectividad del tratamiento antes de administrar inyecciones posteriores. La decisión de continuar con Xolair tras las 16 semanas, o en ocasiones posteriores, debe estar basada en si se observa una notable mejoría en el control global del asma (ver sección 5.1; Valoración global del médico con respecto a la efectividad del tratamiento).
La interrupción del tratamiento con Xolair generalmente da lugar a un retorno de las concentraciones elevadas de IgE libre y de los síntomas asociados. Los valores de IgE total son elevados durante el tratamiento y siguen siéndolo hasta un año después de la interrupción del mismo. Por lo tanto, no puede utilizarse la reevaluación de los valores de IgE durante el tratamiento con Xolair como guía para la determinación de la dosis. La determinación de la dosis tras interrupciones de tratamiento de menos de un año de duración debe basarse en las concentraciones plasmáticas de IgE obtenidas en la determinación de dosis inicial. Si el tratamiento con Xolair se ha interrumpido por más de un año deberán de volver a medirse las concentraciones plasmáticas de IgE total para la determinación de la dosis.
Las dosis deberán ajustarse en caso de variaciones significativas del peso corporal (ver Tablas 2 y 3).
Urticaria crónica espontánea (UCE)
Posología
La dosis recomendada es de 300 mg por inyección subcutánea cada cuatro semanas.
Se recomienda a los prescriptores que reevaluen periódicamente la necesidad de continuar el tratamiento.
La experiencia en ensayos clínicos del tratamiento a largo plazo durante más de 6 meses en esta indicación es limitada.
Poblaciones especiales
Pacientes de edad avanzada (a _partir de 65 años)
Aunque se dispone de datos limitados sobre el uso de Xolair en pacientes mayores de 65 años, no existe evidencia de que los pacientes de edad avanzada requieran una dosis diferente de la de pacientes adultos más jóvenes.
Insuficiencia renal o hepática
No se dispone de estudios sobre el efecto de la insuficiencia renal o hepática en la farmacocinética de omalizumab. Debido a que el aclaramiento de omalizumab a dosis clínicas se lleva a cabo fundamentalmente por el sistema reticuloendotelial (SRE), es improbable que se vea alterado en caso de insuficiencia renal o hepática. Xolair deberá administrarse con precaución en estos pacientes, mientras no se recomiende un ajuste especial de la dosis (ver sección 4.4).
Población pediátrica
En asma alérgica, no se ha establecido la seguridad y eficacia de Xolair en pacientes pediátricos menores de 6 años. No se dispone de datos.
En UCE, no se ha establecido la seguridad y eficacia de Xolair en pacientes pediátricos menores de 12 años.
Forma de administración
Para administración subcutánea únicamente. No administrar por vía intravenosa o intramuscular.
Las inyecciones se administran vía subcutánea en la región deltoidea del brazo. Si por alguna razón no pueden administrarse en esta zona, podrán administrase alternativamente en el muslo.
Existe experiencia limitada con respecto a la autoadministración de Xolair. Por lo tanto, está previsto que el tratamiento sea administrado únicamente por el profesional sanitario.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6 y también la sección “información para el profesional sanitario” del prospecto.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
General
Xolair no está indicado para el tratamiento de las exacerbaciones asmáticas, broncoespasmo o estados asmáticos de carácter agudo.
No se ha estudiado el efecto de Xolair en pacientes con síndrome de hiperinmunoglobulinemia E o aspergilosis broncopulmonar alérgica, ni en la prevención de reacciones anafilácticas, incluyendo las provocadas por alergias alimentarias, dermatitis atópica, o rinitis alérgica. Xolair no está indicado en el tratamiento de estas patologías.
El tratamiento con Xolair tampoco se ha estudiado en pacientes con enfermedades autoinmunes, procesos mediados por inmunocomplejos, o insuficiencia renal o hepática preexistente (ver sección 4.2). Se deberá tener precaución cuando se administre Xolair en esta población de pacientes.
No se recomienda la interrupción brusca de los corticosteroides sistémicos o inhalados tras la iniciación del tratamiento con Xolair. El descenso de los corticosteroides debe realizarse bajo la supervisión directa de un médico y puede ser necesario que se realice gradualmente.
Trastornos del sistema inmulógico
Reacciones alérgicas tipo I
Pueden producirse reacciones alérgicas tipo I locales o sistémicas, incluyendo anafilaxia y shock anafiláctico durante el tratamiento con omalizumab, incluso con inicio tras un tratamiento de larga duración. La mayoría de estas reacciones se produjeron durante las 2 horas posteriores a la primera y siguientes inyecciones de Xolair, pero algunas se iniciaron pasadas las 2 horas e incluso pasadas 24 horas tras la inyección. Por lo tanto, se deberán tener siempre disponibles medicamentos para el tratamiento inmediato de reacciones anafilácticas tras la administración de Xolair. Se deberá informar al paciente de que estas reacciones son posibles y que si se producen deberán solicitar atención médica de inmediato. Un antecedente de anafilaxia no asociado a omalizumab puede suponer un factor de riesgo de sufrir una reacción anafiláctica tras la administración de Xolair.
Se han detectado anticuerpos contra omalizumab en un pequeño número de pacientes en ensayos clínicos (ver sección 4.8). No se conoce bien la relevancia clínica de anticuerpos anti-Xolair.
Enfermedad del suero
Se ha observado enfermedad del suero y reacciones semejantes a la enfermedad del suero, que son reacciones alérgicas tipo III retardadas, en pacientes tratados con anticuerpos monoclonales humanizados incluido omalizumab. El supuesto mecanismo fisiopatológico incluye formación y deposición de inmunocomplejos debido al desarrollo de anticuerpos contra omalizumab. El inicio del cuadro se produce normalmente a los 1-5 días tras la administración de la primera o siguientes inyecciones e incluso tras un tratamiento de larga duración. Los síntomas que sugieren la enfermedad del suero incluyen artritis/artralgias, rash (urticaria u otras formas), fiebre y linfoadenopatía. Los antihistamínicos y corticosteroides pueden ser útiles para prevenir o tratar estas alteraciones y se deberá advertir a los pacientes que notifiquen cualquier síntoma sospechoso.
Síndrome de Churg-Strauss y síndrome hipereosinofílico
Los pacientes con asma grave pueden presentar raramente síndrome hipereosinofílico sistémico o vasculitis granulomatosa eosinofílica alérgica (Síndrome de Churg-Strauss), los cuales son normalmente tratados con corticosteroides sistémicos.
En raras ocasiones, los pacientes en tratamiento con medicamentos antiasmáticos, incluyendo omalizumab, pueden presentar o desarrollar eosinofilia sistémica y vasculitis. Estas reacciones están normalmente asociadas con la reducción del tratamiento con corticosteroides orales.
En estos pacientes, los médicos deberán estar alerta ante el desarrollo de eosinofilia importante, rash vasculítico, empeoramiento de los síntomas pulmonares, anormalidades en el seno paranasal, complicaciones cardíacas, y/o neuropatía.
Deberá considerarse la interrupción del tratamiento con omalizumab en todos aquellos casos graves que cursen con alteraciones del sistema inmune mencionadas anteriormente.
Infecciones parasitarias (helmínticos)
Las IgE pueden estar involucradas en la respuesta inmunológica a algunas infecciones helmínticas. En pacientes con un elevado riesgo crónico de infección helmíntica, un ensayo controlado con placebo en pacientes alérgicos demostró un ligero incremento en la proporción de infección con omalizumab, aunque no se modificó el curso, gravedad y respuesta al tratamiento de la infección. La proporción de infección helmíntica en el programa clínico global, el cual no fue diseñado para detectar este tipo de infecciones, fue inferior a 1 en 1.000 pacientes. Sin embargo, deberá garantizarse precaución en pacientes con elevado riesgo de infección helmíntica, en particular cuando viajen a zonas donde las infecciones helmínticas son endémicas. Si los pacientes no responden al tratamiento antihelmíntico recomendado, deberá considerarse la interrupción del tratamiento con Xolair.
Personas sensibles al látex
La cápsula protectora extraíble de la aguja de esta jeringa precargada contiene un derivado de látex de caucho natural. Hasta la fecha no se ha detectado látex de caucho natural en la cápsula protectora extraíble de la aguja. Sin embargo, no se ha estudiado el uso de Xolair solución inyectable en jeringa precargada en personas sensibles al látex y por lo tanto, existe un riesgo potencial de reacciones de hipersensibilidad, las cuales no se pueden descartar completamente.
4.5 Interacción con otros medicamentos y otras formas de interacción
Dado que las IgE pueden estar relacionadas con la respuesta immunológica a algunas infecciones por helmínticos, Xolair puede reducir indirectamente la eficacia de medicamentos utilizados para el tratamiento de infecciones helmínticas o por otros parásitos (ver sección 4.4).
Las enzimas del citocromo P450, las bombas de eflujo y los mecanismos de unión a proteínas no se hallan implicados en el aclaramiento de omalizumab; por ello, existe un bajo potencial de interacciones farmacológicas. No se han realizado estudios de interacción de otros medicamentos o vacunas con Xolair. No existe un motivo farmacológico para esperar que los medicamentos prescritos frecuentemente en el tratamiento del asma o UCE interaccionen con omalizumab.
Asma alérgica
En los ensayos clínicos Xolair se utilizó frecuentemente asociado a corticosteroides inhalados y orales, beta agonistas inhalados de corta y larga duración, antagonistas de los leucotrienos, teofilinas y antihistamínicos orales. No hubo indicios de que estos medicamentos utilizados habitualmente como antiasmáticos puedan afectar a la seguridad de Xolair. Se dispone de datos limitados sobre el uso de Xolair en combinación con inmunoterapia específica (terapia de hiposensibilización). En un ensayo clínico donde Xolair se administró conjuntamente con inmunoterapia, se observó que la seguridad y eficacia de Xolair en combinación con inmunoterapia específica, no fue diferente a la de Xolair solo.
Urticaria crónica espontánea (UCE)
En ensayos clínicos en UCE, Xolair se utilizó junto con antihistamínicos (anti-H1, anti-H2) y antagonistas del receptor de leucotrieno (ARLTs). No hubo evidencia de que la seguridad de omalizumab se alterase cuando se utilizaba con estos medicamentos en relación a su perfil de seguridad conocido en asma alérgica. Además, un análisis farmacocinético poblacional no mostró un efecto relevante de los antihistamínicos H2 y ARLTs sobre la farmacocinética de omalizumab (ver sección 5.2).
Población pediátrica
Los ensayos clínicos en UCE incluyeron algunos pacientes de 12 a 17 años de edad que usaron Xolair junto con antihistamínicos (anti-H1, anti-H2) y ARLTs. No se realizaron ensayos en niños menores de 12 años.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Hay datos limitados relativos al uso de omalizumab en mujeres embarazadas. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción (ver sección 5.3). Omalizumab atraviesa la barrera placentaria y se desconoce el daño potencial sobre el feto. Omalizumab se ha asociado con descensos de las plaquetas sanguíneas, dependientes de la edad en primates no humanos, con una sensibilidad relativamente superior en animales jóvenes (ver sección 5.3). Xolair no debería utilizarse durante el embarazo excepto si fuese claramente necesario.
Lactancia
Se desconoce si omalizumab se excreta en la leche materna. Los datos disponibles en primates no humanos muestran que omalizumab se excreta en la leche (ver sección 5.3). No se puede excluir el riesgo en recién nacidos/niños. Omalizumab no se debe administrar durante la lactancia.
Fertilidad
No hay datos de fertilidad en humanos para omalizumab. En estudios de fertilidad no clínicos diseñados específicamente en primates no humanos, incluidos los estudios de apareamiento, no se observó empeoramiento de la fertilidad en machos o hembras tras dosis repetidas con omalizumab de hasta 75 mg/kg. Además, no se observaron efectos genotóxicos en estudios separados no clínicos de genotoxicidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Xolair sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Asma alérgica
Resúmen del perfil de seguridad
Las reacciones adversas notificadas más frecuentemente durante los ensayos clínicos en pacientes adultos y adolescentes a partir de 12 años de edad fueron cefalea y reacciones en el lugar de la inyección, que incluían dolor, tumefacción, eritema y prurito. Las reacciones adversas notificadas más frecuentemente en los ensayos clínicos en niños de 6 a <12 años de edad fueron cefalea, pirexia y dolor abdominal superior. La mayoría de las reacciones fueron de gravedad leve a moderada.
Tabla de reacciones adversas
En la Tabla 4 se incluyen las reacciones adversas registradas en la población total de seguridad tratada con Xolair en los ensayos clínicos, por sistema de clasificación de órganos y frecuencia de MedDRA. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Las categorías de frecuencia se definen como: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000) y muy raras (<1/10.000). Las reacciones notificadas en la fase de postcomercialización se enumeran con frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 4: Reacciones adversas en asma alérgica
|
Infecciones e infestaciones | |
|
Poco frecuentes Raras |
Faringitis Infección parasitaria |
|
Trastornos de la sangre y del sistema linfático | |
|
No conocida |
Trombocitopenia idiopática, incluyendo casos graves |
|
Trastornos del sistema inmunológico | |
|
Raras No conocida |
Reacción anafiláctica, otros procesos alérgicos graves, desarrollo de anticuerpos frente a omalizumab Enfermedad del suero que puede cursar con fiebre y linfoadenopatía |
|
Trastornos del sistema nervioso | |
|
Frecuentes Poco frecuentes |
Cefalea* Síncope, parestesia, somnolencia, mareo |
|
Trastornos vasculares | |
|
Poco frecuentes |
Hipotensión postural, rubor |
|
Trastornos respiratorios, torácicos y mediastínicos | |
|
Poco frecuentes Raras No conocida |
Broncoespasmo alérgico, tos Laringoedema Vasculitis granulomatosa alérgica (es decir, síndrome de Churg Strauss) |
|
Trastornos gastrointestinales | |
|
Frecuentes Poco frecuentes |
Dolor abdominal superior** Signos y síntomas dispépticos, diarrea, náuseas |
|
Trastornos de la piel y del tejido subcutáneo | |
|
Poco frecuentes Raras No conocida |
Fotosensibilidad, urticaria, rash, prurito Angioedema Alopecia |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |
|
Raras |
Lupus eritematoso sistémico (LES) |
|
No conocidas |
Artralgia, mialgia, tumefacción de las articulaciones |
|
Trastornos generales y alteraciones en el lugar de administración | |
|
Muy frecuentes Frecuentes Poco frecuentes |
Pirexia** Reacciones en el lugar de la inyección tales como tumefacción, eritema, dolor, prurito Enfermedad pseudo-gripal, brazos hinchados, incremento de peso, fatiga |
*: Muy frecuentes en niños de 6 a <12 años de edad **: En niños de 6 a <12 años de edad
Urticaria crónica espontánea (UCE)
Resúmen del perfil de seguridad
Se investigó la seguridad y tolerancia de omalizumab con dosis de 75 mg, 150 mg y 300 mg cada cuatro semanas en 975 pacientes con UCE, 242 de los cuales recibieron placebo. En global,
733 pacientes se trataron con omalizumab durante 12 semanas y 490 pacientes durante 24 semanas. De estos, 412 pacientes se trataron durante 12 semanas y 333 pacientes se trataron durante 24 semanas a la dosis de 300 mg.
Tabla de reacciones adversas
En otra tabla (Tabla 5) se muestran las reacciones adversas para la indicación de UCE como resultado de las diferentes dosis y poblaciones de tratamiento (con factores de riesgo significativamente diferentes, comorbilidades, medicaciones concomitantes y edades [p.ej. ensayos clínicos en asma que incluyeron niños de 6-12 años de edad]).
En la Tabla 5 se incluyen las reacciones adversas (acontecimientos ocurridos en >1% de los pacientes en cualquiera de los grupos de tratamiento y >2% más frecuentemente en cualquiera de los grupos de tratamiento con omalizumanb que con placebo (después de la revisión médica)) notificadas con la dosis de 300 mg en los tres ensayos de fase III agrupados. Las reacciones adversas presentadas se dividen en dos grupos: las identificadas en los periodos de tratamiento de 12-semanas y de 24-semanas.
Las reacciones adversas se incluyen por sistema de clasificación de órganos de MedDRA. Las reacciones adversas se clasifican por frecuencia dentro de cada sistema de órganos, incluyendo primero las reacciones más frecuentes. Para cada reacción adversa, la correspondiente categoría de frecuencia se basa en la siguiente convención: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 5: Reacciones adversas de la base de datos de seguridad de UCE agrupada (día 1 a semana 24) a la dosis de 300 mg de omalizumab
|
Ensayos con omalizumab 1, 2 y 3 |
Categoría de frecuencia | ||
|
12-Semanas |
agrupados | ||
|
Placebo N=242 |
300 mg N=412 | ||
|
Infecciones e infestaciones | |||
|
Sinusitis |
5 (2,1%) |
20 (4,9%) |
Frecuente |
|
Trastornos del sistema nervioso | |||
|
Cefalea |
7 (2,9%) |
25 (6,1%) |
Frecuente |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |||
|
Artralgia |
1 (0,4%) |
12 (2,9%) |
Frecuente |
|
Trastornos generales y alteraciones en el lugar de administración | |||
|
Reacción en el lugar de la inyección* |
2 (0,8%) |
11 (2,7%) |
Frecuente |
|
Ensayos con omalizumab 1 y 3 agrupados |
Categoría de frecuencia | ||
|
24-Semanas |
Placebo N=163 |
300 mg N=333 | |
|
Infecciones e infestaciones Infección de las vías altas del tracto respiratorio |
5 (3,1%) |
19 (5,7%) |
Frecuente |
* A pesar de no mostrar una diferencia del 2% con respecto a placebo, se incluyeron las reacciones en el lugar de la inyección ya que todos los casos fueron evaluados como relacionados causalmente al tratamiento en estudio.
Descripción de reacciones adversas seleccionadas en relación a las indicaciones de asma alérgica y UCE
No se han obtenido datos relevantes en los ensayos clínicos en UCE que requiriesen una modificación de las secciones siguientes.
Trastornos del sistema inmunológico Para mayor información, ver sección 4.4.
Anafilaxia
Rara vez se observaron reacciones anafilácticas en los ensayos clínicos. Sin embargo, después de una búsqueda acumulada en la base de datos de seguridad, se recogieron un total de 898 casos de anafilaxia post comercialización. En base a una exposición estimada de 566.923 paciente tratado años, resultó en una frecuencia aproximada de 0,20%.
Efectos tromboembólicos arteriales (ETA)
En ensayos clínicos controlados y durante los análisis intermedios de un estudio observacional, se observó un desequilibrio numérico de ETA. La definición de la variable compuesta ETA incluye derrame cerebral, ataque isquémico transitorio, infarto de miocardio, angina inestable y muerte cardiovascular (incluyendo muerte por causa desconocida). En el análisis final del estudio observacional, el índice de ETA por 1.000 pacientes años fue de 7,52 (115/15.286 pacientes años) para los pacientes tratados con Xolair y de 5,12 (51/9.963 pacientes años) para los pacientes control. En un análisis multivariado para controlar los factores basales disponibles de riesgo cardiovascular, la proporción de riesgo fue de 1,32 (intervalo de confianza del 95% 0,91-1,91). En un análisis segregado de un conjunto de ensayos clínicos, el cual incluyó todos los ensayos clínicos aleatorizados, doble ciego, controlados con placebo de 8 semanas o más de duración, el índice de ETA por 1.000 pacientes años fue de 2,69 (5/1.856 pacientes años) para los pacientes tratados con Xolair y de 2,38 (4/1.680 pacientes años) para los pacientes tratados con placebo (tasa de incidencia 1,13, intervalo de confianza del 95% 0,24-5,71).
Plaquetas
En los ensayos clínicos, pocos pacientes presentaron recuentos de plaquetas por debajo del límite inferior del intervalo normal de laboratorio. Ninguno de estos cambios se asoció con episodios hemorrágicos o con una disminución de la hemoglobina. En los seres humanos (pacientes a partir de 6 años de edad), a diferencia de los primates no humanos, no se ha observado ningún patrón de disminución persistente en el recuento de plaquetas (ver sección 5.3), aunque se han notificado casos aislados de trombocitopenia idiopática, incluyendo casos graves, en la fase de postcomercialización.
Infecciones _ parasitarias
En pacientes alérgicos con un elevado riesgo crónico de infección helmíntica, un ensayo controlado con placebo demostró un ligero incremento numérico en la proporción de infección con omalizumab que no fue estadísticamente significativo. No se modificaron el curso, gravedad y respuesta al tratamiento de la infección (ver sección 4.4).
Lupus eritematoso sistémico
En pacientes con asma moderada a grave y urticaria crónica espontánea (UCE) se han notificado casos de lupus eritematoso sistémico (LES) en ensayos clínicos y etapa post comercialización. La patogénesis del lupus eritematoso sistémico no se conoce bien.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
No se ha determinado la dosis máxima tolerada de Xolair. Se han administrado dosis únicas intravenosas de hasta 4.000 mg a pacientes sin evidencia de toxicidad dependiente de la dosis. La mayor dosis acumulada que se administró a los pacientes durante un periodo de 20-semanas fue de 44.000 mg y esta dosis no produjo ningún efecto adverso agudo.
En caso de sospecha de una sobredosis, se deberá monitorizar al paciente para cualquier signo o síntoma anormal. Se deberá buscar e instaurar tratamiento médico adecuado.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: fármacos para enfermedades obstructivas de las vías respiratorias, otros agentes sistémicos para enfermedades obstructivas de las vías respiratorias, código ATC: R03DX05
Omalizumab es un anticuerpo monoclonal humanizado derivado del ADN recombinante que se une selectivamente a la inmunoglobulina E humana (IgE). El anticuerpo es una IgG1 kappa que contiene regiones de estructura humana con regiones determinantes de complementariedad de un anticuerpo de origen murino que se une a la IgE.
Asma alérgica
Mecanismo de acción
Omalizumab se une a la IgE y previene la unión de ésta al FcsRI (receptor de IgE de alta afinidad) en basófilos y mastocitos, reduciendo así la cantidad de IgE libre disponible para desencadenar la cascada alérgica. El tratamiento de pacientes atópicos con omalizumab dio como resultado una disminución acentuada de los receptores FcsRI en los basófilos.
Efectos _ farmacodinámicos
La liberación in vitro de histamina de los basófilos aislados de pacientes que habían sido tratados con Xolair se redujo aproximadamente en un 90% tras la estimulación con un alergeno en comparación con los valores previos al tratamiento.
En los ensayos clínicos en pacientes con asma alérgica, las concentraciones plasmáticas de IgE libre disminuyeron de manera dosis dependiente en la hora posterior a la primera dosis y se mantuvieron reducidas entre dosis. Un año después de interrumpir el tratamiento con Xolair los niveles de IgE volvieron a los niveles previos al tratamiento, sin que se observase efecto de rebote en los niveles de IgE después del periodo de blanqueo del medicamento.
Urticaria crónica espontánea (UCE)
Mecanismo de acción
Omalizumab se une a la IgE y a los niveles bajos de IgE libre. Como consecuencia, disminuyen los receptores de IgE (FCsRI) en las células. No se entiende completamente como esto se traduce en una mejoría de los síntomas de la UCE.
Efectos _ farmacodinámicos
En los ensayos clínicos en pacientes con UCE se observó la supresión máxima de IgE libre tres días después de la primera dosis subcutánea. Después de dosis repetidas una vez cada 4 semanas, los niveles predosis de IgE libre en suero permanecieron estables entre 12 y 24 semanas de tratamiento. Tras la interrupción de Xolair, los niveles de IgE libre incrementaron hacia los niveles pretratamiento tras un periodo de seguimiento superior a 16 semanas libre de tratamiento.
Eficacia clínica y seguridad en asma alérgica
Adultos y adolescentes >12 años de edad
La eficacia y seguridad de Xolair se demostró en un ensayo doble ciego controlado con placebo de 28 semanas (estudio 1) en el que participaron 419 pacientes con asma alérgica grave, de edades comprendidas entre los 12 y 79 años, con función pulmonar reducida (FEVi 40-80% del valor teórico) y un pobre control de los síntomas del asma a pesar de recibir dosis elevadas de corticosteroides inhalados y un agonista beta2 de larga duración. Los pacientes escogidos habían sufrido exacerbaciones asmáticas múltiples habiendo necesitado tratamiento con corticosteroides sistémicos o habiendo sido hospitalizados o atendidos de urgencia debido a una exacerbación asmática grave en el pasado año a pesar del tratamiento continuo con dosis elevadas de corticosteroides inhalados y un agonista beta2 de larga duración. Se administró Xolair vía subcutánea o placebo como tratamiento adicional a >1.000 microgramos de dipropionato de beclometasona (o equivalente) más un agonista beta2 de larga duración. Como tratamiento de mantenimiento se permitieron los corticosteroides orales, la teofilina y los antagonistas de los leucotrienos (22%, 27%, y 35% de pacientes, respectivamente).
Se tomó como variable principal la proporción de exacerbaciones asmáticas que requirieron tratamiento con dosis altas de corticosteroides sistémicos. Omalizumab redujo la proporción de exacerbaciones asmáticas en un 19% (p = 0,153). Posteriores evaluaciones que mostraron significancia estadística (p<0,05) en favor de Xolair incluyeron reducciones de las exacerbaciones graves (donde la función pulmonar de los pacientes se redujo por debajo del 60% del mejor valor personal, necesitando corticosteroides sistémicos) y de las visitas de urgencia relacionadas con el asma (incluyendo hospitalizaciones, sala de urgencias, y visitas al médico no programadas), y mejoría en la valoración global del médico con respecto a la efectividad del tratamiento, calidad de vida relacionada con el asma (AQL), síntomas del asma y función pulmonar.
En un análisis de subgrupos, era más probable que los pacientes con un valor total de IgE >76 Ul/ml previo al tratamiento experimentaran un beneficio clínicamente significativo a Xolair. En estos pacientes en el estudio 1, Xolair redujo la proporción de exacerbaciones asmáticas en un 40% (p = 0,002). Además, un mayor número de pacientes presentaron respuestas clínicamente significativas en la población con un valor total de IgE >76 UI/ml a través de un programa con Xolair de asma grave. La Tabla 6 incluye resultados de la población del estudio 1.
Tabla 6: Resultados del estudio 1
Población total estudio 1 Xolair Placebo
_N=209_N=210
Exacerbaciones asmáticas
Proporción por periodo de 0,74 0,92
28 semanas
Reducción %, valor p de razón de 19,4%, p = 0,153
tasas
Exacerbaciones de asma grave
Proporción por periodo de 0,24 0,48
28 semanas
Reducción %, valor p de razón de 50,1%, p = 0,002
tasas
Visitas de urgencia
Proporción por periodo de 0,24 0,43
28 semanas
Reducción %, valor p de razón de 43,9%, p = 0,038
tasas
Valoración global del médico
42,8%
47,8%
respondedores8 % 60,5%
Valor p9 <0,001
Mejoría AQL
% de pacientes con una mejoría 60,8%
>0,5
Valor p 0,008
En los estudios 3, 4 y 5, los pacientes tratados con Xolair presentaron reducciones en la proporción de exacerbaciones asmáticas del 37,5% (p = 0,027), 40,3% (p<0,001) y 57,6% (p<0,001), respectivamente, comparado con placebo.
En el estudio 6, los pacientes con asma alérgica significativamente más grave tratados con Xolair pudieron reducir su dosis de fluticasona a <500 microgramos/día sin deterioro del control del asma (60,3%) comparado con el grupo placebo (45,8%, p<0,05).
La puntuación de calidad de vida se midió utilizando el cuestionario de calidad de vida relacionada con el asma de Juniper. En los 6 estudios hubo una mejoría estadísticamente significativa con respecto a la puntuación de calidad de vida basal para los pacientes tratados con Xolair con respecto al grupo placebo o control.
Valoración global del médico con respecto a la efectividad del tratamiento:
La valoración global del médico se realizó en cinco de los estudios anteriormente mencionados como una amplia medida del control del asma realizada por el médico que trata al paciente. El médico pudo tener en cuenta el PEF (flujo espiratorio máximo), los síntomas durante el día y la noche, el uso de medicación de rescate, la espirometría y las exacerbaciones. En los cinco estudios una proporción significativamente superior de los pacientes tratados con Xolair declararon haber alcanzado una notable mejoría o un control completo del asma comparado con los pacientes tratados con placebo.
Niños de 6 a <12 años de edad
El soporte principal para la eficacia y seguridad de Xolair en el grupo de edad de 6 a <12 años se obtiene de un ensayo multicéntrico, randomizado, doble ciego, controlado con placebo (estudio 7).
El estudio 7 fue un ensayo controlado con placebo que incluyó un subgrupo específico (n=235) de pacientes tal y como se define en la presente indicación, quienes fueron tratados con corticoides inhalados a dosis altas (>500 pg/día de fluticasona o equivalente) más agonista beta de larga duración.
Según la opinión clínica del investigador, una exacerbación clínicamente significativa se definió como un empeoramiento de los síntomas del asma, la cual requería una dosis doble de corticosteroide inhalado con respecto al valor basal, durante un mínimo de 3 días y/o tratamiento de rescate con corticosteroides sistémicos (oral o intravenoso) durante un mínimo de 3 días.
En el subgrupo específico de pacientes con corticoides inhalados a dosis alta, el grupo de omalizumab tuvo una tasa inferior estadísticamente significativa de exacerbaciones asmáticas clínicamente significativas que el grupo placebo. A las 24 semanas, la diferencia en las tasas entre los grupos de tratamiento representó un descenso del 34% (razón de tasas 0,662, p = 0,047) relativo a placebo para los pacientes con omalizumab. En el segundo periodo de tratamiento doble ciego de 28 semanas la diferencia en las tasas entre los grupos de tratamiento representó un descenso del 63% (razón de tasas 0,37, p<0,001) relativo a placebo para los pacientes con omalizumab.
Durante el periodo de tratamiento doble ciego de 52 semanas (incluyendo la fase de esteroides a dosis fija de 24 semanas y la fase de ajuste de esteroides de 28 semanas) la diferencia en las tasas entre los grupos de tratamiento representó un descenso relativo del 50% (razón de tasas 0,504, p<0,001) en exacerbaciones para los pacientes con omalizumab.
El grupo de omalizumab mostró descensos mayores en el uso de agonistas beta como medicación de rescate que el grupo placebo al final del periodo de tratamiento de 52 semanas, aunque la diferencia entre los grupos de tratamiento no fue estadísticamente significativa. De la evaluación global de la efectividad del tratamiento al final del periodo de tratamiento doble ciego de 52 semanas en el subgrupo de pacientes graves con corticoides inhalados a dosis alta más agonistas beta de larga duración, la proporción de pacientes calificados como que tuvieron una efectividad de tratamiento “excelente” fue superior, y la proporción que tuvo una efectividad de tratamiento “moderada” o “débil” fue inferior en el grupo de omalizumab comparado con el grupo placebo; la diferencia entre grupos fue estadísticamente significativa (p<0,001), mientras que no hubo diferencias entre los grupos de omalizumab y placebo en las valoraciones de calidad de vida subjetiva de los pacientes.
Eficacia clínica y seguridad en urticaria crónica espontánea (UCE)
Se demostró la eficacia y seguridad de Xolair en dos ensayos de fase III aleatorizados, controlados con placebo (ensayos 1 y 2) en pacientes con UCE que permanecieron sintomáticos a pesar del tratamiento antihistamínico H1 a la dosis aprobada. Un tercer ensayo (ensayo 3) evaluó principalmente la seguridad de Xolair en pacientes con UCE que permanecieron sintomáticos a pesar del tratamiento con antihistamínicos H1 hasta cuatro veces la dosis aprobada y antihistamínicos H2 y/o tratamiento con ARLT. Los tres ensayos incluyeron 975 pacientes de edades comprendidas entre 12 y 75 años (edad media 42,3 años; 39 pacientes de 12-17 años, 54 pacientes >65 años; 259 hombres y 716 mujeres). Se requirió que todos los pacientes presentaran un control inadecuado de los síntomas, valorado a través de una escala semanal de la actividad de la urticaria (UAS7, intervalo 0-42) de >16, y una puntuación semanal de la gravedad del prurito (que es un componente de la UAS7; intervalo 0-21) de >8 para los 7 días anteriores a la aleatorización, a pesar de haber usado un antihistamínico durante al menos 2 semanas antes.
En los ensayos 1 y 2, los pacientes presentaron una puntuación semanal media de la gravedad del prurito de entre 13,7 y 14,5 en el periodo basal y una puntuación UAS7 media de 29,5 y 31,7, respectivamente. Los pacientes en el ensayo 3 de seguridad presentaron una puntuación semanal media de la gravedad del prurito de 13,8 y una puntuación UAS7 media de 31,2 en el periodo basal.
En los tres ensayos, los pacientes notificaron que recibían una media de 4 a 6 medicaciones (incluyendo antihistamínicos H1) para los síntomas de la UCE antes de ser incluidos en el ensayo.
Los pacientes recibieron Xolair a las dosis de 75 mg, 150 mg o 300 mg o placebo por inyección subcutánea cada 4 semanas durante 24 y 12 semanas en los ensayos 1 y 2, respectivamente, y 300 mg o placebo por inyección subcutánea cada 4 semanas durante 24 semanas en el ensayo 3. Todos los ensayos tuvieron un periodo de seguimiento libre de tratamiento de 16 semanas.
La variable primaria fue el cambio del periodo basal a la semana 12 en la puntuación semanal de la gravedad del prurito. Omalizumab a la dosis de 300 mg redujo la puntuación semanal de la gravedad del prurito de 8,55 a 9,77 (p <0,0001) comparado con una reducción de 3,63 a 5,14 para placebo (ver Tabla 7). Además se observaron resultados estadísticamente significativos en las tasas de respondedores para UAS7<6 (a la semana 12), las cuales fueron superiores para los grupos de tratamiento de 300 mg, que fueron del 52-66% (p<0,0001) comparado con el 11-19% para los grupos placebo, y se alcanzó la respuesta completa (UAS7=0) en el 34-44% (p<0,0001) de los pacientes tratados con la dosis de 300 mg comparado con el 5-9% de los pacientes en los grupos placebo. Los pacientes en los grupos de tratamiento de 300 mg alcanzaron la proporción media más elevada de días libres de angioedema desde la semana 4 a la semana 12, (91,0-96,1%; p<0,001) comparado con los grupos placebo (88,1-89,2%). El cambio medio del periodo basal a la semana 12 en el DLQI global para los grupos de tratamiento de 300 mg fue superior (p<0,001) que para placebo mostrando una mejora que osciló entre 9,7-10,3 puntos comparado con 5,1-6,1 puntos para los correspondientes grupos placebo.
Tabla 7: Cambio del periodo basal a la semana 12 en la puntuación semanal de la gravedad del prurito, ensayos 1, 2 y 3 (población ITTm10)
|
Placebo |
Omalizumab 300 mg | |
|
Ensayo 1 | ||
|
N |
80 |
81 |
|
Media (DE) |
-3,63 (5,22) |
-9,40 (5,73) |
|
Diferencia en LS media vs. placebo1 |
- |
-5,80 |
|
IC de 95% para diferencia |
- |
-7,49,-4,10 |
|
Valor-P vs. placebo2 |
- |
<0,0001 |
|
Ensayo 2 | ||
|
N |
79 |
79 |
|
Media (DE) |
-5,14 (5,58) |
-9,77 (5,95) |
|
Diferencia en LS media vs. placebo1 |
- |
-4,81 |
|
IC de 95% para diferencia |
- |
-6,49,-3,13 |
|
Valor-P vs. placebo2 |
- |
<0,0001 |
|
Ensayo 3 | ||
|
N |
83 |
252 |
|
Media (DE) |
-4,01 (5,87) |
-8,55 (6,01) |
|
Diferencia en LS media vs. placebo1 |
- |
-4,52 |
|
IC de 95% para diferencia |
- |
-5,97, -3,08 |
|
Valor-P vs. placebo2 |
- |
<0,0001 |
La Figura 1 muestra la puntuación semanal media de la gravedad del prurito a lo largo del tiempo en el ensayo 1. Las puntuaciones semanales medias de la gravedad del prurito descendieron con un efecto máximo alrededor de la semana 12, el cual se mantuvo a lo largo del periodo de tratamiento de 24 semanas. Los resultados fueron similares en el ensayo 3.
En los tres ensayos la puntuación semanal media de la gravedad del prurito se incrementó gradualmente durante el periodo de seguimiento libre de tratamiento de 16 semanas, consistente con la reaparición de los síntomas. Los valores medios al final del periodo de seguimiento fueron similares a los del grupo placebo, pero inferiores a los valores basales medios respectivos.
Figura 2: Puntuación semanal media de la gravedad del prurito a lo largo del tiempo, ensayo 1 (población ITTm)

Omalizumab o placebo administrado
BOCF=observación basal llevada adelante; rTTm=población por intención de tratar modificada
Eficacia después de 24 semanas de tratamiento
La magnitud de los resultados de eficacia observados a la semana 24 de tratamiento fue comparable a la observada a la semana 12:
Para 300 mg, en los ensayos 1 y 3, el descenso medio en relación al periodo basal en la puntuación semanal de la gravedad del prurito fue de 9,8 y 8,6, la proporción de pacientes con UAS7<6 fue del 61,7% y 55,6%, y la proporción de pacientes con respuesta completa (UAS7=0) fue del 48,1% y 42,5%, respectivamente, (todos p<0,0001, cuando se comparó con placebo).
La experiencia clínica en el retratamiento de pacientes con omalizumab es limitada.
Los datos de los ensayos clínicos en adolescentes (12 a 17 años) incluyeron un total de 39 pacientes, de los cuales 11 recibieron la dosis de 300 mg. Se dispone de resultados para la dosis de 300 mg de 9 pacientes a la semana 12 y de 6 pacientes a la semana 24, y mostró una magnitud de respuesta similar al tratamiento con omalizumab comparado a la población adulta. El cambio medio con respecto al valor basal en relación a la puntuación semanal de la gravedad del prurito mostró una reducción de 8,25 a la semana 12 y de 8,95 a la semana 24. Las tasas de respondedores fueron: 33% a la semana 12 y 67% a la semana 24 para UAS7=0, y del 56% a la semana 12 y del 67% a la semana 24 para UAS7<6.
5.2 Propiedades farmacocinéticas
Se ha estudiado la farmacocinética de omalizumab en pacientes adultos y adolescentes con asma alérgica, así como en pacientes adultos y adolescentes con UCE. Las características farmacocinéticas generales de omalizumab son similares en estas poblaciones.
Absorción
Tras la administración subcutánea, omalizumab se absorbe con una biodisponibilidad absoluta media del 62%. Después de administrar una dosis subcutánea única a pacientes adultos y adolescentes con asma o UCE, omalizumab se absorbió lentamente alcanzando concentraciones plasmáticas máximas después de una media de 6-8 días. En pacientes con asma, tras dosis múltiples de omalizumab, las áreas bajo la curva de concentración plasmática-tiempo del Día 0 al Día 14 en estado estacionario fueron de hasta 6-veces las obtenidas tras la primera dosis.
La farmacocinética de omalizumab es lineal a dosis mayores de 0,5 mg/kg. Tras dosis de 75 mg,
150 mg o 300 mg cada 4 semanas en pacientes con UCE, las concentraciones séricas mínimas de omalizumab incrementaron proporcionalmente con el nivel de dosis.
La administración de Xolair fabricado como una formulación liofilizada o líquida dió como resultado perfiles similares de concentración sérica-tiempo de omalizumab.
Distribución
In vitro, omalizumab forma complejos de tamaño limitado con la IgE. No se han observado tanto in vitro como in vivo complejos precipitantes ni complejos con pesos moleculares superiores a un millón de Daltons. En base a la farmacocinética poblacional, la distribución de omalizumab fue similar en pacientes con asma alérgica y en pacientes con UCE. Tras la administración subcutánea el volumen de distribución aparente en los pacientes con asma fue de 78 ± 32 ml/kg.
Eliminación
El aclaramiento de omalizumab comprende procesos de aclaramiento de las IgG, así como, aclaramiento a través de uniones específicas y formación de complejos con su ligando específico, la IgE. La eliminación hepática de IgG incluye una degradación en el sistema reticuloendotelial y células endoteliales. La IgG inalterada también se excreta en la bilis. La vida media de eliminación plasmática de omalizumab en pacientes asmáticos fue de un promedio de 26 días, con un aclaramiento aparente promedio de 2,4 ± 1,1 ml/kg/día. La duplicación del peso corporal hace que el aclaramiento aparente sea de aproximadamente el doble. En pacientes con UCE, en base a las simulaciones de la farmacocinética poblacional, la semivida de eliminación sérica de omalizumab en estado estacionario fue de una media de 24 días y el aclaramiento aparente en estado estacionario para un paciente de 80 kg de peso fue de 3,0 ml/kg/día.
Características demográficas
Pacientes con asma: Se analizó la farmacocinética poblacional de omalizumab con el fin de evaluar los efectos de las características demográficas. El análisis de los escasos datos disponibles indica que no es necesario efectuar un ajuste de la dosis en pacientes con asma en función de la edad (6-76 años), raza/grupo étnico, sexo o índice de masa corporal (ver sección 4.2).
Pacientes con UCE: En base a la farmacocinética poblacional se evaluaron los efectos de las características demográficas y otros factores sobre la exposición de omalizumab. Además, se evaluaron los efectos de covarianza analizando la relación entre las concentraciones de omalizumab y la respuesta clínica. Estos análisis indicaron que no son necesarios ajustes de dosis en pacientes con UCE en función de la edad (12-75 años), raza/grupo étnico, sexo, peso corporal, índice de masa corporal, IgE basal, anticuerpos anti-FcsRI o uso concomitante de antihistamínicos H2 o ARLTs.
Insuficiencia renal y hepática
No se dispone de datos farmacocinéticos o farmacodinámicos en pacientes con asma alérgica o UCE, con insuficiencia renal o hepática (ver secciones 4.2 y 4.4).
5.3 Datos preclínicos sobre seguridad
Se ha estudiado la seguridad de omalizumab en monos cinomolgos, ya que el omalizumab se une a las IgE del cinomolgos y humanas con una afinidad similar. Se detectaron anticuerpos a omalizumab en algunos monos tras la administración subcutánea o intravenosa repetida. Sin embargo, no se observó toxicidad aparente, tales como enfermedad mediada por inmunocomplejos o citotoxicidad dependiente de complemento. No hubo evidencia de una respuesta anafiláctica debido a la desgranulación de los mastocitos en monos cinomolgos.
La administración crónica de omalizumab a niveles de dosis de hasta 250 mg/kg (al menos 14 veces la dosis clínica más alta recomendada en mg/kg de acuerdo con la tabla de dosis recomendada) fue bien tolerada en primates no humanos (tanto en animales adultos como en jóvenes), con la excepción de un descenso en las plaquetas sanguíneas dependiente de la dosis y de la edad, con una mayor sensibilidad en animales jóvenes. La concentración plasmática necesaria para alcanzar un descenso del 50% en las plaquetas con respecto al valor basal en monos adultos cinomolgos fue aproximadamente de 4 a 20 veces más elevada que la concentración plasmática clínica máxima anticipada. Además, se observó inflamación y hemorragia aguda en el lugar de la inyección en monos cinomolgos.
No se han realizado estudios de carcinogenicidad formal con omalizumab.
En los estudios de reproducción en monos cinomolgos, dosis subcutáneas de hasta 75 mg/kg por semana (al menos 8 veces la dosis clínica más alta recomendada en mg/kg durante un periodo de 4-semanas) no provocaron toxicidad materna, embriotoxicidad o teratogenicidad cuando se administró durante toda la organogénesis y no provocó efectos adversos sobre el crecimiento fetal o neonatal cuando se administró durante la fase final de la gestación, parto y lactancia.
Omalizumab se excretó en la leche materna en monos cinomolgos. Los niveles de omalizumab en la leche fueron del 0,15% con respecto a la concentración sérica materna.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Hidrocloruro de L-arginina Hidrocloruro de L-histidina L-histidina Polisorbato 20
Agua para preparaciones inyectables
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros medicamentos.
6.3 Periodo de validez
15 meses.
El periodo de validez incluye posibles desviaciones de temperatura. El medicamento puede conservarse durante un total de 4 horas a 25°C. Si es necesario, el medicamento puede volver a guardarse en la nevera para utilizarlo más tarde, pero esto no debe hacerse más de una vez.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
6.5 Naturaleza y contenido del envase
1 ml de solución en un cilindro de jeringa precargada (vidrio tipo I) con aguja fijada (acero inoxidable), tapón del émbolo (tipo I) y capuchón de la aguja.
Envase conteniendo 1 jeringa precargada, y envases multiples que contienen 4 (4 envases de 1) o 10 (10 envases de 1) jeringas precargadas.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Antes de completar la inyección, evite el contacto con las pinzas de activación del dispositivo para evitar que la aguja quede prematuramente cubierta por el protector.
Uso de la jeringa
1. Manteniendo la jeringa con la aguja mirando hacia arriba, retire con cuidado de la jeringa, la cápsula protectora de la aguja y deséchela. No toque la aguja expuesta. Posteriormente, golpee suavemente la jeringa con su dedo hasta que las burbujas de aire asciendan a la superficie de la jeringa. Presione lentamente el émbolo para forzar la expulsión de las burbujas de aire fuera de la jeringa sin expulsar solución inadvertidamente.
2. Pellizque suavemente la piel en el lugar de la inyección e inserte la aguja.
3. Sosteniendo por la aleta de sujeción, presione lentamente el émbolo hasta donde sea posible. Si gotea solución por el lugar de la inyección, inserte más la aguja.
4. Manteniendo el émbolo completamente presionado, extraiga la aguja cuidadosamente y por completo del lugar de la inyección.
5. Suelte lentamente el émbolo y deje que el protector de la aguja cubra automáticamente la aguja expuesta.
Instrucciones de eliminación
Deseche inmediatamente la jeringa usada en un contenedor para material cortante.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/319/008
EU/1/05/319/009
EU/1/05/319/010
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 25 Octubre 2005 Fecha de la última renovación: 22 Junio 2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante del principio activo biológico
Novartis Pharma S.A.S.
Centre de Biotechnologie 8, rae de lTndustrie F-68330 Huningue Francia
Nombre y dirección del fabricante responsable de la liberación de los lotes
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nuremberg Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado
en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en
cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR EMBALAJE EXTERIOR
1. NOMBRE DEL MEDICAMENTO
Xolair 75 mg polvo y disolvente para solución inyectable omalizumab
2. PRINCIPIO(S) ACTIVO(S)
Un vial contiene 75 mg de omalizumab.
3. LISTA DE EXCIPIENTES
Polvo: sacarosa, L-histidina, hidrocloruro de L-histidina monohidrato y polisorbato 20. Disolvente: agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable Vial, 1 x 75 mg
Ampolla de disolvente, 1 x 2 ml
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Utilizar únicamente como le indique un médico.
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera (entre 2°C y 8°C).
Utilizar inmediatamente después de la reconstitución (puede conservarse durante 8 horas entre 2°C y 8°C o durante 2 horas a 25°C).
No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/319/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Xolair 75 mg
ETIQUETA VIAL_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Xolair 75 mg polvo para solución inyectable
omalizumab
Vía subcutánea
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
75 mg
6. OTROS
Conservar en nevera (entre 2°C y 8°C)
ETIQUETA AMPOLLA_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Disolvente para Xolair
Agua para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN
Utilizar 0,9 ml y desechar el resto.
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
2 ml
6. OTROS
CARTONAJE CONTENIENDO 1 VIAL Y 1 AMPOLLA COMO ENVASE UNITARIO (INCLUYENDO BLUE BOX)_
1. NOMBRE DEL MEDICAMENTO
Xolair 150 mg polvo y disolvente para solución inyectable omalizumab
2. PRINCIPIO(S) ACTIVO(S)
Un vial contiene 150 mg de omalizumab.
3. LISTA DE EXCIPIENTES
Polvo: sacarosa, L-histidina, hidrocloruro de L-histidina monohidrato y polisorbato 20. Disolvente: agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
Vial, 1 x 150 mg
Ampolla de disolvente, 1 x 2 ml
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Utilizar únicamente como le indique un médico.
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera (entre 2°C y 8°C).
Utilizar inmediatamente después de la reconstitución (puede conservarse durante 8 horas entre 2°C y 8°C o durante 2 horas a 25°C).
No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/319/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Xolair 150 mg
CARTONAJE PARA EL ENVASE INTERMEDIO (SIN BLUE BOX) DE LOS ENVASES MÚLTIPLES
1. NOMBRE DEL MEDICAMENTO
Xolair 150 mg polvo y disolvente para solución inyectable omalizumab
2. PRINCIPIO(S) ACTIVO(S)
Un vial contiene 150 mg de omalizumab.
3. LISTA DE EXCIPIENTES
Polvo: sacarosa, L-histidina, hidrocloruro de L-histidina monohidrato y polisorbato 20. Disolvente: agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
Vial, 1 x 150 mg
Ampolla de disolvente, 1 x 2 ml
1 vial y 1 ampolla. Subunidad de un envase múltiple. No puede venderse por separado.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Utilizar únicamente como le indique un médico.
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera (entre 2°C y 8°C).
Utilizar inmediatamente después de la reconstitución (puede conservarse durante 8 horas entre 2°C y 8°C o durante 2 horas a 25°C).
No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/319/003 Envase múltiple conteniendo 4 envases
EU/1/05/319/004 Envase múltiple conteniendo 10 envases
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ETIQUETA DE LA ENVOLTURA DEL ENVASE MÚLTIPLE ENVUELTOS EN UNA LÁMINA (INCLUYENDO BLUE BOX)_
1. NOMBRE DEL MEDICAMENTO
Xolair 150 mg polvo y disolvente para solución inyectable omalizumab
2. PRINCIPIO(S) ACTIVO(S)
Un vial contiene 150 mg de omalizumab.
3. LISTA DE EXCIPIENTES
Polvo: sacarosa, L-histidina, hidrocloruro de L-histidina monohidrato y polisorbato 20. Disolvente: agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
Envase múltiple: 4 envases, cada uno conteniendo 1 vial (150 mg) y 1 ampolla de disolvente (2 ml). Envase múltiple: 10 envases, cada uno conteniendo 1 vial (150 mg) y 1 ampolla de disolvente (2 ml).
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Utilizar únicamente como le indique un médico.
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera (entre 2°C y 8°C).
Utilizar inmediatamente después de la reconstitución (puede conservarse durante 8 horas entre 2°C y 8°C o durante 2 horas a 25°C).
No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/319/003 Envase múltiple conteniendo 4 envases
EU/1/05/319/004 Envase múltiple conteniendo 10 envases
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ETIQUETA VIAL_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Xolair 150 mg polvo para solución inyectable
omalizumab
Vía subcutánea
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
150 mg
6. OTROS
Conservar en nevera (entre 2°C y 8°C)
ETIQUETA AMPOLLA_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Disolvente para Xolair
Agua para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN
Utilizar 1,4 ml y desechar el resto.
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
2 ml
6. OTROS
1. NOMBRE DEL MEDICAMENTO
Xolair 75 mg solución inyectable omalizumab
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa precargada de 0,5 ml de solución contiene 75 mg de omalizumab.
3. LISTA DE EXCIPIENTES
También contiene: hidrocloruro de L-arginina, hidrocloruro de L-histidina, L-histidina, polisorbato 20 y agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable 1 x 0,5 ml
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Utilizar únicamente como le indique un médico.
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Deseche inmediatamente la jeringa usada en un contenedor para material cortante.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/319/005 75 mg solución inyectable
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
1. NOMBRE DEL MEDICAMENTO
Xolair 75 mg solución inyectable omalizumab
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa precargada de 0,5 ml de solución contiene 75 mg de omalizumab.
3. LISTA DE EXCIPIENTES
También contiene: hidrocloruro de L-arginina, hidrocloruro de L-histidina, L-histidina, polisorbato 20 y agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable
Envase múltiple: 4 envases, cada uno conteniendo 1 x 0,5 ml. Envase múltiple: 10 envases, cada uno conteniendo 1 x 0,5 ml.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Utilizar únicamente como le indique un médico.
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Deseche inmediatamente la jeringa usada en un contenedor para material cortante.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/319/006 75 mg solución inyectable (4)
EU/1/05/319/007 75 mg solución inyectable (10)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
1. NOMBRE DEL MEDICAMENTO
Xolair 75 mg solución inyectable omalizumab
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa precargada de 0,5 ml de solución contiene 75 mg de omalizumab.
3. LISTA DE EXCIPIENTES
También contiene: hidrocloruro de L-arginina, hidrocloruro de L-histidina, L-histidina, polisorbato 20 y agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable
1 x 0,5 ml. Subunidad de un envase múltiple. No puede venderse por separado.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Utilizar únicamente como le indique un médico.
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Deseche inmediatamente la jeringa usada en un contenedor para material cortante.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/319/006 75 mg solución inyectable (4)
EU/1/05/319/007 75 mg solución inyectable (10)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLISTER DE LA JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO
Xolair 75 mg solución inyectable
omalizumab
Vía subcutánea.
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETA DE LA JERINGA PRECARGADA_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Xolair 75 mg solución inyectable
omalizumab
Vía subcutánea
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
0,5 ml
6. OTROS
1. NOMBRE DEL MEDICAMENTO
Xolair 150 mg solución inyectable omalizumab
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa precargada de 1 ml de solución contiene 150 mg de omalizumab.
3. LISTA DE EXCIPIENTES
También contiene: hidrocloruro de L-arginina, hidrocloruro de L-histidina, L-histidina, polisorbato 20 y agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable 1 x 1 ml
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Utilizar únicamente como le indique un médico.
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Deseche inmediatamente la jeringa usada en un contenedor para material cortante.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/05/319/008 150 mg solución inyectable
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
1. NOMBRE DEL MEDICAMENTO
Xolair 150 mg solución inyectable omalizumab
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa precargada de 1 ml de solución contiene 150 mg de omalizumab.
3. LISTA DE EXCIPIENTES
También contiene: hidrocloruro de L-arginina, hidrocloruro de L-histidina, L-histidina, polisorbato 20 y agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable
Envase múltiple: 4 envases, cada uno conteniendo 1 x 1 ml. Envase múltiple: 10 envases, cada uno conteniendo 1 x 1 ml.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Utilizar únicamente como le indique un médico.
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Deseche inmediatamente la jeringa usada en un contenedor para material cortante.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
|
EU/1/05/319/009 |
150 mg solución inyectable (4) |
|
EU/1/05/319/010 |
150 mg solución inyectable (10) |
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
1. NOMBRE DEL MEDICAMENTO
Xolair 150 mg solución inyectable omalizumab
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa precargada de 1 ml de solución contiene 150 mg de omalizumab.
3. LISTA DE EXCIPIENTES
También contiene: hidrocloruro de L-arginina, hidrocloruro de L-histidina, L-histidina, polisorbato 20 y agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable
1 x 1 ml. Subunidad de un envase múltiple. No puede venderse por separado.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Utilizar únicamente como le indique un médico.
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Deseche inmediatamente la jeringa usada en un contenedor para material cortante.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
|
EU/1/05/319/009 |
150 mg solución inyectable (4) |
|
EU/1/05/319/010 |
150 mg solución inyectable (10) |
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLISTER DE LA JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO
Xolair 150 mg solución inyectable
omalizumab
Vía subcutánea.
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETA DE LA JERINGA PRECARGADA_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Xolair 150 mg solución inyectable
omalizumab
Vía subcutánea
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
1 ml
6. OTROS
B. PROSPECTO
Prospecto: información para el usuario
Xolair 75 mg polvo y disolvente para solución inyectable
omalizumab
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Xolair y para qué se utiliza
2. Qué necesita saber antes de recibir Xolair
3. Cómo se administra Xolair
4. Posibles efectos adversos
5. Conservación de Xolair
6. Contenido del envase e información adicional
1. Qué es Xolair y para qué se utiliza
El principio activo de Xolair es omalizumab. Omalizumab es una proteina humana, similar a las proteinas naturales producidas por el organismo; pertenece a una clase de medicamentos denominados anticuerpos monoclonales. Se utiliza para prevenir que el asma empeore controlando los síntomas del asma alérgica grave en adultos y adolescentes (12 años de edad y mayores) y niños (a partir de 6 años hasta menores de 12 años de edad) que ya están recibiendo medicamentos para el asma, pero cuyos síntomas no se controlan adecuadamente con medicamentos tales como esteroides inhalados a dosis altas o beta agonistas inhalados.
Xolair actúa bloqueando una sustancia llamada inmunoglobulina E (IgE) que es producida por el organismo. La IgE juega un papel clave como causante del asma alérgica.
2. Qué necesita saber antes de recibir Xolair No debería recibir Xolair
- si es alérgico a omalizumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Si cree que puede ser alérgico a cualquiera de los componentes, informe a su médico ya que no debe recibir Xolair.
Advertencias y precauciones
Xolair contiene una proteína y las proteínas pueden causar reacciones alérgicas graves en algunas personas. Los signos de reacción alérgica incluyen sarpullido, dificultad para respirar, hinchazón o sensación de desmayo. Si usted padece una reacción alérgica después de la administración de Xolair, contacte a su médico tan pronto le sea posible.
En pacientes tratados con Xolair se ha observado un cierto tipo de reacciones alérgicas denominadas enfermedad del suero. Los síntomas de la enfermedad del suero pueden ser uno o más de los descritos a continuación: dolor en las articulaciones con o sin inflamación o rigidez, sarpullido, fiebre, nódulos linfáticos inflamados, dolor muscular. Si sufre cualquiera de estos síntomas, o en particular si sufre una combinación de los mismos, contacte con su médico inmediatamente.
Se ha observado síndrome de Churg-Strauss e Hipereosinofílico en pacientes tratados con Xolair. Los síntomas pueden incluir uno o más de los descritos a continuación: hinchazón, dolor o sarpullido alrededor de los vasos sanguíneos o linfáticos, nivel elevado de un tipo específico de glóbulos blancos (eosinofilia marcada), empeoramiento de los problemas respiratorios, congestión nasal, problemas cardíacos, dolor, adormecimiento, hormigueo en los brazos y piernas. Si sufre cualquiera de estos síntomas, o en particular si sufre una combinación de los mismos, contacte con su médico inmediatamente.
Consulte a su médico antes de recibir Xolair:
- Si tiene problemas de riñón o hígado.
- Si padece una alteración en la que su propio sistema inmune ataca partes de su propio organismo (enfermedad autoinmune).
- Si vive en una región donde las infecciones causadas por parásitos son communes o si viaja a una de estas regiones ya que Xolair puede disminuir su resistencia a dichas infecciones.
Xolair no trata los síntomas del asma agudo, como puede ser un ataque de asma repentino. Por lo tanto Xolair no debe utilizarse para tratar esta clase de síntomas.
Xolair no está destinado para prevenir o tratar otras afecciones de tipo alérgico, como son reacciones alérgicas repentinas, síndrome de hiperinmunoglobulina E (trastorno inmune hereditario), aspergilosis (enfermedad del pulmón causada por un hongo), alergia alimentaria, eczema o fiebre del heno ya que Xolair no se ha estudiado en estas afecciones.
Niños (menores de 6 años de edad)
Xolair no debe administrarse a niños menores de 6 años de edad. No se dispone de datos suficientes en este grupo de edad.
Uso de Xolair con otros medicamentos
Informe a su médico, farmacéutico o enfermero si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Esto es especialmente importante si está utilizando:
- medicamentos para tratar una infección causada por un parásito, ya que Xolair puede reducir el efecto de sus medicamentos,
- corticosteroides inhalados y otros medicamentos para el asma alérgica.
Embarazo y lactancia
No debe recibir Xolair cuando esté embarazada, a no ser que su médico lo considere necesario.
Si planea quedarse embarazada, informe a su médico antes de iniciar el tratamiento con Xolair. Su médico comentará con usted los beneficios y riesgos potenciales de la administración de este medicamento durante el embarazo.
Informe a su médico inmediatamente si queda embarazada mientras está siendo tratada con Xolair.
No debe recibir Xolair cuando esté en periodo de lactancia.
Conducción y uso de máquinas
No es probable que Xolair afecte a su capacidad para conducir y usar máquinas.
3. Cómo se administra Xolair
Las instrucciones sobre como utilizar Xolair se proporcionan en la sección “Información para el profesional sanitario”.
Su médico estimará la cantidad de Xolair que necesita y la frecuencia de administración del mismo. Ello depende de su peso corporal y de los resultados de un análisis de sangre realizado antes de iniciar el tratamiento para determinar la concentración de IgE en la misma.
Xolair se administra, por su médico o enfermero, como una inyección bajo la piel (de forma subcutánea).
Siga detenidamente todas las instrucciones que le haya dado su médico o enfermero.
Cuánto se le administrará
Le administrarán entre 1 y 4 inyecciones al mismo tiempo, cada dos o cuatro semanas.
Continúe tomando su medicación actual para el asma mientras dure el tratamiento con Xolair. No interrumpa ninguna medicación para el asma sin consultarlo con su médico.
Es posible que no perciba una mejoría inmediata del asma antes de iniciar el tratamiento con Xolair. Por lo general, deben transcurrir entre 12 y 16 semanas hasta que el medicamento surta todo su efecto.
Uso en niños y adolescentes
Xolair puede administrarse a niños y adolescentes a partir de 6 años que ya estén recibiendo medicación para el asma, pero cuyos síntomas asmáticos no están bien controlados por medicamentos como dosis elevadas de esteroides inhalados o beta-agonistas inhalados. Su médico le informará qué cantidad de Xolair necesita su hijo y con qué frecuencia se le debe administrar. Esto dependerá del peso del niño y de los resultados obtenidos de los análisis de sangre realizados antes de iniciar el tratamiento para determinar la cantidad de IgE en su sangre.
Si olvidó una dosis de Xolair
Contacte con su médico u hospital tan pronto como sea posible para volver a programar su visita.
Si interrumpe el tratamiento con Xolair
No interrumpa el tratamiento con Xolair a no ser que se lo indique su médico. La interrupción o finalización del tratamiento con Xolair puede causar una recidiva de los síntomas del asma.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no
todas las personas los sufran. Los efectos adversos causados por Xolair son, por lo general, de leves a
moderados pero ocasionalmente pueden ser graves.
Los efectos adversos graves incluyen:
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 pacientes)
• Reacciones alérgicas graves repentinas: si nota signos de alergia graves repentinos o combinación de los mismos, como son, ronchas, picor o erupción cutánea, hinchazón de la cara, labios, lengua, larínge (caja de voz), tráquea u otras partes del cuerpo, ritmo cardíaco rápido, mareo y ligera sensación de vahído, disnea, respiración jadeante o dificultad respiratoria, o cualquier otro síntoma imprevisto, informe inmediatamente a su médico o enfermera. Si tiene antecedentes de reacciones alérgicas graves (anafilaxia) no relacionados con Xolair, puede sufrir mayor riesgo de desarrollar una reacción alérgica grave después del uso de Xolair.
• Lupus eritematoso sistémico (LES). Los síntomas pueden incluir dolor muscular, dolor e hinchazón de las articulaciones y erupción. Usted también puede experimentar otros síntomas como fiebre, pérdida de peso y fatiga.
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
• Desarrollo de uno o más de los síntomas siguientes: hinchazón, dolor o sarpullido alrededor de los vasos sanguíneos o linfáticos, nivel elevado de un tipo específico de glóbulos blancos (eosinofilia marcada), empeoramiento de los problemas respiratorios, congestión nasal, problemas cardíacos, dolor, adormecimiento, hormigueo en los brazos y piernas (signos del también llamado “síndrome de Churg-Strauss o síndrome hipereosinofílico”).
• Recuento de plaquetas sanguíneas bajo con síntomas como sangrado o hematomas que se producen más fácilmente de lo normal.
• Desarrollo de cualquiera de los síntomas siguientes, especialmente si están en combinación: dolor en las articulaciones con o sin inflamación o rigidez, erupción cutánea, fiebre, inflamación de los nódulos linfáticos, dolor muscular (signos de la enfermedad del suero).
Informe inmediatamente a su médico o enfermera si experimenta alguno de los efectos mencionados
anteriormente.
Otros efectos adversos incluyen:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes)
• fiebre (en niños)
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 pacientes)
• reacciones en el lugar de la inyección las cuales incluyen dolor, hinchazón, picor y enrojecimiento
• dolor en la parte superior del estómago (en niños)
• dolor de cabeza (muy frecuente en niños)
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes)
• sensación de vértigo, sueño o cansancio
• hormigueo o entumecimiento de manos o pies
• desmayo, disminución de la tensión arterial al sentarse o ponerse de pie (hipotensión postural), rubefacción
• dolor de garganta, tos, problemas respiratorios agudos
• sensación de mareo (nausea), diarrea, indigestión
• picor, urticaria, sarpullido, mayor sensibilidad de la piel al sol
• aumento de peso
• síntomas de tipo gripal
• brazos hinchados
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 pacientes)
• infección parasitaria
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
• dolor en las articulaciones, dolor muscular y tumefacción de las articulaciones
• pérdida de cabello
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Xolair
- Mantener este medicamento fuera de la vista y del alcance de los niños.
- No utilice este medicamento después de la fecha de caducidad que aparece en el envase. La fecha de caducidad es el último día del mes que se indica.
- Conservar en nevera (entre 2°C y 8°C). No congelar.
6. Contenido del envase e información adicional Composición de Xolair
- El principio activo es omalizumab. Un vial contiene 75 mg de omalizumab. Tras la reconstitución un vial contiene 125 mg/ml de omalizumab (75 mg en 0,6 ml).
- Los demás componentes son sacarosa, L-histidina, hidrocloruro de L-histidina monohidrato y polisorbato 20.
Aspecto del producto y contenido del envase
Xolair 75 mg polvo y disolvente para solución inyectable se presenta en forma de polvo de color blanco a blanquecino en un vial de vidrio pequeño junto con una ampolla que contiene 2 ml de agua para preparaciones inyectables. El polvo se reconstituye en el agua antes de ser inyectado por el médico o enfermero.
Xolair está disponible en envases que contienen un vial de polvo para solución inyectable y una ampolla de 2 ml de agua para preparaciones inyectables.
Xolair también está disponible en viales con 150 mg de omalizumab.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Etarapun Novartis Pharma Services Inc. Tea.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Ceská republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXXáda Novartis (Hellas) A.E.B.E. Tni: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
118 |
Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Suomi/Finland
Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
INFORMACIÓN PARA EL PROFESIONAL SANITARIO
Esta información está destinada únicamente a profesionales del sector sanitario:
El medicamento liofilizado necesita entre 15 y 20 minutos para disolverse, aunque en algunos casos puede requerir más tiempo. El medicamento completamente reconstituido tiene un aspecto transparente a ligeramente opalescente, incoloro a amarillo parduzco claro y puede presentar unas cuantas burbujas pequeñas o espuma alrededor del borde del vial. Debido a la viscosidad del medicamento reconstituido deberá tenerse precaución de extraer todo el medicamento del vial antes de eliminar el aire o el exceso de solución de la jeringa con el fin de obtener los 0,6 ml.
Para preparar los viales de Xolair 75 mg para la administración subcutánea, siga por favor las siguientes instrucciones:
1. Retirar 0,9 ml de agua para inyectables de la ampolla con una jeringa equipada con una aguja gruesa de extracción (calibre 18).
2. Con el vial colocado en posición vertical sobre una superficie plana, insertar la aguja e inocular el agua para inyectables en el vial conteniendo el polvo liofilizado utilizando las técnicas asépticas estándar, dirigiendo el agua para inyectables directamente sobre el polvo.
3. Manteniendo el vial en posición vertical, removerlo vigorosamente (sin agitar) durante 1 minuto aproximadamente para humedecer el polvo uniformemente.
4. Para ayudar a la disolución tras completar el paso 3, remover suavemente el vial durante 5-10 segundos aproximadamente cada 5 minutos con el fin de disolver el polvo restante.
Observe que en ocasiones puede necesitar más de 20 minutos para disolver el polvo completamente. Si este es el caso, repita el paso 4 hasta que desaparezcan las partículas gelatinosas de la solución.
Una vez el medicamento se haya disuelto completamente, no deben quedar partículas gelatinosas visibles en la solución. Las pequeñas burbujas o espuma alrededor del borde del vial son completamente normales. El medicamento reconstituido tiene un aspecto transparente a ligeramente opalescente, incoloro a amarillo parduzco claro. No utilice el producto si observa partículas sólidas.
5. Invertir el vial durante un mínimo de 15 segundos con el fin de que la solución fluya hacia el tapón. Utilizando una jeringa nueva de 3-ml equipada con una aguja gruesa de extracción (calibre 18), insertar la aguja en el vial invertido. Manteniendo el vial en posición invertida colocar el extremo de la aguja al final de la solución en el vial cuando extraiga la solución con la jeringa. Antes de extraer la aguja del vial, tire del émbolo y llévelo hasta el fondo del cilindro de la jeringa con el fin de extraer toda la solución del vial invertido.
6. Reemplazar la aguja de calibre 18 por una de calibre 25 para inyección subcutánea.
7. Eliminar el aire, las burbujas grandes y cualquier exceso de solución con el fin de obtener la dosis requerida de 0,6 ml. Puede quedar una fina capa de pequeñas burbujas en la superficie de la solución contenida en la jeringa. Como la solución es ligeramente viscosa, la administración de la solución por inyección subcutánea puede durar entre 5 y 10 segundos.
El vial proporciona 0,6 ml (75 mg) de Xolair.
8. Las inyecciones se administran por vía subcutánea en la región deltoidea del brazo o en el muslo.
Prospecto: información para el usuario
Xolair 150 mg polvo y disolvente para solución inyectable
omalizumab
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Xolair y para qué se utiliza
2. Qué necesita saber antes de recibir Xolair
3. Cómo se administra Xolair
4. Posibles efectos adversos
5. Conservación de Xolair
6. Contenido del envase e información adicional
1. Qué es Xolair y para qué se utiliza
Xolair se utiliza para el tratamiento del asma alérgica y de la urticaria crónica espontánea (UCE). El principio activo de Xolair es omalizumab. Omalizumab es una proteina humana, similar a las proteinas naturales producidas por el organismo; pertenece a una clase de medicamentos denominados anticuerpos monoclonales. Xolair actúa bloqueando una sustancia llamada inmunoglobulina E (IgE), que es producida por el organismo. La IgE juega un papel clave como causante del asma alérgica o UCE.
Asma alérgica
Este medicamento se utiliza para prevenir que el asma empeore controlando los síntomas del asma alérgica grave en adultos y adolescentes (12 años de edad y mayores) y niños (a partir de 6 años hasta menores de 12 años de edad) que ya están recibiendo medicamentos para el asma, pero cuyos síntomas no se controlan adecuadamente con medicamentos tales como esteroides inhalados a dosis altas o beta agonistas inhalados.
Urticaria crónica espontánea (UCE)
Este medicamento se utiliza para el tratamiento de la urticaria crónica espontánea en adultos y adolescentes (a partir de 12 años de edad) que ya están recibiendo antihistamínicos pero cuyos síntomas de la UCE no están bien controlados por estos medicamentos.
2. Qué necesita saber antes de recibir Xolair
No debería recibir Xolair
- si es alérgico a omalizumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Si cree que puede ser alérgico a cualquiera de los componentes, informe a su médico ya que no debe recibir Xolair.
Advertencias y precauciones
Xolair contiene una proteína y las proteínas pueden causar reacciones alérgicas graves en algunas personas. Los signos de reacción alérgica incluyen sarpullido, dificultad para respirar, hinchazón o sensación de desmayo. Si usted padece una reacción alérgica después de la administración de Xolair, contacte a su médico tan pronto le sea posible.
En pacientes tratados con Xolair se ha observado un cierto tipo de reacciones alérgicas denominadas enfermedad del suero. Los síntomas de la enfermedad del suero pueden ser uno o más de los descritos a continuación: dolor en las articulaciones con o sin inflamación o rigidez, sarpullido, fiebre, nódulos linfáticos inflamados, dolor muscular. Si sufre cualquiera de estos síntomas, o en particular si sufre una combinación de los mismos, contacte con su médico inmediatamente.
Se ha observado síndrome de Churg-Strauss e Hipereosinofílico en pacientes con asma alérgica tratados con Xolair. Los síntomas pueden incluir uno o más de los descritos a continuación: hinchazón, dolor o sarpullido alrededor de los vasos sanguíneos o linfáticos, nivel elevado de un tipo específico de glóbulos blancos (eosinofilia marcada), empeoramiento de los problemas respiratorios, congestión nasal, problemas cardíacos, dolor, adormecimiento, hormigueo en los brazos y piernas. Si sufre cualquiera de estos síntomas, o en particular si sufre una combinación de los mismos, contacte con su médico inmediatamente.
Consulte a su médico antes de recibir Xolair:
- Si tiene problemas de riñón o hígado.
- Si padece una alteración en la que su propio sistema inmune ataca partes de su propio organismo (enfermedad autoinmune).
- Si vive en una región donde las infecciones causadas por parásitos son communes o si viaja a una de estas regiones ya que Xolair puede disminuir su resistencia a dichas infecciones.
Xolair no trata los síntomas del asma agudo, como puede ser un ataque de asma repentino. Por lo tanto Xolair no debe utilizarse para tratar esta clase de síntomas.
Xolair no está destinado para prevenir o tratar otras afecciones de tipo alérgico, como son reacciones alérgicas repentinas, síndrome de hiperinmunoglobulina E (trastorno inmune hereditario), aspergilosis (enfermedad del pulmón causada por un hongo), alergia alimentaria, eczema o fiebre del heno ya que Xolair no se ha estudiado en estas afecciones.
Niños y adolescentes
Asma alérgica
Xolair no está recomendado para niños menores de 6 años de edad.
Urticaria crónica espontánea (UCE)
No administre Xolair a niños menores de 12 años de edad. No se ha estudiado su uso en niños menores de 12 años.
Uso de Xolair con otros medicamentos
Informe a su médico, farmacéutico o enfermero si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Esto es especialmente importante si está utilizando:
- medicamentos para tratar una infección causada por un parásito, ya que Xolair puede reducir el efecto de sus medicamentos,
- corticosteroides inhalados y otros medicamentos para el asma alérgica.
Embarazo y lactancia
No debe recibir Xolair cuando esté embarazada, a no ser que su médico lo considere necesario.
Si planea quedarse embarazada, informe a su médico antes de iniciar el tratamiento con Xolair. Su médico comentará con usted los beneficios y riesgos potenciales de la administración de este medicamento durante el embarazo.
Informe a su médico inmediatamente si queda embarazada mientras está siendo tratada con Xolair. No debe recibir Xolair cuando esté en periodo de lactancia.
Conducción y uso de máquinas
No es probable que Xolair afecte a su capacidad para conducir y usar máquinas.
3. Cómo se administra Xolair
Las instrucciones sobre como utilizar Xolair se proporcionan en la sección “Información para el profesional sanitario”.
Xolair se administra, por su médico o enfermero, como una inyección bajo la piel (de forma subcutánea).
Siga detenidamente todas las instrucciones que le haya dado su médico o enfermero.
Cuánto se le administrará
Asma alérgica
Su médico estimará la cantidad de Xolair que necesita y la frecuencia de administración del mismo. Ello depende de su peso corporal y de los resultados de un análisis de sangre realizado antes de iniciar el tratamiento para determinar la concentración de IgE en la misma.
Le administrarán entre 1 y 4 inyecciones al mismo tiempo, cada dos o cuatro semanas.
Continúe tomando su medicación actual para el asma mientras dure el tratamiento con Xolair. No interrumpa ninguna medicación para el asma sin consultarlo con su médico.
Es posible que no perciba una mejoría inmediata del asma antes de iniciar el tratamiento con Xolair. Por lo general, deben transcurrir entre 12 y 16 semanas hasta que el medicamento surta todo su efecto.
Urticaria crónica espontánea (UCE)
Le administrarán dos inyecciones de 150 mg al mismo tiempo cada cuatro semanas.
Continúe tomando su medicación actual para la UCE durante el tratamiento con Xolair. No interrumpa ningún medicamento sin consultarlo primero con su médico.
Uso en niños y adolescentes
Asma alérgica
Xolair puede administrarse a niños y adolescentes a partir de 6 años que ya estén recibiendo medicación para el asma, pero cuyos síntomas asmáticos no están bien controlados por medicamentos como dosis elevadas de esteroides inhalados o beta-agonistas inhalados. Su médico le informará qué cantidad de Xolair necesita su hijo y con qué frecuencia se le debe administrar. Esto dependerá del peso del niño y de los resultados obtenidos de los análisis de sangre realizados antes de iniciar el tratamiento para determinar la cantidad de IgE en su sangre.
Urticaria crónica espontánea (UCE)
Xolair puede administrarse a adolescentes a partir de 12 años que ya estén recibiendo antihistamínicos pero cuyos síntomas de la UCE no estén bien controlados por estos medicamentos.
Si olvidó una dosis de Xolair
Contacte con su médico u hospital tan pronto como sea posible para volver a programar su visita.
Si interrumpe el tratamiento con Xolair
No interrumpa el tratamiento con Xolair a no ser que se lo indique su médico. La interrupción o finalización del tratamiento con Xolair puede causar una recidiva de los síntomas del asma o UCE.
Sin embargo, si está siendo tratado para la UCE, su médico puede interrumpir el tratamiento con Xolair de vez en cuando para valorar sus síntomas. Siga las instrucciones de su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no
todas las personas los sufran. Los efectos adversos causados por Xolair son, por lo general, de leves a
moderados pero ocasionalmente pueden ser graves.
Los efectos adversos graves incluyen:
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 pacientes)
• Reacciones alérgicas graves repentinas: si nota signos de alergia graves repentinos o combinación de los mismos, como son, ronchas, picor o erupción cutánea, hinchazón de la cara, labios, lengua, larínge (caja de voz), tráquea u otras partes del cuerpo, ritmo cardíaco rápido, mareo y ligera sensación de vahído, disnea, respiración jadeante o dificultad respiratoria, o cualquier otro síntoma imprevisto, informe inmediatamente a su médico o enfermera. Si tiene antecedentes de reacciones alérgicas graves (anafilaxia) no relacionados con Xolair, puede sufrir mayor riesgo de desarrollar una reacción alérgica grave después del uso de Xolair.
• Lupus eritematoso sistémico (LES). Los síntomas pueden incluir dolor muscular, dolor e hinchazón de las articulaciones y erupción. Usted también puede experimentar otros síntomas como fiebre, pérdida de peso y fatiga.
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
• Desarrollo de uno o más de los síntomas siguientes: hinchazón, dolor o sarpullido alrededor de los vasos sanguíneos o linfáticos, nivel elevado de un tipo específico de glóbulos blancos (eosinofilia marcada), empeoramiento de los problemas respiratorios, congestión nasal, problemas cardíacos, dolor, adormecimiento, hormigueo en los brazos y piernas (signos del también llamado “síndrome de Churg-Strauss o síndrome hipereosmofílico”).
• Recuento de plaquetas sanguíneas bajo con síntomas como sangrado o hematomas que se producen más fácilmente de lo normal.
• Desarrollo de cualquiera de los síntomas siguientes, especialmente si están en combinación: dolor en las articulaciones con o sin inflamación o rigidez, erupción cutánea, fiebre, inflamación de los nódulos linfáticos, dolor muscular (signos de la enfermedad del suero).
Informe inmediatamente a su médico o enfermera si experimenta alguno de los efectos mencionados
anteriormente.
Otros efectos adversos incluyen:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes)
• fiebre (en niños)
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 pacientes)
• reacciones en el lugar de la inyección las cuales incluyen dolor, hinchazón, picor y enrojecimiento
• dolor en la parte superior del estómago (en niños)
• dolor de cabeza (muy frecuente en niños)
• infección de las vías altas del tracto respiratorio, como es inflamación de la faringe y resfriado común
• sensación de presión o dolor en las mejillas y la frente (sinusitis, dolor de cabeza sinusal)
• dolor en las articulaciones (artralgia)
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes)
• sensación de vértigo, sueño o cansancio
• hormigueo o entumecimiento de manos o pies
• desmayo, disminución de la tensión arterial al sentarse o ponerse de pie (hipotensión postural), rubefacción
• dolor de garganta, tos, problemas respiratorios agudos
• sensación de mareo (nausea), diarrea, indigestión
• picor, urticaria, sarpullido, mayor sensibilidad de la piel al sol
• aumento de peso
• síntomas de tipo gripal
• brazos hinchados
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 pacientes)
• infección parasitaria
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
• dolor muscular y tumefacción de las articulaciones
• pérdida de cabello
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Xolair
- Mantener este medicamento fuera de la vista y del alcance de los niños.
- No utilice este medicamento después de la fecha de caducidad que aparece en el envase. La fecha de caducidad es el último día del mes que se indica.
- Conservar en nevera (entre 2°C y 8°C). No congelar.
Contenido del envase e información adicional
6.
Composición de Xolair
- El principio activo es omalizumab. Un vial contiene 150 mg de omalizumab. Tras la reconstitución un vial contiene 125 mg/ml de omalizumab (150 mg en 1,2 ml).
- Los demás componentes son sacarosa, L-histidina, hidrocloruro de L-histidina monohidrato y polisorbato 20.
Aspecto del producto y contenido del envase
Xolair 150 mg polvo y disolvente para solución inyectable se presenta en forma de polvo de color blanco a blanquecino en un vial de vidrio pequeño junto con una ampolla que contiene 2 ml de agua para preparaciones inyectables. El polvo se reconstituye en el agua antes de ser inyectado por el médico o enfermero.
Xolair 150 mg polvo y disolvente para solución inyectable está disponible en envases que contienen un viale de polvo para solución inyectable y una ampolla de 2 ml de agua para preparaciones inyectables y en envases múltiples que contienen cuatro o diez envases intermedios, cada uno con un vial de polvo para solución inyectable y una ampolla de 2 ml de agua para preparaciones inyectables. Puede que solamente estén comercializados algunos tamaños de envases.
Xolair también está disponible en viales con 75 mg de omalizumab.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Etarapna
Novartis Pharma Services Inc. Tea.: +359 2 489 98 28
Ceská republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Lietuva
Novartis Pharma Services Inc. Tel: +370 5 269 16 50
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXXáóa Novartis (Hellas) A.E.B.E. Tni: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kórcpog Novartis Pharma Services Inc. Tni: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 Fecha de la última revisión de este prospecto: |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
INFORMACIÓN PARA EL PROFESIONAL SANITARIO
Esta información está destinada únicamente a profesionales del sector sanitario:
El medicamento liofilizado necesita entre 15 y 20 minutos para disolverse, aunque en algunos casos puede requerir más tiempo. El medicamento completamente reconstituido tiene un aspecto transparente aligeramente opalescente, incoloro a amarillo parduzco claro y puede presentar unas cuantas burbujas pequeñas o espuma alrededor del borde del vial. Debido a la viscosidad del medicamento reconstituido deberá tenerse precaución de extraer todo el medicamento del vial antes de eliminar el aire o el exceso de solución de la jeringa con el fin de obtener los 1,2 ml.
Para preparar los viales de Xolair 150 mg para la administración subcutánea, siga por favor las siguientes instrucciones:
1. Retirar 1,4 ml de agua para inyectables de la ampolla con una jeringa equipada con una aguja gruesa de extracción (calibre 18).
2. Con el vial colocado en posición vertical sobre una superficie plana, insertar la aguja e inocular el agua para inyectables en el vial conteniendo el polvo liofilizado utilizando las técnicas asépticas estándar, dirigiendo el agua para inyectables directamente sobre el polvo.
3. Manteniendo el vial en posición vertical, removerlo vigorosamente (sin agitar) durante 1 minuto aproximadamente para humedecer el polvo uniformemente.
4. Para ayudar a la disolución tras completar el paso 3, remover suavemente el vial durante 5-10 segundos aproximadamente cada 5 minutos con el fin de disolver el polvo restante.
Observe que en ocasiones puede necesitar más de 20 minutos para disolver el polvo completamente. Si este es el caso, repita el paso 4 hasta que desaparezcan las partículas gelatinosas de la solución.
Una vez el medicamento se haya disuelto completamente, no deben quedar partículas gelatinosas visibles en la solución. Las pequeñas burbujas o espuma alrededor del borde del vial son completamente normales. El medicamento reconstituido tiene un aspecto transparente a ligeramente opalescente, incoloro a amarillo parduzco claro. No utilice el producto si observa partículas sólidas.
5. Invertir el vial durante un mínimo de 15 segundos con el fin de que la solución fluya hacia el tapón. Utilizando una jeringa nueva de 3-ml equipada con una aguja gruesa de extracción (calibre 18), insertar la aguja en el vial invertido. Manteniendo el vial en posición invertida colocar el extremo de la aguja al final de la solución en el vial cuando extraiga la solución con la jeringa. Antes de extraer la aguja del vial, tire del émbolo y llévelo hasta el fondo del cilindro de la jeringa con el fin de extraer toda la solución del vial invertido.
6. Reemplazar la aguja de calibre 18 por una de calibre 25 para inyección subcutánea.
7. Eliminar el aire, las burbujas grandes y cualquier exceso de solución con el fin de obtener la dosis requerida de 1,2 ml. Puede quedar una fina capa de pequeñas burbujas en la superficie de la solución contenida en la jeringa. Como la solución es ligeramente viscosa, la administración de la solución por inyección subcutánea puede durar entre 5 y 10 segundos.
El vial proporciona 1,2 ml (150 mg) de Xolair. Para obtener una dosis de 75 mg retirar 0,6 ml con la jeringa y desechar la solución restante.
8. Las inyecciones se administran por vía subcutánea en la región deltoidea del brazo o en el muslo.
Prospecto: información para el usuario
Xolair 75 mg solución inyectable
omalizumab
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Xolair y para qué se utiliza
2. Qué necesita saber antes de recibir Xolair
3. Cómo se administra Xolair
4. Posibles efectos adversos
5. Conservación de Xolair
6. Contenido del envase e información adicional
1. Qué es Xolair y para qué se utiliza
El principio activo de Xolair es omalizumab. Omalizumab es una proteina humana, similar a las proteinas naturales producidas por el organismo; pertenece a una clase de medicamentos denominados anticuerpos monoclonales. Se utiliza para prevenir que el asma empeore controlando los síntomas del asma alérgica grave en adultos y adolescentes (12 años de edad y mayores) y niños (a partir de 6 años hasta menores de 12 años de edad) que ya están recibiendo medicamentos para el asma, pero cuyos síntomas no se controlan adecuadamente con medicamentos tales como esteroides inhalados a dosis altas o beta agonistas inhalados.
Xolair actúa bloqueando una sustancia llamada inmunoglobulina E (IgE) que es producida por el organismo. La IgE juega un papel clave como causante del asma alérgica.
2. Qué necesita saber antes de recibir Xolair No debería recibir Xolair
- si es alérgico a omalizumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Si cree que puede ser alérgico a cualquiera de los componentes, informe a su médico ya que no debe recibir Xolair.
Advertencias y precauciones
Xolair contiene una proteína y las proteínas pueden causar reacciones alérgicas graves en algunas personas. Los signos de reacción alérgica incluyen sarpullido, dificultad para respirar, hinchazón o sensación de desmayo. Si usted padece una reacción alérgica después de la administración de Xolair, contacte a su médico tan pronto le sea posible.
En pacientes tratados con Xolair se ha observado un cierto tipo de reacciones alérgicas denominadas enfermedad del suero. Los síntomas de la enfermedad del suero pueden ser uno o más de los descritos a continuación: dolor en las articulaciones con o sin inflamación o rigidez, sarpullido, fiebre, nódulos linfáticos inflamados, dolor muscular. Si sufre cualquiera de estos síntomas, o en particular si sufre una combinación de los mismos, contacte con su médico inmediatamente.
Tenga especial cuidado con Xolair si alguna vez ha presentado una reacción alérgica al látex (el capuchón de la aguja puede contener caucho seco (latex)).
Se ha observado síndrome de Churg-Strauss e Hipereosinofílico en pacientes tratados con Xolair. Los síntomas pueden incluir uno o más de los descritos a continuación: hinchazón, dolor o sarpullido alrededor de los vasos sanguíneos o linfáticos, nivel elevado de un tipo específico de glóbulos blancos (eosinofilia marcada), empeoramiento de los problemas respiratorios, congestión nasal, problemas cardíacos, dolor, adormecimiento, hormigueo en los brazos y piernas. Si sufre cualquiera de estos síntomas, o en particular si sufre una combinación de los mismos, contacte con su médico inmediatamente.
Consulte a su médico antes de recibir Xolair:
- Si tiene problemas de riñón o hígado.
- Si padece una alteración en la que su propio sistema inmune ataca partes de su propio organismo (enfermedad autoinmune).
- Si vive en una región donde las infecciones causadas por parásitos son communes o si viaja a una de estas regiones ya que Xolair puede disminuir su resistencia a dichas infecciones.
Xolair no trata los síntomas del asma agudo, como puede ser un ataque de asma repentino. Por lo tanto Xolair no debe utilizarse para tratar esta clase de síntomas.
Xolair no está destinado para prevenir o tratar otras afecciones de tipo alérgico, como son reacciones alérgicas repentinas, síndrome de hiperinmunoglobulina E (trastorno inmune hereditario), aspergilosis (enfermedad del pulmón causada por un hongo), alergia alimentaria, eczema o fiebre del heno ya que Xolair no se ha estudiado en estas afecciones.
Niños (menores de 6 años de edad)
Xolair no debe administrarse a niños menores de 6 años de edad. No se dispone de datos suficientes en este grupo de edad.
Uso de Xolair con otros medicamentos
Informe a su médico, farmacéutico o enfermero si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Esto es especialmente importante si está utilizando:
- medicamentos para tratar una infección causada por un parásito, ya que Xolair puede reducir el efecto de sus medicamentos,
- corticosteroides inhalados y otros medicamentos para el asma alérgica.
Embarazo y lactancia
No debe recibir Xolair cuando esté embarazada, a no ser que su médico lo considere necesario.
Si planea quedarse embarazada, informe a su médico antes de iniciar el tratamiento con Xolair. Su médico comentará con usted los beneficios y riesgos potenciales de la administración de este medicamento durante el embarazo.
Informe a su médico inmediatamente si queda embarazada mientras está siendo tratada con Xolair.
No debe recibir Xolair cuando esté en periodo de lactancia.
Conducción y uso de máquinas
No es probable que Xolair afecte a su capacidad para conducir y usar máquinas.
3. Cómo se administra Xolair
Las instrucciones sobre como utilizar Xolair se proporcionan en la sección “Información para el profesional sanitario”.
Su médico estimará la cantidad de Xolair que necesita y la frecuencia de administración del mismo. Ello depende de su peso corporal y de los resultados de un análisis de sangre realizado antes de iniciar el tratamiento para determinar la concentración de IgE en la misma.
Xolair se administra, por su médico o enfermero, como una inyección bajo la piel (de forma subcutánea).
Siga detenidamente todas las instrucciones que le haya dado su médico o enfermero.
Cuánto se le administrará
Le administrarán entre 1 y 4 inyecciones al mismo tiempo, cada dos o cuatro semanas.
Continúe tomando su medicación actual para el asma mientras dure el tratamiento con Xolair. No interrumpa ninguna medicación para el asma sin consultarlo con su médico.
Es posible que no perciba una mejoría inmediata del asma antes de iniciar el tratamiento con Xolair. Por lo general, deben transcurrir entre 12 y 16 semanas hasta que el medicamento surta todo su efecto.
Uso en niños y adolescentes
Xolair puede administrarse a niños y adolescentes a partir de 6 años que ya estén recibiendo medicación para el asma, pero cuyos síntomas asmáticos no están bien controlados por medicamentos como dosis elevadas de esteroides inhalados o beta-agonistas inhalados. Su médico le informará qué cantidad de Xolair necesita su hijo y con qué frecuencia se le debe administrar. Esto dependerá del peso del niño y de los resultados obtenidos de los análisis de sangre realizados antes de iniciar el tratamiento para determinar la cantidad de IgE en su sangre.
Si olvidó una dosis de Xolair
Contacte con su médico u hospital tan pronto como sea posible para volver a programar su visita.
Si interrumpe el tratamiento con Xolair
No interrumpa el tratamiento con Xolair a no ser que se lo indique su médico. La interrupción o finalización del tratamiento con Xolair puede causar una recidiva de los síntomas del asma.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no
todas las personas los sufran. Los efectos adversos causados por Xolair son, por lo general, de leves a
moderados pero ocasionalmente pueden ser graves.
Los efectos adversos graves incluyen:
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 pacientes)
• Reacciones alérgicas graves repentinas: si nota signos de alergia graves repentinos o combinación de los mismos, como son, ronchas, picor o erupción cutánea, hinchazón de la cara, labios, lengua, larínge (caja de voz), tráquea u otras partes del cuerpo, ritmo cardíaco rápido, mareo y ligera sensación de vahído, disnea, respiración jadeante o dificultad respiratoria, o cualquier otro síntoma imprevisto, informe inmediatamente a su médico o enfermera. Si tiene antecedentes de reacciones alérgicas graves (anafilaxia) no relacionados con Xolair, puede sufrir mayor riesgo de desarrollar una reacción alérgica grave después del uso de Xolair.
• Lupus eritematoso sistémico (LES). Los síntomas puede incluir dolor muscular, dolor e hinchazón de las articulaciones y picor. Usted también puede experimentar otros síntomas como fiebre, pérdida de peso y fatiga.
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
• Desarrollo de uno o más de los síntomas siguientes: hinchazón, dolor o sarpullido alrededor de los vasos sanguíneos o linfáticos, nivel elevado de un tipo específico de glóbulos blancos (eosinofilia marcada), empeoramiento de los problemas respiratorios, congestión nasal, problemas cardíacos, dolor, adormecimiento, hormigueo en los brazos y piernas (signos del también llamado “síndrome de Churg-Strauss o síndrome hipereosmofílico”).
• Recuento de plaquetas sanguíneas bajo con síntomas como sangrado o hematomas que se producen más fácilmente de lo normal.
• Desarrollo de cualquiera de los síntomas siguientes, especialmente si están en combinación: dolor en las articulaciones con o sin inflamación o rigidez, erupción cutánea, fiebre, inflamación de los nódulos linfáticos, dolor muscular (signos de la enfermedad del suero).
Informe inmediatamente a su médico o enfermera si experimenta alguno de los efectos mencionados
anteriormente.
Otros efectos adversos incluyen:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes)
• fiebre (en niños)
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 pacientes)
• reacciones en el lugar de la inyección las cuales incluyen dolor, hinchazón, picor y enrojecimiento
• dolor en la parte superior del estómago (en niños)
• dolor de cabeza (muy frecuente en niños)
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes)
• sensación de vértigo, sueño o cansancio
• hormigueo o entumecimiento de manos o pies
• desmayo, disminución de la tensión arterial al sentarse o ponerse de pie (hipotensión postural), rubefacción
• dolor de garganta, tos, problemas respiratorios agudos
• sensación de mareo (nausea), diarrea, indigestión
• picor, urticaria, sarpullido, mayor sensibilidad de la piel al sol
• aumento de peso
• síntomas de tipo gripal
• brazos hinchados
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 pacientes)
• infección parasitaria
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
• dolor en las articulaciones, dolor muscular y tumefacción de las articulaciones
• pérdida de cabello
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Xolair
- Mantener este medicamento fuera de la vista y del alcance de los niños.
- No utilice este medicamento después de la fecha de caducidad que aparece en el envase. La
fecha de caducidad es el último día del mes que se indica.
- Conservar en el embalaje original para protegerlo de la luz.
- Conservar en nevera (entre 2°C y 8°C). No congelar.
- No utilizar ningun envase que esté dañado o muestre indicios de deterioro.
6. Contenido del envase e información adicional Composición de Xolair
- El principio activo es omalizumab. Una jeringa de 0,5 ml de solución contiene 75 mg de omalizumab.
- Los demás componentes son hidrocloruro de L-arginina, hidrocloruro de L-histidina, L-histidina, polisorbato 20 y agua para preparaciones inyectables.
- La cápsula protectora de la aguja de la jeringa puede contener caucho seco (látex).
Aspecto del producto y contenido del envase
Xolair solución inyectable se presenta como una solución de transparente a ligeramente opalescente, incolora a color amarillo parduzco claro, en una jeringa precargada.
Xolair 75 mg solución inyectable está disponible en envases que contienen 1 jeringa precargada y en envases múltiples que contienen 4 ó 10 envases intermedios, cada uno de ellos conteniendo 1 jeringa precargada.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Etarapun Novartis Pharma Services Inc. Tea.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Ceská republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXXáda Novartis (Hellas) A.E.B.E. Tni: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Suomi/Finland
Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http: //www.ema.europa.eu
INFORMACIÓN PARA EL PROFESIONAL SANITARIO
Esta información está destinada únicamente a profesionales del sector sanitario:
Antes de utilizar la jeringa, lea detenidamente la siguiente información.
Cada envase de Xolair contiene una jeringa precargada sellada individualmente en una envoltura de plástico.
Partes de la jeringa precargada Cápsula Protector de
protectora de la aguja
la aguja

Ventana visora Etiqueta y fecha de caducidad
Aleta de sujeción
Pinzas de activación
Émbolo

Linea de llenado
Las jeringas de Xolair están destinadas a ser utilizadas únicamente por un profesional sanitario.
La cápsula protectora de la aguja de la jeringa puede contener caucho seco (látex), por lo que las personas sensibles a esta sustancia no deben manejarla.
Preparación de la jeringa para su uso
Antes de completar la inyección, evite el contacto con las pinzas de activación del dispositivo (ver
dibujo anterior) para evitar que la aguja quede prematuramente cubierta por el protector.
1. Saque la caja que contiene la jeringa de la nevera y déjela durante 20 minutos aproximadamente para que así alcance la temperatura ambiente (conserve la jeringa en la caja para protejerla de la luz).
2. Si es necesario, la jeringa puede volver a guardarse en la nevera para utilizarla más tarde, pero esto no debe hacerse más de una vez. El tiempo acumulativo durante el cual la jeringa se conserva a temperatura ambiente (25°C) no debe exceder de 4 horas.
3. Cuando esté listo para utilizar la jeringa, lávese las manos minuciosamente con agua y jabón.
4. Limpie el lugar de la inyección.
5. Extraiga la bandeja de plástico de la caja, quite la cubierta de papel y extraiga la jeringa.
6. Inspeccione la jeringa. NO LA UTILICE si está rota o si el líquido parece turbio o contiene partículas. En todos estos casos, devuelva el envase completo a la farmacia.
7. Manteniendo la jeringa horizontalmente (como se muestra a continuación), mire en la ventana visora para comprobar la dosis (75 mg) del medicamento y la fecha de caducidad impresa en la etiqueta. Nota: Gire la parte interior del ensamblaje de la jeringa tal y como se muestra a continuación de tal forma que la etiqueta se pueda leer en la ventana visora.

NO LA UTILICE si el producto ha caducado o si la dosis es incorrecta. En ambos casos, devuelva el envase completo a la farmacia.
8. Mantenga la jeringa verticalmente con el émbolo hacia arriba y golpee ligeramente el lado de la jeringa contra su dedo para permitir que asciendan las burbujas de aire.
9. Compruebe si el nivel del líquido está en o por encima de la línea de llenado mínima. Si el líquido está por debajo de la línea de llenado, devuelva el envase completo a la farmacia.

Uso de la jeringa

Manteniendo la jeringa con la aguja mirando hacia arriba, retire con cuidado de la jeringa, la cápsula protectora de la aguja y deséchela. No toque la aguja expuesta. Posteriormente, golpee suavemente la jeringa con su dedo hasta que las burbujas de aire asciendan a la superficie de la jeringa. Presione lentamente el émbolo para forzar la expulsión de las burbujas de aire fuera de la jeringa sin expulsar solución inadvertidamente.

Pellizque suavemente la piel en el lugar de la inyección e inserte la aguja.
|
Life |
Sosteniendo por la aleta de sujeción, presione lentamente el émbolo hasta donde sea posible. Si gotea solución por el lugar de la inyección, inserte más la aguja. | |
|
□ feSS^fe1'' //w |
Manteniendo el émbolo completamente presionado, extraiga la aguja cuidadosamente y por completo del lugar de la inyección. | |
|
0 ^ fe \ _\ —i_\ —- |
Suelte lentamente el émbolo y deje que el protector de la aguja cubra automáticamente la aguja expuesta. Sostenga una gasa en el lugar de la inyección durante 30 segundos aproximadamente. |
Instrucciones de eliminación
Deseche inmediatamente la jeringa usada en un contenedor para material cortante. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Prospecto: información para el usuario
Xolair 150 mg solución inyectable
omalizumab
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Xolair y para qué se utiliza
2. Qué necesita saber antes de recibir Xolair
3. Cómo se administra Xolair
4. Posibles efectos adversos
5. Conservación de Xolair
6. Contenido del envase e información adicional
1. Qué es Xolair y para qué se utiliza
Xolair se utiliza para el tratamiento del asma alérgica y de la urticaria crónica espontánea (UCE). El principio activo de Xolair es omalizumab. Omalizumab es una proteina humana, similar a las proteinas naturales producidas por el organismo; pertenece a una clase de medicamentos denominados anticuerpos monoclonales. Xolair actúa bloqueando una sustancia llamada inmunoglobulina E (IgE), que es producida por el organismo. La IgE juega un papel clave como causante del asma alérgica o UCE.
Asma alérgica
Este medicamento se utiliza para prevenir que el asma empeore controlando los síntomas del asma alérgica grave en adultos y adolescentes (12 años de edad y mayores) y niños (a partir de 6 años hasta menores de 12 años de edad) que ya están recibiendo medicamentos para el asma, pero cuyos síntomas no se controlan adecuadamente con medicamentos tales como esteroides inhalados a dosis altas o beta agonistas inhalados.
Urticaria crónica espontánea (UCE)
Este medicamento se utiliza para el tratamiento de la urticaria crónica espontánea en adultos y adolescentes (a partir de 12 años de edad) que ya están recibiendo antihistamínicos pero cuyos síntomas de la UCE no están bien controlados por estos medicamentos.
2. Qué necesita saber antes de recibir Xolair
No debería recibir Xolair
- si es alérgico a omalizumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Si cree que puede ser alérgico a cualquiera de los componentes, informe a su médico ya que no debe recibir Xolair.
Advertencias y precauciones
Xolair contiene una proteína y las proteínas pueden causar reacciones alérgicas graves en algunas personas. Los signos de reacción alérgica incluyen sarpullido, dificultad para respirar, hinchazón o sensación de desmayo. Si usted padece una reacción alérgica después de la administración de Xolair, contacte a su médico tan pronto le sea posible.
En pacientes tratados con Xolair se ha observado un cierto tipo de reacciones alérgicas denominadas enfermedad del suero. Los síntomas de la enfermedad del suero pueden ser uno o más de los descritos a continuación: dolor en las articulaciones con o sin inflamación o rigidez, sarpullido, fiebre, nódulos linfáticos inflamados, dolor muscular. Si sufre cualquiera de estos síntomas, o en particular si sufre una combinación de los mismos, contacte con su médico inmediatamente.
Tenga especial cuidado con Xolair si alguna vez ha presentado una reacción alérgica al látex (el capuchón de la aguja puede contener caucho seco (latex)).
Se ha observado síndrome de Churg-Strauss e Hipereosinofílico en pacientes con asma alérgica tratados con Xolair. Los síntomas pueden incluir uno o más de los descritos a continuación: hinchazón, dolor o sarpullido alrededor de los vasos sanguíneos o linfáticos, nivel elevado de un tipo específico de glóbulos blancos (eosinofilia marcada), empeoramiento de los problemas respiratorios, congestión nasal, problemas cardíacos, dolor, adormecimiento, hormigueo en los brazos y piernas. Si sufre cualquiera de estos síntomas, o en particular si sufre una combinación de los mismos, contacte con su médico inmediatamente.
Consulte a su médico antes de recibir Xolair:
- Si tiene problemas de riñón o hígado.
- Si padece una alteración en la que su propio sistema inmune ataca partes de su propio organismo (enfermedad autoinmune).
- Si vive en una región donde las infecciones causadas por parásitos son communes o si viaja a una de estas regiones ya que Xolair puede disminuir su resistencia a dichas infecciones.
Xolair no trata los síntomas del asma agudo, como puede ser un ataque de asma repentino. Por lo tanto Xolair no debe utilizarse para tratar esta clase de síntomas.
Xolair no está destinado para prevenir o tratar otras afecciones de tipo alérgico, como son reacciones alérgicas repentinas, síndrome de hiperinmunoglobulina E (trastorno inmune hereditario), aspergilosis (enfermedad del pulmón causada por un hongo), alergia alimentaria, eczema o fiebre del heno ya que Xolair no se ha estudiado en estas afecciones.
Niños y adolescentes
Asma alérgica
Xolair no está recomendado para niños menores de 6 años de edad.
Urticaria crónica espontánea (UCE)
No administre Xolair a niños menores de 12 años de edad. No se ha estudiado su uso en niños menores de 12 años.
Uso de Xolair con otros medicamentos
Informe a su médico, farmacéutico o enfermero si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Esto es especialmente importante si está utilizando:
- medicamentos para tratar una infección causada por un parásito, ya que Xolair puede reducir el efecto de sus medicamentos,
- corticosteroides inhalados y otros medicamentos para el asma alérgica.
Embarazo y lactancia
No debe recibir Xolair cuando esté embarazada, a no ser que su médico lo considere necesario.
Si planea quedarse embarazada, informe a su médico antes de iniciar el tratamiento con Xolair. Su médico comentará con usted los beneficios y riesgos potenciales de la administración de este medicamento durante el embarazo.
Informe a su médico inmediatamente si queda embarazada mientras está siendo tratada con Xolair. No debe recibir Xolair cuando esté en periodo de lactancia.
Conducción y uso de máquinas
No es probable que Xolair afecte a su capacidad para conducir y usar máquinas.
3. Cómo se administra Xolair
Las instrucciones sobre como utilizar Xolair se proporcionan en la sección “Información para el profesional sanitario”.
Xolair se administra, por su médico o enfermero, como una inyección bajo la piel (de forma subcutánea).
Siga detenidamente todas las instrucciones que le haya dado su médico o enfermero.
Cuánto se le administrará
Asma alérgica
Su médico estimará la cantidad de Xolair que necesita y la frecuencia de administración del mismo. Ello depende de su peso corporal y de los resultados de un análisis de sangre realizado antes de iniciar el tratamiento para determinar la concentración de IgE en la misma.
Le administrarán entre 1 y 4 inyecciones al mismo tiempo, cada dos o cuatro semanas.
Continúe tomando su medicación actual para el asma mientras dure el tratamiento con Xolair. No interrumpa ninguna medicación para el asma sin consultarlo con su médico.
Es posible que no perciba una mejoría inmediata del asma antes de iniciar el tratamiento con Xolair. Por lo general, deben transcurrir entre 12 y 16 semanas hasta que el medicamento surta todo su efecto.
Urticaria crónica espontánea (UCE)
Le administrarán dos inyecciones de 150 mg al mismo tiempo cada cuatro semanas.
Continúe tomando su medicación actual para la UCE durante el tratamiento con Xolair. No interrumpa ningún medicamento sin consultarlo primero con su médico.
Uso en niños y adolescentes
Asma alérgica
Xolair puede administrarse a niños y adolescentes a partir de 6 años que ya estén recibiendo medicación para el asma, pero cuyos síntomas asmáticos no están bien controlados por medicamentos como dosis elevadas de esteroides inhalados o beta-agonistas inhalados. Su médico le informará qué cantidad de Xolair necesita su hijo y con qué frecuencia se le debe administrar. Esto dependerá del peso del niño y de los resultados obtenidos de los análisis de sangre realizados antes de iniciar el tratamiento para determinar la cantidad de IgE en su sangre.
Urticaria crónica espontánea (UCE)
Xolair puede administrarse a adolescentes a partir de 12 años que ya estén recibiendo antihistamínicos pero cuyos síntomas de la UCE no estén bien controlados por estos medicamentos.
Si olvidó una dosis de Xolair
Contacte con su médico u hospital tan pronto como sea posible para volver a programar su visita.
Si interrumpe el tratamiento con Xolair
No interrumpa el tratamiento con Xolair a no ser que se lo indique su médico. La interrupción o finalización del tratamiento con Xolair puede causar una recidiva de los síntomas del asma o UCE.
Sin embargo, si está siendo tratado para la UCE, su médico puede interrumpir el tratamiento con Xolair de vez en cuando para valorar sus síntomas. Siga las instrucciones de su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no
todas las personas los sufran. Los efectos adversos causados por Xolair son, por lo general, de leves a
moderados pero ocasionalmente pueden ser graves.
Los efectos adversos graves incluyen:
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 pacientes)
• Reacciones alérgicas graves repentinas: si nota signos de alergia graves repentinos o combinación de los mismos, como son, ronchas, picor o erupción cutánea, hinchazón de la cara, labios, lengua, larínge (caja de voz), tráquea u otras partes del cuerpo, ritmo cardíaco rápido, mareo y ligera sensación de vahído, disnea, respiración jadeante o dificultad respiratoria, o cualquier otro síntoma imprevisto, informe inmediatamente a su médico o enfermera. Si tiene antecedentes de reacciones alérgicas graves (anafilaxia) no relacionados con Xolair, puede sufrir mayor riesgo de desarrollar una reacción alérgica grave después del uso de Xolair.
• Lupus eritematoso sistémico (LES). Los síntomas pueden incluir dolor muscular, dolor e hinchazón de las articulaciones y erupción. Usted también puede experimentar otros síntomas como fiebre, pérdida de peso y fatiga.
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
• Desarrollo de uno o más de los síntomas siguientes: hinchazón, dolor o sarpullido alrededor de los vasos sanguíneos o linfáticos, nivel elevado de un tipo específico de glóbulos blancos (eosinofilia marcada), empeoramiento de los problemas respiratorios, congestión nasal, problemas cardíacos, dolor, adormecimiento, hormigueo en los brazos y piernas (signos del también llamado “síndrome de Churg-Strauss o síndrome hipereosmofílico”).
• Recuento de plaquetas sanguíneas bajo con síntomas como sangrado o hematomas que se producen más fácilmente de lo normal.
• Desarrollo de cualquiera de los síntomas siguientes, especialmente si están en combinación: dolor en las articulaciones con o sin inflamación o rigidez, erupción cutánea, fiebre, inflamación de los nódulos linfáticos, dolor muscular (signos de la enfermedad del suero).
Informe inmediatamente a su médico o enfermera si experimenta alguno de los efectos mencionados
anteriormente.
Otros efectos adversos incluyen:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes)
• fiebre (en niños)
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 pacientes)
• reacciones en el lugar de la inyección las cuales incluyen dolor, hinchazón, picor y enrojecimiento
• dolor en la parte superior del estómago (en niños)
• dolor de cabeza (muy frecuente en niños)
• infección de las vías altas del tracto respiratorio, como es inflamación de la faringe y resfriado común
• sensación de presión o dolor en las mejillas y la frente (sinusitis, dolor de cabeza sinusal)
• dolor en las articulaciones (artralgia)
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes)
• sensación de vértigo, sueño o cansancio
• hormigueo o entumecimiento de manos o pies
• desmayo, disminución de la tensión arterial al sentarse o ponerse de pie (hipotensión postural), rubefacción
• dolor de garganta, tos, problemas respiratorios agudos
• sensación de mareo (nausea), diarrea, indigestión
• picor, urticaria, sarpullido, mayor sensibilidad de la piel al sol
• aumento de peso
• síntomas de tipo gripal
• brazos hinchados
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 pacientes)
• infección parasitaria
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
• dolor muscular y tumefacción de las articulaciones
• pérdida de cabello
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Xolair
- Mantener este medicamento fuera de la vista y del alcance de los niños.
- No utilice este medicamento después de la fecha de caducidad que aparece en el envase. La fecha de caducidad es el último día del mes que se indica.
- Conservar en el embalaje original para protegerlo de la luz.
- Conservar en nevera (entre 2°C y 8°C). No congelar.
- No utilizar ningun envase que esté dañado o muestre indicios de deterioro.
Contenido del envase e información adicional
6.
Composición de Xolair
- El principio activo es omalizumab. Una jeringa de 1 ml de solución contiene 150 mg de omalizumab.
- Los demás componentes son hidrocloruro de L-arginina, hidrocloruro de L-histidina, L-histidina, polisorbato 20 y agua para preparaciones inyectables.
- La cápsula protectora de la aguja de la jeringa puede contener caucho seco (látex).
Aspecto del producto y contenido del envase
Xolair solución inyectable se presenta como una solución de transparente a ligeramente opalescente, incolora a color amarillo parduzco claro en una jeringa precargada.
Xolair 150 mg solución inyectable está disponible en envases que contienen 1 jeringa precargada y en envases múltiples que contienen 4 ó 10 envases intermedios, cada uno de ellos conteniendo 1 jeringa precargada.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nuremberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Etarapna
Novartis Pharma Services Inc. Tea.: +359 2 489 98 28
Ceská republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Lietuva
Novartis Pharma Services Inc. Tel: +370 5 269 16 50
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Eesti
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Novartis Pharma Services Inc. Tel: +372 66 30 810
|
EXXáóa Novartis (Hellas) A.E.B.E. T^: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A. Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kórcpog Novartis Pharma Services Inc. Tr(k: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 Fecha de la última revisión de este prospecto: |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http: //www.ema.europa. eu
INFORMACIÓN PARA EL PROFESIONAL SANITARIO
Esta información está destinada únicamente a profesionales del sector sanitario:
Antes de utilizar la jeringa, lea detenidamente la siguiente información.
Cada envase de Xolair contiene una jeringa precargada sellada individualmente en una envoltura de plástico.
Partes de la jeringa precargada Cápsula Protector de
protectora de la aguja
la aguja

Ventana visora Etiqueta y fecha de caducidad
Aleta de sujeción
Pinzas de activación
Émbolo

Linea de llenado
Las jeringas de Xolair están destinadas a ser utilizadas únicamente por un profesional sanitario.
La cápsula protectora de la aguja de la jeringa puede contener caucho seco (látex), por lo que las personas sensibles a esta sustancia no deben manejarla.
Preparación de la jeringa para su uso
Antes de completar la inyección, evite el contacto con las pinzas de activación del dispositivo (ver
dibujo anterior) para evitar que la aguja quede prematuramente cubierta por el protector.
1. Saque la caja que contiene la jeringa de la nevera y déjela durante 20 minutos aproximadamente para que así alcance la temperatura ambiente (conserve la jeringa en la caja para protejerla de la luz).
2. Si es necesario, la jeringa puede volver a guardarse en la nevera para utilizarla más tarde, pero esto no debe hacerse más de una vez. El tiempo acumulativo durante el cual la jeringa se conserva a temperatura ambiente (25°C) no debe exceder de 4 horas.
3. Cuando esté listo para utilizar la jeringa, lávese las manos minuciosamente con agua y jabón.
4. Limpie el lugar de la inyección.
5. Extraiga la bandeja de plástico de la caja, quite la cubierta de papel y extraiga la jeringa.
6. Inspeccione la jeringa. NO LA UTILICE si está rota o si el líquido parece turbio o contiene partículas. En todos estos casos, devuelva el envase completo a la farmacia.
7. Manteniendo la jeringa horizontalmente (como se muestra a continuación), mire en la ventana visora para comprobar la dosis (150 mg) del medicamento y la fecha de caducidad impresa en la etiqueta. Nota: Gire la parte interior del ensamblaje de la jeringa tal y como se muestra a continuación de tal forma que la etiqueta se pueda leer en la ventana visora.

NO LA UTILICE si el producto ha caducado o si la dosis es incorrecta. En ambos casos, devuelva el envase completo a la farmacia.
8. Mantenga la jeringa verticalmente con el émbolo hacia arriba y golpee ligeramente el lado de la jeringa contra su dedo para permitir que asciendan las burbujas de aire.
9. Compruebe si el nivel del líquido está en o por encima de la línea de llenado mínima. Si el líquido está por debajo de la línea de llenado, devuelva el envase completo a la farmacia.

Uso de la jeringa

Manteniendo la jeringa con la aguja mirando hacia arriba, retire con cuidado de la jeringa, la cápsula protectora de la aguja y deséchela. No toque la aguja expuesta. Posteriormente, golpee suavemente la jeringa con su dedo hasta que las burbujas de aire asciendan a la superficie de la jeringa. Presione lentamente el émbolo para forzar la expulsión de las burbujas de aire fuera de la jeringa sin expulsar solución inadvertidamente.

Pellizque suavemente la piel en el lugar de la inyección e inserte la aguja.
|
Life |
Sosteniendo por la aleta de sujeción, presione lentamente el émbolo hasta donde sea posible. Si gotea solución por el lugar de la inyección, inserte más la aguja. | |
|
□ feSS^fe1'' //w |
Manteniendo el émbolo completamente presionado, extraiga la aguja cuidadosamente y por completo del lugar de la inyección. | |
|
0 ^ fe \ _\ —i_\ —- |
Suelte lentamente el émbolo y deje que el protector de la aguja cubra automáticamente la aguja expuesta. Sostenga una gasa en el lugar de la inyección durante 30 segundos aproximadamente. |
Instrucciones de eliminación
Deseche inmediatamente la jeringa usada en un contenedor para material cortante. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
ANEXO IV
CONCLUSIONES CIENTÍFICAS Y MOTIVOS PARA LA MODIFICACIÓN DE LAS CONDICIONES DE LAS AUTORIZACIONES DE COMERCIALIZACIÓN
Conclusiones científicas
Teniendo en cuenta lo dispuesto en el Informe de Evaluación del Comité para la Evaluación de Riesgos en Farmacovigilancia (PRAC) sobre los informes periódicos de seguridad (IPS) para omalizumab, las conclusiones científicas del Comité de Medicamentos de Uso Humano (CHMP) son las siguientes:
Se han notificado casos de lupus eritematoso sistémico (LES) relacionados con el tratamiento con Xolair, incluyendo dos casos en los que se produjo mejoría tras la retirada del medicamento y un caso con mejoría tras la retirada y posterior reintroducción. Aunque en la mayoría de los casos la información era demasiado limitada para permitir una evaluación de causalidad, en los casos restantes existían factores que generaban cofusión tales como lupus preexistente, incluyendo un potencial LES incipiente ,y que la patogénesis del LES/lupus inducido por fármacos todavía no se conoce con claridad y es probablemente multifactorial. Por tanto, parece lógico pensar que Xolair, un medicamento que forma complejos inmunes con IgE con el potencial de inducir daño de complejo inmune y para el que acontecimientos tales como la enfermedad del suero se han notificado en raras ocasiones, pudiera estar implicado en la patogénesis de LES/lupus inducido por fármacos. Después de una evaluación exhaustiva de los datos disponibles, parece existir un argumento razonable para la posibilidad de una relación causal entre Xolair y el lupus eritematoso sistémico.
Por tanto, en vista de los datos presentados en el IPS revisado, el PRAC consideró que los cambios en la información del producto de los medicamentos que contienen omalizumab estaban justificados.
El CHMP está de acuerdo con las conclusiones científicas del PRAC.
Motivos para la modificación de las condiciones de la(s) autorización(es) de comercialización
De acuerdo con las conclusiones científicas para omalizumab, el CHMP considera que el balance beneficio-riesgo del medicamento o medicamentos que contiene(n) omalizumab no se modifica sujeto a los cambios propuestos en la información del producto.
El CHMP recomienda que se modifiquen las condiciones de la(s) autorización(es) de comercialización.
150
notable mejoría o control completo
valor p para la distribución global de valoración
El estudio 2 valoró la eficacia y seguridad de Xolair en una población de 312 pacientes con asma alérgica grave que equiparaban la población del estudio 1. En este estudio abierto, el tratamiento con Xolair condujo a una reducción del 61% en la proporción de exacerbaciones asmáticas que fue clínicamente significativa comparado con el tratamiento actual del asma solo.
Cuatro amplios estudios adicionales de apoyo controlados con placebo de 28 a 52 semanas de duración en 1.722 adultos y adolescentes (estudios 3, 4, 5, 6) valoraron la eficacia y seguridad de Xolair en pacientes con asma persistente grave. La mayoría de los pacientes estuvo inadecuadamente controlado pero recibieron menos tratamiento concomitante del asma que los pacientes en los estudios 1 ó 2. Los estudios 3-5 utilizaron la exacerbación como variable principal, mientras que el estudio 6 evaluó principalmente el ahorro de corticosteroides inhalados.
notable mejoría o control completo
valor p para la distribución global de valoración
Población por intención de tratar modificada (ITTm): incluyó todos los pacientes que fueron aleatorizados y recibieron al menos una dosis de medicación de ensayo.
Se utilizó BOCF (Observación basal llevada adelante) para imputar los datos que faltaban.
1 Se estimó la LS media utilizando un modelo ANCOVA. Los estratos fueron puntuación semanal de la gravedad del prurito basal (<13 vs. >13) y peso basal (<80 kg vs. >80 kg).
2 El valor-p deriva del ensayo-t de ANCOVA.
notable mejoría o control completo
valor p para la distribución global de valoración
El estudio 2 valoró la eficacia y seguridad de Xolair en una población de 312 pacientes con asma alérgica grave que equiparaban la población del estudio 1. En este estudio abierto, el tratamiento con Xolair condujo a una reducción del 61% en la proporción de exacerbaciones asmáticas que fue clínicamente significativa comparado con el tratamiento actual del asma solo.
Cuatro amplios estudios adicionales de apoyo controlados con placebo de 28 a 52 semanas de duración en 1.722 adultos y adolescentes (estudios 3, 4, 5, 6) valoraron la eficacia y seguridad de Xolair en pacientes con asma persistente grave. La mayoría de los pacientes estuvo inadecuadamente controlado pero recibieron menos tratamiento concomitante del asma que los pacientes en los estudios 1 ó 2. Los estudios 3-5 utilizaron la exacerbación como variable principal, mientras que el estudio 6 evaluó principalmente el ahorro de corticosteroides inhalados.
notable mejoría o control completo
valor p para la distribución global de valoración
El estudio 2 valoró la eficacia y seguridad de Xolair en una población de 312 pacientes con asma alérgica grave que equiparaban la población del estudio 1. En este estudio abierto, el tratamiento con Xolair condujo a una reducción del 61% en la proporción de exacerbaciones asmáticas que fue clínicamente significativa comparado con el tratamiento actual del asma solo.
Cuatro amplios estudios adicionales de apoyo controlados con placebo de 28 a 52 semanas de duración en 1.722 adultos y adolescentes (estudios 3, 4, 5, 6) valoraron la eficacia y seguridad de Xolair en pacientes con asma persistente grave. La mayoría de los pacientes estuvo inadecuadamente controlado pero recibieron menos tratamiento concomitante del asma que los pacientes en los estudios 1 ó 2. Los estudios 3-5 utilizaron la exacerbación como variable principal, mientras que el estudio 6 evaluó principalmente el ahorro de corticosteroides inhalados.
Población por intención de tratar modificada (ITTm): incluyó todos los pacientes que fueron aleatorizados y recibieron al menos una dosis de medicación de ensayo.
Se utilizó BOCF (Observación basal llevada adelante) para imputar los datos que faltaban.
1 Se estimó la LS media utilizando un modelo ANCOVA. Los estratos fueron puntuación semanal de la gravedad del prurito basal (<13 vs. >13) y peso basal (<80 kg vs. >80 kg).
2 El valor-p deriva del ensayo-t de ANCOVA.