Xarelto 20 Mg Comprimidos Recubiertos Con Pelicula (100 Comprimidos)
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
▼ Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Xarelto 2,5 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 2,5 mg de rivaroxaban.
Excipiente con efecto conocido:
Cada comprimido recubierto con película contiene 33,92 mg de lactosa (como monohidrato), ver sección 4.4.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
Comprimidos de color amarillo pálido, redondos y biconvexos (6 mm de diámetro y 9 mm de radio de curvatura), con la cruz de BAYER en una cara, y “2.5” y un triángulo en la otra cara.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Xarelto, administrado en combinación con ácido acetilsalicílico (AAS) solo, o con AAS más clopidogrel o ticlopidina, está indicado en la prevención de eventos aterotrombóticos en pacientes adultos tras un síndrome coronario agudo (SCA) con biomarcadores cardiacos elevados (ver secciones 4.3, 4.4 y 5.1).
4.2 Posología y forma de administración
Posología
La dosis recomendada es de 2,5 mg dos veces al día.
Los pacientes deben tomar también una dosis diaria de 75 - 100 mg de AAS, o una dosis diaria de 75 -100 mg de AAS más una dosis diaria de 75 mg de clopidogrel o una dosis diaria estándar de ticlopidina.
El tratamiento deberá ser evaluado de forma regular en cada paciente, valorando el riesgo de eventos isquémicos frente al riesgo de hemorragia. La duración del tratamiento más allá de los 12 meses debe evaluarse individualmente en cada caso, ya que la experiencia hasta los 24 meses es limitada (ver sección 5.1).
El tratamiento con Xarelto debe iniciarse lo antes posible tras la estabilización del acontecimiento de SCA (incluyendo los procedimientos de revascularización), a partir de las 24 horas siguientes a la admisión en el hospital y cuando se interrumpiría normalmente la terapia anticoagulante por vía parenteral.
Cambio de tratamiento con antagonistas de la vitamina K (AVK) aXarelto
Al cambiar el tratamiento con AVK a Xarelto, los valores de INR (International Normalized Ratio) del paciente estarán falsamente elevados después de la toma de Xarelto. El INR no es un parámetro válido para medir la actividad anticoagulante de Xarelto, por lo que no debe utilizarse (ver sección 4.5).
Cambio de tratamiento con Xarelto a antagonistas de la vitamina K (AVK)
Existe la posibilidad de una incorrecta anticoagulación durante la transición de Xarelto a AVK. Debe garantizarse una anticoagulación adecuada y continua durante cualquier transición a un anticoagulante alternativo. Debe señalarse que Xarelto puede contribuir a un aumento del INR.
En los pacientes que cambien de Xarelto a AVK, estos tratamientos deben administrarse simultáneamente hasta que el INR sea > 2,0. Durante los dos primeros días del periodo de cambio se utilizará la dosis inicial estándar de AVK, que se ajustará posteriormente en función de los resultados del INR. Mientras los pacientes estén bajo tratamiento con Xarelto y AVK, el INR puede determinarse a partir de las 24 horas que siguen a la dosis de Xarelto y antes de la siguiente dosis. Una vez interrumpido el tratamiento con Xarelto, el INR puede determinarse con fiabilidad pasadas 24 horas de la última dosis (ver secciones 4.5 y 5.2).
Cambio de tratamiento con anticoagulante parenteral a Xarelto
Los pacientes que están recibiendo un anticoagulante por vía parenteral, deben interrumpir el tratamiento anticoagulante por vía parenteral e iniciar el tratamiento con Xarelto de 0 a 2 horas antes de la siguiente administración programada del medicamento por vía parenteral (p. ej., heparina de bajo peso molecular). En el caso de un anticoagulante parenteral administrado por perfusión continua (p. ej., heparina no fraccionada intravenosa) Xarelto deberá administrarse en el momento de la suspensión del anticoagulante parenteral.
Cambio de tratamiento con Xarelto a anticoagulante parenteral
La primera dosis de anticoagulante parenteral debe administrarse en el momento en que se tomaría la siguiente dosis de Xarelto.
Poblaciones especiales Insuficiencia renal
Los escasos datos clínicos en pacientes con insuficiencia renal grave (aclaramiento de creatinina de 15 a 29 ml/min) indican que las concentraciones plasmáticas de rivaroxaban aumentan significativamente. Por lo tanto, Xarelto se debe usar con precaución en estos pacientes. No se recomienda su uso en pacientes con un aclaramiento de creatinina < 15 ml/min (ver secciones 4.4 y 5.2).
No se requiere ajuste de dosis en pacientes con insuficiencia renal leve (aclaramiento de creatinina de 50 a 80 ml/min) o insuficiencia renal moderada (aclaramiento de creatinina de 30 a 49 ml/min) (Ver sección 5.2).
Insuficiencia hepática
Xarelto está contraindicado en pacientes con hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia incluidos los pacientes cirróticos con Child Pugh B y C (ver secciones 4.3 y 5.2).
Pacientes de edad avanzada
No se requiere ajuste de dosis (ver secciones 4.4 y 5.2).
Peso corporal
No se requiere ajuste de dosis (ver secciones 4.4 y 5.2).
Sexo
No se requiere ajuste de dosis (ver sección 5.2).
Población pediátrica
No se ha establecido la seguridad y eficacia de Xarelto en niños de 0 a 18 años. No se dispone de datos. Por lo tanto, no se recomienda el uso de Xarelto en niños menores de 18 años.
Forma de administración Vía oral.
Xarelto puede tomarse con o sin alimentos (ver secciones 4.5 y 5.2).
Para aquellos pacientes que no puedan tragar el comprimido entero, el comprimido de Xarelto puede triturarse y mezclarse con agua o con puré de manzana inmediatamente antes de su uso y administrarse por vía oral.
El comprimido triturado también se puede administrar a través de sonda gástrica una vez se haya confirmado la colocación correcta de la sonda. El comprimido triturado se administrará diluido con una pequeña cantidad de agua a través de la sonda gástrica, procediendo seguidamente a un lavado adicional de la sonda con agua (ver sección 5.2).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hemorragia activa clínicamente significativa.
Lesión o enfermedad, si se considera que tiene un riesgo significativo de sangrado mayor. Esto puede incluir úlcera gastrointestinal activa o reciente, presencia de neoplasias malignas con alto riesgo de sangrado, traumatismo cerebral o espinal reciente, cirugía cerebral, espinal u oftálmica reciente, hemorragia intracraneal reciente, conocimiento o sospecha de varices esofágicas, malformaciones arteriovenosas, aneurismas vasculares o anomalías vasculares intraespinales o intracerebrales mayores.
Tratamiento concomitante con cualquier otro anticoagulante, p. ej., heparina no fraccionada (HNF), heparinas de bajo peso molecular (enoxaparina, dalteparina, etc.), derivados de la heparina (fondaparinux, etc.), anticoagulantes orales (warfarina, dabigatran etexilato, apixaban, etc.) excepto bajo las circunstancias concretas de cambio de tratamiento anticoagulante (ver sección 4.2) o cuando se administre HNF a las dosis necesarias para mantener un catéter venoso o arterial central abierto (ver sección 4.5).
Tratamiento concomitante del SCA con tratamiento antiagregante en pacientes que han sufrido un ictus o un ataque isquémico transitorio (AIT) previos (ver sección 4.4).
Hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia, incluidos los pacientes cirróticos con Child Pugh B y C (ver sección 5.2).
Embarazo y lactancia (ver sección 4.6).
4.4 Advertencias y precauciones especiales de empleo
Se ha estudiado la eficacia y seguridad de Xarelto en combinación con los medicamentos antiagregantes ácido acetilsalicílico (AAS) y clopidogrel/ticlopidina. No se ha estudiado el tratamiento en combinación con otros medicamentos antiagregantes, como por ejemplo, prasugrel o ticagrelor, por lo que no se recomienda este tipo de combinaciones.
Durante todo el periodo de tratamiento se recomienda una estrecha monitorización clínica del paciente, siguiendo la práctica habitual de anticoagulación.
Riesgo de hemorragia
Al igual que con otros anticoagulantes, los pacientes que toman Xarelto deben ser observados cuidadosamente para detectar cualquier signo de sangrado. Se recomienda utilizar con precaución en
En los ensayos clínicos se observaron con más frecuencia hemorragias a nivel de las mucosas (p.ej. epistaxis, gingival, gastrointestinal, génito-urinaria) y anemia en los pacientes tratados con rivaroxaban a largo plazo además del tratamiento antiagregante unico o doble. Por ello, además de un seguimiento clínico adecuado, las determinaciones de hemoglobina/ hematocrito podrían ser útiles para detectar hemorragias ocultas, cuando se considere apropiado.
Varios subgrupos de pacientes, como se explica a continuación, presentan un mayor riesgo de hemorragia. Por lo tanto, el uso de Xarelto en combinación con la terapia doble antiagregante en pacientes con riesgo aumentado de sangrado conocido debe ser valorado frente al beneficio en cuanto a la prevención de eventos aterotrombóticos. Además, en estos pacientes se debe vigilar cuidadosamente la presencia de signos y síntomas de complicaciones hemorrágicas y de anemia después del inicio del tratamiento (ver sección 4.8).
Cualquier disminución inexplicada de la hemoglobina o de la presión arterial requerirá la búsqueda de una zona de sangrado.
Aunque el tratamiento con rivaroxaban no requiere una monitorización rutinaria de la exposición, la determinación de los niveles de rivaroxaban mediante un ensayo anti-factor Xa cuantitativo calibrado puede ser útil en situaciones excepcionales, en las que el conocimiento de la exposición a rivaroxaban puede ayudar en la toma de decisiones clínicas, como por ejemplo, en caso de sobredosis o cirugía de emergencia (ver secciones 5.1 y 5.2).
Insuficiencia renal
En pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min), las concentraciones plasmáticas de rivaroxaban pueden aumentar de forma significativa (en promedio,
1,6 veces), lo que conllevaría un aumento del riesgo de hemorragia. Xarelto debe utilizarse con precaución en pacientes con un aclaramiento de creatinina de 15 a 29 ml/min. No se recomienda el uso en pacientes con un aclaramiento de creatinina < 15 ml/min (ver secciones 4.2 y 5.2).
En los pacientes con insuficiencia renal moderada (aclaramiento de creatinina 30 - 49 ml/min) que reciban concomitantemente otros medicamentos que aumenten las concentraciones plasmáticas de rivaroxaban, Xarelto se debe utilizar con precaución (ver sección 4.5).
Interacción con otros medicamentos
No se recomienda el uso de Xarelto en pacientes que reciben tratamiento sistémico concomitante con antimicóticos azólicos (p. ej., ketoconazol, itraconazol, voriconazol y posaconazol) o inhibidores de la proteasa del VIH (p. ej., ritonavir). Estos principios activos son inhibidores potentes del CYP3A4 y de la P-gp y por lo tanto pueden aumentar las concentraciones plasmáticas de rivaroxaban hasta un nivel clínicamente relevante (en promedio, 2,6 veces), lo que puede llevar a un aumento del riesgo de hemorragia (ver sección 4.5).
Se debe tener precaución si los pacientes reciben tratamiento concomitante con medicamentos que afectan a la hemostasia, como los antiinflamatorios no esteroideos (AINEs), ácido acetilsalicílico (AAS) o inhibidores de la agregación plaquetaria. Para los pacientes con riesgo de enfermedad gastrointestinal ulcerosa deberá considerarse un tratamiento profiláctico adecuado (ver sección 4.5). Después de un síndrome coronario agudo, los pacientes en tratamiento con Xarelto y AAS o Xarelto y AAS más clopidogrel/ticlopidina sólo deben recibir tratamiento concomitante con AINEs si el beneficio supera el riesgo de hemorragia.
Otros factores de riesgo hemorrágico
Rivaroxaban, al igual que con otros agentes antitrombóticos, no está recomendado en pacientes con un riesgo aumentado de hemorragia, tales como:
• trastornos de la coagulación, congénitos o adquiridos
• hipertensión arterial grave no controlada
• otra enfermedad gastrointestinal sin úlcera activa que pueda producir complicaciones hemorrágicas (por ejemplo, enfermedad inflamatoria intestinal, esofagitis, gastritis o reflujo gastroesofágico)
• retinopatía vascular
• bronquiectasia o antecedentes de hemorragia pulmonar Se debe utilizar con precaución en pacientes con SCA:
• mayores de 75 años, si se administra con AAS solo, o bien con AAS más clopidogrel o ticlopidina.
• con peso corporal bajo (< 60 kg), si se administra con AAS solo, o bien con AAS más clopidogrel o ticlopidina.
Pacientes con ictus o AIT previos
Xarelto 2,5 mg está contraindicado para el tratamiento del SCA en pacientes con un ictus o AIT previos (ver sección 4.3). Se han estudiado pocos pacientes con SCA y un ictus o AIT previos, pero los escasos datos de eficacia disponibles indican que estos pacientes no se benefician del tratamiento
Anestesia espinal/epidural o punción lumbar
Cuando se aplica anestesia neuraxial (anestesia epidural o espinal) o se realiza una punción lumbar o epidural, los pacientes tratados con antitrombóticos para la prevención de complicaciones tromboembólicas tienen riesgo de presentar un hematoma epidural o espinal, que puede causar parálisis a largo plazo o permanente. El riesgo de estos eventos puede estar aumentado por el empleo postoperatorio de catéteres epidurales permanentes o por la administración concomitante de medicamentos que afectan a la hemostasia. El riesgo también puede aumentar por la punción epidural o espinal traumática o repetida. Se debe controlar con frecuencia la presencia de signos y síntomas de deterioro neurológico (p. ej., adormecimiento o debilidad de extremidades inferiores, disfunción intestinal o vesical). Si se observa compromiso neurológico, será necesario un diagnóstico y tratamiento urgente. Antes de la intervención neuraxial, el médico debe valorar el beneficio potencial frente al riesgo en los pacientes con tratamiento anticoagulante o que van a recibir medicamentos anticoagulantes para la tromboprofilaxis. No se dispone de experiencia clínica sobre el uso de 2,5 mg con AAS solo o con AAS más clopidogrel o ticlopidina en estas situaciones.
Para reducir el riesgo potencial de sangrado asociado con el uso concomitante de rivaroxaban y anestesia neuraxial (epidural/espinal) o punción espinal se debe considerar el perfil farmacocinético de rivaroxaban. La colocación o extracción de un catéter epidural o punción lumbar se realiza mejor cuando se estima que el efecto anticoagulante de rivaroxaban es bajo (ver sección 5.2). Sin embargo, se desconoce el momento exacto en el que se alcanza un efecto anticoagulante lo suficientemente bajo en cada paciente. Se deben dejar de administrar los inhibidores de la agregación plaquetaria, siguiendo las indicaciones en la correspondiente Ficha Técnica.
Recomendaciones posológicas antes y después de procedimientos invasivos y de intervenciones quirúrgicas
Si es necesario realizar un procedimiento invasivo o una intervención quirúrgica, se interrumpirá la administración de Xarelto 2,5 mg por lo menos 12 horas antes de la intervención, si es posible y basándose en el criterio clínico del médico. Si un paciente va a someterse a cirugía electiva y no se desea un efecto antiagregante, se interrumpirá la administración de los inhibidores de la agregación plaquetaria siguiendo las instrucciones de la ficha técnica de cada medicamento. Si la intervención no puede retrasarse, debe evaluarse el aumento del riesgo de hemorragia frente a la urgencia de la intervención.
Se debe reiniciar lo antes posible la administración de Xarelto después del procedimiento invasivo o intervención quirúrgica, siempre que la situación clínica lo permita y se haya establecido una hemostasia adecuada, una vez confirmado por el médico que trata al paciente (ver sección 5.2).
Pacientes de edad avanzada
La edad avanzada puede aumentar el riesgo de hemorragia (ver sección 5.2).
Información acerca de los excipientes
Xarelto contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, deficiencia de lactasa de Lapp o malabsorción de glucosa o galactosa no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Inhibidores del CYP3A4 y de la P-gp
La administración concomitante de rivaroxaban con ketoconazol (400 mg una vez al día) o ritonavir (600 mg dos veces al día) produjo un aumento de 2,6 veces / 2,5 veces del AUC media de rivaroxaban, y un aumento de 1,7 veces / 1,6 veces de la Cmax media de rivaroxaban, con aumentos significativos de los efectos farmacodinámicos, lo que puede aumentar el riesgo de hemorragia. Por lo tanto, no se recomienda el uso de Xarelto en pacientes que reciban tratamiento sistémico concomitante con antimicóticos azólicos como ketoconazol, itraconazol, voriconazol y posaconazol o con inhibidores de la proteasa del VIH. Estos principios activos son inhibidores potentes del CYP3A4 y de la P-gp (ver sección 4.4).
Las sustancias activas que inhiben intensamente sólo una de las vías de eliminación de rivaroxaban, el CYP3A4 o la P-gp, pueden aumentar las concentraciones plasmáticas de rivaroxaban en menor grado. La claritromicina (500 mg dos veces al día), por ejemplo, considerada un potente inhibidor del CYP3A4 y un inhibidor moderado de la P-gp, produjo un aumento de 1,5 veces del AUC media de rivaroxaban y un aumento de 1,4 veces de la Cmax. Este aumento no se considera clínicamente relevante. (Pacientes con insuficiencia renal: ver sección 4.4).
La eritromicina (500 mg tres veces al día), que inhibe moderadamente el CYP3A4 y la P-gp, produjo un aumento de 1,3 veces del AUC y de la Cmax medias de rivaroxaban. Este aumento no se considera clínicamente relevante.
En sujetos con insuficiencia renal leve, la eritromicina (500 mg tres veces al día) produjo un aumento de 1,8 veces el AUC media de rivaroxaban y de 1,6 veces en la Cmax, comparado con sujetos con la función renal normal. En sujetos con insuficiencia renal moderada, la eritromicina produjo un aumento de 2.0 veces en el AUC media de rivaroxaban y 1,6 veces en la Cmax, comparado con sujetos con la función renal normal. El efecto de la eritromicina es aditivo al de la insuficiencia renal (ver sección
4.4) .
El fluconazol (400 mg una vez al día), considerado un inhibidor moderado del CYP3A4, produjo un aumento de 1,4 veces del AUC media de rivaroxaban y un aumento de 1,3 veces de la Cmax media.
Este aumento no se consideró clínicamente relevante. (Pacientes con insuficiencia renal: ver sección
4.4) .
Dada la escasa información clínica disponible con dronedarona, se debe evitar la administración concomitante con rivaroxaban.
Anticoagulantes
Después de la administración combinada de enoxaparina (dosis única de 40 mg) con rivaroxaban (dosis única de 10 mg), se observó un efecto aditivo sobre la actividad anti-factor Xa, sin efectos adicionales en las pruebas de coagulación (TP, TTPa). La enoxaparina no afectó a las propiedades farmacocinéticas de rivaroxaban.
Debido al aumento del riesgo de hemorragia, se debe tener precaución si los pacientes reciben tratamiento concomitante con cualquier otro anticoagulante (ver secciones 4.3 y 4.4).
AINEs e inhibidores de la agregación plaquetaria
No se observó una prolongación clínicamente relevante del tiempo de sangrado después de la administración concomitante de rivaroxaban (15 mg) y 500 mg de naproxeno. No obstante, algunas personas pueden tener una respuesta farmacodinámica más pronunciada.
No se observó ninguna interacción farmacocinética ni farmacodinámica clínicamente significativa cuando se administró rivaroxaban concomitantemente con 500 mg de ácido acetilsalicílico.
El clopidogrel (dosis de carga de 300 mg, seguida de una dosis de mantenimiento de 75 mg) no mostró ninguna interacción farmacocinética con rivaroxaban (15 mg); sin embargo, se observó un aumento del tiempo de sangrado en un subgrupo de pacientes, que no se correlacionó con la agregación plaquetaria, las concentraciones de P-selectina o de los receptores GPIIb/IIIa.
Se debe tener precaución si los pacientes reciben tratamiento concomitante con AINEs (incluyendo ácido acetilsalicílico) e inhibidores de la agregación plaquetaria, ya que estos medicamentos aumentan, de por sí, el riesgo de hemorragia (ver sección 4.4).
Warfarina
Los cambios de tratamiento con warfarina (INR de 2,0 a 3,0), un antagonista de la vitamina K, a rivaroxaban (20 mg) o de rivaroxaban (20 mg) a warfarina (INR de 2,0 a 3,0) aumentaron el tiempo de protrombina/INR (Neoplastin) de forma importante (pueden observarse valores individuales del INR de hasta 12), mientras que los efectos sobre el TTPa, la inhibición de la actividad del factor Xa y el potencial de trombina endógena (PTE) fueron aditivos.
Si se desea medir los efectos farmacodinámicos de rivaroxaban durante el periodo de cambio de tratamiento, puede utilizarse la actividad anti-factor Xa, PiCT y Heptest, ya que la warfarina no afecta a estas pruebas. Al cuarto día tras la última dosis de warfarina, todas las pruebas (incluyendo TP, TTPa, inhibición de la actividad del factor Xa y PTE) reflejaron únicamente el efecto de rivaroxaban. Si se desea medir los efectos farmacodinámicos de warfarina durante el periodo de cambio de tratamiento, se puede usar la determinación del INR en la Ctrough de rivaroxaban (24 horas después de su anterior administración), ya que rivaroxaban afecta mínimamente a esta prueba en este punto.
No se observó ninguna interacción farmacocinética entre warfarina y rivaroxaban.
Inductores del CYP3A4
La administración concomitante de rivaroxaban con rifampicina, un potente inductor del CYP3A4, produjo una disminución aproximada del 50% del AUC media de rivaroxaban, con disminuciones paralelas de sus efectos farmacodinámicos. El uso concomitante de rivaroxaban con otros inductores potentes del CYP3A4 (por ejemplo, fenitoína, carbamazepina, fenobarbital o la hierba de San Juan (Hypericum perforatum)) también puede causar una disminución de la concentración plasmática de rivaroxaban. Por tanto, la administración concomitante con inductores potentes del CYP3A4 deberá evitarse a menos que el paciente esté estrechamente monitorizado para detectar signos o síntomas de trombosis.
Otros tratamientos concomitantes
No se observó ninguna interacción farmacocinética o farmacodinámica clínicamente significativa cuando se administró rivaroxaban concomitantemente con midazolam (sustrato del CYP3A4), digoxina (sustrato de la P-gp), atorvastatina (sustrato del CYP3A4 y de la P-gp) u omeprazol (inhibidor de la bomba de protones). Rivaroxaban no inhibe ni induce ninguna isoforma mayor del CYP, como el CYP3A4.
No se observó interacción clínicamente relevante con la toma de alimentos (ver sección 4.2). Parámetros de laboratorio
Los parámetros de la coagulación (p. ej. TP, TTPa, HepTest) se ven afectados de la forma esperada debido al mecanismo de acción de rivaroxaban (ver sección 5.1).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No se ha evaluado la seguridad y eficacia de Xarelto en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3). Debido a la posible toxicidad reproductiva, riesgo intrínseco de hemorragia y la evidencia de que rivaroxaban atraviesa la barrera placentaria, Xarelto está contraindicado durante el embarazo (ver sección 4.3).
Las mujeres en edad fértil deben evitar quedarse embarazadas durante el tratamiento con rivaroxaban.
Lactancia
No se ha evaluado la seguridad y eficacia de Xarelto en mujeres en período de lactancia. Los datos en animales indican que rivaroxaban se excreta en la leche materna. Por lo tanto, Xarelto está contraindicado durante la lactancia (ver sección 4.3). Se debe decidir si es necesario interrumpir la lactancia o bien interrumpir/suspender el tratamiento.
Fertilidad
No se han realizado estudios específicos con rivaroxaban para evaluar los efectos sobre la fertilidad en humanos. En un estudio sobre la fertilidad en ratas macho y hembra no se observó ningún efecto (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Xarelto puede influir ligeramente en la capacidad para conducir y utilizar máquinas. Se han descrito reacciones adversas como síncope (frecuencia: poco frecuente) y mareos (frecuencia: frecuente) (ver sección 4.8). Los pacientes que sufran estas reacciones adversas no deben conducir ni utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Se ha evaluado la seguridad de rivaroxaban en once ensayos clínicos de fase III, que incluyeron 32.625 pacientes expuestos a rivaroxaban (ver la tabla 1).
Tabla 1: Número de pacientes estudiados, dosis máxima diaria y duración del tratamiento en los estudios de fase III.
|
Indicación |
Número de pacientes * |
Dosis máxima diaria |
Duración máxima del tratamiento |
|
Prevención de tromboembolismo venoso (TEV) en pacientes adultos sometidos a cirugía electiva de reemplazo de cadera o rodilla |
6.097 |
10 mg |
39 días |
|
Prevención de tromboembolismo venoso en pacientes encamados |
3.997 |
10 mg |
39 días |
|
Tratamiento de TVP, EP y prevención de las recurrencias de TVP y EP |
4.556 |
Días 1 a 21: 30 mg Día 22 en adelante: 20 mg |
21 meses |
|
Prevención del ictus y de la embolia sistémica en pacientes con fibrilación auricular no valvular |
7.750 |
20 mg |
41 meses |
|
Prevención de eventos en pacientes que han padecido un síndrome coronario agudo (SCA) |
10.225 |
5 mg ó 10 mg respectivamente, co-administrada con AAS o bien con AAS más clopidogrel o ticlopidina |
31 meses |
*Pacientes expuestos por lo menos a una dosis de rivaroxaban.
Las reacciones adversas notificadas con mayor frecuencia en los pacientes que recibieron rivaroxaban fueron hemorragias (ver sección 4.4. y "Descripción de las reacciones adversas seleccionadas” más adelante). Las hemorragias notificadas con mayor frecuencia (> 4%) fueron epistaxis (5,9%) y la hemorragia del tracto gastrointestinal (4,2%).
En total, se notificó la aparición de eventos adversos en aproximadamente un 67% de los pacientes expuestos por lo menos a una dosis de rivaroxaban. Aproximadamente el 22% de los pacientes sufrieron eventos adversos que se consideran relacionados con el tratamiento, según la evaluación de los investigadores. En los pacientes tratados con Xarelto 10 mg sometidos a cirugía electiva de
reemplazo de cadera o rodilla y en pacientes hospitalizados encamados, se produjeron episodios hemorrágicos en aproximadamente 6,8% y 12,6% de los pacientes, respectivamente, y se produjo anemia en aproximadamente un 5,9% y 2,1% de los pacientes, respectivamente. En los pacientes tratados con Xarelto 15 mg dos veces al día y después Xarelto 20 mg una vez al día para el tratamiento de la TVP o EP, o con Xarelto 20 mg una vez al día para la prevención de la recurrencia de la TVP y de la EP, se produjeron episodios hemorrágicos en aproximadamente un 27,8% de los pacientes, y anemia en aproximadamente un 2,2% de los pacientes. En los pacientes tratados para la prevención del ictus y de la embolia sistémica, se notificó hemorragia de cualquier tipo o gravedad, con una tasa de 28 por cada 100 pacientes-años, y anemia con una tasa de 2,5 por cada 100 pacientes-años. En los pacientes tratados para la prevención de eventos aterotrombóticos tras un síndrome coronario agudo (SCA), se notificó hemorragia de cualquier tipo o gravedad con una tasa de 22 eventos por cada 100 pacientes-años. La anemia se notificó con una tasa de 1,4 eventos por cada 100 pacientes-años.
Tabla de reacciones adversas
Las frecuencias de las reacciones adversas notificadas con Xarelto se resumen en la tabla 2, según la clasificación por órganos y sistemas (convención MedDRA) y según las frecuencias.
Las frecuencias se definen como: muy frecuentes (> 1/10) frecuentes (> 1/100 a < 1/10) poco frecuentes (> 1/1.000 a < 1/100) raras (> 1/10.000 a < 1/1.000) muy raras (< 1/10.000)
no conocida: no puede estimarse a partir de los datos disponibles.
Tabla 2: Todas las reacciones adversas observadas con el tratamiento y notificadas en los estudios de fase III
|
Frecuentes |
Poco frecuentes |
Raras |
No conocida |
|
Trastornos de la sangre y del sistema linfático | |||
|
Anemia (incl. respectivos parámetros de laboratorio) |
Trombocitemia (incl. aumento del recuento de plaquetas)A | ||
|
Trastornos del sistema inmunológico | |||
|
Reacción alérgica, dermatitis alérgica | |||
|
Trastornos del sistema nervioso | |||
|
Mareos, cefalea, |
Hemorragia cerebral e intracraneal, síncope | ||
|
Trastornos oculares | |||
|
Hemorragia ocular (incl. hemorragia conjuntival) | |||
|
Trastornos cardiacos | |||
|
Taquicardia | |||
|
Trastornos vasculares | |||
|
Hipotensión, hematoma | |||
|
Trastornos respiratorios, torácicos y mediastínicos | |||
|
Epistaxis, hemoptisis | |||
|
Frecuentes |
Poco frecuentes |
Raras |
No conocida |
|
Trastornos gastrointestinales | |||
|
Sangrado gingival, hemorragia del tracto gastrointestinal (incl. hemorragia rectal), dolor gastrointestinal y abdominal, dispepsia, náuseas, estreñimientoA, diarrea, vómitosA |
Sequedad de boca | ||
|
Trastornos hepatobiliares | |||
|
Alteración de la función hepática |
Ictericia | ||
|
Trastornos de la piel y del tejido subcutáneo | |||
|
Prurito (incl. casos raros de prurito generalizado), exantema, equimosis, hemorragia cutánea y subcutánea |
Urticaria | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |||
|
Dolor en las extremidadesA |
Hemartrosis |
Hemorragia muscular |
Síndrome compartimental secundario a una hemorragia |
|
Trastornos renales y urinarios | |||
|
Hemorragia del tracto urogenital (incl. hematuria y menorragiaB), insuficiencia renal (incl. aumento de creatinina en sangre, aumento de la urea en sangre)A |
Insuficiencia renal /insuficiencia renal aguda secundaria a una hemorragia suficiente para causar hipoperfusión | ||
|
Trastornos generales y alteraciones en el lugar de administración | |||
|
FiebreA, edema periférico, disminución general de la fuerza y la energía (incl. fatiga y astenia) |
Sensación de malestar (incl. malestar general) |
Edema localizadoA | |
|
Exploraciones complementarias | |||
|
Aumento de las transaminasas |
Aumento de la bilirrubina, aumento de la fosfatasa alcalina sanguíneaA, aumento de la LDHA, aumento de la lipasaA, aumento de la amilasaA, aumento de la GGTA |
Aumento de la bilirrubina conjugada (con o sin aumento concomitante de la ALT). | |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos | |||
|
Frecuentes |
Poco frecuentes |
Raras |
No conocida |
|
Hemorragia después de una intervención (incl. anemia postoperatoria y hemorragia de la herida), contusión, secreción de la heridaA |
Pseudoaneurisma vascularC |
A: observado en la prevención del tromboembolismo venoso (TEV) en pacientes adultos sometidos a cirugía electiva de reemplazo de cadera o rodilla
B: observado en el tratamiento de la TVP, EP y prevención de sus recurrencias como muy frecuente en mujeres < 55 años.
C: observado como poco frecuente en la prevención de eventos aterotrombóticos en pacientes que han sufrido un SCA (tras una intervención coronaria percutánea)
Descripción de reacciones adversas seleccionadas
Debido a su mecanismo de acción farmacológica, el uso de Xarelto puede asociarse a un incremento del riesgo de hemorragia oculta o manifiesta en cualquier tejido u órgano, que puede dar lugar a una anemia post-hemorrágica. Los signos, síntomas y gravedad (incluido un posible desenlace mortal) variarán según la localización y el grado o la extensión de la hemorragia, la anemia o ambas (ver sección 4.9 Tratamiento de la hemorragia). En los ensayos clínicos se observaron con más frecuencia hemorragias a nivel de mucosas (p.ej. epistaxis, gingival, gastrointestinal, genito-urinaria) y anemia en los pacientes que recibían rivaroxaban a largo plazo con respecto a los que recibían tratamiento con AVK. Por ello, además de un adecuado seguimiento clínico, las determinaciones de hemoglobina y hematocrito podrían ser útiles para detectar hemorragias ocultas cuando se considere apropiado. El riesgo de hemorragia puede estar aumentado en ciertos grupos de pacientes, como por ejemplo, en pacientes con hipertensión arterial grave no controlada y/o en tratamiento concomitante que afecte a la hemostasia (ver Riesgo de hemorragia en la sección 4.4). El sangrado menstrual puede ser más intenso y/o prolongarse. Las complicaciones hemorrágicas pueden presentarse como debilidad, palidez, mareos, cefalea o tumefacción inexplicada, disnea o shock de causa desconocida. En algunos casos, a consecuencia de la anemia, se han observado síntomas de isquemia cardíaca, como dolor torácico o angina de pecho.
Se han notificado complicaciones conocidas, secundarias a hemorragia intensa, como el síndrome compartimental o insuficiencia renal debida a hipoperfusión. Por lo tanto se debe tener en cuenta la posibilidad de hemorragia al evaluar el estado de cualquier paciente anticoagulado.
Observaciones post-comercialización
Tras la comercialización se han notificado las siguientes reacciones adversas en asociación temporal con el uso de Xarelto. No se ha podido estimar la frecuencia de estas reacciones adversas.
Trastornos del sistema inmunológico: angioedema y edema alérgico.(En los ensayos de fase III agrupados, estos eventos fueron poco frecuentes (> 1/1.000 a <1/100)).
Trastornos hepatobiliares: colestasis, hepatitis (incluyendo lesión hepatocelular). (En los ensayos de fase III agrupados, estos eventos fueron raros (> 1/10.000 a < 1/1.000)).
Trastornos de la sangre y del sistema linfático: trombocitopenia. (En los ensayos de fase III agrupados, estos eventos fueron poco frecuentes (> 1/1.000 a < 1/100)).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se han notificado casos raros de sobredosis de hasta 600 mg sin complicaciones hemorrágicas u otras reacciones adversas. Debido a la escasa absorción a dosis supraterapéuticas de 50 mg de rivaroxaban o superiores, se espera un efecto techo sin un aumento posterior de la exposición plasmática media.
No se dispone de un antídoto específico que antagonice el efecto farmacodinámico de rivaroxaban.
Se puede considerar el uso de carbono activado para reducir la absorción en caso de sobredosis por rivaroxaban.
Tratamiento de la hemorragia
En caso de producirse una complicación hemorrágica en un paciente que recibe tratamiento con rivaroxaban, se deberá retrasar la siguiente administración de rivaroxaban o interrumpir el tratamiento si se considera conveniente. Rivaroxaban tiene una semivida de eliminación de entre 5 y 13 horas (ver sección 5.2). Las medidas terapéuticas deben individualizarse según la gravedad y la localización de la hemorragia. En caso necesario, podría aplicarse el tratamiento sintomático adecuado, como la compresión mecánica (por ejemplo en caso de epistaxis intensa), hemostasia quirúrgica con procedimientos de control de la hemorragia, reemplazo de fluidos y apoyo hemodinámico (concentrado de hematíes o plasma fresco congelado, dependiendo de la anemia o la coagulopatía asociadas) o plaquetas.
Si la hemorragia no se puede controlar con las medidas anteriores, debería plantearse la administración de un agente procoagulante específico para revertir el efecto, como el concentrado de complejo de protrombina (CCP), concentrado de complejo de protrombina activado (CCPA) o factor VIIa recombinante (r-FVIIa). Sin embargo, actualmente hay una experiencia clínica muy limitada con el uso de estos productos en pacientes que reciben rivaroxaban. La recomendación se basa también en datos no clínicos limitados. Deberá plantearse la readministración de factor VIIa recombinante y ajustar la dosis dependiendo de la mejoría de la hemorragia. Dependiendo de la disponibilidad local, en caso de hemorragia mayor debe considerarse consultar a un experto en coagulación (ver sección 5.1).
No se espera que el sulfato de protamina y la vitamina K afecten a la actividad anticoagulante de rivaroxaban. La experiencia con ácido tranexámico es limitada y no hay experiencia con ácido aminocaproico y aprotinina en pacientes tratados con rivaroxaban. No hay una justificación científica sobre la ventaja ni experiencia con el hemostático sistémico desmopresina en pacientes tratados con rivaroxaban. Debido a su elevada fijación a las proteínas plasmáticas, no se espera que rivaroxaban sea dializable.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inhibidores directos del factor Xa, código ATC: B01AF01 Mecanismo de acción
Rivaroxaban es un inhibidor directo del factor Xa altamente selectivo, con biodisponibilidad oral. La inhibición del factor Xa interrumpe las vías intrínseca y extrínseca de la cascada de la coagulación de la sangre, inhibiendo tanto la formación de trombina como la formación de trombos. Rivaroxaban no inhibe la trombina (factor II activado) y no se han demostrado efectos sobre las plaquetas.
Efectos farmacodinámicos
En los seres humanos se ha observado una inhibición de la actividad del factor Xa dosis-dependiente. Rivaroxaban modifica el tiempo de protrombina (TP) de forma dosis-dependiente con una estrecha correlación con las concentraciones plasmáticas (el valor de r es igual a 0,98) si se emplea Neoplastin para el análisis. Otros reactivos proporcionarían unos resultados diferentes. La lectura del TP debe hacerse en segundos, porque el INR sólo se ha calibrado y validado en el caso de los cumarínicos y no puede utilizarse con ningún otro anticoagulante.
En un estudio de farmacología clínica en la reversión de la acción farmacodinámica de rivaroxaban en adultos sanos (n = 22), se evaluaron los efectos de dosis únicas (50 UI/kg) de dos tipos diferentes de CCP, un CCP de 3 factores (factores II, IX y X) y un CCP de 4 factores (factores II, VII, IX y X). El CCP de 3 factores redujo los valores medios del TP (Neoplastina) en aproximadamente 1,0 segundos a los 30 minutos, en comparación con reducciones de, aproximadamente, 3,5 segundos observadas con el CCP de 4 factores. En cambio, el CCP de 3 factores tuvo un efecto global mayor y más rápido en la reversión de los cambios en la generación de trombina endógena que el CCP de 4 factores (ver sección 4.9).
El tiempo de tromboplastina parcial activada (aPTT) y el HepTest también están prolongados de forma dosis-dependiente; sin embargo, no se recomiendan para evaluar el efecto farmacodinámico de rivaroxaban. No es necesario monitorizar los parámetros de la coagulación durante el tratamiento con rivaroxaban en la práctica clínica. Sin embargo, si está indicado clínicamente, se pueden medir los niveles de rivaroxaban mediante ensayos cuantitativos calibrados para la actividad anti-factor Xa (ver sección 5.2).
Eficacia y seguridad clínica
El programa clínico de rivaroxaban fue diseñado para demostrar su eficacia en la prevención de muerte de origen cardiovascular (CV), infarto de miocardio (IM) o ictus en pacientes con un SCA reciente (infarto de miocardio con elevación del segmento ST [IAMCEST], infarto de miocardio sin elevación del segmento ST [IAMSEST] o angina inestable [AI]). En el ensayo pivotal doble ciego ATLAS ACS 2 TIMI 51, 15.526 pacientes fueron asignados aleatoriamente de forma 1:1:1 a uno de los tres grupos de tratamiento: Xarelto 2,5 mg por vía oral dos veces al día, 5 mg por vía oral dos veces al día o placebo dos veces al día, co-administrados con AAS solo o con AAS más una tienopiridina (clopidogrel o ticlopidina). Los pacientes con SCA de menos de 55 años debían tener diabetes mellitus o haber padecido un IM previo. El valor de la mediana del tiempo de tratamiento fue de 13 meses y la duración total del tratamiento fue de casi 3 años. El 93,2% de los pacientes recibieron tratamiento con AAS de forma concomitante más tratamiento con una tienopiridina y el 6,8% sólo AAS. Entre los pacientes que recibieron doble terapia antiagregante, el 98,8% recibieron clopidogrel, un 0,9% ticlopidina y el 0,3% restante prasugrel. Los pacientes recibieron la primera dosis de Xarelto en un mínimo de 24 horas y hasta 7 días (media 4,7 días) tras ser admitidos en el hospital, pero inmediatamente tras la estabilización del SCA, incluyendo los procedimientos de revascularización y una vez concluido el tratamiento anticoagulante por vía parenteral.
Tanto la pauta posológica de 2,5 mg dos veces al día como la de 5 mg dos veces al día de rivaroxaban fueron eficaces en la reducción adicional de la incidencia de eventos CV sobre el tratamiento antiagregante estándar de base. La pauta posológica de 2,5 mg dos veces al día redujo la mortalidad, y hay evidencia de que con la dosis más baja hubo menor riesgo de hemorragia, por lo que, rivaroxaban 2,5 mg dos veces al día, administrado conjuntamente con ácido acetilsalicílico (AAS) solo o con AAS más clopidogrel o ticlopidina se recomienda para la prevención de eventos aterotrombóticos en pacientes adultos tras un SCA con elevación de los biomarcadores cardíacos.
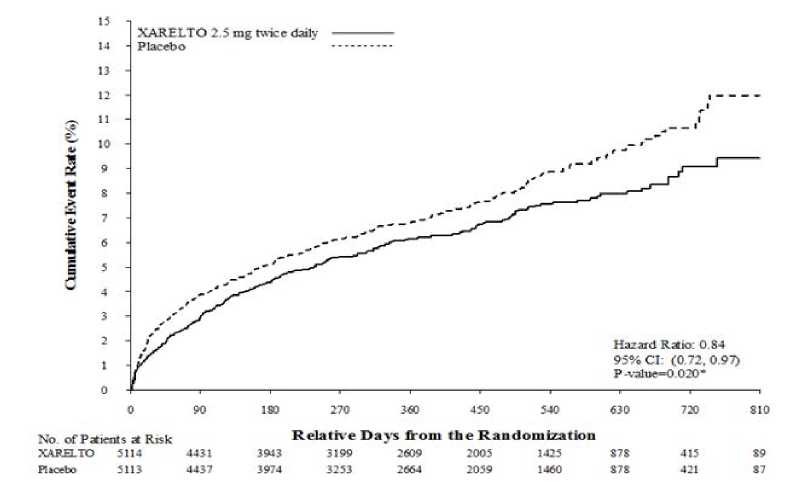
En comparación con placebo, Xarelto redujo significativamente la variable primaria compuesta de muerte de origen cardiovascular, IM o ictus. El beneficio fue debido a la reducción de la muerte de origen CV y MI y se observó inmediatamente con un efecto de tratamiento constante durante todo el período de tratamiento (ver Tabla 3 y Figura 1). También la primera variable secundaria (muerte por cualquier causa, IM o ictus) se redujo significativamente. Un análisis retrospectivo adicional mostró una reducción nominalmente significativa en las tasas de incidencia de trombosis del stent en comparación con placebo (ver Tabla 3). Las tasas de incidencia para la variable principal de seguridad (eventos de hemorragia mayor no-CABG TIMI) fueron superiores en los pacientes tratados con Xarelto en comparación con las de los pacientes que recibieron placebo (ver Tabla 5). Sin embargo, las tasas de incidencia estuvieron equilibradas entre Xarelto y placebo para los componentes de los eventos hemorrágicos fatales, hipotensión que requiere tratamiento con agentes inotrópicos por vía intravenosa e intervención quirúrgica para la hemorragia en curso.
En la Tabla 4 se muestran los resultados de eficacia en pacientes sometidos a una intervención coronaria percutánea (IPC). Los resultados de seguridad de este subgrupo de pacientes sometidos a IPC fueron comparables a los resultados generales de seguridad.
Los pacientes con biomarcadores elevados (troponina o CK-MB) sin antecedente previo de ictus/AIT constituyeron el 80% de la población de estudio. Los resultados en esta población de pacientes también coincidieron con los resultados generales de eficacia y seguridad.
Tabla 3: Resultados de eficacia del estudio de fase III ATLAS ACS 2 TIMI 51
|
Población del estudio |
Pacientes con síndrome coronario agudo reciente a) | |
|
Pauta de tratamiento |
Xarelto 2,5 mg, dos veces al día, N=5.114 n (%) Cociente de riesgos (Hazard Ratio) (95 % CI) valor de p b) |
Placebo N=5.113 n (%) |
|
Muerte de origen cardiovascular, IM o ictus |
313 (6,1 %) 0,84 (0,72, 0,97) p = 0,020* |
376 (7,4 %) |
|
Muerte por todas las causas, IM o ictus |
320 (6,3 %) 0,83 (0,72, 0,97) p = 0,016* |
386 (7,5 %) |
|
Muerte de origen cardiovascular |
94 (1,8 %) 0,66 (0,51, 0,86) p = 0,002** |
143 (2,8 %) |
|
Muerte por todas las causas |
103 (2,0 %) 0,68 (0,53, 0,87) p = 0,002** |
153 (3,0 %) |
|
IM |
205 (4,0 %) 0,90 (0,75, 1,09) p = 0,270 |
229 (4,5 %) |
|
Ictus |
46 (0,9 %) 1,13 (0,74, 1,73) p = 0,562 |
41 (0,8 %) |
|
Trombosis del stent |
61 (1,2 %) 0,70 (0,51, 0,97) p = 0,033** |
87 (1,7 %) |
a) grupo de análisis por intención de tratar modificado (para trombosis del stent)
b) vs. placebo; valor de p Log-Rank * estadísticamente superior
nominalmente significativo
Tabla 4: Resultados de eficacia del estudio de fase III ATLAS ACS 2 TIMI 51 en pacientes sometidos a IPC
|
Población del estudio |
Pacientes con síndrome coronario agudo reciente sometidos a IPC a) | |
|
Pauta de tratamiento |
Xarelto 2,5 mg, dos veces al día, N=3.114 n (%) Cociente de riesgos (Hazard Ratio) (95 % CI) valor de p b) |
Placebo N=3.096 n (%) |
|
Muerte de origen cardiovascular, IM o ictus |
153 (4,9 %) 0,94 (0,75, 1,17) p = 0,572 |
165 (5,3 %) |
|
Muertede origen cardiovascular |
24 (0,8 %) 0,54 (0,33, 0,89) p = 0,013** |
45 (1,5 %) |
|
Muerte por todas las causas |
31 (1,0 %) 0,64 (0,41, 1,01) p = 0,053 |
49 (1,6 %) |
|
IM |
115 (3,7 %) 1,03 (0.79, 1,33) p = 0,829 |
113 (3,6 %) |
|
Ictus |
27 (0.9 %) 1,30 (0.74, 2.31) p = 0.360 |
21 (0,7 %) |
|
Trombosis del stent |
47 (1,5 %) 0,66 (0,46, 0,95) p = 0,026** |
71 (2,3 %) |
a) grupo de análisis por intención de tratar (para trombosis del stent)
b) vs. placebo; valor de p Log-Rank ** nominalmente significativo
Tabla 5: Resultados de seguridad del estudio de fase III ATLAS ACS 2 TIMI 51
|
Población del estudio |
Pacientes con síndrome coronario agudo reciente a) | |
|
Pauta de tratamiento |
Xarelto 2,5 mg, dos veces al día, N=5.115 n (%) Cociente de riesgos (Hazard Ratio) (95 % CI) valor de pb) |
Placebo N=5.125 n (%) |
|
Hemorragia mayor no-CABG TIMI |
65 (1,3 %) 3,46 (2,08, 5,77) p = < 0,001* |
19 (0,4 %) |
|
Hemorragia fatal |
6 (0,1 %) 0,67 (0,24, 1,89) p = 0,450 |
9 (0,2 %) |
|
Hemorragia intracraneal sintomática |
14 (0,3 %) 2,83 (1,02, 7,86) p = 0,037 |
5 (0,1 %) |
|
Hipotensión que requiere tratamiento con agentes inotrópicos por vía intravenosa |
3 (0,1 %) |
3 (0.1 %) |
|
Intervención quirúrgica del sangrado en curso |
7 (0,1 %) |
9 (0,2 %) |
|
Transfusión de 4 o más unidades de sangre durante un periodo de 48 horas |
19 (0,4 %) |
6 (0,1 %) |
|
a) población de seguridad, con tratamiento b) vs. placebo; valor de p Log-Ran * estadísticamente significativo |
k | |
Figura 1: Tiempo hasta la aparición de la primera variable de eficacia primaria (muerte de origen cardiovascular, IM o ictus)
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Xarelto en uno o más grupos de la población pediátrica en el tratamiento de eventos tromboembólicos. La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Xarelto en los diferentes grupos de la población pediátrica en la prevención de eventos tromboembólicos(ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Rivaroxaban se absorbe rápidamente y alcanza concentraciones máximas (Cmax) de 2 a 4 horas después de tomar el comprimido.
La absorción oral de rivaroxaban es casi completa y su biodisponibilidad oral es elevada (80% al 100%) en el caso de la dosis del comprimido de 2,5 mg y de 10 mg, independientemente de las condiciones de ayuno o alimentación. La ingesta de alimentos con rivaroxaban (a la dosis de 2,5 mg o de 10 mg) no afecta al AUC ni a la Cmax. Los comprimidos de 2,5 mg y de 10 mg de rivaroxaban pueden tomarse con o sin alimentos.
Rivaroxaban presenta una farmacocinética lineal hasta, aproximadamente, 15 mg administrados una vez al día. A dosis más altas, rivaroxaban muestra una absorción disminuida, con una reducción de la biodisponibilidad y de la tasa de absorción dosis-dependiente. Este efecto es más marcado en ayunas que después de la ingesta de alimentos.
La variabilidad de la farmacocinética de rivaroxaban es moderada; con una variabilidad interindividual (CV %) entre el 30 y el 40%.
La absorción de rivaroxaban depende del sitio donde se libera en el tracto gastrointestinal. Se ha notificado una disminución del 29% y del 56% en el AUC y la Cmax, en comparación con el comprimido, cuando rivaroxaban en forma de granulado se liberó en el intestino delgado proximal. La exposición se reduce aún más cuando rivaroxaban se libera en el intestino delgado distal o en el colon ascendente. Por lo tanto, debe evitarse la administración de rivaroxaban de forma distal al estómago, ya que esto puede dar lugar a una reducción de la absorción y la correspondiente exposición a rivaroxaban.
La biodisponibilidad (AUC y Cmax) fue comparable para rivaroxaban 20 mg, administrado por vía oral como comprimido triturado y mezclado con puré de manzana o diluido con agua, administrado a través de una sonda gástrica y seguido de una comida líquida, en comparación con el comprimido entero. Dado el perfil farmacocinético predecible, proporcional a la dosis de rivaroxaban, los resultados de biodisponibilidad de este estudio son probablemente aplicables a dosis más bajas de rivaroxaban.
Distribución
La unión a proteínas plasmáticas humanas es alta, del 92 % al 95 % aproximadamente y la albúmina sérica es el principal componente de unión. El volumen de distribución es moderado, con un Vss de 50 litros, aproximadamente.
Biotransformación y eliminación
De la dosis administrada de rivaroxaban se metabolizan aproximadamente 2/3; después, la mitad se elimina por vía renal y la otra mitad por vía fecal. El 1/3 restante de la dosis administrada se excreta directamente por vía renal como principio activo no modificado en la orina, principalmente mediante secreción renal activa.
Rivaroxaban se metaboliza mediante el CYP3A4, el CYP2J2 y mecanismos independientes del CYP. Las principales vías de biotransformación son la degradación oxidativa de la porción de morfolinona y la hidrólisis de los enlaces amida. Según investigaciones in vitro, rivaroxaban es un sustrato de las proteínas transportadoras P-gp (glucoproteína P) y Bcrp (proteína de resistencia al cáncer de mama). Rivaroxaban en forma inalterada es el compuesto más abundante en el plasma humano sin presencia de metabolitos mayores o metabolitos activos circulantes. Con un aclaramiento sistémico de aproximadamente 10 l/h, rivaroxaban puede clasificarse como una sustancia de bajo aclaramiento. Después de la administración por vía intravenosa de una dosis de 1 mg, la semivida de eliminación es de aproximadamente 4,5 horas. Después de la administración por vía oral, la eliminación se ve limitada por la tasa de absorción. En personas jóvenes, la eliminación de rivaroxaban del plasma se produce con una semivida de eliminación de 5 a 9 horas y en personas de edad avanzada, con una semivida de eliminación de 11 a 13 horas.
Poblaciones especiales
Sexo
No hubo ninguna diferencia clínicamente relevante en las propiedades farmacocinéticas y farmacodinámicas entre hombres y mujeres.
Pacientes de edad avanzada
Los pacientes de edad avanzada presentaron concentraciones plasmáticas mayores que los pacientes más jóvenes, con unos valores medios del AUC que fueron aproximadamente 1,5 veces superiores, principalmente debido a la disminución (aparente) del aclaramiento renal y total. No es necesario un ajuste de la dosis.
Peso corporal
Los valores extremos en el peso corporal (< 50 kg ó > 120 kg) tuvieron poco efecto en las concentraciones plasmáticas de rivaroxaban (menos del 25%). No es necesario un ajuste de la dosis.
Origen étnico
No se observaron diferencias interétnicas clínicamente relevantes entre los pacientes de raza blanca, afroamericanos, de origen latinoamericano, japonés o chino, en cuanto a las propiedades farmacocinéticas o farmacodinámicas.
Insuficiencia hepática
Los pacientes cirróticos con insuficiencia hepática leve (clasificados como Child Pugh A), sólo presentaron cambios menores en la farmacocinética de rivaroxaban (aumento medio del AUC de 1,2 veces), lo que fue casi comparable al grupo control de voluntarios sanos. En los pacientes cirróticos con insuficiencia hepática moderada (clasificados como Child Pugh B), el AUC media de rivaroxaban estuvo aumentada significativamente en 2,3 veces, en comparación con los voluntarios sanos. El AUC parcial aumentó 2,6 veces. Estos pacientes también mostraron una disminución de la eliminación renal de rivaroxaban, similar a los pacientes con insuficiencia renal moderada. No hay datos en pacientes con insuficiencia hepática grave.
La inhibición de la actividad del factor Xa se incrementó en un factor de 2,6 en los pacientes con insuficiencia hepática moderada, en comparación con los voluntarios sanos; de manera similar, la prolongación del TP se incrementó en un factor de 2,1. Los pacientes con insuficiencia hepática moderada fueron más sensibles a rivaroxaban, lo que produjo una relación farmacocinética / farmacodinámica más pronunciada entre la concentración y el TP.
Xarelto está contraindicado en pacientes con hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia, incluyendo pacientes cirróticos clasificados como Child Pugh B y C (ver sección 4.3).
Insuficiencia renal
Se observó un aumento de la exposición a rivaroxaban correlacionado con la disminución de la función renal, evaluada mediante las determinaciones del aclaramiento de creatinina. En sujetos con insuficiencia renal leve (aclaramiento de creatinina de 50 - 80 ml/min), moderada (aclaramiento de creatinina de 30 a 49 ml/min) o grave (aclaramiento de creatinina de 15 a 29 ml/min), las concentraciones plasmáticas de rivaroxaban (AUC) aumentaron 1,4, 1,5 y 1,6 veces, respectivamente. Los aumentos correspondientes en los efectos farmacodinámicos fueron más pronunciados. En sujetos con insuficiencia renal leve, moderada y grave, la inhibición total de la actividad del factor Xa aumentó en un factor de 1,5, 1,9 y 2,0 respectivamente, en comparación con voluntarios sanos; de manera similar, la prolongación del TP aumentó en factores de 1,3, 2,2 y 2,4, respectivamente. No hay datos en pacientes con un aclaramiento de creatinina < 15 ml/min.
Debido a la elevada fijación a proteínas plasmáticas, no se espera que rivaroxaban sea dializable.
No se recomienda su uso en pacientes con un aclaramiento de creatinina < 15 ml/min. Xarelto debe utilizarse con precaución en pacientes con un aclaramiento de creatinina de 15 a 29 ml/min (ver sección 4.4).
Datos farmacocinéticos en pacientes
En los pacientes que recibieron rivaroxaban 2,5 mg dos veces al día para la prevención de acontecimientos aterotrombóticos en pacientes con SCA, la concentración media geométrica (intervalo de predicción del 90%) a las 2 - 4 h y a las 12 h aproximadamente después de la dosis (lo que representa aproximadamente las concentraciones máxima y mínima durante el intervalo entre dosis) fue de 47 (13 - 123) y de 9,2 (4,4 - 18) pg/l, respectivamente.
Relación farmacocinética/farmacodinámica
Se ha evaluado la relación farmacocinética/farmacodinámica (PK/PD) entre la concentración plasmática de rivaroxaban y varios criterios de valoración PD (inhibición del factor Xa, tiempo de protrombina (TP, TTPa, Heptest)) después de la administración de un amplio rango de dosis (de 5 a 30 mg dos veces al día). La relación entre la concentración de rivaroxaban y la actividad del factor Xa se describió de manera óptima por un modelo Emax. En el caso del TP, por lo general, el modelo de intersección lineal describió mejor los datos. Dependiendo de los diferentes reactivos usados en el TP, la pendiente varió considerablemente. Con Neoplastin PT, el TP basal fue de aproximadamente 13 seg. y la pendiente fue de alrededor de 3 a 4 seg/(100 ^g/l). Los resultados de los análisis de la relación PK/PD en las fases II y III fueron congruentes con los datos establecidos en los sujetos sanos.
Población pediátrica
No se ha determinado la seguridad y eficacia en niños y adolescentes hasta los 18 años.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad con dosis únicas, fototoxicidad, genotoxicidad, potencial carcinogénico y toxicidad reproductiva.
Los efectos observados en los estudios con dosis repetidas se debieron principalmente a la actividad farmacodinámica incrementada de rivaroxaban. En ratas se observó un aumento de las concentraciones plasmáticas de IgG e IgA a niveles de exposición clínicamente relevantes.
No se observó ningún efecto sobre la fertilidad en las ratas macho o hembra. Los estudios en animales han demostrado una toxicidad reproductiva relacionada con el modo de acción farmacológica de rivaroxaban (p. ej. complicaciones hemorrágicas). A concentraciones plasmáticas clínicamente relevantes se observó toxicidad embriofetal (pérdida después de la implantación, retraso o adelanto de la osificación, varias manchas hepáticas de color claro) y un aumento de la incidencia de malformaciones frecuentes, así como cambios placentarios. En el estudio pre y postnatal en ratas, se observó una disminución de la viabilidad de las crías a dosis que fueron tóxicas para las madres.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido:
Celulosa microcristalina Croscarmelosa sódica Lactosa monohidrato Hipromelosa Laurilsulfato de sodio Estearato de magnesio
Cubierta pelicular:
Macrogol 3350 Hipromelosa
Dióxido de titanio (E 171)
Óxido de hierro amarillo (E 172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Blister de PP / lámina de aluminio, en envases de 14, 28, 30, 56, 60, 98, 168 ó 196 comprimidos recubiertos con película, o blisters precortados unidosis en envases de 10 x 1, 100 x 1 ó en envases múltiples que contienen 100 (10 estuches de 10 x 1) comprimidos recubiertos con película.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
Ninguna especial para su eliminación
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG D-13342 Berlin Alemania
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/025-035
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 30 de septiembre de 2008 Fecha de la última renovación: 22 de mayo de 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
{MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
▼ Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Xarelto 10 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 10 mg de rivaroxaban.
Excipiente con efecto conocido:
Cada comprimido recubierto con película contiene 26,51 mg de lactosa (como monohidrato), ver sección 4.4.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
Comprimidos de color rojo claro, redondos biconvexos (6 mm de diámetro, 9 mm de radio de curvatura), con la cruz de BAYER en una cara, y “10” y un triángulo en la otra cara.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Prevención del tromboembolismo venoso (TEV) en pacientes adultos sometidos a cirugía electiva de reemplazo de cadera o rodilla.
4.2 Posología y forma de administración
Posología
La dosis recomendada es de 10 mg de rivaroxaban, tomado una vez al día. La dosis inicial debe tomarse entre 6 y 10 horas después de la intervención quirúrgica, siempre que se haya establecido la hemostasia.
La duración del tratamiento depende del riesgo individual del paciente de presentar tromboembolismo venoso, que es determinado por el tipo de cirugía ortopédica.
• En los pacientes sometidos a cirugía mayor de cadera, se recomienda una duración de tratamiento de 5 semanas.
• En los pacientes sometidos a cirugía mayor de rodilla, se recomienda una duración de tratamiento de 2 semanas.
Si se olvida una dosis, el paciente debe tomar Xarelto inmediatamente y seguir al día siguiente con la toma una vez al día, como antes.
Cambio de tratamiento con antagonistas de la vitamina K (AVK) a Xarelto
Al cambiar el tratamiento con AVK a Xarelto, los valores de INR (International Normalized Ratio) del paciente estarán falsamente elevados después de la toma de Xarelto. El INR no es un parámetro válido para medir la actividad anticoagulante de Xarelto, por lo que no debe utilizarse (ver sección 4.5).
Cambio de tratamiento con Xarelto a antagonistas de la vitamina K (AVK)
Existe la posibilidad de una incorrecta anticoagulación durante la transición de Xarelto a AVK.
Deberá garantizarse una anticoagulación adecuada y continua durante cualquier transición a un anticoagulante alternativo. Debe señalarse que Xarelto puede contribuir a un aumento del INR.
En los pacientes que cambien de Xarelto a AVK, estos tratamientos deben administrarse simultáneamente hasta que el INR sea > 2,0. Durante los dos primeros días del periodo de cambio se utilizará la dosis inicial estándar de AVK, que se ajustará posteriormente en función de los resultados del INR. Mientras los pacientes están bajo tratamiento con Xarelto y AVK, el INR puede determinarse a partir de las 24 horas que siguen a la dosis de Xarelto y siempre antes de la siguiente dosis. Una vez interrumpido el tratamiento con Xarelto, el INR puede determinarse con fiabilidad pasadas 24 horas de la última dosis (ver secciones 4.5 y 5.2).
Cambio de tratamiento con anticoagulante parenteral a Xarelto
Los pacientes que están recibiendo un anticoagulante por vía parenteral deben interrumpir el tratamiento anticoagulante por vía parenteral e iniciar el tratamiento con Xarelto de 0 a 2 horas antes de la siguiente administración programada del medicamento por vía parenteral (p. ej., heparina de bajo peso molecular). En el caso de un anticoagulante parenteral administrado por perfusión continua (p. ej., heparina no fraccionada intravenosa) Xarelto deberá administrarse en el momento de la suspensión del anticoagulante parenteral.
Cambio de tratamiento con Xarelto a anticoagulante parenteral
La primera dosis de anticoagulante parenteral debe administrarse en el momento en que se tomaría la siguiente dosis de Xarelto.
Poblaciones especiales
Insuficiencia renal
Los escasos datos clínicos sobre los pacientes con insuficiencia renal grave (aclaramiento de creatinina de 15 a 29 ml/min) indican que las concentraciones plasmáticas de rivaroxaban están aumentadas significativamente. Por lo tanto, Xarelto debe usarse con precaución en estos pacientes. No se recomienda su uso en los pacientes con un aclaramiento de creatinina < 15 ml/min (ver secciones 4.4 y 5.2).
No es necesario un ajuste de la dosis en los pacientes con insuficiencia renal leve (aclaramiento de creatinina de 50 a 80 ml/min) o insuficiencia renal moderada (aclaramiento de creatinina de 30 a 49 ml/min) (ver sección 5.2.).
Insuficiencia hepática
Xarelto está contraindicado en los pacientes con hepatopatía asociada a coagulopatía y a riesgo clínicamente relevante de hemorragia incluidos los pacientes cirróticos con Child Pugh B y C (ver secciones 4.3 y 5.2).
Pacientes de edad avanzada
No es necesario ningún ajuste de dosis (ver sección 5.2).
Peso corporal
No es necesario ningún ajuste de dosis (ver sección 5.2).
Sexo
No es necesario ningún ajuste de dosis (ver sección 5.2).
Población pediátrica
No se ha establecido la seguridad y eficacia de Xarelto en niños de 0 a 18 años. No se dispone de datos. Por lo tanto, no se recomienda el uso de Xarelto en niños menores de 18 años.
Forma de administración Vía oral.
Xarelto puede tomarse con o sin alimentos (ver secciones 4.5 y 5.2).
Para aquellos pacientes que no puedan tragar el comprimido entero, el comprimido de Xarelto puede triturarse y mezclarse con agua o con puré de manzana inmediatamente antes de su uso y administrarse por vía oral.
El comprimido triturado también se puede administrar a través de sonda gástrica una vez se haya confirmado la colocación correcta de la sonda. El comprimido triturado se administrará diluido con una pequeña cantidad de agua a través de la sonda gástrica, procediendo seguidamente a un lavado adicional de la sonda con agua (ver sección 5.2).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hemorragia activa, clínicamente significativa.
Lesión o enfermedad, si se considera que tiene un riesgo significativo de hemorragia mayor. Esto puede incluir úlcera gastrointestinal activa o reciente, presencia de neoplasias malignas con alto riesgo de hemorragia, traumatismo cerebral o espinal reciente, cirugía cerebral, espinal u oftálmica reciente, hemorragia intracraneal reciente, conocimiento o sospecha de varices esofágicas, malformaciones arteriovenosas, aneurismas vasculares o anomalías vasculares intraespinales o intracerebrales mayores.
Tratamiento concomitante con cualquier otro anticoagulante, p. ej. heparina no fraccionada (HNF), heparinas de bajo peso molecular (enoxaparina, dalteparina, etc.), derivados de la heparina (fondaparinux, etc.), anticoagulantes orales (warfarina, dabigatran etexilato, apixaban, etc.) excepto bajo las circunstancias concretas de cambio de tratamiento anticoagulante (ver sección 4.2) o cuando se administre HNF a las dosis necesarias para mantener un catéter venoso o arterial central abierto (ver sección 4.5).
Hepatopatía, asociada a coagulopatía y a riesgo clínicamente relevante de hemorragia incluidos los pacientes cirróticos con Child Pugh B y C (ver sección 5.2).
Embarazo y lactancia (ver sección 4.6).
4.4 Advertencias y precauciones especiales de empleo
Riesgo de hemorragia
Varios subgrupos de pacientes, como se explica a continuación, presentan un mayor riesgo de hemorragia. En estos pacientes se debe vigilar cuidadosamente la presencia de signos y síntomas de complicaciones hemorrágicas y anemia después del inicio del tratamiento (ver sección 4.8). Esto puede hacerse mediante exámenes físicos periódicos de los pacientes, una observación estrecha del drenaje de las heridas y determinaciones periódicas de hemoglobina.
Cualquier disminución inexplicada de la hemoglobina o de la presión arterial requerirá la búsqueda de una zona de sangrado.
Aunque durante el tratamiento con rivaroxaban no se necesita una monitorización rutinaria de los parámetros de la coagulación, la determinación de los niveles de rivaroxaban mediante el ensayo antiFactor Xa cuantitativo calibrado puede ser útil en situaciones excepcionales en las que el conocimiento de la exposición a rivaroxaban pueda ayudar en la toma de decisiones desde el punto de vista clínico, como por ejemplo, en caso de sobredosis o cirugía de urgencia (ver secciones 5.1 y 5.2).
Insuficiencia renal
En pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min), las concentraciones plasmáticas de rivaroxaban podrían estar aumentadas significativamente (en promedio, 1,6 veces), lo que conllevaría un aumento del riesgo de hemorragia. Xarelto debe utilizarse con precaución en pacientes con un aclaramiento de creatinina de 15 a 29 ml/min. No se recomienda su uso en pacientes con un aclaramiento de creatinina < 15 ml/min (ver secciones 4.2 y 5.2).
En los pacientes con insuficiencia renal moderada (aclaramiento de creatinina de 30 a 49 ml/min) que reciban concomitantemente otros medicamentos que aumenten las concentraciones plasmáticas de rivaroxaban, Xarelto se debe utilizar con precaución (ver sección 4.5).
Interacción con otros medicamentos
No se recomienda el uso de Xarelto en pacientes que reciben tratamiento sistémico concomitante con antimicóticos azólicos (p. ej., ketoconazol, itraconazol, voriconazol y posaconazol) o inhibidores de la proteasa del VIH (p. ej., ritonavir). Estos principios activos son inhibidores potentes del CYP3A4 y de la P-gp; y pueden, por lo tanto, aumentar las concentraciones plasmáticas de rivaroxaban hasta un grado clínicamente relevante (en promedio, 2,6 veces) que puede llevar a un aumento del riesgo de hemorragia (ver sección 4.5).
Debe tenerse cuidado si los pacientes reciben tratamiento concomitante con medicamentos que afectan a la hemostasia, como los antiinflamatorios no esteroideos (AINEs), ácido acetilsalicílico (AAS) e inhibidores de la agregación plaquetaria. Para los pacientes con riesgo de sufrir una enfermedad gastrointestinal ulcerosa, deberá considerarse un tratamiento profiláctico adecuado (ver sección 4.5).
Otros factores de riesgo hemorrágico
Al igual que otros agentes antitrombóticos, rivaroxaban deberá emplearse con precaución en pacientes con aumento del riesgo de hemorragia, por ejemplo:
• trastornos de la coagulación congénitos o adquiridos
• hipertensión arterial grave y no controlada
• otra enfermedad gastrointestinal sin úlcera activa que pueda producir complicaciones hemorrágicas (por ejemplo, enfermedad inflamatoria intestinal, esofagitis, gastritis o reflujo gastroesofágico)
• retinopatía vascular
• bronquiectasia o antecedentes de hemorragia pulmonar Cirugía de fractura de cadera
No se ha estudiado rivaroxaban en ensayos clínicos intervencionales en pacientes sometidos a cirugía por fractura de cadera para evaluar la eficacia y seguridad.
Anestesia espinal/epidural o punción lumbar
Cuando se aplica anestesia neuraxial (anestesia epidural o espinal) o se realiza una punción lumbar o epidural, los pacientes tratados con antitrombóticos para la prevención de complicaciones tromboembólicas tienen riesgo de presentar un hematoma epidural o espinal, que puede causar parálisis a largo plazo o permanente. El riesgo de estos eventos puede estar aumentado por el empleo postoperatorio de catéteres epidurales permanentes o por la administración concomitante de medicamentos que afectan a la hemostasia. El riesgo también puede aumentar por la punción epidural o espinal traumática o repetida. Se debe controlar con frecuencia la presencia de signos y síntomas de deterioro neurológico (p. ej., adormecimiento o debilidad de extremidades inferiores, disfunción intestinal o vesical). Si se observa compromiso neurológico, será necesario un diagnóstico y tratamiento urgente. Antes de la intervención neuraxial, el médico debe valorar el beneficio potencial frente al riesgo en los pacientes con tratamiento anticoagulante o que van a recibir medicamentos anticoagulantes para la tromboprofilaxis.
Para reducir el riesgo potencial de sangrado asociado con el uso concomitante de rivaroxaban y anestesia neuraxial (epidural/espinal) o punción espinal se debe considerar el perfil farmacocinético de rivaroxaban. La colocación o extracción de un catéter epidural o punción lumbar se realiza mejor cuando se estima que el efecto anticoagulante de rivaroxaban es bajo (ver sección 5.2).
Para retirar un catéter epidural deben haber transcurrido al menos 18 horas desde la última administración de rivaroxaban. Una vez retirado el catéter, deben transcurrir al menos 6 horas para poder administrar la siguiente dosis de rivaroxaban.
Si se produce una punción traumática, la administración de rivaroxaban se deberá retrasar 24 horas.
Recomendaciones posológicas antes y después de procedimientos invasivos y de intervenciones quirúrgicas
Si es necesario realizar un procedimiento invasivo o una intervención quirúrgica, se interrumpirá la administración de Xarelto 10 mg por lo menos 24 horas antes de la intervención, si es posible y basándose en el criterio clínico del médico.
Si la intervención no puede retrasarse, debe evaluarse el aumento del riesgo de hemorragia frente a la urgencia de la intervención.
Se debe reiniciar lo antes posible la administración de Xarelto después del procedimiento invasivo o intervención quirúrgica, siempre que la situación clínica lo permita y se haya establecido una hemostasia adecuada, una vez confirmado por el médico que trata al paciente (ver sección 5.2).
Pacientes de edad avanzada
La edad avanzada puede aumentar el riesgo de hemorragia (ver sección 5.2).
Información acerca de los excipientes
Xarelto contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp o malabsorción de glucosa o galactosa no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Inhibidores del CYP3A4 y de la P-gp
La administración concomitante de rivaroxaban con ketoconazol (400 mg una vez al día) o ritonavir (600 mg dos veces al día) produjo un aumento de 2,6 veces / 2,5 veces del AUC media de rivaroxaban, y un aumento de 1,7 veces / 1,6 veces de la Cmax media de rivaroxaban, con aumentos significativos de los efectos farmacodinámicos, lo que puede aumentar del riesgo de hemorragia. Por lo tanto, no se recomienda el uso de Xarelto en los pacientes que reciban tratamiento sistémico concomitante con antimicóticos azólicos como ketoconazol, itraconazol, voriconazol y posaconazol o con inhibidores de la proteasa del VIH. Estos principios activos son inhibidores potentes del CYP3A4 y de la P-gp (ver sección 4.4).
Las sustancias activas que inhiben intensamente sólo una de las vías de eliminación de rivaroxaban, el CYP3A4 o la P-gp, pueden aumentar las concentraciones plasmáticas de rivaroxaban en menor grado. La claritromicina (500 mg dos veces al día), por ejemplo, considerada un potente inhibidor del CYP3A4 y un inhibidor moderado de la P-gp, produjo un aumento de 1,5 veces del AUC media de rivaroxaban y un aumento de 1,4 veces de la Cmax. Este aumento no se considera clínicamente relevante. (Pacientes con insuficiencia renal: ver sección 4.4).
La eritromicina (500 mg tres veces al día), que inhibe moderadamente el CYP3A4 y la P-gp, produjo un aumento de 1,3 veces de la AUC y la Cmax medias de rivaroxaban. Este aumento no se considera clínicamente relevante.
En sujetos con insuficiencia renal leve, la eritromicina (500 mg tres veces al día) produjo un aumento de 1,8 veces el AUC media de rivaroxaban y de 1,6 veces en la Cmax, comparado con sujetos con la función renal normal. En sujetos con insuficiencia renal moderada, la eritromicina produjo un aumento de 2.0 veces en el AUC media de rivaroxaban y 1,6 veces en la Cmax, comparado con sujetos con la función renal normal. El efecto de la eritromicina es aditivo al de la insuficiencia renal (ver sección
4.4) .
El fluconazol (400 mg una vez al día), considerado un inhibidor moderado del CYP3A4, produjo un aumento de 1,4 veces del AUC media de rivaroxaban y un aumento de 1,3 veces de la Cmáx media.
Este aumento no se consideró clínicamente relevante. (Pacientes con insuficiencia renal: ver sección
4.4) .
Dada la limitada información clínica disponible con dronedarona, debería evitarse la administración concomitante con rivaroxaban.
Anticoagulantes
Después de la administración combinada de enoxaparina (dosis única de 40 mg) con rivaroxaban (dosis única de 10 mg) se observó un efecto aditivo sobre la actividad anti-factor Xa, sin efectos adicionales en las pruebas de coagulación (TP, TTPa). La enoxaparina no afectó a las propiedades farmacocinéticas de rivaroxaban.
Debido al aumento del riesgo de hemorragia, se debe tener precaución si los pacientes reciben tratamiento concomitante con cualquier otro anticoagulante (ver secciones 4.3 y 4.4).
AINEs e inhibidores de la agregación plaquetaria
No se observó una prolongación del tiempo de sangrado clínicamente relevante después de la administración concomitante de rivaroxaban (15 mg) y 500 mg de naproxeno. No obstante, algunas personas pueden tener una respuesta farmacodinámica más pronunciada.
No se observó ninguna interacción farmacocinética ni farmacodinámica clínicamente significativa cuando se administró rivaroxaban concomitantemente con 500 mg de ácido acetilsalicílico.
El clopidogrel (dosis de carga de 300 mg, seguida de una dosis de mantenimiento de 75 mg) no mostró ninguna interacción farmacocinética con rivaroxaban (15 mg); sin embargo, se observó un aumento del tiempo de sangrado en un subgrupo de pacientes, que no se correlacionó con la agregación plaquetaria, las concentraciones de P-selectina o los receptores GPIIb/IIIa.
Se debe tener precaución si los pacientes reciben tratamiento concomitante con AINEs (incluyendo ácido acetilsalicílico) e inhibidores de la agregación plaquetaria, porque estos medicamentos aumentan, de por sí, el riesgo de hemorragia (ver sección 4.4).
Warfarina
Los cambios de tratamiento con warfarina (INR de 2,0 a 3,0), un antagonista de la vitamina K, a rivaroxaban (20 mg) o de rivaroxaban (20 mg) a warfarina (INR de 2,0 a 3,0) aumentaron el tiempo de protrombina/INR (Neoplastin) de forma importante (pueden observarse valores individuales del INR de hasta 12), mientras que los efectos sobre el TTPa, la inhibición de la actividad del factor Xa y el potencial de trombina endógena (PTE) fueron aditivos.
Si se desea medir los efectos farmacodinámicos de rivaroxaban durante el periodo de cambio de tratamiento, puede utilizarse la actividad anti-factor Xa, PiCT y Heptest, ya que la warfarina no afecta a estas pruebas. Al cuarto día tras la última dosis de warfarina, todas las pruebas (incluyendo TP, TTPa, inhibición de la actividad del factor Xa y PTE) reflejaron únicamente el efecto de rivaroxaban. Si se desea medir los efectos farmacodinámicos de warfarina durante el periodo de cambio de tratamiento, se puede usar la determinación del INR en la Ctrough de rivaroxaban (24 horas después de su anterior administración), ya que rivaroxaban afecta mínimamente a esta prueba en este punto.
No se observó ninguna interacción farmacocinética entre warfarina y rivaroxaban.
Inductores del CYP3A4
La administración concomitante de rivaroxaban con rifampicina, un potente inductor del CYP3A4, produjo una disminución aproximada del 50% del AUC media de rivaroxaban, con disminuciones paralelas de sus efectos farmacodinámicos. El uso concomitante de rivaroxaban con otros inductores potentes del CYP3A4 (por ejemplo, fenitoína, carbamazepina, fenobarbital o la hierba de San Juan (Hipericum perforatum)) también puede causar una disminución de la concentración plasmática de rivaroxaban. Por tanto, la administración concomitante con inductores potentes del CYP3A4 deberá evitarse a menos que el paciente esté estrechamente monitorizado para detectar signos o síntomas de trombosis.
Otros tratamientos concomitantes
No se observó ninguna interacción farmacocinética o farmacodinámica clínicamente significativa cuando se administró rivaroxaban concomitantemente con midazolam (sustrato del CYP3A4), digoxina (sustrato de la P-gp), atorvastatina (sustrato del CYP3A4 y de la P-gp) u omeprazol (inhibidor de la bomba de protones). Rivaroxaban no inhibe ni induce ninguna isoforma mayor del CYP, como el CYP3A4.
No se observó ninguna interacción clínicamente relevante con la toma de alimentos (ver sección 4.2).
Parámetros de laboratorio
Los parámetros de la coagulación (p. ej., TP, TTPa, HepTest) se ven afectados de la forma esperada debido al mecanismo de acción de rivaroxaban (ver sección 5.1).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No se ha evaluado la seguridad y eficacia de Xarelto en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3). Debido a la posible toxicidad reproductiva, riesgo intrínseco de hemorragia y la evidencia de que rivaroxaban atraviesa la barrera placentaria, Xarelto está contraindicado durante el embarazo (ver sección 4.3).
Las mujeres en edad fértil deben evitar quedarse embarazadas durante el tratamiento con rivaroxaban.
Lactancia
No se ha evaluado la seguridad y eficacia de Xarelto en mujeres en período de lactancia. Los datos en animales indican que rivaroxaban se excreta en la leche materna. Por lo tanto, Xarelto está contraindicado durante la lactancia (ver sección 4.3). Se debe decidir si es necesario interrumpir la lactancia o bien interrumpir/suspender el tratamiento.
Fertilidad
No se han realizado estudios específicos con rivaroxaban para evaluar los efectos sobre la fertilidad en humanos. En un estudio sobre la fertilidad en ratas macho y hembra no se observó ningún efecto (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Xarelto puede influir ligeramente en la capacidad para conducir y utilizar máquinas. Se han descrito reacciones adversas como síncope (frecuencia: poco frecuente) y mareos (frecuencia; frecuente) (ver sección 4.8). Los pacientes que sufran estas reacciones adversas no deben conducir ni utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Se ha evaluado la seguridad de rivaroxaban en once ensayos clínicos de fase III, que incluyeron 32.625 pacientes expuestos a rivaroxaban (ver la tabla 1).
Tabla 1: Número de pacientes estudiados, dosis máxima diaria y duración del tratamiento en los estudios de fase III.
|
Indicación |
Número de pacientes * |
Dosis máxima diaria |
Duración máxima del tratamiento |
|
Prevención de tromboembolismo venoso (TEV) en pacientes adultos sometidos a cirugía electiva de reemplazo de cadera o rodilla |
6.097 |
10 mg |
39 días |
|
Prevención del tromboembolismo venoso en pacientes encamados |
3.997 |
10 mg |
39 días |
|
Tratamiento de TVP, EP y prevención de sus recurrencias |
4.556 |
Días 1 a 21: 30 mg Día 22 en adelante: 20 mg |
21 meses |
|
Prevención del ictus y de la embolia sistémica en pacientes con fibrilación auricular no valvular |
7.750 |
20 mg |
41 meses |
|
Indicación |
Número de pacientes * |
Dosis máxima diaria |
Duración máxima del tratamiento |
|
Prevención de acontecimientos aterotrombóticos en pacientes que han sufrido un SCA |
10.225 |
5 mg ó 10 mg respectivamente, co-administrada con AAS o bien con AAS más clopidogrel o ticlopidina |
31 meses |
*Pacientes expuestos por lo menos a una dosis de rivaroxaban.
Las reacciones adversas notificadas con mayor frecuencia en los pacientes que recibieron rivaroxaban fueron hemorragias (ver sección 4.4. y “Descripción de las reacciones adversas seleccionadas” más adelante). Las hemorragias notificadas con mayor frecuencia (> 4%) fueron epistaxis (5,9%) y la hemorragia del tracto gastrointestinal (4,2%).
En total, se notificó la aparición de eventos adversos en aproximadamente un 67% de los pacientes expuestos por lo menos a una dosis de rivaroxaban. Aproximadamente el 22% de los pacientes sufrieron eventos adversos que se consideran relacionados con el tratamiento, según la evaluación de los investigadores. En los pacientes tratados con Xarelto 10 mg sometidos a cirugía electiva de reemplazo de cadera o rodilla y en pacientes hospitalizados y encamados, se produjeron episodios hemorrágicos en aproximadamente 6,8% y 12,6% de los pacientes, respectivamente, y se produjo anemia en aproximadamente un 5,9% y 2,1% de los pacientes, respectivamente. En los pacientes tratados con Xarelto 15 mg dos veces al día y después Xarelto 20 mg una vez al día para el tratamiento de la TVP o EP, o con Xarelto 20 mg una vez al día para la prevención de la recurrencia de la TVP y de la EP, se produjeron episodios hemorrágicos en aproximadamente un 27,8% de los pacientes, y anemia en aproximadamente un 2,2% de los pacientes. En los pacientes tratados para la prevención del ictus y de la embolia sistémica, se notificó hemorragia de cualquier tipo o gravedad, con una tasa de 28 por cada 100 pacientes-años, y anemia con una tasa de 2,5 por cada 100 pacientes-años. En los pacientes tratados para la prevención de eventos aterotrombóticos tras un síndrome coronario agudo (SCA), se notificó hemorragia de cualquier tipo o gravedad con una tasa de 22 eventos por cada 100 pacientes-años. La anemia se notificó con una tasa de eventos de 1,4 por 100 pacientes-años.
Tabla de reacciones adversas
Las frecuencias de las reacciones adversas notificadas con Xarelto, se resumen en la tabla 2 según la clasificación de órganos y sistemas (convención MedDRA) y según las frecuencias.
Las frecuencias se definen como: muy frecuentes (> 1/10) frecuentes (> 1/100 a < 1/10) poco frecuentes (> 1/1.000 a < 1/100) raras: (> 1/10.000 a < 1/1.000) muy raras ( < 1/10.000)
no conocida (no puede calcularse a partir de los datos disponibles)
Tabla 2: Todas las reacciones adversas observadas con el tratamiento y notificadas en los estudios de fase III
|
Frecuentes |
Poco frecuentes |
Raras |
No conocida |
|
Trastornos de la sangre y del sistema linfático | |||
|
Anemia (incl. respectivos parámetros de laboratorio) |
Trombocitemia (incl. aumento del recuento de plaquetas)A | ||
|
Frecuentes |
Poco frecuentes |
Raras |
No conocida |
|
Trastornos del sistema inmunológico | |||
|
Reacción alérgica, dermatitis alérgica | |||
|
Trastornos del sistema nervioso | |||
|
Mareos, cefalea, |
Hemorragia cerebral e intracraneal, síncope | ||
|
Trastornos oculares | |||
|
Hemorragia ocular (incl. hemorragia conjuntival) | |||
|
Trastornos cardiacos | |||
|
Taquicardia | |||
|
Trastornos vasculares | |||
|
Hipotensión, hematoma | |||
|
Trastornos respiratorios, torácicos y mediastínicos | |||
|
Epistaxis, hemoptisis | |||
|
Trastornos gastrointestinales | |||
|
Sangrado gingival, hemorragia del tracto gastrointestinal (incl. hemorragia rectal), dolor gastrointestinal y abdominal, dispepsia, náuseas, estreñimientoA, diarrea, vómitosA |
Sequedad de boca | ||
|
Trastornos hepatobiliares | |||
|
Alteración de la función hepática |
Ictericia | ||
|
Trastornos de la piel y del tejido subcutáneo | |||
|
Prurito (incl. casos raros de prurito generalizado), exantema, equimosis, hemorragia cutánea y subcutánea |
Urticaria | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |||
|
Dolor en las extremidadesA |
Hemartrosis |
Hemorragia muscular |
Síndrome compartimental secundario a una hemorragia |
|
Frecuentes |
Poco frecuentes |
Raras |
No conocida |
|
Trastornos renales y urinarios | |||
|
Hemorragia del tracto urogenital (incl. hematuria y menorragiaB), insuficiencia renal (incl. aumento de creatinina en sangre, aumento de la urea en sangre)A |
Insuficiencia renal /insuficiencia renal aguda secundaria a una hemorragia suficiente para causar hipoperfusión | ||
|
Trastornos generales y alteraciones en el lugar de administración | |||
|
FiebreA, edema periférico, disminución general de la fuerza y la energía (incl. fatiga y astenia) |
Sensación de malestar (incl. malestar general), |
Edema localizadoA | |
|
Exploraciones complementarias | |||
|
Aumento de las transaminasas |
Aumento de la bilirrubina, aumento de la fosfatasa alcalina sanguíneaA, aumento de la LDHA, aumento de la lipasaA, aumento de la amilasaA, aumento de la GGTA |
Aumento de la bilirrubina conjugada (con o sin aumento concomitante de la ALT). | |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos | |||
|
Hemorragia después de una intervención (incl. anemia postoperatoria y hemorragia de la herida, contusión), secreción de la heridaA |
Pseudoaneurisma vascularC | ||
A: observado en la prevención del tromboembolismo venoso (TEV) en pacientes adultos sometidos a cirugía electiva de reemplazo de cadera o rodilla.
B: observado en el tratamiento de la TVP, EP y prevención de sus recurrencias como muy frecuente en mujeres < 55 años.
C: observado como poco frecuentes en la prevención de eventos aterotrombóticos en pacientes que han sufrido un SCA (tras una intervención coronaria percutánea).
Descripción de las reacciones adversas seleccionadas
Debido a su mecanismo de acción farmacológica, el uso de Xarelto puede asociarse a un incremento del riesgo de hemorragia oculta o manifiesta en cualquier tejido u órgano que puede dar lugar a una anemia post-hemorrágica. Los signos, síntomas y gravedad (incluido un desenlace mortal) variarán según la localización y el grado o la extensión de la hemorragia, la anemia o ambas (ver sección 4.9 Tratamiento de la hemorragia). En los ensayos clínicos se observaron con más frecuencia hemorragias a nivel de mucosas (p. ej. epistaxis, gingival, gastrointestinal, genito-urinaria) y anemia en los pacientes que recibían rivaroxaban a largo plazo con respecto a los que recibían tratamiento con AVK. Por ello, además de un adecuado seguimiento clínico, las determinaciones de hemoglobina y hematocrito podrían ser útiles para detectar hemorragias ocultas cuando se considere apropiado. El riesgo de hemorragia puede estar aumentado en ciertos grupos de pacientes, como por ejemplo, en pacientes con hipertensión arterial grave no controlada y/o en tratamiento concomitante que afecte a la hemostasia (ver Riesgo de hemorragia en sección 4.4). El sangrado menstrual puede ser más intenso y/o prolongarse. Las complicaciones hemorrágicas pueden presentarse como debilidad, palidez, mareos, cefalea o tumefacción inexplicada, disnea o shock de causa desconocida. En algunos casos, a consecuencia de la anemia, se han observado síntomas de isquemia cardíaca, como dolor torácico o angina de pecho.
Se han notificado complicaciones conocidas, secundarias a hemorragia, como el síndrome compartimental o insuficiencia renal debida a la hipoperfusión. Por lo tanto, se debe tener en cuenta la posibilidad de hemorragia al evaluar el estado de cualquier paciente anticoagulado.
Observaciones post-comercialización
Tras la comercialización se han notificado las siguientes reacciones adversas en asociación temporal con el uso de Xarelto. No se ha podido estimar la frecuencia de estas reacciones adversas.
Trastornos del sistema inmunológico: angioedema y edema alérgico(En los ensayos de fase III agrupados, estos eventos fueron poco frecuentes (> 1/1.000 a <1/100)).
Trastornos hepatobiliares: colestasis, hepatitis (incluyendo lesión hepatocelular). (En los ensayos de fase III agrupados, estos eventos fueron raros (> 1/10.000 a < 1/1.000)).
Trastornos de la sangre y del sistema linfático: trombocitopenia. (En los ensayos de fase III agrupados, estos eventos fueron poco frecuentes (> 1/1.000 a < 1/100)).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se han notificado casos raros de sobredosis de hasta 600 mg sin complicaciones hemorrágicas u otras reacciones adversas. Debido a la escasa absorción a dosis supraterapéuticas de 50 mg de rivaroxaban o superiores, se espera un efecto techo sin un aumento posterior de la exposición plasmática media.
No se dispone de un antídoto específico que antagonice el efecto farmacodinámico de rivaroxaban.
Se puede considerar el uso de carbono activado para reducir la absorción en caso de sobredosis por rivaroxaban.
Tratamiento de la hemorragia
En caso de producirse una complicación hemorrágica en un paciente que recibe tratamiento con rivaroxaban, se deberá retrasar la siguiente administración de rivaroxaban o interrumpir el tratamiento si se considera conveniente. Rivaroxaban tiene una semivida de eliminación de entre 5 y 13 horas (ver sección 5.2). Las medidas terapéuticas deben individualizarse según la gravedad y la localización de la hemorragia. En caso necesario, podría aplicarse el tratamiento sintomático adecuado, como la compresión mecánica (por ejemplo en caso de epistaxis grave), hemostasia quirúrgica con procedimientos de control de la hemorragia, reemplazo de fluidos y apoyo hemodinámico (concentrado de hematíes o plasma fresco congelado, dependiendo de la anemia o la coagulopatía asociadas) o plaquetas.
Si la hemorragia no se puede controlar con las medidas anteriores, debería plantearse la administración de un agente procoagulante específico para revertir el efecto, como el concentrado de complejo de protrombina (CCP), concentrado de complejo de protrombina activado (CCPA) o factor VIIa recombinante (r-FVIIa). Sin embargo, actualmente hay una experiencia clínica muy limitada con el uso de estos productos en pacientes que reciben rivaroxaban. La recomendación se basa también en datos no clínicos limitados. Deberá plantearse la readministración de factor VIIa recombinante y ajustar la dosis dependiendo de la mejoría de la hemorragia. Dependiendo de la disponibilidad local, en caso de hemorragia mayor debe considerarse consultar a un experto en coagulación (ver sección 5.1).
No se espera que el sulfato de protamina y la vitamina K afecten a la actividad anticoagulante de rivaroxaban. La experiencia con ácido tranexámico es limitada y no hay experiencia con ácido
aminocaproico y aprotinina en pacientes tratados con rivaroxaban. No hay una justificación científica sobre la ventaja ni experiencia con el hemostático sistémico desmopresina en pacientes tratados con rivaroxaban. Debido a su elevada fijación a las proteínas plasmáticas, no se espera que rivaroxaban sea dializable.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inhibidores directos del factor Xa, código ATC: B01AF01 Mecanismo de acción
Rivaroxaban es un inhibidor directo del factor Xa, altamente selectivo, con biodisponibilidad oral. La inhibición del factor Xa interrumpe las vías intrínseca y extrínseca de la cascada de la coagulación de la sangre, inhibiendo tanto la formación de trombina como la formación de trombos. Rivaroxaban no inhibe la trombina (factor II activado) y no se han demostrado efectos sobre las plaquetas.
Efectos farmacodinámicos
En los seres humanos se ha observado una inhibición de la actividad del factor Xa dosis-dependiente. Rivaroxaban modifica el tiempo de protrombina (TP) de forma dosis-dependiente, con una estrecha correlación con las concentraciones plasmáticas (el valor de r es igual a 0,98), si se emplea Neoplastin para el análisis. Otros reactivos proporcionarían unos resultados diferentes. La lectura del TP debe hacerse en segundos, porque el INR sólo se ha calibrado y validado en el caso de los cumarínicos y no puede utilizarse con ningún otro anticoagulante. En los pacientes sometidos a una intervención de cirugía ortopédica mayor, los percentiles 5/95 del TP (Neoplastin), de 2 a 4 horas después de tomar el comprimido (es decir, en el momento del efecto máximo), variaron entre 13 y 25 seg (valores basales antes de la intervención 12 a 15 seg.).
En un estudio de farmacología clínica en la reversión de la acción farmacodinámica de rivaroxaban en adultos sanos (n = 22), se evaluaron los efectos de dosis únicas (50 UI/kg) de dos tipos diferentes de CCP, un CCP de 3 factores (factores II, IX y X) y un CCP de 4 factores (factores II, VII, IX y X). El CCP de 3 factores redujo los valores medios del TP (Neoplastina) en aproximadamente 1,0 segundos a los 30 minutos, en comparación con reducciones de, aproximadamente, 3,5 segundos observadas con el CCP de 4 factores. En cambio, el CCP de 3 factores tuvo un efecto global mayor y más rápido en la reversión de los cambios en la generación de trombina endógena que el CCP de 4 factores (ver sección 4.9).
El tiempo de tromboplastina parcial activada (TTPa) y el HepTest también están prolongados de forma dosis-dependiente; sin embargo, no se recomiendan para evaluar el efecto farmacodinámico del rivaroxaban. No es necesario monitorizar los parámetros de la coagulación durante el tratamiento con rivaroxaban en la práctica clínica. Sin embargo, si está indicado clínicamente, se pueden medir los niveles de rivaroxaban mediante ensayos cuantitativos calibrados para la actividad anti-factor Xa (ver sección 5.2).
Eficacia clínica y seguridad
El programa clínico de rivaroxaban fue diseñado para demostrar su eficacia en la prevención de los eventos del tromboembolismo venoso (TEV), es decir, trombosis venosa profunda proximal y distal (TVP), y embolia pulmonar (EP), en pacientes sometidos a cirugía ortopédica mayor de las extremidades inferiores. En ensayos clínicos de fase III, controlados y aleatorizados, el programa RECORD estudió a más de 9.500 pacientes (7.050 con cirugía de reemplazo total de cadera y 2.531 con reemplazo total de rodilla).
El tratamiento con rivaroxaban, a una dosis de 10 mg una vez al día, iniciado no antes de 6 horas después de la intervención quirúrgica, se comparó con enoxaparina, a una dosis de 40 mg una vez al día, administrada 12 horas antes de la intervención.
En los tres ensayos clínicos de fase III (ver tabla 3), rivaroxaban redujo significativamente la tasa de tromboembolismo venoso total (cualquier TVP detectada mediante flebografía o sintomática, EP no mortal y muerte) y la tasa de TEV (TVP, EP no mortal y muerte relacionada con TEV), es decir, las variables principales y secundarias mayores pre-especificadas en la valoración de la eficacia. Además,
en los tres ensayos clínicos, la tasa de TEV sintomático (TVP sintomática, EP no mortal, muerte relacionada con TEV) fue más baja en los pacientes tratados con rivaroxaban, en comparación con los pacientes tratados con enoxaparina.
La variable principal de seguridad, la hemorragia mayor, mostró tasas comparables en los pacientes tratados con 10 mg de rivaroxaban, en comparación con 40 mg de enoxaparina.
Tabla 3: Resultados de eficacia y seguridad de los estudios clínicos de fase III.
|
RECORD 1 |
RECORD 2 |
RECORD 3 | |||
|
Población del estudio |
4.541 pacientes sometidos a cirugía de reemplazo total de cadera |
2.509 pacientes sometidos a cirugía de reemplazo total de cadera |
2.531 pacientes sometidos a cirugía de reemplazo total de rodilla | ||
|
Posología y duración del tratamiento después de la intervención |
Rivaroxaban Enoxaparina P 10 mg una 40 mg una vez al día vez al día 35 ± 4 días 35 ± 4 días |
Rivaroxaban Enoxaparina p 10 mg una 40 mg una vez al día vez al día 35 ± 4 días 12 ± 2 días |
Rivaroxaban 10 mg una vez al día 12 ± 2 días |
Enoxaparina 40 mg una vez al día 12 ± 2 días |
p |
|
TEV Total |
18 (1,1%) 58 (3,7%) < 0,001 |
17 (2,0%) 81 (9,3%) < 0,001 |
79 (9,6%) |
166 (18,9%) |
< 0,001 |
|
TEV mayor |
4 (0,2%) 33 (2,0%) < 0,001 |
6 (0,6%) 49 (5,1%) < 0,001 |
9 (1,0%) |
24 (2,6%) |
0,01 |
|
TEV sintomático |
6 (0,4%) 11 (0,7%) |
3 (0,4%) 15 (1,7 %) |
8 (1,0%) |
24 (2,7%) | |
|
Hemorragias mayores |
6 (0,3%) 2 (0,1%) |
1 (0,1%) 1 (0,1%) |
7 (0,6%) |
6 (0,5%) | |
El análisis de los resultados agrupados de los ensayos clínicos de fase III corroboró los datos obtenidos en los estudios individuales en cuanto a la reducción del TEV total, TEV mayor y TEV sintomático con 10 mg de rivaroxaban, una vez al día, en comparación con 40 mg de enoxaparina, una vez al día.
Además del programa de estudios fase III RECORD, se llevó a cabo un estudio de cohortes, postautorización, de no intervención, abierto (estudio XAMOS) que incluyó 17.413 pacientes sometidos a cirugía ortopédica mayor de cadera o rodilla y en el que se comparó rivaroxaban con el tratamiento tromboprofiláctico estándar, en condiciones de práctica clínica habitual. Se observó TEV sintomático en 57 (0,6%) pacientes del grupo rivaroxaban (n=8.778) y en 88 (1,0%) pacientes del grupo de tratamiento estándar (n=8.635; HR 0,63; IC 95% 0,43-0,91 en la población de seguridad). En cuanto a la hemorragia mayor, se observó en 35 (0,4%) y 29 (0,3%) pacientes en el grupo rivaroxaban y del tratamiento estándar, respectivamente (HR 1,10; IC 95% 0,67-1,80). Estos resultados fueron coherentes con los obtenidos de los ensayos clínicos pivotales aleatorizados.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Xarelto en uno o más grupos de la población pediátrica en el tratamiento de eventos tromboembólicos. La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Xarelto en los diferentes grupos de la población pediátrica en la prevención de eventos tromboembólicos (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Rivaroxaban se absorbe rápidamente y alcanza concentraciones máximas (Cmax) de 2 a 4 horas después de tomar el comprimido.
La absorción oral de rivaroxaban es casi completa y su biodisponibilidad oral es elevada (80% al 100%) en el caso de la dosis del comprimido de 2,5 mg y de 10 mg, independientemente de las condiciones de ayuno o alimentación. La ingesta de alimentos con rivaroxaban (a la dosis de 2,5 mg o de 10 mg) no afecta al AUC ni a la Cmax. Los comprimidos de 2,5 mg y de 10 mg de rivaroxaban pueden tomarse con o sin alimentos.
Rivaroxaban presenta una farmacocinética lineal hasta, aproximadamente, 15 mg administrados una vez al día. A dosis más altas, rivaroxaban muestra una absorción disminuida, con una reducción de la biodisponibilidad y de la tasa de absorción dosis-dependiente. Este efecto es más marcado en ayunas que después de la ingesta de alimentos.
La variabilidad de la farmacocinética de rivaroxaban es moderada; con una variabilidad interindividual (CV %) entre el 30 y el 40%, excepto el día de la intervención quirúrgica y el siguiente día, cuando la variabilidad de la exposición es alta (70%).
La absorción de rivaroxaban depende del sitio donde se libera en el tracto gastrointestinal. Se ha notificado una disminución del 29% y del 56% en el AUC y la Cmax, en comparación con el comprimido, cuando rivaroxaban en forma de granulado se liberó en el intestino delgado proximal. La exposición se reduce aún más cuando rivaroxaban se libera en el intestino delgado distal o en el colon ascendente. Por lo tanto, debe evitarse la administración de rivaroxaban de forma distal al estómago, ya que esto puede dar lugar a una reducción de la absorción y la correspondiente exposición a rivaroxaban.
La biodisponibilidad (AUC y Cmax) fue comparable para rivaroxaban 20 mg, administrado por vía oral como comprimido triturado y mezclado con puré de manzana o diluido con agua, administrado a través de una sonda gástrica y seguido de una comida líquida, en comparación con el comprimido entero. Dado el perfil farmacocinético predecible, proporcional a la dosis de rivaroxaban, los resultados de biodisponibilidad de este estudio son probablemente aplicables a dosis más bajas de rivaroxaban.
Distribución
La unión a las proteínas plasmáticas humanas es alta, del 92% al 95%, aproximadamente, y la albúmina sérica es el principal componente de unión. El volumen de distribución es moderado, con un Vss de 50 litros, aproximadamente.
Biotransformación y eliminación
De la dosis administrada de rivaroxaban, se metaboliza aproximadamente 2/3; después, la mitad se elimina por la vía renal y la otra mitad por vía fecal. El 1/3 restante de la dosis administrada se excreta directamente como principio activo no modificado en la orina, principalmente mediante secreción renal activa.
Rivaroxaban se metaboliza mediante el CYP3A4, el CYP2J2 y mecanismos independientes del CYP. Las principales vías de biotransformación son la degradación oxidativa de la porción de morfolinona y la hidrólisis de los enlaces amida. Según investigaciones in vitro, rivaroxaban es un sustrato de las proteínas transportadoras P-gp (glucoproteína P) y Bcrp (proteína de resistencia al cáncer de mama). Rivaroxaban en forma inalterada es el compuesto más abundante en el plasma humano, sin presencia de metabolitos mayores o metabolitos activos circulantes. Con un aclaramiento sistémico de aproximadamente 10 l/h, rivaroxaban puede clasificarse como una sustancia de bajo aclaramiento. Después de la administración por vía intravenosa de una dosis de 1 mg, la semivida de eliminación es de aproximadamente 4,5 horas. Después de la administración por vía oral, la eliminación se ve limitada por la tasa de absorción. En personas jóvenes, la eliminación de rivaroxaban del plasma se produce con una semivida de eliminación de 5 a 9 horas y en personas de edad avanzada, con una semivida de eliminación de 11 a 13 horas.
Poblaciones especiales
Sexo
No hubo ninguna diferencia clínicamente relevante en las propiedades farmacocinéticas y farmacodinámicas entre pacientes hombres y mujeres.
Pacientes de edad avanzada
Los pacientes de edad avanzada presentaron concentraciones plasmáticas mayores que los pacientes más jóvenes, con unos valores medios del AUC que fueron aproximadamente 1,5 veces superiores, principalmente debido a la disminución (aparente) del aclaramiento renal y total. No es necesario un ajuste de la dosis.
Peso corporal
Los valores extremos en el peso corporal (< 50 kg ó > 120 kg) tuvieron poco efecto en las concentraciones plasmáticas de rivaroxaban (menos del 25%). No es necesario un ajuste de la dosis.
Origen étnico
No se observaron diferencias interétnicas clínicamente relevantes entre los pacientes de raza blanca, afroamericanos, de origen latinoamericano, japonés o chino, en cuanto a las propiedades farmacocinéticas o farmacodinámicas.
Insuficiencia hepática
Los pacientes cirróticos con insuficiencia hepática leve (clasificados como Child Pugh A), sólo presentaron cambios menores en la farmacocinética de rivaroxaban (aumento medio del AUC de 1,2 veces), lo que fue casi comparable al grupo control de voluntarios sanos. En los pacientes cirróticos con insuficiencia hepática moderada (clasificados como Child Pugh B), el AUC media de rivaroxaban estuvo aumentada significativamente en 2,3 veces, en comparación con voluntarios sanos. El AUC parcial aumentó 2,6 veces. Estos pacientes también mostraron una disminución de la eliminación renal de rivaroxaban, similar a los pacientes con insuficiencia renal moderada. No hay datos en pacientes con insuficiencia hepática grave.
La inhibición de la actividad del factor Xa se incrementó en un factor de 2,6 en pacientes con insuficiencia hepática moderada, en comparación con voluntarios sanos; de manera similar, la prolongación del TP se incrementó en un factor de 2,1. Los pacientes con insuficiencia hepática moderada fueron más sensibles a rivaroxaban, lo que produjo una relación farmacocinética / farmacodinámica más pronunciada entre la concentración y el TP.
Xarelto está contraindicado en pacientes con hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia, incluyendo pacientes cirróticos clasificados como Child Pugh B y C (ver sección 4.3).
Insuficiencia renal
Se observó un aumento de la exposición de rivaroxaban, correlacionado con la disminución de la función renal, evaluada mediante las determinaciones del aclaramiento de creatinina. En personas con insuficiencia renal leve (aclaramiento de creatinina de 50 a 80 ml/min), moderada (aclaramiento de creatinina de 30 a 49 ml/min) o grave (aclaramiento de creatinina de 15 a 29 ml/min), las concentraciones plasmáticas de rivaroxaban (AUC) aumentaron 1,4, 1,5 y 1,6 veces, respectivamente. Los aumentos correspondientes de los efectos farmacodinámicos fueron más pronunciados. En sujetos con insuficiencia renal leve, moderada y grave, la inhibición total de la actividad del factor Xa aumentó en factores de 1,5, 1,9 y 2,0 respectivamente, en comparación con voluntarios sanos; de manera similar, la prolongación del TP aumentó en factores de 1,3, 2,2 y 2,4, respectivamente. No hay datos en pacientes con un aclaramiento de creatinina < 15 ml/min.
Debido a la elevada fijación a las proteínas plasmáticas, no se espera que rivaroxaban sea dializable. No se recomienda el uso en pacientes con un aclaramiento de creatinina < 15 ml/min. Xarelto debe utilizarse con precaución en pacientes con un aclaramiento de creatinina de 15 a 29 ml/min (ver sección 4.4).
Datos farmacocinéticos en pacientes
En los pacientes que recibieron rivaroxaban 10 mg una vez al día para la prevención del TEV, la concentración media geométrica (intervalo de predicción del 90%) a las 2-4 h y a las 24 h aproximadamente después de la dosis (lo que representa aproximadamente las concentraciones máxima y mínima durante el intervalo entre dosis) fue de 101 (7-273) y de 14 (4 - 51) ^g/l, respectivamente.
Relación farmacocinética / farmacodinámica
Se ha evaluado la relación farmacocinética / farmacodinámica (PK/PD) entre la concentración plasmática de rivaroxaban y varios criterios de valoración PD (inhibición del factor Xa, tiempo de protrombina (TP), TTPa, Heptest) después de la administración de un amplio rango de dosis (de 5 a 30 mg dos veces al día). La relación entre la concentración de rivaroxaban y la actividad del factor Xa se describió de manera óptima por un modelo Emax. En el caso del TP, por lo general, el modelo de intersección lineal describió mejor los datos. Dependiendo de los diferentes reactivos usados en el TP, la pendiente varió considerablemente. Con Neoplastin PT, el TP basal fue de aproximadamente 13 seg., y la pendiente fue de, aproximadamente, 3 - 4 seg/(100 ^g/l). Los resultados de los análisis de la relación PK/PD en la fase II y III fueron congruentes con los datos establecidos en sujetos sanos. En pacientes, los valores iniciales del factor Xa y del TP se vieron afectados por la intervención quirúrgica y dieron como resultado una diferencia en la pendiente de concentración-TP entre el día después de la intervención y el estado de equilibrio.
Población pediátrica
No se ha determinado la seguridad y eficacia en niños y adolescentes hasta los 18 años.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad con una dosis única, fototoxicidad, genotoxicidad, potencial carcinogénico y toxicidad juvenil.
Los efectos observados en los estudios con dosis repetidas se debieron principalmente a la actividad farmacodinámica incrementada de rivaroxaban. En ratas se observó un aumento de las concentraciones plasmáticas de IgG e IgA, a niveles de exposición clínicamente relevantes.
No se observó ningún efecto sobre la fertilidad en las ratas macho o hembra. Los estudios en animales han demostrado una toxicidad reproductiva relacionada con el modo de acción farmacológica de rivaroxaban (p.ej., complicaciones hemorrágicas). A concentraciones plasmáticas clínicamente relevantes se observó toxicidad embriofetal (pérdida después de la implantación, retraso o adelanto de la osificación, varias manchas hepáticas de color claro) y un aumento de la incidencia de malformaciones frecuentes, así como cambios placentarios. En el estudio pre- y postnatal en ratas, se observó una disminución de la viabilidad de las crías a dosis que fueron tóxicas para las madres.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido:
Celulosa microcristalina Croscarmelosa sódica Lactosa monohidrato Hipromelosa Laurilsulfato de sodio Estearato de magnesio
Cubierta pelicular:
Macrogol 3350
Hipromelosa
Dióxido de titanio (E171)
Óxido de hierro rojo (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Blister de PP / lámina de aluminio o blister de PVC / PVDC / lámina de aluminio en envases de 5, 10 ó 30 comprimidos recubiertos con película, o blisters precortados unidosis en envases de 10 x 1 ó 100 x 1 comprimidos recubiertos con película.
Blisters precortados unidosis en envases múltiples que contienen 100 (10 estuches de 10 x 1) comprimidos recubiertos con película.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación Ninguna especial para su eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlin Alemania
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/001-10, EU/1/08/472/022.
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 30 de septiembre de 2008 Fecha de la última renovación: 22 de mayo de 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
{MES/AÑO}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
▼ Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Xarelto 15 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 15 mg de rivaroxaban.
Excipiente con efecto conocido:
Cada comprimido recubierto con película contiene 24,13 mg de lactosa (como monohidrato), ver sección 4.4.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
Comprimidos de color rojo, redondos y biconvexos (6 mm de diámetro y 9 mm de radio de curvatura), con la cruz de BAYER en una cara, y “15” y un triángulo en la otra cara.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Prevención del ictus y de la embolia sistémica en pacientes adultos con fibrilación auricular no valvular, con uno o más factores de riesgo, como por ejemplo, insuficiencia cardiaca congestiva, hipertensión, edad > 75 años, diabetes mellitus, ictus o ataque isquémico transitorio previos.
Tratamiento de la trombosis venosa profunda (TVP) y de la embolia pulmonar (EP), y prevención de las recurrencias de la TVP y de la EP en pacientes adultos. (Ver en sección 4.4 información sobre pacientes con EP hemodinámicamente inestables.)
4.2 Posología y forma de administración
Posología
Prevención del ictus y de la embolia sistémica
La dosis recomendada es de 20 mg de rivaroxaban una vez al día, que es también la dosis máxima recomendada.
El tratamiento con Xarelto debe continuarse a largo plazo siempre que el beneficio de la prevención del ictus y de la embolia sistémica sea superior al riesgo de hemorragia (ver sección 4.4).
Si se olvida una dosis, el paciente debe tomar inmediatamente Xarelto y seguir al día siguiente con la dosis de una vez al día recomendada. La dosis no debe duplicarse en el mismo día para compensar una dosis olvidada.
Tratamiento de la TVP, tratamiento de la EP y prevención de las recurrencias de la TVP y de la EP La dosis recomendada para el tratamiento inicial de la TVP aguda o de la EP es de 15 mg dos veces al día, durante las tres primeras semanas, seguida de 20 mg una vez al día para el tratamiento continuado así como para la prevención de las recurrencias de la TVP y de la EP, según se indica en la tabla siguiente.
|
Pauta de tratamiento |
Dosis máxima diaria | |
|
Días 1 a 21 |
15 mg dos veces al día |
30 mg |
|
Día 22 en adelante |
20 mg una vez al día |
20 mg |
Para facilitar el cambio de dosis de 15 mg a 20 mg después del Día 21, está disponible un envase para el inicio del tratamiento de Xarelto en las primeras 4 semanas para el tratamiento de la TVP / EP (ver sección 6.5).
La duración del tratamiento debe individualizarse después de una evaluación minuciosa del beneficio del tratamiento frente al riesgo de hemorragia (ver sección 4.4). La duración corta del tratamiento (como mínimo de 3 meses) debe basarse en factores de riesgo transitorios (p.ej. cirugía reciente, traumatismo, inmovilización) y las duraciones más largas deben basarse en factores de riesgo permanente o TVP idiopática, o de EP.
Si el paciente olvida una dosis durante la fase de tratamiento de 15 mg dos veces al día (días 1 a 21), éste deberá tomar inmediatamente Xarelto para garantizar una toma de 30 mg de Xarelto al día. En este caso, se pueden tomar dos comprimidos de 15 mg a la vez y al día siguiente se deberá seguir con la pauta habitual recomendada de 15 mg dos veces al día.
Si el paciente olvida una dosis durante la fase de tratamiento de una vez al día (día 22 en adelante), deberá tomar inmediatamente Xarelto, y seguir al día siguiente con la pauta recomendada de una vez al día. La dosis no debe duplicarse en el mismo día para compensar una dosis olvidada.
Cambio de tratamiento con antagonistas de la vitamina K (AVK) a Xarelto En el caso de pacientes tratados para la prevención del ictus y de la embolia sistémica, deberá interrumpirse el tratamiento con AVK e iniciarse el tratamiento con Xarelto cuando el valor del INR (International Normalized Ratio) sea < 3,0.
En el caso de pacientes tratados por TVP, EP y en la prevención de sus recurrencias, deberá interrumpirse el tratamiento con AVK e iniciarse el tratamiento con Xarelto cuando el valor del INR sea < 2,5.
Al cambiar el tratamiento con AVK a Xarelto, los valores de INR del paciente estarán falsamente elevados después de la toma de Xarelto. El INR no es un parámetro válido para medir la actividad anticoagulante de Xarelto, por lo que no debe utilizarse (ver sección 4.5).
Cambio de tratamiento con Xarelto a antagonistas de la vitamina K (AVK)
Existe la posibilidad de una incorrecta anticoagulación durante la transición de Xarelto a AVK. Debe garantizarse una anticoagulación adecuada y continua durante cualquier transición a un anticoagulante alternativo. Debe señalarse que Xarelto puede contribuir a un aumento del INR.
En los pacientes que cambien de Xarelto a AVK, estos tratamientos deben administrarse simultáneamente hasta que el INR sea > 2,0. Durante los dos primeros días del periodo de cambio se utilizará la dosis inicial estándar de AVK, que se ajustará posteriormente en función de los resultados del INR. Mientras los pacientes están bajo tratamiento con Xarelto y AVK el INR puede determinarse a partir de las 24 horas que siguen a la dosis de Xarelto y siempre antes de la siguiente dosis. Una vez interrumpido el tratamiento con Xarelto, el INR puede determinarse con fiabilidad pasadas 24 horas de la última dosis (ver secciones 4.5 y 5.2).
Cambio de tratamiento con anticoagulante parenteral a Xarelto
Los pacientes que están recibiendo un anticoagulante por vía parenteral, deben interrumpir el tratamiento anticoagulante por vía parenteral e iniciar el tratamiento con Xarelto de 0 a 2 horas antes de la siguiente administración programada del medicamento por vía parenteral (p. ej., heparina de bajo peso molecular). En el caso de un anticoagulante parenteral administrado por perfusión continua (p. ej., heparina no fraccionada intravenosa) Xarelto deberá administrarse en el momento de la suspensión del anticoagulante parenteral.
Cambio de tratamiento con Xarelto a anticoagulante parenteral
La primera dosis de anticoagulante parenteral debe administrarse en el momento en que se tomaría la siguiente dosis de Xarelto.
Poblaciones especiales
Insuficiencia renal
Los escasos datos clínicos en pacientes con insuficiencia renal grave (aclaramiento de creatinina de 15 a 29 ml/min) indican que las concentraciones plasmáticas de rivaroxaban aumentan significativamente. Por lo tanto, Xarelto se debe usar con precaución en estos pacientes. No se recomienda su uso en pacientes con un aclaramiento de creatinina < 15 ml/min (ver secciones 4.4 y 5.2).
En pacientes con insuficiencia renal moderada (aclaramiento de creatinina de 30 a 49 ml/min) o grave (aclaramiento de creatinina de 15 a 29 ml/min) se recomiendan las siguientes pautas posológicas:
- Para la prevención del ictus y de la embolia sistémica en pacientes con fibrilación auricular no valvular, la dosis recomendada es de 15 mg una vez al día (ver sección 5.2).
- Para el tratamiento de la TVP, y de la EP y la prevención de las recurrencias de la TVP y de la EP se debe tratar a los pacientes con 15 mg dos veces al día durante las tres primeras semanas. Después, la dosis recomendada es de 20 mg una vez al día. Deberá considerarse una reducción de la dosis de 20 mg una vez al día a 15 mg una vez al día si el riesgo de sangrado valorado en el paciente supera el riesgo de recurrencia de TVP y de EP. La recomendación para el uso de 15 mg se basa en el modelo farmacocinético que no se ha estudiado en este contexto clínico (ver secciones 4.4, 5.1 y 5.2).
No se requiere ajuste de dosis en pacientes con insuficiencia renal leve (aclaramiento de creatinina de 50 a 80 ml/min) (ver sección 5.2).
Insuficiencia hepática
Xarelto está contraindicado en pacientes con hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia incluidos los pacientes cirróticos con Child Pugh B y C (ver secciones 4.3 y 5.2).
Pacientes de edad avanzada
No se requiere ajuste de dosis (ver sección 5.2).
Peso corporal
No se requiere ajuste de dosis (ver sección 5.2).
Sexo
No se requiere ajuste de dosis (ver sección 5.2).
Población pediátrica
No se ha establecido la seguridad y eficacia de Xarelto en niños de 0 a 18 años. No se dispone de datos. Por lo tanto, no se recomienda el uso de Xarelto en niños menores de 18 años.
Pacientes sometidos a cardioversión
El tratamiento con Xarelto se puede iniciar o continuar en pacientes que requieran cardioversión.
Para una cardioversión guiada por ecocardiografía transesofágica (ETE) en pacientes no tratados previamente con anticoagulantes, el tratamiento con Xarelto debe iniciarse al menos 4 horas antes de la cardioversión para asegurar una anticoagulación adecuada (ver secciones 5.1 y 5.2). En todos los pacientes, se deberá confirmar antes de la cardioversión que el paciente ha tomado Xarelto según lo prescrito. En las decisiones sobre inicio y duración del tratamiento, se tendrán en cuenta las recomendaciones de las guías establecidas para el tratamiento anticoagulante en pacientes sometidos a cardioversión.
Forma de administración Vía oral.
Los comprimidos deben administrarse con alimentos (ver sección 5.2).
Para aquellos pacientes que no puedan tragar el comprimido entero, el comprimido de Xarelto puede triturarse y mezclarse con agua o con puré de manzana inmediatamente antes de su uso y administrarse por vía oral. Inmediatamente después de la administración del comprimido triturado se debe administrar el alimento.
El comprimido triturado también se puede administrar a través de sonda gástrica una vez se haya confirmado la colocación correcta de la sonda. El comprimido triturado se administrará diluido con una pequeña cantidad de agua a través de la sonda gástrica, procediendo seguidamente a un lavado adicional de la sonda con agua. Inmediatamente después de la administración del comprimido triturado se debe administrar el alimento mediante sonda gástrica (ver sección 5.2).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hemorragia activa clínicamente significativa.
Lesión o enfermedad, si se considera que tiene un riesgo significativo de sangrado mayor. Esto puede incluir úlcera gastrointestinal activa o reciente, presencia de neoplasias malignas con alto riesgo de sangrado, traumatismo cerebral o espinal reciente, cirugía cerebral, espinal u oftálmica reciente, hemorragia intracraneal reciente, conocimiento o sospecha de varices esofágicas, malformaciones arteriovenosas, aneurismas vasculares o anomalías vasculares intraespinales o intracerebrales mayores.
Tratamiento concomitante con cualquier otro anticoagulante, p. ej. heparina no fraccionada (HNF), heparinas de bajo peso molecular (enoxaparina, dalteparina, etc.), derivados de la heparina (fondaparinux, etc.), anticoagulantes orales (warfarina, dabigatran etexilato, apixaban, etc.) excepto bajo las circunstancias concretas de cambio de tratamiento anticoagulante (ver sección 4.2) o cuando se administre HNF a las dosis necesarias para mantener un catéter venoso o arterial central abierto (ver sección 4.5).
Hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia, incluidos los pacientes cirróticos con Child Pugh B y C (ver sección 5.2).
Embarazo y lactancia (ver sección 4.6).
4.4 Advertencias y precauciones especiales de empleo
Durante todo el periodo de tratamiento se recomienda una estrecha monitorización clínica del paciente, en línea con la práctica de anticoagulación
Riesgo de hemorragia
Al igual que con otros anticoagulantes, los pacientes que toman Xarelto deben ser observados cuidadosamente para detectar signos de sangrado. Se recomienda utilizar con precaución en condiciones que conlleven un riesgo incrementado de hemorragia. La administración de Xarelto debe interrumpirse si se produce una hemorragia grave.
En los ensayos clínicos se observaron con más frecuencia hemorragias a nivel de mucosas (p.ej. epistaxis, gingival, gastrointestinal, génito-urinaria) y anemia en los pacientes que recibían rivaroxaban a largo plazo respecto a los que recibían tratamiento con AVK. Por ello, además de un seguimiento clínico adecuado, las determinaciones de hemoglobina y hematocrito podrían ser útiles para detectar hemorragias ocultas cuando se considere apropiado.
Varios subgrupos de pacientes, como se explica posteriormente, presentan un mayor riesgo de hemorragia. En estos pacientes se debe vigilar cuidadosamente la presencia de signos y síntomas de complicaciones hemorrágicas y de anemia después del inicio del tratamiento (ver sección 4.8). Cualquier disminución inexplicada de los niveles de hemoglobina o de la presión arterial requerirá la búsqueda de una zona de sangrado.
Aunque el tratamiento con rivaroxaban no requiere una monitorización rutinaria de la exposición, la determinación de los niveles de rivaroxaban mediante un ensayo anti-factor Xa cuantitativo calibrado puede ser útil en situaciones excepcionales, en las que el conocimiento de la exposición a rivaroxaban puede ayudar en la toma de decisiones clínicas, como por ejemplo, en caso de sobredosis o cirugía de emergencia (ver secciones 5.1 y 5.2).
Insuficiencia renal
En pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min), las concentraciones plasmáticas de rivaroxaban podrían aumentar de forma significativa (en promedio,
1,6 veces), lo que conllevaría un aumento del riesgo de hemorragia. Xarelto debe utilizarse con precaución en pacientes con un aclaramiento de creatinina de 15 a 29 ml/min. No se recomienda el uso en pacientes con un aclaramiento de creatinina < 15 ml/min (ver secciones 4.2 y 5.2).
Xarelto debe usarse con precaución en pacientes con insuficiencia renal y que reciben de forma concomitante otros medicamentos que aumenten las concentraciones plasmáticas de rivaroxaban (ver sección 4.5).
Interacción con otros medicamentos
No se recomienda utilizar Xarelto en pacientes que reciben tratamiento sistémico concomitante con antimicóticos azólicos (p. ej., ketoconazol, itraconazol, voriconazol y posaconazol) o inhibidores de la proteasa del VIH (p. ej., ritonavir). Estos principios activos son inhibidores potentes del CYP3A4 y de la P-gp y por lo tanto pueden aumentar las concentraciones plasmáticas de rivaroxaban hasta un nivel clínicamente relevante (en promedio, 2,6 veces), lo que puede llevar a un aumento del riesgo de hemorragia (ver sección 4.5).
Debe tenerse precaución si los pacientes reciben tratamiento concomitante con medicamentos que afectan a la hemostasia, como los antiinflamatorios no esteroideos (AINEs), ácido acetilsalicílico e inhibidores de la agregación plaquetaria. Puede considerarse el uso de un tratamiento profiláctico adecuado en aquellos pacientes con riesgo de enfermedad gastrointestinal ulcerosa, (ver sección 4.5).
Otros factores de riesgo hemorrágico
Al igual que con otros agentes antitrombóticos, rivaroxaban no está recomendado en pacientes con un riesgo aumentado de hemorragia, tales como:
• trastornos de la coagulación, congénitos o adquiridos
• hipertensión arterial grave no controlada
• otra enfermedad gastrointestinal sin úlcera activa que pueda producir complicaciones hemorrágicas (por ejemplo, enfermedad inflamatoria intestinal, esofagitis, gastritis o reflujo gastroesofágico)
• retinopatía vascular
• bronquiectasia o antecedentes de hemorragia pulmonar Pacientes con prótesis valvulares
No se ha estudiado la seguridad y eficacia de Xarelto en pacientes con prótesis valvulares cardiacas; por lo tanto, no hay datos que apoyen que Xarelto 20 mg (15 mg en los pacientes con insuficiencia renal moderada o grave) proporciona una anticoagulación adecuada en esta población. No se recomienda el tratamiento con Xarelto en estos pacientes.
Pacientes con EP hemodinámicamente inestables o pacientes que requieran trombolisis o embolectomía pulmonar
Xarelto no está recomendado como una alternativa a la heparina no fraccionada en pacientes con embolia pulmonar que están hemodinámicamente inestables o que puedan ser sometidos a trombolisis o embolectomía pulmonar, ya que no se ha establecido la seguridad y eficacia de Xarelto en estas situaciones clínicas.
Anestesia espinal/epidural o punción lumbar
Cuando se aplica anestesia neuraxial (anestesia epidural o espinal) o se realiza una punción lumbar o epidural, los pacientes tratados con antitrombóticos para la prevención de complicaciones tromboembólicas tienen riesgo de presentar un hematoma epidural o espinal, que puede causar parálisis a largo plazo o permanente. El riesgo de estos eventos puede estar aumentado por el empleo postoperatorio de catéteres epidurales permanentes o por la administración concomitante de medicamentos que afectan a la hemostasia. El riesgo también puede aumentar por la punción epidural o espinal traumática o repetida. Se debe controlar con frecuencia la presencia de signos y síntomas de deterioro neurológico (p. ej., adormecimiento o debilidad de extremidades inferiores, disfunción intestinal o vesical). Si se observa compromiso neurológico, será necesario un diagnóstico y tratamiento urgente. Antes de la intervención neuraxial, el médico debe valorar el beneficio potencial frente al riesgo en los pacientes con tratamiento anticoagulante o que van a recibir medicamentos anticoagulantes para la tromboprofilaxis. No se dispone de experiencia clínica sobre el uso de rivaroxaban 15 mg en estas situaciones.
Para reducir el riesgo potencial de sangrado asociado con el uso concomitante de rivaroxaban y anestesia neuraxial (epidural/espinal) o punción espinal se debe considerar el perfil farmacocinético de rivaroxaban. La colocación o extracción de un catéter epidural o punción lumbar se realiza mejor cuando se estima que el efecto anticoagulante de rivaroxaban es bajo. Sin embargo, se desconoce el momento exacto en el que se alcanza un efecto anticoagulante lo suficientemente bajo en cada paciente.
En base a las características farmacocinéticas generales, para la extracción de un catéter epidural, debe transcurrir al menos dos veces el tiempo de vida media desde la última administración de rivaroxaban, por ejemplo, 18 horas como mínimo en pacientes jóvenes y 26 horas en pacientes de edad avanzada (ver sección 5.2). Una vez retirado el catéter, deben transcurrir al menos 6 horas para poder administrar la siguiente dosis de rivaroxaban.
Si se produce una punción traumática, la administración de rivaroxaban se deberá retrasar 24 horas.
Recomendaciones posológicas antes y después de procedimientos invasivos y de intervenciones quirúrgicas
Si es necesario realizar un procedimiento invasivo o una intervención quirúrgica, se interrumpirá la administración de Xarelto 15 mg por lo menos 24 horas antes de la intervención, si es posible y basándose en el criterio clínico del médico. Si la intervención no puede retrasarse, debe evaluarse el aumento del riesgo de hemorragia frente a la urgencia de la intervención.
Se debe reiniciar lo antes posible la administración de Xarelto después del procedimiento invasivo o intervención quirúrgica, siempre que la situación clínica lo permita y se haya establecido una hemostasia adecuada una vez confirmado por el médico que trata al paciente (ver sección 5.2).
Pacientes de edad avanzada
La edad avanzada puede aumentar el riesgo de hemorragia (ver sección 5.2).
Información acerca de los excipientes
Xarelto contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, deficiencia de lactasa de Lapp o malabsorción de glucosa o galactosa no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Inhibidores del CYP3A4 y de la P-gp
La administración concomitante de rivaroxaban con ketoconazol (400 mg una vez al día) o ritonavir (600 mg dos veces al día) produjo un aumento de 2,6 veces / 2,5 veces del AUC media de rivaroxaban, y un aumento de 1,7 veces / 1,6 veces de la Cmax media de rivaroxaban, con aumentos significativos de los efectos farmacodinámicos, lo que puede aumentar el riesgo de hemorragia. Por lo tanto, no se recomienda el uso de Xarelto en pacientes que reciban tratamiento sistémico concomitante con antimicóticos azólicos como ketoconazol, itraconazol, voriconazol y posaconazol o con inhibidores de la proteasa del VIH. Estos principios activos son inhibidores potentes del CYP3A4 y de la P-gp (ver sección 4.4).
Las sustancias activas que inhiben intensamente sólo una de las vías de eliminación de rivaroxaban, el CYP3A4 o la P-gp, pueden aumentar las concentraciones plasmáticas de rivaroxaban en menor grado. La claritromicina (500 mg dos veces al día), por ejemplo, considerada un potente inhibidor del CYP3A4 y un inhibidor moderado de la P-gp, produjo un aumento de 1,5 veces del AUC media de rivaroxaban y un aumento de 1,4 veces de la Cmax. Este aumento no se considera clínicamente relevante. (Pacientes con insuficiencia renal: ver sección 4.4).
La eritromicina (500 mg tres veces al día), que inhibe moderadamente el CYP3A4 y la P-gp, produjo un aumento de 1,3 veces del AUC y de la Cmax medias de rivaroxaban. Este aumento no se considera clínicamente relevante.
En sujetos con insuficiencia renal leve, la eritromicina (500 mg tres veces al día) produjo un aumento de 1,8 veces el AUC media de rivaroxaban y de 1,6 veces en la Cmax, comparado con sujetos con la función renal normal. En sujetos con insuficiencia renal moderada, la eritromicina produjo un aumento de 2,0 veces en el AUC media de rivaroxaban y 1,6 veces en la Cmax, comparado con sujetos con la función renal normal. El efecto de la eritromicina es aditivo al de la insuficiencia renal (ver sección
4.4) .
El fluconazol (400 mg una vez al día), considerado un inhibidor moderado del CYP3A4, produjo un aumento de 1,4 veces del AUC media de rivaroxaban y un aumento de 1,3 veces de la Cmax media.
Este aumento no se consideró clínicamente relevante. (Pacientes con insuficiencia renal: ver sección
4.4) .
Dada la limitada información clínica disponible con dronedarona, debería evitarse la administración concomitante con rivaroxaban.
Anticoagulantes
Después de la administración combinada de enoxaparina (dosis única de 40 mg) con rivaroxaban (dosis única de 10 mg), se observó un efecto aditivo sobre la actividad anti-factor Xa, sin efectos adicionales en las pruebas de coagulación (TP, TTPa). La enoxaparina no afectó a las propiedades farmacocinéticas de rivaroxaban.
Debido al aumento del riesgo de hemorragia, se debe tener precaución si los pacientes reciben tratamiento concomitante con cualquier otro anticoagulante (ver secciones 4.3 y 4.4).
AINEs e inhibidores de la agregación plaquetaria
No se observó una prolongación clínicamente relevante del tiempo de sangrado después de la administración concomitante de rivaroxaban (15 mg) y 500 mg de naproxeno. No obstante, algunas personas pueden tener una respuesta farmacodinámica más pronunciada.
No se observó ninguna interacción farmacocinética ni farmacodinámica clínicamente significativa cuando se administró rivaroxaban concomitantemente con 500 mg de ácido acetilsalicílico.
El clopidogrel (dosis de carga de 300 mg, seguida de una dosis de mantenimiento de 75 mg) no mostró ninguna interacción farmacocinética con rivaroxaban (15 mg); sin embargo, se observó un aumento del tiempo de sangrado en un subgrupo de pacientes, que no se correlacionó con la agregación plaquetaria, las concentraciones de P-selectina o los receptores GPIIb/IIIa.
Debe tenerse precaución si los pacientes reciben tratamiento concomitante con AINEs (incluyendo ácido acetilsalicílico) e inhibidores de la agregación plaquetaria, ya que estos medicamentos aumentan, de por sí, el riesgo de hemorragia (ver sección 4.4).
Warfarina
Los cambios de tratamiento con warfarina (INR de 2,0 a 3,0), un antagonista de la vitamina K, a rivaroxaban (20 mg) o de rivaroxaban (20 mg) a warfarina (INR de 2,0 a 3,0) aumentaron el tiempo de protrombina/INR (Neoplastin) de forma importante (pueden observarse valores individuales del INR de hasta 12), mientras que los efectos sobre el TTPa, la inhibición de la actividad del factor Xa y el potencial de trombina endógena (PTE) fueron aditivos.
Si se desea medir los efectos farmacodinámicos de rivaroxaban durante el periodo de cambio de tratamiento, puede utilizarse la actividad anti-factor Xa, PiCT y Heptest, ya que la warfarina no afecta a estas pruebas. Al cuarto día tras la última dosis de warfarina, todas las pruebas (incluyendo TP, TTPa, inhibición de la actividad del factor Xa y PTE) reflejaron únicamente el efecto de rivaroxaban. Si se desea medir los efectos farmacodinámicos de warfarina durante el periodo de cambio de tratamiento, se puede usar la determinación del INR en la Ctrough de rivaroxaban (24 horas después de su anterior administración), ya que rivaroxaban afecta mínimamente a esta prueba en este punto.
No se observó ninguna interacción farmacocinética entre warfarina y rivaroxaban.
Inductores del CYP3A4
La administración concomitante de rivaroxaban con rifampicina, un potente inductor del CYP3A4, produjo una disminución aproximada del 50% del AUC media de rivaroxaban, con disminuciones paralelas de sus efectos farmacodinámicos. El uso concomitante de rivaroxaban con otros inductores potentes del CYP3A4 (por ejemplo, fenitoína, carbamazepina, fenobarbital o la hierba de San Juan (Hypericum perforatum)) también puede causar una disminución de la concentración plasmática de rivaroxaban. Por tanto, la administración concomitante con inductores potentes del CYP3A4 deberá evitarse a menos que el paciente esté estrechamente monitorizado para detectar signos o síntomas de trombosis.
Otros tratamientos concomitantes
No se observó ninguna interacción farmacocinética o farmacodinámica clínicamente significativa cuando se administró rivaroxaban concomitantemente con midazolam (sustrato del CYP3A4), digoxina (sustrato de la P-gp), atorvastatina (sustrato del CYP3A4 y de la P-gp) u omeprazol (inhibidor de la bomba de protones). Rivaroxaban no inhibe ni induce ninguna isoforma mayor del CYP, como el CYP3A4.
Parámetros de laboratorio
Los parámetros de la coagulación (p. ej. TP, TTPa, HepTest) se ven afectados de la forma esperada debido al mecanismo de acción de rivaroxaban (ver sección 5.1).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No se ha evaluado la seguridad y eficacia de Xarelto en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3). Debido a la posible toxicidad reproductiva, riesgo intrínseco de hemorragia y la evidencia de que rivaroxaban atraviesa la barrera placentaria, Xarelto está contraindicado durante el embarazo (ver sección 4.3).
Las mujeres en edad fértil deben evitar quedarse embarazadas durante el tratamiento con rivaroxaban.
Lactancia
No se ha evaluado la seguridad y eficacia de Xarelto en mujeres en período de lactancia. Los datos en animales indican que rivaroxaban se excreta en la leche materna. Por lo tanto, Xarelto está contraindicado durante la lactancia (ver sección 4.3). Se debe decidir si es necesario interrumpir la lactancia o bien interrumpir/suspender el tratamiento.
Fertilidad
No se han realizado estudios específicos con rivaroxaban para evaluar los efectos sobre la fertilidad en humanos. En un estudio sobre la fertilidad en ratas macho y hembra no se observó ningún efecto (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Xarelto puede influir ligeramente en la capacidad para conducir y utilizar máquinas. Se han descrito reacciones adversas como síncope (frecuencia: poco frecuente) y mareos (frecuencia: frecuente) (ver sección 4.8). Los pacientes que sufran estas reacciones adversas no deben conducir ni utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Se ha evaluado la seguridad de rivaroxaban en once ensayos clínicos de fase III, que incluyeron 32.625 pacientes expuestos a rivaroxaban (ver la tabla 1).
Tabla 1: Número de pacientes estudiados, dosis máxima diaria y duración del tratamiento en los estudios de fase III.
|
Indicación |
Número de pacientes * |
Dosis máxima diaria |
Duración máxima del tratamiento |
|
Prevención de tromboembolismo venoso (TEV) en pacientes adultos sometidos a cirugía electiva de reemplazo de cadera o rodilla |
6.097 |
10 mg |
39 días |
|
Prevención de tromboembolismo venoso en pacientes encamados |
3.997 |
10 mg |
39 días |
|
Tratamiento de TVP, EP y prevención de las recurrencias de TVP y EP |
4.556 |
Días 1 a 21: 30 mg Día 22 en adelante: 20 mg |
21 meses |
|
Prevención del ictus y de la embolia sistémica en pacientes con fibrilación auricular no valvular |
7.750 |
20 mg |
41 meses |
|
Prevención de acontecimientos aterotrombóticos en pacientes después de padecer un SCA |
10.225 |
5 mg ó 10 mg respectivamente, co-administrada con AAS o bien con AAS más clopidogrel o ticlopidina |
31 meses |
*Pacientes expuestos por lo menos a una dosis de rivaroxaban.
Las reacciones adversas notificadas con mayor frecuencia en los pacientes que recibieron rivaroxaban fueron hemorragias (ver sección 4.4. y 'Descripción de las reacciones adversas seleccionadas" más adelante. Las hemorragias notificadas con mayor frecuencia (> 4%) fueron epistaxis (5,9%) y la hemorragia del tracto gastrointestinal (4,2%).
En total, se notificó la aparición de eventos adversos en aproximadamente un 67% de los pacientes expuestos por lo menos a una dosis de rivaroxaban. Aproximadamente el 22% de los pacientes sufrieron eventos adversos que se consideran relacionados con el tratamiento, según la evaluación de los investigadores. En los pacientes tratados con Xarelto 10 mg sometidos a cirugía electiva de reemplazo de cadera o rodilla y en pacientes hospitalizados encamados, se produjeron episodios hemorrágicos en aproximadamente un 6,8% y 12,6% de los pacientes, respectivamente, y se produjo anemia en aproximadamente un 5,9% y 2,1% de los pacientes, respectivamente. En los pacientes tratados con Xarelto 15 mg dos veces al día y después Xarelto 20 mg una vez al día para el tratamiento de la TVP o EP, o con Xarelto 20 mg una vez al día para la prevención de la recurrencia de la TVP y de la EP, se produjeron episodios hemorrágicos en aproximadamente un 27,8% de los pacientes, y anemia en aproximadamente un 2,2% de los pacientes. En los pacientes tratados para la prevención del ictus y de la embolia sistémica, se notificó hemorragia de cualquier tipo o gravedad, con una tasa de 28 por cada 100 pacientes-años, y anemia con una tasa de 2,5 por cada 100 pacientes-años. En los pacientes tratados para la prevención de muerte cardiovascular e infarto de miocardio después de un síndrome coronario agudo (SCA), se notificó hemorragia de cualquier tipo o gravedad con una tasa de 22 eventos por cada 100 pacientes-años. La anemia se notificó con una tasa de 1,4 eventos por cada 100 pacientes-años.
Tabla de reacciones adversas
Las frecuencias de las reacciones adversas notificadas con Xarelto se resumen en la tabla 2, según la clasificación por órganos y sistemas (convención MedDRA) y según las frecuencias.
Las frecuencias se definen como: muy frecuentes (> 1/10) frecuentes (> 1/100 a < 1/10) poco frecuentes (> 1/1.000 a < 1/100) raras (> 1/10.000 a < 1/1.000) muy raras ( < 1/10.000)
no conocida (no puede calcularse a partir de los datos disponibles)
Tabla 2: Todas las reacciones adversas observadas con el tratamiento y notificadas en los estudios de fase III
|
Frecuentes |
Poco frecuentes |
Raras |
No conocida |
|
Trastornos de la sangre y del sistema linfático | |||
|
Anemia (incl. respectivos parámetros de laboratorio) |
Trombocitemia (incl. aumento del recuento de plaquetas)A | ||
|
Trastornos del sistema inmunológico | |||
|
Reacción alérgica, dermatitis alérgica | |||
|
Trastornos del sistema nervioso | |||
|
Mareos, cefalea, |
Hemorragia cerebral e intracraneal, síncope | ||
|
Trastornos oculares | |||
|
Hemorragia ocular (incl. hemorragia conjuntival) | |||
|
Trastornos cardiacos | |||
|
Taquicardia | |||
|
Trastornos vasculares | |||
|
Hipotensión, hematoma | |||
|
Trastornos respiratorios, torácicos y mediastínicos | |||
|
Epistaxis, hemoptisis | |||
|
Trastornos gastrointestinales | |||
|
Sangrado gingival, hemorragia del tracto gastrointestinal (incl. hemorragia rectal), dolor gastrointestinal y abdominal, dispepsia, náuseas, estreñimientoA, diarrea, vómitosA |
Sequedad de boca | ||
|
Trastornos hepatobiliares | |||
|
Alteración de la función hepática |
Ictericia | ||
|
Frecuentes |
Poco frecuentes |
Raras |
No conocida |
|
Trastornos de la piel y del tejido subcutáneo | |||
|
Prurito (incl. casos raros de prurito generalizado), exantema, equimosis, hemorragia cutánea y subcutánea |
Urticaria | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |||
|
Dolor en las extremidadesA |
Hemartrosis |
Hemorragia muscular |
Síndrome compartimental secundario a una hemorragia |
|
Trastornos renales y urinarios | |||
|
Hemorragia del tracto urogenital (incl. hematuria y menorragiaB), insuficiencia renal (incl. aumento de creatinina en sangre, aumento de la urea en sangre)A |
Insuficiencia renal /insuficiencia renal aguda secundaria a una hemorragia suficiente para causar hipoperfusión | ||
|
Trastornos generales y alteraciones en el lugar de administración | |||
|
FiebreA, edema periférico, disminución general de la fuerza y la energía (incl. fatiga y astenia) |
Sensación de malestar (incl. malestar general) |
Edema localizadoA | |
|
Exploraciones complementarias | |||
|
Aumento de las transaminasas |
Aumento de la bilirrubina, aumento de la fosfatasa alcalina sanguíneaA, aumento de la LDHA, aumento de la lipasaA, aumento de la amilasaA, aumento de la GGTA |
Aumento de la bilirrubina conjugada (con o sin aumento concomitante de la ALT). | |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos | |||
|
Hemorragia después de una intervención (incl. anemia postoperatoria y hemorragia de la herida), contusión, secreción de la heridaA |
Pseudoaneurisma vascularC | ||
A: observado en la prevención del tromboembolismo venoso (TEV) en pacientes adulto sometidos a cirugía electiva de reemplazo de cadera o rodilla
B: observado en el tratamiento de la TVP, EP y prevención de sus recurrencias como muy frecuente en mujeres < 55 años.
C: observado como poco frecuente en la prevención de eventos aterotrombóticos en pacientes que han sufrido un SCA (tras una intervención coronaria percutánea)
Descripción de reacciones adversas seleccionadas
Debido a su mecanismo de acción farmacológica, el uso de Xarelto puede asociarse a un incremento del riesgo de hemorragia oculta o manifiesta en cualquier tejido u órgano, que puede dar lugar a una anemia post-hemorrágica. Los signos, síntomas y gravedad (incluido un posible desenlace mortal) variarán según la localización y el grado o la extensión de la hemorragia, la anemia o ambas (ver sección 4.9 Tratamiento de la hemorragia). En los ensayos clínicos se observaron con más frecuencia hemorragias a nivel de mucosas (p.ej. epistaxis, gingival, gastrointestinal, genito-urinaria) y anemia en los pacientes que recibían rivaroxaban a largo plazo con respecto a los que recibían tratamiento con AVK. Por ello, además de un adecuado seguimiento clínico, las determinaciones de hemoglobina y hematocrito podrían ser útiles para detectar hemorragias ocultas cuando se considere apropiado.
El riesgo de hemorragia puede estar aumentado en ciertos grupos de pacientes, como por ejemplo, en pacientes con hipertensión arterial grave no controlada y/o en tratamiento concomitante que afecte a la hemostasia (ver Riesgo de hemorragia en la sección 4.4). El sangrado menstrual puede ser más intenso y/o prolongarse. Las complicaciones hemorrágicas pueden presentarse como debilidad, palidez, mareos, cefalea o tumefacción inexplicada, disnea o shock de causa desconocida. En algunos casos, a consecuencia de la anemia, se han observado síntomas de isquemia cardíaca, como dolor torácico o angina de pecho.
Se han notificado complicaciones conocidas, secundarias a hemorragia intensa, como el síndrome compartimental o insuficiencia renal debida a hipoperfusión. Por lo tanto deberá tenerse en cuenta la posibilidad de hemorragia al evaluar el estado de cualquier paciente anticoagulado.
Observaciones post-comercialización
Tras la comercialización se han notificado las siguientes reacciones adversas en asociación temporal con el uso de Xarelto. No se ha podido estimar la frecuencia de estas reacciones adversas.
Trastornos del sistema inmunológico: angioedema y edema alérgico (En los ensayos de fase III agrupados, estos eventos fueron poco frecuentes (> 1/1.000 a <1/100)).
Trastornos hepatobiliares: colestasis, hepatitis (incluyendo lesión hepatocelular). (En los ensayos de fase III agrupados, estos eventos fueron raros (> 1/10.000 a < 1/1.000)).
Trastornos de la sangre y del sistema linfático: trombocitopenia. (En los ensayos de fase III agrupados, estos eventos fueron poco frecuentes (> 1/1.000 a < 1/100)).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se han notificado casos raros de sobredosis de hasta 600 mg sin complicaciones hemorrágicas u otras reacciones adversas. Debido a la limitada absorción a dosis supraterapéuticas de 50 mg de rivaroxaban o superiores se espera un efecto techo sin un aumento posterior de la exposición plasmática media.
No se dispone de un antídoto específico que antagonice el efecto farmacodinámico de rivaroxaban.
Se puede considerar el uso de carbono activado para reducir la absorción en caso de sobredosis por rivaroxaban.
Tratamiento de la hemorragia
En caso de producirse una complicación hemorrágica en un paciente que recibe tratamiento con rivaroxaban, se deberá retrasar la siguiente administración de rivaroxaban o interrumpir el tratamiento si se considera conveniente. Rivaroxaban tiene una semivida de eliminación de entre 5 y 13 horas (ver sección 5.2). Las medidas terapéuticas deben individualizarse según la gravedad y la localización de la hemorragia. En caso necesario, podría aplicarse el tratamiento sintomático adecuado, como la compresión mecánica (por ejemplo en caso de epistaxis intensa), hemostasia quirúrgica con procedimientos de control de la hemorragia, reemplazo de fluidos y apoyo hemodinámico (concentrado de hematíes o plasma fresco congelado, dependiendo de la anemia o la coagulopatía asociadas) o plaquetas.
Si la hemorragia no se puede controlar con las medidas anteriores, debería plantearse la administración de un agente procoagulante específico para revertir el efecto, como el concentrado de complejo de protrombina (CCP), concentrado de complejo de protrombina activado (CCPA) o factor VIIa recombinante (r-FVIIa). Sin embargo, actualmente hay una experiencia clínica muy limitada con el uso de estos productos en pacientes que reciben rivaroxaban. La recomendación se basa también en datos no clínicos limitados. Deberá plantearse la readministración de factor VIIa recombinante y ajustar la dosis dependiendo de la mejoría de la hemorragia. Dependiendo de la disponibilidad local, en caso de hemorragia mayor debe considerarse consultar a un experto en coagulación (ver sección 5.1).
No se espera que el sulfato de protamina y la vitamina K afecten a la actividad anticoagulante de rivaroxaban. La experiencia con ácido tranexámico es limitada y no hay experiencia con ácido aminocaproico y aprotinina en pacientes tratados con rivaroxaban. No hay una justificación científica sobre la ventaja ni experiencia con el hemostático sistémico desmopresina en pacientes tratados con rivaroxaban. Debido a su elevada fijación a las proteínas plasmáticas, no se espera que rivaroxaban sea dializable.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inhibidores directos del factor Xa, código ATC: B01AF01 Mecanismo de acción
Rivaroxaban es un inhibidor directo del factor Xa altamente selectivo, con biodisponibilidad oral. La inhibición del factor Xa interrumpe las vías intrínseca y extrínseca de la cascada de la coagulación de la sangre, inhibiendo tanto la formación de trombina como la formación de trombos. Rivaroxaban no inhibe la trombina (factor II activado) y no se han demostrado efectos sobre las plaquetas.
Efectos farmacodinámicos
En los seres humanos se ha observado una inhibición de la actividad del factor Xa dosis-dependiente. Rivaroxaban modifica el tiempo de protrombina (TP) de forma dosis-dependiente con una estrecha correlación con las concentraciones plasmáticas (el valor de r es igual a 0,98) si se emplea Neoplastin para el análisis. Otros reactivos proporcionarían unos resultados diferentes. La lectura del TP debe hacerse en segundos, porque el INR sólo se ha calibrado y validado en el caso de los cumarínicos y no puede utilizarse con ningún otro anticoagulante.
En pacientes que recibieron rivaroxaban para el tratamiento de la TVP y EP, y para la prevención de sus recurrencias, los percentiles 5/95 del TP (Neoplastin) de 2 a 4 horas después de tomar el comprimido (es decir, en el momento del efecto máximo) variaron de 17 a 32 seg. en el caso de rivaroxaban 15 mg dos veces al día, y de 15 a 30 seg. en el caso de rivaroxaban 20 mg una vez al día. En el momento de la concentración valle (8 - 16 h después de la toma del comprimido) los percentiles 5/95 para la dosis de 15 mg dos veces al día variaron de 14 a 24 seg. y para la dosis de 20 mg una vez al día (18 - 30 h después de la toma del comprimido) variaron de 13 a 20 seg.
En pacientes con fibrilación auricular no valvular que recibieron rivaroxaban para la prevención del ictus y de la embolia sistémica, en el momento del efecto máximo (1 a 4 h después de la toma del comprimido) los percentiles 5/95 del TP (Neoplastin) variaron de 14 a 40 seg. en los pacientes tratados con 20 mg una vez al día, y de 10 a 50 seg. en los pacientes con insuficiencia renal moderada tratados con 15 mg una vez al día. En el momento de la concentración valle (16 - 36 h de la toma del comprimido) los percentiles 5/95 para los pacientes tratados con la dosis de 20 mg una vez al día variaron de 12 a 26 seg. y para los pacientes con insuficiencia renal moderada tratados con la dosis de 15 mg una vez al día variaron de 12 a 26 seg.
En un estudio de farmacología clínica en la reversión de la acción farmacodinámica de rivaroxaban en adultos sanos (n = 22), se evaluaron los efectos de dosis únicas (50 UI/kg) de dos tipos diferentes de CCP, un CCP de 3 factores (factores II, IX y X) y un CCP de 4 factores (factores II, VII, IX y X). El CCP de 3 factores redujo los valores medios del TP (Neoplastina) en aproximadamente 1,0 segundos a los 30 minutos, en comparación con reducciones de, aproximadamente, 3,5 segundos observadas con el CCP de 4 factores. En cambio, el CCP de 3 factores tuvo un efecto global mayor y más rápido en la reversión de los cambios en la generación de trombina endógena que el CCP de 4 factores (ver sección 4.9).
El tiempo de tromboplastina parcial activada (TTPa) y el HepTest también se prolongaron de forma dosis-dependiente; sin embargo, no se recomiendan para evaluar el efecto farmacodinámico de rivaroxaban. No es necesario monitorizar los parámetros de la coagulación durante el tratamiento con rivaroxaban en la práctica clínica. Sin embargo, si está indicado clínicamente, se pueden medir los niveles de rivaroxaban mediante ensayos cuantitativos calibrados para la actividad anti-factor Xa (ver sección 5.2).
Eficacia clínica y seguridad
Prevención del ictus y de la embolia sistémica en pacientes con fibrilación auricular no valvular El programa clínico de Xarelto se diseñó para demostrar la eficacia de Xarelto en la prevención del ictus y de la embolia sistémica en pacientes con fibrilación auricular no valvular.
En el estudio pivotal doble ciego ROCKET AF se aleatorizaron 14.264 pacientes para recibir Xarelto 20 mg una vez al día (Xarelto 15 mg una vez al día en pacientes con un aclaramiento de creatinina de 30 a 49 ml/min) o warfarina ajustada hasta un objetivo de INR de 2,5 (rango terapéutico de 2,0 a 3,0). La mediana del tiempo en tratamiento fue de 19 meses y la duración total del tratamiento fue de hasta 41 meses.
El 34,9% de los pacientes recibió tratamiento con ácido acetilsalicílico y el 11,4% con antiarrítmicos de clase III, incluida la amiodarona.
Xarelto fue no inferior a warfarina para la variable principal de eficacia compuesta de ictus y embolia sistémica fuera del sistema nervioso central. En la población por protocolo y durante el tratamiento se observó ictus o embolia sistémica en 188 pacientes tratados con rivaroxaban (1,71% anual) y en 241 pacientes tratados con warfarina (2,16% anual) (HR 0,79; 95% IC, 0,66-0,96; P<0.001 para no inferioridad). Entre todos los pacientes aleatorizados y analizados por intención de tratar, el número de pacientes que sufrieron un ictus o embolia sistémica fue de 269 en el caso de rivaroxaban (2,12% anual) y de 306 en los tratados con warfarina (2,42% per anual) (HR 0,88; 95% IC, 0,74-1,03;
P<0,001 para no inferioridad; P=0,117 para superioridad). Los resultados para las variables secundarias analizadas en orden de importancia en el análisis de intención por tratar se muestran en la tabla 3.
En los pacientes del grupo tratado con warfarina, los valores del INR estuvieron dentro del rango terapéutico de entre 2,0 y 3,0 un promedio del 55% del tiempo (mediana, 58%; rango intercuartil, 43 a 71). El efecto de rivaroxaban no difirió según el grado de control del TTR (tiempo dentro del rango objetivo de INR entre 2,0-3,0) de los centros en los cuartiles con igual tamaño (P=0,74 para la interacción). En el cuartil más alto con respecto al control de los centros, el cociente de riesgos (hazard ratio) de rivaroxaban con respecto a warfarina fue de 0,74 (95% IC, 0,49 a 1,12).
Las tasas de incidencia de la variable principal de seguridad (episodios de hemorragia mayor y no mayor clínicamente relevante) fueron similares en ambos grupos de tratamiento (ver la tabla 4).
Tabla 3: Resultados de eficacia del estudio de fase III ROCKET AF.
|
Población del estudio |
Análisis ITT de la eficacia en pacientes con fibrilación auricular no valvular | ||
|
Pauta de tratamiento |
Xarelto 20 mg una vez al día (15 mg una vez al día en pacientes con insuficiencia renal moderada) Tasa de acontecimientos (100 pacientes-años) |
Warfarina ajustada hasta un objetivo de INR de 2,5 (rango terapéutico 2,0 a 3,0) Tasa de acontecimientos (100 pacientes-años) |
Cociente de riesgos (hazard ratio) (IC 95%) valor de p, prueba de superioridad |
|
Ictus y embolia sistémica sin afectación del SNC |
269 (2,12) |
306 (2,42) |
0.88 (0.74 - 1.03) 0.117 |
|
Ictus, embolia sistémica |
572 (4.51) |
609 (4.81) |
0.94 |
|
sin afectación del SNC y |
(0.84 - 1.05) | ||
|
muerte vascular |
0.265 | ||
|
Ictus, embolia sistémica |
709 |
0.93 (0.83 - 1.03) 0.158 | |
|
sin afectación del SNC, muerte vascular e infarto de miocardio |
659 (5.24) |
(5.65) | |
|
253 |
281 |
0.90 | |
|
Ictus |
(1.99) |
(2.22) |
(0.76 - 1.07) |
|
0.221 | |||
|
Embolia sistémica sin afectación del SNC |
20 (0.16) |
27 (0.21) |
0.74 (0.42 - 1.32) 0.308 |
|
130 (1.02) |
142 (111) |
0.91 | |
|
Infarto de miocardio |
(0.72 - 1.16) 0.464 |
Tabla 4: Resultados de seguridad del estudio de fase III ROCKET AF.
|
Población del estudio |
Pacientes con fibrilación auricular no valvular^ | ||
|
Pauta de tratamiento |
Xarelto 20 mg una vez al día (15 mg una vez al día en pacientes con insuficiencia renal moderada) |
Warfarina ajustada hasta un objetivo de INR de 2,5 (rango terapéutico de 2,0 a 3,0) |
Cociente de riesgos (hazard ratio) (IC 95%) valor de p |
|
Tasa de acontecimientos (100 pacientes-años) |
Tasa de acontecimientos (100 pacientes-años) | ||
|
Hemorragia mayor y no mayor clínicamente relevante |
1.475 (14,91) |
1.449 (14,52) |
1,03 (0,96 - 1,11) 0,442 |
|
Hemorragia mayor |
395 (3,60) |
386 (3,45) |
1,04 (0,90 - 1,20) 0,576 |
|
Muerte causada por hemorragia* |
27 (0,24) |
55 (0,48) |
0,50 (0,31 - 0,79) 0,003 |
|
Hemorragia en órgano crítico* |
91 (0,82) |
133 (1,18) |
0,69 (0,53 - 0,91) 0,007 |
|
Hemorragia intracraneal* |
55 (0,49) |
84 (0,74) |
0,67 (0,47 - 0,93) 0,019 |
|
Descenso de hemoglobina* |
305 (2,77) |
254 (2,26) |
1,22 (1,03 - 1,44) 0,019 |
|
Transfusión de 2 o más unidades de concentrado de hematíes o sangre total* |
183 (1,65) |
149 (1,32) |
1,25 (1,01 - 1,55) 0,044 |
|
Población del estudio |
Pacientes con fibrilación auricular no valvular^ | ||
|
Pauta de tratamiento |
Xarelto 20 mg una vez al día (15 mg una vez al día en pacientes con insuficiencia renal moderada) Tasa de acontecimientos (100 pacientes-años) |
Warfarina ajustada hasta un objetivo de INR de 2,5 (rango terapéutico de 2,0 a 3,0) Tasa de acontecimientos (100 pacientes-años) |
Cociente de riesgos (hazard ratio) (IC 95%) valor de p |
|
Hemorragia no mayor |
1.185 |
1.151 |
1,04 (0,96 - 1,13) |
|
clínicamente relevante |
(11,80) |
(11,37) |
0,345 |
|
Mortalidad por cualquier |
208 |
250 |
0.85 (0.70 - 1.02) |
|
causa |
(1.87) |
(2.21) |
0.073 |
a) Población de seguridad, durante el tratamiento
* Nominalmente significativo
Además del estudio de fase III ROCKET AF, se ha realizado un estudio prospectivo de cohortes, de un solo brazo, posautorización, no intervencionista, abierto (XANTUS) con adjudicación central de resultados, incluyendo acontecimientos tromboembólicos y hemorragia mayor. Se reclutaron 6.785 pacientes con fibrilación auricular no valvular para la prevención del ictus y de la embolia sistémica fuera del sistema nervioso central (SNC) en condiciones de práctica clínica. En XANTUS, las puntuaciones medias de CHADS2 y HAS-BLED fueron ambas de 2,0, en comparación con la puntuación media de CHADS2 y HAS-BLED de 3,5 y 2,8, respectivamente, en ROCKET AF. Se produjo hemorragia mayor en 2,1 por 100 pacientes-años. Se notificó hemorragia mortal en 0,2 por 100 pacientes-años y hemorragia intracraneal en 0,4 por 100 pacientes-años. Se registró ictus o embolia sistémica fuera del SNC en 0,8 por 100 pacientes-años.
Estas observaciones en condiciones de práctica clínica son consistentes con el perfil de seguridad establecido en esta indicación.
Pacientes sometidos a cardioversión
Se realizó un estudio exploratorio aleatorizado, prospectivo, abierto, multicéntrico con evaluación ciega de las variables (X-VERT) en 1.504 pacientes (con y sin tratamiento anticoagulante oral previo) con fibrilación auricular no valvular programada para cardioversión, para comparar rivaroxaban vs. dosis ajustadas de AVK (aleatorización 2: 1), para la prevención de acontecimientos cardiovasculares. Se utilizaron dos estrategias: cardioversión guiada por ETE (de 1 a 5 días de pre-tratamiento) o cardioversión convencional (mínimo tres semanas de pre-tratamiento). Se produjeron acontecimientos en la variable principal de eficacia (compuesta por todos los ictus, ataque isquémico transitorio, embolia sistémica fuera del SNC, infarto de miocardio y muerte cardiovascular) en 5 pacientes (0,5%) en el grupo de rivaroxaban (n = 978) y 5 pacientes (1,0%) en el grupo de AVK (n = 492; RR 0,50; IC 95%: 0,15 - 1,73; población ITT modificada). Se produjeron acontecimientos en la variable principal de seguridad (hemorragia mayor) en 6 (0,6%) y 4 (0,8%) pacientes en el grupo de rivaroxaban (n = 988) y AVK (n = 499), respectivamente (RR 0,76; IC 95% 0,21-2,67 ; población de seguridad). Este estudio exploratorio mostró una eficacia y seguridad comparables entre los grupos de tratamiento con rivaroxaban y con AVK en la cardioversión.
Tratamiento de la TVP, de la EP y prevención de las recurrencias de la TVP y de la EP
El programa clínico de Xarelto se diseñó para demostrar la eficacia de Xarelto en el tratamiento inicial
y continuado de la TVP aguda y de la EP y en la prevención de sus recurrencias.
En tres estudios clínicos de fase III aleatorizados y controlados (Einstein DVT, Einstein EP y Einstein Extension) se estudiaron más de 9.400 pacientes; adicionalmente, se realizó un análisis agrupado predefinido de los estudios Einstein DVT y Einstein PE. La duración combinada total del tratamiento en todos los estudios fue de 21 meses.
En el estudio Einstein DVT, se estudiaron 3.449 pacientes con TVP aguda para el tratamiento de la TVP y prevención de las recurrencias de la TVP y de la EP (se excluyeron los pacientes que presentaban EP sintomática). La duración del tratamiento fue de 3, 6 ó 12 meses, dependiendo del criterio clínico del investigador.
Para el tratamiento inicial de la TVP aguda se administró rivaroxaban 15 mg dos veces al día durante 3 semanas y a continuación, rivaroxaban 20 mg una vez al día.
En el estudio Einstein PE, se estudiaron 4.832 pacientes con EP aguda para el tratamiento de la EP y para la prevención de las recurrencias de TVP y EP. La duración del tratamiento fue de 3, 6 ó 12 meses, en función del juicio clínico del investigador.
Para el tratamiento inicial de EP aguda, se administró 15 mg de rivaroxaban dos veces al día durante tres semanas. Esta pauta fue seguida por 20 mg de rivaroxaban una vez al día.
En los dos estudios Einstein DVT y Einstein PE, el tratamiento comparador fue enoxaparina administrada durante al menos cinco días, en combinación con un antagonista de la vitamina K hasta que el TP/INR estuviera en rango terapéutico (> 2,0). El tratamiento se continuó con un antagonista de la vitamina K, con un ajuste de dosis para mantener los valores de TP/INR dentro del rango terapéutico de 2,0 a 3,0.
En el estudio Einstein Extension para la prevención de la TVP recurrente o de la EP se estudiaron 1.197 pacientes con TVP o EP. El tratamiento tuvo una duración adicional de 6 ó 12 meses en pacientes que previamente habían completado un periodo de 6 a 12 meses de tratamiento por TEV, en función del criterio clínico del investigador. Se comparó Xarelto 20 mg una vez al día con placebo.
Todos los estudios de fase III usaron las mismas variables principales y secundarias predefinidas de eficacia. La variable principal de eficacia fue el TEV sintomático y recurrente, definido como la combinación de TVP recurrente o bien EP mortal o no mortal. La variable secundaria de eficacia se definió como la combinación de TVP recurrente, EP no mortal y mortalidad por todas las causas.
En el estudio Einstein DVT (ver tabla 5) rivaroxaban demostró ser no inferior a enoxaparina / antagonista de la vitamina K (AVK) para la variable principal de eficacia (p < 0,0001 (prueba de no inferioridad); cociente de riesgos (hazard ratio): 0,680 (0,443 - 1,042), p = 0,076 (prueba de superioridad)). El beneficio clínico neto pre-especificado (variable principal de eficacia más episodios hemorrágicos mayores) mostró un cociente de riesgos (hazard ratio) de 0,67 ((IC 95%: 0,47 - 0,95), valor nominal de p = 0,027) en favor de rivaroxaban. Los valores del INR estuvieron dentro del rango terapéutico un promedio del 60,3% del tiempo para una duración media del tratamiento de 189 días, y del 55,4%, 60,1% y 62,8% del tiempo en los grupos con una duración prevista del tratamiento de 3, 6, y 12 meses, respectivamente. En el grupo de tratamiento con enoxaparina/AVK, no se observó una relación clara entre la media del TTR (tiempo dentro del rango objetivo de INR entre 2,0 - 3,0) del centro en los terciles de igual tamaño y la incidencia de TEV recurrente (p=0,932 para la interacción). En el tercil más alto según el control del centro, el cociente de riesgos (hazard ratio) de rivaroxaban con respecto a warfarina fue de 0,69 (IC 95%: 0,35 - 1,35).
Las tasas de incidencia de la variable principal de seguridad (hemorragia mayor o no mayor clínicamente relevante), así como la variable secundaria de seguridad (hemorragia mayor) fueron similares en ambos grupos de tratamiento.
|
Población del estudio |
3.449 pacientes con trombosis venosa profunda sintomática | |
|
Xareltoa) |
Enoxaparina/AVKb) | |
|
Pauta de tratamiento y duración |
3, 6 ó 12 meses |
3, 6 ó 12 meses |
|
N=1.731 |
N=1.718 | |
|
TEV sintomático recurrente* |
36 (2,1%) |
51 (3,0%) |
|
EP sintomática recurrente |
20 (1,2%) |
18 (1,0%) |
|
TVP sintomática recurrente |
14 (0,8%) |
28 (1,6%) |
|
EP y TVP sintomáticas |
1 (0,1%) |
0 |
|
EP mortal/Muerte en la que no |
4 |
6 |
|
puede descartarse EP |
(0,2%) |
(0,3%) |
|
Hemorragia mayor o no mayor |
139 |
138 |
|
clínicamente relevante |
(8,1%) |
(8,1%) |
|
Eventos hemorrágicos mayores |
14 (0,8%) |
20 (1,2%) |
|
a) Rivaroxaban 15 mg dos veces al día durante 3 semanas, seguido d |
e rivaroxaban 20 mg una vez | |
al día
b) Enoxaparina durante al menos 5 días, solapado con y seguido por AVK
* p <0,0001 (no-inferioridad; cociente de riesgos (hazard ratio) pre-especificado de 2,0); cociente de riesgos (hazard ratio): 0,680 (0,443 - 1,042), p = 0,076 (superioridad)
En el estudio Einstein PE (ver Tabla 6) rivaroxaban demostró ser no inferior a la enoxaparina/AVK para la variable primaria de eficacia (p = 0,0026 (prueba de no inferioridad); cociente de riesgos (hazard ratio): 1,123 (0,749-1,684)). El beneficio clínico neto pre-especificado (resultado de eficacia primaria más eventos de sangrado mayor) se reportó con un cociente de riesgos (hazard ratio) de 0,849 ((IC del 95%: 0,633 a 1,139), valor nominal de p = 0,275). Los valores de INR estuvieron dentro del rango terapéutico de una media de 63% del tiempo para la duración media del tratamiento de 215 días, y el 57%, 62%, y 65% del tiempo en los grupos de duración prevista de tratamiento de 3, 6, y 12 meses, respectivamente. En el grupo de enoxaparina/AVK, no hubo una relación clara entre el nivel de la media TRT del centro (Tiempo en objetivo de INR de 2,0 - 3,0) en los terciles de igual tamaño y la incidencia de la recurrencia de TEV (p = 0,082 para la interacción). En el tercil superior de acuerdo con el centro, el cociente de riesgos (hazard ratio) con rivaroxaban en comparación con warfarina fue 0,642 (IC 95%: 0,277 - 1,484).
Las tasas de incidencia de la variable principal de seguridad (eventos hemorrágicos mayores o no mayores clínicamente relevantes) fueron ligeramente inferiores en el grupo de tratamiento con rivaroxaban (10,3% (249/2412)) frente a las del grupo de tratamiento con enoxaparina/AVK (11,4% (274 / 2405)). La incidencia de las variables secundarias de seguridad (eventos de sangrado mayor) fue inferior en el grupo de rivaroxaban (1,1% (26/2412)) comparado con la de enoxaparina/grupo AVK (2,2% (52/2405)), con un cociente de riesgos (hazard ratio) 0,493 (95 % CI: 0,308 0,789).
|
Población del estudio |
4.832 pacientes con EP sintomática aguda | |
|
Xareltoa) |
Enoxaparina/VKAb) | |
|
Pauta de tratamiento y duración |
3, 6 ó 12 meses |
3, 6 ó 12 meses |
|
N=2.419 |
N=2.413 | |
|
TEV sintomático recurrente* |
50 (2,1%) |
44 (1,8%) |
|
EP sintomática recurrente |
23 (1,0%) |
20 (0,8%) |
|
TVP sintomática recurrente |
18 (0,7%) |
17 (0,7%) |
|
EP y TVP sintomáticas |
0 |
2 (<0,1%) |
|
EP mortal/Muerte en la que no |
11 |
7 |
|
puede descartarse EP |
(0,5%) |
(0,3%) |
|
Hemorragia mayor o no mayor |
249 |
274 |
|
clínicamente relevante |
(10,3%) |
(11,4%) |
|
Eventos hemorrágicos mayores |
26 (1,1%) |
52 (2,2%) |
|
a) Rivaroxaban 15 mg dos veces al día durante 3 semanas, seguido c |
e rivaroxaban 20 mg una vez | |
al día
b) Enoxaparina durante al menos 5 días, solapado con y seguido por AVK
* p <0,0026 (no-inferioridad; cociente de riesgos (hazard ratio) pre-especificado de 2,0); cociente de riesgos (hazard ratio): 1,123 (0,749 - 1,684)
Se realizó un análisis agrupado pre-especificado de los resultados de los estudios Einstein DVT y PE (ver Tabla 7).
Tabla 7: Resultados de eficacia y seguridad del análisis agrupado de los estudios de fase III Einstein DVT y Einstein PE__
|
Población del estudio |
8.281 pacientes con TVP sintomática aguda o EP | |
|
Xareltoa) |
Enoxaparin/VKAb) | |
|
Pauta de tratamiento y duración |
3, 6 ó 12 meses |
3, 6 ó 12 meses |
|
N=4.150 |
N=4.131 | |
|
TEV sintomático recurrente* |
86 (2,1%) |
95 (2,3%) |
|
EP sintomática recurrente |
43 (1,0%) |
38 (0,9%) |
|
TVP sintomática recurrente |
32 (0,8%) |
45 (1,1%) |
|
EP y TVP sintomáticas |
1 (<0,1%) |
2 (<0,1%) |
|
EP mortal/Muerte en la que no |
15 |
13 |
|
puede descartarse EP |
(0,4%) |
(0,3%) |
|
Hemorragia mayor o no mayor |
388 |
412 |
|
clínicamente relevante |
(9,4%) |
(10,0%) |
|
Eventos hemorrágicos mayores |
40 (1,0%) |
72 (1,7%) |
|
a) Rivaroxaban 15 mg dos veces al día durante 3 semanas, seguido d |
e rivaroxaban 20 mg una vez | |
al día
b) Enoxaparina durante al menos 5 días, solapado con y seguido por AVK * p <0,0001 (no-inferioridad; cociente de riesgos (hazard ratio) pre-especificado de 1,75); cociente de riesgos (hazard ratio): 0,886 (0,661 - 1,186)
El beneficio clínico neto pre-especificado (variable primaria de eficacia más eventos de sangrado mayor) del análisis agrupado se reportó con un cociente de riesgos (hazard ratio) de 0,771 ((IC95%: 0,614 - 0,967), valor nominal de p = 0,0244).
En el estudio Einstein Extension (ver tabla 8), rivaroxaban fue superior a placebo en cuanto a las variables principal y secundaria de eficacia. En cuanto a la variable principal de seguridad (hemorragia mayor) hubo una tasa de incidencia numéricamente superior no significativa en los pacientes tratados con rivaroxaban 20 mg una vez al día, en comparación con placebo. La variable secundaria de seguridad (hemorragia mayor o no mayor clínicamente relevante) mostró unas tasas más altas en los pacientes tratados con rivaroxaban 20 mg una vez al día, en comparación con placebo.
Tabla 8: Resultados de eficacia y seguridad del estudio de fase III Einstein Extension.
|
Población del estudio |
1.197 pacientes continuaron el tratamiento y la prevención del TEV recurrente | |
|
Xareltoa) |
Placebo | |
|
Pauta del tratamiento |
6 ó 12 meses |
6 ó 12 meses |
|
N = 602 |
N = 594 | |
|
TEV recurrente y sintomático* |
8 (1,3%) |
42 (7,1%) |
|
EP recurrente y sintomática |
2 (0,3%) |
13 (2,2%) |
|
TVP recurrente y |
5 |
31 |
|
sintomática |
(0,8%) |
(5,2%) |
|
EP mortal / Muerte en la que |
1 |
1 |
|
no se puede descartar EP |
(0,2%) |
(0,2%) |
|
Hemorragia mayor |
4 (0,7%) |
0 (0,0%) |
|
Hemorragia no mayor clínicamente |
32 |
7 |
|
relevante |
(5,4%) |
(1,2%) |
a) Rivaroxaban 20 mg una vez al día
* p < 0,0001 (superioridad), cociente de riesgos (hazard ratio): 0,185 (0,087 - 0,393)
Además del programa de fase III EINSTEIN, se ha realizado un estudio prospectivo de cohortes, no intervencionista, abierto (XALIA) con adjudicación central de resultados, incluyendo TEV recurrente, hemorragia mayor y muerte. Se reclutaron 5.142 pacientes con TVP aguda para investigar la seguridad a largo plazo de rivaroxaban, en comparación con el tratamiento anticoagulante de referencia, en condiciones de práctica clínica. Las tasas de hemorragia mayor, TEV recurrente y mortalidad por cualquier causa para rivaroxaban fueron de 0,7%, 1,4% y 0,5%, respectivamente. Se hallaron diferencias en las características iniciales de los pacientes, incluyendo edad, cáncer e insuficiencia renal. Se realizó un análisis predefinido utilizando el índice de propensión estratificado para ajustar las diferencias en las características iniciales medidas pero, a pesar de esto, la confusión residual puede influir en los resultados. Los cocientes de riesgos (hazard ratios) ajustados que compararon rivaroxaban con el tratamiento de referencia para la hemorragia mayor, TEV recurrente y mortalidad por cualquier causa fueron de 0,77 (IC 95% 0,40 - 1,50), 0,91 (IC 95% 0,54 - 1,54) y 0,51 (IC 95% 0,24 - 1,07), respectivamente.
Estos resultados en condiciones de práctica clínica son consistentes con el perfil de seguridad establecido en esta indicación.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Xarelto en uno o más grupos de la población pediátrica en el tratamiento de eventos tromboembólicos. La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Xarelto en los diferentes grupos de la población pediátrica en la prevención de eventos tromboembólicos (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Rivaroxaban se absorbe rápidamente y alcanza concentraciones máximas (Cmax) de 2 a 4 horas después de tomar el comprimido.
La absorción oral de rivaroxaban es casi completa y su biodisponibilidad oral es elevada (80% al 100%) en el caso de la dosis del comprimido de 2,5 mg y de 10 mg, independientemente de las condiciones de ayuno o alimentación. La ingesta de alimentos con rivaroxaban (a la dosis de 2,5 mg y de 10 mg) no afecta al AUC ni a la Cmax.
Debido a la disminución de la absorción, se determinó una biodisponibilidad del 66% con el comprimido de 20 mg en condiciones de ayuno. Cuando los comprimidos de Xarelto 20 mg se tomaron junto con alimentos, se observaron aumentos del AUC media del 39% en comparación con la toma de comprimidos en condiciones de ayuno, lo que indica una absorción casi completa y una biodisponibilidad oral elevada. Xarelto 15 mg y 20 mg deben tomarse con alimentos (ver sección 4.2). Rivaroxaban presenta una farmacocinética lineal hasta aproximadamente 15 mg administrados una vez al día en ayunas. En condiciones de alimentación reciente, Xarelto en comprimidos de 10 mg, 15 mg y 20 mg demostró proporcionalidad con la dosis. A dosis más altas, rivaroxaban muestra una disolución limitada, con una reducción de la biodisponibilidad y de la tasa de absorción al aumentar la dosis.
La variabilidad de la farmacocinética de rivaroxaban es moderada; con una variabilidad interindividual (CV%) entre el 30% y el 40%.
La absorción de rivaroxaban depende del sitio donde se libera en el tracto gastrointestinal. Se ha notificado una disminución del 29% y del 56% en el AUC y la Cmax, en comparación con el comprimido, cuando rivaroxaban en forma de granulado se liberó en el intestino delgado proximal. La exposición se reduce aún más cuando rivaroxaban se libera en el intestino delgado distal o en el colon ascendente. Por lo tanto, debe evitarse la administración de rivaroxaban de forma distal al estómago, ya que esto puede dar lugar a una reducción de la absorción y la correspondiente exposición a rivaroxaban.
La biodisponibilidad (AUC y Cmax) fue comparable para rivaroxaban 20 mg, administrado por vía oral como comprimido triturado y mezclado con puré de manzana o diluido con agua, administrado a través de una sonda gástrica y seguido de una comida líquida, en comparación con el comprimido entero. Dado el perfil farmacocinético predecible, proporcional a la dosis de rivaroxaban, los resultados de biodisponibilidad de este estudio son probablemente aplicables a dosis más bajas de rivaroxaban.
Distribución
La unión a proteínas plasmáticas humanas es alta, del 92 % al 95 % aproximadamente y la albúmina sérica es el principal componente de unión. El volumen de distribución es moderado, con un Vss de 50 litros, aproximadamente.
Biotransformación y eliminación
De la dosis administrada de rivaroxaban se metabolizan aproximadamente 2/3; después, la mitad se elimina por vía renal y la otra mitad por vía fecal. El 1/3 restante de la dosis administrada se excreta directamente por vía renal como principio activo no modificado en la orina, principalmente mediante secreción renal activa.
Rivaroxaban se metaboliza mediante el CYP3A4, el CYP2J2 y mecanismos independientes del CYP. Las principales vías de biotransformación son la degradación oxidativa de la porción de morfolinona y la hidrólisis de los enlaces amida. Según investigaciones in vitro, rivaroxaban es un sustrato de las proteínas transportadoras P-gp (glucoproteína P) y Bcrp (proteína de resistencia al cáncer de mama). Rivaroxaban en forma inalterada es el compuesto más abundante en el plasma humano sin presencia de metabolitos mayores o metabolitos activos circulantes. Con un aclaramiento sistémico de aproximadamente 10 l/h, rivaroxaban puede clasificarse como una sustancia de bajo aclaramiento. Después de la administración por vía intravenosa de una dosis de 1 mg, la semivida de eliminación es de aproximadamente 4,5 horas. Después de la administración por vía oral, la eliminación se ve limitada por la tasa de absorción. En personas jóvenes, la eliminación de rivaroxaban del plasma se produce con una semivida de eliminación de 5 a 9 horas y en personas de edad avanzada, con una semivida de eliminación de 11 a 13 horas.
Poblaciones especiales
Sexo
No hubo ninguna diferencia clínicamente relevante en las propiedades farmacocinéticas y farmacodinámicas entre hombres y mujeres.
Pacientes de edad avanzada
Los pacientes de edad avanzada presentaron concentraciones plasmáticas mayores que los pacientes más jóvenes, con unos valores medios del AUC que fueron aproximadamente 1,5 veces superiores, principalmente debido a la disminución (aparente) del aclaramiento renal y total. No es necesario un ajuste de la dosis.
Peso corporal
Los valores extremos en el peso corporal (< 50 kg ó > 120 kg) tuvieron poco efecto en las concentraciones plasmáticas de rivaroxaban (menos del 25%). No es necesario un ajuste de la dosis.
Origen étnico
No se observaron diferencias interétnicas clínicamente relevantes entre los pacientes de raza blanca, afroamericanos, de origen latinoamericano, japonés o chino, en cuanto a las propiedades farmacocinéticas o farmacodinámicas.
Insuficiencia hepática
Los pacientes cirróticos con insuficiencia hepática leve (clasificados como Child Pugh A), sólo presentaron cambios menores en la farmacocinética de rivaroxaban (aumento medio del AUC de 1,2 veces), lo que fue casi comparable al grupo control de voluntarios sanos. En los pacientes cirróticos con insuficiencia hepática moderada (clasificados como Child Pugh B), el AUC media de rivaroxaban estuvo aumentada significativamente en 2,3 veces, en comparación con los voluntarios sanos. El AUC parcial aumentó 2,6 veces. Estos pacientes también mostraron una disminución de la eliminación renal de rivaroxaban, similar a los pacientes con insuficiencia renal moderada. No hay datos en pacientes con insuficiencia hepática grave.
La inhibición de la actividad del factor Xa se incrementó en un factor de 2,6 en los pacientes con insuficiencia hepática moderada, en comparación con los voluntarios sanos; de manera similar, la prolongación del TP se incrementó en un factor de 2,1. Los pacientes con insuficiencia hepática moderada fueron más sensibles a rivaroxaban, lo que produjo una relación farmacocinética / farmacodinámica más pronunciada entre la concentración y el TP.
Xarelto está contraindicado en pacientes con hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia, incluyendo pacientes cirróticos clasificados como Child Pugh B y C (ver sección 4.3).
Insuficiencia renal
Se observó un aumento de la exposición a rivaroxaban correlacionado con la disminución de la función renal, evaluada mediante las determinaciones del aclaramiento de creatinina. En sujetos con insuficiencia renal leve (aclaramiento de creatinina de 50 - 80 ml/min), moderada (aclaramiento de creatinina de 30 a 49 ml/min) o grave (aclaramiento de creatinina de 15 a 29 ml/min), las concentraciones plasmáticas de rivaroxaban (AUC) aumentaron 1,4, 1,5 y 1,6 veces, respectivamente. Los aumentos correspondientes en los efectos farmacodinámicos fueron más pronunciados. En sujetos con insuficiencia renal leve, moderada y grave, la inhibición total de la actividad del factor Xa aumentó en un factor de 1,5, 1,9 y 2,0 respectivamente, en comparación con voluntarios sanos; de manera similar, la prolongación del TP aumentó en factores de 1,3, 2,2 y 2,4, respectivamente. No hay datos en pacientes con un aclaramiento de creatinina < 15 ml/min.
Debido a la elevada fijación a proteínas plasmáticas, no se espera que rivaroxaban sea dializable.
No se recomienda su uso en pacientes con un aclaramiento de creatinina < 15 ml/min. Xarelto debe utilizarse con precaución en pacientes con un aclaramiento de creatinina de 15 a 29 ml/min (ver sección 4.4).
Datos farmacocinéticos en pacientes
En los pacientes que recibieron rivaroxaban 20 mg una vez al día para el tratamiento de la TVP aguda, la concentración media geométrica (intervalo de predicción del 90%) a las 2 - 4 h y a las 24 h aproximadamente después de la dosis (lo que representa aproximadamente las concentraciones máxima y mínima durante el intervalo entre dosis) fue de 215 (22 - 535) y de 32 (6 - 239) ^g/l, respectivamente.
Relación farmacocinética / farmacodinámica
Se ha evaluado la relación farmacocinética/farmacodinámica (PK/PD) entre la concentración plasmática de rivaroxaban y varios criterios de valoración PD (inhibición del factor Xa, tiempo de protrombina (TP, TTPa, Heptest)) después de la administración de un amplio rango de dosis (de 5 a 30 mg dos veces al día). La relación entre la concentración de rivaroxaban y la actividad del factor Xa se describió de manera óptima por un modelo Emax. En el caso del TP, por lo general, el modelo de intersección lineal describió mejor los datos. Dependiendo de los diferentes reactivos usados en el TP, la pendiente varió considerablemente. Con Neoplastin PT, el TP basal fue de aproximadamente 13 seg. y la pendiente fue de alrededor de 3 a 4 seg/(100 ^g/l). Los resultados de los análisis de la relación PK/PD en las fases II y III fueron consistentes con los datos establecidos en los sujetos sanos.
Población pediátrica
No se ha determinado la seguridad y eficacia en niños y adolescentes hasta los 18 años.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad con dosis únicas, fototoxicidad, genotoxicidad, potencial carcinogénico y toxicidad juvenil.
Los efectos observados en los estudios con dosis repetidas se debieron principalmente a la actividad farmacodinámica incrementada de rivaroxaban. En ratas se observó un aumento de las concentraciones plasmáticas de IgG e IgA a niveles de exposición clínicamente relevantes.
No se observó ningún efecto sobre la fertilidad en las ratas macho o hembra. Los estudios en animales han demostrado una toxicidad reproductiva relacionada con el modo de acción farmacológica de rivaroxaban (p. ej. complicaciones hemorrágicas). A concentraciones plasmáticas clínicamente relevantes se observó toxicidad embriofetal (pérdida después de la implantación, retraso o adelanto de la osificación, varias manchas hepáticas de color claro) y un aumento de la incidencia de malformaciones frecuentes, así como cambios placentarios. En el estudio pre y postnatal en ratas, se observó una disminución de la viabilidad de las crías a dosis que fueron tóxicas para las madres.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido:
Celulosa microcristalina Croscarmelosa sódica Lactosa monohidrato Hipromelosa Laurilsulfato de sodio Estearato de magnesio
Cubierta pelicular:
Macrogol 3350
Hipromelosa
Dióxido de titanio (E171)
Óxido de hierro rojo (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez 3 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Blister de PP / lámina de aluminio, en envases de 10, 14, 28, 42 ó 98 comprimidos recubiertos con película, o blisters precortados unidosis en envases de 10 x 1, 100 x 1 ó en envases múltiples que contienen 100 (10 estuches de 10 x 1) comprimidos recubiertos con película.
Frascos de HDPE con tapón de rosca de PP que contienen 100 comprimidos recubiertos.
Blisters de PP/ lámina de aluminio, en un envase (con calendario) que contiene 42 comprimidos recubiertos con película de Xarelto 15 mg y 7 comprimidos recubiertos con película de Xarelto 20 mg (envase para el inicio del tratamiento).
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
Ninguna especial para su eliminación
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG D-13342 Berlin Alemania
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/011-016, EU/1/08/472/023, EU/1/08/472/036, EU/1/08/472/038, EU/1/08/472/040
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 30 de septiembre de 2008 Fecha de la última renovación: 22 de mayo de 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
{MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
▼ Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Xarelto 20 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 20 mg de rivaroxaban.
Excipiente con efecto conocido:
Cada comprimido recubierto con película contiene 21,76 mg de lactosa (como monohidrato), ver sección 4.4.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
Comprimidos de color pardo rojizo, redondos y biconvexos (6 mm de diámetro y 9 mm de radio de curvatura), con la cruz de BAYER en una cara, y “20” y un triángulo en la otra cara.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Prevención del ictus y de la embolia sistémica en pacientes adultos con fibrilación auricular no valvular, con uno o más factores de riesgo, como por ejemplo, insuficiencia cardiaca congestiva, hipertensión, edad > 75 años, diabetes mellitus, ictus o ataque isquémico transitorio previos.
Tratamiento de la trombosis venosa profunda (TVP) y de la embolia pulmonar (EP, y prevención de las recurrencias de la TVP y de la EP en pacientes adultos. (Ver en sección 4.4 información sobre pacientes con EP hemodinámicamente inestables.)
4.2 Posología y forma de administración
Posología
Prevención del ictus y de la embolia sistémica
La dosis recomendada es de 20 mg de rivaroxaban una vez al día, que es también la dosis máxima recomendada.
El tratamiento con Xarelto debe continuarse a largo plazo siempre que el beneficio de la prevención del ictus y de la embolia sistémica sea superior al riesgo de hemorragia (ver sección 4.4).
Si se olvida una dosis, el paciente debe tomar inmediatamente Xarelto y seguir al día siguiente con la dosis de una vez al día recomendada. La dosis no debe duplicarse en el mismo día para compensar una dosis olvidada.
Tratamiento de la TVP, tratamiento de la EP y prevención de las recurrencias de la TVP y de la EP La dosis recomendada para el tratamiento inicial de la TVP aguda o de la EP es de 15 mg dos veces al día, durante las tres primeras semanas, seguida de 20 mg una vez al día para el tratamiento continuado así como para la prevención de las recurrencias de la TVP y de la EP, según se indica en la tabla siguiente.
|
Pauta de tratamiento |
Dosis máxima diaria | |
|
Días 1 a 21 |
15 mg dos veces al día |
30 mg |
|
Día 22 en adelante |
20 mg una vez al día |
20 mg |
Para facilitar el cambio de dosis de 15 mg a 20 mg después del Día 21, está disponible un envase para el inicio del tratamiento de Xarelto en las primeras 4 semanas para el tratamiento de la TVP / EP (ver sección 6.5).
La duración del tratamiento debe individualizarse después de una evaluación minuciosa del beneficio del tratamiento frente al riesgo de hemorragia (ver sección 4.4). La duración corta del tratamiento (como mínimo de 3 meses) debe basarse en factores de riesgo transitorios (p.ej. cirugía reciente, traumatismo, inmovilización) y las duraciones más largas deben basarse en factores de riesgo permanente o TVP idiopática o de EP.
Si el paciente olvida una dosis durante la fase de tratamiento de 15 mg dos veces al día (días 1 a 21), éste deberá tomar inmediatamente Xarelto para garantizar una toma de 30 mg de Xarelto al día. En este caso, se pueden tomar dos comprimidos de 15 mg a la vez y al día siguiente se deberá seguir con la pauta habitual recomendada de 15 mg dos veces al día.
Si el paciente olvida una dosis durante la fase de tratamiento de una vez al día (día 22 en adelante), deberá tomar inmediatamente Xarelto, y seguir al día siguiente con la pauta recomendada de una vez al día. La dosis no debe duplicarse en el mismo día para compensar una dosis olvidada.
Cambio de tratamiento con antagonistas de la vitamina K (AVK) a Xarelto En el caso de pacientes tratados para la prevención del ictus y de la embolia sistémica, deberá interrumpirse el tratamiento con AVK e iniciarse el tratamiento con Xarelto cuando el valor del INR (International Normalized Ratio) sea < 3,0.
En el caso de pacientes tratados por TVP, EP y en la prevención de sus recurrencias, deberá interrumpirse el tratamiento con AVK e iniciarse el tratamiento con Xarelto cuando el valor del INR sea < 2,5.
Al cambiar el tratamiento con AVK a Xarelto, los valores de INR del paciente estarán falsamente elevados después de la toma de Xarelto. El INR no es un parámetro válido para medir la actividad anticoagulante de Xarelto, por lo que no debe utilizarse (ver sección 4.5).
Cambio de tratamiento con Xarelto a antagonistas de la vitamina K (AVK)
Existe la posibilidad de una incorrecta anticoagulación durante la transición de Xarelto a AVK. Debe garantizarse una anticoagulación adecuada y continua durante cualquier transición a un anticoagulante alternativo. Debe señalarse que Xarelto puede contribuir a un aumento del INR.
En los pacientes que cambien de Xarelto a AVK, estos tratamientos deben administrarse simultáneamente hasta que el INR sea > 2,0. Durante los dos primeros días del periodo de cambio se utilizará la dosis inicial estándar de AVK, que se ajustará posteriormente en función de los resultados del INR. Mientras los pacientes están bajo tratamiento con Xarelto y AVK el INR puede determinarse a partir de las 24 horas que siguen a la dosis de Xarelto y siempre antes de la siguiente dosis. Una vez interrumpido el tratamiento con Xarelto, el INR puede determinarse con fiabilidad pasadas 24 horas de la última dosis. (ver secciones 4.5 y 5.2).
Cambio de tratamiento con anticoagulante parenteral a Xarelto
Los pacientes que están recibiendo un anticoagulante por vía parenteral, deben interrumpir el tratamiento anticoagulante por vía parenteral e iniciar el tratamiento con Xarelto de 0 a 2 horas antes de la siguiente administración programada del medicamento por vía parenteral (p. ej., heparina de bajo peso molecular). En el caso de un anticoagulante parenteral administrado por perfusión continua (p. ej., heparina no fraccionada intravenosa) Xarelto deberá administrarse en el momento de la suspensión del anticoagulante parenteral.
Cambio de tratamiento con Xarelto a anticoagulante parenteral
La primera dosis de anticoagulante parenteral debe administrarse en el momento en que se tomaría la siguiente dosis de Xarelto.
Poblaciones especiales
Insuficiencia renal
Los escasos datos clínicos en pacientes con insuficiencia renal grave (aclaramiento de creatinina de 15 a 29 ml/min) indican que las concentraciones plasmáticas de rivaroxaban aumentan significativamente. Por lo tanto, Xarelto se debe usar con precaución en estos pacientes. No se recomienda su uso en pacientes con un aclaramiento de creatinina < 15 ml/min (ver secciones 4.4 y 5.2).
En pacientes con insuficiencia renal moderada (aclaramiento de creatinina de 30 a 49 ml/min) o grave (aclaramiento de creatinina de 15 a 29 ml/min) se recomiendan las siguientes pautas posológicas:
- Para la prevención del ictus y de la embolia sistémica en pacientes con fibrilación auricular no valvular, la dosis recomendada es de 15 mg una vez al día (ver sección 5.2).
- Para el tratamiento de la TVP, y de la EP, y la prevención de las recurrencias de la TVP y de la EP se debe tratar a los pacientes con 15 mg dos veces al día durante las tres primeras semanas. Después, la dosis recomendada es de 20 mg una vez al día Deberá considerarse una reducción de la dosis de 20 mg una vez al día a 15 mg una vez al día si el riesgo de sangrado valorado en el paciente supera el riesgo de recurrencia de TVP y de EP. La recomendación para el uso de
15 mg se basa en el modelo farmacocinético que no se ha estudiado en este contexto clínico (ver secciones 4.4, 5.1 y 5.2).
No se requiere ajuste de dosis en pacientes con insuficiencia renal leve (aclaramiento de creatinina de 50 a 80 ml/min) (ver sección 5.2).
Insuficiencia hepática
Xarelto está contraindicado en pacientes con hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia incluidos los pacientes cirróticos con Child Pugh B y C (ver secciones 4.3 y 5.2).
Pacientes de edad avanzada
No se requiere ajuste de dosis (ver sección 5.2).
Peso corporal
No se requiere ajuste de dosis (ver sección 5.2).
Sexo
No se requiere ajuste de dosis (ver sección 5.2).
Población pediátrica
No se ha establecido la seguridad y eficacia de Xarelto en niños de 0 a 18 años. No se dispone de datos. Por lo tanto, no se recomienda el uso de Xarelto en niños menores de 18 años.
Pacientes sometidos a cardioversión
El tratamiento con Xarelto se puede iniciar o continuar en pacientes que requieran cardioversión.
Para una cardioversión guiada por ecocardiografía transesofágica (ETE) en pacientes no tratados previamente con anticoagulantes, el tratamiento con Xarelto debe iniciarse al menos 4 horas antes de la cardioversión para asegurar una anticoagulación adecuada (ver secciones 5.1 y 5.2). En todos los pacientes, se deberá confirmar antes de la cardioversión que el paciente ha tomado Xarelto según lo prescrito. En las decisiones sobre inicio y duración del tratamiento, se tendrán en cuenta las recomendaciones de las guías establecidas para el tratamiento anticoagulante en pacientes sometidos a cardioversión.
Forma de administración Vía oral.
Los comprimidos deben administrarse con alimentos (ver sección 5.2).
Para aquellos pacientes que no puedan tragar el comprimido entero, el comprimido de Xarelto puede triturarse y mezclarse con agua o con puré de manzana inmediatamente antes de su uso y administrarse por vía oral. Inmediatamente después de la administración del comprimido triturado se debe administrar el alimento.
El comprimido triturado también se puede administrar a través de sonda gástrica una vez se haya confirmado la colocación correcta de la sonda. El comprimido triturado se administrará diluido con una pequeña cantidad de agua a través de la sonda gástrica, procediendo seguidamente a un lavado adicional de la sonda con agua. Inmediatamente después de la administración del comprimido triturado se debe administrar el alimento mediante sonda gástrica (ver sección 5.2).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hemorragia activa clínicamente significativa.
Lesión o enfermedad, si se considera que tiene un riesgo significativo de sangrado mayor. Esto puede incluir úlcera gastrointestinal activa o reciente, presencia de neoplasias malignas con alto riesgo de sangrado, traumatismo cerebral o espinal reciente, cirugía cerebral, espinal u oftálmica reciente, hemorragia intracraneal reciente, conocimiento o sospecha de varices esofágicas, malformaciones arteriovenosas, aneurismas vasculares o anomalías vasculares intraespinales o intracerebrales mayores.
Tratamiento concomitante con cualquier otro anticoagulante, p. ej. heparina no fraccionada (HNF), heparinas de bajo peso molecular (enoxaparina, dalteparina, etc.), derivados de la heparina (fondaparinux, etc.), anticoagulantes orales (warfarina, dabigatran etexilato, apixaban, etc.) excepto bajo las circunstancias concretas de cambio de tratamiento anticoagulante (ver sección 4.2) o cuando se administre HNF a las dosis necesarias para mantener un catéter venoso o arterial central abierto (ver sección 4.5).
Hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia, incluidos los pacientes cirróticos con Child Pugh B y C (ver sección 5.2).
Embarazo y lactancia (ver sección 4.6).
4.4 Advertencias y precauciones especiales de empleo
Durante todo el periodo de tratamiento se recomienda una estrecha monitorización clínica del paciente, en línea con la práctica de anticoagulación
Riesgo de hemorragia
Al igual que con otros anticoagulantes, los pacientes que toman Xarelto deben ser observados cuidadosamente para detectar signos de sangrado. Se recomienda utilizar con precaución en condiciones que conlleven un riesgo incrementado de hemorragia. La administración de Xarelto debe interrumpirse si se produce una hemorragia grave.
En los ensayos clínicos se observaron con más frecuencia hemorragias a nivel de mucosas (p.ej. epistaxis, gingival, gastrointestinal, génito-urinaria) y anemia en los pacientes que recibían rivaroxaban a largo plazo respecto a los que recibían tratamiento con AVK. Por ello, además de un seguimiento clínico adecuado, las determinaciones de hemoglobina y hematocrito podrían ser útiles para detectar hemorragias ocultas cuando se considere apropiado.
Varios subgrupos de pacientes, como se explica posteriormente, presentan un mayor riesgo de hemorragia. En estos pacientes se debe vigilar cuidadosamente la presencia de signos y síntomas de complicaciones hemorrágicas y de anemia después del inicio del tratamiento (ver sección 4.8). Cualquier disminución inexplicada de los niveles de hemoglobina o de la presión arterial requerirá la búsqueda de una zona de sangrado.
Aunque el tratamiento con rivaroxaban no requiere una monitorización rutinaria de la exposición, la determinación de los niveles de rivaroxaban mediante un ensayo anti-factor Xa cuantitativo calibrado puede ser útil en situaciones excepcionales, en las que el conocimiento de la exposición a rivaroxaban puede ayudar en la toma de decisiones clínicas, como por ejemplo, en caso de sobredosis o cirugía de emergencia (ver secciones 5.1 y 5.2).
Insuficiencia renal
En pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min), las concentraciones plasmáticas de rivaroxaban podrían aumentar de forma significativa (en promedio,
1,6 veces), lo que conllevaría un aumento del riesgo de hemorragia. Xarelto debe utilizarse con precaución en pacientes con un aclaramiento de creatinina de 15 a 29 ml/min. No se recomienda el uso en pacientes con un aclaramiento de creatinina < 15 ml/min (ver secciones 4.2 y 5.2).
Xarelto debe usarse con precaución en pacientes con insuficiencia renal y que reciben de forma concomitante otros medicamentos que aumenten las concentraciones plasmáticas de rivaroxaban (ver sección 4.5).
Interacción con otros medicamentos
No se recomienda utilizar Xarelto en pacientes que reciben tratamiento sistémico concomitante con antimicóticos azólicos (p. ej., ketoconazol, itraconazol, voriconazol y posaconazol) o inhibidores de la proteasa del VIH (p. ej., ritonavir). Estos principios activos son inhibidores potentes del CYP3A4 y de la P-gp y por lo tanto pueden aumentar las concentraciones plasmáticas de rivaroxaban hasta un nivel clínicamente relevante (en promedio, 2,6 veces), lo que puede llevar a un aumento del riesgo de hemorragia (ver sección 4.5).
Debe tenerse precaución si los pacientes reciben tratamiento concomitante con medicamentos que afectan a la hemostasia, como los antiinflamatorios no esteroideos (AINEs), ácido acetilsalicílico e inhibidores de la agregación plaquetaria. Puede considerarse el uso de un tratamiento profiláctico adecuado en aquellos pacientes con riesgo de enfermedad gastrointestinal ulcerosa, (ver sección 4.5).
Otros factores de riesgo hemorrágico
Al igual que con otros agentes antitrombóticos, rivaroxaban no está recomendado en pacientes con un riesgo aumentado de hemorragia, tales como:
• trastornos de la coagulación, congénitos o adquiridos
• hipertensión arterial grave no controlada
• otra enfermedad gastrointestinal sin úlcera activa que pueda producir complicaciones hemorrágicas (por ejemplo, enfermedad inflamatoria intestinal, esofagitis, gastritis o reflujo gastroesofágico)
• retinopatía vascular
• bronquiectasia o antecedentes de hemorragia pulmonar Pacientes con prótesis valvulares
No se ha estudiado la seguridad y eficacia de Xarelto en pacientes con prótesis valvulares cardiacas; por lo tanto, no hay datos que apoyen que Xarelto 20 mg (15 mg en los pacientes con insuficiencia renal moderada o grave) proporciona una anticoagulación adecuada en esta población. No se recomienda el tratamiento con Xarelto en estos pacientes.
Pacientes con EP hemodinámicamente inestables o pacientes que requieran trombolisis o embolectomía pulmonar
Xarelto no está recomendado como una alternativa a la heparina no fraccionada en pacientes con embolia pulmonar que están hemodinámicamente inestables o que puedan ser sometidos a trombolisis o embolectomía pulmonar, ya que no se ha establecido la seguridad y eficacia de Xarelto en estas situaciones clínicas.
Anestesia espinal/epidural o punción lumbar
Cuando se aplica anestesia neuraxial (anestesia epidural o espinal) o se realiza una punción lumbar o epidural, los pacientes tratados con antitrombóticos para la prevención de complicaciones tromboembólicas tienen riesgo de presentar un hematoma epidural o espinal, que puede causar parálisis a largo plazo o permanente. El riesgo de estos eventos puede estar aumentado por el empleo postoperatorio de catéteres epidurales permanentes o por la administración concomitante de medicamentos que afectan a la hemostasia. El riesgo también puede aumentar por la punción epidural o espinal traumática o repetida. Se debe controlar con frecuencia la presencia de signos y síntomas de deterioro neurológico (p. ej., adormecimiento o debilidad de extremidades inferiores, disfunción intestinal o vesical). Si se observa compromiso neurológico, será necesario un diagnóstico y tratamiento urgente. Antes de la intervención neuraxial, el médico debe valorar el beneficio potencial frente al riesgo en los pacientes con tratamiento anticoagulante o que van a recibir medicamentos anticoagulantes para la tromboprofilaxis. No se dispone de experiencia clínica sobre el uso de rivaroxaban 20 mg en estas situaciones.
Para reducir el riesgo potencial de sangrado asociado con el uso concomitante de rivaroxaban y anestesia neuraxial (epidural/espinal) o punción espinal se debe considerar el perfil farmacocinético de rivaroxaban. La colocación o extracción de un catéter epidural o punción lumbar se realiza mejor cuando se estima que el efecto anticoagulante de rivaroxaban es bajo. Sin embargo, se desconoce el momento exacto en el que se alcanza un efecto anticoagulante lo suficientemente bajo en cada paciente.
En base a las características farmacocinéticas generales, para la extracción de un catéter epidural, debe transcurrir al menos dos veces el tiempo de vida media desde la última administración de rivaroxaban, por ejemplo, 18 horas como mínimo en pacientes jóvenes y 26 horas en pacientes de edad avanzada (ver sección 5.2).
Una vez retirado el catéter, deben transcurrir al menos 6 horas para poder administrar la siguiente dosis de rivaroxaban.
Si se produce una punción traumática, la administración de rivaroxaban se deberá retrasar 24 horas.
Recomendaciones posológicas antes y después de procedimientos invasivos y de intervenciones quirúrgicas
Si es necesario realizar un procedimiento invasivo o una intervención quirúrgica, se interrumpirá la administración de Xarelto 20 mg por lo menos 24 horas antes de la intervención, si es posible y basándose en el criterio clínico del médico. Si la intervención no puede retrasarse, debe evaluarse el aumento del riesgo de hemorragia frente a la urgencia de la intervención.
Se debe reiniciar lo antes posible la administración de Xarelto después del procedimiento invasivo o intervención quirúrgica, siempre que la situación clínica lo permita y se haya establecido una hemostasia adecuada, una vez confirmado por el médico que trata al paciente (ver sección 5.2).
Pacientes de edad avanzada
La edad avanzada puede aumentar el riesgo de hemorragia (ver sección 5.2).
Información acerca de los excipientes
Xarelto contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, deficiencia de lactasa de Lapp o malabsorción de glucosa o galactosa no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Inhibidores del CYP3A4 y de la P-gp
La administración concomitante de rivaroxaban con ketoconazol (400 mg una vez al día) o ritonavir (600 mg dos veces al día) produjo un aumento de 2,6 veces / 2,5 veces del AUC media de rivaroxaban, y un aumento de 1,7 veces / 1,6 veces de la Cmax media de rivaroxaban, con aumentos significativos de los efectos farmacodinámicos, lo que puede aumentar el riesgo de hemorragia. Por lo tanto, no se recomienda el uso de Xarelto en pacientes que reciban tratamiento sistémico concomitante con antimicóticos azólicos como ketoconazol, itraconazol, voriconazol y posaconazol o con inhibidores de la proteasa del VIH. Estos principios activos son inhibidores potentes del CYP3A4 y de la P-gp (ver sección 4.4).
Las sustancias activas que inhiben intensamente sólo una de las vías de eliminación de rivaroxaban, el CYP3A4 o la P-gp, pueden aumentar las concentraciones plasmáticas de rivaroxaban en menor grado. La claritromicina (500 mg dos veces al día), por ejemplo, considerada un potente inhibidor del CYP3A4 y un inhibidor moderado de la P-gp, produjo un aumento de 1,5 veces del AUC media de rivaroxaban y un aumento de 1,4 veces de la Cmax. Este aumento no se considera clínicamente relevante. (Pacientes con insuficiencia renal: ver sección 4.4).
La eritromicina (500 mg tres veces al día), que inhibe moderadamente el CYP3A4 y la P-gp, produjo un aumento de 1,3 veces del AUC y de la Cmax medias de rivaroxaban. Este aumento no se considera clínicamente relevante.
En sujetos con insuficiencia renal leve, la eritromicina (500 mg tres veces al día) produjo un aumento de 1,8 veces el AUC media de rivaroxaban y de 1,6 veces en la Cmax, comparado con sujetos con la función renal normal. En sujetos con insuficiencia renal moderada, la eritromicina produjo un aumento de 2.0 veces en el AUC media de rivaroxaban y 1,6 veces en la Cmax, comparado con sujetos con la función renal normal. El efecto de la eritromicina es aditivo al de la insuficiencia renal (ver sección
4.4) .
El fluconazol (400 mg una vez al día), considerado un inhibidor moderado del CYP3A4, produjo un aumento de 1,4 veces del AUC media de rivaroxaban y un aumento de 1,3 veces de la Cmax media.
Este aumento no se consideró clínicamente relevante. (Pacientes con insuficiencia renal: ver sección
4.4) .
Dada la limitada información clínica disponible con dronedarona, debería evitarse la administración concomitante con rivaroxaban.
Anticoagulantes
Después de la administración combinada de enoxaparina (dosis única de 40 mg) con rivaroxaban (dosis única de 10 mg), se observó un efecto aditivo sobre la actividad anti-factor Xa, sin efectos adicionales en las pruebas de coagulación (TP, TTPa). La enoxaparina no afectó a las propiedades farmacocinéticas de rivaroxaban.
Debido al aumento del riesgo de hemorragia, se debe tener precaución si los pacientes reciben tratamiento concomitante con cualquier otro anticoagulante (ver secciones 4.3 y 4.4).
AINEs e inhibidores de la agregación plaquetaria
No se observó una prolongación clínicamente relevante del tiempo de sangrado después de la administración concomitante de rivaroxaban (15 mg) y 500 mg de naproxeno. No obstante, algunas personas pueden tener una respuesta farmacodinámica más pronunciada.
No se observó ninguna interacción farmacocinética ni farmacodinámica clínicamente significativa cuando se administró rivaroxaban concomitantemente con 500 mg de ácido acetilsalicílico.
El clopidogrel (dosis de carga de 300 mg, seguida de una dosis de mantenimiento de 75 mg) no mostró ninguna interacción farmacocinética con rivaroxaban (15 mg); sin embargo, se observó un aumento del tiempo de sangrado en un subgrupo de pacientes, que no se correlacionó con la agregación plaquetaria, las concentraciones de P-selectina o los receptores GPIIb/IIIa.
Debe tenerse precaución si los pacientes reciben tratamiento concomitante con AINEs (incluyendo ácido acetilsalicílico) e inhibidores de la agregación plaquetaria, ya que estos medicamentos aumentan, de por sí, el riesgo de hemorragia (ver sección 4.4).
Warfarina
Los cambios de tratamiento con warfarina (INR de 2,0 a 3,0), un antagonista de la vitamina K, a rivaroxaban (20 mg) o de rivaroxaban (20 mg) a warfarina (INR de 2,0 a 3,0) aumentaron el tiempo de protrombina/INR (Neoplastin) de forma importante (pueden observarse valores individuales del INR de hasta 12), mientras que los efectos sobre el TTPa, la inhibición de la actividad del factor Xa y el potencial de trombina endógena (PTE) fueron aditivos.
Si se desea medir los efectos farmacodinámicos de rivaroxaban durante el periodo de cambio de tratamiento, puede utilizarse la actividad anti-factor Xa, PiCT y Heptest, ya que la warfarina no afecta a estas pruebas. Al cuarto día tras la última dosis de warfarina, todas las pruebas (incluyendo TP, TTPa, inhibición de la actividad del factor Xa y PTE) reflejaron únicamente el efecto de rivaroxaban. Si se desea medir los efectos farmacodinámicos de warfarina durante el periodo de cambio de tratamiento, se puede usar la determinación del INR en la Ctrough de rivaroxaban (24 horas después de su anterior administración), ya que rivaroxaban afecta mínimamente a esta prueba en este punto.
No se observó ninguna interacción farmacocinética entre warfarina y rivaroxaban.
Inductores del CYP3A4
La administración concomitante de rivaroxaban con rifampicina, un potente inductor del CYP3A4, produjo una disminución aproximada del 50% del AUC media de rivaroxaban, con disminuciones paralelas de sus efectos farmacodinámicos. El uso concomitante de rivaroxaban con otros inductores potentes del CYP3A4 (por ejemplo, fenitoína, carbamazepina, fenobarbital o la hierba de San Juan (Hypericum perforatum)) también puede causar una disminución de la concentración plasmática de rivaroxaban. Por tanto, la administración concomitante con inductores potentes del CYP3A4 deberá evitarse a menos que el paciente esté estrechamente monitorizado para detectar signos o síntomas de trombosis.
Otros tratamientos concomitantes
No se observó ninguna interacción farmacocinética o farmacodinámica clínicamente significativa cuando se administró rivaroxaban concomitantemente con midazolam (sustrato del CYP3A4), digoxina (sustrato de la P-gp), atorvastatina (sustrato del CYP3A4 y de la P-gp) u omeprazol (inhibidor de la bomba de protones). Rivaroxaban no inhibe ni induce ninguna isoforma mayor del CYP, como el CYP3A4.
Parámetros de laboratorio
Los parámetros de la coagulación (p. ej. TP, TTPa, HepTest) se ven afectados de la forma esperada debido al mecanismo de acción de rivaroxaban (ver sección 5.1).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No se ha evaluado la seguridad y eficacia de Xarelto en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3). Debido a la posible toxicidad reproductiva, riesgo intrínseco de hemorragia y la evidencia de que rivaroxaban atraviesa la barrera placentaria, Xarelto está contraindicado durante el embarazo (ver sección 4.3).
Las mujeres en edad fértil deben evitar quedarse embarazadas durante el tratamiento con rivaroxaban.
Lactancia
No se ha evaluado la seguridad y eficacia de Xarelto en mujeres en período de lactancia. Los datos en animales indican que rivaroxaban se excreta en la leche materna. Por lo tanto, Xarelto está contraindicado durante la lactancia (ver sección 4.3). Se debe decidir si es necesario interrumpir la lactancia o bien interrumpir/suspender el tratamiento..
Fertilidad
No se han realizado estudios específicos con rivaroxaban para evaluar los efectos sobre la fertilidad en humanos. En un estudio sobre la fertilidad en ratas macho y hembra no se observó ningún efecto (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Xarelto puede influir ligeramente en la capacidad para conducir y utilizar máquinas. Se han descrito reacciones adversas como síncope (frecuencia: poco frecuente) y mareos (frecuencia: frecuente) (ver sección 4.8). Los pacientes que sufran estas reacciones adversas no deben conducir ni utilizar máquinas.
Resumen del perfil de seguridad
Se ha evaluado la seguridad de rivaroxaban en once ensayos clínicos de fase III, que incluyeron 32.625 pacientes expuestos a rivaroxaban (ver la tabla 1).
Tabla 1: Número de pacientes estudiados, dosis máxima diaria y duración del tratamiento en los estudios de fase III.
|
Indicación |
Número de pacientes * |
Dosis máxima diaria |
Duración máxima del tratamiento |
|
Prevención de tromboembolismo venoso (TEV) en pacientes adultos sometidos a cirugía electiva de reemplazo de cadera o rodilla |
6.097 |
10 mg |
39 días |
|
Prevención de tromboembolismo venoso en pacientes encamados |
3.997 |
10 mg |
39 días |
|
Tratamiento de TVP, EP y prevención de las recurrencias de TVP y EP |
4.556 |
Días 1 a 21: 30 mg Día 22 en adelante: 20 mg |
21 meses |
|
Prevención del ictus y de la embolia sistémica en pacientes con fibrilación auricular no valvular |
7.750 |
20 mg |
41 meses |
|
Prevención de acontecimientos aterotrombóticos en pacientes después de padecer un SCA |
10.225 |
5 mg ó 10 mg respectivamente, co-administrada con AAS o bien de AAS más clopidogrel o ticlopidina |
31 meses |
*Pacientes expuestos por lo menos a una dosis de rivaroxaban.
Las reacciones adversas notificadas con mayor frecuencia en los pacientes que recibieron rivaroxaban fueron hemorragias (ver sección 4.4. y 'Descripción de las reacciones adversas seleccionadas" más adelante. Las hemorragias notificadas con mayor frecuencia (> 4%) fueron epistaxis (5,9%) y la hemorragia del tracto gastrointestinal (4,2%).
En total, se notificó la aparición de eventos adversos en aproximadamente un 67% de los pacientes expuestos por lo menos a una dosis de rivaroxaban. Aproximadamente el 22% de los pacientes sufrieron eventos adversos que se consideran relacionados con el tratamiento, según la evaluación de los investigadores. En los pacientes tratados con Xarelto 10 mg sometidos a cirugía electiva de reemplazo de cadera o rodilla y en pacientes hospitalizados encamados se produjeron episodios hemorrágicos en aproximadamente un 6,8% y 12,6% de los pacientes, respectivamente, y se produjo anemia en aproximadamente un 5,9% y 2,1% de los pacientes, respectivamente. En los pacientes tratados con Xarelto 15 mg dos veces al día y después Xarelto 20 mg una vez al día para el tratamiento de la TVP o EP, o con Xarelto 20 mg una vez al día para la prevención de la recurrencia de la TVP y de la EP, se produjeron episodios hemorrágicos en aproximadamente un 27,8% de los pacientes, y anemia en aproximadamente un 2,2% de los pacientes. En los pacientes tratados para la prevención del ictus y de la embolia sistémica, se notificó hemorragia de cualquier tipo o gravedad, con una tasa de 28 por cada 100 pacientes-años, y anemia con una tasa de 2,5 por cada 100 pacientes-años. En los pacientes tratados para la prevención de muerte cardiovascular e infarto de miocardio después de un síndrome coronario agudo (SCA), se notificó hemorragia de cualquier tipo o gravedad con una tasa de 22 eventos por cada 100 pacientes-años. La anemia se notificó con una tasa de 1,4 eventos por cada 100 pacientes-años.
Las frecuencias de las reacciones adversas notificadas con Xarelto se resumen en la tabla 2, según la clasificación por órganos y sistemas (convención MedDRA) y según las frecuencias.
Las frecuencias se definen como: muy frecuentes (> 1/10) frecuentes (> 1/100 a < 1/10) poco frecuentes (> 1/1.000 a < 1/100) raras (> 1/10.000 a < 1/1.000) muy raras ( < 1/10.000)
no conocida (no puede calcularse a partir de los datos disponibles)
Tabla 2: Todas las reacciones adversas observadas con el tratamiento y notificadas en los estudios de fase III
|
Frecuentes |
Poco frecuentes |
Raras |
No conocida |
|
Trastornos de la sangre y del sistema linfático | |||
|
Anemia (incl. respectivos parámetros de laboratorio) |
Trombocitemia (incl. aumento del recuento de plaquetas) A | ||
|
Trastornos del sistema inmunológico | |||
|
Reacción alérgica, dermatitis alérgica | |||
|
Trastornos del sistema nervioso | |||
|
Mareos, cefalea, |
Hemorragia cerebral e intracraneal, síncope | ||
|
Trastornos oculares | |||
|
Hemorragia ocular (incl. hemorragia conjuntival) | |||
|
Trastornos cardiacos | |||
|
Taquicardia | |||
|
Trastornos vasculares | |||
|
Hipotensión, hematoma | |||
|
Trastornos respiratorios, torácicos y mediastínicos | |||
|
Epistaxis, hemoptisis | |||
|
Trastornos gastrointestinales | |||
|
Sangrado gingival, hemorragia del tracto gastrointestinal (incl. hemorragia rectal), dolor gastrointestinal y abdominal, dispepsia, náuseas, estreñimientoA, diarrea, vómitosA |
Sequedad de boca | ||
|
Trastornos hepatobiliares | |||
|
Alteración de la función hepática |
Ictericia | ||
|
Frecuentes |
Poco frecuentes |
Raras |
No conocida |
|
Trastornos de la piel y del tejido subcutáneo | |||
|
Prurito (incl. casos raros de prurito generalizado), exantema, equimosis, hemorragia cutánea y subcutánea |
Urticaria | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |||
|
Dolor en las extremidadesA |
Hemartrosis |
Hemorragia muscular |
Síndrome compartimental secundario a una hemorragia |
|
Trastornos renales y urinarios | |||
|
Hemorragia del tracto urogenital (incl. hematuria y menorragiaB), insuficiencia renal (incl. aumento de creatinina en sangre, aumento de la urea en sangre)A |
Insuficiencia renal /insuficiencia renal aguda secundaria a una hemorragia suficiente para causar hipoperfusión | ||
|
Trastornos generales y alteraciones en el lugar de administración | |||
|
FiebreA, edema periférico, disminución general de la fuerza y la energía (incl. Fatiga y astenia) |
Sensación de malestar (incl. malestar general) |
Edema localizadoA | |
|
Exploraciones complementarias | |||
|
Aumento de las transaminasas |
Aumento de la bilirrubina, aumento de la fosfatasa alcalina sanguíneaA, aumento de la LDHA, aumento de la lipasaA, aumento de la amilasaA, aumento de la GGTA |
Aumento de la bilirrubina conjugada (con o sin aumento concomitante de la ALT). | |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos | |||
|
Hemorragia después de una intervención (incl. anemia postoperatoria y hemorragia de la herida) contusión, secreción de la heridaA |
Pseudoaneurisma vascularC | ||
A: observado en la prevención del tromboembolismo venoso (TEV) en pacientes adultos sometidos a cirugía electiva de reemplazo de cadera o rodilla
B: observado en el tratamiento de la TVP, EP y prevención de sus recurrencias como muy frecuente en mujeres < 55 años.
C: observado como poco frecuente en la prevención de acontecimientos aterotrombóticos en pacientes que han sufrido un SCA (tras una intervención coronaria percutánea)
Descripción de reacciones adversas seleccionadas
Debido a su mecanismo de acción farmacológica, el uso de Xarelto puede asociarse a un incremento del riesgo de hemorragia oculta o manifiesta en cualquier tejido u órgano, que puede dar lugar a una anemia post-hemorrágica. Los signos, síntomas y gravedad (incluido un posible desenlace mortal) variarán según la localización y el grado o la extensión de la hemorragia, la anemia o ambas (ver sección 4.9 Tratamiento de la hemorragia). En los ensayos clínicos se observaron con más frecuencia hemorragias a nivel de mucosas (p.ej. epistaxis, gingival, gastrointestinal, genito-urinaria) y anemia en los pacientes que recibían rivaroxaban a largo plazo con respecto a los que recibían tratamiento con AVK. Por ello, además de un adecuado seguimiento clínico, las determinaciones de hemoglobina y hematocrito podrían ser útiles para detectar hemorragias ocultas cuando se considere apropiado.
El riesgo de hemorragia puede estar aumentado en ciertos grupos de pacientes, como por ejemplo, en pacientes con hipertensión arterial grave no controlada y/o en tratamiento concomitante que afecte a la hemostasia (ver Riesgo de hemorragia en la sección 4.4). El sangrado menstrual puede ser más intenso y/o prolongarse. Las complicaciones hemorrágicas pueden presentarse como debilidad, palidez, mareos, cefalea o tumefacción inexplicada, disnea o shock de causa desconocida. En algunos casos, a consecuencia de la anemia, se han observado síntomas de isquemia cardíaca, como dolor torácico o angina de pecho.
Se han notificado complicaciones conocidas, secundarias a hemorragia intensa, como el síndrome compartimental o insuficiencia renal debida a hipoperfusión. Por lo tanto deberá tenerse en cuenta la posibilidad de hemorragia al evaluar el estado de cualquier paciente anticoagulado.
Observaciones post-comercialización
Tras la comercialización se han notificado las siguientes reacciones adversas en asociación temporal con el uso de Xarelto. No se ha podido estimar la frecuencia de estas reacciones adversas.
Trastornos del sistema inmunológico: angioedema y edema alérgico. (En los ensayos de fase III agrupados, estos eventos fueron poco frecuentes (> 1/1.000 a <1/100)).
Trastornos hepatobiliares: colestasis, hepatitis (incluyendo lesión hepatocelular). (En los ensayos de fase III agrupados, estos eventos fueron raros (> 1/10.000 a < 1/1.000)).
Trastornos de la sangre y del sistema linfático: trombocitopenia. (En los ensayos de fase III agrupados, estos eventos fueron poco frecuentes (> 1/1.000 a < 1/100)).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se han notificado casos raros de sobredosis de hasta 600 mg sin complicaciones hemorrágicas u otras reacciones adversas. Debido a la limitada absorción a dosis supraterapéuticas de 50 mg de rivaroxaban o superiores se espera un efecto techo sin un aumento posterior de la exposición plasmática media.
No se dispone de un antídoto específico que antagonice el efecto farmacodinámico de rivaroxaban.
Se puede considerar el uso de carbono activado para reducir la absorción en caso de sobredosis por rivaroxaban.
Tratamiento de la hemorragia
En caso de producirse una complicación hemorrágica en un paciente que recibe tratamiento con rivaroxaban, se deberá retrasar la siguiente administración de rivaroxaban o interrumpir el tratamiento si se considera conveniente. Rivaroxaban tiene una semivida de eliminación de entre 5 y 13 horas (ver sección 5.2). Las medidas terapéuticas deben individualizarse según la gravedad y la localización de la hemorragia. En caso necesario, podría aplicarse el tratamiento sintomático adecuado, como la compresión mecánica (por ejemplo en caso de epistaxis intensa), hemostasia quirúrgica con procedimientos de control de la hemorragia, reemplazo de fluidos y apoyo hemodinámico (concentrado de hematíes o plasma fresco congelado, dependiendo de la anemia o la coagulopatía asociadas) o plaquetas.
Si la hemorragia no se puede controlar con las medidas anteriores, debería plantearse la administración de un agente procoagulante específico para revertir el efecto, como el concentrado de complejo de protrombina (CCP), concentrado de complejo de protrombina activado (CCPA) o factor VIIa recombinante (r-FVIIa). Sin embargo, actualmente hay una experiencia clínica muy limitada con el uso de estos productos en pacientes que reciben rivaroxaban. La recomendación se basa también en datos no clínicos limitados. Deberá plantearse la readministración de factor VIIa recombinante y ajustar la dosis dependiendo de la mejoría de la hemorragia. Dependiendo de la disponibilidad local, en caso de hemorragia mayor debe considerarse consultar a un experto en coagulación (ver sección 5.1).
No se espera que el sulfato de protamina y la vitamina K afecten a la actividad anticoagulante de rivaroxaban. La experiencia con ácido tranexámico es limitada y no hay experiencia con ácido aminocaproico y aprotinina en pacientes tratados con rivaroxaban. No hay una justificación científica sobre la ventaja ni experiencia con el hemostático sistémico desmopresina en pacientes tratados con rivaroxaban. Debido a su elevada fijación a las proteínas plasmáticas, no se espera que rivaroxaban sea dializable.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inhibidores directos del factor Xa, código ATC: B01AF01 Mecanismo de acción
Rivaroxaban es un inhibidor directo del factor Xa altamente selectivo, con biodisponibilidad oral. La inhibición del factor Xa interrumpe las vías intrínseca y extrínseca de la cascada de la coagulación de la sangre, inhibiendo tanto la formación de trombina como la formación de trombos. Rivaroxaban no inhibe la trombina (factor II activado) y no se han demostrado efectos sobre las plaquetas.
Efectos farmacodinámicos
En los seres humanos se ha observado una inhibición de la actividad del factor Xa dosis-dependiente. Rivaroxaban modifica el tiempo de protrombina (TP) de forma dosis-dependiente con una estrecha correlación con las concentraciones plasmáticas (el valor de r es igual a 0,98) si se emplea Neoplastin para el análisis. Otros reactivos proporcionarían unos resultados diferentes. La lectura del TP debe hacerse en segundos, porque el INR sólo se ha calibrado y validado en el caso de los cumarínicos y no puede utilizarse con ningún otro anticoagulante.
En pacientes que recibieron rivaroxaban para el tratamiento de la TVP y EP, y para la prevención de sus recurrencias, los percentiles 5/95 del TP (Neoplastin) de 2 a 4 horas después de tomar el comprimido (es decir, en el momento del efecto máximo) variaron de 17 a 32 seg. en el caso de rivaroxaban 15 mg dos veces al día, y de 15 a 30 seg. en el caso de rivaroxaban 20 mg una vez al día. En el momento de la concentración valle (8 - 16 h después de la toma del comprimido) los percentiles 5/95 para la dosis de 15 mg dos veces al día variaron de 14 a 24 seg. y para la dosis de 20 mg una vez al día (18 - 30 h después de la toma del comprimido) variaron de 13 a 20 seg.
En pacientes con fibrilación auricular no valvular que recibieron rivaroxaban para la prevención del ictus y de la embolia sistémica, en el momento del efecto máximo (1 a 4 h después de la toma del comprimido) los percentiles 5/95 del TP (Neoplastin) variaron de 14 a 40 seg. en los pacientes tratados con 20 mg una vez al día, y de 10 a 50 seg. en los pacientes con insuficiencia renal moderada tratados con 15 mg una vez al día. En el momento de la concentración valle (16 - 36 h de la toma del comprimido) los percentiles 5/95 para los pacientes tratados con la dosis de 20 mg una vez al día variaron de 12 a 26 seg. y para los pacientes con insuficiencia renal moderada tratados con la dosis de 15 mg una vez al día variaron de 12 a 26 seg.
En un estudio de farmacología clínica en la reversión de la acción farmacodinámica de rivaroxaban en adultos sanos (n = 22), se evaluaron los efectos de dosis únicas (50 UI/kg) de dos tipos diferentes de CCP, un CCP de 3 factores (factores II, IX y X) y un CCP de 4 factores (factores II, VII, IX y X). El CCP de 3 factores redujo los valores medios del TP (Neoplastina) en aproximadamente 1,0 segundos a los 30 minutos, en comparación con reducciones de, aproximadamente, 3,5 segundos observadas con el CCP de 4 factores. En cambio, el CCP de 3 factores tuvo un efecto global mayor y más rápido en la reversión de los cambios en la generación de trombina endógena que el CCP de 4 factores (ver sección 4.9).
El tiempo de tromboplastina parcial activada (aPTT) y el HepTest también están prolongados de forma dosis-dependiente; sin embargo, no se recomiendan para evaluar el efecto farmacodinámico de rivaroxaban. No es necesario monitorizar los parámetros de la coagulación durante el tratamiento con rivaroxaban en la práctica clínica. Sin embargo, si está indicado clínicamente, se pueden medir los niveles de rivaroxaban mediante ensayos cuantitativos calibrados para la actividad anti-factor Xa (ver sección 5.2).
Eficacia clínica y seguridad
Prevención del ictus y de la embolia sistémica en pacientes con fibrilación auricular no valvular El programa clínico de Xarelto se diseñó para demostrar la eficacia de Xarelto en la prevención del ictus y de la embolia sistémica en pacientes con fibrilación auricular no valvular.
En el estudio pivotal doble ciego ROCKET AF se aleatorizaron 14.264 pacientes para recibir Xarelto 20 mg una vez al día (Xarelto 15 mg una vez al día en pacientes con un aclaramiento de creatinina de 30 a 49 ml/min) o warfarina ajustada hasta un objetivo de INR de 2,5 (rango terapéutico de 2,0 a 3,0). La mediana del tiempo en tratamiento fue de 19 meses y la duración total del tratamiento fue de hasta 41 meses.
El 34,9% de los pacientes recibió tratamiento con ácido acetilsalicílico y el 11,4% con antiarrítmicos de clase III, incluida la amiodarona.
Xarelto fue no inferior a warfarina para la variable principal de eficacia compuesta de ictus y embolia sistémica fuera del sistema nervioso central. En la población por protocolo y durante el tratamiento se observó ictus o embolia sistémica en 188 pacientes tratados con rivaroxaban (1,71% anual) y en 241 pacientes tratados con warfarina (2,16% anual) (HR 0,79; 95% IC, 0,66-0,96; P<0.001 para no inferioridad). Entre todos los pacientes aleatorizados y analizados por intención de tratar, el número de pacientes que sufrieron un ictus o embolia sistémica fue de 269 en el caso de rivaroxaban (2,12% anual) y de 306 en los tratados con warfarina (2,42% per anual) (HR 0,88; 95% IC, 0,74-1,03;
P<0,001 para no inferioridad; P=0,117 para superioridad). Los resultados para las variables secundarias analizadas en orden de importancia en el análisis de intención por tratar se muestran en la tabla 3.
En los pacientes del grupo tratado con warfarina, los valores del INR estuvieron dentro del rango terapéutico de entre 2,0 y 3,0 un promedio del 55% del tiempo (mediana, 58%; rango intercuartil, 43 a 71). El efecto de rivaroxaban no difirió según el grado de control del TTR (tiempo dentro del rango objetivo de INR entre 2,0-3,0) de los centros en los cuartiles con igual tamaño (P=0,74 para la interacción). En el cuartil más alto con respecto al control de los centros, el cociente de riesgos (hazard ratio) de rivaroxaban con respecto a warfarina fue de 0,74 (95% IC, 0,49 a 1,12).
Las tasas de incidencia de la variable principal de seguridad (episodios de hemorragia mayor y no mayor clínicamente relevante) fueron similares en ambos grupos de tratamiento (ver la tabla 4).
Tabla 3: Resultados de eficacia del estudio de fase III ROCKET AF.
|
Población del estudio |
Análisis ITT de la eficacia en pacientes con fibrilación auricular no valvular | ||
|
Pauta de tratamiento |
Xarelto 20 mg una vez al día (15 mg una vez al día en pacientes con insuficiencia renal moderada) Tasa de acontecimientos (100 pacientes-años) |
Warfarina ajustada hasta un objetivo de INR de 2,5 (límites terapéuticos, 2,0 y 3,0) Tasa de acontecimientos (100 pacientes-años) |
Cociente de riesgos (hazard ratio) (IC 95%) valor de p, prueba de superioridad |
|
Ictus y embolia sistémica sin afectación del SNC |
269 (2,12) |
306 (2,42) |
0.88 (0.74 - 1.03) 0.117 |
|
Ictus, embolia sistémica |
572 (4.51) |
609 (4.81) |
0.94 |
|
sin afectación del SNC y |
(0.84 - 1.05) | ||
|
muerte vascular |
0.265 | ||
|
Ictus, embolia sistémica |
709 |
0.93 (0.83 - 1.03) 0.158 | |
|
sin afectación del SNC, muerte vascular e infarto de miocardio |
659 (5.24) |
(5.65) | |
|
253 |
281 |
0.90 | |
|
Ictus |
(1.99) |
(2.22) |
(0.76 - 1.07) |
|
0.221 | |||
|
Embolia sistémica sin afectación del SNC |
20 (0.16) |
27 (0.21) |
0.74 (0.42 - 1.32) 0.308 |
|
130 (1.02) |
142 (111) |
0.91 | |
|
Infarto de miocardio |
(0.72 - 1.16) 0.464 |
Tabla 4: Resultados de seguridad del estudio de fase III ROCKET AF.
|
Población del estudio |
Pacientes con fibrilación auricular no valvular^ | ||
|
Pauta de tratamiento |
Xarelto 20 mg una vez al día (15 mg una vez al día en pacientes con insuficiencia renal moderada) |
Warfarina ajustada hasta un objetivo de INR de 2,5 (rango terapéutico de 2,0 a 3,0) |
Cociente de riesgos (hazard ratio) (IC 95%) valor de p |
|
Tasa de acontecimientos (100 pacientes-años) |
Tasa de acontecimientos (100 pacientes-años) | ||
|
Hemorragia mayor y no mayor clínicamente relevante |
1.475 (14,91) |
1.449 (14,52) |
1,03 (0,96 - 1,11) 0,442 |
|
Hemorragia mayor |
395 (3,60) |
386 (3,45) |
1,04 (0,90 - 1,20) 0,576 |
|
Muerte causada por hemorragia* |
27 (0,24) |
55 (0,48) |
0,50 (0,31 - 0,79) 0,003 |
|
Hemorragia en órgano crítico* |
91 (0,82) |
133 (1,18) |
0,69 (0,53 - 0,91) 0,007 |
|
Hemorragia intracraneal* |
55 (0,49) |
84 (0,74) |
0,67 (0,47 - 0,93) 0,019 |
|
Descenso de hemoglobina* |
305 (2,77) |
254 (2,26) |
1,22 (1,03 - 1,44) 0,019 |
|
Transfusión de 2 o más unidades de concentrado de hematíes o sangre total* |
183 (1,65) |
149 (1,32) |
1,25 (1,01 - 1,55) 0,044 |
|
Población del estudio |
Pacientes con fibrilación auricular no valvular^ | ||
|
Pauta de tratamiento |
Xarelto 20 mg una vez al día (15 mg una vez al día en pacientes con insuficiencia renal moderada) Tasa de acontecimientos (100 pacientes-años) |
Warfarina ajustada hasta un objetivo de INR de 2,5 (rango terapéutico de 2,0 a 3,0) Tasa de acontecimientos (100 pacientes-años) |
Cociente de riesgos (hazard ratio) (IC 95%) valor de p |
|
Hemorragia no mayor |
1.185 |
1.151 |
1,04 (0,96 - 1,13) |
|
clínicamente relevante |
(11,80) |
(11,37) |
0,345 |
|
Mortalidad por cualquier |
208 |
250 |
0.85 (0.70 - 1.02) |
|
causa |
(1.87) |
(2.21) |
0.073 |
a) Población de seguridad, durante el tratamiento
* Nominalmente significativo
Además del estudio de fase III ROCKET AF, se ha realizado un estudio prospectivo de cohortes, de un solo brazo, posautorización, no intervencionista, abierto (XANTUS) con adjudicación central de resultados, incluyendo acontecimientos tromboembólicos y hemorragia mayor. Se reclutaron 6.785 pacientes con fibrilación auricular no valvular para la prevención del ictus y de la embolia sistémica fuera del sistema nervioso central (SNC) en condiciones de práctica clínica. En XANTUS, las puntuaciones medias de CHADS2 y HAS-BLED fueron ambas de 2,0, en comparación con la puntuación media de CHADS2 y HAS-BLED de 3,5 y 2,8, respectivamente, en ROCKET AF. Se produjo hemorragia mayor en 2,1 por 100 pacientes-años. Se notificó hemorragia mortal en 0,2 por 100 pacientes-años y hemorragia intracraneal en 0,4 por 100 pacientes-años. Se registró ictus o embolia sistémica fuera del SNC en 0,8 por 100 pacientes-años.
Estas observaciones en condiciones de práctica clínica son consistentes con el perfil de seguridad establecido en esta indicación.
Pacientes sometidos a cardioversión
Se realizó un estudio exploratorio aleatorizado, prospectivo, abierto, multicéntrico con evaluación ciega de las variables (X-VERT) en 1.504 pacientes (con y sin tratamiento anticoagulante oral previo) con fibrilación auricular no valvular programada para cardioversión, para comparar rivaroxaban vs. dosis ajustadas de AVK (aleatorización 2: 1), para la prevención de acontecimientos cardiovasculares. Se utilizaron dos estrategias: cardioversión guiada por ETE (de 1 a 5 días de pre-tratamiento) o cardioversión convencional (mínimo tres semanas de pre-tratamiento). Se produjeron acontecimientos en la variable principal de eficacia (compuesta por todos los ictus, ataque isquémico transitorio, embolia sistémica fuera del SNC, infarto de miocardio y muerte cardiovascular) en 5 pacientes (0,5%) en el grupo de rivaroxaban (n = 978) y 5 pacientes (1,0%) en el grupo de AVK (n = 492; RR 0,50; IC 95%: 0,15 - 1,73; población ITT modificada). Se produjeron acontecimientos en la variable principal de seguridad (hemorragia mayor) en 6 (0,6%) y 4 (0,8%) pacientes en el grupo de rivaroxaban (n = 988) y AVK (n = 499), respectivamente (RR 0,76; IC 95% 0,21-2,67 ; población de seguridad). Este estudio exploratorio mostró una eficacia y seguridad comparables entre los grupos de tratamiento con rivaroxaban y con AVK en la cardioversión.
Tratamiento de la TVP, de la EP y prevención de las recurrencias de la TVP y de la EP
El programa clínico de Xarelto se diseñó para demostrar la eficacia de Xarelto en el tratamiento inicial
y continuado de la TVP aguda y de la EP y en la prevención de sus recurrencias.
En tres estudios clínicos de fase III aleatorizados y controlados (Einstein DVT, Einstein EP y Einstein Extension) se estudiaron más de 9.400 pacientes; adicionalmente, se realizó un análisis agrupado predefinido de los estudios Einstein DVT y Einstein PE. La duración combinada total del tratamiento en todos los estudios fue de 21 meses.
En el estudio Einstein DVT, se estudiaron 3.449 pacientes con TVP aguda para el tratamiento de la TVP y prevención de las recurrencias de la TVP y de la EP (se excluyeron los pacientes que presentaban EP sintomática). La duración del tratamiento fue de 3, 6 ó 12 meses, dependiendo del criterio clínico del investigador.
Para el tratamiento inicial de la TVP aguda se administró rivaroxaban 15 mg dos veces al día durante 3 semanas y a continuación, rivaroxaban 20 mg una vez al día.
En el estudio Einstein PE, se estudiaron 4.832 pacientes con EP aguda para el tratamiento de la EP y para la prevención de las recurrencias de TVP y EP. La duración del tratamiento fue de 3, 6 ó 12 meses, en función del juicio clínico del investigador.
Para el tratamiento inicial de EP aguda, se administró 15 mg de rivaroxaban dos veces al día durante tres semanas. Esta pauta fue seguida por 20 mg de rivaroxaban una vez al día.
En los dos estudios Einstein DVT y Einstein PE, el tratamiento comparador fue enoxaparina administrada durante al menos cinco días, en combinación con un antagonista de la vitamina K hasta que el TP/INR estuviera en rango terapéutico (> 2,0). El tratamiento se continuó con un antagonista de la vitamina K, con un ajuste de dosis para mantener los valores de TP/INR dentro del rango terapéutico de 2,0 a 3,0.
En el estudio Einstein Extension para la prevención de la TVP recurrente o de la EP se estudiaron 1.197 pacientes con TVP o EP. El tratamiento tuvo una duración adicional de 6 ó 12 meses en pacientes que previamente habían completado un periodo de 6 a 12 meses de tratamiento por TEV, en función del criterio clínico del investigador. Se comparó Xarelto 20 mg una vez al día con placebo.
Todos los estudios de fase III usaron las mismas variables principales y secundarias predefinidas de eficacia. La variable principal de eficacia fue el TEV sintomático y recurrente, definido como la combinación de TVP recurrente o bien EP mortal o no mortal. La variable secundaria de eficacia se definió como la combinación de TVP recurrente, EP no mortal y mortalidad por todas las causas.
En el estudio Einstein DVT (ver tabla 5) rivaroxaban demostró ser no inferior a enoxaparina / antagonista de la vitamina K (AVK) para la variable principal de eficacia (p < 0,0001 (prueba de no inferioridad); cociente de riesgos (hazard ratio): 0,680 (0,443 - 1,042), p = 0,076 (prueba de superioridad)). El beneficio clínico neto pre-especificado (variable principal de eficacia más episodios hemorrágicos mayores) mostró un cociente de riesgos (hazard ratio) de 0,67 ((IC 95%: 0,47 - 0,95), valor nominal de p = 0,027) en favor de rivaroxaban. Los valores del INR estuvieron dentro del rango terapéutico un promedio del 60,3% del tiempo para una duración media del tratamiento de 189 días, y del 55,4%, 60,1% y 62,8% del tiempo en los grupos con una duración prevista del tratamiento de 3, 6, y 12 meses, respectivamente. En el grupo de tratamiento con enoxaparina/AVK, no se observó una relación clara entre la media del TTR (tiempo dentro del rango objetivo de INR entre 2,0 - 3,0) del centro en los terciles de igual tamaño y la incidencia de TEV recurrente (p=0,932 para la interacción). En el tercil más alto según el control del centro, el cociente de riesgos (hazard ratio) de rivaroxaban con respecto a warfarina fue de 0,69 (IC 95%: 0,35 - 1,35).
Las tasas de incidencia de la variable principal de seguridad (hemorragia mayor o no mayor clínicamente relevante), así como la variable secundaria de seguridad (hemorragia mayor) fueron similares en ambos grupos de tratamiento.
|
Población del estudio |
3.449 pacientes con trombosis venosa profunda sintomática | |
|
Xareltoa) |
Enoxaparina/AVKb) | |
|
Pauta de tratamiento y duración |
3, 6 ó 12 meses |
3, 6 ó 12 meses |
|
N=1.731 |
N=1.718 | |
|
TEV sintomático recurrente* |
36 (2,1%) |
51 (3,0%) |
|
EP sintomática recurrente |
20 (1,2%) |
18 (1,0%) |
|
TVP sintomática recurrente |
14 (0,8%) |
28 (1,6%) |
|
EP y TVP sintomáticas |
1 (0,1%) |
0 |
|
EP mortal/Muerte en la que no |
4 |
6 |
|
puede descartarse EP |
(0,2%) |
(0,3%) |
|
Hemorragia mayor o no mayor |
139 |
138 |
|
clínicamente relevante |
(8,1%) |
(8,1%) |
|
Eventos hemorrágicos mayores |
14 (0,8%) |
20 (1,2%) |
|
a) Rivaroxaban 15 mg dos veces al día durante 3 semanas, seguido d |
e rivaroxaban 20 mg una vez | |
al día
b) Enoxaparina durante al menos 5 días, solapado con y seguido por AVK
* p <0,0001 (no-inferioridad; cociente de riesgos (hazard ratio) pre-especificado de 2,0); cociente de riesgos (hazard ratio): 0,680 (0,443 - 1,042), p = 0,076 (superioridad)
En el estudio Einstein PE (ver Tabla 6) rivaroxaban demostró ser no inferior a la enoxaparina/AVK para la variable primaria de eficacia (p = 0,0026 (prueba de no inferioridad); cociente de riesgos (hazard ratio): 1,123 (0,749-1,684)). El beneficio clínico neto pre-especificado (resultado de eficacia primaria más eventos de sangrado mayor) se reportó con un cociente de riesgos (hazard ratio) de 0,849 ((IC del 95%: 0,633 a 1,139), valor nominal de p = 0,275). Los valores de INR estuvieron dentro del rango terapéutico de una media de 63% del tiempo para la duración media del tratamiento de 215 días, y el 57%, 62%, y 65% del tiempo en los grupos de duración prevista de tratamiento de 3, 6, y 12 meses, respectivamente. En el grupo de enoxaparina/AVK, no hubo una relación clara entre el nivel de la media TRT del centro (Tiempo en objetivo de INR de 2,0 - 3,0) en los terciles de igual tamaño y la incidencia de la recurrencia de TEV (p = 0,082 para la interacción). En el tercil superior de acuerdo con el centro, el cociente de riesgos (hazard ratio) con rivaroxaban en comparación con warfarina fue 0,642 (IC 95%: 0,277 - 1,484).
Las tasas de incidencia de la variable principal de seguridad (eventos hemorrágicos mayores o no mayores clínicamente relevantes) fueron ligeramente inferiores en el grupo de tratamiento con rivaroxaban (10,3% (249/2412)) frente a las del grupo de tratamiento con enoxaparina/AVK (11,4% (274 / 2405)). La incidencia de las variables secundarias de seguridad (eventos de sangrado mayor) fue inferior en el grupo de rivaroxaban (1,1% (26/2412)) comparado con la de enoxaparina/grupo AVK (2,2% (52/2405)), con un cociente de riesgos (hazard ratio) 0,493 (95 % CI: 0,308 0,789).
|
Población del estudio |
4.832 pacientes con EP sintomática aguda | |
|
Xareltoa) |
Enoxaparina/VKAb) | |
|
Pauta de tratamiento y duración |
3, 6 ó 12 meses |
3, 6 ó 12 meses |
|
N=2.419 |
N=2.413 | |
|
TEV sintomático recurrente* |
50 (2,1%) |
44 (1,8%) |
|
EP sintomática recurrente |
23 (1,0%) |
20 (0,8%) |
|
TVP sintomática recurrente |
18 (0,7%) |
17 (0,7%) |
|
EP y TVP sintomáticas |
0 |
2 (<0,1%) |
|
EP mortal/Muerte en la que no |
11 |
7 |
|
puede descartarse EP |
(0,5%) |
(0,3%) |
|
Hemorragia mayor o no mayor |
249 |
274 |
|
clínicamente relevante |
(10,3%) |
(11,4%) |
|
Eventos hemorrágicos mayores |
26 (1,1%) |
52 (2,2%) |
|
a) Rivaroxaban 15 mg dos veces al día durante 3 semanas, seguido c |
e rivaroxaban 20 mg una vez | |
al día
b) Enoxaparina durante al menos 5 días, solapado con y seguido por AVK
* p <0,0026 (no-inferioridad; cociente de riesgos (hazard ratio) pre-especificado de 2,0); cociente de riesgos (hazard ratio): 1,123 (0,749 - 1,684)
Se realizó un análisis agrupado pre-especificado de los resultados de los estudios Einstein DVT y PE (ver Tabla 7).
Tabla 7: Resultados de eficacia y seguridad del análisis agrupado de los estudios de fase III Einstein DVT y Einstein PE__
|
Población del estudio |
8.281 pacientes con TVP sintomática aguda o EP | |
|
Xareltoa) |
Enoxaparin/VKAb) | |
|
Pauta de tratamiento y duración |
3, 6 ó 12 meses |
3, 6 ó 12 meses |
|
N=4.150 |
N=4.131 | |
|
TEV sintomático recurrente* |
86 (2,1%) |
95 (2,3%) |
|
EP sintomática recurrente |
43 (1,0%) |
38 (0,9%) |
|
TVP sintomática recurrente |
32 (0,8%) |
45 (1,1%) |
|
EP y TVP sintomáticas |
1 (<0,1%) |
2 (<0,1%) |
|
EP mortal/Muerte en la que no |
15 |
13 |
|
puede descartarse EP |
(0,4%) |
(0,3%) |
|
Hemorragia mayor o no mayor |
388 |
412 |
|
clínicamente relevante |
(9,4%) |
(10,0%) |
|
Eventos hemorrágicos mayores |
40 (1,0%) |
72 (1,7%) |
|
a) Rivaroxaban 15 mg dos veces al día durante 3 semanas, seguido d |
e rivaroxaban 20 mg una vez | |
al día
b) Enoxaparina durante al menos 5 días, solapado con y seguido por AVK * p <0,0001 (no-inferioridad; cociente de riesgos (hazard ratio) pre-especificado de 1,75); cociente de riesgos (hazard ratio): 0,886 (0,661 - 1,186)
El beneficio clínico neto pre-especificado (variable primaria de eficacia más eventos de sangrado mayor) del análisis agrupado se reportó con un cociente de riesgos (hazard ratio) de 0,771 ((IC95%: 0,614 - 0,967), valor nominal de p = 0,0244).
En el estudio Einstein Extension (ver tabla 8), rivaroxaban fue superior a placebo en cuanto a las variables principal y secundaria de eficacia. En cuanto a la variable principal de seguridad (hemorragia mayor) hubo una tasa de incidencia numéricamente superior no significativa en los pacientes tratados con rivaroxaban 20 mg una vez al día, en comparación con placebo. La variable secundaria de seguridad (hemorragia mayor o no mayor clínicamente relevante) mostró unas tasas más altas en los pacientes tratados con rivaroxaban 20 mg una vez al día, en comparación con placebo.
Tabla 8: Resultados de eficacia y seguridad del estudio de fase III Einstein Extension.
|
Población del estudio |
1.197 pacientes continuaron el tratamiento y la prevención del TEV recurrente | |
|
Xareltoa) |
Placebo | |
|
Pauta del tratamiento |
6 ó 12 meses |
6 ó 12 meses |
|
N = 602 |
N = 594 | |
|
TEV recurrente y sintomático* |
8 |
42 |
|
(1,3%) |
(7,1%) | |
|
EP recurrente y sintomática |
2 |
13 |
|
(0,3%) |
(2,2%) | |
|
TVP recurrente y |
5 |
31 |
|
sintomática |
(0,8%) |
(5,2%) |
|
EP mortal / Muerte en la que |
1 |
1 |
|
no se puede descartar EP |
(0,2%) |
(0,2%) |
|
Hemorragia mayor |
4 (0,7%) |
0 (0,0%) |
|
Hemorragia no mayor clínicamente |
32 |
7 |
|
relevante |
(5,4%) |
(1,2%) |
a) Rivaroxaban 20 mg una vez al día
* p < 0,0001 (superioridad), cociente de riesgos (hazard ratio): 0,185 (0,087 - 0,393)
Además del programa de fase III EINSTEIN, se ha realizado un estudio prospectivo de cohortes, no intervencionista, abierto (XALIA) con adjudicación central de resultados, incluyendo TEV recurrente, hemorragia mayor y muerte. Se reclutaron 5.142 pacientes con TVP aguda para investigar la seguridad a largo plazo de rivaroxaban, en comparación con el tratamiento anticoagulante de referencia, en condiciones de práctica clínica. Las tasas de hemorragia mayor, TEV recurrente y mortalidad por cualquier causa para rivaroxaban fueron de 0,7%, 1,4% y 0,5%, respectivamente. Se hallaron diferencias en las características iniciales de los pacientes, incluyendo edad, cáncer e insuficiencia renal. Se realizó un análisis predefinido utilizando el índice de propensión estratificado para ajustar las diferencias en las características iniciales medidas pero, a pesar de esto, la confusión residual puede influir en los resultados. Los cocientes de riesgos (hazard ratios) ajustados que compararon rivaroxaban con el tratamiento de referencia para la hemorragia mayor, TEV recurrente y mortalidad por cualquier causa fueron de 0,77 (IC 95% 0,40 - 1,50), 0,91 (IC 95% 0,54 - 1,54) y 0,51 (IC 95% 0,24 - 1,07), respectivamente.
Estos resultados en condiciones de práctica clínica son consistentes con el perfil de seguridad establecido en esta indicación.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Xarelto en uno o más grupos de la población pediátrica en el tratamiento de eventos tromboembólicos. La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Xarelto en los diferentes grupos de la población pediátrica en la prevención de eventos tromboembólicos (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Rivaroxaban se absorbe rápidamente y alcanza concentraciones máximas (Cmax) de 2 a 4 horas después de tomar el comprimido.
La absorción oral de rivaroxaban es casi completa y su biodisponibilidad oral es elevada (80% al 100%) en el caso de la dosis del comprimido de 2,5 mg y de 10 mg, independientemente de las condiciones de ayuno o alimentación. La ingesta de alimentos con rivaroxaban (a la dosis de 2,5 mg y de 10 mg) no afecta al AUC ni a la Cmax.
Debido a la disminución de la absorción, se determinó una biodisponibilidad del 66% con el comprimido de 20 mg en condiciones de ayuno. Cuando los comprimidos de Xarelto 20 mg se tomaron junto con alimentos, se observaron aumentos del AUC media del 39% en comparación con la toma de comprimidos en condiciones de ayuno, lo que indica una absorción casi completa y una biodisponibilidad oral elevada. Xarelto 15 mg y 20 mg deben tomarse con alimentos (ver sección 4.2). Rivaroxaban presenta una farmacocinética lineal hasta aproximadamente 15 mg administrados una vez al día en ayunas. En condiciones de alimentación reciente, Xarelto en comprimidos de 10 mg, 15 mg y 20 mg demostró proporcionalidad con la dosis. A dosis más altas, rivaroxaban muestra una disolución limitada, con una reducción de la biodisponibilidad y de la tasa de absorción al aumentar la dosis.
La variabilidad de la farmacocinética de rivaroxaban es moderada; con una variabilidad interindividual (CV%) entre el 30% y el 40%.
La absorción de rivaroxaban depende del sitio donde se libera en el tracto gastrointestinal. Se ha notificado una disminución del 29% y del 56% en el AUC y la Cmax, en comparación con el comprimido, cuando rivaroxaban en forma de granulado se liberó en el intestino delgado proximal. La exposición se reduce aún más cuando rivaroxaban se libera en el intestino delgado distal o en el colon ascendente. Por lo tanto, debe evitarse la administración de rivaroxaban de forma distal al estómago, ya que esto puede dar lugar a una reducción de la absorción y la correspondiente exposición a rivaroxaban.
La biodisponibilidad (AUC y Cmax) fue comparable para rivaroxaban 20 mg, administrado por vía oral como comprimido triturado y mezclado con puré de manzana o diluido con agua, administrado a través de una sonda gástrica y seguido de una comida líquida, en comparación con el comprimido entero. Dado el perfil farmacocinético predecible, proporcional a la dosis de rivaroxaban, los resultados de biodisponibilidad de este estudio son probablemente aplicables a dosis más bajas de rivaroxaban.
Distribución
La unión a proteínas plasmáticas humanas es alta, del 92 % al 95 % aproximadamente y la albúmina sérica es el principal componente de unión. El volumen de distribución es moderado, con un Vss de 50 litros, aproximadamente.
Biotransformación y eliminación
De la dosis administrada de rivaroxaban se metabolizan aproximadamente 2/3; después, la mitad se elimina por vía renal y la otra mitad por vía fecal. El 1/3 restante de la dosis administrada se excreta directamente por vía renal como principio activo no modificado en la orina, principalmente mediante secreción renal activa.
Rivaroxaban se metaboliza mediante el CYP3A4, el CYP2J2 y mecanismos independientes del CYP. Las principales vías de biotransformación son la degradación oxidativa de la porción de morfolinona y la hidrólisis de los enlaces amida. Según investigaciones in vitro, rivaroxaban es un sustrato de las proteínas transportadoras P-gp (glucoproteína P) y Bcrp (proteína de resistencia al cáncer de mama). Rivaroxaban en forma inalterada es el compuesto más abundante en el plasma humano sin presencia de metabolitos mayores o metabolitos activos circulantes. Con un aclaramiento sistémico de aproximadamente 10 l/h, rivaroxaban puede clasificarse como una sustancia de bajo aclaramiento. Después de la administración por vía intravenosa de una dosis de 1 mg, la semivida de eliminación es de aproximadamente 4,5 horas. Después de la administración por vía oral, la eliminación se ve limitada por la tasa de absorción. En personas jóvenes, la eliminación de rivaroxaban del plasma se produce con una semivida de eliminación de 5 a 9 horas y en personas de edad avanzada, con una semivida de eliminación de 11 a 13 horas.
Poblaciones especiales
Sexo
No hubo ninguna diferencia clínicamente relevante en las propiedades farmacocinéticas y farmacodinámicas entre hombres y mujeres.
Pacientes de edad avanzada
Los pacientes de edad avanzada presentaron concentraciones plasmáticas mayores que los pacientes más jóvenes, con unos valores medios del AUC que fueron aproximadamente 1,5 veces superiores, principalmente debido a la disminución (aparente) del aclaramiento renal y total. No es necesario un ajuste de la dosis.
Peso corporal
Los valores extremos en el peso corporal (< 50 kg ó > 120 kg) tuvieron poco efecto en las concentraciones plasmáticas de rivaroxaban (menos del 25%). No es necesario un ajuste de la dosis.
Origen étnico
No se observaron diferencias interétnicas clínicamente relevantes entre los pacientes de raza blanca, afroamericanos, de origen latinoamericano, japonés o chino, en cuanto a las propiedades farmacocinéticas o farmacodinámicas.
Insuficiencia hepática
Los pacientes cirróticos con insuficiencia hepática leve (clasificados como Child Pugh A), sólo presentaron cambios menores en la farmacocinética de rivaroxaban (aumento medio del AUC de 1,2 veces), lo que fue casi comparable al grupo control de voluntarios sanos. En los pacientes cirróticos con insuficiencia hepática moderada (clasificados como Child Pugh B), el AUC media de rivaroxaban estuvo aumentada significativamente en 2,3 veces, en comparación con los voluntarios sanos. El AUC parcial aumentó 2,6 veces. Estos pacientes también mostraron una disminución de la eliminación renal de rivaroxaban, similar a los pacientes con insuficiencia renal moderada. No hay datos en pacientes con insuficiencia hepática grave.
La inhibición de la actividad del factor Xa se incrementó en un factor de 2,6 en los pacientes con insuficiencia hepática moderada, en comparación con los voluntarios sanos; de manera similar, la prolongación del TP se incrementó en un factor de 2,1. Los pacientes con insuficiencia hepática moderada fueron más sensibles a rivaroxaban, lo que produjo una relación farmacocinética / farmacodinámica más pronunciada entre la concentración y el TP.
Xarelto está contraindicado en pacientes con hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia, incluyendo pacientes cirróticos clasificados como Child Pugh B y C (ver sección 4.3).
Insuficiencia renal
Se observó un aumento de la exposición a rivaroxaban correlacionado con la disminución de la función renal, evaluada mediante las determinaciones del aclaramiento de creatinina. En sujetos con insuficiencia renal leve (aclaramiento de creatinina de 50 - 80 ml/min), moderada (aclaramiento de creatinina de 30 a 49 ml/min) o grave (aclaramiento de creatinina de 15 a 29 ml/min), las concentraciones plasmáticas de rivaroxaban (AUC) aumentaron 1,4, 1,5 y 1,6 veces, respectivamente. Los aumentos correspondientes en los efectos farmacodinámicos fueron más pronunciados. En sujetos con insuficiencia renal leve, moderada y grave, la inhibición total de la actividad del factor Xa aumentó en un factor de 1,5, 1,9 y 2,0 respectivamente, en comparación con voluntarios sanos; de manera similar, la prolongación del TP aumentó en factores de 1,3, 2,2 y 2,4, respectivamente. No hay datos en pacientes con un aclaramiento de creatinina < 15 ml/min.
Debido a la elevada fijación a proteínas plasmáticas, no se espera que rivaroxaban sea dializable.
No se recomienda su uso en pacientes con un aclaramiento de creatinina < 15 ml/min. Xarelto debe utilizarse con precaución en pacientes con un aclaramiento de creatinina de 15 a 29 ml/min (ver sección 4.4).
Datos farmacocinéticos en pacientes
En los pacientes que recibieron rivaroxaban 20 mg una vez al día para el tratamiento de la TVP aguda, la concentración media geométrica (intervalo de predicción del 90%) a las 2 - 4 h y a las 24 h aproximadamente después de la dosis (lo que representa aproximadamente las concentraciones máxima y mínima durante el intervalo entre dosis) fue de 215 (22 - 535) y de 32 (6 - 239) ^g/l, respectivamente.
Relación farmacocinética / farmacodinámica
Se ha evaluado la relación farmacocinética/farmacodinámica (PK/PD) entre la concentración plasmática de rivaroxaban y varios criterios de valoración PD (inhibición del factor Xa, tiempo de protrombina (TP, TTPa, Heptest)) después de la administración de un amplio rango de dosis (de 5 a 30 mg dos veces al día). La relación entre la concentración de rivaroxaban y la actividad del factor Xa se describió de manera óptima por un modelo Emax. En el caso del TP, por lo general, el modelo de intersección lineal describió mejor los datos. Dependiendo de los diferentes reactivos usados en el TP, la pendiente varió considerablemente. Con Neoplastin PT, el TP basal fue de aproximadamente 13 seg. y la pendiente fue de alrededor de 3 a 4 seg/(100 ^g/l). Los resultados de los análisis de la relación PK/PD en las fases II y III fueron consistentes con los datos establecidos en los sujetos sanos.
Población pediátrica
No se ha determinado la seguridad y eficacia en niños y adolescentes hasta los 18 años.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad con dosis únicas, fototoxicidad, genotoxicidad, potencial carcinogénico y toxicidad juvenil.
Los efectos observados en los estudios con dosis repetidas se debieron principalmente a la actividad farmacodinámica incrementada de rivaroxaban. En ratas se observó un aumento de las concentraciones plasmáticas de IgG e IgA a niveles de exposición clínicamente relevantes.
No se observó ningún efecto sobre la fertilidad en las ratas macho o hembra. Los estudios en animales han demostrado una toxicidad reproductiva relacionada con el modo de acción farmacológica de rivaroxaban (p. ej. complicaciones hemorrágicas). A concentraciones plasmáticas clínicamente relevantes se observó toxicidad embriofetal (pérdida después de la implantación, retraso o adelanto de la osificación, varias manchas hepáticas de color claro) y un aumento de la incidencia de malformaciones frecuentes, así como cambios placentarios. En el estudio pre y postnatal en ratas, se observó una disminución de la viabilidad de las crías a dosis que fueron tóxicas para las madres.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido:
Celulosa microcristalina Croscarmelosa sódica Lactosa monohidrato Hipromelosa Laurilsulfato de sodio Estearato de magnesio
Cubierta pelicular:
Macrogol 3350 Hipromelosa
Dióxido de titanio (E 171)
Óxido de hierro rojo (E 172)
6.2 Incompatibilidades
No procede.
3 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Blister de PP / lámina de aluminio, en envases de 10, 14, 28, ó 98 comprimidos recubiertos con película, o blisters precortados unidosis en envases de 10 x 1, 100 x 1 ó en envases múltiples que contienen 100 (10 estuches de 10 x 1) comprimidos recubiertos con película.
Frascos de HDPE con tapón de rosca de PP que contienen 100 comprimidos recubiertos.
Blisters de PP/ lámina de aluminio, en un envase (con calendario) que contiene 42 comprimidos recubiertos con película de Xarelto 15 mg y 7 comprimidos recubiertos con película de Xarelto 20 mg (envase para el inicio del tratamiento).
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
Ninguna especial para su eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG D-13342 Berlin Alemania
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/017-021, EU/1/08/472/024, EU/1/08/472/037, EU/1/08/472/039, EU/1/08/472/040
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 30 de septiembre de 2008 Fecha de la última renovación: 22 de mayo de 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
{MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
Envase para el inicio del tratamiento
▼ Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Xarelto 15 mg comprimidos recubiertos con película Xarelto 20 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película de 15 mg contiene 15 mg de rivaroxaban.
Cada comprimido recubierto con película de 20 mg contiene 20 mg de rivaroxaban.
Excipiente con efecto conocido:
Cada comprimido recubierto con película de 15 mg contiene 24,13 mg de lactosa (como monohidrato), ver sección 4.4.
Cada comprimido recubierto con película de 20 mg contiene 21,76 mg de lactosa (como monohidrato), ver sección 4.4.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
Comprimido recubierto con película de 15 mg: comprimidos de color rojo, redondos y biconvexos (6 mm de diámetro y 9 mm de radio de curvatura), con la cruz de BAYER en una cara, y “15” y un triángulo en la otra cara.
Comprimido recubierto con película de 20 mg: comprimidos de color pardo rojizo, redondos y biconvexos (6 mm de diámetro y 9 mm de radio de curvatura), con la cruz de BAYER en una cara, y “20” y un triángulo en la otra cara.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento de la trombosis venosa profunda (TVP) y de la embolia pulmonar (EP), y prevención de las recurrencias de la TVP y de la EP en pacientes adultos. (Ver sección 4.4, información sobre pacientes con EP hemodinámicamente inestables.)
4.2 Posología y forma de administración
Posología
Tratamiento de la TVP, tratamiento de la EP y prevención de las recurrencias de la TVP y de la EP La dosis recomendada para el tratamiento inicial de la TVP aguda o de la EP es de 15 mg dos veces al día, durante las tres primeras semanas, seguida de 20 mg una vez al día para el tratamiento continuado así como para la prevención de las recurrencias de la TVP y de la EP, según se indica en la tabla siguiente.
|
Pauta de tratamiento |
Dosis máxima diaria | |
|
Días 1 a 21 |
15 mg dos veces al día |
30 mg |
Día 22 en adelante
20 mg una vez al día
20 mg
Está disponible un envase para el inicio del tratamiento de Xarelto para 4 semanas destinado a pacientes que deben realizar la transición de 15 mg dos veces al día a 20 mg una vez al día a partir del Día 22 (ver sección 6.5)
Para pacientes con insuficiencia renal moderada o grave en los cuales se ha decidido una pauta de 15 mg una vez al día a partir del Día 22, se encuentran disponibles otras presentaciones conteniendo únicamente comprimidos recubiertos con película de 15 mg. (ver a continuación las pautas posológicas en la sección Poblaciones especiales).
La duración del tratamiento debe individualizarse después de una evaluación minuciosa del beneficio del tratamiento frente al riesgo de hemorragia (ver sección 4.4). La duración corta del tratamiento (como mínimo de 3 meses) debe basarse en factores de riesgo transitorios (p.ej. cirugía reciente, traumatismo, inmovilización) y las duraciones más largas deben basarse en factores de riesgo permanente o TVP idiopática, o de EP.
Si el paciente olvida una dosis durante la fase de tratamiento de 15 mg dos veces al día (días 1 a 21), éste deberá tomar inmediatamente Xarelto para garantizar una toma de 30 mg de Xarelto al día. En este caso, se pueden tomar dos comprimidos de 15 mg a la vez y al día siguiente se deberá seguir con la pauta habitual recomendada de 15 mg dos veces al día.
Si el paciente olvida una dosis durante la fase de tratamiento de una vez al día (día 22 en adelante), deberá tomar inmediatamente Xarelto, y seguir al día siguiente con la pauta recomendada de una vez al día. La dosis no debe duplicarse en el mismo día para compensar una dosis olvidada.
Cambio de tratamiento con antagonistas de la vitamina K (AVK) a Xarelto En el caso de pacientes tratados por TVP, EP y en la prevención de sus recurrencias, deberá interrumpirse el tratamiento con AVK e iniciarse el tratamiento con Xarelto cuando el valor del INR sea < 2,5.
Al cambiar el tratamiento con AVK a Xarelto, los valores de INR del paciente estarán falsamente elevados después de la toma de Xarelto. El INR no es un parámetro válido para medir la actividad anticoagulante de Xarelto, por lo que no debe utilizarse (ver sección 4.5).
Cambio de tratamiento con Xarelto a antagonistas de la vitamina K (AVK)
Existe la posibilidad de una incorrecta anticoagulación durante la transición de Xarelto a AVK. Debe garantizarse una anticoagulación adecuada y continua durante cualquier transición a un anticoagulante alternativo. Debe señalarse que Xarelto puede contribuir a un aumento del INR.
En los pacientes que cambien de Xarelto a AVK, estos tratamientos deben administrarse simultáneamente hasta que el INR sea > 2,0. Durante los dos primeros días del periodo de cambio se utilizará la dosis inicial estándar de AVK, que se ajustará posteriormente en función de los resultados del INR. Mientras los pacientes están bajo tratamiento con Xarelto y AVK el INR puede determinarse a partir de las 24 horas que siguen a la dosis de Xarelto y siempre antes de la siguiente dosis. Una vez interrumpido el tratamiento con Xarelto, el INR puede determinarse con fiabilidad pasadas 24 horas de la última dosis (ver secciones 4.5 y 5.2).
Cambio de tratamiento con anticoagulante parenteral a Xarelto
Los pacientes que están recibiendo un anticoagulante por vía parenteral, deben interrumpir el tratamiento anticoagulante por vía parenteral e iniciar el tratamiento con Xarelto de 0 a 2 horas antes de la siguiente administración programada del medicamento por vía parenteral (p. ej., heparina de bajo peso molecular). En el caso de un anticoagulante parenteral administrado por perfusión continua (p. ej., heparina no fraccionada intravenosa) Xarelto deberá administrarse en el momento de la suspensión del anticoagulante parenteral.
Cambio de tratamiento con Xarelto a anticoagulante parenteral
La primera dosis de anticoagulante parenteral debe administrarse en el momento en que se tomaría la siguiente dosis de Xarelto.
Poblaciones especiales
Insuficiencia renal
Los escasos datos clínicos en pacientes con insuficiencia renal grave (aclaramiento de creatinina de 15 a 29 ml/min) indican que las concentraciones plasmáticas de rivaroxaban aumentan significativamente. Por lo tanto, Xarelto se debe usar con precaución en estos pacientes. No se recomienda su uso en pacientes con un aclaramiento de creatinina < 15 ml/min (ver secciones 4.4 y 5.2).
En pacientes con insuficiencia renal moderada (aclaramiento de creatinina de 30 a 49 ml/min) o grave (aclaramiento de creatinina de 15 a 29 ml/min) se recomiendan las siguientes pautas posológicas:
- Para el tratamiento de la TVP, y de la EP y la prevención de las recurrencias de la TVP y de la EP se debe tratar a los pacientes con 15 mg dos veces al día durante las tres primeras semanas. Después, la dosis recomendada es de 20 mg una vez al día. Deberá considerarse una reducción de la dosis de 20 mg una vez al día a 15 mg una vez al día si el riesgo de sangrado valorado en el paciente supera el riesgo de recurrencia de TVP y de EP. La recomendación para el uso de 15 mg se basa en el modelo farmacocinético que no se ha estudiado en este contexto clínico (ver secciones 4.4, 5.1 y 5.2).
No se requiere ajuste de dosis en pacientes con insuficiencia renal leve (aclaramiento de creatinina de 50 a 80 ml/min) (ver sección 5.2).
Insuficiencia hepática
Xarelto está contraindicado en pacientes con hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia incluidos los pacientes cirróticos con Child Pugh B y C (ver secciones 4.3 y 5.2).
Pacientes de edad avanzada
No se requiere ajuste de dosis (ver sección 5.2).
Peso corporal
No se requiere ajuste de dosis (ver sección 5.2).
Sexo
No se requiere ajuste de dosis (ver sección 5.2).
Población pediátrica
No se ha establecido la seguridad y eficacia de Xarelto en niños de 0 a 18 años. No se dispone de datos. Por lo tanto, no se recomienda el uso de Xarelto en niños menores de 18 años.
Forma de administración Vía oral.
Los comprimidos deben administrarse con alimentos (ver sección 5.2).
Para aquellos pacientes que no puedan tragar el comprimido entero, el comprimido de Xarelto puede triturarse y mezclarse con agua o con puré de manzana inmediatamente antes de su uso y administrarse por vía oral. Inmediatamente después de la administración del comprimido triturado se debe administrar el alimento.
El comprimido triturado también se puede administrar a través de sonda gástrica una vez se haya confirmado la colocación correcta de la sonda. El comprimido triturado se administrará diluido con una pequeña cantidad de agua a través de la sonda gástrica, procediendo seguidamente a un lavado adicional de la sonda con agua. Inmediatamente después de la administración del comprimido triturado se debe administrar el alimento mediante sonda gástrica (ver sección 5.2).
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hemorragia activa clínicamente significativa.
Lesión o enfermedad, si se considera que tiene un riesgo significativo de sangrado mayor. Esto puede incluir úlcera gastrointestinal activa o reciente, presencia de neoplasias malignas con alto riesgo de sangrado, traumatismo cerebral o espinal reciente, cirugía cerebral, espinal u oftálmica reciente, hemorragia intracraneal reciente, conocimiento o sospecha de varices esofágicas, malformaciones arteriovenosas, aneurismas vasculares o anomalías vasculares intraespinales o intracerebrales mayores.
Tratamiento concomitante con cualquier otro anticoagulante, p. ej. heparina no fraccionada (HNF), heparinas de bajo peso molecular (enoxaparina, dalteparina, etc.), derivados de la heparina (fondaparinux, etc.), anticoagulantes orales (warfarina, dabigatran etexilato, apixaban, etc.) excepto bajo las circunstancias concretas de cambio de tratamiento anticoagulante (ver sección 4.2) o cuando se administre HNF a las dosis necesarias para mantener un catéter venoso o arterial central abierto (ver sección 4.5).
Hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia, incluidos los pacientes cirróticos con Child Pugh B y C (ver sección 5.2).
Embarazo y lactancia (ver sección 4.6).
4.4 Advertencias y precauciones especiales de empleo
Durante todo el periodo de tratamiento se recomienda una estrecha monitorización clínica del paciente, en línea con la práctica de anticoagulación
Riesgo de hemorragia
Al igual que con otros anticoagulantes, los pacientes que toman Xarelto deben ser observados cuidadosamente para detectar signos de sangrado. Se recomienda utilizar con precaución en condiciones que conlleven un riesgo incrementado de hemorragia. La administración de Xarelto debe interrumpirse si se produce una hemorragia grave.
En los ensayos clínicos se observaron con más frecuencia hemorragias a nivel de mucosas (p.ej. epistaxis, gingival, gastrointestinal, génito-urinaria) y anemia en los pacientes que recibían rivaroxaban a largo plazo respecto a los que recibían tratamiento con AVK. Por ello, además de un seguimiento clínico adecuado, las determinaciones de hemoglobina y hematocrito podrían ser útiles para detectar hemorragias ocultas cuando se considere apropiado.
Varios subgrupos de pacientes, como se explica posteriormente, presentan un mayor riesgo de hemorragia. En estos pacientes se debe vigilar cuidadosamente la presencia de signos y síntomas de complicaciones hemorrágicas y de anemia después del inicio del tratamiento (ver sección 4.8). Cualquier disminución inexplicada de los niveles de hemoglobina o de la presión arterial requerirá la búsqueda de una zona de sangrado.
Aunque el tratamiento con rivaroxaban no requiere una monitorización rutinaria de la exposición, la determinación de los niveles de rivaroxaban mediante un ensayo anti-factor Xa cuantitativo calibrado puede ser útil en situaciones excepcionales, en las que el conocimiento de la exposición a rivaroxaban puede ayudar en la toma de decisiones clínicas, como por ejemplo, en caso de sobredosis o cirugía de emergencia (ver secciones 5.1 y 5.2).
Insuficiencia renal
En pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min), las concentraciones plasmáticas de rivaroxaban podrían aumentar de forma significativa (en promedio,
1,6 veces), lo que conllevaría un aumento del riesgo de hemorragia. Xarelto debe utilizarse con
precaución en pacientes con un aclaramiento de creatinina de 15 a 29 ml/min. No se recomienda el uso en pacientes con un aclaramiento de creatinina < 15 ml/min (ver secciones 4.2 y 5.2).
Xarelto debe usarse con precaución en pacientes con insuficiencia renal y que reciben de forma concomitante otros medicamentos que aumenten las concentraciones plasmáticas de rivaroxaban (ver sección 4.5).
Interacción con otros medicamentos
No se recomienda utilizar Xarelto en pacientes que reciben tratamiento sistémico concomitante con antimicóticos azólicos (p. ej., ketoconazol, itraconazol, voriconazol y posaconazol) o inhibidores de la proteasa del VIH (p. ej., ritonavir). Estos principios activos son inhibidores potentes del CYP3A4 y de la P-gp y por lo tanto pueden aumentar las concentraciones plasmáticas de rivaroxaban hasta un nivel clínicamente relevante (en promedio, 2,6 veces), lo que puede llevar a un aumento del riesgo de hemorragia (ver sección 4.5).
Debe tenerse precaución si los pacientes reciben tratamiento concomitante con medicamentos que afectan a la hemostasia, como los antiinflamatorios no esteroideos (AINEs), ácido acetilsalicílico e inhibidores de la agregación plaquetaria. Puede considerarse el uso de un tratamiento profiláctico adecuado en aquellos pacientes con riesgo de enfermedad gastrointestinal ulcerosa, (ver sección 4.5).
Otros factores de riesgo hemorrágico
Al igual que con otros agentes antitrombóticos, rivaroxaban no está recomendado en pacientes con un riesgo aumentado de hemorragia, tales como:
• trastornos de la coagulación, congénitos o adquiridos
• hipertensión arterial grave no controlada
• otra enfermedad gastrointestinal sin úlcera activa que pueda producir complicaciones hemorrágicas (por ejemplo, enfermedad inflamatoria intestinal, esofagitis, gastritis o reflujo gastroesofágico)
• retinopatía vascular
• bronquiectasia o antecedentes de hemorragia pulmonar
Pacientes con EP hemodinámicamente inestables o pacientes que requieran trombolisis o embolectomía pulmonar
Xarelto no está recomendado como una alternativa a la heparina no fraccionada en pacientes con embolia pulmonar que están hemodinámicamente inestables o que puedan ser sometidos a trombolisis o embolectomía pulmonar, ya que no se ha establecido la seguridad y eficacia de Xarelto en estas situaciones clínicas.
Anestesia espinal/epidural o punción lumbar
Cuando se aplica anestesia neuraxial (anestesia epidural o espinal) o se realiza una punción lumbar o epidural, los pacientes tratados con antitrombóticos para la prevención de complicaciones tromboembólicas tienen riesgo de presentar un hematoma epidural o espinal, que puede causar parálisis a largo plazo o permanente. El riesgo de estos eventos puede estar aumentado por el empleo postoperatorio de catéteres epidurales permanentes o por la administración concomitante de medicamentos que afectan a la hemostasia. El riesgo también puede aumentar por la punción epidural o espinal traumática o repetida. Se debe controlar con frecuencia la presencia de signos y síntomas de deterioro neurológico (p. ej., adormecimiento o debilidad de extremidades inferiores, disfunción intestinal o vesical). Si se observa compromiso neurológico, será necesario un diagnóstico y tratamiento urgente. Antes de la intervención neuraxial, el médico debe valorar el beneficio potencial frente al riesgo en los pacientes con tratamiento anticoagulante o que van a recibir medicamentos anticoagulantes para la tromboprofilaxis. No se dispone de experiencia clínica sobre el uso de rivaroxaban 15 mg o 20 mg en estas situaciones.
Para reducir el riesgo potencial de sangrado asociado con el uso concomitante de rivaroxaban y anestesia neuraxial (epidural/espinal) o punción espinal se debe considerar el perfil farmacocinético de rivaroxaban. La colocación o extracción de un catéter epidural o punción lumbar se realiza mejor cuando se estima que el efecto anticoagulante de rivaroxaban es bajo. Sin embargo, se desconoce el momento exacto en el que se alcanza un efecto anticoagulante lo suficientemente bajo en cada paciente.
En base a las características farmacocinéticas generales, para la extracción de un catéter epidural, debe transcurrir al menos dos veces el tiempo de vida media desde la última administración de rivaroxaban, por ejemplo, 18 horas como mínimo en pacientes jóvenes y 26 horas en pacientes de edad avanzada (ver sección 5.2). Una vez retirado el catéter, deben transcurrir al menos 6 horas para poder administrar la siguiente dosis de rivaroxaban.
Si se produce una punción traumática, la administración de rivaroxaban se deberá retrasar 24 horas.
Recomendaciones posológicas antes y después de procedimientos invasivos y de intervenciones quirúrgicas
Si es necesario realizar un procedimiento invasivo o una intervención quirúrgica, se interrumpirá la administración de Xarelto 15 mg/ Xarelto 20 mg por lo menos 24 horas antes de la intervención, si es posible y basándose en el criterio clínico del médico.
Si la intervención no puede retrasarse, debe evaluarse el aumento del riesgo de hemorragia frente a la urgencia de la intervención.
Se debe reiniciar lo antes posible la administración de Xarelto después del procedimiento invasivo o intervención quirúrgica, siempre que la situación clínica lo permita y se haya establecido una hemostasia adecuada una vez confirmado por el médico que trata al paciente (ver sección 5.2).
Pacientes de edad avanzada
La edad avanzada puede aumentar el riesgo de hemorragia (ver sección 5.2).
Información acerca de los excipientes
Xarelto contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, deficiencia de lactasa de Lapp o malabsorción de glucosa o galactosa no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Inhibidores del CYP3A4 y de la P-gp
La administración concomitante de rivaroxaban con ketoconazol (400 mg una vez al día) o ritonavir (600 mg dos veces al día) produjo un aumento de 2,6 veces / 2,5 veces del AUC media de rivaroxaban, y un aumento de 1,7 veces / 1,6 veces de la Cmax media de rivaroxaban, con aumentos significativos de los efectos farmacodinámicos, lo que puede aumentar el riesgo de hemorragia. Por lo tanto, no se recomienda el uso de Xarelto en pacientes que reciban tratamiento sistémico concomitante con antimicóticos azólicos como ketoconazol, itraconazol, voriconazol y posaconazol o con inhibidores de la proteasa del VIH. Estos principios activos son inhibidores potentes del CYP3A4 y de la P-gp (ver sección 4.4).
Las sustancias activas que inhiben intensamente sólo una de las vías de eliminación de rivaroxaban, el CYP3A4 o la P-gp, pueden aumentar las concentraciones plasmáticas de rivaroxaban en menor grado. La claritromicina (500 mg dos veces al día), por ejemplo, considerada un potente inhibidor del CYP3A4 y un inhibidor moderado de la P-gp, produjo un aumento de 1,5 veces del AUC media de rivaroxaban y un aumento de 1,4 veces de la Cmax. Este aumento no se considera clínicamente relevante. (Pacientes con insuficiencia renal: ver sección 4.4).
La eritromicina (500 mg tres veces al día), que inhibe moderadamente el CYP3A4 y la P-gp, produjo un aumento de 1,3 veces del AUC y de la Cmax medias de rivaroxaban. Este aumento no se considera clínicamente relevante.
En sujetos con insuficiencia renal leve, la eritromicina (500 mg tres veces al día) produjo un aumento de 1,8 veces el AUC media de rivaroxaban y de 1,6 veces en la Cmax, comparado con sujetos con la función renal normal. En sujetos con insuficiencia renal moderada, la eritromicina produjo un aumento de 2,0 veces en el AUC media de rivaroxaban y 1,6 veces en la Cmax, comparado con sujetos con la función renal normal. El efecto de la eritromicina es aditivo al de la insuficiencia renal (ver sección 4.4).
El fluconazol (400 mg una vez al día), considerado un inhibidor moderado del CYP3A4, produjo un aumento de 1,4 veces del AUC media de rivaroxaban y un aumento de 1,3 veces de la Cmax media.
Este aumento no se consideró clínicamente relevante. (Pacientes con insuficiencia renal: ver sección 4.4).
Dada la limitada información clínica disponible con dronedarona, debería evitarse la administración concomitante con rivaroxaban.
Anticoagulantes
Después de la administración combinada de enoxaparina (dosis única de 40 mg) con rivaroxaban (dosis única de 10 mg), se observó un efecto aditivo sobre la actividad anti-factor Xa, sin efectos adicionales en las pruebas de coagulación (TP, TTPa). La enoxaparina no afectó a las propiedades farmacocinéticas de rivaroxaban.
Debido al aumento del riesgo de hemorragia, se debe tener precaución si los pacientes reciben tratamiento concomitante con cualquier otro anticoagulante (ver secciones 4.3 y 4.4).
AINEs e inhibidores de la agregación plaquetaria
No se observó una prolongación clínicamente relevante del tiempo de sangrado después de la administración concomitante de rivaroxaban (15 mg) y 500 mg de naproxeno. No obstante, algunas personas pueden tener una respuesta farmacodinámica más pronunciada.
No se observó ninguna interacción farmacocinética ni farmacodinámica clínicamente significativa cuando se administró rivaroxaban concomitantemente con 500 mg de ácido acetilsalicílico.
El clopidogrel (dosis de carga de 300 mg, seguida de una dosis de mantenimiento de 75 mg) no mostró ninguna interacción farmacocinética con rivaroxaban (15 mg); sin embargo, se observó un aumento del tiempo de sangrado en un subgrupo de pacientes, que no se correlacionó con la agregación plaquetaria, las concentraciones de P-selectina o los receptores GPIIb/IIIa.
Debe tenerse precaución si los pacientes reciben tratamiento concomitante con AINEs (incluyendo ácido acetilsalicílico) e inhibidores de la agregación plaquetaria, ya que estos medicamentos aumentan, de por sí, el riesgo de hemorragia (ver sección 4.4).
Warfarina
Los cambios de tratamiento con warfarina (INR de 2,0 a 3,0), un antagonista de la vitamina K, a rivaroxaban (20 mg) o de rivaroxaban (20 mg) a warfarina (INR de 2,0 a 3,0) aumentaron el tiempo de protrombina/INR (Neoplastin) de forma importante (pueden observarse valores individuales del INR de hasta 12), mientras que los efectos sobre el TTPa, la inhibición de la actividad del factor Xa y el potencial de trombina endógena (PTE) fueron aditivos.
Si se desea medir los efectos farmacodinámicos de rivaroxaban durante el periodo de cambio de tratamiento, puede utilizarse la actividad anti-factor Xa, PiCT y Heptest, ya que la warfarina no afecta a estas pruebas. Al cuarto día tras la última dosis de warfarina, todas las pruebas (incluyendo TP, TTPa, inhibición de la actividad del factor Xa y PTE) reflejaron únicamente el efecto de rivaroxaban. Si se desea medir los efectos farmacodinámicos de warfarina durante el periodo de cambio de tratamiento, se puede usar la determinación del INR en la Ctrough de rivaroxaban (24 horas después de su anterior administración), ya que rivaroxaban afecta mínimamente a esta prueba en este punto.
No se observó ninguna interacción farmacocinética entre warfarina y rivaroxaban.
Inductores del CYP3A4
La administración concomitante de rivaroxaban con rifampicina, un potente inductor del CYP3A4, produjo una disminución aproximada del 50% del AUC media de rivaroxaban, con disminuciones paralelas de sus efectos farmacodinámicos. El uso concomitante de rivaroxaban con otros inductores potentes del CYP3A4 (por ejemplo, fenitoína, carbamazepina, fenobarbital o la hierba de San Juan (Hypericum perforatum)) también puede causar una disminución de la concentración plasmática de rivaroxaban. Por tanto, la administración concomitante con inductores potentes del CYP3A4 deberá evitarse a menos que el paciente esté estrechamente monitorizado para detectar signos o síntomas de trombosis.
Otros tratamientos concomitantes
No se observó ninguna interacción farmacocinética o farmacodinámica clínicamente significativa cuando se administró rivaroxaban concomitantemente con midazolam (sustrato del CYP3A4), digoxina (sustrato de la P-gp), atorvastatina (sustrato del CYP3A4 y de la P-gp) u omeprazol (inhibidor de la bomba de protones). Rivaroxaban no inhibe ni induce ninguna isoforma mayor del CYP, como el CYP3A4.
Parámetros de laboratorio
Los parámetros de la coagulación (p. ej. TP, TTPa, HepTest) se ven afectados de la forma esperada debido al mecanismo de acción de rivaroxaban (ver sección 5.1).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No se ha evaluado la seguridad y eficacia de Xarelto en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3). Debido a la posible toxicidad reproductiva, riesgo intrínseco de hemorragia y la evidencia de que rivaroxaban atraviesa la barrera placentaria, Xarelto está contraindicado durante el embarazo (ver sección 4.3).
Las mujeres en edad fértil deben evitar quedarse embarazadas durante el tratamiento con rivaroxaban.
Lactancia
No se ha evaluado la seguridad y eficacia de Xarelto en mujeres en período de lactancia. Los datos en animales indican que rivaroxaban se excreta en la leche materna. Por lo tanto, Xarelto está contraindicado durante la lactancia (ver sección 4.3). Se debe decidir si es necesario interrumpir la lactancia o bien interrumpir/suspender el tratamiento.
Fertilidad
No se han realizado estudios específicos con rivaroxaban para evaluar los efectos sobre la fertilidad en humanos. En un estudio sobre la fertilidad en ratas macho y hembra no se observó ningún efecto (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Xarelto puede influir ligeramente en la capacidad para conducir y utilizar máquinas. Se han descrito reacciones adversas como síncope (frecuencia: poco frecuente) y mareos (frecuencia: frecuente) (ver sección 4.8). Los pacientes que sufran estas reacciones adversas no deben conducir ni utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Se ha evaluado la seguridad de rivaroxaban en once ensayos clínicos de fase III, que incluyeron 32.625 pacientes expuestos a rivaroxaban (ver la tabla 1).
Tabla 1: Número de pacientes estudiados, dosis máxima diaria y duración del tratamiento en los estudios de fase III.
|
Indicación |
Número de pacientes * |
Dosis máxima diaria |
Duración máxima del tratamiento |
|
Prevención de tromboembolismo venoso (TEV) en pacientes adultos sometidos a cirugía electiva de reemplazo de cadera o rodilla |
6.097 |
10 mg |
39 días |
|
Prevención de tromboembolismo venoso en pacientes encamados |
3.997 |
10 mg |
39 días |
|
Indicación |
Número de pacientes * |
Dosis máxima diaria |
Duración máxima del tratamiento |
|
Tratamiento de TVP, EP y prevención de las recurrencias de TVP y EP |
4.556 |
Días 1 a 21: 30 mg Día 22 en adelante: 20 mg |
21 meses |
|
Prevención del ictus y de la embolia sistémica en pacientes con fibrilación auricular no valvular |
7.750 |
20 mg |
41 meses |
|
Prevención de acontecimientos aterotrombóticos en pacientes después de padecer un SCA |
10.225 |
5 mg ó 10 mg respectivamente, co-administrada con AAS o bien con AAS más clopidogrel o ticlopidina |
31 meses |
*Pacientes expuestos por lo menos a una dosis de rivaroxaban.
Las reacciones adversas notificadas con mayor frecuencia en los pacientes que recibieron rivaroxaban fueron hemorragias (ver sección 4.4. y 'Descripción de las reacciones adversas seleccionadas" más adelante. Las hemorragias notificadas con mayor frecuencia (> 4%) fueron epistaxis (5,9%) y la hemorragia del tracto gastrointestinal (4,2%).
En total, se notificó la aparición de eventos adversos en aproximadamente un 67% de los pacientes expuestos por lo menos a una dosis de rivaroxaban. Aproximadamente el 22% de los pacientes sufrieron eventos adversos que se consideran relacionados con el tratamiento, según la evaluación de los investigadores. En los pacientes tratados con Xarelto 10 mg sometidos a cirugía electiva de reemplazo de cadera o rodilla y en pacientes hospitalizados encamados, se produjeron episodios hemorrágicos en aproximadamente un 6,8% y 12,6% de los pacientes, respectivamente, y se produjo anemia en aproximadamente un 5,9% y 2,1% de los pacientes, respectivamente. En los pacientes tratados con Xarelto 15 mg dos veces al día y después Xarelto 20 mg una vez al día para el tratamiento de la TVP o EP, o con Xarelto 20 mg una vez al día para la prevención de la recurrencia de la TVP y de la EP, se produjeron episodios hemorrágicos en aproximadamente un 27,8% de los pacientes, y anemia en aproximadamente un 2,2% de los pacientes. En los pacientes tratados para la prevención del ictus y de la embolia sistémica, se notificó hemorragia de cualquier tipo o gravedad, con una tasa de 28 por cada 100 pacientes-años, y anemia con una tasa de 2,5 por cada 100 pacientes-años. En los pacientes tratados para la prevención de muerte cardiovascular e infarto de miocardio después de un síndrome coronario agudo (SCA), se notificó hemorragia de cualquier tipo o gravedad con una tasa de 22 eventos por cada 100 pacientes-años. La anemia se notificó con una tasa de 1,4 eventos por cada 100 pacientes-años.
Tabla de reacciones adversas
Las frecuencias de las reacciones adversas notificadas con Xarelto se resumen en la tabla 2, según la clasificación por órganos y sistemas (convención MedDRA) y según las frecuencias.
Las frecuencias se definen como: muy frecuentes (> 1/10) frecuentes (> 1/100 a < 1/10) poco frecuentes (> 1/1.000 a < 1/100) raras (> 1/10.000 a < 1/1.000) muy raras ( < 1/10.000)
no conocida (no puede calcularse a partir de los datos disponibles)
|
Frecuentes |
Poco frecuentes |
Raras |
No conocida |
|
Trastornos de la sangre y del sistema linfático | |||
|
Anemia (incl. respectivos parámetros de laboratorio) |
Trombocitemia (incl. aumento del recuento de plaquetas)A | ||
|
Trastornos del sistema inmunológico | |||
|
Reacción alérgica, dermatitis alérgica | |||
|
Trastornos del sistema nervioso | |||
|
Mareos, cefalea, |
Hemorragia cerebral e intracraneal, síncope | ||
|
Trastornos oculares | |||
|
Hemorragia ocular (incl. hemorragia conjuntival) | |||
|
Trastornos cardiacos | |||
|
Taquicardia | |||
|
Trastornos vasculares | |||
|
Hipotensión, hematoma | |||
|
Trastornos respiratorios, torácicos y mediastínicos | |||
|
Epistaxis, hemoptisis | |||
|
Trastornos gastrointestinales | |||
|
Sangrado gingival, hemorragia del tracto gastrointestinal (incl. hemorragia rectal), dolor gastrointestinal y abdominal, dispepsia, náuseas, estreñimientoA, diarrea, vómitosA |
Sequedad de boca | ||
|
Trastornos hepatobiliares | |||
|
Alteración de la función hepática |
Ictericia | ||
|
Trastornos de la piel y del tejido subcutáneo | |||
|
Prurito (incl. casos raros de prurito generalizado), exantema, equimosis, hemorragia cutánea y subcutánea |
Urticaria | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |||
|
Dolor en las extremidadesA |
Hemartrosis |
Hemorragia muscular |
Síndrome compartimental secundario a una hemorragia |
|
Frecuentes |
Poco frecuentes |
Raras |
No conocida |
|
Trastornos renales y urinarios | |||
|
Hemorragia del tracto urogenital (incl. hematuria y menorragiaB), insuficiencia renal (incl. aumento de creatinina en sangre, aumento de la urea en sangre)A |
Insuficiencia renal /insuficiencia renal aguda secundaria a una hemorragia suficiente para causar hipoperfusión | ||
|
Trastornos generales y alteraciones en el lugar de administración | |||
|
FiebreA, edema periférico, disminución general de la fuerza y la energía (incl. fatiga y astenia) |
Sensación de malestar (incl. malestar general) |
Edema localizadoA | |
|
Exploraciones complementarias | |||
|
Aumento de las transaminasas |
Aumento de la bilirrubina, aumento de la fosfatasa alcalina sanguíneaA, aumento de la LDHA, aumento de la lipasaA, aumento de la amilasaA, aumento de la GGTA |
Aumento de la bilirrubina conjugada (con o sin aumento concomitante de la ALT). | |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos | |||
|
Hemorragia después de una intervención (incl. anemia postoperatoria y hemorragia de la herida), contusión, secreción de la heridaA |
Pseudoaneurisma vascularC | ||
A: observado en la prevención del tromboembolismo venoso (TEV) en pacientes adulto sometidos a cirugía electiva de reemplazo de cadera o rodilla
B: observado en el tratamiento de la TVP, EP y prevención de sus recurrencias como muy frecuente en mujeres < 55 años.
C: observado como poco frecuente en la prevención de eventos aterotrombóticos en pacientes que han sufrido un SCA (tras una intervención coronaria percutánea)
Descripción de reacciones adversas seleccionadas
Debido a su mecanismo de acción farmacológica, el uso de Xarelto puede asociarse a un incremento del riesgo de hemorragia oculta o manifiesta en cualquier tejido u órgano, que puede dar lugar a una anemia post-hemorrágica. Los signos, síntomas y gravedad (incluido un posible desenlace mortal) variarán según la localización y el grado o la extensión de la hemorragia, la anemia o ambas (ver sección 4.9 Tratamiento de la hemorragia). En los ensayos clínicos se observaron con más frecuencia hemorragias a nivel de mucosas (p.ej. epistaxis, gingival, gastrointestinal, genito-urinaria) y anemia en los pacientes que recibían rivaroxaban a largo plazo con respecto a los que recibían tratamiento con AVK. Por ello, además de un adecuado seguimiento clínico, las determinaciones de hemoglobina y hematocrito podrían ser útiles para detectar hemorragias ocultas cuando se considere apropiado.
El riesgo de hemorragia puede estar aumentado en ciertos grupos de pacientes, como por ejemplo, en pacientes con hipertensión arterial grave no controlada y/o en tratamiento concomitante que afecte a la hemostasia (ver Riesgo de hemorragia en la sección 4.4). El sangrado menstrual puede ser más intenso y/o prolongarse. Las complicaciones hemorrágicas pueden presentarse como debilidad, palidez, mareos, cefalea o tumefacción inexplicada, disnea o shock de causa desconocida. En algunos casos, a consecuencia de la anemia, se han observado síntomas de isquemia cardíaca, como dolor torácico o angina de pecho.
Se han notificado complicaciones conocidas, secundarias a hemorragia intensa, como el síndrome compartimental o insuficiencia renal debida a hipoperfusión. Por lo tanto deberá tenerse en cuenta la posibilidad de hemorragia al evaluar el estado de cualquier paciente anticoagulado.
Observaciones post-comercialización
Tras la comercialización se han notificado las siguientes reacciones adversas en asociación temporal con el uso de Xarelto. No se ha podido estimar la frecuencia de estas reacciones adversas.
Trastornos del sistema inmunológico: angioedema y edema alérgico (En los ensayos de fase III agrupados, estos eventos fueron poco frecuentes (> 1/1.000 a <1/100)).
Trastornos hepatobiliares: colestasis, hepatitis (incluyendo lesión hepatocelular). (En los ensayos de fase III agrupados, estos eventos fueron raros (> 1/10.000 a < 1/1.000)).
Trastornos de la sangre y del sistema linfático: trombocitopenia. (En los ensayos de fase III agrupados, estos eventos fueron poco frecuentes (> 1/1.000 a < 1/100)).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se han notificado casos raros de sobredosis de hasta 600 mg sin complicaciones hemorrágicas u otras reacciones adversas. Debido a la limitada absorción a dosis supraterapéuticas de 50 mg de rivaroxaban o superiores se espera un efecto techo sin un aumento posterior de la exposición plasmática media.
No se dispone de un antídoto específico que antagonice el efecto farmacodinámico de rivaroxaban.
Se puede considerar el uso de carbono activado para reducir la absorción en caso de sobredosis por rivaroxaban.
Tratamiento de la hemorragia
En caso de producirse una complicación hemorrágica en un paciente que recibe tratamiento con rivaroxaban, se deberá retrasar la siguiente administración de rivaroxaban o interrumpir el tratamiento si se considera conveniente. Rivaroxaban tiene una semivida de eliminación de entre 5 y 13 horas (ver sección 5.2). Las medidas terapéuticas deben individualizarse según la gravedad y la localización de la hemorragia. En caso necesario, podría aplicarse el tratamiento sintomático adecuado, como la compresión mecánica (por ejemplo en caso de epistaxis intensa), hemostasia quirúrgica con procedimientos de control de la hemorragia, reemplazo de fluidos y apoyo hemodinámico (concentrado de hematíes o plasma fresco congelado, dependiendo de la anemia o la coagulopatía asociadas) o plaquetas.
Si la hemorragia no se puede controlar con las medidas anteriores, debería plantearse la administración de un agente procoagulante específico para revertir el efecto, como el concentrado de complejo de protrombina (CCP), concentrado de complejo de protrombina activado (CCPA) o factor VIIa recombinante (r-FVIIa). Sin embargo, actualmente hay una experiencia clínica muy limitada con el uso de estos productos en pacientes que reciben rivaroxaban. La recomendación se basa también en datos no clínicos limitados. Deberá plantearse la readministración de factor VIIa recombinante y ajustar la dosis dependiendo de la mejoría de la hemorragia. Dependiendo de la disponibilidad local, en caso de hemorragia mayor debe considerarse consultar a un experto en coagulación (ver sección 5.1).
No se espera que el sulfato de protamina y la vitamina K afecten a la actividad anticoagulante de rivaroxaban. La experiencia con ácido tranexámico es limitada y no hay experiencia con ácido aminocaproico y aprotinina en pacientes tratados con rivaroxaban. No hay una justificación científica
sobre la ventaja ni experiencia con el hemostático sistémico desmopresina en pacientes tratados con rivaroxaban. Debido a su elevada fijación a las proteínas plasmáticas, no se espera que rivaroxaban sea dializable.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inhibidores directos del factor Xa, código ATC: B01AF01 Mecanismo de acción
Rivaroxaban es un inhibidor directo del factor Xa altamente selectivo, con biodisponibilidad oral. La inhibición del factor Xa interrumpe las vías intrínseca y extrínseca de la cascada de la coagulación de la sangre, inhibiendo tanto la formación de trombina como la formación de trombos. Rivaroxaban no inhibe la trombina (factor II activado) y no se han demostrado efectos sobre las plaquetas.
Efectos farmacodinámicos
En los seres humanos se ha observado una inhibición de la actividad del factor Xa dosis-dependiente. Rivaroxaban modifica el tiempo de protrombina (TP) de forma dosis-dependiente con una estrecha correlación con las concentraciones plasmáticas (el valor de r es igual a 0,98) si se emplea Neoplastin para el análisis. Otros reactivos proporcionarían unos resultados diferentes. La lectura del TP debe hacerse en segundos, porque el INR sólo se ha calibrado y validado en el caso de los cumarínicos y no puede utilizarse con ningún otro anticoagulante.
En pacientes que recibieron rivaroxaban para el tratamiento de la TVP y EP, y para la prevención de sus recurrencias, los percentiles 5/95 del TP (Neoplastin) de 2 a 4 horas después de tomar el comprimido (es decir, en el momento del efecto máximo) variaron de 17 a 32 seg. en el caso de rivaroxaban 15 mg dos veces al día, y de 15 a 30 seg. en el caso de rivaroxaban 20 mg una vez al día. En el momento de la concentración valle (8 - 16 h después de la toma del comprimido) los percentiles 5/95 para la dosis de 15 mg dos veces al día variaron de 14 a 24 seg. y para la dosis de 20 mg una vez al día (18 - 30 h después de la toma del comprimido) variaron de 13 a 20 seg.
En pacientes con fibrilación auricular no valvular que recibieron rivaroxaban para la prevención del ictus y de la embolia sistémica, en el momento del efecto máximo (1 a 4 h después de la toma del comprimido) los percentiles 5/95 del TP (Neoplastin) variaron de 14 a 40 seg. en los pacientes tratados con 20 mg una vez al día, y de 10 a 50 seg. en los pacientes con insuficiencia renal moderada tratados con 15 mg una vez al día. En el momento de la concentración valle (16 - 36 h de la toma del comprimido) los percentiles 5/95 para los pacientes tratados con la dosis de 20 mg una vez al día variaron de 12 a 26 seg. y para los pacientes con insuficiencia renal moderada tratados con la dosis de 15 mg una vez al día variaron de 12 a 26 seg.
En un estudio de farmacología clínica en la reversión de la acción farmacodinámica de rivaroxaban en adultos sanos (n = 22), se evaluaron los efectos de dosis únicas (50 UI/kg) de dos tipos diferentes de CCP, un CCP de 3 factores (factores II, IX y X) y un CCP de 4 factores (factores II, VII, IX y X). El CCP de 3 factores redujo los valores medios del TP (Neoplastina) en aproximadamente 1,0 segundos a los 30 minutos, en comparación con reducciones de, aproximadamente, 3,5 segundos observadas con el CCP de 4 factores. En cambio, el CCP de 3 factores tuvo un efecto global mayor y más rápido en la reversión de los cambios en la generación de trombina endógena que el CCP de 4 factores (ver sección 4.9).
El tiempo de tromboplastina parcial activada (TTPa) y el HepTest también se prolongaron de forma dosis-dependiente; sin embargo, no se recomiendan para evaluar el efecto farmacodinámico de rivaroxaban. No es necesario monitorizar los parámetros de la coagulación durante el tratamiento con rivaroxaban en la práctica clínica. Sin embargo, si está indicado clínicamente, se pueden medir los niveles de rivaroxaban mediante ensayos cuantitativos calibrados para la actividad anti-factor Xa (ver sección 5.2).
Eficacia clínica y seguridad
Tratamiento de la TVP, de la EP y prevención de las recurrencias de la TVP y de la EP
El programa clínico de Xarelto se diseñó para demostrar la eficacia de Xarelto en el tratamiento inicial y continuado de la TVP aguda y de la EP y en la prevención de sus recurrencias.
En tres estudios clínicos de fase III aleatorizados y controlados (Einstein DVT, Einstein EP y Einstein Extension) se estudiaron más de 9.400 pacientes; adicionalmente, se realizó un análisis agrupado predefinido de los estudios Einstein DVT y Einstein PE. La duración combinada total del tratamiento en todos los estudios fue de 21 meses.
En el estudio Einstein DVT, se estudiaron 3.449 pacientes con TVP aguda para el tratamiento de la TVP y prevención de las recurrencias de la TVP y de la EP (se excluyeron los pacientes que presentaban EP sintomática). La duración del tratamiento fue de 3, 6 ó 12 meses, dependiendo del criterio clínico del investigador.
Para el tratamiento inicial de la TVP aguda se administró rivaroxaban 15 mg dos veces al día durante 3 semanas y a continuación, rivaroxaban 20 mg una vez al día.
En el estudio Einstein PE, se estudiaron 4.832 pacientes con EP aguda para el tratamiento de la EP y para la prevención de las recurrencias de TVP y EP. La duración del tratamiento fue de 3, 6 ó 12 meses, en función del juicio clínico del investigador.
Para el tratamiento inicial de EP aguda, se administró 15 mg de rivaroxaban dos veces al día durante tres semanas. Esta pauta fue seguida por 20 mg de rivaroxaban una vez al día.
En los dos estudios Einstein DVT y Einstein PE, el tratamiento comparador fue enoxaparina administrada durante al menos cinco días, en combinación con un antagonista de la vitamina K hasta que el TP/INR estuviera en rango terapéutico (> 2,0). El tratamiento se continuó con un antagonista de la vitamina K, con un ajuste de dosis para mantener los valores de TP/INR dentro del rango terapéutico de 2,0 a 3,0.
En el estudio Einstein Extension para la prevención de la TVP recurrente o de la EP se estudiaron 1.197 pacientes con TVP o EP. El tratamiento tuvo una duración adicional de 6 ó 12 meses en pacientes que previamente habían completado un periodo de 6 a 12 meses de tratamiento por TEV, en función del criterio clínico del investigador. Se comparó Xarelto 20 mg una vez al día con placebo.
Todos los estudios de fase III usaron las mismas variables principales y secundarias predefinidas de eficacia. La variable principal de eficacia fue el TEV sintomático y recurrente, definido como la combinación de TVP recurrente o bien EP mortal o no mortal. La variable secundaria de eficacia se definió como la combinación de TVP recurrente, EP no mortal y mortalidad por todas las causas.
En el estudio Einstein DVT (ver tabla 3) rivaroxaban demostró ser no inferior a enoxaparina / antagonista de la vitamina K (AVK) para la variable principal de eficacia (p < 0,0001 (prueba de no inferioridad); cociente de riesgos (hazard ratio): 0,680 (0,443 - 1,042), p = 0,076 (prueba de superioridad)). El beneficio clínico neto pre-especificado (variable principal de eficacia más episodios hemorrágicos mayores) mostró un cociente de riesgos (hazard ratio) de 0,67 ((IC 95%: 0,47 - 0,95), valor nominal de p = 0,027) en favor de rivaroxaban. Los valores del INR estuvieron dentro del rango terapéutico un promedio del 60,3% del tiempo para una duración media del tratamiento de 189 días, y del 55,4%, 60,1% y 62,8% del tiempo en los grupos con una duración prevista del tratamiento de 3, 6, y 12 meses, respectivamente. En el grupo de tratamiento con enoxaparina/AVK, no se observó una relación clara entre la media del TTR (tiempo dentro del rango objetivo de INR entre 2,0 - 3,0) del centro en los terciles de igual tamaño y la incidencia de TEV recurrente (p=0,932 para la interacción). En el tercil más alto según el control del centro, el cociente de riesgos (hazard ratio) de rivaroxaban con respecto a warfarina fue de 0,69 (IC 95%: 0,35 - 1,35).
Las tasas de incidencia de la variable principal de seguridad (hemorragia mayor o no mayor clínicamente relevante), así como la variable secundaria de seguridad (hemorragia mayor) fueron similares en ambos grupos de tratamiento.
|
Población del estudio |
3.449 pacientes con trombosis venosa profunda sintomática | |
|
Xareltoa) |
Enoxaparina/AVKb) | |
|
Pauta de tratamiento y duración |
3, 6 ó 12 meses |
3, 6 ó 12 meses |
|
N=1.731 |
N=1.718 | |
|
TEV sintomático recurrente* |
36 (2,1%) |
51 (3,0%) |
|
EP sintomática recurrente |
20 (1,2%) |
18 (1,0%) |
|
TVP sintomática recurrente |
14 (0,8%) |
28 (1,6%) |
|
EP y TVP sintomáticas |
1 (0,1%) |
0 |
|
EP mortal/Muerte en la que no |
4 |
6 |
|
puede descartarse EP |
(0,2%) |
(0,3%) |
|
Hemorragia mayor o no mayor |
139 |
138 |
|
clínicamente relevante |
(8,1%) |
(8,1%) |
|
Eventos hemorrágicos mayores |
14 (0,8%) |
20 (1,2%) |
|
a) Rivaroxaban 15 mg dos veces al día durante 3 semanas, seguido d |
e rivaroxaban 20 mg una vez | |
al día
b) Enoxaparina durante al menos 5 días, solapado con y seguido por AVK
* p <0,0001 (no-inferioridad; cociente de riesgos (hazard ratio) pre-especificado de 2,0); cociente de riesgos (hazard ratio): 0,680 (0,443 - 1,042), p = 0,076 (superioridad)
En el estudio Einstein PE (ver Tabla 4) rivaroxaban demostró ser no inferior a la enoxaparina/AVK para la variable primaria de eficacia (p = 0,0026 (prueba de no inferioridad); cociente de riesgos (hazard ratio): 1,123 (0,749-1,684)). El beneficio clínico neto pre-especificado (resultado de eficacia primaria más eventos de sangrado mayor) se reportó con un cociente de riesgos (hazard ratio) de 0,849 ((IC del 95%: 0,633 a 1,139), valor nominal de p = 0,275). Los valores de INR estuvieron dentro del rango terapéutico de una media de 63% del tiempo para la duración media del tratamiento de 215 días, y el 57%, 62%, y 65% del tiempo en los grupos de duración prevista de tratamiento de 3, 6, y 12 meses, respectivamente. En el grupo de enoxaparina/AVK, no hubo una relación clara entre el nivel de la media TRT del centro (Tiempo en objetivo de INR de 2,0 - 3,0) en los terciles de igual tamaño y la incidencia de la recurrencia de TEV (p = 0,082 para la interacción). En el tercil superior de acuerdo con el centro, el cociente de riesgos (hazard ratio) con rivaroxaban en comparación con warfarina fue 0,642 (IC 95%: 0,277 - 1,484).
Las tasas de incidencia de la variable principal de seguridad (eventos hemorrágicos mayores o no mayores clínicamente relevantes) fueron ligeramente inferiores en el grupo de tratamiento con rivaroxaban (10,3% (249/2412)) frente a las del grupo de tratamiento con enoxaparina/AVK (11,4% (274 / 2405)). La incidencia de las variables secundarias de seguridad (eventos de sangrado mayor) fue inferior en el grupo de rivaroxaban (1,1% (26/2412)) comparado con la de enoxaparina/grupo AVK (2,2% (52/2405)), con un cociente de riesgos (hazard ratio) 0,493 (95 % CI: 0,308 0,789).
|
Población del estudio |
4.832 pacientes con EP sintomática aguda | |
|
Xareltoa) |
Enoxaparina/VKAb) | |
|
Pauta de tratamiento y duración |
3, 6 ó 12 meses |
3, 6 ó 12 meses |
|
N=2.419 |
N=2.413 | |
|
TEV sintomático recurrente* |
50 (2,1%) |
44 (1,8%) |
|
EP sintomática recurrente |
23 (1,0%) |
20 (0,8%) |
|
TVP sintomática recurrente |
18 (0,7%) |
17 (0,7%) |
|
EP y TVP sintomáticas |
0 |
2 (<0,1%) |
|
EP mortal/Muerte en la que no |
11 |
7 |
|
puede descartarse EP |
(0,5%) |
(0,3%) |
|
Hemorragia mayor o no mayor |
249 |
274 |
|
clínicamente relevante |
(10,3%) |
(11,4%) |
|
Eventos hemorrágicos mayores |
26 (1,1%) |
52 (2,2%) |
|
a) Rivaroxaban 15 mg dos veces al día durante 3 semanas, seguido c |
e rivaroxaban 20 mg una vez | |
al día
b) Enoxaparina durante al menos 5 días, solapado con y seguido por AVK
* p <0,0026 (no-inferioridad; cociente de riesgos (hazard ratio) pre-especificado de 2,0); cociente de riesgos (hazard ratio): 1,123 (0,749 - 1,684)
Se realizó un análisis agrupado pre-especificado de los resultados de los estudios Einstein DVT y PE (ver Tabla 5).
Tabla 5: Resultados de eficacia y seguridad del análisis agrupado de los estudios de fase III Einstein DVT y Einstein PE__
|
Población del estudio |
8.281 pacientes con TVP sintomática aguda o EP | |
|
Xareltoa) |
Enoxaparin/VKAb) | |
|
Pauta de tratamiento y duración |
3, 6 ó 12 meses |
3, 6 ó 12 meses |
|
N=4.150 |
N=4.131 | |
|
TEV sintomático recurrente* |
86 (2,1%) |
95 (2,3%) |
|
EP sintomática recurrente |
43 (1,0%) |
38 (0,9%) |
|
TVP sintomática recurrente |
32 (0,8%) |
45 (1,1%) |
|
EP y TVP sintomáticas |
1 (<0,1%) |
2 (<0,1%) |
|
EP mortal/Muerte en la que no |
15 |
13 |
|
puede descartarse EP |
(0,4%) |
(0,3%) |
|
Hemorragia mayor o no mayor |
388 |
412 |
|
clínicamente relevante |
(9,4%) |
(10,0%) |
|
Eventos hemorrágicos mayores |
40 (1,0%) |
72 (1,7%) |
|
a) Rivaroxaban 15 mg dos veces al día durante 3 semanas, seguido d |
e rivaroxaban 20 mg una vez | |
al día
b) Enoxaparina durante al menos 5 días, solapado con y seguido por AVK * p <0,0001 (no-inferioridad; cociente de riesgos (hazard ratio) pre-especificado de 1,75); cociente de riesgos (hazard ratio): 0,886 (0,661 - 1,186)
El beneficio clínico neto pre-especificado (variable primaria de eficacia más eventos de sangrado mayor) del análisis agrupado se reportó con un cociente de riesgos (hazard ratio) de 0,771 ((IC95%: 0,614 - 0,967), valor nominal de p = 0,0244).
En el estudio Einstein Extension (ver tabla 6), rivaroxaban fue superior a placebo en cuanto a las variables principal y secundaria de eficacia. En cuanto a la variable principal de seguridad (hemorragia mayor) hubo una tasa de incidencia numéricamente superior no significativa en los pacientes tratados con rivaroxaban 20 mg una vez al día, en comparación con placebo. La variable secundaria de seguridad (hemorragia mayor o no mayor clínicamente relevante) mostró unas tasas más altas en los pacientes tratados con rivaroxaban 20 mg una vez al día, en comparación con placebo.
Tabla 6: Resultados de eficacia y seguridad del estudio de fase III Einstein Extension.
|
Población del estudio |
1.197 pacientes continuaron el tratamiento y la prevención del TEV recurrente | |
|
Xareltoa) |
Placebo | |
|
Pauta del tratamiento |
6 ó 12 meses |
6 ó 12 meses |
|
N = 602 |
N = 594 | |
|
TEV recurrente y sintomático* |
8 (1,3%) |
42 (7,1%) |
|
EP recurrente y sintomática |
2 (0,3%) |
13 (2,2%) |
|
TVP recurrente y |
5 |
31 |
|
sintomática |
(0,8%) |
(5,2%) |
|
EP mortal / Muerte en la que |
1 |
1 |
|
no se puede descartar EP |
(0,2%) |
(0,2%) |
|
Hemorragia mayor |
4 (0,7%) |
0 (0,0%) |
|
Hemorragia no mayor clínicamente |
32 |
7 |
|
relevante |
(5,4%) |
(1,2%) |
a) Rivaroxaban 20 mg una vez al día
* p < 0,0001 (superioridad), cociente de riesgos (hazard ratio): 0,185 (0,087 - 0,393)
Además del estudio de fase III ROCKET AF, se ha realizado un estudio prospectivo de cohortes, de un solo brazo, posautorización, no intervencionista, abierto (XANTUS) con adjudicación central de resultados, incluyendo acontecimientos tromboembólicos y hemorragia mayor. Se reclutaron 6.785 pacientes con fibrilación auricular no valvular para la prevención del ictus y de la embolia sistémica fuera del sistema nervioso central (SNC) en condiciones de práctica clínica. En XANTUS, las puntuaciones medias de CHADS2 y HAS-BLED fueron ambas de 2,0, en comparación con la puntuación media de CHADS2 y HAS-BLED de 3,5 y 2,8, respectivamente, en ROCKET AF. Se produjo hemorragia mayor en 2,1 por 100 pacientes-años. Se notificó hemorragia mortal en 0,2 por 100 pacientes-años y hemorragia intracraneal en 0,4 por 100 pacientes-años. Se registró ictus o embolia sistémica fuera del SNC en 0,8 por 100 pacientes-años.
Estas observaciones en condiciones de práctica clínica son consistentes con el perfil de seguridad establecido en esta indicación.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Xarelto en uno o más grupos de la población pediátrica en el tratamiento de eventos tromboembólicos. La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Xarelto en los diferentes grupos de la población pediátrica en la prevención de eventos tromboembólicos (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Rivaroxaban se absorbe rápidamente y alcanza concentraciones máximas (Cmax) de 2 a 4 horas después de tomar el comprimido.
La absorción oral de rivaroxaban es casi completa y su biodisponibilidad oral es elevada (80% al 100%) en el caso de la dosis del comprimido de 2,5 mg y de 10 mg, independientemente de las condiciones de ayuno o alimentación. La ingesta de alimentos con rivaroxaban (a la dosis de 2,5 mg y de 10 mg) no afecta al AUC ni a la Cmax.
Debido a la disminución de la absorción, se determinó una biodisponibilidad del 66% con el comprimido de 20 mg en condiciones de ayuno. Cuando los comprimidos de Xarelto 20 mg se tomaron junto con alimentos, se observaron aumentos del AUC media del 39% en comparación con la toma de comprimidos en condiciones de ayuno, lo que indica una absorción casi completa y una biodisponibilidad oral elevada. Xarelto 15 mg y 20 mg deben tomarse con alimentos (ver sección 4.2). Rivaroxaban presenta una farmacocinética lineal hasta aproximadamente 15 mg administrados una vez al día en ayunas. En condiciones de alimentación reciente, Xarelto en comprimidos de 10 mg, 15 mg y 20 mg demostró proporcionalidad con la dosis. A dosis más altas, rivaroxaban muestra una disolución limitada, con una reducción de la biodisponibilidad y de la tasa de absorción al aumentar la dosis.
La variabilidad de la farmacocinética de rivaroxaban es moderada; con una variabilidad interindividual (CV%) entre el 30% y el 40%.
La absorción de rivaroxaban depende del sitio donde se libera en el tracto gastrointestinal. Se ha notificado una disminución del 29% y del 56% en el AUC y la Cmax, en comparación con el comprimido, cuando rivaroxaban en forma de granulado se liberó en el intestino delgado proximal. La exposición se reduce aún más cuando rivaroxaban se libera en el intestino delgado distal o en el colon ascendente. Por lo tanto, debe evitarse la administración de rivaroxaban de forma distal al estómago, ya que esto puede dar lugar a una reducción de la absorción y la correspondiente exposición a rivaroxaban.
La biodisponibilidad (AUC y Cmax) fue comparable para rivaroxaban 20 mg, administrado por vía oral como comprimido triturado y mezclado con puré de manzana o diluido con agua, administrado a través de una sonda gástrica y seguido de una comida líquida, en comparación con el comprimido entero. Dado el perfil farmacocinético predecible, proporcional a la dosis de rivaroxaban, los resultados de biodisponibilidad de este estudio son probablemente aplicables a dosis más bajas de rivaroxaban.
Distribución
La unión a proteínas plasmáticas humanas es alta, del 92 % al 95 % aproximadamente y la albúmina sérica es el principal componente de unión. El volumen de distribución es moderado, con un Vss de 50 litros, aproximadamente.
Biotransformación y eliminación
De la dosis administrada de rivaroxaban se metabolizan aproximadamente 2/3; después, la mitad se elimina por vía renal y la otra mitad por vía fecal. El 1/3 restante de la dosis administrada se excreta directamente por vía renal como principio activo no modificado en la orina, principalmente mediante secreción renal activa.
Rivaroxaban se metaboliza mediante el CYP3A4, el CYP2J2 y mecanismos independientes del CYP. Las principales vías de biotransformación son la degradación oxidativa de la porción de morfolinona y la hidrólisis de los enlaces amida. Según investigaciones in vitro, rivaroxaban es un sustrato de las proteínas transportadoras P-gp (glucoproteína P) y Bcrp (proteína de resistencia al cáncer de mama). Rivaroxaban en forma inalterada es el compuesto más abundante en el plasma humano sin presencia de metabolitos mayores o metabolitos activos circulantes. Con un aclaramiento sistémico de aproximadamente 10 l/h, rivaroxaban puede clasificarse como una sustancia de bajo aclaramiento. Después de la administración por vía intravenosa de una dosis de 1 mg, la semivida de eliminación es de aproximadamente 4,5 horas. Después de la administración por vía oral, la eliminación se ve limitada por la tasa de absorción. En personas jóvenes, la eliminación de rivaroxaban del plasma se produce con una semivida de eliminación de 5 a 9 horas y en personas de edad avanzada, con una semivida de eliminación de 11 a 13 horas.
Poblaciones especiales
Sexo
No hubo ninguna diferencia clínicamente relevante en las propiedades farmacocinéticas y farmacodinámicas entre hombres y mujeres.
Pacientes de edad avanzada
Los pacientes de edad avanzada presentaron concentraciones plasmáticas mayores que los pacientes más jóvenes, con unos valores medios del AUC que fueron aproximadamente 1,5 veces superiores, principalmente debido a la disminución (aparente) del aclaramiento renal y total. No es necesario un ajuste de la dosis.
Peso corporal
Los valores extremos en el peso corporal (< 50 kg ó > 120 kg) tuvieron poco efecto en las concentraciones plasmáticas de rivaroxaban (menos del 25%). No es necesario un ajuste de la dosis.
Origen étnico
No se observaron diferencias interétnicas clínicamente relevantes entre los pacientes de raza blanca, afroamericanos, de origen latinoamericano, japonés o chino, en cuanto a las propiedades farmacocinéticas o farmacodinámicas.
Insuficiencia hepática
Los pacientes cirróticos con insuficiencia hepática leve (clasificados como Child Pugh A), sólo presentaron cambios menores en la farmacocinética de rivaroxaban (aumento medio del AUC de 1,2 veces), lo que fue casi comparable al grupo control de voluntarios sanos. En los pacientes cirróticos con insuficiencia hepática moderada (clasificados como Child Pugh B), el AUC media de rivaroxaban estuvo aumentada significativamente en 2,3 veces, en comparación con los voluntarios sanos. El AUC parcial aumentó 2,6 veces. Estos pacientes también mostraron una disminución de la eliminación renal de rivaroxaban, similar a los pacientes con insuficiencia renal moderada. No hay datos en pacientes con insuficiencia hepática grave.
La inhibición de la actividad del factor Xa se incrementó en un factor de 2,6 en los pacientes con insuficiencia hepática moderada, en comparación con los voluntarios sanos; de manera similar, la prolongación del TP se incrementó en un factor de 2,1. Los pacientes con insuficiencia hepática moderada fueron más sensibles a rivaroxaban, lo que produjo una relación farmacocinética / farmacodinámica más pronunciada entre la concentración y el TP.
Xarelto está contraindicado en pacientes con hepatopatía asociada a coagulopatía y con riesgo clínicamente relevante de hemorragia, incluyendo pacientes cirróticos clasificados como Child Pugh B y C (ver sección 4.3).
Insuficiencia renal
Se observó un aumento de la exposición a rivaroxaban correlacionado con la disminución de la función renal, evaluada mediante las determinaciones del aclaramiento de creatinina. En sujetos con insuficiencia renal leve (aclaramiento de creatinina de 50 - 80 ml/min), moderada (aclaramiento de creatinina de 30 a 49 ml/min) o grave (aclaramiento de creatinina de 15 a 29 ml/min), las concentraciones plasmáticas de rivaroxaban (AUC) aumentaron 1,4, 1,5 y 1,6 veces, respectivamente. Los aumentos correspondientes en los efectos farmacodinámicos fueron más pronunciados. En sujetos con insuficiencia renal leve, moderada y grave, la inhibición total de la actividad del factor Xa aumentó en un factor de 1,5, 1,9 y 2,0 respectivamente, en comparación con voluntarios sanos; de manera similar, la prolongación del TP aumentó en factores de 1,3, 2,2 y 2,4, respectivamente. No hay datos en pacientes con un aclaramiento de creatinina < 15 ml/min.
Debido a la elevada fijación a proteínas plasmáticas, no se espera que rivaroxaban sea dializable.
No se recomienda su uso en pacientes con un aclaramiento de creatinina < 15 ml/min. Xarelto debe utilizarse con precaución en pacientes con un aclaramiento de creatinina de 15 a 29 ml/min (ver sección 4.4).
Datos farmacocinéticos en pacientes
En los pacientes que recibieron rivaroxaban 20 mg una vez al día para el tratamiento de la TVP aguda, la concentración media geométrica (intervalo de predicción del 90%) a las 2 - 4 h y a las 24 h aproximadamente después de la dosis (lo que representa aproximadamente las concentraciones máxima y mínima durante el intervalo entre dosis) fue de 215 (22 - 535) y de 32 (6 - 239) ^g/l, respectivamente.
Relación farmacocinética / farmacodinámica
Se ha evaluado la relación farmacocinética/farmacodinámica (PK/PD) entre la concentración plasmática de rivaroxaban y varios criterios de valoración PD (inhibición del factor Xa, tiempo de protrombina (TP, TTPa, Heptest)) después de la administración de un amplio rango de dosis (de 5 a 30 mg dos veces al día). La relación entre la concentración de rivaroxaban y la actividad del factor Xa se describió de manera óptima por un modelo Emax. En el caso del TP, por lo general, el modelo de intersección lineal describió mejor los datos. Dependiendo de los diferentes reactivos usados en el TP, la pendiente varió considerablemente. Con Neoplastin PT, el TP basal fue de aproximadamente 13 seg. y la pendiente fue de alrededor de 3 a 4 seg/(100 ^g/l). Los resultados de los análisis de la relación PK/PD en las fases II y III fueron consistentes con los datos establecidos en los sujetos sanos.
Población pediátrica
No se ha determinado la seguridad y eficacia en niños y adolescentes hasta los 18 años.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad con dosis únicas, fototoxicidad, genotoxicidad, potencial carcinogénico y toxicidad juvenil.
Los efectos observados en los estudios con dosis repetidas se debieron principalmente a la actividad farmacodinámica incrementada de rivaroxaban. En ratas se observó un aumento de las concentraciones plasmáticas de IgG e IgA a niveles de exposición clínicamente relevantes.
No se observó ningún efecto sobre la fertilidad en las ratas macho o hembra. Los estudios en animales han demostrado una toxicidad reproductiva relacionada con el modo de acción farmacológica de rivaroxaban (p. ej. complicaciones hemorrágicas). A concentraciones plasmáticas clínicamente relevantes se observó toxicidad embriofetal (pérdida después de la implantación, retraso o adelanto de la osificación, varias manchas hepáticas de color claro) y un aumento de la incidencia de malformaciones frecuentes, así como cambios placentarios. En el estudio pre y postnatal en ratas, se observó una disminución de la viabilidad de las crías a dosis que fueron tóxicas para las madres.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido:
Celulosa microcristalina Croscarmelosa sódica Lactosa monohidrato Hipromelosa Laurilsulfato de sodio Estearato de magnesio
Cubierta pelicular:
Macrogol 3350
Hipromelosa
Dióxido de titanio (E171)
Óxido de hierro rojo (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez 3 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Envase para el inicio del tratamiento en las primeras 4 semanas:
Blisters de PP/ lámina de aluminio, en un envase (con calendario) que contiene 49 comprimidos recubiertos con película:
42 comprimidos recubiertos con película de Xarelto 15 mg y 7 comprimidos recubiertos con película de Xarelto 20 mg.
6.6 Precauciones especiales de eliminación
Ninguna especial para su eliminación
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG D-13342 Berlin Alemania
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/040
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 30 de septiembre de 2008 Fecha de la última renovación: 22 de mayo de 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
{MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de los fabricantes responsables de la liberación de los lotes
Bayer Pharma AG 51368 Leverkusen Alemania
Bayer HealthCare Manufacturing Srl.
Via delle Groane, 126 20024 Garbagnate Milanese Italia
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica.
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• - Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:.
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos)
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
• Medidas adicionales de minimización de riesgos
Antes del lanzamiento, el TAC deberá facilitar un pack educacional dirigido a todos los médicos que puedan prescribir/utilizar Xarelto. El material educacional tiene el objetivo de concienciar sobre el
El material educacional para el médico debe contener:
• La Ficha Técnica o Resumen de las Características del Producto
• La Guía de prescripción
• Tarjetas de Alerta para el Paciente [Texto incluido en el Anexo III]
El TAC debe acordar el contenido y el formato de la Guía de Prescripción, junto con un plan de comunicación, con la autoridad nacional competente en cada estado miembro antes de la distribución de dicho pack educacional en su territorio. La Guía de prescripción debe contener los siguientes mensajes clave de seguridad:
• Detalles de las poblaciones con un riesgo potencial de sangrado más alto
• Recomendaciones para la reducción de la dosis en las poblaciones de riesgo
• Consejos para realizar el cambio de/a tratamiento con rivaroxaban
• La necesidad de tomar los comprimidos de 15 mg y 20 mg con alimentos
• Gestión de los casos de sobredosis
• El uso de pruebas de coagulación y su interpretación
• Que se debe aconsejar a todos los pacientes sobre:
- Signos o síntomas de sangrado y cuándo es necesario solicitar la atención de un profesional sanitario.
- La importancia del cumplimiento del tratamiento
- La necesidad de tomar los comprimidos de 15 mg y de 20 mg con alimentos
- La necesidad de llevar la Tarjeta de alerta al paciente que se incluye en cada envase con ellos en todo momento
- La necesidad de informar a los profesionales sanitarios de que están tomando Xarelto si necesitan someterse a cirugía o a un procedimiento invasivo
Asimismo, el TAC deberá incluir una Tarjeta de Alerta para el paciente en cada estuche del medicamento; el texto de la tarjeta de alerta para el paciente está incluido en el Anexo III.
• Obligación de llevar a cabo medidas posautorización
El TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
Descripción |
Fecha límite |
|
Programa de estudios posautorización con el objetivo de analizar la seguridad de rivaroxaban en la prevención secundaria del síndrome coronario agudo fuera del contexto del ensayo clínico, especialmente con respecto a la incidencia, gravedad, gestión y resultados de los eventos hemorrágicos en toda la población y especialmente en pacientes con un mayor riesgo de sangrado. |
• Informes de análisis intermedios a presentar anualmente desde el principio del Q4 2015 hasta la finalización del programa de estudios. • Informe intermedio acumulativo en Q4 2017. • Informes finales de los estudios a presentar hasta Q4 2020. |
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR EMBALAJE EXTERIOR PARA 2,5 mg_
1. NOMBRE DEL MEDICAMENTO
Xarelto 2,5 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 2,5 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
14 comprimidos recubiertos con película 28 comprimidos recubiertos con película 56 comprimidos recubiertos con película 60 comprimidos recubiertos con película 98 comprimidos recubiertos con película 168 comprimidos recubiertos con película 196 comprimidos recubiertos con película 10 x 1 comprimidos recubiertos con película 100 x 1 comprimidos recubiertos con película 30 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE
COMERCIALIZACIÓN
Bayer Pharma AG, 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/025
EU/1/08/472/026
EU/1/08/472/027
EU/1/08/472/028
EU/1/08/472/029
EU/1/08/472/030
EU/1/08/472/031
EU/1/08/472/032
EU/1/08/472/033
EU/1/08/472/035
14 comprimidos recubiertos con película 28 comprimidos recubiertos con película 56 comprimidos recubiertos con película 60 comprimidos recubiertos con película 98 comprimidos recubiertos con película 168 comprimidos recubiertos con película 196 comprimidos recubiertos con película 10 x comprimidos recubiertos con película 100 x 1 comprimidos recubiertos con película 30 x 1 comprimidos recubiertos con película
(blisters de PP/Aluminio) (blisters de PP/Aluminio) (blisters de PP/Aluminio) (blisters de PP/Aluminio) (blisters de PP/Aluminio) (blisters de PP/Aluminio) (blisters de PP/Aluminio) (blisters de PP/Aluminio) (blisters de PP/Aluminio) (blisters de PP/Aluminio)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Xarelto 2,5 mg
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
EMBALAJE EXTERIOR DEL ENVASE MULTIPLE (INCLUIDO BLUE BOX) PARA 2,5 MG
1. NOMBRE DEL MEDICAMENTO
Xarelto 2,5 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 2,5 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Envase múltiple: 100 (10 envases de 10 x 1) comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Bayer Pharma AG 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/034 100 comprimidos recubiertos con película (10 x 10 x 1 (envase múltiple)
(blisters de PP/Alumino)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Xarelto 2,5 mg
1. NOMBRE DEL MEDICAMENTO
Xarelto 2,5 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 2,5 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
10 x 1 comprimidos recubiertos con película
Forma parte de un envase múltiple, no puede venderse por separado.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Bayer Pharma AG 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/034 100 comprimidos recubiertos con película (10 x 10 x 1) (envase múltiple)
(blisters de PP/Alumino)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Xarelto 2,5 mg
1. NOMBRE DEL MEDICAMENTO
Xarelto 2,5 mg comprimidos rivaroxaban
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer (logo)
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLÍSTER DE 10 COMPRIMIDOS PARA 2,5 mg_
1. NOMBRE DEL MEDICAMENTO
Xarelto 2,5 mg comprimidos rivaroxaban
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer (logo)
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
1. NOMBRE DEL MEDICAMENTO
Xarelto 2,5 mg comprimidos rivaroxaban
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer (logo)
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
Lun.
Mar.
Mier.
Jue.
Vie.
Sab.
Dom.
Símbolo del sol Símbolo de la luna
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR EMBALAJE EXTERIOR PARA 10 MG
1. NOMBRE DEL MEDICAMENTO
Xarelto 10 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 10 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
5 comprimidos recubiertos con película 10 comprimidos recubiertos con película 30 comprimidos recubiertos con película 10 x 1 comprimidos recubiertos con película 100 x 1 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO
UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE
COMERCIALIZACIÓN
Bayer Pharma AG, 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/001 5 comprimidos recubiertos con película EU/1/08/472/002 10 comprimidos recubiertos con película EU/1/08/472/003 30 comprimidos recubiertos con película EU/1/08/472/004 100 x 1 comprimidos recubiertos con película EU/1/08/472/005 5 comprimidos recubiertos con película EU/1/08/472/006 10 comprimidos recubiertos con película) EU/1/08/472/007 30 comprimidos recubiertos con película) EU/1/08/472/008 100 x 1 comprimidos recubiertos con película) EU/1/08/472/009 10 x 1 comprimidos recubiertos con película) EU/1/08/472/010 10 x 1 comprimidos recubiertos con película)
(blisters de PVC/PVDC/Aluminio) (blisters de PVC/PVDC/Aluminio) (blisters de PVC/PVDC/Aluminio) (blisters de PVC/PVDC/Aluminio) (blisters de PP/Aluminio) (blisters de PP/Aluminio) (blisters de PP/Aluminio) (blisters de PP/Aluminio) (blisters de PVC/PVDC/Aluminio) (blisters de PP/Aluminio)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Xarelto 10 mg
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
EMBALAJE EXTERIOR DEL ENVASE MULTIPLE (INCLUIDO BLUE BOX) PARA 10 MG
1. NOMBRE DEL MEDICAMENTO
Xarelto 10 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 10 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Envase múltiple: 100 (10 envases de 10 x 1) comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO
UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Bayer Pharma AG, 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/022 100 comprimidos recubiertos con película (10 x 10 x 1) (envase múltiple)
(Blisters de PP/Aluminio)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Xarelto 10 mg
1. NOMBRE DEL MEDICAMENTO
Xarelto 10 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 10 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
10 x 1 comprimidos recubiertos con película.
Forma parte de un envase múltiple, no puede venderse por separado.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO
UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Bayer Pharma AG, 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/022 100 comprimidos recubiertos con película (10 x 10 x 1) (envase múltiple)
(blisters de PP/Aluminio)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Xarelto 10 mg
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLÍSTER PARA 10 MG
|
1. |
NOMBRE DEL MEDICAMENTO |
Xarelto 10 mg comprimidos rivaroxaban
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer (logotipo)
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA PARA EL ENVASE UNITARIO DE 15 MG
1. NOMBRE DEL MEDICAMENTO
Xarelto 15 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 15 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
10 comprimidos recubiertos con película 14 comprimidos recubiertos con película 28 comprimidos recubiertos con película 42 comprimidos recubiertos con película 98 comprimidos recubiertos con película 10 x 1 comprimidos recubiertos con película 100 x 1 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/011
EU/1/08/472/012
EU/1/08/472/013
EU/1/08/472/014
EU/1/08/472/015
EU/1/08/472/016
EU/1/08/472/038
14 comprimidos recubiertos con película (blisters de PP/Alumino)
28 comprimidos recubiertos con película (blisters de PP/Aluminio)
42 comprimidos recubiertos con película (blisters de PP/Aluminio)
98 comprimidos recubiertos con película (blisters de PP/Aluminio)
10 x 1 comprimidos recubiertos con película (blisters de PP/Aluminio) 100 x 1 comprimidos recubiertos con película (blisters PP/Aluminio) 10 comprimidos recubiertos con película (blisters PP/Aluminio)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Xarelto 15 mg
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
EMBALAJE EXTERIOR DEL ENVASE MULTIPLE (INCLUIDO BLUE BOX) PARA 15 MG
1. NOMBRE DEL MEDICAMENTO
Xarelto 15 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 15 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Envase múltiple: 100 (10 envases de 10 x 1) comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Bayer Pharma AG 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/023
100 comprimidos recubiertos con película (10 x 10 x 1 (envase múltiple) listers de PP/Alumino)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Xarelto 15 mg
1. NOMBRE DEL MEDICAMENTO
Xarelto 15 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 15 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
10 x 1 comprimidos recubiertos con película
Forma parte de un envase múltiple, no puede venderse por separado.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Bayer Pharma AG 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/023 100 comprimidos recubiertos con película (10 x 10 x 1) (envase múltiple)
(blisters de PP/Alumino)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Xarelto 15 mg
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS
BLÍSTER UNIDOSIS (10 X 1 COMPRIMIDOS) PARA 15 MG
|
1. |
NOMBRE DEL MEDICAMENTO |
Xarelto 15 mg comprimidos rivaroxaban
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer (logo)
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLÍSTER DE 14 COMPRIMIDOS PARA 15 MG
1. NOMBRE DEL MEDICAMENTO
Xarelto 15 mg comprimidos rivaroxaban
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer (logo)
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
Lun.
Mar.
Mie.
Jue.
Vie.
Sab.
Dom.
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS
BLÍSTER DE 10 COMPRIMIDOS PARA 15 MG
|
1. |
NOMBRE DEL MEDICAMENTO |
Xarelto 15 mg comprimidos rivaroxaban
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer (logo)
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA Y ETIQUETA PARA EL FRASCO DE HDPE PARA 15 MG
1. NOMBRE DEL MEDICAMENTO
Xarelto 15 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 15 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
100 comprimidos recubiertos con película 5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Bayer Pharma AG 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/036
100 comprimidos recubiertos con película (Frasco de HDPE)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Xarelto 15 mg (solo para la caja, no aplica a la etiqueta del frasco)
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA PARA EL ENVASE UNITARIO DE 20 MG
1. NOMBRE DEL MEDICAMENTO
Xarelto 20 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 20 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
10 comprimidos recubiertos con película 14 comprimidos recubiertos con película 28 comprimidos recubiertos con película 98 comprimidos recubiertos con película 10 x 1 comprimidos recubiertos con película 100 x 1 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/017
EU/1/08/472/018
EU/1/08/472/019
EU/1/08/472/020
EU/1/08/472/021
EU/1/08/472/039
14 comprimidos recubiertos con película (blisters de PP/Aluminio)
28 comprimidos recubiertos con película (blisters de PP/Aluminio)
98 comprimidos recubiertos con película (blisters de PP/Aluminio)
10 x 1 comprimidos recubiertos con película (blisters de PP/Aluminio) 100 x 1 comprimidos recubiertos con película (blisters de PP/Aluminio) 10 comprimidos recubiertos con película (blisters de PP/Aluminio)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Xarelto 20 mg
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
EMBALAJE EXTERIOR DEL ENVASE MULTIPLE (INCLUIDO BLUE BOX) PARA 20 MG
1. NOMBRE DEL MEDICAMENTO
Xarelto 20 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 20 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Envase múltiple: 100 (10 envases de 10 x 1) comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Bayer Pharma AG 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/024 100 comprimidos recubiertos con película (10 x10 x 1) (envase múltiple)
(blisters de PP/Aluminio)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Xarelto 20 mg
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA INTERMEDIA DEL ENVASE MULTIPLE (SIN BLUE BOX) PARA 20 MG
1. NOMBRE DEL MEDICAMENTO
Xarelto 20 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 20 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
10 x 1 comprimidos recubiertos con película.
Forma parte de un envase múltiple, no puede venderse por separado.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/024
100 comprimidos recubiertos con película (10 x10 x 1) (envase múltiple) (blisters de PP/Aluminio)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Xarelto 20 mg
1. NOMBRE DEL MEDICAMENTO
Xarelto 20 mg comprimidos rivaroxaban
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer (logo)
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE DEL FABRICANTE
Lot
5. OTROS
1. NOMBRE DEL MEDICAMENTO
Xarelto 20 mg comprimidos rivaroxaban
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer (logo)
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE DEL FABRICANTE
Lot
5. OTROS
Lun.
Mar.
Mie.
Jue.
Vie.
Sab.
Dom.
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS
BLÍSTER DE 10 COMPRIMIDOS PARA 20 MG
|
1. |
NOMBRE DEL MEDICAMENTO |
Xarelto 20 mg comprimidos rivaroxaban
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer (logo)
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE DEL FABRICANTE
Lot
5. OTROS
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA Y ETIQUETA PARA EL FRASCO DE HDPE PARA 20 MG
1. NOMBRE DEL MEDICAMENTO
Xarelto 20 mg comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 20 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
100 comprimidos recubiertos con película 5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/037
100 comprimidos recubiertos con película (Frasco de HDPE)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Xarelto 20 mg (solo para la caja, no aplica a la etiqueta del frasco)
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
EMBALAJE EXTERIOR DEL ENVASE PARA EL INICIO DEL TRATAMIENTO (42 COMPRIMIDOS RECUBIERTOS CON PELÍCULA DE 15 MG Y 7 COMPRIMIDOS RECUBIERTOS CON PELÍCULA DE 20 MG) (INCLUIDO BLUE BOX)_
1. NOMBRE DEL MEDICAMENTO
Xarelto 15 mg Xarelto 20 mg
comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película de color rojo para las semanas 1, 2 y 3 contiene 15 mg de rivaroxaban.
Cada comprimido recubierto con película de color pardo-rojizo para la semana 4 contiene 20 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cada envase de 49 comprimidos recubiertos con película contiene: 42 comprimidos recubiertos con película con 15 mg de rivaroxaban. 7 comprimidos recubiertos con película con 20 mg de rivaroxaban.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
Envase para el inicio del tratamiento
Este envase para el inicio del tratamiento es sólo para las 4 primeras semanas de tratamiento. POSOLOGÍA:
Día 1 a 21: 1 comprimido de 15 mg dos veces al día (un comprimido de 15 mg por la mañana y otro por la noche) con alimentos.
Día 22 en adelante: 1 comprimido de 20 mg una vez al día (tomado cada día a la misma hora) con alimentos.
Día 1 a 21: 15 mg, 1 comprimido dos veces al día (un comprimido de 15 mg por la mañana y otro por la noche) con alimentos.
Día 22 en adelante: 20 mg, 1 comprimido una vez al día (tomado cada día a la misma hora) con alimentos.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlín Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/040 42 comprimidos recubiertos con película con 15 mg de rivaroxaban
7 comprimidos recubiertos con película con 20 mg de rivaroxaban (envase para el inicio del tratamiento)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Xarelto 15 mg Xarelto 20 mg
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
ENVASE CON CALENDARIO DEL ENVASE PARA EL INICIO DEL TRATAMIENTO (42 COMPRIMIDOS RECUBIERTOS CON PELÍCULA DE 15 MG Y 7 COMPRIMIDOS RECUBIERTOS CON PELÍCULA DE 20 MG) (SIN BLUE BOX)_
1. NOMBRE DEL MEDICAMENTO
Xarelto 15 mg Xarelto 20 mg
comprimidos recubiertos con película rivaroxaban
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película de color rojo para las semanas 1, 2 y 3 contiene 15 mg de rivaroxaban.
Cada comprimido recubierto con película de color pardo-rojizo para la semana 4 contiene 20 mg de rivaroxaban.
3. LISTA DE EXCIPIENTES
Contiene lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cada envase de 49 comprimidos recubiertos con película contiene: 42 comprimidos recubiertos con película con 15 mg de rivaroxaban. 7 comprimidos recubiertos con película con 20 mg de rivaroxaban.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
Envase para el inicio del tratamiento
Este envase para el inicio del tratamiento es sólo para las 4 primeras semanas de tratamiento.
Día 1 a 21: 15 mg, 1 comprimido dos veces al día (un comprimido de 15 mg por la mañana y otro por la noche) con alimentos.
Día 22 en adelante: 20 mg, 1 comprimido una vez al día (tomado cada día a la misma hora) con alimentos.
POSOLOGÍA y PAUTA DE TRATAMIENTO:
Día 1 a 21: 1 comprimido de 15 mg dos veces al día (un comprimido de 15 mg por la mañana y otro por la noche).
Día 22 en adelante: 1 comprimido de 20 mg una vez al día(tomado cada día a la misma hora). Tratamiento inicial Xarelto 15 mg dos veces al día Primeras 3 semanas
Tratamiento continuado Xarelto 20 mg una vez al día Semana 4 en adelante Visite a su
médico para asegurar un tratamiento continuado.
Tomar con alimentos.
Xarelto 15 mg Inicio del tratamiento 15 mg
dos veces al día Fecha de inicio
Semana 1, semana 2, semana 3
Día 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
Símbolo del sol Símbolo de la luna
Cambio de dosis Xarelto 20 mg 20 mg
una vez al día
Tomado cada día a la misma hora Fecha del cambio de dosis Semana 4
DÍA 22 DÍA 23 DÍA 24 DÍA 25 DÍA 26 DÍA 27 DÍA 28
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlin Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/472/040 42 comprimidos recubiertos con película con 15 mg de rivaroxaban
7 comprimidos recubiertos con película con 20 mg de rivaroxaban (envase para el inicio del tratamiento)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS
BLÍSTER DEL ENVASE PARA EL INICIO DEL TRATAMIENTO EN ENVASE CON CALENDARIO (42 COMPRIMIDOS RECUBIERTOS CON PELÍCULA DE 15 MG Y 7 COMPRIMIDOS RECUBIERTOS CON PELÍCULA DE 20 MG)_
1. NOMBRE DEL MEDICAMENTO
Xarelto 15 mg comprimidos Xarelto 20 mg comprimidos rivaroxaban
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer (logo)
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE DEL FABRICANTE
Lot
5. OTROS
Tarjeta de alerta para el paciente
Bayer (logo)
Xarelto 2,5 mg Xarelto 15 mg Xarelto 20 mg
♦ Lleve siempre consigo esta tarjeta
♦ Muestre esta tarjeta a todos los médicos o dentistas antes de recibir otro tratamiento
Estoy recibiendo tratamiento anticoagulante con Xarelto (rivaroxaban)
Nombre:
Dirección:
Fecha de nacimiento:
Grupo sanguíneo:
Peso:
Otros medicamentos / afecciones:
En caso de urgencia, por favor, avisen a:
Nombre del médico:
Número de teléfono del médico:
Sello del médico:
Por favor, notifiquen también a:
Nombre:
Teléfono:
Parentesco:
Información para el profesional sanitario:
♦ No deben utilizarse los valores de INR ya que no son una medida válida de la actividad anticoagulante de Xarelto.
¿Qué tengo que saber acerca de Xarelto?
♦ Xarelto hace la sangre más fluida, lo que previene la formación de coágulos de sangre.
♦ Debe tomar Xarelto exactamente como el médico se lo haya indicado. Para asegurar una protección óptima frente a la formación de coágulos, no deje de tomar nunca una dosis.
♦ No deje de tomar Xarelto sin consultar antes con su médico ya que esto aumentaría el riesgo de que se formen coágulos de sangre.
♦ Informe a su médico de cualquier otro medicamento que esté tomando, ha tomado recientemente o vaya a empezar a tomar, antes de tomar Xarelto.
♦ Diga a su médico que está tomando Xarelto antes de una intervención quirúrgica o de un procedimiento invasivo
¿Cuándo debo pedir consejo al médico?
Si toma un anticoagulante como Xarelto es importante que conozca sus posibles efectos secundarios. El efecto secundario más frecuente es el sangrado. No empiece a tomar Xarelto si sabe que tiene riesgo de presentar un sangrado sin hablarlo antes con su médico.
Dígale inmediatamente al médico si usted tiene algún signo o síntoma de sangrado, como por ejemplo
♦ dolor
♦ hinchazón o molestias
♦ dolor de cabeza, mareos o debilidad
♦ moratones anormales, sangrado por la nariz, las encías o cortes que tardan mucho en dejar de sangrar
♦ flujo menstrual o sangrado vaginal más abundante de lo normal
♦ sangre en orina, que puede ser de color rosado o marrón, deposiciones de color rojo o negro
♦ tos con sangre o vómitos de sangre o de un material que tiene aspecto de grumos de café
¿Cómo debo tomar Xarelto?
Para garantizar una protección óptima, Xarelto
♦ 2,5 mg puede tomarse con o sin alimentos.
♦ 15 mg debe tomarse con alimentos.
♦ 20 mg debe tomarse con allimentos.
B. PROSPECTO
PROSPECTO: INFORMACIÓN PARA EL USUARIO
Xarelto 2,5 mg comprimidos recubiertos con película
rivaroxaban
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Xarelto y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Xarelto
3. Cómo tomar Xarelto
4. Posibles efectos adversos
5. Conservación de Xarelto
6. Contenido del envase e información adicional
1. Qué es Xarelto y para qué se utiliza
Se le ha prescrito Xarelto porque se le ha diagnosticado un síndrome coronario agudo (grupo de trastornos que incluyen infarto de miocardio y angina inestable, un tipo de dolor grave en el pecho) y en su análisis de sangre se han encontrado unos resultados elevados en ciertas pruebas del corazón. Xarelto reduce el riesgo de padecer otro infarto de miocardio en adultos, o bien reduce el riesgo de muerte a causa de una enfermedad del corazón o de los vasos sanguíneos.
Xarelto contiene la sustancia activa rivaroxaban, que pertenece a un grupo de medicamentos llamados medicamentos antitrombóticos. Actúa bloqueando un factor de la coagulación (factor Xa) y por lo tanto, reduciendo la tendencia de la sangre a formar coágulos.
Le prescribirán Xarelto junto a otro medicamento. Su médico también le indicará que tome:
• ácido acetilsalicílico (también conocido como aspirina), o bien
• ácido acetilsalicílico más clopidogrel o ticlopidina
2. Qué necesita saber antes de empezar a tomar Xarelto No tome Xarelto
- si es alérgico a rivaroxaban o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6)
- si sangra excesivamente
- si padece una enfermedad o problemas en un órgano del cuerpo que aumente el riesgo de hemorragia grave (por ejemplo, úlcera de estómago, lesión o hemorragia en el cerebro, o una intervención quirúrgica reciente en el cerebro o en los ojos)
- si está tomando medicamentos para prevenir la formación de coágulos sanguíneos (p. ej. warfarina, dabigatran, apixaban o heparina), excepto cuando esté cambiando de tratamiento anticoagulante o mientras se le esté administrando heparina a través de un catéter venoso o arterial, para que éste no se obstruya
- si padece un síndrome coronario agudo y previamente ha sufrido una hemorragia o ha tenido un coágulo sanguíneo en el cerebro (ictus)
- si padece una enfermedad del hígado que pueda aumentar el riesgo de sangrado
- si está embarazada o en período de lactancia.
No tome Xarelto e informe a su médico si alguna de estas circunstancias se aplica a su caso. Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar Xarelto.
Xarelto no se debe utilizar en combinación con otros medicamentos que reduzcan la coagulación de la sangre distintos del ácido acetilsalicílico o clopidogrel/ticlopidina, como por ejemplo, prasugrel o ticagrelor.
Tenga especial cuidado con Xarelto
- Si presenta un riesgo aumentado de sangrado, como puede suceder en las siguientes situaciones:
■ insuficiencia renal grave, ya que el funcionamiento de los riñones puede afectar a la cantidad de medicamento que actúa en su organismo
■ si está tomando otros medicamentos para prevenir la formación de coágulos de sangre (por ejemplo, warfarina, dabigatran, apixaban o heparina), cuando cambie a otro tratamiento anticoagulante o mientras reciba heparina a través de un catéter venoso o arterial, para que éste no se obstruya (ver sección “Uso de Xarelto con otros medicamentos”)
■ enfermedad hemorrágica
■ presión arterial muy alta, no controlada por tratamiento médico
■ enfermedades del estómago o del intestino que puedan causar una hemorragia, como por ejemplo, inflamación del estómago o del intestino, inflamación del esófago (garganta), por ejemplo debido a la enfermedad de reflujo gastroesofágico (enfermedad en la que el ácido del estómago asciende hacia arriba en el esófago)
■ un problema en los vasos sanguíneos de la parte posterior de sus ojos (retinopatía)
■ una enfermedad pulmonar en la que los bronquios están dilatados y llenos de pus (bronquiectasia) o bien, ha sufrido una hemorragia previa en los pulmones
■ tiene más de 75 años
■ su peso es de 60 kg ó menos.
Informe a su médico si presenta alguna de estas situaciones antes de tomar Xarelto. Su médico decidirá si debe ser tratado con este medicamento y si debe mantenerse bajo observación más estrecha.
Si necesita una intervención quirúrgica:
- Es muy importante tomar Xarelto antes y después de la operación, exactamente a las horas en que su médico se lo indique.
- Si su operación requiere la colocación de un catéter o inyección en la columna vertebral (por ejemplo para anestesia epidural o espinal, o reducción del dolor):
■ Es muy importante tomar Xarelto, antes y después de la inyección o de la extracción del catéter, exactamente a las horas que su médico le haya indicado.
■ Informe a su médico inmediatamente si presenta adormecimiento o debilidad en las piernas o problemas en el intestino o en la vejiga al final de la anestesia, porque es necesaria una atención urgente.
Niños y adolescentes
Xarelto no está recomendado en niños y adolescentes menores de 18 años. No se dispone de suficiente información sobre su uso en niños y adolescentes.
Uso de Xarelto con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento, incluso los adquiridos sin receta.
- Si está tomando:
■ algún medicamento para una infección por hongos (p. ej., ketoconazol, itraconazol, voriconazol, posaconazol), salvo si sólo se aplican en la piel
■ algún medicamento antiviral para el VIH / SIDA (p. ej., ritonavir)
■ otros medicamentos para disminuir la coagulación de la sangre (p. ej., enoxaparina, clopidogrel o antagonistas de la vitamina K, como la warfarina o el acenocumarol)
■ antiinflamatorios y medicamentos para aliviar el dolor (p. ej., naproxeno o ácido acetilsalicílico)
■ dronedarona, un medicamento para el tratamiento del latido cardiaco irregular
Si alguna de las circunstancias anteriores le aplica, informe a su médico antes de tomar Xarelto, porque el efecto de Xarelto podría verse aumentado. Su médico decidirá si debe ser tratado con este medicamento y si debe mantenerse bajo observación más estrecha.
Si su médico considera que tiene un mayor riesgo de desarrollar una úlcera gástrica o intestinal, podría recomendarle utilizar además, un tratamiento preventivo.
- Si usted toma:
■ algún medicamento para el tratamiento de la epilepsia (fenitoína, carbamazepina, fenobarbital)
■ hierba de San Juan (Hipericum perforatum) una planta medicinal para el tratamiento de la depresión
■ rifampicina, un antibiótico
Si alguna de las circunstancias anteriores, le aplican, informe a su médico antes de tomar Xarelto, porque el efecto de Xarelto podría verse reducido. Su médico decidirá si debe ser tratado con Xarelto y si debe mantenerse bajo observación más estrecha.
Embarazo y lactancia
Si está embarazada o si está en período de lactancia, no tome Xarelto. Si hay alguna posibilidad de que se quede embarazada, utilice un anticonceptivo fiable mientras toma Xarelto. Si se queda embarazada mientras toma Xarelto, informe inmediatamente a su médico, quien decidirá cómo deberá tratarse.
Conducción y uso de máquinas
Xarelto puede causar mareos (efecto adverso frecuente) o desvanecimientos (efecto adverso poco frecuente) (ver sección 4, “Posibles efectos adversos”). No debe conducir ni utilizar máquinas si está afectado por estos síntomas.
Xarelto contiene lactosa
Si su médico le ha indicado que padece una intolerancia a ciertos azúcares, consulte con él antes de tomar Xarelto.
3. Cómo tomar Xarelto
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Qué dosis tomar
La dosis recomendada es de un comprimido de 2,5 mg dos veces al día. Tome Xarelto a la misma hora cada día (por ejemplo, un comprimido por la mañana y otro por la noche). Este medicamento puede tomarse con o sin alimentos.
Si tiene dificultad para tragar el comprimido entero, consulte a su médico sobre otras formas de tomar Xarelto. El comprimido puede triturarse y mezclarse con agua o con puré de manzana, inmediatamente antes de tomarlo.
Si es necesario, su médico también puede administrarle el comprimido Xarelto triturado a través de una sonda gástrica.
Le prescribirán Xarelto junto a otro medicamento. Su médico le indicará que tome también:
• ácido acetilsalicílico (también conocido como aspirina), o bien
• ácido acetilsalicílico y clopidogrel o ticlopidina
Su médico le indicará la dosis a tomar (generalmente entre 75 y 100 mg de ácido acetilsalicílico al día, o una dosis de 75 a 100 mg de ácido acetilsalicílico más una dosis diaria de 75 mg de clopidogrel o una dosis diaria estándar de ticlopidina).
Cuándo iniciar el tratamiento con Xarelto
El tratamiento con Xarelto debe iniciarse lo antes posible, una vez se ha estabilizado el síndrome coronario agudo, es decir, a partir de las 24 horas tras su admisión en el hospital y en el momento en que finalizaría el tratamiento anticoagulante por vía parenteral (mediante inyección).
El médico decidirá durante cuánto tiempo debe seguir tomando el tratamiento.
Si toma más Xarelto del que debe
Llame inmediatamente a su médico si ha tomado demasiados comprimidos de Xarelto. Tomar demasiado Xarelto aumenta el riesgo de sangrado.
Si olvidó tomar Xarelto
No tome más de un comprimido en un solo día para compensar una dosis olvidada. Si olvida tomar una dosis, tome el siguiente comprimido a la hora habitual.
Si interrumpe el tratamiento con Xarelto
Tome Xarelto de manera regular durante el tiempo que le indique su médico.
No interrumpa el tratamiento con Xarelto sin hablar antes con su médico. Si deja de tomar este medicamento, puede aumentar su riesgo de padecer otro infarto de miocardio, un ictus o morir por una enfermedad relacionada con su corazón o sus vasos sanguíneos.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Al igual que otros medicamentos similares (antitrombóticos) Xarelto puede causar sangrados que pueden poner en peligro la vida del paciente. Un sangrado excesivo puede causar una caída repentina de la presión arterial (shock). En algunos casos el sangrado puede no ser evidente.
Posibles efectos adversos que pueden ser signos de sangrado:
Avise inmediatamente a su médico si experimenta cualquiera de los siguientes síntomas:
- sangrado prolongado o excesivo
- debilidad excepcional, cansancio, palidez, mareos, dolor de cabeza, hinchazón inexplicable, dificultad para respirar, dolor en el pecho o angina de pecho, ya que pueden ser signos de sangrado.
Su médico decidirá entre mantenerle bajo una observación más estrecha o cambiarle el tratamiento.
Lista general de posibles efectos adversos
Frecuentes (pueden afectar hasta 1 de cada 10 pacientes):
- sangrado del estómago o del intestino, hemorragia urogenital (incluyendo sangre en la orina y sangrado menstrual abundante), hemorragia nasal, sangrado de las encías
- sangrado en el ojo (incluyendo sangrado en la parte blanca del ojo)
- sangrado hacia un tejido o cavidad del organismo (hematoma, cardenales)
- tos con sangre
- sangrado de la piel o debajo la piel
- sangrado después de una operación
- supuración de sangre o líquido de una herida quirúrgica
- hinchazón de las extremidades
- dolor de las extremidades
- fiebre
- disminución de los glóbulos rojos que puede causar palidez y debilidad o dificultad para respirar
- dolor de estómago, indigestión, mareo o sensación de mareo, estreñimiento, diarrea
- presión arterial baja (los síntomas pueden ser sensación de mareo o desvanecimiento al ponerse de pie)
- disminución general de la fuerza y la energía (debilidad, cansancio), dolor de cabeza, mareos
- sarpullido, picor de la piel
- alteración de la función de los riñones (puede verse en los análisis realizados por el médico)
- los análisis de sangre pueden mostrar un aumento de algunas enzimas hepáticas
Poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes):
- sangrado en el cerebro o en el interior del cráneo
- sangrado en una articulación, que causa dolor e hinchazón
- desvanecimiento
- sensación de malestar
- sequedad de boca
- aumento de la frecuencia cardiaca
- reacción alérgica, incluyendo reacción alérgica de la piel
- ronchas
- alteración de la función del hígado (puede verse en los análisis realizados por el médico)
- los análisis de sangre pueden mostrar un aumento de la bilirrubina, de algunas enzimas pancreáticas o del número de plaquetas
Raros (pueden afectar hasta 1 de cada 1.000 pacientes):
- sangrado en un músculo
- hinchazón localizada
- coloración amarillenta de la piel y en los ojos (ictericia)
- acumulación de sangre (hematoma) en la ingle como complicación después de una cirugía cardíaca en la que se introduce un catéter en la arteria de la pierna (pseudoaneurisma).
Frecuencia no conocida (la frecuencia no puede estimarse a partir de los datos disponibles):
- aumento de la presión en los músculos de las piernas o de los brazos después de una hemorragia, que causa dolor, hinchazón, alteración de la sensibilidad, adormecimiento o parálisis (síndrome compartimental después de una hemorragia)
- insuficiencia renal después de una hemorragia grave.
Los siguientes efectos adversos han sido comunicados después de la autorización:
- angioedema y edema alérgico (hinchazón de la cara, labios, boca lengua o garganta)
- colestasis (disminución del flujo de la bilis), hepatitis, incluyendo lesión hepatocelular (inflamación o lesión de hígado)
- trombocitopenia (número bajo de plaquetas, células que ayudan a la coagulación de la sangre)
Comunicación de efectos adversos
Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Xarelto
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en cada blíster después de “CAD” o ”EXP”. La fecha de caducidad es el último día del mes que se indica.
Este medicamento no requiere condiciones especiales de conservación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Xarelto
- El principio activo es rivaroxaban. Cada comprimido contiene 2,5 mg de rivaroxaban.
- Los demás componentes son:
Núcleo del comprimido: celulosa microcristalina, croscarmelosa sódica, lactosa monohidrato, hipromelosa, laurilsulfato de sodio, estearato de magnesio.
Recubrimiento pelicular del comprimido: macrogol 3350, hipromelosa, dióxido de titanio (E 171), óxido de hierro amarillo (E 172).
Aspecto del producto y contenido del envase
Los comprimidos recubiertos con película de Xarelto 2,5 mg son de color amarillo pálido, redondos, biconvexos y llevan grabada la cruz BAYER en una cara, y “2.5” y un triángulo en la otra.
Se presentan envasados en blíster, en estuches de 14, 28, 30, 56, 60, 98, 168 ó 196 comprimidos recubiertos con película o en blisters unidosis, en envases de 10 x 1 ó 100 x 1, o en envases múltiples que contienen 10 estuches de 10 x 1 comprimidos recubiertos con película.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Bayer Pharma AG 13342 Berlin Alemania
Responsable de la fabricación
Bayer Pharma AG 51368 Leverkusen Alemania
Bayer HealthCare Manufacturing Srl.
Via delle Groane, 126 20024 Garbagnate Milanese Italia
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie / Belgique / Belgien Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Etarapna Eañep Etarapna EOOfl Tea: +359-(0)2-81 401 01 Ceská republika Bayer s.r.o. Tel: +420-266 101 111 Danmark Bayer A/S Tlf: +45-45 235 000 Deutschland Bayer Vital GmbH Tel: +49-(0)214-30 513 48 Eesti Bayer OÜ Tel: +372-655 85 65 EXláSa Bayer EA7ág ABEE Tn^: +30-210-618 75 00 España Bayer Hispania S.L. Tel: +34-93-495 65 00 France Bayer HealthCare Tél (N° vert): +33-(0)800 87 54 54 Ireland Bayer Limited Tel: +353-(0)1-2999 313 Ísland Icepharma hf. Sími: +354-540 80 00 Italia Bayer S.p.A. Tel: +39-02-3978 1 Kúnpoq NOVAGEM Limited Tn^: +357-22-48 38 58 Latvija SIA Bayer Tel: +371-67 84 55 63 Lietuva UAB Bayer Tel: +370-5-233 68 68 |
Luxembourg / Luxemburg Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Magyarország Bayer Hungária KFT Tel: +36-1-487 4100 Malta Alfred Gera and Sons Ltd. Tel: +356-21 44 62 05 Nederland Bayer B.V. Tel: +31-(0)297-28 06 66 Norge Bayer AS Tlf: +47-23 13 05 00 Osterreich Bayer Austria Ges. m. b. H. Tel: +43-(0)1 -711 460 Polska Bayer Sp. z o.o. Tel: +48-22-572 35 00 Portugal Bayer Portugal, Lda. Tel: +351-21-416 42 00 Romania SC Bayer SRL Tel: +40-(0)21-528 59 00 Slovenija Bayer d. o. o. Tel: +386-(0)1 -58 14 400 Slovenská republika Bayer, spol. s r.o. Tel: +421-(0)2-59 21 31 11 Suomi/Finland Bayer Oy Puh/Tel: +358-(0)20-78521 Sverige Bayer AB Tel: +46-(0)8-580 223 00 United Kingdom Bayer plc Tel: +44-(0)1635-563000 |
Fecha de la última revisión de este prospecto: {MM/AAAA}.
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
Prospecto: Información para el usuario
Xarelto 10 mg comprimidos recubiertos con película
rivaroxaban
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar el medicamento, porque contiene información importante para usted
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted y no debe dárselo a otras personas, aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Xarelto y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Xarelto
3. Cómo tomar Xarelto
4. Posibles efectos adversos
5. Conservación de Xarelto
6. Contenido del envase e información adicional
1. Qué es Xarelto y para qué se utiliza
Xarelto contiene la sustancia activa rivaroxaban y se usa en adultos para prevenir la formación de coágulos de sangre en las venas después de una operación de sustitución de cadera o rodilla. Su médico le ha recetado este medicamento porque después de una operación tiene más riesgo de que se le formen coágulos de sangre.
Xarelto pertenece a un grupo de medicamentos llamados agentes antitrombóticos. Actúa mediante el bloqueo de un factor de la coagulación (factor Xa) y, por lo tanto, reduciendo la tendencia de la sangre a formar coágulos.
2. Qué necesita saber antes de empezar a tomar Xarelto
No tome Xarelto
- si es alérgico a rivaroxaban o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6)
- si sangra excesivamente
- si padece una enfermedad o problemas en un órgano del cuerpo que aumente el riesgo de hemorragia grave (por ejemplo, úlcera de estómago, lesión o hemorragia en el cerebro, o una intervención quirúrgica reciente en el cerebro o en los ojos)
- si está tomando medicamentos para prevenir la formación de coágulos sanguíneos (p. ej. warfarina, dabigatran, apixaban o heparina), excepto cuando esté cambiando de tratamiento anticoagulante o mientras se le esté administrando heparina a través de un catéter venoso o arterial, para que éste no se obstruya
- si padece una enfermedad del hígado que aumente el riesgo de sangrado
- si está embarazada o en período de lactancia
No tome Xarelto e informe a su médico si alguna de estas circunstancias se aplica a su caso.
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar Xarelto.
Tenga especial cuidado con Xarelto
- Si presenta un riesgo aumentado de sangrado, como puede suceder en las siguientes situaciones:
■ insuficiencia renal moderada o grave, ya que el funcionamiento de los riñones puede afectar a la cantidad de medicamento que actúa en su organismo
■ si está tomando otros medicamentos para prevenir la formación de coágulos de sangre (por ejemplo, warfarina, dabigatran, apixaban o heparina), cuando cambie a otro tratamiento anticoagulante o mientras reciba heparina a través de un catéter venoso o arterial, para que éste no se obstruya (ver sección “Uso de Xarelto con otros medicamentos”)
■ enfermedad hemorrágica
■ presión arterial muy alta, no controlada por tratamiento médico
■ enfermedades del estómago o del intestino que puedan causar una hemorragia, como por ejemplo, inflamación del estómago o del intestino, inflamación del esófago (garganta), por ejemplo debido a la enfermedad de reflujo gastroesofágico (enfermedad en la que el ácido del estómago asciende hacia arriba en el esófago)
■ un problema en los vasos sanguíneos de la parte posterior de los ojos (retinopatía)
■ una enfermedad pulmonar en la que los bronquios están dilatados y llenos de pus (bronquiectasia) o bien, una hemorragia previa de los pulmones
Informe a su médico si presenta alguna de estas situaciones antes de tomar Xarelto. Su médico decidirá si debe ser tratado con este medicamento y si debe mantenerse bajo observación más estrecha.
- Si su operación supone la colocación de un catéter o inyección en la columna vertebral (por ejemplo, para anestesia o reducción del dolor epidural o espinal):
■ Es muy importante tomar Xarelto exactamente a las horas en que su médico se lo indique.
■ Informe inmediatamente a su médico si presenta adormecimiento o debilidad en las piernas, o problemas en el intestino o en la vejiga al final de la anestesia, porque necesitaría una atención urgente.
Niños y adolescentes
Xarelto no está recomendado en menores de 18 años. No se dispone de suficiente información sobre su uso en niños y adolescentes.
Uso de Xarelto con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento, incluso los adquiridos sin receta.
Si está tomando:
- algún medicamento para una infección por hongos (p. ej., ketoconazol, itraconazol, voriconazol, posaconazol), a menos que sólo se apliquen en la piel
- algún medicamento antiviral para el VIH / SIDA (p. ej., ritonavir)
- otros medicamentos para disminuir la coagulación de la sangre (por ejemplo, enoxaparina, clopidogrel o antagonistas de la vitamina K, como la warfarina o el acenocumarol)
- antiinflamatorios y medicamentos para aliviar el dolor (p. ej., naproxeno o ácido acetilsalicílico)
- dronedarona, un medicamento para el tratamiento del latido cardíaco irregular
Si alguna de las circunstancias anteriores, le aplica, informe a su médico antes de tomar Xarelto, porque el efecto de Xarelto podría aumentar. Su médico decidirá si debe ser tratado con este medicamento y si debe mantenerse bajo observación más estrecha.
Si su médico considera que tiene un mayor riesgo de desarrollar úlceras estomacales o intestinales, le recomendará utilizar, además, un tratamiento preventivo de las úlceras.
Si usted toma:
- algún medicamento para el tratamiento de la epilepsia (fenitoína, carbamazepina, fenobarbital)
- hierba de San Juan (Hypericum perforatum), una planta medicinal para el tratamiento de la depresión
- rifampicina, un antibiótico
Si alguna de las circunstancias anteriores, le aplican, informe a su médico antes de tomar Xarelto, porque el efecto de Xarelto puede estar disminuido. Su médico decidirá si debe ser tratado con Xarelto y si debe mantenerse bajo observación más estrecha.
Embarazo y lactancia
No tome Xarelto si está embarazada o si está en período de lactancia. Si hay alguna posibilidad de que se quede embarazada, utilice un anticonceptivo fiable mientras toma Xarelto. Si se queda embarazada mientras toma este medicamento, informe a su médico inmediatamente, quien decidirá cómo deberá tratarse.
Conducción y uso de máquinas
Xarelto puede causar mareos (efecto adverso frecuente) o desvanecimientos (efecto adverso poco frecuente) (ver sección 4, “Posibles efectos adversos”). No deberá conducir ni utilizar máquinas si está afectado por estos síntomas.
Xarelto contiene lactosa.
Si su médico le ha indicado que padece una intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento.
3. Cómo tomar Xarelto
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico.
En caso de duda, consulte de nuevo a su médico o farmacéutico.
Qué dosis tomar
La dosis recomendada es un comprimido (10 mg) una vez al día.
Trague el comprimido, preferentemente con agua.
Xarelto puede tomarse con o sin alimentos.
Si tiene dificultad para tragar el comprimido entero, consulte a su médico sobre otras formas de tomar Xarelto. El comprimido puede triturarse y mezclarse con agua o con puré de manzana, inmediatamente antes de tomarlo.
Si es necesario, su médico también puede administrarle el comprimido Xarelto triturado a través de una sonda gástrica.
Cuándo tomar Xarelto
Tome el primer comprimido entre 6 y 10 horas después de la operación.
A continuación, tome un comprimido cada día, hasta que su médico se lo indique.
Trate de tomar un comprimido a la misma hora cada día, para ayudarle a recordarlo.
Si se ha sometido a una cirugía mayor de cadera, generalmente tomará comprimidos durante 5 semanas.
Si se ha sometido a una cirugía mayor de rodilla, generalmente tomará comprimidos durante 2 semanas.
Si toma más Xarelto del que debe
Llame inmediatamente a su médico si ha tomado demasiados comprimidos de Xarelto. Tomar demasiado Xarelto aumenta el riesgo de sangrado.
Si olvidó tomar Xarelto
Si olvidó tomar una dosis, tómela en cuanto se acuerde. Tome el siguiente comprimido al día siguiente y, después, siga tomando un comprimido cada día, como de costumbre.
No tome una dosis doble para compensar las dosis olvidadas.
Si interrumpe el tratamiento con Xarelto
No interrumpa el tratamiento con Xarelto sin hablar primero con su médico, porque Xarelto previene el desarrollo de una afección grave.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, Xarelto puede producir efectos adversos, aunque no todas las personas los sufran.
Al igual que otros medicamentos similares (antitrombóticos), Xarelto puede causar sangrados, que pueden poner en peligro la vida del paciente. El sangrado excesivo puede causar una disminución súbita de la presión arterial (shock). En algunos casos el sangrado puede no ser evidente.
Posibles efectos adversos que pueden ser signos de sangrado:
Avise a su médico inmediatamente si sufre alguno de los siguientes efectos adversos:
• sangrado prolongado o excesivo
• debilidad excepcional, cansancio, palidez, mareos, cefalea, hinchazón inexplicable, dificultad para respirar, dolor en el pecho o angina de pecho, ya que pueden ser signos de sangrado.
Su médico decidirá mantenerle bajo observación más estrecha o modificar su tratamiento.
Lista general de posibles efectos adversos:
Frecuentes (pueden afectar hasta 1 de cada 10 pacientes)
- sangrado del estómago o del intestino, hemorragia urogenital (incluyendo sangre en la orina y sangrado menstrual abundante), hemorragia nasal, sangrado de las encías
- sangrado en el ojo (incluyendo sangrado en la parte blanca del ojo)
- sangrado hacia un tejido o cavidad del organismo (hematoma, cardenales)
- tos con sangre
- sangrado de la piel o debajo de la piel
- sangrado después de una operación
- supuración de sangre o líquido de una herida quirúrgica
- hinchazón de las extremidades
- dolor de las extremidades
- fiebre
- disminución de los glóbulos rojos que puede causar palidez y debilidad o dificultad para respirar
- dolor de estómago, indigestión, mareo o sensación de mareo, estreñimiento, diarrea
- presión arterial baja (los síntomas pueden ser sensación de mareo o desvanecimiento al ponerse de pie)
- disminución general de la fuerza y la energía (debilidad, cansancio), dolor de cabeza, mareos
- sarpullido, picor de la piel
- alteración de la función de los riñones (puede verse en los análisis realizados por el médico)
- los análisis de sangre pueden mostrar un aumento de algunas enzimas del hígado.
Poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes)
- sangrado en el cerebro o en el interior del cráneo
- sangrado en una articulación, que causa dolor e hinchazón
- desvanecimiento
- sensación de malestar
- sequedad de boca
- aumento de la frecuencia cardiaca
- reacción alérgica, incluyendo reacción alérgica de la piel
- ronchas
- alteración de la función del hígado (puede verse en los análisis realizados por el médico)
- los análisis de sangre pueden mostrar un aumento de la bilirrubina, de algunas enzimas pancreáticas o del número de plaquetas
Raros (pueden afectar hasta 1 de cada 1.000 pacientes)
- sangrado en un músculo
- hinchazón localizada
- coloración amarillenta de la piel y en los ojos (ictericia)
- acumulación de sangre (hematoma) en la ingle como complicación después de una cirugía cardíaca en la que se introduce un catéter en la arteria de la pierna (pseudoaneurisma).
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
- aumento de la presión en los músculos de las piernas o de los brazos después de una hemorragia, que causa dolor, hinchazón, alteración de la sensibilidad, adormecimiento o parálisis (síndrome compartimental por hemorragia)
- insuficiencia renal después de una hemorragia grave.
Los siguientes efectos adversos han sido comunicados después de la autorización:
- a ngioedema y edema alérgico (hinchazón de la cara, labios, boca, lengua o garganta)
- colestasis (disminución del flujo de la bilis), hepatitis, incluyendo lesión hepatocelular (inflamación o lesión de hígado)
- trombocitopenia (número bajo de plaquetas, células que ayudan a la coagulación de la sangre). Comunicación de efectos adversos
Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Xarelto
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en cada blíster, después de “CAD” o “EXP”. La fecha de caducidad es el último día del mes que se indica.
No requiere condiciones especiales de conservación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Xarelto
- El principio activo es rivaroxaban. Cada comprimido contiene 10 mg de rivaroxaban.
- Los demás componentes son:
Núcleo del comprimido: celulosa microcristalina, croscarmelosa sódica, monohidrato de lactosa, hipromelosa, laurilsulfato de sodio, estearato de magnesio.
Recubrimiento pelicular del comprimido: macrogol 3350, hipromelosa, dióxido de titanio (E171), óxido de hierro rojo (E172).
Aspecto del producto y contenido del envase
Los comprimidos recubiertos con película de Xarelto 10 mg son redondos biconvexos, de color rojo claro, y llevan grabada la cruz BAYER en una cara, y un “10” y un triángulo en la otra. Se presentan envasados en blíster, en estuches de 5,10 ó 30 comprimidos recubiertos con película, o blisters precortados unidosis en envases de 10 x 1 ó 100 x 1, o en envases múltiples que contienen 10 estuches de 10 x 1 comprimidos recubiertos con película.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Bayer Pharma AG 13342 Berlin Alemania
Responsable de la fabricación
Bayer Pharma AG 51368 Leverkusen Alemania
Bayer HealthCare Manufacturing Srl.
Via delle Groane, 126 20024 Garbagnate Milanese Italia
|
Belgie / Belgique / Belgien Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Etarapna Eañep Etarapna EOOfl Tea: +359-(0)2-81 401 01 Ceská republika Bayer s.r.o. Tel: +420-266 101 111 Danmark Bayer A/S Tlf: +45-45 235 000 Deutschland Bayer Vital GmbH Tel: +49-(0)214-30 513 48 Eesti Bayer OÜ Tel: +372-655 85 65 EXláSa Bayer EAAág ABEE Tn^: +30-210-618 75 00 España Bayer Hispania S.L. Tel: +34-93-495 65 00 France Bayer HealthCare Tél (N° vert): +33-(0) 800 87 54 54 Hrvatska Bayer d.o.o. Tel: + 385-(0)1-6599 900 Ireland Bayer Limited Tel: +353-(0)1-2999 313 Ísland Icepharma hf. Sími: +354-540 80 00 Italia Bayer S.p.A. Tel: +39-02-3978 1 Kúnpoq NOVAGEM Limited Tn^: +357-22-48 38 58 Latvija SIA Bayer Tel: +371-67 84 55 63 |
Lietuva UAB Bayer Tel: +370-5-233 68 68 Luxembourg / Luxemburg Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Magyarország Bayer Hungária KFT Tel: +36-1-487 4100 Malta Alfred Gera and Sons Ltd. Tel: +356-21 44 62 05 Nederland Bayer B.V. Tel: +31-(0)297-28 06 66 Norge Bayer AS Tlf: +47-23 13 05 00 Osterreich Bayer Austria Ges. m. b. H. Tel: +43-(0)1 -711 460 Polska Bayer Sp. z o.o. Tel: +48-22-572 35 00 Portugal Bayer Portugal, Lda Tel: +351-21-416 42 00 Romania SC Bayer SRL Tel: +40-(0)21-528 59 00 Slovenija Bayer d. o. o. Tel: +386-(0)1 -58 14 400 Slovenská republika Bayer, spol. s r.o. Tel: +421-(0)2-59 21 31 11 Suomi/Finland Bayer Oy Puh/Tel: +358-(0)20-78521 Sverige Bayer AB Tel: +46-(0)8-580 223 00 United Kingdom Bayer plc Tel: +44-(0)1635-563000 |
Fecha de la última revisión de este prospecto: {MM/AAAA}.
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Prospecto: Información para el usuario
Xarelto 15 mg comprimidos recubiertos con película Xarelto 20 mg comprimidos recubiertos con película
rivaroxaban
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Xarelto y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Xarelto
3. Cómo tomar Xarelto
4. Posibles efectos adversos
5. Conservación de Xarelto
6. Contenido del envase e información adicional
1. Qué es Xarelto y para qué se utiliza
Xarelto contiene la sustancia activa rivaroxaban y se usa en adultos para:
- prevenir la formación de coágulos de sangre en el cerebro (ictus) o en otros vasos sanguíneos del organismo si padece una forma de ritmo irregular del corazón, denominada fibrilación auricular no valvular.
- tratar los coágulos de sangre en las venas de las piernas (trombosis venosa profunda) y en los vasos sanguíneos de los pulmones (embolia pulmonar), y para prevenir que estos coágulos de sangre vuelvan a aparecer en los vasos sanguíneos de las piernas y/o de los pulmones.
Xarelto pertenece a un grupo de medicamentos llamados agentes antitrombóticos. Actúa bloqueando un factor de la coagulación (factor Xa) y por lo tanto, reduciendo la tendencia de la sangre a formar coágulos.
2. Qué necesita saber antes de empezar a tomar Xarelto No tome Xarelto
- si es alérgico a rivaroxaban o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6)
- si sangra excesivamente
- si padece una enfermedad o problemas en un órgano del cuerpo que aumente el riesgo de sangrado grave (por ejemplo, úlcera de estómago, lesión o hemorragia en el cerebro o una intervención quirúrgica reciente en el cerebro o en los ojos)
- si está tomando medicamentos para prevenir la formación de coágulos en la sangre (p. ej. warfarina, dabigatran, apixaban o heparina), excepto cuando esté cambiando de tratamiento anticoagulante o mientras se le esté administrando heparina a través de un catéter venoso o arterial, para que éste no se obstruya
- si padece una enfermedad del hígado que aumente el riesgo de sangrado
- si está embarazada o en período de lactancia.
No tome Xarelto e informe a su médico si alguna de estas circunstancias se aplica a su caso. Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar Xarelto.
Tenga especial cuidado con Xarelto
- Si presenta un riesgo aumentado de sangrado, como puede suceder en las siguientes situaciones:
■ insuficiencia renal grave, ya que el funcionamiento de los riñones puede afectar a la cantidad de medicamento que actúa en el organismo
■ si está tomando otros medicamentos para prevenir la formación de coágulos de sangre (por ejemplo, warfarina, dabigatran, apixaban o heparina), cuando cambie a otro tratamiento anticoagulante o mientras reciba heparina a través de un catéter venoso o arterial, para que éste no se obstruya (ver sección “Uso de Xarelto con otros medicamentos)
■ enfermedad hemorrágica
■ presión arterial muy alta, no controlada por tratamiento médico
■ enfermedades del estómago o del intestino que puedan causar una hemorragia, como por ejemplo, inflamación intestinal o del estómago, inflamación del esófago (garganta), por ejemplo debido a la enfermedad de reflujo gastroesofágico (enfermedad en la que el ácido del estómago asciende hacia arriba en el esófago)
■ un problema en los vasos sanguíneos de la parte posterior de sus ojos (retinopatía)
■ una enfermedad pulmonar en la que los bronquios están dilatados y llenos de pus (bronquiectasia) o bien, hemorragia previa de los pulmones
- si lleva una prótesis valvular cardiaca
- si su médico determina que su presión arterial es inestable o tiene previsto recibir otro tratamiento o ser sometido a un procedimiento quirúrgico para extraer un coagulo de sangre de sus pulmones.
Informe a su médico si presenta alguna de estas situaciones antes de tomar Xarelto. Su médico decidirá si debe ser tratado con este medicamento y si debe mantenerse bajo observación más estrecha.
Si necesita una intervención quirúrgica:
- Es muy importante tomar Xarelto antes y después de la cirugía, exactamente a las horas en que su médico se lo indique.
- Si su operación requiere la colocación de un catéter o inyección en la columna vertebral (por ejemplo para anestesia epidural o espinal, o reducción del dolor):
■ Es muy importante tomar Xarelto, antes y después de la inyección o de la extracción del catéter, exactamente a las horas que su médico le haya indicado.
■ Informe a su médico inmediatamente si presenta adormecimiento o debilidad en las piernas o problemas en el intestino o en la vejiga al final de la anestesia, porque es necesaria una atención urgente.
Niños y adolescentes
Xarelto no está recomendado en niños y adolescentes menores de 18 años. No se dispone de suficiente información sobre su uso en niños y adolescentes.
Uso de Xarelto con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento, incluso los adquiridos sin receta.
- Si está tomando:
■ algún medicamento para una infección por hongos (p. ej., ketoconazol, itraconazol, voriconazol, posaconazol), salvo si sólo se aplican en la piel
■ algún medicamento antiviral para el VIH / SIDA (p. ej., ritonavir)
■ otros medicamentos para reducir la coagulación de la sangre (p. ej., enoxaparina, clopidogrel o antagonistas de la vitamina K, como la warfarina o el acenocumarol)
■ antiinflamatorios y medicamentos para aliviar el dolor (p. ej., naproxeno o ácido acetilsalicílico)
■ dronedarona, un medicamento para el tratamiento del latido cardiaco irregular
Si alguna de las circunstancias anteriores le aplica, informe a su médico antes de tomar Xarelto, porque el efecto de Xarelto podría verse aumentado. Su médico decidirá si debe ser tratado con este medicamento y si debe mantenerse bajo observación más estrecha.
Si su médico considera que tiene un mayor riesgo de desarrollar una úlcera gástrica o intestinal, podría recomendarle utilizar además, un tratamiento preventivo.
- Si usted toma:
■ algún medicamento para el tratamiento de la epilepsia (fenitoína, carbamazepina, fenobarbital)
■ hierba de San Juan (Hypericum perforatum), una planta medicinal para el tratamiento de la depresión
■ rifampicina, un antibiótico.
Si alguna de las circunstancias anteriores le aplica, informe a su médico antes de tomar Xarelto, porque el efecto de Xarelto podría verse reducido. Su médico decidirá si debe ser tratado con este medicamento y si debe mantenerse bajo observación más estrecha.
Embarazo y lactancia
No tome Xarelto si está embarazada o si está en período de lactancia. Si hay alguna posibilidad de que se quede embarazada, utilice un anticonceptivo fiable mientras toma Xarelto. Si se queda embarazada mientras toma este medicamento, informe a su médico inmediatamente, quien decidirá cómo deberá tratarse.
Conducción y uso de máquinas
Xarelto puede causar mareos (efecto adverso frecuente) o desvanecimientos (efecto adverso poco frecuente) (ver sección 4 “Posibles efectos adversos”). No deberá conducir ni utilizar máquinas si está afectado por estos síntomas.
Xarelto contiene lactosa
Si su médico le ha indicado que padece una intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento.
3. Cómo tomar Xarelto
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Qué dosis tomar
- Para prevenir la formación de coágulos de sangre en el cerebro (ictus) o en otros vasos sanguíneos del organismo
La dosis recomendada es de un comprimido de 20 mg una vez al día.
Si padece problemas en los riñones, la dosis puede disminuirse a un comprimido de 15 mg una vez al día.
- Para tratar los coágulos de sangre en las venas de las piernas y en los vasos sanguíneos de los pulmones, y para prevenir que los coágulos de sangre vuelvan a producirse
La dosis recomendada es de un comprimido de 15 mg dos veces al día durante las 3 primeras semanas. Para el tratamiento después de 3 semanas, la dosis recomendada es de un comprimido de 20 mg una vez al día.
Si padece problemas en los riñones, su médico podría decidir reducirle la dosis del tratamiento a un comprimido de 15 mg una vez al día pasadas 3 semanas si el riesgo de sangrado es superior al riesgo de tener otro coágulo de sangre.
Trague los comprimidos, preferentemente con agua.
Tome Xarelto acompañado de alimentos.
Si tiene dificultad para tragar el comprimido entero, consulte a su médico sobre otras formas de tomar Xarelto. El comprimido puede triturarse y mezclarse con agua o con puré de manzana, inmediatamente antes de tomarlo. A continuación, tome alimento.
Si es necesario, su médico también puede administrarle el comprimido Xarelto triturado a través de una sonda gástrica.
Cuándo tomar Xarelto
Tome los comprimidos cada día, hasta que su médico se lo indique.
Trate de tomar los comprimidos a la misma hora cada día para recordar a qué hora debe tomarlos.
El médico decidirá durante cuánto tiempo debe seguir tomando el tratamiento.
Para evitar la formación de coágulos sanguíneos en el cerebro (ictus) o en otros vasos sanguíneos:
Si es necesario normalizar el latido del corazón mediante un procedimiento denominado cardioversión, tome Xarelto a las horas que su médico le haya indicado.
Si toma más Xarelto del que debe
Llame inmediatamente a su médico si ha tomado demasiados comprimidos de Xarelto. Tomar demasiado Xarelto aumenta el riesgo de sangrado.
Si olvidó tomar Xarelto
- Si está tomando un comprimido de 20 mg o un comprimido de 15 mg una vez al día, y olvidó tomar una dosis, tómela en cuanto se acuerde. No tome más de un comprimido en un solo día para compensar una dosis olvidada. Tome el siguiente comprimido al día siguiente y, después, siga tomando un comprimido cada día.
- Si está tomando un comprimido de 15 mg dos veces al día, y olvidó tomar una dosis, tómela en cuanto se acuerde. No tome más de dos comprimidos de 15 mg en un solo día. Si se olvidó tomar una dosis, puede tomar dos comprimidos de 15 mg a la vez, para obtener un total de dos comprimidos (30 mg) en un día. Al día siguiente deberá seguir tomando un comprimido de
15 mg dos veces al día.
Si interrumpe el tratamiento con Xarelto
No interrumpa el tratamiento con Xarelto sin consultar primero con su médico, porque Xarelto trata y previene afecciones graves.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, Xarelto puede producir efectos adversos, aunque no todas las personas los sufran.
Al igual que otros medicamentos similares (antitrombóticos) Xarelto puede causar sangrados que pueden poner en peligro la vida del paciente. Un sangrado excesivo puede causar una caída repentina de la presión arterial (shock). En algunos casos el sangrado puede no ser evidente.
Posibles efectos adversos que pueden ser signos de sangrado:
Avise inmediatamente a su médico si experimenta cualquiera de los siguientes síntomas:
- sangrado prolongado o excesivo
- debilidad excepcional, cansancio, palidez, mareos, dolor de cabeza, hinchazón inexplicable, dificultad para respirar, dolor en el pecho o angina de pecho, que pueden ser signos de sangrado.
Su médico puede decidir mantenerle bajo una observación más estrecha o cambiarle el tratamiento.
Lista general de posibles efectos adversos:
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 pacientes):
- sangrado del estómago o del intestino, hemorragia urogenital (incluyendo sangre en la orina y sangrado menstrual abundante), hemorragia nasal, sangrado de las encías
- sangrado en el ojo (incluyendo sangrado en la parte blanca del ojo)
- sangrado hacia un tejido o cavidad del organismo (hematoma, cardenales)
- tos con sangre
- sangrado de la piel o debajo la piel
- sangrado después de una operación
- supuración de sangre o líquido de una herida quirúrgica
- hinchazón de las extremidades
- dolor de las extremidades
- fiebre
- disminución de los glóbulos rojos que puede causar palidez y debilidad o dificultad para respirar
- dolor de estómago, indigestión, mareo o sensación de mareo, estreñimiento, diarrea
- presión arterial baja (los síntomas pueden ser sensación de mareo o desvanecimiento al ponerse de pie)
- disminución general de la fuerza y la energía (debilidad, cansancio), dolor de cabeza, mareos,
- sarpullido, picor de la piel
- alteración de la función de los riñones (puede verse en los análisis realizados por el médico)
- los análisis de sangre pueden mostrar un aumento de algunas enzimas hepáticas
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes):
- sangrado en el cerebro o en el interior del cráneo
- sangrado en una articulación, que causa dolor e hinchazón.
- desvanecimiento
- sensación de malestar
- sequedad de boca
- aumento de la frecuencia cardiaca
- reacción alérgica, incluyendo reacción alérgica de la piel
- ronchas
- alteración de la función del hígado (puede verse en los análisis realizados por el médico)
- los análisis de sangre pueden mostrar un aumento de la bilirrubina, de algunas enzimas pancreáticas o del número de plaquetas.
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 pacientes):
- sangrado en un músculo
- hinchazón localizada
- coloración amarillenta de la piel y en los ojos (ictericia)
- acumulación de sangre (hematoma) en la ingle después de una complicación en una cirugía cardíaca en la que se introduce un catéter en la arteria de la pierna (pseudoaneurisma).
Frecuencia no conocida (la frecuencia no puede estimarse a partir de los datos disponibles):
- aumento de la presión en los músculos de las piernas o de los brazos después de una hemorragia, que causa dolor, hinchazón, alteración de la sensibilidad, adormecimiento o parálisis (síndrome compartimental después de una hemorragia)
- insuficiencia renal después de una hemorragia grave.
Los siguientes efectos adversos han sido comunicados después de la autorización:
- angioedema y edema alérgico (hinchazón de la cara, labios, boca, lengua o garganta)
- colestasis (disminución del flujo de la bilis), hepatitis, incluyendo lesión hepatocelular (inflamación o lesión de hígado)
- trombocitopenia (número bajo de plaquetas, células que ayudan a la coagulación de la sangre). Comunicación de efectos adversos
Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Xarelto
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en cada blíster después de “CAD” o ”EXP”.
La fecha de caducidad es el último día del mes que se indica.
No requiere condiciones especiales de conservación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Xarelto
- El principio activo es rivaroxaban. Cada comprimido contiene 15 mg ó 20 mg de rivaroxaban.
- Los demás componentes son:
Núcleo del comprimido: celulosa microcristalina, croscarmelosa sódica, lactosa monohidrato, hipromelosa, laurilsulfato de sodio, estearato de magnesio.
Recubrimiento pelicular del comprimido: macrogol 3350, hipromelosa, dióxido de titanio (E171), óxido de hierro rojo (E 172).
Aspecto del producto y contenido del envase
Los comprimidos recubiertos con película de Xarelto 15 mg son redondos, de color rojo, biconvexos y llevan grabada la cruz BAYER en una cara, y “15” y un triángulo en la otra.
Se presentan envasados en blíster, en estuches de 10, 14, 28, 42 ó 98 comprimidos recubiertos con película o en blisters unidosis, en envases de 10 x 1 ó 100 x 1, o en envases múltiples que contienen 10 estuches de 10 x 1 comprimidos recubiertos con película, o bien, en frascos conteniendo 100 comprimidos recubiertos con película.
Los comprimidos recubiertos con película de Xarelto 20 mg son redondos, de color pardo-rojizo, biconvexos y llevan grabada la cruz BAYER en una cara, y “20” y un triángulo en la otra.
Se presentan envasados en blíster, en estuches de 10, 14, 28, ó 98 comprimidos recubiertos con película o en blisters unidosis, en envases de 10 x 1 ó de 100 x 1, o en envases múltiples que contienen 10 estuches de 10 x 1 comprimidos recubiertos con película, o bien, en frascos conteniendo 100 comprimidos recubiertos con película.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Bayer Pharma AG 13342 Berlin Alemania
Responsable de la fabricación
Bayer Pharma AG 51368 Leverkusen Alemania
Bayer HealthCare Manufacturing Srl.
Via delle Groane, 126 20024 Garbagnate Milanese Italia
|
Belgie / Belgique / Belgien Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Etarapna Eañep Etarapna EOOfl Tea: +359-(0)2-81 401 01 Ceská republika Bayer s.r.o. Tel: +420-266 101 111 Danmark Bayer A/S Tlf: +45-45 235 000 Deutschland Bayer Vital GmbH Tel: +49-(0)214-30 513 48 Eesti Bayer OÜ Tel: +372-655 85 65 EXláSa Bayer EAAág ABEE Tn^: +30-210-618 75 00 España Bayer Hispania S.L. Tel: +34-93-495 65 00 France Bayer HealthCare Tél (N° vert): +33-(0) 800 87 54 54 Hrvatska Bayer d.o.o. Tel: + 385-(0)1-6599 900 Ireland Bayer Limited Tel: +353-(0)1-2999 313 Ísland Icepharma hf. Sími: +354-540 80 00 Italia Bayer S.p.A. Tel: +39-02-3978 1 Kúnpoq NOVAGEM Limited Tn^: +357-22-48 38 58 Latvija SIA Bayer Tel: +371-67 84 55 63 |
Lietuva UAB Bayer Tel: +370-5-233 68 68 Luxembourg / Luxemburg Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Magyarország Bayer Hungária KFT Tel: +36-1-487 4100 Malta Alfred Gera and Sons Ltd. Tel: +356-21 44 62 05 Nederland Bayer B.V. Tel: +31-(0)297-28 06 66 Norge Bayer AS Tlf: +47-23 13 05 00 Osterreich Bayer Austria Ges. m. b. H. Tel: +43-(0)1-711 460 Polska Bayer Sp. z o.o. Tel: +48-22-572 35 00 Portugal Bayer Portugal, Lda. Tel: +351-21-416 42 00 Romania SC Bayer SRL Tel: +40-(0)21-528 59 00 Slovenija Bayer d. o. o. Tel: +386-(0)1 -58 14 400 Slovenská republika Bayer, spol. s r.o. Tel: +421-(0)2-59 21 31 11 Suomi/Finland Bayer Oy Puh/Tel: +358-(0)20-78521 Sverige Bayer AB Tel: +46-(0)8-580 223 00 United Kingdom Bayer plc Tel: +44-(0)1635-563000 |
Fecha de la última revisión de este prospecto: {MM/AAAA}.
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
Prospecto: Información para el usuario
Xarelto 15 mg comprimidos recubiertos con película Xarelto 20 mg comprimidos recubiertos con película
Envase para el inicio del tratamiento
rivaroxaban
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Xarelto y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Xarelto
3. Cómo tomar Xarelto
4. Posibles efectos adversos
5. Conservación de Xarelto
6. Contenido del envase e información adicional
1. Qué es Xarelto y para qué se utiliza
Xarelto contiene la sustancia activa rivaroxaban y se usa en adultos para:
- tratar los coágulos de sangre en las venas de las piernas (trombosis venosa profunda) y en los vasos sanguíneos de los pulmones (embolia pulmonar), y para prevenir que estos coágulos de sangre vuelvan a aparecer en los vasos sanguíneos de las piernas y/o de los pulmones.
Xarelto pertenece a un grupo de medicamentos llamados agentes antitrombóticos. Actúa bloqueando un factor de la coagulación (factor Xa) y por lo tanto, reduciendo la tendencia de la sangre a formar coágulos.
2. Qué necesita saber antes de empezar a tomar Xarelto No tome Xarelto
- si es alérgico a rivaroxaban o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6)
- si sangra excesivamente
- si padece una enfermedad o problemas en un órgano del cuerpo que aumente el riesgo de sangrado grave (por ejemplo, úlcera de estómago, lesión o hemorragia en el cerebro o una intervención quirúrgica reciente en el cerebro o en los ojos)
- si está tomando medicamentos para prevenir la formación de coágulos en la sangre (p. ej. warfarina, dabigatran, apixaban o heparina), excepto cuando esté cambiando de tratamiento anticoagulante o mientras se le esté administrando heparina a través de un catéter venoso o arterial, para que éste no se obstruya
- si padece una enfermedad del hígado que aumente el riesgo de sangrado
- si está embarazada o en período de lactancia.
No tome Xarelto e informe a su médico si alguna de estas circunstancias se aplica a su caso. Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar Xarelto.
Tenga especial cuidado con Xarelto
- Si presenta un riesgo aumentado de sangrado, como puede suceder en las siguientes situaciones:
■ insuficiencia renal grave, ya que el funcionamiento de los riñones puede afectar a la cantidad de medicamento que actúa en el organismo
■ si está tomando otros medicamentos para prevenir la formación de coágulos de sangre (por ejemplo, warfarina, dabigatran, apixaban o heparina), cuando cambie a otro tratamiento anticoagulante o mientras reciba heparina a través de un catéter venoso o arterial, para que éste no se obstruya (ver sección “Uso de Xarelto con otros medicamentos)
■ enfermedad hemorrágica
■ presión arterial muy alta, no controlada por tratamiento médico
■ enfermedades del estómago o del intestino que puedan causar una hemorragia, como por ejemplo, inflamación intestinal o del estómago, inflamación del esófago (garganta), por ejemplo debido a la enfermedad de reflujo gastroesofágico (enfermedad en la que el ácido del estómago asciende hacia arriba en el esófago)
■ un problema en los vasos sanguíneos de la parte posterior de sus ojos (retinopatía)
■ una enfermedad pulmonar en la que los bronquios están dilatados y llenos de pus (bronquiectasia) o bien, hemorragia previa de los pulmones
- si lleva una prótesis valvular cardiaca
- si su médico determina que su presión arterial es inestable o tiene previsto recibir otro tratamiento o ser sometido a un procedimiento quirúrgico para extraer un coágulo de sangre de sus pulmones.
Informe a su médico si presenta alguna de estas situaciones antes de tomar Xarelto. Su médico decidirá si debe ser tratado con este medicamento y si debe mantenerse bajo observación más estrecha.
Si necesita una intervención quirúrgica:
- Es muy importante tomar Xarelto antes y después de la cirugía, exactamente a las horas en que su médico se lo indique.
- Si su operación requiere la colocación de un catéter o inyección en la columna vertebral (por ejemplo para anestesia epidural o espinal, o reducción del dolor):
■ Es muy importante tomar Xarelto, antes y después de la inyección o de la extracción del catéter, exactamente a las horas que su médico le haya indicado.
■ Informe a su médico inmediatamente si presenta adormecimiento o debilidad en las piernas o problemas en el intestino o en la vejiga al final de la anestesia, porque es necesaria una atención urgente.
Niños y adolescentes
Uso de Xarelto con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento, incluso los adquiridos sin receta.
- Si está tomando:
■ algún medicamento para una infección por hongos (p. ej., ketoconazol, itraconazol, voriconazol, posaconazol), salvo si sólo se aplican en la piel
■ algún medicamento antiviral para el VIH / SIDA (p. ej., ritonavir)
■ otros medicamentos para reducir la coagulación de la sangre (p. ej., enoxaparina, clopidogrel o antagonistas de la vitamina K, como la warfarina o el acenocumarol)
■ antiinflamatorios y medicamentos para aliviar el dolor (p. ej., naproxeno o ácido acetilsalicílico)
■ dronedarona, un medicamento para el tratamiento del latido cardiaco irregular
Si alguna de las circunstancias anteriores le aplica, informe a su médico antes de tomar Xarelto, porque el efecto de Xarelto podría verse aumentado. Su médico decidirá si debe ser tratado con este medicamento y si debe mantenerse bajo observación más estrecha.
Si su médico considera que tiene un mayor riesgo de desarrollar una úlcera gástrica o intestinal, podría recomendarle utilizar además, un tratamiento preventivo.
- Si usted toma:
■ algún medicamento para el tratamiento de la epilepsia (fenitoína, carbamazepina, fenobarbital)
■ hierba de San Juan (Hypericum perforatum), una planta medicinal para el tratamiento de la depresión
■ rifampicina, un antibiótico.
Si alguna de las circunstancias anteriores le aplica, informe a su médico antes de tomar Xarelto, porque el efecto de Xarelto podría verse reducido. Su médico decidirá si debe ser tratado con este medicamento y si debe mantenerse bajo observación más estrecha.
Embarazo y lactancia
No tome Xarelto si está embarazada o si está en período de lactancia. Si hay alguna posibilidad de que se quede embarazada, utilice un anticonceptivo fiable mientras toma Xarelto. Si se queda embarazada mientras toma este medicamento, informe a su médico inmediatamente, quien decidirá cómo deberá tratarse.
Conducción y uso de máquinas
Xarelto puede causar mareos (efecto adverso frecuente) o desvanecimientos (efecto adverso poco frecuente) (ver sección 4 “Posibles efectos adversos”). No deberá conducir ni utilizar máquinas si está afectado por estos síntomas.
Xarelto contiene lactosa
Si su médico le ha indicado que padece una intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento.
3. Cómo tomar Xarelto
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Qué dosis tomar
La dosis recomendada es de un comprimido de 15 mg dos veces al día durante las 3 primeras semanas. Para el tratamiento después de 3 semanas, la dosis recomendada es de un comprimido de 20 mg una vez al día.
Este envase para el inicio del tratamiento de Xarelto 15 mg y 20 mg es sólo para las primeras 4 semanas de tratamiento.
Una vez finalizado este envase, se seguirá el tratamiento con Xarelto 20 mg una vez al día, tal y como le ha indicado su médico.
Si padece problemas en los riñones, su médico podría decidir reducirle la dosis del tratamiento a un comprimido de 15 mg una vez al día pasadas 3 semanas si el riesgo de sangrado es superior al riesgo de tener otro coágulo de sangre.
Trague los comprimidos, preferentemente con agua.
Tome Xarelto acompañado de alimentos.
Si tiene dificultad para tragar el comprimido entero, consulte a su médico sobre otras formas de tomar Xarelto. El comprimido puede triturarse y mezclarse con agua o con puré de manzana, inmediatamente antes de tomarlo. A continuación, tome alimento.
Si es necesario, su médico también puede administrarle el comprimido Xarelto triturado a través de una sonda gástrica.
Cuándo tomar Xarelto
Tome los comprimidos cada día, hasta que su médico se lo indique.
Trate de tomar los comprimidos a la misma hora cada día para recordar a qué hora debe tomarlos.
El médico decidirá durante cuánto tiempo debe seguir tomando el tratamiento.
Si toma más Xarelto del que debe
Llame inmediatamente a su médico si ha tomado demasiados comprimidos de Xarelto. Tomar demasiado Xarelto aumenta el riesgo de sangrado.
Si olvidó tomar Xarelto
- Si está tomando un comprimido de 15 mg dos veces al día, y olvidó tomar una dosis, tómela en cuanto se acuerde. No tome más de dos comprimidos de 15 mg en un solo día. Si se olvidó tomar una dosis, puede tomar dos comprimidos de 15 mg a la vez, para obtener un total de dos comprimidos (30 mg) en un día. Al día siguiente deberá seguir tomando un comprimido de
15 mg dos veces al día.
- Si está tomando un comprimido de 20 mg una vez al día, y olvidó tomar una dosis, tómela en cuanto se acuerde. No tome más de un comprimido en un solo día para compensar una dosis olvidada. Tome el siguiente comprimido al día siguiente y, después, siga tomando un comprimido cada día.
Si interrumpe el tratamiento con Xarelto
No interrumpa el tratamiento con Xarelto sin consultar primero con su médico, porque Xarelto trata y previene afecciones graves.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, Xarelto puede producir efectos adversos, aunque no todas las personas los sufran.
Al igual que otros medicamentos similares (antitrombóticos), Xarelto puede causar sangrados que pueden poner en peligro la vida del paciente. Un sangrado excesivo puede causar una caída repentina de la presión arterial (shock). En algunos casos el sangrado puede no ser evidente.
Posibles efectos adversos que pueden ser signos de sangrado:
Avise inmediatamente a su médico si experimenta cualquiera de los siguientes síntomas:
- sangrado prolongado o excesivo
- debilidad excepcional, cansancio, palidez, mareos, dolor de cabeza, hinchazón inexplicable, dificultad para respirar, dolor en el pecho o angina de pecho, que pueden ser signos de sangrado.
Su médico puede decidir mantenerle bajo una observación más estrecha o cambiarle el tratamiento.
Lista general de posibles efectos adversos:
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 pacientes):
- sangrado del estómago o del intestino, hemorragia urogenital (incluyendo sangre en la orina y sangrado menstrual abundante), hemorragia nasal, sangrado de las encías
- sangrado en el ojo (incluyendo sangrado en la parte blanca del ojo)
- sangrado hacia un tejido o cavidad del organismo (hematoma, cardenales)
- tos con sangre
- sangrado de la piel o debajo la piel
- sangrado después de una operación
- supuración de sangre o líquido de una herida quirúrgica
- hinchazón de las extremidades
- dolor de las extremidades
- fiebre
- disminución de los glóbulos rojos que puede causar palidez y debilidad o dificultad para respirar
- dolor de estómago, indigestión, mareo o sensación de mareo, estreñimiento, diarrea
- presión arterial baja (los síntomas pueden ser sensación de mareo o desvanecimiento al ponerse de pie)
- disminución general de la fuerza y la energía (debilidad, cansancio), dolor de cabeza, mareos,
- sarpullido, picor de la piel
- alteración de la función de los riñones (puede verse en los análisis realizados por el médico)
- los análisis de sangre pueden mostrar un aumento de algunas enzimas hepáticas
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes):
- sangrado en el cerebro o en el interior del cráneo
- sangrado en una articulación, que causa dolor e hinchazón.
- desvanecimiento
- sensación de malestar
- sequedad de boca
- aumento de la frecuencia cardiaca
- reacción alérgica, incluyendo reacción alérgica de la piel
- ronchas
- alteración de la función del hígado (puede verse en los análisis realizados por el médico)
- los análisis de sangre pueden mostrar un aumento de la bilirrubina, de algunas enzimas pancreáticas o del número de plaquetas.
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 pacientes):
- sangrado en un músculo
- hinchazón localizada
- coloración amarillenta de la piel y en los ojos (ictericia)
- acumulación de sangre (hematoma) en la ingle después de una complicación en una cirugía cardíaca en la que se introduce un catéter en la arteria de la pierna (pseudoaneurisma).
Frecuencia no conocida (la frecuencia no puede estimarse a partir de los datos disponibles):
- aumento de la presión en los músculos de las piernas o de los brazos después de una hemorragia, que causa dolor, hinchazón, alteración de la sensibilidad, adormecimiento o parálisis (síndrome compartimental después de una hemorragia)
- insuficiencia renal después de una hemorragia grave.
Los siguientes efectos adversos han sido comunicados después de la autorización:
- angioedema y edema alérgico (hinchazón de la cara, labios, boca, lengua o garganta)
- colestasis (disminución del flujo de la bilis), hepatitis, incluyendo lesión hepatocelular (inflamación o lesión de hígado)
Comunicación de efectos adversos
Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Xarelto
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en cada blíster después de “CAD” o ”EXP”.
La fecha de caducidad es el último día del mes que se indica.
No requiere condiciones especiales de conservación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Xarelto
- El principio activo es rivaroxaban. Cada comprimido contiene 15 mg ó 20 mg de rivaroxaban, respectivamente.
- Los demás componentes son:
Núcleo del comprimido: celulosa microcristalina, croscarmelosa sódica, lactosa monohidrato, hipromelosa, laurilsulfato de sodio, estearato de magnesio.
Recubrimiento pelicular del comprimido: macrogol 3350, hipromelosa, dióxido de titanio (E171), óxido de hierro rojo (E 172).
Aspecto del producto y contenido del envase
Los comprimidos recubiertos con película de Xarelto 15 mg son redondos, de color rojo, biconvexos y llevan grabada la cruz BAYER en una cara, y “15” y un triángulo en la otra.
Los comprimidos recubiertos con película de Xarelto 20 mg son redondos, de color pardo-rojizo, biconvexos y llevan grabada la cruz BAYER en una cara, y “20” y un triángulo en la otra.
Envase para el inicio del tratamiento en las primeras 4 semanas: cada envase de 49 comprimidos recubiertos con película para el tratamiento en las primeras 4 semanas contiene:
42 comprimidos recubiertos con película con 15 mg de rivaroxaban y 7 comprimidos recubiertos con película con 20 mg de rivaroxaban en un envase con calendario.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Bayer Pharma AG 13342 Berlin Alemania
Responsable de la fabricación
Bayer Pharma AG 51368 Leverkusen Alemania
Bayer HealthCare Manufacturing Srl. Via delle Groane, 126 20024 Garbagnate Milanese Italia
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie / Belgique / Belgien Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Etarapna Eañep Etarapna EOOfl Tea: +359-(0)2-81 401 01 Ceská republika Bayer s.r.o. Tel: +420-266 101 111 Danmark Bayer A/S Tlf: +45-45 235 000 Deutschland Bayer Vital GmbH Tel: +49-(0)214-30 513 48 Eesti Bayer OÜ Tel: +372-655 85 65 EXláSa Bayer EAAág ABEE Tn^: +30-210-618 75 00 España Bayer Hispania S.L. Tel: +34-93-495 65 00 France Bayer HealthCare Tél (N° vert): +33-(0) 800 87 54 54 Hrvatska Bayer d.o.o. Tel: + 385-(0)1-6599 900 Ireland Bayer Limited Tel: +353-(0)1-2999 313 Ísland Icepharma hf. Sími: +354-540 80 00 Italia Bayer S.p.A. Tel: +39-02-3978 1 Kúnpoq NOVAGEM Limited Tn^: +357-22-48 38 58 Latvija SIA Bayer Tel: +371-67 84 55 63 |
Lietuva UAB Bayer Tel: +370-5-233 68 68 Luxembourg / Luxemburg Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Magyarország Bayer Hungária KFT Tel: +36-1-487 4100 Malta Alfred Gera and Sons Ltd. Tel: +356-21 44 62 05 Nederland Bayer B.V. Tel: +31-(0)297-28 06 66 Norge Bayer AS Tlf: +47-23 13 05 00 Osterreich Bayer Austria Ges. m. b. H. Tel: +43-(0)1 -711 460 Polska Bayer Sp. z o.o. Tel: +48-22-572 35 00 Portugal Bayer Portugal, Lda. Tel: +351-21-416 42 00 Romania SC Bayer SRL Tel: +40-(0)21-528 59 00 Slovenija Bayer d. o. o. Tel: +386-(0)1 -58 14 400 Slovenská republika Bayer, spol. s r.o. Tel: +421-(0)2-59 21 31 11 Suomi/Finland Bayer Oy Puh/Tel: +358-(0)20-78521 Sverige Bayer AB Tel: +46-(0)8-580 223 00 United Kingdom Bayer plc Tel: +44-(0)1635-563000 |
Fecha de la última revisión de este prospecto: {MM/AAAA}.
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
191