Tasigna 150 Mg Capsulas Duras
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Tasigna 150 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una cápsula dura contiene 150 mg de nilotinib (como clorhidrato monohidrato). Excipiente(s) con efecto conocido
Una cápsula dura contiene 117,08 mg de lactosa (como monohidrato).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura
Polvo blanco a amarillento en cápsulas de gelatina dura, opacas, de color rojo, de tamaño 1 con la impresión axial «NVR/BCR» en negro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tasigna está indicado para el tratamiento de pacientes adultos con leucemia mieloide crónica (LMC) cromosoma Filadelfia positivo, de nuevo diagnóstico, en fase crónica.
4.2 Posología y forma de administración
El tratamiento debe iniciarlo un médico con experiencia en el diagnóstico y el tratamiento de pacientes con LMC.
Posología
La dosis recomendada de Tasigna es de 300 mg dos veces al día. El tratamiento debe prolongarse mientras continúe el beneficio para el paciente.
Para la dosis de 400 mg una vez al día (ver ajustes de dosis a continuación), se encuentran disponibles cápsulas duras de 200 mg.
Si el paciente olvida tomar una dosis, no debe tomar una dosis adicional, sino esperar a la siguiente dosis, según la pauta establecida.
Ajustes o modificaciones de la dosis
Puede ser necesario una interrupción temporal y/o una reducción de la dosis de Tasigna por toxicidades hematológicas (neutropenia, trombocitopenia) que no estén relacionadas con la leucemia de base (ver Tabla 1).
Tabla 1 Ajustes de dosis por neutropenia y trombocitopenia
|
LMC de nuevo |
RAN* <1,0 x 109/l y/o recuento |
1. |
Se deberá interrumpir el tratamiento con |
|
diagnóstico en fase |
de plaquetas <50 x 109/l |
Tasigna y controlar los hemogramas. | |
|
crónica a 300 mg |
2. |
Se deberá reanudar el tratamiento a las | |
|
dos veces al día |
2 semanas a la dosis previa cuando RAN >1,0 x 109/l y/o el recuento de plaquetas >50 x 109/l. | ||
|
3. |
Si el hemograma se mantiene bajo, puede necesitarse una reducción de la dosis a 400 mg una vez al día. |
*RAN = Recuento absoluto de neutrófilos
Si se desarrolla toxicidad no hematológica clínicamente significativa, moderada o grave, el tratamiento debe suspenderse, y cuando la toxicidad se haya resuelto puede reanudarse el tratamiento con dosis de 400 mg una vez al día. Si se considera clínicamente adecuado, deberá considerarse el escalado de la dosis a 300 mg dos veces al día.
Aumento de la lipasa sérica: Para elevaciones de lipasa sérica de Grado 3-4, deberá reducirse la dosis a 400 mg una vez al día o bien interrumpir el tratamiento. El nivel de lipasa sérica deberá controlarse mensualmente o según esté indicado clínicamente (ver sección 4.4).
Aumento de la bilirrubina y las transaminasas hepáticas: Para elevaciones de bilirrubina y transaminasas hepáticas de Grado 3-4, deberá reducirse la dosis a 400 mg una vez al día o bien interrumpir el tratamiento. Los niveles de bilirrubina y de transaminasas hepáticas deberán controlarse mensualmente o según esté indicado clínicamente.
Pacientes de edad avanzada
Aproximadamente el 12% de los individuos participantes en el ensayo clínico tenían 65 años ó más. No se observaron diferencias importantes respecto a la seguridad y eficacia en pacientes de >65 años de edad comparado con adultos de entre 18 y 65 años.
Insuficiencia renal
No se han realizado ensayos clínicos en pacientes con insuficiencia renal.
En pacientes con insuficiencia renal no se espera una disminución en el aclaramiento corporal total, puesto que nilotinib y sus metabolitos no se excretan por vía renal.
Insuficiencia hepática
La insuficiencia hepática tiene un efecto moderado sobre la farmacocinética de nilotinib. No se considera necesario un ajuste de dosis en pacientes con insuficiencia hepática. Sin embargo, los pacientes con insuficiencia hepática deberán tratarse con precaución (ver sección 4.4).
Trastornos cardiacos
En los ensayos clínicos, se excluyeron pacientes con enfermedad cardiaca significativa o no controlada (p.ej. infarto de miocardio reciente, insuficiencia cardiaca congestiva, angina inestable o bradicardia clínicamente significativa). Deberá utilizarse con precaución en pacientes con alteraciones cardiacas relevantes (ver sección 4.4).
Se han notificado aumentos en los niveles de colesterol plasmático total con el tratamiento con Tasigna (ver sección 4.4). Se debe determinar el perfil lipídico antes de iniciar el tratamiento con Tasigna, evaluar en el mes 3 y 6 después de iniciar el tratamiento y al menos anualmente durante el tratamiento crónico.
Se han notificado aumentos en los niveles de glucosa en sangre con el tratamiento con Tasigna (ver sección 4.4). Se deben evaluar los niveles de glucosa en sangre antes de iniciar el tratamiento con Tasigna y monitorizar durante el tratamiento.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de Tasigna en niños de menos de 18 años (ver sección 5.1). Por lo tanto, no está recomendado su uso en pacientes pediátricos debido a la ausencia de datos sobre seguridad y eficacia.
Forma de administración
Tasigna debe tomarse dos veces al día con aproximadamente unas 12 horas entre las dos administraciones y no debe tomarse junto con la comida. Las cápsulas duras deben tragarse enteras, con agua. No se debe ingerir alimentos durante las dos horas previas a la administración de la dosis ni durante, al menos, una hora después.
Para pacientes que no puedan tragar las cápsulas duras, el contenido de cada cápsula dura puede dispersarse en una cucharadita de compota de manzana (puré de manzana) y debe tomarse inmediatamente. No debe utilizarse más de una cucharadita de compota de manzana ni ningún otro alimento aparte de compota de manzana (ver secciones 4.4 y 5.2).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Mielosupresión
El tratamiento con Tasigna se ha asociado a trombocitopenia, neutropenia y anemia (Grado 3-4 según el «Common Toxicity Criteria» del National Cancer Institute). Deberán realizarse hemogramas completos cada dos semanas durante los 2 primeros meses y posteriormente cada mes, o con la frecuencia indicada clínicamente. En general, la mielosupresión fue reversible y normalmente se controló suspendiendo la administración de Tasigna de forma temporal o con una reducción de la dosis (ver sección 4.2).
Prolongación del QT
Se ha observado que Tasigna prolonga la repolarización cardiaca ventricular de forma dependiente de la concentración, medida por el intervalo QT del ECG.
En el ensayo de Fase III en pacientes con LMC en fase crónica de nuevo diagnóstico que recibieron 300 mg de nilotinib dos veces al día, el cambio en el tiempo medio del intervalo QTcF respecto al valor basal en el estado estacionario fue de 6 ms. Ningún paciente presentó un QTcF>480 ms. No se observaron episodios de «Torsade de Pointes».
En un ensayo con voluntarios sanos con exposiciones que fueron comparables a las observadas en pacientes, la media de tiempo del cambio del valor de QTcF respecto al valor basal fue de 7 ms (IC ±
4 ms). Ningún individuo presentó un valor de QTcF >450 ms. Además, no se observaron arritmias clínicamente relevantes durante la realización del ensayo. En particular, no se observaron episodios de «Torsade de Pointes» (transitorios o sostenidos).
Puede producirse una prolongación significativa del intervalo QT cuando se administra nilotinib de forma no adecuada con inhibidores potentes de CYP3A4, con fármacos con una capacidad conocida de prolongar el intervalo QT y/o con comida (ver sección 4.5). La presencia de hipopotasemia e hipomagnesemia pueden incrementar este efecto. La prolongación del intervalo QT puede exponer a los pacientes a un riesgo mortal.
Tasigna debe utilizarse con precaución en pacientes que presentan o que tienen un riesgo importante de desarrollar una prolongación del intervalo QTc, tales como:
- con prolongación de QT largo congénita.
- con enfermedad cardíaca significativa o no controlada, incluyendo infarto de miocardio reciente, insuficiencia cardiaca congestiva, angina inestable o bradicardia clínicamente significativa.
- en tratamiento con fármacos antiarrítmicos u otras sustancias que puedan provocar una prolongación del intervalo QTc.
Se recomienda un control cuidadoso del efecto sobre el intervalo QTc y realizar un ECG basal antes de iniciar el tratamiento con Tasigna y según esté clínicamente indicado. La hipopotasemia o hipomagnesemia deberán corregirse antes de la administración de Tasigna y deberán controlarse periódicamente durante el tratamiento.
Muerte súbita
Se han notificado casos poco frecuentes (0,1 a 1%) de muertes súbitas en pacientes con LMC en fase crónica o fase acelerada resistentes o intolerantes a imatinib que tienen antecedentes de enfermedad cardiaca o factores de riesgo cardiaco significativos. También se hallaban presentes frecuentemente co-morbilidades además del proceso maligno de base, así como medicamentos concomitantes. Las alteraciones de la repolarización ventricular pueden ser factores contributivos. No se han notificado casos de muerte súbita en el ensayo de Fase III en pacientes con LMC en fase crónica de nuevo diagnóstico.
Retención de líquidos y edema
Se observaron con poca frecuencia (0,1 a 1%) formas graves de retención de líquidos tales como derrame pleural, edema pulmonar y derrame pericárdico en un ensayo de Fase III en pacientes con LMC de nuevo diagnóstico. Se observaron acontecimientos similares en informes postcomercialización. El aumento rápido e inesperado del peso se debe investigar cuidadosamente. Si aparecen signos de retención grave de líquidos durante el tratamiento con nilotinib, se debe evaluar la etiología y tratar a los pacientes en consecuencia (ver sección 4.2 para instrucciones sobre el control de toxicidades no hematológicas).
Acontecimientos cardiovasculares
Se notificaron acontecimientos cardiovasculares en un ensayo de Fase III aleatorizado en pacientes con LMC de nuevo diagnóstico y se observaron en informes post-comercialización. En este ensayo clínico con una mediana de tiempo en tratamiento de 60,5 meses, los acontecimientos cardiovasculares de grado 3-4 incluyeron: enfermedad arterial oclusiva periférica (1,4% y 1,1% con 300 mg y 400 mg de nilotinib dos veces al día, respectivamente), cardiopatía isquémica (2,2% y 6,1% con 300 mg y 400 mg de nilotinib dos veces al día, respectivamente) y acontecimientos isquémicos cerebrovasculares (1,1% y 2,2% con 300 mg y 400 mg de nilotinib dos veces al día, respectivamente). Se debe advertir a los pacientes que busquen atención médica inmediata si experimentan signos o síntomas agudos de acontecimientos cardiovasculares. Se debe evaluar el estado cardiovascular de los pacientes y monitorizar y controlar activamente los factores de riesgo cardiovasculares durante el tratamiento con Tasigna de acuerdo a las guías generales. Se debe prescribir el tratamiento apropiado para controlar los factores de riesgo cardiovasculares (ver sección 4.2 para instrucciones sobre el control de toxicidades no hematológicas).
Reactivación del virus de la hepatitis B
Se han producido reactivaciones de la hepatitis B en pacientes que son portadores crónicos de este virus después de que los pacientes hayan recibido inhibidores de la tirosina quinasa BCR-ABL. En algunos casos se produjo insuficiencia hepática aguda o hepatitis fulminante que dio lugar a un trasplante de hígado o a un desenlace mortal.
Los pacientes se deben someter a pruebas para detectar la infección por VHB antes de comenzar el tratamiento con Tasigna. Se debe consultar a expertos en enfermedades hepáticas y en el tratamiento de la hepatitis B antes de comenzar el tratamiento en pacientes con una serología positiva para hepatitis B (incluyendo a los pacientes con enfermedad activa) y pacientes que den un resultado positivo en una prueba de infección por VHB durante el tratamiento. Los portadores del VHB que necesiten tratamiento con Tasigna se deben someter a una estrecha monitorización para detectar signos y síntomas de infección activa por VHB a lo largo de todo el tratamiento y durante varios meses después de finalizar el tratamiento (ver sección 4.8).
Pruebas de laboratorio y controles
Lípidos en sangre
En un ensayo de Fase III en pacientes con LMC de nuevo diagnóstico, el 1,1% de los pacientes tratados con 400 mg de nilotinib dos veces al día mostraron un aumento del colesterol total de Grado 3-4; sin embargo no se observaron elevaciones de Grado 3-4 en el grupo de dosis de 300 mg dos veces al día (ver sección 4.8). Se recomienda determinar los perfiles lipídicos antes de iniciar el tratamiento con Tasigna, evaluarlos en el mes 3 y 6 después de iniciar el tratamiento y al menos anualmente durante el tratamiento crónico (ver sección 4.2). Si se necesita un inhibidor de la HMG-CoA reductasa (agente hipolipemiante), ver la sección 4.5 antes de iniciar el tratamiento puesto que algunos inhibidores de la HMG-CoA reductasa también se metabolizan por la vía de CYP3A4.
Glucosa en sangre
En un ensayo de Fase III en pacientes con LMC de nuevo diagnóstico, el 6,9% y el 7,2% de los pacientes tratados con 400 mg de nilotinib y 300 mg de nilotinib dos veces al día, respectivamente, mostraron un aumento de la glucosa en sangre de Grado 3-4. Se recomienda evaluar los niveles de glucosa antes de iniciar el tratamiento con Tasigna y controlarlos durante el tratamiento, según esté clínicamente indicado (ver sección 4.2). Si los resultados de las pruebas justifican el tratamiento, los médicos deben seguir sus estándares de práctica locales y las guías de tratamiento.
Interacciones con otros medicamentos
Debe evitarse la administración de Tasigna con agentes que son inhibidores potentes de CYP3A4 (incluyendo, aunque no de manera exclusiva, ketoconazol, itraconazol, voriconazol, claritromicina, telitromicina, ritonavir). En caso que se requiera el tratamiento con alguno de estos fármacos, se recomienda interrumpir el tratamiento con Tasigna, si es posible (ver sección 4.5). En caso que no sea posible la interrupción temporal del tratamiento, deberá realizarse un control estricto del paciente para la prolongación del intervalo QT (ver secciones 4.2, 4.5 y 5.2).
El uso concomitante de Tasigna con medicamentos que son inductores potentes del CYP3A4 (p.ej. fenitoina, rifampicina, carbamazepina, fenobarbital e hierba de San Juan) es probable que reduzca la exposición a nilotinib en un grado clínicamente relevante. Por lo tanto, en pacientes que reciben tratamiento con Tasigna, deberán elegirse agentes terapéuticos alternativos con menor potencial de inducción de CYP3A4 (ver sección 4.5).
Efecto de los alimentos
La biodisponibilidad de nilotinib aumenta con los alimentos. Tasigna no se debe tomar junto con la comida (ver las secciones 4.2 y 4.5) sino que se debe tomar 2 horas después de una comida. No se debe ingerir ningún alimento durante al menos una hora después de tomar el medicamento. Debe evitarse tomar zumo de pomelo y otros alimentos que se sabe que son inhibidores de CYP3A4. Para pacientes que no puedan tragar las cápsulas duras, el contenido de cada cápsula dura puede dispersarse en una cucharadita de compota de manzana y debe tomarse inmediatamente. No debe utilizarse más de una cucharadita de compota de manzana ni otro alimento aparte de la compota de manzana (ver sección 5.2).
Insuficiencia hepática
La insuficiencia hepática tiene un efecto moderado sobre la farmacocinética de nilotinib. La administración de una dosis única de 200 mg de nilotinib provocó aumentos del AUC de 35%, 35% y 19% en individuos con insuficiencia hepática leve, moderada y grave, respectivamente, en comparación a un grupo control de individuos con función hepática normal. La Cmax prevista del estado estacionario de nilotinib mostró un aumento de 29%, 18% y 22%, respectivamente. Los ensayos clínicos han excluido pacientes con alanino transaminasa (ALT) y/o aspartato transaminasa (AST) >2,5 veces (ó >5, si está relacionado con la enfermedad) al límite superior del intervalo normal y/o bilirrubina total >1,5 veces al límite superior del intervalo normal. El metabolismo de nilotinib es principalmente hepático. Por lo tanto, los pacientes con insuficiencia hepática podrían presentar un aumento de la exposición a nilotinib y deberán tratarse con precaución (ver sección 4.2).
Lipasa sérica
Se han observado elevaciones de la lipasa sérica. Se recomienda precaución en pacientes con antecedentes de pancreatitis. En caso de que las elevaciones de la lipasa estén acompañadas por síntomas abdominales, deberá interrumpirse el tratamiento con Tasigna y deberán considerarse medidas diagnósticas adecuadas para excluir la presencia de pancreatitis.
Gastrectomía total
La biodisponibilidad de nilotinib puede reducirse en pacientes con gastrectomía total (ver sección 5.2). Debe considerarse un seguimiento más frecuente de estos pacientes.
Síndrome de lisis tumoral
Antes de iniciar el tratamiento con Tasigna, se recomienda la corrección de la deshidratación clínicamente significativa y el tratamiento de los niveles altos de ácido úrico, debido a la posible aparición del síndrome de lisis tumoral (SLT) (ver sección 4.8).
Lactosa
Las cápsulas duras de Tasigna contienen lactosa. Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp o problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Tasigna se puede administrar en combinación con factores de crecimiento hematopoyético como eritropoyetina o factor estimulante de las colonias de granulocitos (G-CSF), si está indicado clínicamente. Se puede administrar con hidroxiurea o anagrelida si está clínicamente indicado.
Nilotinib se metaboliza principalmente en el hígado y también es un sustrato de la glicoproteína-P (gp-P), una bomba de eflujo de múltiples fármacos. Por lo tanto, la absorción y subsiguiente eliminación de nilotinib absorbido sistémicamente, pueden verse influenciadas por sustancias que afecten a CYP3A4 y/o a gp-P.
Sustancias que pueden aumentar las concentraciones plasmáticas de nilotinib
La administración conjunta de nilotinib con imatinib (un sustrato e inhibidor de gp-P y de CYP3A4), mostró un ligero efecto inhibidor sobre CYP3A4 y/o gp-P. El AUC de imatinib aumentó entre el 18% y el 39%, y el AUC de nilotinib aumentó entre el 18% y el 40%. Es poco probable que estos cambios sean clínicamente importantes.
La exposición a nilotinib en sujetos sanos aumentó 3 veces cuando se administró conjuntamente con ketoconazol, un inhibidor potente de CYP3A4. Por lo tanto, deberá evitarse el tratamiento concomitante con inhibidores potentes de CYP3A4, incluyendo ketoconazol, itraconazol, voriconazol, ritonavir, claritromicina y telitromicina (ver sección 4.4). También podría esperarse un aumento en la exposición a nilotinib con inhibidores moderados de CYP3A4. Deberán considerarse medicamentos concomitantes alternativos sin inhibición o con una mínima inhibición de CYP3A4.
Sustancias que pueden reducir las concentraciones plasmáticas de nilotinib
La rifampicina, un inductor potente del CYP3A4 disminuye la Cmax de nilotinib un 64% y reduce el AUC de nilotinib un 80%. No deben administrarse conjuntamente rifampicina y nilotinib.
La administración concomitante de otros medicamentos que inducen CYP3A4 (p.ej. fenitoina, carbamazepina, fenobarbital e hierba de San Juan) es asimismo probable que reduzca la exposición a nilotinib a un grado clínicamente relevante. En pacientes para los cuales están indicados los inductores de CYP3A4, deberán utilizarse agentes alternativos con menor potencial de inducción enzimática.
Nilotinib tiene una solubilidad dependiente del pH, con una menor solubilidad a un pH más alto. En individuos sanos que tomaron 40 mg de esomeprazol una vez al día durante 5 días, el pH gástrico aumentó significativamente, pero la absorción de nilotinib sólo disminuyó de forma discreta (disminución de un 27% de la Cmax y disminución de un 34% del AUC0-<»). Nilotinib puede utilizarse de forma concomitante con esomeprazol y otros inhibidores de la bomba de protones, en caso necesario.
En un estudio con sujetos sanos, no se observó ningún cambio significativo en la farmacocinética de nilotinib cuando se administró una dosis única de 400 mg de Tasigna 10 horas después y 2 horas antes de famotidina. Por lo tanto, cuando es necesario el uso concomitante de un bloqueador H2, se puede administrar aproximadamente 10 horas antes y aproximadamente 2 horas después de la dosis de Tasigna.
En el mismo estudio anterior, la administración de un antiácido (hidróxido de aluminio/hidróxido de magnesio/simeticona) 2 horas antes o después de una dosis única de 400 mg de Tasigna tampoco alteró la farmacocinética de nilotinib. Por lo tanto, si es necesario, se puede administrar un antiácido aproximadamente 2 horas antes o aproximadamente 2 horas después de la dosis de Tasigna.
Sustancias cuya concentración plasmática puede verse alterada por nilotinib
In vitro, nilotinib es un inhibidor relativamente potente de CYP3A4, CYP2C8, CYP2C9, CYP2D6 y UGT1A1, con un valor Ki inferior para CYP2C9 (Ki=0,13 microM).
Un estudio de interacción fármaco-fármaco de dosis única en voluntarios sanos con 25 mg de warfarina, un sustrato sensible a CYP2C9, y 800 mg de nilotinib, no dio como resultado ningún cambio en los parámetros farmacocinéticos o farmacodinámicos de la warfarina, medidos como tiempo de protrombina (PT) e índice normalizado internacional (INR). No existen datos en estado estacionario. Este estudio sugiere que una interacción fármaco-fármaco clínicamente significativa entre nilotinib y warfarina es menos probable hasta una dosis de 25 mg de warfarina. Debido a la falta de datos en estado estacionario, se recomienda el control de los marcadores farmacodinámicos de la warfarina (INR o PT) tras el inicio del tratamiento con nilotinib (como mínimo durante las 2 primeras semanas).
En pacientes con LMC, la administración de 400 mg de nilotinib dos veces al día durante 12 días aumentó la exposición sistémica (AUC y Cmax) de midazolam oral (un sustrato del CYP3A4) 2,6 y 2,0 veces, respectivamente. Nilotinib es un inhibidor moderado del CYP3A4. Como resultado, la exposición sistémica de otros fármacos metabolizados principalmente por el CYP3A4 (p.ej. algunos inhibidores de la HMG-CoA reductasa) puede verse aumentada cuando se administren conjuntamente con nilotinib. Una monitorización apropiada y un ajuste de dosis pueden ser necesarios para fármacos que son sustratos del CYP3A4 y que tienen un estrecho margen terapéutico (incluyendo, pero no limitado a alfentanilo, ciclosporina, dihidroergotamina, ergotamina, fentanilo, sirolimus y tacrolimus) cuando se administran conjuntamente con nilotinib.
Medicamentos antiarrítmicos y otras sustancias que pueden prolongar el intervalo QT Nilotinib debe utilizarse con precaución en pacientes que tienen o pueden desarrollar una prolongación del intervalo QT, incluyendo aquellos pacientes en tratamiento con medicamentos antiarrítmicos como amiodarona, disopiramida, procainamida, quinidina y sotalol y otros medicamentos que pueden causar una prolongación del intervalo QT como cloroquina, halofantrina, claritromicina, haloperidol, metadona y moxifloxacina (ver sección 4.4).
Interacciones con alimentos
La absorción y biodisponibilidad de Tasigna aumentan cuando se toma con alimentos, provocando una concentración plasmática más elevada (ver secciones 4.2, 4.4 y 5.2). Debe evitarse tomar zumo de pomelo y otros alimentos que se sabe que son inhibidores de CYP3A4.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar métodos anticonceptivos altamente efectivos durante el tratamiento con Tasigna y hasta dos semanas después de finalizar el tratamiento.
Embarazo
No hay datos o éstos son limitados relativos al uso de nilotinib en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3). Tasigna no debería utilizarse durante el embarazo excepto si la situación clínica de la mujer requiere tratamiento con nilotinib. Si se utiliza durante el embarazo, la paciente debe estar informada del posible riesgo sobre el feto.
Lactancia
Se desconoce si nilotinib se excreta en la leche materna. Los datos toxicológicos disponibles en animales muestran que nilotinib se excreta en la leche (ver sección 5.3). No se puede excluir el riesgo en recién nacidos/lactantes. Tasigna no debe utilizarse durante la lactancia.
Fertilidad
Los estudios en animales no mostraron un efecto sobre la fertilidad en ratas machos y hembras (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Los pacientes que sufran mareos, fatiga, alteraciones de la vista u otros efectos adversos con un posible impacto sobre la capacidad para conducir o utilizar máquinas de forma segura, deberán abstenerse de realizar estas actividades mientras se mantengan estos efectos adversos (ver sección 4.8).
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los datos descritos a continuación reflejan la exposición a Tasigna de 279 pacientes de un ensayo de Fase III aleatorizado en pacientes con LMC Ph+ en fase crónica de nuevo diagnóstico tratados con 300 mg de nilotinib dos veces al día. La duración mediana de la exposición fue de 60,5 meses (intervalo 0,1-70,8 meses).
Las reacciones adversas no hematológicas más frecuentes (>10%) fueron erupción, prurito, cefalea, náuseas, fatiga, alopecia, mialgia y dolor abdominal superior. La mayoría de estas reacciones adversas fueron de intensidad leve a moderada. Se observaron de forma menos frecuente (<10% y >5%) estreñimiento, sequedad de la piel, astenia, espasmos musculares, diarrea, artralgia, dolor abdominal, vómitos y edema periférico, fueron de una intensidad de leve a moderada, manejables y generalmente no requirieron una reducción de la dosis.
La toxicidad hematológica derivada del tratamiento incluyó mielosupresión: trombocitopenia (18%), neutropenia (15%) y anemia (8%). Las reacciones adversas bioquímicas incluyeron aumento de la alanina aminotransferasa (24%), hiperbilirrubinemia (16%), aumento de la aspartato aminotransferasa (12%), aumento de la lipasa (11%), aumento de la bilirrubina sanguínea (10%), hiperglucemia (4%), hipercolesterolemia (3%) e hipertrigliceridemia (<1%). Se observó derrame pleural y pericárdico, independientemente de la causalidad, en un 2% y en <1% de pacientes, respectivamente, en tratamiento con Tasigna 300 mg dos veces al día. Se notificó hemorragia gastrointestinal, independientemente de la causalidad, en un 3% de estos pacientes.
El cambio en el tiempo medio del intervalo QTcF respecto al valor basal en el estado estacionario fue de 6 ms. Ningún paciente presentó un QTcF absoluto >500 ms mientras recibieron medicación en el estudio. En <1% de los pacientes se observó un aumento de QTcF respecto al valor basal que superó los 60 ms, mientras recibieron medicación en el estudio. No se observaron casos de muerte súbita ni episodios de «Torsades de Pointes» (transitorios o sostenidos). No se observó una disminución del valor medio de la fracción de eyección ventricular respecto al valor basal (FEVI) en ningún momento durante el tratamiento. Ningún paciente presentó un valor de FEVI <45% durante el tratamiento ni una reducción absoluta del valor de FEVI de más de 15%.
Se observó una interrupción del tratamiento debida a reacciones adversas en un 10% de los pacientes.
Reacciones adversas notificadas de forma más frecuente en los ensayos clínicos con Tasigna En la Tabla 2 se muestran las reacciones adversas no hematológicas (excepto los valores de laboratorio anormales) que se notificaron en al menos el 5% de los pacientes tratados con 300 mg de nilotinib dos veces al día en el ensayo de Fase III aleatorizado. Estas se ordenan por frecuencias, apareciendo primero las más frecuentes, utilizando una precisión decimal para porcentajes y la siguiente convención: muy frecuentes (>1/10) o frecuentes (>1/100 a <1/10).Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Tabla 2 Reacciones adversas no hematológicas (>5% de todos los pacientes)*
|
Clasificación de órganos del sistema |
Frecuencia |
Reacción adversa |
Todos los grados % |
Grado 3-4 % |
|
Trastornos del sistema nervioso |
Muy frecuentes |
Cefalea |
16 |
2 |
|
Trastornos gastrointestinales |
Muy frecuentes |
Náusea |
14 |
<1 |
|
Muy frecuentes |
Dolor abdominal superior |
10 |
1 | |
|
Frecuentes |
Estreñimiento |
10 |
0 | |
|
Frecuentes |
Diarrea |
9 |
<1 | |
|
Frecuentes |
Dolor abdominal |
6 |
0 | |
|
Frecuentes |
Vómitos |
6 |
0 | |
|
Frecuentes |
Dispepsia |
5 |
0 | |
|
Trastornos de la piel y del tejido subcutáneo |
Muy frecuentes |
Erupción |
33 |
<1 |
|
Muy frecuentes |
Prurito |
18 |
<1 | |
|
Muy frecuentes |
Alopecia |
10 |
0 | |
|
Frecuentes |
Sequedad de la piel |
10 |
0 | |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Muy frecuentes |
Mialgia |
10 |
<1 |
|
Frecuentes |
Espasmos musculares |
9 |
0 | |
|
Frecuentes |
Artralgia |
8 |
<1 | |
|
Frecuentes |
Dolor en las extremidades |
5 |
<1 | |
|
Trastornos generales y alteraciones en el lugar de administración |
Muy frecuentes |
Fatiga |
12 |
0 |
|
Frecuentes |
Astenia |
9 |
<1 | |
|
Frecuentes |
Edema periférico |
5 |
<1 |
*Los porcentajes están redondeados a números enteros para la presentación en esta tabla. Sin embargo, los porcentajes con precisión de un decimal se utilizan para identificar términos con una frecuencia de como mínimo 5% y para clasificar los términos de acuerdo a las categorías de frecuencia.
Las siguientes reacciones adversas se notificaron en el ensayo de Fase III con Tasigna con una frecuencia menor del 5%. También se notifican los valores anormales de laboratorio muy frecuentes (>1/10), no incluidos en la Tabla 2. Estas reacciones adversas se incluyen basadas en la relevancia clínica y se ordenan en orden decreciente de gravedad dentro de cada categoría, utilizando la siguiente convención: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Infecciones e infestaciones:
Frecuentes: foliculitis, infección del tracto respiratorio superior (incluyendo faringitis, nasofaringitis, rinitis).
Frecuencia no conocida: infección por el virus del herpes, candidiasis oral, absceso subcutáneo, absceso anal, tinea pedis, reactivación del virus de la hepatitis B.
Neoplasias benignas, malignas y no especificadas (incluyendo quistes y pólipos) :
Frecuentes: papiloma en la piel.
Frecuencia no conocida: papiloma oral, paraproteinemia.
Trastornos de la sangre y del sistema linfático:
Frecuentes: leucopenia, eosinofilia, linfopenia.
Poco frecuentes: pancitopenia.
Frecuencia no conocida: neutropenia febril.
Trastornos del sistema inmunológico:
Frecuencia no conocida: hipersensibilidad.
Trastornos endocrinos:
Frecuencia no conocida: hiperparatiroidismo secundario.
Trastornos del metabolismo y de la nutrición:
Muy frecuentes: hipofosfatemia (incluyendo disminución del fósforo en la sangre).
Frecuentes: diabetes mellitus, hipercolesterolemia, hiperlipidemia, hipertrigliceridemia, hiperglucemia, disminución del apetito, hipocalcemia, hipopotasemia.
Poco frecuentes: hiperpotasemia, dislipidemia, gota.
Frecuencia no conocida: hiperuricemia, hipoglucemia, alteraciones en el apetito.
Trastornos psiquiátricos:
Frecuentes: insomnio, depresión, ansiedad.
Frecuencia no conocida: amnesia, disforia.
Trastornos del sistema nervioso:
Frecuentes: mareo, hipoestesia, neuropatía periférica.
Poco frecuentes: ictus isquémico, infarto cerebral, migraña, parestesia.
Frecuencia no conocida: accidente cerebrovascular, estenosis de la arteria basilar, síncope, temblor, letargia, disestesia, síndrome de las piernas inquietas, hiperestesia.
Trastornos oculares:
Frecuentes: prurito ocular, conjuntivitis, sequedad ocular (incluyendo xeroftalmia).
Poco frecuentes: edema palpebral, fotopsia, hemorragia conjuntival, hiperemia (escleral, conjuntival, ocular).
Frecuencia no conocida: edema periorbital, blefaritis, dolor ocular, corioretinopatía, conjuntivitis alérgica, enfermedad de la superficie ocular, visión borrosa.
Trastornos del oído y del laberinto:
Frecuentes: vértigo.
Trastornos cardiacos *:
Frecuentes: angina de pecho, arritmia (incluyendo bloqueo atrioventricular, taquicardia, fibrilación auricular, extrasístoles ventriculares, bradicardia), QT prolongado en el electrocardiograma, palpitaciones, infarto de miocardio.
Poco frecuentes: insuficiencia cardiaca, cianosis.
Frecuencia no conocida: disminución de la fracción de eyección, derrame pericárdico, pericarditis, disfunción diastólica, bloqueo de rama izquierda.
*notificados en el brazo de tratamiento de 300 mg dos veces al día y/o 400 mg dos veces al día del ensayo Fase III
Trastornos vasculares:
Frecuentes: hipertensión, sofocos.
Poco frecuentes: claudicación intermitente, enfermedad arterial oclusiva periférica, arteriosclerosis. Frecuencia no conocida: hematoma, estenosis arterial periférica.
Trastornos respiratorios, torácicos y mediastínicos:
Frecuentes: disnea, tos.
Poco frecuentes: derrame pleural.
Frecuencia no conocida: disnea por esfuerzo, pleuresía, epistaxis, dolor orofaríngeo.
Trastornos gastrointestinales:
Frecuentes: distensión abdominal, molestias abdominales, disgeusia, flatulencia.
Poco frecuentes: pancreatitis, gastritis, sensibilidad en los dientes.
Frecuencia no conocida: úlcera esofágica, úlcera gástrica, dolor esofágico, estomatitis, sequedad de la boca, enterocolitis, hemorroides, hernia de hiato, hemorragia rectal, gingivitis.
Trastornos hepatobiliares:
Muy frecuentes: hiperbilirrubinemia (incluyendo aumento de la bilirrubina en la sangre).
Frecuentes: función hepática anormal.
Poco frecuentes: ictericia.
Frecuencia no conocida: hepatitis tóxica.
Trastornos de la piel y del tejido subcutáneo:
Frecuentes: eritema, hiperhidrosis, contusión, acné, dermatitis (incluyendo alérgica, exfoliativa y acneiforme), sudores nocturnos, eczema.
Poco frecuentes: erupción debida al medicamento, dolor en la piel.
Frecuencia no conocida: eritema multiforme, urticaria, ampollas, quistes dérmicos, hiperplasia sebácea, hinchazón en la cara, atrofia de la piel, hipertrofia de la piel, exfoliación de la piel, hiperpigmentación de la piel, decoloración de la piel, hiperqueratosis, psoriasis.
Trastornos musculoesqueléticos y del tejido conjuntivo:
Frecuentes: dolor óseo, dolor de espalda, debilidad muscular.
Poco frecuentes: dolor musculoesquelético, dolor en el costado.
Trastornos renales y urinarios:
Frecuencia no conocida: disuria, polaquiuria, cromaturia.
Trastornos del aparato reproductor y de la mama:
Poco frecuentes: disfunción eréctil.
Frecuencia no conocida: ginecomastia, induración de las mamas, menorragia, hinchazón en el pezón.
Trastornos generales y alteraciones en el lugar de administración:
Frecuentes: pirexia, dolor torácico (incluyendo dolor torácico no cardiaco), molestia torácica.
Poco frecuentes: dolor, escalofríos, sensación de cambio en la temperatura corporal (incluyendo sensación de calor y de frío), malestar.
Frecuencia no conocida: edema facial, edema localizado.
Exploraciones complementarias:
Muy frecuentes: aumento de la alanina aminotransferasa, aumento de aspartato aminotransferasa, aumento de lipasa, aumento del colesterol de lipoproteínas (incluyendo de baja densidad y de alta densidad), aumento del colesterol total, aumento de los triglicéridos sanguíneos.
Frecuentes: disminución de la hemoglobina, aumento de la amilasa sanguínea, aumento de la fosfatasa alcalina sanguínea, aumento de la gamma-glutamiltransferasa, aumento de peso, aumento de la insulina sanguínea, disminución de globulinas.
Frecuencia no conocida: aumento de la hormona paratiroidea sanguínea, disminución de la insulina sanguínea, disminución del péptido C de insulina, disminución de peso.
En la Tabla 3 se presentan los valores de laboratorio anormales clínicamente relevantes o graves encontrados en los controles hematológicos o bioquímicos rutinarios.
Tabla 3 Valores de laboratorio anormales de grado 3-4*
|
n=279 (%) | |
|
Parámetros hematológicos | |
|
Mielosupresión | |
|
- Neutropenia |
12 |
|
- Trombocitopenia |
10 |
|
- Anemia |
4 |
|
Parámetros bioquímicos | |
|
- Creatinina elevada |
0 |
|
- Lipasa elevada |
9 |
|
- SGOT (AST) elevada |
1 |
|
- SGPT (ALT) elevada |
4 |
|
- Hipofosfatemia |
7 |
|
- Bilirrubina elevada (total) |
4 |
|
- Glucosa elevada |
7 |
|
- Colesterol elevado (total) |
0 |
|
- Triglicéridos elevados |
0 |
* Se utilizan porcentajes con precisión de un decimal que están redondeados a números enteros para la presentación en esta tabla.
Descripción de las reacciones adversas seleccionadas
Reactivación de la hepatitis B
Se ha notificado reactivación de la hepatitis B en relación con los inhibidores de la tirosina quinasa BCR-ABL. En algunos casos se ha producido insuficiencia hepática aguda o hepatitis fulminante que ha dado lugar a trasplante de hígado o a un desenlace mortal (ver sección 4.4).
Experiencia post-comercialización
Las siguientes reacciones adversas se han obtenido a partir de la experiencia post-comercialización con Tasigna a través de informes de casos espontáneos, casos descritos en la literatura, programas de acceso expandido, y ensayos clínicos diferentes de los ensayos globales para el registro. Puesto que estas reacciones se han notificado voluntariamente a partir de una población de un tamaño incierto, no siempre es posible estimar de forma fiable o establecer una relación causal con la exposición a nilotinib.
Frecuencia rara: Se han notificado casos de síndrome de lisis tumoral en pacientes tratados con Tasigna.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se han notificado casos aislados de sobredosis intencionada con nilotinib, en que se ingirieron un número no especificado de cápsulas duras de Tasigna combinadas con alcohol y con otros medicamentos. Los acontecimientos incluyeron neutropenia, vómitos y somnolencia. No se notificaron cambios en el ECG o hepatotoxicidad. Las resoluciones de los casos se notificaron como recuperados.
En caso de sobredosis, se deberá mantener al paciente en observación y administrarle el tratamiento de apoyo adecuado.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes antineoplásicos, inhibidores de la proteína quinasa, código ATC: L01XE08
Nilotinib es un inhibidor potente de la actividad de la tirosina cinasa ABL de la oncoproteína BCR-ABL tanto en las líneas celulares como en las células leucémicas primarias cromosoma Filadelfia positivo. La sustancia se une con alta afinidad al lugar de unión del ATP de tal manera que es un inhibidor potente del BCR-ABL de tipo germinal y mantiene la actividad frente a 32/33 formas mutantes de BCR-ABL resistentes a imatinib. Como consecuencia de esta actividad bioquímica, nilotinib inhibe de forma selectiva la proliferación e induce la apoptosis en líneas celulares y en células leucémicas primarias cromosoma Filadelfia positivo, de pacientes con LMC. En modelos de LMC en roedores, nilotinib como agente único reduce la carga del tumor y prolonga la supervivencia después de la administración oral.
Nilotinib tiene poco o ningún efecto sobre la mayoría de las otras proteínas cinasas examinadas, incluyendo Src, excepto para las cinasas de los receptores PDGF, KIT y Ephin, a las que inhibe a concentraciones dentro del intervalo alcanzado tras la administración oral a las dosis terapéuticas recomendadas para el tratamiento de la LMC (ver Tabla 4).
Tabla 4 Perfil de cinasas de nilotinib (fosforilación IC50 nM)
|
BCR-ABL |
PDGFR |
KIT |
|
20 |
69 |
210 |
Ensayos clínicos en LMC de nuevo diagnóstico en fase crónica
Se ha realizado un ensayo Fase III abierto, multicéntrico, aleatorizado para determinar la eficacia de nilotinib frente imatinib en 846 pacientes adultos con LMC cromosoma Filadelfia positivo en fase crónica confirmada citogenéticamente. Los pacientes se encontraban dentro de los seis meses posteriores al diagnóstico y no habían recibido tratamientos anteriores, excepto hidroxiurea y/o anagrelida. Los pacientes fueron aleatorizados 1:1:1 para recibir tratamiento con nilotinib 300 mg dos veces al día (n=282), nilotinib 400 mg dos veces al día (n=281) o imatinib 400 mg una vez al día (n=283). La aleatorización se estratificó por el índice de riesgo Sokal en el momento del diagnóstico.
Las características basales estaban equilibradas entre los tres brazos de tratamiento. La mediana de edad fue de 47 años en los dos brazos de nilotinib y de 46 años en el brazo de imatinib, con un porcentaje de pacientes con una edad >65 años de 12,8%, 10,0% y 12,4% en el grupo de nilotinib 300 mg dos veces al día, de nilotinib 400 mg dos veces al día y de imatinib 400 mg una vez al día, respectivamente. Se incluyeron un número ligeramente superior de pacientes hombres que de mujeres (56,0%, 62,3% y 55,8% en el brazo de nilotinib 300 mg dos veces al día, 400 mg dos veces al día y de imatinib 400 mg una vez al día, respectivamente). Más del 60% de todos los pacientes eran caucásicos y 25% de todos los pacientes eran asiáticos.
El punto para el análisis primario de datos fue cuando todos los 846 pacientes completaron 12 meses de tratamiento (o interrumpieron el tratamiento antes). Los análisis siguientes reflejan datos de cuando los pacientes completaron 24, 36, 48, 60 y 72 meses de tratamiento (o interrumpieron el tratamiento antes). La mediana de tiempo de tratamiento fue aproximadamente de 70 meses en los grupos de tratamiento con nilotinib y 64 meses en el grupo de imatinib. La mediana de las dosis realmente administradas fue de 593 mg/día para nilotinib 300 mg dos veces al día, de 772 mg/día para nilotinib 400 mg dos veces al día y de 400 mg/día para imatinib 400 mg una vez al día. Este estudio continua en marcha.
La variable de eficacia primaria fue la respuesta molecular mayor (RMM) a los 12 meses. La RMM se definió como <0,1% BCR-ABL/ABL% por una escala internacional (EI) medida por RQ-PCR, que corresponde a una reducción de >3 log del tránscrito BCR-ABL respecto al valor basal estandarizado. La tasa de RMM a los 12 meses fue superior de forma estadísticamente significativa para nilotinib 300 mg dos veces al día que para imatinib 400 mg una vez al día (44,3% frente a 22,3%, p<0,0001). La tasa de RMM a los 12 meses también fue superior de forma estadísticamente significativa para nilotinib 400 mg dos veces al día que para imatinib 400 mg una vez al día (42,7% frente a 22,3%,
p<0,0001).
Las tasas de RMM a 3, 6, 9 y 12 meses fueron 8,9%, 33,0%, 43,3% y 44,3% para nilotinib 300 mg dos veces al día, 5,0%, 29,5%, 38,1% y 42,7% para nilotinib 400 mg dos veces al día y 0,7%, 12,0%, 18,0% y 22,3% para imatinib 400 mg una vez al día.
En la Tabla 5 se presenta la tasa de RMM a los 12, 24, 36, 48, 60 y 72 meses.
Tabla 5 Tasa de RMM
|
Tasigna 300 mg dos veces al día n=282 (%) |
Tasigna 400 mg dos veces al día n=281 (%) |
Imatinib 400 mg una vez al día n=283 (%) | |
|
RMM a los 12 meses | |||
|
Respuesta (IC 95%) |
44,3' (38,4; 50,3) |
42J1 (36,8; 48,7) |
22,3 (17,6; 27,6) |
|
RMM a los 24 meses | |||
|
Respuesta (IC 95%) |
61,7' (55,8; 67,4) |
59,1 (53,1; 64,9) |
37,5 (31,8; 43,4) |
|
RMM a los 36 meses2 | |||
|
Respuesta (IC 95%) |
58,54 (52,5; 64,3) |
57,3: (51,3; 63,2) |
38,5 (32,8; 44,5) |
|
RMM a los 48 meses3 | |||
|
Respuesta (IC 95%) |
59,9: (54,0; 65,7) |
55,2 (49,1; 61,1) |
43,8 (38,0; 49,8) |
|
RMM a los 60 meses4 | |||
|
Respuesta (IC 95%) |
62,8 (56,8; 68,4) |
61,2 (55,2; 66,9) |
49,1 (43,2; 55,1) |
|
RMM a los 72 meses5 | |||
|
Respuesta (IC 95%) |
52,5 (46,5; 58,4) |
57,7 (51,6; 63,5) |
41,7 (35,9; 47,7) |
1 Test de Cochran-Mantel-Haenszel (CMH) valor de p para la tasa de respuesta (frente a imatinib
400 mg) <0,0001
2 Sólo se incluyeron los pacientes que estaban en RMM en un punto de tiempo específico como respondedores de este punto de tiempo. Un total de 199 de todos los pacientes (35,2%) no fueron evaluables para la RMM a los 36 meses (87 en el grupo de nilotinib 300 mg dos veces al día y 112 en el grupo de imatinib) debido a unas valoraciones de PCR perdidas/no evaluables (n=17), tránscritos atípicos en el inicio (n=7), o interrupción anterior a los 36 meses (n=175).
3 Sólo se incluyeron pacientes que estaban en RMM en un punto de tiempo específico como respondedores para aquel punto de tiempo. Un total de 305 de todos los pacientes (36,1%) no fueron evaluables para RMM a los 48 meses (98 en el grupo de nilotinib 300 mg dos veces al día, 88 en el grupo de nilotinib 400 mg dos veces al día y 119 en el grupo de imatinib) debido a las valoraciones de PCR no disponibles/no evaluables (n=18), tránscritos atípicos en el punto basal (n=8), o interrupción anterior a los 48 meses (n=279).
4 Sólo se incluyeron pacientes que estaban en RMM en un punto de tiempo específico como respondedores para aquel punto en el tiempo. Un total de 322 (38,1%) de todos los pacientes no fueron evaluables para RMM a los 60 meses (99 en el grupo de nilotinib 300 mg dos veces al día, 93 en el grupo de nilotinib 400 mg dos veces al día y 130 en el grupo de imatinib) debido a las valoraciones de PCR no disponibles/no evaluables (n=9), tránscritos atípicos en el punto basal (n=8) o interrupción anterior a los 60 meses (n=305).
5 Sólo se incluyeron pacientes que estaban en RMM en un punto de tiempo específico como respondedores para aquel punto en el tiempo. Un total de 395 de todos los pacientes (46,7%) no fueron evaluables para RMM a los 72 meses (130 en el grupo de nilotinib 300 mg dos veces al día,
110 en el grupo de nilotinib 400 mg dos veces al día y 155 en el grupo de imatinib) debido a las valoraciones de PCR no disponibles/no evaluables (n=25), transcritos atípicos en el punto basal (n=8) o interrupción anterior a los 72 meses (n=362).
Se presentan las tasas de RMM en diferentes puntos de tiempo (incluyendo pacientes que alcanzaron la RMM en estos puntos de tiempo o antes como respondedores) en la incidencia acumulada de RMM (ver Figura 1).
Figura 1 Incidencia acumulada de RMM
incidencia acumulada de RMM, %
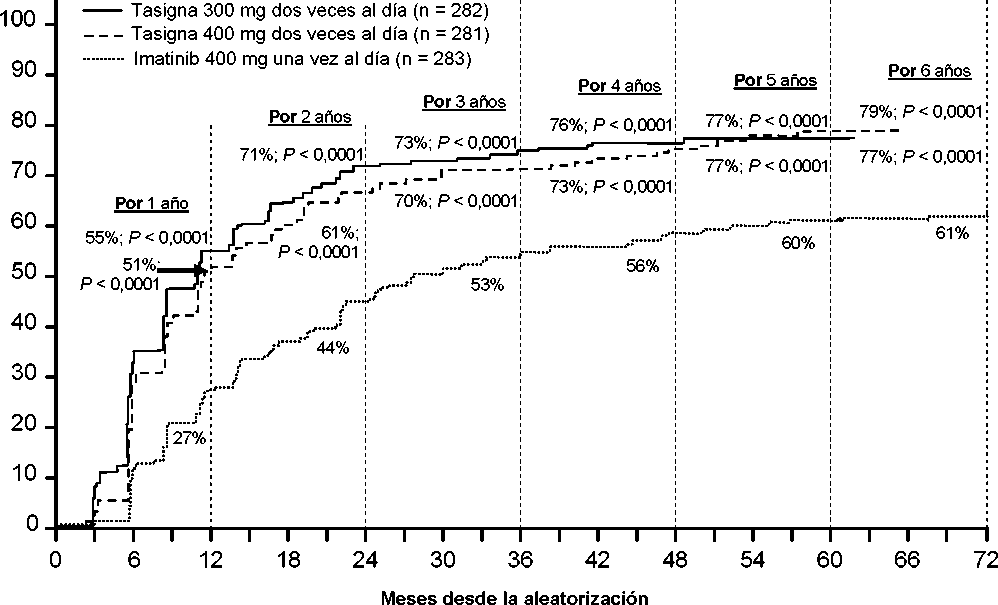
Para todos los grupos de riesgo Sokal, las tasas de RMM en todos los puntos de tiempo se mantuvieron de forma constante superiores en los dos grupos de nilotinib respecto al grupo de imatinib.
En un análisis retrospectivo, el 91% de los pacientes (234/258) tratados con nilotinib 300 mg dos veces al día alcanzaron los niveles de BCR-ABL <10% a los 3 meses de tratamiento comparado al 67% de los pacientes (176/264) tratados con imatinib 400 mg una vez al día. Los pacientes con niveles de BCR-ABL <10% a los 3 meses de tratamiento mostraron una supervivencia global a los 72 meses comparado a los que no alcanzaron este nivel de respuesta molecular (94,5% frente a 77,1% respectivamente [p=0,0005].
En base al análisis de Kaplan-Meier del tiempo hasta la primera RMM la probabilidad de alcanzar una RMM a diferentes puntos de tiempo fue superior para ambos grupos de nilotinib a 300 mg y 400 mg dos veces al día que para imatinib 400 mg una vez al día (HR=2,17, y orden logarímico (log-rank) estratificado p<0,0001 entre nilotinib 300 mg dos veces al día e imatinib 400 mg una vez al día, HR=1,88 y orden logarítmico estratificado p<0,0001 entre nilotinib 400 mg dos veces al día e imatinib 400 mg una vez al día).
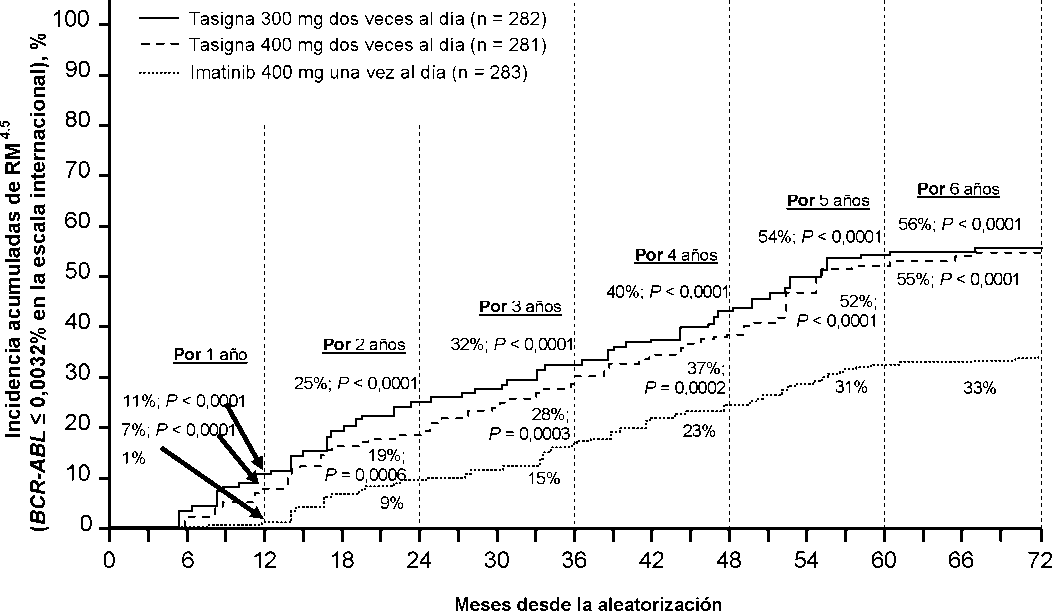
La proporción de pacientes que presentaron una respuesta molecular de <0,01% y <0,0032% por EI a diferentes puntos de tiempo se presentan en la Tabla 6 y la proporción de pacientes que presentaron una respuesta molecular de <0,01% y <0,0032% por EI por diferentes puntos de tiempo se presentan en las Figuras 2 y 3. Las respuestas moleculares de <0,01% y <0,0032% por EI corresponde a una reducción de >4 log y >4,5 log respectivamente de los tránscritos de BCR-ABL a partir del valor basal.
Tabla 6 Proporciones de pacientes que presentaron respuesta molecular de <0,01% (reducción de 4 log) y <0,0032% (reducción de 4,5 log)
|
Ta 300 mg do n= |
signa s veces al día 282 '%) |
Ta 400 mg do n= |
signa s veces al día =281 '%) |
Im 400 mg u n= |
atinib na vez al día =283 í%) | |
|
<0,01% |
<0,0032% |
<0,01% |
< 0,0032% |
<0,01% |
<0,0032% | |
|
A los 12 meses |
11,7 |
4,3 |
8,5 |
4,6 |
3,9 |
0,4 |
|
A los 24 meses |
24,5 |
12,4 |
22,1 |
7,8 |
10,2 |
2,8 |
|
A los 36 meses |
29,4 |
13,8 |
23,8 |
12,1 |
14,1 |
8,1 |
|
A los 48 meses |
33,0 |
16,3 |
29,9 |
17,1 |
19,8 |
10,2 |
|
A los 60 meses |
47,9 |
32,3 |
43,4 |
29,5 |
31,1 |
19,8 |
|
A los 72 meses |
44,3 |
31,2 |
45,2 |
28,8 |
27,2 |
18,0 |
Figura 2 Incidencia acumulada de respuesta molecular de <0,01% (reducción de 4-log)
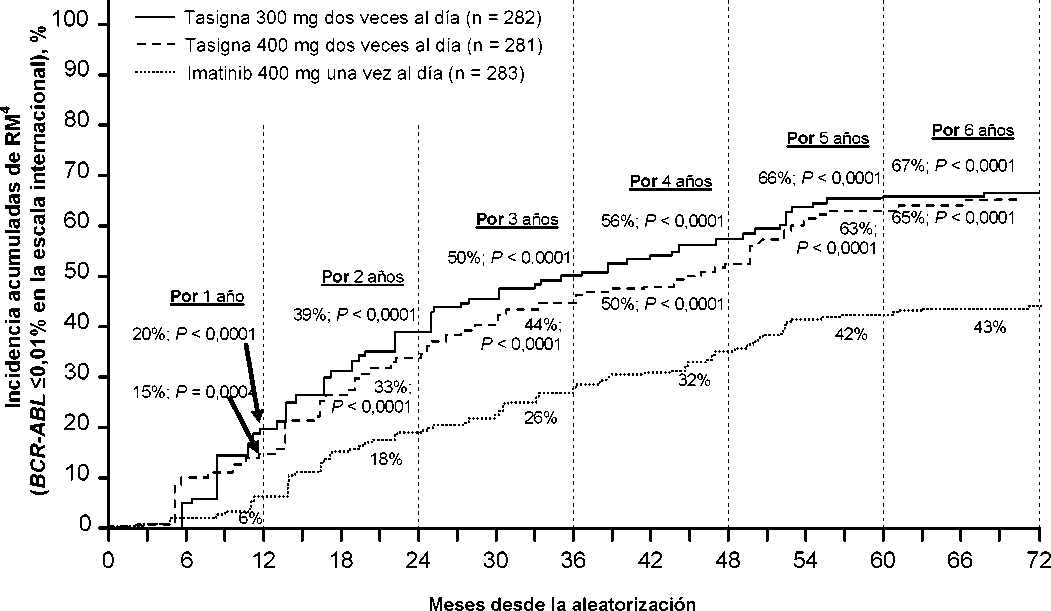
Figura 3 Incidencia acumulada de respuesta molecular de <0,0032% (reducción de 4,5 log)
En base a la estimación de Kaplan-Meier de la duración de la primera RMM, la proporción de pacientes que mantuvieron la respuesta durante 72 meses entre los pacientes que alcanzaron una RMM fueron 92,5% (IC 95: 88,6-96,4%) en el grupo de nilotinib 300 mg dos veces al día, 92,2% (IC 95%: 88,5-95,9%) en el grupo de nilotinib 400 mg dos veces al día y 88,0% (IC 95%: 83,0-93,1%) en el grupo de imatinib 400 mg una vez al día.
La respuesta citogenética completa (RCC) se definió como un 0% de metafases en la médula ósea basado en un mínimo de 20 metafases evaluadas. La mejor tasa de RCC a los 12 meses (incluyendo pacientes que alcanzaron la RCC a los 12 meses o antes como respondedores) fue estadísticamente superior para ambos grupos de nilotinib 300 mg y 400 mg dos veces al día comparado con imatinib 400 mg una vez al día, ver Tabla 7.
La tasa de RCC hasta los 24 meses (incluye pacientes que alcanzaron RCC a los 24 meses o antes como respondedores) fue superior estadistica y significativamente para ambos grupos nilotinib 300 mg dos veces al día y 400 mg dos veces al día en comparación con el grupo de imatinib 400 mg una vez al día.
Tabla 7 Tasa de mejor respuesta citogenética completa (RCC)
|
Tasigna (nilotinib) 300 mg dos veces al día n=282 (%) |
Tasigna (nilotinib) 400 mg dos veces al día n=281 (%) |
Glivec (imatinib) 400 mg una vez al día n=283 (%) | |
|
Hasta los 12 meses | |||
|
Respuesta (IC 95%) |
80,1 (75,0; 84,6) |
77,9 (72,6; 82,6) |
65,0 (59,2; 70,6) |
|
Sin respuesta |
19,9 |
22,1 |
35,0 |
|
Test CMH valor de p para la tasa de respuesta (frente a imatinib 400 mg una vez al día) |
<0,0001 |
0,0005 | |
|
Hasta los 24 meses | |||
|
Respuesta (IC 95%) |
86,9 (82,4; 90,6) |
84,7 (79,9; 88,7) |
77,0 (71,7; 818) |
|
Sin respuesta |
13,1 |
15,3 |
23,0 |
|
Test CMH valor de p para la tasa de respuesta (frente a imatinib 400 mg una vez al día) |
0,0018 |
0,0160 |
En base a la estimación de Kaplan-Meier, la proporción de pacientes que mantuvieron la respuesta durante 72 meses entre los pacientes que alcanzaron una RCC fueron 99,1% (IC 95%: 97,9-100%) en el grupo de nilotinib 300 mg dos veces al día, 98,7% (IC 95%: 97,1-100%) en el grupo de nilotinib 400 mg dos veces al día y 97,0% (IC 95%: 94,7-99,4%) en el grupo de imatinib 400 mg una vez al día.
La progresión a fase acelerada (FA) o crisis blástica (CB) durante el tratamiento se define como el tiempo desde la fecha de la aleatorización hasta la primera progresión documentada de la enfermedad a fase acelerada o crisis blástica o muerte relacionada con la LMC. Se observó una progresión a fase acelerada o crisis blástica durante el tratamiento en un total de 17 pacientes: 2 pacientes en el grupo de nilotinib 300 mg dos veces al día, 3 pacientes en el grupo de nilotinib 400 mg dos veces al día y 12 pacientes en el grupo de imatinib 400 mg una vez al día. Las tasas estimadas de pacientes sin progresión a fase acelerada o crisis blástica a los 72 meses fueron de 99,3%, 98,7% y 95,2%, respectivamente (HR=0,1599 y orden logarítmico (log-rank) estratificado p=0,0059 entre nilotinib 300 mg dos veces al día e imatinib una vez al día, HR=0,2457 y orden logarítmico (log-rank) estratificado p=0,0185 entre nilotinib 400 mg dos veces al día e imatinib una vez al día). No se notificaron nuevos eventos de progresión a FA/CB durante el tratamiento desde el análisis de los 2 años.
Incluyendo la evolución clonal como criterio de progresión, un total de 25 pacientes progresaron a fase acelerada o crisis blástica durante el tratamiento en la fecha de corte (3 en el grupo de nilotinib 300 mg dos veces al día, 5 en el grupo de nilotinib 400 mg dos veces al día y 17 en el grupo de imatinib 400 mg una vez al día). Las tasas estimadas de pacientes sin progresión a fase acelerada o crisis blástica incluyendo evolución clonal a los 72 meses fueron 98,7%, 97,9% y 93,2%, respectivamente (HR=0,1626 y orden logarítmico (log-rank) estratificado p=0,0009 entre nilotinib 300 mg dos veces al día e imatinib una vez al día, HR=0,2848 y orden logarítmico (log-rank) estratificado p=0,0085 entre nilotinib 400 mg dos veces al día e imatinib una vez al día).
Un total de 55 pacientes fallecieron durante el tratamiento o durante el seguimiento después de la interrupción del tratamiento. (21 en el grupo de nilotinib 300 mg dos veces al día, 11 en el grupo de nilotinib 400 mg dos veces al día y 23 en el grupo de imatinib 400 mg una vez al día). Veintiséis (26) de estas 55 muertes estuvieron relacionadas con la LMC (6 en el grupo de nilotinib 300 mg dos veces al día, 4 en el grupo de nilotinib 400 mg dos veces al día y 16 en el grupo de imatinib 400 mg una vez al día). Las tasas estimadas de pacientes vivos a los 72 meses fueron 91,6%, 95,8% y 91,4%, respectivamente (HR=0,8934 y orden logarítmico (log-rank) estratificado p=0,7085 entre nilotinib 300 mg dos veces al día e imatinib, HR=0,4632 y orden logarítmico (log-rank) estratificado p=0,0314 entre nilotinib 400 mg dos veces al día e imatinib). Considerando sólo las muertes relacionadas con la LMC como eventos, las tasas estimadas de supervivencia global a los 72 meses fueron 97,7%, 98,5% y 93,9%, respectivamente (HR=0,3694 y orden logarítmico (log-rank) estratificado p=0,0302 entre nilotinib 300 mg dos veces al día e imatinib, HR=0,2433 y orden logarítmico (log-rank) estratificado p=0,0061 entre nilotinib 400 mg dos veces al día e imatinib).
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Tasigna en pacientes pediátricos desde el nacimiento hasta menos de 18 años en el tratamiento de la leucemia mieloide crónica cromosoma Filadelfia positivo (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Se alcanzaron concentraciones pico de nilotinib 3 horas después de la administración oral. La absorción de nilotinib después de la administración oral fue de aproximadamente el 30%. No se ha determinado la biodisponibilidad absoluta de nilotinib. Comparado con una solución oral (pH de 1,2 a 1,3), la biodisponibilidad relativa de nilotinib cápsulas es aproximadamente del 50%. En voluntarios sanos, cuando Tasigna se administró junto con la comida aumentaron la Cmax y el area bajo la curva de concentración plasmática-tiempo (AUC) de nilotinib en un 112% y un 82%, respectivamente, en comparación a las condiciones de ayuno. La administración de Tasigna 30 minutos o 2 horas después de la comida aumentó la biodisponibilidad de nilotinib en un 29% o un 15%, respectivamente (ver secciones 4.2, 4.4 y 4.5).
La absorción de nilotinib (biodisponibilidad relativa) puede reducirse en aproximadamente un 48% y un 22% en pacientes con gastrectomía total y gastrectomía parcial, respectivamente.
Distribución
La relación sangre - plasma de nilotinib es 0,71. La unión a proteínas plasmáticas es de aproximadamente el 98% en base a los experimentos in vitro.
Biotransformación
Las principales vías metabólicas identificadas en voluntarios sanos son la oxidación y la hidroxilación. Nilotinib es el principal componente que circula en el plasma. Ninguno de los metabolitos contribuye significativamente a la actividad farmacológica de nilotinib. Nilotinib se metaboliza principalmente por CYP3A4, con una posible contribución menor de CYP2C8.
Eliminación
Tras una dosis única de nilotinib marcado radiactivamente en voluntarios sanos, más del 90% de la dosis se eliminó dentro de los siguientes 7 días, principalmente por las heces (94% de la dosis). Nilotinib inalterado supuso el 69% de la dosis.
La semivida de eliminación aparente estimada a partir de la farmacocinética a dosis múltiples con dosis diarias fue de aproximadamente 17 horas. La variabilidad interpaciente en la farmacocinética de nilotinib fue de moderada a alta.
Linealidad/No linealidad
La exposición a nilotinib en el estado estacionario fue dependiente de la dosis, con aumentos menores a los aumentos proporcionales a la dosis en la exposición sistémica a dosis superiores a 400 mg administrados como única dosis diaria. La exposición sistémica diaria a nilotinib con dosis de 400 mg dos veces al día en el estado estacionario fue un 35% superior que con una dosis de 800 mg una vez al día. La exposición sistémica (AUC) de nilotinib en el estado estacionario a un nivel de dosis de 400 mg dos veces al día fue aproximadamente un 13,4% superior a la de la dosis de 300 mg dos veces al día. La media de concentraciones de nilotinib valle y pico durante 12 meses fueron aproximadamente 15,7% y 14,8% superiores tras la administración de 400 mg dos veces al día comparado con 300 mg dos veces al día. No se observó un aumento relevante en la exposición a nilotinib cuando se aumentó la dosis de 400 mg dos veces al día a 600 mg dos veces al día.
Las condiciones en el estado estacionario se alcanzaron en el día 8. Se observó un aumento en la exposición plasmática a nilotinib entre la primera dosis y el estado estacionario de 2 veces para la dosis diaria y de 3,8 veces para la dosis dos veces al día.
Estudios de biodisponibilidad/bioequivalencia
La administración de una dosis única de 400 mg de nilotinib, utilizando 2 cápsulas duras de 200 mg en los que el contenido de cada cápsula dura se dispersó en una cucharadita de compota de manzana, mostró que era bioequivalente con una administración única de 2 cápsulas duras intactas de 200 mg.
5.3 Datos preclínicos sobre seguridad
Se han realizado estudios con nilotinib para evaluar la farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad, toxicidad reproductiva, fototoxicidad y carcinogenicidad (ratas y ratones).
Nilotinib no mostró efectos sobre las funciones del SNC o respiratorias. Estudios de seguridad cardiaca in vitro mostraron una señal preclínica de prolongación de QT, basadas en el bloqueo de las corrientes hERG y prolongación de la duración del potencial de acción en corazones de conejo aislados, por nilotinib. No se observaron efectos en las medidas del ECG en perros o monos tratados hasta 39 semanas o en un estudio telemétrico especial en perros.
Estudios de toxicidad a dosis repetidas en perros de hasta 4 semanas de duración y en monos cynomolgus de hasta 9 meses de duración mostraron que el órgano diana de toxicidad causada por nilotinib era el hígado. Las alteraciones incluyeron un aumento de la actividad alanina aminotransferasa y fosfatasa alcalina y hallazgos histopatológicos (principalmente hiperplasia/hipertrofia de la célula sinusoidal o célula Kupffer, hiperplasia del conducto biliar y fibrosis periportal). En general los cambios en la química clínica fueron completamente reversibles después de un periodo de recuperación de cuatro semanas y las alteraciones histológicas mostraron una reversibilidad parcial. Las exposiciones a los niveles de dosis más bajos a las que se observaron efectos sobre el hígado fueron menores que la exposición en humanos a una dosis de 800 mg/día. En ratones o ratas tratadas durante un máximo de 26 semanas sólo se observaron alteraciones hepáticas menores. En ratas, perros y monos se observó un aumento del nivel de colesterol, mayoritariamente reversible.
Los estudios de genotoxicidad en sistemas bacterianos in vitro y en sistemas de mamíferos in vitro e in vivo con y sin activación metabólica no revelaron ninguna evidencia de potencial mutagénico para nilotinib.
En el estudio de carcinogenicidad de 2 años en ratas, el órgano diana más importante para lesiones no neoplásicas fue el útero (dilatación, ectasia vascular, hiperplasia celular endotelial, inflamación y/o hiperplasia epitelial). No se encontró evidencia de carcinogenicidad tras la administración de nilotinib a 5, 15 y 40 mg/kg/día. La exposición (en términos de AUC) a la dosis más alta representó aproximadamente de 2 a 3 veces la exposición diaria humana a nilotinib en el estado estacionario (en base a la AUC) a la dosis de 800 mg/día.
En el estudio Tg.rasH2 de 26 semanas sobre carcinogenicidad en ratones, en los que se administró 30, 100 y 300 mg/kg/día de nilotinib, se detectaron papilomas cutáneos/carcinomas a 300 mg/kg, lo que representa aproximadamente de 30 a 40 veces (en términos de AUC) la dosis máxima aprobada en humanos de 800 mg/día (administrada como 400 mg dos veces al día). El Nivel de Efecto-No-Observado para las lesiones neoplásicas de piel fue de 100 mg/kg/día, lo que representa aproximadamente de 10 a 20 veces la dosis máxima aprobada en humanos de 800 mg/día (administrada como 400 mg dos veces al día). Los principales órganos afectados de lesiones no neoplásicas fueron la piel (hiperplasia epidérmica), el crecimiento de los dientes (degeneración/atrofia del órgano del esmalte de los incisivos superiores y la inflamación de la encía/epitelio odontogénico de los incisivos) y el timo (aumento de la incidencia y/o la gravedad de la disminución de los linfocitos).
Nilotinib no indujo teratogenicidad, pero mostró embrio y fetotoxicidad a dosis que también mostraron toxicidad materna. Se observó un aumento en las pérdidas post implantación en estudios de fertilidad, con tratamiento en machos y hembras, y en el estudio de embriotoxicidad, con tratamiento de hembras. En los estudios de embriotoxicidad se observó letalidad embriológica y efectos fetales (principalmente disminución del peso fetal, fusión prematura de los huesos faciales (fusión huesos maxilar superior/cigomático) cambios viscerales y esqueléticos) en ratas y un aumento de la resorción de fetos y modificaciones esqueléticas en conejos. En un estudio de desarrollo pre y postnatal en ratas, la exposición materna a nilotinib causó una reducción en el peso corporal de las crías con cambios asociados en los parámetros de desarrollo físico así como una reducción en los índices de apareamiento y fertilidad de las crías. La exposición a nilotinib en hembras a Niveles de No Observación de Efectos Adversos fue generalmente menor o igual a la de los humanos a dosis de 800 mg/día.
En un estudio de desarrollo juvenil, se administró nilotinib por vía oral mediante una sonda a ratas jóvenes desde la primera semana post parto hasta que eran adultos jóvenes (día 70 post parto) a dosis de 2, 6 y 20 mg/kg/día. Además de los parámetros estándar del estudio, se llevaron a cabo evaluaciones de elementos de referencia del desarrollo, efectos sobre el SNC, el apareamiento y la fertilidad. En base a una reducción del peso corporal en ambos géneros y un retraso en la separación prepucial en machos (que puede asociarse con una reducción del peso), el Nivel Sin Efectos Observados en ratas jóvenes se consideró que era 6 mg/kg/día. Los animales jóvenes no mostraron una sensibilidad aumentada a nilotinib comparado con los adultos. Además, el perfil de toxicidad en ratas jóvenes fue comparable al observado en ratas adultas.
No se observaron efectos sobre el recuento/movilidad espermática ni sobre la fertilidad en ratas macho o hembra hasta la dosis más alta probada, aproximadamente 5 veces la dosis recomendada en humanos.
Se observó que nilotinib absorbe la luz en el rango de UV-B y UV-A, se distribuye en la piel y muestra un potencial fototóxico in vitro, pero no se observaron efectos in vivo. Por lo tanto, se considera que el riesgo de que nilotinib cause fotosensibilidad en los pacientes es bajo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula dura Lactosa monohidrato Crospovidona Poloxamero 188 Sílice coloidal anhidra Estearato de magnesio
Cubierta de la cápsula dura Gelatina
Dióxido de titanio (E171)
Rojo óxido de hierro (E172)
Amarillo, óxido de hierro (E172)
Tinta de impresión Shellac
Negro óxido de hierro (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30°C.
Conservar en el embalaje original para protegerlo de la humedad.
6.5 Naturaleza y contenido del envase Blísteres de PVC/PVDC/Alu.
Tasigna está disponible en los siguientes formatos:
• Envases unitarios que contienen 28 cápsulas duras (7 blísteres diarios, cada uno conteniendo 4 cápsulas duras) o 40 cápsulas duras (5 blísteres, cada uno conteniendo 8 cápsulas duras).
• Envases múltiples que contienen 112 cápsulas duras (4 envases de 28), 120 cápsulas duras (3 envases de 40) o 392 cápsulas duras (14 envases de 28).
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación Ninguna especial para su eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/422/005-006
EU/1/07/422/009-010
EU/1/07/422/013
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 19 de Noviembre de 2007 Fecha de la última renovación: 19 de Noviembre de 2012
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
1. NOMBRE DEL MEDICAMENTO
Tasigna 200 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una cápsula dura contiene 200 mg de nilotinib (como clorhidrato monohidrato). Excipiente(s) con efecto conocido
Una cápsula dura contiene 156,11 mg de lactosa (como monohidrato).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura
Polvo blanco a amarillento en cápsulas de gelatina dura, opacas, de color amarillo claro, de tamaño 0 con la impresión axial «NVR/TKI» en rojo.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tasigna está indicado para el tratamiento de pacientes adultos con:
- leucemia mieloide crónica (LMC) cromosoma Filadelfia positivo, de nuevo diagnóstico, en fase crónica,
- LMC cromosoma Filadelfia positivo en fase crónica y en fase acelerada, con resistencia o intolerancia a un tratamiento previo, incluido imatinib. No se dispone de datos de eficacia en pacientes con LMC en crisis blástica.
4.2 Posología y forma de administración
El tratamiento debe iniciarlo un médico con experiencia en el diagnóstico y el tratamiento de pacientes con LMC.
Posología
La dosis recomendada de Tasigna es:
- 300 mg dos veces al día en pacientes con LMC de nuevo diagnóstico en fase crónica,
- 400 mg dos veces al día en pacientes con LMC en fase crónica o fase acelerada con resistencia o intolerancia al tratamiento previo.
El tratamiento debe prolongarse mientras continúe el beneficio para el paciente.
Para la dosis de 300 mg dos veces al día, se encuentran disponibles cápsulas duras de 150 mg.
Si el paciente olvida tomar una dosis, no debe tomar una dosis adicional, sino esperar a la siguiente dosis, según la pauta establecida.
Ajustes o modificaciones de la dosis
Puede ser necesario una interrupción temporal y/o una reducción de la dosis de Tasigna por toxicidades hematológicas (neutropenia, trombocitopenia) que no estén relacionadas con la leucemia de base (ver Tabla 1).
Tabla 1 Ajustes de dosis por neutropenia y trombocitopenia
|
LMC de nuevo diagnóstico en fase crónica a 300 mg dos veces al día y LMC en fase crónica resistente o intolerante a imatinib a 400 mg dos veces al día |
RAN* <1,0 x 109/l y/o recuento de plaquetas <50 x 109/l |
1. Se deberá interrumpir el tratamiento con Tasigna, y controlar los hemogramas. 2. Se deberá reanudar el tratamiento a las 2 semanas a la dosis previa cuando RAN >1,0 x 109/l y/o el recuento de plaquetas >50 x 109/l. 3. Si el hemograma se mantiene bajo, puede necesitarse una reducción de la dosis a 400 mg una vez al día. |
|
LMC en fase acelerada resistente o intolerante a imatinib a 400 mg dos veces al día |
RAN* <0,5 x 109/l y/o recuento de plaquetas <10 x 109/l |
1. Se deberá interrumpir el tratamiento con Tasigna, y controlar los hemogramas. 2. Se deberá reanudar el tratamiento a las 2 semanas a la dosis previa cuando RAN >1,0 x 109/l y/o el recuento de plaquetas >20 x 109/l. 3. Si el hemograma se mantiene bajo, puede necesitarse una reducción de la dosis a 400 mg una vez al día. |
*RAN = Recuento absoluto de neutrófilos
Si se desarrolla toxicidad no hematológica clínicamente significativa, moderada o grave, el tratamiento debe suspenderse, y cuando la toxicidad se haya resuelto puede reanudarse el tratamiento con dosis de 400 mg una vez al día. Si se considera clínicamente adecuado, deberá considerarse el escalado de la dosis a la dosis inicial de 300 mg dos veces al día en pacientes con LMC de nuevo diagnóstico en fase crónica o a 400 mg dos veces al día en pacientes con LMC en fase crónica y fase acelerada resistentes o intolerantes a imatinib.
Aumento de la lipasa sérica: Para elevaciones de lipasa sérica de Grado 3-4, deberá reducirse la dosis a 400 mg una vez al día o bien interrumpir el tratamiento. El nivel de lipasa sérica deberá controlarse mensualmente o según esté indicado clínicamente (ver sección 4.4).
Aumento de la bilirrubina y las transaminasas hepáticas: Para elevaciones de bilirrubina y transaminasas hepáticas de Grado 3-4, deberá reducirse la dosis a 400 mg una vez al día o bien interrumpir el tratamiento. Los niveles de bilirrubina y de transaminasas hepáticas deberán controlarse mensualmente o según esté indicado clínicamente.
Pacientes de edad avanzada
Aproximadamente el 12% de los individuos del ensayo de Fase III en pacientes con LMC de nuevo diagnóstico en fase crónica y aproximadamente el 30% de los individuos del ensayo de Fase II en pacientes con LMC resistentes o intolerantes a imatinib en fase crónica y fase acelerada tenían 65 años ó más. No se observaron diferencias importantes respecto a la seguridad y eficacia en pacientes de >65 años de edad comparado con adultos de entre 18 y 65 años.
Insuficiencia renal
No se han realizado ensayos clínicos en pacientes con insuficiencia renal.
En pacientes con insuficiencia renal no se espera una disminución en el aclaramiento corporal total, puesto que nilotinib y sus metabolitos no se excretan por vía renal.
Insuficiencia hepática
La insuficiencia hepática tiene un efecto moderado sobre la farmacocinética de nilotinib. No se considera necesario un ajuste de dosis en pacientes con insuficiencia hepática. Sin embargo, los pacientes con insuficiencia hepática deberán tratarse con precaución (ver sección 4.4).
Trastornos cardiacos
En los ensayos clínicos, se excluyeron pacientes con enfermedad cardiaca significativa o no controlada (p.ej. infarto de miocardio reciente, insuficiencia cardiaca congestiva, angina inestable o bradicardia clínicamente significativa). Deberá utilizarse con precaución en pacientes con alteraciones cardiacas relevantes (ver sección 4.4).
Se han notificado aumentos en los niveles de colesterol plasmático total con el tratamiento con Tasigna (ver sección 4.4). Se debe determinar el perfil lipídico antes de iniciar el tratamiento con Tasigna, evaluar en el mes 3 y 6 después de iniciar el tratamiento y al menos anualmente durante el tratamiento crónico.
Se han notificado aumentos en los niveles de glucosa en sangre con el tratamiento con Tasigna (ver sección 4.4). Se deben evaluar los niveles de glucosa en sangre antes de iniciar el tratamiento con Tasigna y monitorizar durante el tratamiento.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de Tasigna en niños de menos de 18 años (ver sección 5.1). Por lo tanto, no está recomendado su uso en pacientes pediátricos debido a la ausencia de datos sobre seguridad y eficacia.
Forma de administración
Tasigna debe tomarse dos veces al día con aproximadamente unas 12 horas entre las dos administraciones y no debe tomarse junto con la comida. Las cápsulas duras deben tragarse enteras, con agua. No se debe ingerir alimentos durante las dos horas previas a la administración de la dosis ni durante, al menos, una hora después.
Para pacientes que no puedan tragar las cápsulas duras, el contenido de cada cápsula dura puede dispersarse en una cucharadita de compota de manzana (puré de manzana) y debe tomarse inmediatamente. No debe utilizarse más de una cucharadita de compota de manzana ni ningún otro alimento aparte de compota de manzana (ver secciones 4.4 y 5.2).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Mielosupresión
El tratamiento con Tasigna se ha asociado a trombocitopenia, neutropenia y anemia (Grado 3-4 según el «Common Toxicity Criteria» del National Cancer Institute). Se produce con más frecuencia en pacientes con LMC resistentes o intolerantes a imatinib, en particular en pacientes con LMC en fase acelerada. Deberán realizarse hemogramas completos cada dos semanas durante los 2 primeros meses y posteriormente cada mes, o con la frecuencia indicada clínicamente. En general, la mielosupresión fue reversible y normalmente se controló suspendiendo la administración de Tasigna de forma temporal o con una reducción de la dosis (ver sección 4.2).
Prolongación del QT
Se ha observado que Tasigna prolonga la repolarización cardiaca ventricular de forma dependiente de la concentración, medida por el intervalo QT del ECG.
En el ensayo de Fase III en pacientes con LMC en fase crónica de nuevo diagnóstico que recibieron 300 mg de nilotinib dos veces al día, el cambio en el tiempo medio del intervalo QTcF respecto al valor basal en el estado estacionario fue de 6 ms. Ningún paciente presentó un QTcF>480 ms. No se observaron episodios de «Torsade de Pointes».
En el ensayo de Fase II en pacientes con LMC en fase crónica y acelerada resistentes e intolerantes a imatinib, que recibieron 400 mg de nilotinib dos veces al día, el cambio en el tiempo medio del intervalo QTcF respecto al valor basal en el estado estacionario fue de 5 y 8 ms, respectivamente. Se observó un QTcF de >500 ms en <1% de estos pacientes. No se observaron episodios de «Torsade de Pointes» en los ensayos clínicos.
En un ensayo con voluntarios sanos con exposiciones que fueron comparables a las observadas en pacientes, la media de tiempo del cambio del valor de QTcF respecto al valor basal fue de 7 ms (IC ±
4 ms). Ningún individuo presentó un valor de QTcF >450 ms. Además, no se observaron arritmias clínicamente relevantes durante la realización del ensayo. En particular, no se observaron episodios de «Torsade de Pointes» (transitorios o sostenidos).
Puede producirse una prolongación significativa del intervalo QT cuando se administra nilotinib de forma no adecuada con inhibidores potentes de CYP3A4, con fármacos con una capacidad conocida de prolongar el intervalo QT y/o con comida (ver sección 4.5). La presencia de hipopotasemia e hipomagnesemia pueden incrementar este efecto. La prolongación del intervalo QT puede exponer a los pacientes a un riesgo mortal.
Tasigna debe utilizarse con precaución en pacientes que presentan o que tienen un riesgo importante de desarrollar una prolongación del intervalo QTc, tales como:
- con prolongación de QT largo congénita.
- con enfermedad cardíaca significativa o no controlada, incluyendo infarto de miocardio reciente, insuficiencia cardiaca congestiva, angina inestable o bradicardia clínicamente significativa.
- en tratamiento con fármacos antiarrítmicos u otras sustancias que puedan provocar una prolongación del intervalo QTc.
Se recomienda un control cuidadoso del efecto sobre el intervalo QTc y realizar un ECG basal antes de iniciar el tratamiento con Tasigna y según esté clínicamente indicado. La hipopotasemia o hipomagnesemia deberán corregirse antes de la administración de Tasigna y deberán controlarse periódicamente durante el tratamiento.
Muerte súbita
Se han notificado casos poco frecuentes (0,1 a 1%) de muertes súbitas en pacientes con LMC en fase crónica o fase acelerada resistentes o intolerantes a imatinib que tienen antecedentes de enfermedad cardiaca o factores de riesgo cardiaco significativos. También se hallaban presentes frecuentemente co-morbilidades además del proceso maligno de base, así como medicamentos concomitantes. Las alteraciones de la repolarización ventricular pueden ser factores contributivos. No se han notificado casos de muerte súbita en el ensayo de Fase III en pacientes con LMC en fase crónica de nuevo diagnóstico.
Retención de líquidos y edema
Se observaron con poca frecuencia (0,1 a 1%) formas graves de retención de líquidos tales como derrame pleural, edema pulmonar y derrame pericárdico en un ensayo de Fase III en pacientes con LMC de nuevo diagnóstico. Se observaron acontecimientos similares en informes postcomercialización. El aumento rápido e inesperado del peso se debe investigar cuidadosamente. Si aparecen signos de retención grave de líquidos durante el tratamiento con nilotinib, se debe evaluar la etiología y tratar a los pacientes en consecuencia (ver sección 4.2 para instrucciones sobre el control de toxicidades no hematológicas).
Acontecimientos cardiovasculares
Se notificaron acontecimientos cardiovasculares en un ensayo de Fase III aleatorizado en pacientes con LMC de nuevo diagnóstico y se observaron en informes post-comercialización. En este ensayo clínico con una mediana de tiempo en tratamiento de 60,5 meses, los acontecimientos cardiovasculares de grado 3-4 incluyeron: enfermedad arterial oclusiva periférica (1,4% y 1,1% con 300 mg y 400 mg de nilotinib dos veces al día, respectivamente), cardiopatía isquémica (2,2% y 6,1% con 300 mg y 400 mg de nilotinib dos veces al día, respectivamente) y acontecimientos isquémicos cerebrovasculares (1,1% y 2,2% con 300 mg y 400 mg de nilotinib dos veces al día, respectivamente). Se debe advertir a los pacientes que busquen atención médica inmediata si experimentan signos o síntomas agudos de acontecimientos cardiovasculares. Se debe evaluar el estado cardiovascular de los pacientes y monitorizar y controlar activamente los factores de riesgo cardiovasculares durante el tratamiento con Tasigna de acuerdo a las guías generales. Se debe prescribir el tratamiento apropiado para controlar los factores de riesgo cardiovasculares (ver sección 4.2 para instrucciones sobre el control de toxicidades no hematológicas).
Reactivación del virus de la hepatitis B
Se han producido reactivaciones de la hepatitis B en pacientes que son portadores crónicos de este virus después de que los pacientes hayan recibido inhibidores de la tirosina quinasa BCR-ABL. En algunos casos se produjo insuficiencia hepática aguda o hepatitis fulminante que dio lugar a un trasplante de hígado o a un desenlace mortal.
Los pacientes se deben someter a pruebas para detectar la infección por VHB antes de comenzar el tratamiento con Tasigna. Se debe consultar a expertos en enfermedades hepáticas y en el tratamiento de la hepatitis B antes de comenzar el tratamiento en pacientes con una serología positiva para hepatitis B (incluyendo a los pacientes con enfermedad activa) y pacientes que den un resultado positivo en una prueba de infección por VHB durante el tratamiento. Los portadores del VHB que necesiten tratamiento con Tasigna se deben someter a una estrecha monitorización para detectar signos y síntomas de infección activa por VHB a lo largo de todo el tratamiento y durante varios meses después de finalizar el tratamiento (ver sección 4.8).
Pruebas de laboratorio y controles
Lípidos en sangre
En un ensayo de Fase III en pacientes con LMC de nuevo diagnóstico, el 1,1% de los pacientes tratados con 400 mg de nilotinib dos veces al día mostraron un aumento del colesterol total de Grado 3-4; sin embargo no se observaron elevaciones de Grado 3-4 en el grupo de dosis de 300 mg dos veces al día (ver sección 4.8). Se recomienda determinar los perfiles lipídicos antes de iniciar el tratamiento con Tasigna, evaluarlos en el mes 3 y 6 después de iniciar el tratamiento y al menos anualmente durante el tratamiento crónico (ver sección 4.2). Si se necesita un inhibidor de la HMG-CoA reductasa (agente hipolipemiante), ver la sección 4.5 antes de iniciar el tratamiento puesto que algunos inhibidores de la HMG-CoA reductasa también se metabolizan por la vía de CYP3A4.
Glucosa en sangre
En un ensayo de Fase III en pacientes con LMC de nuevo diagnóstico, el 6,9% y el 7,2% de los pacientes tratados con 400 mg de nilotinib y 300 mg de nilotinib dos veces al día, respectivamente, mostraron un aumento de la glucosa en sangre de Grado 3-4. Se recomienda evaluar los niveles de glucosa antes de iniciar el tratamiento con Tasigna y controlarlos durante el tratamiento, según esté clínicamente indicado (ver sección 4.2). Si los resultados de las pruebas justifican el tratamiento, los médicos deben seguir sus estándares de práctica locales y las guías de tratamiento.
Interacciones con otros medicamentos
Debe evitarse la administración de Tasigna con agentes que son inhibidores potentes de CYP3A4 (incluyendo, aunque no de manera exclusiva, ketoconazol, itraconazol, voriconazol, claritromicina, telitromicina, ritonavir). En caso que se requiera el tratamiento con alguno de estos fármacos, se recomienda interrumpir el tratamiento con Tasigna, si es posible (ver sección 4.5). En caso que no sea posible la interrupción temporal del tratamiento, deberá realizarse un control estricto del paciente para la prolongación del intervalo QT (ver secciones 4.2, 4.5 y 5.2).
El uso concomitante de Tasigna con medicamentos que son inductores potentes del CYP3A4 (p.ej. fenitoina, rifampicina, carbamazepina, fenobarbital e hierba de San Juan) es probable que reduzca la exposición a nilotinib en un grado clínicamente relevante. Por lo tanto, en pacientes que reciben tratamiento con Tasigna, deberán elegirse agentes terapéuticos alternativos con menor potencial de inducción de CYP3A4 (ver sección 4.5).
Efecto de los alimentos
La biodisponibilidad de nilotinib aumenta con los alimentos. Tasigna no se debe tomar junto con la comida (ver las secciones 4.2 y 4.5) sino que se debe tomar 2 horas después de una comida. No se debe ingerir ningún alimento durante al menos una hora después de tomar el medicamento. Debe evitarse tomar zumo de pomelo y otros alimentos que se sabe que son inhibidores de CYP3A4. Para pacientes que no puedan tragar las cápsulas duras, el contenido de cada cápsula dura puede dispersarse en una cucharadita de compota de manzana y debe tomarse inmediatamente. No debe utilizarse más de una cucharadita de compota de manzana ni otro alimento aparte de la compota de manzana (ver sección 5.2).
Insuficiencia hepática
La insuficiencia hepática tiene un efecto moderado sobre la farmacocinética de nilotinib. La administración de una dosis única de 200 mg de nilotinib provocó aumentos del AUC de 35%, 35% y 19% en individuos con insuficiencia hepática leve, moderada y grave, respectivamente, en comparación a un grupo control de individuos con función hepática normal. La Cmax prevista del estado estacionario de nilotinib mostró un aumento de 29%, 18% y 22%, respectivamente. Los ensayos clínicos han excluido pacientes con alanino transaminasa (ALT) y/o aspartato transaminasa (AST) >2,5 veces (ó >5, si está relacionado con la enfermedad) al límite superior del intervalo normal y/o bilirrubina total >1,5 veces al límite superior del intervalo normal. El metabolismo de nilotinib es principalmente hepático. Por lo tanto, los pacientes con insuficiencia hepática podrían presentar un aumento de la exposición a nilotinib y deberán tratarse con precaución (ver sección 4.2).
Lipasa sérica
Se han observado elevaciones de la lipasa sérica. Se recomienda precaución en pacientes con antecedentes de pancreatitis. En caso de que las elevaciones de la lipasa estén acompañadas por síntomas abdominales, deberá interrumpirse el tratamiento con Tasigna y deberán considerarse medidas diagnósticas adecuadas para excluir la presencia de pancreatitis.
Gastrectomía total
La biodisponibilidad de nilotinib puede reducirse en pacientes con gastrectomía total (ver sección 5.2). Debe considerarse un seguimiento más frecuente de estos pacientes.
Síndrome de lisis tumoral
Antes de iniciar el tratamiento con Tasigna, se recomienda la corrección de la deshidratación clínicamente significativa y el tratamiento de los niveles altos de ácido úrico, debido a la posible aparición del síndrome de lisis tumoral (SLT) (ver sección 4.8).
Lactosa
Las cápsulas duras de Tasigna contienen lactosa. Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp o problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Tasigna se puede administrar en combinación con factores de crecimiento hematopoyético como eritropoyetina o factor estimulante de las colonias de granulocitos (G-CSF), si está indicado clínicamente. Se puede administrar con hidroxiurea o anagrelida si está clínicamente indicado.
Nilotinib se metaboliza principalmente en el hígado y también es un sustrato de la glicoproteína-P (gp-P), una bomba de eflujo de múltiples fármacos. Por lo tanto, la absorción y subsiguiente eliminación de nilotinib absorbido sistémicamente, pueden verse influenciadas por sustancias que afecten a CYP3A4 y/o a gp-P.
Sustancias que pueden aumentar las concentraciones plasmáticas de nilotinib La administración conjunta de nilotinib con imatinib (un sustrato e inhibidor de gp-P y de CYP3A4), mostró un ligero efecto inhibidor sobre CYP3A4 y/o gp-P. El AUC de imatinib aumentó entre el 18% y el 39%, y el AUC de nilotinib aumentó entre el 18% y el 40%. Es poco probable que estos cambios sean clínicamente importantes.
La exposición a nilotinib en sujetos sanos aumentó 3 veces cuando se administró conjuntamente con ketoconazol, un inhibidor potente de CYP3A4. Por lo tanto, deberá evitarse el tratamiento concomitante con inhibidores potentes de CYP3A4, incluyendo ketoconazol, itraconazol, voriconazol, ritonavir, claritromicina y telitromicina (ver sección 4.4). También podría esperarse un aumento en la exposición a nilotinib con inhibidores moderados de CYP3A4. Deberán considerarse medicamentos concomitantes alternativos sin inhibición o con una mínima inhibición de CYP3A4.
Sustancias que pueden reducir las concentraciones plasmáticas de nilotinib
La rifampicina, un inductor potente del CYP3A4 disminuye la Cmax de nilotinib un 64% y reduce el AUC de nilotinib un 80%. No deben administrarse conjuntamente rifampicina y nilotinib.
La administración concomitante de otros medicamentos que inducen CYP3A4 (p.ej. fenitoina, carbamazepina, fenobarbital e hierba de San Juan) es asimismo probable que reduzca la exposición a nilotinib a un grado clínicamente relevante. En pacientes para los cuales están indicados los inductores de CYP3A4, deberán utilizarse agentes alternativos con menor potencial de inducción enzimática.
Nilotinib tiene una solubilidad dependiente del pH, con una menor solubilidad a un pH más alto. En individuos sanos que tomaron 40 mg de esomeprazol una vez al día durante 5 días, el pH gástrico aumentó significativamente, pero la absorción de nilotinib sólo disminuyó de forma discreta (disminución de un 27% de la Cmax y disminución de un 34% del AUC0-<»). Nilotinib puede utilizarse de forma concomitante con esomeprazol y otros inhibidores de la bomba de protones, en caso necesario.
En un estudio con sujetos sanos, no se observó ningún cambio significativo en la farmacocinética de nilotinib cuando se administró una dosis única de 400 mg de Tasigna 10 horas después y 2 horas antes de famotidina. Por lo tanto, cuando es necesario el uso concomitante de un bloqueador H2, se puede administrar aproximadamente 10 horas antes y aproximadamente 2 horas después de la dosis de Tasigna.
En el mismo estudio anterior, la administración de un antiácido (hidróxido de aluminio/hidróxido de magnesio/simeticona) 2 horas antes o después de una dosis única de 400 mg de Tasigna tampoco alteró la farmacocinética de nilotinib. Por lo tanto, si es necesario, se puede administrar un antiácido aproximadamente 2 horas antes o aproximadamente 2 horas después de la dosis de Tasigna.
Sustancias cuya concentración plasmática puede verse alterada por nilotinib
In vitro, nilotinib es un inhibidor relativamente potente de CYP3A4, CYP2C8, CYP2C9, CYP2D6 y UGT1A1, con un valor Ki inferior para CYP2C9 (Ki=0,13 microM).
Un estudio de interacción fármaco-fármaco de dosis única en voluntarios sanos con 25 mg de warfarina, un sustrato sensible a CYP2C9, y 800 mg de nilotinib, no dio como resultado ningún cambio en los parámetros farmacocinéticos o farmacodinámicos de la warfarina, medidos como tiempo de protrombina (PT) e índice normalizado internacional (INR). No existen datos en estado estacionario. Este estudio sugiere que una interacción fármaco-fármaco clínicamente significativa entre nilotinib y warfarina es menos probable hasta una dosis de 25 mg de warfarina. Debido a la falta de datos en estado estacionario, se recomienda el control de los marcadores farmacodinámicos de la warfarina (INR o PT) tras el inicio del tratamiento con nilotinib (como mínimo durante las 2 primeras semanas).
En pacientes con LMC, la administración de 400 mg de nilotinib dos veces al día durante 12 días aumentó la exposición sistémica (AUC y Cmax) de midazolam oral (un sustrato del CYP3A4) 2,6 y 2,0 veces, respectivamente. Nilotinib es un inhibidor moderado del CYP3A4. Como resultado, la exposición sistémica de otros fármacos metabolizados principalmente por el CYP3A4 (p.ej. algunos inhibidores de la HMG-CoA reductasa) puede verse aumentada cuando se administren conjuntamente con nilotinib. Una monitorización apropiada y un ajuste de dosis pueden ser necesarios para fármacos que son sustratos del CYP3A4 y que tienen un estrecho margen terapéutico (incluyendo, pero no limitado a alfentanilo, ciclosporina, dihidroergotamina, ergotamina, fentanilo, sirolimus y tacrolimus) cuando se administran conjuntamente con nilotinib.
Medicamentos antiarrítmicos y otras sustancias que pueden prolongar el intervalo QT Nilotinib debe utilizarse con precaución en pacientes que tienen o pueden desarrollar una prolongación del intervalo QT, incluyendo aquellos pacientes en tratamiento con medicamentos antiarrítmicos como amiodarona, disopiramida, procainamida, quinidina y sotalol y otros medicamentos que pueden causar una prolongación del intervalo QT como cloroquina, halofantrina, claritromicina, haloperidol, metadona y moxifloxacina (ver sección 4.4).
Interacciones con alimentos
La absorción y biodisponibilidad de Tasigna aumentan cuando se toma con alimentos, provocando una concentración plasmática más elevada (ver secciones 4.2, 4.4 y 5.2). Debe evitarse tomar zumo de pomelo y otros alimentos que se sabe que son inhibidores de CYP3A4.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar métodos anticonceptivos altamente efectivos durante el tratamiento con Tasigna y hasta dos semanas después de finalizar el tratamiento.
Embarazo
No hay datos o éstos son limitados relativos al uso de nilotinib en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3). Tasigna no debería utilizarse durante el embarazo excepto si la situación clínica de la mujer requiere tratamiento con nilotinib. Si se utiliza durante el embarazo, la paciente debe estar informada del posible riesgo sobre el feto.
Lactancia
Se desconoce si nilotinib se excreta en la leche materna. Los datos toxicológicos disponibles en animales muestran que nilotinib se excreta en la leche (ver sección 5.3). No se puede excluir el riesgo en recién nacidos/lactantes. Tasigna no debe utilizarse durante la lactancia.
Fertilidad
Los estudios en animales no mostraron un efecto sobre la fertilidad en ratas machos y hembras (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Los pacientes que sufran mareos, fatiga, alteraciones de la vista u otros efectos adversos con un posible impacto sobre la capacidad para conducir o utilizar máquinas de forma segura, deberán abstenerse de realizar estas actividades mientras se mantengan estos efectos adversos (ver sección 4.8).
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los datos descritos a continuación reflejan la exposición a Tasigna de un total de 717 pacientes de un ensayo de Fase III aleatorizado en pacientes con LMC Ph+ en fase crónica de nuevo diagnóstico tratados a la dosis recomendada de 300 mg dos veces al día (n=279) y de un ensayo abierto y multicéntrico de Fase II en pacientes con LMC resistentes o intolerantes a imatinib en fase crónica (n=321) y en fase acelerada (n=137) tratados a la dosis recomendada de 400 mg dos veces al día.
En pacientes con LMC en fase crónica de nuevo diagnóstico
La duración mediana de la exposición fue de 60,5 meses (intervalo 0,1-70,8 meses).
Las reacciones adversas no hematológicas más frecuentes (>10%) fueron erupción, prurito, cefalea, náuseas, fatiga, alopecia, mialgia y dolor abdominal superior. La mayoría de estas reacciones adversas fueron de intensidad leve a moderada. Se observaron de forma menos frecuente (<10% y >5%) estreñimiento, sequedad de la piel, astenia, espasmos musculares, diarrea, artralgia, dolor abdominal, vómitos y edema periférico, fueron de una intensidad de leve a moderada, manejables y generalmente no requirieron una reducción de la dosis.
La toxicidad hematológica derivada del tratamiento incluyó mielosupresión: trombocitopenia (18%), neutropenia (15%) y anemia (8%). Las reacciones adversas bioquímicas incluyeron aumento de la alanina aminotransferasa (24%), hiperbilirrubinemia (16%), aumento de la aspartato aminotransferasa (12%), aumento de la lipasa (11%), aumento de la bilirrubina sanguínea (10%), hiperglucemia (4%), hipercolesterolemia (3%) e hipertrigliceridemia (<1%). Se observó derrame pleural y pericárdico, independientemente de la causalidad, en un 2% y en <1% de pacientes, respectivamente, en tratamiento con Tasigna 300 mg dos veces al día. Se notificó hemorragia gastrointestinal, independientemente de la causalidad, en un 3% de estos pacientes.
El cambio en el tiempo medio del intervalo QTcF respecto al valor basal en el estado estacionario fue de 6 ms. Ningún paciente presentó un QTcF absoluto >500 ms mientras recibieron medicación en el estudio. En <1% de los pacientes se observó un aumento de QTcF respecto al valor basal que superó los 60 ms, mientras recibieron medicación en el estudio. No se observaron casos de muerte súbita ni episodios de «Torsades de Pointes» (transitorios o sostenidos). No se observó una disminución del
valor medio de la fracción de eyección ventricular respecto al valor basal (FEVI) en ningún momento durante el tratamiento. Ningún paciente presentó un valor de FEVI <45% durante el tratamiento ni una reducción absoluta del valor de FEVI de más de 15%.
Se observó una interrupción del tratamiento debida a reacciones adversas en un 10% de los pacientes.
En pacientes con LMC en fase crónica y fase acelerada resistente o intolerante a imatinib Los datos descritos a continuación reflejan la exposición a Tasigna de 458 pacientes en un ensayo abierto y multicéntrico de Fase II en pacientes con LMC resistentes o intolerantes a imatinib en fase crónica (n=321) y fase acelerada (n=137) tratados a la dosis recomendada de 400 mg dos veces al día.
Las reacciones adversas no hematológicas más frecuentes (>10%) relacionadas con el fármaco fueron erupción, prurito, náuseas, fatiga, cefalea, vómitos, mialgia, estreñimiento y diarrea. La mayoría de estas reacciones adversas fueron de intensidad leve a moderada. De forma menos frecuente (<10% y >5%) se observaron alopecia, espasmos musculares, disminución del apetito, artralgia, dolor abdominal, dolor óseo, edema periférico, astenia, dolor abdominal superior, sequedad en la piel, eritema y dolor en las extremidades y fueron de intensidad leve a moderada (Grado 1 o 2). Se produjo una interrupción del tratamiento debida a reacciones adversas en el 16% de los pacientes en fase crónica y en el 10% de los pacientes en fase acelerada.
La toxicidad hematológica derivada del tratamiento incluyó mielosupresión: trombocitopenia (31%), neutropenia (17%) y anemia (14%). En <1% de los pacientes en tratamiento con Tasigna apareció derrame pleural y pericárdico así como complicaciones derivadas de la retención de líquidos. Se observó insuficiencia cardiaca en un <1% de los pacientes. Se notificó hemorragia gastrointestinal y del SNC en el 1% y <1% de los pacientes, respectivamente.
Se observó un valor de QTcF superior a 500 ms en <1% de los pacientes. No se observaron episodios de «Torsades de Pointes» (transitorios o sostenidos).
Reacciones adversas notificadas de forma más frecuente en los ensayos clínicos con Tasigna En la Tabla 2 se muestran las reacciones adversas no hematológicas (excepto los valores de laboratorio anormales) que se notificaron en al menos el 5% de los pacientes en los ensayos clínicos con Tasigna. Estas se ordenan por frecuencias, apareciendo primero las más frecuentes, utilizando una precisión decimal para porcentajes y la siguiente convención: muy frecuentes (>1/10) o frecuentes (>1/100 a <1/10). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Tabla 2 Reacciones adversas no hematológicas (>5% de todos los pacientes)*
|
LMC-FC de nuevo diagnóstico 300 mg dos veces al día n=279 |
LMC-FC y LMC-FA resistente o intolerante a imatinib 400 mg dos veces al día n=458 | |||
|
Análisis a los 60 meses |
Análisis a los 24 meses | |||
|
LMC- |
LMC- | |||
|
Clasificación |
Frecuencia Todos Grado |
Frecuencia Todos Grado |
FC |
FA |
|
de órganos del |
los 3-4 |
los 3-4 |
n=321 |
n=137 |
|
sistema/ |
grados |
grados |
Grado |
Grado |
|
Reacción |
3-4 |
3-4 | ||
|
adversa | ||||
|
% % |
% % |
% |
% | |
|
Trastornos del metabolismo y de la nutrición | ||||||||
|
Disminución del apetito** |
Frecuentes |
4 |
0 |
Frecuentes |
8 |
<1 |
<1 |
0 |
|
Trastornos del sistema nervioso | ||||||||
|
Cefalea |
Muy frecuentes |
16 |
2 |
Muy frecuentes |
15 |
1 |
2 |
<1 |
|
Trastornos gastrointestinales | ||||||||
|
Náuseas |
Muy frecuentes |
14 |
<1 |
Muy frecuentes |
20 |
<1 |
<1 |
<1 |
|
Estreñimiento |
Frecuentes |
10 |
0 |
Muy frecuentes |
12 |
<1 |
<1 |
0 |
|
Diarrea |
Frecuentes |
9 |
<1 |
Muy frecuentes |
11 |
2 |
2 |
<1 |
|
Vómitos |
Frecuentes |
6 |
0 |
Muy frecuentes |
10 |
<1 |
<1 |
0 |
|
Dolor abdominal superior |
Muy frecuentes |
10 |
1 |
Frecuentes |
5 |
<1 |
<1 |
0 |
|
Dolor abdominal |
Frecuentes |
6 |
0 |
Frecuentes |
6 |
<1 |
<1 |
<1 |
|
Dispepsia |
Frecuentes |
5 |
0 |
Frecuentes |
3 |
0 |
0 |
0 |
|
Trastornos de la piel y del tejido subcutáneo | ||||||||
|
Erupción |
Muy frecuentes |
33 |
<1 |
Muy frecuentes |
28 |
1 |
2 |
0 |
|
Prurito |
Muy frecuentes |
18 |
<1 |
Muy frecuentes |
24 |
<1 |
<1 |
0 |
|
Alopecia |
Muy frecuentes |
10 |
0 |
Frecuentes |
9 |
0 |
0 |
0 |
|
Sequedad de la piel |
Frecuentes |
10 |
0 |
Frecuentes |
5 |
0 |
0 |
0 |
|
Eritema |
Frecuentes |
3 |
0 |
Frecuentes |
5 |
<1 |
<1 |
0 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | ||||||||
|
Mialgia |
Muy frecuentes |
10 |
<1 |
Muy frecuentes |
10 |
<1 |
<1 |
<1 |
|
Espasmos musculares |
Frecuentes |
9 |
0 |
Frecuentes |
8 |
<1 |
<1 |
0 |
|
Artralgia |
Frecuentes |
8 |
<1 |
Frecuentes |
7 |
<1 |
1 |
0 |
|
Dolor óseo |
Frecuentes |
4 |
0 |
Frecuentes |
6 |
<1 |
<1 |
0 |
|
Dolor en las extremidades |
Frecuentes |
5 |
<1 |
Frecuentes |
5 |
<1 |
<1 |
<1 |
|
Trastornos generales y alteraciones en el lugar de administración | ||||||||
|
Fatiga |
Muy frecuentes |
12 |
0 |
Muy frecuentes |
17 |
1 |
1 |
<1 |
|
Astenia |
Frecuentes |
9 |
<1 |
Frecuentes |
6 |
<1 |
0 |
<1 |
|
Edema periférico |
Frecuentes |
5 |
0 |
Frecuentes |
6 |
0 |
0 |
0 |
*Los porcentajes están redondeados a números enteros para la presentación en esta tabla. Sin embargo, los porcentajes con precisión de un decimal se utilizan para identificar términos con una frecuencia de como mínimo 5% y para clasificar los términos de acuerdo a las categorías de frecuencia.
** También incluye el término anorexia
Las siguientes reacciones adversas se notificaron en los ensayos clínicos con Tasigna con una frecuencia menor del 5%. También se notifican los valores anormales de laboratorio muy frecuentes (>1/10), no incluidos en la Tabla 2. Estas reacciones adversas se incluyen basadas en la relevancia clínica y se ordenan en orden decreciente de gravedad dentro de cada categoría, utilizando la siguiente convención: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Infecciones e infestaciones:
Frecuentes: foliculitis, infección del tracto respiratorio superior (incluyendo faringitis, nasofaringitis, rinitis)
Poco frecuentes: neumonía, infección del tracto urinario, gastroenteritis, bronquitis, infección por el virus del herpes, candidiasis (incluyendo candidiasis oral).
Frecuencia no conocida: sepsis, absceso subcutáneo, absceso anal, furúnculo, tinea pedis, reactivación del virus de la hepatitis B.
Neoplasias benignas, malignas y no especificadas (incluyendo quistes y pólipos) :
Frecuentes: papiloma en la piel.
Frecuencia no conocida: papiloma oral, paraproteinemia.
Trastornos de la sangre y del sistema linfático:
Frecuentes: leucopenia, eosinofilia, neutropenia febril, pancitopenia, linfopenia.
Poco frecuentes: trombocitopenia, leucocitosis.
Trastornos del sistema inmunológico:
Frecuencia no conocida: hipersensibilidad.
Trastornos endocrinos:
Poco frecuentes: hipertiroidismo, hipotiroidismo.
Frecuencia no conocida: hiperparatiroidismo secundario, tiroiditis.
Trastornos del metabolismo y de la nutrición:
Muy frecuentes: hipofosfatemia (incluyendo disminución del fósforo en la sangre).
Frecuentes: desequilibrio de electrolitos (incluyendo hipomagnesemia, hiperpotasemia, hipopotasemia, hiponatremia, hipocalcemia, hipercalcemia, hiperfosfatemia), diabetes mellitus, hiperglucemia, hipercolesterolemia, hiperlipidemia, hipertrigliceridemia.
Poco frecuentes: deshidratación, aumento del apetito, gota, dislipidemia.
Frecuencia no conocida: hiperuricemia, hipoglucemia.
Trastornos psiquiátricos:
Frecuentes: depresión, insomnio, ansiedad.
Frecuencia no conocida: desorientación, estado confusional, amnesia, disforia.
Trastornos del sistema nervioso:
Frecuentes: mareo, neuropatía periférica, hipoestesia, parestesia.
Poco frecuentes: hemorragia intracraneal, ictus isquémico, ataque isquémico transitorio, infarto cerebral, migraña, pérdida de conciencia (incluyendo síncope), temblor, trastornos en la atención, hiperestesia.
Frecuencia no conocida: accidente cerebrovascular, edema cerebral, neuritis óptica, letargia, disestesia, síndrome de las piernas inquietas.
Trastornos oculares:
Frecuentes: hemorragia ocular, edema periorbital, prurito ocular, conjuntivitis, sequedad ocular (incluyendo xeroftalmia).
Poco frecuentes: alteración visual, visión borrosa, hemorragia conjuntival, reducción de la agudeza visual, edema palpebral, fotopsia, hiperemia (escleral, conjuntival, ocular), irritación ocular. Frecuencia no conocida: papiloedema, corioretinopatía, diplopía, fotofobia, hinchazón en los ojos, blefaritis, dolor ocular, conjuntivitis alérgica, enfermedad de la superficie ocular.
Trastornos del oído y del laberinto:
Frecuentes: vértigo.
Frecuencia no conocida: trastornos de audición, dolor de oído, tinnitus.
Trastornos cardiacos:
Frecuentes: angina de pecho, arritmia (incluyendo bloqueo atrioventricular, flutter cardiaco, extrasístoles, taquicardia, fibrilación auricular, bradicardia), palpitaciones, QT prolongado en el electrocardiograma.
Poco frecuentes: insuficiencia cardiaca, infarto de miocardio, enfermedad arterial coronaria, soplo cardiaco, derrame pericárdico, cianosis.
Frecuencia no conocida: disfunción ventricular, pericarditis, disminución de la fracción de eyección.
Trastornos vasculares:
Frecuentes: hipertensión, sofocos, estenosis arterial periférica.
Poco frecuentes: crisis hipertensiva, enfermedad arterial oclusiva periférica, claudicación intermitente, extremidad con estenosis arterial, hematoma, arteriosclerosis.
Frecuencia no conocida: shock hemorrágico, hipotensión, trombosis.
Trastornos respiratorios, torácicos y mediastínicos:
Frecuentes: disnea, disnea por esfuerzo, epistaxis, tos, disfonía.
Poco frecuentes: edema pulmonar, derrame pleural, enfermedad pulmonar intersticial, dolor pleural, pleuresía, dolor faringolaríngeo, irritación de garganta.
Frecuencia no conocida: hipertensión pulmonar, respiración sibilante, dolor orofaríngeo.
Trastornos gastrointestinales:
Frecuentes: pancreatitis, molestias abdominales, distensión abdominal, disgeusia, flatulencia.
Poco frecuentes: hemorragia gastrointestinal, melena, úlceras en la boca, reflujo gastroesofágico, estomatitis, dolor esofágico, boca seca, gastritis, sensibilidad en los dientes.
Frecuencia no conocida: perforación de úlcera gastrointestinal, hemorragia retroperitoneal, hematemesis, úlcera gástrica, esofagitis ulcerativa, subileus, enterocolitis, hemorroides, hernia de hiato, hemorragia rectal, gingivitis.
Trastornos hepatobiliares:
Muy frecuentes: hiperbilirrubinemia (incluyendo aumento de la bilirrubina en la sangre).
Frecuentes: función hepática anormal.
Poco frecuentes: hepatotoxicidad, hepatitis tóxica, ictericia.
Frecuencia no conocida: colestasis, hepatomegalia.
Trastornos de la piel y del tejido subcutáneo:
Frecuentes: sudores nocturnos, eczema, urticaria, hiperhidrosis, contusión, acné, dermatitis (incluyendo alérgica, exfoliativa y acneiforme).
Poco frecuentes: erupción exfoliativa, erupción debida al medicamento, dolor en la piel, equimosis, hinchazón facial.
Frecuencia no conocida: eritema multiforme, eritema nodoso, úlceras en la piel, síndrome de eritrodisestesia palmar-plantar, petequias, fotosensibilidad, ampollas, quistes dérmicos, hiperplasia sebácea, atrofia de la piel, decoloración de la piel, exfoliación de la piel, hiperpigmentación de la piel, hipertrofia de la piel, hiperqueratosis, psoriasis.
Trastornos musculoesqueléticos y del tejido conjuntivo:
Frecuentes: dolor torácico músculoesquelético, dolor musculoesquelético, dolor de espalda, dolor en el costado, dolor en el cuello, debilidad muscular.
Poco frecuentes: rigidez musculoesquelética, hinchazón articular.
Frecuencia no conocida: artritis.
Trastornos renales y urinarios:
Frecuentes: polaquiuria.
Poco frecuentes: disuria, urgencia miccional, nocturia.
Frecuencia no conocida: insuficiencia renal, hematuria, incontinencia urinaria, cromaturia.
Trastornos del aparato reproductor y de la mama:
Poco frecuentes: dolor en las mamas, ginecomastia, disfunción eréctil.
Frecuencia no conocida: induración de las mamas, menorragia, hinchazón en el pezón.
Trastornos generales y alteraciones en el lugar de administración:
Frecuentes: dolor en el pecho (incluyendo dolor torácico no cardiaco), dolor, pirexia, molestia torácica, malestar.
Poco frecuentes: edema facial, edema gravitacional, síndrome pseudogripal, escalofríos, sensación de cambio en la temperatura corporal (incluyendo sensación de calor y de frío).
Frecuencia no conocida: edema localizado.
Exploraciones complementarias:
Muy frecuentes: aumento de la alanina aminotransferasa, aumento de aspartato aminotransferasa, aumento de lipasa, aumento del colesterol de lipoproteínas (incluyendo de baja densidad y de alta densidad), aumento del colesterol total, aumento de los triglicéridos sanguíneos.
Frecuentes: disminución de la hemoglobina, aumento de la amilasa sanguínea, aumento de la fosfatasa alcalina sanguínea, aumento de la gamma-glutamiltransferasa, aumento de la creatinina fosfoquinasa sanguínea, disminución de peso, aumento de peso, aumento de la insulina sanguínea, disminución de globulinas.
Poco frecuentes: aumento de la lactato deshidrogenasa sanguínea, disminución de la glucosa sanguínea, aumento de la urea sanguínea.
Frecuencia no conocida: aumento de la troponina, aumento de la bilirrubina no conjugada en la sangre, disminución de la insulina sanguínea, disminución del péptido C de insulina, aumento de la hormona paratiroidea sanguínea.
En la Tabla 3 se presentan los valores de laboratorio anormales clínicamente relevantes o graves encontrados en los controles hematológicos o bioquímicos rutinarios.
Tabla 3 Valores de laboratorio anormales de grado 3-4*
|
LMC-FC de nuevo diagnóstico 300 mg dos veces al día |
LMC-FC y LMC-FA con resistencia o intolerancia a imatinib 400 mg dos veces al día | ||
|
N=279 (%) |
LMC-FC n=321 (%) |
LMC-FA n=137 (%) | |
|
Parámetros hematológicos | |||
|
Mielosupresión | |||
|
- Neutropenia |
12 |
31 |
42 |
|
- Trombocitopenia |
10 |
30 |
42 |
|
- Anemia |
4 |
11 |
27 |
|
Parámetros bioquímicos | |||
|
- Creatinina aumentada |
0 |
1 |
<1 |
|
- Lipasa aumentada |
9 |
18 |
18 |
|
- SGOT (AST) aumentada |
1 |
3 |
2 |
|
- SGPT (ALT) aumentada |
4 |
4 |
4 |
|
- Hipofosfatemia |
7 |
17 |
15 |
|
- Bilirrubina aumentada (total) |
4 |
7 |
9 |
|
- Glucosa elevada |
7 |
12 |
6 |
|
- Colesterol elevado (total) |
0 |
** |
** |
|
- Triglicéridos elevados |
0 |
** |
** |
* Se utilizan porcentajes con precisión de un decimal que están redondeados a números enteros para la presentación en esta tabla.
** Parámetros no recogidos.
Descripción de las reacciones adversas seleccionadas
Muerte súbita
Se han notificado casos poco frecuentes (0,1 a 1%) de muertes súbitas en los ensayos clínicos y/o en los programas de uso compasivo con Tasigna en pacientes con LMC en fase crónica o fase acelerada con resistencia o intolerancia a imatinib con antecendentes de enfermedad cardiaca o factores de riesgo cardiacos significativos (ver sección 4.4).
Reactivación de la hepatitis B
Se ha notificado reactivación de la hepatitis B en relación con los inhibidores de la tirosina quinasa BCR-ABL. En algunos casos se ha producido insuficiencia hepática aguda o hepatitis fulminante que ha dado lugar a trasplante de hígado o a un desenlace mortal (ver sección 4.4).
Experiencia post-comercialización
Las siguientes reacciones adversas se han obtenido a partir de la experiencia post-comercialización con Tasigna a través de informes de casos espontáneos, casos descritos en la literatura, programas de acceso expandido, y ensayos clínicos diferentes de los ensayos globales para el registro. Puesto que estas reacciones se han notificado voluntariamente a partir de una población de un tamaño incierto, no siempre es posible estimar de forma fiable o establecer una relación causal con la exposición a nilotinib.
Frecuencia rara: Se han notificado casos de síndrome de lisis tumoral en pacientes tratados con Tasigna.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se han notificado casos aislados de sobredosis intencionada con nilotinib, en que se ingirieron un número no especificado de cápsulas duras de Tasigna combinadas con alcohol y con otros medicamentos. Los acontecimientos incluyeron neutropenia, vómitos y somnolencia. No se notificaron cambios en el ECG o hepatotoxicidad. Las resoluciones de los casos se notificaron como recuperados.
En caso de sobredosis, se deberá mantener al paciente en observación y administrarle el tratamiento de apoyo adecuado.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes antineoplásicos, inhibidores de la proteína quinasa, código ATC: L01XE08
Nilotinib es un inhibidor potente de la actividad de la tirosina cinasa ABL de la oncoproteína BCR-ABL tanto en las líneas celulares como en las células leucémicas primarias cromosoma Filadelfia positivo. La sustancia se une con alta afinidad al lugar de unión del ATP de tal manera que es un inhibidor potente del BCR-ABL de tipo germinal y mantiene la actividad frente a 32/33 formas mutantes de BCR-ABL resistentes a imatinib. Como consecuencia de esta actividad bioquímica, nilotinib inhibe de forma selectiva la proliferación e induce la apoptosis en líneas celulares y en células leucémicas primarias cromosoma Filadelfia positivo, de pacientes con LMC. En modelos de LMC en roedores, nilotinib como agente único reduce la carga del tumor y prolonga la supervivencia después de la administración oral.
Nilotinib tiene poco o ningún efecto sobre la mayoría de las otras proteínas cinasas examinadas, incluyendo Src, excepto para las cinasas de los receptores PDGF, KIT y Ephin, a las que inhibe a concentraciones dentro del intervalo alcanzado tras la administración oral a las dosis terapéuticas recomendadas para el tratamiento de la LMC (ver Tabla 4).
Tabla 4 Perfil de cinasas de nilotinib (fosforilación IC50 nM)
|
BCR-ABL |
PDGFR |
KIT |
|
20 |
69 |
210 |
Ensayos clínicos en LMC de nuevo diagnóstico en fase crónica
Se ha realizado un ensayo Fase III abierto, multicéntrico, aleatorizado para determinar la eficacia de nilotinib frente imatinib en 846 pacientes adultos con LMC cromosoma Filadelfia positivo en fase crónica confirmada citogenéticamente. Los pacientes se encontraban dentro de los seis meses posteriores al diagnóstico y no habían recibido tratamientos anteriores, excepto hidroxiurea y/o anagrelida. Los pacientes fueron aleatorizados 1:1:1 para recibir tratamiento con nilotinib 300 mg dos veces al día (n=282), nilotinib 400 mg dos veces al día (n=281) o imatinib 400 mg una vez al día (n=283). La aleatorización se estratificó por el índice de riesgo Sokal en el momento del diagnóstico.
Las características basales estaban equilibradas entre los tres brazos de tratamiento. La mediana de edad fue de 47 años en los dos brazos de nilotinib y de 46 años en el brazo de imatinib, con un porcentaje de pacientes con una edad >65 años de 12,8%, 10,0% y 12,4% en el grupo de nilotinib 300 mg dos veces al día, de nilotinib 400 mg dos veces al día y de imatinib 400 mg una vez al día, respectivamente. Se incluyeron un número ligeramente superior de pacientes hombres que de mujeres (56,0%, 62,3% y 55,8% en el brazo de nilotinib 300 mg dos veces al día, 400 mg dos veces al día y de imatinib 400 mg una vez al día, respectivamente). Más del 60% de todos los pacientes eran caucásicos y 25% de todos los pacientes eran asiáticos.
El punto para el análisis primario de datos fue cuando todos los 846 pacientes completaron 12 meses de tratamiento (o interrumpieron el tratamiento antes). Los análisis siguientes reflejan datos de cuando los pacientes completaron 24, 36, 48, 60 y 72 meses de tratamiento (o interrumpieron el tratamiento antes). La mediana de tiempo de tratamiento fue aproximadamente de 70 meses en los grupos de tratamiento con nilotinib y 64 meses en el grupo de imatinib. La mediana de las dosis realmente administradas fue de 593 mg/día para nilotinib 300 mg dos veces al día, de 772 mg/día para nilotinib 400 mg dos veces al día y de 400 mg/día para imatinib 400 mg una vez al día. Este estudio continua en marcha.
La variable de eficacia primaria fue la respuesta molecular mayor (RMM) a los 12 meses. La RMM se definió como <0,1% BCR-ABL/ABL% por una escala internacional (EI) medida por RQ-PCR, que corresponde a una reducción de >3 log del tránscrito BCR-ABL respecto al valor basal estandarizado. La tasa de RMM a los 12 meses fue superior de forma estadísticamente significativa para nilotinib 300 mg dos veces al día que para imatinib 400 mg una vez al día (44,3% frente a 22,3%, p<0,0001). La tasa de RMM a los 12 meses también fue superior de forma estadísticamente significativa para nilotinib 400 mg dos veces al día que para imatinib 400 mg una vez al día (42,7% frente a 22,3%,
p<0,0001).
Las tasas de RMM a 3, 6, 9 y 12 meses fueron 8,9%, 33,0%, 43,3% y 44,3% para nilotinib 300 mg dos veces al día, 5,0%, 29,5%, 38,1% y 42,7% para nilotinib 400 mg dos veces al día y 0,7%, 12,0%, 18,0% y 22,3% para imatinib 400 mg una vez al día.
En la Tabla 5 se presenta la tasa de RMM a los 12, 24, 36, 48, 60 y 72 meses.
Tabla 5 Tasa de RMM
|
Tasigna 300 mg dos veces al día n=282 (%) |
Tasigna 400 mg dos veces al día n=281 (%) |
Imatinib 400 mg una vez al día n=283 (%) | |
|
RMM a los 12 meses | |||
|
Respuesta (IC 95%) |
44,3' (38,4; 50,3) |
42J1 (36,8; 48,7) |
22,3 (17,6; 27,6) |
|
RMM a los 24 meses | |||
|
Respuesta (IC 95%) |
61,7' (55,8; 67,4) |
59,1 (53,1; 64,9) |
37,5 (31,8; 43,4) |
|
RMM a los 36 meses2 | |||
|
Respuesta (IC 95%) |
58,54 (52,5; 64,3) |
57,3: (51,3; 63,2) |
38,5 (32,8; 44,5) |
|
RMM a los 48 meses3 | |||
|
Respuesta (IC 95%) |
59,9: (54,0; 65,7) |
55,2 (49,1; 61,1) |
43,8 (38,0; 49,8) |
|
RMM a los 60 meses4 | |||
|
Respuesta (IC 95%) |
62,8 (56,8; 68,4) |
61,2 (55,2; 66,9) |
49,1 (43,2; 55,1) |
|
RMM a los 72 meses5 | |||
|
Respuesta (IC 95%) |
52,5 (46,5; 58,4) |
57,7 (51,6; 63,5) |
41,7 (35,9; 47,7) |
1 Test de Cochran-Mantel-Haenszel (CMH) valor de p para la tasa de respuesta (frente a imatinib
400 mg) <0,0001
2 Sólo se incluyeron los pacientes que estaban en RMM en un punto de tiempo específico como respondedores de este punto de tiempo. Un total de 199 de todos los pacientes (35,2%) no fueron evaluables para la RMM a los 36 meses (87 en el grupo de nilotinib 300 mg dos veces al día y 112 en el grupo de imatinib) debido a unas valoraciones de PCR perdidas/no evaluables (n=17), tránscritos atípicos en el inicio (n=7), o interrupción anterior a los 36 meses (n=175).
3 Sólo se incluyeron pacientes que estaban en RMM en un punto de tiempo específico como respondedores para aquel punto de tiempo. Un total de 305 de todos los pacientes (36,1%) no fueron evaluables para RMM a los 48 meses (98 en el grupo de nilotinib 300 mg dos veces al día, 88 en el grupo de nilotinib 400 mg dos veces al día y 119 en el grupo de imatinib) debido a las valoraciones de PCR no disponibles/no evaluables (n=18), tránscritos atípicos en el punto basal (n=8), o interrupción anterior a los 48 meses (n=279).
4 Sólo se incluyeron pacientes que estaban en RMM en un punto de tiempo específico como respondedores para aquel punto en el tiempo. Un total de 322 (38,1%) de todos los pacientes no fueron evaluables para RMM a los 60 meses (99 en el grupo de nilotinib 300 mg dos veces al día, 93 en el grupo de nilotinib 400 mg dos veces al día y 130 en el grupo de imatinib) debido a las valoraciones de PCR no disponibles/no evaluables (n=9), tránscritos atípicos en el punto basal (n=8) o interrupción anterior a los 60 meses (n=305).
5 Sólo se incluyeron pacientes que estaban en RMM en un punto de tiempo específico como respondedores para aquel punto en el tiempo. Un total de 395 de todos los pacientes (46,7%) no fueron evaluables para RMM a los 72 meses (130 en el grupo de nilotinib 300 mg dos veces al día,
110 en el grupo de nilotinib 400 mg dos veces al día y 155 en el grupo de imatinib) debido a las valoraciones de PCR no disponibles/no evaluables (n=25), transcritos atípicos en el punto basal (n=8) o interrupción anterior a los 72 meses (n=362).
Se presentan las tasas de RMM en diferentes puntos de tiempo (incluyendo pacientes que alcanzaron la RMM en estos puntos de tiempo o antes como respondedores) en la incidencia acumulada de RMM (ver Figura 1).
Figura 1 Incidencia acumulada de RMM
incidencia acumulada de RMM, %
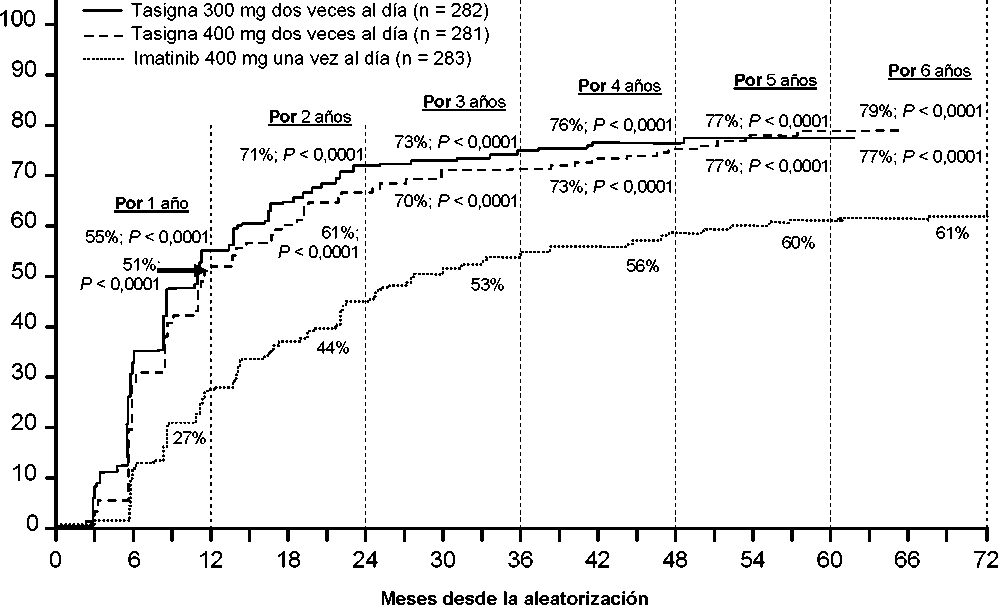
Para todos los grupos de riesgo Sokal, las tasas de RMM en todos los puntos de tiempo se mantuvieron de forma constante superiores en los dos grupos de nilotinib respecto al grupo de imatinib.
En un análisis retrospectivo, el 91% de los pacientes (234/258) tratados con nilotinib 300 mg dos veces al día alcanzaron los niveles de BCR-ABL <10% a los 3 meses de tratamiento comparado al 67% de los pacientes (176/264) tratados con imatinib 400 mg una vez al día. Los pacientes con niveles de BCR-ABL <10% a los 3 meses de tratamiento mostraron una supervivencia global a los 72 meses comparado a los que no alcanzaron este nivel de respuesta molecular (94,5% frente a 77,1% respectivamente [p=0,0005].
En base al análisis de Kaplan-Meier del tiempo hasta la primera RMM la probabilidad de alcanzar una RMM a diferentes puntos de tiempo fue superior para ambos grupos de nilotinib a 300 mg y 400 mg dos veces al día que para imatinib 400 mg una vez al día (HR=2,17, y orden logarímico (log-rank) estratificado p<0,0001 entre nilotinib 300 mg dos veces al día e imatinib 400 mg una vez al día, HR=1,88 y orden logarítmico estratificado p<0,0001 entre nilotinib 400 mg dos veces al día e imatinib 400 mg una vez al día).
La proporción de pacientes que presentaron una respuesta molecular de <0,01% y <0,0032% por EI a diferentes puntos de tiempo se presentan en la Tabla 6 y la proporción de pacientes que presentaron una respuesta molecular de <0,01% y <0,0032% por EI por diferentes puntos de tiempo se presentan en las Figuras 2 y 3. Las respuestas moleculares de <0,01% y <0,0032% por EI corresponde a una reducción de >4 log y >4,5 log respectivamente de los tránscritos de BCR-ABL a partir del valor basal.
Tabla 6 Proporciones de pacientes que presentaron respuesta molecular de <0,01% (reducción de 4 log) y <0,0032% (reducción de 4,5 log)
|
Ta 300 mg do n= |
signa s veces al día 282 '%) |
Ta 400 mg do n= |
signa s veces al día =281 '%) |
Im 400 mg u n= |
atinib na vez al día =283 í%) | |
|
<0,01% |
<0,0032% |
<0,01% |
< 0,0032% |
<0,01% |
<0,0032% | |
|
A los 12 meses |
11,7 |
4,3 |
8,5 |
4,6 |
3,9 |
0,4 |
|
A los 24 meses |
24,5 |
12,4 |
22,1 |
7,8 |
10,2 |
2,8 |
|
A los 36 meses |
29,4 |
13,8 |
23,8 |
12,1 |
14,1 |
8,1 |
|
A los 48 meses |
33,0 |
16,3 |
29,9 |
17,1 |
19,8 |
10,2 |
|
A los 60 meses |
47,9 |
32,3 |
43,4 |
29,5 |
31,1 |
19,8 |
|
A los 72 meses |
44,3 |
31,2 |
45,2 |
28,8 |
27,2 |
18,0 |
Figura 2 Incidencia acumulada de respuesta molecular de <0,01% (reducción de 4-log)
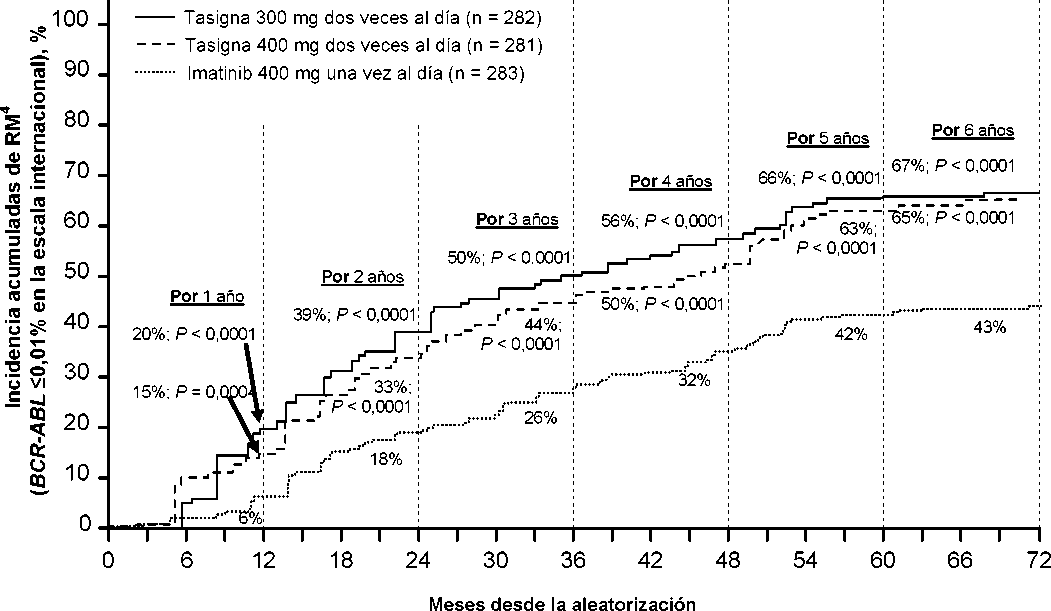

En base a la estimación de Kaplan-Meier de la duración de la primera RMM, la proporción de pacientes que mantuvieron la respuesta durante 72 meses entre los pacientes que alcanzaron una RMM fueron 92,5% (IC 95: 88,6-96,4%) en el grupo de nilotinib 300 mg dos veces al día, 92,2% (IC 95%: 88,5-95,9%) en el grupo de nilotinib 400 mg dos veces al día y 88,0% (IC 95%: 83,0-93,1%) en el grupo de imatinib 400 mg una vez al día.
La respuesta citogenética completa (RCC) se definió como un 0% de metafases en la médula ósea basado en un mínimo de 20 metafases evaluadas. La mejor tasa de RCC a los 12 meses (incluyendo pacientes que alcanzaron la RCC a los 12 meses o antes como respondedores) fue estadísticamente superior para ambos grupos de nilotinib 300 mg y 400 mg dos veces al día comparado con imatinib 400 mg una vez al día, ver Tabla 7.
La tasa de RCC hasta los 24 meses (incluye pacientes que alcanzaron RCC a los 24 meses o antes como respondedores) fue superior estadistica y significativamente para ambos grupos nilotinib 300 mg dos veces al día y 400 mg dos veces al día en comparación con el grupo de imatinib 400 mg una vez al día.
Tabla 7 Tasa de mejor respuesta citogenética completa (RCC)
|
Tasigna (nilotinib) 300 mg dos veces al día n=282 (%) |
Tasigna (nilotinib) 400 mg dos veces al día n=281 (%) |
Glivec (imatinib) 400 mg una vez al día n=283 (%) | |
|
Hasta los 12 meses | |||
|
Respuesta (IC 95%) |
80,1 (75,0; 84,6) |
77,9 (72,6; 82,6) |
65,0 (59,2; 70,6) |
|
Sin respuesta |
19,9 |
22,1 |
35,0 |
|
Test CMH valor de p para la tasa de respuesta (frente a imatinib 400 mg una vez al día) |
<0,0001 |
0,0005 | |
|
Hasta los 24 meses | |||
|
Respuesta (IC 95%) |
86,9 (82,4; 90,6) |
84,7 (79,9; 88,7) |
77,0 (71,7; 818) |
|
Sin respuesta |
13,1 |
15,3 |
23,0 |
|
Test CMH valor de p para la tasa de respuesta (frente a imatinib 400 mg una vez al día) |
0,0018 |
0,0160 |
En base a la estimación de Kaplan-Meier, la proporción de pacientes que mantuvieron la respuesta durante 72 meses entre los pacientes que alcanzaron una RCC fueron 99,1% (IC 95%: 97,9-100%) en el grupo de nilotinib 300 mg dos veces al día, 98,7% (IC 95%: 97,1-100%) en el grupo de nilotinib 400 mg dos veces al día y 97,0% (IC 95%: 94,7-99,4%) en el grupo de imatinib 400 mg una vez al día.
La progresión a fase acelerada (FA) o crisis blástica (CB) durante el tratamiento se define como el tiempo desde la fecha de la aleatorización hasta la primera progresión documentada de la enfermedad a fase acelerada o crisis blástica o muerte relacionada con la LMC. Se observó una progresión a fase acelerada o crisis blástica durante el tratamiento en un total de 17 pacientes: 2 pacientes en el grupo de nilotinib 300 mg dos veces al día, 3 pacientes en el grupo de nilotinib 400 mg dos veces al día y 12 pacientes en el grupo de imatinib 400 mg una vez al día. Las tasas estimadas de pacientes sin progresión a fase acelerada o crisis blástica a los 72 meses fueron de 99,3%, 98,7% y 95,2%, respectivamente (HR=0,1599 y orden logarítmico (log-rank) estratificado p=0,0059 entre nilotinib 300 mg dos veces al día e imatinib una vez al día, HR=0,2457 y orden logarítmico (log-rank) estratificado p=0,0185 entre nilotinib 400 mg dos veces al día e imatinib una vez al día). No se notificaron nuevos eventos de progresión a FA/CB durante el tratamiento desde el análisis de los 2 años.
Incluyendo la evolución clonal como criterio de progresión, un total de 25 pacientes progresaron a fase acelerada o crisis blástica durante el tratamiento en la fecha de corte (3 en el grupo de nilotinib 300 mg dos veces al día, 5 en el grupo de nilotinib 400 mg dos veces al día y 17 en el grupo de imatinib 400 mg una vez al día). Las tasas estimadas de pacientes sin progresión a fase acelerada o crisis blástica incluyendo evolución clonal a los 72 meses fueron 98,7%, 97,9% y 93,2%, respectivamente (HR=0,1626 y orden logarítmico (log-rank) estratificado p=0,0009 entre nilotinib 300 mg dos veces al día e imatinib una vez al día, HR=0,2848 y orden logarítmico (log-rank) estratificado p=0,0085 entre nilotinib 400 mg dos veces al día e imatinib una vez al día).
Un total de 55 pacientes fallecieron durante el tratamiento o durante el seguimiento después de la interrupción del tratamiento. (21 en el grupo de nilotinib 300 mg dos veces al día, 11 en el grupo de nilotinib 400 mg dos veces al día y 23 en el grupo de imatinib 400 mg una vez al día). Veintiséis (26) de estas 55 muertes estuvieron relacionadas con la LMC (6 en el grupo de nilotinib 300 mg dos veces al día, 4 en el grupo de nilotinib 400 mg dos veces al día y 16 en el grupo de imatinib 400 mg una vez al día). Las tasas estimadas de pacientes vivos a los 72 meses fueron 91,6%, 95,8% y 91,4%, respectivamente (HR=0,8934 y orden logarítmico (log-rank) estratificado p=0,7085 entre nilotinib 300 mg dos veces al día e imatinib, HR=0,4632 y orden logarítmico (log-rank) estratificado p=0,0314 entre nilotinib 400 mg dos veces al día e imatinib). Considerando sólo las muertes relacionadas con la LMC como eventos, las tasas estimadas de supervivencia global a los 72 meses fueron 97,7%, 98,5% y 93,9%, respectivamente (HR=0,3694 y orden logarítmico (log-rank) estratificado p=0,0302 entre nilotinib 300 mg dos veces al día e imatinib, HR=0,2433 y orden logarítmico (log-rank) estratificado p=0,0061 entre nilotinib 400 mg dos veces al día e imatinib).
Ensayos clínicos en LMC en fase crónica y fase acelerada resistente o intolerante a imatinib Se ha realizado un ensayo de fase II, multicéntrico, no controlado y abierto para determinar la eficacia de Tasigna en pacientes con LMC resistente o intolerante a imatinib con brazos de tratamiento separados para la enfermedad en fase crónica y en fase acelerada. La eficacia se basó en los 321 pacientes en FC y 137 pacientes en FA incluidos. La mediana de duración del tratamiento fue de 561 días para pacientes en FC y 264 días para pacientes en FA (ver Tabla 8). Tasigna se administró de forma continuada (dos veces al día, 2 horas después de una comida y sin ingerir alimentos durante al menos una hora después de la administración) excepto en los casos de evidencia de una respuesta no adecuada o de progresión de la enfermedad. La dosis fue de 400 mg dos veces al día y se permitió el escalado de dosis a 600 mg dos veces al día.
Tabla 8 Duración de la exposición a Tasigna
|
Fase crónica |
Fase acelerada | |
|
n=321 |
n=137 | |
|
Mediana de duración del tratamiento en |
561 |
264 |
|
días |
(196-852) |
(115-595) |
|
(percentiles 25°-75°) |
La resistencia a imatinib incluyó el fallo en alcanzar una respuesta hematológica completa (en 3 meses), una respuesta citogenética completa (en 6 meses) o una respuesta citogenética mayor (en 12 meses) o la progresión de la enfermedad después de una respuesta citogenética o hematológica previa. La intolerancia a imatinib incluye pacientes que abandonaron el tratamiento con imatinib debido a la toxicidad y que no tenían respuesta citogenética mayor en el momento de la entrada en el ensayo.
En conjunto, el 73% de los pacientes fueron resistentes a imatinib, mientras que el 27% fueron intolerantes a imatinib. La mayoría de pacientes presentaban una larga historia de LMC que incluía un amplio tratamiento previo con otros agentes antineoplásicos, incluyendo imatinib, hidroxiurea, interferon, y algunos incluso habían fallado a un trasplante hematopoyético (Tabla 9). La mediana de dosis más alta de imatinib previa había sido 600 mg/día. La dosis más alta de imatinib previa fue >600 mg/día en el 74% del total de pacientes, con un 40% de pacientes que recibieron dosis previas de imatinib >800 mg/día.
Tabla 9 Características de los antecedentes de la LMC
|
Fase crónica |
Fase acelerada | |
|
(n=321) |
(n=137)* | |
|
Mediana de tiempo desde el diagnóstico |
58 |
71 |
|
en meses (intervalo) |
(5-275) |
(2-298) |
|
Imatinib | ||
|
Resistente |
226 (70%) |
109 (80%) |
|
Intolerante sin RCM |
95 (30%) |
27 (20%) |
|
Mediana de tiempo de tratamiento con |
975 |
857 |
|
imatinib en días (percentil 25°-75°) |
(519-1.488) |
(424-1.497) |
|
Hidroxiurea previa |
83% |
91% |
|
Interferon previo |
58% |
50% |
|
Trasplante de médula ósea previo |
7% |
8% |
* Para un paciente falta la información sobre el estado de resistencia/intolerancia a imatinib
El objetivo primario en los pacientes en FC fue la respuesta citogenética mayor (RCM), definido como la eliminación (RCC, respuesta citogenética completa) o la reducción significativa a <35% de las metafases Ph+ (respuesta citogenética parcial) de las células hematopoyéticas Ph+. Como objetivo secundario se evaluó la respuesta hematológica completa (RHC) en los pacientes en FC. El objetivo primario en los pacientes en FA fue la respuesta hematológica (RH) confirmada total, definida bien como respuesta hematológica completa, sin evidencia de leucemia o bien retorno a la fase crónica.
Fase crónica
La tasa de RCM en 321 pacientes en FC fue del 51%. La mayoría de respondedores consiguieron una RCM rápidamente dentro de los 3 meses (2,8 meses de mediana) del inicio del tratamiento con Tasigna y esta se mantuvo. La mediana de tiempo para alcanzar la RCM fue justo tras 3 meses (mediana 3,4 meses). De los pacientes que alcanzaron la RCM, el 77% (IC95%: 70% - 84%) mantuvieron la respuesta a los 24 meses. No se ha alcanzado la mediana de duración de la RCM. De los pacientes que alcanzaron la RCC el 85% (IC95%: 78% - 93%) mantuvieron la respuesta a los 24 meses. No se ha alcanzado la mediana de duración de la RCC. Los pacientes con una RHC basal alcanzaron una RCM más rápidamente (1,9 meses frente a 2,8 meses). De los pacientes en FC sin una RHC basal, el 70% consiguieron una RHC la mediana de tiempo hasta la RHC fue de 1 mes y la duración mediana de la RHC fue de 32,8 meses. La tasa de supervivencia global estimada a 24 meses en pacientes con LMC-FC fue de 87%.
Fase acelerada
La tasa de RH confirmada total en 137 pacientes en FA fue del 50%. La mayoría de los respondedores alcanzaron una RH temprana con el tratamiento con Tasigna (mediana 1,0 meses) y éstas han sido duraderas (la duración mediana de la RH confirmada fue de 24,2 meses). De los pacientes que alcanzaron RH, el 53% (IC95%: 39% - 67%) mantuvieron la respuesta a los 24 meses. La tasa de RCM fue del 30% con una mediana de tiempo hasta la respuesta de 2,8 meses. De los pacientes que alcanzaron RCM, el 63% (IC95%: 45% - 80%) mantuvieron la respuesta a los 24 meses. La mediana de duración de la RCM fue de 32,7 meses. La tasa de supervivencia global estimada a 24 meses en pacientes con LMC-FA fue del 70%.
1 114 pacientes en FC presentaron una RHC basal y no fueron, por lo tanto, evaluables para la respuesta hematológica completa
Tabla 10 Respuesta en LMC
|
(Mejor Tasa de Respuesta) |
Fase Crónica |
Fase Acelerada | ||||
|
Intolerantes (n=95)_ |
Resistentes (n=226) |
Total (n=321) |
Intolerantes (n=27)_ |
Resistentes (n=109) |
Total* (n=137) | |
|
Respuesta hematológica (%) | ||||||
|
Global (95%IC) Completa NEL Retorno a la FC |
87 (74-94) |
65 (56-72) |
701 (63-76) |
48 (29-68) 37 7 4 |
51 (42-61) 28 10 13 |
50 (42-59) 30 9 11 |
|
Respuesta citogenética (%) | ||||||
|
Mayor (IC95%) Completa Parcial |
57 (46-67) 41 16 |
49 (42-56) 35 14 |
51 (46-57) 37 15 |
33 (17-54) 22 11 |
29 (21-39) 19 10 |
30 (22-38) 20 10 |
|
NEL = no evidencia de leucemia/respuesta méd |
ula | |||||
* Para un paciente falta la información sobre el estado de resistencia/intolerancia a imatinib.
No se dispone todavía de datos de eficacia en pacientes con LMC-CB. En el ensayo de Fase II también se incluyeron brazos separados de tratamiento para investigar Tasigna en un grupo de pacientes en FC y FA que habían sido ampliamente pre-tratados con múltiples tratamientos incluyendo un agente inhibidor de la tirosina cinasa además de imatinib. De estos pacientes, 30/36 (83%) fueron resistentes al tratamiento no intolerantes. En 22 pacientes en FC evaluados para eficacia, Tasigna indujo una tasa del 32% de RCM y una tasa del 50% de RHC. En 11 pacientes en FA, evaluados para eficacia, el tratamiento indujo una tasa de RH total del 36%.
Tras el fallo de imatinib, se observaron 24 mutaciones BCR-ABL diferentes en el 42% de pacientes con LMC en fase crónica y el 54% de pacientes con LMC en fase acelerada, en los que se evaluaron las mutaciones. Tasigna demostró eficacia en los pacientes con una variedad de mutaciones BCR-ABL asociadas a resistencia a imatinib, excepto para T315I.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Tasigna en pacientes pediátricos desde el nacimiento hasta menos de 18 años en el tratamiento de la leucemia mieloide crónica cromosoma Filadelfia positivo (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Se alcanzaron concentraciones pico de nilotinib 3 horas después de la administración oral. La absorción de nilotinib después de la administración oral fue de aproximadamente el 30%. No se ha determinado la biodisponibilidad absoluta de nilotinib. Comparado con una solución oral (pH de 1,2 a 1,3), la biodisponibilidad relativa de nilotinib cápsulas es aproximadamente del 50%. En voluntarios sanos, cuando Tasigna se administró junto con la comida aumentaron la Cmax y el area bajo la curva de concentración plasmática-tiempo (AUC) de nilotinib en un 112% y un 82%, respectivamente, en comparación a las condiciones de ayuno. La administración de Tasigna 30 minutos o 2 horas después de la comida aumentó la biodisponibilidad de nilotinib en un 29% o un 15%, respectivamente (ver secciones 4.2, 4.4 y 4.5).
La absorción de nilotinib (biodisponibilidad relativa) puede reducirse en aproximadamente un 48% y un 22% en pacientes con gastrectomía total y gastrectomía parcial, respectivamente.
Distribución
La relación sangre - plasma de nilotinib es 0,71. La unión a proteínas plasmáticas es de aproximadamente el 98% en base a los experimentos in vitro.
Biotransformación
Las principales vías metabólicas identificadas en voluntarios sanos son la oxidación y la hidroxilación. Nilotinib es el principal componente que circula en el plasma. Ninguno de los metabolitos contribuye significativamente a la actividad farmacológica de nilotinib. Nilotinib se metaboliza principalmente por CYP3A4, con una posible contribución menor de CYP2C8.
Eliminación
Tras una dosis única de nilotinib marcado radiactivamente en voluntarios sanos, más del 90% de la dosis se eliminó dentro de los siguientes 7 días, principalmente por las heces (94% de la dosis). Nilotinib inalterado supuso el 69% de la dosis.
La semivida de eliminación aparente estimada a partir de la farmacocinética a dosis múltiples con dosis diarias fue de aproximadamente 17 horas. La variabilidad interpaciente en la farmacocinética de nilotinib fue de moderada a alta.
Linealidad/No linealidad
La exposición a nilotinib en el estado estacionario fue dependiente de la dosis, con aumentos menores a los aumentos proporcionales a la dosis en la exposición sistémica a dosis superiores a 400 mg administrados como única dosis diaria. La exposición sistémica diaria a nilotinib con dosis de 400 mg dos veces al día en el estado estacionario fue un 35% superior que con una dosis de 800 mg una vez al día. La exposición sistémica (AUC) de nilotinib en el estado estacionario a un nivel de dosis de 400 mg dos veces al día fue aproximadamente un 13,4% superior a la de la dosis de 300 mg dos veces al día. La media de concentraciones de nilotinib valle y pico durante 12 meses fueron aproximadamente 15,7% y 14,8% superiores tras la administración de 400 mg dos veces al día comparado con 300 mg dos veces al día. No se observó un aumento relevante en la exposición a nilotinib cuando se aumentó la dosis de 400 mg dos veces al día a 600 mg dos veces al día.
Las condiciones en el estado estacionario se alcanzaron en el día 8. Se observó un aumento en la exposición plasmática a nilotinib entre la primera dosis y el estado estacionario de 2 veces para la dosis diaria y de 3,8 veces para la dosis dos veces al día.
Estudios de biodisponibilidad/bioequivalencia
La administración de una dosis única de 400 mg de nilotinib, utilizando 2 cápsulas duras de 200 mg en los que el contenido de cada cápsula dura se dispersó en una cucharadita de compota de manzana, mostró que era bioequivalente con una administración única de 2 cápsulas duras intactas de 200 mg.
5.3 Datos preclínicos sobre seguridad
Se han realizado estudios con nilotinib para evaluar la farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad, toxicidad reproductiva, fototoxicidad y carcinogenicidad (ratas y ratones).
Nilotinib no mostró efectos sobre las funciones del SNC o respiratorias. Estudios de seguridad cardiaca in vitro mostraron una señal preclínica de prolongación de QT, basadas en el bloqueo de las corrientes hERG y prolongación de la duración del potencial de acción en corazones de conejo aislados, por nilotinib. No se observaron efectos en las medidas del ECG en perros o monos tratados hasta 39 semanas o en un estudio telemétrico especial en perros.
Estudios de toxicidad a dosis repetidas en perros de hasta 4 semanas de duración y en monos cynomolgus de hasta 9 meses de duración mostraron que el órgano diana de toxicidad causada por nilotinib era el hígado. Las alteraciones incluyeron un aumento de la actividad alanina aminotransferasa y fosfatasa alcalina y hallazgos histopatológicos (principalmente hiperplasia/hipertrofia de la célula sinusoidal o célula Kupffer, hiperplasia del conducto biliar y fibrosis periportal). En general los cambios en la química clínica fueron completamente reversibles después de un periodo de recuperación de cuatro semanas y las alteraciones histológicas mostraron una reversibilidad parcial. Las exposiciones a los niveles de dosis más bajos a las que se observaron efectos sobre el hígado fueron menores que la exposición en humanos a una dosis de 800 mg/día. En ratones o ratas tratadas durante un máximo de 26 semanas sólo se observaron alteraciones hepáticas menores. En ratas, perros y monos se observó un aumento del nivel de colesterol, mayoritariamente reversible.
Los estudios de genotoxicidad en sistemas bacterianos in vitro y en sistemas de mamíferos in vitro e in vivo con y sin activación metabólica no revelaron ninguna evidencia de potencial mutagénico para nilotinib.
En el estudio de carcinogenicidad de 2 años en ratas, el órgano diana más importante para lesiones no neoplásicas fue el útero (dilatación, ectasia vascular, hiperplasia celular endotelial, inflamación y/o hiperplasia epitelial). No se encontró evidencia de carcinogenicidad tras la administración de nilotinib a 5, 15 y 40 mg/kg/día. La exposición (en términos de AUC) a la dosis más alta representó aproximadamente de 2 a 3 veces la exposición diaria humana a nilotinib en el estado estacionario (en base a la AUC) a la dosis de 800 mg/día.
En el estudio Tg.rasH2 de 26 semanas sobre carcinogenicidad en ratones, en los que se administró 30, 100 y 300 mg/kg/día de nilotinib, se detectaron papilomas cutáneos/carcinomas a 300 mg/kg, lo que representa aproximadamente de 30 a 40 veces (en términos de AUC) la dosis máxima aprobada en humanos de 800 mg/día (administrada como 400 mg dos veces al día). El Nivel de Efecto-No-Observado para las lesiones neoplásicas de piel fue de 100 mg/kg/día, lo que representa aproximadamente de 10 a 20 veces la dosis máxima aprobada en humanos de 800 mg/día (administrada como 400 mg dos veces al día). Los principales órganos afectados de lesiones no neoplásicas fueron la piel (hiperplasia epidérmica), el crecimiento de los dientes (degeneración/atrofia del órgano del esmalte de los incisivos superiores y la inflamación de la encía/epitelio odontogénico de los incisivos) y el timo (aumento de la incidencia y/o la gravedad de la disminución de los linfocitos).
Nilotinib no indujo teratogenicidad, pero mostró embrio y fetotoxicidad a dosis que también mostraron toxicidad materna. Se observó un aumento en las pérdidas post implantación en estudios de fertilidad, con tratamiento en machos y hembras, y en el estudio de embriotoxicidad, con tratamiento de hembras. En los estudios de embriotoxicidad se observó letalidad embriológica y efectos fetales (principalmente disminución del peso fetal, fusión prematura de los huesos faciales (fusión huesos maxilar superior/cigomático) cambios viscerales y esqueléticos) en ratas y un aumento de la resorción de fetos y modificaciones esqueléticas en conejos. En un estudio de desarrollo pre y postnatal en ratas, la exposición materna a nilotinib causó una reducción en el peso corporal de las crías con cambios asociados en los parámetros de desarrollo físico así como una reducción en los índices de
apareamiento y fertilidad de las crías. La exposición a nilotinib en hembras a Niveles de No Observación de Efectos Adversos fue generalmente menor o igual a la de los humanos a dosis de 800 mg/día.
En un estudio de desarrollo juvenil, se administró nilotinib por vía oral mediante una sonda a ratas jóvenes desde la primera semana post parto hasta que eran adultos jóvenes (día 70 post parto) a dosis de 2, 6 y 20 mg/kg/día. Además de los parámetros estándar del estudio, se llevaron a cabo evaluaciones de elementos de referencia del desarrollo, efectos sobre el SNC, el apareamiento y la fertilidad. En base a una reducción del peso corporal en ambos géneros y un retraso en la separación prepucial en machos (que puede asociarse con una reducción del peso), el Nivel Sin Efectos Observados en ratas jóvenes se consideró que era 6 mg/kg/día. Los animales jóvenes no mostraron una sensibilidad aumentada a nilotinib comparado con los adultos. Además, el perfil de toxicidad en ratas jóvenes fue comparable al observado en ratas adultas.
No se observaron efectos sobre el recuento/movilidad espermática ni sobre la fertilidad en ratas macho o hembra hasta la dosis más alta probada, aproximadamente 5 veces la dosis recomendada en humanos.
Se observó que nilotinib absorbe la luz en el rango de UV-B y UV-A, se distribuye en la piel y muestra un potencial fototóxico in vitro, pero no se observaron efectos in vivo. Por lo tanto, se considera que el riesgo de que nilotinib cause fotosensibilidad en los pacientes es bajo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula dura Lactosa monohidrato Crospovidona Poloxamero 188 Sílice coloidal anhidra Estearato de magnesio
Cubierta de la cápsula dura Gelatina
Dióxido de titanio (E171)
Amarillo, óxido de hierro (E172)
Tinta de impresión Shellac (E904)
Rojo óxido de hierro (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30°C.
Conservar en el embalaje original para protegerlo de la humedad.
6.5 Naturaleza y contenido del envase
Blisteres de PVC/PVDC/Alu y PA/Alu/PVC/Alu.
Tasigna está disponible en los siguientes formatos:
• Envases unitarios que contienen 28 cápsulas duras en un estuche.
• Envases unitarios que contienen 28 cápsulas duras (7 blísteres diarios, cada uno conteniendo
4 cápsulas duras) o 40 cápsulas duras (5 blísteres, cada uno conteniendo 8 cápsulas duras).
• Envases múltiples que contienen 112 cápsulas duras (4 estuches de 28).
• Envases múltiples que contienen 112 cápsulas duras (4 envases de 28), 120 cápsulas duras
(3 envases de 40) o 392 cápsulas duras (14 envases de 28).
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación Ninguna especial para su eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/422/001-004
EU/1/07/422/007-008
EU/1/07/422/011-012
EU/1/07/422/014
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 19 de Noviembre de 2007 Fecha de la última renovación: 19 de Noviembre de 2012
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes
Novartis Pharma GmbH RoonstraBe 25 D-90429 Nürnberg Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia en la Unión (lista EURD), prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Medidas adicionales de minimización de riesgos
El Titular de la Autorización de Comercialización (TAC) debe asegurar que, antes del lanzamiento, todos los médicos que se espera que prescriban este medicamento, y todos los farmacéuticos que puedan dispensar este medicamento, reciben un paquete informativo para profesionales sanitarios que contenga lo siguiente:
• Folleto educativo
• Resumen de Características del Producto (Ficha Técnica), Prospecto y Etiquetado
Elementos clave a incluir en el folleto educativo
• Breve resumen sobre Tasigna, su indicación y posología autorizadas
• Información sobre los riesgos cardíacos asociados con el uso de Tasigna
o Que Tasigna puede provocar la prolongación del intervalo QT y que Tasigna se debe utilizar con precaución en pacientes que tienen una prolongación del QTc o que tienen un riesgo importante de desarrollarlo. El uso concomitante de Tasigna con antiarrítmicos u otros medicamentos que pueden prolongar el intervalo QT se debe realizar con precaución. o Precaución al prescribir Tasigna a pacientes con antecedentes o que presenten factores de riesgo para una enfermedad cardiaca coronaria o Que Tasigna puede causar retención de líquidos, insuficiencia cardiaca y edema pulmonar
• Que Tasigna es metabolizado por CYP3A4 y que los inhibidores potentes o los inductores de esta enzima pueden afectar de forma significativa la exposición a Tasigna.
o Que los inhibidores pueden aumentar el potencial de aparición de reacciones adversas, especialmente la prolongación del intervalo QT. o Advertencia a los pacientes sobre los medicamentos adquiridos sin receta médica, especialmente hierba de San Juan.
• La necesidad de informar a los pacientes sobre los efectos de la comida sobre Tasigna
o No ingerir comida durante dos horas antes y una hora después de tomar Tasigna
o La necesidad de evitar alimentos como el zumo de pomelo, que inhiben las enzimas de CYP3A4
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Tasigna 150 mg cápsulas duras Nilotinib
2. PRINCIPIO(S) ACTIVO(S)
Una cápsula dura contiene 150 mg de nilotinib (como clorhidrato monohidrato).
3. LISTA DE EXCIPIENTES
Contiene lactosa - Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsulas duras
28 cápsulas duras 40 cápsulas duras
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
No conservar a temperatura superior a 30°C.
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/422/005 28 cápsulas duras
EU/1/07/422/009 40 cápsulas duras
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Tasigna 150 mg
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLÍSTERS
1. NOMBRE DEL MEDICAMENTO
Tasigna 150 mg cápsulas duras Nilotinib
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
1. NOMBRE DEL MEDICAMENTO
Tasigna 150 mg cápsulas duras Nilotinib
2. PRINCIPIO(S) ACTIVO(S)
Una cápsula dura contiene 150 mg de nilotinib (como clorhidrato monohidrato).
3. LISTA DE EXCIPIENTES
Contiene lactosa - Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsulas duras
Envase múltiple: 112 cápsulas duras (4 envases de 28). Envase múltiple: 120 cápsulas duras (3 envases de 40). Envase múltiple: 392 cápsulas duras (14 envases de 28).
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
No conservar a temperatura superior a 30°C.
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/422/006
EU/1/07/422/010
EU/1/07/422/013
112 cápsulas duras 120 cápsulas duras 392 cápsulas duras
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Tasigna 150 mg
1. NOMBRE DEL MEDICAMENTO
Tasigna 150 mg cápsulas duras Nilotinib
2. PRINCIPIO(S) ACTIVO(S)
Una cápsula dura contiene 150 mg de nilotinib (como clorhidrato monohidrato).
3. LISTA DE EXCIPIENTES
Contiene lactosa - Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsulas duras
28 cápsulas duras. Componente de un envase múltiple. No se vende de forma separada. 40 cápsulas duras. Componente de un envase múltiple. No se vende de forma separada.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
No conservar a temperatura superior a 30°C.
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/422/006
EU/1/07/422/010
EU/1/07/422/013
112 cápsulas duras 120 cápsulas duras 392 cápsulas duras
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Tasigna 150 mg
1. NOMBRE DEL MEDICAMENTO
Tasigna 200 mg cápsulas duras Nilotinib
2. PRINCIPIO(S) ACTIVO(S)
Una cápsula dura contiene 200 mg de nilotinib (como clorhidrato monohidrato).
3. LISTA DE EXCIPIENTES
Contiene lactosa - Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsulas duras
28 cápsulas duras 40 cápsulas duras
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
No conservar a temperatura superior a 30°C.
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/422/001
EU/1/07/422/002
EU/1/07/422/007
EU/1/07/422/011
PVC/PVDC/Alu [en estuche] 28 cápsulas duras PA/Aul/PVC/Alu [en estuche] 28 cápsulas duras PVC/PVDC/Alu [en caja] 28 cápsulas duras PVC/PVDC/Alu [en caja] 40 cápsulas duras
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Tasigna 200 mg
1. NOMBRE DEL MEDICAMENTO
Tasigna 200 mg cápsulas duras Nilotinib
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
CAJA DEL ENVASE MÚLTIPLE (ESTUCHE) (INCLUYENDO EL BLUE BOX) CAJA DEL ENVASE MÚLTIPLE (CAJA) (INCLUYENDO EL BLUE BOX)
1. NOMBRE DEL MEDICAMENTO
Tasigna 200 mg cápsulas duras Nilotinib
2. PRINCIPIO(S) ACTIVO(S)
Una cápsula dura contiene 200 mg de nilotinib (como clorhidrato monohidrato).
3. LISTA DE EXCIPIENTES
Contiene lactosa - Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsulas duras
Envase múltiple: 112 cápsulas duras (4 estuches de 28). Envase múltiple: 112 cápsulas duras (4 envases de 28). Envase múltiple: 120 cápsulas duras (3 envases de 40). Envase múltiple: 392 cápsulas duras (14 envases de 28).
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
No conservar a temperatura superior a 30°C.
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/422/003
EU/1/07/422/004
EU/1/07/422/008
EU/1/07/422/012
EU/1/07/422/014
PVC/PVDC/Alu [en estuche] 112 cápsulas duras PA/Alu/PVC/Alu [en estuche] 112 cápsulas duras PVC/PVDC/Alu [en caja] 112 cápsulas duras PVC/PVDC/Alu [en caja] 120 cápsulas duras PVC/PVDC/Alu [en caja] 392 cápsulas duras
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Tasigna 200 mg
ESTUCHE INTERMEDIO DEL ENVASE MÚLTIPLE (SIN EL BLUE BOX) CAJA INTERMEDIA DEL ENVASE MÚLTIPLE (SIN EL BLUE BOX)
1. NOMBRE DEL MEDICAMENTO
Tasigna 200 mg cápsulas duras Nilotinib
Una cápsula dura contiene 200 mg de nilotinib (como clorhidrato monohidrato).
3. LISTA DE EXCIPIENTES
Contiene lactosa - Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsulas duras
28 cápsulas duras. Componente de un envase múltiple que contiene 4 estuches. No se vende de forma separada.
28 cápsulas duras. Componente de un envase múltiple que contiene 4 cajas. No se vende de forma separada.
40 cápsulas duras. Componente de un envase múltiple que contiene 3 cajas. No se vende de forma separada.
28 cápsulas duras. Componente de un envase múltiple que contiene 14 cajas. No se vende de forma separada.
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30°C.
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/422/003
EU/1/07/422/004
EU/1/07/422/008
EU/1/07/422/012
EU/1/07/422/014
PVC/PVDC/Alu [en estuche] 112 cápsulas duras PA/Alu/PVC/Alu [en estuche] 112 cápsulas duras PVC/PVDC/Alu [en caja] 112 cápsulas duras PVC/PVDC/Alu [en caja] 120 cápsulas duras PVC/PVDC/Alu [en caja] 392 cápsulas duras
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Tasigna 200 mg
B. PROSPECTO
Prospecto: información para el paciente
Tasigna 150 mg cápsulas duras
Nilotinib
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Tasigna y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Tasigna
3. Cómo tomar T asigna
4. Posibles efectos adversos
5. Conservación de Tasigna
6. Contenido del envase e información adicional
1. Qué es Tasigna y para qué se utiliza Qué es Tasigna
Tasigna es un medicamento que contiene un principio activo denominado nilotinib.
Para qué se utiliza Tasigna
Tasigna se utiliza para tratar un tipo de leucemia llamada leucemia mieloide crónica cromosoma Filadelfia positivo (LMC Ph-positivo). La LMC es un cáncer de la sangre que provoca que el organismo produzca un exceso de glóbulos blancos anómalos.
Tasigna se utiliza en pacientes con LMC de nuevo diagnóstico.
Cómo actúa Tasigna
En pacientes con LMC, un cambio en el DNA (material genético) genera una señal que hace que el organismo produzca glóbulos blancos anómalos. Tasigna bloquea esta señal y por tanto interrumpe la producción de estas células.
Control del tratamiento con Tasigna
Durante el tratamiento le van a realizar controles de forma regular, incluyendo análisis de sangre. Estos análisis van a controlar:
- la cantidad de células sanguíneas en el organismo (glóbulos blancos, glóbulos rojos y plaquetas) para comprobar si Tasigna es bien tolerado.
- la función del páncreas y del hígado de su organismo para comprobar si Tasigna es bien tolerado.
- los electrolitos de su cuerpo (potasio, magnesio). Estos son importantes en el funcionamiento de su corazón.
- el nivel de azúcar y grasas en su sangre.
También se controlará su frecuencia cardiaca utilizando una máquina que mide la actividad eléctrica del corazón (una prueba llamada “ECG”).
Si tiene cualquier pregunta sobre cómo funciona Tasigna o la causa por la que le han prescrito el fármaco, consulte con su médico.
2. Qué necesita saber antes de empezar a tomar Tasigna
Siga cuidadosamente todas las instrucciones de su médico, aunque éstas sean diferentes de la
información general contenida en este prospecto.
No tome Tasigna
- si es alérgico al nilotinib o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
Si cree que puede ser alérgico, informe a su médico antes de tomar Tasigna.
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar Tasigna:
- si ha sufrido con anterioridad acontecimientos cardiovasculares como un ataque al corazón, dolor en el pecho (angina), problemas con el aporte de sangre a su cerebro (ictus) o problemas con el flujo de sangre a su pierna (claudicación) o si tiene factores de riesgo para enfermedades cardiovasculares como presión sanguínea alta (hipertensión), diabetes o problemas con el nivel de grasas en su sangre (alteraciones de los lípidos).
- si tiene una alteración del corazón, como una señal eléctrica anómala llamada «prolongación del intervalo QT».
- si recibe tratamiento con medicamentos que afectan el ritmo cardíaco (antiarrítmicos) o el hígado (ver Uso de Tasigna con otros medicamentos).
- si sufre falta de potasio o magnesio.
- si tiene una alteración del hígado o del páncreas.
- si tiene síntomas como facilidad de aparición de hematomas, sensación de cansancio o dificultad al respirar o ha presentado infecciones de forma repetida.
- si ha sufrido una intervención quirúrgica que ha supuesto la extirpación del estómago completo (gastrectomía total).
- si alguna vez ha tenido o podría tener en este momento una infección por el virus de la hepatitis B. Esto se debe a que Tasigna podría hacer que la hepatitis B se volviese activa de nuevo, lo que puede resultar mortal en algunos casos. El médico deberá comprobar atentamente si hay signos de esta infección antes de comenzar el tratamiento.
Si alguno de estos casos le es aplicable, informe a su médico.
Durante el tratamiento con Tasigna
- si sufre un desmayo (pérdida de consciencia) o tiene un ritmo cardíaco irregular mientras está tomando Tasigna, informe a su médico inmediatamente pues esto puede ser un signo de un problema grave del corazón. La prolongación del intervalo QT o bien un ritmo cardiaco irregular pueden provocar la muerte súbita. Se han notificado casos poco frecuentes de muerte súbita en pacientes que toman Tasigna.
- si sufre palpitaciones repentinas del corazón, debilidad muscular grave o parálisis, convulsiones o cambios repentinos de comportamiento o nivel de alerta, informe a su médico inmediatamente puesto que puede ser un signo de una rotura rápida de células cancerosas denominado síndrome de lisis tumoral. Se han notificado casos raros de síndrome de lisis tumoral en pacientes tratados con Tasigna.
- si desarrolla dolor en el pecho o malestar, entumecimiento o debilidad, problemas al caminar o con el habla, dolor, decoloración o sensación de frío en una extremidad, informe a su médico inmediatamente pues esto puede ser un signo de un acontecimiento cardiovascular. Se han comunicado casos de acontecimientos cardiovasculares graves incluyendo problemas con el flujo de sangre a la pierna (enfermedad arterial oclusiva periférica), enfermedad isquémica del corazón y problemas con el aporte de sangre al cerebro (enfermedad isquémica cerebrovascular) en pacientes que toman Tasigna. Su médico debe evaluar el nivel de grasas (lípidos) y azúcar en su sangre antes de comenzar el tratamiento con Tasigna y durante el tratamiento con Tasigna.
- si desarrolla hinchazón de los pies o de las manos, hinchazón generalizada o aumento rápido de peso, informe a su médico pues éstos pueden ser signos de retención grave de líquidos. Se han comunicado casos poco frecuentes de retención grave de líquidos en pacientes tratados con Tasigna.
Uso de Tasigna con otros medicamentos
Tasigna puede interferir con algunos medicamentos.
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o pudiera tener que
utilizar cualquier otro medicamento. Estos incluyen, en particular:
- antiarrítmicos - utilizados para tratar el ritmo cardíaco irregular;
- cloroquina, halofantrina, claritromicina, haloperidol, metadona, moxifloxacina - medicamentos que pueden tener un efecto no deseado sobre la función del corazón;
- ketoconazol, itraconazol, voriconazol, claritromicina, telitromicina - utilizados para tratar infecciones;
- ritonavir - un medicamento del grupo de las «antiproteasas» utilizado para tratar el VIH;
- carbamazepina, fenobarbital, fenitoina - utilizados para tratar la epilepsia;
- rifampicina - utilizada para tratar la tuberculosis;
- Hierba de San Juan - un producto derivado de las plantas utilizado para tratar la depresión y otras situaciones (también conocido como Hypericum perforatum);
- midazolam - utilizado para aliviar la ansiedad antes de la cirugía;
- alfentanilo y fentanilo - utilizados para el tratamiento del dolor y como sedantes antes o durante la cirugía o procedimientos médicos;
- ciclosporina, sirolimus y tacrolimus - medicamentos que suprimen la habilidad de “autodefensa” del cuerpo y luchan contra infecciones y que se usan frecuentemente para prevenir el rechazo a órganos trasplantados tales como el hígado, corazón o riñón;
- dihidroergotamina y ergotamina - utilizadas para tratar la demencia;
- lovastatina, simvastatina - utilizadas para tratar los niveles elevados de grasas en sangre;
- warfarina - utilizado para tratar alteraciones de la coagulación (como coágulos en la sangre o trombosis);
- astemizol, terfenadina, cisaprida, pimozida, quinidina, bepridil o alcaloides ergóticos (ergotamina, dihidroergotamina).
Deberá evitarse el uso de estos medicamentos durante el tratamiento con Tasigna. Si está tomando
alguno de estos fármacos, su médico podrá recetarle otros medicamentos alternativos.
Además, informe a su médico o farmacéutico antes de tomar Tasigna si está tomando cualquier antiácido, que son medicamentos contra la acidez de estómago. Estos medicamentos se deben tomar separadamente de Tasigna:
- bloqueadores de H2, que disminuyen la producción de ácido en el estómago. Los bloqueadores H2 se deben tomar aproximadamente 10 horas antes y aproximadamente 2 horas después de tomar Tasigna;
- antiácidos como los que contienen hidróxido de aluminio, hidróxido de magnesio y simeticona, que neutraliza la elevada acidez en el estómago. Estos antiácidos se deben tomar aproximadamente 2 horas antes o aproximadamente 2 horas después de tomar Tasigna.
También debe informar a su médico si ya está tomando Tasigna y le recetan un nuevo medicamento que no ha tomado anteriormente durante el tratamiento con Tasigna.
Toma de Tasigna con alimentos y bebidas
No tome Tasigna junto con las comidas. Los alimentos pueden aumentar la absorción de Tasigna y por lo tanto aumentar la cantidad de Tasigna en la sangre, posiblemente hasta un nivel peligroso. No debe beber zumo de pomelo o comer pomelo. Puede aumentar la cantidad de Tasigna en la sangre, probablemente hasta un nivel peligroso.
Ancianos (65 años o mayores)
Tasigna puede utilizarse por pacientes de 65 años o mayores a la misma dosis que el resto de adultos. Embarazo, lactancia y fertilidad
- No se recomienda el uso de Tasigna durante el embarazo a menos que sea claramente necesario. Si está embarazada o piensa que pudiera estarlo, informe a su médico, quien comentará con usted si puede tomar Tasigna durante el embarazo.
- Las mujeres que pueden quedarse embarazadas deberán utilizar medidas muy efectivas de anticoncepción durante el tratamiento y hasta dos semanas después de finalizar el tratamiento.
- No se recomienda la lactancia durante el tratamiento con Tasigna. Informe a su médico si está en período de lactancia.
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Conducción y uso de máquinas
Si nota efectos adversos (como mareos o problemas de visión) que puedan influir en la capacidad de conducir de forma segura o utilizar herramientas o máquinas después de tomar Tasigna, deberá evitar realizar estas actividades hasta que haya desaparecido el efecto.
Tasigna contiene lactosa
Este medicamento contiene lactosa (también conocido como azúcar de la leche). Si su médico le ha indicado que padece una intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento.
3. Cómo tomar Tasigna
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Qué cantidad de Tasigna tomar
- La dosis recomendada es de 600 mg cada día. Esta dosis se consigue tomando dos cápsulas de 150 mg dos veces al día.
Su médico puede prescribirle una dosis más baja dependiendo de cómo responda al tratamiento.
Cuándo tomar Tasigna
Tome las cápsulas duras:
- dos veces al día (aproximadamente cada 12 horas);
- al menos 2 horas después de ingerir alimentos;
- después esperar 1 hora antes de comer otra vez.
Consulte con su médico o farmacéutico si tiene dudas sobre cuándo tomar Tasigna. La toma de Tasigna cada día a la misma hora le ayudará a recordar cuándo debe tomar las cápsulas duras.
Cómo tomar Tasigna
- Tragar las cápsulas duras enteras con agua.
- No tomar ningún alimento junto con las cápsulas duras.
- No abrir las cápsulas duras excepto si no puede tragarlas. En este caso, puede espolvorear el contenido de cada cápsula dura en una cucharadita de compota de manzana y tomarlo inmediatamente. No utilice más que una cucharadita de compota de manzana para cada cápsula dura y no utilice ningún otro alimento aparte de compota de manzana.
Durante cuánto tiempo tomar Tasigna
Tome Tasigna cada día durante el tiempo que le indique su médico. Este es un tratamiento a largo plazo. Su médico controlará periódicamente su situación para comprobar que el tratamiento está teniendo el efecto deseado.
Si tiene dudas sobre cuánto tiempo debe tomar Tasigna, consulte con su médico.
Si toma más Tasigna del que debe
Si ha tomado más Tasigna del que debe, o si otra persona accidentalmente toma sus cápsulas duras, contacte con un médico u hospital rápidamente. Muestre la caja de las cápsulas duras y este prospecto. Puede necesitar tratamiento médico.
Si olvidó tomar Tasigna
Si se ha olvidado de tomar una dosis, tome la siguiente dosis a su hora habitual. No tome una dosis doble para compensar la cápsula dura olvidada.
Si interrumpe el tratamiento con Tasigna
No interrumpa el tratamiento con Tasigna a menos que se lo indique su médico. Interrumpir el tratamiento con Tasigna sin que se lo haya recomendado su médico le sitúa a usted en un riesgo de empeoramiento de su enfermedad que podría tener consecuencias mortales. Asegúrese de comentarlo con su médico, enfermera y/o farmacéutico si está pensando en interrumpir el tratamiento con Tasigna.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. La mayoría de las reacciones adversas son de leves a moderadas y normalmente desaparecen después de unos pocos días o semanas de tratamiento.
Algunas reacciones adversas pueden ser graves.
Estas reacciones adversas son frecuentes (pueden afectar hasta 1 de cada 10 pacientes), poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes) o se han notificado en muy pocos pacientes.
- rápido aumento de peso, hinchazón de las manos, tobillos, pies o cara (signos de retención de agua)
- dolor en el pecho, aumento de la presión arterial, ritmo cardíaco irregular, coloración azul de los labios, lengua o piel (signos de alteraciones del corazón)
- dificultades para respirar, tos, estertores con o sin fiebre (sonidos que se producen al respirar), hinchazón de pies o piernas (signos de alteraciones de pulmón)
- fiebre, facilidad para tener hematomas, infecciones frecuentes (signos de alteraciones de la sangre)
- visión borrosa, pérdida de visión, sangre en el ojo (signos de alteraciones en el ojo)
- hinchazón y dolor en una parte del cuerpo (signos de coágulos en una vena)
- dolor abdominal, náusea, estreñimiento, hinchazón del abdomen (signos de alteraciones gastrointestinales)
- dolor abdominal superior grave (signo de pancreatitis)
- color amarillo en la piel y los ojos, náusea, pérdida de apetito, orina de color oscuro (signos de alteración del hígado)
- erupción, bultos rojos dolorosos, dolor en las articulaciones y músculos (signos de alteraciones en la piel)
- sed excesiva, aumento en la eliminación de orina, aumento del apetito con pérdida de peso, cansancio (signos de un alto nivel de azúcar en la sangre)
- náuseas, dificultad para respirar, latido cardíaco irregular, orina turbia, cansancio y/o molestias en las articulaciones asociados con resultados anormales de los análisis de sangre (como niveles elevados de potasio, ácido úrico y fósforo y bajos niveles de calcio)
- dolor, molestias, debilidad o calambres en los músculos de las piernas, que pueden ser debidos a la disminución del riego sanguíneo, úlceras en las piernas o brazos que cicatrizan lentamente o no cicatrizan y cambios apreciables en el color (azul o pálido) o la temperatura (enfriamiento) de las piernas o los brazos, pues estos síntomas podrían ser signos de bloqueo de las arterias en el miembro afectado (pierna o brazo) y los dedos (de los pies o de las manos)
- recurrencia (reactivación) de la infección por el virus de la hepatitis B si ha tenido hepatitis B en el pasado (una infección del hígado).
Si le ocurre alguno de estos acontecimientos, informe a su médico inmediatamente.
Algunas reacciones adversas son muy frecuentes (pueden afectar más de 1 de cada 10 pacientes)
- dolor de cabeza
- cansancio
- dolor muscular
- picor, erupción, ronchas
- náuseas
- pérdida de pelo
- nivel de bilirrubina alto en la sangre (función del hígado)
- nivel de lipasa alto en la sangre (función del páncreas)
Si alguno de estos acontecimientos le afecta de forma grave, informe a su médico.
Algunas reacciones adversas son frecuentes (pueden afectar hasta 1 de cada 10 pacientes)
- diarrea, vómitos, malestar abdominal, malestar del estómago después de las comidas, flatulencia, hinchazón del abdomen
- dolor en los huesos, dolor en las articulaciones, espasmos musculares, dolor en las extremidades, dolor de espalda, dolor o molestia en el costado del cuerpo
- irritación del ojo, hinchazón, secreción, picor o enrojecimiento, sequedad del ojo (signos de alteraciones del ojo)
- enrojecimiento de la piel, sequedad de la piel, acné, verrugas, disminución en la sensibilidad de la piel
- pérdida de apetito, alteración en el sentido del gusto, aumento de peso
- insomnio, ansiedad
- sudores nocturnos, excesiva sudoración, sofocos
- mareo, sensación de dar vueltas
- palpitaciones (sensación de latido cardiaco rápido)
Si alguno de estos acontecimientos le afecta de forma grave, informe a su médico.
Algunas reacciones adversas son poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes)
- dolor en la piel
- hinchazón en los párpados
- sangrado de la nariz
- síntomas gripales
- hormigueo o adormecimiento
- alteraciones visuales
- sensación de cambio de temperatura en el cuerpo (incluyendo sensación de calor, de frío)
- manchas engrosadas de color rojo/plateado en la piel (signos de psoriasis)
- sensibilidad en los dientes
Si alguno de estos acontecimientos le afecta de forma grave, informe a su médico.
En muy pocos pacientes tratados con Tasigna, se han notificado las siguientes reacciones adversas:
- pérdida de memoria, alteración en el estado de ánimo o ánimo depresivo, falta de energía, sensación general de malestar
- llagas bucales, infección bacteriana de la piel
- ampollas, quistes en la piel, piel aceitosa, adelgazamiento de la piel, manchas oscuras de la piel, decoloración de la piel
- aumento de la sensibilidad de la piel
- hemorragia, encías sensibles o aumentadas
- rinitis, estornudos
- boca seca, dolor de garganta, llagas en la boca
- temblores
- dolor o enrojecimiento en los ojos, dolor, picor en los párpados
- articulaciones dolorosas y con hinchazón (gota), debilidad muscular
- pérdida de conocimiento
- dificultad y dolor al orinar, sensación exagerada de necesidad de orinar
- orinar frecuentemente, color de la orina anormal
- hemorroides
- sensación de endurecimiento de los pechos, periodos menstruales fuertes, hinchazón en el pezón
- alteración del apetito, disminución de peso
- dolor de cabeza grave, normalmente acompañado de náuseas, vómitos y sensibilidad a la luz
- ardor de estómago
- aumento de mamas en hombres
- síntomas del síndrome de piernas inquietas (una urgencia irresistible a mover una parte del cuerpo, generalmente una pierna, acompañado por sensaciones incómodas)
Si alguno de estos le afecta de forma grave, informe a su médico.
Durante el tratamiento con Tasigna también puede presentar algunos resultados anómalos en los análisis de sangre tales como un nivel bajo de células de la sangre (glóbulos blancos, glóbulos rojos, plaquetas), un nivel alto de lipasa o amilasa en la sangre (función del páncreas), nivel alto de bilirrubina en la sangre (función del hígado) o nivel alto de creatinina en la sangre (función del riñón), nivel bajo o alto de insulina en la sangre (una hormona reguladora del nivel de azúcar en la sangre), nivel bajo o alto de azúcar, o nivel alto de grasas en la sangre.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Tasigna
- Mantener este medicamento fuera de la vista y del alcance de los niños.
- No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en el
blister. La fecha de caducidad es el último día del mes que se indica.
- No conservar a temperatura superior a 30°C
- Conservar en el embalaje original para protegerlo de la humedad.
- No utilice este medicamento, si observa que el envase esté dañado o muestre signos de manipulación.
- Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Tasigna
- El principio activo es nilotinib. Cada cápsula dura contiene 150 mg de nilotinib (como clorhidrato monohidrato).
- Los demás componentes son lactosa monohidrato, crospovidona, poloxamero 188, sílice coloidal anhidra, estearato de magnesio. La cubierta de la cápsula dura se compone de gelatina, dióxido de titanio (E171), rojo y amarillo óxido de hierro (E172), shellac y negro óxido de hierro (E172) para la impresión.
Aspecto de Tasigna y contenido del envase
Tasigna se presenta como cápsulas duras. Las cápsulas duras son de color rojo. Cada cápsula dura tiene una impresión negra («NVR/BCR»).
Tasigna está disponible en envases que contiene 28 o 40 cápsulas duras y en envases múltiples de 112 cápsulas duras (conteniendo 4 cajas, cada una con 28 cápsulas duras), 120 cápsulas duras (conteniendo 3 cajas, cada una con 40 cápsulas duras) o 392 cápsulas duras (conteniendo 14 cajas, cada una con 28 cápsulas duras).
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH RoonstraBe 25 D-90429 Nürnberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Etarapna Novartis Pharma Services Inc. Tea.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Ceská republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 555 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXXáóa Novartis (Hellas) A.E.B.E. Tni: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A. Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
Ísland
Vistor hf.
Sími: +354 535 7000 Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Slovenská republika
Novartis Slovakia s.r.o.
Tel: +421 2 5542 5439
Suomi/Finland
Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu/, y en la página web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) (http://www.aemps.gob.es/). También existen enlaces a otras páginas web sobre enfermedades raras y medicamentos huérfanos.
Prospecto: información para el paciente
Tasigna 200 mg cápsulas duras
Nilotinib
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Tasigna y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Tasigna
3. Cómo tomar T asigna
4. Posibles efectos adversos
5. Conservación de Tasigna
6. Contenido del envase e información adicional
1. Qué es Tasigna y para qué se utiliza Qué es Tasigna
Tasigna es un medicamento que contiene un principio activo denominado nilotinib.
Para qué se utiliza Tasigna
Tasigna se utiliza para tratar un tipo de leucemia llamada leucemia mieloide crónica cromosoma Filadelfia positivo (LMC Ph-positivo). La LMC es un cáncer de la sangre que provoca que el organismo produzca un exceso de glóbulos blancos anómalos.
Tasigna se utiliza en pacientes con LMC de nuevo diagnóstico o en pacientes con LMC que ya no obtienen beneficios con el tratamiento anterior, incluyendo imatinib. También se utiliza en pacientes que han sufrido efectos adversos graves con el tratamiento anterior y que no lo pueden seguir usando.
Cómo actúa Tasigna
En pacientes con LMC, un cambio en el DNA (material genético) genera una señal que hace que el organismo produzca glóbulos blancos anómalos. Tasigna bloquea esta señal y por tanto interrumpe la producción de estas células.
Control del tratamiento con Tasigna
Durante el tratamiento le van a realizar controles de forma regular, incluyendo análisis de sangre.
Estos análisis van a controlar:
- la cantidad de células sanguíneas en el organismo (glóbulos blancos, glóbulos rojos y plaquetas) para comprobar si Tasigna es bien tolerado.
- la función del páncreas y del hígado de su organismo para comprobar si Tasigna es bien tolerado.
- los electrolitos de su cuerpo (potasio, magnesio). Estos son importantes en el funcionamiento de su corazón.
- el nivel de azúcar y grasas en su sangre.
También se controlará su frecuencia cardiaca utilizando una máquina que mide la actividad eléctrica
del corazón (una prueba llamada “ECG”).
Si tiene cualquier pregunta sobre cómo funciona Tasigna o la causa por la que le han prescrito el
fármaco, consulte con su médico.
2. Qué necesita saber antes de empezar a tomar Tasigna
Siga cuidadosamente todas las instrucciones de su médico, aunque éstas sean diferentes de la
información general contenida en este prospecto.
No tome Tasigna
- si es alérgico al nilotinib o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
Si cree que puede ser alérgico, informe a su médico antes de tomar Tasigna.
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar Tasigna:
- si ha sufrido con anterioridad acontecimientos cardiovasculares como un ataque al corazón, dolor en el pecho (angina), problemas con el aporte de sangre a su cerebro (ictus) o problemas con el flujo de sangre a su pierna (claudicación) o si tiene factores de riesgo para enfermedades cardiovasculares como presión sanguínea alta (hipertensión), diabetes o problemas con el nivel de grasas en su sangre (alteraciones de los lípidos).
- si tiene una alteración del corazón, como una señal eléctrica anómala llamada «prolongación del intervalo QT».
- si recibe tratamiento con medicamentos que afectan el ritmo cardíaco (antiarrítmicos) o el hígado (ver Uso de Tasigna con otros medicamentos).
- si sufre falta de potasio o magnesio.
- si tiene una alteración del hígado o del páncreas.
- si tiene síntomas como facilidad de aparición de hematomas, sensación de cansancio o dificultad al respirar o ha presentado infecciones de forma repetida.
- si ha sufrido una intervención quirúrgica que ha supuesto la extirpación del estómago completo (gastrectomía total).
- si alguna vez ha tenido o podría tener en este momento una infección por el virus de la hepatitis B. Esto se debe a que Tasigna podría hacer que la hepatitis B se volviese activa de nuevo, lo que puede resultar mortal en algunos casos. El médico deberá comprobar atentamente si hay signos de esta infección antes de comenzar el tratamiento.
Si alguno de estos casos le es aplicable, informe a su médico.
Durante el tratamiento con Tasigna
- si sufre un desmayo (pérdida de consciencia) o tiene un ritmo cardíaco irregular mientras está tomando Tasigna, informe a su médico inmediatamente pues esto puede ser un signo de un problema grave del corazón. La prolongación del intervalo QT o bien un ritmo cardiaco irregular pueden provocar la muerte súbita. Se han notificado casos poco frecuentes de muerte súbita en pacientes que toman Tasigna.
- si sufre palpitaciones repentinas del corazón, debilidad muscular grave o parálisis, convulsiones o cambios repentinos de comportamiento o nivel de alerta, informe a su médico inmediatamente puesto que puede ser un signo de una rotura rápida de células cancerosas denominado síndrome de lisis tumoral. Se han notificado casos raros de síndrome de lisis tumoral en pacientes tratados con Tasigna.
- si desarrolla dolor en el pecho o malestar, entumecimiento o debilidad, problemas al caminar o con el habla, dolor, decoloración o sensación de frío en una extremidad, informe a su médico inmediatamente pues esto puede ser un signo de un acontecimiento cardiovascular. Se han comunicado casos de acontecimientos cardiovasculares graves incluyendo problemas con el flujo de sangre a la pierna (enfermedad arterial oclusiva periférica), enfermedad isquémica del corazón y problemas con el aporte de sangre al cerebro (enfermedad isquémica cerebrovascular) en pacientes que toman Tasigna. Su médico debe evaluar el nivel de grasas (lípidos) y azúcar en su sangre antes de comenzar el tratamiento con Tasigna y durante el tratamiento con Tasigna.
- si desarrolla hinchazón de los pies o de las manos, hinchazón generalizada o aumento rápido de peso, informe a su médico pues éstos pueden ser signos de retención grave de líquidos. Se han comunicado casos poco frecuentes de retención grave de líquidos en pacientes tratados con Tasigna.
Uso de Tasigna con otros medicamentos
Tasigna puede interferir con algunos medicamentos.
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o pudiera tener que
utilizar cualquier otro medicamento. Estos incluyen, en particular:
- antiarrítmicos - utilizados para tratar el ritmo cardíaco irregular;
- cloroquina, halofantrina, claritromicina, haloperidol, metadona, moxifloxacina - medicamentos que pueden tener un efecto no deseado sobre la función del corazón;
- ketoconazol, itraconazol, voriconazol, claritromicina, telitromicina - utilizados para tratar infecciones;
- ritonavir - un medicamento del grupo de las «antiproteasas» utilizado para tratar el VIH;
- carbamazepina, fenobarbital, fenitoina - utilizados para tratar la epilepsia;
- rifampicina - utilizada para tratar la tuberculosis;
- Hierba de San Juan - un producto derivado de las plantas utilizado para tratar la depresión y otras situaciones (también conocido como Hypericum perforatum);
- midazolam - utilizado para aliviar la ansiedad antes de la cirugía;
- alfentanilo y fentanilo - utilizados para el tratamiento del dolor y como sedantes antes o durante la cirugía o procedimientos médicos;
- ciclosporina, sirolimus y tacrolimus - medicamentos que suprimen la habilidad de “autodefensa” del cuerpo y luchan contra infecciones y que se usan frecuentemente para prevenir el rechazo a órganos trasplantados tales como el hígado, corazón o riñón;
- dihidroergotamina y ergotamina - utilizadas para tratar la demencia;
- lovastatina, simvastatina - utilizadas para tratar los niveles elevados de grasas en sangre;
- warfarina - utilizado para tratar alteraciones de la coagulación (como coágulos en la sangre o trombosis);
- astemizol, terfenadina, cisaprida, pimozida, quinidina, bepridil o alcaloides ergóticos (ergotamina, dihidroergotamina).
Deberá evitarse el uso de estos medicamentos durante el tratamiento con Tasigna. Si está tomando
alguno de estos fármacos, su médico podrá recetarle otros medicamentos alternativos.
Además, informe a su médico o farmacéutico antes de tomar Tasigna si está tomando cualquier antiácido, que son medicamentos contra la acidez de estómago. Estos medicamentos se deben tomar separadamente de Tasigna:
- bloqueadores de H2, que disminuyen la producción de ácido en el estómago. Los bloqueadores H2 se deben tomar aproximadamente 10 horas antes y aproximadamente 2 horas después de tomar Tasigna;
- antiácidos como los que contienen hidróxido de aluminio, hidróxido de magnesio y simeticona, que neutraliza la elevada acidez en el estómago. Estos antiácidos se deben tomar aproximadamente 2 horas antes o aproximadamente 2 horas después de tomar Tasigna.
También debe informar a su médico si ya está tomando Tasigna y le recetan un nuevo medicamento que no ha tomado anteriormente durante el tratamiento con Tasigna.
Toma de Tasigna con alimentos y bebidas
No tome Tasigna junto con las comidas. Los alimentos pueden aumentar la absorción de Tasigna y por lo tanto aumentar la cantidad de Tasigna en la sangre, posiblemente hasta un nivel peligroso. No debe beber zumo de pomelo o comer pomelo. Puede aumentar la cantidad de Tasigna en la sangre, probablemente hasta un nivel peligroso.
Ancianos (65 años o mayores)
Tasigna puede utilizarse por pacientes de 65 años o mayores a la misma dosis que el resto de adultos. Embarazo, lactancia y fertilidad
- No se recomienda el uso de Tasigna durante el embarazo a menos que sea claramente necesario. Si está embarazada o piensa que pudiera estarlo, informe a su médico, quien comentará con usted si puede tomar Tasigna durante el embarazo.
- Las mujeres que pueden quedarse embarazadas deberán utilizar medidas muy efectivas de anticoncepción durante el tratamiento y hasta dos semanas después de finalizar el tratamiento.
- No se recomienda la lactancia durante el tratamiento con Tasigna. Informe a su médico si está en período de lactancia.
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Conducción y uso de máquinas
Si nota efectos adversos (como mareos o problemas de visión) que puedan influir en la capacidad de conducir de forma segura o utilizar herramientas o máquinas después de tomar Tasigna, deberá evitar realizar estas actividades hasta que haya desaparecido el efecto.
Tasigna contiene lactosa
Este medicamento contiene lactosa (también conocido como azúcar de la leche). Si su médico le ha indicado que padece una intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento.
3. Cómo tomar Tasigna
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Qué cantidad de Tasigna tomar
- La dosis recomendada es de 800 mg cada día. Esta dosis se consigue tomando dos cápsulas de 200 mg dos veces al día.
Su médico puede prescribirle una dosis más baja dependiendo de cómo responda al tratamiento.
Cuándo tomar Tasigna
Tome las cápsulas duras:
- dos veces al día (aproximadamente cada 12 horas);
- al menos 2 horas después de ingerir alimentos;
- después esperar 1 hora antes de comer otra vez.
Consulte con su médico o farmacéutico si tiene dudas sobre cuándo tomar Tasigna. La toma de Tasigna cada día a la misma hora le ayudará a recordar cuándo debe tomar las cápsulas duras.
Cómo tomar Tasigna
- Tragar las cápsulas duras enteras con agua.
- No tomar ningún alimento junto con las cápsulas duras.
- No abrir las cápsulas duras excepto si no puede tragarlas. En este caso, puede espolvorear el contenido de cada cápsula dura en una cucharadita de compota de manzana y tomarlo inmediatamente. No utilice más que una cucharadita de compota de manzana para cada cápsula dura y no utilice ningún otro alimento aparte de compota de manzana.
Durante cuánto tiempo tomar Tasigna
Tome Tasigna cada día durante el tiempo que le indique su médico. Este es un tratamiento a largo plazo. Su médico controlará periódicamente su situación para comprobar que el tratamiento está teniendo el efecto deseado.
Si tiene dudas sobre cuánto tiempo debe tomar Tasigna, consulte con su médico.
Si toma más Tasigna del que debe
Si ha tomado más Tasigna del que debe, o si otra persona accidentalmente toma sus cápsulas duras, contacte con un médico u hospital rápidamente. Muestre la caja de las cápsulas duras y este prospecto. Puede necesitar tratamiento médico.
Si olvidó tomar Tasigna
Si se ha olvidado de tomar una dosis, tome la siguiente dosis a su hora habitual. No tome una dosis doble para compensar la cápsula dura olvidada.
Si interrumpe el tratamiento con Tasigna
No interrumpa el tratamiento con Tasigna a menos que se lo indique su médico. Interrumpir el tratamiento con Tasigna sin que se lo haya recomendado su médico le sitúa a usted en un riesgo de empeoramiento de su enfermedad que podría tener consecuencias mortales. Asegúrese de comentarlo con su médico, enfermera y/o farmacéutico si está pensando en interrumpir el tratamiento con Tasigna.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. La mayoría de las reacciones adversas son de leves a moderadas y normalmente desaparecen después de unos pocos días o semanas de tratamiento.
Algunas reacciones adversas pueden ser graves.
Estas reacciones adversas son frecuentes (pueden afectar hasta 1 de cada 10 pacientes), poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes) o se han notificado en muy pocos pacientes.
- rápido aumento de peso, hinchazón de las manos, tobillos, pies o cara (signos de retención de agua)
- dolor en el pecho, aumento de la presión arterial, ritmo cardíaco irregular, coloración azul de los labios, lengua o piel (signos de alteraciones del corazón)
- dificultades para respirar, tos, estertores con o sin fiebre, (sonidos que se producen al respirar), hinchazón de pies o piernas (signos de alteraciones de pulmón)
- fiebre, facilidad para tener hematomas, infecciones frecuentes (signos de alteraciones de la sangre)
- debilidad o parálisis de los miembros o la cara, dificultad para hablar, dolor de cabeza grave, ver, sentir o escuchar cosas que no son reales (signos de alteración del sistema nervioso)
- sed, sequedad de la piel, irritabilidad, color oscuro de la orina, descenso en la cantidad de orina (signos de alteración en los riñones)
- visión borrosa, pérdida de visión, sangre en el ojo (signos de alteraciones en el ojo)
- hinchazón y dolor en una parte del cuerpo (signos de coágulos en una vena)
- dolor abdominal, náusea, vómitos de sangre, heces negras, estreñimiento, hinchazón del abdomen (signos de alteraciones gastrointestinales)
- dolor abdominal superior grave (signo de pancreatitis)
- color amarillo en la piel y los ojos, náusea, pérdida de apetito, orina de color oscuro (signos de alteración del hígado)
- erupción, bultos rojos dolorosos, dolor en las articulaciones y músculos (signos de alteraciones en la piel)
- sed excesiva, aumento en la eliminación de orina, aumento del apetito con pérdida de peso, cansancio (signos de un alto nivel de azúcar en la sangre)
- ritmo cardiaco rápido, ojos saltones, pérdida de peso, hinchazón en la parte delantera del cuello (signos de aumento de actividad de la glándula tiroides)
- náuseas, dificultad para respirar, latido cardíaco irregular, orina turbia, cansancio y/o molestias en las articulaciones asociados con resultados anormales de los análisis de sangre (como niveles elevados de potasio, ácido úrico y fósforo y bajos niveles de calcio)
- dolor, molestias, debilidad o calambres en los músculos de las piernas, que pueden ser debidos a la disminución del riego sanguíneo, úlceras en las piernas o brazos que cicatrizan lentamente o no cicatrizan y cambios apreciables en el color (azul o pálido) o la temperatura (enfriamiento) de las piernas o los brazos, pues estos síntomas podrían ser signos de bloqueo de las arterias en el miembro afectado (pierna o brazo) y los dedos (de los pies o de las manos)
- recurrencia (reactivación) de la infección por el virus de la hepatitis B si ha tenido hepatitis B en el pasado (una infección del hígado).
Si le ocurre alguno de estos acontecimientos, informe a su médico inmediatamente.
Algunas reacciones adversas son muy frecuentes (pueden afectar más de 1 de cada 10 pacientes)
- diarrea
- dolor de cabeza
- cansancio
- dolor muscular
- picor, erupción, ronchas
- náuseas
- vómitos
- pérdida de pelo
- nivel de bilirrubina alto en la sangre (función del hígado)
- nivel de lipasa alto en la sangre (función del páncreas)
Si alguno de estos acontecimientos le afecta de forma grave, informe a su médico.
Algunas reacciones adversas son frecuentes (pueden afectar hasta 1 de cada 10 pacientes)
- molestia abdominal, malestar del estómago después de las comidas, flatulencia, hinchazón del abdomen
- dolor en los huesos, dolor en las articulaciones, espasmos musculares,
- dolor, incluyendo dolor de espalda, dolor en el cuello y dolor en las extremidades, dolor o molestia en el costado del cuerpo
- irritación del ojo, hinchazón, secreción, picor o enrojecimiento, sequedad del ojo (signos de alteraciones del ojo)
- enrojecimiento de la piel, sequedad de la piel, acné, verrugas, disminución de la sensibilidad de la piel
- pérdida de apetito, alteración en el sentido del gusto, aumento o disminución de peso
- insomnio, depresión, ansiedad
- sudores nocturnos, excesiva sudoración, sofocos
- mareo, sensación general de malestar, sensación de dar vueltas
- hormigueo o adormecimiento
- alteración de la voz
- sangrado por la nariz
- necesidad de orinar de forma frecuente
- palpitaciones (sensación de latido cardiaco rápido)
Si alguno de estos acontecimientos le afecta de forma grave, informe a su médico.
Algunas reacciones adversas son poco frecuentes (pueden afectar hasta 1 de cada 100 pacientes)
- aumento de la sensibilidad de la piel, dolor en la piel
- hinchazón en los párpados
- sequedad de boca, dolor de garganta, llagas en la boca
- ardor del estómago
- dolor en las mamas
- aumento del apetito
- alteración de la atención
- dificultad y dolor al orinar, sensación exagerada de necesidad de orinar
- incapacidad de alcanzar o mantener una erección
- aumento de las mamas en los hombres
- síntomas gripales, debilidad muscular
- temblores
- disminución de la agudeza visual
- dolor de cabeza grave, muchas veces acompañado de náusea, vómitos y sensibilidad a la luz
- alteraciones visuales
- úlceras bucales o vaginales
- rigidez de músculos y articulaciones
- pérdida de conciencia
- aumento de peso
- sensación de cambio de temperatura en el cuerpo (incluyendo sensación de calor, sensación de frío)
- manchas engrosadas de color rojo/plateado en la piel (signos de psoriasis)
- sensibilidad en los dientes
Si alguno de estos acontecimientos le afecta de forma grave, informe a su médico.
En muy pocos pacientes tratados con Tasigna, se han notificado las siguientes reacciones adversas:
- confusión, desorientación, pérdida de memoria, alteración en el estado de ánimo, falta de energía
- infección bacteriana de la piel
- ampollas, quistes en la piel, piel aceitosa, adelgazamiento de la piel, manchas oscuras de la piel, decoloración de la piel
- hemorragia, encías sensibles o aumentadas
- rinitis, estornudos
- enrojecimiento y/o hinchazón y posible descamación de las palmas y plantas (también llamado síndrome mano-pie)
- aumento de la sensibilidad de los ojos o de la piel a la luz
- dolor o enrojecimiento en los ojos, dolor, picor en los párpados
- dificultad para oír, dolor de oído, ruidos (zumbido) en los oídos
- articulaciones dolorosas y con hinchazón (gota)
- sangre en orina, color anormal en la orina, incontinencia urinaria
- hemorroides
- sensación de endurecimiento de los pechos, periodos menstruales fuertes, hinchazón en el pezón
- síntomas del síndrome de piernas inquietas (una urgencia irresistible a mover una parte del cuerpo, generalmente una pierna, acompañado por sensaciones incómodas)
Si alguno de estos le afecta de forma grave, informe a su médico.
Durante el tratamiento con Tasigna también puede presentar algunos resultados anómalos en los análisis de sangre tales como un nivel bajo de células de la sangre (glóbulos blancos, glóbulos rojos, plaquetas), un nivel alto de lipasa o amilasa en la sangre (función del páncreas), nivel alto de bilirrubina en la sangre (función del hígado) o nivel alto de creatinina en la sangre (función del riñón), bajo o alto nivel de insulina en la sangre (una hormona reguladora del nivel de azúcar en la sangre), nivel bajo o alto de azúcar, o nivel alto de grasas en la sangre.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Tasigna
- Mantener este medicamento fuera de la vista y del alcance de los niños.
- No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en el
blister. La fecha de caducidad es el último día del mes que se indica.
- No conservar a temperatura superior a 30°C
- Conservar en el embalaje original para protegerlo de la humedad.
- No utilice este medicamento, si observa que el envase esté dañado o muestre signos de manipulación.
- Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de Tasigna
- El principio activo es nilotinib. Cada cápsula dura contiene 200 mg de nilotinib (como clorhidrato monohidrato).
- Los demás componentes son lactosa monohidrato, crospovidona, poloxamero 188, sílice coloidal anhidra, estearato de magnesio. La cubierta de la cápsula dura se compone de gelatina, dióxido de titanio (E171), amarillo óxido de hierro (E172), shellac (E904) y rojo óxido de hierro (E172) para la impresión.
Aspecto de Tasigna y contenido del envase
Tasigna se presenta como cápsulas duras. Las cápsulas duras son de color amarillo claro. Cada
cápsula dura tiene una impresión roja («NVR/TKI»).
Tasigna está disponible en un estuche que contiene 28 cápsulas duras y en una caja que contiene 28 o
40 cápsulas duras.
Tasigna también está disponible en envases múltiples de:
- 112 cápsulas duras (4 estuches de 28).
- 112 cápsulas duras (4 envases de 28).
- 120 cápsulas duras (3 envases de 40).
- 392 cápsulas duras (14 envases de 28).
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH RoonstraBe 25 D-90429 Nürnberg Alemania
Pueden solicitar más información respecto a este titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Btnrapun
Novartis Pharma Services Inc.
Tea.: +359 2 489 98 28
Ceská republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc.
Tel: +372 66 30 810
EkXúóa
Novartis (Hellas) A.E.B.E.
Tni: +30 210 281 17 12
España
Novartis Farmacéutica, S.A.
Tel: +34 93 306 42 00
France
Novartis Pharma S.A.S.
Tél: +33 1 55 47 66 00 medicamento dirigiéndose al representante local del
Lietuva
Novartis Pharma Services Inc.
Tel: +370 5 269 16 50
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc.
Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 555
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Osterreich
Novartis Pharma GmbH Tel: +43 1 86 6570
Polska
Novartis Poland Sp. z o.o.
Tel.: +48 22 375 4888
Portugal
Novartis Farma - Produtos Farmacéuticos, S.A. Tel: +351 21 000 8600
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kúnpoq Novartis Pharma Services Inc. Tr(k: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu/, y en la página web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) (http://www.aemps.gob.es/). También existen enlaces a otras páginas web sobre enfermedades raras y medicamentos huérfanos.
96