Tafinlar 50Mg Capsulas Duras
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Tafinlar 50 mg cápsulas duras Tafinlar 75 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Tafinlar 50 mg cápsulas duras
Cada cápsula dura contiene dabrafenib mesilato equivalente a 50 mg de dabrafenib. Tafinlar 75 mg cápsulas duras
Cada cápsula dura contiene dabrafenib mesilato equivalente a 75 mg de dabrafenib. Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura (cápsula).
Tafinlar 50 mg cápsulas duras
Cápsulas opacas de color rojo oscuro, de aproximadamente 18 mm de longitud, impresas con ‘GS TEW’ y ‘50 mg’.
Tafinlar 75 mg cápsulas duras
Cápsulas opacas de color rosa oscuro, de aproximadamente 19 mm de longitud, impresas con ‘GS LHF’ y ‘75 mg’.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Dabrafenib en monoterapia o en combinación con trametinib está indicado para el tratamiento de pacientes adultos con melanoma no resecable o metastásico con mutación BRAF V600 (ver las secciones 4.4 y 5.1).
4.2 Posología y forma de administración
El tratamiento con dabrafenib debe iniciarse y ser supervisado por un médico especializado en el uso de medicamentos anticancerígenos.
Antes de comenzar el tratamiento con dabrafenib, los pacientes deben tener un diagnóstico de mutación BRAF V600 positiva en el tumor, confirmado por un test validado.
No se ha establecido la eficacia y seguridad de dabrafenib en pacientes con melanoma BRAF de tipo nativo y por lo tanto dabrafenib no se debe utilizar en pacientes con melanoma BRAF de tipo nativo (ver secciones 4.4 y 5.1).
Posología
La dosis recomendada de dabrafenib, tanto en monoterapia como en combinación con trametinib, es de 150 mg (dos cápsulas de 75 mg) dos veces al día (equivalente a una dosis diaria total de 300 mg). La dosis recomendada de trametinib, cuando se utiliza en combinación con dabrafenib, es de 2 mg una vez al día.
Duración del tratamiento
El tratamiento debe de continuar hasta que el paciente no obtenga beneficio clínico del tratamiento o cuando desarrolle una toxicidad intolerable (ver Tabla 2).
Dosis olvidadas
Si olvida tomar una dosis de dabrafenib, no debe volver a tomar el medicamento si quedan menos de 6 horas hasta la próxima toma.
Si olvida tomar una dosis de trametinib, cuando se utiliza en combinación con dabrafenib, tome la dosis olvidada de trametinib solo en caso de que falten más de 12 horas hasta la siguiente dosis.
Modificaciones de dosis
Se dispone de dos tipos de cápsulas con concentraciones de dabrafenib de 50 mg y 75 mg para poder ajustar de manera efectiva las modificaciones de dosis necesarias.
El manejo de las reacciones adversas puede requerir la interrupción del tratamiento, la reducción de dosis o la suspensión del tratamiento (ver Tablas 1 y 2).
No se recomienda realizar modificaciones o interrupciones del tratamiento por reacciones adversas de Carcinoma Cutáneo de Células Escamosas (CCE) o nuevo melanoma primario (ver sección 4.4).
El tratamiento se debe interrumpir si la temperatura del paciente es >38,5°C. Se debe examinar a los pacientes en busca de signos y síntomas de infección (ver sección 4.4).
No es necesario modificar la dosis en caso de uveítis, siempre y cuando los tratamientos eficaces locales puedan controlar la inflamación ocular. Si la uveítis no responde al tratamiento ocular local suspender dabrafenib hasta la resolución de la inflamación ocular y luego reinicie con dabrafenib reducido en un nivel de dosis (ver sección 4.4).
Tabla 1 Reducciones de dosis recomendadas
|
Nivel de dosis |
Dosis de Dabrafenib Utilizado en monoterapia o en combinación con dabrafenib |
Dosis de Trametinib* Unicamente cuando se utiliza en combinación con trametinib |
|
Dosis de inicio |
150 mg dos veces al día |
2 mg una vez al día |
|
1a reducción de dosis |
100 mg dos veces al día |
1,5 mg una vez al día |
|
2a reducción de dosis |
75 mg dos veces al día |
1 mg una vez al día |
|
3a reducción de dosis |
50 mg dos veces al día |
1 mg una vez al día |
|
No se recomienda realizar modificaciones de dosis por debajo de 50 mg de dabrafenib dos veces al día, cuando se utiliza como monoterapia o en combinación con trametinib. No se recomienda realizar modificaciones de dosis por debajo de 1 mg de trametinib una vez al día, cuando se utiliza en combinación con dabrafenib. | ||
|
*Por favor, para instrucciones de dosis en monoterapia con trametinib, consultar la sección “Posología y forma de administración” de la Ficha Técnica de trametinib. | ||
Tabla 2
Esquema de modificaciones de dosis en función de los Acontecimientos Adversos (AA) de cualquier grado
|
Grado (CTC-AA)* |
Modificaciones de dosis recomendadas Utilizado en monoterapia o en combinación con trametinib |
|
Grado 1 o Grado 2 (Tolerable) |
Continuar el tratamiento y monitorizar a los pacientes en función de la clínica. |
|
Grado 2 (Intolerable) o Grado 3 |
Interrumpir el tratamiento hasta que la toxicidad sea de Grado 0 a 1 y reducir la dosis un nivel cuando se reinicie el tratamiento. |
|
Grado 4 |
Suspender o interrumpir el tratamiento hasta que la toxicidad sea de Grado 0 a 1 y reducir un nivel la dosis cuando se reinicie el tratamiento. |
|
* Grado de intensidad de acontecimientos adversos clínicos según los criterios de Common Terminology Criteria for Adverse Events (CTC-AE) v4.0 | |
Cuando una reacción adversa individual se maneja de manera efectiva, se puede considerar realizar un re-escalado de dosis, siguiendo las mismas pautas posológicas empleadas para las reducciones de dosis. La pauta posológica de dabrafenib no debe exceder de 150 mg dos veces al día.
Si apareciera toxicidad relacionada con el tratamiento cuando se utiliza en combinación con trametinib se debe suspender o interrumpir o reducir la dosis de los dos tratamientos simultáneamente. Solo en los casos de pirexia, de uveítis, de cáncer no cutáneo con mutación RAS positiva, de reducción en la fracción de eyección del ventrículo izquierdo (FEVI), de oclusión de las venas retinianas (OCV), de desprendimiento del epitelio pigmentario retiniano (DEPR) y de enfermedad pulmonar intersticial (EPI) / Pneumonitis (principalmente relacionado con trametinib), podría ser necesario que solo se modificara la dosis de uno de los dos tratamientos.
Pirexia
Si la temperatura del paciente es >38,5°C se debe interrumpir el tratamiento con dabrafenib cuando se utiliza solo o en combinación con trametinib (por favor refiérase a la Tabla 2 sobre el esquema de modificaciones de dosis). Se debe continuar con la misma dosis de trametinib. Se debe iniciar un tratamiento antipirético con ibuprofeno o acetaminofeno/paracetamol. En aquellos casos en los que los antipiréticos no son suficientes se debe considerar utilizar corticoides orales. Se deben evaluar los signos y síntomas de infección y en caso necesario, se deberían tratar de acuerdo con las prácticas locales (ver sección 4.4).
Una vez resuelta la pirexia, puede reiniciarse dabrafenib con un tratamiento antipirético adecuado profiláctico incluso 1) a la misma dosis o 2) a un nivel de dosis inferior si la pirexia fuera recurrente y/o se acompañara de otros síntomas como la deshidratación, hipotensión o fallo renal.
Uveitis
Si el tratamiento local puede controlar la inflamación ocular, no es necesario hacer ningún ajuste de dosis para la uveítis. En el caso que no respondiera al tratamiento local ocular, se debe suspender dabrafenib hasta que se resuelva la inflamación ocular y se debe reiniciar con dabrafenib reducido en un nivel de dosis. No es necesario modificar la dosis de trametinib cuando se toma en combinación con dabrafenib (ver sección 4.4).
Cáncer no-cutáneo con mutación-RAS-positiva
En pacientes con un cáncer no cutáneo con mutación RAS positivo sopesar los beneficios y riesgos antes de continuar con el tratamiento con dabrafenib. No es necesario modificar la dosis de trametinib cuando se toma en combinación con dabrafenib.
Reducción en la fracción de eyección del ventrículo izquierdo (FEVI)/Disfunción del ventrículo izquierdo
Si dabrafenib se utiliza en combinación con trametinib y apareciera una reducción absoluta de >10% de la FEVI en comparación con la situación basal y que está por debajo del límite inferior normal establecido, por favor consulte la Ficha Técnica de trametinib (ver sección 4.2) para ver las instrucciones de modificación de dosis de trametinib. No es necesario modificar la dosis de dabrafenib cuando se toma en combinación con trametinib.
Oclusión de las venas retinianas (OCV) y desprendimiento del epitelio pigmentario retiniano (DEPR)
Si durante el tratamiento combinado con darafenib y trametinib los pacientes notifican nuevas alteraciones en la visión, como una disminución de la visión central, visión borrosa o pérdida de visión, por favor consulte la Ficha Técnica de trametinib (ver sección 4.2) para ver las instrucciones de modificación de dosis de trametinib. En los casos confirmados de OCV o DEPR no es necesario modificar la dosis de dabrafenib cuando se toma en combinación con trametinib.
Enfermedad pulmonar intersticial (EPI) / Pneumonitis
Los pacientes en tratamiento con dabrafenib en combinación con trametinib con sospechas de padecer EPI o pneumonitis, incluyendo pacientes que presenten síntomas pulmonares nuevos o progresivos y signos de tos, disnea, hipoxia, derrame pleural o infiltrados, por favor consulte la Ficha Técnica de trametinib (ver sección 4.2) para ver las instrucciones de modificación de dosis de trametinib. En los casos de EPI o pneumonía no es necesario modificar la dosis de dabrafenib cuando se toma en combinación con trametinib.
Pacientes no Caucásicos
No se ha establecido la eficacia y seguridad de dabrafenib en pacientes no caucásicos. No se dispone de datos.
No se requieren ajustes de la dosis inicial en pacientes >65 años de edad.
Insuficiencia renal
No se requieren ajustes de dosis en pacientes con insuficiencia renal leve o moderada. No existen datos clínicos en sujetos con insuficiencia renal grave y no se ha podido determinar la posible necesidad de ajuste de dosis (ver sección 5.2). Dabrafenib se debe utilizar con precaución en pacientes con insuficiencia renal grave cuando se administre como monoterapia o en combinación con trametinib.
Insuficiencia hepática
No se requieren ajustes de dosis en pacientes con insuficiencia hepática leve. No existen datos clínicos en sujetos con insuficiencia hepática de moderada a grave y no se ha podido determinar la posible necesidad de ajuste de dosis (ver sección 5.2). El metabolismo hepático y la secreción biliar son las principales rutas de eliminación de dabrafenib y sus metabolitos, por lo que los pacientes con insuficiencia hepática de moderada a grave pueden presentar un aumento de la exposición. Dabrafenib se debe utilizar con precaución en pacientes con insuficiencia hepática moderada o grave cuando se administre como monoterapia o en combinación con trametinib.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de dabrafenib en niños y adolescentes (<18 años de edad). No se dispone de datos clínicos. En los estudios de dabrafenib en animales jóvenes aparecieron efectos adversos, los cuales no fueron observados en animales adultos (ver sección 5.3).
Forma de administración
Las cápsulas de dabrafenib se deben tragar enteras con agua. Las cápsulas no se deben masticar o abrir y no se deben mezclar con alimentos o líquidos, debido a la inestabilidad química de dabrafenib.
Se recomienda que la dosis de dabrafenib se tome a la misma hora cada día, dejando un intervalo de aproximadamente 12 horas entre dosis. Cuando se toma dabrafenib en combinación con trametinib, la dosis diaria de trametinib se debe tomar a la vez que la dosis matutina o la dosis vespertina de dabrafenib.
Dabrafenib se debe tomar al menos una hora antes o 2 horas después de las comidas.
Si el paciente vomita después de tomar dabrafenib, no debe volver a tomar la dosis, y debe esperar a la siguiente toma.
Por favor, consulte la Ficha Técnica de trametinib para información sobre la forma de administración cuando se toma en combinación con dabrafenib.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Cuando se da dabrafenib en combinación con trametinib, se debe consultar la Ficha Técnica de trametinib antes de comenzar el tratamiento. Por favor, consultar la Ficha Técnica de trametinib para información adicional sobre las advertencias y precauciones asociados al tratamiento con trametinib.
Test mutación BRAF V600
No se ha establecido la eficacia y seguridad de dabrafenib en pacientes con melanoma BRAF de tipo nativo y por lo tanto, dabrafenib no se debe utilizar en pacientes con melanoma BRAF de tipo nativo (ver secciones 4.2 y 5.1).
Dabrafenib en combinación con trametinib, en pacientes que han progresado con un inhibidor BRAF
Existen pocos datos de pacientes en combinación de dabrafenib con trametinib que han progresado a un tratamiento previo con un inhibidor de BRAF. Estos datos muestran que la eficacia de la combinación es menor en estos pacientes (ver sección 5.1). Por tanto se deben considerar otras opciones terapéuticas antes de tratar con la combinación a esta población tratada previamente con un inhibidor de BRAF. No se ha establecido la secuencia de tratamientos tras progresión con un tratamiento inhibidor de BRAF.
Trametinib en combinación con dabrafenib en pacientes con metástasis en el cerebro
La seguridad y eficacia de la combinación de dabrafeniby trametinib no se ha evaluado en pacientes con melanoma BRAF v600 positivo con metástasis en el cerebro.
Nuevos cánceres
Pueden aparece nuevos cánceres, cutáneos y no cutáneos, cuando dabrafenib se utiliza en combinación con trametinib.
Carcinoma cutáneo de células escamosas (CCE)
Se han notificado casos de CCE (incluido queratoacantoma) en pacientes tratados con dabrafenib solo o en combinación con trametinib (ver sección 4.8). En el estudio Fase III MEK115306, CCE apareció en el 3%(6/209) de los pacientes que recibieron trametinib en combinación con dabrafenib y en un 10% (22/211) de los pacientes que recibieron solo dabrafenib. En el estudio Fase III MEK116513, CEE apareció en el 1% (5/350) de los pacientes que recibieron trametinib en combinación con dabrafenib y en un 18% (63/349) de los pacientes que recibieron solo vemurafenib. El tiempo medio de dianóstico del primer CCE en el estudio MEK115306 fue de 223 días (de 56 a 510 días) en el brazo de tratamiento combinado y de 60 días (de 9 a 653 días) en el brazo de dabrafenib en monoterapia.
Antes de empezar el tratamiento con dabrafenib, se recomienda realizar exámenes cutáneos para detectar CCE. Durante todo el tratamiento y durante los 6 meses posteriores a la finalización del tratamiento las revisiones se realizaran mensualmente. Se debe monitorizar a los pacientes durante un periodo de 6 meses tras la suspensión del tratamiento con dabrafenib o hasta la iniciación de otra terapia antineoplásica.
Los casos de CCE se deben tratar mediante extirpación dermatológica o, si se toma en combinación, dabrafenib y trametinib, el tratamiento con dabrafenib se debe continuar sin ajustes de dosis. Se debe indicar a los pacientes que informen inmediatamente a su médico si desarrollan nuevas lesiones.
En los ensayos clínicos se han notificado casos de nuevos melanomas primarios en pacientes tratados con dabrafenib. Estos casos fueron identificados durante los primeros 5 meses de tratamiento con dabrafenib en monoterapia. Los nuevos casos de melanoma primario se pueden tratar mediante extirpación sin la necesidad de realizar modificaciones en el tratamiento. Se deben monitorizar las lesiones cutáneas tal y como se ha descrito anteriormente para casos de CCE.
Neoplasia maligna no cutánea secundaria/recurrente
Los experimentos in vitro han demostrado señales de activación paradójica de la proteína quinasa activada por mitógenos (MAP-kinasa) en células BRAF nativas con mutaciones RAS, cuando fueron expuestas a inhibidores BRAF. Esto puede provocar un aumento del riesgo de aparición de neoplasias malignas no cutáneas relacionadas con la exposición a dabrafenib (ver sección 4.8) cuando existen mutaciones en RAS. Se han notificado neoplasias malignas asociadas a mutaciones RAS en los ensayos clínicos, en pacientes tratados con otro inhibidor de BRAF (leucemia mielomonocítica crónica y carcinoma no cutáneo de células escamosas en cabeza y cuello), asi como en pacientes tratados con dabrafenib en monoterapia (adenocarcinoma pancreático, adenomacarcinoma del conducto biliar), y con dabrafenib en combinación con trametinib, un inhibidor de MEK (cáncer colorectal, cáncer de páncreas).
Antes de iniciar el tratamiento, los pacientes se deben someter a exploraciones de cabeza y cuello, con inspección visual de la mucosa oral y palpación de los nódulos linfáticos, así como realizar un escaner por tomografía computerizada (TC) de tórax y abdomen. Durante el tratamiento, se debe monitorizar a los pacientes según se indique clínicamente, incluyendo exploraciones de cabeza y cuello cada 3 meses y TC de tórax y abdomen cada 6 meses. Se recomienda realizar exploraciones anales y pélvicas (en mujeres) antes y al final del tratamiento o cuando se considere clínicamente necesario. Se debe realizar un recuento completo de células sanguíneas cuando esté indicado clínicamente.
Antes de administrar dabrafenib a pacientes con un cáncer previo o simultaneo, asociado a mutaciones RAS, se ha de considerar cuidadosamente los beneficios y los riesgos. No es necesario modificar la dosis de trametinib cuando se toma en combinación con dabrafenib.
Tras la suspensión del tratamiento con dabrafenib, se debe continuar la monitorización de neoplasias no cutáneas secundarias/recurrentes durante 6 meses o hasta el inicio de otro tratamiento antineoplásico. Cualquier resultado anómalo debe ser tratado de acuerdo a la práctica clínica.
Hemorragias
Se han dado casos de hemorragias, incluidas hemorragias graves y mortales, en pacientes en tratamiento con dabrafenib en combinación con trametininb (ver sección 4.8). Para más información, por favor consultar la Ficha Técnica de trametinib (ver sección 4.4).
Alteraciones visuales
En los ensayos clínicos se han notificado reacciones oftalmológicas, incluyendo uveitis, iriociclitis e iritis en pacientes tratados con dabrafenib en monoterapia y en combinación con trametinib. Se debe monitorizar a los pacientes de manera rutinaria durante el tratamiento, para detectar signos y síntomas oculares (del tipo, cambios en la visión, fotofobia y dolor ocular).
No es necesario modificar la dosis, siempre y cuando los tratamientos eficaces locales puedan controlar la inflamación ocular. Si la uveítis no responde al tratamiento ocular local suspender dabrafenib hasta la resolución de la inflamación ocular y luego reinicie con dabrafenib reducido en un nivel de dosis. Tras el diagnóstico de uveitis, no es necesario modificar la dosis de trametinib cuando se toma en combinación con dabrafenib.
Con dabrafenib en combinación con trametinib puede aparecer DEPR y OCV. Por favor consulte la Ficha Técnica de trametinib (ver sección 4.4). Tras el diagnóstico de OCV o DEPR, no es necesario modificar la dosis de dabrafenib cuando se toma en combinación con trametinib.
Pirexia
En los ensayos clínicos con dabrafenib en monoterapia y en combinación con trametinib se han notificado episodios de fiebre (ver sección 4.8). El 1 % de los pacientes de los ensayos clínicos con dabrafenib en monoterapia, presentaron eventos febriles graves no infecciosos, definidos como fiebre acompañada de escalofríos graves, deshidratación, hipotensión y/o insuficiencia renal aguda de origen pre-renal en sujetos con una función renal normal (ver sección 4.8). El inicio de estos eventos febriles graves no infecciosos se produjo principalmente durante el primer mes de tratamiento. Los pacientes con eventos febriles graves no infecciosos respondieron bien a las reducciones y/o interrupciones de dosis y a los cuidados complementarios.
La incidencia y gravedad de la pirexia aumenta con el tratamiento combinado. En el estudio MEK115306, se notificó pirexia en un 57% (119/209) de los pacientes del brazo de la combinación, un 7% fueron de Grado 3 en comparación con el brazo de dabrafenib en monoterapia, con un 33% (69/211) de los pacientes que notificaron pirexia, siendo el 2% de Grado 3.
En pacientes que recibieron dabrafenib en combinación con trametinib y que desarrollaron pirexia, aproximadamente la mitad de las primeras apariciones de pirexia sucedieron en el primer mes de tratamiento y aproximadamente una tercera parte de los pacientes tuvieron 3 o más episodios.
Se debe interrumpir el tratamiento con dabrafenib si la temperatura del pacientes es >38,5°C (por favor consulte la Tabla 2 de esquema de modificación de dosis). Se debe examinar a los pacientes en busca de signos y síntomas de infección. El tratamiento con dabrafenib se puede reiniciar cuando la fiebre remita mediante el uso profiláctico adecuado de antiinflamatorios no esteroideos o paracetamol. Si la fiebre se asocia con otros signos o síntomas graves, se debe reinciar el tratamiento con dabrafenib a una dosis reducida una vez que la fiebre remita y de acuerdo a la clínica del paciente (ver sección 4.2). No es necesario modificar la dosis de trametinib cuando se toma en combinación con dabrafenib
Reducción de la FEVI/Disfunción del ventrículo izquierdo
Se ha notificado que dabrafenib en combinación con trametinib disminuye la FEVI (ver sección 4.8). Para información adicional, por favor consulte la Ficha Técnica de trametinib (ver sección 4.4). No es necesario modificar la dosis de dabrafenib cuando se toma en combinación con trametinib.
Fallo renal
Se han identificado casos de fallo renal en <1% de los pacientes tratados con dabrafenib solo y en <1% de los pacientes tratados con dabrafenib en combinación. Los casos observados estuvieron asociados generalmente a pirexia y deshidratación, y respondieron bien a interrupciones de dosis y con medidas generales complementarias. Se ha noficiado nefritis granulomatosa (ver sección 4.8). Se deben monitorizar periódicamente los niveles de creatinina sérica en los pacientes que estén en tratamiento.
Si se producen aumentos de los niveles de creatinina, podría ser necesario interrumpir el tratamiento con dabrafenib cuando sea clínicamente apropiado. No se ha estudiado el uso de dabrafenib en pacientes con insuficiencia renal (definida por niveles de creatinina >1,5 x LSN), por lo tanto, se debe utilizar con precaución en este grupo de pacientes (ver sección 5.2).
Acontecimientos hepáticos
En ensayos clínicos con dabrafenib en combinación con trametinib, se han notificado acontecimientos adversos hepáticos (ver sección 4.8). Se recomienda realizar una monitorización de la función hepática cada cuatro semanas durante 6 meses tras iniciar el tratamiento con dabrafenib en combinación con trametinib. A partir de entonces, se ha de monitorizar de acuerdo a la práctica clínica. Por favor, consulte la Ficha Técnica de trametinib para información adicional.
Hipertensión
Se han notificado elevaciones de la presión arterial asociadas al uso de dabrafenib en combinación con trametinib, en pacientes con y sin hipertensión preexistente (ver sección 4.8). Por favor, consulte la Ficha Técnica de trametinib para información adicional.
Enfermedad pulmonar intersticial (EPI) /Pneumonitis
En los estudios clínicos con dabrafenib en combinación con trametinib se han notificado casos de pneumonitis o EPI. Por favor, consulte la Ficha Técnica de trametinib para información adicional. Si dabrafenib se utilizara en combinación con trametinib, podría continuar con el tratamiento con dabrafenib a la misma dosis.
Erupción
Se han observado erupciones aproximadamente en un 25% de los pacientes en los ensayos clínicos cuando se utiliza dabrafenib en combinación con trametinib. Por favor, consulte la sección 4.4 de la Ficha Técnica de trametinib para información adicional.
Rabdomiolisis
Se ha observado rabdomiolisis en pacientes tratados con dabrafenib en combinación con trametinib (ver sección 4.8). Por favor, consulte la sección 4.4 de la Ficha Técnica de trametinib para información adicional.
Pancreatitis
Se han notificado casos de pancreatitis en <1% de los sujetos tratados con dabrafenib en monoterapia y en combinación con trametinib. Uno de los eventos ocurrió en el primer día de tratamiento con dabrafenib y volvió a aparecer tras administrar una dosis reducida. Se debe investigar cuanto antes la aparición de dolor abdominal inexplicable y realizar un análisis de amilasa y lipasa en suero. Se debe monitorizar detenidamente a los pacientes que reinicien el tratamiento con dabrafenib tras un episodio de pancreatitis.
Trombosis venosa profunda / embolismo pulmonar
Cuando dabrafenib se utiliza en monoterapia o en combinación con trametinib, puede aparecer embolismo pulmonar o trombosis venosa profunda. Si el paciente desarrolla síntomas de embolismo pulmonar o trombosis venosa profunda tales como respiración entrecortada, dolor en el pecho, hinchazón de brazos o piernas, debe buscar atención médica urgente. Interrumpir trametinib y dabrafenib de manera permanente por riesgo de muerte por embolismo pulmonar.
Efectos de otros medicamentos sobre dabrafenib
Dabrafenib es un sustrato de CYP2C8 y CYP3A4. Siempre que sea posible, se debe evitar el uso concomitante con inductores potentes de estas enzimas, ya que estos agentes pueden disminuir la eficacia de dabrafenib (ver sección 4.5).
Siempre que sea posible, se debe evitar el uso concomitante con agentes que aumentan el pH gástrico, ya que pueden disminuir la biodisponibilidad de dabrafenib (ver sección 4.5).
Dabrafenib es un inductor del metabolismo enzimático que puede provocar la pérdida de eficacia de muchos medicamentos que se utilizan de forma habitual (ver ejemplos en la sección 4.5). Es importante realizar una revisión de la utilización de medicamentos que usa el paciente cuando se inicia el tratamiento con dabrafenib. Se debe evitar el uso concomitante de dabrafenib con medicamentos que son sustratos sensibles a ciertas enzimas metabolizadoras o transportadoras (ver sección 4.5), si no puede realizarse una monitorización de la eficacia y de los ajustes de la dosis.
La administración concomitante de dabrafenib con warfarina provoca una disminución de la exposición a warfarina. Se debe tener precaución y se recomienda un mayor control del INR (International Normalized Ratio) cuando se utilice dabrafenib de forma concomitante con warfarina y cuando se suspenda el tratamiento de dabrafenib (ver sección 4.5).
La administración concomitante de dabrafenib con digoxina puede provocar una disminuación de la exposición a digoxina. Se debe tener precaución y se recomienda una mayor monitorización cuando digoxina (un transportador de sustrato) se use simultaneamente con dabrafenib y cuando se suspenda el tratamiento de dabrafenib (ver sección 4.5).
4.5 Interacción con otros medicamentos y otras formas de interacción
Efectos de otros medicamentos sobre dabrafenib
Dabrafenib es un sustrato de las enzimas metabólicas CYP2C8 y CYP3A4, mientras que los metabolitos activos, hidróxi-dabrafenib y desmetil-dabrafenib son sustratos de CYP3A4. Los medicamentos que actúan como inhibidores o inductores potentes de CYP2C8 o CYP3A4, tienden a aumentar o disminuir las concentraciones de dabrafenib, respectivamente. Por ello, y si es posible, durante el tratamiento con dabrafenib se debe considerar la administración de agentes alternativos. Se debe tener precaución cuando se administren inhibidores potentes (por ejemplo, ketoconazol, gemfibrozilo, nefazodona, claritromicina, ritonavir, saquinavir, telitromicina, itraconazol, voriconazol, posaconazol, atazanavir) con dabrafenib. Se debe evitar la coadministración de dabrafenib con inductores potentes de CYP2C8 o CYP3A4 (por ejemplo, rifampicinia, fenitoina, carbamazepina, fenobarbital, o Hierba de San Juan (Hypericum perforatum)) con dabrafenib.
La administración de 400 mg de ketoconazol (un inhibidor de CYP3A4) una vez al día, con 75 mg de dabrafenib dos veces al día, produjo un incremento en el AUC de dabrafenib del 71%, y un incremento en la Cmax de dabrafenib del 33%, en relación con la administración de 75 mg de dabrafenib dos veces al día en monoterapia. La administración concomitante produjo un incremento del AUC de hidroxi-dabrafenib y desmetil-dabrafenib (incrementos del 82% y del 68%, respectivamente). Se observó una disminución en el AUC de carboxi-dabrafenib de un 16%.
La administración de 600 mg de gemfibrozilo (un inhibidor de CYP2C8), dos veces al día, con 75 mg de dabrafenib dos veces al día, produjo un incremento en el AUC de dabrafenib del 47%, pero no alteró la Cmax de dabrafenib, en relación con la administración de 75 mg de dabrafenib dos veces al día en monoterapia. Gemfibrozilo no tuvo efectos clínicamente relevantes sobre la exposición sistémica de los metabolitos de dabrafenib (<13%).
La solubilidad de dabrafenib es pH dependiente con disminuciones de la solubilidad cuanto mayor sea el pH. Los medicamentos como los inhibidores de la bomba de protones, que inhiben la secrección gástrica elevando el pH gástrico, pueden disminuir la solubilidad de dabrafenib y reducir su biodisponibilidad. No se ha llevado a cabo ningún ensayo clínico para evaluar el efecto del pH en la farmacocinética de dabrafenib. Debido al riesgo teórico de que los agentes que elevan el pH puedan disminuir la biodisponibilidad oral y la exposición a dabrafenib, se debe evitar en la medida de lo posible el uso de medicamentos que aumenten el pH gástrico durante el tratamiento con dabrafenib.
Dabrafenib es un inductor enzimático que incrementa la síntesis de enzimas que metabolizan medicamentos entre las que se incluyen CYP3A4, CYP2Cs y CYP2B6. También puede incrementar la síntesis de transportadores. Este efecto provoca una reducción de los niveles plasmáticos de medicamentos metabolizados por estas enzimas, y puede afectar a algunos medicamentos transportados. La reducción de las concentraciones plasmáticas puede provocar una pérdida o una reducción del efecto clínico de estos medicamentos. Existe también el riesgo de un aumento en la formación de metabolitos activos de estos medicamentos. Entre las enzimas que pueden ser inducidas se incluyen CYP3A presente en el hígado e intestino, CYP2B6, CYP2C8, CYP2C9, CYP2C19 y UGTs (enzimas glucurónido conjugadas). El transportador de proteína Pgp puede también ser inducido al igual que otros transportadores, por ejemplo MRP-2, BCRP y OATP1B1/1B3.
In vitro, dabrafenib produce incrementos en CYP2B6 y CYP3A4 de manera dosis dependiente. En un estudio de interacción de fármacos, la Cmax y el AUC de midazolam administrado por vía oral (un sustrato de CYP3A4) disminuyó un 61% y un 74% respectivamente, al administrarlo junto con dosis repetidas de dabrafenib, usando una formulación con menor biodisponibildad que la formulación de dabrafenib.
La administración de 150 mg de dabrafenib dos veces al día con warfarina, produjo una disminución en el AUC de S-warfarina y R-warfarina de un 37% y un 33% respectivamente, en comparación con la administración de warfarina en monoterapia. La Cmax de S-warfarina y R-warfarina, se incrementó un 18% y un 19%, respectivamente.
Es de esperar que existan interacciones con muchos medicamentos que se eliminen a través del metabolismo o por transporte activo. Estos medicamentos se deben evitar o se deben utilizar con precaución, si su efecto terapéutico es de gran importancia para el paciente y si los ajustes de dosis no se pueden realizar con facilidad en función de la eficacia o las concentraciones plasmáticas. Se sospecha que el riesgo de daño hepático tras la administración de paracetamol es mayor en pacientes tratados de forma concomitante con inductores enzimáticos.
Es de esperar que el número de medicamentos afectados sea grande, aunque la magnitud de la interacción puede variar. Entre el grupo de medicamentos que pueden verse afectados se incluyen los siguientes, pero no están limitados sólo a estos:
• Analgésicos (p. ej. fentanilo, metadona)
• Antibióticos (p. ej. claritromicina, doxiciclina)
• Medicamentos anticancerígenos (p. ej.cabazitaxel)
• Anticoagulantes (p. ej. acenocumarol, warfarina, ver sección 4.4)
• Antiepilépticos (p. ej. carbamazepina, fenitoína, primidona, ácido valproico)
• Antipsicóticos (p. ej. haloperidol)
• Bloqueantes de canales de cálcio (p. ej. diltiazem, felodipino, nicardipino, nidefipino, verapamilo)
• Glucosidos cardiacos (p. ej. digoxina, ver sección 4.4)
• Corticosteroides (p. ej. dexametasona, metilprednisolona)
• Antivirales para el VIH (p. ej. amprenavir, atazanavir, darunavir, delavirdina, efavirenz, fosamprenavir, indinavir, lopinavir, saquinavir, tipranavir)
• Anticonceptivos hormonales (ver sección 4.6)
• Hipnóticos (p. ej. diazepam, midazolam, zolpidem)
• Inmunosupresores (p. ej. ciclosporina, tacrolimus, sirolimus)
• Estatinas metabolizadas por CYP3A4 (p. ej. atorvastatina, simvastatina)
Es probable que el inicio de la inducción ocurra después de 3 días de tratamiento repetido con dabrafenib. Cuando se suspende el tratamiento con dabrafenib la inducción es contrarestada de forma gradual, puediendo incrementarse las concentraciones susceptibles de CYP3A4, CYP2B6, CYP2C8, CYP2C9 y CYP2C19, UDP glucuronosil transferasa (UGT) y los trasportadores de sustratos; por ello, los pacientes deben ser monitorizados en caso de toxicidad y la pauta posológica de estos medicamentos debe ser ajustada.
In vitro, dabrafenib es un inhibidor del mecanismo de CYP3A4. Por lo tanto, durante los primeros días de tratamiento se pueden observar inhibición transitoria de CYP3A4.
Efectos de dabrafenib sobre los sistemas transportadores de sustancias
Dabrafenib es un inhibidor in vitro del polipéptido transportador de aniones orgánicos humanos (OATP 1B1 y OATP 1B3), por lo tanto la relevancia clínica no se puede descartar. Se recomienda precaución cuando se administren conjuntamente dabrafenib y sustratos de OATP1B1 y OATP1B3, como las estatinas.
Aunque dabrafenib y sus metabolitos, hidroxi-dabrafenib, carboxi-dabrafenib y desmetil-dabrafenib, son inhibidores del transportador de aniones orgánicos humanos OAT1 y OAT3 in vitro, el riesgo de interacciones entre medicamentos es mínimo basándose en la exposición clínica. Dabrafenib y desmetil-dabrafenib también demostraron ser inhibidores moderados de la proteína humana de resistencia al cáncer de mama (BCRP); sin embargo, basándose en la exposición clínica, el riesgo de interacción entre medicamentos es mínimo.
Combinación con tra.met.inih
La administración concomitante a dosis repetidas de 2 mg de trametinib una vez al día y de 150 mg dos veces al día no dio lugar a cambios clínicamente significativos en la Cmax y AUC de trametinib o de dabrafenib, con incrementos del 16 y 23% en la Cmax y AUC respectivamente de dabrafenib. Utilizando un análisis farmacocinético poblacional se observó una pequeña disminución en la biodisponibilidad de trametinib, correspondiente a una disminución del AUC del 12%, cuando trametinib se administraba en combinación con dabrafenib, un inductor CYP3A4.
Cuando dabrafenib se utiliza en combinación con trametinib consulte las interacciones del medicamento que se encuentran en las secciones 4.4 y 4.5 de la ficha técnica de dabrafenib y trametinib.
Efectos de los alimentos sobre dabrafenib
Los pacientes deben de tomar dabrafenib en monoterapia o en combinación con trametinib al menos una hora antes o dos horas después de las comidas debido al efecto de los alimentos sobre la absorción de dabrafenib (ver sección 5.2).
Población pediátrica
Los estudios de interacción sólo se han realizado en adultos.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil /Anticoncepción en mujeres
Las mujeres en edad fértil deben utilizar un método anticonceptivo eficaz durante el tratamiento y durante las 4 semanas siguientes a la suspensión del tratamiento de dabrafenib y 4 meses tras la última dosis de trametinib, cuando se ha dado en combinación con debrafenib. Dabrafenib puede disminuir la eficacia de los anticonceptivos hormonales, por lo que se debe utilizar otro método anticonceptivo alternativo, tales como un método de barrera (ver sección 4.5).
Embarazo
No hay datos relativos al uso de dabrafenib en mujeres embarazadas. En estudios en animales se ha observado toxicidad en la reproducción y toxicidades en el desarrollo embriofetal, incluyendo efectos teratogénicos (ver sección 5.3). No se debe administrar dabrafenib a mujeres embarazadas a no ser que los beneficios para la madre superen los posibles riesgos para el feto. Si la paciente se queda embarazada durante el tratamiento con dabrafenib, se le debe informar del posible riesgo para el feto. Por favor consulte la Ficha Técnica de trametinib (sección 4.6) cuando se utilice en combinación con dabrafenib.
Lactancia
Se desconoce si dabrafenib se excreta en la leche materna. Debido a que muchos medicamentos se excretan en la leche materna, no se puede descartar la existencia de riesgo para los lactantes. Se debe decidir si es necesario suspender la lactancia o suspender el tratamiento con dabrafenib, tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Fertilidad
No hay datos en seres humanos de dabrafenib en monoterapia o en combinación con trametinib. En animales, se ha observado que dabrafenib puede afectar a la fertilidad de machos y hembras como un efecto adverso sobre los órganos reproductores masculinos y femeninos. Se debe informar a los pacientes varones que toman dabrafenib en monoterapia o en combinación con trametinib del posible riesgo de deterioro de la espermatogénesis, que puede ser irreversible.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de dabrafenib sobre la capacidad para conducir y utilizar máquinas es pequeña. A la hora de considerar la capacidad del paciente para realizar tareas que requieran juicio,habilidades motoras o cognitivas, se deben tener en cuenta tanto el estado clínico del paciente como el perfil de reacciones adversas de dabrafenib. Los pacientes deberán ser conscientes de la posibilidad de padecer fatiga o problemas oculares que afectan a estas actividades.
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad de dabrafenib en monoterapia se basa en los datos procedentes de cinco estudios clínicos e incluyen 578 pacientes con melanoma. Las reacciones adversas más frecuentes (>15%) notificadas con dabrafenib fueron, hiperqueratosis, cefalea, pirexia, artralgia, fatiga, náusea, papiloma, alopecia, erupción cutánea y vómitos.
La seguridad de dabrafenib en combinación con trametinib se ha evaluado en 2 estudios fase III,
MEK115306 y MEK116513, donde se ha realizado un análisis de la seguridad de dabrafenib en combinación con trametinib en 209 y 350 pacientes, respectivamente, con melanoma no resecable o metastásico con mutación BRAF V600 que recibieron tratamiento combinado (ver información sobre el tratamiento combinado en la sección 5.1) de dabrafenib (150 mg dos veces al día) con trametinib (2 mg una vez al día). Las reacciones adversas más frecuentes (>20%) del tratamiento combinado de dabrafenib y trametinib son pirexia, fatiga, naúseas, cefalea, escalofríos, diarrea, prurito, artralgia, hipertensión, vómitos y tos.
Tabla de reacciones adversas
Las reacciones adversas que fueron notificadas se enumeran bajo la clasificación de órganos del sistema MedDRA, por frecuencia y por nivel de gravedad. La siguiente clasificación se ha utilizada para ordenarlas por frecuencia:
Muy frecuentes: Frecuentes:
Poco frecuentes: Raras:
No conocida:
>1/10
>1/100 a <1/10 >1/1.000 a <1/100 >1/10.000 a <1/1.000
(no se pueden estimar a partir de los datos disponibles)
Dabrafenib en monoterapia
Tabla 3 Reacciones adversas notificadas en los ensayos clínicos en melanoma
|
Sistema de clasificación de órganos |
Frecuencia (todos los grados) |
Reacciones adversas |
|
Neoplasias benignas, malignas y no especificadas (incluyendo quistes y pólipos) |
Muy frecuentes |
Papiloma |
|
Frecuentes |
Carcinoma cutáneo de células escamosas | |
|
Queratosis seborreica | ||
|
Acrocordón | ||
|
Carcinoma de células basales | ||
|
Poco frecuentes |
Nuevo melanoma primario | |
|
Trastornos del sistema inmunológico |
Poco frecuentes |
Hipersensibilidad |
|
Trastornos del metabolismo y de la nutrición |
Muy frecuentes |
Disminución del apetito |
|
Frecuentes |
Hipofosfatemia | |
|
Hiperglucemia | ||
|
Trastornos del sistema nervioso |
Muy frecuentes |
Cefalea |
|
Trastornos oculares |
Poco frecuentes |
Uveitis |
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy frecuentes |
Tos |
|
Trastornos gastrointestinales |
Muy frecuentes |
Náusea |
|
Vómitos | ||
|
Diarrea | ||
|
Frecuentes |
Estreñimiento | |
|
Poco frecuentes |
Pancreatitis | |
|
Trastornos de la piel y del tejido subcutáneo |
Muy frecuentes |
Hiperqueratosis |
|
Alopecia | ||
|
Erupción cutánea | ||
|
Síndrome de eritrodisestesia palmoplantar | ||
|
Frecuentes |
Piel seca | |
|
Prurito | ||
|
Queratosis actínica | ||
|
Lesión en la piel | ||
|
Eritema | ||
|
Poco frecuentes |
Paniculitis |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Muy frecuentes |
Artralgia |
|
Mialgia | ||
|
Dolor en las extremidades | ||
|
Trastornos renales y urinarios |
Poco frecuentes |
Fallo renal, fallo renal agudo |
|
Nefritis | ||
|
Pirexia | ||
|
Trastornos generales y |
Muy frecuentes |
Fatiga |
|
alteraciones en el lugar de |
Escalofríos | |
|
administración |
Astenia | |
|
Frecuentes |
Enfermedad parecida a la gripe | |
|
Exploraciones |
Frecuentes |
Disminución de la FEVI |
|
complementarias |
Poco frecuentes |
Prolongación intervalo QT |
Tratamiento combinado de dabrafenib y trametinib
Tabla 4 Reacciones adversas que aparecieron en los estudios Fase III randomizados de la combinación MEK115306 (n=209) y MEK116513a (n=350)
|
Sistema de Clasificación de Órganos |
Frecuencia (todos los grados) |
Reacciones adversas |
|
Infecciones e infestaciones |
Muy frecuentes |
Infecciones del tracto urinario |
|
Nasofaringitis | ||
|
Frecuentes |
Celulitis | |
|
Foliculitis | ||
|
Paroniquia | ||
|
Erupción pustular | ||
|
Neoplasias benignas, malignas y no especificadas (incl quistes y pólipos) |
Frecuentes |
Carcinoma de células escamosasb |
|
Papilomac | ||
|
Queratosis seborreica | ||
|
Acrocordón (marcas en la piel) | ||
|
Poco frecuentes |
Nuevo melanoma primario | |
|
Trastornos de la sangre y del sistema linfático |
Muy frecuentes |
Neutropenia |
|
Frecuentes |
Anemia | |
|
T rombocitopenia | ||
|
Leucopenia | ||
|
Trastornos del sistema inmunológico |
Poco frecuentes |
Hipersensibilidad a medicamentos |
|
Trastornos del metabolismo y la nutrición |
Muy frecuentes |
Disminución del apetito |
|
Frecuentes |
Deshidratación | |
|
Hiponatremia | ||
|
Hipofosfatemia | ||
|
Hiperglucemia | ||
|
Trastornos del sistema nervioso |
Muy frecuentes |
Cefalea |
|
Mareo | ||
|
Trastornos oculares |
Frecuentes |
Visión borrosa |
|
Alteraciones visuales | ||
|
Poco frecuentes |
Coriorretinopatía | |
|
Uveitis | ||
|
Desprendimiento de retina | ||
|
Edema periorbital |
|
Trastornos cardiacos |
Frecuentes |
Disminución de la fracción de eyección |
|
Bradicardia | ||
|
Trastornos vasculares |
Muy frecuentes |
Hipertensión |
|
Hemorragiad | ||
|
Frecuentes |
Hipotensión | |
|
Poco frecuentes |
Limfoedemaa | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy frecuentes |
Tos |
|
Frecuentes |
Disnea | |
|
Poco frecuentes |
Pneumonitis | |
|
Trastornos gastrointestinales |
Muy frecuentes |
Dolor abdominal |
|
Estreñimiento | ||
|
Diarrea | ||
|
Nausea | ||
|
Vómitos | ||
|
Frecuentes |
Sequedad de boca | |
|
Estomatitis | ||
|
Poco frecuentes |
Pancreatitis | |
|
Trastornos hepatobiliares |
Muy frecuentes |
Alanina aminotransferasa elevada |
|
Aspartato aminotransferase elevada | ||
|
Frecuentes |
Fosfatasa alcalina en sangre elevada | |
|
Gamma-glutamiltransferasa elevada | ||
|
Trastornos de la piel y del tejido subcutáneo |
Muy frecuentes |
Piel seca |
|
Prurito | ||
|
Erupción | ||
|
Dermatitis acneiforme | ||
|
Frecuentes |
Eritema | |
|
Queratosis actinica | ||
|
Sudores nocturnos | ||
|
Hiperqueratosis | ||
|
Alopecia | ||
|
Síndrome de eritrodisestesia palmoplantar | ||
|
Lesiones de piel | ||
|
Hiperhidrosis | ||
|
Paniculitis | ||
|
Fisuras de la piel | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Muy frecuentes |
Artralgia |
|
Mialgia | ||
|
Dolor en las extremidades | ||
|
Frecuentes |
Espasmos musculares3 | |
|
Creatina fosfoquinasa en sangre elevada | ||
|
Trastornos renales y urinarios |
Poco frecuentes |
Fallo renala |
|
Nefritis |
|
Trastornos generales y alteraciones en el lugar de administración |
Muy frecuentes |
Fatiga |
|
Escalofríos | ||
|
Astenia | ||
|
Edema periférico | ||
|
Pirexia | ||
|
Frecuentes |
Inflamación de la mucosa | |
|
Síntomas gripales | ||
|
Edema facial | ||
|
Exploraciones complementarias |
Frecuentes |
Disminución del ritmo cardiaco |
|
a El perfil de seguridad de MEK116513 es en general similar al de MEK115306 con las siguientes excepciones: 1) Las siguientes reacciones adversas se presentan en mayor frecuencia en comparación con MEK115306: espasmos musculares (/> muy frecuentes); fallo renal y linfoedema (frecuentes); fallo renal agudo (poco frecuente); 2) Las siguientes reacciones adversas aparecieron en MEK116513 pero no en MEK115306: fallo cardiaco, disfunción del ventrículo izquierdo, enfermedad pulmonar intersticial, rabdomiolisis (poco frecuente). b:CCE: CCE de la piel, CCE in situ (enfermedad de Bowen) y queratoacantoma c Papiloma, papiloma de piel d Sangrado de varios sitios, incluido hemorragia intracranial y sangrado grave y mortal | ||
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
Descripción de las reacciones adversas seleccionadas
Carcinoma cutáneo de células escamosas(CCE)
En el 9% de los pacientes tratados con dabrafenib en monoterapia de la población de seguridad y en el 3% de los pacientes tratados con dabrafenib en combinación con trametinib en el estudio MEK115306, aparecieron casos de carcinoma cutáneo de células escamosas (incluyendo aquellos clasificados como queratoacantomas o subtipos mixtos de queratoacantomas). Con dabrafenib en monoterapia, aproximandamente el 70% de los eventos se produjo durante las primeras 12 semanas de tratamiento, con una mediana del tiempo de aparición de 8 semanas. Los pacientes que recibieron dosis de dabrafenib combinado con trametinib, los eventos se produjeron más tarde, con tiempo de aparición medio de 22 semanas. El 96% de los pacientes con dabrafenib en monoterapia de la población de seguridad y de todos los pacientes en tratamiento combinado de los estudios Fase III que desarrollaron CCE continuaron el tratamiendo sin modificaciones de la pauta posológica.
Nuevo melanoma primario
En los ensayos clínicos con dabrafenib en monoterapia y en combinación con trametinib se han notificado casos de nuevos melanomas primarios. Estos casos fueron tratados mediante extirpación y no fueron necesarias modificaciones del tratamiento (ver sección 4.4).
La activación de la señalización de MAP-kinasa en células BRAF nativas que fueron expuestas a inhibidores BRAF, incluyendo aquellas con mutaciones RAS, puede conducir a un aumento de riesgo de aparición de neoplasias malignas no cutáneas (ver sección 4.4). En los ensayos clínicos se notificaron neoplasias malignas no cutáneas en 1% (6/586) de los pacientes con dabrafenib en monoterapia y 1% (3(209) de los pacientes en el estudio MEK115306 y <1% (3/350) de los pacientes en el estudio MEK116513 con dabrafenib en combinación con trametinib. Se han visto casos de neoplasias malignas en pacientes trados con dabrafenib en monoterapia y en combinación con trametinib, provocados por mutaciones RAS. Se debe monitorizar a los pacientes en función de la clínica.
Hemorragias
Se han dado casos de hemorragias, incluidas hemorragias graves y mortales, en pacientes en tratamiento con dabrafenib en combinación con trametinib. Por favor consulte la Ficha Técnica de trametinib.
Reducción de la FEVI/Disfunción del ventrículo izquierdo
En la problación de seguridad se han notificado casos de disminución de la FEVI en el 1% de los pacientes tratados con dabrafenib en monoterapia, y del 6 al 8% de los pacientes tratados con dabrafenib en combinación con trametinib en dos ensayos clínicos Fase III, siendo en la mayoría de los casos sintomática y reversible. Los pacientes con FEVI por debajo del límite inferior normal no fueron incluidos en los ensayos clínicos con dabrafenib. Dabrafenib en combinación con trametinib se debe usar con precaución en pacientes con afecciones que puedan alterar la función del ventriculo izquierdo.
Pirexia
En los ensayos clínicos con dabrafenib en monoterapia y en combinación con trametinib se ha notificado fiebre; sin embargo, la incidencia y gravedad de la pirexia aumenta con el tratamiento combinado (ver sección 4.4). En pacientes que recibieron dabrafenib en combinación con trametinib y desarrollaron pirexia, aproximadamente, la mitad de las primeras apariciones de pirexia sucedieron en el primer mes de tratamiento y aproximadamente una tercera parte de los pacientes tuvieron 3 o más episodios. El 1% de los pacientes con dabrafenib en monoterapia de la población de seguridad de los ensayos clínicos, presentaron eventos febriles graves no infecciosos, definidos como fiebre acompañada de escalofríos graves, deshidratación, hipotensión y/o insuficiencia renal aguda de origen pre-renal en sujetos con una función renal normal (ver sección 4.8). El inicio de estos eventos febriles graves no infecciosos se produjo principalmente durante el primer mes de tratamiento. Los pacientes con eventos febriles graves no infecciosos respondieron bien a las reducciones y/o interrupciones de dosis y a los cuidados complementarios.
Acontecimientos hepáticos
En ensayos clínicos con dabrafenib en monoterapia y en combinación con trametinib, se han notificado acontecimientos adversos hepáticos. Por favor consulte la Ficha Técnica de trametinib.
Hipertensión
Se han notificado elevaciones de la presión arterial asociadas al uso de dabrafenib en combinación con trametinib, en pacientes con y sin hipertensión preexistente. Se debe medir la presión arterial al inicio del tratamiento, llevar a cabo una monitorización durante el tratamiento, y cuando proceda controlar la hipertensión con un tratamiento estándar.
Los casos de artralgia notificados en los ensayos clínicos con dabrafenib en monoterapia y en combinación con trametinib han sido muy frecuentes (aproximadamente 25%), aunque estos fueron principalmente clasificados de gravedad Grado 1 y 2, siendo poco frecuentes los casos de Grado 3 (<1%), y no se notificó ningún caso de Grado 4.
Hipofosfatemia
En la población de seguridad de los ensayos clínicos con dabrafenib en monoterapia se han notificado muy frecuentemente casos de hipofosfatemia (7%) y en los ensayos clínicos Fase III en combinación con trametinib (3 al 4%). Se debe tener en cuenta que aproximadamente la mitad de estos casos con dabrafenib en monoterapia (4%) y <1% con dabrafenib en combinación con trametinib presentaron una gravedad de Grado 3.
Pancreatitis
Se han notificado casos de pancreatitis con dabrafenib en monoterapia y en combinación con trametinib. Se debe investigar cuanto antes la aparición de dolor abdominal de origen desconocido y realizar un análisis de amilasa y lipasa séricas. Se debe monitorizar detenidamente a los pacientes que reinicien el tratamiento con dabrafenib tras un episodio de pancreatitis (ver sección 5.2).
Fallo renal
Los casos de fallo renal debidos a pirexia asociada a azotemia prerrenal o a nefritis granulomatosa fueron poco frecuentes. Sin embargo, no se han estudiado los efectos de dabrafenib en pacientes con insuficiencia renal (definida por niveles de creatinina >1,5 x LSN). Se debe tener precaución en estos pacientes (ver sección 4.4).
Poblaciones especiales
Pacientes de avanzada edad
Del número total de pacientes incluidos en los estudios de dabrafenib (N=578), el 22% eran mayores de 65 años, y un 6% eran mayores de 75 años. En comparación con sujetos más jóvenes (<65 años), hubo un mayor número de pacientes >65 años que presentaron reacciones adversas que condujeron a reducciones de dosis (22% vs 12%) o interrupciones (39% vs 27%). Además, los pacientes de mayor edad experimentaron más reacciones adversas graves en comparación con los pacientes jóvenes (41% vs 22%). No se encontraron diferencias globales de eficacia entre estos sujetos y los sujetos más jóvenes.
Los pacientes >65 años que participaron en los estudios de Fase III, MEK115306 (n=209) y MEK116513 (n=350) con dabrafenib en combinación con trametinib en pacientes con melanoma no resecable o metastásico, fueron 56 pacientes (27%) y 77 pacientes (22%), respectivamente; los pacientes >75 años fueron 11 pacientes (5%) y 21 pacientes (6%), respectivamente. En ambos estudios, la proporción de pacientes que experimentaron acontecimientos adversos fue similar para los <65 años que para los >65 años. Sin embargo los pacientes >65 años fueron más propensos a sufrir efectos adversos, incluso tuvieron que interrumpir el tratamiento de forma permanente, reducir o interrumpir su dosis debido a los efectos adversos, que los pacientes <65 años.
4.9 Sobredosis
No existe un tratamiento específico para tratar la sobredosis de dabrafenib. Si se produce una sobredosis, el paciente debe ser tratado con medidas complementarias y una apropiada monitorización según sea necesario.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agente antineoplásico, inhibidor de la proteína quinasa, código ATC: L01XE23
Mecanismo de acción
Dabrafenib es un inhibidor de quinasas RAF. Las mutaciones oncogénicas en BRAF conducen a la activación constitutiva de la vía RAS/RAF/MEK/ERK. Las mutaciones BRAF se han identificado de manera muy frecuente en tipos de cáncer específicos, siendo aproximadamente el 50% melanomas. La mutación BRAF observada con mayor frecuencia es la V600E que representa aproximadamente el 90% de las mutaciones BRAF observadas en melanoma.
Los datos preclínicos generados en ensayos bioquímicos muestran que dabrafenib inhibe las quinasas BRAF que contienen mutaciones activadas del codón 600 (ver Tabla 5).
Tabla 5 Actividad inhibidora de dabrafenib contra quinasas RAF
|
Quinasa |
Concentración inhibitoria 50 (nM) |
|
BRAF V600E |
0,65 |
|
BRAF V600K |
0,50 |
|
BRAF V600D |
1,8 |
|
BRAF WT |
3,2 |
|
CRAF WT |
5,0 |
In vitro, y en modelos animales, dabrafenib demostró inhibición de la cascada farmacodinámica de ERK fosforilado, e inhibición del crecimiento celular de líneas celulares de melanoma con mutación BRAF V600.
En sujetos con melanoma con mutación BRAF V600 positiva, la administración de dabrafenib provocó la inhibición de ERK fosforilado del tumor, respecto a la situación basal.
Combinación con trametinib
Trametinib es un inhibidor alostérico, reversible, altamente selectivo, de la señal extracelular activada por mitógenos que regula la activación y la actividad quinasa, de las quinasas MEK1 y MEK2. Las proteínas MEK con componentes de la vía de señalización extracelular relacionada con quinasas (ERK). Por tanto, trametinib y dabrafenib inhiben dos quinasas de la misma vía, MEK y RAF, por lo que la combinación proporciona una doble inhibición de esta vía. La combinación de trametinib con dabrafenib ha demostado actividad antitumoral in vitro en las lineas celulares con melanoma con la mutación BRAF V600 y ha retrasado la aparición de resistencias in vivo en injertos con melanoma con la mutación BRAF V600.
Determinación del estado de la mutación BRAF
Antes de empezar el tratamiento con dabrafenib o en combinación con trametinib, se debe confirmar que los pacientes tienen mutación positiva V600 de BRAF en el tumor, mediante la realización de un test validado. En los ensayos clínicos Fase II y Fase III se realizó un cribado para determinar la elegibilidad. Para el cribado se requería realizar un test para identificar la mutación BRAF V600, el cual se realizó mediante un ensayo de detección de mutación BRAF que se llevó a cabo en las muestras más recientes de tumor disponibles. Los tumores primarios o tumores procedentes de un lugar donde se haya producido metástasis fueron analizados mediante un ensayo de uso exclusivo en investigación (IUO). El IUO es un ensayo de identificación de un alelo específico mediante la reacción de la cadena de la polimerasa (PCR) realizado en ADN extraído de tejido tumoral fijado con formalina y embebido en parafina (FFPE). Esta prueba ha sido especialmente diseñada para diferenciar las mutaciones V600E y V600K. Solamente aquellos sujetos con mutación positiva BRAF V600E o V600K fueron candidatos a participar en el estudio.
Posteriormente, todas las muestras de pacientes fueron testadas de nuevo utilizando el ensayo validado bioMerieux (bMx) THxID BRAF, que posee marcado CE. El ensayo bMx THxID BRAF es un ensayo de identificación de un alelo específico de PCR realizado sobre ADN extraído de tejido tumoral en FFPE. Este ensayo se diseñó para detectar con mayor sensibilidad las mutaciones BRAF V600E y V600K (menos de un 5% de secuencias V600E y V600K, sobre un panel de secuencias de tipo nativo a partir de ADN extraído de un tejido tumoral en FFPE). Los análisis de secuenciación retrospectivos y bidireccionales realizados por el método Sanger en estudios clínicos y preclínicos, han demostrado que el ensayo también detecta las mutaciones menos frecuentes BRAF V600D y V600E/K601E con menor sensibilidad. De las muestras procedentes de estudios preclínicos y clínicos (n=876) que presentaron mutación positiva por el test THxIB BRAF y que posteriormente fueron secuenciadas utilizando el método de referencia, la especificidad del ensayo fue del 94%.
Eficacia clínica y seguridad
Dabrafenib en combinación con trametinib Pacientes que no habían recibido un tratamiento previo
La seguridad y eficacia de trametinib (2 mg una vez al día) con dabrafenib (150 mg dos veces al día) en pacientes adultos con melanoma no resecable o metastásico con mutación BRAF V600 se ha estudiado en dos estudios Fase III y un estudio Fase I/II de soporte.
MEK115306 (COMBI-d):
MEK115306 es un estudio Fase III, aleatorizado, doble ciego que compara la combinación de dabrafenib y trametinib con dabrafenib y placebo en primera línea para pacientes con melanoma no resecable (estadío IIIC) o mestatásico (estadío IV) con mutación BRAF V600. La variable primaria del estudio fue la supervivencia libre de progresión (SLP) y la variable secundaria fue la supervivencia global (SG). Los pacientes se clasificaron por niveles de lactato de deshidrogenasa (LDH) (>vs < del límite superior normal) y por tipo de mutación BRAF (V600E vs V600K).
Se aleatorizaron 423 pacientes en una relación 1:1, N=211, en el grupo de la combinación y N=212 en el grupo de dabrafenib. La mayoría de los sujetos fueron de raza caucásica (>99%) y varones (53%), con una edad media de 56 años (28% fueron >65 años). La mayoría se encontraban en un estadío IVM1c (67%) de la enfermedad. Al inicio, la mayoría tenían un LDH < del límite superior normal (65%), un estado de desarrollo ECOG de 0 (72%) y con enfermedad visceral (73%). La mayoría tenían mutación BRAF V600E (85%). Los sujetos con metástasis cerebrales no se incluyeron en el ensayo.
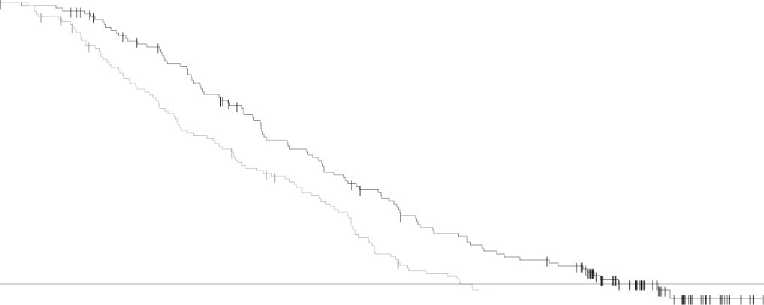
El análisis final de la supervivencia global (a 12 de enero de 2015) demostró una mejoría estadísticamente significativa de la SG con la combinación en comparación con dabrafenib en monoterapia (Figura 1). Se estima una mayor supervivencia global para el brazo de la combinación (74% al año y 51% a los 2 años) que en dabrafenib en monoterapia (68% y 42% , respectivamente).
Page 1 of 1
Figura 1 Curvas Kaplan-Meier de Supervivencia Global del estudio MEK115306 (Población por intención de tratar (ITT)
Dabrafenib + Trametinib Dabrafenib + Placebo
1.0
0.9
0.7
0.4
0.3
0.2
Dabrafenib + Trametinib
Dabrafenib + Placebo
Number at risk 211 208
212 206
ni mui mu ih i—i-
|
Dabrafenib + trametinib (N=211) |
Dabrafenib + placebo (N=212) | |||||
|
Supervivencia Global 12 Enero 2015 | ||||||
|
Número de eventos (%) |
99 (47%) |
123 (58%) | ||||
|
SG media (meses) |
25,1 |
18,7 | ||||
|
Hazard Ratio ajustado (95% CI) |
0,71 (0,55, 0,92) | |||||
|
Valor P Rango-log estratificado |
0,011 | |||||
200
191
187
175
174
159
159
147
10
144
138
135
127
124
111
112
104
106
95
103
88
i
22
88
70
24
53
42
26
21
10
28
12 14 16 18 20
Time from Randomization (Months)
3
30
32
Tabla 6 Resultados de eficacia del estudio MEK115306 (COMBI-d)
|
Objetivo |
Dabrafenib + |
Dabrafenib + |
Dabrafenib + |
Dabrafenib + |
|
Trametinib |
Placebo |
Trametinib |
Placebo | |
|
(N=211) |
(N=212) |
(N=211) |
(N=212) | |
|
Fecha de corte de |
26 Agosto2013 |
12 Enero 2015 | ||
|
datos | ||||
|
SLPa | ||||
|
Progresión de la enfermedad o muerte, n (%) |
102 (48) |
109 (51) |
139 (66) |
162 (76) |
|
SLP medio (meses) |
9,3 |
8,8 |
11,0 |
8,8 |
|
(95% IC) |
(7,7, 11,1) |
(5,9, 10,9) |
(8,0, 13,9) |
(5,9, 9,3) |
|
HR (Hazard Ratio) (95% IC) |
0,75 (0,57, 0,99) |
0,67 (0,53, 0,84) | ||
|
valor P |
0,035 |
<0,001 | ||
|
TRGb |
67 |
51 |
69 |
53 |
|
(95% IC) |
(59,9, 73,0) |
(44,5, 58,4) |
(61,8,74,8) |
(46,3, 60,2) |
|
Diferencia TRG |
15 |
e |
15e | |
|
(95% IC) |
(5,9, 24,5) |
(6.0, 24,5) | ||
|
valor P |
0,0015 |
0.0014 | ||
|
DdRc medio (meses) |
9,2d |
10,2d |
12,9 |
10,6 |
|
(95% CI) |
(7,4, NA) |
(7,5, NA) |
(9,4,19,5) |
(9,1, 13,8) |
|
a - Supervivencia libre de progresión (valorado por el investigador) b - Tasa de respuesta global (TRG) =Respuesta completa + Respuesta parcial c - Duración de la respuesta (DdR) d - Estaba en marcha en el momento de recogida de la mayoría de las respuestas evaluadas por el investigador (>59%) e - diferencia de TRG calculada a partir de los resultados de TRG sin redondear | ||||
|
NA = No alcanzado | ||||
MEK116513 (COMBI-v):
MEK116513 es un estudio abierto Fase III, aleatorizado, que compara la combinación de dabrafenib y trametinib con vemurafenib en monoterapia en pacientes con melanoma metastásico con mutación BRAF V600. La variable primaria del estudio fue la supervivencia global con SLP, como variable secundaria. Los pacientes se clasificaron por niveles de lactato de deshidrogenasa (LDH) (>vs < del límite superior normal) y por tipo de mutación BRAF (V600E vs V600K).
Se aleatorizaron 704 pacientes en una relación 1:1 tanto en el grupo de la combinación como en el grupo de vemurafenib. La mayoría de los sujetos fueron de raza caucásica (>96%) y varones (55%), con una edad media de 55 años (24% fueron >65 años). La mayoría se encontraban en un estadío IVM1c (61%) de la enfermedad. Al inicio, la mayoría tenían un LDH < del límite superior normal (67%), un estado de desarrollo ECOG de 0 (70%) y con enfermedad visceral (78%). La mayoría, 54% de los pacientes, teían <3 enfermedades al inicio. La mayoría tenían mutación BRAF V600E (89%). Los sujetos con metástasis cerebrales no se incluyeron en el ensayo.
El análisis final actualizado de SG (13 de marzo de 2015) demostró una mejoría estadísticamente significativa de la SG con la combinación en comparación con vemurafenib en monoterapia (Figura 2). A los 12 meses, se estima una supervivencia global del 72% para el tratamiento de la combinación y de un 65% para vemurafenib.
Figura 2 Curvas Kaplan-Meier del análisis de SG actualizado para el estudio MEK116513' f 1
Proportion Alive
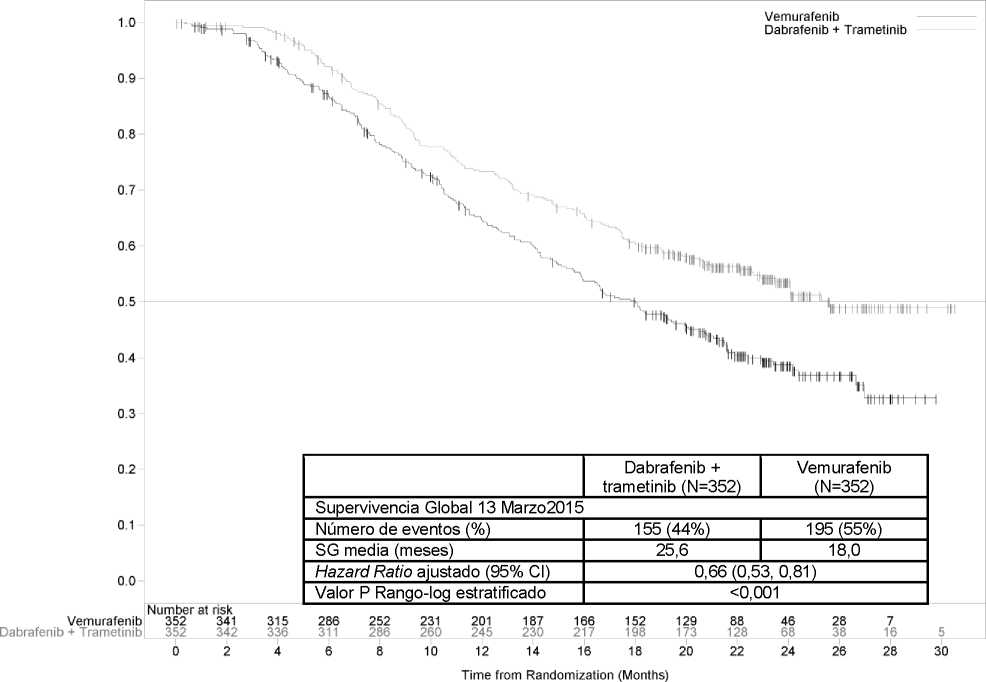
Tabla 7 Resultados de eficacia del estudio MEK116513 (COMBI-v)
|
Objetivo |
Dabrafenib + Trametinib (N=352) |
Vemurafenib (N=352) |
|
SLP | ||
|
Progresión de la enfermedad o |
166 (47) |
217 (62) |
|
muerte, n (%) | ||
|
SLP medio (meses) (95% IC) |
11,4 |
7,3 |
|
(9,9, 14,9) |
(5,8, 7,8) | |
|
HR (Hazard Ratio) |
0,56 | |
|
(95% IC) |
(0,46, 0,69) | |
|
valor P |
<0,001 | |
|
TRGb |
226 (64) |
180 (51) |
|
(95% IC) |
(59,1, 69,4) |
(46,1, 56,8) |
|
Diferencia TRG |
13 | |
|
(95% IC) |
(5,7, 20,2) | |
|
valor P |
0,0005 | |
|
DdRc medio (meses) | ||
|
(95% CI) |
13,8 |
7,5 |
|
(11,0, NR) |
(7,3, 9,3) | |
Tratamiento previo con un inhibidor BRAF
Hay pocos datos de pacientes que tomaran la combinación de dabrafenib con trametinib que hayan progresado con un inhibidor BRAF previo.
Se evaluó la seguridad, la farmacocinética, farmacodinamia y eficacia clínica de la combinación de dabrafenib y trametinib, en pacientes con melanoma con mutación BRAF en un estudio abierto Fase I/II (BRF113220). Este studio constó de cuatro partes, A-D. La Parte A un estudio de interacción de medicamento (n=8), Parte B, un estudio de escalado y expansión de dosis (n=80), Parte C, un estudio aleatorio de rango de dosis (n=162) y, una Parte D de evaluación farmacocinética y seguridad de las cápsulas de dabrafenib HPMC (n=109).
La Parte B del estudio BRF113220 (incluida la cohorte de 26 pacientes que progresaron con un inhibidor BRAF). La combinación de 2 mg de trametibib una vez al día y 150 mg de dabrafenib dos veces al día demostró actividad clínica limitada en pacientes que habían progresado con un inhibidor BRAF. La evaluación del investigador confirmó la tasa de respuesta del 15% (95% IC: 4,4, 34,9) y la SLP fue de 3,6 meses (95% IC: 1,9, 5,2). Se vieron resultados similares en 45 pacientes que pasaron de dabrafenib en monoterapia a la combinación 2 mg de trametinib una vez al día y 150 mg de dabrafenib dos veces al día en la Parte C del estudio. En estos pacientes un 13% (95% IC: 5,0, 27.0) confirmó la tasa de respuesta con un SLP media de 3,6 meses (95% IC: 2, 4).
La eficacia de dabrafenib en el tratamiento de pacientes adultos con melanoma metastásico o irresecable con mutación BRAF V600 positiva, fue evaluada en 3 estudios (BRF113683 [BREAK-3], BRF113929 [BREAK-MB], y BRF113710 [BREAK-2]) que incluyeron pacientes con mutaciones BRAF V600E y/o V600K.
En número total de sujetos incluidos en estos estudios fue de 402 sujetos con mutación BRAF V600E y 49 sujetos con mutación BRAF V600K. Los pacientes con melanoma provocado por mutaciones BRAF diferentes a V600E fueron excluidos del ensayo clínico confirmatorio y en relación a los pacientes con mutación V600K en estudios de un solo brazo, la actividad parece ser menor en estos tumores que en tumores V600E.
No se dispone de datos de pacientes que tuviesen melanoma con mutaciones BRAF V600 diferentes a V600E y V600K. No se ha investigado la eficacia de dabrafenib en sujetos previamente tratados con un inhibidor de tirosina quinasa.
Pacientes no tratados previamente (resultados del estudio Fase III [BREAK-3])
La eficacia y seguridad de dabrafenib fue evaluada en un estudio Fase III aleatorizado, abierto, donde se comparaba dabrafenib frente a dacarbazina (DTIC) en pacientes con melanoma avanzado (Estadío III no resecable) o metastásico (Estadío IV) con mutación BRAF V600E positiva, no tratados previamente. Los pacientes con melanoma provocado por mutaciones BRAF distintas a V600E fueron excluidos.
El objetivo primario de este estudio fue evaluar la eficacia de dabrafenib frente a DTIC con respecto a la Supervivencia Libre de Progresión (SLP) mediante la evaluación del investigador. A los pacientes del brazo de DTIC, tras confirmación de progresión inicial de enfermedad mediante radiografía independiente, se les permitió cambiar de brazo para recibir tratamiento con dabrafenib. Las características basales entre ambos grupos de tratamiento fueron equilibradas. El 60% de los pacientes eran varones y el 99,6% eran caucásicos. La mediana de la edad fue de 52 años, con un 21% de pacientes >65 años, el 98,4% con estado ECOG de 0 ó 1, y el 97% de los pacientes presentaban enfermedad metastásica.
En el análisis pre-específico con fecha de corte de datos 19 de diciembre de 2011, se alcanzó una mejora significativa en la variable primaria SLP (HR=0,30; IC 95% 0,18 - 0,51; p<0,0001). En la Tabla 8 se presenta un resumen de los resultados de eficacia procedentes del análisis primario y de los análisis post-hoc con un seguimiento adicional de 6 meses. Los datos de supervivencia global de un análisis post-hoc posterior se muestran en la Figura 3.
Tabla 8 Eficacia en pacientes no tratados previamente (Estudio BREAK-3, 25 Junio 2012)
|
Resultados a 19 de diciembre 2011 |
Resultados a 25 de junio de 2012 | |||
|
Dabrafenib N=187 |
DTIC N=63 |
Dabrafenib N=187 |
DTIC N=63 | |
|
Supervivencia Libre de Progresión | ||||
|
Mediana, meses (IC 95%) HR (IC 95%) |
5,1 (4,9, 6.9) 0,30 (0, P<0. |
2,7 (1,5, 3.2) 8, 0,51) 0001 |
6,9 (5.2,9.0) 0,37 (0,2 P<0. |
2,7 (1.5,32) 14, 0,58) 0001 |
|
Respuesta globala | ||||
|
% (IC 95%) |
53 (45,5, 60,3) |
19 (10,2, 30,9) |
59 (51,4, 66,0) |
24 (14, 36,2) |
|
Duración de la respuesta | ||||
|
Mediana, meses (IC 95%) |
N=99 5,6 (4,8, NR) |
N=12 NR (5,0, NR) |
N=110 8,0 (6,6, 11.5) |
N=15 7,6 (5,0, 9,7) |
|
Abreviaturas: IC: intervalo de confianza; DTIC: dacarbazina; HR: hazard ratio; NR: sin respuesta. a Definida como respuesta completa confirmada + respuesta parcial. | ||||
A fecha de corte del 25 de Junio 2012, treinta y cinco sujetos (55,6%) de los 63 aleatorizados para recibir DTIC cambiaron de brazo para recibir dabrafenib. El 63% de los sujetos aleatorizados para recibir dabrafenib y el 79% de los sujetos aleatorizados para recibir DTIC, progresaron o murieron. La mediana de la SLP después de cambiar de brazo de tratamiento fue de 4,4 meses.
Tabla 9 Datos de supervivencia procedentes del análisis primario y análisis post-hoc.
|
Fecha de corte |
Tratamiento |
Número de muertes (%) |
Hazard Ratio (95% CI) |
|
19 de diciembre 2011 |
DTIC |
9 (14%) |
0,61 (0,25, 1,48) (a) |
|
dabrafenib |
21 (11%) | ||
|
25 de junio de 2012 |
DTIC |
21 (33%) |
0,75 (0,44, 1,29) (a) |
|
dabrafenib |
55 (29%) | ||
|
18 de diciembre de 2012 |
DTIC |
28 (44%) |
0,76 (0,48, 1,21) (a) |
|
dabrafenib |
78 (42%) |
(a) Los pacientes no fueron censurados cuando se realizó el cruce
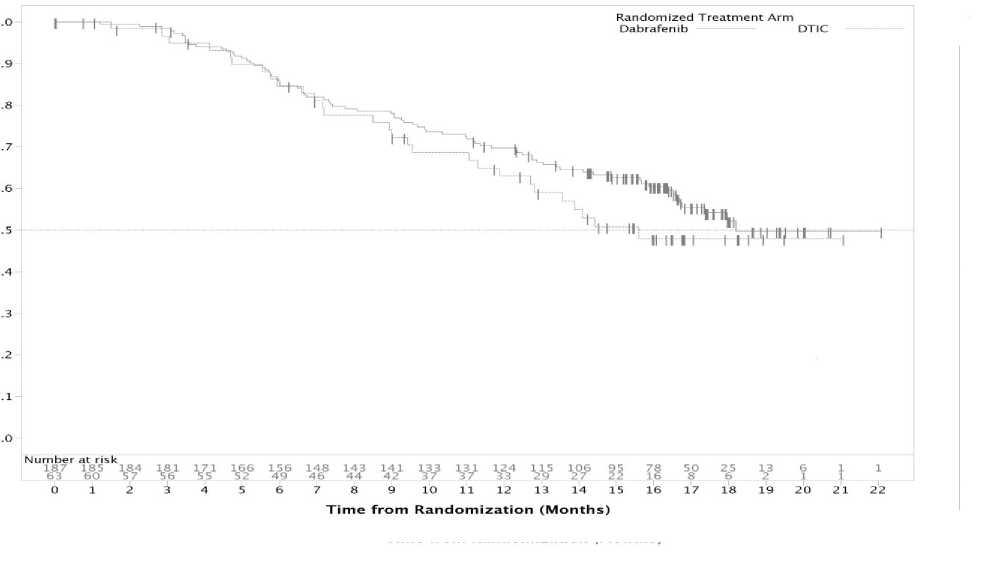
Los datos de SG procedentes de un análisis post-hoc basado en los datos de corte de fecha 18 de diciembre de 2012, mostraron una tasa de SG del 63% y el 70% para DTIC y dabrafenib respectivamente.
Figura 3 Curvas Kaplan-Meier de Supervivencia global (BREAK-3) (18 Diciembre 2012)
Proportion Alive
DAB
DTIC
Pacientes con metástasis cerebrales (Resultados del estudio Fase II [BREAK-MB])
El estudio BREAK-MB es un estudio Fase II, abierto, multicéntrico, de 2 cohortes, diseñado para evaluar la respuesta intracraneal de dabrafenib en sujetos con melanoma metastático en el cerebro (Estadío IV) y con confirmación histológica de mutación BRAF positiva (V600E o V600K). Los sujetos fueron reclutados en la Cohorte A (sujetos que no habían recibido tratamiento local previo para metástasis cerebral) o en la Cohorte B (sujetos que habían recibido tratamiento local previo para metástasis cerebral).
La variable primaria del estudio fue la tasa de respuesta global intracraneal (OIRR) en la población de pacientes con mutación V600E, según la evaluación de los investigadores. En la Tabla 10 se muestran los resultados según la evaluación del investigador de la OIRR confirmada y otros resultados de eficacia.
Tabla 10 Datos de la eficacia en pacientes con metástasis cerebrales (Estudio BREAK-MB)
|
Población de sujetos tratados | ||||
|
BRAF V600E (Primaria) |
BRAF V600K | |||
|
Cohorte A N=74 |
Cohorte B N=65 |
Cohorte A N=15 |
Cohorte B N=18 | |
|
Tasa de respuesta global intracraneal, % (I |
IC 95%)a | |||
|
39% (28,0, 51,2) P<0,001b |
31% (19,9, 43,4) P<0,001b |
7% (0,2, 31,9) |
22% (6,4, 47,6) | |
|
Duración de la respuesta intracraneal, met |
iana, meses (IC95%) | |||
|
N=29 4,6 (2,8, NR) |
N=20 6,5 (4,6, 6,5) |
N=1 2,9 (NR, NR) |
N=4 3,8 (NR, NR) | |
|
Respuesta global, % (IC 95 %)a | ||||
|
38% (26,8, 49,9) |
31% (19,9, 43,4) |
0 (0, 21,8) |
28% (9,7, 53,5) | |
|
Duración de la respuesta, mediana, meses ( |
IC 95%) | |||
|
N=28 5,1 (3,7, NR) |
N=20 4,6 (4,6, 6,5) |
NA |
N=5 3,1 (2,8, NR) | |
|
Supervivencia libre de progresión, mediana, meses (IC 95%) | ||||
|
3,7 (3,6, 5.0) |
3,8 (3,6, 5.5) |
1,9 (0,7, 3,7) |
3,6 (1,8, 5,2) | |
|
Supervivencia global, mediana, meses (IC 95%) | ||||
|
Mediana, meses |
7,6 (5,9, NR) |
7,2 (5,9, NR) |
3,7 (1,6, 5,2) |
5,0 (3,5, NR) |
|
Abreviaturas: IC: intervalo de confianza; NR: respuesta no alcanzada; NA: no aplicable a - Respuesta confirmada. b -Este estudio fue diseñado para apoyar o rechazar la hipótesis nula de OIRR <10% (basada en resultados históricos) a favor de la hipótesis alternativa de OIRR >30% en sujetos con mutación BRAF V600E positiva. | ||||
Pacientes que no fueron tratados previamente o que progresaron a una tratamiento sistémico previo (Resultados del estudio Fase II [BREAK-2])
El estudio BRF113710 (BREAK-2) fue un estudio multicéntrico, de un solo brazo, donde se reclutaron 92 sujetos con melanoma metastásico (Estadío IV) con mutación BRAF V600E o V600K confirmada.
La evaluación del investigador confirmó una tasa de respuesta del 59% (IC 95%: 48,2; 70,3) en pacientes con melanoma metastásico con mutación BRAF V600E (n=76) y la mediana de duración de la respuesta fue de 5,2 meses (IC 95%: 3,9; no calculada) en base a la mediana del peridodo de seguimiento de 6,5 meses. En pacientes con melanoma metastásico con mutación BRAF V600K (n=16), la tasas de respuesta fue del 13% (IC 95%: 0,0; 28,7), con una mediana de duración de la respuecta de 5,3 meses (IC 95%: 3,7; 6,8). A pesar de que los resultados son limitados debido al pequeño número de pacientes, la mediana de la SG parece ser consistente con los datos de pacientes con tumores BRAF V600E positivos.
Prolongación del intervalo QT
El peor caso de prolongación del intervalo QTc fue >60 milisegundos (mseg), que fue observado en el 3% de los sujetos tratados con dabrafenib (un caso >500 mseg integrado en la población de seguridad). En el estudio Fase III de MEK115306 no hubo ningún paciente tratado con trametinib en combinación con dabrafenib con el peor caso de prolongación del intervalo QTcB >500 msec; el 1% (3/209) de los pacientes, aumentó el intervalo QTcB en más de 60 msec respecto al inicio. En el estudio Fase III MEK116513, cuatro pacientes (1%) tratados con trametinib en combinación con dabrafenib, tuvo un aumento del intervalo QTcB de Grado 3 (>500 msec). Dos de estos pacientes que tuvieron un aumento del intervalo QTcB de Grado 3 (>500 msec) también presentaron un incremento de >60 msec respecto al inicio.
El efecto potencial de dabrafenib sobre la prolongación del intervalo QT fue evaluado en un estudio específico de dosis múltiple. Una dosis supraterapéutica de 300 mg de dabrafenib dos veces al día fue administrada a 32 sujetos con tumores BRAF V600E positivos. No se observó ningún efecto clínicamente relevante de dabrafenib o sus metabolitos en el intervalo QTc.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la oblicación de presentar los resultados de los ensayos realizados con dabrafenib en uno o más grupos de la población pediátrica en melanoma (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Dabrafenib se absorbe por vía oral con una mediana de tiempo hasta alcanzar el pico de concentración plásmática de 2 horas tras la administración de una dosis. La biodisponibilidad media absoluta de dabrafenib por vía oral es del 95% (IC 90%: 81; 110%). La exposición a dabrafenib (Cmáx y AUC) se incrementó de forma proporcional a la dosis entre 12 y 300 mg tras la administración de una única dosis, pero este incremento fue menor que el incremento proporcional de la dosis, tras la administración de dosis repetidas dos veces al día. Se observó una disminución de la exposición a dosis repetidas, probablemente debida a la inducción de su propio metabolismo. La tasa de acumulación media, AUC Día 18/ Día 1, fue de 0,73. Tras la administración de 150 mg dos veces al día, la media geométrica de la Cmax, AUC (0-T) y la concentración antes de la dosis (Cx) fue de 1.478 ng/ml, 4.341 ng*hr/ml y 26 ng/ml, respectivamente.
La administración de dabrafenib con alimentos reduce la biodisponibilidad (Cmax y AUC disminuye un 51% y 31% respectivamente) y retrasa la absorción de las cápsulas de dabrafenib al compararlo con la administración en ayunas.
Distribución
Dabrafenib se une a proteínas plasmáticas en un 99,7%. El volumen de distribución en estado estacionario tras la administración intravenosa de una microdosis es de 46 L.
Dabrafenib es un sustrato de la glicoproteína-P humana (Pgp) y de murina BCRP in vitro. Sin embargo, estos trasportadores tienen un impacto mínimo en la biodisponibilidad y eliminación de dabrafenib administrado por vía oral, y el riesgo de interacciones farmacológicas clínicamente relevantes con inhibidores de Pgp o BCRP es bajo. Dabrafenib no es un sustrato de los transportadores OATP1B1, OATP1B3 o OATP2B1 in vitro.
Ni dabrafenib ni sus 3 principales metabolitos demostraron ser inhibidores de Pgp in vitro. Biotransformación
El metabolismo de dabrafenib está mediado principalmente por CYP2C8 y CYP3A4, para formar hidroxi-dabrafenib, el cual es oxidado posteriormente vía CYP3A4 para formar carboxi-dabrafenib. El carboxi-dabrafenib puede ser descarboxilado mediante un proceso no enzimático para formar desmetil-dabrafenib. El carboxi-dabrafenib se excreta por la bilis y la orina. El desmetil-dabrafenib se puede formar también en el intestino y ser reabsorbido. El desmetil-dabrafenib se metaboliza mediante CYP3A4 en metabolitos oxidativos. La vida media terminal de hidroxi-dabrafenib es paralela a la del fármaco original con una vida media de 10 horas, mientras que los metabolitos carboxi-dabrafenib y desmetil-dabrafenib presentan vidas medias más largas (21-22 horas). Tras la administración de dosis repetidas, la media de metabolitos con respecto a los ratios del AUC del fármaco original fueron 0,9;
11 y 0,7 para hidroxi, carboxi y desmetil-dabrafenib, respectivamente. En base a la exposición, a la potencia relativa y a las propiedades farmacocinéticas, es probable que tanto hidroxi-dabrafenib como
desmetil-dabrafenib contribuyan a la actividad clínica de dabrafenib, mientras que es poco probable que la actividad de carboxi-dabrafenib sea significativa.
Eliminación
La vida media terminal tras la administración intravenosa de una única microdosis es de 2,6 horas. La vida media terminal de dabrafenib tras la administración oral de una dosis única es de 8 horas debido a la absorción limitada durante la eliminación (flip-flop farmacocinética). El aclaramiento plasmático tras la administración intravenosa es de 12 l/h.
Después de una dosis oral, la principal ruta de eliminación de dabrafenib es metabólica, mediada vía CYP3A4 y CYP2C8. Los productos de degradación relacionados con dabrafenib se excretan principalmente a través de las heces. El 71% de la dosis se recupera en las heces en forma de metabolitos y sólo el 23% en la orina.
Poblaciones especiales
Insuficiencia hepática
Un análisis farmacocinético poblacional indicó que niveles ligeramente elevados de bilirrubina y/o AST (según la clasificación del National Cancer Institute [NCI]) no afectaron significativamente al aclaramiento de dabrafenib cuando se administra por vía oral. Además, la insuficiencia hepática leve, definida según los niveles de bilirrubina y AST, no provocó un efecto significativo en la concentración de metabolitos de dabrafenib en plasma. No hay datos disponibles de pacientes con insuficiencia hepática de moderada a grave. Debido a que el metabolismo hepático y la secrección biliar son las principales rutas de eliminación de dabrafenib y sus metabolitos, la administración de dabrafenib se debe realizar con precaución en pacientes con insuficiencia hepática de moderada a grave (ver sección 4.2).
Insuficiencia renal
Un análisis farmacocinético poblacional indicó que la insuficiencia renal leve no afecta al aclaramiento de dabrafenib cuando se administra por vía oral. Aunque los datos en insuficiencia renal moderada son limitados, estos pueden indicar que no existe un efecto clínico relevante. No hay datos disponibles en sujetos con insuficiencia renal grave (ver sección 4.2).
Pacientes de edad avanzada
En base al análisis farmacocinético poblacional, la edad no tiene un efecto significativo en la farmacocinética de dabrafenib. Tener una edad superior a 75 años, es un indicador significativo de las concentraciones plasmáticas de carboxi-dabrafenib y desmetil-dabrafenib, con exposiciones mayores del 40% en sujetos >75 años, en comparación con sujetos <75 años.
Peso corporal y género
Basándose en el análisis farmacocinético poblacional, se determinó que el género y el peso influyen el aclaramiento de dabrafenib cuando se administra por vía oral. El peso también impactó sobre el volumen de distribución y la distribución del aclaramiento de dabrafenib cuando se administra por vía oral. Estas diferencias farmacocinéticas no fueron consideradas clínicamente relevantes.
Raza
Los datos que hay son insuficientes para evaluar el posible efecto de la raza en la farmacocinética de dabrafenib.
No se han realizado estudios para investigar la farmacocinética de dabrafenib en pacientes pediátricos.
5.3 Datos preclínicos sobre seguridad
No se han realizado estudios de carcinogenicidad con dabrafenib. Los test in vitro realizados en bacterias y cultivos celulares de mamíferos y el ensayo in vivo en micronúcleos de roedores, mostraron que dabrafenib no fue mutagénico o clastogénico.
En los estudios combinados de fertilidad femenina, estudios en embriones tempranos y de desarrollo embriofetal en ratas, el número de cuerpos lúteos en el ovario se redujo en las hembras preñadas a dosis de 300 mg/Kg/día (aproximadamente 3 veces la exposición clínica en humanos en relación al AUC), pero no hubo efectos en el ciclo estral, en el apareamiento, ni en los índices de fertilidad. Con dosis de 300 mg/Kg/día se observó toxicidad en el desarrollo incluyendo muerte embrionaria y defectos en el septo ventricular, y a dosis >20 mg/Kg/día (>0,5 veces la exposición clínica en humanos en relación al AUC) se observó retraso en el desarrollo esquelético y reducción de peso corporal fetal.
No se han realizado estudios de fertilidad en machos con dabrafenib. Sin embargo, en estudios a dosis repetidas en ratas y perros, se observó degeneración/depleción testicular (>0,2 veces la exposición clínica en humanos en relación al AUC). Los cambios testiculares en ratas y perros siguieron presentes durante un periodo de recuperación de 4 semanas (ver sección 4.6).
En perros, se observaron efectos cardiovasculares, incluyendo degeneración/necrosis de la arteria coronaria y/o hemorragia, hipertrofia/hemorragia de la válvula auriculo-ventricular cardiaca y proliferación fibrovascular auricular (>2 veces la exposición clínica en relación al AUC). En ratones se observó inflamación focal arterial/perivascular en varios tejidos, y en ratas se observó un incremento de la incidencia de la degeneración arterial hepática y degeneración espontánea de cardiomiocitos con inflamación (cardiomiopatía espontánea) (>0,5 y 0,6 veces la exposición clínica en ratas y ratones, respectivamente). En ratones se observaron efectos hepáticos, incluyendo necrosis hepatocelular e inflamación (>0,6 veces la exposición clínica). En algunos perros se observó inflamación bronco-alveolar en los pulmones a dosis >20 mg/Kg/día (>9 veces la exposición clínica en humanos en relación al AUC) y fue asociada con respiración poco profunda y/o entrecortada.
Se han observados efectos hematológicos reversibles en perros y ratas a las que se administró dabrafenib. En estudios en perros y ratas de hasta 13 semanas de duración, se observó una disminución en el recuento de reticulocitos y/o glóbulos rojos (>10 y 1,4 veces la exposición clínica, respectivamente).
En estudios de toxicidad en ratas jóvenes se observaron efectos (>0,2 veces la exposición clínica en humanos en relación al AUC) en el crecimiento (huesos largos de menor longitud), toxicidad renal (depósitos tubulares, aumento de la incidencia de quistes corticales y basofilia tubular e incrementos reversibles en las concentraciones de urea y/o creatinina) y toxicidad testicular (degeneración y dilatación tubular).
En un ensayo in vitro realizado en fibroblasos de ratón 3T3 mediante el método Neutral Red Uptake (NRU) y en un estudio in vivo de fototoxicidad oral con ratones sin pelo a dosis >100 mg/kg (>44 veces la exposición clínica en relación a la Cmax), dabrafenib fue fototóxico.
Combinación con trametinib
En un estudio en perros a los que se les dio trametinib y dabrafenib en combinación durante 4 semanas se observó signos de toxicidad gastrointestinasl y disminución de las células linfoides del timo a niveles inferiores que los perros con solo trametinib. Por lo contrario, se observaron misma toxicidad es que los estudios en monoterapia.
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Contenido de la cápsula Celulosa microcristalina Estearato de magnesio Dióxido de sílice coloidal
Cubierta de la cápsula Óxido de hierro rojo (E172)
Dióxido de titanio (E171)
Hipromelosa (E464)
Tinta de impresión Óxido de hierro negro (E172)
Shellac
Propilenglicol
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
2 años.
6.4 Precauciones especiales de conservación
Este medicamento no requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Frasco de polietileno de alta densidad (PEAD) de color blanco opaco con un tapón de rosca de polipropileno y un desecante de silica gel.
Cada frasco puede contener 28 o 120 cápsulas duras.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Tafínlar 50 mg cápsulas duras
EU/1/13/865/001
EU/1/13/865/002
Tafinlar 75 mg cápsulas duras
EU/1/13/865/003
EU/1/13/865/004
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
26 de agosto de 2013 10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de los fabricantes responsables de la liberación de los lotes
GLAXO WELLCOME, S.A.
Avda. Extremadura, 3, Pol. Ind. Allendeduero 09400, Aranda de Duero (Burgos)
España
Novartis Pharmaceuticals UK Limited
Frimley Business Park
Frimley
Camberley, Surrey GU16 7SR Reino Unido
Novartis Pharma GmbH RoonstraBe 25 D-90429 Nuremberg Alemania
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 quater, (párrafo 7), de la Directiva 2001/83/CE y cualquier modificación posterior publicada en el portal web europeo sobre medicamentos.
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo
acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de
Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Medidas adicionales de minimización de riesgos
No aplica.
• Obligación de llevar a cabo medidas post-autorización
No aplica.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
ESTUCHE
1. NOMBRE DEL MEDICAMENTO
Tafínlar 50 mg cápsulas duras dabrafenib
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene dabrafenib mesilato equivalente a 50 mg de dabrafenib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsula, dura
28 cápsulas 120 cápsulas
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Contiene un desecante, no se debe retirar ni tragar.
8. FECHA DE CADUCIDAD
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/865/001 28 cápsulas
EU/1/13/865/002 120 cápsulas
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
tafinlar 50 mg
ETIQUETA DEL FRASCO
1. NOMBRE DEL MEDICAMENTO
Tafínlar 50 mg cápsulas duras dabrafenib
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene dabrafenib mesilato equivalente a 50 mg de dabrafenib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsula, dura
28 cápsulas 120 cápsulas
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/865/001 28 cápsulas
EU/1/13/865/002 120 cápsulas
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ESTUCHE
1. NOMBRE DEL MEDICAMENTO
Tafínlar 75 mg cápsulas duras dabrafenib
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene dabrafenib mesilato equivalente a 75 mg de dabrafenib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsula, dura
28 cápsulas 120 cápsulas
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Contiene un desecante, no se debe retirar ni tragar.
8. FECHA DE CADUCIDAD
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/865/003 28 cápsulas
EU/1/13/865/004 120 cápsulas
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
tafinlar 75 mg
ETIQUETA DEL FRASCO
1. NOMBRE DEL MEDICAMENTO
Tafínlar 75 mg cápsulas duras dabrafenib
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene dabrafenib mesilato equivalente a 75 mg de dabrafenib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsula, dura
28 cápsulas 120 cápsulas
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/865/003 28 cápsulas
EU/1/13/865/004 120 cápsulas
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
B. PROSPECTO
Prospecto: información para el paciente
Tafinlar 50 mg cápsulas duras Tafinlar 75 mg cápsulas duras
dabrafenib
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Tafinlar y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Tafinlar
3. Cómo tomar Tafinlar
4. Posibles efectos adversos
5. Conservación de Tafinlar
6. Contenido del envase e información adicional
1. Qué es Tafinlar y para qué se utiliza
Tafinlar es un medicamento que contiene el principio activo dabrafenib. Se utiliza solo o en combinación con otro medicamento que contiene trametiniben adultos para tratar un tipo de cáncer de piel llamado melanoma,
• que tiene un cambio (mutación) concreto en un gen llamado “BRAF”, y
• que se ha extendido a otras partes del cuerpo o no puede ser eliminado por cirugía.
Esta mutación en el gen, puede haber causado el desarrollo del melanoma. Tafinlar actúa sobre las proteínas producidas por el gen BRAF modificado y ralentiza o detiene el desarrollo del cáncer.
2. Qué necesita saber antes de empezar a tomar Tafinlar
Tafinlar sólo se puede utilizar para tratar melanomas que tengan ese cambio (mutación) en el gen BRAF, por lo que su médico en primer lugar, tomará muestras de tejido del tumor para determinar si Tafinlar es adecuado para usted.
Si su médico decidiera que tome el tratamiento combinado de Tafinlar y trametinib, lea detenidamente el prospecto de dabrafenib así como este prospecto.
No tome Tafinlar:
• si es alérgico a dabrafenib o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6). Si cree que esto le aplica, consulte con su médico.
Advertencias y precauciones
Consulte a su médico antes de empezar a tomar Tafinlar. Su médico necesita saber si:
• tiene problemas de hígado.
• tiene o ha tenido alguna vez problemas de riñón.
Puede que mientras esté en tratamiento con Tafinlar, su médico necesite tomar muestras de sangre para comprobar el funcionamiento de su hígado y riñones.
• ha tenido otro tipo de cáncer distinto al melanoma, ya que puede tener mayor riesgo de desarrollar otros cánceres que no sean de piel, mientras esté tomando Tafinlar.
Antes de tomar Tafinlar en combinación con trametinib, su médico debe saber:
• Si tiene problemas de corazón como fallo cardiaco o problemas con la forma en que late el corazón
• Si tiene problemas oculares, como bloqueo de las venas que irrigan el ojo (oclusión de las venas de la retina) o hinchazón del ojo que se pueda deber a un bloqueo de fluido (coriorretinopatía).
• Si tiene o ha tenido algúna vez problemas de respiración o pulmonares, comodificultad al respirar frecuentemente acompañado de tos seca, respiración entrecortada y fatiga.
Consulte con su médico si piensa que alguna de las circustancias anteriores le aplica.
Enfermedades a las que tiene que estar atento
Algunas personas que toman Tafinlar desarrollan otras enfermedades que pueden ser graves. Necesita saber a qué signos y síntomas debe prestar atención mientras está tomando este medicamento. En esta sección, se mencionan brevemente algunos de estos síntomas (sangrados, fiebre, cambios en la piel y problemas en los ojos), pero encontrará información más detallada en la sección 4, “Posibles efectos adversos”.
Sangrados
Tomar Tafinlar en combinación con trametinib puede causar sangrados graves en el cerebro, sistema digestivo (tales como estómago, recto o intestino), pulmones y otros órganos, que pueden provocarle la muerte. Los síntomas pueden ser:
• dolor de cabeza, mareo o sentirse sin fuerzas
• que pase sangre a las heces o heces negras
• que pase sangre a la orina
• dolor de estómago
• tos /vómitos de sangre
Infome lo antes posible a su médico si siente estos síntomas Fiebre (temperatura elevada)
Este medicamento puede causar fiebre (ver también la sección 4). En algunos casos, hay personas con fiebre que desarrollan tensión sanguinea baja, mareo y otros síntomas. Si mientras toma este medicamento tiene fiebre, consulte ínmediatamente con su médico, farmacéutico o enfermero.
Alteraciones del corazón
Tafinlar puede causar problemas en el corazón, o puede hacer que problemas existentes empeoren en personas que tomen Tafinlar con trametinib.
Informe a su médico si presenta algún problema en el corazón. Antes de empezar el tratamiento y durante el tratamiento con Tafinlar en combinación con Mekinist, su médico le realizará pruebas para comprobar si su corazón funciona adecuadamente. Informe a su médico inmediatamente si siente que: su corazón late con fuerza, se le acelera el corazón o late a un ritmo irregular, si presenta mareos, se siente cansado, aturdido, le falta el aire o se le hinchan las piernas. Si es necesario, su médico puede decidir interrumpir el tratamiento o suspenderlo.
Cambios en la piel durante y después del tratamiento
Su médico revisará su piel antes de empezar el tratamiento con este medicamento, y de forma regular mientras lo esté tomando.
Consulte a su médico inmediatamente si nota cualquier cambio en su piel mientras esté tomando este medicamento o después del tratamiento (ver también la sección 4).
Alteraciones en los ojos
Su médico debe examinar sus ojos mientras esté tomando este medicamento.
Consulte a su médico inmediatamente si tiene enrojecimiento e irritación de los ojos, visión borrosa, dolor en los ojos u otros cambios en la visión durante el tratamiento (ver también la sección 4). Tafinlar cuando se da en combinación con trametinib puede causar problemas en los ojos, incluso ceguera. Trametinin no está recomendado si alguna vez ha tenido un bloqueo en las venas que drenan los ojos (oclusión de las venas retinianas). Informe a su médico inmediatamente si durante el tratamiento presenta los siguientes síntomas relacionados con problemas en los ojos: visión borrosa, pérdida de visión u otros cambios en la visión, si ve puntos de colores o halos (visión borrosa alrededor de objetos). Si es necesario, su médico puede decidir interrumpir el tratamiento o suspenderlo.
Lea la información sobre la fiebre, los cambios en la piel y las alteraciones en los ojos en la sección 4 de este prospecto. Comunique a su médico, farmacéutico o enfermero si tiene alguno de estos signos o síntomas.
Problemas de hígado
Tafinlar en combinación con trametinib puede causar problemas en su hígado que puede desarrollar enfermedades como hepatitis o fallo hepático, que puede ser mortal. Su médico le controlará periodicamente. Los signos de que su hígado no funciona adecuadamente son:
• pérdida de apetito
• encontrase mal (náuseas)
• sentirse enfermo (vómitos)
• dolor en el estómado (abdomen)
• amarilleo de su piel o del blanco de los ojos (ictericia)
• orina de color oscuro
• picores de piel
Infome lo antes posible a su médico si siente estos síntomas Dolor muscular
Tafinlar en combinación con trametinib puede provocarle deterioro de los músculos (rabdomiolisis). Informe a su médico lo antes posible si nota alguno de estos síntomas:
• dolor muscular
• orina oscura debido al daño en riñones
Si fuera necesario, su médico podría decidir interrumpir su tratamiento o detenerlo por completo. Niños y adolescentes
Tafinlar no está recomendado en niños y adolescentes. No se conocen los efectos de Tafinlar en personas menores de 18 años.
Toma de Tafinlar con otros medicamentos
Antes de empezar el tratamiento, informe a su médico, farmacéutico o enfermero si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento. Esto incluye medicamentos adquiridos sin receta médica.
Algunos medicamentos pueden afectar la forma de actuar de Tafinlar, o hacer que sea más probable
que tenga efectos adversos. Tafinlar también puede afectar la forma de actuar de otros medicamentos.
Entre ellos se incluyen:
• medicamentos para el control de la natalidad (anticonceptivos) que contengan hormonas, como la píldora, las inyecciones o los parches
• warfarina y acenocumarol, medicamentos utilizados para hacer la sangre más fluida
• digoxina, utilizado para tratar enfermedades del corazón
• medicamentos para tratar infecciones por hongos, como ketoconazol, itraconazol, voriconazol y posaconazol
• algunos bloqueantes de los canales de calcio, utilizados para tratar la tensión arterial alta, como diltiazem, felodipino, nicardipino, nifedipino o verapamilo
• medicamentos para el tratamiento del cáncer, como cabazitaxel
• algunos medicamentos que disminuyen la grasa (lípidos) en sangre, como el gemfibrozilo
• algunos medicamentos para tratar ciertas enfermedades psiquiátricas, como haloperidol
• algunos antibióticos, como claritromicina, doxiciclina y telitromicina
• algunos medicamentos utilizados para la tuberculosis, como la rifampicina
• algunos medicamentos para reducir los niveles de colesterol, como atorvastatina y simvastatina
• algunos medicamentos inmunosupresores, como ciclosporina, tacrolimus y sirolimus
• medicamentos que reducen la acidez de estómago, como omeprazol
• algunos medicamentos antiinflamatorios, como dexametasona y metilprednisolona
• algunos medicamentos para tratar el VIH, como ritonavir, amprenavir, indinavir, darunavir,
delavirdina, efavirenz, fosamprenavir, lopinavir, nelfinavir, tipranavir, saquinavir y atazanavir
• algunos medicamentos utilizados para el alivio del dolor, como fentanilo o metadona
• medicamentos para tratar convulsiones (crisis epilepticas), como fenitoína, fenobarbital, primidona, ácido valpróico o carbamacepina
• medicamentos antidepresivos como nefazodona y la Hierba de San Juan (Hypericum perforatum).
Consulte con su médico, farmacéutico o enfermero si está tomando cualquiera de estos medicamentos (o si no está seguro). Su médico puede considerar ajustar su dosis.
Mantenga una lista con los medicamentos que este tomando para poder enseñársela a su médico,
farmacéutico o enfermero.
Embarazo, lactancia y fertilidad
No se recomienda utilizar Tafinlar durante el embarazo.
• Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico, farmacéutico antes de utilizar este medicamento. No se recomienda utilizar Tafinlar durante el embarazo, ya que posiblemente podría dañar al feto.
• Si es mujer, y puede quedarse embarazada, debe utilizar un método de control de natalidad fiable mientras esté tomando Tafinlar y hasta 4 semanas después de haber terminado el tratamiento y 4 meses después de la última dosis de trametinib cuando se de en combinación con Tafinlar.
• Los métodos anticonceptivos que contienen hormonas (como la píldora, las inyecciones o los parches) pueden no ser tan eficaces cuando se está tomando Tafinlar o el tratmiento combinado (Tafinlar así como trametinib). Por ello, mientras esté tomando este medicamento, necesita utilizar otro método anticonceptivo, como un método de barrera (p.ej. los preservativos) para no quedarse embarazada. Consulte con su médico, farmacéutico o enfermero.
• Si se queda embarazada mientras está tomando este medicamento, consulte con su médico inmediatamente.
No se recomienda utilizar Tafinlar durante la lactancia
Se desconoce si los componentes de este medicamento pueden pasar a la leche materna.
Si está en periodo de lactancia, o planea dar el pecho, debe indicárselo a su médico. Usted y su médico decidirán si toma este medicamento o dar el pecho.
Fertilidad - ambos, hombres y mujeres
Los estudios realizados en animales han demostrado que la sustancia activa dabrafenib puede reducir permanentemente la fertilidad de los hombres. Así mismo, los hombres que toman Tafinlar pueden ver reducido su recuento de esperma y puede que su esperma no vuelva a los niveles normales hasta que haya dejado de tomar este medicamento.
Antes de empezar el tratamiento con Tafinlar, consulte con su médico sobre las opciones para mejorar las oportunidades de tener hijos en el futuro.
Tomar Tafinlar con trametinib: trametinib puede afectar a la fertilidad tanto de hombres y mujeres.
Si tiene más preguntas sobre los efectos de este medicamento sobre el recuento de esperma, pregunte a su médico, farmacéutico o enfermero.
Conducción y uso de máquinas
Tafinlar puede provocar efectos adversos que pueden afectar a su capacidad de conducir o utilizar máquinas.
Evite conducir o utilizar máquinas si tiene problemas de visión o si se siente cansado o débil, o si siente que le falta energía.
La descripción de los efectos adversos puede encontrarse en las secciones 2 y 4 de este prospecto.
Si no está seguro, hable con su médico, farmacéutico o enfermero. Su capacidad para conducir y utilizar máquinas se puede ver afectada incluso por la propia enfermedad, los síntomas o el tratamiento.
3. Cómo tomar Tafinlar
Tome siempre Tafinlar exactamente como se lo haya indicado su médico, farmacéutico o enfermero. En caso de duda pregunte a su médico, farmacéutico o enfermero.
Cuánto tomar
La dosis recomendada de Tafinlar, tanto se utilice solo o en combinación con trametinib es de 2 cápsulas de 75 mg dos veces al día (lo que equivale a una dosis diaria de 300 mg). La dosis recomendada de trametinib, tanto se utilice solo o en combinación con Tafinlar, es de 2 mg una vez al día.
Su médico decidirá si necesita tomar una dosis menor según los efectos adversos que tenga.
Tafinlar también está disponible en cápsulas de 50 mg para aquellos casos en los que se recomiende una reducción de la dosis.
No tome más Tafinlar del que le ha recomendado su médico, ya que podría aumentar el riesgo de tener efectos adversos.
Cómo tomarlo
Tome las cápsulas enteras, con agua, una tras otra.
No mastique o rompa las cápsulas, ya que perderán su efecto.
Tome Tafinlar dos veces al día, con el estómago vacío. Es decir:
• una vez que haya tomado Tafinlar, debe esperar por lo menos 1 hora antes de comer, o
• si ha comido, debe esperar por lo menos 2 horas antes de tomar Tafinlar.
Tome Tafinlar por la mañana y por la noche, con una separación de unas 12 horas. Tome sus dosis de la mañana y de la noche a la misma hora del día, todos los días. Esto aumentará la probabilidad de que recuerde tomar las cápsulas.
No tome las dosis de la mañana y de la noche a la vez.
Si toma más Tafinlar del que debe
Si toma demasiadas cápsulas de Tafinlar, contacte con su médico, farmacéutico o enfermero. Si le
es posible, enséñeles el envase de Tafinlar con este prospecto.
Si olvidó tomar Tafinlar
Si han transcurrido menos de 6 horas desde la hora habitual a la que debería haber tomado Tafinlar, tómelo tan pronto como se acuerde.
Si han transcurrido más de 6 horas desde la hora habitual a la que debería haber tomado Tafinlar, sáltese esta dosis y tome la siguiente a su hora habitual. Luego, continúe tomando las cápsulas a sus horas habituales.
No tome una dosis doble para compensar las dosis olvidadas.
Si interrumpe el tratamiento con Tafinlar
Tome Tafinlar durante el tiempo que le haya recomendado su médico. No deje de usarlo a menos que su médico, farmacéutico o enfermero se lo recomienden.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
Cómo debe tomar Tafinlar en combinación con trametinib
• Tome Tafinlar en combinación con trametinib exactamente como le ha dicho su médico, enfermera o farmacéutico. No cambie la dosis o deje de tomar Tafinlar o trametinib a menos que su médico, enfermera o farmacéutico se lo indique.
• Tome Tafinlar dos veces al día y tome trametinib una vez al día. Sería bueno para usted que tuviera el hábito de tomarse los dos medicamentos a la vez cada día. Las dosis de Tafinlar se deben tomar con una diferencia de 12 horas. Trametinib cuando se toma en combinación con Tafinlar se puede tomar tanto con la dosis matutina de Tafinlar o con la dosis nocturna de Tafinlar.
• Tome Tafinlar y trametinib con el estómago vacío, al menos una hora antes o dos horas depués de una comida. Tómeselos enteros con un vaso lleno de agua.
• Si se le olvida una dosis de Tafinlar o de trametinib, tómesela tan pronto se acuerde. No compense la dosis olvidada y tómese la siguiente dosis cuando le tocara:
o Si quedan menos de 6 horas hasta la siguiente dosis de Tafinlar, que se toma dos veces al día.
o Si quedan menos de 12 horas hasta la siguiente dosis de trametinib, que se toma una al día.
• Si toma más Tafinlar o trametinib, contacte inmediatamente con su médico, enfermero o farmacéutico. Llévese las cápsulas Tafinlar y los comprimidos de trametinib si puede. Si fuera posible, enseñe el envase de Tafinlar y de trametinib con el prospecto.
• Si presenta efectos adversos su médico puede decidir darle una dosis inferior de Tafinlar y de trametinib. Tóme las dosis de Tafinlar y trametinib exactamente como le ha dicho su médico, enfermera o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos graves
Problemas de sangrado
Tafinlar puede causar problemas de sangrado graves, especialmente en el cerebro cuando se toma en combinación con trametinib. Contacte con su médico o enfermero, y obtenga atención médica inmediata si presenta signos inusuales de sangrado, entre los que se incluyen:
• dolor de cabeza, mareo o debilidad
• tos con sangre o tos con coágulos de sangre
• si vomita sangre o si el vómito tiene aspecto de “granos de café”
• si presenta heces de color rojo, o de color negro con aspecto de alquitrán.
Fiebre (temperatura elevada)
Más de 1 de cada 10 personas que toman Tafinlar puede tener fiebre. Si tiene fiebre mientras esté tomando este medicamento (temperatura de 38,5°C o más alta), consulte con su médico, farmacéutico o enfermero inmediatamente. Le realizarán pruebas para saber si existen otras causas que puedan provocar la fiebre, y le administrarán un tratamiento.
En algunos casos, la gente que tiene fiebre puede tener bajadas de tensión y mareo. Si la fiebre es grave, su médico podría recomendarle que deje de tomar Tafinlar mientras le tratan la fiebre con otros medicamentos. Una vez que la fiebre esté controlada, su médico le recomendará que comience a tomar Tafinlar de nuevo.
Afecciones del corazón
Tafinlar puede afectar al funcionamiento de su corazón cuando se toma en combinación con trametinib. Es más probable que afecte a personas con un problema de corazón existente. Durante el tratamiento con Tafinlar en combinación con trametinib le realizarán pruebas de corazón. Entre los signos y síntomas relacionados con problemas de corazón se incluyen:
• palpitaciones, aumento del ritmo del corazón o latidos irregulares
• mareos
• cansancio
• sentirse aturdido
• falta de aire
• piernas hinchadas
Si experimenta cualquiera de estos síntomas, informe a su médico tan pronto como sea posible, tanto si los experimenta por primera vez como si considera que están empeorando.
Cambios en la piel
Si nota cualquier cambio en su piel mientras está tomando este medicamento, consulte con su médico, farmacéutico o enfermero tan pronto como le sea posible.
Hasta 1 de cada 10 personas que toman Tafinlar pueden desarrollar un tipo diferente de cáncer de piel llamado Carcinoma Cutáneo de Células Escamosas(CCE). Otros, pueden desarrollar un tipo de cáncer llamado Carcinoma Basocelular (CBC). Habitualmente, estos cambios sólo afectan a la piel de forma local y pueden ser eliminados con cirugía y continuar el tratamiento con Tafinlar sin necesidad de interrumpirlo.
Algunas personas que toman Tafinlar pueden notar que tienen nuevos melanomas. Estos melanomas son eliminados habitualmente mediante cirugía y continuar el tratamiento con Tafinlar sin necesidad de interrumpirlo.
Su médico examinará su piel antes de empezar el tratamiento con Tafinlar. Posteriormente su médico examinará su piel mensualmente durante todo el tratamiento y durante los 6 meses posteriores a la finalización del tratamiento. El motivo de estas revisiones es buscar lesiones cancerígenas nuevas.
Su médico también examinará su cabeza, cuello, boca, ganglios linfáticos y le realizarán regularmente un escáner (Tomografía Computerizada) de pecho y de la zona abdominal. También puede que le realicen análisis de sangre. Estas revisiones sirven para detectar si ha desarrollado otros cánceres, incluyendo carcinoma de células escamosas. Se recomienda que tanto al principio como al final del tratamiento se realice un examen pélvico (en mujeres) y anal.
Revise regularmente su piel mientras toma Tafinlar
Si nota cualquiera de los siguientes:
• verrugas nuevas
• llagas en la piel o bultos enrojecidos que sangran o no se curan
• cambios en el tamaño o color de los lunares
Consulte con su médico, farmacéutico o enfermero tan pronto como le sea posible
tanto si los síntomas aparecen por primera vez como si empeoran.
Puede aparecerle reacciones cutáneas (erupción) mientras tome Tafinlar en combinación con trametinib. Consulte con su médico si le aparece erupción cutánea mientras tome Tafinlar en combinación con trametinib.
Alteraciones en los ojos
Hasta 1 de cada 100 personas que toman Tafinlar solo o en combinación con trametinib pueden desarrollar una alteración en los ojos llamada uveítis, que si no se trata puede dañar su visión. La uveítis puede desarrollarse rápidamente, y los síntomas incluyen:
• enrojecimiento e irritación de los ojos
• visión borrosa
• dolor de ojos
• aumento en la sensibilidad a la luz
• manchas flotantes
Consulte con su médico, farmacéutico o enfermero inmediatamente si tiene cualquiera de estos síntomas.
Tafinlar puede causar problemas en los ojos cuando se tome en combinación con trametinib. No se recomienda tomar trametinib si alguna vez ha tenido una obstrucción en las venas que drenan los ojos (oclusión de las venas retinianas). Su médico le aconsejará que se realice un chequeo de los ojos antes de empezar el tratamiento con Tafinlar en combinación con trametinib y durante el tratamiento. Su médico puede pedirle que deje de tomar trametinib o derivarle a un especialista, si experimenta signos y síntomas en la visión, entre los que se incluyen:
• pérdida de visión
• enrojecimiento e irritación en los ojos
• si ve puntos de colores
• si ve un halo (visión borrosa alrededor de objetos)
• visión borrosa.
Consulte con su médico, farmacéutico o enfermero inmediatamente si aparecen estos síntomas.
Es muy importante que se ponga en contacto inmediatamente con su médico farmacéutico o enfermero si desarrolla estos síntomas, especialmente si tiene dolor en los ojos y enrojecimiento que no mejoran rápidamente. En este caso, deberá acudir a un especialista (oftalmólogo) para que le realice un examen completo de los ojos.
Otros efectos adversos
Además de los efectos adversos mencionados anteriormente, las personas que toman Tafinlar pueden tener otros efectos adversos. Estos efectos adversos incluyen:
Efectos adversos muy frecuentes que pueden afectar a más de 1 de cada 10 personas:
• engrosamiento de las capas externas de la piel
• efectos en la piel como erupción, crecimientos de la piel similares a verrugas, o enrojecimiento e inflamación de las palmas de las manos, dedos, y las plantas de los pies (ver “Cambios en la piel” en la sección 4)
• dolor de cabeza
• malestar (náusea), vómitos, diarrea
• disminución del apetito
• escalofríos
• sensación de debilidad
• falta de energía
• fiebre (ver “Fiebre (temperatura elevada) en la sección 4)
• dolor en las articulaciones, dolor muscular, o dolor en las manos o pies
• tos
• pérdida inusual del cabello o cabello fino.
Efectos adversos frecuentes que pueden afectar hasta 1 de cada 10 personas:
• estreñimiento
• síntomas parecidos a los de la gripe
• niveles bajos de fósforo en la sangre, observados en los análisis de sangre
• aumento del azúcar (glucosa) en sangre, observado en análisis de sangre
• cambios en la forma en la que el corazón bombea la sangre
• efectos en la piel incluyendo parches de piel rugosos y con escamas, engrosamiento de la piel de color marrón o amarillento, acrocordones, piel seca, bultos de aspecto brillante, llagas abiertas, picor o enrojecimiento de la piel (ver “cambios en la piel” en la sección 4).
Efectos adversos poco frecuentes que pueden afectar hasta 1 de cada 100 personas:
• inflamación de los ojos (uveítis, ver “Alteraciones en los ojos” en la sección 4)
• inflamación del páncreas (que causa dolor abdominal fuerte)
• inflamación de la capa grasa bajo la piel cuyos síntomas incluyen la aparición de nódulos en la
piel
• reacción alérgica
• nuevo melanoma
• alteraciones en el riñón, fallo renal, observado en los análisis de sangre
• alteraciones en el ritmo cardiaco
Efectos adversos cuando Tafinlar se toma junto con trametinib
Cuando Tafinlar se toma junto con trametinib puede sentir cualquiera de los efectos adversos incluido antes, aunque su frecuencia puede cambiar (aumentar o disminuir)
También podría aparecerle nuevos efectos adversos debido a tomar trametinib a la vez con Tafilar, que se incluyen a continuación.
Informe a su médico tan pronto como le sea posible si nota tanto que los síntomas aparecen por primera vez como si empeoran.
Lea el prospecto de trametinib para más detalles sobre los efectos adversos o que podrían aparecerle mientras toma este medicamento.
Los efectos adversos que puede verse mientras tome Tafinlar en combinación con trametinib son los siguientes:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
• mareo
• escalofríos
• temperatura elevada
• erupción, piel seca, picor, problemas similares al acné
• disminución del apetito
• dolor de cabeza
• tensión sanguínea elevada
• tos
• dolor de estómago
• sentirse mal (náusea), estar mal (vómitos)
• diarrea
• estreñimiento
• dolor en las articulaciones, dolor muscular o dolor en las manos o pies
• falta de energía, sentirse débil
• manos y pies hinchados
• inflamación de la nariz y de la garganta
• sangrados (hemorragia)
• infección del sistema urinario
Efectos adversos muy frecuentes que pueden aparecer en los análisis de sangre:
• niveles bajos de glóbulos blancos
• resultados anormales en sangre de las pruebas de hígado
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
• tensión sanguínea baja
• sudoración excesiva
• efectos en la piel incluyendo parches de piel rugosos y con escamas, erupciones en la piel con ampollas de pus, engrosamiento de la piel de color marrón o amarillento, marcas en la piel, grietas, crecimiento de verrugas o enrojecimiento en las palmas de las manos y pies, cáncer de piel de células escamosas (un tipo de cáncer de piel), inflamación de la capa grasa de la piel, papiloma (un tipo de tumor que normalmente no es dañino), infección de la piel (celulitis), inflamación de los folículos pilosos
• alteraciones en las uñas, como los cambios en el lecho de las uñas, dolor en las uñas, infección e inflamación de las cutículas
• pérdida inusual de pelo o de volumen
• visión borrosa, problemas de la vista
• respiración entrecortada
• úlceras o dolor en la boca, inflamación de la mucosa
• sequedad de boca
• síntomas gripales
• espasmos musculares
• hinchazón de la cara
• sudores nocturnos
• bombeo del corazón menos eficiente
• el ritmo del corazón es inferior de lo normal y/o existe una disminución en el ritmo cardíaco
Efectos adversos frecuentes que pueden aparecer en los análisis de sangre:
• disminución de las plaquetas en sangre (células que ayudan a la sangre a coagular)
• disminución de glóbulos rojos (Anemia) y un tipo de células blancas (leucopenia)
• niveles bajos de sal en sangre
• aumento de algunas sustancias (enzimas) producidas por el hígado
• aumento de la creatinina fosfoquinasa, una enzima que se encuentra principalmente en el corazón, cerebro y músculo esquelético
• aumento de los niveles de azúcar en sangre
• niveles bajos de fosfato en sangre
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
• reacciones alérgicas
• cambios en el ojo, como hinchazón debido al acumulación de líquido (coriorretinopatía), inflamación del ojo (uveítis), separación de la membrana sensible a la luz en la parte posterior del ojo (la retina) de sus capas de soporte (desprendimiento de retina) e hinchazón alrededor de los ojos
• hinchazón de la cara, localizado hinchazón de los tejidos
• inflamación del páncreas
• fallo renal, inflamación de los riñones
• inflamación del pulmón
• nuevo melanoma primario
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Tafinlar
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el frasco y en el envase, después de la abreviatura CAD. La fecha de caducidad es el último día del mes que se indica.
Este medicamento no requiere condiciones especiales de almacenamiento.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Tafinlar
- El principio activo es dabrafenib. Cada cápsula dura contiene dabrafenib mesilato, equivalente a 50 mg o 75 mg de dabrafenib.
- Los demás componentes son: celulosa microcristalina, estearato de magnesio, dióxido de silicio coloidal, óxido de hierro rojo (E-172), dióxido de titanio (E-171), e hipromelosa (E-464). Además, las cápsulas están impresas con tinta negra que contiene óxido de hierro negro (E-172), shellac y propilenglicol.
Aspecto del producto y contenido del envase
Las cápsulas de Tafinlar 50 mg cápsulas duras, son de color rojo oscuro opaco y van impresas con “GS TEW” y “50 mg”.
Las cápsulas de Tafinlar 75 mg cápsulas duras, son de color rosa oscuro opaco y van impresas con “GS LHF” y “75 mg”.
Los frascos son de plástico, color blanco opacoscon tapones de rosca de plástico.
Los frascos incluyen un desecante con silica gel en un pequeño contenedor cilíndrico. Este desecante se debe dejar dentro del frasco y no se debe ingerir.
Tafinlar 50 mg y 75 mg cápsulas duras están disponibles en envases con 28 o 120 cápsulas. Puede que no todos los envases estén comercializados en su país.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Glaxo Wellcome, S.A., Avda. Extremadura, 3, 09400 Aranda de Duero, Burgos, España Novartis Pharmaceuticals UK Limited, Frimley Business Park, Frimley, Camberley, Surrey GU16 7SR, Reino Unido
Novartis Pharma GmbH, RoonstraBe 25, D-90429 Nuremberg, Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien Lietuva
Novartis Pharma N.V. Novartis Pharma Services Inc.
Tél/Tel: +32 2 246 16 11 Tel: +370 5 269 16 50
Etarapun
Novartis Pharma Services Inc. Tea: +359 2 489 98 28
Ceská republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 555
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
|
ELXáóa Novartis (Hellas) A.E.B.E. Tnk: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kúnpoq Novartis Pharma Services Inc. Tnk: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
En la página web de la Agencia Europea de Medicamentos puede encontrarse este prospecto en todas las lenguas de la Unión Europea/Espacio Económico Europeo.
61