Spiriva 18 Microgramos, Polvo Para Inhalacion
.Uí1.
"I
an
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
SPIRIVA 18 microgramos, polvo para inhalación
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula contiene 22,5 microgramos de bromuro de tiotropio monohidrato, equivalentes a 18 microgramos de tiotropio.
La dosis liberada (la dosis que se libera de la boquilla del dispositivo HandiHaler) es de 10 microgramos de tiotropio.
Excipiente: Lactosa monohidrato.
Para la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para inhalación, contenido en cápsulas duras.
Cápsulas duras de color verde claro con el código de producto TI 01 y el logotipo de la empresa impresos en la cápsula.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
El tiotropio está indicado como tratamiento broncodilatador de mantenimiento para aliviar los síntomas en pacientes con enfermedad pulmonar obstructiva crónica (EPOC).
4.2 Posología y forma de administración
Posología
Este medicamento está destinado únicamente para uso por inhalación.
La dosificación recomendada de bromuro de tiotropio es la inhalación del contenido de una cápsula mediante el dispositivo HandiHaler, una vez al día y a la misma hora.
No debe superarse la dosis recomendada.
Las cápsulas de bromuro de tiotropio son para inhalación exclusivamente y no para la ingesta oral.
Las cápsulas de bromuro de tiotropio no deben ingerirse.
El bromuro de tiotropio sólo debe inhalarse con el dispositivo HandiHaler.
Poblaciones especiales
Los pacientes geriátricos pueden utilizar el bromuro de tiotropio a la dosis recomendada.
Los pacientes con insuficiencia renal pueden utilizar el bromuro de tiotropio a la dosis recomendada. Para pacientes con insuficiencia renal de moderada a grave (aclaramiento de creatinina < 50 ml/min) ver sección 4.4 y sección 5.2.
Los pacientes con insuficiencia hepática pueden utilizar el bromuro de tiotropio a la dosis recomendada (ver sección 5.2).
Población pediátrica
EPOC
No existe un uso relevante en población pediátrica (menores de 18 años) para la indicación contenida en la sección 4.1.
Fibrosis quística
¿lili»;
ÍTTI
No se ha establecido la seguridad y la eficacia de Spiriva 18 microgramos en niños y adolescentes. No se dispone de datos.
Forma de administración
Para asegurar una correcta administración del medicamento el paciente debe ser instruído por un médico u otro profesional sanitario en cómo usar el inhalador.
Instrucciones de uso y manipulación

Recuerde seguir cuidadosamente las instrucciones de su médico para utilizar SPIRIVA.
El HandiHaler está especialmente diseñado para SPIRIVA. No debe utilizarlo para ningún otro medicamento. Puede utilizar su HandiHaler durante un período de hasta un año para su medicación.
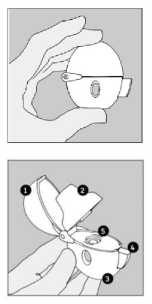
El HandiHaler
1 Capuchón protector
2 Boquilla
3 Base
4 Botón perforador
5 Cámara central
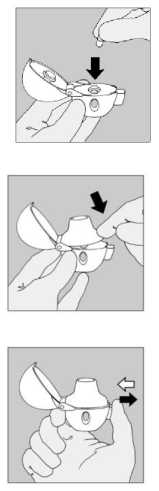
1) Para abrir el capuchón protector apretar el botón perforador hasta el fondo y soltar.
2) Abrir el capuchón protector completamente levantándolo hacia arriba y hacia detrás. Después abrir la boquilla levantándola hacia arriba y hacia detrás.
3) Extraer una cápsula de SPIRIVA del blister (sólo inmediatamente antes de usar, ver manejo del blister) y colocarla en la cámara central (5), tal como se indica en la figura. No importa la posición en que esté la cápsula dentro de la cámara.
4) Cerrar la boquilla firmemente hasta oír un clic, dejando abierto el capuchón protector.
5) Coger el dispositivo HandiHaler con la boquilla hacia arriba, presionar a fondo el botón perforador una sola vez y soltarlo. Esta maniobra perfora la cápsula y permite que se libere el medicamento cuando se aspira.
•ítp
Í-*
am
hH
j --
6)
Espirar a fondo. Importante: nunca se debe espirar dentro de la boquilla.
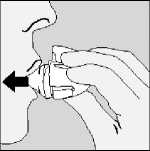
7) Llevar el HandiHaler a la boca y cerrar los labios fuertemente alrededor de la boquilla. Mantener la cabeza en posición derecha y aspirar lenta y profundamente pero de forma suficiente como para oír o notar vibrar la cápsula. Aspirar hasta que los pulmones estén llenos; después mantener la respiración durante unos momentos y, al mismo tiempo, retirar el HandiHaler de la boca. Continuar respirando normalmente. Repetir los pasos 6 y 7 una vez más; esto vaciará la cápsula completamente.
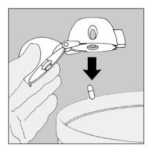
8) Abrir la boquilla otra vez. Sacar la cápsula utilizada y tirarla. Cerrar la boquilla y el capuchón protector para guardar el dispositivo HandiHaler.
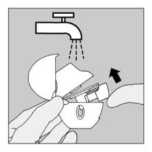
Limpieza del HandiHaler
Limpiar el HandiHaler una vez al mes. Abrir el capuchón protector y la boquilla. Después abrir la base levantando el botón perforador. Enjuagar todo el inhalador con agua caliente para eliminar todo el polvo. Secar bien el HandiHaler, eliminando el exceso de agua con una toallita de papel y dejando secar posteriormente al aire, dejando abiertos el capuchón protector, la boquilla y la base. Debido a que tarda 24 horas en secarse al aire, se debe limpiar justo después de utilizarlo y así estará preparado para la próxima utilización. En caso necesario, el exterior de la boquilla se puede limpiar con un pañuelo de papel húmedo pero no mojado.
Manejo del blister
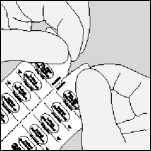
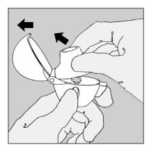
A. Separar las tiras del blister rasgando por la línea de puntos.
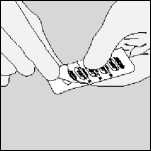
B. Desprender la lámina de aluminio (sólo inmediatamente antes de usar) levantando la lengüeta, hasta que sea completamente visible una cápsula.
Si accidentalmente otra cápsula queda expuesta al aire no debe utilizarse.
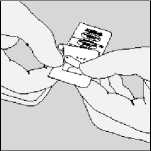
C. Extraer la cápsula.
La cantidad de polvo contenido en las cápsulas de SPIRIVA es pequeña por lo que la cápsula está parcialmente llena.
4.3 Contraindicaciones
El polvo para inhalación de bromuro de tiotropio está contraindicado en pacientes con hipersensibilidad al bromuro de tiotropio, a la atropina o sus derivados, p.ej. ipratropio u oxitropio, o al excipiente lactosa monohidrato que contiene proteínas de la leche.
4.4 Advertencias y precauciones especiales de empleo
El bromuro de tiotropio, como broncodilatador de mantenimiento de administración una vez al día, no debería ser utilizado para el tratamiento inicial de los episodios agudos de broncoespasmo, es decir, como tratamiento de rescate.
Después de la administración de bromuro de tiotropio, polvo para inhalación, pueden aparecer reacciones de hipersensibilidad inmediata.
Dada su actividad anticolinérgica, el bromuro de tiotropio debe utilizarse con precaución en pacientes con glaucoma de ángulo estrecho, hiperplasia prostática u obstrucción del cuello de la vejiga. (Ver sección 4.8)
Los medicamentos inhalados pueden provocar broncoespasmo inducido por la inhalación.
El tiotropio se debe utilizar con precaución en pacientes con: infarto de miocardio reciente, hace menos de 6 meses; cualquier arritmia inestable o que ponga en riesgo la vida; arritmia cardíaca que requiera intervención o un cambio en el tratamiento farmacológico; hospitalización debido a fallo cardíaco (New York Heart Association (NYHA) Clase III o IV) en el año previo. Estos pacientes se excluyeron de los ensayos clínicos y pueden verse afectados por el mecanismo de acción anticolinérgico.
En pacientes con insuficiencia renal de moderada a grave (aclaramiento de creatinina < 50 ml/min), el bromuro de tiotropio sólo debe utilizarse si el beneficio esperado supera el riesgo potencial, ya que la concentración plasmática aumenta cuando la función renal está disminuida. No existe experiencia a largo plazo en pacientes con insuficiencia renal grave (ver sección 5.2.).
Debe advertirse a los pacientes que eviten la introducción del polvo del medicamento en los ojos. Se les debe indicar que ello puede provocar o empeorar un glaucoma de ángulo estrecho, dolor o molestia ocular, visión borrosa transitoria, halos visuales o imágenes coloreadas, junto con enrojecimiento ocular por congestión de la conjuntiva y edema de la córnea. Si aparece cualquier combinación de estos síntomas oculares, los pacientes deben interrumpir el uso de bromuro de tiotropio y consultar inmediatamente un especialista.
La sequedad de boca, observada con el tratamiento anticolinérgico, a largo plazo puede asociarse con caries dental.
El bromuro de tiotropio no debe utilizarse con una frecuencia superior a una vez al día (ver sección 4.9).
Las cápsulas de SPIRIVA contienen 5,5 mg de lactosa monohidrato.
4 de 14 MINISTWODE
SANIDAD, POLITICA SOCIAL E IGUALDAD Agencia es pañosa de medicamentos y productos san-lanos
ÍTTI
4.5 Interacción con otros medicamentos y otras formas de interacción
Aunque no se han llevado a cabo estudios formales de interacción con otros fármacos, el polvo para inhalación de bromuro de tiotropio ha sido utilizado conjuntamente con otros medicamentos sin evidencia clínica de interacciones. Estos fármacos incluyen los broncodilatadores simpaticomiméticos, metilxantinas y corticoides orales e inhalados, utilizados habitualmente para el tratamiento de la EPOC.
El uso de agonistas P de acción prolongada o corticosteroides inhalados (LABA o ICS) no se ha visto que alteren la exposición a tiotropio.
La administración simultánea de bromuro de tiotropio con otros medicamentos que contienen anticolinérgicos no ha sido estudiada y, por tanto, no se recomienda.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Se dispone de una cantidad muy limitada de datos del uso de tiotropio en mujeres embarazadas. Los estudios en animales no indican efectos nocivos directos o indirectos respecto a toxicidad reproductiva a dosis clínicamente relevantes (ver sección 5.3). Como medida de precaución, es preferible evitar el uso de Spiriva durante el embarazo.
Lactancia
Se desconoce si el bromuro de tiotropio se excreta en la leche materna. Aunque los estudios en roedores muestran que la excreción del bromuro de tiotropio en la leche materna es en pequeña cantidad, no se recomienda usar Spiriva durante la lactancia. El bromuro de tiotropio es un compuesto de acción prolongada. La decisión en cuanto a continuar/suspender la lactancia o continuar/suspender el tratamiento con Spiriva debe tomarse considerando el beneficio de la lactancia para el niño y el beneficio del tratamiento con Spiriva para la mujer.
Fertilidad
No se dispone de datos clínicos de fertilidad para tiotropio. Un ensayo preclínico realizado con tiotropio no mostró ninguna indicación de efecto adverso sobre la fertilidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios sobre la capacidad para conducir y utilizar máquinas. La aparición de mareos, visión borrosa o cefalea pueden influir en la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas Resumen del perfil de seguridad
Muchas de las reacciones adversas listadas pueden atribuirse a las propiedades anticolinérgicas de SPIRIVA.
Resumen tabulado de reacciones adversas
Las frecuencias asignadas a las reacciones adversas que se listan a continuación se basan en las tasas de incidencia bruta de las reacciones adversas (es decir, acontecimientos atribuidos a tiotropio) observadas en el grupo tratado con tiotropio (9.647 pacientes) obtenidas de un conjunto de 28 ensayos clínicos controlados con placebo, con periodos de tratamiento de 4 semanas a cuatro años.
La frecuencia se define utilizando el siguiente convenio:
ílMt
ÍTTI
Muy frecuentes (> 1/10); frecuentes (> 1/100, < 1/10); poco frecuentes (> 1/1.000, < 1/100); raras (>1/10.000, < 1/1.000); muy raras (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Clasificación por Órganos y Sistemas /Término preferente MedDRA |
Frecuencia |
|
Trastornos del metabolismo y de la nutrición Deshidratación |
No conocida |
|
Trastornos del sistema nervioso Mareo Cefalea Alteraciones del gusto Insomnio |
Poco frecuente Poco frecuente Poco frecuente Rara |
|
Trastornos oculares Visión borrosa Glaucoma Aumento de la presión intraocular |
Poco frecuente Rara Rara |
|
Trastornos cardiacos Fibrilación auricular Taquicardia supraventricular Taquicardia Palpitaciones |
Poco frecuente Rara Rara Rara |
Trastornos respiratorios, torácicos y mediastínicos
|
Faringitis |
Poco frecuente |
|
Disfonía |
Poco frecuente |
|
Tos |
Poco frecuente |
|
Broncoespasmo |
Rara |
|
Epistaxis |
Rara |
|
Laringitis |
Rara |
|
Sinusitis |
Rara |
|
Trastornos gastrointestinales | |
|
Sequedad de boca |
Frecuente |
|
Reflujo gastroesofágico |
Poco frecuente |
|
Estreñimiento |
Poco frecuente |
|
Candidiasis orofaríngea |
Poco frecuente |
|
Obstrucción intestinal, incluyendo íleo paralítico |
Rara |
|
Gingivitis |
Rara |
|
Glositis |
Rara |
|
Disfagia |
Rara |
|
Estomatitis |
Rara |
|
Náuseas |
Rara |
|
Caries dental |
No conocida |
Trastornos de la piel y del tejido subcutáneo, trastornos del sistema inmunológico
Exantema Poco frecuente
"I
an
Rara
Rara
Rara
Rara
No conocida No conocida No conocida
No conocida
Poco frecuente Poco frecuente Rara
Urticaria
Prurito
Hipersensibilidad (incluyendo reacciones inmediatas)
Angioedema
Reacción anafiláctica
Infección cutánea, úlcera cutánea
Sequedad de piel
Trastornos musculoesqueléticos y del tejido conjuntivo
Tumefacción de las articulaciones
Trastornos renales y urinarios
Disuria
Retención de orina Infección del tracto urinario
Descripción de reacciones adversas seleccionadas
En ensayos clínicos controlados, las reacciones adversas observadas generalmente fueron efectos adversos anticolinérgicos como la sequedad de boca, que ocurrió en aproximadamente un 4% de los pacientes.
En 28 ensayos clínicos, la sequedad de boca provocó el abandono en 18 de los 9.647 pacientes tratados con tiotropio (0,2%).
Reacciones adversas graves relacionadas con los efectos anticolinérgicos incluyen glaucoma, estreñimiento y obstrucción intestinal, incluyendo íleo paralítico así como retención de orina.
Otra población especial
Con la edad pueden aumentar los efectos anticolinérgicos.
Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: https://www.notificaram.es
4.9 Sobredosis
Las dosis elevadas de bromuro de tiotropio pueden provocar la aparición de signos y síntomas anticolinérgicos.
No obstante, después de la administración de una dosis única inhalada de hasta 340 microgramos de bromuro de tiotropio en voluntarios sanos, no se observaron efectos adversos anticolinérgicos sistémicos. Adicionalmente, después de la administración de dosis de hasta 170 microgramos de bromuro de tiotropio durante 7 días a voluntarios sanos, no se observaron efectos adversos relevantes aparte de la sequedad de boca. En un estudio de dosis múltiple llevado a cabo en pacientes con EPOC, con una dosis diaria máxima de 43 microgramos de bromuro de tiotropio durante un período de cuatro semanas, no se observaron reacciones adversas significativas.
La intoxicación aguda por ingestión oral accidental de cápsulas de bromuro de tiotropio es poco probable, debido a su baja biodisponibilidad oral.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Otros agentes contra padecimientos obstructivos de las vías respiratorias,
inhalatorios, anticolinérgicos
^ de 14 MINISTKIODE
SANIDAD, POLITICA SOCIAL E IGUALDAD Agencia españoiaóe medie amentos y proouctos san-tanos
R03B B04
Código ATC: Mecanismo de acción
El bromuro de tiotropio es un antagonista específico de los receptores muscarínicos de acción prolongada, denominado con frecuencia anticolinérgico en la práctica clínica. El bromuro de tiotropio inhibe los efectos colinérgicos (broncoconstrictores) de la acetilcolina, liberada de las terminaciones nerviosas parasimpáticas, uniéndose a los receptores muscarínicos de la musculatura lisa bronquial. Tiene una afinidad similar por los diferentes subtipos de receptores muscarínicos, M1 a M5. En las vías aéreas, el bromuro de tiotropio antagoniza, de forma competitiva y reversible, los receptores M3 provocando relajación de la musculatura lisa bronquial. El efecto fue dependiente de la dosis y duró más de 24 horas.
Es probable que la duración prolongada del efecto sea debida a su disociación muy lenta de los receptores M3, presentando una vida media de disociación significativamente más prolongada que la del ipratropio. Como anticolinérgico N-cuaternario, el bromuro de tiotropio es tópicamente (bronco-) selectivo cuando se administra por inhalación, demostrando un rango terapéutico aceptable antes de que aparezcan efectos anticolinérgicos sistémicos.
Efectos farmacodinámicos
La broncodilatación es principalmente un efecto local (sobre las vías aéreas), no un efecto sistémico.
La disociación de los receptores M2 es más rápida que la de los receptores M3, hecho que en los estudios funcionales in vitro fue considerado como una selectividad (controlado cinéticamente) del subtipo de receptor, M3 sobre M2. La elevada potencia y la lenta disociación del receptor mostraron su correlación clínica en forma de una broncodilatación significativa y de larga duración en los pacientes con EPOC.
Electrofisiología cardíaca
Electrofisiología: En un estudio específico sobre el intervalo QT con 53 voluntarios sanos, la administración de 18 mcg y 54 mcg de SPIRIVA (es decir, tres veces la dosis terapéutica) durante 12 días no prolongó de manera significativa los intervalos QT del ECG.
Eficacia clínica y seguridad
El programa de desarrollo clínico incluyó cuatro estudios de un año y dos de seis meses de duración, aleatorizados y a doble ciego, en 2663 pacientes (1308 recibieron bromuro de tiotropio). El programa de un año consistió en dos estudios controlados con placebo y dos con un control activo (ipratropio). Los dos estudios de seis meses fueron ambos controlados con salmeterol y placebo. Estos estudios incluyeron determinaciones de la función pulmonar y mediciones de los resultados de salud por lo que respecta a la disnea, exacerbaciones de la enfermedad y calidad de vida relacionada con la salud.
Función pulmonar
El bromuro de tiotropio, administrado una vez al día, produjo una mejoría significativa de la función pulmonar (volumen espiratorio forzado en un segundo, FEV1, y capacidad vital forzada, FVC) en los 30 minutos posteriores a la administración de la primera dosis, que se mantuvo durante un período de 24 horas. El estado de equilibrio farmacodinámico se alcanzó en el plazo de una semana, observándose la mayor broncodilatación al tercer día. El bromuro de tiotropio mejoró de forma significativa el PEFR matinal y nocturno (tasa del flujo espiratorio máximo) medido en los registros diarios de los pacientes. Los efectos broncodilatadores del bromuro de tiotropio se mantuvieron durante todo el período de estudio, de un año de duración, sin que se observaran signos de tolerancia.
Un ensayo clínico aleatorizado y controlado con placebo, en 105 pacientes con EPOC, demostró que la broncodilatación se mantenía durante todo el intervalo de dosificación de 24 horas en comparación con el placebo, independientemente de si se administraba el medicamento por la mañana o por la noche.
Estudios a largo plazo (6 meses y un año)
Disnea, tolerancia al ejercicio
ÍTTI
El bromuro de tiotropio mejoró de forma significativa la disnea (tal como se evaluó utilizando el Índice de Transición de Disnea de Mahler). Esta mejoría se mantuvo durante todo el período de tratamiento.
El impacto de la mejora de la disnea sobre la tolerancia al ejercicio, se evaluó en dos estudios aleatorizados, a doble ciego, controlados con placebo en los que participaron 433 pacientes con EPOC de moderada a grave. En estos estudios, seis semanas de tratamiento con SPIRIVA, mejoró significativamente los síntomas que limitan el tiempo de tolerancia al ejercicio en un 19,7% (Estudio A) y en un 28,3% (Estudio B) comparado con placebo.
Calidad de vida relacionada con la salud
En un ensayo clínico aleatorizado, doble ciego, controlado con placebo, de 9 meses de duración y con 492 pacientes, SPIRIVA mejoró la calidad de vida relacionada con la salud, según la puntuación total del St. George’s Respiratory Questionary (SGRQ). La proporción de pacientes tratados con SPIRIVA que alcanzaron una mejora significativa en la puntuación total de SGQR (es decir, > 4 unidades) fue un 10,9% mayor que en el grupo tratado con placebo (59,1% en el grupo tratado con SPIRIVA frente al 48,2% en el grupo tratado con placebo (p = 0,029)). La diferencia promedio entre los grupos fue de 4,19 unidades (p = 0,001; intervalo de confianza: 1,69-6,68). Las mejoras obtenidas en los distintos dominios del SGRQ fueron de 8,19 unidades en el de “síntomas”, 3,91 unidades en el de “actividad” y 3,61 unidades en el de “impacto en la vida diaria”. La mejora de todos estos dominios fue estadísticamente significativa.
Exacerbaciones de la EPOC
En un ensayo clínico aleatorizado, doble ciego, controlado con placebo y con 1.829 pacientes con EPOC de moderada a muy grave, el bromuro de tiotropio redujo de forma estadísticamente significativa la proporción de pacientes que experimentaron exacerbaciones de la EPOC (de 32,2% a 27,8%) y redujo de forma estadísticamente significativa el número de exacerbaciones en un 19% (1,05 a 0,85 eventos por paciente y año de exposición). Además, el 7,0% de los pacientes en el grupo de bromuro de tiotropio y el 9,5% de los pacientes en el grupo de placebo fueron hospitalizados debido a una exacerbación de la EPOC (p=0,056). El número de hospitalizaciones debidas a la EPOC se redujo en un 30% (de 0,25 a 0,18 eventos por paciente y año de exposición).
En un ensayo aleatorizado, doble ciego, doble simulación, de grupos paralelos, de un año de duración, se comparó el efecto del tratamiento con 18 microgramos de SPIRIVA una vez al día y con 50 microgramos de salmeterol HFA pMDI dos veces al día en la incidencia de exacerbaciones moderadas y graves en 7.376 pacientes con EPOC y una historia de exacerbaciones en el año previo.
Tabla 1: Resumen de las variables de valoración de exacerbaciones
9 de 14 MINISTWIODE
SANIDAD, POLITICA SOCIAL E IGUALDAD Agencia españoiade medie amentos y proouctos sabíanos
.Uí1.
"I ■"
¡m
|
Variable de valoración |
SPIRIVA 18 microgramos (HandiHaler) N = 3.707 |
Salmeterol 50 microgramos (HFA pMDI) N = 3.669 |
Cociente (95% IC) |
Valor de probabilidad |
|
Tiempo [días] hasta la primera exacerbación |
187 |
145 |
0,83 (0,77 - 0,90) |
<0,001 |
|
Tiempo hasta la primera exacerbación grave (hospitalización)§ |
- |
- |
0,72 (0,61 - 0,85) |
<0,001 |
|
Pacientes con >1 exacerbación, n (%)* |
1.277 (34,4) |
1.414 (38,5) |
0,90 (0,85 - 0,95) |
<0,001 |
|
Pacientes con >1 exacerbación grave (hospitalización), n (%)* |
262 (7,1) |
336 (9,2) |
0,77 (0,66 - 0,89) |
<0,001 |
f Tiempo [días] se refiere al primer cuartil de pacientes. El análisis del tiempo hasta el evento se realizó utilizando el modelo de regresión de riesgos proporcionales de Cox con el centro y tratamiento como covariables (combinados); el cociente se refiere al cociente de riesgos.
§ El análisis del tiempo hasta el evento se realizó utilizando el modelo de regresión de riesgos proporcionales de Cox con el centro y tratamiento como covariables (combinados); el cociente se refiere al cociente de riesgos. El tiempo [días] para el primer cuartil de pacientes no puede ser calculado dado que la proporción de pacientes con exacerbaciones graves es demasiado baja.
* El número de pacientes con un acontecimiento se analizaron utilizando el test de Cochran-Mantel-Haenszel estratificado por centros combinados; el cociente se refiere al cociente de riesgos.
Comparado con salmeterol, SPIRIVA aumentó el tiempo hasta la primera exacerbación (187 días frente a 145 días), con una reducción del riesgo del 17 % (cociente de riesgos, 0,83; intervalo de confianza 95%
[IC], 0,77 a 0,90; P<0,001). SPIRIVA también aumentó el tiempo hasta la primera exacerbación grave (hospitalización) (cociente de riesgos, 0,72; IC 95% , 0,61 a 0,85; P<0,001).
Estudios a largo plazo (más de 1 año y hasta 4 años)
En un ensayo clínico aleatorizado, doble ciego, controlado con placebo, de 4 años de duración y con 5.993 pacientes aleatorizados (3.006 con placebo y 2.987 con Spiriva) la mejora del FEV1 con Spiriva, en comparación con placebo, permaneció constante durante los 4 años. Una proporción mayor de pacientes completó 45 meses o más de tratamiento en el grupo de Spiriva comparado con el grupo de placebo (63,8% frente a 55,4%, p<0,001). La tasa anual de disminución del FEV1 en comparación con placebo fue similar entre Spiriva y placebo. Durante el tratamiento se produjo una reducción del 16% del riesgo de muerte. La incidencia de muerte fue del 4,79 por 100 pacientes-año en el grupo placebo frente al 4,10 por 100 pacientes-año en el grupo de tiotropio (cociente de riesgos (tiotropio/placebo) = 0,84, IC 95% = 0,73, 0,97). El tratamiento con tiotropio redujo el riesgo de insuficiencia respiratoria (recogido a través de la notificación de acontecimientos adversos) en un 19% (2,09 frente a 1,68 casos por 100 pacientes-año, riesgo relativo (tiotropio/placebo) = 0,81, IC 95% = 0,65, 0,999).
Estudio de tiotropio con control activo
Se ha llevado a cabo un estudio aleatorizado, doble ciego, con control activo, a largo plazo y a gran escala, con un periodo de seguimiento de hasta 3 años para comparar la eficacia y seguridad de Spiriva HandiHaler y Spiriva Respimat (5.694 pacientes recibieron Spiriva HandiHaler; 5.711 pacientes recibieron Spiriva Respimat). Las variables principales fueron el tiempo hasta la primera exacerbación de la EPOC, el tiempo hasta mortalidad por cualquier causa y en un subestudio (906 pacientes) el FEV1 valle (pre-dosis).
•m
El tiempo hasta la primera exacerbación de la EPOC fue numéricamente similar durante el estudio de Spiriva HandiHaler y Spiriva Respimat (cociente de riesgos (Spiriva HandiHaler /Spiriva Respimat) 1,02 con un IC 95% de 0,97 a 1,08). El número medio de días hasta la primera exacerbación de EPOC fue de 719 días para Spiriva HandiHaler y de 756 días para Spiriva Respimat.
El efecto broncodilatador de Spiriva HandiHaler se mantuvo durante las 120 semanas, y fue similar a Spiriva Respimat. La diferencia promedio en el FEV1 valle de Spiriva HandiHaler versus Spiriva Respimat fue -0.010L (IC 95%: -0,018-0,038 mL).
En el estudio post-comercialización TIOSPIR comparando Spiriva Respimat y Spiriva HandiHaler, la mortalidad por cualquier causa incluyendo el seguimiento del estado vital fue similar durante el estudio con Spiriva HandiHaler y Spiriva Respimat (coeficiente de riesgos (Spiriva HandiHaler /Spiriva Respimat) 1,04 con IC 95% de 0,91 a 1,19).
Población pediátrica
La Agencia Europea de Medicamentos ha eximido de la obligación de presentar los resultados de los estudios con Spiriva en todos los subgrupos de población pediátrica en EPOC y fibrosis quística (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
a) Introducción general
El bromuro de tiotropio es un compuesto de amonio cuaternario no quiral y escasamente soluble en agua. El bromuro de tiotropio se administra por inhalación del polvo seco. Generalmente cuando se administra por vía inhalatoria, la mayor parte de la dosis liberada se deposita en el tracto gastrointestinal y, en menor grado, se deposita en el órgano diana, es decir, en el pulmón. Muchos de los datos farmacocinéticos que se describen a continuación, se obtuvieron con dosis superiores a las recomendadas para el tratamiento.
b) Características generales del principio activo después de la administración del medicamento
Absorción: Después de la inhalación del polvo seco en voluntarios jóvenes sanos, la biodisponibilidad absoluta del 19,5% sugiere que la fracción que alcanza el pulmón es altamente biodisponible. Las soluciones orales de tiotropio tienen una biodisponibiliad absoluta del 2-3%.
Se observaron concentraciones plasmáticas máximas de tiotropio 5-7 minutos después de la inhalación.
En el estado de equilibrio, los niveles plasmáticos máximos de tiotropio en pacientes con EPOC fueron de
12,9 pg/ml y disminuyeron rápidamente según un modelo multicompartimental. Las concentraciones plasmáticas valle en el estado de equilibrio fueron de 1,71 pg/ml.
La exposición sistémica tras la inhalación de tiotropio a través del dispositivo HandiHaler fue similar a la de tiotropio inhalado a través del dispositivo Respimat.
Distribución: Tiotropio tiene una unión a proteínas plasmáticas del 72 % y un volumen de distribución de 32 l/kg. Se desconocen las concentraciones locales en el pulmón pero la forma de administración sugiere concentraciones sustancialmente superiores en este órgano. Los estudios en ratas han mostrado que el bromuro de tiotropio no atraviesa la barrera hematoencefálica en un grado significativo.
Biotransformación: El grado de biotransformación es bajo. Ello se hace evidente por una excreción urinaria del 74% de fármaco inalterado, después de una dosis intravenosa en voluntarios jóvenes sanos. El éster del bromuro de tiotropio se fragmenta de manera no enzimática al alcohol (N-metilescopina) y al ácido (ácido ditienilglicólico), que son inactivos sobre los receptores muscarínicos. Los ensayos in vitro en microsomas hepáticos humanos y hepatocitos humanos sugieren que alguna proporción adicional del fármaco (< 20% de la dosis después de administración intravenosa) se metaboliza mediante oxidación
*2
Jll^a
sm
dependiente del citocromo P450 (CYP) y posterior conjugación con glutation, a varios metabolitos de Fase II.
Los estudios in vitro en microsomas hepáticos revelan que la vía enzimática puede ser inhibida por los inhibidores del CYP 2D6 (y 3A4), quinidina, ketoconazol y gestodeno. Así pues, el CYP 2D6 y 3A4 están implicados en la vía metabólica que es responsable de la eliminación de una fracción inferior de la dosis. Incluso a concentraciones supraterapéuticas, el bromuro de tiotropio no inhibe el citocromo CYP 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 o 3A en microsomas hepáticos humanos.
Eliminación: La vida efectiva de tiotropio en pacientes con EPOC oscila entre 27 y 45 h. El aclaramiento total fue de 880 ml/min después de la administración de una dosis intravenosa a voluntarios jóvenes sanos. El tiotropio administrado intravenosamente se excreta inalterado principalmente en la orina (74%). Después de la inhalación del polvo seco por pacientes con EPOC hasta el estado estacionario, la excreción urinaria es del 7% (1,3 pg) del fármaco inalterado durante 24 horas, permaneciendo el resto del fármaco sin absorber en el intestino, eliminándose por las heces. El aclaramiento renal de tiotropio es superior al aclaramiento de creatinina, indicando la existencia de una secreción a la orina. Después de una inhalación una vez al día por pacientes con EPOC en tratamiento crónico, se alcanzó el estado de equilibrio farmacocinético al cabo del día 7, sin que se produjera una acumulación posterior.
Linealidad/No linealidad: El tiotropio muestra una farmacocinética lineal en el intervalo terapéutico independientemente de la formulación.
c) Características en pacientes
Pacientes geriátricos: Tal como era de esperar para todos los fármacos excretados predominantemente por vía renal, una edad más avanzada se asoció a una reducción del aclaramiento renal de tiotropio (de 365 ml/min en pacientes con EPOC < 65 años hasta 271 ml/min en pacientes con EPOC > 65 años). Esto no dio lugar a un correspondiente incremento en la AUCo-6,ss o Cmax,ss
Pacientes con insuficiencia renal: Después de la administración por inhalación una vez al día de tiotropio hasta el estado de equilibrio en pacientes con EPOC, la insuficiencia renal leve (CLcr 50-80 ml/min) resultó en un ligero aumento del AUC0-6,ss (entre 1,8-30% más alto) y valores similares de Cmax,ss comparado con pacientes con función renal normal (CLcr > 80 ml/min).
En los pacientes con EPOC y con una insuficiencia renal de moderada a grave (CLcr < 50 ml/min), la administración intravenosa de tiotropio resultó en el doble de la exposición total (82% más alto de la AUC0-4h y 52% más alto de Cmax) comparado con pacientes con EPOC con función renal normal, lo que fue confirmado por las concentraciones plasmáticas después de la inhalación del polvo seco.
Pacientes con insuficiencia hepática: No se espera que la insuficiencia hepática tenga ninguna influencia importante sobre la farmacocinética de tiotropio. El tiotropio se aclara predominantemente por eliminación renal (un 74% en los voluntarios jóvenes sanos) y por una fragmentación simple, no enzimática del éster a productos farmacológicamente inactivos.
Pacientes japoneses con EPOC: En ensayos comparativos cruzados, las concentraciones plasmáticas pico medias 10 minutos tras la dosificación en el estadio estacionario fueron del 20% al 70% superiores en los pacientes con EPOC japoneses que en los caucásicos tras la inhalación de tiotropio, pero no hubo signos de mayor mortalidad o riesgo cardíaco en los pacientes japoneses en comparación con los pacientes caucásicos. Hay datos farmacocinéticos insuficientes de otras etnias o razas.
Pacientes pediátricos: Ver sección 4.2.
d) Relación (es) farmacocinética/farmacodinamia
No existe relación directa entre la farmacocinética y la farmacodinamia.
ÍTT1
5.3 Datos preclínicos sobre seguridad
Muchos de los efectos observados en los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas y toxicidad para la reproducción podrían explicarse por las propiedades anticolinérgicas del bromuro de tiotropio. En animales normalmente se observó una reducción del consumo de alimentos, inhibición del aumento de peso corporal, sequedad de boca y nariz, reducción del lagrimeo y la salivación, midriasis y aumento de la frecuencia cardiaca. Otros efectos relevantes observados en los estudios de toxicidad de dosis repetidas fueron: irritación leve del tracto respiratorio en ratas y ratones, puesta de manifiesto por aparición de rinitis y cambios en el epitelio de la cavidad nasal y la laringe, y prostatitis junto con depósitos de proteínas y litiasis de la vejiga en ratas.
Los efectos nocivos sobre la gravidez, desarrollo embrional/fetal, parto o desarrollo posnatal únicamente se pudieron demostrar en los niveles de dosis tóxicas para las madres. El bromuro de tiotropio no fue teratogénico en ratas ni conejos. En un estudio general de reproducción y fertilidad en ratas, no hubo ninguna indicación de ningún efecto adverso sobre la fertilidad o el apareamiento de los progenitores tratados o de sus crías a ninguna dosis.
Los cambios respiratorios (irritación) y urogenitales (prostatitis) y la toxicidad sobre la reproducción, fueron observados con la exposición local o sistémica a dosis cinco veces superiores a la dosis terapéutica. Los estudios sobre genotoxicidad y potencial carcinogénico no mostraron un peligro especial para el ser humano.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Lactosa monohidrato (conteniendo proteínas de la leche)
6.2 Incompatibilidades No procede.
6.3 Periodo de validez
2 años
Después de la primera apertura del blister: 9 días.
Desechar el dispositivo HandiHaler 12 meses después de la primera utilización
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 25°C.
No congelar.
6.5 Naturaleza y contenido del envase
Tiras blister de aluminio / PVC / aluminio que contienen 10 cápsulas.
El HandiHaler es un dispositivo de inhalación de dosis única compuesto de materiales plásticos (ABS) y acero inoxidable.
Tamaño de envases y dispositivos disponibles:
• Envase con 30 cápsulas (3 tiras blister)
• Envase con 60 cápsulas (6 tiras blister)
• Envase con 90 cápsulas (9 tiras blister)
• Envase con un dispositivo HandiHaler y 10 cápsulas (1 tira blister)
• Envase con un dispositivo HandiHaler y 30 cápsulas (3 tiras blister)
• Envase clínico: caja que contiene 5 envases con 30 cápsulas más un dispositivo HandiHaler
• Envase clínico: caja que contiene 5 envases con 60 cápsulas
El dispositivo HandiHaler está envasado/disponible en un envase.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Boehringer Ingelheim International GmbH Binger Strasse 173 D-55216 Ingelheim am Rhein Alemania
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
64.796
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 23/mayo/2002
Fecha de la renovación de la autorización: 4/noviembre/2006
10. FECHA DE LA REVISIÓN DEL TEXTO
Noviembre 2014
14 de 14
de 14 MINISTERIO DE
SANIDAD, POLITICA SOCIAL E IGUALDAD Agencia españciaóe medie amentos y proouctos san-tanos