Somavert 30 Mg Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
NOMBRE DEL MEDICAMENTO
1.
Somavert 10 mg polvo y disolvente para solución inyectable Somavert 15 mg polvo y disolvente para solución inyectable Somavert 20 mg polvo y disolvente para solución inyectable Somavert 25 mg polvo y disolvente para solución inyectable Somavert 30 mg polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Somavert 10 mg polvo y disolvente para solución inyectable Cada vial contiene 10 mg de pegvisomant.
Después de la reconstitución, 1 ml de solución contiene 10 mg de pegvisomant.* Excipiente con efecto conocido
Cada vial de polvo de 10 mg del medicamento contiene 0,4 mg de sodio.
Somavert 15 mg polvo y disolvente para solución inyectable Cada vial contiene 15 mg de pegvisomant.
Después de la reconstitución, 1 ml de solución contiene 15 mg de pegvisomant.* Excipiente con efecto conocido
Cada vial de polvo de 15 mg del medicamento contiene 0,4 mg de sodio.
Somavert 20 mg polvo y disolvente para solución inyectable Cada vial contiene 20 mg de pegvisomant.
Después de la reconstitución, 1 ml de solución contiene 20 mg de pegvisomant.* Excipiente con efecto conocido
Cada vial de polvo de 20 mg del medicamento contiene 0,4 mg de sodio.
Somavert 25 mg polvo y disolvente para solución inyectable Cada vial contiene 25 mg de pegvisomant.
Después de la reconstitución, 1 ml de solución contiene 25 mg de pegvisomant.* Excipiente con efecto conocido
Cada vial de polvo de 25 mg del medicamento contiene 0,5 mg de sodio.
Somavert 30 mg polvo y disolvente para solución inyectable Cada vial contiene 30 mg de pegvisomant.
Después de la reconstitución, 1 ml de solución contiene 30 mg de pegvisomant.* Excipiente con efecto conocido
Cada vial de polvo de 30 mg del medicamento contiene 0,6 mg de sodio. *producido en células de Escherichia coli por tecnología ADN recombinante. Para consultar la lista completa de excipientes, ver sección 6.1.
FORMA FARMACÉUTICA
3.
Polvo y disolvente para solución inyectable (polvo para inyectable). El polvo es de color blanco a blanquecino.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento de pacientes adultos con acromegalia que no hayan respondido de forma adecuada a tratamiento con cirugía y/o radiación y en los que un adecuado tratamiento médico con análogos de la somatostatina no haya normalizado las concentraciones del factor de crecimiento insulínico tipo I (IGF-I) o no haya sido tolerado.
4.2 Posología y forma de administración
El tratamiento debe ser iniciado bajo la supervisión de un médico con experiencia en el tratamiento de la acromegalia.
Posología
Se debe administrar una dosis inicial de 80 mg de pegvisomant por vía subcutánea bajo supervisión médica. A continuación, se administrarán por vía subcutánea una vez al día SOMAVERT 10 mg reconstituido en 1 ml de disolvente.
Los ajustes de dosis deben realizarse de acuerdo a los niveles séricos de IGF-I. Se deben medir las concentraciones séricas de IGF-I cada 4 a 6 semanas y el ajuste de la dosis adecuado debe realizarse con incrementos de 5 mg/día para mantener la concentración sérica de IGF-I dentro del rango normal adecuado a la edad y al mantenimiento de una óptima respuesta terapéutica.
La dosis máxima no debe exceder de 30 mg/día.
Para las diferentes pautas posológicas, se dispone de las siguientes presentaciones: SOMAVERT 10 mg, SOMAVERT 15 mg, SOMAVERT 20 mg, SOMAVERT 25 mg y SOMAVERT 30 mg.
Población pediátrica
No se ha establecido la seguridad y eficacia de SOMAVERT en niños de 0 a 17 años. No se dispone de datos.
Pacientes de edad avanzada No es necesario un ajuste de dosis.
Insuficiencia hepática o renal
La seguridad y eficacia de SOMAVERT en pacientes con insuficiencia renal o hepática no ha sido establecida.
Forma de administración
Pegvisomant se debe administrar por vía subcutánea.
El lugar de la inyección debe ser diferente cada día para ayudar a prevenir la lipohipertrofia.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
Contraindicaciones
4.3
Tumores secretores de hormona de crecimiento
En ocasiones, los tumores de la glándula pituitaria secretora de la hormona de crecimiento pueden extenderse y causar graves complicaciones (p.ej., defectos en el campo visual). El tratamiento con pegvisomant no reduce el tamaño del tumor. Todos los pacientes con este tipo de tumores deben ser monitorizados cuidadosamente para evitar cualquier eventual progresión en el tamaño del tumor en tratamiento.
Monitorización de las concentraciones séricas de IGF-I
Pegvisomant es un potente antagonista de la acción de la hormona de crecimiento. La administración de este medicamento puede causar un estado deficiente de hormona de crecimiento, a pesar de la presencia de niveles elevados de hormona de crecimiento en suero. Las concentraciones séricas de IGF-I deben ser monitorizadas y mantenidas dentro del rango normal ajustado a la edad, ajustando la dosis de pegvisomant.
Aumentos de ALT o AST
Las concentraciones séricas de alanin-aminotransferasa (ALT) y aspartato transaminasa (AST) deben ser monitorizadas en intervalos de 4 a 6 semanas durante los primeros seis meses de tratamiento con pegvisomant, o en cualquier momento en aquellos pacientes que presenten síntomas que puedan sugerir hepatitis. En pacientes con aumento de ALT y AST o con antecedentes previos de tratamiento con cualquier análogo de somatostatina, debe descartarse evidencia de trastorno obstructivo del tracto biliar. La administración de pegvisomant se debe suspender si persisten los síntomas de trastorno hepático.
Hipoglucemia
El estudio realizado con pegvisomant en pacientes diabéticos tratados con insulina o con hipoglucemiantes orales, reveló riesgo de hipoglucemia en esta población. Por tanto, puede ser necesario un ajuste de la dosis de insulina o de los hipoglucemiantes orales en pacientes acromegálicos con diabetes mellitus (ver sección 4.5).
Aumento de la fertilidad
Los beneficios terapéuticos de una reducción en la concentración de IGF-I cuyo resultado es una mejoría en la condición clínica del paciente, pueden incrementar potencialmente la fertilidad en las mujeres. Se debe advertir a los pacientes para que utilicen medios anticonceptivos adecuados si fuera necesario. Pegvisomant no está recomendado durante el embarazo (ver sección 4.6).
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacción. Se debe considerar la continuidad del tratamiento con análogos de somatostatina. El uso de este medicamento en combinación con otros medicamentos para el tratamiento de la acromegalia no ha sido ampliamente investigado.
Los pacientes tratados con insulina o con hipoglucemiantes orales pueden necesitar una reducción de la dosis de estos principios activos debido al efecto de pegvisomant en la sensibilidad a la insulina (ver sección 4.4).
Pegvisomant tiene una importante similitud estructural con la hormona de crecimiento que provoca
una reacción cruzada en los ensayos disponibles de hormona de crecimiento. Dado que las concentraciones séricas de dosis terapéuticamente eficaces de este medicamento, son normalmente de 100 a 1000 veces más elevadas que las concentraciones séricas de hormona de crecimiento en pacientes acromegálicos, los valores de las concentraciones séricas de hormona de crecimiento resultarán erróneos en los análisis de hormona de crecimiento. Por tanto, el tratamiento con pegvisomant no debe ser monitorizado o ajustado de acuerdo a las concentraciones séricas de hormona de crecimiento cuantificadas en dichos análisis.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil.
Ver sección 4.4.
Embarazo
No se dispone de datos clínicos sobre embarazos de riesgo para pegvisomant.
Los estudios en animales son insuficientes para determinar las reacciones en el embarazo, desarrollo embrional/fetal, parto o desarrollo posnatal (ver sección 5.3). Se desconoce el riesgo potencial en seres humanos.
SOMAVERT no debe utilizarse durante el embarazo excepto si fuese claramente necesario (ver sección 4.4).
Lactancia
No se ha estudiado en animales la excreción de pegvisomant en la leche materna. Los datos clínicos son demasiado limitados (un caso notificado) como para extraer conclusiones sobre la excreción de pegvisomant en la leche materna. Por tanto, pegvisomant no se debe utilizar en mujeres en periodo de lactancia. Sin embargo, el periodo de lactancia puede continuarse en caso de que se suspenda el tratamiento con este medicamento: esta decisión debe tener en cuenta tanto el beneficio del tratamiento con pegvisomant para la madre como el beneficio de la lactancia para el niño.
Fertilidad
No se dispone de datos acerca del efecto de pegvisomant sobre la fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas Resumen del perfil de seguridad
La relación que se incluye a continuación contiene las reacciones adversas observadas en ensayos clínicos con SOMAVERT.
En los ensayos clínicos realizados en pacientes tratados con pegvisomant (n = 550), la mayoría de las reacciones adversas con pegvisomant fueron de intensidad leve a moderada, de duración limitada y no fue necesario suspender el tratamiento.
Las reacciones adversas notificadas con más frecuencia, ocurridas en > 10% de los pacientes con acromegalia tratados con pegvisomant durante los ensayos clínicos realizados, fueron cefalea 25%, artralgia 16% y diarrea 13%.
El listado que se indica a continuación contiene las reacciones adversas notificadas en los ensayos clínicos o que fueron notificadas de manera espontánea, clasificadas según el sistema de clasificación de órgano y sistemas.
Las reacciones adversas se clasifican de acuerdo con las siguientes categorías:
Muy frecuentes: >1/10
Frecuentes: >1/100 a <1/10
Poco frecuentes: >1/1.000 a <1/100
No conocidas (no se puede estimar la frecuencia a partir de los datos disponibles)
|
Clasificación de órganos y sistemas |
Muy frecuentes (>1/10) |
Frecuentes (>1/100 a <1/10) |
Poco frecuentes (>1/1.000 a < 1/100) |
No conocidas (no se puede estimar la frecuencia a partir de los datos disponibles) |
|
Trastornos de la sangre y del sistema linfático |
trombocitopenia, leucopenia, leucocitosis, diátesis hemorrágica | |||
|
Trastornos del sistema inmunológico |
reacciones de hipersensibilidadb |
reacción anafilácticab, reacción anafilactoideb | ||
|
Trastornos del metabolismo y de la nutrición |
hipercolesterole mia, hiperglucemia, hipoglucemia, aumento de peso |
hipertrigliceridemia | ||
|
Trastornos psiquiátricos |
sueños anormales |
ataque de pánico, pérdida de memoria a corto plazo, apatía, confusión, trastorno del sueño, aumento de la líbido |
irritabilidad | |
|
Trastornos del sistema nervioso |
cefalea |
somnolencia, temblores, mareo, hipoestesia |
narcolepsia, migraña, disgeusia | |
|
Trastornos oculares |
dolor ocular |
astenopía | ||
|
Trastornos del oído y del laberinto |
enfermedad de Meniere | |||
|
Trastornos cardiacos |
edema periférico | |||
|
Trastornos vasculares |
hipertensión | |||
|
Trastornos respiratorios, torácicos y mediastínicos |
disnea |
laringoespasmo b |
|
Clasificación de órganos y sistemas |
Muy frecuentes (>1/10) |
Frecuentes (>1/100 a <1/10) |
Poco frecuentes (>1/1.000 a < 1/100) |
No conocidas (no se puede estimar la frecuencia a partir de los datos disponibles) |
|
Trastornos gastrointestinales |
diarrea |
vómitos, estreñimiento, náuseas, distensión abdominal, dispepsia, flatulencia |
hemorroides, aumento de la salivación, sequedad de boca, trastornos dentales | |
|
Trastornos hepatobiliares |
pruebas de función hepática anormales (ej. aumento de transaminasas) (ver sección 4.4) | |||
|
Trastornos de la piel y del tejido subcutáneo |
hiperhidrosis, contusión, pruritob, exantemab |
edema facial, sequedad de piel, tendencia a hematomas, sudoración nocturna, eritemab, urticariab |
angioedemab | |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
artralgia |
mialgia, artritis | ||
|
Trastornos renales y urinarios |
hematuria |
proteinuria, poliuria, insuficiencia renal | ||
|
Trastornos generales y alteraciones en el lugar de administración |
reacción en el lugar de la inyección (incluyendo hipersensibilidad en el lugar de la inyección), hematomas o hemorragia en el lugar de la inyección, hipertrofia en el lugar de la inyección (por ejemplo lipohipertrofia)a, síndrome gripal, fatiga, astenia, pirexia |
sensación anómala, alteración de la cicatrización, hambre |
a Ver más abajo la descripción de las reacciones adversas señaladas b Reacción adversa relacionada con la reacción de hipersensibilidad
La mayoría de las reacciones en el lugar de la inyección se caracterizaron por eritemas y dolor localizados, que se resolvieron de forma espontánea con tratamiento sintomático local, sin necesidad de interrumpir el tratamiento con pegvisomant. Se han observado casos de hipertrofia en el lugar de administración incluyendo lipohipertrofia.
Se observó el desarrollo de anticuerpos aislados de baja titulación para la hormona de crecimiento en el 16,9% de los pacientes tratados con pegvisomant. Se desconoce la relevancia clínica de estos anticuerpos.
Durante la experiencia poscomercialización se han notificado reacciones sistémicas de hipersensibilidad incluyendo reacciones anafilácticas/anafilectoides, laringoespasmo, angioedema, reacciones cutáneas generalizadas (exantema, eritema, prurito, urticaria). Algunos pacientes requirieron hospitalización. Los síntomas no volvieron a aparecer en todos los pacientes tras reiniciar el tratamiento.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Existe una experiencia limitada en la sobredosis con pegvisomant. En el único incidente notificado de sobredosis aguda, en el que se administraron 80 mg/día durante 7 días, el paciente presentó un ligero aumento de la fatiga y sequedad de boca. En la semana siguiente a la suspensión del tratamiento las reacciones adversas observadas fueron: insomnio, aumento de la fatiga, edema periférico, temblor y aumento del peso. Dos semanas después de haber suspendido el tratamiento, se observaron leucocitosis y sangrado moderado en los lugares de inyección y punción venosa, que se consideraron posiblemente relacionados con pegvisomant.
En caso de sobredosis, se debe interrumpir la administración de este medicamento y no reanudarla hasta que los niveles de IGF-I estén dentro o por debajo del rango normal.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Otras hormonas del lóbulo anterior de la hipófisis y sus análogos, código ATC: H01AX01.
Mecanismo de acción
Pegvisomant es un análogo de la hormona de crecimiento humana modificado genéticamente para ser un antagonista del receptor de la hormona de crecimiento. Pegvisomant se une a la superficie celular de los receptores de la hormona de crecimiento y bloquea la unión de la hormona de crecimiento, por lo que interfiere con la señal de transducción intracelular de la hormona de crecimiento. Pegvisomant es altamente selectivo para el receptor de la GH (hormona de crecimiento) y no provoca reacciones cruzadas con otros receptores de las citoquinas, incluida la prolactina.
Efectos farmacodinámicos
La inhibición de la acción de la hormona de crecimiento con pegvisomant lleva a una disminución de las concentraciones séricas del factor de crecimiento insulínico tipo I (IGF-I), así como de otras
proteínas séricas sensibles a la hormona de crecimiento, como IGF-I libre, subunidad ácido-lábil de IGF-I (ALS) y proteína trasportadora del factor de crecimiento insulínico tipo 3 (IGFBP-3).
Eficacia clínica y seguridad
Pacientes acromegálicos (n = 112) fueron tratados en un estudio aleatorizado, doble ciego, multicéntrico comparativo con placebo y pegvisomant durante 12 semanas. Dependiendo de la dosis, se observaron reducciones estadísticamente significativas en IGF-I medio (p<0,0001), IGF-I libre (p<0,05), IGFBP-3 (p<0,05) y ALS (p<0,05) en todas las visitas post-basal en el grupo de tratamiento con pegvisomant. El factor IGF-I en suero se normalizó al final del estudio (semana 12) en el 9,7%, 38,5%, 75% y 82% de los pacientes tratados con placebo, 10 mg/día, 15 mg/día o 20 mg/día de pegvisomant respectivamente.
Se observaron diferencias estadísticamente significativas (p<0,05) en la mejora de la puntuación total de signos y síntomas en todos los grupos con distintas dosis comparados con el grupo de placebo.
Se realizó el seguimiento de un estudio de cohorte a largo plazo, abierto, de determinación de dosis, de al menos 12 meses consecutivos en 38 pacientes acromegálicos con tratamiento diario con pegvisomant (media: 55 semanas). La concentración sérica media de IGF-I en esta cohorte cayó de 917 ng/ml a 299 ng/ml con un 92% que alcanzaron la concentración normal de IGF-I (ajustada a la edad).
5.2 Propiedades farmacocinéticas
Absorción
La absorción de pegvisomant después de la administración subcutánea es lenta y prolongada, y los picos de concentración en suero no se alcanzan generalmente hasta las 33-77 horas de su administración. La absorción media de una dosis subcutánea fue de 57% en relación con una dosis intravenosa.
Distribución
El volumen aparente de distribución de pegvisomant es relativamente pequeño (7 - 12 l). Biotransformación
No se ha estudiado el metabolismo de pegvisomant. Eliminación
Se estima que el aclaramiento medio sistémico total de pegvisomant después de múltiples dosis es de 28 ml/h en dosis subcutáneas en un intervalo de 10 a 20 mg/día. El aclaramiento renal de pegvisomant es insignificante e inferior a un 1% del aclaramiento total. Pegvisomant es eliminado del suero lentamente, con una estimación media de semivida que va generalmente de 74 a 172 horas tanto después de administración única como múltiple.
Linealidad/No linealidad
Después de la administración única subcutánea de pegvisomant, no se observa linealidad al aumentar la dosis de 10, 15 ó 20 mg. Se observa una farmacocinética aproximadamente lineal en estado estacionario en la población de los estudios farmacocinéticos. Los datos de 145 pacientes pertenecientes a dos estudios a largo plazo, que habían recibido diariamente dosis subcutáneas de 10, 15 ó 20 mg, muestran que las concentraciones séricas medias (±DS) de pegvisomant fueron aproximadamente de 8800 ± 6300, 13200 ± 8000 y 15600 ± 10300 ng/ml, respectivamente.
La farmacocinética de pegvisomant es similar en voluntarios sanos y en pacientes acromegálicos,
9
aunque los individuos más corpulentos tienden a tener un aclaramiento total de pegvisomant más elevado que los individuos más delgados, y por tanto se pueden necesitar dosis más altas de pegvisomant.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios realizados sobre toxicidad a dosis repetidas en ratas y monos. No obstante, debido a la acusada respuesta farmacológica en monos, no se estudiaron exposiciones sistémicas superiores a las alcanzadas en pacientes a dosis terapéuticas. A excepción de un test de segmento II en conejos, no se han realizado otros estudios sobre toxicidad reproductora.
Se observaron histiocitomas fibrosos malignos asociados con fibrosis y con inflamación histiocítica en los puntos de inyección en las ratas macho del estudio de carcinogenicidad a unos niveles de exposición equivalentes a tres veces la exposición en humanos de acuerdo a las concentraciones plasmáticas medias en dos estudios a largo plazo a una dosis diaria de 30 mg. Actualmente se desconoce la relevancia de esta respuesta en humanos.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato
Disolvente:
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 3 años.
Después de la reconstitución el producto debe utilizarse inmediatamente.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C). No congelar. Conservar el/los vial(es) y la(s) jeringa(s) precargada(s) en el embalaje exterior para protegerlo de la luz.
Después de la reconstitución:
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
10 mg o 15 mg o 20 mg o 25 mg o 30 mg de pegvisomant en polvo en un vial (vidrio flint tipo I) con un tapón (goma de clorobutilo) y 1 ml de disolvente (agua para preparaciones inyectables) en una jeringa precargada (vidrio de borosilicato tipo I) con un tapón de émbolo (de goma de bromobutilo) y un tapón en el extremo (de goma de bromobutilo). El color del precinto protector de plástico es específico de la concentración del producto.
Somavert 10 mg y 15 mg
Tamaño de envase de 30 viales, jeringas precargadas y agujas de seguridad.
Somavert 20 mg, 25 mg y 30 mg
Tamaños de envase de 1 y 30 vial(es), jeringa(s) precargada(s) y aguja(s) de seguridad.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
La jeringa y la aguja de seguridad utilizadas para administrar la inyección se proporcionan con el medicamento.
Antes de colocar la aguja de seguridad suministrada, es necesario retirar la tapa de la jeringa precargada. Para ello tire de la tapa hacia fuera. La jeringa debe mantenerse en posición vertical para evitar fugas y el extremo de la jeringa no debe entrar en contacto con nada.

El polvo debe reconstituirse con 1 ml de disolvente. Al añadir el disolvente contenido en la jeringa, el vial y la jeringa deben mantenerse en ángulo como se indica en el siguiente diagrama.
Añada el disolvente al vial que contiene el polvo. Añada el disolvente al vial lentamente para evitar la formación de espuma. Esto haría que el medicamento quedara inutilizable. Disuelva suavemente el polvo con un movimiento rotatorio lento. No agite violentamente ya que esto puede causar la desnaturalización del principio activo.
Después de la reconstitución, inspeccione visualmente la solución reconstituida para observar si existe alguna partícula ajena (o extraña) o alguna variación de aspecto físico antes de su administración. En caso de observar alguna de estas circunstancias, deseche el medicamento.
Antes de extraer la solución de Somavert, invierta el vial con la jeringa todavía insertada en él y asegúrese de que puede verse el espacio en el tapón como se indica en el siguiente diagrama:
Tire de la aguja hacia abajo de manera que la punta de la aguja esté en el punto más bajo en el líquido. Tire del émbolo de la jeringa lentamente para extraer el medicamento del vial. Si observa aire en la jeringa, presione el cilindro de la jeringa para que las burbujas se deplacen hacia arriba y luego empuje las burbujas despacio hacia el vial.
Antes de desechar la jeringa y la aguja, doble el protector de la aguja sobre la aguja y asegúrese de que encaja en su lugar. La jeringa y la aguja no se deben reutilizar nunca.
Para un solo uso. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
8. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/001 10 mg 30 viales EU/1/02/240/002 15 mg 30 viales EU/1/02/240/004 20 mg 1 vial EU/1/02/240/003 20 mg 30 viales EU/1/02/240/009 25 mg 1 vial EU/1/02/240/010 25 mg 30 viales EU/1/02/240/011 30 mg 1 vial EU/1/02/240/012 30 mg 30 viales
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 13/noviembre/2002 Fecha de la última renovación: 20/septiembre/2007
10. FECHA DE LA REVISIÓN DEL TEXTO
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del (de los) fabricante(s) del (de los) principio(s) activo(s) biológico(s)
Pfizer Health AB Mariefredsvagen 37 645 41 Strangnas Suecia
Pfizer Ireland Pharmaceuticals
Grange Castle Business Park
Clondalkin
Dublin 22
Irlanda
Nombre y dirección del (de los) fabricante(s) responsable(s) de la liberación de los lotes
Pfizer Manufacturing Belgium NV Rijksweg 12 B-2870, Puurs Bélgica
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado
en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en
cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
SOMAVERT 10 mg polvo y disolvente para solución inyectable pegvisomant
Un vial contiene 10 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 10 mg de pegvisomant
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato Agua para preparaciones inyectables
Polvo y disolvente para solución inyectable 30 viales de polvo
30 jeringas precargadas de disolvente 30 agujas para inyección
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 10 mg
SOMAVERT 10 mg polvo para solución inyectable pegvisomant
Un vial contiene 10 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 10 mg de pegvisomant
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN_
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO, CUANDO CORRESPONDA
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 10 mg
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
VIAL DE POLVO
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
SOMAVERT 10 mg
polvo para inyectablepegvisomant
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DEL LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
10 mg
6. OTROS
SOMAVERT 15 mg polvo y disolvente para solución inyectable pegvisomant
Un vial contiene 15 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 15 mg de pegvisomant
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato Agua para preparaciones inyectables
Polvo y disolvente para solución inyectable 30 viales de polvo
30 jeringas precargadas de disolvente 30 agujas para inyección
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 15 mg
SOMAVERT 15 mg polvo para solución inyectable pegvisomant
Un vial contiene 15 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 15 mg de pegvisomant
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN_
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 15 mg
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
VIAL DE POLVO
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
SOMAVERT 15 mg
polvo para inyectablepegvisomant
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DEL LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
15 mg
6. OTROS
SOMAVERT 20 mg polvo y disolvente para solución inyectable pegvisomant
Un vial contiene 20 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 20 mg de pegvisomant
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato Agua para preparaciones inyectables
Polvo y disolvente para solución inyectable 30 viales de polvo
30 jeringas precargadas de disolvente 30 agujas para inyección
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 20 mg
SOMAVERT 20 mg polvo para solución inyectable pegvisomant
Un vial contiene 20 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 20 mg de pegvisomant
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN_
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 20 mg
1. NOMBRE DEL MEDICAMENTO
SOMAVERT 20 mg polvo y disolvente para solución inyectable pegvisomant
2. PRINCIPIO(S) ACTIVO(S)
Un vial contiene 20 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 20 mg de pegvisomant
3. LISTA DE EXCIPIENTES
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato Agua para preparaciones inyectables
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable 1 vial de polvo
1 jeringa precargada de disolvente 1 aguja para inyección
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/004
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 20 mg
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
VIAL DE POLVO
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
SOMAVERT 20 mg
polvo para inyectablepegvisomant
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DEL LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
20 mg
6. OTROS
SOMAVERT 25 mg polvo y disolvente para solución inyectable pegvisomant
Un vial contiene 25 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 25 mg de pegvisomant
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato Agua para preparaciones inyectables
Polvo y disolvente para solución inyectable 30 viales de polvo
30 jeringas precargadas de disolvente 30 agujas para inyección
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/010
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 25 mg
SOMAVERT 25 mg polvo para solución inyectable pegvisomant
Un vial contiene 25 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 25 mg de pegvisomant
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN_
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/010
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 25 mg
SOMAVERT 25 mg polvo y disolvente para solución inyectable pegvisomant
Un vial contiene 25 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 25 mg de pegvisomant
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato Agua para preparaciones inyectables
Polvo y disolvente para solución inyectable 1 vial de polvo
1 jeringa precargada de disolvente 1 aguja para inyección
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE
COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/009
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 25 mg
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
VIAL DE POLVO
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
SOMAVERT 25 mg
polvo para inyectablepegvisomant
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DEL LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
25 mg
6. OTROS
SOMAVERT 30 mg polvo y disolvente para solución inyectable pegvisomant
Un vial contiene 30 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 30 mg de pegvisomant
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato Agua para preparaciones inyectables
Polvo y disolvente para solución inyectable 30 viales de polvo
30 jeringas precargadas de disolvente 30 agujas para inyección
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/012
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 30 mg
SOMAVERT 30 mg polvo para solución inyectable pegvisomant
Un vial contiene 30 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 30 mg de pegvisomant
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN_
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/012
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 30 mg
SOMAVERT 30 mg polvo y disolvente para solución inyectable pegvisomant
Un vial contiene 30 mg de pegvisomant
Después de la reconstitución, 1 ml de solución contiene 30 mg de pegvisomant
Glicina
Manitol (E421)
Hidrogenofosfato de disodio anhidro Dihidrogenofosfato de sodio monohidrato Agua para preparaciones inyectables
Polvo y disolvente para solución inyectable 1 vial de polvo
1 jeringa precargada de disolvente 1 aguja para inyección
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
Utilizar inmediatamente después de su reconstitución. Para un solo uso.
Conservar en nevera. No congelar.
Conservar el envase en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/240/011
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
SOMAVERT 30 mg
VIAL DE POLVO
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
SOMAVERT 30 mg
polvo para inyectablepegvisomant
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DEL LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
30 mg
6. OTROS
JERINGA PRECARGADA DE DISOLVENTE
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Disolvente para SOMAVERT SC
|
2. |
FORMA DE ADMINISTRACIÓN |
|
3. |
FECHA DE CADUCIDAD |
|
CAD | |
|
4. |
NÚMERO DE LOTE |
|
Lote | |
|
5. |
CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES |
1 ml de agua para preparaciones inyectables
6. OTROS
B. PROSPECTO
Prospecto: información para el usuario
inyectable
inyectable
inyectable
inyectable
inyectable
SOMAVERT 10 mg polvo y disolvente para solución SOMAVERT 15 mg polvo y disolvente para solución SOMAVERT 20 mg polvo y disolvente para solución SOMAVERT 25 mg polvo y disolvente para solución SOMAVERT 30 mg polvo y disolvente para solución
Pegvisomant
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es SOMAVERT y para qué se utiliza
2. Qué necesita saber antes de empezar a usar SOMAVERT
3. Cómo usar SOMAVERT
4. Posibles efectos adversos
5. Conservación de SOMAVERT
6. Contenido del envase e información adicional
1. Qué es SOMAVERT y para qué se utiliza
SOMAVERT se usa para el tratamiento de la acromegalia, un trastorno hormonal resultante del aumento de la secreción de la hormona de crecimiento (HC) y del IGF-I (factores de crecimiento tipo insulina), y se caracteriza por sobrecrecimiento de los huesos, engrosamiento de los tejidos blandos, enfermedad del corazón y trastornos relacionados.
El principio activo de SOMAVERT, pegvisomant, es conocido como un antagonista del receptor de la hormona de crecimiento. Estas sustancias reducen la acción de la HC y los niveles de IGF-I que circulan en sangre.
2. Qué necesita saber antes de empezar a usar SOMAVERT
No use SOMAVERT:
• Si es alérgico a pegvisomant o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a usar SOMAVERT.
- Si nota trastornos en la visión o dolores de cabeza mientras está utilizando SOMAVERT, debe decírselo a su médico inmediatamente.
- Su médico o enfermera controlará los niveles de IGF-I (factores de crecimiento tipo insulina) que circulan en sangre y si es necesario ajustará la dosis de SOMAVERT.
- Su médico debe también controlar su adenoma (tumor benigno).
- Su médico o enfermera controlará los niveles de enzimas hepáticas en sangre cada 4-6 semanas durante los primeros seis meses de tratamiento con SOMAVERT. La administración de SOMAVERT debe suspenderse si persisten los síntomas de la enfermedad hepática.
- Si es diabético, su médico puede necesitar ajustar la cantidad de insulina o de otros medicamentos que esté utilizando.
- Las pacientes deben emplear métodos anticonceptivos ya que la fertilidad puede aumentar.
Véase también el apartado sobre Embarazo que aparece más adelante.
Uso de SOMAVERT con otros medicamentos
Debe decirle a su médico si ha usado anteriormente otro medicamento para el tratamiento de la acromegalia o algún medicamento para el tratamiento de la diabetes.
Informe a su médico o farmacéutico si está utilizando o ha utilizado recientemente cualquier otro medicamento. Como parte de su tratamiento, puede ser tratado con otros medicamentos. Es importante que siga utilizando todos los medicamentos incluido SOMAVERT a menos que reciba otra indicación por parte de su médico, farmacéutico o enfermero.
Embarazo, lactancia y fertilidad
Se desconocen los efectos de SOMAVERT en mujeres embarazadas por lo que no se recomienda la utilización de este medicamento en mujeres embarazadas. Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
No se sabe si pegvisomant pasa a la leche materna. No deberá dar el pecho mientras esté tomando SOMAVERT a menos que lo haya discutido con su médico.
Conducción y uso de máquinas
No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas. SOMAVERT contiene sodio
Este medicamento contiene menos de 1 mg de sodio por dosis, esto es esencialmente “exento de sodio”.
3. Cómo usar SOMAVERT
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Su médico le administrará por vía subcutánea (debajo de la piel) una dosis inicial de 80 mg de pegvisomant. Después, la dosis diaria habitual es de 10 mg de pegvisomant administrada por inyección subcutánea (debajo de la piel).
Cada 4-6 semanas, su médico realizará los ajustes de dosis necesarios aumentando la dosis en 5 mg de pegvisomant por día, de acuerdo con sus niveles en sangre del ya mencionado IGF-I, con el fin de obtener una respuesta terapéutica óptima.
Forma y vía de administración
SOMAVERT se inyecta bajo la piel. La inyección puede ponérsela usted mismo u otra persona, como su médico o ayudante. Debe seguir las instrucciones detalladas sobre el proceso de inyección, que se incluyen al final de este prospecto. Deberá continuar inyectándose este medicamento durante todo el tiempo que le indique su médico.
Este medicamento debe disolverse antes de su uso. La inyección no debe mezclarse en la misma jeringa o vial con otro medicamento.
El tejido graso de la piel puede aumentarse en el lugar de la inyección. Para evitarlo, utilice puntos de inyección ligeramente diferentes cada vez, tal y como se describe en el paso 2 de la sección de este prospecto “Instrucciones para la preparación y administración de una inyección de Somavert”. Así le dará tiempo a la piel y a la zona bajo la piel a recuperarse entre una inyección y otra antes de volver a inyectarse en el mismo lugar.
Si tiene la impresión de que el efecto de este medicamento es demasiado fuerte o demasiado débil, hable con su médico, farmacéutico o enfermero.
Si se inyecta más SOMAVERT del que debe
Si accidentalmente se ha inyectado más cantidad de SOMAVERT de la que le dijo su médico, no es probable que esto sea grave pero deberá indicárselo inmediatamente a su médico, farmacéutico o enfermero.
Si olvidó usar SOMAVERT
Si olvidó ponerse una inyección, deberá inyectarse la dosis siguiente tan pronto lo recuerde y deberá seguir inyectándose SOMAVERT tal como le ha sido prescrito por su médico. No se inyecte una dosis doble para compensar las dosis olvidadas.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Se han notificado reacciones alérgicas (anafilácticas) de leves a graves en algunos pacientes que usan SOMAVERT. Los síntomas de una reacción alérgica grave pueden incluir uno o más de los siguientes síntomas: hinchazón de la cara, de la lengua, de los labios o de la garganta; pitidos o dificultad para respirar (espasmo de la laringe); erupción generalizada de la piel, urticaria o picor; o mareo. Póngase en contacto inmediatamente con su médico si presenta alguno de estos síntomas.
Muy frecuentes: pueden afectar a más de 1 de cada 10 personas:
- Dolor de cabeza.
- Diarrea.
- Dolor de las articulaciones.
Frecuentes: pueden afectar hasta a 1 de cada 10 personas:
- Dificultad para respirar.
- Aumentos en los niveles de las sustancias que determinan la función del hígado. Pueden verse en los resultados de los análisis de sangre.
- Sangre en la orina.
- Aumento de la tensión arterial.
- Estreñimiento, malestar, sensación de estar enfermo, sensación de estar hinchado, indigestión, flatulencia.
- Mareo, somnolencia, temblor incontrolado, disminución del sentido del tacto.
- Cardenales o sangrado en el lugar de la inyección, dolor o hinchazón en el lugar de la inyección, aumento del tejido graso bajo la piel en el lugar de la inyección, hinchazón de las extremidades, debilidad, fiebre.
- Sudoración, picor, erupción, tendencia a tener cardenales.
- Dolor en los musculos, artritis.
- Aumento del colesterol en sangre, aumento de peso, aumento de la glucosa en sangre, descenso de
la glucosa en sangre.
- Síntomas gripales, fatiga.
- Sueños anormales.
- Dolor en los ojos.
Poco frecuentes: pueden afectar hasta a 1 de cada 100 personas:
- Reacción alérgica tras la administración (fiebre, erupción, prurito y, en casos graves, dificultad para respirar, hinchazón rápida de piel, que requieren atención médica urgente). Pueden ocurrir inmediatamente o varios días después de la administración.
- Proteínas en la orina, aumento de la cantidad de orina, problemas en los riñones.
- Ausencia de interés, sensación de confusión, aumento de la libido, ataques de pánico, pérdida de memoria, dificultades para dormir.
- Reducción de plaquetas en sangre, aumento o reducción de leucocitos en la sangre, tendencia a sangrar.
- Sensación anormal, alteración en la cicatrización.
- Pesadez de ojos, problemas en el oído interno.
- Hinchazón de la cara, sequedad de la piel, sudoración nocturna, enrojecimiento de la piel (eritema), picor y ronchas elevadas en la piel (urticaria).
- Aumento de sustancias grasas en sangre, aumento del apetito.
- Sequedad de boca, aumento de la salivación, problemas dentales, hemorroides.
- Sentido del gusto anormal, migrañas.
No conocidas: no se puede estimar la frecuencia a partir de los datos disponibles
- Irritabilidad.
- Dificultad para respirar grave (laringoespasmo).
- Hinchazón rápida de la piel, el tejido subyacente y el revestimiento interno (mucosa) de los órganos (angioedema).
Aproximadamente un 17% de los pacientes desarrollarán anticuerpos frente a la hormona del crecimiento durante el tratamiento. Parece que los anticuerpos no afectan a la acción de este medicamento.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de SOMAVERT
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en los viales y en el envase después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2°C y 8°C). No congelar. Conservar el/los vial(es) y la(s) jeringa(s) precargada(s) en el embalaje exterior para protegerlo de la luz.
Después de preparar la solución de SOMAVERT, ésta debe ser utilizada inmediatamente.
No utilice este medicamento si observa que la solución está turbia o contiene partículas.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo
deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de SOMAVERT
- El principio activo es pegvisomant.
- SOMAVERT 10 mg: un vial de polvo contiene 10 mg de pegvisomant. Después de la reconstitución con 1 ml de disolvente, 1 ml de solución contiene 10 mg de pegvisomant.
- SOMAVERT 15 mg: un vial de polvo contiene 15 mg de pegvisomant. Después de la reconstitución con 1 ml de disolvente, 1 ml de solución contiene 15 mg de pegvisomant.
- SOMAVERT 20 mg: un vial de polvo contiene 20 mg de pegvisomant. Después de la reconstitución con 1 ml de disolvente, 1 ml de solución contiene 20 mg de pegvisomant.
- SOMAVERT 25 mg: un vial de polvo contiene 25 mg de pegvisomant. Después de la reconstitución con 1 ml de disolvente, 1 ml de solución contiene 25 mg de pegvisomant.
- SOMAVERT 30 mg: un vial de polvo contiene 30 mg de pegvisomant. Después de la reconstitución con 1 ml de disolvente, 1 ml de solución contiene 30 mg de pegvisomant.
- Los demás componentes son glicina, manitol (E-421), hidrogeno fosfato de sodio anhidro, dihidrógeno fosfato de sodio monohidrato
- El disolvente es agua para preparaciones inyectables.
Aspecto del producto y contenido del envase
SOMAVERT se presenta en forma de polvo y disolvente para inyección (en un vial de 10 mg, 15 mg, 20 mg, 25 mg o 30 mg de pegvisomant y 1 ml de disolvente en una jeringa precargada). Tamaños de envase de 1 y/o 30. Puede que solamente estén comercializados algunos tamaños de envases. El polvo es de color blanco y el disolvente es transparente e incoloro.
Titular de la autorización de comercialización y responsable de la fabricación:
Titular de la autorización de comercialización
Pfizer Limited Ramsgate Road Sandwich,
Kent CT13 9NJ Reino Unido
Responsable de la fabricación
Pfizer Manufacturing Belgium NV Rijksweg 12 2870 Puurs Bélgica
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Pfizer S.A./N.V.
Tél/Tel: +32 (0)2 554 62 11
Luxembourg/Luxemburg
Pfizer S.A.
Tél/Tel: +32 (0)2 554 62 11
Magyarország
Pfizer Kft.
Tel.: + 36 1 488 37 00
EtnrapHH
n^aroep .HroKceMÓypr CAPH, KnoH Etnrapna
Ten.: +359 2 970 4333
|
Ceská republika Pfizer PFE, spol. s. r.o. Tel: +420 283 004 111 |
Malta V.J. Salomone Pharma Ltd. Tel: + 356 21 22 01 74 |
|
Danmark Pfizer ApS Tlf: +45 44 20 11 00 |
Nederland Pfizer bv Tel: +31 (0)10 406 43 01 |
|
Deutschland Pfizer Pharma GmbH Tel: +49 (0)30 550055 51000 |
Norge Pfizer Norge AS Tlf: +47 67 52 61 00 |
|
Eesti Pfizer Luxembourg SARL Eesti filiaal Tel: +372 666 7500 |
Osterreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0 |
|
EXXáSa PFIZER EAAAI A.E. TnL +30 210 6785800 |
Polska Pfizer Polska Sp. z o.o. Tel.: +48 22 335 61 00 |
|
España Pfizer S.L. Tel: +34 91 490 99 00 |
Portugal Pfizer Biofarmaceutica, Sociedade Unipessoal Lda Tel: +351 21 423 5500 |
|
France Pfizer Tél: +33 (0)1 58 07 34 40 |
Romania Pfizer Romania S.R.L. Tel: +40 (0)21 207 28 00 |
|
Hrvatska Pfizer Croatia d.o.o. Tel: + 385 1 3908 777 |
Slovenija Pfizer Luxembourg SARL, Pfizer, podruznica za svetovanje s podrocja farmacevtske dejavnosti, Ljubljana Tel: + 386 (0)1 52 11 400 |
|
Ireland Pfizer Healthcare Ireland Tel: 1800 633 363 (toll free) +44 (0)1304 616161 |
Slovenská republika Pfizer Luxembourg SARL, organizacná zlozka Tel: +421 2 3355 5500 |
|
Ísland Icepharma hf. Sími: + 354 540 8000 |
Suomi/Finland Pfizer Oy Puh/Tel: +358 (0)9 43 00 40 |
|
Italia Pfizer S.r.l. Tel: +39 06 33 18 21 |
Sverige Pfizer Innovations AB Tel: +46 (0)8 550 520 00 |
|
Kúnpoq PFIZER EAAAE A.E. (Cyprus Branch) Tn^: +357 22 817690 Latvija Pfizer Luxembourg SARL filiale Latvija Tel: +371 670 35 775 |
United Kingdom Pfizer Limited Tel: +44 (0)1304 616161 |
Lietuva
Pfizer Luxembourg SARL filíalas Lietuvoje Tel. +3705 2514000
Fecha de la última revisión de este prospecto:.
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu. También existen enlaces a otras páginas web sobre enfermedades raras y medicamentos huérfanos.
INSTRUCCIONES DE USO
Somavert polvo en vial con disolvente en una jeringa precargada
pegvisomant para solución inyectable Unicamente para inyección subcutánea Vial de dosis única
Somavert se presenta en un vial como un bloque de polvo blanco. Debe mezclar Somavert con un líquido (disolvente) antes de poder usarlo.
El líquido se presenta en una jeringa precargada con la etiqueta “Disolvente para SOMAVERT”. No mezcle Somavert con ningún otro líquido.
Es importante que no intente administrarse a usted mismo o a otra persona una inyección sin haber recibido formación de su profesional sanitario.
Conserve el envase completo en la nevera entre 2°C y 8°C y lejos de la luz solar directa. Mantener fuera del alcance de los niños.
1. Qué necesita
Un envase de Somavert con:
• Un vial de Somavert polvo
• Una jeringa precargada con disolvente
• Una aguja de seguridad También necesita:
• Una torunda de algodón
• Un algodón con alcohol
• Un contenedor de objetos punzantes adecuado
Vial
tapón del vial (sin la tapa del vial)
espacio del tapón
cilindro
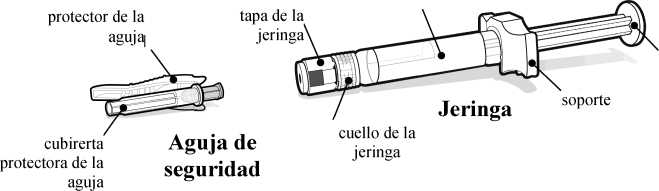
varilla del émbolo
2. Preparación
Antes de empezar:
• Mezcle Somavert con el disolvente únicamente cuando esté preparado para inyectarse la dosis.
• Saque un único envase de Somavert de la nevera y deje que alcance la temperatura ambiente de manera natural en un lugar seguro.
58
• Lávese las manos con agua y jabón, y séqueselas bien.
• Abra el envoltorio de la jeringa y la aguja de seguridad para que sea más fácil coger cada elemento mientras se prepara para la inyección.
• No use la jeringa o el vial si:
o están dañados o defectuosos; o la fecha de caducidad se ha sobrepasado;
o la jeringa se ha congelado, incluso si se ha descongelado a continuación (únicamente la jeringa).
3. Elija una zona de inyección
3
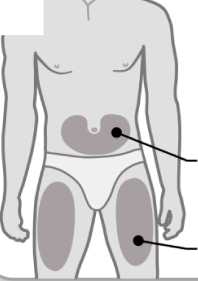
Brazos o zona lumbar:
Zona superior posterior de los brazos (únicamente el
Abdomen:
Mantenga una distancia de al menos 5 cm de su ombligo
Muslos
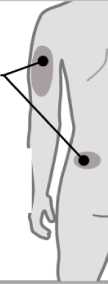

• Elija un lugar diferente dentro de cada área para la inyección.
• Evite las zonas óseas, enrojecidas, dolorosas o duras, o que tengan cardenales, cicatrices o enfermedades de la piel.
• Limpie la zona de inyección con el algodón con alcohol como le haya indicado su profesional sanitario.
• Espere a que la zona de inyección se seque.
4. Retire la tapa del vial
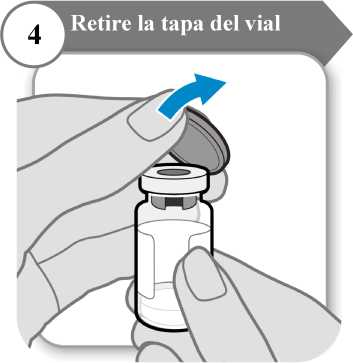
Retire la tapa del vial.
Deseche la tapa; no se necesita de nuevo.
Precaución: No deje que nada toque el tapón del vial.
5. Retire la tapa de la jeringa
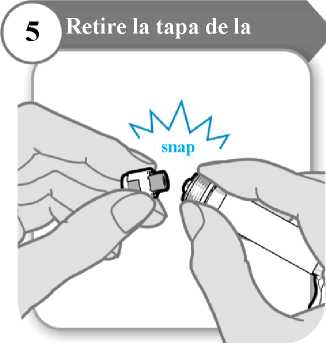
• Desprenda la tapa de la jeringa. Puede que necesite más fuerza de la que cabría esperar.
• Deseche la tapa de la jeringa; no se necesita de nuevo.
• Mantenga la jeringa en posición vertical para evitar fugas.
Precaución: No deje que el extremo de la jeringa toque nada una vez que haya retirado la tapa.
6. Coloque la aguja de seguridad
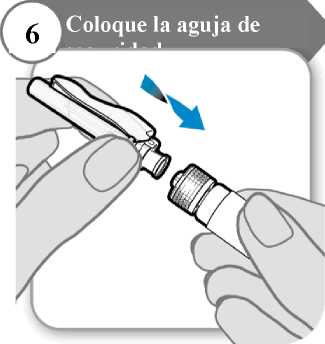
• Coloque la aguja de seguridad en la jeringa girando firmemente tanto como pueda.
7. Retire la cubierta protectora de la aguja

Retire la cubierta .protectora de la aguja
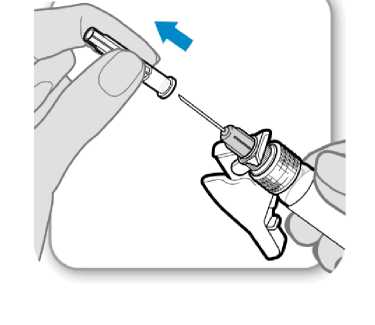
• Doble hacia afuera el protector de la aguja apartándolo de la cubierta protectora de la aguja.
• Con cuidado, tire de la cubierta protectora de la aguja directamente hacia afuera.
• Deseche la cubierta protectora de la aguja; no se necesita de nuevo.
Precaución: No deje que la aguja toque nada.
8. Inserte la aguja
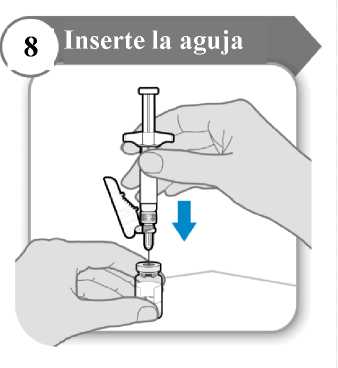
• Empuje la aguja a través del centro del tapón del vial como se indica.
• Sostenga la jeringa mientras la aguja esté insertada en el tapón del vial para evitar que la aguja se doble.
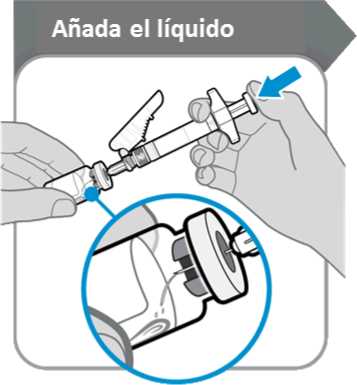
9. Añada el líquido
Añada el líquido
• Incline el vial y la jeringa formando un ángulo como se indica.
• Empuje la varilla del émbolo lentamente hasta que todo el líquido se encuentre dentro del vial.
• Precaución: Procure que el líquido no caiga directamente sobre el polvo, ya que eso formaría espuma. La espuma hace que el medicamento quede inutilizable.
• No retire la aguja todavía.
10. Haga girar el vial
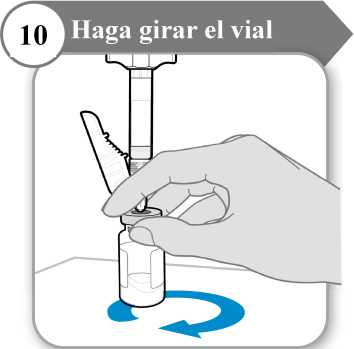
• Sostenga la jeringa y el vial con una mano como se indica.
• Haga girar el líquido suavemente deslizando el vial con un movimiento cicular sobre una superficie plana.
• Continúe haciendo girar el líquido hasta que todo el polvo se haya disuelto por completo.
Nota: Esto puede tardar hasta 5 minutos.
11. Examine el medicamento

Examine el medicamento
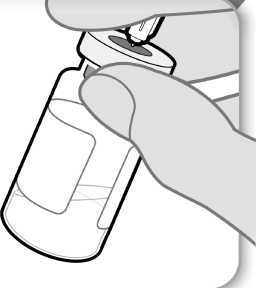
• Con la aguja todavía insertada en el vial, inspeccione el medicamento atentamente. Éste debe ser transparente y sin partículas.
• No lo use si:
o el medicamento está turbio u oscuro;
o el medicamento tiene algún color;
o contiene partículas o hay una capa de espuma en el vial.
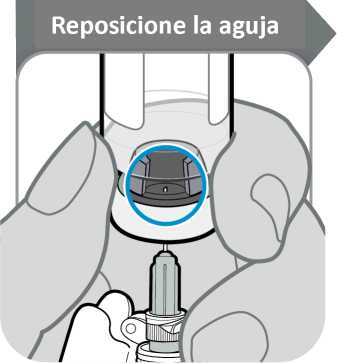
12. Reposicione la aguja
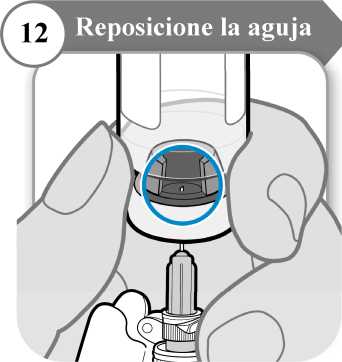
• Gire el vial de manera que pueda ver el espacio en el tapón del vial, como se indica.
• Tire de la aguja hacia abajo de manera que la punta de la aguja esté en el punto más bajo en el líquido. Esto le ayudará a extraer tanto líquido como sea posible.
• Compruebe que la varilla del émbolo no se ha movido. Si lo ha hecho, empuje para volver a introducirlo por completo en la jeringa. Esto asegura que todo el aire haya salido de la jeringa antes de extraer la dosis.
13. Extraiga la dosis
Extraiga la dosis
• Tire de la varilla del émbolo despacio para extraer tanta cantidad de medicamento del vial como sea posible.
Nota: Si observa aire en la jeringa, presione el cilindro de la jeringa para que las burbujas se desplacen hacia arriba, y luego empuje las burbujas despacio hacia el vial.
• Retire la aguja del vial.
14. Inserte la aguja
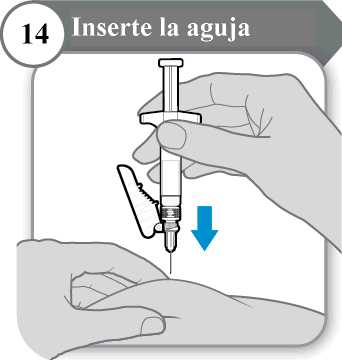
• Con cuidado, pellizque la piel en la zona de inyección.
• Inserte la aguja por completo en la piel pellizcada.
15. Inyecte el medicamento
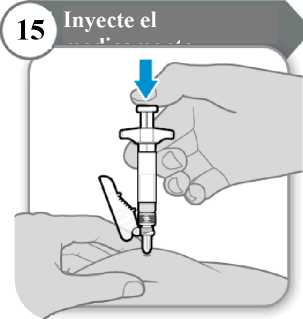
• Empuje la varilla del émbolo hacia abajo despacio hasta que la jeringa esté vacía. Nota: Asegúrese de que la aguja esté insertada por completo.
• Suelte la piel pellizcada y extraiga la aguja en línea recta.
16. Asegure la aguja
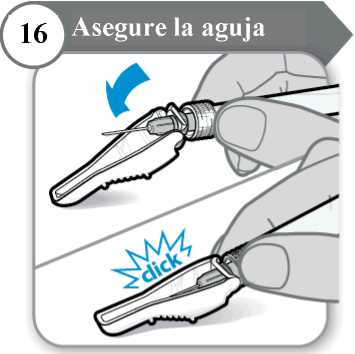
• Doble el protector de la aguja sobre la aguja.
• Con cuidado, presione contra una superficie dura para cerrar el protector de la aguja. Nota: Escuchará un clic cuando el protector de la aguja se cierre.
17. Desechar
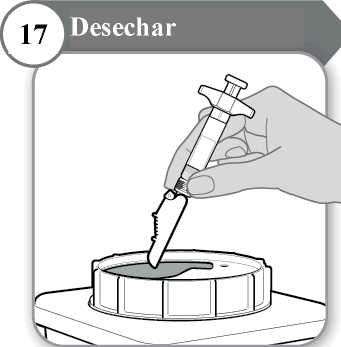
• La jeringa y la aguja no se deben reutilizar NUNCA. Deseche la aguja y la jeringa como le haya indicado su médico, enfermero o farmacéutico y según las directrices sanitarias locales y la legislación en materia de seguridad.
18. Después de la inyección
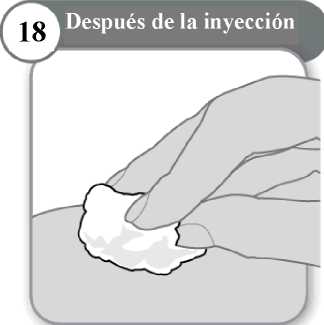
• Si es necesario, presione ligeramente con una torunda de algodón limpio en la zona de inyección.
• No frote la zona.
PREGUNTAS Y RESPUESTAS ¿Qué debo hacer si algo ha tocado accidentalmente el tapón del vial?
• Limpie el tapón del vial con una toallita de alcohol nueva, y deje que se seque por completo. Si es incapaz de limpiar el tapón, no utilice el vial.
¿Qué debo hacer con la jeringa si se ha caído?
• No la use, aunque parezca que no está dañada. Deseche la jeringa de la misma manera que desecha una jeringa usada. Necesitará otra jeringa.
¿Cuántas veces puedo insertar de forma segura la aguja en el tapón del vial?
• Sólo una vez. Extraer y reinsertar la aguja aumenta considerablemente el riesgo de daño a la aguja y puede quedar roma. Esto puede causar incomodidad y aumentar el riesgo de daño a la piel e infección. También existe el riesgo de que se pierda parte del medicamento.
¿Está bien agitar el vial si el polvo no se disuelve?
• No, nunca agite el vial. Las sacudidas pueden inutilizar el medicamento y formaría espuma. El polvo puede tardar unos minutos en disolverse por completo, así que continúe moviendo el vial despacio con movimientos circulares hasta que el líquido sea completamente transparente.
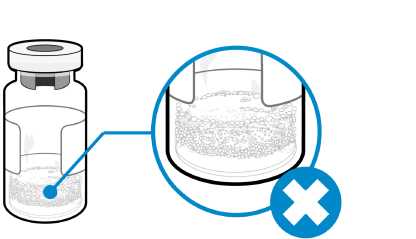
¿Cómo puedo saber si hay espuma en el vial?
• La espuma aparece como una masa de pequeñas burbujas que flota formando una capa sobre el líquido. No inyecte Somavert si se ha formado espuma.
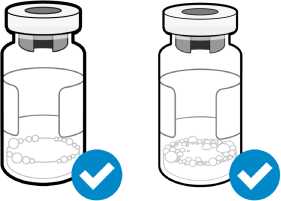
Las pequeñas burbujas de aire son aceptables
Una capa de espuma no es aceptable
¿Cómo puedo evitar que el medicamento forme espuma?
• Empuje el émbolo muy despacio de manera que el líquido fluya suavemente hacia el interior del vial. No deje caer el líquido directamente sobre el polvo, ya que eso forma espuma. Esta técnica también reducirá el tiempo necesario para mezclar el medicamento y permitirá extraer más medicamento.
Puedo ver algo de aire en la jeringa. ¿Está bien?
• Las pequeñas burbujas de aire en el líquido son normales y la inyección es segura. No obstante, es posible aspirar accidentalmente algo de aire dentro de la jeringa, que debe ser eliminado antes de la inyección. Las burbujas o espacios de aire que flotan sobre el líquido se deben expulsar hacia el vial.
¿Por qué no puedo extraer todo el medicamento del vial?
• La forma del vial hace que una pequeña cantidad del medicamento quede en el vial. Esto es normal. Para asegurarse de que sólo una pequeña cantidad del medicamento queda en el vial, asegúrese cuando extraiga la dosis, de que la punta de la aguja está introducida dentro del vial tanto como sea posible.
¿Qué debo hacer si tengo alguna duda sobre el medicamento?
• Todas las preguntas deben ser dirigidas a un médico, enfermero o farmacéutico con experiencia con SOMAVERT.
67