Sandostatin Lar 20 Mg Polvo Y Disolvente Para Suspension Inyectable
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
SANDOSTATIN LAR 10 mg polvo y disolvente para suspensión inyectable SANDOSTATIN LAR 20 mg polvo y disolvente para suspensión inyectable SANDOSTATIN LAR 30 mg polvo y disolvente para suspensión inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
2.2 Composición cualitativa y cuantitativa
Sandostatin LAR 10 mg: Un vial contiene 10 mg de octreotida (como octreotida acetato).
Sandostatin LAR 20 mg: Un vial contiene 20 mg de octreotida (como octreotida acetato).
Sandostatin LAR 30 mg: Un vial contiene 30 mg de octreotida (como octreotida acetato).
2.2.1 Excipiente(s) con efecto conocido
Contiene menos de 1 mmol (23 mg) de sodio por dosis, por lo que se considera esencialmente “exento de sodio”.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para suspensión inyectable.
Polvo: Polvo blanco o blanco con un tono amarillento.
Disolvente: solución transparente, incolora a ligeramente amarilla o marrón.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento de pacientes con acromegalia para los que la cirugía no es adecuada o no es eficaz, o en el periodo intermedio hasta que la radioterapia sea completamente eficaz (ver sección 4.2).
Tratamiento de pacientes con síntomas asociados con tumores gastroenteropancreáticos funcionales p.ej. tumores carcinoides con características del síndrome carcinoide (ver sección 5.1).
Tratamiento de pacientes con tumores neourendocrinos avanzados del intestino o tumor primario de origen desconocido donde se han excluido los lugares de origen no intestinal.
Tratamiento de adenomas de la pituitaria secretores de TSH:
• cuando la secreción no se ha normalizado después de la cirugía y/o la radioterapia;
• en pacientes para los que la cirugía no es adecuada;
• en pacientes irradiados, hasta que la radioterapia sea eficaz.
4.2 Posología y forma de administración
4.2.1 Posología
Acromegalia
Se recomienda iniciar el tratamiento con la administración de 20 mg de Sandostatin LAR a intervalos de 4 semanas durante 3 meses. Los pacientes en tratamiento con Sandostatin s.c. pueden iniciar el tratamiento con Sandostatin LAR el día después de la última dosis de Sandostatin s.c.. El posterior ajuste de dosis se debe basar en las concentraciones séricas de la hormona de crecimiento (GH) y del factor de crecimiento I tipo insulina/somatomedina C (IGF-1) y en los síntomas clínicos.
Para los pacientes que, durante este periodo de 3 meses, los síntomas clínicos y los parámetros bioquímicos (GH; IGF-1) no estén totalmente controlados (concentraciones de GH todavía por encima de
2,5 microgramos/L), se puede aumentar la dosis a 30 mg cada 4 semanas. Si después de 3 meses, GH, IGF-1, y/o los síntomas no están adecuadamente controlados a una dosis de 30 mg, se puede aumentar la dosis a 40 mg cada 4 semanas.
Los pacientes con concentraciones de GH por debajo de 1 microgramo/L de forma continuada, con concentraciones séricas de IGF-1 normalizadas, y en los que la mayoría de signos/síntomas reversibles de la acromegalia hayan desaparecido después de 3 meses de tratamiento con 20 mg, se puede administrar 10 mg de Sandostatin LAR cada 4 semanas. Sin embargo, especialmente en este grupo de pacientes, es recomendable controlar estrechamente las concentraciones séricas de GH e IGF-1, y los signos/síntomas clínicos a esta dosis baja de Sandostatin LAR.
Para pacientes en una dosis estable de Sandostatin LAR, se debe realizar una evaluación de GH e IGF-1 cada 6 meses.
Tumores endocrinos gastroenteropancreaticos
Tratamiento de pacientes con síntomas asociados con tumores neuroendocrinos gastroenteropancreaticos funcionales
Se recomienda iniciar el tratamiento con la administración de 20 mg de Sandostatin LAR a intervalos de 4 semanas. Los pacientes en tratamiento con Sandostatin s.c. deben continuar con la dosis efectiva previamente durante 2 semanas después de la primera inyección de Sandostatin LAR.
En pacientes en los que los síntomas y los marcadores biológicos están bien controlados después de 3 meses de tratamiento, se puede reducir la dosis a 10 mg de Sandostatin LAR cada 4 semanas.
En pacientes en los que los síntomas están controlados sólo parcialmente después de 3 meses de tratamiento, se puede aumentar la dosis a 30 mg de Sandostatin LAR cada 4 semanas.
Para los días en los que los síntomas asociados con tumores gastroenteropancreaticos pueden aumentar durante el tratamiento con Sandostatin LAR, se recomienda la administración adicional de Sandostatin s.c. a la dosis anterior al tratamiento con Sandostatin LAR. Esto puede suceder principalmente los primeros
2 meses del tratamiento hasta que se alcancen las concentraciones terapéuticas de octreotida.
Tratamiento de pacientes con tumores neuroendocrinos avanzados del intestino o de origen primario desconocido en que se han excluido los lugares de origen no intestinal
La dosis recomendada de Sandostatin LAR es de 30 mg administrados cada 4 semanas (ver sección 5.1). El tratamiento con Sandostatin LAR para el control del tumor se debe continuar en ausencia de progresión del tumor.
Tratamiento de adenomas secretores de TSH
El tratamiento con Sandostatin LAR se debe iniciar a la dosis de 20 mg a intervalos de 4 semanas durante
3 meses antes de considerar un ajuste de dosis. Después se ajusta la dosis en base a la TSH y la respuesta de la hormona tiroidea.
Uso en pacientes con insuficiencia renal
La insuficiencia renal no afectó la exposición total (AUC) a octreotida cuando se administró como Sandostatin s.c.. Por lo tanto, no es necesario un ajuste de dosis con Sandostatin LAR.
Uso en pacientes con insuficiencia hepática
En un estudio con Sandostatin administrado por vía s.c. e i.v. se demostró que la capacidad de eliminación puede verse reducida en pacientes con cirrosis hepática, pero no en pacientes con enfermedad hepática grasa. En algunos casos los pacientes con insuficiencia hepática pueden requerir un ajuste de dosis.
Uso en _pacientes de edad avanzada
En un estudio con Sandostatin administrado s.c., no fue necesario un ajuste de dosis en pacientes de >65 años. Por tanto, no es necesario un ajuste de dosis en este grupo de pacientes con Sandostatin LAR.
Uso en niños
Existe una experiencia limitada con el uso de Sandostatin LAR en niños.
4.2.2 Forma de administración
Sandostatin LAR sólo se puede administrar por inyección intramuscular profunda. Se debe alternar el lugar de inyecciones intramusculares repetidas entre el músculo glúteo izquierdo y derecho (ver sección 6.6).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
General
Debido a que los tumores pituitarios secretores de GH pueden, en algunas ocasiones, expandirse produciendo graves complicaciones (p.ej. defectos del campo visual), es esencial que todos los pacientes sean controlados cuidadosamente. Si aparece evidencia de expansión del tumor, son aconsejables procedimientos alternativos.
En mujeres con acromegalia los beneficios terapéuticos de una reducción en los niveles de la hormona del crecimiento (GH) y de la normalización del factor de crecimiento 1 tipo insulina (IGF-1) pueden potencialmente restaurar la fertilidad. Se debe advertir a las pacientes con posibilidad de quedarse embarazadas que tomen las medidas anticonceptivas necesarias durante el tratamiento con octreotida (ver sección 4.6).
En pacientes que reciben un tratamiento prolongado con octreotida se deberá controlar la función tiroidea. Durante el tratamiento con octreotida se deberá controlar la función hepática.
Efectos cardiovasculares
Se han notificado casos frecuentes de bradicardia. Puede ser necesario ajustar la dosis de medicamentos como beta-bloqueantes, bloqueantes de canales de calcio, o agentes para controlar el equilibrio de líquidos y electrolitos (ver sección 4.5).
Vesícula biliar y efectos relacionados
Octreotida inhibe la secreción de colecistocinina, lo que supone una contractibilidad reducida de la vesicular biliar y un aumento del riesgo de sedimento y de formación de cálculos. Se ha notificado el
desarrollo de cálculos biliares en un 15 a 30% de pacientes en tratamiento a largo plazo con Sandostatin s.c. La prevalencia en la población general (de 40 a 60 años) es de alrededor del 5 al 20%. La exposición a largo plazo a Sandostatin LAR de pacientes con acromegalia o tumores endocrinos gastroenteropancreaticos sugiere que el tratamiento con Sandostatin LAR no aumenta la incidencia de formación de cálculos biliares, comparado con el tratamiento s.c.. Por lo tanto, se recomienda un examen ecográfico de la vesícula biliar antes y a intervalos de 6 meses durante el tratamiento con Sandostatin LAR. Si aparecen cálculos biliares, estos son normalmente asintomáticos; los cálculos sintomáticos se deben tratar o bien con terapia de disolución con ácidos biliares o con cirugía.
Metabolismo de la glucosa
Debido a su acción inhibidora sobre la hormona del crecimiento, glucagón e insulina, Sandostatin LAR puede afectar la regulación de la glucosa. Se puede alterar la tolerancia a la glucosa postprandial. Al igual que se ha notificado en pacientes tratados con Sandostatin s.c., en algunos casos,como consecuencia de la administración crónica, se puede inducir un estado de hiperglucemia persistente. También se han notificado casos de hipoglucemia.
En pacientes con diabetes mellitus Tipo I concomitante, es probable que Sandostatin LAR afecte la regulación de glucosa, y se pueden reducir los requisitos de insulina. En pacientes no diabéticos y en pacientes diabéticos de tipo II con reservas de insulina intactas, la administración de Sandostatin s.c. puede suponer un aumento de la glucemia postprandial. Por lo tanto, se recomienda controlar la tolerancia a la glucosa y el tratamiento antidiabético.
En pacientes con insulinomas, octreotida puede aumentar la intensidad y prolongar la duración de la hipoglucemia, debido a su potencia superior relativa para inhibir la secreción de GH y glucagón con respecto a insulina, y debido a la duración de acción más corta de su acción inhibitoria sobre la insulina. Se debe controlar estrechamente a estos pacientes.
Nutrición
Octreotida puede alterar la absorción de las grasas de la dieta en algunos pacientes.
En algunos pacientes que reciben tratamiento con octreotida se ha observado una reducción del nivel de vitamina B12 y resultados anormales en el test de Schilling. Se recomienda controlar los niveles de vitamina B12 durante el tratamiento con Sandostatin LAR en pacientes con antecedentes de déficit de vitamina B12.
Contenido en sodio
Sandostatin LAR contiene menos de 1 mmol (23 mg) de sodio por dosis, por lo que se considera esencialmente “exento de sodio”
4.5 Interacción con otros medicamentos y otras formas de interacción
Pueden ser necesarios ajustes de dosis de medicamentos como beta bloqueantes, antagonistas de canales de calcio, o agentes que controlan el equilibrio de líquidos y electrolitos, cuando se administran de forma concomitante con Sandostatin LAR (ver sección 4.4).
Pueden ser necesarios ajustes de dosis de insulina y medicamentos antidiabéticos cuando se administra Sandostatin LAR de forma concomitante (ver sección 4.4).
Se ha observado que octreotida reduce la absorción intestinal de ciclosporina y retrasa la de cimetidina.
La administración concomitante de octreotida y bromocriptina aumenta la biodisponibilidad de bromocriptina.
Datos publicados limitados indican que los análogos de somatostatina podrían disminuir el aclaramiento metabólico de las sustancias que se sabe que se metabolizan mediante los enzimas del citocromo P450, que puede ser debido a la supresión de la hormona del crecimiento. Dado que no se puede excluir que octreotida pueda tener este efecto, se deben utilizar con precaución otros fármacos metabolizados principalmente por CYP3A4 y que tienen un bajo índice terapéutico (p.ej. quinidina, terfenadina).
4.6 Fertilidad, embarazo y lactancia
4.6.1 Embarazo
Hay datos limitados (datos en menos de 300 embarazos) relativos al uso de octreotida en mujeres embarazadas, y en aproximadamente un tercio de los casos se desconoce el desenlace del embarazo. La mayoría de las notificaciones se recibieron después del uso post-comercialización de octreotida y más de un 50% de embarazos expuestos se notificaron en pacientes con acromegalia. La mayoría de mujeres se expusieron a octreotida durante el primer trimestre del embarazo a dosis que oscilaban entre 100-1.200 microgramos/día de Sandostatin por vía s.c. o 10-40 mg/mes de Sandostatin LAR. Se notificaron anomalías congénitas en aproximadamente un 4% de casos de embarazo, de los cuales se conoce el desenlace. No se sospecha una relación con octreotida para estos casos.
Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción (ver sección 5.3).
Como medida de precaución, es preferible evitar el uso de Sandostatin LAR durante el embarazo (ver sección 4.4).
4.6.2 Lactancia
Se desconoce si octreotida se excreta en la leche materna. Los estudios en animales muestran que octreotida se excreta en la leche. Las pacientes no deben amamantar durante el tratamiento con Sandostatin LAR.
4.6.3 Fertilidad
Se desconoce si octreotida tiene algún efecto sobre la fertilidad humana. Se observó un retraso en el descenso de los testículos para las crías macho de madres tratadas durante la gestación y la lactancia. Sin embargo, octreotida no alteró la fertilidad en ratas machos y hembras a dosis de hasta 1 mg/kg de peso corporal por día (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Sandostatin LAR sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Se debe advertir a los pacientes que tengan precaución al conducir o utilizar máquinas si presentan mareo, astenia/fatiga, o cefalea durante el tratamiento con Sandostatin LAR.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Las reacciones adversas más frecuentes notificadas durante el tratamiento con octreotida incluyen alteraciones gastrointestinales, alteraciones del sistema nervioso, alteraciones hepatobiliares y alteraciones del metabolismo y la nutrición.
Las reacciones adversas notificadas de forma más frecuente en los ensayos clínicos con administración de octreotida fueron diarrea, dolor abdominal, náuseas, flatulencia, cefalea, colelitiasis, hiperglucemia y estreñimiento. Otras reacciones adversas notificadas de forma frecuente fueron mareo, dolor localizado, sedimento biliar, alteración tiroidea (p.ej. disminución de la hormona estimulante del tiroides [TSH],
disminución de T4 total, y disminución de T4 libre), heces líquidas, tolerancia a la glucosa alterada, vómitos, astenia e hipoglucemia.
Lista tabulada de reacciones adversas
Las siguientes reacciones adversas, listadas en la Tabla 1, se han recogido de los ensayos clínicos con octreotida:
Las reacciones adversas (Tabla 1) se presentan agrupadas por frecuencias, la más frecuente primero, utilizando la siguiente convención: muy frecuentes (>1/10); frecuentes (> 1/100, <1/10); poco frecuentes (>1/1.000, <1/100); raras (>1/10.000, <1/1.000) muy raras (<1/10.000), incluyendo casos aislados. Dentro de cada frecuencia, se ordenan las reacciones adversas en orden decreciente de gravedad.
Tabla 1 Reacciones adversas notificadas en los ensayos clínicos
|
Trastornos gastrointestinales Muy frecuentes: Frecuentes: |
Diarrea, dolor abdominal, náuseas, estreñimiento, flatulencia. Dispepsia, vómitos, hinchazón abdominal, esteatorrea, deposiciones líquidas, coloración de las heces. |
|
Trastornos del sistema nervioso | |
|
Muy frecuentes: |
Cefalea. |
|
Frecuentes: |
Mareo. |
|
Trastornos endocrinos | |
|
Frecuentes: |
Hipotiroidismo, disfunción tiroidea (p.ej. disminución de TSH, |
|
disminución de T4 total, y disminución de T4 libre). | |
|
Trastornos hepatobiliares | |
|
Muy frecuentes: |
Colelitiasis. |
|
Frecuentes: |
Colecistitis, depósitos biliares, hiperbilirubinemia. |
|
Trastornos del metabolismo y de la nutrición | |
|
Muy frecuentes: |
Hiperglucemia. |
|
Frecuentes: |
Hipoglucemia, tolerancia alterada a la glucosa, anorexia. |
|
Poco frecuentes: |
Deshidratación. |
|
Trastornos generales y alteraciones en el lugar de administración | |
|
Muy frecuentes: |
Reacciones en el lugar de inyección. |
|
Frecuentes: |
Astenia. |
|
Exploraciones complementarias | |
|
Frecuentes: |
Niveles elevados de transaminasas. |
|
Trastornos de la piel y del tejido subcutáneo | |
|
Frecuentes: |
Prurito, exantema, alopecia. |
|
Trastornos respiratorios, torácicos y mediastínicos | |
|
Frecuentes: |
Disnea. |
|
Trastornos cardiacos | |
|
Frecuentes: |
Bradicardia. |
|
Poco frecuentes: |
Taquicardia. |
Postcomercialización
Las reacciones adversas notificadas de forma espontánea, presentadas en la Tabla 2, se notifican voluntariamente y no siempre es posible establecer de forma fiable la frecuencia o la relación causal con la exposición al fármaco.
Tabla 2 Reacciones adversas derivadas de notificaciones espontáneas
Trastornos del sistema inmunológico
Anafilaxis, alergia/reacciones de hipersensibilidad._
Trastornos de la piel y del tejido subcutáneo
Urticaria_
Trastornos hepatobiliares
Pancreatitis aguda, hepatitis aguda sin colestasis, hepatitis colestásica, colestasis, ictericia, ictericia
colestásica._
Trastornos cardiacos
Arritmias._
Exploraciones complementarias
Aumento del nivel de fosfatasa alcalina, aumento del nivel de gama glutamil transferasa._
Descripción de reacciones adversas seleccionadas
Trastornos gastrointestinales
En raras ocasiones, las reacciones adversas gastrointestinales pueden parecer una obstrucción intestinal aguda con distensión abdominal progresiva, dolor epigástrico intenso, sensibilidad abdominal y resistencia.
Se sabe que la frecuencia de las reacciones adversas gastrointestinales disminuye a lo largo del tiempo con el tratamiento adecuado.
Reacciones en el lugar de inyección
Se notificaron de forma frecuente reacciones asociadas al lugar de inyección que incluyen dolor, quemazón, enrojecimiento, hematoma, hemorragia, prurito o hinchazón en pacientes que reciben Sandostatin LAR; sin embargo, estos efectos no requirieron ninguna intervención clínica en la mayoría de los casos.
Trastornos del metabolismo y de la nutrición
Aunque puede aumentar la excreción medible de la grasa fecal, no existe evidencia hasta la fecha de que el tratamiento a largo plazo con octreotida produzca una deficiencia nutricional debida a una malabsorción.
Enzimas pancreáticas
En casos muy raros, se ha notificado pancreatitis aguda durante las primeras horas o días del tratamiento con Sandostatin s.c. y revirtieron con la retirada del fármaco. Además, se ha descrito pancreatitis inducida por colelitiasis en pacientes en tratamiento prolongado con Sandostatin s.c..
Trastornos cardiacos
En pacientes acromegálicos y en pacientes con síndrome carcinoide se han observado cambios en el ECG como prolongación del intervalo QT, desviaciones de los ejes, repolarización precoz, voltaje bajo, transición R/S, progresión precoz de la onda R, cambios no específicos de la onda ST-T. No se ha establecido la relación de estos acontecimientos con octreotida acetato, debido a que muchos de estos pacientes tienen enfermedades cardiacas subyacentes (ver sección 4.4).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es.
4.9 Sobredosis
Se han notificado un número limitado de casos de sobredosis accidentales de Sandostatin LAR. Las dosis oscilaron entre 100 mg y 163 mg/mes de Sandostatin LAR. El único efecto adverso notificado fueron sofocos.
Se han notificado casos de pacientes con cáncer que recibieron dosis de Sandostatin LAR de hasta 60 mg/mes y de hasta 90 mg/2 semanas. Estas dosis fueron en general bien toleradas; sin embargo se notificaron las siguientes reacciones adversas: micción frecuente, fatiga, depresión, ansiedad, y falta de concentración.
El tratamiento de la sobredosis es sintomático.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Somatostatina y análogos, código ATC: H01CB02
Octreotida es un octapéptido sintético derivado de la somatostatina de origen natural con efectos farmacológicos similares, pero con una duración de acción considerablemente superior. Inhibe la secreción aumentada patológicamente de la hormona de crecimiento (GH) y de los péptidos y la serotonina producidos en el sistema endocrino GEP.
En animales, octreotida es un inhibidor más potente de la liberación de GH, glucagón e insulina que la somatostatina, con una mayor selectividad para la supresión de GH y glucagón.
En individuos sanos, octreotida, como somatostatina, ha mostrado inhibición de:
• liberación de GH estimulada por arginina, ejercicio e hipoglucemia inducida por insulina,
• liberación postprandial de insulina, glucagón, gastrina, otros péptidos del sistema endocrino GEP, y liberación de insulina y glucagón,
• respuesta de la hormona estimulante del tiroides (TSH) inducida por la hormona liberadora de la tirotropina (TRH).
A diferencia de la somatostatina, octreotida inhibe la secreción de GH preferentemente sobre la insulina y su administración no está seguida por una hipersecreción de hormonas de rebote (p.ej. GH en pacientes con acromegalia).
En pacientes con acromegalia, Sandostatin LAR, una formulación galénica de octreotida adecuada para la administración repetida a intervalos de 4 semanas, libera concentraciones séricas de octreotida constantes y terapéuticas, disminuyendo de forma consistente la GH y normalizando las concentraciones séricas IGF 1 en la mayoría de pacientes. En la mayoría de pacientes, Sandostatin LAR reduce notablemente los síntomas clínicos de la enfermedad, como cefalea, transpiración, parestesia, fatiga, osteoartralgia y síndrome del túnel carpiano. En pacientes con acromegalia no tratados previamente con adenoma pituitario secretor de GH, el tratamiento con Sandostatin LAR supuso una reducción del volumen del tumor de >20% en una proporción significativa (50%) de pacientes.
En pacientes individuales con adenoma pituitario secretor de GH, se ha notificado que Sandostatin LAR condujo a una disminución del tamaño del tumor (antes de la cirugía). Sin embargo, no se debe retrasar la cirugía.
En pacientes con tumores funcionales del sistema endocrino gastroenteropancreatico, el tratamiento con Sandostatin LAR aporta un control continuado de los síntomas relacionados con la enfermedad subyacente. El efecto de octreotida en diferentes tipos de tumores gastroenteropancreaticos es el siguiente:
Tumores carcinoides
La administración de octreotida puede producir una mejoría de los síntomas, especialmente de los sofocos y la diarrea. En muchos casos, esto está acompañado por una disminución en la serotonina plasmática y una reducción de la excreción urinaria de ácido 5-hidroxiindol acético.
VIPomas
La característica bioquímica de estos tumores es la sobreproducción de péptido intestinal vasoactivo (VIP). En la mayoría de casos, la administración de octreotida produce un alivio de la diarrea secretora grave típica de esta situación, con la consiguiente mejoría en la calidad de vida. Esto va acompañado por una mejoría de las alteraciones electrolíticas asociadas, p.ej. hipocalemia, permitiendo la retirada de líquidos enterales y parenterales y suplementos de electrolitos. En algunos pacientes, la tomografía computerizada sugiere un retraso o una parada en la progresión del tumor o incluso una reducción del tumor, particularmente de las metástasis hepáticas. La mejoría clínica normalmente está acompañada por una reducción en los niveles plasmáticos de VIP, que pueden descender hasta los valores de referencia normales.
Glucagonomas
La administración de octreotida produce en la mayoría de los casos una mejoría relevante de la urticaria migratoria necrolítica que es característica de esta situación. El efecto de octreotida sobre el estado de la diabetes mellitus moderada que frecuentemente aparece, no es muy marcada y, en general, no supone una reducción de los requisitos de insulina o de agentes antidiabéticos orales. Octreotida produce una mejoría de la diarrea, y por tanto aumento de peso, en los pacientes afectados. Aunque la administración de Sandostatin produce frecuentemente una reducción inmediata en los niveles de glucagón plasmático, esta disminución no se mantiene generalmente durante un periodo prolongado de administración, a pesar de la mejoría sintomática continuada.
Gastrinomas/síndrome Zollinger-Ellison
El tratamiento con inhibidores de la bomba de protones o con agentes bloqueantes del receptor H2 generalmente controla la hipersecreción ácida gástrica. Sin embargo, la diarrea, que es un síntoma importante, puede que no se alivie adecuadamente con inhibidores de la bomba de protones o agentes que bloquean receptores H2. Sandostatin LAR puede ayudar en la reducción mayor de la hipersecreción ácida gástrica y mejorar los síntomas, incluyendo diarrea, ya que supone una supresión de niveles de gastrina elevados, en algunos pacientes.
Insulinomas
La administración de octreotida produce una disminución de la insulina inmunoreactiva circulante. En pacientes con tumores operables, octreotida puede ayudar a restaurar y mantener la normoglucemia precirugía. En pacientes con tumores benignos o malignos no operables, se puede mejorar el control glucémico sin una reducción concomitante sostenida en los niveles de insulina circulante.
Tumores neuroendocrinos avanzados del intestino o de origen primario desconocido donde se han excluido los lugares de origen que no son del intestino.
Un estudio de Fase III, aleatorizado, doble ciego, controlado con placebo (PROMID) demostró que Sandostatin LAR inhibe el crecimiento del tumor en pacientes con tumores neuroendocrinos avanzados del intestino.
Se aleatorizaron 85 pacientes para recibir tratamiento con Sandostatin LAR 30 mg cada 4 semanas (n=42) o placebo (n=43) durante 18 meses, o hasta la progresión del tumor o muerte.
Los principales criterios de inclusión fueron: no haber recibido tratamiento previo; confirmación histológica; localmente inoperable o metastásico bien diferenciado; tumores/carcinomas neuroendocrinos activos funcionalmente o inactivos; con tumor primario localizado en el intestino o de origen desconocido que se cree que pueda ser de origen intestinal si se ha excluido un tumor primario en el páncreas, el tórax o en alguna otra localización.
La variable principal fue tiempo hasta la progresión del tumor o muerte relacionada con el tumor (TTP).
En la población de análisis por intención de tratar (ITT) (todos los pacientes aleatorizados), se observaron 26 y 41 muertes por progresión o relacionadas con el tumor en los grupos de Sandostatin LAR y placebo, respectivamente (HR = 0,32; IC95% , 0,19 a 0,55; valor p =0,000015).
En la población de análisis por ITT conservador (ITTc) en la que 3 pacientes fueron censurados en la aleatorización, se observaron 26 y 40 muertes por progresión o relacionadas con el tumor en los grupos de Sandostatin LAR y placebo, respectivamente (HR= 0,34; IC95%, 0,20 a 0.59; valor p=0,000072; Fig 1). La mediana de tiempo hasta la progresión del tumor fue de 14,3 meses (IC95%, 11,0 a 28,8 meses) en el grupo de Sandostatin LAR y 6,0 meses (IC95%, 3,7 a 9,4 meses) en el grupo de placebo.
En la población de análisis por protocolo (PP) en el que se censuraron pacientes adicionales al final del tratamiento del ensayo, se observaron muertes por progresión de tumor o relacionadas con el tumor en 19 y 38 pacientes que recibían Sandostatin LAR y placebo, respectivamente (HR = 0,24; IC95% , 0,13 a 0,45; valor p =0,0000036).
Figura 1 Valores Kaplan-Meier estimados de TTP comparando Sandostatin LAR con placebo (población ITT conservadora)

<u
13
Placebo: 40 eventos Mediana 6,0 meses Octreotida LAR 26 eventos Mediana 14,3 meses
Tiempo desde la aleatorización (meses)
Pacientes a riesgo
Test Logrank estratificado por actividad funcional: p=0,000072, HR= 0,34 [IC95%: 0,20-0,59]
Tabla 3 Resultados TTP por poblaciones de análisis
|
Eventos TTP |
Mediana de meses TTP [I.C. 95% ] |
HR [I.C. 95%1 Valor p * | |||
|
Sandostatin LAR |
Placebo |
Sandostatin LAR |
Placebo | ||
|
ITT |
26 |
41 |
NR |
NR |
0,32 [95% CI, 0,19 a 0,551 P=0,000015 |
|
cITT |
26 |
40 |
14.3 [IC95%, 11,0 a 28,81 |
6.0 [IC95%, 3,7 a 9,41 |
0,34 [IC95%, 0,20 a 0,59] P=0,000072 |
|
PP |
19 |
38 |
NR |
NR |
0,24 [IC95%, 0,13 a 0,45] P=0,0000036 |
NR=no reportado; HR=hazard ratio; TTP=tiempo hasta la progresión del tumor; ITT=intención de tratar; cITT=ITT conservador; PP=por protocolo
*Test Logrank estratificado por actividad funcional_
El efecto del tratamiento fue similar en pacientes con tumores activos funcionalmente (HR = 0,23; IC95%, 0,09 a 0,57) y con tumores inactivos (HR = 0,25; IC95%, 0,10 a 0,59).
Después de 6 meses de tratamiento, se observó enfermedad estable en un 66% de pacientes en el grupo de Sandostatin LAR y 37% de pacientes en el grupo de placebo.
En base al beneficio clínico significativo de Sandostatin LAR observado en este análisis intermedio preestablecido se interrumpió el reclutamiento.
La seguridad de Sandostatin LAR en este ensayo fue consistente con el perfil de seguridad ya establecido. Tratamiento de adenomas pituitarios secretores de TSH
Sandostatin LAR, en inyección i.m. cada 4 semanas, ha mostrado que suprime las hormonas tiroideas elevadas, para normalizar la TSH y para mejorar los signos y síntomas clínicos del hipertiroidismo en pacientes con adenomas secretores de TSH. Los efectos del tratamiento de Sandostatin LAR consiguieron significación estadística respecto al valor basal después de 28 días y un beneficio del tratamiento continuado durante hasta 6 meses.
5.2 Propiedades farmacocinéticas
Después de inyecciones i.m. únicas de Sandostatin LAR, las concentraciones plasmáticas de octreotida alcanzan un pico inicial transitorio 1 hora después de la administración, seguido de una disminución progresiva hasta un nivel de octreotida indetectable al cabo de 24 horas. Después de este pico inicial en el día 1, octreotida se mantiene en niveles subterapéuticos en la mayoría de pacientes durante los 7 días siguientes. Posteriormente, las concentraciones de octreotida aumentan de nuevo, y alcanzan un nivel meseta alrededor del día 14 y se mantienen relativamente constantes durante 3 a 4 semanas. El nivel del pico durante el día 1 es inferior a los niveles durante la fase meseta y durante el día 1 no se produce más del 0,5% de la liberación total del fármaco. Después del día 42 aproximadamente, las concentraciones de octreotida disminuyen lentamente, coincidiendo con la fase de degradación terminal de la matriz del polímero de la forma farmacéutica.
En pacientes con acromegalia, las concentraciones meseta después de dosis únicas de 10 mg, 20 mg y 30 mg de Sandostatin LAR alcanzan 358 ng/L, 926 ng/L, y 1.710 ng/L, respectivamente. Las concentraciones séricas de octreotida en estado estacionario, tras 3 inyecciones a intervalos de 4 semanas,
son superiores en un factor de aproximadamente 1,6 a 1,8 y suponen 1.557 ng/L y 2.384 ng/L después de inyecciones múltiples de 20 mg y 30 mg de Sandostatin LAR, respectivamente.
En pacientes con tumores carcinoides, la media (y mediana) de las concentraciones séricas de octreotida en el estado estacionario después de inyecciones múltiples de 10 mg, 20 mg y 30 mg de Sandostatin LAR administradas a intervalos de 4 semanas también aumentaron linealmente con la dosis y fueron 1.231 (894) ng/L, 2.620 (2.270) ng/L y 3.928 (3,010) ng/L, respectivamente.
No se produjo acumulación de octreotida fuera de lo esperado de los perfiles de liberación superpuestos durante un periodo de hasta 28 inyecciones mensuales de Sandostatin LAR.
El perfil farmacocinético de la octreotida después de la inyección de Sandostatin LAR refleja el perfil de liberación de la matriz del polímero y su biodegradación. Una vez liberada en la circulación sistémica, la octreotida se distribuye de acuerdo con sus propiedades farmacocinéticas conocidas, como se han descrito para la administración subcutánea. El volumen de distribución de la octreotida en estado estacionario es de 0,27 l/kg y el aclaramiento total de 160 ml/min. La unión a proteínas plasmáticas asciende al 65% y fundamentalmente no hay fármaco unido a las células sanguíneas.
En pacientes pediátricos, de 7 a 17 años de edad, con obesidad hipotalámica que recibían Sandostatin LAR 40 mg una vez al mes, se obtuvieron datos farmacocinéticos a partir de la extracción limitada de muestras de sangre que mostraron medias de las concentraciones plasmáticas valle de octreotida de 1395 ng/L tras la primera inyección y de 2973 ng/L en el estado estacionario. Se observó una elevada variabilidad interindividual.
Las concentraciones plasmáticas valle en el estado estacionario no correlacionaban con la edad y con el IMC, pero si correlacionaron moderadamente con el peso corporal (52,3-133 kg) y fueron significativamente diferentes entre hombres y mujeres, es decir aproximadamente un 17% superiores para la población de pacientes femenina.
5.3 Datos preclínicos sobre seguridad
Estudios en animales de toxicología aguda y de dosis repetidas, de genotoxicidad, carcinogenicidad y toxicología de la reproducción revelaron que no existe ningún tema de seguridad específico para humanos.
Los estudios de reproducción en animales no revelaron ninguna evidencia de efectos teratogénicos, embriofetales u otros efectos sobre la reproducción debidos a octreotida a dosis en los progenitores de hasta 1 mg/kg/día. Se observó algún retraso en el crecimiento fisiológico de las crías de las ratas que fue transitorio y atribuible a la inhibición de GH originada por un exceso de actividad farmacodinámica (ver sección 4.6).
No se llevaron a cabo estudios específicos en ratas jóvenes. En los estudios de desarrollo pre y post natal, se observó una reducción en el crecimiento y la maduración en las crías F1 de madres a las que se administró octreotida durante el embarazo completo y el periodo de lactancia. Se observó un retraso en el descenso de los testículos para las crías F1 macho, pero se mantuvo normal la fertilidad de las crías F1 macho afectadas. Por tanto, las observaciones mencionadas anteriormente fueron transitorias y se consideraron consecuencia de una inhibición de la GH.
6 . DATOS FARMACÉUTICOS 6.1 Lista de excipientes
Polvo (Vial):
Poli (DL-láctido-co-glicólido) Manitol (E421)
Disolvente (Jeringa precargada): Carboximetilcelulosa sódica Manitol (E421)
Poloxamer 188
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no se debe mezclar con otros medicamentos.
6.3 Periodo de validez
3 años
El medicamento no se debe conservar después de la reconstitución (se debe utilizar inmediatamente).
6.4 Precauciones especiales de conservación
Conservar en el envase original para protegerlo de la luz.
Conservar en nevera (entre 2 y 8 °C). No congelar.
Sandostatin LAR puede conservarse por debajo de 25°C durante el día de la inyección.
Para consultar las condiciones después de la reconstitución, ver la sección 6.3.
6.5 Naturaleza y contenido del envase
Envases unitarios que contienen un vial de vidrio de 6 ml con un tapón de goma (bromobutilo), sellado con una lengüeta de aluminio, que contienen el polvo para suspensión inyectable y una jeringa precargada de vidrio incoloro de 3 ml con un tapón frontal y un tapón émbolo (goma clorobutilo) con 2 ml de disolvente, envasado conjuntamente en una bandeja blister sellada con un adaptador al vial y una aguja de inyección de seguridad.
Envases múltiples de tres envases unitarios, cada envase unitario conteniendo: un vial de vidrio de 6 ml con tapón de goma (bromobutilo), sellado con una lengüeta de aluminio, que contiene el polvo para suspensión inyectable y una jeringa precargada de vidrio incoloro de 3 ml con un tapón frontal y un tapón émbolo (goma clorobutilo) con 2 ml de disolvente, envasado conjuntamente en una bandeja blister sellada con un adaptador al vial y una aguja de inyección de seguridad.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
Instrucciones para la preparación y la inyección intramuscular de Sandostatin LAR.
SÓLO PARA INYECCIÓN INTRAMUSCULAR
Componentes del kit de inyección:
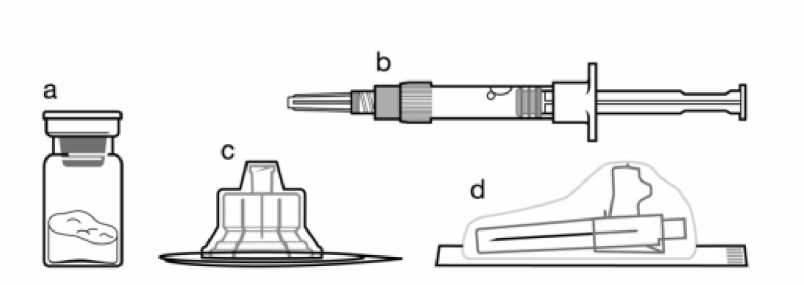
a Un vial que contiene el polvo de Sandostatin LAR b Una jeringa precargada que contiene el disolvente para la reconstitución
c Un adaptador al vial para la reconstitución del medicamento d Una aguja de inyección de seguridad
Siga cuidadosamente las instrucciones que se indican a continuación para asegurar la reconstitución de Sandostatin LAR antes de la inyección intramuscular profunda
Hay tres pasos críticos en el proceso de reconstitución de Sandostatin LAR. Si no se realizan correctamente, podría suponer que no se administre el medicamento de forma adecuada.
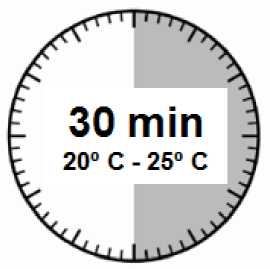
• El kit de inyección debe alcanzar la temperatura ambiente. Sacar el kit de inyección de la nevera y dejar que el kit alcance la temperatura ambiente durante un mínimo de 30 minutos antes de la reconstitución, pero sin que se superen las 24 h.
• Después de añadir la solución del diluyente, dejar reposar el vial durante 5 minutos para asegurar que el polvo queda completamente saturado.
• Después de la saturación, agitar el vial moderadamente en dirección horizontal durante un mínimo de 30 segundos, hasta que se forme una suspensión uniforme. La suspensión de Sandostatin LAR sólo se debe preparar inmediatamente antes de la administración.
Sandostatin LAR debe ser administrado sólo por un profesional sanitario experto.
PASO 1
• Sacar de la nevera el kit de inyección de Sandostatin LAR.
ATENCIÓN: es esencial iniciar el proceso de reconstitución sólo después de que el kit de inyección haya alcanzado la temperatura ambiente. Dejar que el kit alcance la temperatura ambiente durante un mínimo de 30 minutos antes de la reconstitución pero no exceder las 24 h.
Nota: El Kit de inyección puede refrigerarse de nuevo, en caso necesario.
PASO 2
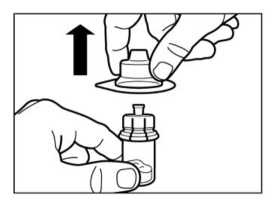
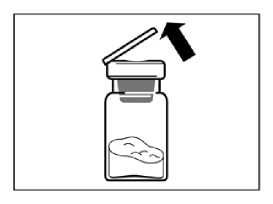
Retirar el tapón de plástico del vial y limpiar el tapón de caucho del vial con una torunda de algodón con alcohol.
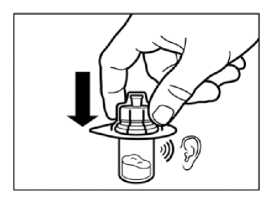
• Retirar la lámina protectora del envoltorio que contiene el adaptador del vial. NO extraer el adaptador del vial de su envoltorio.
• Sujetando el adaptador del vial por su envoltorio, colocarlo en la parte superior del vial y apretarlo completamente hacia abajo para que encaje encima del vial. Esto lo confirmará cuando escuche un “click”.
• Retirar el envoltorio del adaptador del vial con un movimiento vertical.
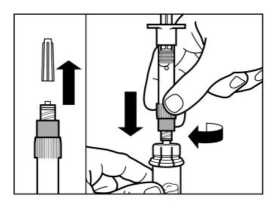
PASO 3
• Retirar el tapón de la jeringa precargada que contiene el disolvente y enroscar la jeringa en el adaptador de vial.
• Presionar lentamente el émbolo hasta el fondo hasta transferir toda la solución diluyente al vial.
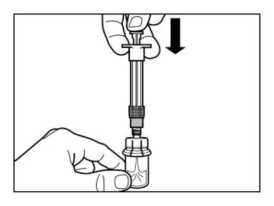
PASO 4
ATENCIÓN: Es esencial dejar reposar el vial durante 5 minutos
para asegurar que el disolvente ha saturado completamente el polvo.
Nota: es normal que el émbolo se desplace hacia arriba, ya que puede haber una sobrepresión en el vial.
• En este punto, preparar al paciente para administrarle la inyección.
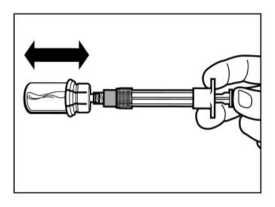
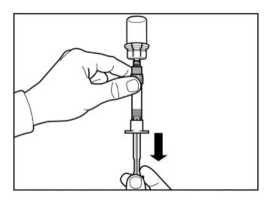
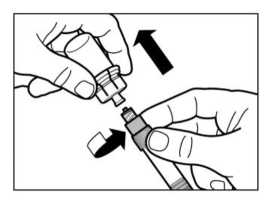
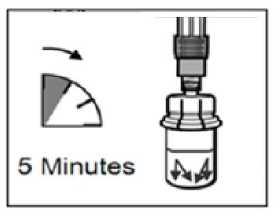
PASO 5
• Después del periodo de saturación, asegurarse que se presiona el émbolo hacia el fondo de la jeringa.
ATENCIÓN: Mantener el émbolo presionado y agitar el vial moderadamente en sentido horizontal durante un mínimo de 30 segundos para que el polvo esté completamente suspendido en el disolvente (suspensión uniforme de consistencia lechosa). Repetir la agitación moderada durante otros 30 segundos más si el polvo no está completamente suspendido.
PASO 6
• Girar la jeringa y el vial boca abajo, y lentamente tirar del émbolo hasta transferir el contenido completo del vial a la jeringa.
• Desenroscar la jeringa del adaptador del vial.
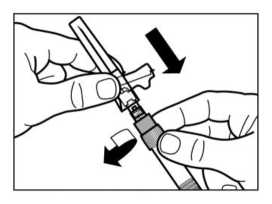
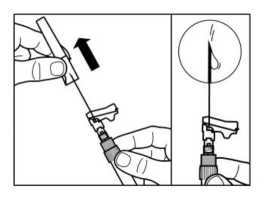
PASO 7
• Enroscar la aguja de inyección de seguridad en la jeringa.
• Si se retrasa la administración inmediata, re-agitar moderadamente la jeringa para asegurar la suspensión uniforme, de consistencia lechosa.
• Preparar el lugar de inyección con una torunda de alcohol
• Retirar la cubierta protectora de la aguj a.
• Golpear suavemente la jeringa para desplazar cualquier burbuja visible y expulsarla fuera de la jeringa.
• Pasar inmediatamente a la Etapa 8 para la administración al paciente. Cualquier retraso en la administración puede producir una sedimentación.
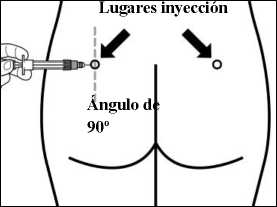
PASO 8
• Sandostatin LAR debe administrarse sólo mediante inyección intramuscular profunda, NUNCA se debe administrar por vía intravenosa.
• Insertar la aguja completamente en el glúteo derecho o izquierdo, formando un ángulo de 90° con la piel.
• Lentamente aspirar con el émbolo para comprobar que no se ha penetrado un vaso sanguíneo (si fuera así, cambiar la zona de punción).
• Utilizando una presión constante sobre el émbolo, administrar lentamente hasta que la jeringa esté vacía. Extraer la aguja del lugar de inyección y activar el mecanismo de seguridad (tal como se muestra en el Paso 9, a continuación).
PASO 9
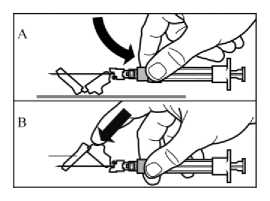
• Activar el mecanismo de seguridad sobre la aguja, utilizando uno de los dos métodos siguientes:
o bien presionando la sección articulada del dispositivo de seguridad hacia abajo sobre una superficie rígida (figura A)
o bien presionando la parte articulada con un dedo (figura B)
• El sonido de un “click” confirma la activación adecuada.
• Desechar la jeringa inmediatamente (en un contenedor de objetos punzantes).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Farmacéutica, S.A.
Gran Via de les Corts Catalanes, 764 08013 Barcelona
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
Sandostatin LAR 10 mg, polvo y disolvente para suspensión inyectable: 62.139 Sandostatin LAR 20 mg, polvo y disolvente para suspensión inyectable: 62.140 Sandostatin LAR 30 mg, polvo y disolvente para suspensión inyectable: 62.141
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
15 Octubre 1998
10. FECHA DE LA REVISIÓN DEL TEXTO 03/2016
La información detallada de este medicamento está disponible en la página web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.gob.es/
18 de 18