Rixubis 500Ui/Vial Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8 en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
RIXUBIS 250 UI Polvo y disolvente para solución inyectable RIXUBIS 500 UI Polvo y disolvente para solución inyectable RIXUBIS 1000 UI Polvo y disolvente para solución inyectable RIXUBIS 2000 UI Polvo y disolvente para solución inyectable RIXUBIS 3000 UI Polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
RIXUBIS 250 UI Polvo y disolvente para solución inyectable
Un vial contiene nominalmente 250 UI de nonacog gamma, factor IX humano de coagulación recombinante (ADNr), correspondiente a una concentración de 50 UI/ml tras su reconstitución con 5 ml de disolvente.
RIXUBIS 500 UI Polvo y disolvente para solución inyectable
Un vial contiene nominalmente 500 UI de nonacog gamma, factor IX humano de coagulación recombinante (ADNr), correspondiente a una concentración de 100 UI/ml tras su reconstitución con 5 ml de disolvente.
RIXUBIS 1000 UI Polvo y disolvente para solución inyectable
Un vial contiene nominalmente 1000 UI de nonacog gamma, factor IX humano de coagulación recombinante (ADNr), correspondiente a una concentración de 200 UI/ml tras su reconstitución con 5 ml de disolvente.
RIXUBIS 2000 UI Polvo y disolvente para solución inyectable
Un vial contiene nominalmente 2000 UI de nonacog gamma, factor IX humano de coagulación recombinante (ADNr), correspondiente a una concentración de 400 UI/ml tras su reconstitución con 5 ml de disolvente.
RIXUBIS 3000 UI Polvo y disolvente para solución inyectable
Un vial contiene nominalmente 3000 UI de nonacog gamma, factor IX humano de coagulación recombinante (ADNr), correspondiente a una concentración de 600 UI/ml tras su reconstitución con 5 ml de disolvente.
La potencia (UI) se determina utilizando el ensayo de coagulación de una fase de la Farmacopea Europea. La actividad específica de RIXUBIS es de aproximadamente 200-390 UI/mg de proteína.
Nonacog gamma (factor de coagulación recombinante IX) es una glicoproteína purificada de una sola cadena que contiene 415 aminoácidos. Se produce mediante tecnología de ADN recombinante en una línea celular de ovario de hámster chino (CHO).
Excipiente(s) con efecto conocido:
Un vial contiene 19 mg de sodio.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
El polvo es de color blanco o casi blanco. El disolvente es transparente e incoloro.
DATOS CLÍNICOS
4.
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia B (deficiencia congénita de factor IX).
RIXUBIS está indicado en pacientes de todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento se debe realizar bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Supervisión del tratamiento
Durante el tratamiento, se recomienda la determinación adecuada de los niveles de factor IX para calcular la dosis que se debe administrar y la frecuencia de las perfusiones repetidas. Los pacientes individuales pueden diferir en su respuesta al factor IX con diferentes semividas y recuperaciones.
La dosis basada en el peso corporal puede requerir un ajuste en pacientes con bajo peso o sobrepeso. En el caso particular de intervenciones quirúrgicas importantes, es indispensable una supervisión precisa de la terapia de sustitución mediante análisis de la coagulación (actividad del factor IX de plasma).
Para garantizar que se ha alcanzado el nivel plasmático de actividad de factor IX deseado, se aconseja realizar un control exhaustivo utilizando un ensayo adecuado de actividad de factor IX y, si es necesario, se deben aplicar los ajustes adecuados a la dosis y la frecuencia de las perfusiones repetidas. Al utilizar el ensayo in vitro de coagulación en una etapa basado en el tiempo de la tromboplastina (aPTT) para determinar la actividad del factor IX en muestras sanguíneas de pacientes, los resultados de actividad del factor IX pueden verse significativamente afectados por el tipo de reactivo de aPTT y el estándar de referencia utilizado en el ensayo. Esto es importante especialmente al cambiar el laboratorio y/o los reactivos utilizados en el ensayo.
Posología
La dosis y la duración de la terapia de sustitución depende de la gravedad de la deficiencia de factor IX, de la ubicación y la extensión de la hemorragia, así como el estado clínico, la edad y los parámetros farmacocinéticos de factor IX del paciente, como la recuperación incremental y semivida.
El número de unidades de factor IX administradas se expresa en unidades internacionales (UI), que están relacionadas con el estándar actual de la OMS para productos de factor IX. La actividad de factor IX en el plasma se expresa como un porcentaje (relativo al plasma humano normal) o en unidades internacionales (relativas a un estándar internacional para el factor IX en el plasma).
Una unidad internacional (UI) de actividad de factor IX es equivalente a la cantidad de factor IX existente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor IX se basa en el hallazgo empírico de que 1 unidad internacional (UI) de factor IX por kg de peso corporal incrementa la actividad de factor IX del plasma en 0,9 UI/dl (intervalo de 0,5 a 1,4 UI/dl) o 0,9% de actividad normal en pacientes de 12 años de edad y mayores (para obtener información adicional, ver sección 5.2).
La dosis necesaria se determina utilizando la siguiente fórmula:
Pacientes de 12 años de edad y mayores
Unidades = peso corporal x aumento deseado de x recíproco de
requeridas (kg) factor IX recuperación observada
(%) o (UI/dl) (dl/kg)
Para una recuperación incremental de 0,9 UI/dl por UI/kg, la dosis se calcula de la siguiente manera:
Unidades = peso corporal x aumento deseado de x 1,1 dl/kg
requeridas (kg) factor IX
(%) o (UI/dl)
La cantidad que se debe administrar y la frecuencia de la administración deben estar siempre orientadas a la eficacia clínica en el caso concreto.
En el caso de los episodios hemorrágicos siguientes, la actividad de factor IX no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. Se puede utilizar la siguiente tabla como guía de dosificación en episodios hemorrágicos y cirugía:
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de factor IX requerido (%) o (UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia Hemartrosis incipiente o hemorragia muscular u oral |
20 - 40 |
Repetir cada 24 horas. Al menos 1 día, hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma |
30 - 60 |
Repetir perfusión cada 24 horas durante 3 - 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir perfusión cada 8 a 24 horas hasta superar el peligro. |
|
Cirugía Cirugía menor incluyendo extracción dental |
30 - 60 |
Cada 24 horas, al menos 1 día, hasta lograr la curación. |
|
Operación importante |
80 - 100 (pre y postoperatorio) |
Repetir perfusión cada 8 a 24 horas hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor IX del 30% al 60% (UI/dl). |
Es especialmente importante una supervisión cuidadosa de la terapia de sustitución en los casos de operación importante o hemorragia potencialmente mortal.
Profilaxis
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia B grave, las dosis normales son de 40 a 60 UI de factor IX por kilogramo de peso corporal a intervalos de 3 a 4 días para los pacientes de 12 años de edad y mayores. En algunos casos, dependiendo de los parámetros farmacocinéticos, la edad, el fenotipo de hemorragia y la actividad física del paciente, es posible que se necesiten intervalos de dosificación más cortos o dosis más altas.
Perfusión continua
No administre RIXUBIS mediante perfusión continua.
Población _ pediátrica Tratamiento a demanda:
El cálculo de la dosis de factor IX requerida se basa en el hallazgo empírico de que 1 unidad internacional (UI) de factor IX por kg de peso corporal incrementa la actividad de factor IX del plasma en 0,7 UI/dl (intervalo de 0,31 a 1,0 UI/dl) o 0,7% de la actividad normal en pacientes de menos de 12 años de edad (para obtener información adicional, ver sección 5.2).
La dosificación requerida se determina mediante la siguiente fórmula:
Pacientes de menos de 12 años de edad:
Unidades = peso corporal (kg) x aumento deseado de x recíproco de recuperación requeridas factor IX observada (dl/kg)
(%) o (UI/dl)
Para una recuperación incremental de 0,7 UI/dl por UI/kg, la dosis se calcula de la siguiente manera:
Unidades = peso corporal (kg) x aumento deseado de x 1,4 dl/kg requeridas factor IX
(%) o (UI/dl)
Se puede utilizar la misma tabla para los adultos como guía de dosificación en episodios hemorrágicos y cirugía (ver anterior).
Profilaxis:
El intervalo de dosis recomendadas para pacientes pediátricos de menos de 12 años de edad es de 40 a 80 UI/kg a intervalos de 3 a 4 días. En algunos casos, dependiendo de los parámetros farmacocinéticos, la edad, el fenotipo de la hemorragia y la actividad física del paciente, es posible que se necesiten intervalos de dosificación más cortos o dosis más altas.
Forma de administración
Vía intravenosa.
En caso de autoadministración o administración por un cuidador, es necesario realizar un entrenamiento adecuado.
RIXUBIS se debe administrar a una velocidad que garantice la comodidad del paciente, hasta un máximo de 10 ml/min.
Después de la reconstitución, la solución es transparente, incolora y libre de partículas extrañas y tiene un pH de entre 6,8 y 7,2. La osmolalidad es superior a 240 m osmol/kg.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
Solo se deben utilizar jeringas luer lock de plástico con este producto.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Reacción alérgica conocida a la proteína de hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad:
Con RIXUBIS se han notificado reacciones de hipersensibilidad de tipo alérgico. El medicamento contiene trazas de proteínas de hámster. Se debe informar a los pacientes o sus cuidadores de que, en caso de que ocurran síntomas de hipersensibilidad, deben interrumpir el tratamiento y consultar con su médico de forma inmediata. Se debe informar a los pacientes de los primeros signos de las reacciones de hipersensibilidad de tipo inmediato que incluyen habón urticarial, urticaria generalizada, tirantez en el pecho, sibilancia, hipotensión y anafilaxia.
El riesgo es mayor durante las primeras fases de la exposición inicial a los concentrados de factor IX en pacientes sin tratamiento previo, en particular en pacientes con mutaciones de un gen de alto riesgo. En las publicaciones médicas se ha informado de casos que muestran una asociación entre la aparición de un inhibidor de factor IX y reacciones alérgicas, en particular en pacientes con mutaciones de un gen de alto riesgo. Por tanto, se debe evaluar la presencia de un inhibidor en los pacientes que experimenten reacciones alérgicas.
En caso de que se produzca un shock, se deben seguir las pautas médicas estándar para su tratamiento. Inhibidores:
Tras el tratamiento repetido con productos de factor IX humano de coagulación recombinante (ADNr), debe controlarse a los pacientes para ver si han desarrollado anticuerpos neutralizantes (inhibidores) que deben cuantificarse en unidades Bethesda (BU) mediante ensayos biológicos adecuados.
En las publicaciones médicas se ha informado de casos que muestran una correlación entre la aparición de un inhibidor de factor IX y reacciones alérgicas. Por tanto, se debe evaluar la presencia de un inhibidor en los pacientes que experimenten reacciones alérgicas. Debe señalarse que los pacientes con inhibidores de factor IX pueden tener un mayor riesgo de anafilaxia con la exposición posterior al factor IX.
Debido al riesgo de reacciones alérgicas con concentrados de factor IX, las administraciones iniciales de factor IX deben, según el criterio del médico encargado del tratamiento, realizarse bajo observación médica en un lugar donde puedan tomarse las medidas médicas adecuadas para reacciones alérgicas.
Síndrome nefrótico:
Se ha notificado síndrome nefrótico tras intentos de inducción de inmunotolerancia en pacientes con hemofilia B con inhibidores de factor IX.
Tromboembolia:
Dado el posible riesgo de complicaciones trombóticas, debe establecerse la vigilancia clínica adecuada para detectar los primeros signos de coagulopatía de consumo y trombótica con los ensayos biológicos adecuados cuando se administre este producto a pacientes con trastornos hepáticos, pacientes tras una intervención quirúrgica, recién nacidos o pacientes con riesgo de fenómenos trombóticos o CID.
En cada una de estas situaciones, el beneficio del tratamiento con RIXUBIS se debe sopesar frente al riesgo de estas complicaciones.
Eventos cardiovasculares
En pacientes con factores de riesgo cardiovascular, la terapia de sustitución con FIX puede aumentar el riesgo cardiovascular.
Complicaciones relacionadas con el catéter
Si se requiere un dispositivo de acceso venoso (CVAD) se debe considerar el riesgo de complicaciones relacionadas con CVAD incluyendo infecciones locales, bacteremia y trombosis del lado del catéter.
Consideraciones relativas al excipiente
Después de la reconstitución, este medicamento contiene 0,83 mmol de sodio (19 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre RIXUBIS a un paciente para poder mantener una relación entre el paciente y el lote del medicamento.
Población pediátrica
Las advertencias y precauciones incluidas se aplican tanto a adultos como a niños.
Personas de edad avanzada
Los ensayos clínicos de RIXUBIS no incluyeron sujetos con 65 años o más. Se desconoce si estos sujetos responden de manera diferente a los sujetos de menor edad. Al igual que ocurre con todos los pacientes, la selección de la dosis para un paciente de edad avanzada se debe individualizar.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han notificado interacciones de productos de factor IX humano de coagulación (ADNr) con otros medicamentos.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor IX. Dados los raros casos de hemofilia B en mujeres, no se dispone de experiencia relacionada con el uso de factor IX durante el embarazo y el periodo de lactancia. Por lo tanto, solo debe utilizarse factor IX durante el embarazo y el periodo de lactancia si está claramente indicado.
No hay información sobre los efectos de factor IX sobre la fertilidad.
4.7 Efectos sobre la capacidad de conducir y utilizar máquinas
La influencia de RIXUBIS sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas Resumen del perfil de seguridad
La hipersensibilidad y las reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el lugar de infusión, escalofríos, rubefacción, urticaria generalizada, dolor de cabeza, habón urticarial, hipotensión, letargia, náuseas, inquietud, taquicardia, tirantez en el pecho, cosquilleo, vómitos, sibilancia) se observaron con poca frecuencia pero, en algunos casos, pueden progresar a anafilaxia grave (incluyendo shock). En algunos casos, estas reacciones han progresado a anafilaxia grave y se han producido con una estrecha asociación temporal con el desarrollo de inhibidores de factor IX (ver también sección 4.4).
Se ha notificado síndrome nefrótico tras intentos de inducción de inmunotolerancia en pacientes con hemofilia B con inhibidores de factor IX y antecedentes de reacciones alérgicas.
Se ha observado de forma muy rara el desarrollo de anticuerpos a la proteína de hámster con reacciones de hipersensibilidad relacionadas.
Los pacientes con hemofilia B pueden desarrollar anticuerpos neutralizantes (inhibidores) frente al factor IX. Si aparecen tales inhibidores, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia.
Existe un posible riesgo de episodios tromboembólicos tras la administración de productos de factor IX, con un mayor riesgo para las preparaciones de menor pureza. El uso de productos de factor IX de baja pureza se ha asociado a casos de infarto de miocardio, coagulación intravascular diseminada, trombosis venosa y embolia pulmonar. El uso de factor IX de alta pureza raramente está asociado con tales reacciones adversas.
Tabla de reacciones adversas
Los ensayos clínicos con RIXUBIS incluyeron 99 sujetos con al menos una exposición a RIXUBIS y registraron un total de 5 reacciones adversas. La tabla que se incluye a continuación sigue la clasificación de órganos del sistema MedDRA (COS y Nivel de término preferido).
Las frecuencias se han evaluado utilizando el siguiente criterio: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raros (>1/10.000 a <1/1.000), muy raros (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Reacciones adversas al medicamento, de ensayos clínicos y comunicaciones espontáneas | ||
|
Clasificación de órganos del sistema MedDRA |
Reacciones adversas |
Frecuencia por paciente |
|
Trastornos del sistema inmunológico |
Hipersensibilidad a) |
Frecuencia no conocida |
|
Trastornos del sistema nervioso |
Disgeusia |
Frecuentes |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor en una extremidad |
Frecuentes |
a) RAM explicadas en la sección siguiente.
Descripción de reacciones adversas seleccionadas
Hipersensibilidad
Se han manifestado reacciones de tipo alérgico por disnea, prurito, urticaria generalizada y erupción. Población pediátrica
Se espera que la frecuencia, el tipo y la gravedad de las reacciones adversas en niños sean las mismas que las de los adultos. No obstante, no se disponen de datos sobre pacientes sin tratamiento previo, ya que en los estudios clínicos únicamente participaron pacientes previamente tratados; no se realizó por tanto ninguna investigación sobre inmunogenicidad o desarrollo de inhibidores en esta población en riesgo.
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación pertinente incluido en el Anexo V.
4.9 Sobredosis
No se han caracterizado los efectos de dosis de RIXUBIS superiores a las recomendadas.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos, factor IX de coagulación. Código ATC: B02BD04.
RIXUBIS contiene factor IX de coagulación recombinante (nonacog gamma). El factor IX es una glicoproteína de una sola cadena con un peso molecular de aproximadamente 68.000 Dalton. Es un factor de coagulación dependiente de la vitamina K y lo sintetiza el hígado. El factor IX lo activa el factor XIa en la vía de coagulación intrínseca y el complejo factor VII/factor de tejido en la vía de coagulación extrínseca. El factor IX activado, en combinación con el factor VIII activado, activa el factor X. El factor X activado convierte la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina, con lo que se forma un coágulo.
La hemofilia B es un trastorno hereditario de la coagulación sanguínea ligada al sexo y producida por la disminución de los niveles de factor IX que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, se aumentan los niveles de factor IX del plasma, consiguiendo una corrección temporal de la deficiencia de factor y una corrección de las tendencias hemorrágicas.
Eficacia clínica y seguridad:
Profilaxis y control de las hemorragias en pacientes previamente tratados de 12 años de edad y mayores:
La eficacia de RIXUBIS se evaluó en la parte abierta y no controlada de un estudio combinado de fases 1/3, donde se administró RIXUBIS a un total de 73 pacientes masculinos previamente tratados de edades entre 12 y 59 años, para la profilaxis y/o el tratamiento de episodios hemorrágicos a demanda. Todos los sujetos presentaban hemofilia B grave (nivel de factor IX < 1%) o moderadamente grave (nivel de factor IX < 2%). Se administró RIXUBIS a cincuenta y nueve pacientes previamente tratados para profilaxis. Cincuenta y seis de estos pacientes previamente tratados a los que se administró RIXUBIS durante un mínimo de 3 meses se incluyeron en la evaluación de la eficacia para la profilaxis. Se administró RIXUBIS a 14 pacientes previamente tratados adicionales únicamente para el tratamiento de episodios hemorrágicos. Los sujetos de la cohorte a demanda debían tener como mínimo 12 episodios hemorrágicos documentados que necesitasen tratamiento en un plazo de 12 meses previo a la inscripción. La duración media del tratamiento en la cohorte a demanda fue de 3,5±1,00 meses (mediana 3,4, oscilando entre 1,2 y 5,1 meses), la tasa de hemorragia anual media total fue de 33,9±17,37 con una mediana de 27,0, oscilando entre 12,9 y 73,1.
La mediana de la tasa de hemorragia anual para la profilaxis con RIXUBIS para todas las hemorragias fue de 2,0, para las hemorragias espontáneas 0,0 y para las hemorragias articulares 0,0. 24 sujetos (42,9%) no experimentaron ninguna hemorragia.
Se trataron un total de 249 episodios hemorrágicos con RIXUBIS, de los cuales 197 fueron hemorragias articulares y 52 hemorragias no articulares (tejido blando, músculo, cavidad corporal, intracraneal y otras). De un total de 249 episodios hemorrágicos, 163 fueron moderados, 71 fueron menores y 15 fueron mayores. El tratamiento se individualizó en función de la gravedad, la causa y el lugar de la hemorragia. De los 249 episodios hemorrágicos, la mayoría (211; 84,7%) se trataron con 1-2 perfusiones. La eficacia hemostática en la resolución de la hemorragia se consideró excelente o buena en el 96% de todos los episodios hemorrágicos tratados.
Profilaxis y control de las hemorragias en pacientes previamente tratados de menos de 12 años de edad:
La eficacia de RIXUBIS se evaluó en un estudio combinado de fases 2/3, en el que se administró RIXUBIS a un total de 23 pacientes masculinos previamente tratados de edades entre 1,8 y 11,8 años (mediana de la edad, 7,10 años) con 11 pacientes < 6 años, para la profilaxis y el control de episodios hemorrágicos. Todos los sujetos presentaban hemofilia B grave (nivel de factor IX < 1%) o moderadamente grave (nivel de factor IX < 2%). Los 23 sujetos recibieron tratamiento profiláctico con RIXUBIS durante un mínimo de 3 meses y se incluyeron en la evaluación de la eficacia para la profilaxis.
La mediana de la tasa de hemorragia anual fue de 2,0, para las hemorragias espontáneas 0,0 y para las hemorragias articulares 0,0.
Nueve sujetos (39,1%) no experimentaron ninguna hemorragia.
Se trataron un total de 26 episodios hemorrágicos con RIXUBIS, de los cuales 23 hemorragias se debieron a lesiones, 2 espontáneas y 1 de origen desconocido. 19 hemorragias fueron no articulares (tejido blando, músculo, cavidad corporal, intracraneal y otras) y 7 fueron hemorragias articulares de las que 1 fue una hemorragia en una articulación de destino. De los 26 episodios hemorrágicos, 15 fueron menores, 9 moderados y 2 mayores. El tratamiento se individualizó en función de la gravedad, la causa y el lugar de la hemorragia. La mayoría (23; 88,5%) se trataron con 1-2 perfusiones. La eficacia hemostática en la resolución de la hemorragia se consideró excelente o buena en el 96,2% de todos los episodios hemorrágicos tratados.
Control perioperatorio:
La seguridad y la eficacia en el entorno perioperatorio se evaluaron en un estudio de fase3 no controlado, al descubierto, prospectivo y realizado en varios centros con pacientes masculinos previamente tratados con hemofilia B grave y moderadamente grave mediante RIXUBIS. El análisis de eficacia según el protocolo incluye 37 intervenciones quirúrgicas realizadas en 27 pacientes de edades entre 17 y 57 años sometidos a intervenciones quirúrgicas mayores o menores, dentales y otros procedimientos quirúrgicos invasivos. Veinte procedimientos fueron mayores, incluidas 13 intervenciones ortopédicas y 3 dentales. 17 procedimientos, incluidas 10 extracciones dentales, se consideraron menores. Los pacientes sometidos a intervenciones quirúrgicas mayores tuvieron que realizar una evaluación farmacocinética. A todos los pacientes se les administró una dosis basada en su recuperación incremental individual más reciente. La dosis de carga inicial recomendada de RIXUBIS se estableció para garantizar que durante la intervención se mantenían niveles de actividad de factor IX de 80-100% en el caso de intervenciones quirúrgicas mayores y 30-60% para intervenciones quirúrgicas menores. RIXUBIS se administró mediante perfusión en bolo.
La hemostasia se mantuvo durante toda la duración del estudio.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con RIXUBIS en pacientes sin tratamiento previo para el tratamiento y la profilaxis de las hemorragias en pacientes con hemofilia B (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Pacientes previamente tratados de 12 años de edad y mayores:
Se llevó a cabo un estudio de farmacocinética aleatorizado, ciego, controlado y con grupos cruzados de RIXUBIS y un comparador con sujetos masculinos sin hemorragias (edad > 15 años) como parte del estudio esencial combinado de fases 1/3. Se administró a los sujetos uno de los productos mediante una única perfusión intravenosa. La media (± DE) y la mediana de la dosis de RIXUBIS en el conjunto de análisis según el protocolo (n = 25) fue de 74,69 ± 2,37 y 74,25 UI/kg, respectivamente, con un intervalo de 71,27 a 79,38 UI/kg. Los parámetros farmacocinéticos se calcularon a partir de las medidas de actividad de factor IX en muestras de sangre obtenidas en un plazo máximo de 72 horas después de cada perfusión.
La evaluación de la farmacocinética se repitió en un estudio al descubierto y no controlado con RIXUBIS en sujetos masculinos que participaron en el estudio inicial de farmacocinética con grupos cruzados y a los que se administró profilaxis con RIXUBIS durante 26 ± 1 semanas (media ± desviación estándar) y acumularon como mínimo 30 días de exposición a RIXUBIS. El intervalo de dosis de RIXUBIS en el estudio de farmacocinética de repetición fue de 64,48 a 79,18 UI/kg (n = 23).
En la tabla siguiente se indican los parámetros farmacocinéticos de los sujetos evaluables (análisis según el protocolo).
|
Parámetro |
RIXUBIS Estudio inicial con grupos cruzados (N = 25) |
RIXUBIS Evaluación repetida (N = 23) |
|
Área bajo la curvao_72 h (UI.h/dl)a Media ± DE Mediana (intervalo) |
1067,81±238,42 1108,35 (696,07-1571,16) |
1156,15±259,44 1170,26 (753,85-1626,81) |
|
Recuperación incremental a Cmáx (UI/dl:UI/kg)b Media ± DE Mediana (intervalo) |
0,87±0,22 0,88 (0,53-1,35) |
0,95±0,25 0,93 (0,52-1,38) |
|
Semivida (h) Media ± DE Mediana (intervalo) |
26,70±9,55 24,58 (15,83-52,34) |
25,36±6,86 24,59 (16,24-42,20) |
|
Cmáx (UI/dl) Media ± DE Mediana (intervalo) |
66,22±15,80 68,10 (41,70-100,30) |
72,75±19,73 72,40 (38,50-106,30) |
|
Tiempo de residencia medio (h) Media ± DE Mediana (intervalo) |
30,82±7,26 28,93 (22,25-47,78) |
29,88±4,16 29,04 (21,32-37,52) |
|
Vssc (dl/kg) Media ± DE Mediana (intervalo) |
2,02±0,77 1,72 (1,10-3,94) |
1,79±0,45 1,74 (1,12-2,72) |
|
Aclaramiento (dl/(kg.h)) Media ± DE Mediana (intervalo) |
0,0644±0,0133 0,0622 (0,0426-0,0912) |
0,0602±0,0146 0,0576 (0,0413-0,0945) |
|
a Área bajo la curva de concentración en plasma-tiempo desde e |
l momento 0-72 horas tras la | |
b
perfusión.
Calculada como (Cmáx valor basal de factor IX) dividida por la dosis en UI/kg, donde Cmáx es la medida máxima de factor IX tras la perfusión.
Volumen de distribución en estado estacionario
c
La recuperación incremental 30 minutos después de la perfusión se determinó para todos los sujetos del estudio combinado de fases 1/3 en el día de exposición 1, en las visitas de las semanas 5, 13 y 26, y en el momento de la terminación o finalización del estudio, si no coincidía con la visita de la semana 26. Los datos demuestran que la recuperación incremental es constante con el tiempo (ver la siguiente tabla).
|
Día de exposición 1 (N = 73) |
Semana 5 (N = 71) |
Semana 13 (N = 68) |
Semana 26 (N = 55) |
En la finalización / terminación del estudiob (N = 23) | |
|
Recuperación incremental 30 min después de la perfusión (UI/dl: UI/kg)a Media ± DE Mediana (intervalo) |
0,79±0,20 0,78 (0,26-1,35) |
0,83±0,21 0,79 (0,46-1,48) |
0,85±0,25 0,83 (0,14-1,47) |
0,89±0,12 0,88 (0,52-1,29) |
0,87±0,20 0,89 (0,52-1,32) |
a
b
Calculada como (C30 min valor basal de factor IX) dividida por la dosis en Ul/kg, donde C30 mm es la medida de factor IX 30 minutos después de la perfusión.
Si no coincide con la visita de la semana 26.
Población pediátrica (pacientes previamente tratados de menos de 12 años de edad)
Los 23 sujetos masculinos se sometieron a una evaluación farmacocinética inicial de RIXUBIS en un estado sin hemorragias como parte del estudio pediátrico combinado de fases 2/3. Los sujetos se aleatorizaron a una de dos secuencias de recogida de sangre para reducir la carga de las frecuentes extracciones de sangre de los diferentes sujetos. La media (± DE) y la mediana de la dosis de RIXUBIS en el conjunto de análisis completo (n = 23) fue de 75,50 ± 3,016 y 75,25 UI/kg, respectivamente, con un intervalo de 70,0 a 83,6 UI/kg. Los parámetros farmacocinéticos se calcularon a partir de las medidas de actividad de factor IX en muestras de sangre obtenidas en un plazo máximo de 72 horas después de la perfusión.
En la tabla siguiente se indican los parámetros farmacocinéticos de todos los sujetos (conjunto de análisis completo).
|
Parámetro |
< 6 años (N = 11) |
6 - < 12 años (N = 12) |
Todos (N = 23) |
|
Área bajo la curvainf (UI.h/dl)a Media ± DE Mediana (intervalo) |
723,7 ± 119,00 717,2 (488-947) |
886,0 ± 133,66 863,7 (730-1138) |
808,4 ± 149,14 802,9 (488-1138) |
|
Semivida (h) Media ± DE Mediana (intervalo) |
27,67 ± 2,66 27,28 (24,0-32,2) |
23,15 ± 1,58 22,65 (21,8-27,4) |
25,31 ± 3,13 24,48 (21,8-32,2) |
|
Tiempo de residencia medio (h) Media ± DE Mediana (intervalo) |
30,62 ±3,27 30,08 (26,2-36,2) |
25,31 ± 1,83 24,74 (23,7-30,3) |
27,85 ± 3,73 26,77 (23,7-36,2) |
|
Vssb (dl/kg) Media ± DE Mediana (intervalo) |
3,22 ± 0,52 3,16 (2,65-4,42) |
2,21 ± 0,32 2,185 (1,70-2,70) |
2,7 ± 0,67 2,69 (1,70-4,42) |
|
Aclaramiento (dl/(kg.h)) Media ± DE Mediana (intervalo) |
0,1058 ± 0,01650 0,1050 (0,081-0,144) |
0,0874 ± 0,01213 0,0863 (0,069-0,108) |
0,0962 ± 0,01689 0,0935 (0,069-0,144) |
Área bajo la curva de concentración en plasma-tiempo desde el momento 0 hasta el infinito.
b
Volumen de distribución en estado estacionario
La recuperación incremental 30 minutos después de la perfusión se determinó para todos los sujetos del estudio combinado de fases 2/3 en la evaluación farmacocinética inicial (día de exposición 1), en las visitas de las semanas 5, 13 y 26, y en el momento de la terminación o finalización del estudio, si no coincidía con la visita de la semana 26. Los datos demuestran que la recuperación incremental es constante con el tiempo en todos los grupos de edad pediátrica. Ver tablas siguientes.
Recuperación incremental para RIXUBIS 30 minutos después de la perfusión, en ambos grupos de edad pediátrica:
|
Recuperación incremental 30 minutos después de la perfusión |
PK (ED 1) Todos (N = 22) |
Semana 5 Todos (N = 23) |
Semana 13 Todos (N = 21) |
Semana 26 Todos (N = 21) |
|
(UI/dl: UI/kg)a Media ± DE Mediana (intervalo) |
0,67 ±0,16 0,69 (0,31 - 1,00) |
0,68 ± 0,12 0,66 (0,48 - 0,92) |
0,71 ± 0,13 0,66 (0,51-1,00) |
0,72 ± 0,15 0,734 (0,51-1,01) |
|
a Calculada como (C30 min valor basal de la medida de factor IX 30 minutos desp Recuperación incremental para RIXUBIS 30 < 6 años: |
actor IX) dividida por la dosis en UI/kg, donde C30 min es ués de la perfusión. minutos después de la perfusión, pacientes pediátricos | |||
|
Recuperación incremental 30 minutos después de la perfusión |
PK (ED 1) Todos (N = 10) |
Semana 5 Todos (N = 11) |
Semana 13 Todos (N = 10) |
Semana 26 Todos (N = 10) |
|
(UI/dl: UI/kg)a Media ± DE Mediana (intervalo) |
0,59 ± 0,13 0,59 (0,31-0,75) |
0,63 ± 0,10 0,6 (0,49-0,80) |
0,68 ± 0,12 0,66 (0,51-0,84) |
0,65 ± 0,13 0,61 (0,51-0,84) |
|
a Calculada como (C30 min valor basal de la medida de factor IX 30 minutos desp Recuperación incremental para RIXUBIS 30 de 6 a < 12 años: |
actor IX) dividida por la dosis en UI/kg, donde C30 min es ués de la perfusión. minutos después de la perfusión, pacientes pediátricos | |||
|
Recuperación incremental 30 minutos después de la perfusión |
PK (ED 1) Todos (N = 12) |
Semana 5 Todos (N = 12) |
Semana 13 Todos (N = 11) |
Semana 26 Todos (N = 11) |
|
(UI/dl: UI/kg)a Media ± DE Mediana (intervalo) |
0,73 ± 0,16 0,71 (0,51-1,00) |
0,73 ± 0,13 0,70 (0,48-0,92) |
0,73 ± 0,14 0,70 (0,54 - 1,00) |
0,8 ± 0,14 0,78 (0,56-1,01) |
|
a Calculada como (C30 min valor basal de |
actor IX) dividida por la dosis en UI/kg, donde C30 min es | |||
la medida de factor IX 30 minutos después de la perfusión.
5.3 Datos preclínicos sobre seguridad
RIXUBIS no tuvo efectos trombogénicos a una dosis de 750 UI/kg en un modelo de estasis de conejo (test de Wessler).
RIXUBIS no causó ninguna reacción adversa clínica, respiratoria o cardiovascular hasta 450 UI/kg en monos cynomolgus.
No se han llevado a cabo investigaciones sobre la carcinogenicidad, alteración de la fertilidad y desarrollo fetal.
RIXUBIS fue bien tolerado en los estudios de toxicidad en una sola dosis y dosis repetidas realizados en ratones, ratas y monos cynomolgus con dosis de hasta 7500 UI/kg (una sola dosis) y 750 UI/kg (aplicación repetida).
6. DATOS FARMACÉUTICOS 6.1 Lista de excipientes
Polvo
Sacarosa
Manitol
Cloruro de sodio
Cloruro de calcio L-histidina Polisorbato 80
Disolvente
Agua esterilizada para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
Solo se deben utilizar jeringas luer lock de plástico con este producto. Se puede producir una dosificación incorrecta como consecuencia de la adsorción de factor IX de coagulación humano por las superficies internas de algunos equipos de perfusión.
6.3 Período de validez
3 años.
La estabilidad química y física necesaria para su uso se ha demostrado durante 3 horas a una temperatura no superior a 30 °C. Desde el punto de vista microbiológico, a menos que el método de reconstitución descarte el riesgo de contaminación microbiana, el producto debe utilizarse inmediatamente. Si no se utiliza de inmediato, las condiciones y los tiempos de almacenamiento necesarios para su uso serán responsabilidad del usuario. No refrigerar.
6.4 Precauciones especiales de conservación
Conservar por debajo de 30°C.
No congelar.
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase y equipo especial para su utilización
Un envase contiene un vial de polvo (vidrio tipo I) con un tapón (caucho de butilo) y un sello desprendible, un vial de disolvente de 5 ml (vidrio tipo I) con un tapón (caucho de butilo) y un sello desprendible, y un equipo de reconstitución sin agujas (BAXJECT II).
Tamaño de envase de 1.
6.6 Precauciones especiales de eliminación y otras manipulaciones
RIXUBIS se debe administrar por vía intravenosa después de la reconstitución del polvo con el disolvente suministrado.
- Para la reconstitución, utilice únicamente el disolvente y el dispositivo de reconstitución (BAXJECT II) incluidos en el envase.
- Para la administración se requiere el uso de una jeringa luer lock.
- No utilizar si el equipo BAXJECT II, el sistema estéril de protección o su envase está dañado o muestra algún signo de deterioro.
Reconstitución Utilizar técnica aséptica:
1. Si el medicamento se encuentra en la nevera, sacar de la nevera los viales de polvo y de disolvente de RIXUBIS y esperar a que alcancen la temperatura ambiente (entre 15 °C y 30 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Con BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo RIXUBIS. El vacío hará que el disolvente penetre en el vial de polvo RIXUBIS (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. El medicamento se disuelve rápidamente (en unos 2 minutos). Asegúrese de que RIXUBIS esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. Deben inspeccionarse visualmente los medicamentos reconstituidos para detectar partículas
o decoloración antes de la administración. La solución debe ser transparente o ligeramente opalescente. No utilizar soluciones que estén turbias o tengan depósitos.
Fig. a
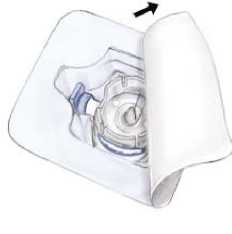
Fig. b

Fig. c

No refrigerar la preparación después de la reconstitución. Utilizar inmediatamente.
Administración Utilizar técnica aséptica:
1. Quitar el protector azul del equipo BAXJECT II. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II (Fig. d).
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente (Fig. e).
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar por vía intravenosa.
La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto.
Fig. d
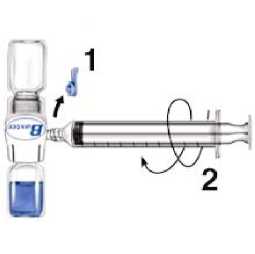
Fig. e

Siempre que sea posible, anote el nombre del medicamento y el número de lote cada vez que utilice RIXUBIS (por ejemplo, en su diario) para mantener un registro de los medicamentos y los lotes que ha utilizado.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxalta Innovations GmbH Industriestrasse 67 A-1221 Vienna Austria
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/970/001
EU/1/14/970/002
EU/1/14/970/003
EU/1/14/970/004
EU/1/14/970/005
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 19 Diciembre 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante del principio activo biológico
Baxter AG Uferstrasse 15 A-2304 Orth an der Donau Austria
Nombre y dirección de los fabricantes responsables de la liberación de los lotes
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines Bélgica
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Bélgica
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
RIXUBIS 250 UI Polvo y disolvente para solución inyectable Nonacog gamma (factor IX humano de coagulación recombinante)
1 vial: 250 UI de nonacog gamma, aprox. 50 UI/ml después de la reconstitución con 5 ml de disolvente.
Excipiente con efectos conocidos: sodio (de cloruro de sodio)
Excipientes: sacarosa, cloruro de calcio, histidina, manitol, polisorbato 80.
Disolvente: agua esterilizada para preparaciones inyectables
Para mayor información consultar el prospecto
Polvo y disolvente para solución inyectable
Contenido: 1 vial de polvo, 1 vial de disolvente, 1 equipo BAXJECT II
Leer el prospecto antes de utilizar este medicamento. Para uso por vía intravenosa y un solo uso.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Conservar por debajo de 30°C. No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxalta Innovations GmbH A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/970/001
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
RIXUBIS 250
ETIQUETA DEL VIAL DE POLVO
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
RIXUBIS 250 UI polvo para inyección Nonacog gamma Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Inyección de un solo uso.
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
250 UI
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
CAD:
4. NÚMERO DE LOTE_
Lote:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES_
5 ml
6. OTROS
RIXUBIS 500 UI Polvo y disolvente para solución inyectable Nonacog gamma (factor IX humano de coagulación recombinante)
1 vial: 500 UI de nonacog gamma, aprox. 100 UI/ml después de la reconstitución con 5 ml de disolvente.
Excipiente con efectos conocidos: sodio (de cloruro de sodio)
Excipientes: sacarosa, cloruro de calcio, histidina, manitol, polisorbato 80.
Disolvente: agua esterilizada para preparaciones inyectables
Para mayor información consultar el prospecto
Polvo y disolvente para solución inyectable
Contenido: 1 vial de polvo, 1 vial de disolvente, 1 equipo BAXJECT II
Leer el prospecto antes de utilizar este medicamento. Para uso por vía intravenosa y un solo uso.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Conservar por debajo de 30°C. No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxalta Innovations GmbH A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/970/002
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
RIXUBIS 500
ETIQUETA DEL VIAL DE POLVO
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
RIXUBIS 500 UI polvo para inyección Nonacog gamma Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Inyección de un solo uso.
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
500 UI
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
CAD:
4. NÚMERO DE LOTE_
Lote:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES_
5 ml
6. OTROS
RIXUBIS 1000 UI Polvo y disolvente para solución inyectable Nonacog gamma (factor IX humano de coagulación recombinante)
1 vial: 1000 UI de nonacog gamma, aprox. 200 UI/ml después de la reconstitución con 5 ml de disolvente.
Excipiente con efectos conocidos: sodio (de cloruro de sodio)
Excipientes: sacarosa, cloruro de calcio, histidina, manitol, polisorbato 80.
Disolvente: agua esterilizada para preparaciones inyectables
Para mayor información consultar el prospecto
Polvo y disolvente para solución inyectable
Contenido: 1 vial de polvo, 1 vial de disolvente, 1 equipo BAXJECT II
Leer el prospecto antes de utilizar este medicamento. Para uso por vía intravenosa y un solo uso.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Conservar por debajo de 30°C. No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxalta Innovations GmbH A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/970/003
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
RIXUBIS 1000
ETIQUETA DEL VIAL DE POLVO
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
RIXUBIS 1000 UI polvo para inyección Nonacog gamma Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Inyección de un solo uso.
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
1000 UI
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
CAD:
4. NÚMERO DE LOTE_
Lote:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES_
5 ml
6. OTROS
RIXUBIS 2000 UI Polvo y disolvente para solución inyectable Nonacog gamma (factor IX humano de coagulación recombinante)
1 vial: 2000 UI de nonacog gamma, aprox. 400 UI/ml después de la reconstitución con 5 ml de disolvente.
Excipiente con efectos conocidos: sodio (de cloruro de sodio)
Excipientes: sacarosa, cloruro de calcio, histidina, manitol, polisorbato 80.
Disolvente: agua esterilizada para preparaciones inyectables
Para mayor información consultar el prospecto
Polvo y disolvente para solución inyectable
Contenido: 1 vial de polvo, 1 vial de disolvente, 1 equipo BAXJECT II
Leer el prospecto antes de utilizar este medicamento. Para uso por vía intravenosa y un solo uso.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Conservar por debajo de 30°C. No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxalta Innovations GmbH A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/970/004
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
RIXUBIS 2000
ETIQUETA DEL VIAL DE POLVO
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
RIXUBIS 2000 UI polvo para inyección Nonacog gamma Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Inyección de un solo uso.
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
2000 UI
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
CAD:
4. NÚMERO DE LOTE_
Lote:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES_
5 ml
6. OTROS
RIXUBIS 3000 UI Polvo y disolvente para solución inyectable Nonacog gamma (factor IX humano de coagulación recombinante)
1 vial: 3000 UI de nonacog gamma, aprox. 600 UI/ml después de la reconstitución con 5 ml de disolvente.
Excipiente con efectos conocidos: sodio (de cloruro de sodio)
Excipientes: sacarosa, cloruro de calcio, histidina, manitol, polisorbato 80.
Disolvente: agua esterilizada para preparaciones inyectables
Para mayor información consultar el prospecto
Polvo y disolvente para solución inyectable
Contenido: 1 vial de polvo, 1 vial de disolvente, 1 equipo BAXJECT II
Leer el prospecto antes de utilizar este medicamento. Para uso por vía intravenosa y un solo uso.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Conservar por debajo de 30°C. No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxalta Innovations GmbH A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/970/005
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
RIXUBIS 3000
ETIQUETA DEL VIAL DE POLVO
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
RIXUBIS 3000 UI polvo para inyección Nonacog gamma Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Inyección de un solo uso.
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
3000 UI
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
CAD:
4. NÚMERO DE LOTE_
Lote:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES_
5 ml
6. OTROS
B. PROSPECTO
Prospecto: información para el usuario
RIXUBIS 250 UI Polvo y disolvente para solución inyectable RIXUBIS 500 UI Polvo y disolvente para solución inyectable RIXUBIS 1000 UI Polvo y disolvente para solución inyectable RIXUBIS 2000 UI Polvo y disolvente para solución inyectable RIXUBIS 3000 UI Polvo y disolvente para solución inyectable
Nonacog gamma (factor IX humano de coagulación recombinante)
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas, aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es RIXUBIS y para qué se utiliza
2. Qué necesita saber antes de empezar a usar RIXUBIS
3. Cómo usar RIXUBIS
4. Posibles efectos adversos
5. Conservación de RIXUBIS
6. Contenido del envase e información adicional
1. Qué es RIXUBIS y para qué se utiliza
RIXUBIS contiene el principio activo nonacog gamma y es un factor IX humano de coagulación.
El Factor IX es un componente normal de la sangre humana necesario para su correcta coagulación, RIXUBIS se utiliza en pacientes con hemofilia B (enfermedad de Christmas, un trastorno hereditario de la sangre causado por una deficiencia de factor IX). Actúa sustituyendo el factor IX que falta para permitir que la sangre del paciente se coagule.
RIXUBIS se utiliza para el tratamiento y la prevención de hemorragias en pacientes con hemofilia B de todos los grupos de edad.
2. Qué necesita saber antes de empezar a usar RIXUBIS No use RIXUBIS
- si es alérgico a nonacog gamma o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6)
- si es alérgico a las proteínas de hámster
Advertencias y precauciones
Es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico con RIXUBIS. Detenga la perfusión y póngase en contacto inmediatamente con su médico o busque atención médica urgente si experimenta los primeros signos de las reacciones de hipersensibilidad/alergia como habón
urticarial, erupción, tirantez en el pecho, sibilancia, presión arterial baja o anafilaxia (reacción alérgica grave que puede ocasionar dificultad para tragar y/o respirar, cara y/o manos rojas o hinchadas).
Es posible que su médico deba tratarle inmediatamente en caso de estas reacciones. Es posible que su médico también le realice un análisis de sangre para comprobar si ha desarrollado anticuerpos neutralizantes de la actividad (inhibidores) frente al medicamento, ya que los inhibidores pueden desarrollarse junto con las alergias. Los pacientes con inhibidores de factor IX pueden tener un mayor riesgo de anafilaxia durante el tratamiento posterior con factor IX.
Consulte inmediatamente a su médico si la hemorragia no se detiene de la manera esperada o si experimenta un aumento significativo del uso de RIXUBIS para controlar una hemorragia. Su médico le realizará un análisis de sangre para comprobar si ha desarrollado anticuerpos neutralizantes de la actividad (inhibidores) frente a RIXUBIS. El riesgo de desarrollar inhibidores es mayor en pacientes a los que no se ha tratado anteriormente con un medicamento sustituto de factor IX o en las primeras fases del tratamiento, es decir, en el caso de niños pequeños.
La producción de factor IX en el cuerpo la controla el gen de factor IX. Los pacientes que tienen mutaciones específicas de su gen de factor IX, como por ejemplo una eliminación mayor, tal vez tengan más probabilidades de tener inhibidores de factor IX y una reacción alérgica en las primeras fases con cualquier concentrado de factor IX. Por tanto, si se sabe que usted tiene tal mutación, su médico le controlará con mayor cuidado para detectar signos de una reacción alérgica.
Si padece enfermedad hepática o cardiaca, o si se ha sometido recientemente a una intervención quirúrgica importante, informe a su médico, ya que existe un mayor riesgo de complicaciones en la coagulación de la sangre.
Se han notificado casos de trastornos renales (síndrome nefrótico) tras la administración de dosis elevadas de factor IX en pacientes con hemofilia B que tenían inhibidores de factor IX y antecedentes de reacciones alérgicas.
Siempre que sea posible, anote el nombre del medicamento y el número de lote cada vez que utilice RIXUBIS (por ejemplo, en su diario) para mantener un registro de los medicamentos y los lotes que ha utilizado.
Uso de RIXUBIS con otros medicamentos
Informe a su médico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento. No se han notificado interacciones de RIXUBIS con otros medicamentos.
Embarazo, lactancia y fertilidad
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento. La hemofilia B aparece en muy raras ocasiones en mujeres.
Conducción y uso de máquinas
La influencia de RIXUBIS sobre la capacidad para conducir y utilizar máquinas es nula.
RIXUBIS contiene sodio
Este medicamento contiene 19 mg de sodio por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
3. Cómo usar RIXUBIS
El tratamiento con RIXUBIS lo iniciará por un médico con experiencia en el tratamiento de pacientes con hemofilia B.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico.
Su médico decidirá la dosis de RIXUBIS que se le administrará. Esta dosis y la duración dependerán de la gravedad de su deficiencia de factor IX, de la ubicación y la extensión de la hemorragia, así como de su estado clínico, la edad y la rapidez con la que su cuerpo consume el factor IX, que se debe comprobar con regularidad.
Su médico o enfermero le administrarán RIXUBIS mediante perfusión intravenosa (IV) después de reconstituir el polvo con el disolvente suministrado. Usted o cualquier otra persona pueden también administrar la inyección de RIXUBIS, pero únicamente después de recibir la formación adecuada.
Reconstitución y administración
- Para la reconstitución, utilice únicamente el disolvente y el dispositivo de reconstitución (BAXJECT II) incluidos en el envase.
- Para la administración se requiere el uso de una jeringa luer lock.
- No utilizar si el equipo BAXJECT II, el sistema estéril de protección o su envase está dañado o muestra algún signo de deterioro.
Reconstitución Utilizar técnica aséptica:
1. Si el medicamento se encuentra en la nevera, sacar de la nevera los viales de polvo y de disolvente de RIXUBIS y esperar a que alcancen la temperatura ambiente (entre 15 °C y 30 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Con BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo RIXUBIS. El vacío hará que el disolvente penetre en el vial de polvo RIXUBIS (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. El medicamento se disuelve rápidamente (en unos 2 minutos). Asegúrese de que RIXUBIS esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. Deben inspeccionarse visualmente los medicamentos reconstituidos para detectar partículas
o decoloración antes de la administración. La solución debe ser transparente o ligeramente opalescente. No utilizar soluciones turbias o con depósitos.
Fig. a
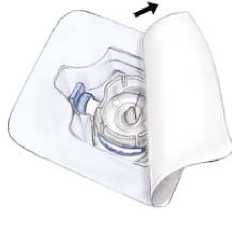
Fig. b


No refrigerar la preparación después de la reconstitución. Utilizar inmediatamente.
Administración Utilizar técnica aséptica:
1. Quitar el protector azul del equipo BAXJECT II. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II (Fig. d).
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente (Fig. e).
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar por vía intravenosa.
La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto.
Fig. d
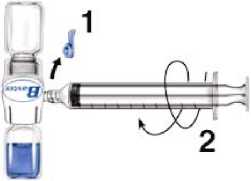
Fig. e

Siempre que sea posible, anote el nombre del medicamento y el número de lote cada vez que utilice RIXUBIS (por ejemplo, en su diario) para mantener un registro de los medicamentos y los lotes que ha utilizado.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Si usa más RIXUBIS del que debe
Siga exactamente las instrucciones de administración de RIXUBIS indicadas por su médico. En caso de duda, consulte de nuevo a su médico. Si se inyecta una dosis mayor de RIXUBIS de la recomendada, consulte con su médico lo antes posible.
Si olvidó usar RIXUBIS
No se inyecte una dosis doble para compensar las dosis olvidadas. Adminístrese la siguiente inyección como está establecido y continúe como le había indicado su médico.
Si interrumpe el tratamiento con RIXUBIS
No deje de usar RIXUBIS sin consultarlo con su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico con RIXUBIS. Entre estas reacciones se incluyen sensaciones de ardor y punzadas en el lugar de perfusión, escalofríos, rubefacción, letargia, inquietud, cosquilleo, habón urticarial, picor y sarpullido, presión arterial baja, frecuencia cardiaca rápida, tirantez en el pecho, sibilancia, hinchazón de la garganta, anafilaxia (reacción alérgica grave), dolor de cabeza, náuseas y vómitos. Consulte inmediatamente a su médico si experimenta estos signos. Es posible que su médico deba tratarle inmediatamente en caso de estas reacciones (ver sección 2 ‘Advertencias y precauciones’).
Se han observado los siguientes efectos adversos con RIXUBIS:
Efectos adversos frecuentes (pueden afectar hasta a 1 de cada 10 pacientes)
- alteración del gusto
- dolor en las extremidades.
Efectos adversos con frecuencia desconocida (no se puede estimar la frecuencia a partir de los datos disponibles)
- reacciones alérgicas (hipersensibilidad).
No se han observado con este medicamento problemas ocasionados por una coagulación excesiva de la sangre (episodios tromboembólicos), pero pueden ocurrir con cualquier producto de factor IX. Entre estos, se incluyen ataques al corazón, coágulos de sangre en las venas o en el pulmón.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de RIXUBIS
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el embalaje exterior y la etiqueta del vial después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar por debajo de 30°C.
No congelar.
Utilice la solución reconstituida inmediatamente.
No utilice RIXUBIS si la solución no es incolora y transparente.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de RIXUBIS
- El principio activo es nonacog gamma (factor IX humano de coagulación recombinante). Cada vial contiene nominalmente 250, 500, 1000, 2000 o 3000 UI, correspondientes a una concentración de 50, 100, 200, 400 o 600 UI/ml después de la reconstitución con 5 ml de disolvente.
- Los demás componentes del polvo son sacarosa, manitol, cloruro de sodio, cloruro de calcio, L-histidina, polisorbato 80.
Vial de disolvente: 5 ml de agua esterilizada para preparaciones inyectables.
Aspecto del producto y contenido del envase
RIXUBIS se proporciona como polvo y disolvente para solución inyectable.
El contenido del envase es el siguiente:
• un vial de polvo RIXUBIS 250, 500, 1000, 2000 o 3000 UI en un vial de vidrio con un tapón de caucho
• un vial de 5 ml de agua esterilizada para preparaciones inyectables en un vial de vidrio con un tapón de caucho
• un BAXJECT II (equipo de reconstitución sin agujas)
Titular de la autorización de comercialización
Baxalta Innovations GmbH Industriestrasse 67 A-1221 Vienna
Responsable de la fabricación
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines Bélgica
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Bélgica
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00
Lietuva
UAB "Baxter Lithuania''
Tel: +370 5 269 16 90/+370 5 252 71 00
BtarapHH
EaKCTep Etarapna EOOfl Tea.: +359 2 9808482
Ceská republika
Baxter Czech spol.s.r.o. Tel.: +420 225774111
Luxembourg/Luxemburg
Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00
Magyarország Baxter Hungary Kft.
Tel.: +361 202 19 80
Danmark
Baxalta Denmark A/S Tlf: +45 32 70 12 00
Malta
Baxalta UK Limited Tel.: +44 1 635 798 777
Deutschland
Baxalta Deutschland GmbH Tel: +49 89 262077-011
Eesti
OÜ Baxter Estonia Tel.: +372 6 515 120
Nederland
Baxalta Netherlands B.V. Tel: +31 30 799 27 77
Norge
Baxalta AS
Tlf: +47 22 585 000
|
ELLáóa Baxter (Hellas) E.n.E. TnL: +30 210 28 80 000 |
Osterreich Baxalta Osterreich GmbH Tel.: +43 1 20100-0 |
|
España Baxalta Spain S.L. Tel: +34 91 790 42 22 |
Polska Baxter Polska Sp. z o.o. Tel.: +48 22 4883 777 |
|
France Baxalta France S.A.S. Tél: +33 1 70 96 06 00 |
Portugal Baxalta Portugal, Unipessoal, Lda. Tel: +351 21 122 03 00 |
|
Hrvatska Baxter d.o.o. Tel: +386 1 420 16 80 |
Romania FARMACEUTICA REMEDIA SA Tel.: +40 21 321 16 40 |
|
Ireland Baxalta UK Limited Tel: +44 1 635 798 777 |
Slovenija Baxter d.o.o. Tel.: +386 1 420 16 80 |
|
Ísland Lyfjaver ehf. Sími: +354 533 6100 |
Slovenská republika Baxter Slovakia s.r.o. Tel: +421 2 3210 1150 |
|
Italia Baxalta Italy S.r.l. Tel: +39 06 45224 600 |
Suomi/Finland Baxalta Finland Oy Puh/Tel: +358 201478200 |
|
Kúnpoq Baxter (Hellas) E.n.E. TnL: +30 210 28 80 000 |
Sverige Baxalta Sweden AB Tel: +46 8 50 53 26 00 |
|
Latvija SIA BAXTER Latvia Tel.: +371 67 784 784 |
United Kingdom Baxalta UK Limited Tel: +44 1 635 798 777 |
Fecha de la última revisión de este prospecto MM/AAAA.
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Esta información está destinada únicamente a profesionales del sector sanitario:
Supervisión del tratamiento
Durante el tratamiento, se recomienda la determinación adecuada de los niveles de factor IX para calcular la dosis que se debe administrar y la frecuencia de las perfusiones repetidas. Los pacientes individuales pueden diferir en su respuesta al factor IX con diferentes semividas y recuperaciones. La dosis basada en el peso corporal puede requerir un ajuste en pacientes con bajo peso o sobrepeso. En el caso particular de intervenciones quirúrgicas importantes, es indispensable una supervisión precisa de la terapia de sustitución mediante análisis de la coagulación (actividad del factor IX de plasma).
Para garantizar que se ha alcanzado el nivel plasmático de actividad de factor IX deseado, se aconseja realizar un control exhaustivo utilizando un ensayo adecuado de actividad de factor IX y, si es necesario, se deben aplicar los ajustes adecuados a la dosis y la frecuencia de las perfusiones repetidas. Al utilizar el ensayo in vitro de coagulación en una etapa basado en el tiempo de la tromboplastina (aPTT) para determinar la actividad del factor IX en muestras sanguíneas de pacientes, los resultados de actividad del factor IX pueden verse significativamente afectados por el tipo de reactivo de aPTT y el estándar de referencia utilizado en el ensayo. Esto es importante especialmente al cambiar el laboratorio y/o los reactivos utilizados en el ensayo.
Posología
La dosis y la duración de la terapia de sustitución depende de la gravedad de la deficiencia de factor IX, de la ubicación y la extensión de la hemorragia, así como el estado clínico, la edad y los parámetros farmacocinéticos de factor IX del paciente, como la recuperación incremental y semivida.
El número de unidades de factor IX administradas se expresa en unidades internacionales (UI), que están relacionadas con el estándar actual de la OMS para productos de factor IX. La actividad de factor IX en el plasma se expresa como un porcentaje (relativo al plasma humano normal) o en unidades internacionales (relativas a un estándar internacional para el factor IX en el plasma).
Una unidad internacional (UI) de actividad de factor IX es equivalente a la cantidad de factor IX existente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor IX se basa en el hallazgo empírico de que 1 unidad internacional (UI) de factor IX por kg de peso corporal incrementa la actividad de factor IX del plasma en 0,9 UI/dl (intervalo de 0,5 a 1,4 UI/dl) o 0,9% de actividad normal en pacientes de 12 años de edad y mayores (para obtener información adicional, ver sección 5.2).
La dosis necesaria se determina utilizando la siguiente fórmula:
Pacientes de 12 años de edad y mayores
Unidades = peso corporal (kg) x aumento deseado de x recíproco de recuperación requeridas factor IX observada (dl/kg)
(%) o (UI/dl)
Para una recuperación incremental de 0,9 UI/dl por UI/kg, la dosis se calcula de la siguiente manera:
Unidades = peso corporal (kg) x aumento deseado de x 1,1 dl/kg requeridas factor IX
(%) o (UI/dl)
La cantidad que se debe administrar y la frecuencia de la administración deben estar siempre orientadas a la eficacia clínica en el caso concreto.
En el caso de los episodios hemorrágicos siguientes, la actividad de factor IX no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. Se puede utilizar la siguiente tabla como guía de dosificación en episodios hemorrágicos y cirugía:
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de factor IX requerido (%) o (UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia Hemartrosis incipiente o hemorragia muscular u oral |
20 - 40 |
Repetir cada 24 horas. Al menos 1 día, hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma |
30 - 60 |
Repetir perfusión cada 24 horas durante 3 - 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir perfusión cada 8 a 24 horas hasta superar el peligro. |
|
Cirugía Cirugía menor incluyendo extracción dental |
30 - 60 |
Cada 24 horas, al menos 1 día, hasta lograr la curación. |
|
Operación importante |
80 - 100 (pre y postoperatorio) |
Repetir perfusión cada 8 a 24 horas hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor IX del 30% al 60% (UI/dl). |
Es especialmente importante una supervisión cuidadosa de la terapia de sustitución en los casos de operación importante o hemorragia potencialmente mortal.
Profilaxis
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia B grave, las dosis normales son de 40 a 60 UI de factor IX por kilogramo de peso corporal a intervalos de 3 a 4 días para los pacientes de 12 años de edad y mayores. En algunos casos, dependiendo de los parámetros farmacocinéticos, la edad, el fenotipo de hemorragia y la actividad física del paciente, es posible que se necesiten intervalos de dosificación más cortos o dosis más altas.
Perfusión continua
No administre RIXUBIS mediante perfusión continua.
Población pediátrica Tratamiento a demanda:
El cálculo de la dosis de factor IX requerida se basa en el hallazgo empírico de que 1 unidad internacional (UI) de factor IX por kg de peso corporal incrementa la actividad de factor IX del plasma en 0,7 UI/dl (intervalo de 0,31 a 1,0 UI/dl) o 0,7% de la actividad normal en pacientes de menos de 12 años de edad (para obtener información adicional, ver sección 5.2).
La dosificación requerida se determina mediante la siguiente fórmula:
Pacientes de menos de 12 años de edad:
Unidades = peso corporal requeridas (kg)
x aumento deseado de factor IX x recíproco de recuperación
(%) o (UI/dl)
observada (dl/kg)
Para una recuperación incremental de 0,7 UI/dl por UI/kg, la dosis se calcula de la siguiente manera:
Unidades
requeridas
peso corporal (kg) x aumento deseado de factor IX x 1,4 dl/kg
(%) o (UI/dl)
Se puede utilizar la misma tabla para los adultos como guía de dosificación en episodios hemorrágicos y cirugía (ver anterior).
Profilaxis:
El intervalo de dosis recomendadas para pacientes pediátricos de menos de 12 años de edad es de 40 a 80 UI/kg a intervalos de 3 a 4 días. En algunos casos, dependiendo de los parámetros farmacocinéticos, la edad, el fenotipo de la hemorragia y la actividad física del paciente, es posible que se necesiten intervalos de dosificación más cortos o dosis más altas.
52